

Acinetobacter baumannii causes a wide range of nosocomial infections. This pathogen is considered a threat to human health due to the increasingly frequent isolation of multidrug-resistant strains.
KEYWORDS: Acinetobacter, lipid A, virulence
ABSTRACT
Acinetobacter baumannii causes a wide range of nosocomial infections. This pathogen is considered a threat to human health due to the increasingly frequent isolation of multidrug-resistant strains. There is a major gap in knowledge on the infection biology of A. baumannii, and only a few virulence factors have been characterized, including lipopolysaccharide. The lipid A expressed by A. baumannii is hepta-acylated and contains 2-hydroxylaurate. The late acyltransferases controlling the acylation of lipid A have been already characterized. Here, we report the characterization of A. baumannii LpxO, which encodes the enzyme responsible for the 2-hydroxylation of lipid A. By genetic methods and mass spectrometry, we demonstrate that LpxO catalyzes the 2-hydroxylation of the laurate transferred by A. baumannii LpxL. LpxO-dependent lipid A 2-hydroxylation protects A. baumannii from polymyxin B, colistin, and human β-defensin 3. LpxO contributes to the survival of A. baumannii in human whole blood and is required for pathogen survival in the waxmoth Galleria mellonella. LpxO also protects Acinetobacter from G. mellonella antimicrobial peptides and limits their expression. Further demonstrating the importance of LpxO-dependent modification in immune evasion, 2-hydroxylation of lipid A limits the activation of the mitogen-activated protein kinase Jun N-terminal protein kinase to attenuate inflammatory responses. In addition, LpxO-controlled lipid A modification mediates the production of the anti-inflammatory cytokine interleukin-10 (IL-10) via the activation of the transcriptional factor CREB. IL-10 in turn limits the production of inflammatory cytokines following A. baumannii infection. Altogether, our studies suggest that LpxO is a candidate for the development of anti-A. baumannii drugs.
INTRODUCTION
Lipopolysaccharide (LPS), located in the outer leaflet of the outer membrane (OM), is composed of three regions: the lipid A moiety, the core oligosaccharide, and the O-polysaccharide. Hexa-acylated lipid A, such as the canonical lipid A structure expressed by Escherichia coli K-12 (1), is recognized by the Toll-like receptor 4 (TLR4)-MD2 complex, resulting in the activation of downstream signaling cascades leading to cytokine production and subsequent inflammation (2). The lipid A structure can be modified by pathogens in response to different conditions (1, 3). Well-characterized modifications are the addition or removal of fatty acids, alteration of the phosphate groups, or additions of other chemical groups, such as amino acids and sugars (1, 3). These modifications may result in resistance to cationic antimicrobial peptides (CAMPs), reduced or increased activation of inflammatory responses, or greater protection against disturbances in pH or desiccation (2, 3).
Acinetobacter baumannii is an opportunistic Gram-negative pathogen causing a wide range of nosocomial infections, although the most common clinical manifestations comprise ventilator-associated pneumonia and central line-associated bloodstream infections (4). A. baumannii is considered a global threat to human health due to the increasing isolation of multidrug-, extensively drug-, and even pan-drug-resistant strains. Colistin and tigecycline have become the last-line treatment for multidrug-resistant A. baumannii (5). Despite its clinical relevance, there is a major knowledge gap on the infection biology of A. baumannii, and only a few virulence factors have been characterized, including LPS (6). A. baumannii synthesizes lipooligosaccharide (LOS), rather than LPS, due to the absence of the O-polysaccharide. Structurally, A. baumannii lipid A consists of a β(1′-6)-linked disaccharide of glucosamine phosphorylated at the 1 and 4′ positions with positions 2, 3, 2′, and 3′ acylated with R-3-hydroxymyristoyl groups. The 2 and 3′ R-3-hydroxymyristoyl groups are further acylated with laurate (C12) through the action of the late acyltransferase LpxM, whereas the 2’ R-3-hydroxymyristoyl group is acylated with laurate (C12) through the action of the late acyltransferase LpxL. Thus, in contrast to many Gram-negative pathogens, lipid A of A. baumannii is predominantly hepta-acylated (7–11). The evidence demonstrates that hepta-acylation of A. baumannii lipid A promotes resistance to CAMPs, contributes to desiccation survival, and is important for A. baumannii virulence (7). This lipid A is recognized by the TLR4/MD2 receptor complex and triggers the activation of NF-κB and mitogen-activated protein kinases (MAPKs) to induce inflammation and antibacterial molecules such as defensins (7, 8).
A. baumannii can modify its lipid A with phosphoethanolamine and galactosamine, and these modifications are implicated in colistin resistance (9–11). PmrC/EptA is responsible for the addition of phosphoethanolamine to lipid A (9), whereas NaxD is required for the modification with galactosamine (10). The PmrAB two-component system governs expression of PmrC and NaxD, and there are reports of clinical isolates harboring mutations in PmrA or PmrB leading to the upregulation of the system with a concomitant increase in the replacement of the lipid A with phosphoethanolamine and galactosamine (9–11). Colistin resistance is also mediated through complete loss of the LOS by loss-of-function mutations within genes essential for lipid A biosynthesis (lpxA, lpxC, or lpxD) (12). However, LOS-deficient strains of A. baumannii show a dramatic reduction in virulence, and not all strains possess the ability to mutate early-stage lipid A biosynthetic enzymes (13).
All of the published studies on A. baumannii lipid A report the presence of 2-hydroxylaurate (7–11). Few Gram-negative bacteria have been shown to produce lipid A species containing hydroxylated secondary acyl chains, and the evidence suggests that this lipid A modification enables the pathogens to successfully infect their hosts. In Vibrio cholerae, the late acyltransferase LpxN is responsible for transferring the hydroxylated laurate to the lipid A domain (14), whereas in Bordetella bronchiseptica, Salmonella enterica serovar Typhimuirum, and Klebsiella pneumoniae, the incorporation of a hydroxyl group in a fatty acid is catalyzed by the enzyme LpxO (15–17). Interestingly, in silico analysis of A. baumannii genomes revealed the presence of an lpxO homolog, suggesting that A. baumannii LpxO is responsible for the lipid A modification with 2-hydroxylaurate. In this work, we characterized A. baumannii lpxO, which is required for the hydroxylation of lipid A. This study demonstrates that LpxO plays an important role in A. baumannii infection biology, since LpxO-dependent lipid A modification protects the pathogen from CAMPs, mediates resistance to phagocytosis, and limits the activation of host defense responses in invertebrates and mammalian cells.
RESULTS
A. baumannii LpxO 2-hydroxylates lipid A.
In silico analyses of A. baumannii genomes revealed that this pathogen carries one homolog of K. pneumoniae and S. enterica serovar Typhimurium lpxO. Analysis of the genome of A. baumannii strain ATCC 17978 showed that LpxO (locus tag A1S_0308) is 62 and 59% identical to K. pneumoniae and Salmonella enterica serovar Typhimurium LpxO, respectively (Fig. 1A). To confirm that the identified loci are indeed responsible for the 2-hydroxylation of lipid A, A. baumannii strain ATCC 17978 lpxO was mutated by double recombination. Control experiments showed that the wild type and lpxO mutant had similar growth kinetics in both rich and minimal media (see Fig. S1 in the supplemental material). Lipid A was extracted from the wild type and the lpxO mutant using an ammonium hydroxide-isobutyric acid method and subjected to negative-ion matrix-assisted laser desorption ionization–time of flight (MALDI-TOF) mass spectrometry. Lipid A produced by the wild type contained the species with mass-to-charge ratios (m/z) of 1,910, 1,728, and 1,530 (Fig. 1B). These molecular species have been previously found in lipid A of A. baumannii strains (7–11), and the proposed chemical structures are shown in Fig. 1E. In contrast, the lpxO mutant produced a lipid A which contained the species m/z 1,894, 1,712, and 1,514, an m/z difference of 16 compared to the wild-type species (i.e., m/z 1,910, 1,728, and 1,530, respectively), which corresponded with the lack of ions containing 2-hydroxylaurate (C12:OH) (Fig. 1C). Complementation of the mutant restored the production of wild-type lipid A (Fig. 1D), demonstrating that lpxO is responsible for the 2-hydroxylation of A. baumannii lipid A.
FIG 1.

A. baumannii LpxO 2-hydroxylates lipid A. (A) A. baumannii encodes an LpxO homolog with 62% and 59% sequence similarity to K. pneumoniae and S. enterica serovar Typhimurium LpxO, respectively. Shown are negative-ion MALDI-TOF mass spectrometry spectra of lipid A purified from A. baumannii ATCC 17978 (B), A. baumannii ΔlpxO mutant (ΔlpxO) (C), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO) (D). Data represent the mass-to-charge ratios (m/z) of each lipid A species detected and are representative of three extractions. (E) Proposed lipid A structures of the species produced by wild-type A. baumannii based on previous publications (7–11).
We sought to determine whether other membrane lipids were affected in the membrane of the lpxO mutant. However, the analysis of the lipid composition by thin-layer chromatography did not reveal any differences in the levels of cardiolipin, phosphatidylglycerol, or phosphoethanolamine between the membranes of the wild type and lpxO mutant (Fig. S2A).
To assess whether the changes in the lipid A structure observed in the lpxO mutant affect the outer membrane permeability to hydrophobic agents, we measured the partition of the hydrophobic fluorescent probe (1-N-phenylnaphthylamine; NPN) into the cell membrane. NPN is excluded by the intact bacterial OM but exhibits increased fluorescence after partitioning into disrupted OMs (18). Thus, an increase in fluorescence indicates alterations in the OM. However, we did not observe any differences in the uptake of NPN between the wild type and the lpxO mutant (Fig. S2B), suggesting that no major functional alterations occur in the OM of the lpxO mutant.
A. baumannii LpxO hydroxylates the laurate linked to the 2’ R-3-hydroxymyristoyl group of lipid A.
Recently, Boll and coworkers (7) have shown that 2-hydroxylation in A. baumannii lipid A is absent from an lpxLAb mutant, suggesting that LpxO hydroxylates the 2’-R-hydroxylaurate group. To validate this hypothesis, we used a genetic approach by expressing LpxO in E. coli BN1 (19). This E. coli strain produces the canonical hexa-acylated lipid A of m/z 1,797 (Fig. 2A), corresponding to a β(1′-6)-linked disaccharide of glucosamine phosphorylated at the 1 and 4′ positions, with positions 2, 3, 2′, and 3′ being acylated with R-3-hydroxymyristoyl groups. The 2′ and 3′ R-3-hydroxymyristoyl groups are further acylated with laurate (C12) and myristate (C14) (19). Expression of A. baumannii LpxO in E. coli BN1 resulted in the hydroxylation of the hexa-acylated lipid A species to give m/z 1,813 (Fig. 2B). In contrast, LpxO-mediated 2-hydroxylation was not observed when LpxO was expressed in the E. coli BN1 lpxL mutant (Fig. 2C), confirming that LpxO hydroxylates the laurate fatty acid chain transferred by LpxL.
FIG 2.

A. baumannii LpxO modifies the LpxL-transferred lauryl group. Shown are negative-ion MALDI-TOF mass spectrometry spectra of lipid A purified from E. coli BN1 (A), E. coli BN1::Tn7lpxO mutant (BN1::Tn7lpxO) (B), and E. coli BN1-ΔlpxL::Tn7lpxO mutant (BN1- ΔlpxL::Tn7lpxO) (C). Data represent the m/z of each lipid A species detected and are representative of three extractions.
LpxO-dependent lipid A modification increases A. baumannii resistance to antimicrobial peptides.
Polymyxins B and E (also known as colistin) interact with LPS by electrostatic interactions, causing a disruption of the OM barrier (20, 21). Polymyxin B is most often used as a topical ointment, whereas colistin is becoming the antibiotic of last resort to treat multidrug-resistant infections, including those triggered by A. baumannii. We sought to determine whether LpxO-dependent lipid A modification protects A. baumannii from polymyxins. Results displayed in Fig. 3 demonstrate that the lpxO mutant was more susceptible than the wild type to both polymyxin B and colistin. Complementation restored wild-type levels of resistance, suggesting that LpxO-dependent hydroxylation contributes to A. baumannii resistance to polymyxins.
FIG 3.

Deletion of lpxO decreases A. baumannii resistance to CAMPs. Shown is percent survival of A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO) following 1 h of exposure to polymyxin B (A), colistin (B), and human β-defensin 3 (C). Values are presented as the means ± standard deviations (SD) from three independent experiments measured in duplicate. P values were <0.01 (***) and <0.001 (****) for the indicated comparisons using one-way ANOVA with Bonferroni contrasts; n.s., not significant.
Human antimicrobial peptides such as defensins are weapons of the innate immune system against infections. They do share with polymyxins the initial interaction with the LPS (20), and we therefore hypothesized that LpxO-mediated lipid A modification increases resistance to human antimicrobial peptides. To test this hypothesis, we exposed A. baumannii strains to β-defensin 3 and determined the survival after 1 h of incubation. Human β-defensin 3 is active against multidrug-resistant bacteria, and it has been reported to increase severalfold in the lungs upon infection (22–24). We observed increased killing of the lpxO mutant by β-defensin 3 (Fig. 3C). The enhanced killing was abrogated in the complemented strain, indicating that LpxO-mediated lipid A modification is associated with resistance to β-defensin 3.
Altogether, our findings demonstrate that LpxO-governed 2-hydroxylation of A. baumannii lipid A promotes resistance to human antimicrobial peptides and clinically relevant polymyxins.
LpxO contributes to survival of A. baumannii in human whole blood.
Bacterial killing assays in whole blood allow the ex vivo assessment of the interaction between pathogens and professional phagocytes. We then sought to determine whether LpxO contributes to A. baumannii survival in whole human blood. The lpxO mutant was recovered in significantly lower numbers than the wild type (Fig. 4), whereas there were not differences between the complemented and wild-type strains. These results demonstrate that the decreased survival of the lpxO mutant in whole human blood is associated with the lack of 2-hydroxylation in lipid A.
FIG 4.
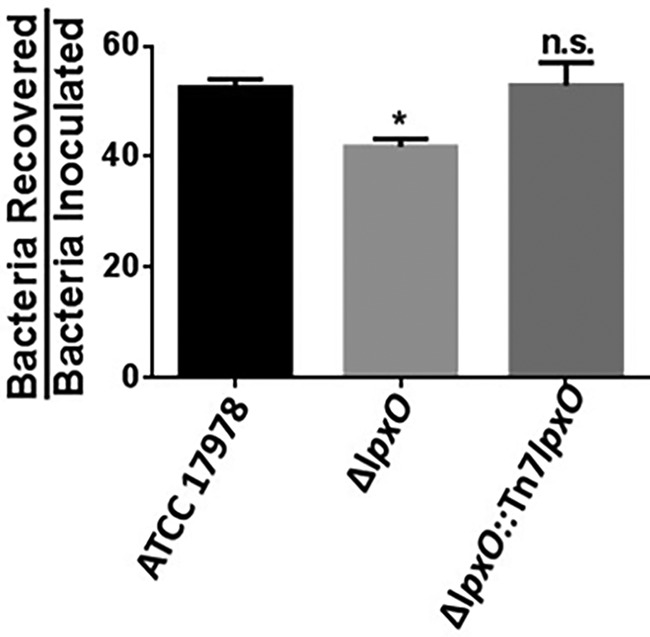
Deletion of lpxO increases human phagocyte-mediated killing of A. baumannii. Three hundred μl of fresh human blood (from three different donors) was mixed with 1 × 107 CFU of A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO) and incubated at 37°C for 3 h. The bacterial counts recovered were then divided by the initial counts. Experiments were performed with duplicate samples on three independent occasions. *, P < 0.05 versus A. baumannii ATCC 17978, determined using one-way ANOVA with Bonferroni contrasts; n.s., not significant.
A. baumannii lpxO is attenuated in the Galleria mellonella infection model.
The G. mellonella infection model is widely used to assess the virulence of A. baumannii (25). Moreover, there is good correlation between virulence in G. mellonella and that in the mouse model (26, 27). Equal numbers of CFU of all three strains were injected into G. mellonella, and survival of the larvae was monitored over several days. Inoculation with sterile PBS into the larvae resulted in no mortality (Fig. 5). Three days postinoculation, only 10% of the Galleria organisms survived when challenged with the wild-type and complemented strains; in contrast, 50% of the larvae survived when inoculated with the lpxO mutant (Fig. 5).
FIG 5.

A. baumannii lpxO mutant displays decreased virulence in the G. mellonella waxworm infection model. (A) Percent survival of G. mellonella over 72 h postinfection with 5 × 104 organisms of A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO). Thirty larvae were infected in each group. Level of significance was determined using the log-rank (Mantel-Cox) test with Bonferroni correction for multiple comparisons. (B) Percent survival of A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO) following 1 h of exposure to G. mellonella hemolymph obtained from larvae challenged with heat-killed E. coli. (C to E) G. mellonella antimicrobial peptide expression determined after 12 h of infection with A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO) by reverse transcriptase quantitative real‐time PCR. Three larvae per group were infected, and values are presented as the means ± SD from two independent cDNA preparations measured in duplicate. In panels B to E, P values were <0.05 (*), 0.01 (**), <0.001 (***), and <0.0001 (****) versus A. baumannii ATCC 17978, determined using one-way ANOVA with Bonferroni contrasts.
CAMPs are part of the G. mellonella defenses activated upon infection (28). A few hours after infection, multiple CAMPs are synthesized and released into the hemolymph to neutralize bacterial infection (28–30). Therefore, we sought to determine whether the lpxO mutant is susceptible to these antimicrobial peptides. G. mellonella was challenged with heat‐killed Escherichia coli to boost the expression of antimicrobial peptides (29, 30), and after 24 h, the hemolymph rich in CAMPs was collected and used to assess the susceptibility of the A. baumannii strains by a broth microdilution assay. Results shown in Fig. 5B demonstrate that the lpxO mutant was more susceptible to G. mellonella peptides than the wild type. Complementation restored the wild-type levels of resistance to the hemolymph peptides, indicating that the 2-hydroxylation of A. baumannii lipid A promotes resistance to a repertoire of CAMPs produced in response to infections.
We have previously demonstrated that there is a correlation between virulence of Gram-negative pathogens and the expression of G. mellonella antimicrobial peptides (29, 30). The reduced virulence of the lpxO mutant led us to assess the expression of CAMPs in G. mellonella larvae infected with the lpxO mutant. After 12 h of infection, the expression levels of gallerimycin, galiomycin, and lysozyme were significantly higher in larvae infected with the lpxO mutant than in larvae infected with the wild-type strain (Fig. 5C to E). Complementation restored the expression of the peptides to wild-type levels, suggesting that the 2-hydroxylation of A. baumannii lipid A is associated with an attenuated expression of G. mellonella CAMPs.
Altogether, our findings provide evidence that LpxO-mediated 2-hydroxylation of A. baumannii lipid A is necessary for the virulence of this pathogen in G. mellonella by limiting the production of CAMPs and promoting resistance to them.
Hydroxylated A. baumannii lipid A reduces inflammatory responses in macrophages.
The lipid A pattern is recognized by the TLR4/MD2 complex, leading to the activation of innate signaling pathways and resulting in inflammation and clearance of the infection. However, many pathogens alter the LPS molecular pattern to evade detection by the TLR4/MD2 complex to reduce inflammation (2). Therefore, we sought to investigate whether LpxO-dependent lipid A modification helps A. baumannii to limit the activation of inflammatory responses. Figure 6A shows that the expression of tnfα was higher in immortalized bone marrow-derived macrophages (iBMDMs) infected with the lpxO mutant than in iBMDMs infected with the wild type. As anticipated, the lpxO mutant induced higher levels of tumor necrosis factor alpha (TNF-α) than the wild type (Fig. 6B). Complementation restored the expression and production of cytokines to wild-type levels, indicating that the heightened inflammation induced by the lpxO mutant is due to the lack of 2-hydroxylation in lipid A.
FIG 6.
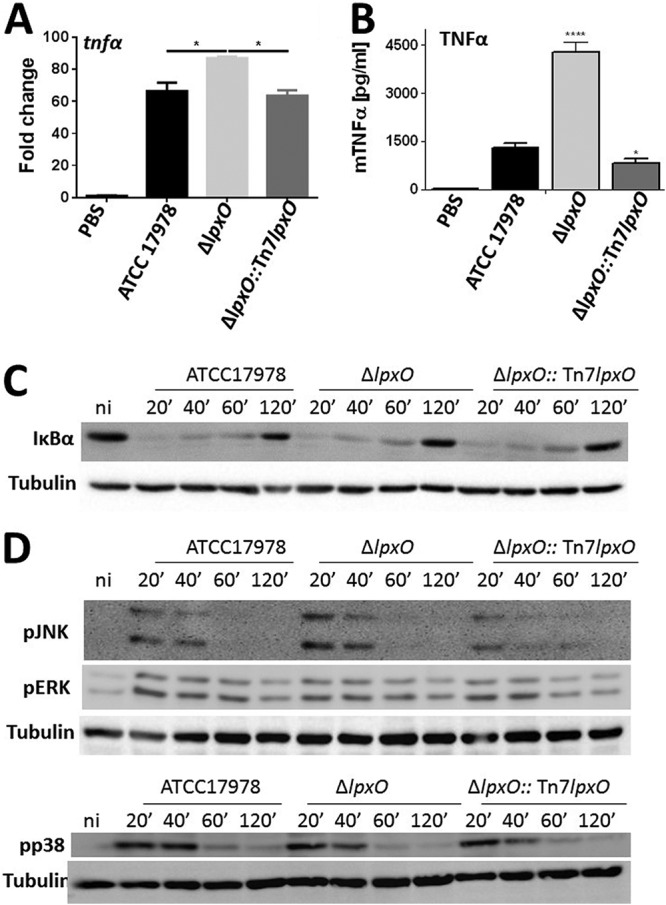
lpxO deletion in A. baumannii results in upregulation of inflammatory responses in macrophages upon infection. (A) tnfα expression in iBMDMs infected for 5 h with UV-killed A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO) by reverse transcriptase quantitative real‐time PCR. Values are presented as the means ± SD from three independent cDNA preparations measured in duplicate. (B) TNF-α secretion by iBMDMs stimulated for 5 h with UV-killed A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO). In panels A and B, P values were <0.05 (*) and <0.0001 (****) versus A. baumannii ATCC 17978, determined using one-way ANOVA with Bonferroni contrasts. (C) Immunoblot analysis of IκBα and tubulin levels in lysates of iBMDMs infected with UV-killed A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO) for the indicated times. (D) Immunoblot analysis of phospho‐JNK (pJNK), phospho‐ERK (pERK), phosphor-p38 (pp38), and tubulin levels in lysates of iBMDMs infected with UV-killed A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO). In panels C and D, data are representative of at least three independent experiments.
The activation of NF-κB and MAPK governed inflammatory responses following infection. We then sought to determine the signaling pathways activated by the lpxO mutant. iBMDMs were infected with the wild type, the lpxO mutant, and the complemented strain, with the activation of NF‐κB and MAPKs assessed by immunoblotting. In the canonical NF‐κB activation pathway, nuclear translocation of NF‐κB is preceded by phosphorylation and subsequent degradation of IκBα. All strains triggered the degradation of IκBα (Fig. 6C), and no differences were observed between strains. Whereas the three strains triggered the phosphorylation of the MAPKs p38, extracellular signal-regulated kinase (ERK), and Jun N-terminal protein kinase (JNK) (Fig. 6B), the phosphorylation of JNK was increased in macrophages infected with the lpxO mutant. Image analysis showed a 22 and 281% increase in the phosphorylation of JNK 20 and 40 min postinfection with the lpxO mutant, respectively, compared to the phosphorylation induced by the wild type. The complemented strain induced phosphorylation of JNK similar to that of the wild type at all time points. Control experiments showed that A. baumannii induction of TNF-α is dependent on the activation of JNK, because the levels of this cytokine were significantly lower in the supernatants of infected macrophages treated with the JNK inhibitor SP600125 (Fig. S3). Collectively, these results indicate that the 2-hydroxylation of lipid A limits the activation of MAPK JNK to attenuate inflammatory responses.
Interestingly, the expression of the anti-inflammatory cytokine il10 was significantly reduced in iBMDMs infected with the lpxO mutant, whereas we did not observe any difference in il10 levels triggered by the wild-type and complemented strains (Fig. 7A). These results suggested that the 2-hydroxylation of A. baumannii lipid A contributes to the induction of the anti-inflammatory cytokine interleukin-10 (IL-10). To investigate whether the reduced levels of IL-10 triggered by the lpxO mutant could be responsible for the increased inflammatory response activated by this mutant, we infected il10−/− iBMDMs and assessed inflammatory responses. No differences were observed in the levels of inflammatory cytokines induced by the wild type and lpxO mutant (Fig. 7B), supporting the notion that the increased inflammation triggered by the lpxO mutant is due to a decrease in the levels of the anti-inflammatory cytokine IL-10.
FIG 7.
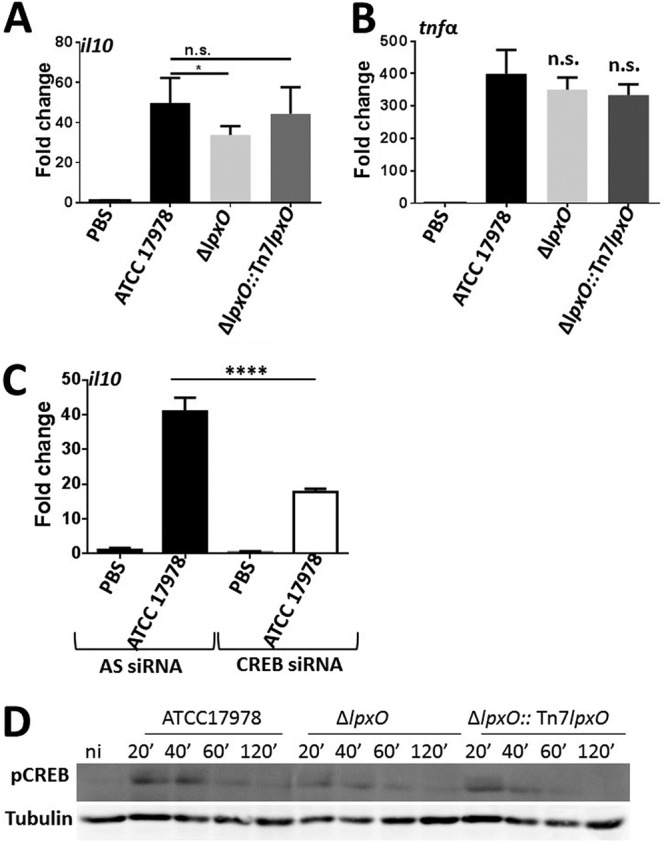
lpxO deletion in A. baumannii results in a decrease in il10 expression. (A) il10 expression in iBMDMs infected for 5 h with UV-killed A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO) by reverse transcriptase quantitative real‐time PCR. Values are presented as the means ± SD from three independent cDNA preparations measured in duplicate. (B) tnfα expression in il10−/− iBMDMs infected for 5 h with UV-killed A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO) by reverse transcriptase quantitative real‐time PCR. Values are presented as the means ± SD from two independent cDNA preparations measured in duplicate. (C) il10 expression in control (AS) and CREB siRNA-transfected iBMDMs infected for 5 h with UV-killed A. baumannii ATCC 17978 by reverse transcriptase quantitative real‐time PCR. Values are presented as the means ± SD from two independent cDNA preparations measured in duplicate. (D) Immunoblot analysis of phosphor-CREB (pCREB) and tubulin levels in lysates of iBMDMs infected with UV-killed A. baumannii ATCC 17978, A. baumannii ΔlpxO mutant (ΔlpxO), and A. baumannii ΔlpxO::Tn7lpxO mutant (ΔlpxO::Tn7lpxO). Data are representative of at least three independent experiments. In panels A to C, P values were <0.05 (*) and <0.0001 (****) versus A. baumannii ATCC 17978, determined using one-way ANOVA with Bonferroni contrasts. n.s., not significant.
We next sought to determine the signaling pathway governing the production of IL-10 in A. baumannii-infected macrophages. The transcriptional factor CREB controls the expression of IL-10 (31), and MAPK p38 and ERK have been shown to cooperate in IL-10 induction following infection (32). Since we have already shown that A. baumanii induced the activation of p38 and ERK (Fig. 6D), we focused on investigating the potential role of CREB to control IL-10 production in A. baumanii-infected cells. Suppression of endogenous expression of CREB using CREB-specific short interfering RNA (siRNA) led to a significant decrease in the levels of il10 in macrophages infected with the wild-type strain (Fig. 7C), indicating that CREB indeed does govern A. baumannii induction of IL-10. Remarkably, immunoblot analysis revealed that the phosphorylation of CREB was decreased in macrophages infected with the lpxO mutant, in sharp contrast to that of cells infected with the wild type (Fig. 7D). Image analysis showed a 19 and 29% decrease in the phosphorylation of CREB 20 and 40 min postinfection with the lpxO mutant compared to that triggered by the wild type. Complementation of the lpxO mutant restored the phosphorylation of CREB to wild-type levels (Fig. 7D).
Altogether, these findings demonstrate that the 2-hydroxylation of A. baumannii lipid A mediates the production of the anti-inflammatory cytokine IL-10 via the activation of CREB. IL-10, in turn, limits the production of inflammatory cytokines following A. baumannii infection.
DISCUSSION
The work described in this study demonstrates that A. baumannii encodes a lipid A hydroxylase, LpxO, which mediates the 2-hydroxylation of the laurate transferred by LpxLAb. This study demonstrates that LpxO-mediated 2-hydroxylation of lipid A protects the pathogen from CAMPs, mediates resistance to phagocytosis, and limits the activation of inflammatory responses by macrophages. Our findings also show that the lpxO mutant is attenuated in the G. mellonella infection model and that it induces a heightened antimicrobial response. Altogether, this evidence supports that LpxO is a bona fide immune evasion factor of A. baumannii.
Few Gram-negative bacteria are known to produce lipid A containing hydroxylated fatty acids, including pathogens such as Salmonella, Klebsiella, Bordetella, Pseudomonas, Legionella, and V. cholerae (14–17, 33–35), and also environmental bacteria such as Marinomonas and symbionts such as Vibrio fischeri (36–38). Mechanistically, lipid A hydroxylation is achieved either by the hydroxylation of fatty acids by the hydrolase catalyzed by LpxO (15, 16) or by transfer of a hydroxylated fatty acid by the acyl transferase LpxN (14). A. baumannii encodes an LpxO homolog which hydroxylates the laurate linked to the 2′ R-3-hydroxymyristoyl position of lipid A. Although our results and those of Boll et al. (7) conclusively demonstrate the function of A. baumannii LpxO, there is diversity on the lipid A position and fatty acid modified by other LpxO homologs. K. pneumoniae LpxO hydroxylates the myristate linked to the same position (16), whereas Salmonella LpxO hydroxylates the myristate linked to the 3′ R-3-hydroxymyristoyl position (15). The fact that lipid A hydroxylation seems to play a conserved role counteracting immune defenses independently of how it is achieved strongly suggests that incorporation of the hydroxyl group to lipid A is of the utmost importance here.
Our results clearly show that the 2-hydroxylation of lipid A is another of the mechanisms of A. baumannii to avoid the action of CAMPs and polymyxins. Of note, the hydroxylation of lipid A also mediates resistance to CAMPs in Klebsiella and V. cholerae (14, 16, 29), suggesting that this is an evolutionarily conserved mechanism of resistance against these antimicrobial agents. The fact that A. baumannii lipid A is constitutively hydroxylated supports the notion that LpxO-mediated lipid A modification can be considered part of the intrinsic resistome of this pathogen against these antimicrobials. Likewise, LpxM-dependent hepta-acylation of lipid A is also part of the A. baumannii intrinsic resistome (7). In contrast, the modification of A. baumannii lipid A with phosphoethanolamine and galactosamine is an inducible mechanism of resistance so far only found in clinical isolates as a result of colistin treatment (9–11). Future studies are warranted to assess the relative contribution of these mechanisms to counteract antimicrobial peptides and polymyxins. An extreme mechanism of inducible resistance is the loss of LOS due to mutations in lipid A biosynthetic genes (12). However, these strains increase the expression of other OM molecules, polysaccharides, and transporters, which may also play a role in the resistance to CAMPs (13, 39).
Another novel finding of this study is that LpxO-mediated lipid A hydroxylation attenuates the activation of defense responses. In G. mellonella, infection with the lpxO mutants triggered an upregulation of the expression of CAMPs, whereas in macrophages the lpxO mutant elicited a heightened inflammatory response. These results reinforce the notion that G. mellonella is a suitable surrogate infection model to assess the activation of early innate immune responses. The findings of this work are consistent with our earlier studies investigating K. pneumoniae LpxO-mediated lipid A modification (29, 30, 40), suggesting that the presence of a hydroxyl group on a lipid A secondary acyl chain is a conserved microbial immune evasion mechanism.
TLR4 signaling is essential for initiating inflammatory responses in vitro and in vivo to clear Acinetobacter infections (8, 41). Therefore, it is tempting to conclude that 2-hydroxylation of A. baumannii lipid A limits recognition by TLR4. However, our data showed that LpxO-mediated hydroxylation contributes to the expression of IL-10 in macrophages following infection. The fact that no differences were found in the immune responses activated by the wild type and lpxO mutant in il10−/− macrophages strongly suggests that there are no differences in the recognition of LPS with or without hydroxylation. Collectively, this evidence is consistent with the notion that A. baumannii LpxO-mediated lipid A modification is an immune evasion mechanism dependent on the production of the anti-inflammatory cytokine IL-10. Providing additional support to this notion, the activation of the transcriptional factor CREB, necessary for the expression of IL-10 in macrophages (32), is significantly reduced in macrophages infected with the lpxO mutant. It is exciting to speculate that the induction of anti-inflammatory responses upon recognition of hydroxylated lipid A is an evolutionarily conserved mechanism to blunt immune responses. Future studies investigating other Gram-negative pathogens synthesizing hydroxylated lipid A are required to validate this hypothesis.
The World Health Organization has recently included A. baumannii in the list of pathogens for which new therapeutics are urgently needed. Acinetobacter infections are particularly troublesome within the health care setting and intensive care unit due to the organism's ability to resist disinfectants and to develop antibiotic resistance. The emergence of even pan-drug-resistant strains emphasizes the urgent need of developing alternative therapeutics. Among other possibilities and based on the results of this work, we put forward the idea that antivirulence therapeutics should be considered a viable option. Compounds directed to block LpxO may render A. baumannii susceptible to host CAMPs and may limit the attenuation of inflammatory responses. Interestingly, this approach might be useful to treat other infections caused by other multidrug-resistant pathogens producing hydroxylated lipid A, including Pseudomonas and Klebsiella.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
All strains and plasmids used in this study are listed in Table 1. Strains were grown on lysogeny broth (LB) agar from frozen glycerol stocks at 37°C. Isolated colonies were used to inoculate LB broth at 37°C on an orbital shaker (180 rpm). When indicated, antibiotics were used at the following concentrations: carbenicillin (Carb), 50 μg/ml; kanamycin (Km), 25 μg/ml; chloramphenicol (Cm), 10 μg/ml; trimethoprim (Tp), 100 μg/ml; tetracycline (Tet), 12.5 μg/ml.
TABLE 1.
Strains and plasmids used in this study
| Bacterial strain or plasmid | Genotype or comment(s) | Source or reference |
|---|---|---|
| Strains | ||
| A. baumannii | ||
| ATCC 17978 | Wild-type strain | American Type Culture Collection |
| ATCC 17978-ΔlpxO | ATCC 17978 with the lpxO gene inactivated | This study |
| ATCC 17978-ΔlpxO::Tn7lpxO | ATCC 17978, ΔlpxO; Tn7Km_17978lpxOCom integrated into attTn7 site, Kmr | This study |
| E. coli | ||
| C600 | thi thr leuB tonA lacY supE | Laboratory collection |
| GT115 | F− mcrA Δ(mrr-hsdRMS-mcrBC) φ80lacZΔM15 ΔlacX74 recA1 rpsL (strA) endA1 Δdcm uidA(ΔmluI)::pir-116 ΔsbcC-sbcD | InvivoGen |
| β2163 | F RP4-2-Tc::Mu-d dapA::(erm-pir), Kmr, Emr | 43 |
| MG1655 | F− λ− ilvG rfb-50 rph-1 | Laboratory collection |
| BN1 | W3110 ΔeptA ΔlpxT ΔpagP | 19 |
| BN1 ΔlpxL | BN1 ΔlpxL::FRT; the lpxL gene was inactivated | 40 |
| BN1 ΔlpxL::Tn7lpxO | BN1 ΔlpxL::FRT; Tn7Km_17978lpxOCom integrated into attTn7 site, Kmr | This study |
| Plasmids | ||
| pGEM-T Easy | Cloning plasmid, Ampr | Promega |
| pGPI-SceI-2 | Suicide vector, R6Kgamma origin of replicaion, Mob−, carries an I-SceI endonuclease site, Tpr | 42 |
| pDAI-SceI-SacB | Expresses the I-SceI endonuclease gene, sacB, Tetr | 42 |
| pTNS2 | T7 transposase expression vector, oriR6K, Ampr | 49 |
| pUC18R6kT-mini-Tn7TKm | pUC18R6KT-mini-Tn7TKm complementation vector, Ampr, Kmr | 44 |
| pGEMΔlpxO | pGEM-T Easy containing ΔlpxO; Ampr | This study |
| pGPI-SceiI-2ΔlpxO | pGPI-SceI-2 containing ΔlpxO, Tpr | This study |
| pUC18R6kT-mini-Tn7TKm-17978lpxO | pUC18R6kT-mini-Tn7TKm containing lpxO gene for complementation, Ampr, Kmr | This study |
To assess growth of A. baumannii strains, bacteria were grown as stated previously, and 5 μl of this culture was used to inoculate 250 μl of fresh LB. Absorbance readings of the optical density at 600 nm (OD600) were measured and recorded over a 24-h period with readings at 20-min intervals using a Bioscreen C automated microbial growth analyzer (MTX Lab Systems, Vienna, VA, USA). A total of 10 independent growth curves were obtained per strain.
A. baumannii mutant construction.
Primers for mutant construction (see Table S1 in the supplemental material) were designed using the whole-genome sequence of A. baumannii ATCC 17978 (GenBank accession no. CP000521.1). The primer pairs lpxO_UPFWD, lpxO-UPRVS, lpxO_DOWNFWD, and lpxO_DOWNRVS (Table S1) were used in two separate PCRs to amplify fragments flanking A1S_0308 (A. baumannii lpxO) using Ex Taq polymerase (TaKaRa). Each amplicon had internal BamHI restriction sites added internally to the flanking regions. Gel-extracted lpxO UP and DOWN fragments were polymerized and amplified to generate a single PCR amplicon of the lpxO gene using primers lpxO_UPFWD and lpxO_DOWNRVS. This 1,655-bp amplicon was cloned into pGEM-T Easy (Promega) to obtain pGEMΔlpxO and transformed into E. coli C600 cells. After EcoRI digestion, the 1,655-bp amplicon was gel purified, cloned into EcoRI-digested Antarctic Phosphatase (New England Biolabs)-treated pGPI-SceI-2 suicide vector (42) to obtain pGPI-SceI-2ΔlpxO, and transformed into E. coli GT115 cells. pGPI-SceI-2ΔlpxO was transformed into E. coli β2163 (43), a diaminopimelate (DAP) auxotrophic donor strain with conjugative pili. pGPI-SceI-2ΔlpxO was mobilized into A. baumannii by conjugation. The cointegrant clones were selected using LB agar supplemented with Tp at 37°C. A second crossover event was performed by conjugating the pDAI-SceI-SacB plasmid (42) into an overnight culture grown to mid-exponential phase containing up to three Tp-resistant cointegrant clones. Exconjugants were selected with LB agar containing Tet at 37°C. Candidate clone colonies were screened for susceptibility to Tp on LB agar supplemented with Tp. Additionally, colony PCRs were performed using the lpxO_ UPFWD and lpxO_DOWNRVS primers. sacB allows curing of the pDAI-SceI-SacB vector by passaging the Tet-resistant, Tp-sensitive, PCR-confirmed colonies onto 6% sucrose LB agar without NaCl for 24 h at 30°C. The resulting colonies were screened for susceptibility to both Tet and Tp and confirmed with the aforementioned primers. This mutant was named the A. baumannii ΔlpxO mutant.
Complementation of A. baumannii lpxO mutant.
To complement the A. baumannii ΔlpxO mutant, a DNA fragment comprising the coding and promoter regions of lpxO was amplified using Phusion high-fidelity DNA polymerase (New England Biolabs) and primers Ab_lpxO_UP_F1 and Ab_lpxO_DWN_R1 (Table S1). The amplicon was gel extracted and cloned into ScaI-digested (New England Biolabs), Antarctic Phosphatase-treated pUC18R6KT-mini-Tn7Km plasmid (44) to generate pUC18R6KT-mini-Tn7Km_17978lpxOCom. The resulting plasmid was then transformed into E. coli GT115 and subsequently into E. coli β2163. The transposase-containing pTNS2 and pUC18R6KT-mini-Tn7Km_17978lpxOCom were mobilized into the A. baumannii ΔlpxO mutant by triparental conjugation and, once recovered on LB DAP agar, were serially diluted on LB Km agar; the pTNS2-thermosensitive helper plasmid was cured by incubating for 6 h at 42°C, followed by overnight incubation at 37°C. Candidate colonies were screened for sensitivity to Tp. Correct integration of the Tn7 transposon was confirmed by PCR using the primers Tn7-R and glmSF1. The presence of lpxO was confirmed by PCR using the primers lpxO_UPFWD and lpxO_DOWNRVS. This strain was named the A. baumannii ΔlpxO::Tn7lpxO mutant.
pUC18R6KT-mini-Tn7Km_17978lpxOCom was also conjugated into E. coli BN1 and the BN1 ΔlpxL mutant as we have previously described (40).
Lipid extraction and analysis.
Total lipids were extracted as described by Bligh and Dyer (45) and analyzed on silica gel 60 high-performance thin-layer chromatography (HPTLC) plates (Merck, Darmstadt, Germany). The plates were prewashed by solvent migration with a chloroform-methanol-acetic acid mixture (13:5:2) and dried thoroughly, samples were applied to the plate, and chromatography was performed in the same mixture of solvents. Plates were developed by charring with 15% sulfuric acid in ethanol at 180°C. Spots corresponding to lipids were quantified by densitometry using a GS-800 calibrated densitometer and Quantity One software (Bio-Rad).
Fluorimetry.
The fluorescent probe N-phenyl-1-naphthylamine (NPN) (Sigma) has been used in outer membrane permeability studies. Fluorimetric assays were carried out as described by Martínez de Tejada et al. (46), with minor modifications. Exponentially growing bacteria were resuspended in 1 mM KCN–10 mM HEPES (pH 7.2) at an optical density at 600 nm of 0.1 and transferred to fluorimetric cuvettes. Fluorescence was monitored with an FLS920 Edinburgh fluorimeter with the following settings: excitation wavelength, 350 nm; emission wavelength, 420 nm; slit width, 5.0 nm. The results were expressed in relative fluorescence units.
Lipid A extraction.
Lipid A was extracted using an isobutyric acid-ammonium hydroxide method (47) and analyzed with negative-ion MALDI-TOF mass spectrometry. Briefly, 10 ml of bacteria was grown to mid-exponential phase and washed three times with equal volumes of 10 mM phosphate buffer. The pellet was washed twice in 400 μl of chloroform-methanol (1:2 [vol/vol]) in a screw-cap test tube and centrifuged (2,000 × g for 15 min); the resulting pellet was resuspended in 400 μl of chloroform-methanol-water (3:2:0.25 [vol/vol/vol]) and centrifuged (2,000 × g for 15 min). The pellet was treated with 400 μl of isobutyric acid–1 M ammonium hydroxide (5:3 [vol/vol]) and incubated for 2 h at 100°C with occasional vortexing to hydrolyze the 3-deoxy-d-manno-octulosonic acid bond. The lipid A suspensions were then cooled in ice water and centrifuged (2,000 × g for 15 min), and the supernatant was transferred to a fresh screw-cap tube with an equal volume of deionized, sterile water and lyophilized. The lyophilized material was washed twice with 400 μl of absolute methanol and centrifuged (2,000 × g for 15 min). Lipid A was solubilized in 60 to 100 μl of chloroform-methanol-water (3:1.5:0.25 [vol/vol/vol]). A small volume of the solubilized lipid A was transferred to fresh 1.5-ml microcentrifuge tubes, desalted with a few grains of ion exchange resin (H+; Dowex 50W-X8), and briefly centrifuged. A 1-μl aliquot was deposited on a polished steel target plate for the dried droplet method. An equal volume of 2,5-dihydroxybenzoic acid (2,5-DHB) matrix (Bruker Daltonics, Inc.) was saturated in 100 mM citric acid (Sigma-Aldrich) or acetonitrile–0.1% trifluoroacetic acid (1:2 [vol/vol]) and allowed to air dry. Lipid A structural spectra were generated with a Bruker Autoflex Speed TOF/TOF mass spectrometer (Bruker Daltons Inc.) in negative reflectron mode with delayed extraction. All spectra were achieved with ion-accelerating voltage set at 20 kV, and resulting spectra were generated with an average of 300 shots. A peptide calibration standard (Bruker Daltonics, Inc.) was used to calibrate the MALDI-TOF mass spectrometer prior to analysis of each sample. Lyophilized E. coli MG1655 lipid A grown in LB broth at 37°C with identical extraction was used as an internal calibrant. Spectra are representative of at least three independent lipid A extractions.
CAMP susceptibility assays.
To assess A. baumannii susceptibility to CAMPs, we performed a modified version of the sensitivity assay described by Llobet and coworkers (48). Each strain was grown to mid-exponential phase in LB broth, washed once with PBS, and diluted in liquid test medium composed of 1% tryptone soy broth (Oxoid), 10 mM phosphate buffer (pH 6.5), 100 mM sodium chloride (Sigma-Aldrich) to approximately 3 × 104 CFU/ml. Aliquots (25 μl) of each strain were then mixed with 5 μl of either PBS or PBS containing antimicrobial peptides in 0.2-ml tubes. After 1 h of incubation at 37°C, 15 μl of the suspension was spread onto LB agar and incubated overnight at 37°C. Percent survival of the bacterial strains exposed to the antimicrobial peptides was calculated through comparison with the unexposed PBS controls. All assays were performed in duplicate on three independent experiments.
Whole-blood phagocytosis assay.
Bacteria were grown at 37°C in 5 ml of LB broth on an orbital shaker (180 rpm) until mid-exponential phase and then harvested (3,000 × g, 15 min). Bacteria were washed once with sterile PBS and adjusted to an OD600 of 1.00. Three hundred microliters of fresh human blood (used within 30 min of collection with 10% citrate dextrose as an anticoagulant) was mixed with ∼1 × 107 CFU/100 μl bacterial suspension and incubated on an orbital shaker at 37°C (180 rpm) for 3 h. Following incubation, serial dilutions were performed in sterile PBS and spread on LB agar. Bacterial counts recovered were then divided by the initial counts. Experiments were performed using blood from three individual donors, and for each blood sample the strains were tested in duplicate. Donors were all adult males, with no existing medical conditions or infections; none of the donors had used any anti-inflammatory medication for at least 24 h prior to blood withdrawal. Ethical approval for the use of blood from healthy volunteers to study bacterial killing was obtained from the Research Ethics Committee of the School of Medicine, Dentistry, and Biomedical Sciences (Queen’s Univeristy, Belfast, Ireland).
Infection of G. mellonella larvae.
G. mellonella larvae were obtained from UK Waxworm Limited and stored at 12°C in the dark without dietary supplementation. All experiments were performed within 7 days of receipt, comprising randomly selected larvae with a healthy, nonmelanized appearance and within a weight range of 250 to 350 mg.
Infections were performed as described previously, with minor modifications (30). Briefly, bacteria were grown in 5 ml LB broth until mid-exponential phase (37°C, 180 rpm) and centrifuged (3,000 × g, 15 min), washed once with sterile PBS, and adjusted to 5 × 106 CFU/ml. Larvae were surface disinfected with 70% (vol/vol) ethanol and then injected with 10 μl of the bacterial suspension, containing 5 × 104 CFU, in the rear right proleg with a Hamilton syringe with a 27-gauge needle. A group of 10 larvae was also injected with 10 μl of sterile PBS to ensure death was not due to mechanical trauma. Larvae were placed in a 9.2-cm petri dish, kept at 37°C in the dark, and monitored every 24 h. Galleria organisms were considered dead when they were unresponsive to physical stimuli. Larvae were examined for pigmentation, and time of death was recorded. Pupa formation was observed among control larvae after 3 days, and experiments were ceased from this time point. The virulence assay was performed in triplicate (30 larvae total per strain).
G. mellonella RNA extraction and RT-qPCR.
Larvae were infected with ∼ 5 × 104 CFU of the three A. baumannii strains, incubated at 37°C for 12 h, and homogenized in 1 ml TRIzol reagent (Ambion) using a VDI 12 tissue homogenizer (VWR). Total RNA was extracted according to the manufacturer’s guidelines. Further purification was achieved by treating 1 μg of RNA with DNase I (Thermo Scientific) at 37°C for 30 min and then 65°C for 10 min in the presence of 50 mM EDTA. RNA was quantified using a Nanovue Plus spectrophotometer (GE Healthcare Life Sciences). cDNA was generated by retrotranscription of 1 μg of total RNA using Moloney murine leukemia virus (M-MLV) reverse transcriptase (Invitrogen) and random primers (Invitrogen). Ten nanograms of cDNA was used as the template in a 5-μl reaction mixture from a KAPA SYBR FAST quantitative PCR (qPCR) kit (Kapa Biosystems) and target primer mix (Table S1). Reverse transcription-qPCR (RT-qPCR) was performed using a Rotor-Gene Q (Qiagen) with the following thermocycling conditions: 95°C for 3 min for hot-start polymerase activation, followed by 40 cycles of 95°C for 5 s and 60°C for 20 s. Fluorescence of SYBR green dye was measured at 510 nm. Relative quantities of mRNAs were obtained using the comparative threshold cycle (ΔΔCT) method by normalization to the amount of 18S rRNA.
G. mellonella hemolymph microbroth assay.
Larvae were infected with ∼ 1 × 106 heat-killed (65°C for 20 min) E. coli MG1655 organisms to prime the antimicrobial factors in the hemolymph and incubated at 37°C for 24 h. Hemolymphs from ten larvae were pooled in an ice-cold microcentrifuge tube containing 10 μl of 1 mg/ml N-phenylthiourea (Sigma). Five microliters of the hemolymph suspension was then used in the antimicrobial peptide assay previously described.
Generation of iBMDM-derived macrophages.
To isolate BMDMs (bone marrow-derived macrophages), tibias and femurs from C57BL/6 or IL-10−/− mice (C57BL/6 background) were removed using sterile techniques, and the bone marrow was flushed with fresh medium. To obtain macrophages, cells were plated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 20% filtered L929 cell supernatant (a source of macrophage colony-stimulating factor) and maintained at 37°C in a humidified atmosphere of 5% CO2. Medium was replaced with fresh supplemental medium after 1 day. After 5 days, BMDMs were immortalized by exposing them for 24 h to the J2 CRE virus (carrying v-myc and v-Raf/v-Mil oncogenes, kindly donated by Avinash R. Shenoy, Imperial College London). This step was repeated 2 days later (day 7), followed by continuous culture in DMEM supplemented with 20% (vol/vol) filtered L929 cell supernatant for 4 to 6 weeks. The presence of a homogeneous population of macrophages was accessed by flow cytometry using antibodies for CD11b (clone M1/70; catalog number 17-0112-82; eBioscience) and CD11c (clone N418; catalog number 48-0114-82; eBioscience).
Macrophage infections.
iBMDMs (cell line derived from wild-type NR9456 mice; BEI Resources, NIAID, NIH) were grown in DMEM (catalog number 41965; Gibco) supplemented with heat-inactivated fetal calf serum, 100 U/ml penicillin, and 0.1 mg/ml streptomycin (Gibco) at 37°C in a humidified 5% CO2 incubator. Cells were routinely tested for Mycoplasma contamination. For infections, iBMDMs were seeded to a density of 2.5 × 104/well in 96-well plates, 5 × 105/well in 12-well plates, and 1 × 106/well in 6-well plates.
Bacteria were grown in 5 ml LB broth until mid-exponential phase at 37°C on an orbital shaker (180 rpm), recovered by centrifugation (3,000 × g, 15 min), and adjusted to an OD600 of 1.00 in sterile PBS corresponding to ∼1.2 × 108 CFU/ml. Bacterial suspensions were serially diluted in PBS to count input of the bacteria and then UV irradiated at 15 J for 30 min, with confirmation of bacterial cell death by plating on LB agar. Infections were performed at a multiplicity of infection (MOI) of 20 bacteria per iBMDM. Synchronization of the infection was performed by centrifugation (200 × g for 5 min). All infections were performed in duplicate and repeated three independent times.
Macrophage RNA isolation and RT-qPCR.
Infections were performed in 6-well plates with UV-killed A. baumannii for 5 h. Cells were washed three times with prewarmed sterile PBS, and total RNA was extracted from the cells in 1 ml of TRIzol reagent (Ambion) according to the manufacturer’s instructions. Extracted RNA was treated with DNase I (Roche) and precipitated with sodium acetate (Ambion) and ethanol. RNA was quantified using a Nanovue Plus spectrophotometer (GE Healthcare Life Sciences).
cDNA was generated by retrotranscription of 1 μg of total RNA using M-MLV reverse transcriptase (Invitrogen) and random primers (Invitrogen). Ten nanograms of cDNA was used as a template in a 5-μl reaction mixture from a KAPA SYBR FAST qPCR kit (Kapa Biosystems) and target primer mix (Table S1). RT-qPCR was performed using a Rotor-Gene Q (Qiagen) with the following thermocycling conditions: 95°C for 3 min for hot-start polymerase activation, followed by 40 cycles of 95°C for 5 s and 60°C for 20 s. Fluorescence of SYBR green dye was measured at 510 nm. Relative quantities of mRNAs were obtained using the ΔΔCT method by using hypoxanthine phosphoribosyltransferase 1 (hprt) gene normalization.
Transfection conditions.
For transfection of siRNAs, ∼1 × 106 iBMDMs (6-well format) were transfected in suspension with 20 nM siRNA using Lipofectamine and Opti-MEM I (ThermoFisher) in a final volume of 2 ml. AllStars negative-control siRNA (Qiagen) or ON-TARGETplus SMARTpool siRNA targeting CREB1 (no. L-040959-01; Dharmacon) was used to transfect cells for 16 h. Cells then were mock infected with PBS or infected with UV-killed A. baumannii ATCC 17978 as described previously. RNA was collected and RT-qPCR analyses were performed as previously described. Knockdown efficiency (Fig. S4) was determined using primers for creb (Table S1).
Immunoblots.
Proteins were resolved on standard 10% SDS-PAGE gels and electroblotted in a semidry manner onto nitrocellulose membranes. Membranes were subsequently blocked with 3% (wt/vol) bovine serum albumin in TBS-Tween (TBST), and specific antibodies were used to detect protein using chemiluminescence reagents and a G:BOX Chemi XRQ chemiluminescence imager (Syngene).
All antibodies were probed with a secondary goat anti-rabbit/mouse antibody conjugated to IgG horseradish peroxidase, diluted 1:5,000 (no. 170-6515/170-6516; Bio-Rad). Antibodies used in this study were anti-IκBα (anti-rabbit, 1:1,000; no. 4812S; Cell Signaling), anti-phospho-JNK (anti-rabbit, 1:1,000; no. 9251S; Cell Signaling), anti-phospho-ERK (anti-rabbit, 1:1,000; no. 9101; Cell Signaling), anti-phospho-CREB (anti-rabbit, 1:1,000; no. sc-7978-R; Santa Cruz Biotechnology), and anti-phospho-p38 (anti-rabbit, 1:1,000; no. 4511; Cell Signaling). To ensure that equal amounts of proteins were loaded, blots were reprobed with α-tubulin (anti-mouse, 1:4,000; no. T5168; Sigma-Aldrich).
To detect multiple proteins, membranes were reprobed after stripping of previously used antibodies using a pH 2.2 glycine‐HCl–SDS buffer.
Bands were analyzed using ImageJ with the Histogram analysis tool, and results were expressed as a percentage of the intensity of the band found in the samples from cells infected with the wild-type strain.
Quantification of cytokines.
Infections were performed in 12-well plates (5 × 105 cells per well) at an MOI of 20:1 UV-killed bacteria. After 5 h of infection, supernatants were collected and spun down at 12,000 × g for 5 min to remove any debris. TNF-α in the supernatants was determined using a murine TNF-α standard TMB enzyme-linked immunosorbent assay development kit (no. 900-T54; PeproTech) according to the manufacturer’s instructions. All experiments were performed in duplicate, and three independent experiments were conducted.
Statistical analysis.
Statistical analyses were performed using one-way analysis of variance (ANOVA) with Bonferroni corrections, the one-tailed t test, or, when the requirements were not met, the Mann-Whitney U test. P values of <0.05 were considered statistically significant. Normality and equal variance assumptions were tested with the Kolmogorov-Smirnov test and the Brown-Forsythe test, respectively. Survival analyses were undertaken using the log‐rank (Mantel-Cox) test with Bonferroni correction for multiple comparisons (α = 0.008). All analyses were performed using GraphPad Prism for Windows (version 6.01) software.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the J.A.B. laboratory for their thoughtful discussions and support with this project.
T.L.B. is the recipient of a Ph.D. fellowship funded by the Department for Employment and Learning (Northern Ireland, UK). T.J.K. is the recipient of an ERS‐EU RESPIRE2 Marie Skłodowska‐Curie Postdoctoral Research Fellowship–MC RESPIRE2 1st round 4571‐2013 and a National Health and Medical Research Council Early Career Fellowship (GNT1088448). The research leading to these results has received funding from the People Program of the European Union's Seventh Framework Program (FP7/2007‐2013) under REA grant agreement 600368. Work at Departamento de Microbiología y Parasitología, Universidad de Navarra, was supported by Instituto de Salud Tropical funders (Obra Social la CAIXA, Fundaciones Caja Navarra and Roviralta, PROFAND, Ubesol, ACUNSA and Artai). This work was supported by Biotechnology and Biological Sciences Research Council (BBSRC, BB/L007223/1, and BB/P006078/1) and Queen’s University Belfast startup funds to J.A.B.
T.L.B. and J.A.B. conceived the study and wrote the first draft of the manuscript. T.L.B., T.J.K., J.S.P., and R.C.A. performed the experiments and contributed data for this work. T.L.B., T.J.K., J.S.P., R.C.A., and J.A.B. contributed to and approved the final version of the manuscript. We declare that we have no conflicts of interest.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00066-19.
REFERENCES
- 1.Raetz CR, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maeshima N, Fernandez RC. 2013. Recognition of lipid A variants by the TLR4-MD-2 receptor complex. Front Cell Infect Microbiol 3:3. doi: 10.3389/fcimb.2013.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Needham BD, Trent MS. 2013. Fortifying the barrier: the impact of lipid A remodeling on bacterial pathogenesis. Nat Rev Microbiol 11:467–481. doi: 10.1038/nrmicro3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peleg AY, Seifert H, Paterson DL. 2008. Acinetobacter baumannii: emergence of a successful pathogen. Clin Microbiol Rev 21:538–582. doi: 10.1128/CMR.00058-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gordon NC, Wareham DW. 2010. Multidrug-resistant Acinetobacter baumannii: mechanisms of virulence and resistance. Int J Antimicrob Agents 35:219–226. doi: 10.1016/j.ijantimicag.2009.10.024. [DOI] [PubMed] [Google Scholar]
- 6.Harding CM, Hennon SW, Feldman MF. 2018. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat Rev Microbiol 16:91–102. doi: 10.1038/nrmicro.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boll JM, Tucker AT, Klein DR, Beltran AM, Brodbelt JS, Davies BW, Trent MS. 2015. Reinforcing lipid A acylation on the cell surface of Acinetobacter baumannii promotes cationic antimicrobial peptide resistance and desiccation survival. mBio 6:e00478-15. doi: 10.1128/mBio.00478-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.March C, Regueiro V, Llobet E, Moranta D, Morey P, Garmendia J, Bengoechea JA. 2010. Dissection of host cell signal transduction during Acinetobacter baumannii-triggered inflammatory response. PLoS One 5:e10033. doi: 10.1371/journal.pone.0010033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beceiro A, Llobet E, Aranda J, Bengoechea JA, Doumith M, Hornsey M, Dhanji H, Chart H, Bou G, Livermore DM, Woodford N. 2011. Phosphoethanolamine modification of lipid A in colistin-resistant variants of Acinetobacter baumannii mediated by the pmrAB two-component regulatory system. Antimicrob Agents Chemother 55:3370–3379. doi: 10.1128/AAC.00079-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chin CY, Gregg KA, Napier BA, Ernst RK, Weiss DS. 2015. A PmrB-regulated deacetylase required for lipid A modification and polymyxin resistance in Acinetobacter baumannii. Antimicrob Agents Chemother 59:7911–7914. doi: 10.1128/AAC.00515-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pelletier MR, Casella LG, Jones JW, Adams MD, Zurawski DV, Hazlett KRO, Doi Y, Ernst RK. 2013. Unique structural modifications are present in the lipopolysaccharide from colistin-resistant strains of Acinetobacter baumannii. Antimicrob Agents Chemother 57:4831–4840. doi: 10.1128/AAC.00865-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moffatt JH, Harper M, Harrison P, Hale JD, Vinogradov E, Seemann T, Henry R, Crane B, St Michael F, Cox AD, Adler B, Nation RL, Li J, Boyce JD. 2010. Colistin resistance in Acinetobacter baumannii is mediated by complete loss of lipopolysaccharide production. Antimicrob Agents Chemother 54:4971–4977. doi: 10.1128/AAC.00834-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boll JM, Crofts AA, Peters K, Cattoir V, Vollmer W, Davies BW, Trent MS. 2016. A penicillin-binding protein inhibits selection of colistin-resistant, lipooligosaccharide-deficient Acinetobacter baumannii. Proc Natl Acad Sci U S A 113:E6228–E6237. doi: 10.1073/pnas.1611594113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hankins JV, Madsen JA, Giles DK, Childers BM, Klose KE, Brodbelt JS, Trent MS. 2011. Elucidation of a novel Vibrio cholerae lipid A secondary hydroxy-acyltransferase and its role in innate immune recognition. Mol Microbiol 81:1313–1329. doi: 10.1111/j.1365-2958.2011.07765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gibbons HS, Lin S, Cotter RJ, Raetz CR. 2000. Oxygen requirement for the biosynthesis of the S-2-hydroxymyristate moiety in Salmonella typhimurium lipid A. Function of LpxO, A new Fe2+/alpha-ketoglutarate-dependent dioxygenase homolog. J Biol Chem 275:32940–32949. doi: 10.1074/jbc.M005779200. [DOI] [PubMed] [Google Scholar]
- 16.Llobet E, Martinez-Moliner V, Moranta D, Dahlstrom KM, Regueiro V, Tomas A, Cano V, Perez-Gutierrez C, Frank CG, Fernandez-Carrasco H, Insua JL, Salminen TA, Garmendia J, Bengoechea JA. 2015. Deciphering tissue-induced Klebsiella pneumoniae lipid A structure. Proc Natl Acad Sci U S A 112:E6369–E6378. doi: 10.1073/pnas.1508820112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacArthur I, Jones JW, Goodlett DR, Ernst RK, Preston A. 2011. Role of pagL and lpxO in Bordetella bronchiseptica lipid A biosynthesis. J Bacteriol 193:4726–4735. doi: 10.1128/JB.01502-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bengoechea JA, Diaz R, Moriyon I. 1996. Outer membrane differences between pathogenic and environmental Yersinia enterocolitica biogroups probed with hydrophobic permeants and polycationic peptides. Infect Immun 64:4891–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Needham BD, Carroll SM, Giles DK, Georgiou G, Whiteley M, Trent MS. 2013. Modulating the innate immune response by combinatorial engineering of endotoxin. Proc Natl Acad Sci U S A 110:1464–1469. doi: 10.1073/pnas.1218080110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nizet V. 2006. Antimicrobial peptide resistance mechanisms of human bacterial pathogens. Curr Issues Mol Biol 8:11–26. [PubMed] [Google Scholar]
- 21.Nikaido H. 2003. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev 67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moranta D, Regueiro V, March C, Llobet E, Margareto J, Larrarte E, Larrate E, Garmendia J, Bengoechea JA. 2010. Klebsiella pneumoniae capsule polysaccharide impedes the expression of beta-defensins by airway epithelial cells. Infect Immun 78:1135–1146. doi: 10.1128/IAI.00940-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ishimoto H, Mukae H, Date Y, Shimbara T, Mondal MS, Ashitani J, Hiratsuka T, Kubo S, Kohno S, Nakazato M. 2006. Identification of hBD-3 in respiratory tract and serum: the increase in pneumonia. Eur Respir J 27:253–260. doi: 10.1183/09031936.06.00105904. [DOI] [PubMed] [Google Scholar]
- 24.Maisetta G, Batoni G, Esin S, Florio W, Bottai D, Favilli F, Campa M. 2006. In vitro bactericidal activity of human beta-defensin 3 against multidrug-resistant nosocomial strains. Antimicrob Agents Chemother 50:806–809. doi: 10.1128/AAC.50.2.806-809.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Peleg AY, Jara S, Monga D, Eliopoulos GM, Moellering RC Jr, Mylonakis E. 2009. Galleria mellonella as a model system to study Acinetobacter baumannii pathogenesis and therapeutics. Antimicrob Agents Chemother 53:2605–2609. doi: 10.1128/AAC.01533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gebhardt MJ, Gallagher LA, Jacobson RK, Usacheva EA, Peterson LR, Zurawski DV, Shuman HA. 2015. Joint transcriptional control of virulence and resistance to antibiotic and environmental stress in Acinetobacter baumannii. mBio 6:e01660-15. doi: 10.1128/mBio.01660-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang N, Ozer EA, Mandel MJ, Hauser AR. 2014. Genome-wide identification of Acinetobacter baumannii genes necessary for persistence in the lung. mBio 5:e01163-14. doi: 10.1128/mBio.01163-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wojda I. 2017. Immunity of the greater wax moth Galleria mellonella. Insect Sci 24:342–357. doi: 10.1111/1744-7917.12325. [DOI] [PubMed] [Google Scholar]
- 29.Kidd TJ, Mills G, Sa-Pessoa J, Dumigan A, Frank CG, Insua JL, Ingram R, Hobley L, Bengoechea JA. 2017. A Klebsiella pneumoniae antibiotic resistance mechanism that subdues host defenses and promotes virulence. EMBO Mol Med 9:430–447. doi: 10.15252/emmm.201607336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Insua JL, Llobet E, Moranta D, Perez-Gutierrez C, Tomas A, Garmendia J, Bengoechea JA. 2013. Modeling Klebsiella pneumoniae pathogenesis by infection of the wax moth Galleria mellonella. Infect Immun 81:3552–3565. doi: 10.1128/IAI.00391-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mellett M, Atzei P, Jackson R, O'Neill LA, Moynagh PN. 2011. Mal mediates TLR-induced activation of CREB and expression of IL-10. J Immunol 186:4925–4935. doi: 10.4049/jimmunol.1002739. [DOI] [PubMed] [Google Scholar]
- 32.Saraiva M, O'Garra A. 2010. The regulation of IL-10 production by immune cells. Nat Rev Immunol 10:170–181. doi: 10.1038/nri2711. [DOI] [PubMed] [Google Scholar]
- 33.Gibbons HS, Reynolds CM, Guan Z, Raetz CR. 2008. An inner membrane dioxygenase that generates the 2-hydroxymyristate moiety of Salmonella lipid A. Biochemistry 47:2814–2825. doi: 10.1021/bi702457c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kulshin VA, Zahringer U, Lindner B, Jager KE, Dmitriev BA, Rietschel ET. 1991. Structural characterization of the lipid A component of Pseudomonas aeruginosa wild-type and rough mutant lipopolysaccharides. Eur J Biochem 198:697–704. doi: 10.1111/j.1432-1033.1991.tb16069.x. [DOI] [PubMed] [Google Scholar]
- 35.Zahringer U, Knirel YA, Lindner B, Helbig JH, Sonesson A, Marre R, Rietschel ET. 1995. The lipopolysaccharide of Legionella pneumophila serogroup 1 (strain Philadelphia 1): chemical structure and biological significance. Prog Clin Biol Res 392:113–139. [PubMed] [Google Scholar]
- 36.Krasikova IN, Kapustina NV, Isakov VV, Dmitrenok AS, Dmitrenok PS, Gorshkova NM, Solov'eva TF. 2004. Detailed structure of lipid A isolated from lipopolysaccharide from the marine proteobacterium Marinomonas vaga ATCC 27119. Eur J Biochem 271:2895–2904. doi: 10.1111/j.1432-1033.2004.04212.x. [DOI] [PubMed] [Google Scholar]
- 37.Vorob'eva EV, Dmitrenok AS, Dmitrenok PS, Isakov VV, Krasikova IN, Solov'eva TF. 2005. The structure of uncommon lipid A from the marine bacterium Marinomonas communis ATCC 27118T. Bioorg Khim 31:404–413. [PubMed] [Google Scholar]
- 38.Phillips NJ, Adin DM, Stabb EV, McFall-Ngai MJ, Apicella MA, Gibson BW. 2011. The lipid A from Vibrio fischeri lipopolysaccharide: a unique structure bearing a phosphoglycerol moiety. J Biol Chem 286:21203–21219. doi: 10.1074/jbc.M111.239475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Henry R, Vithanage N, Harrison P, Seemann T, Coutts S, Moffatt JH, Nation RL, Li J, Harper M, Adler B, Boyce JD. 2012. Colistin-resistant, lipopolysaccharide-deficient Acinetobacter baumannii responds to lipopolysaccharide loss through increased expression of genes involved in the synthesis and transport of lipoproteins, phospholipids, and poly-beta-1,6-N-acetylglucosamine. Antimicrob Agents Chemother 56:59–69. doi: 10.1128/AAC.05191-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mills G, Dumigan A, Kidd T, Hobley L, Bengoechea JA. 2017. Identification and characterization of two Klebsiella pneumoniae lpxL lipid A late acyltransferases and their role in virulence. Infect Immun 85:e00068-17. doi: 10.1128/IAI.00068-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Knapp S, Wieland CW, Florquin S, Pantophlet R, Dijkshoorn L, Tshimbalanga N, Akira S, van der Poll T. 2006. Differential roles of CD14 and toll-like receptors 4 and 2 in murine Acinetobacter pneumonia. Am J Respir Crit Care Med 173:122–129. doi: 10.1164/rccm.200505-730OC. [DOI] [PubMed] [Google Scholar]
- 42.Aubert DF, Hamad MA, Valvano MA. 2014. A markerless deletion method for genetic manipulation of Burkholderia cenocepacia and other multidrug-resistant gram-negative bacteria. Methods Mol Biol 1197:311–327. doi: 10.1007/978-1-4939-1261-2_18. [DOI] [PubMed] [Google Scholar]
- 43.Demarre G, Guérout A-M, Matsumoto-Mashimo C, Rowe-Magnus DA, Marlière P, Mazel D. 2005. A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPalpha) conjugative machineries and their cognate Escherichia coli host strains. Res Microbiol 156:245–255. doi: 10.1016/j.resmic.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 44.Choi KH, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. 2005. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods 2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- 45.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/o59-099. [DOI] [PubMed] [Google Scholar]
- 46.Martínez de Tejada G, Pizarro-Cerdá J, Moreno E, Moriyón I. 1995. The outer membranes of Brucella spp. are resistant to bactericidal cationic peptides. Infect Immun 63:3054–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.El Hamidi A, Tirsoaga A, Novikov A, Hussein A, Caroff M. 2005. Microextraction of bacterial lipid A: easy and rapid method for mass spectrometric characterization. J Lipid Res 46:1773–1778. doi: 10.1194/jlr.D500014-JLR200. [DOI] [PubMed] [Google Scholar]
- 48.Llobet E, Campos MA, Gimenez P, Moranta D, Bengoechea JA. 2011. Analysis of the networks controlling the antimicrobial-peptide-dependent induction of Klebsiella pneumoniae virulence factors. Infect Immun 79:3718–3732. doi: 10.1128/IAI.05226-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Crepin S, Harel J, Dozois CM. 2012. Chromosomal complementation using Tn7 transposon vectors in Enterobacteriaceae. Appl Environ Microbiol 78:6001–6008. doi: 10.1128/AEM.00986-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.