

Abstract
The missing phenylalanine at position 508, located in nucleotide-binding domain (NBD1) of the cystic fibrosis transmembrane regulator (CFTR), is the most common cystic fibrosis mutation. Severe disease-causing mutations also occur in NBD2. To provide information on potential therapeutic strategies for mutations in NBD2, we used a combination of biochemical, cell biological and electrophysiological approaches and newly created cell lines to study two disease-causing NBD2 mutants, N1303K and S1235R. We observed that neither was sensitive to E64, a cysteine protease inhibitor. However, further investigation showed that when treated with a combination of correctors, C4 + C18, both mutants also responded to E64. Further exploration to assess aggresome throughput using the autophagy regulator LC3 as a marker showed that, in the absence of correctors, N1303K showed a stalled throughput of LC3-II to the aggresome. The throughput became active again after treatment with the corrector combination C4 + C18. Confocal microscopic studies showed that the N1303K and S1235R mutant proteins both co-localized with LC3, but this co-localization was abolished by the corrector combination and, to a lesser extent, by VX-809. Both the corrector combination and VX-809 increased the CFTR chloride channel function of both mutants. We conclude that correctors have a dual effect, particularly on N1303K: they improve trafficking and function at the plasma membrane and reduce the association with autophagosomes. After treatment with correctors persistent degradation by the autophagosome may limit restoration of function. Thus, mutations in NBD2 of CFTR, in contrast to ΔF508-CFTR, may require additional personalized strategies to rescue them.
Keywords: Autophagy, Mutations, CFTR, Degradation, Correctors, Western blot, Short circuit current
1. Introduction
One of the best-studied genetic disorders, cystic fibrosis (CF), is caused by mutations in the CF transmembrane conductance regulator (CFTR), an ATP-binding cassette protein (ABC, sub-family C, member 7) composed of two transmembrane domains, two nucleotide-binding domains, and a unique regulatory domain [1]. The most common clinical manifestations of CF are recurrent pulmonary infection, pancreatic insufficiency, diabetes mellitus, and male infertility [2].
Over 2000 disease-causing mutations in the CFTR have been described; the most common is a deletion of phenylalanine at position 508 (ΔF508) in the NBD1 domain [3], which results in severe CF. ΔF508-CFTR, located in NBD1, is well-known as a Class II mutation. Class II mutations are misfolded and misprocessed, leading to premature degradation in the ER [4]. ΔF508-CFTR is retained in the ER, incompletely glycosylated, and subsequently degraded in the proteasome [5]. A number of chaperone molecules bind to ΔF508-CFTR and prevent it from aggregating [6] and facilitate its degradation in the proteasome.
Although this mutation was once thought to be uncorrectable, restoration of ΔF508-CFTR trafficking has been achieved. Early studies using phenylbutyrate showed rescue [7] of ΔF508-CFTR both in vitro and in patients [8–10]. More recently, VX-809, a small molecule identified from a high-throughput screen, was shown to rescue ΔF508-CFTR processing and trafficking [11]. This compound, known as lumacaftor, had only limited clinical efficacy when utilized alone in a Phase IIa clinical trial in ΔF508-homozygous patients [11]. However, Orkambi, a combination of lumacaftor with the potentiator VX-770 (Ivacaftor) [12], does produce therapeutic results and has achieved approval as a drug from the FDA [7].
Although there seems to be continued success with ΔF508-CFTR, the question arises as to whether therapies developed for mutations in NBD1 are also effective for use with mutations in NBD2. In a previous report, we focused on two NBD2 mutations, N1303K and S1235R. N1303K has a high incidence in Europe. For example, it is the second most common mutation in Italy (4%) and is even more frequently seen (7.8%) in the southwest of France [13, 14]. N1303K is classified as a severe mutation, causing pancreatic insufficiency and diabetes mellitus and associated with pneumothorax in the lungs [15]. In contrast, the incidence of S1235R is approximately 1% (3.3% in Eastern Europe), significantly higher than that of other rare mutations, but its symptoms are milder and mainly related to chronic pancreatitis, with sweat chloride test results that are on the borderline of what is considered to be typical of CF [16, 17].
We previously studied these two NBD2 mutants using a combination of biochemical approaches and newly created cell lines. We found that both of these mutants are processing mutants with unique characteristics. We previously observed [18] that unlike ΔF508-CFTR whose processing is enhanced when cells are grown at low temperature, the processing of N1303K and S1235R is profoundly different, although all three appear to be trafficking mutants. We did find however, that MG-132, the non-selective proteasome inhibitor [19], rescued the immature B and mature C bands of both N1303K and S1235R, indicating that degradation occurs via proteasomes. Thus, on one had the impact of the mutation on the trafficking of the disease causing mutation, N1303K, appeared to be more severe compared to ΔF508-CFTR, the protein could still be degraded by the proteasome.
Several years ago, Kopito and colleagues recognized that ΔF508-CFTR formed aggregates [20] and might actually interfere with proteasomal degradation. Aggregated proteins accumulate and trigger a process referred to as autophagy which means “self eating”. Autophagy is a process that clears the cells of cellular components and misfolded proteins that cannot be degraded by the proteasome [21]. Aggregated proteins move to the microtubule organizing center facilitated by histone deacetylase 6, HDAC6, where they form aggresomes [22]. These are processed by the phagophor to from autophagosomes, double-membrane structures that ultimately latter fuse with lysosomes to degrade the misfolded proteins [23]. Luciani and colleagues [24] shown that ΔF508-CFTR causes defects in autophagy which ultimately contribute to accumulation of protein aggregates and lung inflammation. To understand further how NBD2 mutants are processed we undertook this study to determine whether they are processed by autophagy.
2. Methods
2.1. Cell culture
CFBE41o− cells stably transfected with the NBD2 mutants (a gift of Dieter Gruenert [25]) were maintained in Eagle’s minimum essential medium (MEM, Invitrogen) with 10% FBS, penicillin/streptomycin, L-glutamate (200 mΜ), and puromycin (5 μg/mL, Sigma). To generate the stably transfected cells, WT, N1303K and S1235R were subcloned into the pcDNA5-FRT expression vector using Flp-In technology, to generate stably transfected cells as described previously [26]. Cells were cultured at 37° or 27 °C.
2.2. Treatment
Small-molecule correctors were applied for 16 h to assess their ability to rescue CFTR. C4 and C18 were obtained from the CFFT panel library (www.cftrfolding.org). VX-809 was purchased from Selleck Chemical, LLC. The compounds were used alone or in combinations of two. E64 was purchased from Calbiochem, and tubacin from Sigma Corporation.
2.3. Immunoblotting
The cells were harvested and solubilized in a lysis buffer described previously [27]. CFTR protein was detected with monoclonal anti-human CFTR antibody (217; 1:1000; provided by Dr. J. Riordan, Department of Biochemistry and Biophysics and Cystic Fibrosis Center of North Carolina). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), used as a loading control, was detected with a monoclonal antibody (1:10,000; US Biological). A rabbit polyclonal anti-human polyclonal antibody MAPILC3A/B was used to detect LC3 (BioRad). The protein expression levels were determined by Western blotting and quantified by densitometry.
2.4. Short-circuit currents
The short-circuit currents (Isc) were measured in Ussing chambers (Physiological Instruments; San Diego, CA) as described previously [28]. Confluent CF bronchial epithelial cells (CFBE) stably expressing the NBD2 mutants were seeded onto 12 mm-diameter Costar® Snapwell™ cell culture inserts (Corning Costar, Acton, MA; 3801) and cultured for 7 days to establish polarized monolayers. Inserts were mounted in a Ussing-type chamber and bathed in solutions maintained at 37 °C, as mentioned previously [28]. Data are expressed as the CFTRinh172 sensitive short-circuit current (ΔIsc), calculated by subtracting the Isc after CFTRinh172 treatment from the peak forskolin-genistein-stimulated Isc.
2.5. Microscopy
A Zeiss LSM 510 laser scanning system and 63× oil-immersion lens were used. CFBE41o-cells were seeded onto cover glasses. The following steps were done at room temperature to prepare slides for assessment: After two washes with cold DPBS, the cells were fixed with 4% paraformaldehyde for 15 min. Subsequently, the cells were permeabilized with 0.3% Triton X-100 in DPBS for 5–7 min and blocked with 3% bovine albumin serum (BSA) for 45 min. After blocking, the cells were washed once with DPBS and then incubated with primary antibody in 3% BSA for 1 h. N1303K or S1235R were detected using anti-CFTR antibody 769 and either AlexaFluor 488 goat anti-mouse or AlexaFluor 594 goat anti-mouse secondary antibody. LC3 was detected using an anti-LC3 antibody and either AlexaFluor 488 goat anti-rabbit or AlexaFluor 594 goat anti-rabbit secondary antibody. The cells were incubated with a 1:1000 dilution of the nuclear stain, DAPI for 5 min and washed three times with DPBS, then mounted using ProLong Gold Antifade (Invitrogen). All image processing was performed using Imaris Imaging.
2.6. Statistical assays
Statistical comparisons were made by using an unpaired Student’s t-test (GraphPad Software). All data are presented as means ± SEM. All measurements were done at least three times, and values were considered significant at P < 0.05.
3. Results
As a first step in understanding how N1303K is degraded, we created a CFBE41o− cell line stably expressing the mutant Fig. 1A, and then evaluated the steady-state levels of the mutant CFTR protein. Fig. 1A shows that neither the lysosomal inhibitor, E64 nor tubacin, the HDAC6 inhibitor [18], when applied alone, did not induce any changes in the steady-state levels of the N1303K product. These results suggest that the N1303K gene product is not likely to be degraded by autophagy. Next we studied the effect of two CFTR correctors, one class I and one class II: C18, developed by the Vertex Corp [29], and C4, identified by Pedemonte and collaborators [30]. C18 is a class I corrector that is thought to stabilize NBD1-TMD1/2 interfaces. C4 is assigned to class II and is hypothesized to restore the stability of NBD2 or its interfaces with other domains of CFTR [31]. Adding a combination of the two correctors produced a large increase in the steady-state levels of the immature B band and a small increase in C band of N1303K, indicating that the combined correctors C4 + C18 stabilized the B band and produces some processing to the mature C band. These data are identical to those that we have previously published for N1303K [18]. It is interesting, and important, to observe what happens when the trafficking to the aggresome and aggresome-mediated degradation are inhibited in the presence of the corrector combination. When either E64 was applied in the presence of the corrector combination (Fig. 1A), we saw a large increase in the steady-state level of the B band of N1303K. This sensitivity to the E64 suggests that in the presence of the correctors, N1303K protein is most likely degraded to a larger extent by the aggresomal degradation pathway after treatment. A smaller but not significant increase was also noted following tubacin treatment.
Fig. 1.
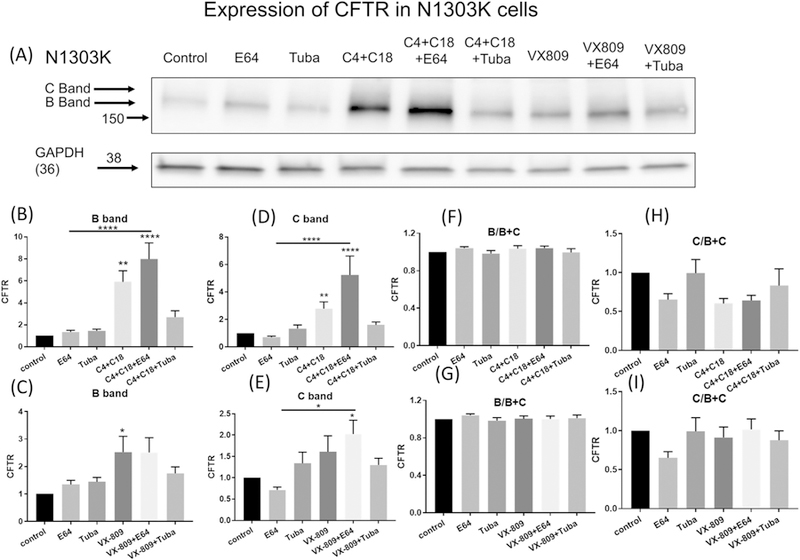
The degradation of N1303K. (1A) Western blot of CFBE41o− cells stably expressing N1303K represents the data using anti-CFTR antibody (lane 1). Note that N1303K protein is present mostly as the B band. E64 treatment alone (lane 2) has no effect on N1303K steady-state protein levels, suggesting that the lysosome does not play a role in the degradation of N1303K. Tubacin treatment alone (lane 3) also has no effect on N1303K steady-state protein levels. Note that the combination of correctors C4 + C18 increases band B and C bands of N1303K (lane 4) and shows considerable synergy when combined with E64 (lane 5), Interesting tubacin in combination with the correctors C3 + C4 reduces the steady state levels of N1303K compared to the combination treatment alone (lane 6). VX-809 treatment in contrast has very little effect on N1303K, either applied alone (lane 7) or in combination with E64 (lane 8) or tubacin (lane 9). Summary data are normalized to the absence of treatment (1C–I). (1B & C) depict the B and (1D & E) the C band summary data respectively. Data are also represented as the ratio of B/B + C (1F & G) or C/B + C (1H & I). Note that treatment with combination of correctors C4 + C18 alone or together with E64 increases both B and C band expression but the ratio of the B relative to the C band remains the same indicating that both increase proportionally. Methods: The CFBE cell line stably expressing N1303K protein was treated for 16 h with: C4 + C18 at 10 μM each, tubacin at 10 μM, or E64 at 10 μM. The protein expression level was determined by Western blotting and quantified of B band by densitometry. Data are expressed as the mean ± SEM of 3 independent experiments. Unpaired Student’s t-tests were performed for all experiments in the figures; *(P > 0.05) **(P > 0.01); ***(P > 0.001); ****(P > 0.0001).
Another Class II corrector, lumacaftor (VX-809), in combination with the potentiator Ivacaftor (VX-770), is on the market as an approved drug called Orkambi [7]. VX-809 alone increased the B band of N1303K (Fig. 1A). The steady-state levels of N1303K protein were further increased in the presence of both VX-809 and E64, but not in the presence of both VX-809 and tubacin. These data suggest that although the responses to VX-809 were similar, they were less marked (not statistically significant) than those produced by C4 + C18, the combination of correctors described in Fig. 1A suggesting as noted previously that a combination of correctors is required to get rescue of N1303K [18]. The data are summarized in Figs. 1B-I.
To further assess the role of autophagy, we evaluated the microtubule-associated light chain 3 protein (LC3) [32]. LC3 is rapidly processed by proteolytic digestion after its conserved glycine into the active cytosolic LC3-I protein, then modified further into LC3-II, which is located in the autophagosome membrane. Thus, the amount of LC3-II is a measure of the number of autophagosomes ([32, 33]). It is known that LC3-II not only associates with the autophagosomes but is degraded by them. Thus, the degree of autophagy is commonly assessed by measuring the increase in either the absolute amount of LC3-II or the ratio of LC3-II/I following inhibition of lysosomal degradation by the cysteine protease inhibitor E64 [33]. In our work, we also evaluated the effect of tubacin, the specific inhibitor of HDAC6. HDAC6, by binding to ubiquitinated proteins, facilitates degradation by the so-called “quality control autophagy” because it facilitates the degradation of severely misfolded proteins such as the N1303K protein. HDAC6 is involved in the fusion of autophagosomes to lysosomes [34, 35]; it still allows autophagosomes to form, but they cannot fuse with lysosomes, leading to an accumulation of LC3-II [35].
In order to assess whether autophagy is involved in N1303K protein degradation, we evaluated the ratio of LC3-II/I and the absolute amount of LC3-II. We saw no statistically significant increase in either the ratio of LC3-II/I or the absolute amount of LC3-II when E64 or tubacin was applied in the absence of the C4 + C18 corrector combination (Fig. 2A, B–C). Also, the corrector combination alone also had no statistically significant effect. In sharp contrast, was an increase in the ratio of LC3-II/I when the corrector combination was applied along with E64 or tubacin. This surprising result indicates that the rate of autophagy is slow in the CFBE 41o− cells containing N1303K. An increase in the ratio of LC3-II /I in the presence of the corrector combination + E64 or tubacin indicated that the correctors are increasing autophagy, which causes the increase in E64-dependent degradation of N1303K protein (Fig. 2A, D–E). Taken together, these data indicate that the combination of correctors, by increasing the level of the B band, makes N1303K more susceptible to degradation by autophagosomes and thus increases autophagy.
Fig. 2.
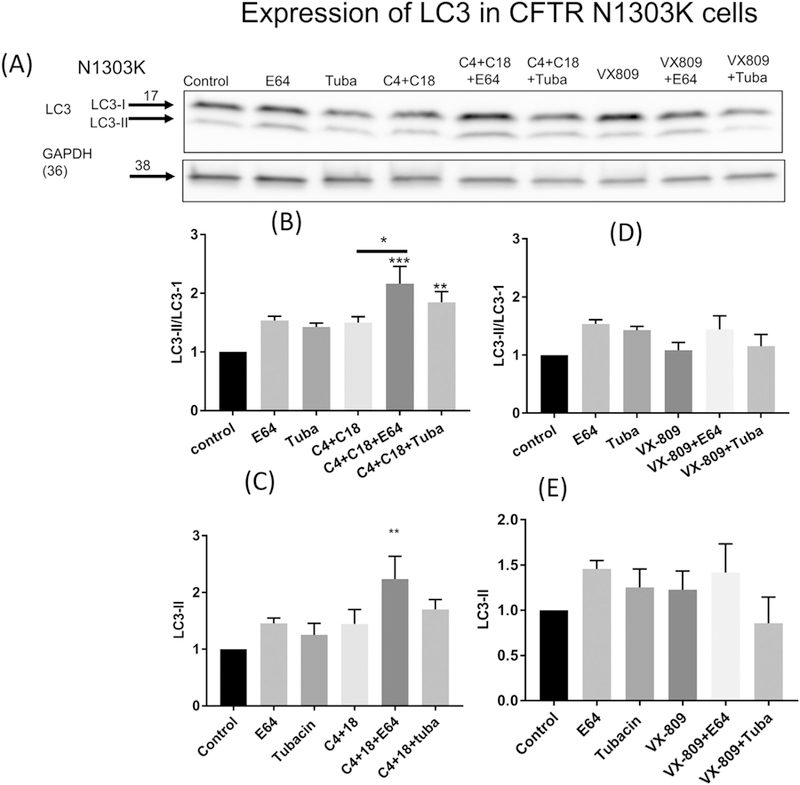
Autophagy and N1303K. (2A). Western blot of LC3 I&II measured in CFBE41o− cells stably expressing N1303K. Note that treatment with E64, tubacin or the combination of correctors C4 + C18 has little effect on the ratio of LC3II/I (2B) or absolute amount of LC3-II (lanes 1–4) (2C). In contrast, following treatment with the combination of correctors and E64 (lane 5) there is a significant increase in the LC3II/I ratio and absolute amount of LC3II and (2B–C). Tubacin treatment has a small effect in combination with C4 + C18 (Lane 6) (4B–C). In contrast VX-809 does not influence LC3II or the ratio either alone or in combination with E64 or tubacin (lanes 7–9) (2D–C). Data are normalized to the absence of treatment. Data are expressed as the mean ± SEM of 3 independent experiments. Methods as described for Fig. 1.
VX-809 alone and in the presence of E64 or tubacin did not cause an increase in the LC3-I/II ratio. Neither did VX-809 significantly increase the steady-state level of LC3-II when it was applied alone or in combination with tubacin (Fig. 2 D–E). These data indicate that VX-809 is not as effective in stimulating the flux of LC3 through autophagy as the combination of correctors.
Fig. 3A shows the effect on the milder mutation S1235R when stably expressed in CFBE 41o− cells. Consistent with S1235R being a mild mutation, there was a larger amount of mature C band in S1235R compared to B band. As was seen for N1303K, E64 alone had no effect, whereas tubacin alone increased the steady-state levels of S1235R protein. These data are in sharp contrast to the results observed in the presence of the correctors which alone increase the steady state levels of S1235R. E64 and tubacin, applied individually in the presence of the C4 + C18 corrector combination, resulted in a significant increases in the steady-state levels of the S1235R protein, suggesting the occurrence of lysosomal degradation in the presence of the correctors.
Fig. 3.
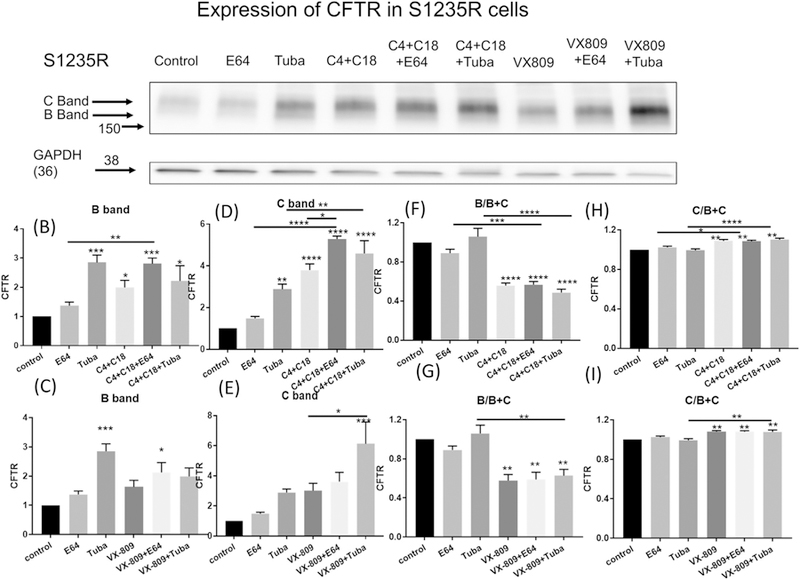
The degradation of S1235R. (3A) Western blot of CFBE41o− cells stably expressing S1235R represents the data using anti-CFTR antibody (lane 1). Note that S1235R protein is present most as the C band. E64 treatment alone (lane 2) has no effect on S1235R steady-state protein levels, suggesting that the lysosome does not play a role in the degradation of S1235R. Tubacin treatment alone (lane 3) increases S1235R steady-state protein levels. Note that the combination of correctors C4 + C18 increases band B and C of S1235R (lane 4) and shows considerable synergy when combined with E64 (lane 5), Interesting tubacin in combination with the correctors C3 + C4 increases the steady state levels of S1235R compared to the corrector combination treatment alone (lane 6). VX-809 treatment in contrast has a smaller effect on S1235R compared to the corrector combination C4 + C18 when applied alone (lane 7) but does show an increase in combination with E64 (lane 8) or tubacin (lane 9). Summary data are normalized to the absence of treatment (3B–I). (3B & C) depict the B and (3D & E) the C band summary data respectively. Data are also represented as the ratio of B/B + C (3F & G) or C/B + C (3H & I). Note that treatment with combination of correctors C4 + C18 alone or together with E64 or tubacin increases both B and C band expression and the ratio of the C relative to the total B + C bands indicating that an increase in processing of B to C bands. In contrast there was a decrease in the amount of B band relative to the total of B + C bands.
VX-809 has a similar pattern of effect on S1235R as the combination. However, the effects were smaller. All the data are summarized in Figs. 3B–I.
These results were considerably strengthened by our examination of the effect on the LC-II/I ratio and the absolute amount of LC3-II (Fig. 4 A–E). In contrast to N1303K, E64 or tubacin treatment in cells containing S1235R causes significant increases in the LC3-II/I ratio. E64 treatment also caused an increase in the absolute amount of LC3-II. The combination of correctors alone had no effect on either the LC3-II/I or absolute amount of LC3-II. However, the corrector combination, together with E64 caused a large increase in the LC3-II/I ratio and the absolute amount of LC3-II. However, the increase was not different than what is observed without correctors (first compare the magnitude of the change from control following E64 treatment in the absence of corrector treatment and then compare the magnitude of the same change following the corrector combination). Small effects were noted when the cells were treated with the combination of correctors + tubacin. Both the increase in the ratio of LC3-II/I and in the absolute amount of LC3-II when E64 was applied alone suggest that, unlike N1303K, the milder S1235R does not stall autophagy.
Fig. 4.
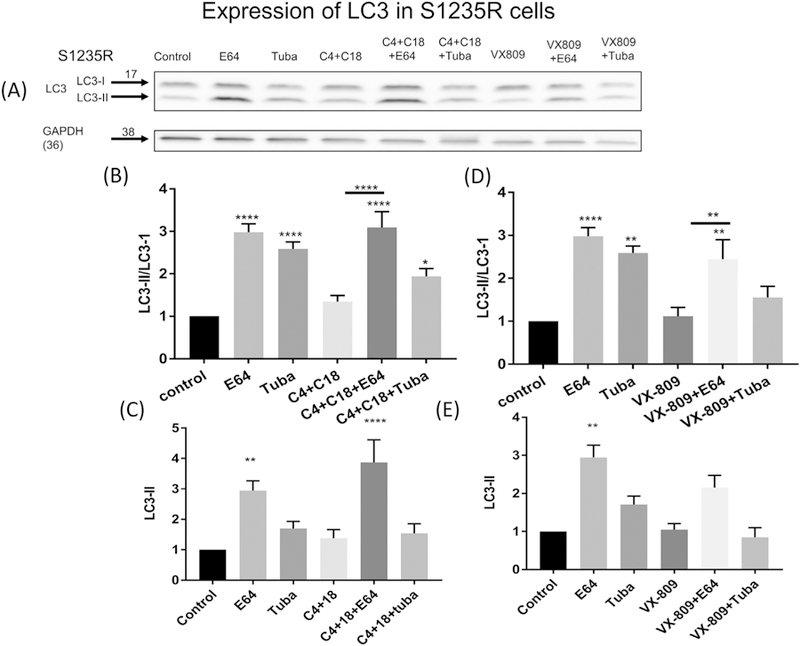
Autophagy and S1235R. (4A). Western blot of LC3 I&II measured in CFBE41o− cells stably expressing S1235R (Lane 1). Note that treatment with E64 (Lane 2), or tubacin (Lane 3) has an effect on the ratio of LC3II/I (4B & D) and on the absolute amount of LC3-II (4C & E) which is significant only for the E64 treatment. Treatment with the combination of C4 + C18 (Lane 4) had no significant effect either on the LC3II/I ratio (4B) or the absolute amount of LC3II (4C). In contrast, following treatment with the combination of correctors and E64 (lane 5) there is a significant increase in LC3II and the LC3II/I ratio (4B–D). Tubacin treatment has a small effect in combination with C4 + C18 (Lane 6). VX-809 has a similar albeit smaller effect compared to the combination of correctors (lane 6–8) (4C–D). Data are normalized to the absence of treatment Data are expressed as the mean ± SEM of 3 independent experiments. Methods as described for Fig. 1.
Treatment of VX809 alone also did not affect the LC3-II/I ratio (Fig.4A & D) or the amount of LC3-II (Fig. 4A & E). There was an increase in the LC3-II/I ratio (Fig. 4C) and the amount of LC3-II (Fig. 4D in the presence of both VX-809 and E64 not in the amount of LC3-II (Fig. 4E). However, the increase was not different than what is observed without VX-809. In contrast, there was no effect of the combination of VX-809 and tubacin over untreated controls (Fig. 4D & E). We conclude that VX-809 has a minor effect on autophagy in the presence of the S1235R mutation.
For comparison we studied CFBE41o− cells containing wt-CFTR. Supplemental Fig. 1A shows the protein expression of wt-CFTR when stably expressed in CFBE 41o− cells. Please note that direct comparison of band intensity among the gels expressing the mutants and wt-CFTR are not possible because band intensity of each gel is adjusted to give relatively consistent band intensities among the different experiments. Consistent with wt-CFTR, there was a larger amount of mature C band. E64 treatment alone had no effect, whereas tubacin alone increased the steady-state levels of wt protein. These data are in sharp contrast to the results observed in the presence of the correctors which alone increase the steady state levels of wt-CFTR. E64 and tubacin, applied individually in the presence of the C4 + C18 corrector combination, resulted in a significant increases in the steady-state levels of the wt-CFTR protein, suggesting the occurrence of lysosomal degradation in the presence of the correctors. VX-809 has a similar pattern of effect on wt-CFTR as the combination of correctors. All the data are summarized in Supplemental Figs. 1B–I.
Next we examined the LC-II/I ratio and the absolute amount of LC3-II (Supplemental Fig. 2A–E) in cells containing wt-CFTR. In contrast to N1303K but similar to S1235R, E64 or tubacin treatment in cells containing wt-CFTR causes significant increases in the LC3-II/I ratio. The combination of correctors alone did increase the LC3-II/I or absolute amount of LC3-II. The corrector combination, together with E64 caused an increase in the LC3-II/I ratio and the absolute amount of LC3-II. However, the increase was not more than that observed without the corrector combination (first compare the magnitude of the change from control following E64 treatment in the absence of corrector treatment and then compare the magnitude of the same change following the corrector combination). Increases were noted when the cells were treated with the combination of correctors + tubacin. Both the increase in the ratio of LC3-II/I and in the absolute amount of LC3-II when E64 was applied alone suggest that, unlike N1303K, the wt-CFTR does not stall autophagy.
Treatment of VX809 alone also did not affect the LC3-II/I ratio (Supplemental Fig. 2A & D) or the amount of LC3-II (Supplemental Fig. 2A & E). There was an increase in the LC3-II/I ratio (Supplemental Fig. 2D) and the amount of LC3-II (Supplemental Fig. 2E in the presence of both VX-809 and E64. However, the increase was not different than what is observed without corrector treatment (first compare the magnitude of the change from control following E64 treatment in the absence of corrector treatment and then compare the magnitude of the same change following the corrector combination). In contrast, there was no effect of the combination of VX-809 and tubacin over untreated controls (Supplemental Fig. 2D & E). We conclude that VX-809 has a minor effect on autophagy in cells containing wt-CFTR.
As a control experiment, we show the results of corrector treatment in the parental CFBE41o− cells which express ΔF508-CFTR but at barely detectable levels as a result none of the treatments has a detectable effect on the endogenous ΔF508-CFTR (Supplemental Fig. 3A–I). For comparison we again measured the LC3II/I ratio and the total amount of LC3-II (Supplemental Fig. 4A–E). Similar to wt containing cells, E64 or tubacin treatment in parental CFBE41o− cells cause significant increases in the LC3-II/I ratio. The combination of correctors alone did increase the LC3-II/I ratio. The corrector combination, together with E64 caused a further increase in the LC3-II/I ratio and the absolute amount of LC3-II. However, the increase was not different than what is observed without the correctors (first compare the magnitude of the change from control following E64 treatment in the absence of corrector treatment and then compare the magnitude of the same change following the corrector combination). Increases were noted when the cells were treated with the combination of correctors + tubacin. Both the increase in the ratio of LC3-II/I and in the absolute amount of LC3-II when E64 was applied alone suggest that, unlike N1303K, in the parental cells autophagy is still functioning.
Treatment of VX809 alone also did not affect the LC3-II/I ratio (Supplemental Fig. 4A & D) or the amount of LC3-II (Supplemental Fig. 4A & E). There was an increase in the LC3-II/I ratio (Supplemental Fig. 4C) but not the amount of LC3-II (Supplemental Fig. 4D in the presence of both VX-809 and E64 (Supplemental Fig. 4E). However, the increase was not different than what is observed without VX-809 (first compare the magnitude of the change from control following E64 treatment in the absence of corrector treatment and then compare the magnitude of the same change following the corrector combination). In contrast, there was no effect of the combination of VX-809 and tubacin over untreated controls (Supplemental Fig. 4D & E). We conclude that VX-809 has a minor effect on autophagy in the parental cells.
To take our investigation one-step further, we performed confocal experiments to evaluate the degree of co-localization between the NBD mutants and LC3 (as a marker for autophagosomes). Because LC3-II is associated with autophagosomes and LC3–1 is cytosolic, we argued that co-localization between LC3 and N1303K, a membrane protein, would represent to degree to which both proteins reside in the autophagsome membrane. Confocal microscopy indeed revealed the co-localization of N1303K with LC3 (Fig. 5A). Note that there was significant overlap in the localization of LC3 and N1303K in untreated cells. Fig. 5B shows that the co-localization was reduced by treatment with the combination of C4 + C18. We have shown previously that treatment of cells containing N1303K with the combination of correctors, C3 + C4, increases the surface expression of N1303K approximately 3-fold [18]. The published data showing that N1303K moves to the plasma membrane following correction are consistent with a reduction in its colocalization with LC3 we observe here following correction with the closely related combination, C4 + C18. Fig. 5C shows the effect of VX-809. As compared to the cells shown in Fig. 5B, there was still an overlap in the localization of N1303K and LC3 expression in the presence of VX-809 that was clearly lower than that observed in its absence.
Fig. 5.
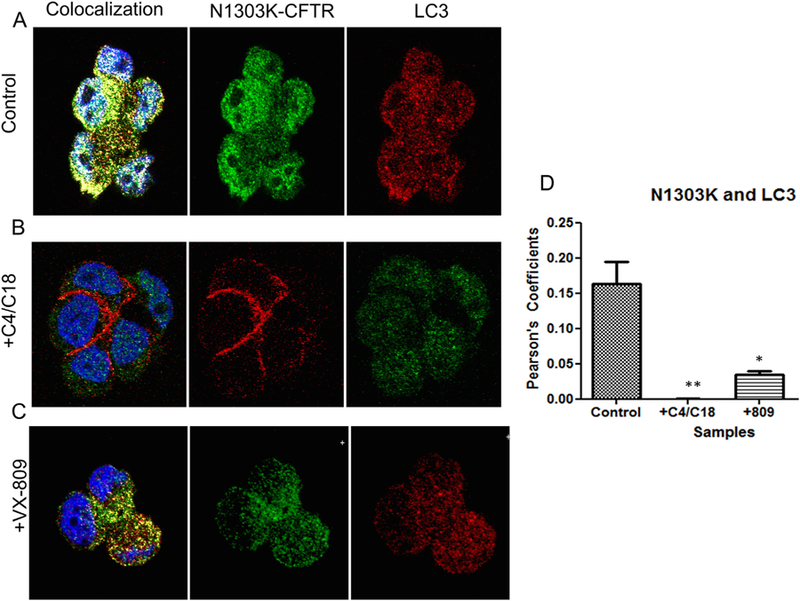
Co-localization of LC3 with N1303K CFTR. (5A). Confocal images depicting the individual staining of N1303K protein (middle panel) and LC3 (right panel). The left panel shows considerable overlap between the N1303K protein and LC3, indicating that the N1303K protein is highly associated with aggresomes. (5B) Confocal images depicting the individual staining of the N1303K protein (middle panel) and LC3 (right panel). The left panel shows almost no overlap between the N1303K protein and LC3 when the cells are treated with the corrector combination, indicating that the N1303K protein is no longer associated with aggresomes. (5C). Confocal images depicting the individual staining of the N1303K protein (middle panel) and LC3 (right panel) in cells treated with VX-809. The left panel shows less overlap between the N1303K protein and LC3, indicating that there is less N1303K protein associated with aggresomes after treatment. (5D) Summary data. In the control, there is significant overlap between the N1303K protein and LC3, suggesting that the N1303K protein is stalled in the aggresomes. The combination of correctors C4 + C18 relieves this stalling almost completely. VX-809, on the other hand, is not as effective, indicating that it is not fully adequate by itself to rescue N1303K (n = 3). Methods: CFBE 41o− cells stably expressing N1303K-CFTR were plated on coverslips. Cells were treated with the corrector combination of C4 and C18 or VX-809 for 12 h. Co-localization was established by the presence of a yellow signal and changes in associated Pearson’s coefficients (A–E). Nuclei are stained blue by the DAPI stain. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
The summary data are presented in Fig. 5D. In absence of C4 + C18, cells expressing N1303K displayed considerable co-localization of N1303K and LC3; on the other hand, treatment with this combination of correctors virtually abolished the overlap. A similar reduction occurred with VX-809 alone, although it was not as dramatic as that seen for the corrector combination. Given that cells expressing N1303K showed reduced degradation of LC3-II, indicative of defective autophagy (see Fig. 2), the large overlap with LC3 indicated that N1303K was highly associated with autophagosomes but could not be degraded by them. As a result, in the absence of correctors, autophagy was inhibited; LC3 and N1303K accumulated in the autophagosome membrane. On the other hand, the corrector combination C18 + C4 or VX-809 reduced the co-localization and increased both the steady-state levels of N1303K product and its degradation by lysosomes.
Next we conducted similar experiments on S1235R. Again, we saw a large amount of overlap in localization between S1235R and LC3, which was reduced dramatically by treating the cells with C4 + C18 (Fig. 6). Fig. 6C shows that treating the cells with VX-809 reduced the overlap, but not as effectively as did the corrector combination.
Fig. 6.
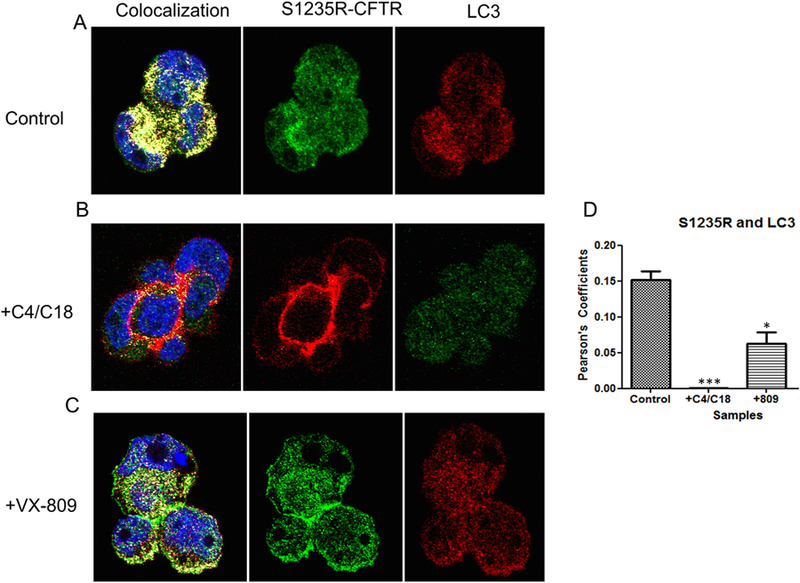
Co-localization of LC3 with S1235R CFTR. (6A). Confocal images depicting the individual staining of the S1235R protein (middle panel) and LC3 (right panel). The left panel shows considerable overlap between the S1235R protein and LC3, indicating that the S1235R protein is highly associated with aggresomes. (6B) Confocal images depicting the individual staining of the S1235R protein (middle panel) and LC3 (right panel). The left panel shows some overlap between the S1235R protein and LC3 when the cells are treated with the corrector combination, indicating that S1235R CFTR’s association with aggresomes is reduced by the corrector combination. (6C) Confocal images depict the individual staining of the S1235R protein (middle panel) and LC3 (right panel) in cells treated with VX-809. The left panel shows less overlap between the S1235R protein and LC3, indicating that there is less S1235R protein associated with the aggresomes after treatment (6D) Summary data (n = 3). In the control, there is significant overlap between the S1235R protein and LC3, suggesting that the S1235R protein is also associated with aggresomes. The combination of correctors relieves the overlap almost completely. X-809, on the other hand, is not as effective, indicating that it is not fully adequate by itself to rescue S1235R.
The summary data for S1235R are shown in Fig. 6D. There was a significant overlap in localization for LC3 and S1235R, which was surprising since this mutation is considered mild [36]. The overlap was reduced dramatically by treatment with C4 + C18, but less so by treatment with VX-809.
In order to evaluate the effect of the corrector compounds on CFTR function, we measured short-circuit currents for N1303K and S1236R in response to C4 + C18 and to VX-809. In the case of N1303K (Fig. 7), the corrector C4 or the combination of C4 + C18 increased the Isc by approximately 4-fold. In the case of S1235R, either the combination of C4 + C18 or VX-809 alone had a similar effect, increasing the Isc by approximately 3-fold.
Fig. 7.
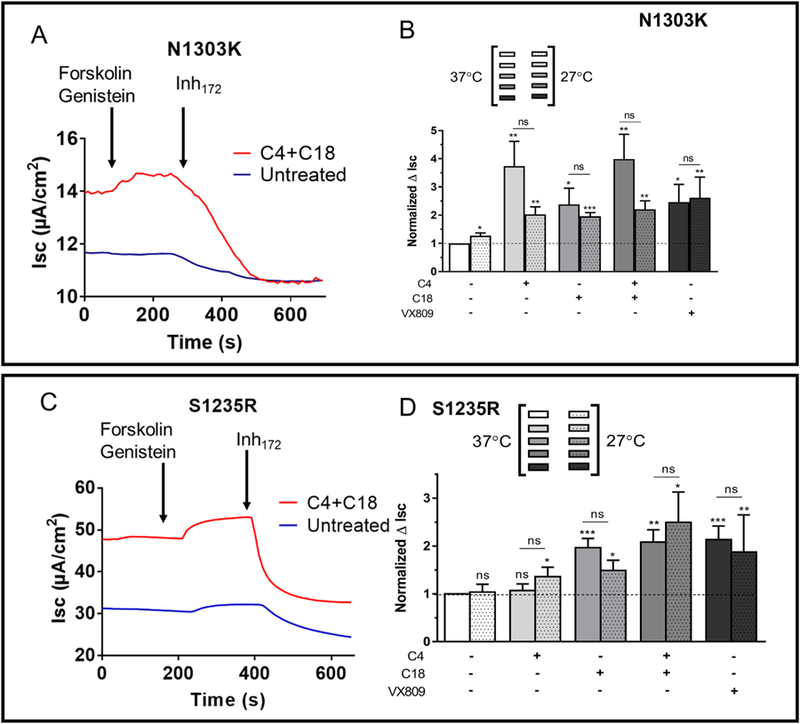
Ussing chamber experiments performed to evaluate rescue of N1303K and S1235R. (7A) Representative tracing of N1303K containing cells grown at 37 °C and treated with the combination of correctors C4 + C18. Cells were grown at 37 °C or 27 °C, as indicated. (7B) Summary of short-circuit currents in N1303K-containing untreated cells or after incubation with C4 + C18 or VX-809 (10 μM, 16 h), as indicated. From the left, data are normalized to the untreated control (bar graph, pair 1). Note that C1 + C18 combination corrector treatment results (bar graph, pair 4) are somewhat higher than when the correctors are applied individually, especially when compared to C18 treatment alone (bar graph, pairs 2 and 3). VX-809 alone is also less effective than the corrector combination (bar graph, pair 5). N1303K, however, is not rescued by growing the cells at low temperature. (7C) Representative tracing of S1235R containing cells grown at 37 °C and treated with the combination of correctors C4 + C18. (7D) Summary of short-circuit currents in S1235R-containing cells for untreated cells or after incubation with C4 + C18 or VX-809 (10 μM, 16 h), as indicated. From the left, data are normalized to the untreated control (bar graph, pair 1). Note that all the corrector treatments have less of an effect on S1235R than they do on N1303K. S1235R is also not rescued by growing the cells at low temperature. Y axis represents the ΔIsc following exposure of the cells to CFTR inh172 for the no treatment control divided into ΔIsc obtained in each of the other experimental procedures and thus reported as normalized to control. Statistical significance is presented as follows: ns, no significant difference; *P < 0.05; **P < 0.01; ***P < 0.001 (n = 8–13 for each condition) when compared with the control condition (n = 12). Amiloride (100 μM) was present during the whole experiment to avoid interference by ENaC-mediated Na+ currents.
Growing cells at a lower temperature did not increase the CFTR-dependent chloride currents for either N1303K or S1235R, nor did it increase the currents in response to the correctors. We have previously shown that neither the basal or the corrector-induced increase in the steady-state levels of the proteins is enhanced by low temperature [18].
4. Discussion
Several compounds have been developed to rescue ΔF508-CFTR, and one combination of a corrector and a potentiator, Orkambi, is already an approved drug [7]. The challenge is to extend the correction of ΔF508-CFTR to include other mutants. Our long-term goal is to understand how amino acid changes resulting from the genetic mutations other than ΔF508-CFTR translate into malfunctioning and mistrafficked proteins. We have focused here on N1303K, a severe disease-causing mutation [36] in NBD2. We compared it to a milder mutant, S1235R, in the same domain [3]. We show here that autophagy is altered both by the presence of N1303K mutants and by the correctors that are designed to rescue them.
One of the nagging questions regarding studies performed in vitro, such as this one, is whether the noted affects and resulting conclusions have in vivo relevance or whether they are merely caused by the overexpression of misfolded proteins in cell lines. To address this problem, we utilized a CFBE 41o− cell line that was originally derived from a CF patient [25] to stably express the two NBD2 mutants using Flp-In technology, which inserts exogenous cDNAs into chromosomal locations at low copy number. Therefore, as we have shown previously [18], the expression levels are low when compared to other cell lines derived from CFBE41o− expressing either wild-type or ΔF508 CFTR. However, the epithelium still forms a confluent monolayer on permeable supports and generates CFTR-dependent chloride currents.
We show here that the two NBD2 mutants associate avidly with the autophagosome and that N1303K stalls autophagy. It has been demonstrated previously that ΔF508-CFTR down-regulates beclin 1 and increases the expression of p62, resulting in defective autophagy [37, 38]. Beclin 1, a tumor suppressor protein, plays an essential role in autophagsome formation [39]. p62 (sequestosome-1) is a multifunctional protein with an ability to bind and regulate many cellular processes, including inhibition of autophagy [40]. Interestingly, restoring beclin 1 levels or reducing p62 levels rescued both autophagy and the trafficking of ΔF508-CFTR to the cell surface [37]. How restoring autophagy increases the trafficking of ΔF508-CFTR is unclear. One possible explanation is that inhibition of autophagy causes a misregulation of the cell’s ability to handle misfolded proteins by both the autophagsome and proteasome, leading to lung inflammation as suggested by Luciani and colleagues [37]. Thus, correcting defective autophagy might be expected to restore ΔF508-CFTR by increasing the cell’s ability to handle misfolded proteins.
It is well known that the immature B band of ΔF508-CFTR is rapidly degraded by the proteasome [41]. ΔF508-CFTR trafficking and processing is temperature-sensitive [28]. For example, growing cells at a reduced temperature leads to significant increases in the B band and maturation to the C band. However, when the temperature is restored to 37 °C, the C band of ΔF508-CFTR is unstable and is rapidly degraded by proteasomes [42]. Therefore, although cells containing ΔF508-CFTR exhibit defective autophagy, it is clear that their ER quality control and proteasomal degradation are operating to degrade the misfolded proteins.
N1303K and S1235R have characteristics that differ from those of ΔF508-CFTR. One notable difference is that their processing and trafficking are not temperature-sensitive. We have shown previously that the proteins produced by both mutants are degraded by proteasomes, suggesting that this pathway is working normally in cells bearing these NBD2 mutants [18]. However, we did notice that when the cells are treated with cycloheximide to inhibit protein synthesis, the B band of ΔF508-CFTR rapidly disappears, consistent with its inherent instability, but the immature B band of N1303K is remarkably stable [27]. We posit that the stability of the immature B band of N1303K represents its association with aggresomes. Given that autophagy is not occurring, the immature B band on N1303K remains un-degraded for an extended period.
We previously demonstrated [18] that application of a combination of correctors from Classes I and II increases the presence of the C band of N1303K by approximately four-fold and that of S1235R by approximately three-fold, in a pattern virtually identical to that for the increase in Isc noted in Fig. 7. The reason that there is less of an increase with S1235R is that there is already a large amount of mature C band at the surface prior to treatment. We showed that there are a large number of chaperones associated with ERAD that bind to N1303K protein, leading to its productive degradation by the proteasome. Treatment with the corrector combination of C4 + C18 considerably reduces chaperone binding. The net results of our previous experiments and those presented here indicate that the corrector combination can rescue N1303K by increasing its stability, reducing chaperone binding and degradation, and improving its chloride channel function.
The results presented here show that the N1303K and S1235R proteins associate less with the aggresome after treatment with the corrector combination than they do before treatment. These results are entirely consistent with our previous findings that showed an approximately 50% reduction in the binding of HDAC6 to the N1303K mutant protein [18]. Given that HDAC6 is involved in the transport of misfolded proteins to the aggresome [43], a reduction in the binding induced by the corrector combination is also indicative that the correctors are increasing N1303K protein stability.
The conundrum is that when treated with the corrector combination, the N1303K become more sensitive to E64 and lysosomal degradation via autophagy. This is another difference between N1303K and ΔF508-CFTR, in which the rescued protein is still degraded by mechanisms associated with the proteasome after trafficking occurs [42]. This increased lysosomal degradation explains why the corrector combination increases the residence time of the mature C band in cells treated with the corrector combination but still does not bring it close to that of wild-type CFTR.
The challenge in the future will be to develop therapeutically effective combined corrector cocktails, as shown here, that will be effective in treating patients bearing the most common CFTR mutations. Using a combination of correctors and growing cells at reduced temperature, we have shown that ΔF508-CFTR can be rescued to wild-type levels [28], suggesting that it is feasible to develop a corrector combination for the effective treatment of patients bearing the ΔF508-CFTR mutation. Our data here show that corrector combinations are certainly effective in rescuing N1303K. However, a further challenge is to develop effective treatment for trafficking mutants such as N1303K that engage the cell’s quality control mechanism in ways inherently different from ΔF508-CFTR.
Supplementary Material
Acknowledgments
The authors thank Dr. Deborah McClellan for editing the manuscript.
Funding
The U.S. Cystic Fibrosis Foundation (GUGGIN14XX0) and the NHLBI (R01 HL122267) funded this work.
Footnotes
Competing interests
The authors declare that W.B.G. has a consultant agreement with the Vertex Corporation. L.C. has a license agreement with the Vertex Corporation for mutant cell lines. Neither of these relationships influenced the work conducted in this manuscript.
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jcf.2018.05.016.
References
- [1].Fuller CM, Benos DJ. CFTR. Am J Physiol Cell Physiol 1992;263: C267–86. [DOI] [PubMed] [Google Scholar]
- [2].Boucher RC. An overview of the pathogenesis of cystic fibrosis lung disease. Adv Drug Deliv Rev 2002;54:1359–71. [DOI] [PubMed] [Google Scholar]
- [3].Sosnay PR, Castellani C, Corey M, Dorfman R, Zielenski J, Karchin R, et al. Evaluation of the disease liability of CFTR variants. Methods Mol Biol 2011;742:355–72. [DOI] [PubMed] [Google Scholar]
- [4].Riordan JR. Cystic fibrosis as a disease of misprocessing of the cystic fibrosis transmembrane conductance regulator glycoprotein. Am J Hum Genet 1999;64:1499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gregory RJ, Cheng SH, Rich DP, Marshall J, Paul S, Hehir K, et al. Expression and characterization of the cystic fibrosis transmembrane conductance regulator. Nature 1990;347:382–6. [DOI] [PubMed] [Google Scholar]
- [6].Skach WR. Pharmacological chaperoning: two ‘hits’ are better than one. Biochem J 2007;406:e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mayer M Lumacaftor-ivacaftor (Orkambi) for cystic fibrosis: behind the ‘breakthrough’. Evid Based Med 2016;21:83–6. [DOI] [PubMed] [Google Scholar]
- [8].Rubenstein RC, Egan ME, Zeitlin PL. In vitro pharmacologic restoration of CFTR-mediated chloride transport with sodium 4-phenylbutyrate in cystic fibrosis epithelial cells containing DeltaF508-CFTR. J Clin Invest 1997;100:2457–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rubenstein RC, Zeitlin PL. Use of protein repair therapy in the treatment of cystic fibrosis. Curr Opin Pediatr 1998;10:250–5. [DOI] [PubMed] [Google Scholar]
- [10].Rubenstein RC, Zeitlin PL. A pilot clinical trial of oral sodium 4-phenylbutyrate (Buphenyl) in deltaF508-homozygous cystic fibrosis patients: partial restoration of nasal epithelial CFTR function. Am J Respir Crit Care Med 1998;157:484–90. [DOI] [PubMed] [Google Scholar]
- [11].Van Goor F, Hadida S, Grootenhuis PD, Burton B, Stack JH, Straley KS, et al. Correction of the F508del-CFTR protein processing defect in vitro by the investigational drug VX-809. Proc Natl Acad Sci U S A 2011;108: 18843–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Van Goor F, Hadida S, Grootenhuis PD, Burton B, Cao D, Neuberger T, et al. Rescue of CF airway epithelial cell function in vitro by a CFTR potentiator, VX-770. Proc Natl Acad Sci U S A 2009;106:18825–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Cotellessa M, Minicucci L, Diana MC, Prigione F, Di FL, Gagliardini R, et al. Phenotype/genotype correlation and cystic fibrosis related diabetes mellitus (Italian Multicenter Study). J Pediatr Endocrinol Metab 2000;13: 1087–93. [DOI] [PubMed] [Google Scholar]
- [14].Scotet V, Barton DE, Watson JB, Audrezet MP, McDevitt T, McQuaid S, et al. Comparison of the CFTR mutation spectrum in three cohorts of patients of Celtic origin from Brittany (France) and Ireland. Hum Mutat 2003;22:105. [DOI] [PubMed] [Google Scholar]
- [15].Osborne L, Santis G, Schwarz M, Klinger K, Dork T, McIntosh I, et al. Incidence and expression of the N1303K mutation of the cystic fibrosis (CFTR) gene. Hum Genet 1992;89:653–8. [DOI] [PubMed] [Google Scholar]
- [16].Castellani C, Gomez LM, Frulloni L, Delmarco A, Marzari M, Bonizzato A, et al. Analysis of the entire coding region of the cystic fibrosis transmembrane regulator gene in idiopathic pancreatitis. Hum Mutat 2001; 18:166. [DOI] [PubMed] [Google Scholar]
- [17].Feldmann D, Couderc R, Audrezet MP, Ferec C, Bienvenu T, Desgeorges M, et al. CFTR genotypes in patients with normal or borderline sweat chloride levels. Hum Mutat 2003;22:340. [DOI] [PubMed] [Google Scholar]
- [18].Rapino D, Sabirzhanova I, Lopes-Pacheco M, Grover R, Guggino WB, Cebotaru L. Rescue of NBD2 mutants N1303K and S1235R of CFTR by small-molecule correctors and transcomplementation. PLoS One 2015;10: e0119796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jensen TJ, Loo MA, Pind S, Williams DB, Goldberg AL, Riordan JR. Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 1995;83:129–35. [DOI] [PubMed] [Google Scholar]
- [20].Bence NF, Sampat RM, Kopito RR. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001;292:1552–5. [DOI] [PubMed] [Google Scholar]
- [21].Hyttinen JM, Amadio M, Viiri J, Pascale A, Salminen A, Kaarniranta K. Clearance of misfolded and aggregated proteins by aggrephagy and implications for aggregation diseases. Ageing Res Rev 2014;18:16–28. [DOI] [PubMed] [Google Scholar]
- [22].Rodriguez-Gonzalez A, Lin T, Ikeda AK, Simms-Waldrip T, Fu C, Sakamoto KM. Role of the aggresome pathway in cancer: targeting histone deacetylase 6–dependent protein degradation. Cancer Res 2008; 68:2557–60. [DOI] [PubMed] [Google Scholar]
- [23].Ohsumi Y Molecular dissection of autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol 2001;2:211–6. [DOI] [PubMed] [Google Scholar]
- [24].Luciani A, Villella VR, Esposito S, Gavina M, Russo I, Silano M, et al. Targeting autophagy as a novel strategy for facilitating the therapeutic action of potentiators on ΔF508 cystic fibrosis transmembrane conductance regulator. Autophagy 2012;8:1657–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Illek B, Maurisse R, Wahler L, Kunzelmann K, Fischer H, Gruenert DC. Cl transport in complemented CF bronchial epithelial cells correlates with CFTR mRNA expression levels. Cell Physiol Biochem 2008;22: 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sabirzhanova I, Lopes Pacheco M, Rapino D, Grover R, Handa JT, Guggino WB, et al. Rescuing trafficking mutants of the ATP-binding cassette protein, ABCA4, with small molecule correctors as a treatment for Stargardt eye disease. J Biol Chem 2015;290:19743–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Cebotaru L, Rapino D, Cebotaru V, Guggino WB. Correcting the cystic fibrosis disease mutant, A455E CFTR. PLoS One 2014;9:e85183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lopes-Pacheco M, Boinot C, Sabirzhanova I, Morales MM, Guggino WB, Cebotaru L. Combination of correctors rescue ΔF508-CFTR by reducing its association with Hsp40 and Hsp27. J Biol Chem 2015;290:25636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Van GF, Straley KS, Cao D, Gonzalez J, Hadida S, Hazlewood A, et al. Rescue of DeltaF508-CFTR trafficking and gating in human cystic fibrosis airway primary cultures by small molecules. Am J Physiol Lung Cell Mol Physiol 2006;290:L1117–30. [DOI] [PubMed] [Google Scholar]
- [30].Pedemonte N, Lukacs GL, Du K, Caci E, Zegarra-Moran O, Galietta LJ, et al. Small-molecule correctors of defective DeltaF508-CFTR cellular processing identified by high-throughput screening. J Clin Invest 2005; 115:2564–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Okiyoneda T, Veit G, Dekkers JF, Bagdany M, Soya N, Xu H, et al. Mechanism-based corrector combination restores DeltaF508-CFTR folding and function. Nat Chem Biol 2013;9:444–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008;451:1069–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy 2007;3:542–5. [DOI] [PubMed] [Google Scholar]
- [34].Lee JH, Yao Y, Mahendran A, Ngo L, Venta-Perez G, Choy ML, et al. Development of a histone deacetylase 6 inhibitor and its biological effects. PNAS 2013;110(39):15704–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Lee JY, Koga H, Kawaguchi Y, Tang W, Wong E, Gao YS, et al. HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J 2010;29:969–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Sosnay PR, Siklosi KR, Van GF, Kaniecki K, Yu H, Sharma N, et al. Defining the disease liability of variants in the cystic fibrosis transmembrane conductance regulator gene. Nat Genet 2013;45:1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina D, Settembre C, et al. Defective CFTR induces aggresome formation and lung inflammation in cystic fibrosis through ROS-mediated autophagy inhibition. Nat Cell Biol 2010;12:863–75. [DOI] [PubMed] [Google Scholar]
- [38].Luciani A, Villella VR, Esposito S, Brunetti-Pierri N, Medina DL, Settembre C, et al. Cystic fibrosis: a disorder with defective autophagy. Autophagy 2011;7:104–6. [DOI] [PubMed] [Google Scholar]
- [39].Kang R, Zeh H, Lotze M, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011;18:571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Moscat J, Diaz-Meco MT. p62: a versatile multitasker takes on cancer. Trends Biochem Sci 2012;37:230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lukacs GL, Chang XB, Bear C, Kartner N, Mohamed A, Riordan JR, et al. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem 1993;268:21592–8. [PubMed] [Google Scholar]
- [42].Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, et al. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 2010;329:805–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Yan J Interplay between HDAC6 and its interacting partners: essential roles in the aggresome-autophagy pathway and neurodegenerative diseases. DNA Cell Biol 2014;33:567–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.