

SUMMARY
Transcriptional induction of heat shock protein (HSP) genes is accompanied by dynamic changes in their 3D structure and spatial organization, yet the molecular basis for these phenomena remains unknown. Using chromosome conformation capture and single-cell imaging, we show that genes transcriptionally activated by Hsf1 specifically interact across chromosomes and coalesce into diffraction-limited intranuclear foci. Genes activated by the alternative stress regulators Msn2/Msn4, in contrast, do not interact among themselves nor with Hsf1 targets. Likewise, constitutively expressed genes, even those interposed between HSP genes, show no detectable interaction. Hsf1 forms discrete subnuclear puncta when stress activated, and these puncta dissolve in concert with transcriptional attenuation, paralleling the kinetics of HSP gene coalescence and dissolution. Nuclear Hsf1 and RNA Pol II are both necessary for intergenic HSP gene interactions, while DNA-bound Hsf1 is necessary and sufficient to drive heterologous gene coalescence. Our findings demonstrate that Hsf1 can dynamically restructure the yeast genome.
Graphical Abstract
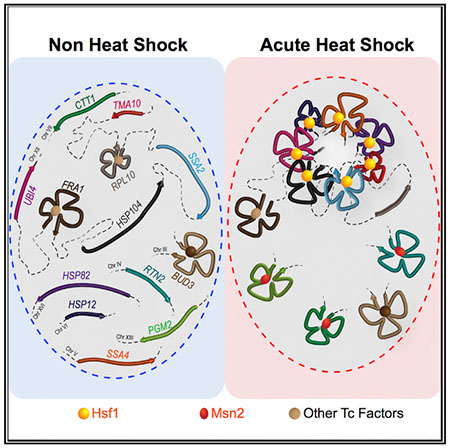
In Brief
While gene repositioning is thought to be a general feature of transcription, Chowdhary et al. provide evidence that argues against this concept. The authors demonstrate that Hsf1-regulated genes in Saccharomyces cerevisiae distinctively coalesce into intranuclear foci upon their transcriptional activation, while those activated by alternative transcription factors do not.
INTRODUCTION
Increasing evidence suggests that nuclear processes such as transcription, recombination, and repair can be influenced not only by local chromatin structure but also by three-dimensional (3D) genome architecture. Genomes of higher eukaryotes are compartmentalized into discrete structural and regulatory units called topologically associating domains (TADs) (Wendt and Grosveld, 2014). Genes located within these structures tend to have similar expression states and epigenetic signatures, and the perturbation of TAD integrity may lead to aberrant activation (Hnisz et al., 2018). Recent studies using genome-wide chromosome conformation capture (3C)-based techniques have unveiled spatial genomic structures that are analogous to mammalian TADs in budding yeast (Eser et al., 2017; Hsieh et al., 2015).
DNA looping has been directly implicated in transcriptional control. For example, the DNA between enhancers and cognate promoters, typically 20–50 kb in mammals, loops out, permitting physical contact between these regulatory regions. Analogous, albeit smaller, enhancer (upstream activation sequence [UAS])-promoter loops have been observed in Saccharomyces cerevisiae (Chowdhary et al., 2017; Dobi and Winston, 2007). Physical contacts between the 5′ and 3′ ends of actively transcribed genes, as well as between their regulatory elements and coding sequences, have been observed in both yeast (Chowdhary et al., 2017; Hampsey et al., 2011) and mammals (Beagrie et al., 2017; Lee et al., 2015).
DNA loops tend to be dynamic, and such dynamism facilitates long-range chromosomal interactions. For example, both proximally and distally located mammalian genes engage in frequent contacts that may contribute to their co-regulation (Fanucchi et al., 2013; Li et al., 2012; Rao et al., 2014). Moreover, activated mammalian genes have been observed to reposition themselves into discrete sites of intense RNA synthesis called transcription factories (Papantonis et al., 2012; Park et al., 2014; Schoenfelder et al., 2010). In these and other examples, it is thought that increased transcription is fostered by high local concentrations of RNA Pol II and pre-mRNA processing factors present in pre-existing stable substructures (Mitchell and Fraser, 2008). However, a single-molecule analysis using super-resolution microscopy indicated that Pol II clusters form transiently, with their mean lifetime increasing upon transcriptionally stimulating conditions (Cisse et al., 2013). Therefore, whether all transcriptionally active genes cluster, whether such clustering is the cause or consequence of transcription, what factors mediate clustering, and to what extent this mode of transcriptional control exists in eukaryotes other than mammals remain unknown.
A powerful model with which to study dynamic genome restructuring is the heat shock (HS)-responsive family of genes in S. cerevisiae. Many of these genes, including those encoding molecular chaperones and cytoprotective heat shock proteins (HSPs), are under the regulation of heat shock factor 1 (Hsf1), an evolutionarily conserved, gene-specific activator (Gomez-Pastor et al., 2018). Genes under the regulation of Hsf1 undergo dramatic transformations in chromatin structure upon their activation. These alterations include the gene-wide disassembly of nucleosomes (Zhao et al., 2005) and substantial increases in Hsf1, Mediator, SAGA, and Pol II occupancy (Fan et al., 2006; Kim and Gross, 2013; Vinayachandran et al., 2018). In addition, prominent intragenic and intergenic chromosomal contacts accompany HSP gene activation. These alterations include DNA looping between UAS and promoter, between promoter and terminator, and between regulatory regions and coding sequences. Activated HSP genes also engage in frequent cis- and trans-interactions with one another, dynamically coalescing into diffraction-limited foci (Chowdhary et al., 2017). It is unknown what underlies these genomic rearrangements. It is also unclear whether gene clustering is the default state for transcriptional control in budding yeast, as suggested for mammalian cells.
Here, we demonstrate that stress-activated Hsf1 is a key determinant driving interactions between yeast HSP gene loci during heat shock. Thermal stress-responsive genes activated by alternative activators Msn2 and Msn4 do not detectably cluster, nor do coordinately regulated ribosomal protein genes. While high levels of transcription are necessary for coalescence, they are not sufficient. Our results argue against the idea that gene repositioning is a general feature of transcriptional activation and instead point to activators such as Hsf1 as the drivers of global genome restructuring.
RESULTS
HSP Genes Engage in Robust Intergenic Interactions that Exclude Genes Interposed between Them
HSP genes engage in extensive intra- and interchromosomal interactions upon their heat shock-induced activation (Chowdhary et al., 2017). If HSP gene coalescence is biologically significant, then one might predict that non-HSP genes would be excluded from such clustering, even those residing in close linear proximity. To test this, we used a modified version of 3C to investigate intergenic interactions within a 35-kb domain on Chr. XII (TaqI-3C; see STAR Methods). Three HSP genes—UBI4, HSP104, and SSA2—lie within this domain, and the UAS of each is inducibly occupied by Hsf1 in cells exposed to acute heat shock (30°C–39°C shift for 5 min; Figure 1A). Under non-heat shock (NHS) conditions, no 3C interactions above background could be detected between these genes (Figure 1B, blue matrices; gene regions defined in Figure S1), which is consistent with their low basal transcription and previous nucleosome-resolution chromatin contact analysis, indicating that they lie within separate chromosome interaction domains (CIDs) (Hsieh et al., 2015).
Figure 1. Hsf1-Regulated Genes Engage in Highly Specific Intergenic Interactions.

(A) Hsf1 ChIP-seq profile of a 50-kb region on the left arm of Chr. XII in NHS and 5 min heat shock (HS) cells. Genes subjected to TaqI-3C analysis in are highlighted. RPKM, reads per kilobase per million mapped reads.
(B) Left: contact frequencies between the indicated regions of UBI4, FRA1, HSP104, and SSA2 in strain BY4741 grown under NHS conditions (30°C), as determined by TaqI-3C. Values indicate normalized interaction frequencies, determined as described in Experimental Model and Subject Details. Those ≤0.1 are indistinguishable from background. Gene regions are defined in Figures S1A and S1B. Right: the same as left, except that chromatin was isolated from cells exposed to a 10-min 39°C HS. HSP104-PAU17 contact frequencies are shown below triangulated analysis. Intensity of color is proportional to the frequency of interaction. Data are derived from two independent biological replicates (qPCR = 4 for each primer combination).
(C) Physical map of Chr. XII. The rDNA repeats and TMA10 are located on the right arm of Chr. XII, 600 kb and 300 kb from TEL12R, respectively. Coordinates are numbered relative to the left telomere and do not take into account the presence of rDNA.
(D) Hsf1 occupancy profile of the indicated ~20 kb region on the right arm of Chr. XII. Hsf1 ChIP-seq analysis presented as in (A).
(E) Intergenic interaction frequencies between HSP104-TMA10 and SSA2-TMA10 in NHS and 10 min HS cells. For each pairwise test, n = 2 and qPCR = 4.
(F) Fixed-cell fluorescence microscopy of diploid strain ASK727 (HSP104-lacO256 TMA10-tetO200 GFP-LacI TetR-mCherry). Top:schematic of lacO-tagged HSP104 and tetO-tagged TMA10 loci (filled circle, centromere). Bottom left: micrographs of representative cells under NHS and 10 min HS conditions. Scale bar, 2 μm. Bottom right: the percentage of cells exhibiting coalescence; 50–80 cells were evaluated per sample at each time point. n = 2; **p < 0.01 (calculated using one-tailed t test).
See also Figures S1, S2, and S4.
However, following heat shock, not only did neighboring HSP104 and SSA2 engage in intense interactions but UBI4 also frequently contacted both genes (Figure 1B, red matrices). This occurred despite the fact that UBI4 is separated from HSP104 by 25 kb and from SSA2 by 33 kb, distances encompassing 6 and 8 CIDs, respectively (Figure S2A). By contrast, the constitutively active and looped FRA1 gene, located between UBI4 and HSP104, engaged in no detectable interactions with the HSP genes under either condition (Figures 1A and 1B). Likewise, PAU17, a non-HSP gene interposed between HSP104 and SSA2, failed to engage in physical interactions with either gene.
We next wished to know whether cis-intergenic interactions are spatially confined by chromosomal location. Toward this end, we asked whether HSP104 and SSA2, residing on the left arm of Chr. XII, would physically interact with TMA10, a gene located on the distal right arm and inducibly occupied by Hsf1 (Figures 1C and 1D). Genome-wide 3C-based analyses have indicated that the left and right ends of Chr. XII are physically isolated from each other, due to a “near absolute” barrier conferred by the 100–200 rDNA repeats that assemble into the nucleolus (Cournac et al., 2012; Duan et al., 2010; Rutledge et al., 2015). Consistent with these prior studies, TaqI-3C failed to detect above-background interaction between HSP104-TMA10 or SSA2-TMA10 in non-induced cells (Figure 1E, left). However, following a 10-min heat shock, physical interactions between these Hsf1 targets were readily detectable (Figure 1E, right).
To validate this result using an orthogonal approach, we performed single-cell microscopy analysis of a heterozygous diploid bearing chromosomally linked lacO-tagged HSP104 and tetO-tagged TMA10 genes and co-expressing GFP-LacI and TetR-mCherry (Figure 1F). Under NHS conditions, the two genes rarely co-localized (in <10% of cells), a frequency that is likely attributable to coincidental overlap. However, following acute heat shock, the frequency of HSP104-lacO256-TMA10-tetO200 co-localization increased to 35%, which is consistent with 3C analysis. Fluorescence imaging of a red fluorescent protein (RFP)-tagged nucleolar protein suggests that nucleolar integrity is maintained in acutely heat-shocked cells (Figure S2B). These results demonstrate that HSP gene interactions are not only specific but are also sufficiently robust to circumvent the physical barrier imposed by the nucleolus.
Heat Shock-Induced Intergenic Interactions Are Distinct to Hsf1 Target Genes
While a large number of genes are transcriptionally induced by heat shock, only a small fraction are dependent on Hsf1, as revealed by transcriptome-wide sequencing (RNA sequencing [RNA-seq]) of Hsf1+ versus Hsf1− cells (Figure 2A) (Hsf1− cells conditionally depleted of nuclear Hsf1; see below). Therefore, are intergenic interactions a general feature of heat shock-activated genes? Of particular interest are genes whose thermal-responsive regulation is under the control of Msn2 (and its paralog Msn4). Msn2/Msn4 (hereafter referred to as Msn2/4) regulates the transcription of 200–300 genes in response to a variety of environmental stresses, including heat, oxidative, osmotic, and salt (Elfving et al., 2014). We selected three genes—CTT1, PGM2, and RTN2—whose thermal stress-induced transcription is dependent on Msn2/4 and independent of Hsf1 (Figures 2A, 2B, and S3A).
Figure 2. Msn2/4-Regulated Genes Are Strongly Induced by Heat Shock, yet Fail to Engage in Intergenic Interactions.
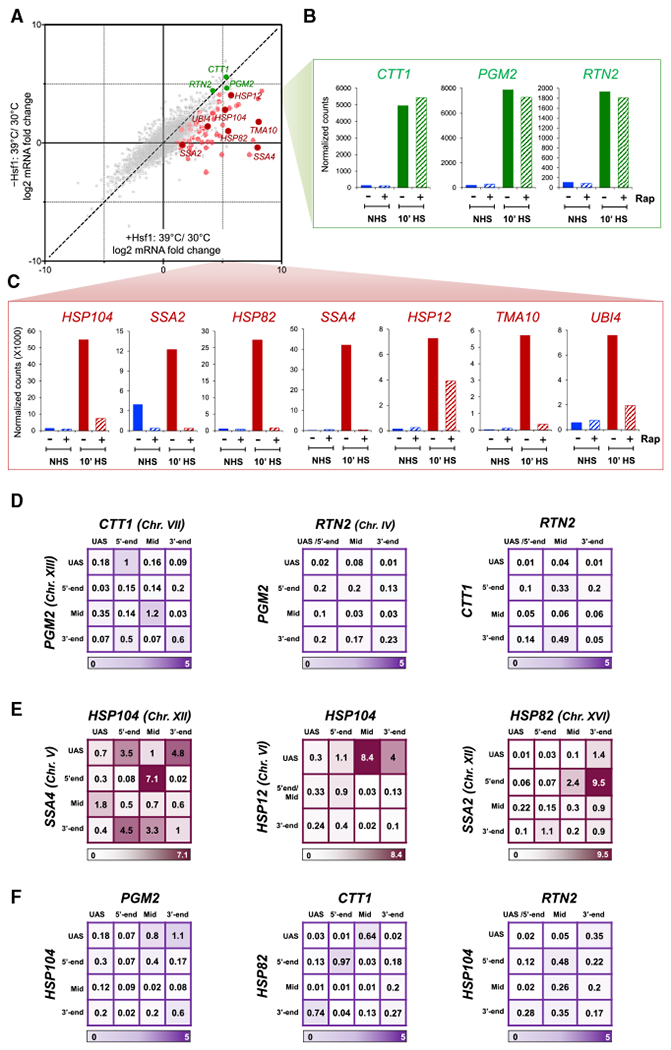
(A) Transcriptome-wide HS/NHS fold change in expression in the presence and absence of nuclear Hsf1 using the Hsf1 anchor away system (Hsf1-AA). Plotted are RNA-seq count ratios determined for every gene. Categories: red and pink, Hsf1-occupied genes in acutely heat-shocked cells (Pincus et al., 2018); green, Msn2/4-dependent, Hsf1-independent genes (as defined in text); gray, all others.
(B) Transcript levels (normalized RNA-seq reads) of select Msn2/4-dependent genes under each of the indicated conditions. Rap, rapamycin.
(C) As in B, except for select Hsf1-dependent genes.
(D) Matrix summaries of intergenic interaction frequencies between the indicated Msn2/4-target genes in 10 min HS cells (analysis and presentation as in Figure 1B). For each pairwise test, n = 2 and qPCR = 4.
(E) As in (D), except that pairwise tests were conducted between the indicated Hsf1-target genes.
(F) As in (D), except that intergenic interactions between Msn2/4- and Hsf1-regulated genes were determined.
See also Figures S1, S3, and S4.
TaqI-3C analysis revealed heat shock-dependent intragenic looping interactions within all three Msn2/4-target genes (Figure S3B; gene maps in Figure S1C), which is consistent with the notion that such restructuring is characteristic of actively transcribed genes (Chowdhary et al., 2017). Nonetheless, using primers corresponding to the UAS, 5’ end, mid-open reading frame (ORF) and 3’ end of each gene, we were unable to detect above-background interactions between CTT1, PGM2, and RTN2 upon their transcriptional activation (Figure 2D), despite the presence of readily detectable interactions between Hsf1-target genes (Figure 2E) in the same heat-shocked cells. Moreover, we did not detect any interactions between heat shock-induced Msn2/4-target genes and Hsf1-target genes (Figure 2F). For all three categories, no interaction was detectable under NHS conditions (Figure S3C).
We next asked whether constitutively active genes physically interact and tested intergenic interactions between two coordinately regulated ribosomal protein genes, RPL10 and RPL22A, as well as between two unrelated genes, FAS2 and RPL10. Each gene is heavily transcribed under NHS conditions, as determined by both RNA-seq and nascent RNA measurements (Figure S4A; Pincus et al., 2018), yet no above-background interactions could be detected (Figure S4B). These data suggest that neither heat shock-inducible Msn2/4-regulated genes nor other highly expressed, coordinately regulated genes interact either among themselves or with Hsf1-target genes.
Single-cell imaging corroborated these findings. We observed that HSP104-lacO256 and HSP12-lacO128, residing on Chr. XII and Chr. VI, respectively, coalesced into single diffraction-limited foci in >30% of cells under acute heat shock conditions (2.5 or 10 min heat shock), and the frequency of such coalescence was significantly higher than in either the control or the 30-min heat-shocked state (Figure 3A, solid bars). This is in agreement with our previous findings that HSP gene interactions are highly dynamic, detectable within 60 s of heat shock, yet evanescent (Chowdhary et al., 2017). In contrast, only background levels of coalescence were observed between Hsf1-regulated HSP104 and Msn2-regulated PGM2 (Figure 3A, striped bars). Moreover, while the distance between HSP104 and HSP12 was normally distributed under NHS conditions, the distribution became skewed toward shorter distances during the acute stages of heat shock (Figure 3B), which is consistent with interchromosomal clustering of the two loci. No such change was observed between HSP104 and PGM2 in identically treated cells. Collectively, 3C and microscopy analyses suggest that (1) Msn2/4-regulated genes do not coalesce, either with themselves or with Hsf1-targets, in response to heat shock, and (2) Hsf1 targets do not coalesce with other transcriptionally active genes, even those induced by heat shock. These observations argue that coalescence may be a distinguishing feature of Hsfl-activated genes.
Figure 3. Single-Cell Analysis Reveals Preferential, Dynamic Coalescence between Hsf1-Target Genes Paralleled by Hsf1 Puncta Formation.
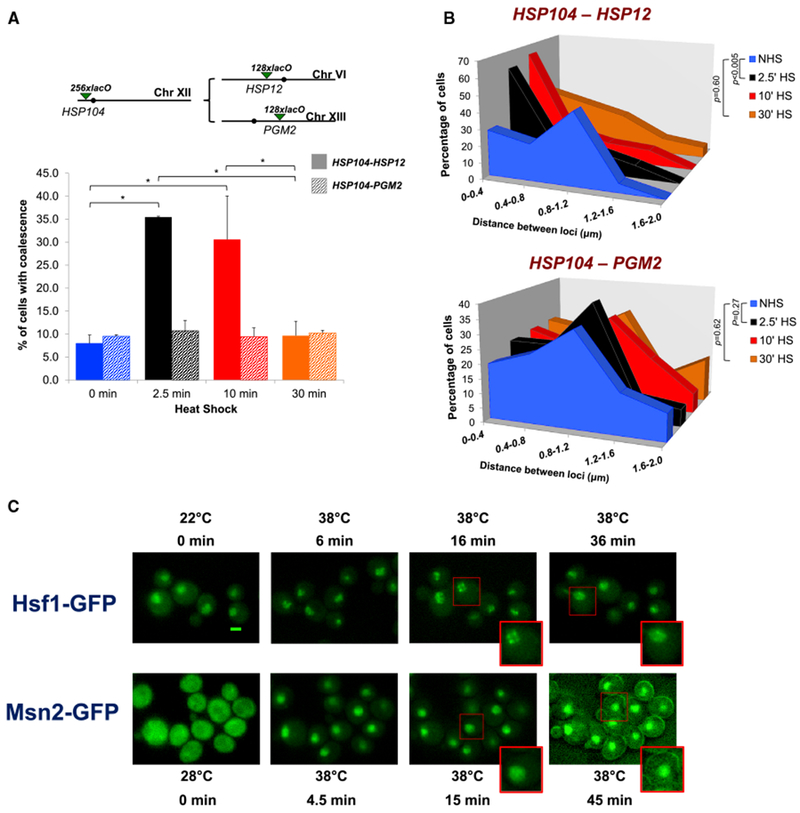
(A) Top: schematic of lacO-tagged loci present in HSP104/HSP12 or HSP104/PGM2 heterozygous diploids (filled circles, centromeres). Bottom: the percentage of cells bearing tagged HSP104-HSP12 (solid bars) or HSP104-PGM2 (shaded bars) exhibiting coalescence as determined by fixed-cell fluorescence microscopy. For HSP104-HSP12, 50–70 cells were evaluated per sample at each time point (n = 2); for HSP104-PMA2, 50–100 cells were evaluated (n = 2). *p < 0.05 (calculated by one-way ANOVA followed by Tukey’s post hoc analysis). Data for HSP104-HSP12 coalescence are from Chowdhary et al.,2017 and are used with permission.
(B) Distribution of distances between HSP104 and HSP12 (top) or HSP104 and PGM2 (bottom) in cells subjected to the indicated conditions. Depicted is the percentage of cells with tagged loci within the indicated distance (binned at intervals of 0.4 μm) in a given population of fixed cells. This analysis encompasses only those cells in which two spots lie in the same plane and thus represents a subset of cells analyzed in (A). The distance across multiple planes could not be accurately measured due to the lower resolution in the Z direction. p values were calculated by Wilcoxon rank sum test.
(C) Fluorescence microscopy of live cells expressing Hsf1-GFP (top) or Msn2-GFP (bottom) before or following the application of heat for the times and temperatures indicated. Images are presented at the same magnification; cells boxed in red are enlarged at the bottom of the respective images. Scale bar, 2 μm.
See also Figures S2 and S3.
Hsf1 Forms Discrete Intranuclear Puncta in Cells Exposed to Thermal Stress
To address the possibility that Hsf1 itself coalesces upon activation, we imaged live cells harboring Hsf1-GFP. As shown in Figure 3C, Hsf1 is largely nuclear and diffusely localized under NHS conditions. Following brief heat shock (6–16 min), the protein forms discrete nuclear puncta. However, by later time points (36–66 min), Hsf1 puncta dissolve, and the distribution of intranuclear Hsf1 returns to a diffuse state, closely paralleling the kinetics of HSP gene coalescence and dissociation described above. If the formation of Hsf1 puncta reflects the coalescence of its gene targets, then it might be predicted that Msn2, despite strongly activating transcription in response to heat shock (Figures 2B and S3A), will not form nuclear puncta. As shown in Figure 3C, Msn2-GFP, largely cytoplasmic in NHS cells (0 min), translocates to the nucleus under acutely stressful conditions (4.5 min), which is consistent with previous reports (Chi et al., 2001). In contrast to Hsf1, the intranuclear distribution of Msn2 remains diffuse throughout the heat shock time course (4.5–45 min; Figure 3C). Therefore, the ability or inability to form nuclear puncta may be an inherent property of Hsf1 and Msn2 activation.
Hsf1 and Pol II Are Necessary to Drive Interactions between HSP Genes during Heat Shock
To directly test the importance of Hsf1 in driving changes in HSP gene conformation and 3D nuclear organization, we conditionally depleted it from the nucleus using the anchor away technique (Haruki et al., 2008). Growth of HSF1-FRB cells on rapamycin demonstrates that Hsf1 is essential for viability, even at 30°C (Figure 4A), which is consistent with previous observations (Sorger and Pelham, 1988). Cytoplasmic sequestration of Hsf1-FRB depleted its occupancy of representative HSP genes following a subsequent 10-min heat shock (Figure 4B, left). Consistent with the central role of Hsf1 in regulating these genes (Pincus et al., 2018; Solís et al., 2016), Pol II occupancy was likewise severely reduced in rapamycin-treated cells (Figure 4B, right), as was transcript accumulation (Figure 2C). Concomitant with this reduction in transcription, the formation of 5′-3′ gene loops was obviated, as were other intragenic interactions, including UAS-promoter looping (Figure 4C, pink bars). In conjunction, intergenic coalescence was reduced to background levels (Figure 4D). As expected, the intragenic looping of constitutively expressed BUD3 was unaffected by this perturbation (Figure S4C). Therefore, Hsf1 is required to drive its target genes into a looped and physically interactive state in response to heat shock.
Figure 4. Nuclear Hsf1 and Pol II Are Necessary for Driving Intergenic HSP Gene Interactions.

(A) Spot dilution analysis of wild-type (WT) (BY4742) and Hsf1-AA (BY4742-HSF1-AA) cells.
(B) Hsf1 and Pol II (Rpb1) ChIP analysis of HSP104. Hsf1-AA cells were pretreated with rapamycin for 90 min or not, as indicated, then subjected to a 10-min HS and processed for ChIP. Depicted are means + SD (n = 2; qPCR = 4). **p < 0.01; ***p < 0.001 (calculated using one-tail t test).
(C and D) TaqI-3C analysis of HSP intragenic (C) and intergenic (D) interactions using the indicated primer pairs in Hsf1-AA cells pretreated with rapamycin followed by 10 min HS. Depicted are means + SD (n = 2; qPCR = 4). *p < 0.05; **p < 0.01 (calculated as in B).
(E) Spot dilution analysis of Rpb1-AA cells (yFR1324).
(F) Rpb1 and Hsf1 ChIP analysis of HSP82. Rpb1-AA cells were pretreated with rapamycin for 60 min or not, as indicated, followed by a 10-min HS. Depicted are means + SD (n = 2; qPCR = 4). *p < 0.05; **p < 0.01; n.s. (not significant), p > 0.05 (as in B).
(G and H) TaqI-3C analysis of HSP intragenic (G) and intergenic (H) interactions in Rpb1-AA cells pretreated with rapamycin or not, as indicated, followed by a 10-min HS. Depicted are means + SD (n = 2; qPCR = 4). *p < 0.05; **p < 0.01; ***p < 0.001 (as in B).
See also Figures S1 and S4.
We next asked whether Pol II, in particular its largest subunit (Rpb1), is likewise required for the changes observed in HSP gene conformation and nuclear organization. Rapamycin-induced cytoplasmic sequestration of Rpb1-FRB rendered cells inviable on solid medium (Figure 4E). In cells pre-exposed to rapamycin, Rpb1-FRB occupancy of heat shock-induced HSP gene promoters and coding regions was significantly reduced, although this nuclear depletion had little or no effect on Hsf1 occupancy (Figure 4F). Nonetheless, all of the intragenic interactions tested were greatly diminished by prior removal of Rpb1, including UAS-promotor looping (Figure 4G, first pairwise test), implicating Pol II in the stable formation of such loops. Concomitant with the loss of intragenic looping was the loss of all tested intergenic interactions (Figure 4H, pink bars), implicating Pol II and/or transcription in the interactions among HSP genes. As expected, intragenic looping interactions at BUD3 were also diminished by Rpb1 depletion (Figure S4C). Therefore, Pol II is critical for the formation of novel intergenic interactions that are characteristic of activated HSP genes, yet even high levels of it—as inferred from intragenic looping, chromatin immunoprecipitation (ChIP), and expression assays (Figures S3A, S3B, S4A, and 5C–5E)—are not sufficient.
Figure 5. DNA-Bound Hsf1 Is Both Necessary and Sufficient to Drive Coalescence of a Pol II Gene.

(A) Left: physical map of HSP12 depicting the HSE and STREs within its upstream region (DNA motifs from www.yeastract.com); location of the deletion in hsp12-ΔHSE is indicated. Coordinates are numbered with respect to ATG. Right: Hsf1 ChIP-seq profile of the indicated region on Chr. VI in NHS and 5 min HS cells.
(B) Hsf1 ChIP analysis was conducted on HSP12 and hsp12-ΔHSE cells (strains AJ303 and AJ305, respectively) maintained at 30°C (NHS) or subjected to a 10-min HS as indicated. Depicted are means + SD (n = 2; qPCR = 4). *p < 0.05.
(C) Rpb1 ChIP analysis conducted as in (B). n.s., p > 0.05.
(D) Intragenic contact frequencies of HSP12 and hsp12-ΔHSE following 10 min HS as deduced by TaqI-3C (n = 2; qPCR = 4). n.s., p > 0.05.
(E) Intergenic contact frequencies between HSP12 or hsp12-ΔHSE and the indicated gene loci following 10 min HS. **p < 0.01.
(F) TaqI-3C analysis of HSP12 in Hsf1-AA cells pre-treated with 1 μM rapamycin for 90 min or not (+Rap or −Rap, respectively) followed by 10 min HS. Depicted are normalized frequencies of representative intragenic and intergenic interactions. *p < 0.05; n.s., p > 0.05.
(G) Physical map of the chromosomal transgene UASHS-BUD3.
(H) Hsf1 occupancy of BUD3 and UASHS-BUD3 (strains BY4741 and ASK804, respectively) under NHS and 10 min HS states. Analysis as in (B). **p < 0.05.
(I) Summary of intragenic interactions detected within UASHS-BUD3 in NHS and 10 min HS states (n = 2; qPCR = 4).
(J) Intergenic contact frequencies between BUD3 or UASHS-BUD3 and the indicated HSP genes in 10 min HS cells (n = 2; qPCR = 4).
See also Figures S1, S4, and S5.
Preventing Hsf1 DNA Binding Uncouples HSP12 Coalescence from Transcription
Can intergenic HSP interactions be decoupled from transcription? To address this question, we analyzed the HSP12 gene as a test case since it is regulated by both Hsf1 and Msn2/4. HSP12 has an Hsf1 binding site (heat shock element [HSE]) consisting of TTCn-NNNNN-nTTCn-NNNNN-nTTC, lying ~800 bp upstream of its transcription start site (TSS) and inducibly occupied by Hsf1 (Figure 5A). It also has seven stress-response elements (STREs; CCCCY motifs) recognized by Msn2/4 located between the HSE and TSS. Conditional nuclear depletion of Hsf1 caused a moderate reduction in both heat shock-induced HSP12 transcription (Figure 2C) and intragenic looping (Figures 5F and S5A). By contrast, Hsf1 depletion obviated HSP12 interactions with HSP104, HSP82, and SSA2 (Figures 5F and S5A). Thus, although HSP12 transcription is only mildly affected by Hsf1 depletion, its interchromosomal interactions with other HSP genes are abolished. This observation suggests that HSP gene transcription, while strongly correlated with intragenic looping, can be uncoupled from intergenic coalescence.
To further strengthen this point, we performed the complementary experiment by chromosomally excising the HSE upstream of HSP12, creating an allele termed hsp12-ΔHSE (Figure 5A). This manipulation reduced Hsf1 occupancy to near-background levels under both NHS and 10-min heat shock conditions (Figure 5B). However, consistent with the Hsf1 anchor away results, there was little effect on either Pol II occupancy or intragenic looping (Figures 5C and 5D), which is in line with the notion that Hsf1 is largely dispensable for HSP12 transcription under these conditions. Despite this, the HSE deletion suppressed hsp12 interaction with HSP104, HSP82, and SSA2 (Figure 5E, compare solid versus striped bars). As expected, other interchromosomal HSP gene interactions were unaffected (Figure S5B). These observations demonstrate that even when Hsf1 is largely dispensable for stimulating transcription, it is necessary for driving a target gene into physical interactions with other HSP genes.
Ectopic Targeting of Hsf1 Is Sufficient to Drive Intergenic Association of a Heterologous Gene
Finally, to test whether DNA-bound Hsf1 is sufficient to cause an otherwise unrelated gene to coalesce with HSP genes, we chromosomally integrated a high-affinity Hsf1 binding site upstream of BUD3, creating an allele called UASHS-BUD3 (Figures 5G and S1D). As shown in Figure 5H, Hsf1 strongly occupied UASHS-BUD3 but not BUD3+ in response to a 10-min heat shock. Moreover, UASHS-BUD3 exhibited increased levels of intragenic looping interactions following heat shock (Figure 5I), consistent with its increased transcription (A.S.K. and D.S.G., unpublished observation). Most significantly, UASHS-BUD3 engaged in novel intergenic interactions with both HSP104 and HSP82 (Figure 5J, right), in marked contrast to the wild-type (WT) gene (Figure 5J, left). Therefore, DNA-bound Hsf1 is sufficient to direct a heterologous gene into novel interactions with HSP genes in response to heat shock.
DISCUSSION
Hsf1 Target Genes Distinctively Interact among Themselves upon Activation
We present evidence using both molecular and single-cell imaging approaches that physical interactions between heat shock-activated Hsf1-target genes, located on the same or different chromosomes, are specific and robust. Genes interposed between Hsf1 targets are excluded from these interactions, yet HSP genes spatially segregated by rDNA repeats that make up the nucleolus readily interact. The HSP genes tested were located at a variety of chromosomal latitudes (Table S1), thereby rendering it unlikely that the interchromosomal interactions we detect reflect fortuitous alignment of these genes along Rabl-organized chromosomes. Not all heavily transcribed genes coalesce, even those whose transcription is activated by alternative thermal stress-responsive activators. Likewise, coordinately regulated ribosomal protein genes show no detectable interaction, despite the fact that the genes tested lie on the same chromosome and are separated by only 20 kb. The latter observation is consistent with genome-wide 3C analyses that failed to uncover significant interactions between Pol II genes across the yeast genome under the control condition (NHS) of our experiments (Duan et al., 2010; Hsieh et al., 2015; Rutledge et al., 2015).
What distinguishes a coalescing from a non-coalescing gene, therefore, is not whether it is coordinately regulated, transcribed at a high level, or induced by heat shock. What dictates coalescence is whether a gene is regulated by Hsf1. Using a combination of conditional Hsf1 nuclear depletion, ectopic Hsf1 targeting, and genome editing, we have demonstrated that Hsf1 is both necessary and sufficient to drive the interaction of a transcriptionally competent Pol II gene with other Hsf1-regulated genes. Particularly compelling are two complementary observations: (1) de novo interaction of an otherwise non-heat shock-responsive gene converted to thermal responsiveness and spatial reorganization by ectopic targeting of Hsf1, and (2) abrogation of interactions between hsp12-ΔHSE and other HSP genes in thermally stressed cells, despite continued robust transcription of hsp12-ΔHSE (paralleling similar observations of HSP12+ in Hsf1-depleted cells).
It is worth emphasizing that intergenic interactions between Hsf1-regulated genes go well beyond classic transvection phenomena (enhancer-promoter interactions). The discovery of coding region interactions between Hsf1-regulated genes, but not of other actively transcribed genes, constitutes an important finding of the present study and our earlier study (Chowdhary et al., 2017). We are unaware of other yeast activators that possess comparable activity, although our experiments do not rule out their existence. The closest example may be that of an erythroid-specific transcription factor, Klf1. Using a combination of 3C, ChIP-3C, fluorescence in situ hybridization (FISH), and immunofluorescence, Fraser and colleagues have shown that in mouse erythroid cells, Klf1-regulated globin genes relocate into transcription factories, where they engage in preferential (although not exclusive) interchromosomal associations with other Klf1-regulated genes (Schoenfelder et al., 2010). Thus, Klf1 drives preferential physical interactions between its target genes in response to a developmental signal; evidence reported here indicates that Hsf1 drives preferential interactions between its target genes in response to an environmental signal. A genome-wide analysis will be required to show whether such interactions are exclusive to the Hsf1 regulon.
Relationship to Other Examples of Gene Clustering and Repositioning of Active Genes to the Nuclear Periphery
As alluded to above, a particularly striking aspect of our study is that constitutively active genes FRA1 and PAU17, despite being located in close linear proximity to HSP genes, do not interact with them. Such specificity contrasts with a recent report of methionine-responsive genes in yeast that engage in intrachromosomal clustering upon their induction as assessed by 3C, yet unlike what we observed here, unrelated neighboring genes also tended to interact (Du et al., 2017). More similar to the specificity and selectivity of Hsf1-target gene interactions are observations that tumor necrosis factor-α (TNF-α)-responsive genes in human endothelial cells engage in intrachromosomal interactions upon cytokine stimulation (Papantonis et al., 2012), whereas an actively transcribed gene interposed between them, and located nearby to one of them, is excluded from such colocalization (Fanucchi et al., 2013).
In addition, microscopy and biochemical analyses have shown that yeast GAL genes relocate to the nuclear pore complex (NPC) upon galactose induction (Casolari et al., 2004). Such repositioning has been reported to be accompanied by sustained clustering of GAL alleles located on homologous chromosomes, initially at the NPC and subsequently in the nucleoplasm, as detected by a microscopy-based analysis (resolution of ~500 nm) (Brickner et al., 2016). However, no evidence of GAL1-10 allelic interaction was seen in galactose-induced diploids using either Hi-C (Kim et al., 2017) or wide-field fluorescence localization microscopy (Backlund et al., 2014). Therefore, it is unclear whether repositioning of the GAL locus to the NPC or its interallelic clustering is related to the robust and intricate physical interactions that we detect between HSP genes using 3C, whose resolution is ~1–5 nm (Dekker and Mirny, 2016). In a similar vein, earlier studies on the effect of heat shock on human nuclear substructure reported the existence of “stress bodies.” As these stress bodies appear to be arrays of HSF1 bound to repetitive, heterochromatic DNA sequences that are spatially independent from HSP gene transcription (Jolly et al., 1997), they are unlikely to be related to the concerted coalescence of HSP genes reported here.
Evanescent interactions between Hsf1-target genes contrast with models suggesting that actively transcribed genes relocate into statically assembled substructures (Mitchell and Fraser, 2008). Instead, our observations resemble the dynamic assembly of Pol II clusters in serum-stimulated human cells (Cisse et al., 2013) or the dynamic sorting of immunoglobulin genes residing on different chromosomes into transcription factories during mouse B cell development (Park et al., 2014).
Is HSP Gene Coalescence an Example of Phase Separation?
Recently, phase separation of multi-molecular assemblies has been suggested as a mechanism for transcriptional control (Chong et al., 2018; Hnisz et al., 2017; Sabari et al., 2018). We have described observations that are consistent with the HSP regulon undergoing a liquid-liquid phase separation-like process in response to heat shock. In particular, we have observed that genes sharing in common only the identity of the DNA-bound transcription factor coalesce into diffraction-limited foci under activating conditions. While such coalescence accompanies heightened expression of these genes—and Pol II transcription is indeed required for HSP gene interactions, as we demonstrate here—the intensity of transcription cannot be the only parameter dictating foci formation. RNA measurements in acutely heat-shocked cells reveal that Msn2/4-regulated CTT1 and PGM2 are expressed at levels that equal or exceed several Hsf1 targets studied here, including HSP12, UBI4, and TMA10 (Figures 2A–2C). However, CTT1 and PGM2 do not detectably interact with each other, nor with representative HSP genes.
Why then do Hsfl-regulated genes interact with one another, while Msn2/4-regulated genes do not? One possibility is the presence of low complexity domains (LCDs) or intrinsically disordered regions (IDRs) in Hsf1. IDRs have been postulated to contribute to the phase separation of membraneless organelles such as stress granules (Protter and Parker, 2016); those in the abundant nuclear protein HP1 appear to contribute to the phase separation of constitutive heterochromatin in both insects and mammals (Larson et al., 2017; Strom et al., 2017). While such low complexity, intrinsically disordered structures in Hsf1 may contribute to HSP gene coalescence, it cannot be the only reason, since most gene-specific transcription factors, including Msn2/4, also possess IDRs. Hsf1 DNA binding in the absence of transcription does not trigger genes to interact in trans (Figures 4F and 4H). Therefore, Hsf1 may recruit a distinct set and/or quantity of coactivators and Pol II-associated machinery, with Mediator, which is prodigiously recruited to Hsf1-regulated genes (Fan et al., 2006; Kim and Gross, 2013), being a prime candidate (Cho et al., 2018; Sabari et al., 2018). Our future studies will address this and other intriguing possibilities.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. David Gross (dgross@lsuhsc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast Strains
The experimental model used in this study was Saccharomyces cerevisiae (budding yeast). The diploid strains, ASK702 (HSP104-lacO256 HSP12-lacO128 GFP-LacI) and IGY101 (HSP104-lacO256 PGM2-lacO128 GFP-LacI), were created by crossing a MATα derivative of DBY255 with ICY33 and DBY646, respectively (haploid strains generously provided by D. G. Brickner and J. H. Brickner, Northwestern University). ASK706 is a derivative of ASK702 bearing POM34 C-terminally tagged with mCherry. For fluorescence microscopy of fixed cells, strains ASK706 and IGY101 were used. The diploid strain, SCY712 (NOP56-mRFP POM34-GFP), was made by crossing ATY1513 with SCY711.
To construct the diploid strain ASK727 (HSP104-lacO256 TMA10-tetO200 GFP-LacI TetR-mCherry), we first excised KAN-MX from ASK701 (MATα HSP104-lacO256 SEC63-Myc13::KAN-MX), creating VPY101. KAN-MX was then integrated downstream of TMA10, creating ASK721. Plasmid pSR14, containing tetO200::LEU2 (gift of S. Gasser, Friedrich Miescher Institute for Biomedical Research), was linearized using AscI (generates homologous ends to KAN-MX). This linearized DNA was integrated at the TMA10-KAN-MX locus, creating ASK722. To create a MATa TetR-mCherry expressing strain, we crossed YAM1269 (MATa leu2::TetR-mCherry::hphMX::leu2; gift of A. MacQueen, Wesleyan University) with W303-1B (MATα), sporulated the resultant diploid, and isolated the desired spore (MATa leu2::TetR-mCherry::hphMX::leu2 and lacking other markers present in YAM1269; see Table S2) termed ASK726. ASK726 was then crossed with ASK722 to obtain the diploid ASK727. We note that TetR-mCherry contains an N-terminal nuclear localization signal (A. MacQueen, personal communication).
To construct ASK804, a strain bearing the UASHS-BUD3 transgene, a loxP-flanked KAN-MX containing fragment was amplified from plasmid pUG6 (Euroscarf) using primers BUD3F_loxpF and AdaptR_loxpR (primer sequences are provided in Table S3). Employing BY4741 genomic DNA as template, UASHS (an 86 bp sequence spanning HSEs 1 - 3 of HSP82 (Pincus et al., 2018)) was amplified using primers AdaptF_HSE1F and BUD3R_HSE3R. The two amplicons were then combined in a PCR overlap extension reaction to generate a DNA fragment containing loxP-kanMX-loxP-3xHSE flanked by ~40 nt of BUD3 sequence centered at −182 (with respect to ATG [+1]). This DNA fragment was gel purified and transformed into BY4741; transformants were screened using genomic PCR. The selection marker, KAN-MX, was subsequently excised from the desired integrant by ectopic expression of Cre recombinase. The insertion and flanking chromosomal sequence were validated by DNA sequencing (Eurofins Genomics).
To construct AJ305, a strain bearing a deletion of the heat shock element (HSE) upstream of HSP12, we used BY4741 trp1Δ::KAN-MX as the parent. AJ303, a derivative bearing C-terminally tagged HSP12-Mycx9, was transformed with a PCR amplicon containing loxp-LEU2-loxp in place of the 127 bp DNA sequence spanning −930 → −804 with respect to the ATG of HSP12. The approximate midpoint of this deleted sequence corresponds to the HSE occupied by Hsf1 (see Figure 5A, right). The template for overlap extension was pUG73, a plasmid bearing loxP-flanked KlLEU2. Following transformation, the desired loxp-LEU2-loxp integrant (ΔHSE::loxp-LEU2-loxP::hsp12-Mycx9::TRP1) was identified and named AJ304. AJ304 was then transformed with pSH47, a URA3-CEN plasmid containing GAL1-CRE. LEU2 was excised upon expression of Cre recombinase and pSH47 counter selected using standard procedures. The DNA sequence of the ΔHSE-hsp12-Mycx9::TRP1 locus (referred to as “hsp12-ΔHSE”) was confirmed and the resultant strain named AJ305.
A complete list of strains is provided in Table S2. PCR primer sequences are provided in Table S3.
Culture Conditions
For 3C analysis, cells (strain BY4741 except where noted otherwise) were grown at 30°C in YPDA (YPD [yeast extract-peptone-dextrose] supplemented with 0.002% adenine) to a mid-log density (A600 = 0.65-0.8). A portion of the culture was maintained at 30°C (control conditions; non-heat-shocked (NHS) sample) while the remainder (heat-shocked sample) was subjected to an instantaneous 30°C to 39°C thermal upshift for the indicated duration as previously described (Chowdhary et al., 2017).
Conditional depletion of select nuclear proteins was conducted using the Anchor Away system (Haruki et al., 2008). BY4742-HSF1-AA and yFR1324 (Rpb1-AA) were grown in YPDA at 30°C. To anchor-away FRB-tagged Hsf1 and Rpb1 proteins, rapamycin (LC Laboratories) was added to a final concentration of 1 μg/ml to early log cultures (A600 = 0.4-0.5). BY4742-HSF1-AA cells were incubated for 90 min in the presence of the drug (except where noted below), while yFR1324 cells were incubated for 60 min. At this point, cells were subjected to an instantaneous heat shock at 39°C for 10 min, and then processed for 3C or ChIP analysis. In the case of RT-qPCR analysis (Figure S3A), a heat shock time course (0, 2.5, 15, 45 min) was employed following rapamycin pre-treatment. RNA-seq analysis was conducted using strain DPY438 as described below.
For spot dilution analysis, cells were grown to stationary phase in YPDA. Master suspensions for each strain were prepared by diluting the saturated culture to a uniform cell density (A600 = 0.5) and were transferred to a 96-well microtiter plate. These were then serially diluted five-fold and 6 μL were transferred onto a solid YPDA or YPDA + 1 μg/ml rapamycin. Cells were grown at either 30° or 37°C for 2-3 days.
METHOD DETAILS
Chromosome Conformation Capture
TaqI-3C was conducted essentially as previously described (Chowdhary et al., 2017). Cells, cultivated to early log phase at 30°C, were either maintained at 30°C or heat-shocked at 39°C for 10 min, and then crosslinked with 1% formaldehyde. Crosslinked cells were harvested from a 50 mL culture and then subjected to glass bead lysis in FA lysis buffer (50 mM HEPES pH 7.9, 140 mM NaCl, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM EDTA, 1 mM PMSF) for two cycles (20 min each) of vortexing at 4°C. A 10% fraction of the crude chromatin lysate was digested with 200 U of TaqI (New England Biolabs) at 60°C for 7 h. TaqI was heat-inactivated (80°C for 20 min) in the presence of SDS (final concentration of 1%). Excess SDS was quenched via addition of Triton X-100 (final concentration of 1%). The digested chromatin fragments were centrifuged, and the pellet was resuspended in 100 μL of 10 mM Tris-HCl (pH 7.5). Proximity ligation was performed with 7-fold diluted TaqI-digested chromatin and 10,000 cohesive end units of Quick T4 DNA ligase (New England Biolabs) at 25°C for 2h. The ligated sample was then digested with RNase (final concentration of 30 ng/μl; Sigma Aldrich) at 37°C for 20 min. Proteinase K (final concentration of 70 ng/μl; Sigma Aldrich) digestion was performed at 65°C for 12 h in the presence of 0.1% SDS. The 3C DNA template was extracted using phenol-chloroform and precipitated in the presence of glycogen.
Quantitative PCR was performed on a 7900HT Fast Real-Time PCR system (Applied Biosystems) using Power SYBR Green PCR master mix (Fisher Scientific). Anchor primers were paired with primers abutting TaqI sites lying on the same or another gene, to enable detection of intragenic or intergenic interactions, respectively. Locations of 3C primers used in this study are illustrated in Figure S1; primer sequences are provided in Table S3.
A notable feature of our procedure was the use of a 4 bp cutter, TaqI, which recognizes sites located within the UAS/promoter, 5′ end, coding region and 3′UTR/terminator regions of most genes evaluated in this study (see Figure S1). In addition, to circumvent typical problems associated with 3C-based techniques, such as the difficulty in normalizing raw contact frequencies of different chromosomal regions, we incorporated multiple controls. The most important of these was to normalize each restriction site tested to percent digestion to account for the potential variation in accessibility of local chromatin structure under each physiological condition or genetic context (see Figure S6 for representative examples). This step, which to our knowledge is unique to our procedure, greatly alleviates an under-appreciated problem endemic with all 3C-based procedures.
Additional controls incorporated into TaqI-3C included the following: (i) normalization to purified genomic DNA (gDNA) similarly cleaved and ligated to account for variation in primer pair efficiencies; (ii) normalization to a no-template control to account for primer dimer background; (iii) normalization to a non-cut region of the genome (internal recovery control) to account for variation in the recovery of 3C templates; and (iv) normalization to a no-ligation control to ensure a ligation-dependent 3C signal. See below for algorithms used to calculate normalized 3C interaction frequencies.
We note that for genes with closely related paralogs (HSP82 and SSA2, whose coding regions bear 92% and 96% sequence homology to HSC82 and SSA1, respectively), we typically used primers with multiple (and/or 3′ end) mismatches to the paralogous sequence to maximize specificity of the tested interaction. At certain TaqI sites, adequate mismatches within nearby DNA sequence did not exist (e.g., HSP82 +986, HSP82 +1838, SSA2 +855, SSA2 +1905) and these loci were not evaluated.
Chromatin Immunoprecipitation (ChIP)
For ChIP analysis, cells were grown, heat-shocked at 39°C (or maintained at 30°C) and crosslinked with 1% formaldehyde. Crosslinked cells were harvested from a 50 mL culture and subjected to glass bead lysis in lysis buffer (50 mM HEPES pH 7.5, 140 mM NaCl, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM EDTA, 2 mM PMSF, and 250 μg/ml cOmplete, EDTA-free Protease Inhibitor Cocktail for 30 min of vortexing at 4°C. The crude chromatin lysate was sonicated to an average size of ~0.25 kb using 40 cycles of sonication (30 s on/off pulses on High-Power setting; Diagenode Biorupter Plus). A 20% fraction of the sonicated chromatin was incubated with 1 μl of anti-Rpb1 or anti-Hsf1 antiserum (Chowdhary et al., 2017) for 16 h at 4°C. Antibody-chromatin complexes were immobilized on Protein A-Sepharose beads (GE Healthcare) for 16 h at 4°C. These were then subjected to sequential washes with lysis buffer, high salt buffer (50 mM HEPES pH 7.5, 500 mM NaCl, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM EDTA), wash buffer (10 mM Tris pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA) and 1× TE (10 mM Tris-HCl pH 8.0, 0.5 mM EDTA). Chromatin was eluted from beads by incubation in elution buffer (50 mM Tris pH 8.0, 1% SDS, 10 mM EDTA) at 65°C for 30 min. RNA and proteins were removed in the presence of DNase-free RNase (final concentration of 200 μg/ml, 37°C for 1 h) and Proteinase K (final concentration of 50 μg/ml, 60°C for 16 h). The ChIP template was then extracted using phenol-chloroform, precipitated in the presence of ethanol, and quantified by qPCR. The quantity of ChIP DNA for each primer pair combination was deduced from interpolation of a standard curve generated using genomic DNA template. Primer sequences are provided in Table S3.
To correct for variation in the yield of soluble chromatin, PCR signal for each primer combination was normalized to the corresponding signal from the input DNA. The input DNA was prepared from a 10% fraction of the total sonicated crosslinked chromatin subjected to Proteinase K treatment, phenol-chloroform extraction and ethanol precipitation.
ChIP-Seq
ChIP-seq was conducted similarly to ChIP, except that chromatin was isolated from a 600 mL early log culture of BY4741 cells (either NHS or 5 min heat-shocked) and sonicated for 60 cycles. A 10% fraction of the total chromatin was immunoprecipitated using the anti-Hsf1 antibody. The Hsf1-ChIP libraries were generated using 5 ng of the purified ChIP DNA (ChIP-seq Sample Prep Kit, New England Biolabs). Libraries were sequenced using an Illumina MiSeq genome sequencer. To generate normalized UCSC tracks, we combined the replicate aligned BAM files and called peaks with MACS2 using the -B option. Since these were paired-end libraries (-BAMPE), the fragment size was measured directly from the data and MACS2 reported the pileup aligned reads using the insert size from the paired-end BAM file. As a default, the bedGraphs were not normalized by read depth but each bedGraph coordinate intensity was multiplied by a normalization factor (10 million divided by total fragments in the library).
Reverse Transcription-qPCR (RT-qPCR)
Cells were grown and subjected to a heat shock time course (per Figure S3A). At the appropriate time, 20 mM sodium azide was added to terminate transcription. Total RNA was isolated using RNeasy kit (QIAGEN). A 0.5-2 μg aliquot of the purified RNA template was used to synthesize cDNA (High-Capacity cDNA Reverse Transcription Kit, Applied Biosystems). The purified RNA template was incubated in the Reverse Transcription master mix at 37°C for 2 h. This reaction mix was diluted 20-fold, and 5 μl of this dilution were used in each qPCR reaction. Relative cDNA levels were calculated using the ΔΔCt method (see Chowdhary et al., 2017).
To correct for variation in the recovery of cDNA templates, PCR signal from SCR1 Pol III transcript was used as a normalization control. Relative fold change per minute in mRNA levels was calculated by dividing mean mRNA levels (derived from two independent biological samples for each time point) by that of the previous time point, and then normalizing by the time elapsed in minutes. Primer combinations used are listed in Table S3.
RNA-seq
RNA-seq read counts were obtained from ribosomal RNA-depleted total RNA which was isolated from W303-derived Hsf1-AA cells (DPY438) cultivated at 30°C. Rapamycin was added to a final concentration of 1 μM for 45 min (Hsf1-nuclear depleted cells) or not (Hsf1-containing cells) prior to continued incubation at 30°C or heat shock at 39°C for 10 min. Reads were aligned using TopHat and quantified with HTseq-count; DEseq2 was used to normalize counts across co-sequenced libraries.
Fluorescence Microscopy
For fixed-cell imaging, cells were grown at 30°C to early log phase in YPDA, subjected to instantaneous heat shock at 39°C for the indicated times, and then fixed in 1% formaldehyde for 10 min. Cells were harvested from 1.5 mL of culture and the cell pellet was washed with 1 mL of phosphate-buffered saline (PBS) (pH 7.4). A small quantity was transferred to a patch of 2% agarose (prepared in PBS) on a glass slide. Images (binned 2 × 2) were acquired across 9 planes on the z axis with an interplanar distance of 0.5 μm. For analysis of the diploid strain ASK727 (HSP104-lacO256 TMA10-tetO200 GFP-LacI TetR-mCherry), we used an Olympus UPlanFl 100/1.3-NA objective attached to a Photometrics Prime 95B CMOS camera. All other images were taken using a CoolSNAP HQ Charge coupled device camera. The 89021 filter set (Chroma Technology) was used for imaging GFP and mCherry. Slidebook, version 4 or 6 (Intelligent Imaging Innovations), was used to control camera acquisition and the z axis stepping motor (Ludl Electronic products). Post-acquisition analysis of images was done using ImageJ (1.48v).
For coalescence analysis, cells with large buds typically had one or both genes replicated, as indicated by more than two green fluorescent spots. Such cells were not used in our analysis. Nine planes in the z direction, covering the entire depth of nuclei, were inspected for location of tagged genomic loci. A cell was scored positive for coalescence if the two green spots were not resolvable in all nine planes (Figure 3A) or the distance between the centroids of green and red spots was < 0.4 μm (Figure 1F).
For Hsf1-GFP and Msn2-GFP live cell imaging, cells were grown in synthetic dextrose complete (SDC) medium containing 2% glucose and supplemented with 0.1 mg/ml adenine until early log phase. 1 mL of culture was harvested and washed with 1 mL of SDC media. An aliquot of cells was pipetted onto a 3% agarose pad made with SDC medium, covered with a coverslip and then imaged. Samples were subjected to heat shock by heating the objective from room temperature to 38°C using a Bioptechs objective heater system. The 89021 filter set was used for imaging GFP. Images were taken through an Olympus Ach 100/1.25-numeric-aperture (NA) objective using a CoolSNAP HQ charge-coupled-device camera. All other manipulations were as above.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantification of 3C
The percent digestion efficiency at each TaqI site was determined by amplifying a region across the restriction site using the corresponding pair of convergent primers (sequences provided in Table S3). Cycle threshold values (Ct) for undigested (UND) and digested only (DO) crosslinked chromatin templates were determined for each primer combination and incorporated into the following formula:
where CtR is defined as the cycle threshold quantification of the DO or UND templates, and CtARS504 is defined as the cycle threshold quantification of the ARS504 locus (a region lacking a TaqI site) for either DO or UND templates as indicated.
For measurement of intragenic or intergenic interactions, anchor primers were paired with primers abutting TaqI sites lying on either the same or another gene. To circumvent the possibility of 3C products arising from crosslink-independent ligation, all 3C primers were designed in tandem orientation (sequences of 3C primers are provided in Table S3). Cycle threshold values (Ct) for digested only (DO3C) and ligated (Lig3C) templates for crosslinked chromatin were determined for each tandem primer combination. Likewise, Ct values were determined for ligated and digested only genomic DNA (LiggDNA and DOgDNA, respectively) and incorporated into the following formula:
Note (i): ΔCt values were obtained by subtracting Ct (no-template) from those of Lig3C, DO3C, LiggDNA or DOgDNA templates as indicated.
Note (ii): 2−ΔCt/ARS504 are the fold-over signals normalized to ARS504 locus.
Note (iii): Ligation-dependent signals were calculated as the ratio of fold-over normalized signals of Lig3C and DO3C templates (2−ΔCtLig3C/ARS504Lig3C)/(2−CtDO3C/ARS504DO3C); the same applies to the gDNA control.
Note (iv): Ligation-dependent signals were corrected for variation in TaqI digestion efficiencies of sites 1 and 2 (as described above).
Note (v): The Normalized Frequency of Interaction is defined as the ratio of ligation-dependent signals of 3C and gDNA control templates after correcting for differences in their digestion efficiencies.
Note (vi): Normalized interaction frequencies ≤ 0.1 are indistinguishable from background.
Statistical tests used
Student’s t test (one-tailed) was used to calculate statistical significance between all pairwise comparisons (as assumptions of parametric distributions were fulfilled) with two exceptions. First, in Figure 3A, one-way ANOVA followed by Tukey’s post hoc analysis was used as more than two groups were compared. Second, in Figure 3B, Wilcoxon Rank Sum test was used as the population distributions were skewed.
Each pairwise comparison was done using means of two independent biological samples, with significance indicated as: n.s., p > 0.05; *, p < 0.05; **, p < 0.01; and ***, p < 0.001.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE122666.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit Polyclonal anti-Hsf1 | Gross Lab | N/A |
| Rabbit Polyclonal anti-Rpb1 | Gross Lab | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| cOmplete, EDTA-free Protease Inhibitor Cocktail | Sigma Aldrich | Cat#11873580001 |
| Formaldehyde | Fisher Scientific | Cat#F79-500 |
| Power SYBR Green PCR Master Mix | Fisher Scientific | Cat#4367660 |
| Protein A Sepharose Beads | GE Life Sciences | Cat#17096303 |
| Proteinase K | Sigma Aldrich | Cat#P2308-100G |
| Quick T4 DNA Ligase | New England BioLabs | Cat#M2200L |
| Rapamycin | LC Laboratories | Cat#R5000 |
| RNase DNase Free | Sigma Aldrich | Cat#11579681001 |
| Taqα I | New England BioLabs | Cat#R0149 |
| Critical Commercial Assays | ||
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Cat#4368814 |
| NEBNext Ultra DNA Library Prep Kit for Illumina | New England BioLabs | Cat#E7370 |
| NEBNext Ultra RNA Library Prep Kit | New England BioLabs | Cat#E7530S |
| RNeasy MinElute Cleanup Kit | QIAGEN | Cat#74204 |
| Deposited Data | ||
| Hsf1 ChIP-seq | (Pincus et al., 2018) | GEO# GSE117653 |
| Hsf1-AA RNA-seq | This paper | GEO# GSE122666 |
| Experimental Models: Organisms/Strains | ||
| Saccharomyces cerevisiae | This paper | N/A |
| Strain background: BY4741 and W303 | ||
| Yeast strains, see Table S2 | ||
| Oligonucleotides | ||
| Primers for 3C analysis, see Table S3 | This paper | N/A |
| Primers for ChIP analysis, see Table S3 | This paper | N/A |
| Primers for strain construction, see Table S3 | This paper | N/A |
| Primers for RT-qPCR, see Table S3 | This paper | N/A |
| Recombinant DNA | ||
| Plasmid pSH47 | Euroscarf | P30119 |
| Plasmid pSR14 | S. Gasser Lab | N/A |
| Plasmid pUG6 | Euroscarf | P30114 |
| Plasmid pUG73 | Euroscarf | P30118 |
| Software and Algorithms | ||
| Bowtie2 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml | N/A |
| DESeq2 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html | N/A |
| HTSeq-Count | https://www-huber.embl.de/HTSeq | N/A |
| ImageJ 1.48v | https://imagej.nih.gov/ij/ | N/A |
| MACS2 | https://pypi.org/pypi/MACS2/ | N/A |
| Slidebook Software, v4 and v6 | https://www.intelligent-imaging.com/slidebook | N/A |
| Tophat | http://ccb.jhu.edu/software/tophat/index.shtml | N/A |
| Other | ||
| Bioptechs Objective Heater System | Bioptechs | Cat#150803 and Cat#150819-19 |
| Bioruptor Plus | Diagenode | Cat#B01020001 |
Highlights.
Hsf1, but not Msn2/Msn4, drives gene coalescence in response to heat shock
Hsf1-target genes coalesce, while genes interposed between them do not
Ectopic targeting of Hsf1 drives coalescence of a heterologous gene
Preventing Hsf1 binding to a natural HSP gene selectively obviates its coalescence
ACKNOWLEDGMENTS
We thank Jayamani Anandhakumar and Michael Guertin for experimental and bioinformatics assistance, respectively; KellyTatchell for assistance with fluorescence microscopy; Rini Ravindran, Ishita Ghosh, and Vickky Pandit for strain construction; and Jason and Donna Brickner, Susan Gasser, Frank Holstege, Amy MacQueen, Anne Cornelis Meinema, Francois Robert, and Kelly Tatchell for generous gifts of strains and plasmids. This work was supported by grants from the National Science Foundation (MCB-1025025, MCB-1518345) and the NIH (R15GM128065) to D.S.G., Ike Muslow predoctoral fellowships to S.C. and A.S.K., and an NIH Early Independence Award (DP5 OD017941) to D.P.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.034.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Jiang and Pugh (2009).
REFERENCES
- Backlund MP, Joyner R, Weis K, and Moerner WE (2014). Correlations of three-dimensional motion of chromosomal loci in yeast revealed by the double-helix point spread function microscope. Mol. Biol. Cell 25, 3619–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beagrie RA, Scialdone A, Schueler M, Kraemer DC, Chotalia M, Xie SQ, Barbieri M, de Santiago I, Lavitas LM, Branco MR, et al. (2017). Complex multi-enhancer contacts captured by genome architecture mapping. Nature 543, 519–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner DG, Sood V, Tutucci E, Coukos R, Viets K, Singer RH, and Brickner JH (2016). Subnuclear positioning and interchromosomal clustering of the GAL1–10 locus are controlled by separable, interdependent mechanisms. Mol. Biol. Cell 27, 2980–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casolari JM, Brown CR, Komili S, West J, Hieronymus H, and Silver PA (2004). Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell 117, 427–439. [DOI] [PubMed] [Google Scholar]
- Chi Y, Huddleston MJ, Zhang X, Young RA, Annan RS, Carr SA, and Deshaies RJ (2001). Negative regulation of Gcn4 and Msn2 transcription factors by Srb10 cyclin-dependent kinase. Genes Dev 15, 1078–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho WK, Spille JH, Hecht M, Lee C, Li C, Grube V, and Cisse II (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong S, Dugast-Darzacq C, Liu Z, Dong P, Dailey GM, Cattoglio C, Heckert A, Banala S, Lavis L, Darzacq X, et al. (2018). Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361, eaar2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhary S, Kainth AS, and Gross DS (2017). Heat shock protein genes undergo dynamic alteration in their three-dimensional structure and genome organization in response to thermal stress. Mol. Cell. Biol 37, e00292–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisse II, Izeddin I, Causse SZ, Boudarene L, Senecal A, Muresan L, Dugast-Darzacq C, Hajj B, Dahan M, and Darzacq X (2013). Real-time dynamics of RNA polymerase II clustering in live human cells. Science 341, 664–667. [DOI] [PubMed] [Google Scholar]
- Cournac A, Marie-Nelly H, Marbouty M, Koszul R, and Mozziconacci J (2012). Normalization of a chromosomal contact map. BMC Genomics 13, 436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekker J, and Mirny L (2016).The3D genome as moderator of chromosomal communication. Cell 164, 1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobi KC, and Winston F (2007). Analysis of transcriptional activation at a distance in Saccharomyces cerevisiae. Mol. Cell. Biol 27, 5575–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du M, Zhang Q, and Bai L (2017). Three distinct mechanisms of long-distance modulation of gene expression in yeast. PLoS Genet 13, e1006736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, and Noble WS (2010). A three-dimensional model of the yeast genome. Nature 465, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elfving N, Chereji RV, Bharatula V, Björklund S, Morozov AV, and Broach JR (2014).Adynamic interplay of nucleosome and Msn2 binding regulates kinetics of gene activation and repression following stress. Nucleic Acids Res 42, 5468–5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eser U, Chandler-Brown D, Ay F, Straight AF, Duan Z, Noble WS, and Skotheim JM (2017). Form and function of topologically associating genomic domains in budding yeast. Proc. Natl. Acad. Sci. USA 114, E3061–E3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X, Chou DM, and Struhl K (2006). Activator-specific recruitment of Mediator in vivo. Nat. Struct. Mol. Biol 13, 117–120. [DOI] [PubMed] [Google Scholar]
- Fanucchi S, Shibayama Y, Burd S, Weinberg MS, and Mhlanga MM (2013). Chromosomal contact permits transcription between coregulated genes. Cell 155, 606–620. [DOI] [PubMed] [Google Scholar]
- Gomez-Pastor R, Burchfiel ET, and Thiele DJ (2018). Regulation of heat shock transcription factors and their roles in physiology and disease. Nat. Rev. Mol. Cell Biol 19, 4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampsey M, Singh BN, Ansari A, Lainé JP, and Krishnamurthy S (2011). Control of eukaryotic gene expression: gene loops and transcriptional memory. Adv. Enzyme Regul 51, 118–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H, Nishikawa J, and Laemmli UK (2008). The anchor-away technique: rapid, conditional establishment of yeast mutant phenotypes. Mol. Cell 31, 925–932. [DOI] [PubMed] [Google Scholar]
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, and Sharp PA (2017). A phase separation model for transcriptional control. Cell 169, 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Schuijers J, Li CH, and Young RA (2018). Regulation and dys-regulation of chromosome structure in cancer. Annu. Rev. Cancer Biol 2, 21–40. [Google Scholar]
- Hsieh TH, Weiner A, Lajoie B, Dekker J, Friedman N, and Rando OJ (2015). Mapping nucleosome resolution chromosome folding in yeast by Micro-C. Cell 162, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, and Pugh BF (2009). A compiled and systematic reference map of nucleosome positions across the Saccharomyces cerevisiae genome. Genome Biol 10, R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly C, Morimoto R, Robert-Nicoud M, and Vourc’h C (1997). HSF1 transcription factor concentrates in nuclear foci during heat shock: relationship with transcription sites. J. Cell Sci 110, 2935–2941. [DOI] [PubMed] [Google Scholar]
- Kim S, and Gross DS (2013). Mediator recruitment to heat shock genes requires dual Hsf1 activation domains and Mediator tail subunits Med15 and Med16. J. Biol. Chem 288, 12197–12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Liachko I, Brickner DG, Cook K, Noble WS, Brickner JH, Shendure J, and Dunham MJ (2017). The dynamic three-dimensional organization of the diploid yeast genome. eLife 6, e23623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson AG, Elnatan D, Keenen MM, Trnka MJ, Johnston JB, Burlingame AL, Agard DA, Redding S, and Narlikar GJ (2017). Liquid droplet formation by HP1a suggests a role for phase separation in heterochromatin. Nature 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Hsiung CC, Huang P, Raj A, and Blobel GA (2015). Dynamic enhancer-gene body contacts during transcription elongation. Genes Dev 29, 1992–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Ruan X, Auerbach RK, Sandhu KS, Zheng M, Wang P, Poh HM, Goh Y, Lim J, Zhang J, et al. (2012). Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148, 84–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell JA, and Fraser P (2008). Transcription factories are nuclear sub-compartments that remain in the absence of transcription. Genes Dev 22, 20–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papantonis A, Kohro T, Baboo S, Larkin JD, Deng B, Short P, Tsutsumi S, Taylor S, Kanki Y, Kobayashi M, et al. (2012). TNFα signals through specialized factories where responsive coding and miRNA genes are transcribed. EMBO J 31, 4404–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Xiang Y, Feng X, and Garrard WT (2014). Pronounced cohabitation of active immunoglobulin genes from three different chromosomes in transcription factories during maximal antibody synthesis. Genes Dev 28, 1159–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pincus D, Anandhakumar J, Thiru P, Guertin MJ, Erkine AM, and Gross DS (2018). Genetic and epigenetic determinants establish a continuum of Hsf1 occupancy and activity across the yeast genome. Mol. Biol. Cell 29,3168–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Protter DSW, and Parker R (2016). Principles and properties of stress granules. Trends Cell Biol 26, 668–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutledge MT, Russo M, Belton JM, Dekker J, and Broach JR (2015). The yeast genome undergoes significant topological reorganization in quiescence. Nucleic Acids Res 43, 8299–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabari BR, Dall’Agnese A, Boija A, Klein IA, Coffey EL, Shrinivas K, Abraham BJ, Hannett NM, Zamudio AV, Manteiga JC, et al. (2018). Co-activator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfelder S, Sexton T, Chakalova L, Cope NF, Horton A, Andrews S, Kurukuti S, Mitchell JA, Umlauf D, Dimitrova DS, et al. (2010). Preferential associations between co-regulated genes reveal a transcriptional interactome in erythroid cells. Nat. Genet 42, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solís EJ, Pandey JP, Zheng X, Jin DX, Gupta PB, Airoldi EM, Pincus D, and Denic V (2016). Defining the essential function of yeast Hsf1 reveals a compact transcriptional program for maintaining eukaryotic proteostasis. Mol. Cell 63, 60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorger PK, and Pelham HRB (1988). Yeast heat shock factor is an essential DNA-binding protein that exhibits temperature-dependent phosphorylation. Cell 54, 855–864. [DOI] [PubMed] [Google Scholar]
- Strom AR, Emelyanov AV, Mir M, Fyodorov DV, Darzacq X, and Karpen GH (2017). Phase separation drives heterochromatin domain formation. Nature 547, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayachandran V, Reja R, Rossi MJ, Park B, Rieber L, Mittal C, Mahony S, and Pugh BF (2018). Widespread and precise reprogramming of yeast protein-genome interactions in response to heat shock. Genome Res 28, 357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendt KS, and Grosveld FG (2014). Transcription in the context of the 3D nucleus. Curr. Opin. Genet. Dev 25, 62–67. [DOI] [PubMed] [Google Scholar]
- Zhao J, Herrera-Diaz J, and Gross DS (2005). Domain-wide displacement of histones by activated heat shock factor occurs independently of Swi/Snf and is not correlated with RNA polymerase II density. Mol. Cell. Biol 25, 8985–8999. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA-seq data reported in this paper is GEO: GSE122666.