

Current primary cell models for HIV latency correlate poorly with the reactivation behavior of patient cells. We have developed a new model, called QUECEL, which generates a large and homogenous population of latently infected CD4+ memory cells. By purifying HIV-infected cells and inducing cell quiescence with a defined cocktail of cytokines, we have eliminated the largest problems with previous primary cell models of HIV latency: variable infection levels, ill-defined polarization states, and inefficient shutdown of cellular transcription. Latency reversal in the QUECEL model by a wide range of agents correlates strongly with RNA induction in patient samples. This scalable and highly reproducible model of HIV latency will permit detailed analysis of cellular mechanisms controlling HIV latency and reactivation.
KEYWORDS: cell quiescence, HIV latency, HIV reservoir, P-TEFb, human immunodeficiency virus
ABSTRACT
The latent HIV reservoir is generated following HIV infection of activated effector CD4 T cells, which then transition to a memory phenotype. Here, we describe an ex vivo method, called QUECEL (quiescent effector cell latency), that mimics this process efficiently and allows production of large numbers of latently infected CD4+ T cells. Naïve CD4+ T cells were polarized into the four major T cell subsets (Th1, Th2, Th17, and Treg) and subsequently infected with a single-round reporter virus which expressed GFP/CD8a. The infected cells were purified and coerced into quiescence using a defined cocktail of cytokines, including tumor growth factor beta, interleukin-10 (IL-10), and IL-8, producing a homogeneous population of latently infected cells. Flow cytometry and transcriptome sequencing (RNA-Seq) demonstrated that the cells maintained the correct polarization phenotypes and had withdrawn from the cell cycle. Key pathways and gene sets enriched during transition from quiescence to reactivation include E2F targets, G2M checkpoint, estrogen response late gene expression, and c-myc targets. Reactivation of HIV by latency-reversing agents (LRAs) closely mimics RNA induction profiles seen in cells from well-suppressed HIV patient samples using the envelope detection of in vitro transcription sequencing (EDITS) assay. Since homogeneous populations of latently infected cells can be recovered, the QUECEL model has an excellent signal-to-noise ratio and has been extremely consistent and reproducible in numerous experiments performed during the last 4 years. The ease, efficiency, and accuracy of the mimicking of physiological conditions make the QUECEL model a robust and reproducible tool to study the molecular mechanisms underlying HIV latency.
INTRODUCTION
Patients adhering to combination antiretroviral (cART) regimens have minimal viremia, but HIV persists due to a pool of transcriptionally silenced but replication-competent proviruses found in a small population of resting memory CD4+ T cells (1 to 100 per 106 cells) accumulating in the peripheral blood (1) and tissues (2). Since silenced proviruses produce only minimal viral RNA and proteins, they are refractory to antiviral drugs and effectively evade immune surveillance.
Recent studies of proviral integration sites have provided strong evidence for the clonal expansion of memory T cells carrying silenced proviruses (3, 4). These data suggest that the reservoir is in a pseudo-steady state with persistent low-level rates of viral reactivation and cell death counterbalanced by homeostatic expansion of latent clones (3–6). These combined processes result in a very stable reservoir with an apparent half-life of 44 months in the presence of cART (1, 7, 8). The persistent reservoir almost invariably rebounds within 2 to 8 weeks when cART is interrupted (9, 10). HIV latency therefore remains the major obstacle to a functional cure for HIV infections. An attractive approach for eradicating the latent reservoir is the “shock-and-kill” strategy. In this strategy, latency-reversing agents (LRAs) (11, 12) are used to make latently infected cells visible to the immune system (shock) and target them for elimination (kill).
It is currently unknown whether HIV targets activated effector CD4 T cells, which then enter quiescence after the removal of the immune stimuli, and their transition to a memory phenotype (13, 14), or whether HIV enters already quiescent cells (15, 16). Analysis of HIV transcription in resting cells demonstrates that quiescence leads to suppression of both the initiation of HIV transcription and its elongation (17). Several key transcription initiation factors, including NF-κB and NFAT, are sequestered in the cytoplasm of quiescent cells (17). The positive transcription elongation factor b (P-TEFb), which is an essential host cofactor for the HIV transactivator protein, Tat, is also absent from quiescent cells due to degradation of the CycT1 subunit (18–21). Additionally, epigenetic silencing events ensure that proviral latency is maintained (17). T-cell receptor activation reverses each of these restrictions and stimulates HIV transcription, replication, and spread. One reason why the switch to productive transcription when resting cells are activated is so efficient is that multiple signaling pathways are activated, and HIV transcription is selectively enhanced by a positive feedback mechanism fueled by the viral trans-activator protein Tat and P-TEFb (17).
Although latency can be established in transformed T cell lines, such as Jurkat T cells (22–26), where P-TEFb is constitutively activated (27), this can lead to potentially unrepresentative mechanisms of HIV latency (26, 28). A more physiologically relevant approach is to infect primary cells isolated from healthy donors (29, 30), and a wide variety of primary cell models of HIV latency have been developed (14, 29–33). Unfortunately, results in these models typically do not correlate well between themselves and with RNA induction measurements in patient cells (26).
Bosques and Planelles (30, 34, 35) introduced methods for infection of an effector cell population derived from naive T cells that is then allowed to enter quiescence. Based on their work, we have developed a refined polarized cell model of HIV latency, called the QUECEL (quiescent effector cell latency) model. A key innovation in the QUECEL model is the use of a defined cocktail of cytokines, including tumor growth factor beta (TGF-β), to force cells into quiescence and yield a highly enriched latent HIV-infected cell population. The QUECEL model has proved to be highly consistent and reproducible in numerous experiments performed during the last 4 years, and activation of HIV by LRAs closely mimics RNA induction profiles seen in cells from well-suppressed HIV patient samples.
RESULTS
QUECEL model for HIV latency in primary cell helper subsets.
The QUECEL (quiescent effector cell latency) model (Fig. 1) is a refinement of the model of Bosque and Planelles (30, 35). Briefly, naive helper T cells are activated, polarized, and then infected using a single-round vesicular stomatitis virus (VSV)-pseudotyped reporter virus (Fig. 1A) (22, 23, 36). The reporter carries a CD8-enhanced green fluorescent protein (EGFP) fusion protein that permits purification of infected cells with anti-CD8 magnetic beads. Three days after infection, CD8a-expressing cells were isolated and then allowed to expand up to 50-fold for 7 to 10 days.
FIG 1.

Overview of HIV latency model in effector CD4 T cells. (A) Genome organization of the HIV-1 pNL4-3 delta gag CD8a/GFP-IRES-Nef reporter virus. (B) Method. Naïve T cells were isolated from healthy donors and stimulated through the TCR or by ConA. Activated CD4+ T cells were polarized into specific effector CD4+ T cell subsets with cytokine cocktails at day 0. Effector cells were infected at day 7 with HIV-GFP/CD8a, sorted by magnetic bead separation, and allowed to enter quiescence, resulting in latently infected cells. Important steps in protocol and relative cytokine levels are indicated on the timeline. (C) Flow cytometry. Expression of HIV Nef, GFP, Ki67, CDK9 pSer175, cyclin D3, and cyclin B1, EDU incorporation, and DNA levels were monitored at each stage of the protocol.
To induce quiescence, the expansion cytokines were reduced 4-fold to simulate natural cytokine conditions developed in vivo in the absence of inflammation (37), and a silencing cocktail containing TGF-β, interleukin-10 (IL-10), and IL-8 was added (Fig. 1B). Expression of the cellular activation markers CDK9 pSer175 (P-TEFb) (38, 39) and Ki67 (14, 26) was high during the expansion phase, while the quiescence conditions resulted in a dramatic decrease of CDK9 pSer175 and Ki67. Cyclin D3 and B1 levels were also reduced drastically upon cellular quiescence, indicative of the cells leaving the cell cycle. This was confirmed by checking EDU incorporation and total DNA content using 4′,6-diamidino-2-phenylindole (DAPI) (Fig. 1C).
Once in quiescence, the level of HIV Nef protein expression is reduced to almost undetectable levels (1%), which is indicative of HIV latency. GFP levels also decline, but because of the high stability of the CD8a-GFP fusion protein (see Fig. S1A and B in the supplemental material), GFP persists in the quiescent cells, albeit at a lower intensity (Fig. 1C). Upon stimulation through the T cell receptor (TCR) (α-CD3/α-CD28 Dynal magnetic beads), the vast majority of cells (>82%) express Nef, and there is a concomitant increase in GFP, Ki67, cyclin B1, cyclin D3, and CDK9 pSer175 levels (Fig. 1C). HIV RNA is also detected in the majority of reactivated cells by RNA fluorescent in situ hybridization (FISH) (Fig. S1C). A useful variation of the QUECEL protocol includes the use of Thy1.2 for purification of the infected cell population and replacement of the CD8a-GFP reporter with a GFP or red fluorescent protein (RFP) reporter (Fig. S1D). The experimental basis for each of the steps in this protocol are described in detail below. For most of the experiments that follow, we have concentrated on the Th17-polarized cells, since there is very limited variation between the different effector subtypes and the Th17 cells have slightly higher viability.
CD8a/GFP persists in quiescent QUECEL cells. (A) Flow cytometry analysis of d2EGFP levels in Th17 cells infected with HIV expressing either the membrane-bound CD8a/GFP fusion protein or cytoplasmic d2EGFP. Cells were analyzed before and after treatment with 10 μg/ml cycloheximide (24 h). (B) Half-life of CD8a/GFP fusion protein and Nef during the establishment of quiescence. Solid lines, positive cells (%); dashed lines, mean fluorsescent intensity. (C) Immunofluorescence and RNA FISH analysis of Th17 cells reactivated through the TCR. Yellow, RNA detected by Stellaris RNA FISH (Biosearch Technologies) using probes spanning the U3 and U5 regions of the provirus. Green, GFP. Blue, DAPI. (D) Flow cytometry analysis of d2EGFP in Th17 cells latently infected with an HIV lentiviral vector expressing a Thy1.2-P2A-d2EGFP reporter gene (top), which permits decay of the cytoplasmic d2EGFP signal during quiescence. (Bottom) d2EGFP induction after stimulation by the TCR, 500 nM SAHA, and 100 ng/ml TNF-α. Download FIG S1, TIF file, 3.3 MB (3.3MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Efficient ex vivo polarization of Th1, Th2, Th17, and Treg effector CD4 T cell subsets.
A series of in vitro polarization conditions were developed to generate the four most abundant effector CD4 T cell subsets: Th1, Th2, Th17, and Treg (Fig. 1B, Table 1, and the supplemental material). For the Th1 and Th2 subsets, naive CD4 T cells were stimulated through the TCR, and unwanted cytokines were neutralized with monoclonal antibodies to IL-4, IL-12, or gamma interferon (IFN-γ) (40). Th17 cells were polarized in the presence of TGF-β, neutralizing antibodies to IL-4, and the cytokines IFN-γ, IL-6, IL-1β, and IL-23 (41, 42). Regulatory CD4 T cells were generated using TGF-β and neutralizing antibodies to IL-4 and IFN-γ (30).
TABLE 1.
Polarization conditions
| CD4 T cell subset | Cytokine | Concn (ng/ml) | Antibody | Antibody concn (µg/ml) |
|---|---|---|---|---|
| T helper 1 | TGF-β | 5 | α-Human IL-4 | 500 |
| T helper 2 | TGF-β | 5 | α-Human IFN-γ | 500 |
| T helper 17 | TGF-β | 5 | α-Human IL-4 | 500 |
| IL-23 | 50 | α-Human IFN-γ | 500 | |
| IL-6 | 10 | |||
| T regulatory cells | TGF-β | 5 | α-Human IL-4 | 500 |
| α-Human IFN-γ | 500 |
Successful cell polarization was initially analyzed using quantitative PCR (qPCR) mRNA expression arrays (Fig. S2) and confirmed by comprehensive transcriptome sequencing (RNA-Seq) analyses (Fig. 2) of exponentially growing cells (day 6). We were able to detect abundant mRNA transcripts that distinguish each T cell subset, including IFNG and STAT4 production in the Th1 subset, IL-4 and GATA2 in the Th2 subset, IL-17A and RORC in the Th17 subset, and FOXP3 and IL-2 in the regulatory T cell subset (Fig. S2) (43).
FIG 2.

RNA-Seq analysis of cells at the effector, quiescent, and reactivated stages of the QUECEL method. (A) Pathway analysis indicates that pathways most enriched during reactivation in QUECEL (Th17 cells) are also induced in H80 (BioProject accession number PRJNA376596) and Planelles (BioProject accession number PRJNA322599) ex vivo primary cell models after reactivation. Genes differentially expressed following reactivation of quiescent cells were identified in all three data sets through pairwise comparison of RNA-Seq expression patterns of the quiescent versus reactivated cells and matched to the HALLMARK gene set of the mSigDB using the GSEA tool. The false discovery rate values were used to draw the heat map while controlling for the direction of enrichment. PI3K, phosphatitylinositol 3 kinase. (B) Principal component analysis of expanding, quiescent, and reactivated infected T helper cell subsets and expanding uninfected control using RNA-Seq data. (C) RORγC mRNA expression in each of the T helper cell subsets. Normalized read histograms of RNA-Seq data from uninfected cells are shown. The position of features in nucleotides on hg38 chromosome 1 and the position of the introns and exons of the two isoforms of RORC are shown at the top. (D) HIV mRNA expression in expanding, quiescent, and reactivated infected Th17 T cells and the uninfected control. For panels C and D, the scales to the left of the histograms represent normalized RNA-Seq reads. For uninfected expanding and infected quiescent cells, the scales have been 5× expanded to visualize the small number of mapped reads. The HIV construct used in this study is shown at the top.
QUECEL T cell subsets express cell specific mRNAs. (A) Heat maps showing relative qPCR expression levels of 10 T cell subset-specific mRNAs (top) and the corresponding RNA-Seq levels (bottom). (B) Correlation plot of polarized Th17s compared to peripheral Th17s and fresh total CD4 memory T cells using the RT2 Profiler PCR array human Th17 response array (330231; Qiagen) (left) or RT2 Profiler PCR array human T helper cell differentiation array (330231; Qiagen) (right). Download FIG S2, TIF file, 1.8 MB (1.8MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
To gain further insight into the overall pattern of gene expression among the four polarized cells in exponentially growing, quiescent, and reactivated states, we performed a principal-component analysis of RNA-Seq data sets (Fig. 2B). This objective analysis of the gene expression profiles clearly demonstrated the close similarity of each of the cell growth states independent of the cell polarization conditions. However, within each cluster, Th17 cells were the most distinct for each of the 4 activation/infection states (Fig. 2B).
The polarization phenotypes were further confirmed using flow cytometry to measure cytokine expression (Fig. S3A and B) and specific transcription factors (Fig. S3C) (44). As expected, the vast majority of cells expressed the expected transcription factors and cytokines that were appropriate for their polarization phenotypes: Th1 (Tbet, 80%), Th2 (GATA3, 95.6%), Th17 (RORC and Tbet, 97%), and Tregs (FOXP3, 61%). RORγC is an important transcription factor only expressed in Th17 cells, and this transcript is only found in the Th17 subset (Fig. 2C) (45).
Polarized QUECEL T cells express subset-specific cytokines and transcription factors. (A) Flow cytometry of subset-specific transcription factors in unstimulated memory T cells isolated from PBMCs. (B) Cytokine expression levels in all T cell subsets. (C) Transcription factor expression in the cells shown in panel B. Download FIG S3, TIF file, 3.3 MB (3.4MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We also compared a qPCR Th17-specific inflammation transcription array of RNA from cells generated in vitro to patient-derived peripheral Th17 cells (41, 42, 46). The polarized Th17 cell transcripts correlated to peripheral Th17 with an R2 value of 0.98336 (Fig. S2B).
The RNA-Seq data sets also allowed us to measure the HIV transcripts in expanding, quiescent, and reactivated populations (Fig. 2D). Analysis of the pattern of RNA-Seq reads mapping to the infected HIV construct indicated that the expanding infected and reactivated populations contained high expression levels of transcripts for the entire HIV genome, but these were largely absent from the quiescent cell population (the scale has been expanded 5-fold to show the residual reads). As expected, uninfected controls showed no HIV transcripts.
Efficient induction of quiescence by cytokines.
To minimize the amount of cell death, subsets were gradually weaned of their expansion cytokines over a period of a week and then placed under reduced/protective cytokine conditions for 1 to 2 weeks. The cell cycle analyses shown in Fig. 3A documents that during this period, cells progressively lose cyclin B1 and D3 and enter the G0 phase (47).
FIG 3.

Induction of quiescence by cytokines. (A) Cell cycle analysis of cyclin D3, cyclin B1, and total DNA content (PI) during the expansion phase as well as the quiescence phase of Th17-polarized cells. (B) Cytokine secretion by H80 cells. (C) Loss of cell cycle markers in polarized Th17 cells exposed to different combinations of cytokines for 7 days and 10 ng/ml TGF-β.
Although infected polarized T cells will spontaneously senesce and drive HIV into latency (30, 35), this process was inefficient and highly variable in our experience. In our original primary cell model for HIV latency (29), we used H80 feeder cells (48) to gradually induce HIV silencing while promoting CD4+ T cell survival. Analysis of cytokine production by H80 cells showed that they overexpressed IL-2, IL-8, and IL-10 (Fig. 3B). A systematic analysis of the ability of the effector T-cells to enter quiescence when cultured for up to 4 weeks in combinations of IL-6, IL-8, and IL-10 in the presence and absence of TGF-β, a cytokine known to suppress activation of T cells (49, 50) is shown in Fig. 3C (with TGF-β) and Fig. S4A (without TGF-β). The combination of IL-8 and IL-10 strongly enhanced entry of cells into quiescence and latency, whereas IL-6 blocked entry into quiescence (Fig. 3C). Entry into quiescence was greatly enhanced by the addition of TGF-β, allowing the entire cell population to enter latency when cells are cultured for 3 weeks in the presence of TGF-β, IL-8, and IL-10 (Fig. 3C). There is a gradual loss of cells, due to senescence and apoptosis, throughout the shutdown process. Approximately 60% viable cells are recovered at 3 weeks, and there is no significant difference in cell viability in the presence of the different cytokine cocktails.
The H13L Tat mutant does not affect reactivation of latent HIV. (A) Cyclin B1 and D3 levels in Th17 cells treated with multiple cytokines in the absence of TGF-β. (B) Reactivation of latently infected Th17 cells carrying either WT, H13L Tat, or the inactivated C22G Tat mutant in response to a panel of activators. (C) Reactivation of latently infected Jurkat clones carrying either H13L Tat (2D10, G5) or WT Tat (E4). Download FIG S4, TIF file, 1.8 MB (1.8MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Entry into latency is driven primarily by the quiescence of the host cells. Reporter viruses carrying the H13L and wild-type (WT) Tat genes showed identical shutdown and reactivation profiles in this system (Fig. S4B). Similarly, in the Jurkat system, the reactivation profile of the H13L Tat gene was indistinguishable from that of the wild type (Fig. S4C).
Quiescent cells have a unique RNA transcript profile.
Pathway analysis performed on the RNA-Seq data sets demonstrated a decline in the expression of genes associated with expanding cells in the quiescent cell population. The expression of these pathways is restored in quiescent cells treated with TCR agonists (Fig. 2A and Fig. S5A). The differentially enriched pathways included those associated with cell division (G2-M checkpoint and mitotic spindle), global induction of transcription and translation (MYC targets and MTORC1 signaling), and induction of metabolic states associated with rapidly expanding T cells (glycolysis and hypoxia) (Fig. S6). All of the pathways enriched in the QUECEL model are also enriched in RNA-Seq data sets obtained from cells that have entered quiescence after culturing on H80 feed cells (51), or by the Bosque and Planelles method (52), and the three data sets are highly correlated (Fig. 2A and Fig. S5). However, additional pathways associated with senescence and innate immune responses also become activated in the H80 and Bosque and Planelles models (Fig. 2A and Fig. S5A).
Differential gene expression pattern following reactivation in QUECEL versus two other ex vivo primary cell models. (A) Table of pathways and gene sets enriched during transition from quiescence to reactivation in Th17 cells. A false discovery rate cutoff value of 0.1 was used to identify gene sets and pathways that were significantly enriched. (B) Genes induced following reactivation in QUECEL show significant overlap those upregulated in H80 and Planelles ex vivo models, with downregulated genes showing a smaller amount of overlap. (C) Pairwise scatterplots indicate a strong overall correlation of changes in gene expression pattern following reactivation between the three ex vivo primary cell models. In order to compare gene expression data obtained from similar populations of cells between the three datasets, all four polarized cells in the QUECEL method (Th1, Th2, Th17, and Treg) have been used in aggregate to generate the lists of QUECEL up- and downregulated genes in panels B and C. Download FIG S5, TIF file, 2.1 MB (2.2MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Heat maps of gene sets corresponding to the top enriched pathways after TCR stimulation of quiescent cells (24 h). Data indicate the strong induction of genes involved in metabolic, transcriptional, and translational activation of cells. The values graphed in the heatmaps correspond to the differential expression value (in log2 units) obtained by pairwise comparison of quiescent versus TCR-stimulated cells. Note that while the shown pathways were originally found to be enriched using only the Th17 polarized cells, the panels in this figure have been generated using the aggregate of all four polarized cells in the QUECEL method (Th1, Th2, Th17, and Treg), indicating the reproducibility and generality of these results across the effector T cell subsets. Download FIG S6, TIF file, 2.4 MB (2.5MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Latently infected proviruses are restricted for transcription initiation and elongation.
To demonstrate RNA polymerase II (RNAP II) recruitment to the HIV long terminal repeats (LTRs) after TCR activation of the latently infected cells, chromatin immunoprecipitation (ChIP) assays were performed using primers that specifically amplified either the 5′ LTR, 3′ LTR, or the promoter region of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a control (23) (Fig. S7). In contrast to Jurkat cells (23, 27, 53), initiation is highly restricted, and only a minimal level of paused, promoter-proximal RNAP II is seen at the LTR in the unstimulated cells. This can only be detected using an antibody against total RNAP II, suggesting that it is hypophosphorylated (54). Within 0.5 h of TCR stimulation there is a significant recruitment of pSer5 RNAP II at the 5′ LTR of HIV and a significant increase in both pSer5 RNAP II and pSer2 RNAP II at the 3′ LTR at the 16-h time point due full Tat-dependent elongation of the provirus (23). As an additional control, we also evaluated levels of H3K27me3, a methylated histone that is known to accumulate on latent proviruses and is removed during activation (28, 36). A high level of H3mK27me3 is present at the 5′ LTR of the unstimulated cells, and this is lost upon activation (Fig. S7B).
Chromatin immunoprecipitation (ChIP) analysis of the latent provirus in Th17 cells. (A) Location of primers used in the ChIP assays to specifically analyze the 5′ and 3′ HIV LTRs. (B) ChIP assays. Cells were stimulated through the TCR for 30 min or 16 h (n = 3). (Top) Total RNAPII; RNAP II (pSer5). (Bottom) RNAP II (pSer2) and methylated histone H3 (H3K27me3). GAPDH was used as a control. Download FIG S7, TIF file, 1.4 MB (1.4MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The QUECEL model accurately reflects the behavior of LRAs in patient cells.
Because the QUECEL model allows recovery of large homogeneous populations of latently infected cells, it is highly suitable for the screening of latency-reversing agents (LRAs). In addition to induction of Nef, we routinely monitor pSer175 CDK9, which is a marker of P-TEFb reactivation status (38). In quiescent cells, there are minimal levels of both HIV Nef and pSer175 CDK9 (<1%), and any increase in their levels is indicative of reactivation of the latent HIV provirus (Fig. 1C).
To determine the reproducibility of the system, we tracked HIV Nef expression between multiple donors (n = 3) over many separate experiments (n = 6) performed during the last 4 years. For each donor, cells were reactivated using either TCR, suberoylanilid hydroxamic acid (SAHA), or TNF-α, and the total amount of Nef expression was scored (Fig. 4A). The same stimulation conditions were also tested in all four T helper subsets over multiple experiments (n = 5) (Fig. 4B). The responses of the different subsets to the latency-reversing agents was virtually identical. However, we prefer to work with the Th17 cells, since they have the lowest basal activation levels and highest viability, most likely due to their reliance on IL-23, compared to IL-2 alone for the other T helper subsets (Fig. 4B).
FIG 4.
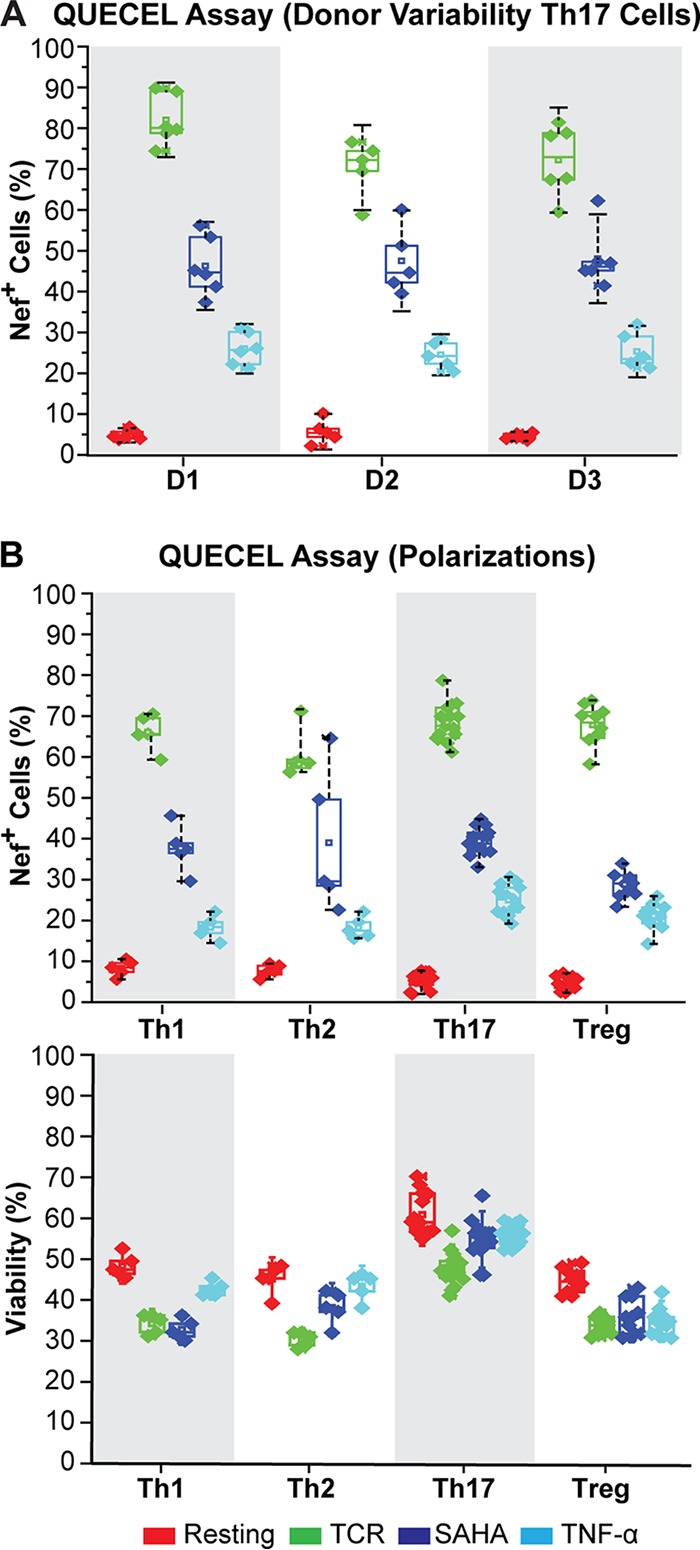
Reproducibility of the QUECEL model. (A) Donor variation (Th17 cells). Induction of HIV Nef expression in latently infected Th17 cells stimulated with TCR beads, SAHA, or TNF-α was measured in separate experiments using cells derived from three donors (donor 1, n = 7; donor 2, n = 6; donor 3, n = 6). (B) Induction of HIV Nef expression in each of 4 different T helper subsets stimulated with TCR, SAHA, and TNF-α (Th1, n = 4; Th2, n = 4; Th17, n = 10; Treg, n = 8). (Top) Cells were obtained from 3 different donors. (Bottom) Viability of cells measured by light scatter.
When LRAs are combined, synergistic activation conditions can be demonstrated (Fig. 5A). It is especially noteworthy that a combination of IL-15 and SAHA resulted in activation levels that are statistically indistinguishable from those of TCR stimulation (Fig. S8B). The amount of synergism was measured by the coefficient of drug interaction (CDI), where a value of 1 is considered additivity and anything below 0.7 is considered synergistic. IL-15 synergized best with SAHA, followed by TNF-α and Ingenol (Fig. 5A).
FIG 5.

Correlation between the QUECEL model and RNA induction in patient cells. (A) Synergistic reactivation of HIV in Th17 cells (% Nef+, pS175 CDK9+ cells after 24 h). Green color intensity represents BLISS scores demonstrating additive or synergistic (below 1) activation. (B) Correlation between reactivation of HIV in patient cells (EDITS, vertical axis) and QUECEL model (Th17 cells, horizontal axis) for a wide number of activation conditions. Data were fitted with a linear model (R2 = 0.941; n = 18). (C) Heatmap of responses of QUECEL (Th17 cells) to a panel of primary cell latency models analyzed by Spina et al. (26). Note that the QUECEL model clusters mostly with the QVOA and has a broader dynamic range than the other primary cell models of latency. Normalized values were calculated relative to the highest activation within each model and expressed as a percentage of maximal stimulation. DMSO, dimethyl sulfoxide. PHA-M, phytohemagglutinin-M; PMA, phorbol myristate acetate.
Synergistic proviral reactivation in the QUECEL assay. (A) Representative flow plots of HIV Nef+ and pS175 CDK9+ Th17 cells treated with IL-15 (10 ng/ml) alone or in combination with SAHA (500 nM) and TNF-α (10 ng/ml). (B) Correlation between the QUECEL (Th17) model (top, n = 6) and RNA induction in patient cells (EDITS assay, bottom, n = 10). (C) HIV reactivation in Th17 cells treated with IL-15 in combination with other LRAs (n = 3). (D) Heat map comparison of QUECEL and QVOA reactivation to latently infected Jurkat cell clones from the Karn laboratory (2D10, E4, and G5) and the Verdin laboratory (JLat 5A8, JLat 8.4, JLat 6.3, and JLat 11.1). Values were normalized compared to highest stimuli within each model and expressed as percentage of maximal stimulation. Download FIG S8, TIF file, 2.0 MB (2.1MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The QUECEL results were compared to the activation profiles of well-suppressed HIV-infected patient CD4 memory cells (n = 11) using the EDITS assay (55) (Fig. 5B). The hierarchy of responses was comparable for a wide range of different activators, and the data were highly correlated. However, basal activation levels are higher in the QUECEL system than in the patient cells, and the patient cells are generally more restricted in their responses to LRAs. Notably, the synergy between IL-15 and SAHA is maintained, and latency reversal under these conditions is comparable to that seen after TCR activation.
HIV reactivation in the QUECEL model was also compared to that of a wide variety of previously tested latency models using the same reactivation conditions, as described by Spina et al. (26) (Fig. 5C). It is notable from the heat map that the QUECEL data clustered most closely with the quantitative viral outgrowth assay (QVOA) data (Fig. 5C) and showed the broadest range of responses to the various activators, including hexamethylene bisacetamide (HMBA) and SAHA. As expected, there is a poor correlation between the responses of Jurkat cells (E4 [wild-type Tat], 2D10 [H13L Tat], and G5 [H13L Tat and Nef]) and the QUECEL model (Fig. S8D) due to the dependence of Jurkat cells on NF-κB for HIV transcription (17) and their comparatively poor responses to TCR stimulation (22).
HIV reactivation in primary cells is dependent primarily upon NFAT.
The small-molecule inhibitors cyclosporine and the IKK inhibitor IV, which block activation of NFAT and IKK, respectively, were used to assess the relative contributions of NFAT and NF-κB to proviral reactivation in the QUECEL model (Fig. S9). The cells were pretreated with these compounds for 2 h and then subsequently stimulated with TCR, TNF-α, or SAHA. An Amnis ImageStream analysis of the nuclear localization of NF-κB and NFATc1 after stimulation with LRAs confirmed that there was a significant increase in both NF-κB (50%) and NFAT (68%) after TCR stimulation but no significant activation of these factors by SAHA (Fig. S9A). Inhibition of NFAT induction by cyclosporine resulted in an 8-fold decrease in Nef and pS175 CDK9 double-positive cells after TCR stimulation and a 3-fold decrease after SAHA stimulation (Fig. S9B and C). However, there was no inhibition of the TNF-α-treated cells. In contrast, blocking of NF-κB had a marginal effect on TCR and SAHA stimulation but a 6-fold effect on TNF-α stimulation. TNF-α resulted in a significant increase in NF-κβ recruitment to the nuclei, even though there was limited HIV Nef expression under these conditions. Thus, in sharp contrast to the Jurkat cell system, there is relatively low contribution of NF-κB to proviral reactivation compared to that of NFAT (30, 56, 57).
NFAT is the dominant transcription initiation factor regulating HIV latency in the QUECEL model. (A) Amnis ImageStream analysis of NFκB-p65 and NFATC1 localization in quiescent Th17 cells treated with LRAs. (B) HIV reactivation in quiescence stimulated through the T-cell receptor in the presence of cyclosporin (CsA) (1 µM) or IKK (1 µM). (Top) Resting cells. (Bottom) TCR-treated cells (24 h). (C) HIV reactivation in Th17 cells treated with CsA and IKK prior to activation with TCR, SAHA, and TNF-α (n = 3). Download FIG S9, TIF file, 3.5 MB (3.6MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
DISCUSSION
Entry into quiescence forces HIV into latency.
The most likely explanation for the generation of latently infected cells in patients is that HIV-1 initiates infection in activated CD4+ T cells, which are highly permissive for infection and support active viral gene transcription, but the virus then becomes silenced as host cells transition to a quiescent (resting) state (13, 14). In the QUECEL model, we recapitulate the transition from effector cells to memory cells under highly defined conditions.
One key innovation of the QUECEL model is the use of effector cells polarized into each of the major helper subsets. Helper cells with a Th17 (58) or Th2 (59) phenotype specialize in antifungal, antihelminthic, and antitumor immune defense, while Th1-polarized cells are predominantly directed against viral pathogens (51). The Lichterfeld group (52) has recently demonstrated that clonal proliferation of replication-competent virus can be observed in each of these polarization subsets but especially in cells with a Th1 or Th17 phenotype. The Th17 polarization phenotype, in which we have successfully modeled HIV latency for the first time, represents the most abundant effector T cell population in the lamina propria of the gastrointestinal tract (60, 61) and a target for HIV infection (62).
We have rigorously characterized each of these polarization states by flow cytometry assays of transcription factor and cytokine expression and comprehensive RNA-Seq analyses. Although each phenotype is distinct, comparison of the RNA-Seq data sets obtained from the QUECEL and two related primary cell models of HIV latency demonstrate clearly that the dominant driver of HIV latency is entry of cells into quiescence (63–66). However, in contrast to White et al. (66), we did not see enrichment for the p53 pathway in the data set. Instead, the dominant factors controlling entry into quiescence appear to be associated with c-myc-dependent pathways. Nonetheless, there are subtle differences in the utilization of signaling pathways between the different polarized cell types, and this may influence the responses of HIV to exogenous stimulation.
Previous investigators have relied on cellular exhaustion and/or relaxation for cells to undergo the transition to a quiescent state (13, 29, 30, 33, 35, 48, 67). In our initial primary cell model for HIV latency, cells were cocultured with H80 cells to permit them to gradually enter quiescence (29, 48, 63). However, this process took many weeks and was variable. In an effort to develop more defined conditions to force cells into quiescence, we analyzed cytokine production by H80 cells and found that they overproduced IL-10 and IL-8. Although these cytokines inefficiently promoted entry of cells into quiescence, they were synergistic with TGF-β, and this combination allowed us to force cells into quiescence. To confirm that the cells are fully quiescent, we routinely performed cell cycle analysis experiments, which show absence of cell cycle-dependent cyclins D3 and B1 and severe restrictions on the expression of CD69, CD25, and P-TEFb.
The main function of IL-10 is to induce an anti-inflammatory response. IL-10 is elevated during HIV, hepatitis C virus, and hepatitis B virus infections (68). The IL-10/IL-10R pathway is a key regulator of viral persistence, and blockade of IL-10R by anti-IL-10 monoclonal antibodies clears the chronic lymphocytic choriomeningitis virus (LCMV) infection in a mouse model (69). TGF-β is another anti-inflammatory/profibrotic cytokine which remains persistently elevated in both untreated and virally suppressed HIV-infected persons (70). The antiproliferative effects of TGF-β on TCR-activated cells is well documented (71, 72). TGF-β also inhibits IL-7-induced proliferation in memory, but not in naïve, human CD4+ T cells by interfering with c-myc induction (73, 74). Notably, in our model, c-myc pathways are highly repressed in the quiescent cells. IL-8, a CXC chemokine, which is also raised in the peripheral circulation of HIV-1-infected individuals (75), also interferes with HIV replication and suppresses HIV induction in the U1 cell model of HIV latency (76, 77). The fact that all these cytokines are elevated in HIV-infected individuals suggests that they play a role in maintaining and reseeding the HIV reservoir.
The QUECEL model accurately recapitulates the transcriptional behavior of patient cells.
There is an excellent correlation between a wide range of latency-reversing agents in the QUECEL model and RNA induction in highly active antiretroviral therapy-treated, well-suppressed HIV patient cells, as measured by the EDITS assay. This was true not only for the very potent activators, such as TCR stimulation, but also for less potent activators, such as SAHA, and synergistic agents, such as SAHA used in combination with IL-15.
In contrast, the responses of the Jurkat cell model system to latency-reversing agents generally shows a poor correlation to RNA induction in patient cells. One key molecular difference is that P-TEFb is constitutively expressed in Jurkat T cells, whereas induction of P-TEFb plays a crucial role in the reactivation of HIV latency in primary cells (39). Reactivation of cells in the QUECEL model is strictly correlated with reactivation of PTEF-b and phosphorylation of pSer175 on CDK9, which is a prerequisite for efficient P-TEFb binding to Tat (38, 39). In contrast to Jurkat cells, which are highly dependent upon NF-κB activation (27, 53), reactivation in the QUECEL model is primarily dependent upon NFAT activation and NF-κB is largely dispensable.
Because of highly variable protocols, there is also a poor correlation between different primary cell latency models and how closely they mimic what is occurring in the patient samples (26). For example, TNF-α and SAHA were ineffective in the Bosque and Planelles model, but both are partially effective in the QUECEL model and patient cells.
In summary, we have developed methods to establish HIV latency in primary effector cells forced to enter quiescence by exposure to a defined cocktail of cytokines. The QUECEL model gives highly reproducible results using multiple donors and many different experiments. Since homogeneous populations of latently infected cells can be recovered, there is an excellent signal-to-noise ratio and no need to normalize for variations in infection efficiency. We therefore believe the QUECEL model will become an invaluable tool to study the molecular mechanisms underlying HIV latency.
MATERIALS AND METHODS
Polarization of CD4 T cell subsets.
A detailed protocol is provided in the supplemental material. Briefly, naive CD4 T cells were isolated using a RoboSep CD4 Naïve T cell negative selection kit (Stemcell). Naive CD4 T cells (2 × 106) were resuspended in 10 ml RPMI medium and stimulated with 10 µg/ml concanavalin A (ConA) (EMD Milipore) in the presence of subset-specific cytokines (Table 1). Cells were cultured for 72 h at 37°C, followed by addition of 10 ml of fresh medium, additional 10 µg/ml ConA, polarization cocktail cytokines, and 120 IU/ml of IL-2. After 6 days, the cells were washed and placed into primary cell RPMI medium with growth cytokines of IL-23 (50 ng/ml) and IL-2 (60 IU/ml) for Th17 cells and IL-2 (60 IU/ml) for Th1, Th2, and Treg cells.
Infection of polarized CD4+ T cells.
The polarized CD4 T cells were infected at a multiplicity of infection (MOI) of 2.0 at 5 × 106 cells per ml using VSV glycoprotein-pseudotyped virus (22), in the presence of cell subset cytokines, in a 24-well plate. Cells were spinoculated at 2,000 × g for 1.5 h at room temperature and then placed in an incubator overnight. The cells were adjusted to 1 × 106 per ml in the presence of cell subset-appropriate growth cytokines. After 48 h, infection efficiency was determined by GFP expression.
Isolation of HIV-infected cells using magnetic isolation technology.
After transduction, HIV-infected cells were isolated using RoboSep mouse CD8a positive selection kit II (Stemcell). Cells (50 × 106 per ml) were preincubated with 50 µl/ml of antibody cocktail and 40 µl/ml of magnetic beads and diluted into 2.5 ml RoboSep buffer. The positive cells were recovered by magnetic bead separation, suspended in 1 ml of medium, and vortexed to release the cells and beads from the tube wall. Positively selected cells were placed in primary cell RPMI medium with cell type-specific growth cytokines at 1 × 106 in upright flasks and allowed to expand for a week.
Quiescence of polarized CD4 T cell subsets.
The number of cytokines was slowly reduced by incubating cells for 1 week without medium changes. Cell were resuspended in IL-2 (15 IU/ml), IL-23 (12.5 ng/ml), TGF-β (10 ng/ml), IL-10 (10 ng/ml), and IL-8 (50 ng/ml) for Th17 cells or IL-2 (15 IU/ml), TGF-β (10 ng/ml), and IL-8 (50 ng/ml) for Th1, Th2, and Treg cells. Entry of cells into quiescence was measured by the reduction in cyclin D3, pSer175 CDK9, and HIV Nef levels. This takes anywhere from one to two additional weeks depending on donor and experimental variation.
Stimulation procedure and inhibitor panel.
Quiescent cells (2 × 105) were stimulated for 24 to 48 h in a U-bottom 96-well plate in the presence of either 1 TCR Dynabead per cell (Life Technologies), 500 nM SAHA (Caymen Chemical), 10 ng/ml TNF-α, 10 ng/ml IL-15, or 5 ng/ml IL-7 (all from Peprotech). For the transcription inhibitor panel, 2 × 105 cells were pretreated for 2 h prior to stimulation with one of the following: 1 µM cyclosporine (Sigma-Aldrich), 1 µM Go6976 (Sigma-Aldrich), or 1 µM IKK inhibitor IV (Santa Cruz Biotechnologies).
Partially automated chromatin immunoprecipitation.
Quiescent Th17 cells were stimulated either for 30 min or overnight using Dynabeads in a 96-well U-bottom plate. ChIP was performed on 1 × 106 cells using our previously described partially automated ChIP protocol (23). Antibodies used for immunoprecipitations were anti-RNAP II (sc-899; Santa Cruz Biotechnologies), anti-RNAP II pSer5 (ab5131; Abcam), anti-RNAP II pSer2 (ab5095; Abcam), and anti-H3K27me3 (17-622; Millipore). DNA was amplified using HIV-specific primers (see Fig. S7 in the supplemental material), and each sample was barcoded for Ion Torrent sequencing with Ioncode barcodes. Sequences were separated by barcode, trimmed, and mapped to a pHR′-Nef-GFP/CD8a sequence.
EDITS analysis from HIV-infected individuals.
Well-suppressed cART-treated patient peripheral blood mononuclear cells (PBMCs), from a UCSF patient cohort, were tested using the EDITS assay as previously described (55).
Flow cytometry.
Samples were fixed in 4% formaldehyde (Electron Microscope Sciences) for 15 min and permeabilized with a Saponin-based buffer (Cytofix/Cytoperm; BD Biosciences) for 5 min. An antibody cocktail containing AF647-HIV EH1 Nef (Jim Hoxie laboratory), AF750-pSer175 CDK9 (generated by Covance for our laboratory), and tetramethyl rhodamine isothiocyanate-cyclin D3 (Santa Cruz Biotechnologies) was added to each sample for 20 min. Samples were washed twice and run on a BD LSR Fortessa instrument. Flow cytometry analysis was performed using Winlist 9.0.
RNA-Seq library preparation and analysis.
Total cellular RNA was extracted from Th1-, Th2-, Treg-, and Th17-polarized exponentially growing cells (day 6) prior to infection with the HIV construct and 72 h postinfection. Additional samples were taken for each polarized cell population after the full induction of quiescence and HIV latency (day 28) and 24 h after TCR agonist-mediated stimulation of quiescent cells to induce reactivation. After RNA samples from all time points were taken, library preparation was performed using the TruSeq RNA sample preparation and single-end cluster generation kits from Illumina, which include a poly(A)+ enrichment step. ERCC Exfold RNA spike-ins (Life Technologies) were added according to the manufacturer’s instructions. High-throughput sequencing was performed on an Illumina HiSeq2000 instrument.
Data availability.
The RNA-Seq data sets, comprising two replicate studies performed over a year apart, have been submitted to the SRA database (accession number SRP145508). All RNA-Seq data sets passed the quality control step, which was performed using FastQC (Babraham Bioinformatics).
Detailed protocol. Download Text S1, PDF file, 0.3 MB (290.2KB, pdf) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Footnotes
Citation Dobrowolski C, Valadkhan S, Graham AC, Shukla M, Ciuffi A, Telenti A, Karn J. 2019. Entry of polarized effector cells into quiescence forces HIV latency. mBio 10:e00337-19. https://doi.org/10.1128/mBio.00337-19.
Contributor Information
Stephen P. Goff, Columbia University.
Melanie Ott, Gladstone Institutes University of California San Francisco.
Andrew Henderson, Pennsylvania State University.
Celsa Spina, UCSD School of Medicine.
REFERENCES
- 1.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 2.Yukl SA, Gianella S, Sinclair E, Epling L, Li Q, Duan L, Choi AL, Girling V, Ho T, Li P, Fujimoto K, Lampiris H, Hare CB, Pandori M, Haase AT, Gunthard HF, Fischer M, Shergill AK, McQuaid K, Havlir DV, Wong JK. 2010. Differences in HIV burden and immune activation within the gut of HIV-positive patients receiving suppressive antiretroviral therapy. J Infect Dis 202:1553–1561. doi: 10.1086/656722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, Spindler J, Ferris AL, Mellors JW, Kearney MF, Coffin JM, Hughes SH. 2014. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345:179–183. doi: 10.1126/science.1254194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wagner TA, McLaughlin S, Garg K, Cheung CY, Larsen BB, Styrchak S, Huang HC, Edlefsen PT, Mullins JI, Frenkel LM. 2014. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345:570–573. doi: 10.1126/science.1256304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel M-R, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy J-P, Haddad EK, Sékaly R-P. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Z, Gurule EE, Brennan TP, Gerold JM, Kwon KJ, Hosmane NN, Kumar MR, Beg SA, Capoferri AA, Ray SC, Ho YC, Hill AL, Siliciano JD, Siliciano RF. 2018. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc Natl Acad Sci U S A 115:e2575–e2584. doi: 10.1073/pnas.1720665115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siliciano JD, Kajdas J, Finzi D, Quinn TC, Chadwick K, Margolick JB, Kovacs C, Gange SJ, Siliciano RF. 2003. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat Med 9:727–728. doi: 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- 8.Crooks AM, Bateson R, Cope AB, Dahl NP, Griggs MK, Kuruc JD, Gay CL, Eron JJ, Margolis DM, Bosch RJ, Archin NM. 2015. Precise quantitation of the latent HIV-1 reservoir: implications for eradication strategies. J Infect Dis 212:1361–1365. doi: 10.1093/infdis/jiv218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chun TW, Davey RT Jr, Engel D, Lane HC, Fauci AS. 1999. Re-emergence of HIV after stopping therapy. Nature 401:874–875. doi: 10.1038/44755. [DOI] [PubMed] [Google Scholar]
- 10.Colby DJ, Trautmann L, Pinyakorn S, Leyre L, Pagliuzza A, Kroon E, Rolland M, Takata H, Buranapraditkun S, Intasan J, Chomchey N, Muir R, Haddad EK, Tovanabutra S, Ubolyam S, Bolton DL, Fullmer BA, Gorelick RJ, Fox L, Crowell TA, Trichavaroj R, O’Connell R, Chomont N, Kim JH, Michael NL, Robb ML, Phanuphak N, Ananworanich J. 2018. Rapid HIV RNA rebound after antiretroviral treatment interruption in persons durably suppressed in Fiebig I acute HIV infection. Nat Med 24:923–926. doi: 10.1038/s41591-018-0026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deeks SG, Autran B, Berkhout B, Benkirane M, Cairns S, Chomont N, Chun TW, Churchill M, Mascio MD, Katlama C, Lafeuillade A, Landay A, Lederman M, Lewin SR, Maldarelli F, Margolis D, Markowitz M, Martinez-Picado J, Mullins JI, Mellors J, Moreno S, O'Doherty U, Palmer S, Penicaud MC, Peterlin M, Poli G, Routy JP, Rouzioux C, Silvestri G, Stevenson M, Telenti A, Lint CV, Verdin E, Woolfrey A, Zaia J, Barre-Sinoussi F. 2012. Towards an HIV cure: a global scientific strategy. Nat Rev Immunol 12:607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siliciano RF, Greene WC. 2011. HIV latency. Cold Spring Harb Perspect Med 1:a007096. doi: 10.1101/cshperspect.a007096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shan L, Deng K, Gao H, Xing S, Capoferri AA, Durand CM, Rabi SA, Laird GM, Kim M, Hosmane NN, Yang HC, Zhang H, Margolick JB, Li L, Cai W, Ke R, Flavell RA, Siliciano JD, Siliciano RF. 2017. Transcriptional reprogramming during effector-to-memory transition renders CD4(+) T cells permissive for latent HIV-1 infection. Immunity 47:766–775.e3. doi: 10.1016/j.immuni.2017.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cameron PU, Saleh S, Sallmann G, Solomon A, Wightman F, Evans VA, Boucher G, Haddad EK, Sekaly RP, Harman AN, Anderson JL, Jones KL, Mak J, Cunningham AL, Jaworowski A, Lewin SR. 2010. Establishment of HIV-1 latency in resting CD4+ T cells depends on chemokine-induced changes in the actin cytoskeleton. Proc Natl Acad Sci U S A 107:16934–16939. doi: 10.1073/pnas.1002894107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saleh S, Solomon A, Wightman F, Xhilaga M, Cameron PU, Lewin SR. 2007. CCR7 ligands CCL19 and CCL21 increase permissiveness of resting memory CD4+ T cells to HIV-1 infection: a novel model of HIV-1 latency. Blood 110:4161–4164. doi: 10.1182/blood-2007-06-097907. [DOI] [PubMed] [Google Scholar]
- 16.Rezaei SD, Lu HK, Chang JJ, Rhodes A, Lewin SR, Cameron PU. 2018. The pathway to establishing HIV latency is critical to how latency is maintained and reversed. J Virol 92:e02225-17. doi: 10.1128/JVI.02225-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mbonye U, Karn J. 2017. The molecular basis for human immunodeficiency virus latency. Annu Rev Virol 4:261–285. doi: 10.1146/annurev-virology-101416-041646. [DOI] [PubMed] [Google Scholar]
- 18.Liou LY, Herrmann CH, Rice AP. 2002. Transient induction of cyclin T1 during human macrophage differentiation regulates human immunodeficiency virus type 1 Tat transactivation function. J Virol 76:10579–10587. doi: 10.1128/JVI.76.21.10579-10587.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghose R, Liou LY, Herrmann CH, Rice AP. 2001. Induction of TAK (cyclin T1/P-TEFb) in purified resting CD4(+) T lymphocytes by combination of cytokines. J Virol 75:11336–11343. doi: 10.1128/JVI.75.23.11336-11343.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garriga J, Peng J, Parreno M, Price DH, Henderson EE, Grana X. 1998. Upregulation of cyclin T1/CDK9 complexes during T cell activation. Oncogene 17:3093–3102. doi: 10.1038/sj.onc.1202548. [DOI] [PubMed] [Google Scholar]
- 21.Natarajan M, Schiralli-Lester GM, Lee C, Missra A, Wasserman GA, Steffen M, Gilmour DS, Henderson AJ. 2013. NELF coordinates RNA polymerase II pausing, premature termination and chromatin remodeling to regulate HIV transcription. J Biol Chem 288:25995–26003. doi: 10.1074/jbc.M113.496489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pearson R, Kim YK, Hokello J, Lassen K, Friedman J, Tyagi M, Karn J. 2008. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J Virol 82:12291–12303. doi: 10.1128/JVI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jadlowsky JK, Wong JY, Graham AC, Dobrowolski C, Devor RL, Adams MD, Fujinaga K, Karn J. 2014. The negative elongation factor (NELF) is required for the maintenance of proviral latency but does not induce promoter proximal pausing of RNAP II on the HIV LTR. Mol Cell Biol 34:1911–1928. doi: 10.1128/MCB.01013-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jordan A, Bisgrove D, Verdin E. 2003. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22:1868–1877. doi: 10.1093/emboj/cdg188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. 2006. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J 25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, Mau M, Ruelas D, Saleh S, Shirakawa K, Siliciano RF, Singhania A, Soto PC, Terry VH, Verdin E, Woelk C, Wooden S, Xing S, Planelles V. 2013. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathog 9:e1003834. doi: 10.1371/journal.ppat.1003834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim YK, Mbonye U, Hokello J, Karn J. 2011. T-cell receptor signaling enhances transcriptional elongation from latent HIV proviruses by activating P-TEFb through an ERK-dependent pathway. J Mol Biol 410:896–916. doi: 10.1016/j.jmb.2011.03.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nguyen K, Das B, Dobrowolski C, Karn J. 2017. Multiple histone lysine methyltransferases are required for the establishment and maintenance of HIV-1 latency. mBio 8:e00133-17. doi: 10.1128/mBio.00133-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tyagi M, Pearson RJ, Karn J. 2010. Establishment of HIV latency in primary CD4+ cells is due to epigenetic transcriptional silencing and P-TEFb restriction. J Virol 84:6425–6437. doi: 10.1128/JVI.01519-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bosque A, Planelles V. 2009. Induction of HIV-1 latency and reactivation in primary memory CD4+ T cells. Blood 113:58–65. doi: 10.1182/blood-2008-07-168393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hakre S, Chavez L, Shirakawa K, Verdin E. 2012. HIV latency: experimental systems and molecular models. FEMS Microbiol Rev 36:706–716. doi: 10.1111/j.1574-6976.2012.00335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lassen KG, Hebbeler AM, Bhattacharyya D, Lobritz MA, Greene WC. 2012. A flexible model of HIV-1 latency permitting evaluation of many primary CD4 T-cell reservoirs. PLoS One 7:e30176. doi: 10.1371/journal.pone.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xing S, Bullen CK, Shroff NS, Shan L, Yang HC, Manucci JL, Bhat S, Zhang H, Margolick JB, Quinn TC, Margolis DM, Siliciano JD, Siliciano RF. 2011. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J Virol 85:6060–6064. doi: 10.1128/JVI.02033-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martins LJ, Bonczkowski P, Spivak AM, De Spiegelaere W, Novis CL, DePaula-Silva AB, Malatinkova E, Trypsteen W, Bosque A, Vanderkerckhove L, Planelles V. 2016. Modeling HIV-1 latency in primary T cells using a replication-competent virus. AIDS Res Hum Retrovir 32:187–193. doi: 10.1089/aid.2015.0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bosque A, Famiglietti M, Weyrich AS, Goulston C, Planelles V. 2011. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog 7:e1002288. doi: 10.1371/journal.ppat.1002288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman J, Cho WK, Chu CK, Keedy KS, Archin NM, Margolis DM, Karn J. 2011. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2 (EZH2). J Virol 85:9078–9089. doi: 10.1128/JVI.00836-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Card CM, Ball TB, Fowke KR. 2013. Immune quiescence: a model of protection against HIV infection. Retrovirology 10:141. doi: 10.1186/1742-4690-10-141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mbonye UR, Gokulrangan G, Datt M, Dobrowolski C, Cooper M, Chance MR, Karn J. 2013. Phosphorylation of CDK9 at Ser175 enhances HIV transcription and is a marker of activated P-TEFb in CD4(+) T lymphocytes. PLoS Pathog 9:e1003338. doi: 10.1371/journal.ppat.1003338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mbonye U, Wang B, Gokulrangan G, Shi W, Yang S, Karn J. 2018. Cyclin-dependent kinase 7 (CDK7)-mediated phosphorylation of the CDK9 activation loop promotes P-TEFb assembly with Tat and proviral HIV reactivation. J Biol Chem 293:10009–10025. doi: 10.1074/jbc.RA117.001347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rumble J, Segal BM. 2014. In vitro polarization of T-helper cells. Methods Mol Biol 1193:105–113. doi: 10.1007/978-1-4939-1212-4_11. [DOI] [PubMed] [Google Scholar]
- 41.de Wit J, Souwer Y, van Beelen AJ, de Groot R, Muller FJ, Klaasse Bos H, Jorritsma T, Kapsenberg ML, de Jong EC, van Ham SM. 2011. CD5 costimulation induces stable Th17 development by promoting IL-23R expression and sustained STAT3 activation. Blood 118:6107–6114. doi: 10.1182/blood-2011-05-352682. [DOI] [PubMed] [Google Scholar]
- 42.Valmori D, Raffin C, Raimbaud I, Ayyoub M. 2010. Human RORgammat+ TH17 cells preferentially differentiate from naive FOXP3+Treg in the presence of lineage-specific polarizing factors. Proc Natl Acad Sci U S A 107:19402–19407. doi: 10.1073/pnas.1008247107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reiner SL. 2007. Development in motion: helper T cells at work. Cell 129:33–36. doi: 10.1016/j.cell.2007.03.019. [DOI] [PubMed] [Google Scholar]
- 44.Chang JT, Wherry EJ, Goldrath AW. 2014. Molecular regulation of effector and memory T cell differentiation. Nat Immunol 15:1104–1115. doi: 10.1038/ni.3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. 2006. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- 46.O'Shea JJ, Steward-Tharp SM, Laurence A, Watford WT, Wei L, Adamson AS, Fan S. 2009. Signal transduction and Th17 cell differentiation. Microbes Infect 11:599–611. doi: 10.1016/j.micinf.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darzynkiewicz Z, Gong J, Juan G, Ardelt B, Traganos F. 1996. Cytometry of cyclin proteins. Cytometry 25:1–13. doi:. [DOI] [PubMed] [Google Scholar]
- 48.Sahu GK, Lee K, Ji J, Braciale V, Baron S, Cloyd MW. 2006. A novel in vitro system to generate and study latently HIV-infected long-lived normal CD4+ T-lymphocytes. Virology 355:127–137. doi: 10.1016/j.virol.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 49.Tu E, Chia CPZ, Chen W, Zhang D, Park SA, Jin W, Wang D, Alegre ML, Zhang YE, Sun L, Chen W. 2018. T cell receptor-regulated TGF-beta type I receptor expression determines T cell quiescence and activation. Immunity 48:745–759. doi: 10.1016/j.immuni.2018.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruegemer JJ, Ho SN, Augustine JA, Schlager JW, Bell MP, McKean DJ, Abraham RT. 1990. Regulatory effects of transforming growth factor-beta on IL-2- and IL-4-dependent T cell-cycle progression. J Immunol 144:1767–1776. [PubMed] [Google Scholar]
- 51.Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, Kaech SM. 2011. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity 35:633–646. doi: 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee GQ, Orlova-Fink N, Einkauf K, Chowdhury FZ, Sun X, Harrington S, Kuo HH, Hua S, Chen HR, Ouyang Z, Reddy K, Dong K, Ndung'u T, Walker BD, Rosenberg ES, Yu XG, Lichterfeld M. 2017. Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J Clin Investig 127:2689–2696. doi: 10.1172/JCI93289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Williams SA, Kwon H, Chen LF, Greene WC. 2007. Sustained induction of NF-kappaB is required for efficient expression of latent human immunodeficiency virus type 1. J Virol 81:6043–6056. doi: 10.1128/JVI.02074-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim YK, Bourgeois CF, Pearson R, Tyagi M, West MJ, Wong J, Wu SY, Chiang CM, Karn J. 2006. Recruitment of TFIIH to the HIV LTR is a rate-limiting step in the emergence of HIV from latency. EMBO J 25:3596–3604. doi: 10.1038/sj.emboj.7601248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Das B, Dobrowolski C, Luttge B, Valadkhan S, Chomont N, Johnston R, Bacchetti P, Hoh R, Gandhi M, Deeks SG, Scully E, Karn J. 2018. Estrogen receptor-1 is a key regulator of HIV-1 latency that imparts gender-specific restrictions on the latent reservoir. Proc Natl Acad Sci U S A 115:e7795–e7804. doi: 10.1073/pnas.1803468115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cron RQ, Bartz SR, Clausell A, Bort SJ, Klebanoff SJ, Lewis DB. 2000. NFAT1 enhances HIV-1 gene expression in primary human CD4 T cells. Clin Immunol 94:179–191. doi: 10.1006/clim.1999.4831. [DOI] [PubMed] [Google Scholar]
- 57.Bosque A, Planelles V. 2011. Studies of HIV-1 latency in an ex vivo model that uses primary central memory T cells. Methods 53:54–61. doi: 10.1016/j.ymeth.2010.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Muranski P, Restifo NP. 2013. Essentials of Th17 cell commitment and plasticity. Blood 121:2402–2414. doi: 10.1182/blood-2012-09-378653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bamias G, Cominelli F. 2015. Role of type 2 immunity in intestinal inflammation. Curr Opin Gastroenterol 31:471–476. doi: 10.1097/MOG.0000000000000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. 2008. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim CJ, McKinnon LR, Kovacs C, Kandel G, Huibner S, Chege D, Shahabi K, Benko E, Loutfy M, Ostrowski M, Kaul R. 2013. Mucosal Th17 cell function is altered during HIV infection and is an independent predictor of systemic immune activation. J Immunol 191:2164–2173. doi: 10.4049/jimmunol.1300829. [DOI] [PubMed] [Google Scholar]
- 62.El Hed A, Khaitan A, Kozhaya L, Manel N, Daskalakis D, Borkowsky W, Valentine F, Littman DR, Unutmaz D. 2010. Susceptibility of human Th17 cells to human immunodeficiency virus and their perturbation during infection. J Infect Dis 201:843–854. doi: 10.1086/651021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mohammadi P, di Iulio J, Munoz M, Martinez R, Bartha I, Cavassini M, Thorball C, Fellay J, Beerenwinkel N, Ciuffi A, Telenti A. 2014. Dynamics of HIV latency and reactivation in a primary CD4+ T cell model. PLoS Pathog 10:e1004156. doi: 10.1371/journal.ppat.1004156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Golumbeanu M, Cristinelli S, Rato S, Munoz M, Cavassini M, Beerenwinkel N, Ciuffi A. 2018. Single-cell RNA-Seq reveals transcriptional heterogeneity in latent and reactivated HIV-infected cells. Cell Rep 23:942–950. doi: 10.1016/j.celrep.2018.03.102. [DOI] [PubMed] [Google Scholar]
- 65.Rato S, Rausell A, Munoz M, Telenti A, Ciuffi A. 2017. Single-cell analysis identifies cellular markers of the HIV permissive cell. PLoS Pathog 13:e1006678. doi: 10.1371/journal.ppat.1006678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.White CH, Moesker B, Beliakova-Bethell N, Martins LJ, Spina CA, Margolis DM, Richman DD, Planelles V, Bosque A, Woelk CH. 2016. Transcriptomic analysis implicates the p53 signaling pathway in the establishment of HIV-1 latency in central memory CD4 T cells in an in vitro model. PLoS Pathog 12:e1006026. doi: 10.1371/journal.ppat.1006026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Marini A, Harper JM, Romerio F. 2008. An in vitro system to model the establishment and reactivation of HIV-1 latency. J Immunol 181:7713–7720. doi: 10.4049/jimmunol.181.11.7713. [DOI] [PubMed] [Google Scholar]
- 68.Wilson EB, Brooks DG. 2011. The role of IL-10 in regulating immunity to persistent viral infections. Curr Top Microbiol Immunol 350:39–65. doi: 10.1007/82_2010_96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brooks DG, Trifilo MJ, Edelmann KH, Teyton L, McGavern DB, Oldstone MB. 2006. Interleukin-10 determines viral clearance or persistence in vivo. Nat Med 12:1301–1309. doi: 10.1038/nm1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Theron AJ, Anderson R, Rossouw TM, Steel HC. 2017. The role of transforming growth factor beta-1 in the progression of HIV/AIDS and development of non-AIDS-defining fibrotic disorders. Front Immunol 8:1461. doi: 10.3389/fimmu.2017.01461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Delisle JS, Giroux M, Boucher G, Landry JR, Hardy MP, Lemieux S, Jones RG, Wilhelm BT, Perreault C. 2013. The TGF-beta-Smad3 pathway inhibits CD28-dependent cell growth and proliferation of CD4 T cells. Genes Immun 14:115–126. doi: 10.1038/gene.2012.63. [DOI] [PubMed] [Google Scholar]
- 72.Yoshimura A, Muto G. 2011. TGF-beta function in immune suppression. Curr Top Microbiol Immunol 350:127–147. doi: 10.1007/82_2010_87. [DOI] [PubMed] [Google Scholar]
- 73.Nguyen TP, Sieg SF. 2017. TGF-beta inhibits IL-7-induced proliferation in memory but not naive human CD4(+) T cells. J Leukoc Biol 102:499–506. doi: 10.1189/jlb.3A1216-520RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Frederick JP, Liberati NT, Waddell DS, Shi Y, Wang XF. 2004. Transforming growth factor beta-mediated transcriptional repression of c-myc is dependent on direct binding of Smad3 to a novel repressive Smad binding element. Mol Cell Biol 24:2546–2559. doi: 10.1128/MCB.24.6.2546-2559.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Matsumoto T, Miike T, Nelson RP, Trudeau WL, Lockey RF, Yodoi J. 1993. Elevated serum levels of IL-8 in patients with HIV infection. Clin Exp Immunol 93:149–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mackewicz CE, Ortega H, Levy JA. 1994. Effect of cytokines on HIV replication in CD4+ lymphocytes: lack of identity with the CD8+ cell antiviral factor. Cell Immunol 153:329–343. doi: 10.1006/cimm.1994.1032. [DOI] [PubMed] [Google Scholar]
- 77.Tiemessen CT, Kilroe B, Martin DJ. 2000. Interleukin-8 fails to induce human immunodeficiency virus-1 expression in chronically infected promonocytic U1 cells but differentially modulates induction by proinflammatory cytokines. Immunology 101:140–146. doi: 10.1046/j.1365-2567.2000.00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
CD8a/GFP persists in quiescent QUECEL cells. (A) Flow cytometry analysis of d2EGFP levels in Th17 cells infected with HIV expressing either the membrane-bound CD8a/GFP fusion protein or cytoplasmic d2EGFP. Cells were analyzed before and after treatment with 10 μg/ml cycloheximide (24 h). (B) Half-life of CD8a/GFP fusion protein and Nef during the establishment of quiescence. Solid lines, positive cells (%); dashed lines, mean fluorsescent intensity. (C) Immunofluorescence and RNA FISH analysis of Th17 cells reactivated through the TCR. Yellow, RNA detected by Stellaris RNA FISH (Biosearch Technologies) using probes spanning the U3 and U5 regions of the provirus. Green, GFP. Blue, DAPI. (D) Flow cytometry analysis of d2EGFP in Th17 cells latently infected with an HIV lentiviral vector expressing a Thy1.2-P2A-d2EGFP reporter gene (top), which permits decay of the cytoplasmic d2EGFP signal during quiescence. (Bottom) d2EGFP induction after stimulation by the TCR, 500 nM SAHA, and 100 ng/ml TNF-α. Download FIG S1, TIF file, 3.3 MB (3.3MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
QUECEL T cell subsets express cell specific mRNAs. (A) Heat maps showing relative qPCR expression levels of 10 T cell subset-specific mRNAs (top) and the corresponding RNA-Seq levels (bottom). (B) Correlation plot of polarized Th17s compared to peripheral Th17s and fresh total CD4 memory T cells using the RT2 Profiler PCR array human Th17 response array (330231; Qiagen) (left) or RT2 Profiler PCR array human T helper cell differentiation array (330231; Qiagen) (right). Download FIG S2, TIF file, 1.8 MB (1.8MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Polarized QUECEL T cells express subset-specific cytokines and transcription factors. (A) Flow cytometry of subset-specific transcription factors in unstimulated memory T cells isolated from PBMCs. (B) Cytokine expression levels in all T cell subsets. (C) Transcription factor expression in the cells shown in panel B. Download FIG S3, TIF file, 3.3 MB (3.4MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The H13L Tat mutant does not affect reactivation of latent HIV. (A) Cyclin B1 and D3 levels in Th17 cells treated with multiple cytokines in the absence of TGF-β. (B) Reactivation of latently infected Th17 cells carrying either WT, H13L Tat, or the inactivated C22G Tat mutant in response to a panel of activators. (C) Reactivation of latently infected Jurkat clones carrying either H13L Tat (2D10, G5) or WT Tat (E4). Download FIG S4, TIF file, 1.8 MB (1.8MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Differential gene expression pattern following reactivation in QUECEL versus two other ex vivo primary cell models. (A) Table of pathways and gene sets enriched during transition from quiescence to reactivation in Th17 cells. A false discovery rate cutoff value of 0.1 was used to identify gene sets and pathways that were significantly enriched. (B) Genes induced following reactivation in QUECEL show significant overlap those upregulated in H80 and Planelles ex vivo models, with downregulated genes showing a smaller amount of overlap. (C) Pairwise scatterplots indicate a strong overall correlation of changes in gene expression pattern following reactivation between the three ex vivo primary cell models. In order to compare gene expression data obtained from similar populations of cells between the three datasets, all four polarized cells in the QUECEL method (Th1, Th2, Th17, and Treg) have been used in aggregate to generate the lists of QUECEL up- and downregulated genes in panels B and C. Download FIG S5, TIF file, 2.1 MB (2.2MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Heat maps of gene sets corresponding to the top enriched pathways after TCR stimulation of quiescent cells (24 h). Data indicate the strong induction of genes involved in metabolic, transcriptional, and translational activation of cells. The values graphed in the heatmaps correspond to the differential expression value (in log2 units) obtained by pairwise comparison of quiescent versus TCR-stimulated cells. Note that while the shown pathways were originally found to be enriched using only the Th17 polarized cells, the panels in this figure have been generated using the aggregate of all four polarized cells in the QUECEL method (Th1, Th2, Th17, and Treg), indicating the reproducibility and generality of these results across the effector T cell subsets. Download FIG S6, TIF file, 2.4 MB (2.5MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Chromatin immunoprecipitation (ChIP) analysis of the latent provirus in Th17 cells. (A) Location of primers used in the ChIP assays to specifically analyze the 5′ and 3′ HIV LTRs. (B) ChIP assays. Cells were stimulated through the TCR for 30 min or 16 h (n = 3). (Top) Total RNAPII; RNAP II (pSer5). (Bottom) RNAP II (pSer2) and methylated histone H3 (H3K27me3). GAPDH was used as a control. Download FIG S7, TIF file, 1.4 MB (1.4MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Synergistic proviral reactivation in the QUECEL assay. (A) Representative flow plots of HIV Nef+ and pS175 CDK9+ Th17 cells treated with IL-15 (10 ng/ml) alone or in combination with SAHA (500 nM) and TNF-α (10 ng/ml). (B) Correlation between the QUECEL (Th17) model (top, n = 6) and RNA induction in patient cells (EDITS assay, bottom, n = 10). (C) HIV reactivation in Th17 cells treated with IL-15 in combination with other LRAs (n = 3). (D) Heat map comparison of QUECEL and QVOA reactivation to latently infected Jurkat cell clones from the Karn laboratory (2D10, E4, and G5) and the Verdin laboratory (JLat 5A8, JLat 8.4, JLat 6.3, and JLat 11.1). Values were normalized compared to highest stimuli within each model and expressed as percentage of maximal stimulation. Download FIG S8, TIF file, 2.0 MB (2.1MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
NFAT is the dominant transcription initiation factor regulating HIV latency in the QUECEL model. (A) Amnis ImageStream analysis of NFκB-p65 and NFATC1 localization in quiescent Th17 cells treated with LRAs. (B) HIV reactivation in quiescence stimulated through the T-cell receptor in the presence of cyclosporin (CsA) (1 µM) or IKK (1 µM). (Top) Resting cells. (Bottom) TCR-treated cells (24 h). (C) HIV reactivation in Th17 cells treated with CsA and IKK prior to activation with TCR, SAHA, and TNF-α (n = 3). Download FIG S9, TIF file, 3.5 MB (3.6MB, tif) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Detailed protocol. Download Text S1, PDF file, 0.3 MB (290.2KB, pdf) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The RNA-Seq data sets, comprising two replicate studies performed over a year apart, have been submitted to the SRA database (accession number SRP145508). All RNA-Seq data sets passed the quality control step, which was performed using FastQC (Babraham Bioinformatics).
Detailed protocol. Download Text S1, PDF file, 0.3 MB (290.2KB, pdf) .
Copyright © 2019 Dobrowolski et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.