

Chronic and fatal infections caused by Staphylococcus aureus are sometimes associated with biofilm formation. Secreted proteins and cell wall-anchored proteins (CWAPs) are important for the development of polysaccharide-independent biofilms, but functional relationships between these proteins are unclear.
KEYWORDS: CWAP, Eap, SasG, Staphylococcus aureus, biofilms, infection model, secreted protein, silkworm
ABSTRACT
Chronic and fatal infections caused by Staphylococcus aureus are sometimes associated with biofilm formation. Secreted proteins and cell wall-anchored proteins (CWAPs) are important for the development of polysaccharide-independent biofilms, but functional relationships between these proteins are unclear. In the present study, we report the roles of the extracellular adherence protein Eap and the surface CWAP SasG in S. aureus MR23, a clinical methicillin-resistant isolate that forms a robust protein-dependent biofilm and accumulates a large amount of Eap in the extracellular matrix. Double deletion of eap and sasG, but not single eap or sasG deletion, reduced the biomass of the formed biofilm. Mutational analysis demonstrated that cell wall anchorage is essential for the role of SasG in biofilm formation. Confocal laser scanning microscopy revealed that MR23 formed a rugged and thick biofilm; deletion of both eap and sasG reduced biofilm ruggedness and thickness. Although sasG deletion did not affect either of these features, eap deletion reduced the ruggedness but not the thickness of the biofilm. This indicated that Eap contributes to the rough irregular surface structure of the MR23 biofilm and that both Eap and SasG play roles in biofilm thickness. The level of pathogenicity of the Δeap ΔsasG strain in a silkworm larval infection model was significantly lower (P < 0.05) than those of the wild type and single-deletion mutants. Collectively, these findings highlight the redundant and distinct roles of a secreted protein and a CWAP in biofilm formation and pathogenicity of S. aureus and may inform new strategies to control staphylococcal biofilm infections.
INTRODUCTION
Biofilms are recognized as the dominant form of life of microbes on Earth (1, 2). Pathogenic and commensal bacteria that form biofilms in the human body or artificial implants can cause chronic infections (3, 4). Since bacteria embedded within biofilms acquire tolerance to host immunity and antibacterial drugs (5), biofilm-associated infections are difficult to treat, can be life-threatening, and can increase treatment costs in the clinical setting (6). To combat biofilm-associated issues, understanding the molecular mechanisms of biofilm formation and developing strategies to control biofilm formation based on the gained mechanistic insights are pivotal.
Staphylococcus aureus is a commensal bacterium carried by approximately 30% of the healthy population (7, 8). It is an opportunistic pathogen that causes various infectious diseases, from superficial skin infections to invasive infections (9, 10). S. aureus can also cause chronic infections associated with biofilms (11). Within biofilms, bacterial cells are embedded in an extracellular matrix (ECM) comprised of DNA, polysaccharides, and/or proteins (12), but the amount of each component differs depending on the strain and culture conditions (13). Extracellular DNA (eDNA) is important for the primary attachment of cells to the substratum and contributes to the maintenance of biofilm structure in both Gram-negative bacteria and Gram-positive bacteria, including S. aureus (14–16). Specific polysaccharides, i.e., polysaccharide intercellular adhesin (PIA) or poly-N-acetylglucosamine, play an important role in S. aureus biofilms (17–19). On the other hand, certain strains produce PIA-independent and protein-dependent biofilms (20), mainly relying on either secreted proteins or cell wall-anchored proteins (CWAPs) (12).
Extracellular adherence protein (Eap), also known as major histocompatibility complex (MHC) class II analog protein (Map), is an S. aureus-specific secreted protein (21). Eap contributes to the virulence of S. aureus by facilitating interactions between the bacterial cell surface and several plasma proteins, thus promoting adherence to the host endothelium and internalization into human fibroblasts and epithelial cells (22, 23). Eap plays an important role in biofilm formation under certain growth conditions (24, 25). Disruption of the eap gene in S. aureus Newman leads to a slight reduction of biofilm formation under low-iron conditions (24). Furthermore, deletion of eap remarkably reduces biofilm formation by S. aureus SA113 (ATCC 35556) derived from the laboratory strain NCTC8325 under iron-replete conditions in the presence of human serum (25).
CWAPs are classified into four distinct groups (26). The first group belongs to the microbial surface component-recognizing adhesive matrix molecules and includes clumping factors (ClfA and ClfB) and fibronectin binding proteins (FnBPA and FnBPB). Proteins from the second group harbor the near-iron transporter motif and include iron-regulated surface determinant proteins (IsdA, IsdB, and IsdH). The third group of proteins contains tandemly repeated three-helical bundles, e.g., protein A. The fourth group comprises the G5-E repeat family proteins, including S. aureus surface protein G (SasG). All CWAPs contain a characteristic 5-amino-acid structure called the LPXTG motif (Leu-Pro-any amino acid-Thr-Gly) (27). The LPXTG motif is recognized by the membrane protein sortase A (SrtA) (27), which cleaves the peptide bond between threonine and glycine residues and covalently bridges the threonine to lipid II, a precursor of peptidoglycan (28). Although extensive efforts have been made to demonstrate the individual importance of these proteins in biofilm formation and pathogenesis, functional relationships among them are still largely unknown.
SasG, also known as Aap in Staphylococcus epidermidis, is one of the extensively characterized CWAPs in S. aureus and promotes cell-cell interactions during biofilm formation (29, 30). SasG/Aap is comprised of the N-terminal secretion signal, the A domain, and repeated B domains harboring two short G5-E repeats, followed by the C-terminal wall/membrane-spanning regions containing the LPXTG motif (31). Biofilm promotion is mainly achieved by interactions between B domains of two SasG/Aap molecules in a Zn2+-dependent fashion (32). Atomic force microscopy demonstrated that homophilic interactions between B domains of two SasG/Aap molecules are involved in biofilm promotion activity (33). On the other hand, it is still unclear whether cell wall anchorage is essential for SasG/Aap function in biofilm promotion.
In this study, we aimed to define the roles of Eap and SasG in biofilm formation. We show that the secreted protein Eap and the CWAP SasG compensate for one another in biofilm formation, but only Eap plays a key role in the ruggedness of biofilm structure. In addition, the loss of both proteins significantly reduces the pathogenicity of S. aureus in a silkworm larval infection model. These findings provide insight into the multicellular behaviors and pathogenicity of S. aureus and emphasize the importance of developing antibiofilm therapies that target multiple biofilm components.
RESULTS
Eap and CWAPs play similar roles in biofilm biomass determination.
In this study, S. aureus strains were cultured at 37°C in brain heart infusion (BHI) medium supplemented with 1% (wt/vol) glucose (BHIG medium), since a large variety of S. aureus strains produce substantial biofilms under these conditions (34). We previously showed that MR23, a clinical methicillin-resistant isolate of S. aureus, forms a robust protein-dependent biofilm in BHIG medium that is dispersed by proteinase K (see Fig. S1 in the supplemental material) (35). To identify proteins that are important for biofilm formation, known biofilm-associated genes were deleted in MR23 by using the in-frame deletion method (36, 37). Although the MR23 ECM contains large amounts of Eap and Eap promotes biofilm formation in strains that do not produce substantial biofilms (35), deletion of eap did not affect the biomass of the MR23 biofilm (Fig. 1A), similarly to S. aureus SA113 Δeap grown in BHIG medium (25). This suggested that Eap does not contribute to biofilm formation in MR23 and/or that other molecules, including proteins, eDNA, and PIA, compensate for the loss of Eap function. To address the latter possibility, we treated a preformed MR23 Δeap biofilm with enzymes that degrade major biofilm components. The biofilm was destroyed by proteinase K but not by DNase I or PIA-degrading dispersin B (Fig. S1A). This indicated that MR23 Δeap formed a protein-dependent biofilm and that other proteins contributed to biofilm formation.
FIG 1.
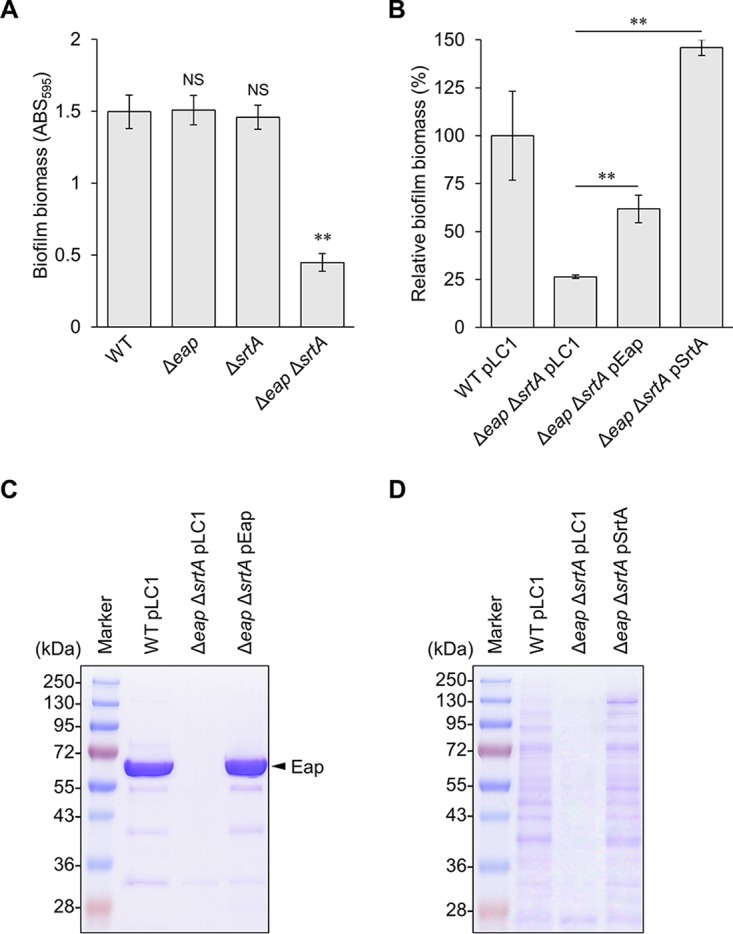
Eap and SrtA play redundant roles in biofilm formation. (A) Biomasses of biofilms formed by MR23 wild-type (WT), Δeap, ΔsrtA, and Δeap ΔsrtA strains grown in BHIG medium at 37°C for 24 h were determined by crystal violet staining. (B) Reduced biomass of Δeap ΔsrtA strain biofilm was complemented by providing plasmid-encoded Eap or SrtA. An empty vector (pLC1) was introduced into the WT and Δeap ΔsrtA strains as positive and negative controls, respectively. The data are presented relative to the positive control, which was designated 100%. For panels A and B, the means and standard deviations of biofilm biomasses from three independent experiments are shown. **, P < 0.01; NS, not significant. (C) Protein profiles of ECM isolated from WT/pLC1, Δeap ΔsrtA/pLC1, Δeap ΔsrtA/pEap, and Δeap ΔsrtA/pSrtA strains were analyzed by SDS-PAGE with Coomassie brilliant blue (CBB) staining. Prominent bands indicated by an arrowhead correspond to Eap (35). (D) Cell wall fractions of the indicated strains were analyzed by SDS-PAGE with CBB staining. The images are representative of results from at least three independent analyses.
CWAPs are important for biofilm formation in various bacteria (38–42). We therefore asked whether CWAPs play a role in biofilm formation in MR23 Δeap. Sortase A covalently links the CWAP LPXTG motif to peptidoglycan, anchoring CWAPs to the cell wall (27). Therefore, the role of CWAPs can be investigated by deleting srtA. To test whether Eap played a role in biofilm formation, srtA was disrupted in wild-type MR23 and MR23 Δeap. Although the biofilm biomass of the ΔsrtA mutant was the same as that of the wild type, the Δeap ΔsrtA strain formed significantly less biofilm than other strains (Fig. 1A). In addition, the biomass of the Δeap ΔsrtA strain biofilm was restored by expressing either Eap or SrtA from the respective plasmids (Fig. 1B). Expression of Eap and SrtA was confirmed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) of the ECM (Fig. 1C) and cell wall fractions (Fig. 1D). Drastically reduced levels of CWAPs in the Δeap ΔsrtA mutant were recovered by the expression of exogenous SrtA (Fig. 1D). These observations indicated that Eap and a certain CWAP(s) play redundant roles in the formation of the substantial protein-dependent biofilm of MR23.
Eap and SasG play redundant roles in biofilm formation.
To identify the CWAP that compensated for the loss of Eap in biofilm formation by MR23, we disrupted major CWAP-encoding genes belonging to different groups (26) in the MR23 wild-type and Δeap mutant strains. Simultaneous deletion of eap and sasG resulted in a significant reduction of the biofilm biomass, while combined deletions of eap and other CWAP genes did not (Fig. 2A). In addition, the biomass of the ΔsasG mutant biofilm was similar to those of the wild-type and Δeap strains (Fig. 2A). The reduced biomass of the Δeap ΔsasG strain biofilm was restored by the expression of either Eap or SasG from the respective plasmids (Fig. 2B). Expression of Eap and SasG was confirmed by SDS-PAGE (Fig. 2C) and Western blotting (Fig. 2D). Of note, the amount of SasG produced by a plasmid-borne gene was larger than that of the wild-type strain harboring the empty vector pLC1 (Fig. 2D), which may account for a slight increase in the biomass of biofilm formed by the Δeap ΔsasG mutant harboring pSasGWT (Fig. 2B). These observations revealed that Eap and SasG play redundant roles in the formation of substantial biofilm by MR23.
FIG 2.
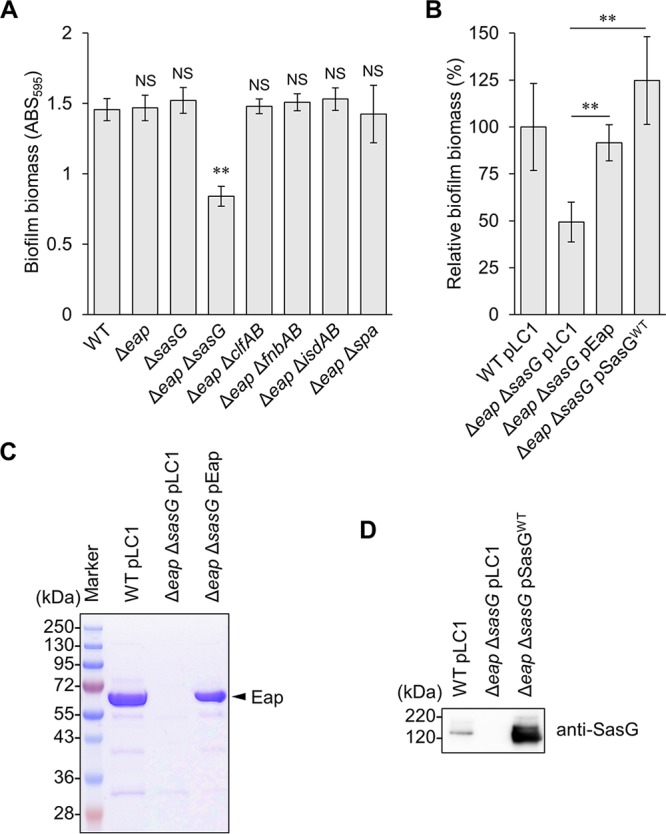
Identification of CWAPs responsible for biofilm formation in MR23. (A and B) Biomasses of biofilms produced by the indicated strains were quantified as described in the legend of Fig. 1. The means and standard deviations of biofilm biomasses from three independent experiments are shown. **, P < 0.01; NS, not significant. (C) Protein profiles of ECM isolated from the indicated strains were analyzed by SDS-PAGE with CBB staining. (D) Proteins in the cell wall fractions of the indicated strains were subjected to SDS-PAGE with CBB staining or Western blotting with anti-SasG antibody.
MR23 is a hyper-Eap-producing strain compared with other clinical isolates, as recently reported (34). In addition, the protein level of SasG in MR23 was similar to or higher than those in most other strains (Fig. S2). Therefore, the redundant roles of Eap and SasG might be specific for certain strains that overproduce Eap and SasG. As shown in Fig. S3A in the supplemental material, single knockouts of eap and sasG slightly reduced the biofilm biomass of RN4220, a restriction-deficient strain of S. aureus derived from the laboratory strain NCTC8325 (43), while double knockout of both genes did so more effectively. Overproduction of either Eap or SasG drastically stimulated biofilm formation of RN4220 Δeap ΔsasG. The biofilm biomass of RN4220 Δeap ΔsasG co-overexpressing Eap and SasG was similar to those of cells overexpressing each protein (Fig. S3B). Under the conditions tested, the amount of Eap expressed from pEap-SasG was smaller than that expressed from pEap, while the amount of SasG produced from pEap-SasG was almost the same as that produced from pSasG (Fig. S3C). Supplementation of high concentrations of anhydrotetracycline (aTc), an inducer, did not stimulate biofilm formation of RN4220 Δeap ΔsasG/pEap-SasG (data not shown). Therefore, we added purified Eap into the biofilm culture of RN4220 Δeap ΔsasG/pEap-SasG. Exogenously added Eap did not promote biofilm formation of RN4220 Δeap ΔsasG/pEap-SasG, although it stimulated that of RN4220 Δeap ΔsasG/pLC1 in a dose-dependent manner (Fig. S3D). Analysis of the extracellular matrix confirmed that a sufficient amount of Eap was used in this experiment, since the concentration of Eap in the culture of RN4220 Δeap ΔsasG/pEap was estimated to be 90.9 ± 2.9 nM (n = 3). These results indicate that redundancy between Eap and SasG is likely specific for certain strains that overproduce Eap and SasG, as in the case of MR23.
SasG is a DNA binding protein and is capable of stabilizing eDNA in the biofilm.
The biofilms formed by wild-type and Δeap strains were resistant to DNase I treatment, whereas those formed by the ΔsasG and Δeap ΔsasG strains were slightly and remarkably sensitive, respectively (Fig. 3A and Fig. S1), suggesting two possibilities, that Eap and SasG interact with and protect eDNA from nucleases and that the importance of eDNA is masked by these proteins. Recently, it was reported that Eap can bind to DNA and is capable of blocking neutrophil extracellular trap (NET) formation (44). However, the DNA binding capacity of SasG has not yet been elucidated. Therefore, we performed a gel shift assay to examine the interaction between SasG and DNA. As shown in Fig. 3B, purified SasG bound to purified lambda DNA in a dose-dependent manner. Next, we examined effect of SasG on the stability of DNA. Although lambda DNA was digested by DNase I rapidly, SasG protected it from degradation under the tested conditions (Fig. 3C). Taken together, these results indicate that SasG is a DNA binding protein and is capable of stabilizing eDNA in the biofilm.
FIG 3.
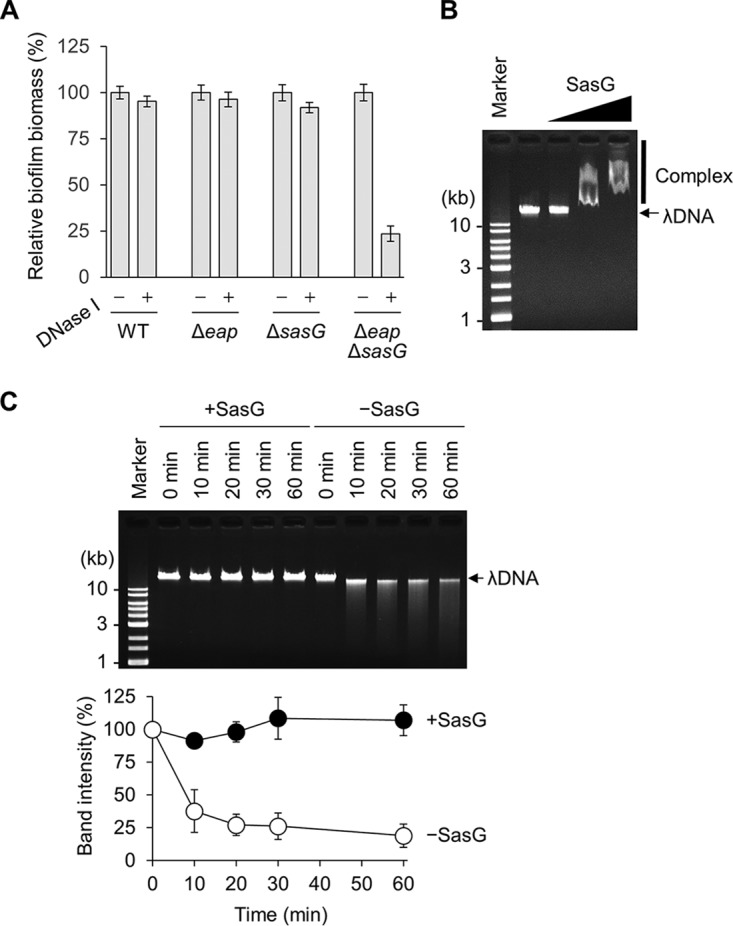
SasG is a DNA binding protein. (A) DNase I sensitivities of the biofilms formed by the indicated strains. Relative biofilm biomasses are shown (nontreated biofilms are defined as 100%). Original data are shown in Fig. S1 in the supplemental material. −, absence; +, presence. (B) The DNA binding capacity of SasG was analyzed by a gel shift assay. Purified SasG (0.1, 0.5, and 1 μM) was mixed with lambda DNA (λDNA) prior to agarose gel electrophoresis. (C) Degradation of λDNA by DNase I was analyzed in the presence and absence of purified SasG. After treatment, DNase I and SasG were degraded by proteinase K, and the residual DNA was analyzed by agarose gel electrophoresis. Band intensities were measured using an LAS-4000 image analyzer, and the relative intensities are shown in the graph.
Cell wall anchorage of SasG is essential for biofilm formation.
SasG contains an N-terminal signal peptide, an A domain, repeated B domains, and an LPXTG motif (28). DNA sequencing revealed that the MR23 sasG gene encodes a protein of approximately 108 kDa, containing four B domains (Fig. 4A). SasG promotes adhesion of bacterial cells during biofilm formation, and while interactions between the B domains are important for adhesion, the A domain is dispensable (31). However, it was unclear whether cell wall anchorage was essential for SasG function in biofilm development. To address this, we removed the SasG LPXTG motif and tested whether this affected biofilm formation by MR23. Plasmid-encoded wild-type SasG (SasGWT) restored the biofilm biomass of the Δeap ΔsasG strain to wild-type levels, whereas expression of the protein lacking the LPXTG motif (SasGΔL) did not (Fig. 4B). As expected, SasGWT was present in the cell wall fraction, but SasGΔL was not (Fig. 4C). In contrast, a large amount of SasGΔL was detected in the culture supernatant (Fig. 4D). These observations indicated that cell wall anchorage is essential for the key role of SasG in biofilm formation. This was consistent with the reduced biofilm biomass of the Δeap ΔsrtA strain (Fig. 1A), in which SasG was not tethered to the cell wall.
FIG 4.
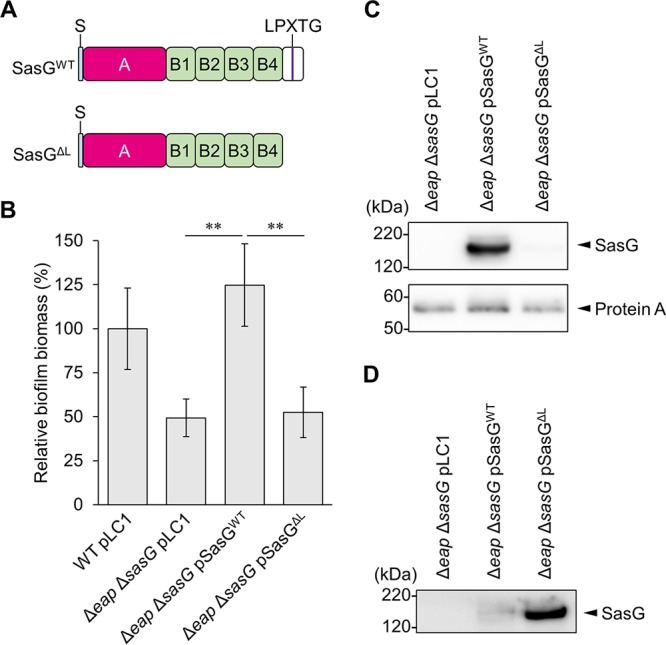
The LPXTG motif in SasG plays a role in compensating for the loss of Eap in biofilm formation. (A) The domain structure of MR23 SasG. S, signal peptide; A, A domain; B, B domain; LPXTG, LPXTG motif. Plasmids for the expression of SasGWT and SasGΔL were constructed in the present study (see Table S1 in the supplemental material). (B) Biomasses of biofilms produced by the indicated strains were determined as described in the legend of Fig. 1. The means and standard deviations from three independent experiments are shown. **, P < 0.01. (C and D) SasG proteins in the cell wall fractions (C) and culture supernatants (D) were analyzed by Western blotting using anti-SasG antibody.
Eap plays an important role in rugged biofilm formation.
Eap is a major component of the MR23 ECM. Eap contributes to biofilm formation by promoting bacterial cohesion and bacterial cell surface interactions (30, 38), whereas SasG promotes bacterial cell-cell adhesion (31). To determine whether these proteins affected the three-dimensional structure of MR23 biofilm, we analyzed biofilms formed by MR23 wild-type and derived mutant strains by using confocal laser scanning microscopy (CLSM). The wild-type strain formed a thick biofilm with a highly rugged surface (Fig. 5). The ΔsasG strain formed a biofilm whose thickness and ruggedness were similar to those of the wild-type biofilm. The Δeap strain formed a biofilm that had a similar thickness to but was smoother than those of wild-type and ΔsasG strain biofilms. On the other hand, the Δeap ΔsasG strain formed a thin and smooth biofilm. These observations indicated that Eap and SasG play similar roles in determining biofilm biomass and thickness but different roles in determining biofilm ruggedness.
FIG 5.

Three-dimensional structure of bacterial biofilms. (A) Biofilms formed by the indicated strains were stained with thioflavin T and analyzed using CLSM. Typical top oblique and side views of the biofilms are shown. (B) Thicknesses of the biofilms formed in three independent dishes were determined using ImageJ software. The line in each box-and-whisker plot represents the median thickness of biofilms formed by the indicated strains. O, outlying values; **, P < 0.01; NS, not significant.
We then analyzed biofilms at an initial phase of formation (4-h biofilms) by using atmospheric scanning electron microscopy (ASEM). This enabled the visualization of biofilms in solution at a higher resolution than that afforded by CLSM and with minimal artifacts caused by dehydration (45). The MR23 wild-type biofilm contained highly aggregated cell clusters (Fig. S4). In contrast, the biofilm formed by the Δeap ΔsasG strain spread on the surface of ASEM dish, and the formed cell clusters were smaller and less numerous than those of the wild-type strain. In addition, both focused and defocused cells were observed in the wild-type strain, while focused cells were predominant in the Δeap ΔsasG strain. This indicated that the former formed a multilayer biofilm while the latter formed a monolayer biofilm after 4 h of cultivation. These differences might be associated with the three-dimensional structures of the biofilms observed using CLSM (Fig. 5).
Double deletion of eap and sasG reduces pathogenicity of S. aureus.
We next examined the impact of the deletion of eap and sasG on the pathogenicity of S. aureus in vivo, using silkworm larvae (Bombyx mori) as a model of human-pathogenic bacterial infection (46). All silkworm larvae survived for at least 60 h after injection of a 0.6% NaCl solution, but 70% of larvae died within that period after injection of 1 × 107 cells of the MR23 wild-type strain (Fig. 6). No statistically significant difference in the survival rates of silkworm larvae infected with the Δeap strain and the wild type were apparent. Similarly, no statistically significant differences were observed in the rates of mortality of larvae infected with the ΔsasG and wild-type strains. In contrast, the survival rate of silkworm larvae infected with the Δeap ΔsasG strain was significantly higher than that of larvae infected with the wild type (Fig. 6). Since proliferation of S. aureus in silkworm larvae is required for the lethality of infection (46), we also evaluated the survival of the tested strains in silkworm larvae. After a 24-h infection, no significant differences in the retrieved CFU per milliliter were apparent between the strains (Fig. S5). Since the rate of mortality of larvae infected with the Δeap ΔsasG strain was significantly lower than that of larvae infected with the wild type, this indicated that the effect was not associated with a reduction of bacterial cell numbers in silkworm larvae but rather was associated with the strain’s biofilm-forming capacity in vitro.
FIG 6.
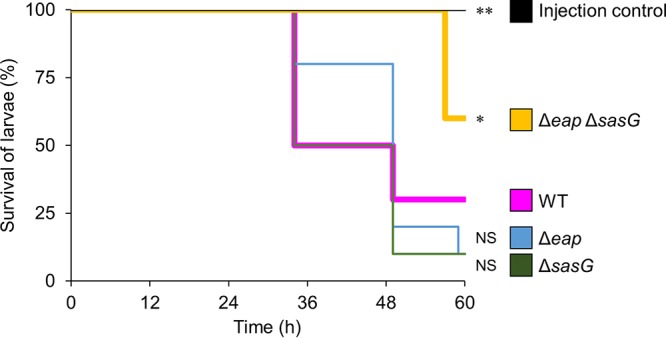
Evaluation of the pathogenicity of S. aureus strains in silkworm larvae. Ten silkworm larvae were injected with diluted cultures (1 × 107 CFU) of the indicated strains grown overnight. Larval survival was monitored at 25°C for 60 h. The curves are representative of data from at least three independent experiments. The differences between the WT and Δeap ΔsasG strains or between the WT and the injection control (0.6% NaCl solution) were statistically significant (*, P < 0.05, and **, P < 0.01, respectively). In contrast, no statistical difference was apparent between WT and Δeap strain treatments or between WT and ΔsasG strain treatments (P > 0.05).
DISCUSSION
In this study, genetic and microscopic analyses and silkworm infection experiments revealed that the secreted protein Eap and the CWAP SasG play redundant and distinct roles in biofilm development and pathogenicity of S. aureus. To the best of our knowledge, this is the first-ever report describing the functional relationship between a secreted protein and a bona fide CWAP in the multicellular behavior and pathogenesis of an opportunistic pathogen.
Mutational analyses revealed that simultaneous disruption of eap and srtA and that of eap and sasG resulted in a significant reduction of the biomass of MR23 biofilm (Fig. 1 and 2). However, the biomass of the Δeap ΔsrtA strain biofilm was lower than that of the Δeap ΔsasG strain biofilm (Fig. 1 and 2), indicating that an additional CWAP(s) might be responsible for biofilm formation by the Δeap ΔsasG strain. In addition, the Δeap ΔsrtA strain formed a small but significant amount of biofilm (approximately 25% of that produced by the wild type) that was sensitive to proteinase K and DNase I but not dispersin B (see Fig. S1 in the supplemental material), suggesting that other non-CWAP proteins and eDNA contribute slightly to biofilm formation.
SasG promotes cell-cell interactions during biofilm formation. This is mainly achieved by interactions between B domains of two SasG molecules anchored to different S. aureus cells, in a Zn2+-dependent fashion (32). Although at least five B domains were shown to be indispensable for SasG to promote biofilm formation, the number of B domains varies between 2 and 10 (29). In this study, we confirmed that MR23 SasG harbors four B domains (DDBJ accession number LC388387) and yet it contributes to biofilm formation of MR23 (Fig. 1). The slight contradiction between the previous study and our study regarding the number of B repeats required for biofilm formation could be due to the different strains and experimental conditions. Eap harbors four to six tandem repeats of a characteristic domain, the EAP domain, comprised of an alpha-helix positioned diagonally across a five-stranded, mixed beta-sheet (47). DNA sequencing revealed that MR23 Eap has five EAP domains (DDBJ accession number LC388386). Revisiting the minimum number of B domains in SasG and determining the role of the EAP domain in biofilm formation will provide further insights into molecular mechanisms of biofilm formation mediated by these proteins.
Previously, the involvement of a secreted protein, Sbp, and a cell wall-anchored protein, Aap, in biofilm formation by S. epidermidis was analyzed (48). Those authors reported that Sbp, but not Aap, is involved in biofilm formation by S. epidermidis strain 1457, a PIA-dependent biofilm producer. They noticed that reduced biofilm formation in the Δsbp strain was due to the downregulation of icaA transcription. Subsequently, those authors revisited the roles of Sbp and Aap in an ica-negative mutant of strain 1457. Overproduction of the B domains of Aap and supplementation of recombinant Sbp promoted biofilm formation of the ica-negative strain in a dose-dependent manner. It should be emphasized that those authors used only the B domain of Aap (amino acids 596 to 1507) that was not covalently linked to the cell wall and did not show the importance of cell wall-anchored Aap. Interestingly, we found that cell wall anchorage was essential for SasG function in MR23 biofilm and that the role of SasG was reinforced in the absence of Eap (Fig. 4). On the other hand, S. aureus protein A and Listeria monocytogenes internalin A do not require cell wall anchoring to promote biofilm development (39, 49). The requirement of cell wall anchorage for biofilm promotion depends on the CWAP. Protein A and internalin A may bind to the cell surface even in the absence of the LPXTG motif and are expected to promote bacterial cell-cell interactions via protein-protein or protein-other component interactions. Data presented here indicated that SasGΔL was present in the culture supernatant, but no or very little protein was present in the cell surface fraction (Fig. 4). This suggested that SasG was unable to associate tightly with the bacterial cell surface in the absence of the LPXTG motif. Reduced biofilm formation by the Δeap ΔsrtA strain (Fig. 1) supported this notion, since LPTXG-containing proteins cannot be covalently bridged to the cell wall in this strain.
CLSM analysis revealed that Δeap and ΔsasG strains produced a thick biofilm, similar to that of the wild type, and that the Δeap ΔsasG strain formed a thinner biofilm than the wild type (Fig. 5). These observations were consistent with the results of conventional crystal violet staining (Fig. 2A). Interestingly, deletion of eap significantly reduced the ruggedness of the MR23 biofilm, whereas that of sasG did not (Fig. 5), indicating that Eap contributes to the ruggedness of the biofilm. ASEM analysis indicated that Eap is important for bacterial cohesion, leading to the formation of highly aggregated cell clusters at the initial stage of biofilm formation (Fig. S4). This property of Eap appeared to be associated with the roughness of biofilms.
We also examined the impacts of eap and sasG deletions on S. aureus pathogenicity in vivo, using the silkworm larva model. No statistically significant differences between the survival rates of silkworm larvae infected with wild-type and those infected Δeap strains were apparent; however, the survival rate of silkworm larvae infected with the Δeap strain was slightly higher than that of larvae infected with the wild-type and ΔsasG strains (Fig. 6). Eap contributes to the virulence of S. aureus by interacting with the bacterial cell surface and several host plasma proteins (22, 23). The role of SasG in virulence is still unclear. It was reasonable to assume that the effect of Eap on pathogenicity is greater than that of SasG and that biofilm biomass quantified in vitro correlated with pathogenicity. Indeed, the pathogenicity of S. aureus in the silkworm larva model is associated with adhesion to host cells but not toxin production (50). Therefore, it is not surprising that key players in biofilm formation also contribute to pathogenicity in silkworm larvae.
How does MR23 produce large amounts of Eap and SasG, and how does such a strain emerge? Our preliminary data revealed that agrC and one-third of agrA are spontaneously deleted in MR23 (data not shown). Dysfunction of agr leads to upregulation of the expression of surface proteins, including Spa and FnBPA, at the transcriptional (51–53) and protein (54, 55) levels. Although regulation of SasG expression is largely unknown, a previous report suggested that the transcription of sasG was increased in agr-dysfunctional isolates (56). In addition, surface proteins are stabilized due to downregulation of the expression of extracellular proteases in agr mutants (57). These data suggest that agr dysfunction may be involved in the enhanced expression and accumulation of SasG in MR23. Previously, it was reported that S. aureus Newman produced a large amount of Eap via enhanced activity of SaeS, a positive regulator of Eap, due to a unique mutation in SaeS (Leu18 to Pro18) (58). The same mutation was not found in MR23, but another mutation was detected in the C-terminal part of SaeS (Val299 to Leu299). It would be interesting to determine whether the latter mutation is also involved in the overproduction of Eap. Glucose was shown to repress SaeS, leading to downregulation of Eap (59); however, MR23 still produced a large amount of Eap even in the presence of glucose (34). Therefore, other factors, apart from SaeS, should also be involved in the extremely large amount of Eap in MR23. We found a single-base substitution in the 5′ untranslated region of eap in MR23 compared with other strains (data not shown). This base substitution may stabilize eap mRNA and/or promote its translation, which could account for the extremely large amount of Eap in MR23.
Taken together, the presented findings highlight the functional relationship between a secreted protein and a CWAP in S. aureus biofilm formation and pathogenicity. There may be similar redundancies between Eap and SasG, between another secretion protein(s) and another CWAP(s), between proteins and eDNA, and between polysaccharides and other extracellular substances in other strains and bacteria. In fact, we found redundancy between proteins (Eap and SasG) and eDNA, as the DNase I-resistant biofilm of MR23 became DNase I sensitive when eap and sasG were deleted (Fig. 3A and Fig. S1). In addition, our results indicate for the first time that SasG binds to and stabilizes DNA (Fig. 3B and C). The present paper will provide an important avenue to consider that different biofilm adhesins may play overlapping roles in both biofilm formation and infection. This knowledge may contribute to the development of antibiofilm therapies targeting multiple biofilm components.
MATERIALS AND METHODS
Bacterial strains and culture media.
Bacterial strains used in this study are listed in Table S1 in the supplemental material. S. aureus strains were grown at 37°C in BHI medium (Becton, Dickinson, Franklin Lakes, NJ), BHIG medium (Wako, Osaka, Japan), or mannitol salt agar (Merck, Darmstadt, Germany). Escherichia coli strains were grown at 37°C in Luria-Bertani (LB) medium containing 1% (wt/vol) tryptone (Becton, Dickinson), 0.5% (wt/vol) yeast extract (Becton, Dickinson), and 1% (wt/vol) NaCl. When required, appropriate antibiotics (100 μg/ml ampicillin and 5 μg/ml chloramphenicol; Nacalai Tesque, Kyoto, Japan) and an inducer (100 ng/ml aTc; Sigma, St. Louis, MO) were added to the media.
Plasmid construction.
Mutant strains of S. aureus MR23 were constructed using the E. coli-S. aureus shuttle vector pKOR1 (37), as described previously by Chiba et al. (60). Briefly, sequences approximately 500 bp upstream and downstream of each target gene were PCR amplified from MR23 genomic DNA using KOD Plus ver. 2 DNA polymerase (Toyobo, Osaka, Japan) and the appropriate primer sets (Table S2). The fragments were connected by splicing by overlap extension PCR (36). The generated PCR products were cloned into pKOR1 using the Gateway BP Clonase II enzyme mix (Life Technologies, Palo Alto, CA); the resulting plasmids are described in Table S2 in the supplemental material.
Plasmids for the complementation of the respective gene deletions were constructed using the E. coli-S. aureus shuttle vector pLC1 as previously described (61). Briefly, eap, srtA, sasGWT, sasGΔL, and eap-sasGWT genes were PCR amplified from genomic DNA using KOD Plus ver. 2 DNA polymerase and primers listed in Table S2 in the supplemental material. The amplified fragments were cloned into pLC1 linearized by inverse PCR with primers pLC1-F and pLC1-R (Table S2) using the GeneArt seamless cloning and assembly kit (Life Technologies) according to the manufacturer’s instructions.
To overproduce recombinant N-terminally His6-tagged SasG (His-SasG) in E. coli, the fragment encoding MR23 SasG lacking the signal sequence and the C-terminal portion with the LPXTG motif (amino acid residues 51 to 954) was PCR amplified from the MR23 genome using KOD Plus Neo DNA polymerase (Toyobo) and the primers pCold-SasG-F and pCold-SasG-R (Table S2). The amplified fragment was cloned into pCold I (TaKaRa, Otsu, Japan) using the GeneArt seamless cloning and assembly kit, as described above. The resultant plasmid was named pCold-SasG (Table S1).
Oligonucleotide primers (Table S2) were synthesized by Life Technologies.
Construction of deletion mutants.
Plasmids derived from pKOR1, as described above, were used to transform S. aureus RN4220 by electroporation (62). After purification, the plasmids were introduced into strain MR23 by electroporation, and the target genes were deleted from the MR23 genome by in-frame deletion as described previously (32, 45) (Table S1). Similarly, RN4220 isogenic mutants were generated.
Biofilm formation.
S. aureus cultures grown overnight in BHI medium at 37°C were diluted 1,000 times in BHIG medium. Next, 200-μl suspensions were cultured at 37°C for 24 h in 96-well polystyrene flat-bottom plates (Corning, Corning, NY). When required, aTc (100 ng/ml), an inducer, and chloramphenicol (5 μg/ml), a selective agent, were added to the culture medium from the onset of biofilm formation. Biofilms formed on a plastic surface were washed twice with 200 μl of phosphate-buffered saline and stained with 200 μl of 0.05% (wt/vol) crystal violet for 5 min at 25°C. After staining, biofilms were washed once with 200 μl of phosphate-buffered saline, and their masses were quantified by measuring the absorbance at 595 nm (ABS595) using an Infinite F200 Pro microplate reader (Tecan, Männedorf, Switzerland). The limit of the microplate reader was an ABS595 of 4.0.
To analyze biofilm susceptibility to enzymes, proteinase K (100 μg/ml) (Sigma), DNase I (100 U/ml) (Roche, Mannheim, Germany), or dispersin B (20 μg/ml) (Kane Biotech, Winnipeg, MB, Canada) (63) was added to 24-h biofilms, and the mixture was incubated for 1 h at 37°C. Biofilm biomass was then quantified as described above.
Isolation of ECM, cell wall, and culture supernatant fractions.
The ECM was isolated from bacteria grown under biofilm-forming conditions as previously reported (60). Briefly, cultures grown overnight were diluted 1,000 times in 10 ml of BHIG medium in 15-ml conical tubes (Becton, Dickinson) and statically incubated at 37°C for 24 h. After incubation, the conical tubes were centrifuged at 8,000 × g for 10 min at 25°C to separate bacterial cells from the culture supernatant. To extract ECM components, cell pellets were suspended in 100 μl of a 1.5 M NaCl solution. The suspensions were centrifuged at 5,000 × g for 10 min at 25°C. The supernatants (ECM fractions) were then transferred to new test tubes. Cell pellets were suspended in 100 μl of a 25% (wt/vol) sucrose (Nacalai Tesque) solution containing 10 mM Tris-HCl (pH 8.0) (Wako) and a protease inhibitor cocktail (Nacalai Tesque). The suspensions were treated with lysostaphin (200 μg/ml) (Wako) for 30 min at 37°C and then centrifuged at 15,000 × g for 10 min at 25°C. The supernatants were collected as the cell wall fractions. To concentrate the culture supernatants, the supernatants (600 μl) were mixed with an equal amount of 20% (wt/vol) trichloroacetic acid (Nacalai Tesque) and incubated for 30 min on ice. After centrifugation at 10,000 × g for 10 min at 4°C, the pellets were washed with 1 ml of acetone (Wako) and centrifuged (10,000 × g for 10 min at 4°C). The pellets were suspended in 60 μl of SDS sample buffer (125 mM Tris-HCl [pH 6.8], 4% [wt/vol] SDS, 20% [wt/vol] glycerol, 10% [vol/vol] 2-mercaptoethanol) and used as concentrated (10×) culture supernatant fractions.
Purification of recombinant SasG.
His-SasG was overexpressed from pCold-SasG in E. coli BLR(DE3) cells (Table S1), which were grown at 30°C in 1 liter of LB medium containing 100 μg/ml ampicillin. Expression of His-SasG was induced by the addition of isopropyl β-d-1-thiogalactopyranoside (1 mM) and incubation at 15°C for 24 h. Cells were harvested by centrifugation and resuspended in 50 ml of buffer A (20 mM Tris-HCl [pH 8.0] and 300 mM NaCl) supplemented with a protease inhibitor cocktail (Nacalai Tesque). After sonication on ice, cell lysates were centrifuged at 8,500 × g for 30 min at 4°C, and the supernatant was loaded onto a 2-ml bed volume of Talon resin (TaKaRa) that had been washed with buffer A supplemented with 5 mM imidazole. Recombinant proteins were eluted using 250 mM imidazole. Eluted fractions were dialyzed against buffer B (20 mM Tris-HCl [pH 8.0], 1 mM dithiothreitol, and 20% [wt/vol] glycerol) using Slide-A-Lyzer dialysis cassettes (Thermo Fisher, Waltham, MA) and purified by chromatography using a HiTrap Q column (GE Healthcare, Pittsburgh, PA) and a 0 to 1 M NaCl gradient in buffer C (20 mM Tris-HCl [pH 8.0], 1 mM dithiothreitol, and 10% [wt/vol] glycerol). Purified His-SasG was pooled and quantified using a Bradford assay kit (Bio-Rad Laboratories, Hercules, CA).
Characterization of proteins in ECM, cell wall, and culture supernatant fractions.
The amount of proteins in the ECM fractions was standardized to the wet weight of bacterial pellets before ECM fraction isolation. Protein concentrations in the cell wall fractions were determined using the Pierce protein assay reagent (Thermo Fisher). Standardized amounts of proteins were resolved on 15% (wt/vol) polyacrylamide gels (Atto, Tokyo, Japan). After SDS-PAGE, the gels were stained with Coomassie brilliant blue (CBB) (Nacalai Tesque) or used for Western blotting.
Western blotting.
After SDS-PAGE, proteins were transferred to a polyvinylidene difluoride membrane using the iBlot 2 dry blotting system (Thermo Fisher). The membrane was treated for 30 min at 25°C with 1% (wt/vol) skimmed milk (Wako) dissolved in Tris-buffered saline composed of 10 mM Tris-HCl (pH 7.4) (Wako), 100 mM NaCl, and 0.1% (vol/vol) Tween 20 (TBS-T). After gentle washing with TBS-T, the membrane was probed with anti-SasG primary rabbit polyclonal antibody (developed by Eurofins Genomics [Tokyo, Japan] using purified His-SasG as the antigen), diluted 5,000 times in CanGet signal 1 (Toyobo), for 1 h at 25°C. The membrane was washed twice with TBS-T and subsequently incubated with a secondary goat anti-rabbit IgG antibody conjugated with horseradish peroxidase (Bio-Rad Laboratories), diluted 100,000 times in CanGet signal 2 (Toyobo), for 1 h at 25°C. After washing three times with TBS-T, the signal was detected using the ECL prime Western blotting detection reagent (GE Healthcare) and the LAS-4000 image analyzer (GE Healthcare).
Gel shift assay.
Purified SasG (0.1, 0.5, and 1 μM) was mixed with purified lambda DNA (15 μg/ml; TaKaRa) in a buffer containing 0.5 mM Tris-HCl (pH 8.0) and 0.05 mM EDTA. After incubation at 25°C for 1 h, the samples were analyzed by agarose gel electrophoresis with ethidium bromide staining.
DNA protection assay.
Purified lambda DNA (15 μg/ml; TaKaRa) was preincubated with or without purified SasG (1 μM) in a buffer containing 40 mM Tris-HCl (pH 7.9), 10 mM NaCl, 6 mM MgCl2, and 1 mM CaCl2 at 25°C for 30 min. DNase I (1 U/ml) was then added to the mixture. At the indicated time points, small aliquots of the mixture were taken, mixed with proteinase K (1 mg/ml), and incubated at 25°C for 10 min to digest DNase I and SasG. The residual DNA was analyzed by agarose gel electrophoresis. Band intensities were measured using the LAS-4000 image analyzer.
CLSM.
Bacterial cultures grown overnight in BHI medium at 37°C were diluted 1,000 times in BHIG medium and incubated at 37°C for 24 h on a glass-bottomed dish (35-mm diameter; Matsunami Glass, Osaka, Japan). Biofilms were fixed with 1% (wt/vol) glutaraldehyde for 10 min at 25°C (Wako). After glutaraldehyde removal, 50 mM ammonium chloride (Kanto Chemical, Tokyo, Japan) was added to quench the residual glutaraldehyde. The fixed biofilms were stained with 25 μM thioflavin T (AAT Bioquest, Sunnyvale, CA), which binds to bacterial RNA and is used to visualize bacterial cells (64). Three-dimensional biofilm structures were observed using an LSM880 confocal laser scanning microscope with a 63× oil lens objective (Carl Zeiss, Oberkochen, Germany). Thioflavin T fluorescence was detected using excitation at 458 nm and emission at 470 to 510 nm. All z-sections were collected at 0.25-μm intervals, and three-dimensional structures were reconstructed using the free microscope software ZEN for Zeiss microscopy (Carl Zeiss).
Evaluation of S. aureus pathogenicity in the silkworm model.
Fifth-instar silkworm larvae of B. mori (S30 × xe5) were obtained from the Institute of Genetic Resources Faculty of Agriculture (Kyusyu University, Fukuoka, Japan). To prepare bacterial suspensions, 700 μl of cultures grown overnight was centrifuged at 8,000 × g for 5 min at 25°C. The pellets were washed with 700 μl of a 0.6% NaCl solution, centrifuged at 8,000 × g for 5 min at 25°C, and resuspended in 700 μl of a 0.6% NaCl solution. Next, 50 μl of bacterial suspensions was injected (1 × 107 CFU) into the hemolymph through the dorsal vessel using a 30-gauge syringe (Becton, Dickinson). Pressure was immediately applied to the injection site for 20 s to stop leakage of the body fluid. As a control, larvae were injected with 50 μl of a 0.6% NaCl solution. Larval survival was observed at 25°C for 60 h without feeding.
To evaluate bacterial survival in silkworm larvae, the rear legs of injected silkworms were cut with scissors, and the body fluid was collected 24 h after injection of bacteria. After 10-fold serial dilution, samples were spread on mannitol salt agar plates selective for S. aureus and incubated at 37°C overnight, and the colonies were counted.
Statistical analyses.
Statistical analysis of biofilm biomass was performed using EZR software (65). Analysis of variance (ANOVA) and Student’s t test were used to assess significant differences in biofilm formation between bacterial strains and in enzyme susceptibility between treatments. For multiple-group comparisons, Kruskal-Wallis and Mann-Whitney U tests with Bonferroni correction were used to determine whether any of the groups exhibited statistically significant different thicknesses of biofilms. The log rank test was used to assess significant differences in the pathogenicities of bacterial strains in the silkworm model. For all statistical analyses, a P value of <0.05 was considered significant.
Accession number(s).
Nucleotide sequences of eap and sasG from S. aureus MR23 determined in the present study have been deposited in the DDBJ database under the accession numbers LC388386 and LC388387, respectively.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge T. Bae for providing pKOR1, L. Cui for gifting pLC1, Anne-Aurelie Lopes for critical reading of the manuscript, and all members of the Department of Bacteriology in Jikei University for stimulating discussions.
This work was supported in part by a grant-in-aid for young scientists (A) from the JSPS (number 15H05619 to S.S.), a grant-in-aid for scientific research (B) from the JSPS (number 26293100 to Y.M.), a grant from the MEXT-Supported Program for the Strategic Research Foundation at Private Universities, 2012–2016 (to Y.M.), and JST ERATO (number JPMJER1502 to S.S.).
We declare no conflicts of interest.
A.C., S.S., and Y.M. planned the project. K.Y., A.C., and S.S. designed the experiments. K.Y., A.C., and S.S. performed the experiments and analyzed the data. C.S. developed nanogold and heavy metal labeling for biofilms and assisted during ASEM analysis. K.Y., A.C., and S.S. wrote the paper, with input from M.S., Y.K., K.M., and Y.M.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00894-18.
REFERENCES
- 1.Flemming H-C, Neu TR, Wozniak DJ. 2007. The EPS matrix: the “house of biofilm cells.” J Bacteriol 189:7945–7947. doi: 10.1128/JB.00858-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hall-Stoodley L, Costerton JW, Stoodley P. 2004. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2:95–108. doi: 10.1038/nrmicro821. [DOI] [PubMed] [Google Scholar]
- 3.Costerton JW, Stewart PS, Greenberg EP. 1999. Bacterial biofilms: a common cause of persistent infections. Science 284:1318–1322. doi: 10.1126/science.284.5418.1318. [DOI] [PubMed] [Google Scholar]
- 4.Lebeaux D, Ghigo J-M, Beloin C. 2014. Biofilm-related infections: bridging the gap between clinical management and fundamental aspects of recalcitrance toward antibiotics. Microbiol Mol Biol Rev 78:510–543. doi: 10.1128/MMBR.00013-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mah T-FC, O’Toole GA. 2001. Mechanisms of biofilm resistance to antimicrobial agents. Trends Microbiol 9:34–39. doi: 10.1016/S0966-842X(00)01913-2. [DOI] [PubMed] [Google Scholar]
- 6.Römling U, Balsalobre C. 2012. Biofilm infections, their resilience to therapy and innovative treatment strategies. J Intern Med 272:541–561. doi: 10.1111/joim.12004. [DOI] [PubMed] [Google Scholar]
- 7.Kluytmans J, van Belkum A, Verbrugh H. 1997. Nasal carriage of Staphylococcus aureus: epidemiology, underlying mechanisms, and associated risks. Clin Microbiol Rev 10:505–520. doi: 10.1128/CMR.10.3.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wertheim HF, Melles DC, Vos MC, van Leeuwen W, van Belkum A, Verbrugh HA, Nouwen JL. 2005. The role of nasal carriage in Staphylococcus aureus infections. Lancet Infect Dis 5:751–762. doi: 10.1016/S1473-3099(05)70295-4. [DOI] [PubMed] [Google Scholar]
- 9.Arciola CR, Campoccia D, Speziale P, Montanaro L, Costerton JW. 2012. Biofilm formation in Staphylococcus implant infections. A review of molecular mechanisms and implications for biofilm-resistant materials. Biomaterials 33:5967–5982. doi: 10.1016/j.biomaterials.2012.05.031. [DOI] [PubMed] [Google Scholar]
- 10.Tong SYC, Davis JS, Eichenberger E, Holland TL, Fowler VG. 2015. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharya M, Wozniak DJ, Stoodley P, Hall-Stoodley L. 2015. Prevention and treatment of Staphylococcus aureus biofilms. Expert Rev Anti Infect Ther 13:1499–1516. doi: 10.1586/14787210.2015.1100533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flemming H-C, Wingender J. 2010. The biofilm matrix. Nat Rev Microbiol 8:623–633. doi: 10.1038/nrmicro2415. [DOI] [PubMed] [Google Scholar]
- 13.O’Neill E, Pozzi C, Houston P, Smyth D, Humphreys H, Robinson DA, O’Gara JP. 2007. Association between methicillin susceptibility and biofilm regulation in Staphylococcus aureus isolates from device-related infections. J Clin Microbiol 45:1379–1388. doi: 10.1128/JCM.02280-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. 2002. Extracellular DNA required for bacterial biofilm formation. Science 295:1487. doi: 10.1126/science.295.5559.1487. [DOI] [PubMed] [Google Scholar]
- 15.Mann EE, Rice KC, Boles BR, Endres JL, Ranjit D, Chandramohan L, Tsang LH, Smeltzer MS, Horswill AR, Bayles KW. 2009. Modulation of eDNA release and degradation affects Staphylococcus aureus biofilm maturation. PLoS One 4:e5822. doi: 10.1371/journal.pone.0005822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Das T, Sehar S, Manefield M. 2013. The roles of extracellular DNA in the structural integrity of extracellular polymeric substance and bacterial biofilm development. Environ Microbiol Rep 5:778–786. doi: 10.1111/1758-2229.12085. [DOI] [PubMed] [Google Scholar]
- 17.Cramton SE, Ulrich M, Gotz F, Doring G. 2001. Anaerobic conditions induce expression of polysaccharide intercellular adhesin in Staphylococcus aureus and Staphylococcus epidermidis. Infect Immun 69:4079–4085. doi: 10.1128/IAI.69.6.4079-4085.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vergara-Irigaray M, Maira-Litrán T, Merino N, Pier GB, Penadés JR, Lasa I. 2008. Wall teichoic acids are dispensable for anchoring the PNAG exopolysaccharide to the Staphylococcus aureus cell surface. Microbiology 154:865–877. doi: 10.1099/mic.0.2007/013292-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arciola CR, Campoccia D, Ravaioli S, Montanaro L. 2015. Polysaccharide intercellular adhesin in biofilm: structural and regulatory aspects. Front Cell Infect Microbiol 5:7. doi: 10.3389/fcimb.2015.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.O’Gara JP. 2007. ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol Lett 270:179–188. doi: 10.1111/j.1574-6968.2007.00688.x. [DOI] [PubMed] [Google Scholar]
- 21.Hussain M, von Eiff C, Sinha B, Joost I, Herrmann M, Peters G, Becker K. 2008. eap gene as novel target for specific identification of Staphylococcus aureus. J Clin Microbiol 46:470–476. doi: 10.1128/JCM.01425-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haggar A, Hussain M, Lonnies H, Herrmann M, Norrby-Teglund A, Flock J-I. 2003. Extracellular adherence protein from Staphylococcus aureus enhances internalization into eukaryotic cells. Infect Immun 71:2310–2317. doi: 10.1128/IAI.71.5.2310-2317.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chavakis T, Wiechmann K, Preissner KT, Herrmann M. 2005. Staphylococcus aureus interactions with the endothelium. The role of bacterial “secretable expanded repertoire adhesive molecules” (SERAM) in disturbing host defense systems. Thromb Haemost 94:278–285. doi: 10.1160/TH05-05-0306. [DOI] [PubMed] [Google Scholar]
- 24.Johnson M, Cockayne A, Morrissey JA. 2008. Iron-regulated biofilm formation in Staphylococcus aureus Newman requires ica and the secreted protein Emp. Infect Immun 76:1756–1765. doi: 10.1128/IAI.01635-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thompson KM, Abraham N, Jefferson KK. 2010. Staphylococcus aureus extracellular adherence protein contributes to biofilm formation in the presence of serum. FEMS Microbiol Lett 305:143–147. doi: 10.1111/j.1574-6968.2010.01918.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foster TJ, Geoghegan JA, Ganesh VK, Höök M. 2014. Adhesion, invasion and evasion: the many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol 12:49–62. doi: 10.1038/nrmicro3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazmanian SK, Ton-That H, Schneewind O. 2001. Sortase-catalysed anchoring of surface proteins to the cell wall of Staphylococcus aureus. Mol Microbiol 40:1049–1057. doi: 10.1046/j.1365-2958.2001.02411.x. [DOI] [PubMed] [Google Scholar]
- 28.Roche FM, Massey R, Peacock SJ, Day NPJ, Visai L, Speziale P, Lam A, Pallen M, Foster TJ. 2003. Characterization of novel LPXTG-containing proteins of Staphylococcus aureus identified from genome sequences. Microbiology 149:643–654. doi: 10.1099/mic.0.25996-0. [DOI] [PubMed] [Google Scholar]
- 29.Corrigan RM, Rigby D, Handley P, Foster TJ. 2007. The role of Staphylococcus aureus surface protein SasG in adherence and biofilm formation. Microbiology 153:2435–2446. doi: 10.1099/mic.0.2007/006676-0. [DOI] [PubMed] [Google Scholar]
- 30.Kuroda M, Ito R, Tanaka Y, Yao M, Matoba K, Saito S, Tanaka I, Ohta T. 2008. Staphylococcus aureus surface protein SasG contributes to intercellular autoaggregation of Staphylococcus aureus. Biochem Biophys Res Commun 377:1102–1106. doi: 10.1016/j.bbrc.2008.10.134. [DOI] [PubMed] [Google Scholar]
- 31.Geoghegan JA, Corrigan RM, Gruszka DT, Speziale P, O’Gara JP, Potts JR, Foster TJ. 2010. Role of surface protein SasG in biofilm formation by Staphylococcus aureus. J Bacteriol 192:5663–5673. doi: 10.1128/JB.00628-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Conrady DG, Brescia CC, Horii K, Weiss AA, Hassett DJ, Herr AB. 2008. A zinc-dependent adhesion module is responsible for intercellular adhesion in staphylococcal biofilms. Proc Natl Acad Sci U S A 105:19456–19461. doi: 10.1073/pnas.0807717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Formosa-Dague C, Speziale P, Foster TJ, Geoghegan JA, Dufrêne YF. 2016. Zinc-dependent mechanical properties of Staphylococcus aureus biofilm-forming surface protein SasG. Proc Natl Acad Sci U S A 113:410–415. doi: 10.1073/pnas.1519265113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sugimoto S, Sato F, Miyakawa R, Chiba A, Onodera S, Hori S, Mizunoe Y. 2018. Broad impact of extracellular DNA on biofilm formation by clinically isolated methicillin-resistant and -sensitive strains of Staphylococcus aureus. Sci Rep 8:2254. doi: 10.1038/s41598-018-20485-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sugimoto S, Iwamoto T, Takada K, Okuda K, Tajima A, Iwase T, Mizunoe Y. 2013. Staphylococcus epidermidis Esp degrades specific proteins associated with Staphylococcus aureus biofilm formation and host-pathogen interaction. J Bacteriol 195:1645–1655. doi: 10.1128/JB.01672-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 37.Bae T, Schneewind O. 2006. Allelic replacement in Staphylococcus aureus with inducible counter-selection. Plasmid 55:58–63. doi: 10.1016/j.plasmid.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 38.Lévesque CM, Voronejskaia E, Huang YC, Mair RW, Ellen RP, Cvitkovitch DG. 2005. Involvement of sortase anchoring of cell wall proteins in biofilm formation by Streptococcus mutans. Infect Immun 73:3773–3777. doi: 10.1128/IAI.73.6.3773-3777.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Franciosa G, Maugliani A, Scalfaro C, Floridi F, Aureli P. 2009. Expression of internalin A and biofilm formation among Listeria monocytogenes clinical isolates. Int J Immunopathol Pharmacol 22:183–193. doi: 10.1177/039463200902200121. [DOI] [PubMed] [Google Scholar]
- 40.Reardon-Robinson ME, Wu C, Mishra A, Chang C, Bier N, Das A, Ton-That H. 2014. Pilus hijacking by a bacterial coaggregation factor critical for oral biofilm development. Proc Natl Acad Sci U S A 111:3835–3840. doi: 10.1073/pnas.1321417111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sanchez CJ, Shivshankar P, Stol K, Trakhtenbroit S, Sullam PM, Sauer K, Hermans PWM, Orihuela CJ. 2010. The pneumococcal serine-rich repeat protein is an intraspecies bacterial adhesin that promotes bacterial aggregation in vivo and in biofilms. PLoS Pathog 6:e1001044. doi: 10.1371/journal.ppat.1001044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rohde H, Burdelski C, Bartscht K, Hussain M, Buck F, Horstkotte MA, Knobloch JKM, Heilmann C, Herrmann M, Mack D. 2005. Induction of Staphylococcus epidermidis biofilm formation via proteolytic processing of the accumulation-associated protein by staphylococcal and host proteases. Mol Microbiol 55:1883–1895. doi: 10.1111/j.1365-2958.2005.04515.x. [DOI] [PubMed] [Google Scholar]
- 43.Kreiswirth BN, Löfdahl S, Betley MJ, O’Reilly M, Schlievert PM, Bergdoll MS, Novick RP. 1983. The toxic shock syndrome exotoxin structural gene is not detectably transmitted by a prophage. Nature 305:709–712. doi: 10.1038/305709a0. [DOI] [PubMed] [Google Scholar]
- 44.Eisenbeis J, Saffarzadeh M, Peisker H, Jung P, Thewes N, Preissner KT, Herrmann M, Molle V, Geisbrecht BV, Jacobs K, Bischoff M. 2018. The Staphylococcus aureus extracellular adherence protein Eap is a DNA binding protein capable of blocking neutrophil extracellular trap formation. Front Cell Infect Microbiol 8:235. doi: 10.3389/fcimb.2018.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sugimoto S, Okuda K, Miyakawa R, Sato M, Arita-Morioka K, Chiba A, Yamanaka K, Ogura T, Mizunoe Y, Sato C. 2016. Imaging of bacterial multicellular behaviour in biofilms in liquid by atmospheric scanning electron microscopy. Sci Rep 6:25889. doi: 10.1038/srep25889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kaito C, Akimitsu N, Watanabe H, Sekimizu K. 2002. Silkworm larvae as an animal model of bacterial infection pathogenic to humans. Microb Pathog 32:183–190. doi: 10.1006/mpat.2002.0494. [DOI] [PubMed] [Google Scholar]
- 47.Geisbrecht BV, Hamaoka BY, Perman B, Zemla A, Leahy DJ. 2005. The crystal structures of EAP domains from Staphylococcus aureus reveal an unexpected homology to bacterial superantigens. J Biol Chem 280:17243–17250. doi: 10.1074/jbc.M412311200. [DOI] [PubMed] [Google Scholar]
- 48.Decker R, Burdelski C, Zobiak M, Büttner H, Franke G, Christner M, Saß K, Zobiak B, Henke HA, Horswill AR, Bischoff M, Bur S, Hartmann T, Schaeffer CR, Fey PD, Rohde H. 2015. An 18 kDa scaffold protein is critical for Staphylococcus epidermidis biofilm formation. PLoS Pathog 11:e1004735. doi: 10.1371/journal.ppat.1004735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Merino N, Toledo-Arana A, Vergara-Irigaray M, Valle J, Solano C, Calvo E, Lopez JA, Foster TJ, Penadés JR, Lasa I. 2009. Protein A-mediated multicellular behavior in Staphylococcus aureus. J Bacteriol 191:832–843. doi: 10.1128/JB.01222-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyazaki S, Matsumoto Y, Sekimizu K, Kaito C. 2012. Evaluation of Staphylococcus aureus virulence factors using a silkworm model. FEMS Microbiol Lett 326:116–124. doi: 10.1111/j.1574-6968.2011.02439.x. [DOI] [PubMed] [Google Scholar]
- 51.Pohl K, Francois P, Stenz L, Schlink F, Geiger T, Herbert S, Goerke C, Schrenzel J, Wolz C. 2009. CodY in Staphylococcus aureus: a regulatory link between metabolism and virulence gene expression. J Bacteriol 191:2953–2963. doi: 10.1128/JB.01492-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saravia-Otten P, Müller HP, Arvidson S. 1997. Transcription of Staphylococcus aureus fibronectin binding protein genes is negatively regulated by agr and an agr-independent mechanism. J Bacteriol 179:5259–5263. doi: 10.1128/jb.179.17.5259-5263.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xiong Y-Q, Bayer AS, Yeaman MR, Van Wamel W, Manna AC, Cheung AL. 2004. Impacts of sarA and agr in Staphylococcus aureus strain Newman on fibronectin-binding protein A gene expression and fibronectin adherence capacity in vitro and in experimental infective endocarditis. Infect Immun 72:1832–1836. doi: 10.1128/IAI.72.3.1832-1836.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolz C, McDevitt D, Foster TJ, Cheung AL. 1996. Influence of agr on fibrinogen binding in Staphylococcus aureus Newman. Infect Immun 64:3142–3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rom JS, Atwood DN, Beenken KE, Meeker DG, Loughran AJ, Spencer HJ, Lantz TL, Smeltzer MS. 2017. Impact of Staphylococcus aureus regulatory mutations that modulate biofilm formation in the USA300 strain LAC on virulence in a murine bacteremia model. Virulence 8:1776–1790. doi: 10.1080/21505594.2017.1373926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferreira FA, Souza RR, de Sousa Moraes B, de Amorim Ferreira AM, Américo MA, Fracalanzza SEL, dos Santos Silva Couceiro JN, Sá Figueiredo AM. 2013. Impact of agr dysfunction on virulence profiles and infections associated with a novel methicillin-resistant Staphylococcus aureus (MRSA) variant of the lineage ST1-SCCmec IV. BMC Microbiol 13:93. doi: 10.1186/1471-2180-13-93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Houston P, Rowe SE, Pozzi C, Waters EM, O’Gara JP. 2011. Essential role for the major autolysin in the fibronectin-binding protein-mediated Staphylococcus aureus biofilm phenotype. Infect Immun 79:1153–1165. doi: 10.1128/IAI.00364-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mainiero M, Goerke C, Geiger T, Gonser C, Herbert S, Wolz C. 2010. Differential target gene activation by the Staphylococcus aureus two-component system saeRS. J Bacteriol 192:613–623. doi: 10.1128/JB.01242-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Harraghy N, Kormanec J, Wolz C, Homerova D, Goerke C, Ohlsen K, Qazi S, Hill P, Herrmann M. 2005. sae is essential for expression of the staphylococcal adhesins Eap and Emp. Microbiology 151:1789–1800. doi: 10.1099/mic.0.27902-0. [DOI] [PubMed] [Google Scholar]
- 60.Chiba A, Sugimoto S, Sato F, Hori S, Mizunoe Y. 2015. A refined technique for extraction of extracellular matrices from bacterial biofilms and its applicability. Microb Biotechnol 8:392–403. doi: 10.1111/1751-7915.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cui L, Neoh H, Iwamoto A, Hiramatsu K. 2012. Coordinated phenotype switching with large-scale chromosome flip-flop inversion observed in bacteria. Proc Natl Acad Sci U S A 109:E1647–E1656. doi: 10.1073/pnas.1204307109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schenk S, Laddaga RA. 1992. Improved method for electroporation of Staphylococcus aureus. FEMS Microbiol Lett 94:133–138. doi: 10.1111/j.1574-6968.1992.tb05302.x. [DOI] [PubMed] [Google Scholar]
- 63.Kaplan JB, Ragunath C, Ramasubbu N, Fine DH. 2003. Detachment of Actinobacillus actinomycetemcomitans biofilm cells by an endogenous β-hexosaminidase activity. J Bacteriol 185:4693–4698. doi: 10.1128/JB.185.16.4693-4698.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sugimoto S, Arita-Morioka K, Mizunoe Y, Yamanaka K, Ogura T. 2015. Thioflavin T as a fluorescence probe for monitoring RNA metabolism at molecular and cellular levels. Nucleic Acids Res 43:e92. doi: 10.1093/nar/gkv338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kanda Y. 2013. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant 48:452–458. doi: 10.1038/bmt.2012.244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.