

Abstract
Cubane was recently validated as a phenyl ring (bio)isostere, but highly strained caged carbocyclic systems lack π character, which is often critical for mediating key biological interactions. This electronic property restriction associated with cubane has been addressed herein with cyclooctatetraene (COT), using known pharmaceutical and agrochemical compounds as templates. COT either out performed or matched cubane in multiple cases suggesting that versatile complementarity exists between the two systems for enhanced bioactive molecule discovery.
Keywords: agrochemicals, pharmaceuticals, cubane, cyclooctatetraene, bioisostere
Graphical Abstract
COTton on: the potential of cyclooctatetraene (COT) as a bioactive motif was assessed using a portfolio of six pharmaceutical and agrochemical templates. The protean nature and π character of COT was found to compliment the attributes of the already established cubane (bio)isostere concept, and the facile conversion of the latter to the former inaugurate the pair as an effective tool for deployment in bioactive molecule discovery.
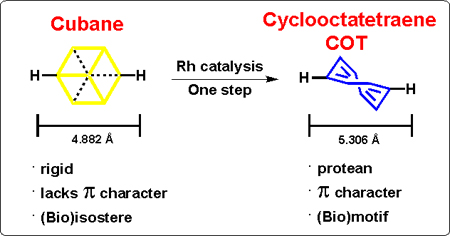
Eaton’s 1992 conjecture,[1] that cubane (1) (Scheme 1) can act as a phenyl ring (bio)isostere,[2] was recently validated by our group using a variety of known pharmaceutical and agrochemical compounds.[3] Considering that cage hydrocarbon motifs (e.g. 1), generally only provide steric replacement attributes,[4] and obviously lack π character, this aspect places substantial limits on bioactive compound discovery efforts using this approach i.e. due to a loss in potential π interactions[5] and/or even critical metabolic reactivity.[6] This situation is further compounded by the fact that contemporary bioactive molecule discovery is attempting to deviate from “flatland” chemical space[7] (i.e. highly planar aromatic compounds arising from ease of sp2 coupling[8]) by adopting cage hydrocarbons for their physical attributes,[9] which introduce increased three dimensionality, increased lipophilicity and outright novelty.
Scheme 1.

Space filling model comparisons of cubane (1), and the ring inversion isomers (D2d 3) of cyclooctatetraene (2, COT shown as one of two π-bond shift isomers); bond distances given in angstroms [M06–2X/6–311+G(d,p)], and angles in degrees. These values are consistent with the available X-ray crystallographic data (see SI). Valence isomer of COT (i.e. 4), and rhodium catalyzed valence tautomerization of cubane to COT, via the intermediate tricyclo[4.2.0.02,5]octa-3,7-diene (5) is also shown
In search of a readily accessible moiety that might deliver both steric and π components, 1,3,5,7-cyclooctatetraene (COT, 2),[10] was identified as a motif that potentially fulfills the desired bimodal criteria. In addition, because of the dynamic equilibrium (shape shifting) that exists between ring inversion (e.g. 3), π-bond shifts and valence tautomerisation (i.e. bicyclo[4.2.0]octatriene 4),[11,12] COT offers a unique protean capability i.e. to potentially access an optimum conformation to engage biological targets in a “skeleton key” fashion (Scheme 1).
A number of synthetic protocols give access to the COT (2) ring system,[13] but a convenient method derives from the rhodium catalyzed valence isomerization of cubane (1),[1] which our group has extensively explored (Scheme 1).[14] This synthetic aspect is not only highly convenient, but allows simultaneous exploration of both cubane and COT frameworks for those working in any bioactive molecule discovery program.
Herein, is disclosed a broad examination of COT as a bioactive motif, using a wide range of known pharmaceutical and agrochemical templates (i.e. for blood coagulation, depression, pain, cancer, parasites, and agricultural pests), with direct comparison to the corresponding cubane system.
Warfarin (6)[15] was initially chosen to explore the COT concept. Although, warfarin is the most widely-used oral anticoagulant for the prevention and treatment of thrombosis in humans globally, it is ranked among the top 10 drugs with serious adverse drug consequences.[16] In addition, controversy over treating individual genotypes of the target enzyme vitamin K epoxide reductase (VKOR),[17] and the fact that warfarin resistance is emerging,[18] make this a crucial pharmaceutical template for investigation.
Authentic warfarin along with cubyl-warfarin (7) and COT-warfarin (8) were prepared (Figure 1) (see SI, Schemes S1–S2). For the cubane (7) and COT (8) analogs both enantiomers were obtained separately (also for warfarin) by preparative enantioselective chromatography. In solution (1H NMR, CDCl3) the hemi-ketal form was partially observed for the cubane case, whereas the COT example existed solely in the hemi-ketal form (i.e. 8) in CDCl3 and a mixture of DMSO:D2O (3:7). Initial VKOR inhibition evaluation was performed using FIXgla-PC/HEK293 reporter cells.[19] Results showed that the potency of inhibition of VKOR activity by (S)-warfarin was about 8-fold higher than (R)-warfarin, which was consistent with reported data.[20] The (S)-cubane analogue demonstrated ca. 3-fold lower activity than the (R)-enantiomer, but overall ca. 10-fold less activity than the warfarin enantiomers (. Pleasingly, however, the racemic COT derivative exhibited a ca. 2-fold increase in activity beyond the more active enantiomer [(S)–isomer] of warfarin (see SI, Figure S4). A subsequent evaluation showed that the (S)-enantiomer of COT-warfarin was 4-fold more potent than the (R)-enantiomer (see SI, Figure S5). Considering that the genetic variation in the VKOR gene is one of the largest factors contributing to the difficulty in the clinical use of warfarin,[16c] the performance of the racemic COT analogue (8) was explored against the 27 naturally occurring human VKOR mutants using VKOR/VKORC1L1 double gene knockout reporter cells[18a,c]. It was found that 8 and warfarin displayed a similar pattern of resistance to all naturally occurring VKOR mutants, suggesting they might bind to the same site in VKOR. Gratifyingly, however, the resistant variation of the COT surrogate to all the naturally occurring mutants was ca. 4-fold less than warfarin(Figure 1, see also SI, Figure S6).
Figure 1:

Comparison of the resistance of warfarin (6) and COT-warfarin (8) against the 27 naturally occurring VKOR mutants. The potency of warfarin and COT-warfarin (8) on VKOR activity inhibition expressed as IC50 (concentrations required for 50% inhibition of VKOR activity) and normalized (resistance for wild-type VKOR is normalized as 1).
Overall, these initial observations suggested that the π rich COT motif had potential to deliver improved activity, in a system where the cubane analogue failed to produce phenyl (bio)isosterism.
Other Pharmaceuticals and Pesticides.
Although the warfarin case provided promising evidence, additional examples were required to validate COT as a viable bioactive motif. Three pharmaceuticals [moclobemide (9), pravadoline (10) and SAHA (11)], an acaracide [benzyl benzoate (12)], and a pesticide [diflubenzuron (13)], were subjected to COT editing (Figure 2).
Figure 2: Additional pharmaceuticals and pesticides investigated.

Moclobemide, pravadoline, benzyl benzoate, diflubenzuron and SAHA and the corresponding cubane (yellow) and COT (blue) analogues. For synthetic procedures and characterization data see the SI.
Synthesis of the COT analogues was simply achieved by first acquiring the cubane analog (14-20), or advanced precursor, followed by treatment with rhodium(I) norbornadiene chloride dimer {[Rh(nbd)Cl]2},[14] which gave the corresponding COT derivatives (21-27) via valence isomerization (see SI, Schemes S3–S7).
The third-generation antidepressant moclobemide (9) was next pursued, as it displays a good selectivity profile towards monoamine oxidase A (MAO-A) i.e. important for patients with dietary requirements.[21] An open field test (OFT) was used to assess the efficacy of 9, 14 and 21 using adult male C57BL6/J mice[22] (see SI, Figures S27–S28). The locomotor profiles of cubane analog 14 were compared with those of 9, where both were observed to decrease the total distance travelled by the same amount compared to the vehicle, indicating (in the OFT model of anxiolytic activity) that the cubane analogue 14 performed equally well as the clinically established antidepressant moclobemide. In comparison, the COT example (21) had a locomotor profile that was indistinguishable from the vehicle (water).
Pravadoline (10) was also evaluated (even though it did not proceed to clinical development beyond Phase 1 clinical trials[23]), because the pharmacological characteristics include an interesting dual mechanism of analgesia (i.e. both cyclooxygenase inhibitory and cannabinoid receptor agonist properties).[24] The cubane and COT analogues (15 and 22) were examined using the Freund’s Complete Adjuvant (FCA) rat model of inflammatory pain in the hindpaw, and both were found to evoke similar pain relief to that of pravadoline itself (see SI, Figures S29–S30).
The histone deacetylase inhibitor SAHA (11, suberanilohydroxamic acid),[25] which is used clinically to treat cutaneous T-cell lymphoma (CTCL),[26] was chosen as the third pharmaceutical comparator. Evaluation of cubane derivative 16 and the COT analogue (23) against breast cancer (MCF7) and melanoma (MM96L) cell lines respectively, showed that 23 (IC50 37±3 and 73±8 ng/ml respectively) was ca. 2-fold more active that 16 (IC50 53±4 and 137±5 ng/ml respectively), although ca. 2-fold less active than SAHA (17±1 and 26±5 ng/ml respectively) (see SI, Figures S31–S34). That said, our earlier in vivo mouse studies showed that SAHA and the cubane derivative 16 had identical activity.[3]
The acaracide benzyl benzoate (12), which is used as a topical treatment of an infectious skin disease caused by scabies mites,[27] previously resulted in considerable losses in activity when interrogated using cubane.[3] All cubane analogs of benzyl benzoate (17-19) were converted in one step into the corresponding COT examples (24-26) (see SI, Figures S35–S37). Benzyl benzoate (12) caused 100% mite mortality within 5 min. At the same concentration, cubane surrogates caused substantially less mortality, even after an exposure time of 24 h. Although still not as effective as 12, the COT analogs (24-26) displayed much higher mortality rates than the cubane analogues (17-19). Specifically, COT replacement of the benzoyl fragment resulted in the highest level of mortality (i.e. 75% at 8 h for COT 26).
When exposed to late instar larvae of laboratory-cultured T. castaneum (rust-red flour beetle; a major pest of stored grain), the cubane analogue (20)[3] outperformed diflubenzuron (13).[28] However, when examined against field strain beetles it was found that COT replacement (i.e. 27) did not outperform diflubenzuron, and was not as effective as cubane (see SI, Figures S38–S39).
Summarizing the six exemplars evaluated. COT-Warfarin (8) outperformed the corresponding cubane counterpart as did COT-SAHA (23) and the benzyl benzoate COT system 26. However, both the COT and cubane surrogates (i.e. 22 and 15) of pravadoline (10) showed essentially equal activity. Of the two remaining cases (i.e. moclobemide and diflubenzuron) the cubane surrogates (i.e. 14 and 20) outperformed the corresponding COT derivatives, but performed equally well against the benzene comparator.
Although the above results demonstrate a compelling case for COT as a bioactive complement to cubane, the remaining questions to address for pharmaceutical or agrochemical discovery concern the physical and metabolic properties of COT.
Lipophilicity.
Log P,[29] and associated lipophilicity measures,[9] are crucial assessment criteria for lead compounds and/or drug candidates. Although we previously used the clog P method,[3] for this study the traditional octanol water partition coefficient determination method was used for increased accuracy (see SI, Figure S1). In the warfarin (6, log P = 2.73; 7, log P = 4.62; 8, log P = 2.82), moclobemide (9, 1.85; 14, 2.53; 21, 1.95) and benzyl benzoate (12, 3.36; 19, 4.22; 26, 3.34) examples, the log P difference between COT and benzene counterparts was minimal (i.e. <0.10), and much less than that seen for the cubane replacements. For pravadoline (10, 3.19; 15, 1.73; 22, 2.52), SAHA (11, 0.99; 16, 1.52; 23, 1.38) and diflubenzuron (13, 3.33; 20, 3.31; 27, 4.36) COT substitution resulted in slightly increased log P values (i.e. 0.39–1.03), but in the case of 10 both COT and cubane systems had a lower log P than the corresponding benzene counterpart. Although no general trend regarding COT replacement can be claimed in relation to lowering or increasing log P, COT and cubane are certainly complementary in that one or the other appears to closely match the benzene counterpart, depending on the molecular and/or functional group context.
Metabolic Stability and Protein Plasma Binding.
For COT editing to be considered viable in bioactive discovery an appreciation of Phase I drug metabolism was required.[30] Therefore, a metabolic stability study was undertaken using both enantiomers of warfarin and the corresponding enantiomers of the COT system (i.e. 8) in the presence of human liver microsomes (see SI, Figures S7–S10). The warfarin enantiomers showed little metabolism in a standard 60 min incubation, with (S)-warfarin having an observed half-life of 210±120 min, and (R)-warfarin showing no significant change. In the case of the two enantiomers of 8 these had similar metabolic stabilities [i.e. (S)-COT had a half-life of 110±38 mins, and the (R)-COT 130±20 mins]. Although the COT enantiomers appear slightly more metabolically labile, warfarin is known to have a long half-life (i.e. 36 h in the human body), whereas other clinically relevant warfarin analogues[31] are more rapidly metabolized, e.g., acenocoumarol (10 h)[32]. Further information on the metabolism of the (S)- and (R)-COT enantiomers was available through coupled liquid chromatography mass spectrometric analysis of the stored human liver microsomal incubates (see SI, Figures S14–S26). Analysis revealed substantial peaks exhibiting an M+16 (oxygen atom addition) in the case of (S)-COT warfarin, which is typical of Phase I cytochrome P450 metabolism and could correspond to a simple hydroxylated metabolite or an epoxide. Warfarin is known to be primarily metabolized by a variety of cytochrome P450 isoforms to monohydroxylated metabolites, although different isoforms process the different enantiomers.[31] Carbonyl reductases also reduce the side chain moiety to the corresponding alcohol in warfarin[31] and significant quantities of an M+2 metabolite were also observed in the metabolism of (R)-COT warfarin, as were M+18 peaks (carbonyl reduction plus hydroxylation). However, any M+34 peak (epoxidation followed by epoxide hydration) was small, consistent with little epoxidation, or interception by glutathione (GSH) etc., as observed for (S)-COT warfarin. Certainly, epoxides of COT[33] are stable, and in the view that non-catalyzed ring opening of epoxides by thiols requires elevated temperatures,[34] these results suggest that the COT ring system is not a particular metabolic liability. It is possible that the variation in metabolic stability is due to differential binding of 8 to a cytochrome P450 (CYP2C9 is responsible for warfarin metabolism) promoting increased hydroxylation of the coumarin ring system, as is the case for acenocoumarol.[32] In addition, the enantiomers of warfarin and COT 8 were observed to bind human plasma proteins similarly (see SI, Tables S1–S8). The extent of binding was similar at 200 and 2000 ng/mL for most of the analytes. Lastly, the pharmacokinetics of substituted COT derivatives certainly warrant continued investigation, however, these results provide optimism in its ability to perform as a complementary motif.
Chemical Synthesis and Stability.
Although, a number of synthetic methods are available,[13] as mentioned above a convenient route to access the COT ring system is via rhodium catalyzed valence isomerization of cubane (1) (Scheme 1), which we recently demonstrated is broad in scope.[14] Furthermore, the COT system is also readily accessible via photochemical reaction of benzene with substituted acetylenes, as successfully utilized by us previously.[13] In terms of chemical stability from a bioactive COT development perspective,1H NMR analysis of COT-warfarin (8), COT-moclobemide (21), and COT diflubenzorun (27) samples, that had been stored at 8 °C for approximately two years, showed no signs of decomposition (see SI). Although, the COT ring system can be converted to semi-bullvalene on UV irradiation in acetone,[35] the photochemical aspects associated with COT synthesis[13] suggest concerns regarding light sensitivity are likely premature. Unlike cubane, COT can form cation−π interactions,[36] but very seldom reported (e.g. alkaline earth metals).[37]
Conformational aspects.
Cubane has a single rigid conformation, whereas COT is in dynamic equilibrium with multiple conformations and isomers (Scheme 1). This protean feature is especially relevant when COT is 1,4-disubstituted i.e. enabling comparison to both 1,3- and 1,4-disubstituted cubane. Inspection of the energy minimized geometries optimized using DFT (see SI, Figures S2–S3) suggests that the conformation of COT with substituents anti is a fair approximation for 1,4-disubstituted cubane, whilst syn is a better approximation for 1,3-disubstituted cubane (Figure 3). Gas-phase free energy calculations place the syn-isomer 0.34 kcal/mol lower in energy than the anti-isomer. This corresponds to a 2:1 ratio at equilibrium (from the Arrhenius equation), which is consistent with the ratio observed experimentally for 1,4-di-t-butyl COT (2:1 syn:anti).[38–40] The free energy difference is such that while syn is slightly preferred at room temperature, both isomers are readily accessible due to facile interconversion by bond-shifting, enabling sampling of both functional group arrangements e.g. in an enzyme active site. A further utility of COT in this context lies in providing an easily accessible alternative to 1,3-disubstituted (or meta) cubanes, which are synthetically demanding.
Figure 3:

A) Energy minimized geometries [M06–2X/6–311+G(d,p)] with selected distances and angles comparing 1,4-disubstituted COT with that of 1,3- and 1,4- disubstituted cubane. Bond distances are in angstroms, angles are in degrees; B) Face centered view of 1,4-COT; C) Angle between C1–C2–C3; D) Dihedral angle of C1 and C4 along the C2–C3 axis. Methyl hydrogen atoms have been omitted for clarity.
Conclusion
In an era of multiple drug discovery platforms, the deployment of (bio)isostere replacement, and the sister concept of scaffold hopping, is prevalent. The benzene, or phenyl, (bio)isostere landscape has been dominated by saturated caged hydrocarbons, which lack the ability to provide π bond donation capabilities. The cyclooctatetraene (COT) motif was examined as a potential π containing complement to cubane (bio)isosterism using the known pharmaceutical and agrichemical templates, warfarin, moclobemide, pravadoline, SAHA, benzyl benzoate and diflubenzuron. The dynamic equilibrium and conformational isomer attributes possessed by COT provide both electronic and steric properties unmatched by classical saturated hydrocarbon systems if a π component is required by the biological partner for optimum binding. This was most evident in the case of the popular anti-coagulant warfarin, and with the synthetic cannabinoid pravadoline responding well to COT editing. In the case of moclobemide it was clear that cubane outperformed COT, further reinforcing complementarity between the two systems i.e. if cubane fails then investigate COT or vice versa. The demonstration herein that the COT (bio)motif acts in concert with cubane, opens additional opportunities to explore chemical space, rejuvenate pioneer drugs, fight resistance and revitalize failed clinical and agrochemical candidates.
Supplementary Material
Acknowledgements
The authors thank The University of Queensland (UQ), University of North Carolina at Chapel Hill, the QIMR Berghofer Medical Research Institute and the CSIRO (Melbourne) for financial support; S.G and A.K were supported financially by Therapeutic Innovation Australia (TIA), and TIA is supported by the Australian Government through the National Collaborative Research Infrastructure Strategy (NCRIS) program. CIPDD research infrastructure was purchased using investment funds from the Queensland Government Smart State Research Facilities Fund as well as from TIA. The Australian Research Council for a Future Fellowship award (grant number FT110100851) to C.M.W. is also gratefully acknowledged, in addition, to the National Health and Medical Research Council of Australia (NHMRC). S.D.H. gratefully acknowledges the Northcote Trust and Britain-Australia Society for their award of the Northcote Graduate Scholarship. J.M.B is indebted to The University of Queensland Research Computing Centre, the Queensland Cyber Infrastructure Foundation (QCIF) and the National Computational Infrastructure (NCI, supported by the Australian Government) for access to supercomputing resources. We thank A/Prof. Bryan Fry (School of Biological Sciences, UQ), Prof. Jenny Martin (University of Newcastle) and Ms. Pauline Ko (Queensland Brain Institute, UQ) for valuable discussions. Dr Kate Mounsey, from the University of Sunshine Coast, who facilitated access to pig mites at QASP-UQ, Gatton, and provided help with mite bioassays for the scabies study.
Footnotes
Conflict of interest
J.T., G. P. S., and C. M. W. have formal and informal commercial relationships with companies developing and supplying cubane intermediates.
Dedicated to Professors Armin de Meijere and Henning Hopf for their extraordinary contributions to hydrocarbon chemistry
References
- [1].a) Eaton PE, Angew. Chem. Int. Ed 1992, 31, 1421–1436; [Google Scholar]; b) Eaton PE, Angew. Chem 1992, 104, 1447–1462; [Google Scholar]; c) Biegasiewicz KF, Griffiths JR, Savage GP, Tsanaktsidis J, Priefer R, Chem. Rev 2015, 115, 6719–6745; [DOI] [PubMed] [Google Scholar]; d) Griffin GW, Marchand AP, Chem. Rev 1989, 89, 997–1010. [Google Scholar]
- [2].a) Meanwell NA, J. Med. Chem 2011, 54, 2529–2591; [DOI] [PubMed] [Google Scholar]; b) Patani GA, LaVoie EJ, Chem. Rev 1996, 96, 3147–3176; [DOI] [PubMed] [Google Scholar]; c) Marson CM, Chem. Soc. Rev 2011, 40, 5514–5533; [DOI] [PubMed] [Google Scholar]; d) Brown N, Mol. Inf 2014, 33, 458–462. [DOI] [PubMed] [Google Scholar]
- [3].a) Chalmers BA, Xing H, Houston S, Clark C, Ghassabian S, Kuo A, Cao B, Reitsma A, Murray C-EP, Stok JE, Boyle GM, Pierce CJ, Littler SW, Winkler DA, Bernhardt PV, Pasay C, De Voss JJ, McCarthy J, Parsons PG, Walter GH, Smith MT, Cooper HM, Nilsson SK, Tsanaktsidis J, Savage GP, Williams CM, Angew. Chem. Int. Ed 2016, 55, 3580–3585; Angew. Chem. 2016, 128, 3644–3649; [DOI] [PubMed] [Google Scholar]; b) See also: Auberson YP, Brocklehurst C, Furegati M, Fessard TC, Koch G, Decker A, LaVecchia L, Briard E, ChemMedChem 2017, 12, 590–598. [DOI] [PubMed] [Google Scholar]
- [4].The lipophilic (bio)isostere adamantane has been reported to mimick a π–π πstacking interaction, see:; Yusuff N, Doré M, Joud C, Visser M, Springer C, Xie X, Herlihy K, Porter D, Touré BB, ACS Med. Chem. Lett 2012, 3, 579–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Mahadevi AS, Sastry GN, Chem. Rev 2013, 113, 2100–2138. [DOI] [PubMed] [Google Scholar]
- [6].Stok JE, Chow S, Krenske EH, Soto CF, Matyas C, Poirier RA, Williams CM, De Voss JJ, Chem. Eur. J 2016, 22, 4408–4412. [DOI] [PubMed] [Google Scholar]
- [7].a) Lovering F, Bikker J and Humblet C, J. Med. Chem 2009, 52, 6752–6756; [DOI] [PubMed] [Google Scholar]; b) Lovering F, Med. Chem. Commun 2013, 4, 515–519. [Google Scholar]
- [8].Ritchie TJ, Macdonald SJF, J. Med. Chem 2014, 57, 7206–7215. [DOI] [PubMed] [Google Scholar]
- [9].a) Stockdale TP, Williams CM, Chem. Soc. Rev 2015, 44, 7737–7763; [DOI] [PubMed] [Google Scholar]; b) Wanka L, Iqbal K, Schreiner PR, Chem. Rev 2013, 113, 3516–3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Willstätter R, Waser E, Ber. Dtsch. Chem. Ges 1911, 44, 3423–3445; [Google Scholar]; b) Hopf H, Classics in hydrocarbon chemistry: syntheses, concepts, perspectives, Vol. 1, Wiley-VCH, Weinheim; New York, 2000. [Google Scholar]
- [11].Paquette LA, Acc. Chem. Res 1993, 26, 57–62. [Google Scholar]
- [12].Klärner F-G, Angew. Chem. Int. Ed 2001, 40, 3977–3981. [PubMed] [Google Scholar]
- [13].Grange RL, Gallen MJ, Schill H, Johns JP, Dong L, Parsons PG, Reddell PW, Gordon VA, Bernhardt PV, Williams CM, Chem. Eur. J 2010, 16, 8894–8903. [DOI] [PubMed] [Google Scholar]
- [14].Houston SD, Xing H, Bernhardt PV, Vanden Berg TJ, Tsanaktsidis J, Savage GP, Williams CM, Chem. Eur. J 2018, DOI: 10.1002/chem.201805124. [DOI] [PubMed] [Google Scholar]
- [15].a) Wardrop D, Keeling D, Br. J. Haematol 2008, 141, 757–763; [DOI] [PubMed] [Google Scholar]; b) L KP, Circulation 1959, 19, 97–107. [DOI] [PubMed] [Google Scholar]
- [16] a).Navarro JL, Cesar JM, Fernández MA, Fontcuberta J, Reverter JC, Gol-Freixa J, Rev. Esp. Cardiol 2007, 60, 1226–1232; [DOI] [PubMed] [Google Scholar]; b) Institute for Safe Medication Practices (2012) Anticoagulants the Leading Reported Drug Risk in 2011. http://www.ismp.org/QuarterWatch/pdfs/2011Q4.pdf;; c) Lund K, Gaffney D, Spooner R, Etherington AM, Tansey P, Tait RC, Br. J. Haematol 2012, 158, 256–261. [DOI] [PubMed] [Google Scholar]
- [17].a) Verhoef TI, Ragia G, de Boer A, Barallon R, Kolovou G, Kolovou V, Konstantinides S, Le Cessie S, Maltezos E, van der Meer FJM, Redekop WK, Remkes M, Rosendaal FR, van Schie RMF, Tavridou A, Tziakas D, Wadelius M, Manolopoulos VG, Maitland-van der Zee AH, New Engl. J. Med 2013, 369, 2304–2312; [DOI] [PubMed] [Google Scholar]; b) Kimmel SE, French B, Kasner SE, Johnson JA, Anderson JL, Gage BF, Rosenberg YD, Eby CS, Madigan RA, McBane RB, Abdel-Rahman SZ, Stevens SM, Yale S, Mohler ER, Fang MC, Shah V, Horenstein RB, Limdi NA, Muldowney JAS, Gujral J, Delafontaine P, Desnick RJ, Ortel TL, Billett HH, Pendleton RC, Geller NL, Halperin JL, Goldhaber SZ, Caldwell MD, Califf RM, Ellenberg JH, N. Engl. J. Med 2013, 369, 2283–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].a) Tie J-K, Jin D-Y, Tie K, Stafford DW, J. Thromb. Haemost 2013, 11, 1556–1564; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rost S, Fregin A, Ivaskevicius V, Conzelmann E, Hörtnagel K, Pelz H-J, Lappegard K, Seifried E, Scharrer I, Tuddenham EGD, Müller CR, Strom TM, Oldenburg J, Nature 2004, 427, 537; X. Chen, D.-Y. Jin, D. W. Stafford, J.-K. Tie, Blood 2018, 10.1182/blood-2018-05-846592. [DOI] [PubMed] [Google Scholar]
- [19].Tie J-K, Jin D-Y, Straight DL, Stafford DW, Blood 2011, 117, 2967–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Choonara IA, Haynes BP, Cholerton S, Breckenridge AM, Park BK, Br. J. Clin. Pharmacol 1986, 22, 729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Bonnet U, CNS Drug Rev. 2003, 9, 97–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Prut L, Belzung C, Eur. J. Pharmacol 2003, 463, 3–33. [DOI] [PubMed] [Google Scholar]
- [23].Everett RM, Descotes G, Rollin M, Greener Y, Bradford JC, Benziger DP, Ward SJ, Fundam. Appl. Toxicol 1993, 21, 59–65. [DOI] [PubMed] [Google Scholar]
- [24].D’Ambra TE, Estep KG, Bell MR, Eissenstat MA, Josef KA, Ward SJ, Haycock DA, Baizman ER, Casiano FM, J. Med. Chem 1992, 35, 124–135. [DOI] [PubMed] [Google Scholar]
- [25].Richon VM, Br. J. Cancer 2006, 95, S2. [Google Scholar]
- [26].Grant S, Easley C, Kirkpatrick P, Nat. Rev. Drug. Discov 2007, 6, 21. [DOI] [PubMed] [Google Scholar]
- [27].Currie BJ, McCarthy JS, New Engl. J. Med 2010, 362, 717–725. [DOI] [PubMed] [Google Scholar]
- [28].Merzendorfer H, Kim HS, Chaudhari SS, Kumari M, Specht CA, Butcher S, Brown SJ, Robert Manak J, Beeman RW, Kramer KJ, Muthukrishnan S, Insect Biochem. Mol. Biol 2012, 42, 264–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lipinski CA, Lombardo F, Dominy BW, Feeney PJ, Adv. Drug Deliv. Rev 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- [30].Hodgson J, Nat. Biotechnol 2001, 19, 722–726. [DOI] [PubMed] [Google Scholar]
- [31].Ufer M, Clin. Pharmacokinet 2005, 44, 1227–1246. [DOI] [PubMed] [Google Scholar]
- [32].Barcellona D, Vannini ML, Fenu L, Balestrieri C, Marongiu F, Thromb. Haemost 1998, 80, 899–902. [PubMed] [Google Scholar]
- [33].Murray RW, Singh M, Rath NP, Tetrahedron 1999, 55, 4539–4558. [Google Scholar]
- [34].Mukherjee C, Maiti GH, Misra AK, ARKIVOC 2008, 2008, 44–65. [Google Scholar]
- [35].Zimmerman HE, Iwamura H, J. Am. Chem. Soc 1968, 90, 4763–4764. [Google Scholar]
- [36].Mahadevi AS, Sastry GN, Chem. Rev 2013, 113, 2100–2138. [DOI] [PubMed] [Google Scholar]
- [37].He L, Cheng J, Wang T, Li C, Gong Z, Liu H, Zeng B, Jiang H, Zhu W, Chem. Phys. Lett 2008, 462, 45–48. [Google Scholar]
- [38].a) Lyttle MH, Streitwieser A Jr., Kluttz RQ, J. Am. Chem. Soc 1981, 103, 3232–3233; [Google Scholar]; b) Miller MJ, Lyttle MH, Streitwieser A Jr., J. Org. Chem 1981, 46, 1977–1984; [Google Scholar]; c) Paquette LA, Hefferon GJ, Samodral R, Hanzawa Y, J. Org. Chem 1983, 48, 1262–1266. [Google Scholar]
- [39].We thank the reviewer for drawing our attention to references [38].
- [40].Variations in ratios are observed depending on the functional group and substitution pattern, see [14].
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.