

Abstract
This paper describes the synthesis and characterization of a class of highly stretchable and degradable semiconducting polymers. These materials are multi-block copolymers (BCPs) in which the semiconducting blocks are based on the diketopyrrolopyrrole (DPP) unit flanked by furan rings and the insulating blocks are poly(ε-caprolactone) (PCL). The combination of stiff conjugated segments with flexible aliphatic polyesters produces materials that can be stretched >100%. Remarkably, BCPs containing up to 90 wt% of insulating PCL have the same field-effect mobility as the pure semiconductor. Spectroscopic (ultraviolet-visible absorption) and morphological (atomic force microscopic) evidence suggests that the semiconducting blocks form aggregated and percolated structures with increasing content of the insulating PCL. Both PDPP and PCL segments in the BCPs degrade under simulated physiological conditions. Such materials could find use in wearable, implantable, and disposable electronic devices.
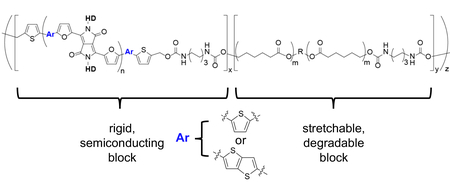
Table of Contents Figure. For table of contents use only.
Introduction
Semiconducting polymers and biomacromolecules share few characteristics beyond the fact that their molecular structures contain carbon and thus that they share the classification of “organic.” Electronic materials that combine semiconducting properties with properties inspired by biological tissue, however, have the potential to enable a new class of wearable, implantable, and disposable devices. Mechanical compliance commensurate with that of the skin and internal organs could permit seamless interfaces between synthetic materials and these biological structures.1–4 Moreover, the property of biodegradability could eliminate the need for surgeries to remove implanted devices, and could reduce the environmental impact of their disposal. Currently, the most successful approach to endowing medical devices with stretchability and biodegradability is based on silicon nanomembranes and metallic interconnects.5 Semiconducting polymers, on the other hand, offer the possibilities of synthetic tunability, oxide-free interfaces, mechanical toughness, detection of a range of physiological signals, and the potential for scalable manufacturing by printing.6 This article describes highly stretchable and biodegradable multi-block copolymers based on diketopyrrolopyrrole (DPP)—a component of semiconducting polymers that have achieved high charge-carrier mobilities (~1 cm2 V−1 s−1) in organic field-effect transistors (OFETs)7,8—and poly(ε-caprolactone) (PCL), an aliphatic polyester that degrades under simulated physiological conditions.9 Remarkably, one of the materials synthesized exhibits no decrease in charge-carrier mobility in OFET even with insulating PCL content up to 90 wt%. The invariance in mobility suggests that stretchability and biodegradability can be incorporated into semiconducting polymers described.
One of the long-purported advantages of organic semiconductors is mechanical compliance, i.e., flexibility and stretchability. Semiconducting polymers in particular are logical candidates for applications demanding deformability because polymer chains form entanglements, which produce the extensibility and toughness characteristics of plastics. The rigidity of the π-conjugated units, the semi-crystalline microstructure, and the generally high glass transition temperatures, however, are usually incompatible with mechanical softness. Several approaches have been used to circumvent the apparent incompatibility of semiconducting behavior and mechanical softness.10 One goal of this research is to achieve stretchability ≥50%, which is similar to that of human skin.11 One approach to improving the mechanical robustness of polymer semiconductors is to blend amorphous insulating polymer with semiconducting polymers.12–15 Blending, however, leads to large-scale phase separation, which may limit charge transport. Alternatively, polymer semiconductors can be synthetically modified to improve their mechanical properties (e.g., engineering the backbone and side chain) to produce single-component materials not susceptible to phase separation.16–19
Devices that degrade into harmless byproducts have the potential to reduce surgeries for removal, which will be desirable given the risk of hospital-borne illness. Moreover, materials that degrade in the environment have the potential to decrease e-waste. Recently, the Bao laboratory reported a degradable low-bandgap semiconducting polymer with a high charge-carrier mobility.20 This material was condensed through imine linkages, which require acidic conditions to hydrolyze. Moreover, the mechanical properties of these materials were not reported.
In this work, we designed a new class of stretchable and biodegradable semiconductors whose defining structural motif is a segmented copolymer with semiconducting conjugated segments linked by soft, biodegradable PCL segments. We measured the mechanical properties, field-effect mobility, and biodegradability of these polymers in phosphate buffers solution. By tuning the structure of the semiconducting segment, it was possible to retain interconnected pathways for charge transport even with high loadings of the insulating fraction. The approach shown in this work could be extended to various conjugated polymers that are synthesized by coupling polymerizations.21,22
Experimental
Synthesis of Conjugated Blocks.
For the BCP-T blocks, DPP-Br (499 mg, 0.570 mmol), 2,5-bis(trimethylstannyl)-thiophene (T) (247 mg, 0.600 mmol), 5-bromo-2-thiophenemethanol (T-OH) (11.9 mg, 0.060 mmol), tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3, 2.7 mol %), and tri(o-tolyl)phosphine (P(o-tol)3, 10.7 mol %) were charged within a 25 mL 3-necked flask, cycled with nitrogen, and subsequently dissolved in 10 mL of degassed chlorobenzene (CB). The mixture was stirred for 20 h at 110 °C. The reaction mixture was allowed to cool to 25 °C, 5 mL of CHCl3 were added, and the polymer precipitated into methanol (200 mL). The precipitate was purified via Soxhlet extraction for 4 h with methanol and 6 h with acetone, followed by collection in CHCl3. The polymer T-0 was obtained as a dark blue solid (428 mg). A similar procedure for the BCP-TT blocks is reported in the Supporting Information.
Synthesis of Soft Blocks.
ε-Caprolactone (ε-CL) (2.28 g, 20.0 mmol) and 1,6-hexanediol (HDO) (11.8 mg, 0.10 mmol), were charged within a 25 mL round-bottom flask, cycled with nitrogen and subsequently dissolved in 5 mL of degassed dichloromethane (CH2Cl2). Then, a 1.0 wt% CH2Cl2-containing solution of trifluoromethanesulfonimide (HNTf2) (2.811mg, 0.010mmol) was added. The mixture was stirred for 68 h at 25 °C. After that, 5 mL of CHCl3 were added to the reaction mixture, and the polymer precipitated into methanol (90 mL). The precipitate was collected via centrifugation. The polymer PCL was obtained as a white solid (428 mg).
Synthesis of Segmented Copolymer.
For T-50: T-0 (75 mg, 3.7 mmol) and PCL (75 mg, 4.1 mmol) were charged within a 10 mL round-bottom flask, cycled with nitrogen, and subsequently dissolved in 1.5 mL of degassed CHCl3. Then, a 0.5 wt% CHCl3-containing solution of hexamethylene diisocyanate (HMDI) (1.30 mg, 7.8 mmol) was added. The mixture was stirred for 190 h at 50 °C. The polymer was precipitated into methanol (80 mL). The precipitate was collected via centrifugation. The polymer T-50 was obtained as a dark blue solid (144 mg). The remaining BCPs were obtained by changing the weight fraction of PCL and/or using TT-0.
Preparation of Substrates for Mechanical Testing.
Glass slides, cut into squares (1 in × 1 in) with a diamond-tipped scribe, were used as substrates for the polymer thin films. The slides were thoroughly cleaned in an ultrasonic bath in the following sequence of 10 min steps: powdered Alconox detergent dissolved in DI water, DI water only, acetone, and then IPA. After that, the slides were dried with compressed (house) air and then treated with plasma (30 W) for 5 min at a base pressure of 200 mTorr of air to remove residual organic debris and improve surface wettability.
Preparation of Films.
Solutions of BCPs in CHCl3 (10 mg mL−1) were prepared and allowed to stir overnight. Prior to use, all solutions were slightly heated (~15 s) with a heat gun to promote dissolution of the polymer; the solutions were then filtered with 0.45 μm PTFE filters, immediately after which they were spin-coated (Headway Research PWM32) onto the cleaned glass substrates at 1000 rpm (ramping at 500 rpm s−1) for 120 s. These conditions produced films of thicknesses ranging from 100 to 150 nm as determined by profilometry (Dektak 150 Surface Profiler). Absorption maxima for the BCP-T polymers: λmax T-0 = 798 nm, λmax T-50 = 796 nm, λmax T-75 = 791 nm, λmax T-90 = 789 nm. Absorption maxima for the BCP-TT polymers: λmax TT-0 = 794 nm, λmax TT-50 = 794 nm, λmax TT-75 = 794 nm, λmax TT-90 = 794 nm. Full absorption spectra are available in Figure S38.
Testing of Mechanical Properties.
We examined cracking behavior of thin film using a linear actuator and optical microscopy. Spin-coated thin films were removed from plasma-treated glass substrates and floated on a water surface. The films on water were transferred to stretchable PDMS (Sylgard 184, Dow Corning) substrates. PDMS was prepared according to the manufacturer’s instructions at a ratio of 10:1 (base:crosslinker) and cured at room temperature for 36 to 48 h prior to use for mechanical testing. For our stress-strain curves, we use the film-on-water technique previously described by Kim and coworkers.28,29
Measurement of Field-Effect Mobility.
Highly doped n-type Si wafers (0.001–0.005 Ω-cm) with 300 nm of thermal oxide were sonicated for 10 min each in detergent solution (2% Alconox in deionized (DI) water), DI water only, and isopropyl alcohol (IPA). The substrates were then treated with plasma (30 W) for 5 min at a base pressure of 200 mTorr, then placed in a 0.33 vol.% solution of octadecyltrichlorosilane (OTS) in cyclohexane for 18 min. Samples were rinsed with cyclohexane and then sonicated for 10 min in chloroform (CHCl3) before spinning a 10 mg/mL solution of PDPP-b-PCL in CHCl3 at 1500 rpm for 30 seconds. Samples were annealed at 100 °C for 1 h. 40 nm thick Au electrodes were then thermally evaporated with a length of 1000 μm and width of 30 μm. Samples were tested on a Keithley 4200.
Results and Discussion
For the conjugated segments, we chose a polymer based on the N-substituted DPP unit flanked by two furan rings (Figure 1a).23 The DPP unit and furan rings are susceptible to decomposition by acids, peroxides, and enzymes.20,24 We chose PCL for the flexible segment since PCL exhibits high elongation at break (>1,000%) and biodegradability. Our strategy to synthesize BCPs involved connecting PDPP and PCL (PDPP-b-PCL) at the respective end-hydroxyl units using diisocyanate linking agents (Scheme S1, Supporting Information). First, hydroxyl end-capped PDPP were synthesized by Stille polymerization of dibromo DPP with thiophene (T) or thienothiophene (TT) bis-stannane in the presence of 5-bromo-2-thiophenemethanol as an end-capping agent.25 Next, PCL was synthesized by ring-opening polymerization using 1,6-hexanediol to afford hydroxyl-terminated chains.26 The molecular weight of the PDPPs and PCL we obtained were comparable. Finally, the PDPP and PCL segments connected with hexamethylene diisocyanate to form urethane linkages.27 Since formation of the urethane linkage does not generate by-products nor requires a catalyst, the BCPs were easily purified. After final reaction, we confirmed the increase of weight-average molecular weight by GPC using both refractive index and UV-Vis detectors, the formation of urethane linkages by NMR and the ratio of PDPP to PCL by NMR (Table S1 and Figures S22 to S36, Supporting Information). The synthetic strategy does not exclude the formation of polymer chains arising from homocoupling.
Figure 1.

(a) Determination of the effect of PCL content in PDPP-b-PCL on mechanical properties. Stress-strain curves were obtained from films suspended on water. The photographs illustrate fracture behavior, scale bar, 1 cm. (b) Optical micrographs of the cracking behavior of BCP and blend films on PDMS at 100% strain, 100 cycles, scale bar, 500 μm.
The tensile response of organic semiconductors is an important predictor of their durability in actual devices.10 In particular, a high elongation at break increases the robustness of devices against fracture.19 To measure the mechanical properties of the BCPs, we first used a “film-on-water” (FOW) technique originally described by Kim and coworkers.28,29 Spin-coated films were removed from a glass substrate, floated on the surface of water, and subjected to tensile pull testing to obtain the stress-strain curves shown in Figure 1b. The improvement in mechanical robustness with increasing PCL content is clearly observed; the toughness increased by a factor of 22 for the sample containing 75 wt% of PCL compared to T-0 (Table S2, Supporting Information). Furthermore, images of the breaking point, shown in the inset of Figure 1b, demonstrate that increasing the PCL content leads to fracture with greater ductile character. Importantly, the films of T-50 and T-75 could withstand over 50% elongation without fracturing.
Since semiconducting materials are fabricated on elastomeric substrates in stretchable devices,1 we also measured the micro-crack behavior on poly(dimethylsiloxane) (PDMS, Figure 1c). Thin, spin-coated films were removed from a glass substrate, floated on the surface of water, transferred to PDMS, and dried with compressed air. The fracture behavior of BCPs on elastomers is correlated with the film-on-water measurements, and T-50 and T-75 can be effectively stretched to 100% strain on PDMS without resulting in any visible cracks. In comparison, films of physical blends of T-0 and PCL 50/50 wt% (T-blend-50) show pervasive cracking at 100% strain. These results reflect the superiority of BCPs (i.e., covalently bonded polymers)—as opposed to physical blends—for improved mechanical properties. We also applied this synthetic strategy to another DPP segment in which the thiophene unit (T) was replaced with the fused thienothiophene (TT) unit (TT-0). DPP units coupled through the TT unit usually show higher charge carrier mobility than those coupled through the T unit, but are less flexible.30 Using our approach, we again observed significant improvement in stretchability, with homogeneous films achieving over 100% strain without cracking (TT-50).
The charge-carrier mobility of semiconducting materials is an important metric for device performance. Unfortunately, many molecular design strategies that improve the stretchability of materials often reduce their mobility.10 We measured charge-carrier mobility of our materials using top-contact, bottom-gate field-effect transistors (FETs) after annealing at 100 °C for 1 h (Figure 2a). For the devices based on BCP-T, we observed that the mobility declined with increasing PCL content: the mobility of T-0 was an order of magnitude higher than that of T-90 (90 wt% PCL). In contrast, the devices fabricated with BCP-TTs showed the remarkable result of no appreciable change in mobility at any fraction up to 90 wt% PCL. We hypothesized that this difference in electronic performance between the segmented BCPs was due to the morphological differences between the materials.31
Figure 2.

Characterization of electronic performance and morphology. (a) Schematic illustration of OFET structure and relationships between charge-carrier mobility and PCL content, standard deviations of three distinct measurements. (b) AFM height images of thin films after immersion in 0.5 M NaOH solution, scale bar, 300 nm. (c) UV-vis spectra showing the absorbance of BCPs thin films with different PCL content after annealing at 100 °C for 1 h, normalized to the A0–1 peak of the pure conjugated block (T-0 or TT-0), (d) Relationship between relative heights of vibronic peak (A0–0/A0–1) and PCL content.
To determine what morphological differences could account for such different electronic behavior with high loadings of PCL, we characterized the morphology of the films using atomic force microscopy (AFM). Since the surface morphologies of as-spun films resulted in images in which the two phases were poorly differentiated, we observed micro-phase separation behavior of PDPP segments after selectively etching the PCL segments by exposing the BCP films to a solution of 0.5 M sodium hydroxide in water/methanol = 60/40 (volume/volume)% (0.5 M NaOH solution). The removal of PCL was monitored by1H NMR spectroscopy. As shown in Figure 2b, comparison of the AFM height images revealed that the PDPP segments in TT-75 thin film formed more percolated, fiber-like networks than did the segments in T-75 films, which formed disjointed globular phases. This morphological observation is consistent with the differences in electronic behavior of the two BCP systems and can, in part, be attributed to an increase in the stiffness of the PDPP segments containing TT units.
Charge transport through the PDPP phase is strongly influenced by the ordering of PDPP segments. This molecular-scale ordering can be correlated to the state of photophysical aggregation, which is observable through ultraviolet-visible (UV-vis) absorption spectroscopy. To analyze the aggregation of PDPP segments in BCPs, we measured the UV-vis spectra of BCP films after annealing at 100 °C for 1 h (Figure 2c, d). As we increased PCL content in the BCP-TTs, we observed a red shift in absorbance of PDPP segments and an increase in the relative height of the 0−0 to the 0−1 vibronic peaks (A0–0/A0–1), which correlated with the strength of intrachain J-aggregation.32,33 This trend implies that the aggregation of PDPP segments in the BCP-TTs was enhanced by PCL. On the contrary, the BCP-Ts show a decrease in A0–0/A0–1 as well as a blue shift in absorbance, indicating a disturbance of aggregation of PDPP segments by the addition of PCL.
In vitro degradation experiments were performed to investigate the degradation rate of BCP-T (Figure 3a). The amount of residual PCL was calculated by1H NMR by comparing the peak integration of PCL segments (4.1 ppm) to that of hexyldecyl groups in PDPP segments (0.8 ppm). The PCL segments in T-50 were completely degraded in a 0.5 M NaOH solution in less than 3 days, confirming that PCL segments in the BCP-Ts can be fully degraded. However, immersion of T-50 in phosphate buffered saline (PBS, pH = 7.4)34 at physiological temperature (37 °C) only degraded the PCL segments of T-50 to 48% of their original content after 12 weeks.
Figure 3.

Characterization of BCP-films degradability. In vitro degradation of a T-50 thin film for different immersion times in PBS at 37 °C and in 0.5 M NaOH solution at 25°C. (a) Plot of residual PCL over time, (b) UV-vis spectra of T-0 and T-50 in chloroform for pristine films (solid line) and after 12 weeks of immersion in PBS (dotted line) normalized to the A0–1 peak of the pristine film, with the A0–0 peak present at 798 nm and the A0–1 peak present at 720 nm, (c) AFM height images of thin films before and after 12 weeks of immersion in PBS. Ra is the average surface roughness of the scanned area, scale bar, 1 μm.
While the purpose of incorporating the hydrolyzable polyester segments was to confine the degradation to these flexible units, we could not discount the possibility that the conjugated PDPP units were also degraded by PBS. To quantify the degradation of the conjugated segments, we performed the following experiment. First, we immersed a thin film of T-0 in PBS for 12 weeks. We then dissolved the film in chloroform and obtained UV-vis spectra. We found that the ratio of the absorbance peaks, A0–0/A0–1, decreased by 6% after this treatment (Figure 3b). This result suggests that the conjugated unit itself was partially degraded under these conditions. Although we were not able to determine the identity of the products of degradation, we hypothesize that this degradation is the result of the reaction between the furan units and singlet oxygen generated from light and oxygen.35 Again using the ratio A0–0/A0–1 as a metric, degradation of the conjugated units of T-50 was twice as great as for T-0. The difference may be because of faster degradation kinetics of the conjugated units due to an increase in surface area of the PDPP segments upon decomposition of the PCL.
AFM height images further supported our observations regarding degradation of PDPP and PCL segments (Figure 3c). The roughness of T-0 films increased after 12 weeks of immersion in PBS; we attribute the increase in roughness to the degradation of PDPP segments. On the other hand, the surface morphology of T-50 films dramatically changed compared to T-0 films due to the decomposition of PCL segments. The average surface roughness (Ra) of T-50 films increased to a similar value as the Ra of T-50 films etched with 0.5 M NaOH solution (Figure S3, Supporting Information).
Conclusion
We reported a new class of stretchable and degradable semiconducting polymers based on multi-block copolymers of PDPP and PCL. Given that our approach to stretchability and biodegradability is based on the addition of PCL segments, we believe this approach is extendable to other conjugated polymers. Furthermore, the retention of field-effect mobility in these materials, particularly the BCP-TT series, suggests that biologically inspired properties need not come at the expense of electronic properties. These materials could lead to devices that are not only stretchable, degradable, and disposable, but also those having other properties inspired by biological tissue, namely self-repair.
Supplementary Material
Scheme 1.

Synthetic strategy for BCPs containing DPP-based polymers and an aliphatic polyester. (a) Stille coupling polymerization of DPP-Br, stannane compound, and end-modifier. (b) Ring-opening polymerization of ε-caprolactone (ε-CL) with HDO. (c) Synthesis of BCPs with diisocyanate linker.
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health Director’s New Innovator Award, grant 1DP2EB022358–02 to D.J.L. Additional support was provided by a gift from JSR. This work was performed in part at the San Diego Nanotechnology Infrastructure (SDNI), a member of the National Nanotechnology Coordinated Infrastructure, which is supported by the National Science Foundation (grant ECCS-1542148).
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI:
Synthetic procedure, polymer characterization, additional mechanical properties, AFM images, photograph of cracking behavior, and characterization of OFET (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Chortos A; Bao Z Skin-Inspired Electronic Devices. Mater. Today 2014, 17 (7), 321–331. [Google Scholar]
- (2).Wagner S; Bauer S Materials for Stretchable Electronics. MRS Bull 2012, 37 (03), 207–213. [Google Scholar]
- (3).Keum H; Mccormick M; Liu P; Zhang Y; Omenetto FG Epidermal Electronics. Science (80-.) 2011, 333 (6044), 838–844. [DOI] [PubMed] [Google Scholar]
- (4).Shyu TC; Damasceno PF; Dodd PM; Lamoureux A; Xu L; Shlian M; Shtein M; Glotzer SC; Kotov NA A Kirigami Approach to Engineering Elasticity in Nanocomposites through Patterned Defects. Nat. Mater 2015, 14 (8), 785–789. [DOI] [PubMed] [Google Scholar]
- (5).Kang S; Murphy RKJ; Hwang S; Lee SHSM; Harburg DV; Krueger NA; Shin J; Gamble P; Cheng H; Yu S; et al. Bioresorbable Silicon Electronic Sensors for the Brain. Nature 2016, 530 (7588), 71–76. [DOI] [PubMed] [Google Scholar]
- (6).Yan H; Chen Z; Zheng Y; Newman C; Quinn JR; Dötz F; Kastler M; Facchetti A A High-Mobility Electron-Transporting Polymer for Printed Transistors. Nature 2009, 457 (7230), 679–686. [DOI] [PubMed] [Google Scholar]
- (7).Li J; Zhao Y; Tan HS; Guo Y; Di C-A; Yu G; Liu Y; Lin M; Lim SH; Zhou Y; et al. A Stable Solution-Processed Polymer Semiconductor with Record High-Mobility for Printed Transistors. Sci. Rep 2012, 2, 754–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Kang I; Yun HJ; Chung DS; Kwon SK; Kim YH Record High Hole Mobility in Polymer Semiconductors via Side-Chain Engineering. J. Am. Chem. Soc 2013, 135 (40), 14896–14899. [DOI] [PubMed] [Google Scholar]
- (9).Oh SH; Park IK; Kim JM; Lee JH In Vitro and in Vivo Characteristics of PCL Scaffolds with Pore Size Gradient Fabricated by a Centrifugation Method. Biomaterials 2007, 28 (9), 1664–1671. [DOI] [PubMed] [Google Scholar]
- (10).Root SE; Savagatrup S; Printz AD; Rodriquez D; Lipomi DJ Mechanical Properties of Organic Semiconductors for Stretchable, Highly Flexible, and Mechanically Robust Electronics. Chem. Rev 2017, 117 (9), 6467–6499. [DOI] [PubMed] [Google Scholar]
- (11).Amjadi M; Yoon YJ; Park I Ultra-Stretchable and Skin-Mountable Strain Sensors Using Carbon Nanotubes-Ecoflex Nanocomposites. Nanotechnology 2015, 26 (37), 375501. [DOI] [PubMed] [Google Scholar]
- (12).Song E; Kang B; Choi HH; Sin DH; Lee H; Lee WH; Cho K Stretchable and Transparent Organic Semiconducting Thin Film with Conjugated Polymer Nanowires Embedded in an Elastomeric Matrix. Adv. Electron. Mater 2016, 2 (1), 1500250. [Google Scholar]
- (13).Shin M; Oh JY; Byun KE; Lee YJ; Kim B; Baik HK; Park JJ; Jeong U Polythiophene Nanofibril Bundles Surface-Embedded in Elastomer: A Route to a Highly Stretchable Active Channel Layer. Adv. Mater 2015, 27 (7), 1255–1261. [DOI] [PubMed] [Google Scholar]
- (14).Xu J; Wang S; Wang G-JN; Zhu C; Luo S; Jin L; Gu X; Chen S; Feig VR; To JWF; et al. Highly Stretchable Polymer Semiconductor Films through the Nanoconfinement Effect. Science (80-.) 2017, 355 (6320), 59–64. [DOI] [PubMed] [Google Scholar]
- (15).Scott JI; Xue X; Wang M; Kline RJ; Hoffman BC; Dougherty D; Zhou C; Bazan G; O’Connor BT Significantly Increasing the Ductility of High Performance Polymer Semiconductors through Polymer Blending. ACS Appl. Mater. Interfaces 2016, 8 (22), 14037–14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lipomi DJ; Chong H; Vosgueritchian M; Mei J; Bao Z Toward Mechanically Robust and Intrinsically Stretchable Organic Solar Cells: Evolution of Photovoltaic Properties with Tensile Strain. Sol. Energy Mater. Sol. Cells 2012, 107, 355–365. [Google Scholar]
- (17).Bérenger R; Savagatrup S; De Los Santos NV; Hagemann O; Carlé JE; Helgesen M; Livi F; Bundgaard E; Søndergaard RR; Krebs FC; et al. Mechanical Properties of a Library of Low-Band-Gap Polymers. Chem. Mater 2016, 28 (7), 2363–2373. [Google Scholar]
- (18).Wang GJN; Shaw L; Xu J; Kurosawa T; Schroeder BC; Oh JY; Benight SJ; Bao Z Inducing Elasticity through Oligo-Siloxane Crosslinks for Intrinsically Stretchable Semiconducting Polymers. Adv. Funct. Mater 2016, 26 (40), 7254–7262. [Google Scholar]
- (19).Oh JY; Rondeau-Gagné S; Chiu Y-C; Chortos A; Lissel F; Wang G-JN; Schroeder BC; Kurosawa T; Lopez J; Katsumata T; et al. Intrinsically Stretchable and Healable Semiconducting Polymer for Organic Transistors. Nature 2016, 539 (7629), 411–415. [DOI] [PubMed] [Google Scholar]
- (20).Lei T; Guan M; Liu J; Lin H-C; Pfattner R; Shaw L; McGuire AF; Huang T-C; Shao L; Cheng K-T; et al. Biocompatible and Totally Disintegrable Semiconducting Polymer for Ultrathin and Ultralightweight Transient Electronics. Proc. Natl. Acad. Sci 2017, 114 (20), 5107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Carsten B; He F; Son HJ; Xu T; Yu L Stille Polycondensation for Synthesis of Functional Materials. Chem. Rev 2011, 111 (3), 1493–1528. [DOI] [PubMed] [Google Scholar]
- (22).Miyaura N; Suzuki A Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev 1995, 95 (7), 2457–2483. [Google Scholar]
- (23).Woo CH; Beaujuge PM; Holcombe TW; Lee OP; Fréchet JMJ Incorporation of Furan into Low Band-Gap Polymers for Efficient Solar Cells. J. Am. Chem. Soc 2010, 132 (44), 15547–15549. [DOI] [PubMed] [Google Scholar]
- (24).Carrette LLG; Gyssels E; De Laet N; Madder A Furan Oxidation Based Cross-Linking: A New Approach for the Study and Targeting of Nucleic Acid and Protein Interactions. Chem. Commun 2016, 52 (8), 1539–1554. [DOI] [PubMed] [Google Scholar]
- (25).Robb MJ; Montarnal D; Eisenmenger ND; Ku S; Chabinyc ML; Hawker CJ A One-Step Strategy for End-Functionalized Donor − Acceptor Conjugated Polymers. Macromolecules 2013, 46 (16), 6431–6438. [Google Scholar]
- (26).Kakuchi R; Tsuji Y; Chiba K; Fuchise K; Sakai R Controlled Ring-Opening Polymerization of δ-Valerolactone Using Triflylimide as an Efficient Cationic Organocatalyst. Macromolecules 2010, 43 (17), 7090–7094. [Google Scholar]
- (27).Wang W; Ping P; Yu H; Chen X; Jing Xi. Synthesis and Characterization of a Novel Biodegradable, Thermoplastic Polyurethane Elastomer. J. Polym. Sci. Part A Polym. Chem 2006, 44 (19), 5505–5512. [Google Scholar]
- (28).Kim J-H; Nizami A; Hwangbo Y; Jang B; Lee H-J; Woo C-S; Hyun S; Kim T-S Tensile Testing of Ultra-Thin Films on Water Surface. Nat. Commun 2013, 4, 2520. [DOI] [PubMed] [Google Scholar]
- (29).Kim T; Kim J-H; Kang TE; Lee C; Kang H; Shin M; Wang C; Ma B; Jeong U; Kim T-S; et al. Flexible, Highly Efficient All-Polymer Solar Cells. Nat. Commun 2015, 6 (May), 8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li Y; Singh SP; Sonar P A High Mobility P-Type DPP-Thieno[3,2-b]Thiophene Copolymer for Organic Thin-Film Transistors. Adv. Mater 2010, 22 (43), 4862–4866. [DOI] [PubMed] [Google Scholar]
- (31).Müller C; Goffri S; Breiby DW; Andreasen JW; Chanzy HD; Janssen RAJ; Nielsen MM; Radano CP; Sirringhaus H; Smith P; et al. Tough, Semiconducting Polyethylene-Poly(3-Hexylthiophene) Diblock Copolymers. Adv. Funct. Mater 2007, 17 (15), 2674–2679. [Google Scholar]
- (32).Clark J; Chang J; Spano FC; Friend RH; Silva C; Clark J; Chang J; Spano FC; Friend RH; Silva C Determining Exciton Bandwidth and Film Microstructure in Polythiophene Films Using Linear Absorption Spectroscopy Determining Exciton Bandwidth and Film Microstructure in Polythiophene. Appl. Phys. Lett 2009, 94, 163306. [Google Scholar]
- (33).Bencheikh F; Duché D; Ruiz CM; Simon JJ; Escoubas L Study of Optical Properties and Molecular Aggregation of Conjugated Low Band Gap Copolymers: PTB7 and PTB7-Th. J. Phys. Chem. C 2015, 119 (43), 24643–24648. [Google Scholar]
- (34).Tsuji H; Ikada Y Blends of Aliphatic Polyesters. II. Hydrolysis of Solution-Cast Blends from Poly (L -Lactide) and Poly (e-Caprolactone) in Phosphate-Buffered Solution. J. Appl. Polym. Sci 1998, 67, 405–415. [Google Scholar]
- (35).Cao H; Rupar PA Recent Advances in Conjugated Furans. Chem. - A Eur. J 2017, 23 (59), 14670–14675. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.