

SUMMARY
Using proteomic approaches, we uncovered a DNA damage response (DDR) function for peroxisome proliferator activated receptor γ (PPAR γ) through its interaction with the DNA damage sensor MRE11-RAD50-NBS1 (MRN) and the E3 ubiquitin ligase UBR5. We show that PPAR γ promotes ATM signaling and is essential for UBR5 activity targeting ATM interactor (ATMIN). PPARγ depletion increases ATMIN protein independent of transcription and suppresses DDR-induced ATM signaling. Blocking ATMIN in this context restores ATM activation and DNA repair. We illustrate the physiological relevance of PPARγ DDR functions by using pulmonary arterial hypertension (PAH) as a model that has impaired PPARγ signaling related to endothelial cell (EC) dysfunction and unresolved DNA damage. In pulmonary arterial ECs (PAECs) from PAH patients, we observed disrupted PPARγ-UBR5 interaction, heightened ATMIN expression, and DNA lesions. Blocking ATMIN in PAH PAEC restores ATM activation. Thus, impaired PPARγ DDR functions may explain the genomic instability and loss of endothelial homeostasis in PAH.
In Brief
Li et al. identify PPARγ interactions with MRN and UBR5. PPARγ promotes UBR5-mediated ATMIN degradation, necessary for ATM activation upon DNA damage. Pulmonary arterial hypertension (PAH) endothelial cells exhibit genomic instability and disrupted PPARγ-UBR5 interaction. Blocking ATMIN restores ATM signaling in these cells, highlighting the significance of the PPARγ-ATMIN axis.
Graphical Abstract
INTRODUCTION
Peroxisome proliferator activated receptor γ (PPARγ) is a member of the nuclear receptor family that interacts with canonical retinoic acid receptors (RXR) (Chandra et al., 2008) and other co-factors as a transcription factor complex in multiple cell types, including vascular cells (Alastalo et al., 2011). Aberrant PPARγ-mediated transcription has been implicated in disease conditions, including obesity, diabetes, cancer, inflammation, and vascular disorders (Ahmadian et al., 2013; Rabinovitch, 2010) that include atherosclerosis (Duval et al., 2002), aortic aneurysm (Hamblin et al., 2010), and pulmonary arterial hyper-tension (PAH) (Rabinovitch, 2010). Endothelial dysfunction is a feature of all these vascular diseases, and in PAH, it is associated with the obliteration and loss of microvessels that increase resistance to pulmonary blood flow and can culminate in heart failure and the need for a lung transplant (Rabinovitch, 2012).
Mice with PPARγ deleted in endothelial cells (ECs) (Tie2-Pparγ−/−) develop pulmonary hypertension that persists upon re-exposure to room air after hypoxia (Guignabert et al., 2009). In human pulmonary arterial ECs (PAECs), an interaction between PPARγ and β-catenin co-regulates the gene expression of apelin, a major factor that promotes PAEC survival and suppresses smooth muscle cell proliferation (Alastalo et al., 2011). This interaction is disrupted by rosiglitazone, an agonist previously used to treat type II diabetes (Alastalo et al., 2011). These observations reinforce the need to discover interactions between PPARγ and other proteins that are perturbed in PAH and other vascular disorders and have pharmacologic relevance.
Here, we report the results of a proteomic approach using affinity purification with mass spectrometry (AP-MS) to identify PPARγ nuclear interacting proteins. These studies uncovered PPARγ interactions with the DNA damage sensor MRN (MRE11-RAD50-NBS1) and the E3 ubiquitin ligase UBR5 and a role for PPARγ in the DNA damage response (DDR) pathway. We showed that PPARγ promotes UBR5 ubiquitin ligase activity and regulates ATM interactor (ATMIN) levels, thereby permitting efficient ATM phosphorylation and the initiation of DNA repair upon DNA damage. Perturbation of this axis is observed in PAH and can account for unresolved DNA damage that is associated with impaired endothelial functions (de Jesus Perez et al., 2014; Diebold et al., 2015).
RESULTS
AP-MS Identified PPARγ Interactions with MRN and UBR5 Independent of RXRα
We transiently transfected 293T cells with a FLAG-tagged PPARγ1 construct and isolated nuclear extracts in the presence of micrococcal nuclease for affinity purification using a FLAG antibody. We used 293T cells for their high transfection efficiency that permitted efficient pull-down of FLAG-PPARγ and detection of interactors. The quadruplicate AP-MS screen revealed 352 proteins that co-purified with FLAG-PPARγ with a log2 fold change (Log2FC) of >1.5 and an adjusted p value (adj. P) ≤ 0.05 (Figure S1A). Not surprisingly, we detected known PPARγ interactors, such as mediator of RNA polymerase II transcription subunit 1 and 24 (MED1 and MED24, respectively), promyelocytic leukemia protein (PML), p53, and others. We ranked 87 proteins as high-confidence PPARγ-interacting proteins, and those included the canonical partners RXRα and β (Figure S1A; Table S1). Using databases of published physical and functional interactions, we constructed and analyzed networks of high confidence proteins for enriched biological functions. In addition to cellular metabolism, we observed DDR and DNA replication among the most enriched functions (Figure 1A; Table S2). From the DDR network, four interactions were verified by co-immunoprecipitation, i.e., the components of the DNA damage sensing complex MRN (MRE11-RAD50-NBS1) and p53 (Figure S1B).
Figure 1. PPARγ Interacts with the MRE11-RAD50-NBS1 (MRN) Complex and UBR5 in 293T Cells.

(A) A network of PPARγ and its interactors (yellow) in the DNA damage response (DDR) and DNA replication pathway. Red connections denote interactions obtained from the protein complex enrichment analysis tool (COMPLEAT) database.
(B) Silver staining shows gel fragments containing proteins (identified by MS) sequentially co-purified with tagged PPARγ (−2xStrep, S) and NBS1 (-FLAG, F) but not with green fluorescent protein (GFP-SF).
(C) Representative immunoblots of interactions between the PPARγ-NBS1 complex with RXRα and UBR5 upon hydroxyurea (HU) treatment (24 h).
(D) The 293T cells expressing FLAG-PPARγ were pretreated with GW9662 (5 μM, 1 h) and treated with SR10221 (5 μM, 24 h) (GW+SR). Controls included are cells treated with DMSO (vehicle), GW9662, or SR10221 only. Cells expressing FLAG-vector were used as the immunoprecipitation negative control. Representative immunoblots show effects of GW9662 pretreatment together with SR10221 on interactions between PPARγ and UBR5/NBS1.
See also Figures S1 and S2 and Tables S1–S4.
MRN initiates the DDR pathway using NBS1 to recruit proteins necessary for DNA repair (Reinhardt and Yaffe, 2013). We hypothesize that PPARγ binds to MRN via NBS1. To test this, we used tandem affinity purification (TAP) of PPARγ−2x Streptavidin (PPARγ−2xStrep) and FLAG-NBS1 in 293T cells, and the crosslinking agent bis(sulfosuccinimidyl)suberate (BS3) was added on beads before elution. The crosslinked immunocomplexes were analyzed by mass spectrometry (XL-MS) (Figure S1C). XL-MS identified three PPARγ peptides crosslinked to NBS1 (Figure S1D), demonstrating a direct interaction. Using structural mapping based on PPARγ crystal structure (Chandra et al., 2008), we located two of the three peptides in the zinc-finger motif within the PPARγ DNA-binding domain (DBD) and one in the ligand-binding domain (LBD) (Figures S1E and S1F). These data suggest that NBS1 binding might interfere with PPARγ transcription factor function. We used size-exclusion chromatography of nuclear extracts overexpressing PPARγ−2xStrep and FLAG-NBS1 and showed that PPARγ exists in multiple pools: a higher molecular weight (MW, approximated >1,500 kDa) pool, a lower MW (approximated 67–440 kDa) pool, and a monomeric pool (from overexpression, <67 kDa). NBS1 and RXRα reside in the high and low MW PPARγ pools, respectively, supporting mutually exclusive PPARγ interactions with NBS1 or RXRα (Figure S2A). In the absence of NBS1, we also found that PPARγ and three out of the seven PPARγ target genes were upregulated (Figure S2B). The requirement of PPARγ-LBD for MRN interactions was confirmed using mutagenesis (Figure S2C). These data suggest that upon MRN binding, PPARγ undergoes structural changes, which can interfere with its transcription factor property, implicating an independent function for PPARγ.
To investigate PPARγ functions in relation to MRN binding, we performed initial silver staining of the TAP elution from unperturbed cell lysates and identified all components of MRN but not RXRα (Figure 1B), supporting our XL-MS and size-exclusion chromatography results. Silver-stained gel fragments from the TAP elution also identified TR150 (thyroid hormone receptor-associated protein 3, encoded by THRAP3) and the ubiquitin ligase UBR5 co-purifying with the PPARγ-MRN complex (Figure 1B). Under conditions of DNA damage induced by hydroxyurea (HU), TAP-MS revealed associations of UBR5 and TR150 with the PPARγ-MRN complex (Figure 1C; Tables S3 and S4). We performed nuclear co-immunoprecipitation (co-IP) of endogenous UBR5 and NBS1 and showed that both UBR5 and NBS1 bind strongly to PPARγ but weakly to each other (Figure S2D). This was confirmed by co-IP of UBR5 and PPARγ in the absence of NBS1 (Figure S2E). To verify the specificity of these PPARγ interactions, we altered PPARγ conformations by using the pharmacological modulator SR10221, which destabilizes helix 12 in the PPARγ LBD (Marciano et al., 2015). SR10221 disrupted PPARγ interactions with MRN and UBR5, which were restored by pre-treatment with GW9662, which blocks the SR10221 target site (Figure 1D). Our proteomic and biochemical data suggest that PPARγ interactions with MRN and UBR5 implicate a potential role for PPARγ in the DDR pathway.
PPARγ Promotes the Initiation of ATM Signaling
The MRN complex (Lee and Paull, 2004) and UBR5 (Zhang et al., 2014) are required for ATM activity, which is necessary for DNA repair induced by genotoxic agents. In this study, we activated ATM signaling using doxorubicin (DoxR), which intercalates DNA and generates double-strand breaks (Kurz et al., 2004), and HU, which induces replication fork collapse and a progressive accumulation of double-strand breaks (Cuadrado et al., 2006). We first verified endogenous nuclear PPARγ interactions with UBR5 and MRN at baseline and in response to DoxR or HU (Figure 2A). To determine if PPARγ is necessary for ATM activation, we depleted PPARγ using small interfering RNA (siRNA) and induced damage using HU and DoxR. The loss of PPARγ and UBR5 reduced HU-mediated ATM phosphorylation (pATM, Ser1981) and its targets KAP1 (Ser824) (Ziv et al., 2006), γH2AX (Ser139) (Burma et al., 2001), and SMC1 (Ser966) (Yazdi et al., 2002) (which was not affected by siUBR5) (Figure 2B; densitometry in Figure S3A). PPARγ/UBR5-dependent ATM signaling was also evident in response to DoxR treatment (Figure S3B). We further investigated the role of PPARγ in HU-induced DNA damage because replication stress damage is relevant to PAH (de Jesus Perez et al., 2014).
Figure 2. PPARγ Promotes ATM Signaling by Increasing UBR5-Mediated ATMIN Ubiquitination in 293T Cells.

(A) Representative immunoblots of endogenous nuclear PPARγ interactions with MRN and UBR5 at baseline and upon DNA damage induced by HU and doxorubicin (DoxR).
(B) Representative immunoblots of HU-induced pATM and its targets with PPARγ or UBR5 depletion.
(C) Representative immunoblots of reduced UBR5 binding to ubiquitinated proteins with PPARγ depletion.
(D and E) Representative immunoblots of ATMIN and pRPA2 levels with PPARγ (D) or UBR5 (E) depletion upon HU treatments.
(F) Cells were transfected with HA-tagged ubiquitin and subsequently the siRNA as indicated. Cells were treated with the proteasome inhibitor MG132 (MG) for 2 h before lysis in a denaturing buffer. Endogenous ATMIN was immunoprecipitated to determine its polyubiquitinated form. Representative immunoblots show effects of PPARγ or UBR5 depletion on endogenous ATMIN ubiquitination detected by anti-hemagglutinin (HA) antibody.
(G) Quantitative real-time PCR shows effects of PPARγ or UBR5 depletion by the respective siRNA on ATMIN mRNA levels (normalized to β-actin mRNA).
siC, siControl; siPg, siPPARγ; siU5, siUBR5; Veh; vehicle. Error bars, mean ± SEM.
See also Figure S3.
PPARγ and UBR5 Modulate ATMIN Protein Levels through Ubiquitination
To understand how PPARγ and UBR5 regulate ATM signaling, we determined whether PPARγ is required for UBR5 E3 ubiquitin ligase activity. Indeed, PPARγ depletion inhibited UBR5-mediated ubiquitination, judging by a decrease in ubiquitinated proteins immunoprecipitated with UBR5 (Figure 2C). We further investigated whether PPARγ depletion affects ATMIN levels, an UBR5 substrate that regulates ATM phosphorylation. Previous studies indicated that UBR5 ubiquitinates ATMIN upon ionizing radiation to release and allow ATM activation (Zhang et al., 2014; Zhang et al., 2012). In contrast, other studies have shown the opposite with replication stress, i.e., that loss of ATMIN suppresses ATM activation (Schmidt et al., 2014).
Here, we observed that upon depletion of PPARγ or UBR5, ATMIN levels were elevated both at baseline and in response to HU in association with the suppression of the ATM target pRPA2 (Ser4/8) (Liu et al., 2012) (Figures 2D and 2E; densitometry in Figures S3C and S3D). Consistent with the function for PPARγ related to UBR5 ubiquitin ligase activity, elevated ATMIN protein in the absence of PPARγ or UBR5 was accompanied by a decrease in its ubiquitination (Figure 2F). Moreover, ubiquitination of ATMIN was associated with its degradation since the proteasome inhibitor MG132 maintains ATMIN protein levels (Figure 2F, input panel). In the absence of UBR5, PPARγ remained bound to the truncated FLAG-ATMIN (aa1–354), supporting UBR5 as downstream of PPARγin ATMIN regulation (Figure S3E). In addition, both UBR5 and PPARγ bind to FLAG-ATMIN with and without HU, with UBR5 binding more sustained upon HU treatment (Figure S3F). The effects of PPARγ depletion on protein degradation was further evident judging by the reduced cellular lysine (K)48-linked ubiquitins, which represent protein degradative signals (Glickman and Ciechanover, 2002). This reduction was restored by overexpressing siRNA-resistant PPARγ (siResPPARγ) (Figure S3G). Since PPARγ is a transcription factor, we confirmed that ATMIN mRNA levels were not significantly altered by the depletion of PPARγ or of UBR5 (Figure 2G). Taken together, our data indicate that the loss of PPARγ alters cellular protein degradative signals and, specifically, it increases ATMIN levels by suppressing UBR5-mediated ubiquitination, and that this function is not related to PPARγ-mediated transcription.
PPARγ-DDR Function Is Conserved in ECs
We and others showed that PPARγ promotes endothelial survival and regeneration (Alastalo et al., 2011; Vattulainen-Collanus et al., 2016). In a transgenic mouse with deficient endothelial PPARγ, pulmonary hypertension and adverse vascular remodeling did not reverse following re-exposure to room air after chronic hypoxia (Guignabert et al., 2009). As impaired PPARγ function and chromosomal instability related to persistent DNA damage are features of PAECs from patients with PAH (Aldred et al., 2010), we determined if PPARγ functions in DDR are compromised in PAH and could contribute to the loss of vascular homeostasis.
We first verified nuclear PPARγ and UBR5 interactions in primary human PAECs (Figure 3A). Consistent with our findings in 293T cells, PPARγ depletion in PAECs also led to reduced pATM, pRPA2, and γH2AX upon prolonged HU treatment (Figure 3B; densitometry, Figures S4A and S4B). To confirm the specificity of PPARγ-ATM signaling, we restored pATM in human umbilical venous ECs (HUVECs) by overexpressing siResPPARγ (Figure 3C; densitometry, Figure S4C). HUVECs were used to withstand the cytotoxicity from DNA and siRNA sequential transfections. Verifying ATMIN regulation of PPARγ-dependent ATM signaling in ECs, we depleted ATMIN in addition to PPARγ and observed that this restored pATM and its target pKAP1 (Figure 3D; densitometry, Figure S4D). Although ATMIN regulation of ATM signaling is highly context dependent (Leszczynska et al., 2016; Schmidt et al., 2014; Zhang et al., 2014), our results demonstrate that in the absence of PPARγ, abnormal accumulation of ATMIN suppresses ATM activation in response to DNA damage.
Figure 3. PPARγ-ATMIN Regulation of ATM Signaling Is Conserved in Primary Human Endothelial Cells.

(A) Representative immunoblots of endogenous nuclear PPARγ interaction with UBR5 in primary pulmonary arterial endothelial cells (PAECs) isolated from controls (Table S5).
(B) Representative immunoblots of HU-induced pATM expression with PPARγ depletion in PAECs.
(C) Representative immunoblots of restoration of HU-induced pATM expression with siRNA (siPPARγ#9)-resistant PPARγ overexpression in human umbilical venous ECs (HUVECs).
(D) Representative immunoblots of HU-induced pATM and pKAP1 with PPARγ or/and ATMIN depletions in PAECs.
(E) Confocal microscopy of PAECs shows effects of PPARγ depletion on pATM foci with hypoxia (<0.1% O2, 24 h) and reoxygenation (10 min). The line in the box of the box and whisker plots marks the median and whiskers correspond to the 10th to 90th percentiles. Unpaired Student t test was used. ****p <0.0001. Scale bars, 20 μm.
siC, siControl; siPγ, siPPARγ. See also Figure S4.
We also verified the inhibitory effects of siPPARγ on pATM and γH2AX foci by using immunofluorescence in PAEC (Figures S4E and S4F). This response was replicated with three individual siRNAs targeting PPARγ (Figure S4G). Importantly, the reduced ATM signaling upon PPARγ depletion was not due to altered cell cycle progression (Figure S4H). Since elevated oxidative stress has been implicated in PAH pathogenesis (Diebold et al., 2015) and ATM signaling is activated by oxidative stress (Hammond et al., 2003), we investigated if PPARγ also promotes ATM signaling upon oxidant injury. By exposing PAECs to hypoxia (<0.1% O2, 24 h) and reoxygenation (10 min), we detected the presence of 8-oxo-2′-deoxyguanosine (8-oxo-dG) foci (S4I), a marker for oxidative damage DNA (Cheng et al., 1992). We showed that PPARγ depletion also suppressed oxidative stress-induced pATM (Figure 3E; replicates, Figure S4J).
The PPARγ-ATMIN Axis Is Required for Endothelial DNA Repair and Homeostasis
We now showed that PPARγ is necessary to initiate the DDR, and we hypothesize that it is also important for DNA repair. We used the comet assay and demonstrated that PPARγ depletion did not affect the magnitude of DNA damage, as judged by comet tails assessed after a 6-h exposure to HU (Figure 4A; replicates, Figure S5A), but the capacity to repair DNA was reduced, as judged by persistent comet tails after a 24-h recovery period. We also examined levels of pRPA2 and γH2AX damage foci during recovery (24–72 h), as evidence of unrepaired DNA lesions. These foci were resolved in the control cells but were sustained in PPARγ-depleted PAECs (Figure 4B; replicates, Figure S5B). We validated that ATMIN also functions in PPARγ-dependent DNA repair by demonstrating that depletion of ATMIN in addition to PPARγ resolved pRPA2 foci during recovery (Figures 4C and 4D; densitometry and replicates, Figures S5C and S5D).
Figure 4. PPARγ Promotes DNA Repair through ATMIN in Primary Human ECs.

(A) Comet assay shows effects of PPARγ depletion on comet tail lengths after 24-h recovery (Rec 24h) from 6 h of HU (HU 6h) treatment.
(B) Confocal microscopy shows effects of PPARγ depletion on unresolved γH2AX and pRPA2 foci over 72 h after recovery (Rec 72h) from 24 h of HU (HU 24h) treatment. Recovery time points are as indicated.
(C) Quantification of pRPA2 foci with PPARγ or/and ATMIN depletion. Cells were fixed and analyzed using confocal microscopy at 72 h after recovery from 24 h of HU treatment.
(D) Representative immunoblots of ATMIN and pRPA2 levels from the same experimental as in (C).
(E) Confocal microscopy shows staining of γH2AX foci in pulmonary ECs (labeled by vWF) in Tie2 -PPARγ−/− mice and wild-type littermates subjected to three weeks of hypoxia (Hy) (10% O2) and four weeks of recovery in room air (n = 5). Arrowheads indicate cells in insets.
siC, siControl; siPγ, siPPARγ. The line in the box of the box and whisker plots marks the median and whiskers correspond to the 10th to 90th percentiles (A, B, C, and E). Kruskal-Wallis ANOVA test with Dunn’s test (A–C). Two-tailed Mann-Whitney test (E). *p < 0.05; **p < 0.01; ****p < 0.0001, ns., not significant. Scale bars, 50 μm (A and B); 20 mm (E). See also Figure S5.
We then determined whether unresolved DNA damage accompanied the pulmonary hypertension that did not reverse in mice with PPARγ depleted in ECs (Tie2-PPARγ−/−) that were re-exposed to room air after chronic hypoxia (Guignabert et al., 2009). Lung sections from Tie2-PPARγ−/− mice and wild-type littermates were co-stained with von Willebrand factor (vWF) antibody to detect ECs and γH2AX antibody. Confocal microscopy revealed increased γH2AX in the ECs of the mutant versus control mice previously studied following re-exposure to room air (Figure 4E). These data further supported our mechanistic studies in cultured PAECs that link PPARγ to regulation of DNA damage sensing and repair.
Reduced PPARγ-UBR5 Interaction, Elevated ATMIN, and Impaired DDR in PAH-PAEC
The loss of genome integrity and an increased propensity for apoptosis and transformation are key features of PAECs from PAH patients (PAH-PAECs) (Aldred et al., 2010; Hopper et al., 2016; Ranchoux et al., 2015; Sa et al., 2016). We, therefore, assessed evidence of unrepaired DNA damage in PAH versus unused donor control lung sections and in cultured PAECs harvested from explanted PAH lungs and from control lungs. Demographic information related to controls (unused donor) and PAH-PAECs is provided in Table S5. Representative cell images indicating healthy, actively proliferating primary PAEC cultures are shown in Figure S6A.
Increased γH2AX foci were evident in PAH versus control PAECs in lung tissue sections (Figure 5A), and in cell cultures, there were more extended comet tails (Figure 5B) in PAH-PAECs compared with control-PAECs. Upon HU treatment, PAHPAECs showed reduced pATM foci compared with control-PAECs (Figure 5C; replicates, Figure S6B).
Figure 5. The PPARγ-ATMIN Axis Is Impaired in PAH-PAECs with Genomic Instability.

(A) Confocal microscopy shows representative staining of γH2AX foci in PAECs (labeled by vWF) in lung tissue sections from pulmonary arterial hypertension (PAH) patients (18 vessels, 5 subjects) and controls (19 vessels, 6 subjects). Arrowheads indicate cells in insets.
(B) Representative comet assay of comet tail lengths in control and PAH-PAECs (n = 16 and 22, respectively).
(C) Confocal microscopy shows pATM foci in controls and PAH-PAECs with HU treatment (24 h).
(D) Representative immunoblots show nuclear endogenous interactions of PPARγ and UBR5 in controls and PAH-PAECs.
(E) Representative immunoblots show elevated ATMIN levels in PAH-PAECs compared to controls.
(F) Confocal microscopy shows pATM foci in controls and PAH-PAECs with siATMIN after HU treatment (24 h).
siC, siControl. The line in the box of the box and whisker plots marks the median and whiskers correspond to the 10th to 90th percentiles (A–C and F). Two-tailed Mann-Whitney test (A and B). Kruskal–Wallis ANOVA test with Dunn’s test (C and F). *p < 0.05; ****p < 0.0001, ns., not significant. Scale bars, 5 μm (A); 20 mm (B, C, and E). See also Figure S6.
Impaired ATM signaling in PAH-PAECs suggested that the PPARγ-UBR5-ATMIN axis may be dysfunctional in these cells. Indeed, reduced interactions between PPARγ and UBR5 in PAH-PAECs were evident when compared to control-PAECs (Figure 5D). This was independent of PPARγ levels that were similar in controls and PAH-PAECs (Figure 5D, input panel). A possible explanation could be PPARγ or UBR5 post-translational modifications (PTMs), which can confer structural changes that alter protein-protein interactions (Choi et al., 2014a). Consistent with the disruption of the PPARγ-UBR5 complex related to UBR5 ubiquitin ligase activity, we found heightened ATMIN expression in PAH versus control PAECs (Figure 5E; densitometry, Figure S6C). We confirmed that reducing ATMIN levels in PAH-PAECs restored HU-induced pATM foci formation to a level comparable to control-PAECs (Figure 5F; replicates, Figure S6D).
DISCUSSION
Our data obtained from cultured cells, transgenic mice, and clinical samples reveal a non-canonical role for PPARγ in the DDR and, subsequently, in DNA repair. Through its interactions with MRN and UBR5 independent of RXRα, PPARγ promotes ATM signaling in response to genotoxic stimuli. We propose a model in Figure 6, suggesting that the PPARγ DDR complex regulates the ATMIN-ATM interaction necessary for the activation of ATM in response to DNA damage. We provide data showing that PPARγ interaction with UBR5 is required for UBR5-mediated ubiquitination of multiple substrates, including ATMIN. A disrupted PPARγ-UBR5 complex in PAECs from PAH patients results in elevated ATMIN, impaired ATM signaling, and persistent DNA damage. Under these circumstances, reducing ATMIN can restore the DDR and result in efficient DNA repair.
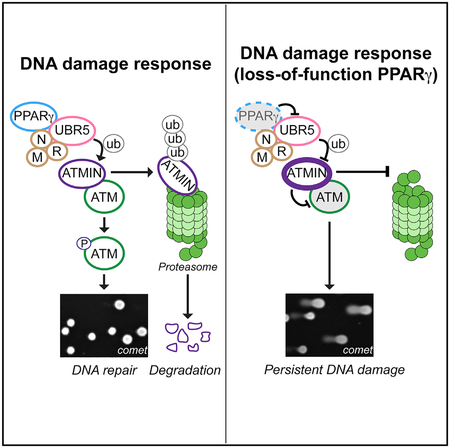
Figure 6. Proposed Model for PPARγ-Mediated DNA Damage Response Signaling.

In response to DNA damage, ubiquitination of ATMIN is increased, leading to its proteasomal degradation to release ATM. ATM binds to NBS1 and is autophosphorylated (pATM) and recruited to the DNA lesions where ATM phosphorylates its targets, such as H2AX (γH2AX) and RPA2 (pRPA2) to facilitate DNA repair. Where there is a loss of function of PPARγ, ATMIN ubiquitination by UBR5 is inhibited; hence, ATMIN accumulates. This suppresses ATM activation and its signaling, eventually leading to persistent DNA lesions and genomic instability. P, phosphorylation; ub, ubiquitination.
There is much known about PPARγ function related to its transcriptional targets associated with adipocyte differentiation and lipid metabolism and their perturbation in obesity and diabetes (Ahmadian et al., 2013). Our previous work showing differences in the response to PPARγ agonists in endothelial (Alastalo et al., 2011) and smooth muscle cells (Hansmann et al., 2008) led to a more comprehensive investigation of proteins interacting with PPARγ. Using an unbiased proteomic approach in 293T cells, we detected RXRα and RXRβ but not other known PPARγ transcriptional co-factors, such as the nuclear receptor coactivators and corepressors (NCOAs and NCORs) (Koppen and Kalkhoven, 2010) or β-catenin (Alastalo et al., 2011). Since these interactions were established in adipocytes and ECs, they might be cell-type specific and undetectable in 293T cells. We uncovered previously unknown interactions with MRN and UBR5 that are relevant to 293T and ECs and, hence, are likely of biological significance in other PPARγ-expressing cell types. Supporting this contention is evidence that PPARγ synthetic ligands synergize with platinum-based drugs by activating the DDR pathway and inducing apoptosis of non-small-cell lung cancer cells (Girnun et al., 2007; Khandekar et al., 2018).
PPARγ DDR functions implicated by its binding partners MRN and UBR5 support the notion that novel cellular functions can be uncovered by understanding protein-protein interactions. Understanding the NBS1-PPARγ binding interface by using XL-MS and biochemical studies indicates that PPARγ DDR functions would require its DBD and LBD, similar to its non-canonical function in degrading nuclear factor kB (NF-κB)/p65 (Hou et al., 2012). We detected recently described PPARγ interactors, TR150 (Choi et al., 2014a; Khandekar et al., 2018). TR150 is part of the mediator complex, potentially involved in chromatin remodeling (Fondell et al., 1996). It also promotes PPARγ-mediated gene transcription (Choi et al., 2014a), as well as functions in RNA processing (Beli et al., 2012). Further study is warranted to investigate whether PPARγ is related to any of these functions.
DDR activation requires layers of control, including the ubiquitination pathway, to ensure rapid modifications and trans-localization of proteins (Mirzoeva and Petrini, 2001, 2003; Polo and Jackson, 2011). UBR5 belongs to the HECT (homology to E6-AP carboxyl terminus) family of E3 ubiquitin ligases that maintains its substrates at optimal levels for effective signaling transduction. Some of the UBR5 substrates include an ubiquitin ligase, RNF168 (Gudjonsson et al., 2012), a pro-apoptotic protein, MOAP-1 (Matsuura et al., 2017), and an ATM modulator, ATMIN (Zhang et al., 2014), which act independently in the DDR pathway. Here, we demonstrated that PPARγ is necessary for UBR5 ubiquitin ligase activity and potentially has a broad effect on other UBR5 substrates.
We focused on ATMIN because of its relationship with ATM, the nature of which has been context dependent (Liu et al., 2017; Schmidt et al., 2014; Zhang et al., 2014). We used prolonged HU treatment to induce replication stress-dependent double-strand breaks and, hence, ATM signaling. HU-induced damage also closely resembles chronic replication-induced genotoxic insults associated with genomic instability in vascular ECs from PAH patients (Aldred et al., 2010; de Jesus Perez et al., 2014). In both 293T and ECs, increased ATMIN resulting from silencing PPARγ or UBR5 inhibited ATM signaling. Importantly, depleting ATMIN in this context restored pATM and DNA repair. We demonstrated that PPARγ or UBR5-mediated ATMIN ubiquitination is associated with its proteasomal degradation. Others have shown that ionizing radiation-induced ATMIN ubiquitination (via UBR5) does not lead to degradation (Zhang et al., 2014). We propose that in response to the nature and duration of the DNA damage stimulus, the type and amount of ATMIN ubiquitination might vary, producing either degradative or a non-degradative response. This “ubiquitin threshold” model has been previously proposed (Swatek and Komander, 2016) and could account for our observations linking PPARγ and UBR5 to ATMIN ubiquitination and degradation. In addition, the C-terminal ATMIN SQ or TQ motif cluster domain could be highly modified, especially in response to DNA damage (Jurado et al., 2010). We postulate that the aberrant increase in ATMIN protein and possibly its modifications in PPARγ- and UBR5-depleted cells sterically inhibit ATM activation in response to DNA damage (Figure 6). Determining precisely how this occurs could lead to opportunities to selectively modulate the DDR pathway.
ATMIN was first identified as a transcription factor for DYNLL1. Both ATMIN and DYNLL1 are required for the initiation of lung budding during lung organogenesis (Goggolidou et al., 2014; Jurado et al., 2010). Distinguishing between ATMIN developmental and DDR functions by defining its targets of transcription or interacting partners would provide a greater understanding of ATMIN biology. Upstream of ATMIN, both PPARγ, and UBR5 knockout mice die in early embryonic life with developmental defects in the vasculature (Barak et al., 1999; Saunders et al., 2004). In the context of PAH, we postulate that the disrupted interactions between PPARγ and UBR5 would modulate the expression and activities of other substrates, which could be critical for EC function.
Our previous observations related to the role of PPARγ in the maintenance of endothelial homeostasis (Alastalo et al., 2011) and now in the DDR, coupled with an increasing body of evidence showing genomic instability and DNA damage in PAECs and smooth muscle cells from patients with PAH (Aldred et al., 2010; Meloche et al., 2014), led us to investigate whether PPARγ-mediated DNA damage sensing was impaired. PAH is a progressive disease associated with severe vascular occlusion owing to EC dysfunction, judged by propensity to apoptosis, inability to form tubes in culture (Sa et al., 2016), and cellular transformation (Hopper et al., 2016; Ranchoux et al., 2015). Our study indicates that the PPARγ-ATMIN axis is indeed perturbed in PAECs from PAH patients, with high ATMIN levels related to impaired DNA damage sensing and repair.
The common response of 293T cells and primary ECs further strengthens the notion that, perturbations in the PPARγ-UBR5-ATMIN axis could potentially occur in multiple cell types where PPARγ is expressed and, hence, would be applicable to a wide range of disease mechanisms.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Marlene Rabinovitch (marlener@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
293T cells (ATCC) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM, GIBCO) supplemented with 10% fetal bovine serum (FBS, GIBCO) and penicillin/streptomycin (GIBCO). Commercially available primary human PAECs (PromoCell) and umbilical vein ECs (HUVEC) (Angio-Proteomie) were cultured in complete EC medium (ScienCell). Primary human PAEC were purified from explanted lungs of patients undergoing transplantation due to PAH, or from controls (unused donors) obtained with approval from the Pulmonary Hypertension Breakthrough Initiative (PHBI) Network (see Acknowledgments). The PAEC were isolated by scraping the endothelial layer of pulmonary arteries and cultured in complete EC medium. Once PAEC cultures were established from ex-planted lungs, EC were further purified by incubating the cell suspension in complete EC medium with CD31 Dynabeads (Thermo Fisher Scientific) for 10 min at room temperature, followed by three washes in phosphate-buffered saline (PBS, GIBCO), resuspended and re-cultured in complete EC medium. Demographic information for patient and control (unused donor) lungs is provided in Table S5. In this study, PAEC isolated from small (< 1 mm) and large pulmonary arteries were used between passages 4–8 with similar distribution when comparing controls and PAH. All cultures were tested negative for mycoplasma contamination. Further details can be found in Supplemental Information.
METHOD DETAILS
siRNAs
ON-TARGETplus SMARTpool siRNA targeting PPARγ (L-003436-00-0005; Dharmacon) was used to deplete PPARγ for all experiments. For verifying γH2AX suppression, individual siRNA of the pool were used (#7, J-003436-07-0005; #8, J-003436-08-0005; #9, J-003436-09-0005, Dharmacon). For depleting UBR5, we used siGENOME SMARTpool siRNA targeting UBR5 (M-007189-02-0010, Dharmacon). For depleting ATMIN, we used ON-TARGETplus SMARTpool siRNA targeting ATMIN (L-020304-01-0010, Dharmacon). ON-TARGETplus non-targeting pool (D-001810-10-05, Dharmacon) was used for all siControl transfections.
Plasmids
An amino-terminal Flag tagged-PPARγ1 (courtesy of Dr. László Nagy, University of Debrecen, Hungary) was mutagenized to achieve 100% identity to the published sequence (NM_005037) using site-directed mutagenesis via a quick-change PCR (QC-PCR) protocol. Deletion of the PPARγ LBD was generated using the QC-PCR protocol. Flag-PPARγ plasmids were amplified using PfuUltra (Agilent) and primers that introduce a stop codon to delete PPARγ LBD (Table S6). Products were DpnI (NEB) digested then transformed into One Shot TOP10 cells (Thermo Fisher Scientific). For generating the carboxyl-terminal 2xStrep tagged-PPARγ, the PPARγ open reading frame was amplified with primers to create EcoRI and NotI flanking sites (Table S6). PCR product was inserted into the pcDNA 2xStrep vector following the In-Fusion HD Cloning Kit protocol (Clontech). For generating the Flag-PPARγ siRNA resistant construct, primers were used to generate three silent mutations in the PPARγ siRNA#9 targeted region: GACAGCGACTTGGCAATAT (Table S6). The silent mutations introduced are underlined: GACAGCGATCTCGCAATAT. The carboxyl-terminal StrepFlag-tagged GFP, 2xStrep pcDNA plasmids and the amino-terminal HA-tagged ubiquitin were kindly provided to us by Dr. Nevan Krogan, UCSF. The amino-terminal Flag tagged-NBS1 was kindly provided by Dr. Hui-Kuan Lin, University of Texas MD Anderson Cancer Center. The amino-terminal Flag tagged-ATMIN (amino acids 1–354) was kindly provided by Dr. Axel Behrens, King’s College London. pCMV2-Flag (Sigma) was used as vector control.
Transient transfections
Plasmids were transfected into 293T cells using PolyJet at 3:1 (polyjet:DNA) according to the manufacturer’s instructions (SignaGen Laboratories). siRNAs were transfected into 293T using Lipofectamine RNAiMAX (Invitrogen). In primary PAEC, siRNAs or plasmid DNA were transfected using P5 Primary Cell 4D-Nucleofector X Kit (Lonza) according to manufacturer’s instructions (Lonza).
Nuclear extraction
Cells were washed and scraped in ice-cold Tris-buffered saline (TBS, Corning). Cell pellets were resuspended in hypotonic buffer [10 mM HEPES, pH 7.9, 10 mM KCl, 1.5 mM MgCl2, Halt protease and phosphatase inhibitor (Thermo Scientific)], and homogenized 10 times using a dounce homogenizer (pestle B) (Kimble Chase). The nuclei were then pelleted by centrifugation at 13,000 rpm at 4°C for 5 min, and lysed in high salt buffer [20 mM HEPES, pH 7.9, 0.42 M NaCl, 25% glycerol, 5 mM CaCl2, 1.5 mM MgCl2, 0.1% Nonidet P-40 (NP-40), 0.2 mM EDTA, Halt protease and phosphatase inhibitor]. Lysates were homogenized 20 times using a dounce homogenizer (pestle B), and incubated with micrococcal nuclease (Thermo Scientific) at room temperature for 15 min to further digest chromatin. Nuclear extracts were collected by centrifugation at 14,000 rpm at 4°C for 20 min. For immunoprecipitation, lysates were diluted with 3× volumes of low detergent buffer (20 mM HEPES, pH 7.9, 1 mM EDTA, 0.2% NP-40, Halt protease and phosphatase inhibitor).
Whole cell extraction
For whole-cell extract for immunoprecipitation, cells were washed in ice-cold TBS, lysed in 0.2% NP-40 buffer (50 mM Tris-HCl, pH8.0, 150 mM NaCl, 5 mM CaCl2, 1 mM EDTA, 0.2% NP-40, Halt protease and phosphatase inhibitor), and incubated on ice for 15 min. Lysates were homogenized 20 times using a dounce homogenizer (pestle B), and incubated with micrococcal nuclease as described above. Cell extracts were collected by centrifugation at 14,000 rpm at 4°C for 20 min. For protein analyses, cells were washed in ice-cold TBS, and lysed in RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 5 mM CaCl2, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, Halt protease and phosphatase inhibitor). Cell extracts were incubated with micrococcal nuclease at room temperature for 15 min, and collected as described above.
Immunoprecipitation
Diluted nuclear extracts or undiluted whole cell extracts were incubated with antibodies overnight at 4°C with rotation. The next day, Protein-G Dynabeads (Thermo Fisher Scientific) were added to cell extracts containing antibodies, and incubated for 3 h at 4°C with rotation. After incubation, beads were washed three times in ice-cold wash buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.05% NP-40). Proteins were eluted in acid using IgG elution buffer (Thermo Scientific) at room temperature for 10 min on gentle vortex. The final elution was collected and neutralized with 1/10 volume of 1 M Tris-HCl, pH 9.0.
Denaturing immunoprecipitation
293T cells were preincubated with MG132 (10 μM) for 2 hours. Cells were then washed in ice-cold TBS containing N-ethylmaleimide (10 mM, Thermo Fisher Scientific). Cells were immediately lysed in boiled 1% SDS buffer (50 mM Tris-HCl, pH 8.0 and 1% SDS), and incubated at 95 C for 20 min. Cell extracts were collected by centrifugation at 14,000 rpm for 20 min. Cell extracts were diluted with 3× volumes of dilution buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, Halt protease and phosphatase inhibitor), and proceeded with immunoprecipitation protocol described above. Proteins were eluted by incubating beads in 2× Laemmli sample buffer (Bio-Rad) containing TCEP, at 95°C for 10 min.
Immunoblotting
Equal amounts of proteins, measured by BCA assay (Thermo Scientific), were mixed with sample buffers (NuPAGE LDS Sample Buffer, Thermo Fisher Scientific; Laemmli Protein Sample Buffer, Bio-Rad) containing TCEP (Pierce) and were separated by SDS-PAGE on 4%–12% Bis-Tris gels (Thermo Fisher Scientific) for 15–70 kDa proteins or 4%–20% Tris-Glycine gels (Bio-Rad) for 30–350 kDa proteins and transferred onto PVDF membranes (Bio-Rad) using the wet transfer system (Bio-Rad). Bis-Tris gels were transferred in NuPAGE transfer buffer (Novex) containing 20% methanol. Tris-Glycine gels were transferred in Tris-Glycine buffer containing 5% methanol and 0.01% SDS. Membranes were blocked with 5% bovine serum albumin (BSA; Research Products International) in TBST (0.1% (v/v) Tween-20) at room temperature for 1 h. Primary antibody incubations were carried out in the blocking buffer at 4°C overnight, and secondary antibody incubations in the blocking buffer at room temperature for 2 h. Proteins were visualized with Amersham ECL Western Blotting Detection Reagent (GE), Clarity ECL Western Blotting Substrate (Bio-Rad) or SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific) and imaged using Image Lab software (Bio-Rad) on ChemiDoc XRS System (Bio-Rad).
Size-exclusion chromatography
Nuclear extracts were collected and diluted as described for immunoprecipitation. Lysates were incubated overnight at 4°C and centrifuged through a 0.45 μm Ultrafree-MC HV Centrifugal Filter (UFC30HVNB, EMD Millipore) before being applied to a pre-equilibrated Superose 6 10/300 GL column (17-5172-01, GE Healthcare Life Sciences). Fractions were collected and separated by SDS-PAGE on 4%–20% Tris-Glycine gels. The Superose 6 standard curve supplied by manufacturer was used to estimate molecular weight range for the collected fractions.
RNA extraction and quantitative PCR
Total RNA was extracted and purified from cells using the Quick-RNA MiniPrep Kit (Zymo Research, Irvine, CA). Equal amounts of RNA were reverse transcribed using High Capacity RNA to cDNA Kit (Applied Biosystems, Foster City, CA) according to the manufacturer’s instructions. Real-time PCR reactions were prepared with PowerUp SYBR Green Master Mix (Applied Biosystems, Foster City, CA). mRNA levels were normalized to the house-keeping gene, β-actin.
Immunoprecipitation for mass spectrometry analyses
For AP-MS, cell extracts were pre-cleared with Protein-G Dynabeads for 1 h at 4°C before primary antibody incubation. For TAP-MS, whole cell extracts were incubated with Strep-Tactin Sepharose (IBA) at 4°C overnight. Cell extracts were washed three times in wash buffer and incubated in 1× Strep-tag elution buffer (IBA) for 10 min at room temperature on gentle vortex. The eluted solution was diluted with five times wash buffer and incubated with EZview red Flag M2 affinity gel (Sigma) for 4 h at 4°C. Beads were washed three times and eluted in wash buffer containing 150 μg/mL Flag peptide (Sigma) for 30 min at room temperature on gentle vortex.
Mass spectrometry
MS sample preparation and analyses were performed by the Stanford University Mass Spectrometry facility. In brief, for gel-free MS analysis, the final elutions from immunoprecipitations were solubilized and digested using the filter aided sample preparation (FASP) protocol (Wi sniewski et al., 2009). For gel-based analysis, final elutions were separated by 4%–20% SDS-PAGE (Bio-Rad) followed by silver staining (Thermo Scientific). Gel fragments were excised and cut into 1 mm3 cubes, reduced with 5 mM DTT and alkylated with acrylamide. Trypsin/Lys-C Mix (Promega) was used as the protease for protein digestion. Peptides were extracted and dried using a speed-vac prior to reconstitution and analysis.
Nano reverse-phase HPLC was performed using either an Eksigent 2D nanoLC (Eksigent) or Waters nanoAcquity (Waters) with mobile phase A consisting of 0.1% formic acid in water and mobile phase B consisting of 0.1% formic acid in acetonitrile. A fused silica column self packed with duragel. C18 (Peeke) matrix was used with a linear gradient from 2% B to 40% B at a flow rate of 600 nL/minute. The nanoHPLC was interfaced with a Bruker/Michrom Advance Captive spray source for nanoESI into either a LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) or an Orbitrap Elite (Thermo Fisher Scientific) operating in data-dependent acquisition mode to perform MS/MS on the top twelve most intense multiply charged cations.
Statistical analysis for AP-MS and TAP-MS
PPARγ AP-MS RAW data were searched with MaxQuant v. 1.5.0 (Cox and Mann, 2008) using default options, enabling matching between runs, against the reviewed version of the human reference proteome (07/30/2013). Then, the MS-1 peak intensities were Log2-transformed and their distributions were median-centered across all runs. Missing intensities for peptides in a given run were imputed by setting their value to the mean minimal intensity across all runs, as an estimate for the under limit of detectability by MS. The normalized dataset was then analyzed by fitting a mixed effects model per protein in MSstats (v. 2.3.4, available on msstats.org) (Choi et al., 2014b) using the model without interaction terms, unequal feature variance and restricted scope of technical and biological replication. Pathway analyses were performed using COMPLEAT (Vinayagam et al., 2013) and Ingenuity Pathway Analysis (QIAGEN) and further curated manually based on published literature. Finally, known interactions between the 47 high confidence interactors of the DDR and replication pathway were computed by mining the COMPLEAT dataset and represented using Cytoscape(v.2.8.3) (Smoot et al., 2011).
For analysis of potential crosslinked peptides, data were searched using Byonic v3.1.0 (Protein Metrics), allowing for crosslinks between PPARγ and NBS1, assuming that peptides were tryptic with up to two missed cleavages and linked by BS3. The resulting spectral assignments were further analyzed using Byologic v3.2–38 (Protein Metrics) to identify and qualitatively assess crosslinked spectra at a chromatographic, MS1, and MS/MS level as described previously (https://www.ncbi.nlm.nih.gov/pubmed/28431242). Following this qualitative assessment, potential crosslinked peptides were compared against structural constraints based on the crystallography source PDB ID: 3DZU (Chandra et al., 2008) using the PyMOL Molecular Graphics System v.2.2.2 (Schrödinger, LLC).
EdU incorporation assay for cell cycle analysis
Cell cycle analysis was performed using the Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s protocol. EdU (5-ethynyl-2′-deoxyuridine, 10 μM) was added to cells 2 h before harvest. Cells were co-stained with propidium iodide to quantitate DNA content.
Alkaline comet assay
DNA breaks were monitored using the CometAssay Reagent Kit for Single Cell Gel Electrophoresis Assay (Trevigen) according to the manufacturer’s protocol. DNA was stained with SYBR-gold (Thermo Fisher Scientific). The comet tail lengths (defined as the length from the center of the DNA head to the end of the DNA tail) were measured by counting 100–150 cells for each sample and analyzed using ImageJ (v. 2.0) with a Comet Assay Plugin, downloaded from https://www.med.unc.edu/microscopy/resources/imagej-plugins-and-macros/comet-assay. The box in box-and-whiskers plots corresponds to the 25th to 75th percentiles. The line in the box marks the median and whiskers correspond to the 10th to 90th percentiles. Data points represent average comet tail length per sample. The outliers are represented as dots outside the whiskers.
Hypoxia and Reoxygenation
Cells were seeded on coverslips overnight and transferred to a Baker Ruskinn Concept anaerobic (< 0.1% O2) chamber. Cells were incubated for 24 hours before exposing to room air for 10 min to induce oxidative damage. Cells were fixed according to the immunofluorescence protocol.
Immunofluorescence staining and confocal microscopy
After staining cultured cells, to avoid bias coverslips were randomlyassigned with a number. The investigator was blinded to the randomization during data acquisition and analyses. Cultured cells were seeded on coverslips pre-coated with mouse Collagen IV (Corning). For staining of pATM (S1981), pRPA2 (S4/8), γH2AX and 8-oxo-dG foci, cells were pre-extracted with ice-cold 0.25% Triton in TBS at 4°C for 10 min with gentle rocking. Cells were then washed in TBS and fixed with 4% paraformaldehyde (Electron Microscopy Sciences) at room temperature for 10 min. After fixation, cells were blocked with 3% BSA in TBST at room temperature for 1 h. Primaryantibody incubations were carried out in the blocking buffer at 4°C overnight, and secondary antibody incubations in the blocking buffer at room temperature for 1 h. Cells were mounted with DAPI Fluoromount-G (DAPI, 4,6-diamidino-2-phenylindole) (SoutherBiotech). Stained cells were imaged using Leica Application Suite X software on a Leica CTR 6500 (Leica).
For staining lung tissue sections, the genotypes of the lung sections were blinded to the investigator before the staining procedures were carried out, and during the data acquisition. Lungs tissues were fixed with 10% Neutral Buffered Formalin (Thermo Scientific) and embedded in paraffin (Leica). Sections were first deparaffinized and rehydrated. For antigen retrieval, sections were incubated in sub-boiling buffer (0.25 mM EDTA, pH 8.0) for 50 min. After cooling to room temperature, sections were incubated in 3% H2O2 for 10 min. Sections were incubated with the blocking buffer [3% normal goat serum (Jackson ImmunoResearch), 2% BSA, 0.2% Triton] at room temperature for 1 h. Primary antibody incubation was then carried out at 4°C overnight, followed by secondary antibody incubation at room temperature for 1 h. Sections were mounted with VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories). Stained lung sections were imaged using the FV10-ASW4.3 software on a Fluoview 1000 confocal microscope.
Immunofluorescence image analyses
For pATM (S1981), pPRA2 (S4/8), γH2AX and 8-oxo-dG imaging, 1–3 fields were acquired with the 20X objective to obtain 100–150 cells per sample. Nuclear fluorescence intensities were measured using ImageJ (Schneider et al., 2012), and box-and-whiskers graphs generated as described above. Each data point represents nuclear fluorescence per cell. Each graph is representative of one out of three independent experiments.
For evaluating γH2AX in mouse lung sections, 7 – 11 arteries (based on vWF staining and location) per animal (5 animals per group) were imaged. For evaluating γH2AX in clinical lung sections, 3 – 6 arteries (based on vWF staining and location) per subject were imaged (control: 6 and PAH: 5 subjects). Z stacked images were acquired with the 60X and 40X objectives for the mouse and human samples respectively. Nuclear γH2AX fluorescence was measured using the FociCounter software. The box-and-whiskers plots were generated as described above. Each data point represents average nuclear γH2AX fluorescence (after normalization to cell number) per artery. Details can be found in Supplemental Information.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism 6.0 (GraphPad Software). Experimental data were first subjected to the Shapiro-Wilk normality test to determine if the data are of a Gaussian distribution. The following tests were then selected for further statistical analyses. For pairwise comparison, unpaired t test (parametric) or two-tailed Mann-Whitney test (nonparametric) was used. For one-way ANOVA, Fisher’s LSD test was used for pairwise comparison. For two-way ANOVA, Fisher’s LSD test was used for pairwise comparison. For comparing immunofluorescence signals in more than two groups, Kruskal–Wallis ANOVA test followed by Dunn’s test was used. P values are indicated as *, p < 0.05; **, p < 0.01; ***, p < 0.001; ****, p < 0.0001.
DATA AND SOFTWARE AVAILABILITY
The accession number for the mass spectrometry data generated from this study is Mass Spectrometry Interactive Virtual Environment (MassIVE) database: MSV000083257.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG® M2 antibody | Sigma-Aldrich | Cat#F1804, RRID:AB_262044 |
| Anti-Strep Tag antibody [GT661] | Abcam | Cat#ab184224 |
| Mre11 Antibody | Novus | Cat#NB100-142, RRID:AB_10077796 |
| Rad50 (13B3/2C6) antibody | Santa Cruz Biotechnology | Cat#sc-56209, RRID:AB_785402 |
| NBS1 Antibody | Novus | Cat#NB100-221, RRID:AB_10001212 |
| RXRalpha (D-20) antibody | Santa Cruz Biotechnology | Cat#sc-553, RRID:AB_2184874 |
| UBR5/EDD1 Antibody | Bethyl | Cat#A300-573A, RRID:AB_2210189 |
| Anti-ATM Protein Kinase pS1981 (MOUSE) Monoclonal Antibody | Rockland | Cat#200-301-400, RRID:AB_217868 |
| ATM antibody | Abcam | Cat#ab2631, RRID:AB_2062948 |
| PPAR (D69) Antibody | Cell Signaling Technology | Cat#2430, RRID:AB_823599 |
| Anti-ASCIZ/ATMIN antibody | Millipore | Cat#AB3271, RRID:AB_2243333 |
| Phospho RPA32 (S4/S8) Antibody | Bethyl | Cat#A300-245A, RRID:AB_210547 |
| RPA32 Antibody | Bethyl | Cat#A300-244A, RRID:AB_185548 |
| Phospho KAP-1 (S824) Antibody | Bethyl | Cat# A300-767A, RRID:AB_669740 |
| KAP-1 Antibody | Bethyl | Cat#A303-838A, RRID:AB_2620189 |
| Phospho SMC1 (S966) Antibody | Bethyl | Cat#A300-050A, RRID:AB_67578 |
| SMC1 Antibody | Bethyl | Cat#A303-834A, RRID:AB_2620185 |
| Mouse Anti-HA.11 Monoclonal Antibody, Unconjugated, Clone 16B12 | Covance Research Products Inc | Cat#MMS-101 P-500, RRID:AB_291261 |
| Mouse Anti-Histone H2A.X, phospho (Ser139) Monoclonal antibody, Unconjugated, Clone jbw301 | Millipore | Cat#05-636, RRID:AB_309864 |
| H2AX Antibody | Bethyl | Cat#A300-083A, RRID:AB_203289 |
| Von Willebrand Factor antibody | Abcam | Cat#ab6994, RRID:AB_305689 |
| PPARG / PPAR Gamma Antibody (phospho- Ser245/273) | LS Bio | Cat#LS-C209422 |
| K48-linkage Specific Polyubiquitin Antibody | Cell Signaling Technology | Cat#4289, RRID:AB_10557239 |
| Mouse Anti-8-oxo-dG Monoclonal Antibody, Unconjugated, Clone 200 | R and D Systems | Cat#4354-MC-050, RRID:AB_1857195 |
| Lamin B1 (S-20) antibody | Santa Cruz Biotechnology | Cat#sc-30264, RRID:AB_2136305 |
| Mouse Anti-Glyceraldehyde-3-PDH (GAPDH) Monoclonal antibody, Unconjugated | Millipore | Cat#MAB374, RRID:AB_2107445 |
| Rat Anti-ORC2 Monoclonal Antibody, Unconjugated, Clone 3G6 | Cell Signaling Technology | Cat#4736, RRID:AB_2157716 |
| Mouse Anti-alpha-Tubulin Monoclonal Antibody, Unconjugated, Clone B-5-1-2 | Sigma-Aldrich | Cat#T6074, RRID:AB_477582 |
| Rabbit anti-Goat IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat#81-1620, RRID:AB_2534006) |
| Goat anti-Rat IgG (H+L) Secondary Antibody, HRP | Thermo Fisher Scientific | Cat#31470, RRID:AB_228356 |
| Peroxidase-IgG Fraction Monoclonal Mouse AntiRabbit IgG, Light Chain Specific (min X Bov,Gt,Arm Hms,Hrs,Hu,Ms,Rat,Shp Ig) antibody | Jackson ImmunoResearch Labs | Cat#211-032-171, RRID:AB_2339149 |
| Peroxidase-AffiniPure Goat Anti-Mouse IgG, Light Chain* Specific (min X Bov,Gt,Hrs,Hu,Rb,Rat,Shp Ig) antibody | Jackson ImmunoResearch Labs | Cat#115-035-174, RRID:AB_2338512 |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#A-21202, RRID:AB_141607 |
| Donkey anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#A-21207, RRID:AB_141637 |
| Biological Samples | ||
| Human control and PAH patient specimens (tissues and cells) | The Pulmonary Hypertension Breakthrough Initiative (PHBI) Network | Table S5 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PolyJet In Vitro DNA Transfection Reagent | SignaGen Laboratories | Cat#SL100688 |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Fisher Scientific | Cat#13778150 |
| GW9662 | Sigma-Aldrich | Cat#M6191 |
| SR10221 | Drs. Patrick Griffin, Theodore Kamenecka, Scripps Research Institute, Florida | Marciano et al., 2015 |
| Sigma-Aldrich | Cat#D1515 | |
| HU | Sigma-Aldrich | Cat#H8627 |
| BS3 (bis(sulfosuccinimidyl)suberate) | Thermo Fisher Scientific | Cat#21580 |
| ChromPure Rabbit IgG, whole molecule | Jackson ImmunoResearch Labs | Cat#011-000-003 |
| ChromPure Rabbit IgG, whole molecule | Jackson ImmunoResearch Labs | Cat#015-000-003 |
| MG-132 | Calbiochem | Cat#474790 |
| NEM (N-ethylmaleimide) | Thermo Fisher Scientific | Cat#23030 |
| Flag Peptide | Sigma-Aldrich | Cat#F3290 |
| Critical Commercial Assays | ||
| Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit | Thermo Fisher Scientific | Cat#C10634 |
| CometAssay Reagent Kit for Single Cell Gel Electrophoresis Assay | Trevigen | Cat#4250-050-K |
| P5 Primary Cell 4D-Nucleofector X Kit L | Lonza | Cat#V4XP-5024 |
| Deposited Data | ||
| Raw mass spectrometry data | https://massive.ucsd.edu/ProteoSAFe/dataset.jsp?task=d597007ca8d7436587ccc4a7d4848842 | Accession number: MSV000083257 |
| PPARg and RXRa structure | Chandra et al., 2008 | PDB ID: 3DZU |
| Experimental Models: Cell Lines | ||
| Human: 293T | ATCC | Cat#CRL-3216 |
| Human PAECs | PromoCell | Cat#C-12241 |
| Human umbilical vein ECs | Angio-Proteomie | Cat#cAP-0001GFP |
| Experimental Models: Organisms/Strains | ||
| Mouse: Tie2-PPARγ−/− tissue specimens | Guignabert et al., 2009 | Guignabert et al., 2009 |
| Oligonucleotides | ||
| siRNA targeting sequence (pool): PPARγ | Dharmacon | Cat#L-003436-00-0005 |
| siRNA targeting sequence (#9): PPARγ | Dharmacon | Cat#J-003436-09-0005 |
| siRNA targeting sequence (pool): UBR5 | Dharmacon | Cat#M-007189-02-0010 |
| siRNA targeting sequence (pool): ATMIN | Dharmacon | Cat#L-020304-01-0010 |
| siRNA targeting sequence (pool): NBS1 | Dharmacon | Cat#M-009641-02-0005 |
| siRNA targeting sequence (pool): Non-targeting | Dharmacon | Cat#D-001810-10 |
| Primers for quantitative real-time PCR and cloning | This paper | Table S6 |
| Recombinant DNA | ||
| Plasmid: pCMV2-Flag-PPARγ1 | This paper | N/A |
| Plasmid: pCMV2-Flag-PPARγ1-LBD | This paper | N/A |
| Plasmid: pcDNA-PPARγ1-2xStrep | This paper | N/A |
| Plasmid: pcDNA3-NBS1 | Dr. Hui-Kuan Lin, University of Texas MD Anderson Cancer Center | Wu etal., 2012 |
| Plasmid: pCMV2-siRNA-resistant-Flag-PPARγ1 (siRES-PPARγ) | This paper | N/A |
| Plasmid: pCMVtag2B-Flag-ATMIN (1-354) | Dr. Axel Behrens, King’s College London | Zhang et al., 2014 |
| Plasmid: pCMV2-siRNA-resistant-Flag-PPARγ1 (siRES-PPARγ) | This paper | N/A |
| Plasmid: pcDNA-GFP-StrepFlag | Dr. Nevan Krogan, UCSF | N/A |
| Plasmid: HA-ubiquitin | Dr. Nevan Krogan, UCSF | N/A |
| Software and Algorithms | ||
| MaxQuant | Cox and Mann, 2008 | http://www.coxdocs.org/doku.php?id=maxquant:common:download_and_installation#download |
| MSstats | Choi et al., 2014b | https://www.bioconductor.org/packages/release/bioc/html/MSstats.html |
| COMPLEAT | Vinayagam et al., 2013 | http://www.flyrnai.org/compleat/ |
| Ingenuity Pathway Analysis | QIAGEN | https://www.qiagenbioinformatics.com/products/ingenuity-pathway-analysis/ |
| Cytoscape | Smoot et al., 2011 | https://cytoscape.org/ |
| Byonic | Protein Metrics | https://www.proteinmetrics.com/products/byonic/ |
| Byologic | Protein Metrics | https://www.proteinmetrics.com/products/byologic/ |
| PyMOL Molecular Graphical System | Schrodinger, LCC | https://pymol.org/2/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Comet Assay Plugin for ImageJ | Dr. Robert Bagnell, University of North Carolina | https://www.med.unc.edu/microscopy/resources/imagej-plugins-and-macros/comet-assay |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
Highlights.
PPARγ proteomics identifies interactions with the MRE11-RAD50-NBS1 complex and UBR5
Upon DNA damage, PPARγ promotes UBR5-mediatedATMIN degradation to activate ATM
PPARγ-UBR5 interaction is disrupted in endothelial cells isolated from PAH patients
Depleting ATMIN in PAH endothelial cells restores ATM signaling upon DNA damage
ACKNOWLEDGMENTS
We thank A. Giaccia for helpful comments on the manuscript. We thank P. Griffin and T. Kamenecka (Scripps Research Institute, Florida) for providing PPARγ modulators; L. Nagy (University of Debrecen, Hungary), C. Craik (UCSF), N. Krogan (UCSF), H.-K. Lin (MD Anderson Cancer Center), and A. Behrens (King’s College London) for providing plasmids; T. Desai (Stanford) for the use of the Leica confocal microscope; P.A. del Rosario (Stanford) for providing clinical information of the PHBI samples; and M. Fox and M. Roof (Stanford) for administrative assistance. We thank members of the Rabinovitch Lab and the Department of Radiation Oncology (Stanford) for their support. Mass spectrometry was performed at the Vincent Coates Foundation Mass Spectrometry Laboratory, Stanford University. Mass spectrometry was funded by award number S10RR027425 from the National Center For Research Resources. The Pulmonary Hypertension Breakthrough Initiative (PHBI) Network is funded by NIH/National Heath, Lung, and Blood Institute (NHLBI) R24 HL123767 and the Cardiovascular Medical Research and Education Fund (CMREF) grant UL1RR024986. This work was supported by NIH/NHLBI grant R01 HL087118 (M.R.), fellowships from the Deutsche Forschungsgemeinschaft (He 6855/1–1 to J.K.H. and Ni 1456/1–1 to N.P.N.), and from the Deutsche Herzstiftung e.V. (S/06/11 to I.D.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and six tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2019.01.013.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, and Evans RM (2013). PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med 19, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alastalo TP, Li M, de. J. Perez V, Pham D, Sawada H, Wang JK, Koskenvuo M, Wang L, Freeman BA, Chang HY, and Rabinovitch M (2011). Disruption of PPARγ/β-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J. Clin. Invest 121, 3735–3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldred MA, Comhair SA, Varella-Garcia M, Asosingh K, Xu W, Noon GP, Thistlethwaite PA, Tuder RM, Erzurum SC, Geraci MW, and Coldren CD (2010). Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med 182, 1153–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, Koder A, and Evans RM (1999). PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol. Cell 4, 585–595. [DOI] [PubMed] [Google Scholar]
- Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, Mann M, Jackson SP, and Choudhary C (2012). Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol. Cell 46, 212–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burma S, Chen BP, Murphy M, Kurimasa A, and Chen DJ (2001). ATM phosphorylates histone H2AX in response to DNA double-strand breaks.J. Biol. Chem 276, 42462–42467. [DOI] [PubMed] [Google Scholar]
- Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, Burris TP, and Rastinejad F (2008). Structure of the intact PPAR-gamma-RXR- nuclear receptor complex on DNA. Nature 456, 350–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng KC, Cahill DS, Kasai H, Nishimura S, and Loeb LA (1992). 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes G– –T and A– –C substitutions. J. Biol. Chem 267, 166–172. [PubMed] [Google Scholar]
- Choi JH, Choi SS, Kim ES, Jedrychowski MP, Yang YR, Jang HJ, Suh PG, Banks AS, Gygi SP, and Spiegelman BM (2014a). Thrap3 docks on phosphoserine 273 of PPARγ and controls diabetic gene programming. Genes Dev 28, 2361–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Chang CY, Clough T, Broudy D, Killeen T, MacLean B, and Vitek O (2014b). MSstats: an R package for statistical analysis of quantitative mass spectrometry-based proteomic experiments. Bioinformatics 30, 2524–2526. [DOI] [PubMed] [Google Scholar]
- Cox J, and Mann M (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- Cuadrado M, Martinez-Pastor B, Murga M, Toledo LI, Gutierrez-Martinez P, Lopez E, and Fernandez-Capetillo O (2006). ATM regulates ATR chromatin loading in response to DNA double-strand breaks. J. Exp. Med 203, 297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jesus Perez VA, Yuan K, Lyuksyutova MA, Dewey F, Orcholski ME, Shuffle EM, Mathur M, Yancy L Jr., Rojas V, Li CG, et al. (2014). Whole-exome sequencing reveals TopBP1 as a novel gene in idiopathic pulmonary arterial hypertension. Am. J. Respir. Crit. Care Med 189, 1260–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold I, Hennigs JK, Miyagawa K, Li CG, Nickel NP, Kaschwich M, Cao A, Wang L, Reddy S, Chen PI, et al. (2015). BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab 21, 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duval C, Chinetti G, Trottein F, Fruchart JC, and Staels B (2002). The role of PPARs in atherosclerosis. Trends Mol. Med 8, 422–430. [DOI] [PubMed] [Google Scholar]
- Fondell JD, Ge H, and Roeder RG (1996). Ligand induction of a transcriptionally active thyroid hormone receptor coactivator complex. Proc. Natl. Acad. Sci. USA 93, 8329–8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girnun GD, Naseri E, Vafai SB, Qu L, Szwaya JD, Bronson R, Alberta JA, and Spiegelman BM (2007). Synergy between PPARγamma ligands and platinum-based drugs in cancer. Cancer Cell 11, 395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman MH, and Ciechanover A (2002). The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol. Rev 82, 373–428. [DOI] [PubMed] [Google Scholar]
- Goggolidou P, Stevens JL, Agueci F, Keynton J, Wheway G, Grimes DT, Patel SH, Hilton H, Morthorst SK, DiPaolo A, et al. (2014). ATMIN is a transcriptional regulator of both lung morphogenesis and ciliogenesis. Development 141, 3966–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudjonsson T, Altmeyer M, Savic V, Toledo L, Dinant C, Grøfte M, Bart-kova J, Poulsen M, Oka Y, Bekker-Jensen S, et al. (2012). TRIP12 and UBR5 suppress spreading of chromatin ubiquitylation at damaged chromosomes. Cell 150, 697–709. [DOI] [PubMed] [Google Scholar]
- Guignabert C, Alvira CM, Alastalo TP, Sawada H, Hansmann G, Zhao M, Wang L, El-Bizri N, and Rabinovitch M (2009). Tie2-mediated loss of peroxisome proliferator-activated receptor-gamma in mice causes PDGF receptor-beta-dependent pulmonary arterial muscularization. Am. J. Physiol. Lung Cell. Mol. Physiol 297, L1082–L1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamblin M, Chang L, Zhang H, Yang K, Zhang J, and Chen YE (2010). Vascular smooth muscle cell peroxisome proliferator-activated receptor-γ deletion promotes abdominal aortic aneurysms. J. Vasc. Surg 52, 984–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond EM, Dorie MJ, and Giaccia AJ (2003). ATR/ATM targets are phosphorylated by ATR in response to hypoxia and ATM in response to reoxygenation. J. Biol. Chem 278, 12207–12213. [DOI] [PubMed] [Google Scholar]
- Hansmann G, de Jesus Perez VA, Alastalo TP, Alvira CM, Guignabert C, Bekker JM, Schellong S, Urashima T, Wang L, Morrell NW, and Rabinovitch M (2008). An antiproliferative BMP-2/PPARγamma/apoE axis in human and murine SMCs and its role in pulmonary hypertension. J. Clin. Invest 118, 1846–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopper RK, Moonen JR, Diebold I, Cao A, Rhodes CJ, Tojais NF, Hennigs JK, Gu M, Wang L, and Rabinovitch M (2016). In pulmonary arterial hypertension, reduced BMPR2 promotes endothelial-to-mesenchymal transition via HMGA1 and its target slug. Circulation 133, 1783–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y, Moreau F, and Chadee K (2012). PPARγ is an E3 ligase that induces the degradation of NFkB/p65. Nat. Commun 3, 1300. [DOI] [PubMed] [Google Scholar]
- Jurado S, Smyth I, van Denderen B, Tenis N, Hammet A, Hewitt K, Ng JL, McNees CJ, Kozlov SV, Oka H, et al. (2010). Dual functions of ASCIZ in the DNA base damage response and pulmonary organogenesis. PLoS Genet 6, e1001170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khandekar MJ, Banks AS, Laznik-Bogoslavski D, White JP, Choi JH, Kazak L, Lo JC, Cohen P, Wong KK, Kamenecka TM, et al. (2018). Noncanonical agonist PPARγ ligands modulate the response to DNA damage and sensitize cancer cells to cytotoxic chemotherapy. Proc. Natl. Acad. Sci. USA 115, 561–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppen A, and Kalkhoven E (2010). Brown vs white adipocytes: the PPARγamma coregulator story. FEBS Lett 584, 3250–3259. [DOI] [PubMed] [Google Scholar]
- Kurz EU, Douglas P, and Lees-Miller SP (2004). Doxorubicin activates ATM-dependent phosphorylation of multiple downstream targets in part through the generation of reactive oxygen species. J. Biol. Chem 279, 53272–53281. [DOI] [PubMed] [Google Scholar]
- Lee JH, and Paull TT (2004). Direct activation of the ATM protein kinase by the Mre11/Rad50/Nbs1 complex. Science 304, 93–96. [DOI] [PubMed] [Google Scholar]
- Leszczynska KB, Göttgens EL, Biasoli D, Olcina MM, Ient J, Anbalagan S, Bernhardt S, Giaccia AJ, and Hammond EM (2016). Mechanisms and consequences of ATMIN repression in hypoxic conditions: roles for p53 and HIF-1. Sci. Rep 6, 21698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Opiyo SO, Manthey K, Glanzer JG, Ashley AK, Amerin C, Troksa K, Shrivastav M, Nickoloff JA, and Oakley GG (2012). Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res 40, 10780–10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, King A, Hoch NC, Chang C, Kelly GL, Deans AJ, and Heierhorst J (2017). ASCIZ/ATMIN is dispensable for ATM signaling in response to replication stress. DNA Repair (Amst.) 57, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marciano DP, Kuruvilla DS, Boregowda SV, Asteian A, Hughes TS, Garcia-Ordonez R, Corzo CA, Khan TM, Novick SJ, Park H, et al. (2015). Pharmacological repression of PPARγ promotes osteogenesis. Nat. Commun 6, 7443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura K, Huang NJ, Cocce K, Zhang L, and Kornbluth S (2017). Downregulation of the proapoptotic protein MOAP-1 by the UBR5 ubiquitin ligase and its role in ovarian cancer resistance to cisplatin. Oncogene 36, 1698–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meloche J, Pflieger A, Vaillancourt M, Paulin R, Potus F, Zervopoulos S, Graydon C, Courboulin A, Breuils-Bonnet S, Tremblay E, et al. (2014). Role for DNA damage signaling in pulmonary arterial hypertension. Circulation 129, 786–797. [DOI] [PubMed] [Google Scholar]
- Mirzoeva OK, and Petrini JH (2001). DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol. Cell. Biol 21, 281–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzoeva OK, and Petrini JH (2003). DNA replication-dependent nuclear dynamics of the Mre11 complex. Mol. Cancer Res 1, 207–218. [PubMed] [Google Scholar]
- Polo SE, and Jackson SP (2011). Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev 25, 409–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovitch M (2010). PPARγamma and the pathobiology of pulmonary arterial hypertension. Adv. Exp. Med. Biol 661, 447–458. [DOI] [PubMed] [Google Scholar]
- Rabinovitch M (2012). Molecular pathogenesis of pulmonary arterial hyper-tension. J. Clin. Invest 122, 4306–4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranchoux B, Antigny F, Rucker-Martin C, Hautefort A, Péchoux C, Bogaard HJ, Dorfmüller P, Remy S, Lecerf F, Planté S, et al. (2015). Endothelial-to-mesenchymal transition in pulmonary hypertension. Circulation 131, 1006–1018. [DOI] [PubMed] [Google Scholar]
- Reinhardt HC, and Yaffe MB (2013). Phospho-Ser/Thr-binding domains: navigating the cell cycle and DNA damage response. Nat. Rev. Mol. Cell Biol 14, 563–580. [DOI] [PubMed] [Google Scholar]
- Sa S, Gu M, Chappell J, Shao NY, Ameen M, Elliott KA, Li D, Grubert F, Li CG, Taylor S, et al. (2016). iPSC model of pulmonary arterial hypertension reveals novel gene expression and patient specificity. Am. J. Respir. Crit. Care Med 195, 930–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders DN, Hird SL, Withington SL, Dunwoodie SL, Henderson MJ, Biben C, Sutherland RL, Ormandy CJ, and Watts CK (2004). Edd, the murine hyperplastic disc gene, is essential for yolk sac vascularization and chorioallantoic fusion. Mol. Cell. Biol 24, 7225–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt L, Wiedner M, Velimezi G, Prochazkova J, Owusu M, Bauer S, and Loizou JI (2014). ATMIN is required for the ATM-mediated signaling and recruitment of 53BP1 to DNA damage sites upon replication stress. DNA Repair (Amst.) 24, 122–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoot ME, Ono K, Ruscheinski J, Wang PL, and Ideker T (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, and Komander D (2016). Ubiquitin modifications. Cell Res 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vattulainen-Collanus S, Akinrinade O, Li M, Koskenvuo M, Li CG, Rao SP, de Jesus Perez V, Yuan K, Sawada H, Koskenvuo JW, et al. (2016). Loss of PPARγ in endothelial cells leads to impaired angiogenesis. J. Cell Sci 129, 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinayagam A, Hu Y, Kulkarni M, Roesel C, Sopko R, Mohr SE, and Perrimon N (2013). Protein complex-based analysis framework for high-throughput data sets. Sci. Signal 6, rs5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wi sniewski JR, Zougman A, Nagaraj N, and Mann M (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. [DOI] [PubMed] [Google Scholar]
- Wu J, Zhang X, Zhang L, Wu CY, Rezaeian AH, Chan CH, Li JM, Wang J, Gao Y, Han F, et al. (2012). Skp2 E3 ligase integrates ATM activation and homologous recombination repair by ubiquitinating NBS1. Mol. Cell 46, 351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdi PT, Wang Y, Zhao S, Patel N, Lee EY, and Qin J (2002). SMC1 is a downstream effector in the ATM/NBS1 branch of the human S-phase checkpoint. Genes Dev 16, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Penicud K, Bruhn C, Loizou JI, Kanu N, Wang ZQ, and Behrens A (2012). Competition between NBS1 and ATMIN controls ATM signaling pathway choice. Cell Rep 2, 1498–1504. [DOI] [PubMed] [Google Scholar]
- Zhang T, Cronshaw J, Kanu N, Snijders AP, and Behrens A (2014). UBR5-mediated ubiquitination of ATMIN is required for ionizing radiation- induced ATM signaling and function. Proc. Natl. Acad. Sci. USA 111, 12091–12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv Y, Bielopolski D, Galanty Y, Lukas C, Taya Y, Schultz DC, Lukas J, Bekker-Jensen S, Bartek J, and Shiloh Y (2006). Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat. Cell Biol 8, 870–876. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the mass spectrometry data generated from this study is Mass Spectrometry Interactive Virtual Environment (MassIVE) database: MSV000083257.