

Abstract
As human population density and antibiotic exposure increase, specialised bacterial subtypes have begun to emerge. Arising among species that are common commensals and infrequent pathogens, antibiotic-resistant ‘high-risk clones’ have evolved to better survive in the modern human. Here, we show that the major matrix porin (OmpK35) of Klebsiella pneumoniae is not required in the mammalian host for colonisation, pathogenesis, nor for antibiotic resistance, and that it is commonly absent in pathogenic isolates. This is found in association with, but apparently independent of, a highly specific change in the co-regulated partner porin, the osmoporin (OmpK36), which provides enhanced antibiotic resistance without significant loss of fitness in the mammalian host. These features are common in well-described ‘high-risk clones’ of K. pneumoniae, as well as in unrelated members of this species and similar adaptations are found in other members of the Enterobacteriaceae that share this lifestyle. Available sequence data indicate evolutionary convergence, with implications for the spread of lethal antibiotic-resistant pathogens in humans.
Author summary
Klebsiella pneumoniae is a Gram-negative enteric bacterium and a significant cause of human disease. It is a frequent agent of pneumonia, and systemic infections can have high mortality rates (60%). OmpK35 and OmpK36 are the major co-regulated outer membrane porins of K. pneumoniae. OmpK36 absence has been related to antibiotic resistance but also decreased bacterial fitness and diminished virulence. A mutation that constricts the porin channel (Gly134Asp135 duplication in loop 3 of the porin, OmpK36GD) has been previously observed and suggested as a solution to the fitness cost imposed by loss of OmpK36. In the present study we constructed isogenic mutants to verify this and test the impact of these porin changes on antimicrobial resistance, fitness and virulence. Our results show that loss of OmpK35 has no significant impact on bacterial survival in both nutrient-rich environments and in the mammalian host, which is consistent with a predicted role outside that niche. When directly compared with the complete loss of the partner osmoporin OmpK36, we found that isogenic OmpK36GD strains maintain high levels of antibiotic resistance and that the GD duplication significantly reduces neither gut colonisation nor pathogenicity in a pneumonia mouse model. These changes are widespread in unrelated genomes. Our data provide evidence of specific variations in the loop 3 of OmpK36 and the absence of OmpK35 in K. pneumoniae clinical isolates that are examples of successful adaptation to human colonization/infection and antibiotic pressure, and are features of a fundamental evolutionary shift in this important human pathogen.
Introduction
Host adaptation and niche specialisation are well described in bacteria. As human population density rises, commensals and pathogens among the Enterobacteriaceae are transmitted directly from human to human and increasingly exposed to antibiotics. K. pneumoniae is now a common cause of healthcare-associated infections and is one of the most important agents of human sepsis [1]. High morbidity and mortality are associated with acquired antibiotic resistance, most importantly by horizontal transfer of genes encoding extended-spectrum β-lactamases (ESBL) [2] and plasmid-mediated AmpC β-lactamases (pAmpC) [3]. Carbapenem antibiotics have been effective against such isolates for decades, but resistance to these antibiotics is increasingly common in turn [4] and in February 2017, carbapenem resistant Enterobacteriaceae were listed among the highest (‘critical’) research priorities by the World Health Organisation. Acquired genes encoding efficient carbapenem hydrolysing enzymes [5] typically require phenotypic augmentation by permeability reduction to be clinically relevant in the Enterobacteriaceae. Indeed, clinically significant carbapenem resistance may even be seen with the less specialised AmpC or ESBL enzymes in strains with sufficiently reduced outer membrane permeability [6,7].
K. pneumoniae expresses two major nonspecific porins (OmpK35 and OmpK36) through which nutrients and other hydrophilic molecules such as β-lactams diffuse into the cell [8,9]. The expression of these two major porins in K. pneumoniae is strongly linked with β-lactam susceptibility [6,7] and strains lacking both porins exhibit high levels of resistance [10]. K. pneumoniae is commonly present in the human gut [1] but also grows in low-nutrient and low-osmolarity conditions, with decreased expression of the ‘osmoporin’, OmpK36, and increased expression of the ‘matrix porin’, OmpK35, which has greater general permeability. In the mammalian host in vivo, and in nutritious media in vitro, OmpK36 is the principal general porin and the gateway for β-lactam antibiotics, which are the most frequently prescribed antibiotic class in humans and the cornerstone of therapy for serious infections.
The fitness cost of certain antibiotic resistance mutations is well described [11,12,13,14]. Significantly reduced expression of porins provides some protection from β-lactam antibiotics but may incur a considerable metabolic cost as vital nutrients are simultaneously excluded [15]. Outer membrane permeability is thus a balance between self-defence and competitive fitness [16,17]. Global antibiotic restriction policies are founded on the premise of an inverse relationship between competitive fitness and resistance to antibiotics [18] and the expectation that antibiotic-resistant mutants will fail to successfully compete with their antibiotic-susceptible ancestors [19]. However, analysis of the principal porin relevant to infection in the mammalian host, OmpK36, revealed a key role for a transmembrane β-strand loop (loop3, L3) in the porin inner channel (‘eyelet’), which is electronegative at physiological pH. Minor changes in this region have been observed that are expected to be relatively permissive of small nutrient molecule diffusion but which may exclude more bulky anionic carbapenem and cephalosporin antibiotics [20].
Highly antibiotic-resistant K. pneumoniae is both a critical threat pathogen and a model of adaptation in a world with increasing human density and antibiotic exposure. The aim of this study was therefore to understand the pathogenesis and antimicrobial resistance implications of common changes in major porins that diminish membrane permeability.
Materials and methods
Bacterial strains, plasmids, primers and growth conditions
The bacterial strains, plasmids and primers used in this study are listed in Table 1 and S1 Table. Porin mutants were constructed in three antibiotic-susceptible K. pneumoniae strains (ATCC 13883, and clinical isolates 10.85 and 11.76 from our laboratory). Bacterial isolates were stored at -80°C in Nutrient broth (NB) with 20% glycerol and recovered on LB agar plates. Unless otherwise indicated, strains were routinely grown in Mueller-Hinton broth (MHB, BD Diagnostics, Franklin lakes, NJ, USA) or Luria-Bertani (LB, Life Technologies, Carlsbad, CA, USA). E. coli and K. pneumoniae strains carrying the chloramphenicol-resistant plasmids pKM200 and pCACtus were grown at 30°C on LB agar or in LB broth supplemented with 20 μg/ml chloramphenicol (Sigma-Aldrich, St. Louis, MO, USA). The growth of bacterial cells was determined by measuring the optical density at 600 nm (OD600) in an Eppendorf Biophotometer (Eppendorf AG, Hamburg, Germany).
Table 1. Bacterial strains used in this study.
| Strain | Relevant characteristic(s)a | Source or referenceb |
|---|---|---|
| K. pneumoniae | ||
| ATCC13883 | Klebsiella pneumoniae, ATCC 13883 | ATCC |
| ATCCΔOmpK35 | OmpK35 deletion strain of ATCC 13883; Tetr | TS |
| ATCCOmpK36GD | OmpK36 L3 GD strain of ATCC 13883 | TS |
| ATCCΔOmpK36 | OmpK36 deletion strain of ATCC 13883; Kmr | TS |
| ATCCΔOmpK35OmpK36GD | OmpK35 deletion strain with OmpK36 L3 GD; Tetr | TS |
| ATCCΔOmpK35ΔOmpK36 | OmpK35 and OmpK36 deletion strain of ATCC 13883; Tetr:Kmr | TS |
| 10.85 | Wild-type Klebsiella pneumoniae, clinical isolate | [21] |
| 10.85ΔOmpK35 | OmpK35 deletion strain of 10.85; Tetr | TS |
| 10.85OmpK36GD | OmpK36 L3 GD strain of ATCC 13883 | TS |
| 10.85ΔOmpK36 | OmpK36 deletion strain of ATCC 13883; Kmr | TS |
| 10.85ΔOmpK35OmpK36GD | OmpK35 deletion strain with OmpK36 L3 GD; Tetr | TS |
| 10.85ΔOmpK35ΔOmpK36 | OmpK35 and OmpK36 deletion strain of ATCC 13883; Tetr:Kmr | TS |
| 10.85ΔOmpK35ΔOmpK36+ pACYC184 | OmpK35 and OmpK36 double deletion mutant of the 10.85 clinical strain with the pACYC184 control plasmid; Tetr:Kmr | TS |
| 10.85ΔOmpK35ΔOmpK36+ pJIQQ-1 | OmpK35 and OmpK36 double deletion mutant of the 10.85 clinical strain complemented with ompK36; Tetr:Kmr | TS |
| 10.85ΔOmpK35ΔOmpK36+ pJIQQ-2 | OmpK35 and OmpK36 double deletion mutant of the 10.85 clinical strain complemented with ompK36GD; Tetr:Kmr | TS |
| 11.76 | Wild-type Klebsiella pneumoniae, clinical isolate | [21] |
| 11.76ΔOmpK35 | OmpK35 deletion strain of ATCC 13883; Tetr | TS |
| 11.76OmpK36GD | OmpK36 L3 GD strain of ATCC 13883 | TS |
| 11.76ΔOmpK36 | OmpK36 deletion strain of ATCC 13883; Kmr | TS |
| 11.76ΔOmpK35OmpK36GD | OmpK35 deletion strain with OmpK36 L3 GD; Tetr | TS |
| 11.76ΔOmpK35ΔOmpK36 | OmpK35 and OmpK36 deletion strain of ATCC 13883; Tetr:Kmr | TS |
| JIE1333 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE1334 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE1335 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE1348 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE1383 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE1462 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE1474 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE1482 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE2038 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE2055 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE2218 | OmpK36 L3 GD K. pneumoniae, clinical isolate | [21] |
| JIE4101 | OmpK36 L3 TD K. pneumoniae, clinical isolate | WH |
| JIE4111 | OmpK36 L3 TD K. pneumoniae, clinical isolate | WH |
| JIE4212 | OmpK36 L3 GD K. pneumoniae, clinical isolate | WH |
| JIE4609 | OmpK36 L3 TD K. pneumoniae, clinical isolate | WH |
| JIE4656 | OmpK36 L3 TD K. pneumoniae, clinical isolate | WH |
| JIE4735 | OmpK36 L3 TD K. pneumoniae, clinical isolate | WH |
| JIE2771 | OmpK35/OmpK36 deletion K. pneumoniae, clinical isolate. | [22] |
| JIE2771 + pACYC184 | K. pneumoniae JIE2771 harbouring pACYC184 | TS |
| JIE2771+pJIQQ-1 | K. pneumoniae JIE2771 complemented with ompK36 | TS |
| JIE2771+pJIQQ-2 | K. pneumoniae JIE2771 complemented with ompK36GD | TS |
| E. coli | ||
| DH5α | hsdR17 recAl; high efficiency transformation strain | [23] |
| S17 λpir | λpir lysogen of S17 (Tpr Smr thi pro ΔhsdR hsdM+ recA RP4::2-Tc::Mu-km::Tn7) | [24] |
aL3 GD: Gly+Asp duplication in loop 3, ATCC, American Type Culture collection, Tetr; Tetracycline resistant, Kmr; Kanamycin resistant
bTS, This study. WH, Westmead Hospital.
Construction of porin mutants
Chemical transformation, conjugation and electroporation were carried out using standard protocols. Platinum pfx DNA polymerase (Invitrogen, USA) was used to amplify blunt-ended PCR products. All PCR products were purified (PureLink Quick PCR Purification Kit; Invitrogen, USA). PCR and Sanger sequencing were used to confirm all constructs. Genomic DNA extractions were performed using a DNeasy Blood and Tissue kit (Qiagen, Valencia, CA, USA) and plasmid DNA using a PureLink Quick Plasmid Miniprep kit (Life Technologies, Carlsbad, CA, USA) or a HiSpeed Plasmid Midi Kit (Qiagen, Valencia, CA, USA).
Porin deletions mutants of K. pneumoniae ATCC 13883, 10.85 and 11.76 were created by introduction of tetA (tetracycline-resistance) or aphA-3 (kanamycin-resistance) into unique sites in ompK35 and ompK36 (HincII and StuI, respectively) which had been previously cloned into pGEM-T easy (Promega, Madison, WI, USA). The disrupted porin genes were then cloned into the pCACtus temperature-sensitive suicide vector (pJIAF-7 to pJIAF-12) to replace the respective chromosomal genes by homologous recombination [25]. Confirmation of correct single-copy chromosomal mutations were finally verified by PCR (S1 Table).
OmpK36GD mutants were obtained by amplification of OmpK36 from each parental strain using K36GD1 / K36GD2 and K36GD3 / K36GD4 primers (S1 Table). The amplicon, containing a GD duplication in L3, was cloned first in pGEM-T easy and after digestion with SphI and SacI (New England Biolabs, MA, USA) was introduced into pCACtus. The pCACtus-based constructs (pJIAF-13 to pJIAF-18) were transformed into S17λpir and conjugated into K. pneumoniae ΔOmpK36 (kanamycin-resistance mutant) in which the interrupted gene was replaced by OmpK36 porin with GD duplication in L3 by homologous recombination. Mutants were selected by loss of kanamycin resistance and confirmed by PCR and sequencing.
Double mutants (ΔOmpK35ΔOmpK36 and ΔOmpK35OmpK36GD) were constructed using Lambda Red-mediated recombineering as described previously [26,27], with some modifications. A tetracycline cassette flanked by OmpK35 deletion (~2.5 kb in size) was PCR amplified from an OmpK35 deletion mutant (ΔOmpK35; tetracycline resistant-previously obtained) using primers ompK35X-F and ompK35X-R (S1 Table), and the PCR products were purified. The Red helper plasmid pKM200 was electroporated into ΔOmpK36 or OmpK36GD single mutants. ompK35:tetA fragments were electroporated into ΔOmpK36 or OmpK36GD clones carrying pKM200. Bacteria were grown at 30°C for 2 h with agitation (225 rpm) followed by overnight incubation at 37°C. Different dilutions of the electroporated cells were spread on LB agar plates containing 10 μg/ml tetracycline to select for transformants at 37°C. The correct structure was confirmed by sequencing of PCR amplicons (primers ompK35F1 and ompK35R2, S1 Table).
All the engineered strains were verified by whole genome sequencing.
Complementation of porin mutations
The ompK36 gene with its predicted ribosomal binding site and transcriptional terminator was PCR amplified using 5’-GACAAGCTTTAAAAGGCATATAACAAACAG-3’ (forward) and 5’-CTGGGATCCAGCGAGGTTAAACCGG-3’ (reverse, S1 Table). Genomic DNA from K. pneumoniae ATCC 13883 wild-type strain (Table 1) was used as template. To generate ompK36 PCR product with the L3 GD mutation, the ATCC OmpK36GD strain was used as template DNA (Table 1). DNA inserts containing ompK36 and ompK36GD were cloned into the low-copy number pACYC184 vector [28] at the HindIII/BamHI restriction sites to generate pJIQQ-1 (pACYC184-OmpK36) and pJIQQ-2 (pACYC184-OmpK36GD) plasmids (S1 Table), respectively.
Two K. pneumoniae strains, 10.85ΔOmpK35ΔOmpK36 and JIE2771 (a clinical strain with naturally-occurring lesions in ompK35 and ompK36), were grown overnight in LB broth. On the next day, the strains were centrifuged and washed three times with ice-cold 10% glycerol. pACYC184, pJIQQ-1 and pJIQQ-2 were electroporated into the electrocompetent cells to generate the strains shown in Table 1. Sanger sequencing was performed to verify the absence of unintended non-synonymous mutations in the coding regions of ompK36 and ompK36GD.
Antimicrobial susceptibility tests
Susceptibilities to cefazolin (CFZ, Sigma-Aldrich, St. Louis, MO, USA), cephalothin (CEF, Sigma-Aldrich, St. Louis, MO, USA), cefoxitin (FOX, Sigma-Aldrich, St. Louis, MO, USA), cefuroxime (CXM, Sigma-Aldrich, St. Louis, MO, USA), cefotaxime (CTX, A.G. Scientific, Inc., San Diego, CA, USA), ceftazidime (CAZ, Sigma-Aldrich, St. Louis, MO, USA), ertapenem (ETP, Sigma-Aldrich, St. Louis, MO, USA), imipenem (IPM, Sigma-Aldrich, St. Louis, MO, USA), meropenem (MEM, A.G Scientific, Inc, San Diego, CA, USA) and ampicillin (MEM, A.G Scientific, Inc, San Diego, CA, USA) were performed by broth microdilution in cation-adjusted Mueller-Hinton (MH) broth (Becton Dickinson) with inocula of 5 x 105 CFU/ml in accordance with CLSI MO7-A9 recommendations [29]. All MICs were determined in triplicate at least on three separate occasions to obtain at least 9 discrete data points and compared with EUCAST and CLSI clinical breakpoints for all antibiotics [30,31]. E. coli (ATCC 25922) and Pseudomonas aeruginosa (ATCC 27853) were included in each experiment as quality controls.
For the in trans complemented strains, the susceptibilities of plasmid-bearing mutants of 10.85ΔOmpK35ΔOmpK36 to CEF, CFZ and FOX, as well as the susceptibilities of the plasmid-bearing mutants of JIE2771 to ETP, IPM and MEM, were performed in cation-adjusted Mueller-Hinton broth with chloramphenicol at a concentration of 25 μg/ml. Subsequent procedures follow those used for all other bacterial strains in this study.
Transfer of resistance genes
The filter mating method [32] was used to transfer plasmids from clinical isolates carrying blaCTX-M-15, (pJIE143) [33] blaIMP-4 (pEl1573) [34] and blaKPC-2 (pJIE2543-1) [22] to K. pneumoniae ATCC 13883 and porin mutants (ΔOmpK35, ΔOmpK36, OmpK36GD, ΔOmpK35ΔOmpK36 and ΔOmpK35OmpK36GD). The presence of resistance genes in transconjugants was confirmed by PCR [22,35,36] and the presence of plasmids of the expected size confirmed by S1 nuclease pulsed-field gel electrophoresis (S1 Fig) (Promega, Madison, WI, USA) [37,38].
Outer membrane porin investigation
Isolates were grown overnight under different temperatures (37°C, 30°C and 25°C) and different nutrient concentrations (MH and MH 1:10). Bacteria were disrupted by sonication and outer membrane porins (OMPs) isolated with sarcosyl (Sigma-Aldrich, St. Louis, MO, USA), as previously described [21,39]. Samples were boiled, analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) (12% separating gels), and stained with Imperial Protein Stain (Thermo Scientific, Rockford, IL, USA), following the manufacturer’s instructions. K. pneumoniae ATCC 13883, which produces both porins (OmpK35 and OmpK36) was used as a control [40]. Colour prestained protein standard, broad range (11–245 kDA) (New England Biolabs, MA, USA) was used as size marker.
Real-time reverse transcription-PCR
The expression levels of the different porins were measured by real-time RT-PCR. Cells were harvested in logarithmic phase at an OD600 of 0.5–0.6. Total RNA was isolated using RNeasy system (Qiagen). RNA was treated with DNase (TURBO DNA-free Kit, Ambion). cDNA was synthesized by high-capacity cDNA reverse transcriptase kit (Applied Biosystems). One microgram of the initially isolated RNA was used in each reverse transcription reaction. cDNA was diluted 1:10 and 2 μl were used for the real-time reaction. Three biological replicates, each with three technical replicates, were used in each of the assays. The relative levels of expression were calculated using the threshold cycle (2−ΔΔCT) method [41]. The expression of rpoD was used to normalize the results. The primers used are listed in S1 Table.
Determination of growth rate
Growth rates were determined as previously described [42]. Overnight broth cultures were diluted 1:1000. Six aliquots of 200 μl per dilution were transferred into 96-well microtiter plates (Corning Incorporated, Durham, NC, USA). Samples were incubated at 37°C and shaken before measurement of OD600 in a Vmax Kinetic microplate reader (Molecular Devices, Sunnyvale, CA, USA). Growth rates and generation times were calculated on OD600 values between 0.02–0.09. The relative growth rate was calculated by dividing the generation time of each mutant by the generation time of the parental strain (K. pneumoniae ATCC 13883, 10.85 or 11.76), which was included in every experiment. Experiments were performed in six technical replicates in three independent cultures on three different occasions. Results are expressed as means ± standard errors of the means.
For complemented strains, K. pneumoniae plasmid-bearing mutants of 10.85 ΔOmpK35ΔOmpK36 and JIE 2771 were streaked on Mueller-Hinton agar with 25 μg/ml chloramphenicol and incubated overnight at 37°C. On the next day, bacterial cells were resuspended in 0.85% saline to a turbidity of 0.5 McFarland. The inoculum was diluted 1:400 in cation-adjusted Mueller-Hinton broth, which were then transferred in six aliquots of 200 μl into 96-well microtiter plates (Corning Incorporated, Durham, NC, USA) and incubated with continuous gentle orbital shaking at 37°C in a SpectraMax iD5 Hybrid Multi-Mode Microplate Reader (Molecular Devices, San Jose, CA, USA). Measurements of OD600 were obtained every 5 minutes. Subsequent procedures follow those used for all other bacterial strains in this study.
In vitro competition experiments
Competition experiments were carried out as described previously [43]. Viable cell counts were obtained by plating every 24 h on antibiotic-free LB agar and on LB agar supplemented with antibiotic (kanamycin 20 μg/ml or tetracycline 10 μg/ml) to distinguish between mutants and wild-type cells. PCR (with primer pair K36GD4 / K36GD11 or K36GD12 / K36GD13 primers, S1 Table) was performed for the calculation of the competition results between the parental strain and OmpK36GD mutant (in this particular experiments, bacteria were diluted in fresh media every 24 h and PCR on 100 viable colonies of each replicate was performed every 48 h). All experiments were carried out in triplicate with three independent cultures. Mean values of three independent experiments ± standard deviation were plotted.
Mouse model of gastrointestinal tract colonization (GI) and competition experiments
Five to six week-old female BALB/c mice (Animal Resources Centre (ARC), Sydney, Australia) were used for GI colonization [44,45,46] and competition experiments. Mice were caged in groups of three and had unrestricted access to food and drinking water. Faecal samples were collected and screened for the presence of indigenous K. pneumoniae before inoculation. For the colonization study, three mice were inoculated with the parental strain or a porin mutant (1 x 1010 CFU / mouse), suspended in 20% (w/v) sucrose. For individual colonization, ampicillin was added to drinking water on day 4 (0.5 g / L) after an inoculation [47]. For the competition experiment, equal volumes of the parental strain and each mutant or equal volume of different mutants (1x 1010 CFU / mouse) were mixed and suspended in 20% (w/v) sucrose. Colonization was maintained with ampicillin 0.5 g / L throughout the experiment [48,49,50]. Faeces samples were collected every second day, emulsified in 0.9% NaCl and appropriate serial dilutions plated on MacConkey-inositol-carbenicillin agar, which selectively recovers K. pneumoniae [51]. Animal experiments were approved by the Western Sydney Local Health District Animal Ethics Committee (AEC Protocol no. 4205.06.13).
Mouse model of virulence: Intranasal infection
Five-six week-old female BALB/c mice [Animal Resources Centre (ARC), Sydney, Australia] used in the inhalation (pneumonia) model [52,53,54] were exposed to ATCC 13883 and 10.85 and their isogenic ΔOmpK35OmpK36GD mutants. Overnight bacterial cultures were harvested, washed and resuspended at 109 CFU in 20 μl of saline and inoculated into the nasal passages. A control group of mice was inoculated with saline. Following infection, survival studies were performed (10 mice per strain). At the same time and using the same inoculum, organ (lung and spleen) and blood infection burdens were also assessed at various points throughout the infection period, by plating out blood and homogenised tissue onto LB agar, and counting CFU (5 mice per strain, per time point). Animal experiments were approved by the Western Sydney Local Health District Animal Ethics Committee (AEC Protocol no. 4275.06.17).
Structural modelling of OmpK36 variants
Tri-dimensional structural models of ATCC 13883 OmpK36 and its mutated variant OmpK36GD were computed with ProMod3 Version 1.1.0 on the SWISS-MODEL online server [55] using the target–template alignment method. The best scoring model used as a template was 5nupA (93.84% sequence identity, with a QMEAN equal to -2.29 and -2.14, respectively for both sequences). For comparison purposes, models were also computed using the second best OmpK36 structure available in PDB (1osmA). All predicted models were evaluated using MolProbity [56,57] and Verify3D [58,59], with Ramachandran plots generated by MolProbity indicating for all computed models that at least >98% of residues were in allowed regions. Predicted structures were displayed by PyMol software (version 2.1.1) [60].
Additionally, the specific impact of two amino-acids insertions was also investigated by altering the OmpK36 structure under PDB accession 5nupA, adding either the amino-acids GD-, TD- or SD-, after position G113 and modeling the resulting variant sequences in the same manner as mentioned above.
Genome sequencing and comparative analysis
All isolates used in final experiments were subjected to whole genome sequencing to verify their altered sequence and ensure that no additional mutations had arisen. Genomic DNA was extracted from 2 ml overnight cultures using the DNeasy Blood and Tissue kit (Qiagen). Paired-end multiplex libraries were prepared using the Illumina Nextera kit in accordance with the manufacturer’s instructions. Whole genome sequencing was performed on Illumina NextSeq 500 (150bp paired-end) at the Australian Genome Research Facility (AGRF) and at Professor Vitali Sintchenko’s laboratory (Translational Public Health Bacterial Genomics Group, Centre for Infectious Diseases and Microbiology (CIDM) Public Health, Westmead Hospital, NSW, Australia). Raw sequence reads are available on NCBI under Bioproject accession number PRJNA430457. Reads were quality-checked, trimmed and assembled using the Nullarbor pipeline v.1.20 (available at: https://github.com/tseemann/nullarbor), as previously described [61], but with the exception of the assembly step which was performed using Shovill (available at: https://github.com/tseemann/shovill), a genome assembler pipeline wrapped around SPAdes v.3.9.0 [62] which includes post-assembly correction. Assemblies were also reordered against reference strain K. pneumoniae 30660/NJST258_1 (accession number CP006923) using progressive Mauve v.2.4.0 [63] prior to annotation with Prokka [64] and screened for antibiotic resistance genes using Abricate v.0.6 (available at: https://github.com/tseemann/abricate).
Population analysis
To investigate the significance of OmpK35 and OmpK36 mutations in a wider population, we collected a total of 1,557 draft and complete K. pneumoniae genomes publicly available in Genbank (Feb 2017, S2 Table). Sequences were typed using Kleborate v0.1.0 [65] to identify MLST (S3 Table) and minimum spanning trees were generated using Bionumerics v.7.60. Presence and absence of porins were assessed in the pangenome using Roary v3.6.0 [66] with default parameters, and mutations in loop 3 (L3) identified using BLAST. The 2,253,033 bp core genes alignment predicted by Roary was used to build a maximum-likelihood tree using IQ-TREE v1.6.1 [67], with a GTR+G+I nucleotide substitution model and branch supports assessed with ultrafast bootstrap approximation (1,000 replicates). Trees were visualized alongside contextual information with Phandango [68].
Statistical analysis was performed using Chi-squared test and Wilcoxon test, to determine associations between ST, porin defects and antibiotic resistance genes. Extended mosaic plots were used to assess the distribution of OmpK35 and OmpK36 with or without GD/TD insertion across i) ST, ii) country of origin and iii) year of isolation. Extended mosaic plots offer a convenient way to visualize the relative frequencies of a set of categorical data using proportional areas, as well as the fit of a log-linear model (assuming independence). Areas are thus colored according to the direction and magnitude of standardized deviation from the expected frequency (Pearson residual). Cut-offs of +/- 2 and 4 are defined heuristically on the assumption that the Pearson residuals approximate a standard normal distribution, and can be approximated to the statistical significance alpha = 0.05 and and alpha = 0.001 levels, respectively [69]. All statistical analyses were performed in R version 3.5.1 and “vcd” package. Relevant R scripts were also made available at https://github.com/nbenzakour/Klebsiella_antibiotics_paper.
Statistical analysis
For doubling time and Real Time RT-PCR, the results were analysed using the Student t test to determine their significance. For survival studies the results were analysed using Long-rank (Mantel-Cox) test and Gehan-Breslow-Wilcoxon test. To compare bacterial load in organs during lung infection, results were compared using Mann-Whitney unpaired t test. The analyses were performed using Prism7 (GraphPad Software).
Results
Outer membrane porins and resistance to beta-lactam antibiotics
Minimal inhibitory concentrations for commonly used carbapenems (ertapenem and meropenem), third-generation cephalosporins (ceftazidime, cefotaxime and ceftriaxone), cephamycins (also called ‘second generation cephalosporins’, cefoxitin and cefuroxime), first generation cephalosporins (cephalothin and cefazolin), and the semi-synthetic penicillin ampicillin were determined in three K. pneumoniae strains and their isogenic porin mutants, with representative results in Table 2 (for complete results, see S4 Table). The SHV enzyme characteristically expressed by K. pneumoniae hydrolyses ampicillin very effectively, providing high MICs to ampicillin [70,71,72,73], but does not provide clinically important resistance to cephalosporins or carbapenems in the setting of normal membrane permeability.
Table 2. Antibiotic MICs K. pneumoniae ATCC 13883 and porin mutants.
| Antibiotics, MIC (mg/L)a | ||||||
|---|---|---|---|---|---|---|
| Strain | ETP | MEM | CAZ | CTX | FOXb | CEFb |
| ATCC 13883 | 0.015 | 0.03 | 0.5 | 0.06 | 8 | 8 |
| ΔK35 | 0.03 | 0.06 | 1 | 0.125 | 16 | 8–16 |
| ΔK36 | 0.0625 | 0.06 | 1 | 0.25 | 16–32 | 32 |
| K36GD | 0.03–0.06 | 0.03–0.06 | 1 | 0.25 | 16–32 | 16–32 |
| ΔK35ΔK36 | 1 | 0.125–0.25 | 1 | 0.5 | 64 | 64 |
| ΔK35K36GD | 0.25 | 0.06 | 1 | 0.5 | 64 | 32–64 |
MIC. Minimal Inhibitory Concentration. ETP, Ertapenem (S ≤ 0.5, R > 1). MEM,
Meropenem (S ≤ 2, R > 8). CAZ, Ceftazidime (S ≤ 1, R > 4). CTX, Cefotaxime (S ≤ 1, R > 2). FOX, Cefoxitin (S ≤ 8, R ≥ 32). CEF, Cephalothin (S ≤ 8, R ≥ 32).
Loss of OmpK36 (ΔK36 in Table 2 and S4 Table) is associated with a minor increase in MIC for carbapenems and cephalosporins (Table 2 and S4 Table), with a lesser impact from OmpK36GD mutations, consistent with an important role for OmpK36 in the nutritious growth media (MHB) normally used for standardised MIC determinations (Table 2 and S4 Table).
OmpK35 loss (ΔK35 in Table 2 and S4 Table) has little impact alone but further increases MICs for most antibiotics in the presence of OmpK36 lesions (e.g. ΔK35ΔK36 and ΔK35ΔK36GD). In addition to ertapenem non-susceptibility, ΔOmpK35ΔOmpK36 and ΔOmpK35OmpK36GD strains are clinically resistant to first (e.g. cephalothin, CEF) and second generation cephalosporins/ cephamycins (e.g. cefoxitin, FOX) (Table 2 and S4 Table).
Naturally occurring plasmids from other K. pneumoniae strains encoding a common ESBL (blaCTX-M-15) [33], a metallo-carbapenemase (blaIMP-4) [34] and a serine-carbapenemase (blaKPC-2) [22] were transferred into ATCC 13883 and its isogenic mutants by conjugation, with transfer verified by PCR (S1 Table) and S1/PFGE (S1 Fig). Even the common ESBL CTX-M-15 confers reduced susceptibility to ETP in the presence of an OmpK36 deletion or inner channel mutation (GD duplication), especially if accompanied by an OmpK35 defect (Table 3). Expression of the specialised carbapenemases IMP and KPC from their naturally occurring plasmids resulted in greatly increased carbapenem MICs (Table 3), with the double porin mutants being highly resistant to all carbapenems tested.
Table 3. Carbapenem MICs against ATCC 13883 and porin mutants with blaCTX-M-15, blaIMP-4 or blaKPC.
| MIC (mg/L) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| CTX-M-15 | IMP-4 | KPC | |||||||
| Strain | ETP | MEM | IPM | ETP | MEM | IPM | ETP | MEM | IPM |
| ATCC 13883 | 0.25 | 0.125 | 1 | 8 | 8 | 4 | 16 | 8 | 8 |
| ΔK35 | 0.5 | 0.125 | 1 | 8 | 8 | 4 | 32 | 32 | 16 |
| ΔK36 | 1 | 0.25 | 1 | 8 | 8 | 4 | 32 | 32 | 32 |
| K36GD | 1 | 0.25 | 1 | 8 | 8 | 4 | 32 | 16 | 16 |
| ΔK35ΔK36 | 8 | 2 | 1 | 64 | 32 | 64 | 128 | 128 | 128 |
| ΔK35K36GD | 4 | 1 | 1 | 32 | 32 | 16 | 128 | 128 | 64 |
MIC. Minimal Inhibitory Concentration. ETP, Ertapenem (S ≤ 0.5, R > 1). MEM, Meropenem (S ≤ 2, R > 8). IPM, Imipenem (S ≤ 2, R > 8).
aBoldface numbers indicate at least 4-fold increase between the MICs of the parental strain (K. pneumoniae ATCC 13883) and the porin mutants. The underlined numbers mean intermediate or resistant based on EUCAST breakpoints [74].
K pneumoniae JIE2771 is a clinical isolate of K pneumoniae carrying blaKPC and a natural double mutant of ompK35 and ompK36 [22]. As expected, attenuation of the resistance phenotype was evident in this wild-type double mutant and susceptibility restored to the constructed 10.85 double mutant by in trans complementation with ompK36 but not ompK36GD (S5 Table).
Altered expression of other common porins in ΔOmpK35, ΔOmpK36, and OmpK36GD
Other porins may compensate for the loss of major outer membrane porins in K. pneumoniae [76,77,78,79]. Expression of ompK35, ompK36, ompK37, phoE, ompK26 and lamB was measured in isogenic porin mutants of ATCC 13883 and 10.85 K. pneumoniae strains (Fig 1, S6 Table). Neither the introduction of a GD duplication into the OmpK36 inner channel (OmpK36GD) nor the loss of OmpK35 (ΔOmpK35 and ΔOmpK35OmpK36GD) affected expression of OmpK36 in MH broth. Loss of OmpK36, however, was associated with increased OmpK35 expression in MH broth, in which OmpK36, but not OmpK35, is ordinarily expressed (S2 Fig). Restitution of OmpK36 by replacing the interrupted gene with ompK36GD directly in the chromosome restored normal porin regulation (Fig 1, S6 Table). Loss of both of these major porins (ΔOmpK35ΔOmpK36) resulted in increased expression of phoE and lamB. (Fig 1, S6 Table).
Fig 1. Real-time RT-PCR in K. pneumoniae ATCC 13883 and 10.85 porin mutants.

The expression of rpoD was used to normalize results. The levels of expression of each mutant are shown relative to the wild type strain ATCC 13883 or 10.85.
Relative fitness costs of major porin lesions
Exponential phase growth in MH broth was only affected when both major porins were absent (ΔOmpK35ΔOmpK36, S7A Table). In trans complementation with either ompK36 or ompK36GD resulted in amelioration of the growth defect in JIE2771 wild-type double mutant and the constructed 10.85ΔOmpK35ΔOmpK36 (S7B Table).
The ability of ΔOmpK35 strains to directly compete against their intact isogenic parents in MH broth was little affected over seven-day growth (Fig 2A1 and 2A2 and S3 Fig). However, any ΔOmpK35 mutant was rapidly outcompeted by its isogenic parent in nutrient-limited conditions (S4A1 and S4B1 Fig). Furthermore, competition experiments clearly illustrate the importance of OmpK36 in high osmolarity highly nutritious media (Fig 2B1 and 2B2 and S3 Fig) but not in low nutrient conditions (S4B1 and S4B2 Fig). OmpK36GD strains are clearly much more able than ΔOmpK36 strains to compete with their isogenic parent strains (Fig 2C1 vs 2B1 and 2C2 vs 2B2). For ATCC 13883, at day 3, the OmpK36GD population was still 40% of the total combined population (Fig 2C1), while ΔOmpK36 fell to 20% in the same period (Fig 2B1). This difference was more marked in the presence of an OmpK35 lesion but ΔOmpK35OmpK36GD populations were still clearly more able than ΔOmpK35ΔOmpK36 to compete with the intact parent strain (Fig 2F1 vs 2E1). In fact, the introduction of an OmpK36GD mutation had no detectable cost at all in K. pneumoniae 10.85 (Fig 2C2 vs 2B2 and 2F2 vs 2E2), with ΔOmpK35OmpK36GD competing very successfully against the isogenic parent 10.85 (Fig 2F2: 37±4% and 26±15% of the total population represented by ΔOmpK35OmpK36GD on days 6 and 7 respectively). Finally, as expected, directly competing OmpK36GD with ΔOmpK36 (and ΔOmpK35OmpK36GD with ΔOmpK35ΔOmpK36) further illustrates the competitive advantage, with OmpK36GD strains quickly displacing isogenic ΔOmpK36 strains in MH broth (Fig 2D1 and 2G1).
Fig 2. In vitro competition experiments in K. pneumoniae ATCC 13883 and 10.85 porin mutants.
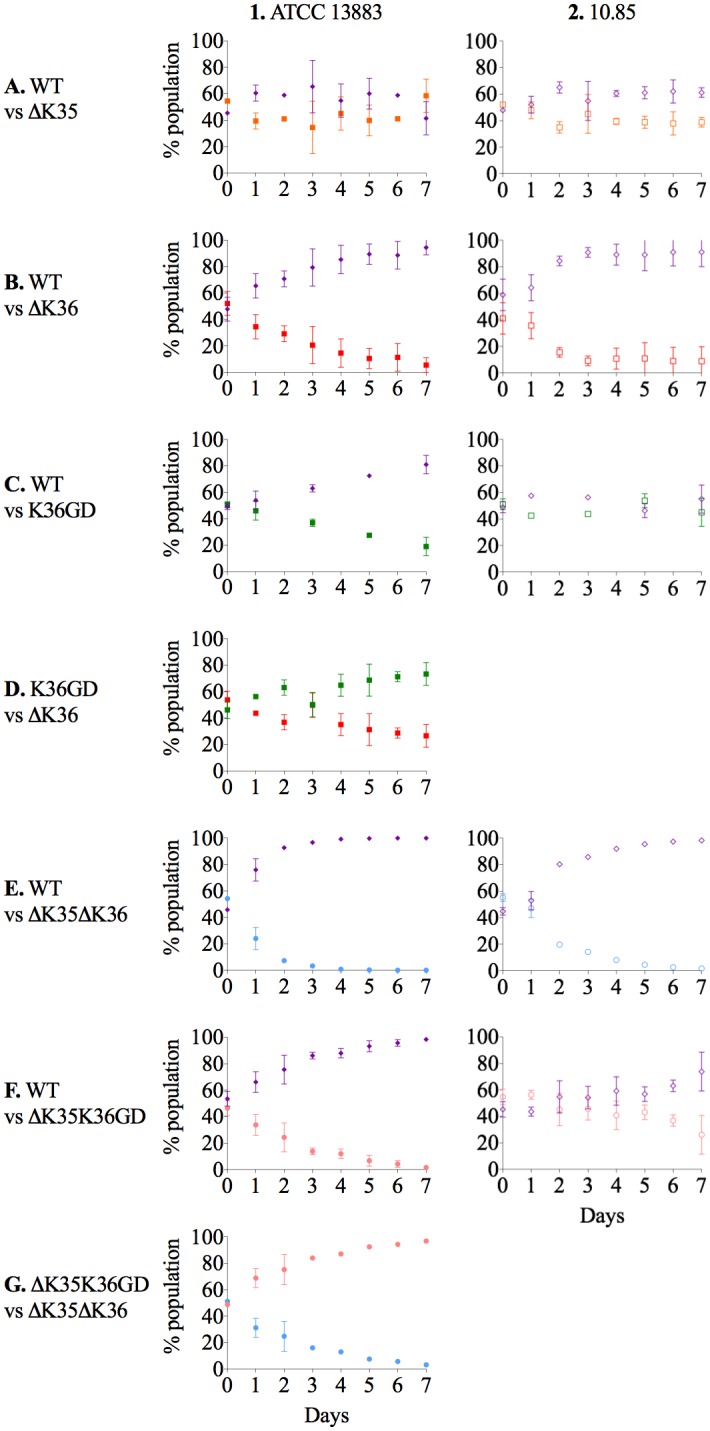
The relative fitness of porin mutants in comparison with parental strain (ATCC 13883 or 10.85) or between porin mutants was determined by competition experiments in co-cultures and expressed as a percentage of the mutant or wild type cells versus total population at each time point. In vitro growth conditions, MH broth with continuous shaking at 37°C. Violet diamond, ATCC 13883 or 10.85 wild type strains. Orange square, ΔOmpK35. Red square, ΔOmpK36 mutant. Green square, OmpK36GD mutant. Blue circle, ΔOmpK35ΔOmpK36 mutant. Pink circle, ΔOmpK35OmpK36GD mutant.
Mouse gut colonizing studies yielded similar results (Fig 3). Mice were confirmed not to harbor indigenous K. pneumoniae on arrival [51], and stable colonisation at ∼109 CFU/g faeces was achieved (S5 Fig). OmpK35 deficient mutants (ΔOmpK35) were not disadvantaged (Fig 3A3) and OmpK36GD strains strongly outperformed OmpK36 strains in competition with their isogenic parents (Fig 3A1 vs 3B1 and 3A2 vs 3B2). Similarly, direct in vivo competition confirmed a clear fitness advantage of OmpK36GD over ΔOmpK36 (Fig 3C1 and 3C2).
Fig 3. In vivo competition experiments in K. pneumoniae ATCC 13883 and porin mutants.

The relative fitness of porin mutants in comparison with parental strain (ATCC 13883) or between porin mutants was determined by competition experiments in co-cultures. The values for each mouse are represented individually. Violet diamond, ATCC 13883 wild type strains. Orange square, ΔOmpK35 mutant. Red square, ΔOmpK36 mutant. Green square, OmpK36GD mutant. Blue circle, ΔOmpK35ΔOmpK36 mutant. Pink circle, ΔOmpK35OmpK36GD mutant.
Pathogenicity is not attenuated in ΔOmpK35OmpK36GD strains
In a mouse pneumonia model [52,53,54], we showed no difference in lethality between a wild type strain and its isogenic mutant ΔOmpK35/OmpK36GD (Fig 4). Intranasal inoculation of mice with 10.85 and ATCC 13883 ΔOmpK35/OmpK36GD strains showed that these mutations had no significant impact on virulence, with equivalent mortality curves (Fig 4A1 and 4A2) and similar viable counts developing in lung, blood and spleen over the course of infection compared with their isogenic wild-type strains 10.85 and ATCC 13883, respectively (Fig 4B to 4D). As OmpK36 deletion mutants have been clearly shown by other studies to be attenuated in vivo [80,81], and we also demonstrate this completely predictable virulence cost by in vitro and in vivo competition assays, experiments in the acute pneumonia model were confined to these two isolates and their key isogenic ompK36 variants in order to minimize the use of animals in experimentation.
Fig 4. Lung infection experiments in K. pneumoniae ATCC 13883 and 10.85 as well as their isogenic porin mutants ΔOpmK35/OmpK36GD.

Survival curves after intranasal infection are represented in panels A1 (for ATCC 13883) and A2 (for 10.85). Organ burden after 24, 48 or 72h is represented in panels B1 to D1 for ATCC 13883 and B2 to D2 for 10.85. Violet diamond, ATCC 13883 or 10.85 wild type strains. Pink circle, ΔOmpK35OmpK36GD mutant. For survival studies the results were analysed using Long-rank (Mantel-Cox) test and Gehan-Breslow-Wilcoxon test. To compare bacterial load in organs during lung infection, results were compared using Mann-Whitney unpaired t tests. The analyses were performed using Prism7 (GraphPad Software). The differences between the wild type and the porin mutants were not statistically significant (P < 0.05) in any case.
Structural impact of OmpK36 loop L3 mutations
Two crystal structures of native OmpK36 available in the Protein Data Bank under accession number 5nup (2.9 Å, Xray) and 1osm (3.2 Å, Xray) were evaluated as templates for structural modelling of OmpK36 and OmpK36GD from ATCC 13883, with targets and templates sharing around 93% nucleotide sequence identity. While Ramachandran plots analysis for all predicted models show at least 98% of residues in allowed regions, other metrics such as QMEAN and Molprobity score were marginally better for ATCC 13883 OmpK36 and OmpK36GD models based on the 5nup structure (Fig 5, S8 Table). Although several differences can be observed in the final alignment (Fig 5D), the most prominent differences between the original structure (Fig 5A) and the ATCC 13883 OmpK36 model lie within the loop L6, which can be seen in yellow, slightly obstructing the outmost channel of the porin (Fig 5B). Much more striking is the impact of single two amino-acid -GD insertion within loop L3, which is expected to further constrict the porin channel (Fig 5C) and is likely responsible for the difference in phenotype between the two variants.
Fig 5. Channel restriction of OmpK36 variants.

Comparison of the reference OmpK36 structure under PDB accession 5nupA (A) against predicted structural models of OmpK36 (B) and OmpK36GD mutant (C) from ATCC 13883, showing progressive restriction of the porin channel. The conformation visualised in panel B, in particular the loop 6 in yellow which can be seen partially obstructing the channel, is not associated with a carbapenem resistance phenotype, contrary to the GD mutant shown in panel C. Panel D consists of the multiple alignment of the 3 corresponding sequences, along with a representation of the predicted secondary structures designated as follows; B for barrel, T for turn, and L for loop. Signal peptide is not shown in 5nupA sequence (Panel D).
OmpK35 loss and convergent evolution of OmpK36GD
The successful antibiotic resistance, colonisation and pathogenicity phenotypes of ΔOmpK35OmpK36GD strains should be reflected in their representation among strains causing human infection. Of 165 unique K. pneumoniae ompK36 sequences in GenBank, 16% varied from the consensus L3 inner channel motif (PEFGGD). The most common was the GD duplication (PEFGGDGD, in 14 of 26 OmpK36 L3 variants identified), along with 6 additional variants: PEFGGDD, PEFGGDSD, PEFGGDTD, PEFGGDTYD, PEFGGDTYG and PEFGGDTYGSD (S6 Fig, showing modeled secondary structures based on previous studies [82,83], including of the pore eyelet region [84]). Using the native OmpK36 structure 5nup as a template, modelled structures of mutants, namely GD-, TD- and SD- insertions in L3, were computed as previously described, and showed similar restriction of the porin channel, slightly greater in the case of a bulkier amino-acid such as Threonine (S7 Fig). Inspection of their corresponding nucleotide sequences suggests that these variants originated from various combinations of short in-frame duplications, combined with additional point mutations in rare cases (S8 Fig). Similar variations in L3 of OmpK36 homologues were found when analysing ompK36 sequences in other Enterobacteriaceae (S9 Fig).
To investigate OmpK36 among clinical isolates without specialised carbapenemases, we specifically analysed L3 variation in all such K. pneumoniae isolates with an Ertapenem MIC > 1 in our local clinical collection (Table 1) by PCR and sequencing (S1 Table). Of (n = 51), 17 strains (33%) were identified: all revealed either the previously described GD or TD mutation in the L3 loop of ompK36 on sequencing and these encoded up to 6 distinct beta-lactamases. These isolates were genetically diverse but belonged to major epidemic clones found elsewhere in the world: i.e. ST14, ST16, ST101, ST147, with as many as 6 distinct ompK35 mutations, all of which introduced disrupting frame-shifts and all of which were relatively lineage-specific (S10 Fig).
Finally, all K. pneumoniae (complete and draft) genomes available from Genbank, i.e. 1,557 entries (as of February 2017) were examined: the two common (GD and TD) variants are shown in a minimum spanning tree built using MLST profiles (Fig 6) to be distributed across the whole spectrum of diversity of K. pneumoniae, including in most major epidemic clones, e.g. ST258 and its derivative ST512, ST11, ST101, ST147, ST14 and ST37.
Fig 6. Minimum spanning tree of 1,557 K. pneumoniae strains based on their MLST profile.

Each circle corresponds to a distinct ST, with its size being proportional to the number of strains of that particular ST (for scale, ST258 contains 552 isolates). Within an ST, the proportion of strains harbouring either a GD or TD insertion in the loop L3 of ompK36 is shown as a sector coloured in red and pink, respectively. STs carrying these mutations are also circled in grey.
A maximum likelihood phylogeny using a 2,253,033 bp core genome alignment of all 1,557 genomes was computed to contextualize variations in ompK36 and ompK35, with metadata relative to the population (year, source, geographical region of isolation, as well as major beta-lactamases genes) (Fig 7). Those genes most relevant to a carbapenem resistance phenotype are shown, and the expected clustering of some of these is as expected (e.g. blaCTX-M-15 with OXA-1 and TEM-1b). Major associations with other genes not affected by porin changes are not shown (e.g. aminoglycoside resistance due to 16S methylase genes that are common companions of blaNDM, other class I integron cassettes from the array in which blaIMP-4 is found, etc). The predominance of ompK36 variations in L3 compared to its loss or disruption is evident at a glance, as is the common loss or disruption of ompK35 in unrelated strains. There is no obvious relationship between ompK36 L3 variations and the presence of blaKPC but there is strong clustering of these variations in certain types (ST258, 512 etc).
Fig 7. Maximum likelihood tree of 1,557 K. pneumoniae strains.

A phylogenetic tree was built using a 2,253,033 bp long core alignment. Contextual information relevant to the collection was visualized using Phandango and includes ST (of which the major ones are indicated on the tree); GD or TD insertion in the loop L3 of ompK36, in black and red, respectively; presence or absence of ompK36, in orange and purple, respectively; presence or absence of ompK35, in orange and purple, respectively. Additional metadata include year(date) of isolation, in a gradient from purple to yellow; source and geographical region of isolation in a rainbow gradient; and presence of major beta-lactamases (bla) alleles identified, in dark blue.
As expected for a gene so clearly linked to fitness and virulence, ompK36 is highly conserved across the dataset (present in 1,499 out of 1,577), and we found no statistical evidence of ST-dependence (Chisq = 207.51, df = 227, p-value = 0.8188). Conversely, ompK35 (evidently dispensable in the host) is disrupted in nearly a third of all strains (Fig 6), with statistically significant association with ST (Chisq = 603.7, df = 227, p-value = 5.748e-36). Three-way comparison of the distributions of frequencies of presence/absence of ompK35, mutations in ompK36, and ST (considering only those STs harbouring ompK36 GD/TD variants) was performed using an extended mosaic plot (S11 Fig). Standard Pearson’s residuals were calculated and displayed on the mosaic plot to identify over-represented categories (residuals [2,4] and >4) and under-represented categories (residuals [–2,–4] and <-4). We found statistically significant evidence (residual cut-off 2 and 4 equivalent to p < 0.05 and p < 0.001) that i) some STs prevalently have both ompK36 and ompK35 intact (mainly ST15, ST16, ST17); ii) others prevalently have intact ompK36 with ompK35 disrupted (ST129, ST258); and iii) some STs prevalently have ompK36 (GD/TD) variants combined with ompK35 disrupted (ST11, ST14, ST147, ST258 and ST37). Furthermore, we found statistically significant evidence for more disruptions of ompK35 in i) strains from the USA compared to other countries (S12A Fig) and ii) strains from 2011 and 2014 (S13A Fig). We also observed over-representation of ompK36 (GD/TD) variants in i) China, Greece, Germany, Italy and India (S12B Fig) and ii) in 2011 (S13B Fig). Finally, we looked at associations between the number of resistance genes and porin defects in major STs, and found that the presence of ompK36 GD/TD variants did not correlate with a higher number of resistance genes (with the exception of OmpK36GD in ST14). In fact, successful clones such as ST258 and ST11 harbouring OmpK36GD encoded significantly less resistance genes (p<0.001, Wilcoxon test) (S14 Fig). It should be noted that due to the inherent opportunistic nature of the sampling present in Genbank (e.g. USA), our conclusions are only applicable to this dataset. More sampling would be required to assess the significance of porin mutations in an unbiased K. pneumoniae population.
Discussion
β-lactam antibiotics are among the most commonly prescribed for severe infections [85,86] and the emergence of β-lactam resistance in K. pneumoniae has become a global health threat [87,88]. In general, E. coli and K. pneumoniae carrying transmissible β-lactam resistance genes have predictable and normally distributed β-lactam MICs [21] but carbapenem MICs in K. pneumoniae are bimodally distributed with higher MICs correlating with OmpK36 defects [21]. OmpK36 loss or mutation is not uncommonly reported in highly resistant clinical isolates producing KPC, ESBL and AmpC β-lactamases [20,89,90].
Diffusion of β-lactam antibiotics through non-specific porins such as OmpK35 and OmpK36 is dependent on size, charge and hydrophobicity [91,92], with bulky negatively charged compounds diffusing at a lower rate than small zwitterions of the same molecular weight [93]. OmpK35 is much less expressed in high osmolarity nutrient-rich conditions than OmpK36, which has the narrower porin channel of the two (S2 Fig) [9] and large negatively charged β-lactams such as third-generation cephalosporins and carbapenems diffuse more efficiently through OmpK35 than OmpK36 [80,94]. Here we confirm the significantly increased MICs, commonly attributed to mutations in these two major porins [10,95,96] in three K. pneumoniae strains (the widely-published ATCC strain 13883 and two locally isolated clinical strains (Table 2 and S4 Table) and unequivocally identify the primary role of OmpK36 in carbapenem resistance.
Comparable MIC changes in single (OmpK36GD and ΔOmpK36) and double (ΔOmpK35OmpK36GD and ΔOmpK35ΔOmpK36) mutants indicate that duplication of a glycine aspartate (GD) pair in a critical position in the porin eyelet region (loop 3) is almost as effective as a complete deletion of the porin in excluding large anionic antibiotics. Both single and double porin mutants were susceptible to extended-spectrum cephalosporins (cefotaxime and ceftazidime) in the absence of acquired hydrolysing enzymes, demonstrating the impotence of the naturally occurring chromosomal SHV enzymes [70,71,72] against these compounds [95].
Differences relating to porin permeability in K pneumoniae are most striking and important in the presence of acquired carbapenemases and it is clear that these permeability changes greatly enhance the associated resistance phenotypes. The common Ambler Class A serine protease KPC-2 and Class B metalloenzyme IMP-4 expressed from their natural plasmids produce only borderline resistance against meropenem and the smaller zwitterionic imipenem in the presence of the ‘wild type’ OmpK36 osmoporin (Table 3) but MICs that exceed therapeutic tissue levels [97,98] are the rule in strains of the commonly occurring ΔOmpK35OmpK36GD genotype.
We also show here that the OmpK35 matrix porin has little or no relevance in vivo or in vitro conditions that reliably predict antibiotic efficacy in the clinic (MICs and competitive fitness in Mueller-Hinton broth). Consistent with this, a high percentage of clinical isolates whose genomes have been lodged with GenBank appear to have lost their ability to express OmpK35 altogether (Fig 7). Increased production of the larger channel OmpK35 is expected under low-temperature, low-osmolarity and low nutrient conditions (S2 Fig). These favour survival outside the mammalian host and we show that ΔOmpK35 strains fail to compete successfully with their isogenic parents in nutrient-limited conditions (S4 Fig). We confirm that OmpK35 is not naturally expressed at significant levels in optimal growth conditions nor in the mammalian host, as previously described [78,80]. As expected, competition experiments, the most sensitive and direct measures of comparative fitness, evince no discernible disadvantage from the loss of OmpK35 in vivo [19,99].
Loss of OmpK36 trades off nutrient influx for antibiotic resistance [42,80], and we show that these more resistant bacteria cannot compete successfully with the antibiotic-susceptible populations from which they arise once antibiotic selection ceases to operate (Fig 2). Double porin mutants (ΔOmpK35ΔOmpK36) are the most antibiotic-resistant (Table 2 and S4 Table) but this resistance comes at the cost of a 10% relative growth reduction in nutritious media (S7 Table). OmpK36, the main porin normally expressed in vivo, is responsible for most of this fitness cost (Figs 2 and 3 and S3 Fig). The less permeable phosphoporin PhoE and maltodextrin channel LamB, most important in the usual compensatory response when OmpK35 is not available, are not efficient substitutes (Fig 1 and S6 Table). Defects in these porins have been implicated in carbapenem resistance in association with only an AmpC-type enzyme [42,76,79,100], but other defects are ill-defined and the fitness cost may be high as such strains are rarely described. By contrast, ΔOmpK35OmpK36GD mutants are little disadvantaged in vivo or in optimal growth conditions in vitro (Figs 2 and 3 and S7 Table). Expression of OmpK36 is unaffected (Fig 1 and S6 Table) as is that of other porins such as OmpK35 (Fig 1 and S6 Table), presumably because OmpK36 ‘rescue’ is not required.
The precise loop 3 variation in OmpK36 is best explained by a convergent evolutionary process, as a range of different variants occur within genetically distant K. pneumoniae populations, all with an extra negatively charged aspartate (D) residue that significantly constricts the inner channel (Fig 5). The most common solution is the extra glycine and aspartate (PEFGGD to PEFGGDGD in the critical region) which we recreated in isogenic mutants for our experiments. The next most frequent, an extra TD (rather than GD), is similarly likely to spontaneously arise (S8 Fig) but is much less common, including in STs in which both GD and TD are found (Figs 6 and 7), implying a less optimal conformation. A recent survey of nearly 500 ertapenem-resistant Klebsiellae lacking specialised carbapenemases [101] supports our own finding of the extra aspartate in that position, most commonly as a GD pair, with TD and SD much less often, and other variants being rare. We found no examples of similarly acidic (glutamate) residues occurring in this position, perhaps reflecting the fact that even simple sequence changes (here, GAY to GAR) add an additional step to a simple duplication event, or the fact that glutamate’s extra carbon makes it slightly less compact than an aspartate in this position.
Other Enterobacteria face the same challenge of excluding bulky anionic carbapenem antibiotics in order to survive high concentrations, even in the presence of a specialist carbapenemase. High level antimicrobial resistance has been ascribed to similar variations in L3 of OmpK36 homologues in Enterobacter aerogenes, Escherichia coli (S9 Fig) and Neisseria gonorrhoeae [102,103,104,105,106]. In comparison with their E. coli homologues (OmpF and OmpC), OmpK35 and OmpK36 permit greater diffusion of β-lactams [107]. Specifically, OmpK35 appears to be highly permeable to third-generation cephalosporins such as cefotaxime due to its particular L3 domain, which is also seen in Omp35 in E. aerogenes but not in other species, and has been proposed as an explanation for the high proportion of K. pneumoniae clinical isolates that lack this porin [84,107]. Our findings of increased MICs in OmpK35 mutants are consistent with those of others [107] but we show here that the more permeable OmpK35 is not important in the mammalian host. Rather, the much less permeable OmpK36 (equivalent to E coli OmpC) [107] is the bottleneck for large anionic antibiotics.
The term ‘high risk clone’ [108,109] is given to host-adapted/pathogenic strains that dominate the epidemiology of (antibiotic resistant) infections, presumably because they are more transmissible, more pathogenic and/or more tolerant of host-associated stresses (including antibiotics). Here, we see a range of unrelated clonal groups already identifiable as high-risk clones that are dispensing with the OmpK35 porin (Fig 7). The minimal antibiotic resistance advantage in nutritious media is only evident with carbapenems and is unlikely to arise in the presence of an existing OmpK36 loss mutation because the fitness cost is substantial. The loss of OmpK35 through low-level carbapenem exposure in environmental conditions is possible [110] but has a marked fitness cost and the exposure to carbapenems in the environment is expected to be limited, as they are a still a minority class of prescribed antibiotics and are not yet as common in environmental waters as the sulfonamides, quinolones, macrolides, tetracyclines and other beta-lactams [111].
A recent review of antibiotic resistance in Klebsiella pointed out that “The exact role of porins in antimicrobial resistance is difficult to determine because other mechanisms…are commonly present …” [112]. We suggest that host-adaptation in K. pneumoniae is widespread and that many K. pneumoniae have dispensed with the OmpK35 matrix porin required for an environmental life cycle. Bacteria are expected to adapt effectively to major stress such as antibiotic pressure or high concentrations of bile salts in the intestinal lumen [113]. Our hypothesis of adaptive loss of OmpK35 is based on results presented in this study and on strong evidence from others: i) toxic agents as antibiotics and bile salts diffuse better through the larger OmpF channel (homolog of OmpK35) than the narrower OmpC (equivalent to OmpK36 in K. pneumoniae) [114]; ii) high osmolarity, high temperature, low pH and anaerobiosis (typical conditions in gut environment) induce the production of OmpK36 but inhibit the expression of ompK35 [115] [116] [117] and iii) E. coli mutants with reduced permeability (decreased ompF and increased ompC mRNA and protein levels compared with parental strain) can be easily recovered from intestinal gut of germ-free mice after few days of colonization [118]. In addition, we have shown that the highly specific variation in the inner channel of OmpK36 provides carbapenem resistance at no cost to colonising ability, competitiveness or pathogenicity and can be expected to be an increasingly common feature of host-adapted ‘high-risk clones’.
There are three direct and immediate implications. Firstly, efforts to control the spread of such strains will be facilitated to some extent by the loss of environmental hardiness resulting from OmpK35 deletion, and should shift slightly more toward managing interpersonal transmission. Secondly, K. pneumoniae can be expected to become more antibiotic resistant overall, and organisms expressing currently circulating plasmid-borne carbapenemases will more commonly be untreatable with carbapenem antibiotics (e.g. ST258 strains with blaKPC); the second (higher MIC) peak in the bimodal distribution of carbapenem MICs in K. pneumoniae populations will become more prominent. Finally, the mobile carbapenemase gene pool can be expected to flourish in the protected niche provided by host-adapted K. pneumoniae populations under strong carbapenem selection pressure in human hosts, thereby increasing the general availability of highly transmissible carbapenem resistance plasmids among host-adapted pathogens in the Enterobacteriaceae.
Supporting information
White arrows show the plasmids in original host isolates and transconjugants. MW; Mid-range PFG Marker.
(TIF)
Wild type strains ATCC 13883, 10.85 and 11.76 were cultured under different temperatures (37°C, 30°C and 25°C) and different nutrient concentrations (MH and MH 1:10). Blue arrow, OmpK35. Black arrow, OmpK36. Red arrow, OmpA.
(TIF)
The relative fitness of deletion porin mutants in comparison with parental strain (11.76) was performed by competition experiments in co-cultures and expressed as a percentage of the mutant or wild type cells versus total population at each time point. In vitro growth conditions, MH broth, 37°C. Violet diamond, 11.76 wild type strains. Orange square, ΔOmpK35 mutant. Red square, ΔOmpK36 mutant.
(TIFF)
The relative fitness of porin mutants in comparison with parental strain ATCC 13883 was performed by competition experiments in co-cultures and expressed as a percentage of the mutant or wild type cells versus total population at each time point. In vitro growth conditions: A, MH 1:10 broth, 25°C; B, MH 1:10 broth, 37°C. Violet diamond, ATCC 13883. Orange square, ΔOmpK35. Red square, ΔOmpK36.
(TIF)
K. pneumoniae intestinal colonization in a mouse model. CFU counts of K. pneumoniae ATCC 13883 and porin mutants from mice faecal sample. Bacterial inoculum at day 0 is 1x1010 CFU /mouse. Addition of ampicillin 0.5 g / L in the drinking water on day 4. Violet diamond, ATCC 13883 wild type strains. Orange square, ΔOmpK35 mutant. Red square, ΔOmpK36 mutant. Blue circle, ΔOmpK35ΔOmpK36 mutant. Green square, OmpK36GD mutant. Pink circle, ΔOmpK35OmpK36GD mutant.
(TIF)
26 unique sequences with OmpK36 L3 variants from GenBank were compared with OmpK36 of NTUH_K2044. Isolates with wild-type L3 sequence are not included. Dot line, signal peptide. Black line, beta strands. Red line, loops. Blue line, alpha helix. Green squares, turns. OmpK36 secondary structure based on previous studies [82,83]. Red boxes, residues involved in the pore eyelet based on [84]. Black box, L3 variants.
(TIF)
Comparison of the reference OmpK36 structure under PDB accession 5nup1A (WT, wild type) against predicted structural models of mutants harbouring a two amino-acid insertion in loop 3 after G113, namely GGDGD, GGDTD and GGDSD. For each predicted structure, the 2 most protruding amino-acids resulting from the insertion were marked and coloured according to their backbone structure (carbons in yellow, oxygens in red and nitrogens in blue).
(TIF)
Based on observations of the codon sequences, the extra–SD and–SYG following GGD likely result from a combination of duplication followed by point mutation.
(TIF)
A. Alignment of E. coli OmpC_L3 variants. 11 unique Omp36_L3 variants from GenBank were compared with L3 of OmpC of K-12 MG1655 (NP_416719). Black boxes, residues different from NP_416719. B. Alignment of E. aerogenes Omp36_L3 variants. Four unique Omp36_L3 variants from GenBank were compared with L3 of Omp36 from ATCC 13048 (AF335467). Black boxes, residues different from AF335467. Isolates with wild-type L3 sequence are not included. Black line, loop 3. OmpK36 L3 location based on previous studies. (82, 83). Red boxes, residues involved in the pore eyelet based on (84).
(TIF)
Metadata includes year of isolation; MIC levels for ETP: ertapenem, IMP: imipenem, and MEM: meropenem; ompK36 L3 mutation; ompK35 disrupted mutations (as listed in S7 Table); ST: sequence type; number of predicted resistance genes encoded; carbapenamase gene encoded; ESBL: extended-spectrum beta-lactamase gene encoded.
(TIF)
The mosaic plot shows the relationships between 3 variables; ST (in purple) and presence/absence of ompK35 (in black) on the x-axis; and presence/absence and mutations of ompK36 (in grey) on the y-axis. The size of each plot tile is proportional to counts. Plot tiles are colored according to their standardized Pearson residuals, as determined by a log-linear model. Deeper shades of red and blue corresponding to a standardized residual less than -4 or greater than +4, respectively, can be interpreted as combinations observed significantly less or more than expected (under the assumptions that proportions have equal levels).
(TIF)
The mosaic plots show the relationships between 2 variables; A) country of isolation on the x-axis and presence/absence of ompK35 on the y-axis; B) country of isolation on the x-axis, and presence/absence and mutations of ompK36 on the y-axis. The size of each plot tile is proportional to counts. Plot tiles are colored according to their standardized Pearson residuals, as determined by a log-linear model. Deeper shades of red and blue corresponding to a standardized residual less than -4 or greater than +4, respectively, can be interpreted as combinations observed significantly less or more than expected (under the assumptions that proportions have equal levels).
(TIF)
The mosaic plots show the relationships between 2 variables; A) year of isolation on the x-axis and presence/absence of ompK35 on the y-axis; B) year of isolation on the x-axis, and presence/absence and mutations of ompK36 on the y-axis. The size of each plot tile is proportional to counts. Plot tiles are colored according to their standardized Pearson residuals, as determined by a log-linear model. Deeper shades of red and blue corresponding to a standardized residual less than -4 or greater than +4, respectively, can be interpreted as combinations observed significantly less or more than expected (under the assumptions that proportions have equal levels).
(TIF)
Boxplots were used to display the distribution of resistance genes identified with Abricate within each ST with the following OmpK36 variants, namely isolates with–GD in bright red,–TD in brown, or no insertion (–) in grey. Mean comparison p-values are also shown for each ST (Wilcoxon test, with ‘-’ used as a reference group; ns: p > 0.05; *: p < = 0.05; **: p < = 0.01; ***: p < = 0.001; ****: p < = 0.0001). In addition, the corresponding underlying isolate population is also visualised as individual points, coloured according to OmpK35 type, (1) intact in turquoise or (0) disrupted in coral.
(TIF)
(DOC)
(XLSX)
(XLSX)
(DOC)
(DOC)
The expression of rpoD was used to normalize the results. The levels of expression of each mutant are shown relative to the wild type strain ATCC 13883 or 10.85.
(DOC)
(DOC)
(XLSX)
Acknowledgments
The authors thank Ali Khalid and Muhammad Kamruzzaman for their helpful advice on various technical and scientific issues examined in this work.
Data Availability
Data availability statement has been added at the end of the manuscript after Supporting Information. Raw sequence reads are available on NCBI under Bioproject accession number PRJNA430457. Relevant R scripts were also made available at https://github.com/nbenzakour/Klebsiella_antibiotics_paper.
Funding Statement
This work, including the efforts of all the authors was funded by Department of Health, National Health and Medical Research Council (NHMRC) (G1046886 and G1104232). This work, including the efforts of Alex Agyekum, was funded by Ghana Education Trust Fund (GETFUND). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Podschun R, Ullmann U (1998) Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin Microbiol Rev 11: 589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Canton R, Coque TM (2006) The CTX-M β-lactamase pandemic. Curr Opin Microbiol 9: 466–475. 10.1016/j.mib.2006.08.011 [DOI] [PubMed] [Google Scholar]
- 3.Jacoby GA (2009) AmpC β-lactamases. Clin Microbiol Rev 22: 161–182. 10.1128/CMR.00036-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nordmann P, Dortet L, Poirel L (2012) Carbapenem resistance in Enterobacteriaceae: here is the storm! Trends Mol Med 18: 263–272. 10.1016/j.molmed.2012.03.003 [DOI] [PubMed] [Google Scholar]
- 5.Nordmann P, Naas T, Poirel L (2011) Global spread of carbapenemase-producing Enterobacteriaceae. Emerg Infect Dis 17: 1791–1798. 10.3201/eid1710.110655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Padilla E, Alonso D, Domenech-Sanchez A, Gomez C, Perez JL, et al. (2006) Effect of porins and plasmid-mediated AmpC β-lactamases on the efficacy of β-lactams in rat pneumonia caused by Klebsiella pneumoniae. Antimicrob Agents Chemother 50: 2258–2260. 10.1128/AAC.01513-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Agyekum A, Fajardo-Lubian A, Ai X, Ginn AN, Zong Z, et al. (2016) Predictability of phenotype in relation to common β-lactam resistance mechanisms in Escherichia coli and Klebsiella pneumoniae. J Clin Microbiol 54: 1243–1250. 10.1128/JCM.02153-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alberti S, Rodriquez-Quinones F, Schirmer T, Rummel G, Tomas JM, et al. (1995) A porin from Klebsiella pneumoniae: sequence homology, three-dimensional model, and complement binding. Infect Immun 63: 903–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hernández-Allés S, Albertí S, Alvarez D, Doménech-Sánchez A, Martínez-Martínez L, et al. (1999) Porin expression in clinical isolates of Klebsiella pneumoniae. Microbiology 145: 673–679. 10.1099/13500872-145-3-673 [DOI] [PubMed] [Google Scholar]
- 10.Domenech-Sánchez A, Pascual A, Suarez AI, Alvarez D, Benedí VJ, et al. (2000) Activity of nine antimicrobial agents against clinical isolates of Klebsiella pneumoniae producing extended-spectrum β-lactamases and deficient or not in porins. J Antimicrob Chemother 46: 858–859. [DOI] [PubMed] [Google Scholar]
- 11.Andersson DI, Levin BR (1999) The biological cost of antibiotic resistance. Curr Opin Microbiol 2: 489–493. [DOI] [PubMed] [Google Scholar]
- 12.Guo B, Abdelraouf K, Ledesma KR, Nikolaou M, Tam VH (2012) Predicting bacterial fitness cost associated with drug resistance. J Antimicrob Chemother 67: 928–932. 10.1093/jac/dkr560 [DOI] [PubMed] [Google Scholar]
- 13.Marcusson LL, Frimodt-Møller N, Hughes D (2009) Interplay in the selection of fluoroquinolone resistance and bacterial fitness. PLoS Pathog 5: e1000541 10.1371/journal.ppat.1000541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Choi MJ, Ko KS (2015) Loss of hypermucoviscosity and increased fitness cost in colistin-resistant Klebsiella pneumoniae sequence type 23 strains. Antimicrob Agents Chemother 59: 6763–6773. 10.1128/AAC.00952-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pagès JM, James CE, Winterhalter M (2008) The porin and the permeating antibiotic: a selective diffusion barrier in Gram-negative bacteria. Nat Rev Microbiol 6: 893–903. 10.1038/nrmicro1994 [DOI] [PubMed] [Google Scholar]
- 16.King T, Ishihama A, Kori A, Ferenci T (2004) A regulatory trade-off as a source of strain variation in the species Escherichia coli. J Bacteriol 186: 5614–5620. 10.1128/JB.186.17.5614-5620.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferenci T (2005) Maintaining a healthy SPANC balance through regulatory and mutational adaptation. Mol Microbiol 57: 1–8. 10.1111/j.1365-2958.2005.04649.x [DOI] [PubMed] [Google Scholar]
- 18.Ferenci T, Phan K (2015) How porin heterogeneity and trade-offs affect the antibiotic susceptibility of Gram-negative bacteria. Genes (Basel) 6: 1113–1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andersson DI, Hughes D (2010) Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol 8: 260–271. 10.1038/nrmicro2319 [DOI] [PubMed] [Google Scholar]
- 20.Crowley B, Benedi VJ, Domenech-Sanchez A (2002) Expression of SHV-2 β-lactamase and of reduced amounts of OmpK36 porin in Klebsiella pneumoniae results in increased resistance to cephalosporins and carbapenems. Antimicrob Agents Chemother 46: 3679–3682. 10.1128/AAC.46.11.3679-3682.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Agyekum A, Fajardo-Lubi A, Xiaoman A, Ginn AN, Zong Z, et al. (2016) Predictability of phenotype in relation to common beta-lactam resistance mechanisms in Escherichia coli and Klebsiella pneumoniae. J Clin Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Partridge SR, Ginn AN, Wiklendt AM, Ellem J, Wong JS, et al. (2015) Emergence of blaKPC carbapenemase genes in Australia. Int J Antimicrob Agents 45: 130–136. 10.1016/j.ijantimicag.2014.10.006 [DOI] [PubMed] [Google Scholar]
- 23.Woodcock DM, Crowther PJ, Doherty J, Jefferson S, DeCruz E, et al. (1989) Quantitative evaluation of Escherichia coli host strains for tolerance to cytosine methylation in plasmid and phage recombinants. Nucleic Acids Res 17: 3469–3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crepin S, Harel J, Dozois CM (2012) Chromosomal complementation using Tn7 transposon vectors in Enterobacteriaceae. Appl Environ Microbiol 78: 6001–6008. 10.1128/AEM.00986-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ogunniyi AD, Kotlarski I, Morona R, Manning PA (1997) Role of SefA subunit protein of SEF14 fimbriae in the pathogenesis of Salmonella enterica serovar Enteritidis. Infect Immun 65: 708–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Datsenko KA, Wanner BL (2000) One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97: 6640–6645. 10.1073/pnas.120163297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murphy KC, Campellone KG (2003) Lambda Red-mediated recombinogenic engineering of enterohemorrhagic and enteropathogenic E. coli. BMC Mol Biol 4: 11 10.1186/1471-2199-4-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang AC, Cohen SN (1978) Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J Bacteriol 134: 1141–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.CLSI (2012) Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically: Clinical Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 30.EUCAST (2015) Breakpoint tables for the interpretation of MICs and zone diameters: European Committee on Antimicrobial Susceptibility Testing, Váxjö, Sweden.
- 31.CLSI (2014) Performance standards for antimicrobial susceptibility testing; Twenty-fourth informational supplement. [Google Scholar]
- 32.Valenzuela JK, Thomas L, Partridge SR, van der Reijden T, Dijkshoorn L, et al. (2007) Horizontal gene transfer in a polyclonal outbreak of carbapenem-resistant Acinetobacter baumannii. J Clin Microbiol 45: 453–460. 10.1128/JCM.01971-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Partridge SR, Ellem JA, Tetu SG, Zong Z, Paulsen IT, et al. (2011) Complete sequence of pJIE143, a pir-type plasmid carrying ISEcp1-blaCTX-M-15 from an Escherichia coli ST131 isolate. Antimicrob Agents Chemother 55: 5933–5935. 10.1128/AAC.00639-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Partridge SR, Ginn AN, Paulsen IT, Iredell JR (2012) pEl1573 carrying blaIMP-4, from Sydney, Australia, is closely related to other IncL/M plasmids. Antimicrob Agents Chemother 56: 6029–6032. 10.1128/AAC.01189-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zong Z, Partridge SR, Thomas L, Iredell JR (2008) Dominance of blaCTX-M within an Australian extended-spectrum β-lactamase gene pool. Antimicrob Agents Chemother 52: 4198–4202. 10.1128/AAC.00107-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Espedido BA, Partridge SR, Iredell JR (2008) blaIMP-4 in different genetic contexts in Enterobacteriaceae isolates from Australia. Antimicrob Agents Chemother 52: 2984–2987. 10.1128/AAC.01634-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barton BM, Harding GP, Zuccarelli AJ (1995) A general method for detecting and sizing large plasmids. Anal Biochem 226: 235–240. 10.1006/abio.1995.1220 [DOI] [PubMed] [Google Scholar]
- 38.Partridge SR, Zong Z, Iredell JR (2011) Recombination in IS26 and Tn2 in the evolution of multiresistance regions carrying blaCTX-M-15 on conjugative IncF plasmids from Escherichia coli. Antimicrob Agents Chemother 55: 4971–4978. 10.1128/AAC.00025-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Carlone GM, Thomas ML, Rumschlag HS, Sottnek FO (1986) Rapid microprocedure for isolating detergent-insoluble outer membrane proteins from Haemophilus species. J Clin Microbiol 24: 330–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rasheed JK, Anderson GJ, Yigit H, Queenan AM, Domenech-Sanchez A, et al. (2000) Characterization of the extended-spectrum β-lactamase reference strain, Klebsiella pneumoniae K6 (ATCC 700603), which produces the novel enzyme SHV-18. Antimicrob Agents Chemother 44: 2382–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-DDC(T)) Method. Methods 25: 402–408. 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- 42.Knopp M, Andersson DI (2015) Amelioration of the fitness costs of antibiotic resistance due to reduced outer membrane permeability by upregulation of alternative porins. Mol Biol Evol 32: 3252–3263. 10.1093/molbev/msv195 [DOI] [PubMed] [Google Scholar]
- 43.Olivares J, Alvarez-Ortega C, Linares JF, Rojo F, Kohler T, et al. (2012) Overproduction of the multidrug efflux pump MexEF-OprN does not impair Pseudomonas aeruginosa fitness in competition tests, but produces specific changes in bacterial regulatory networks. Environ Microbiol 14: 1968–1981. 10.1111/j.1462-2920.2012.02727.x [DOI] [PubMed] [Google Scholar]
- 44.Nevola JJ, Stocker BA, Laux DC, Cohen PS (1985) Colonization of the mouse intestine by an avirulent Salmonella typhimurium strain and its lipopolysaccharide-defective mutants. Infect Immun 50: 152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Favre-Bonte S, Licht TR, Forestier C, Krogfelt KA (1999) Klebsiella pneumoniae capsule expression is necessary for colonization of large intestines of streptomycin-treated mice. Infect Immun 67: 6152–6156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schjorring S, Struve C, Krogfelt KA (2008) Transfer of antimicrobial resistance plasmids from Klebsiella pneumoniae to Escherichia coli in the mouse intestine. J Antimicrob Chemother 62: 1086–1093. 10.1093/jac/dkn323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schjørring S, Struve C, Krogfelt KA (2008) Transfer of antimicrobial resistance plasmids from Klebsiella pneumoniae to Escherichia coli in the mouse intestine. J Antimicrob Chemother 62: 1086–1093. 10.1093/jac/dkn323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chaves J, Ladona MG, Segura C, Coira A, Reig R, et al. (2001) SHV-1 beta-lactamase is mainly a chromosomally encoded species-specific enzyme in Klebsiella pneumoniae. Antimicrob Agents Chemother 45: 2856–2861. 10.1128/AAC.45.10.2856-2861.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haeggman S, Lofdahl S, Paauw A, Verhoef J, Brisse S (2004) Diversity and evolution of the class A chromosomal beta-lactamase gene in Klebsiella pneumoniae. Antimicrob Agents Chemother 48: 2400–2408. 10.1128/AAC.48.7.2400-2408.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bush K, Fisher JF (2011) Epidemiological expansion, structural studies, and clinical challenges of new b-lactamases from Gram-negative bacteria. Annu Rev Microbiol 65: 455–478. 10.1146/annurev-micro-090110-102911 [DOI] [PubMed] [Google Scholar]
- 51.Tomás JM, Ciurana B, Jofre JT (1986) New, simple medium for selective, differential recovery of Klebsiella spp. Appl Environ Microbiol 51: 1361–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lawlor MS, Hsu J, Rick PD, Miller VL (2005) Identification of Klebsiella pneumoniae virulence determinants using an intranasal infection model. Mol Microbiol 58: 1054–1073. 10.1111/j.1365-2958.2005.04918.x [DOI] [PubMed] [Google Scholar]
- 53.Struve C, Bojer M, Krogfelt KA (2008) Characterization of Klebsiella pneumoniae type 1 fimbriae by detection of phase variation during colonization and infection and impact on virulence. Infect Immun 76: 4055–4065. 10.1128/IAI.00494-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Renois F, Jacques J, Guillard T, Moret H, Pluot M, et al. (2011) Preliminary investigation of a mice model of Klebsiella pneumoniae subsp. ozaenae induced pneumonia. Microbes Infect 13: 1045–1051. 10.1016/j.micinf.2011.05.013 [DOI] [PubMed] [Google Scholar]
- 55.Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, et al. (2014) SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res 42: W252–258. 10.1093/nar/gku340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, et al. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res 35: W375–383. 10.1093/nar/gkm216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, et al. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66: 12–21. 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bowie JU, Luthy R, Eisenberg D (1991) A method to identify protein sequences that fold into a known three-dimensional structure. Science 253: 164–170. [DOI] [PubMed] [Google Scholar]
- 59.Luthy R, Bowie JU, Eisenberg D (1992) Assessment of protein models with three-dimensional profiles. Nature 356: 83–85. 10.1038/356083a0 [DOI] [PubMed] [Google Scholar]
- 60.Schrodinger L (2015) The PyMOL Molecular Graphics System, Version 2.1.1.
- 61.Lee RS, Seemann T, Heffernan H, Kwong JC, Goncalves da Silva A, et al. (2018) Genomic epidemiology and antimicrobial resistance of Neisseria gonorrhoeae in New Zealand. J Antimicrob Chemother 73: 353–364. 10.1093/jac/dkx405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19: 455–477. 10.1089/cmb.2012.0021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Darling AE, Mau B, Perna NT (2010) progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5: e11147 10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Seemann T (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069. 10.1093/bioinformatics/btu153 [DOI] [PubMed] [Google Scholar]
- 65.Lam MMC, Wick RR, Wyres KL, Gorrie C, Judd LM, et al. (2017) Frequent emergence of pathogenic lineages of Klebsiella pneumoniae via mobilisation of yersiniabactin and colibactin. bioRxiv. [Google Scholar]
- 66.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, et al. (2015) Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31: 3691–3693. 10.1093/bioinformatics/btv421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nguyen LT, Schmidt HA, von Haeseler A, Minh BQ (2015) IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol Biol Evol 32: 268–274. 10.1093/molbev/msu300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hadfield J, Croucher NJ, Goater RJ, Abudahab K, Aanensen DM, et al. (2017) Phandango: an interactive viewer for bacterial population genomics. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Achim Zeileis DM, Kurt Hornik (2007) Residual-based shadings for visualizing (conditional) independence. Jpurnal of Computational and Graphical statistics 16 (3): 507–525. [Google Scholar]
- 70.Haanperä M, Forssten SD, Huovinen P, Jalava J (2008) Typing of SHV extended-spectrum β-lactamases by pyrosequencing in Klebsiella pneumoniae strains with chromosomal SHV β-lactamase. Antimicrob Agents Chemother 52: 2632–2635. 10.1128/AAC.01259-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Babini GS, Livermore DM (2000) Are SHV β-lactamases universal in Klebsiella pneumoniae? Antimicrob Agents Chemother 44: 2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hæggman S, Löfdahl S, Paauw A, Verhoef J, Brisse S (2004) Diversity and evolution of the class A chromosomal β-lactamase gene in Klebsiella pneumoniae. Antimicrob Agents Chemother 48: 2400–2408. 10.1128/AAC.48.7.2400-2408.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chaves J, Ladona MG, Segura C, Coira A, Reig R, et al. (2001) SHV-1 β-lactamase is mainly a chromosomally encoded species-specific enzyme in Klebsiella pneumoniae. Antimicrob Agents Chemother 45: 2856–2861. 10.1128/AAC.45.10.2856-2861.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.EUCAST (2018) The European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretation of MICs and zone diameters. Version 8.0. www.eucast.org. European Committee on Antimicrobial Susceptibility Testing.
- 75.CLSI (2014) M100-S24. Performance Standards for Antimicrobial Susceptibility Testing; Twenty-Fourth Informational Supplement. Clinical and Laboratory Standards Institute; Wayne, PA. [Google Scholar]
- 76.Garcia-Sureda L, Juan C, Domenech-Sanchez A, Alberti S (2011) Role of Klebsiella pneumoniae LamB Porin in antimicrobial resistance. Antimicrob Agents Chemother 55: 1803–1805. 10.1128/AAC.01441-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kaczmarek FM, Dib-Hajj F, Shang W, Gootz TD (2006) High-level carbapenem resistance in a Klebsiella pneumoniae clinical isolate is due to the combination of blaACT-1 β-lactamase production, porin OmpK35/36 insertional inactivation, and down-regulation of the phosphate transport porin PhoE. Antimicrob Agents Chemother 50: 3396–3406. 10.1128/AAC.00285-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Doménech-Sánchez A, Martínez-Martínez L, Hernández-Allés S, del Carmen Conejo M, Pascual A, et al. (2003) Role of Klebsiella pneumoniae OmpK35 porin in antimicrobial resistance. Antimicrob Agents Chemother 47: 3332–3335. 10.1128/AAC.47.10.3332-3335.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garcia-Sureda L, Domenech-Sanchez A, Barbier M, Juan C, Gasco J, et al. (2011) OmpK26, a novel porin associated with carbapenem resistance in Klebsiella pneumoniae. Antimicrob Agents Chemother 55: 4742–4747. 10.1128/AAC.00309-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tsai YK, Fung CP, Lin JC, Chen JH, Chang FY, et al. (2011) Klebsiella pneumoniae outer membrane porins OmpK35 and OmpK36 play roles in both antimicrobial resistance and virulence. Antimicrob Agents Chemother 55: 1485–1493. 10.1128/AAC.01275-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.March C, Cano V, Moranta D, Llobet E, Perez-Gutierrez C, et al. (2013) Role of bacterial surface structures on the interaction of Klebsiella pneumoniae with phagocytes. PLoS One 8: e56847 10.1371/journal.pone.0056847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cowan SW, Schirmer T, Rummel G, Steiert M, Ghosh R, et al. (1992) Crystal structures explain functional properties of two E. coli porins. Nature 358: 727–733. 10.1038/358727a0 [DOI] [PubMed] [Google Scholar]
- 83.Domenech-Sanchez A, Hernandez-Alles S, Martinez-Martinez L, Benedi VJ, Alberti S (1999) Identification and characterization of a new porin gene of Klebsiella pneumoniae: its role in beta-lactam antibiotic resistance. J Bacteriol 181: 2726–2732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bornet C, Saint N, Fetnaci L, Dupont M, Davin-Regli A, et al. (2004) Omp35, a new Enterobacter aerogenes porin involved in selective susceptibility to cephalosporins. Antimicrob Agents Chemother 48: 2153–2158. 10.1128/AAC.48.6.2153-2158.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pitout JD, Sanders CC, Sanders WE Jr. (1997) Antimicrobial resistance with focus on β-lactam resistance in Gram-negative bacilli. Am J Med 103: 51–59. [DOI] [PubMed] [Google Scholar]
- 86.Fridkin SK, Steward CD, Edwards JR, Pryor ER, McGowan JE Jr., et al. (1999) Surveillance of antimicrobial use and antimicrobial resistance in United States hospitals: project ICARE phase 2. Project Intensive Care Antimicrobial Resistance Epidemiology (ICARE) hospitals. Clin Infect Dis 29: 245–252. 10.1086/520193 [DOI] [PubMed] [Google Scholar]
- 87.Hidron AI, Edwards JR, Patel J, Horan TC, Sievert DM, et al. (2008) NHSN annual update: antimicrobial-resistant pathogens associated with healthcare-associated infections: annual summary of data reported to the National Healthcare Safety Network at the Centers for Disease Control and Prevention, 2006–2007. Infect Control Hosp Epidemiol 29: 996–1011. 10.1086/591861 [DOI] [PubMed] [Google Scholar]
- 88.Wollheim C, Guerra IM, Conte VD, Hoffman SP, Schreiner FJ, et al. (2011) Nosocomial and community infections due to class A extended-spectrum beta-lactamase (ESBLA)-producing Escherichia coli and Klebsiella spp. in southern Brazil. Braz J Infect Dis 15: 138–143. [PubMed] [Google Scholar]
- 89.Doumith M, Ellington MJ, Livermore DM, Woodford N (2009) Molecular mechanisms disrupting porin expression in ertapenem-resistant Klebsiella and Enterobacter spp. clinical isolates from the UK. J Antimicrob Chemother 63: 659–667. 10.1093/jac/dkp029 [DOI] [PubMed] [Google Scholar]
- 90.Chudácková E, Bergerová T, Fajfrlík K, Cervená D, Urbásková P, et al. (2010) Carbapenem-nonsusceptible strains of Klebsiella pneumoniae producing SHV-5 and/or DHA-1 β-lactamases in a Czech hospital. FEMS Microbiol Lett 309: 62–70. 10.1111/j.1574-6968.2010.02016.x [DOI] [PubMed] [Google Scholar]
- 91.Nikaido H, Rosenberg EY (1981) Effect on solute size on diffusion rates through the transmembrane pores of the outer membrane of Escherichia coli. J Gen Physiol 77: 121–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jaffe A, Chabbert YA, Semonin O (1982) Role of porin proteins OmpF and OmpC in the permeation of β-lactams. Antimicrob Agents Chemother 22: 942–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nitzan Y, Deutsch EB, Pechatnikov I (2002) Diffusion of β-lactam antibiotics through oligomeric or monomeric porin channels of some Gram-negative bacteria. Curr Microbiol 45: 446–455. 10.1007/s00284-002-3778-6 [DOI] [PubMed] [Google Scholar]
- 94.Sugawara E, Kojima S, Nikaido H (2016) Klebsiella pneumoniae major porins OmpK35 and OmpK36 allow more efficient diffusion of β-lactams than their Escherichia coli homologs OmpF and OmpC. J Bacteriol 198: 3200–3208. 10.1128/JB.00590-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen JH, Siu LK, Fung CP, Lin JC, Yeh KM, et al. (2010) Contribution of outer membrane protein K36 to antimicrobial resistance and virulence in Klebsiella pneumoniae. J Antimicrob Chemother 65: 986–990. 10.1093/jac/dkq056 [DOI] [PubMed] [Google Scholar]
- 96.Tsai YK, Liou CH, Fung CP, Lin JC, Siu LK (2013) Single or in combination antimicrobial resistance mechanisms of Klebsiella pneumoniae contribute to varied susceptibility to different carbapenems. PLoS One 8: e79640 10.1371/journal.pone.0079640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bedikian A, Okamoto MP, Nakahiro RK, Farino J, Heseltine PN, et al. (1994) Pharmacokinetics of meropenem in patients with intra-abdominal infections. Antimicrob Agents Chemother 38: 151–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Condon RE, Walker AP, Hanna CB, Greenberg RN, Broom A, et al. (1997) Penetration of meropenem in plasma and abdominal tissues from patients undergoing intraabdominal surgery. Clin Infect Dis 24 Suppl 2: S181–183. [DOI] [PubMed] [Google Scholar]
- 99.Hughes D, Andersson DI (2015) Evolutionary consequences of drug resistance: shared principles across diverse targets and organisms. Nat Rev Genet 16: 459–471. 10.1038/nrg3922 [DOI] [PubMed] [Google Scholar]
- 100.Kaczmarek FM, Dib-Hajj F, Shang W, Gootz TD (2006) High-level carbapenem resistance in a Klebsiella pneumoniae clinical isolate is due to the combination of blaACT-1 b-lactamase production, porin OmpK35/36 insertional inactivation, and down-regulation of the phosphate transport porin phoE. Antimicrob Agents Chemother 50: 3396–3406. 10.1128/AAC.00285-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wise MG, Horvath E, Young K, Sahm DF, Kazmierczak KM (2018) Global survey of Klebsiella pneumoniae major porins from ertapenem non-susceptible isolates lacking carbapenemases. J Med Microbiol 67: 289–295. 10.1099/jmm.0.000691 [DOI] [PubMed] [Google Scholar]
- 102.De E, Baslé A, Jaquinod M, Saint N, Malléa M, et al. (2001) A new mechanism of antibiotic resistance in Enterobacteriaceae induced by a structural modification of the major porin. Mol Microbiol 41: 189–198. [DOI] [PubMed] [Google Scholar]
- 103.Olesky M, Zhao S, Rosenberg RL, Nicholas RA (2006) Porin-mediated antibiotic resistance in Neisseria gonorrhoeae: ion, solute, and antibiotic permeation through PIB proteins with penB mutations. J Bacteriol 188: 2300–2308. 10.1128/JB.188.7.2300-2308.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lou H, Chen M, Black SS, Bushell SR, Ceccarelli M, et al. (2011) Altered antibiotic transport in OmpC mutants isolated from a series of clinical strains of multi-drug resistant E. coli. PLoS One 6: e25825 10.1371/journal.pone.0025825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bredin J, Saint N, Mallea M, De E, Molle G, et al. (2002) Alteration of pore properties of Escherichia coli OmpF induced by mutation of key residues in anti-loop 3 region. Biochem J 363: 521–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Thiolas A, Bornet C, Davin-Regli A, Pagès JM, Bollet C (2004) Resistance to imipenem, cefepime, and cefpirome associated with mutation in Omp36 osmoporin of Enterobacter aerogenes. Biochem Biophys Res Commun 317: 851–856. 10.1016/j.bbrc.2004.03.130 [DOI] [PubMed] [Google Scholar]
- 107.Sugawara E, Kojima S, Nikaido H (2016) Klebsiella pneumoniae major porins OmpK35 and OmpK36 allow more efficient diffusion of b-Lactams than their Escherichia coli homologs OmpF and OmpC. J Bacteriol 198: 3200–3208. 10.1128/JB.00590-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Woodford N, Turton JF, Livermore DM (2011) Multiresistant Gram-negative bacteria: the role of high-risk clones in the dissemination of antibiotic resistance. FEMS Microbiol Rev 35: 736–755. 10.1111/j.1574-6976.2011.00268.x [DOI] [PubMed] [Google Scholar]
- 109.Wright LL, Turton JF, Livermore DM, Hopkins KL, Woodford N (2015) Dominance of international ‘high-risk clones’ among metallo-beta-lactamase-producing Pseudomonas aeruginosa in the UK. J Antimicrob Chemother 70: 103–110. 10.1093/jac/dku339 [DOI] [PubMed] [Google Scholar]
- 110.Andersson DI, Hughes D (2014) Microbiological effects of sublethal levels of antibiotics. Nat Rev Microbiol 12: 465–478. 10.1038/nrmicro3270 [DOI] [PubMed] [Google Scholar]
- 111.Yang Y, Song W, Lin H, Wang W, Du L, et al. (2018) Antibiotics and antibiotic resistance genes in global lakes: A review and meta-analysis. Environ Int 116: 60–73. 10.1016/j.envint.2018.04.011 [DOI] [PubMed] [Google Scholar]
- 112.Pulzova L, Navratilova L, Comor L (2017) Alterations in outer membrane permeability favor drug-resistant phenotype of Klebsiella pneumoniae. Microb Drug Resist 23: 413–420. 10.1089/mdr.2016.0017 [DOI] [PubMed] [Google Scholar]
- 113.Elena SF, Lenski RE (2003) Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet 4: 457–469. 10.1038/nrg1088 [DOI] [PubMed] [Google Scholar]
- 114.Harder KJ, Nikaido H, Matsuhashi M (1981) Mutants of Escherichia coli that are resistant to certain beta-lactam compounds lack the ompF porin. Antimicrob Agents Chemother 20: 549–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hasegawa Y, Yamada H, Mizushima S (1976) Interactions of outer membrane proteins O-8 and O-9 with peptidoglycan sacculus of Escherichia coli K-12. J Biochem 80: 1401–1409. [DOI] [PubMed] [Google Scholar]
- 116.Matsubara M, Kitaoka SI, Takeda SI, Mizuno T (2000) Tuning of the porin expression under anaerobic growth conditions by his-to-Asp cross-phosphorelay through both the EnvZ-osmosensor and ArcB-anaerosensor in Escherichia coli. Genes Cells 5: 555–569. [DOI] [PubMed] [Google Scholar]
- 117.Heyde M, Portalier R (1987) Regulation of major outer membrane porin proteins of Escherichia coli K 12 by pH. Mol Gen Genet 208: 511–517. [DOI] [PubMed] [Google Scholar]
- 118.Giraud A, Arous S, De Paepe M, Gaboriau-Routhiau V, Bambou JC, et al. (2008) Dissecting the genetic components of adaptation of Escherichia coli to the mouse gut. PLoS Genet 4: e2 10.1371/journal.pgen.0040002 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
White arrows show the plasmids in original host isolates and transconjugants. MW; Mid-range PFG Marker.
(TIF)
Wild type strains ATCC 13883, 10.85 and 11.76 were cultured under different temperatures (37°C, 30°C and 25°C) and different nutrient concentrations (MH and MH 1:10). Blue arrow, OmpK35. Black arrow, OmpK36. Red arrow, OmpA.
(TIF)
The relative fitness of deletion porin mutants in comparison with parental strain (11.76) was performed by competition experiments in co-cultures and expressed as a percentage of the mutant or wild type cells versus total population at each time point. In vitro growth conditions, MH broth, 37°C. Violet diamond, 11.76 wild type strains. Orange square, ΔOmpK35 mutant. Red square, ΔOmpK36 mutant.
(TIFF)
The relative fitness of porin mutants in comparison with parental strain ATCC 13883 was performed by competition experiments in co-cultures and expressed as a percentage of the mutant or wild type cells versus total population at each time point. In vitro growth conditions: A, MH 1:10 broth, 25°C; B, MH 1:10 broth, 37°C. Violet diamond, ATCC 13883. Orange square, ΔOmpK35. Red square, ΔOmpK36.
(TIF)
K. pneumoniae intestinal colonization in a mouse model. CFU counts of K. pneumoniae ATCC 13883 and porin mutants from mice faecal sample. Bacterial inoculum at day 0 is 1x1010 CFU /mouse. Addition of ampicillin 0.5 g / L in the drinking water on day 4. Violet diamond, ATCC 13883 wild type strains. Orange square, ΔOmpK35 mutant. Red square, ΔOmpK36 mutant. Blue circle, ΔOmpK35ΔOmpK36 mutant. Green square, OmpK36GD mutant. Pink circle, ΔOmpK35OmpK36GD mutant.
(TIF)
26 unique sequences with OmpK36 L3 variants from GenBank were compared with OmpK36 of NTUH_K2044. Isolates with wild-type L3 sequence are not included. Dot line, signal peptide. Black line, beta strands. Red line, loops. Blue line, alpha helix. Green squares, turns. OmpK36 secondary structure based on previous studies [82,83]. Red boxes, residues involved in the pore eyelet based on [84]. Black box, L3 variants.
(TIF)
Comparison of the reference OmpK36 structure under PDB accession 5nup1A (WT, wild type) against predicted structural models of mutants harbouring a two amino-acid insertion in loop 3 after G113, namely GGDGD, GGDTD and GGDSD. For each predicted structure, the 2 most protruding amino-acids resulting from the insertion were marked and coloured according to their backbone structure (carbons in yellow, oxygens in red and nitrogens in blue).
(TIF)
Based on observations of the codon sequences, the extra–SD and–SYG following GGD likely result from a combination of duplication followed by point mutation.
(TIF)
A. Alignment of E. coli OmpC_L3 variants. 11 unique Omp36_L3 variants from GenBank were compared with L3 of OmpC of K-12 MG1655 (NP_416719). Black boxes, residues different from NP_416719. B. Alignment of E. aerogenes Omp36_L3 variants. Four unique Omp36_L3 variants from GenBank were compared with L3 of Omp36 from ATCC 13048 (AF335467). Black boxes, residues different from AF335467. Isolates with wild-type L3 sequence are not included. Black line, loop 3. OmpK36 L3 location based on previous studies. (82, 83). Red boxes, residues involved in the pore eyelet based on (84).
(TIF)
Metadata includes year of isolation; MIC levels for ETP: ertapenem, IMP: imipenem, and MEM: meropenem; ompK36 L3 mutation; ompK35 disrupted mutations (as listed in S7 Table); ST: sequence type; number of predicted resistance genes encoded; carbapenamase gene encoded; ESBL: extended-spectrum beta-lactamase gene encoded.
(TIF)
The mosaic plot shows the relationships between 3 variables; ST (in purple) and presence/absence of ompK35 (in black) on the x-axis; and presence/absence and mutations of ompK36 (in grey) on the y-axis. The size of each plot tile is proportional to counts. Plot tiles are colored according to their standardized Pearson residuals, as determined by a log-linear model. Deeper shades of red and blue corresponding to a standardized residual less than -4 or greater than +4, respectively, can be interpreted as combinations observed significantly less or more than expected (under the assumptions that proportions have equal levels).
(TIF)
The mosaic plots show the relationships between 2 variables; A) country of isolation on the x-axis and presence/absence of ompK35 on the y-axis; B) country of isolation on the x-axis, and presence/absence and mutations of ompK36 on the y-axis. The size of each plot tile is proportional to counts. Plot tiles are colored according to their standardized Pearson residuals, as determined by a log-linear model. Deeper shades of red and blue corresponding to a standardized residual less than -4 or greater than +4, respectively, can be interpreted as combinations observed significantly less or more than expected (under the assumptions that proportions have equal levels).
(TIF)
The mosaic plots show the relationships between 2 variables; A) year of isolation on the x-axis and presence/absence of ompK35 on the y-axis; B) year of isolation on the x-axis, and presence/absence and mutations of ompK36 on the y-axis. The size of each plot tile is proportional to counts. Plot tiles are colored according to their standardized Pearson residuals, as determined by a log-linear model. Deeper shades of red and blue corresponding to a standardized residual less than -4 or greater than +4, respectively, can be interpreted as combinations observed significantly less or more than expected (under the assumptions that proportions have equal levels).
(TIF)
Boxplots were used to display the distribution of resistance genes identified with Abricate within each ST with the following OmpK36 variants, namely isolates with–GD in bright red,–TD in brown, or no insertion (–) in grey. Mean comparison p-values are also shown for each ST (Wilcoxon test, with ‘-’ used as a reference group; ns: p > 0.05; *: p < = 0.05; **: p < = 0.01; ***: p < = 0.001; ****: p < = 0.0001). In addition, the corresponding underlying isolate population is also visualised as individual points, coloured according to OmpK35 type, (1) intact in turquoise or (0) disrupted in coral.
(TIF)
(DOC)
(XLSX)
(XLSX)
(DOC)
(DOC)
The expression of rpoD was used to normalize the results. The levels of expression of each mutant are shown relative to the wild type strain ATCC 13883 or 10.85.
(DOC)
(DOC)
(XLSX)
Data Availability Statement
Data availability statement has been added at the end of the manuscript after Supporting Information. Raw sequence reads are available on NCBI under Bioproject accession number PRJNA430457. Relevant R scripts were also made available at https://github.com/nbenzakour/Klebsiella_antibiotics_paper.