

Abstract
Aims
Elamipretide (ELAM), an aromatic–cationic tetrapeptide, interacts with cardiolipin and normalizes dysfunctional mitochondria of cardiomyocytes. This study examined the effects of ELAM on skeletal muscle mitochondria function in dogs with chronic heart failure (HF).
Methods and results
Studies were performed in skeletal muscle biopsy specimens obtained from normal dogs (n = 7) and dogs with chronic intracoronary microembolization‐induced HF (n = 14) treated with subcutaneous ELAM 0.5 mg/kg (HF + ELAM, n = 7) or vehicle (normal saline control, HF‐CON, n = 7). After 3 months of therapy, triceps skeletal muscle samples were obtained from all dogs, and the proportion of type 1 and type 2 fibres was assessed. Mitochondria isolated from myofibrils of the vastus lateralis skeletal muscle exposed in vitro to ELAM for 1 h were used to assess mitochondrial function. The proportion of skeletal muscle type 1 fibres was lower in HF‐CON dogs compared with normal dogs (23 ± 4 vs. 32 ± 5%, P < 0.05). Treatment with ELAM restored a near‐normal fibre‐type composition (31 ± 7%, P < 0.05 vs. HF‐CON). Skeletal muscle mitochondria showed significantly lower levels of adenosine diphosphate‐dependent mitochondrial respiration (100 ± 9 vs. 164 ± 15 natom O/min/mg protein, P < 0.05), mitochondrial membrane potential (0.17 ± 0.03 vs. 0.53 ± 0.03 red/green fluorescence ratio, P < 0.05), mitochondrial permeability transition pore (38 ± 3 vs. 62 ± 2 relative light units, P < 0.05), maximum rate of adenosine triphosphate synthesis (3284 ± 418 vs. 8835 ± 423 RLU/μg protein, P < 0.05), and cytochrome c oxidase activity (1390 ± 108 vs. 2459 ± 210 natom O/min/mg protein, P < 0.05) compared with normal dogs. Exposure of skeletal muscle myofibrillar mitochondria from HF dogs to ELAM showed a dose‐dependent improvement/normalization of all measures of mitochondrial function. In mitochondria from skeletal muscle of HF dogs exposed to 0.10 μM ELAM, adenosine diphosphate‐dependent mitochondrial respiration increased to 183 ± 18 natom O/min/mg protein, membrane potential increased to 0.30 ± 0.03 red/green fluorescence ratio, mitochondrial permeability transition pore increased to 54 ± 4 RLU, maximum rate of adenosine triphosphate synthesis increased to 4423 ± 414, and cytochrome c oxidase activity increased to 2033 ± 191 natom O/min/mg protein. Exposure of skeletal muscle myofibrillar mitochondria from normal dogs to ELAM had no effect on mitochondrial function parameters.
Conclusions
The results indicate that ELAM, previously shown to positively influence mitochondrial function of the failing heart, can also positively impact mitochondrial function of skeletal muscle and potentially help restore skeletal muscle function and improve exercise tolerance.
Keywords: Heart failure, Mitochondria, Skeletal muscle, Energy metabolism, Animal models, Exercise tolerance
Introduction
Functional mitochondria are necessary for optimal energy production and organ function, and their dysfunction can have a profound effect on many serious disease states that include chronic heart failure (HF) and skeletal muscle myopathies with associated fatigue and exercise intolerance.1, 2 In the healthy heart, optimal generation of adenosine triphosphate (ATP) by mitochondria sustains excitation–contraction coupling and supports normal cardiac function.3 Dysfunctional mitochondria in HF reduce the capacity for on‐demand cardiac ATP generation, leading to an energy‐deficit state and dysfunction of both cardiac and skeletal muscle.3 Dysfunctional mitochondria also lead to increased formation of reactive oxygen species (ROS)4, 5 that can potentially cause further injury to mitochondrial membrane structures and lead to further worsening of the bioenergetic state believed to be a primary factor in HF progression.
Exercise intolerance is a hallmark of chronic HF. A complete understanding of the possible haemodynamic, structural, and molecular abnormalities that lead to exercise intolerance in HF is lacking. The observation of a poor correlation between central haemodynamics and exercise intolerance in patients with HF shifted interest to peripheral factors that include skeletal muscle structure and metabolism and to other factors that include iron deficiency, muscle atrophy and loss, abnormal muscle function, decreased capillary density, and endothelial dysfunction.6, 7, 8, 9, 10, 11 Along these lines, exercise intolerance in HF has been attributed to skeletal muscle atrophy, a shift from slow‐twitch type 1 (oxidative) to fast‐twitch type 2 (glycolytic) muscle fibres, mitochondrial abnormalities, and increased expression of inducible nitric oxide synthase (iNOS) with the resulting increase in nitric oxide, causing a decrease in mitochondrial creatine kinase, a key enzyme necessary for the transfer of high‐energy phosphates from mitochondria to cytosol.12, 13, 14, 15, 16
Elamipretide (ELAM, also known as MTP‐131, SS‐31) is an aromatic–cationic tetrapeptide that readily crosses the plasma membrane and localizes to the inner membrane of mitochondria via its selective, transient binding to cardiolipin.17 Through interaction with cardiolipin, ELAM increases the electron flux through cytochrome c and reduces electron leak.18 Elamipretide has also been shown to improve mitochondrial respiration, reduces the production of reactive oxygen species, and increases the maximum rate of ATP synthesis in dysfunctional mitochondria while having no effect on normal healthy mitochondria.17, 19, 20, 21, 22 The present study was designed to evaluate the effects of ELAM on skeletal muscle fibre‐type composition and mitochondrial function in dogs with chronic HF.
Methods
All studies were performed in skeletal muscle biopsy specimens obtained from normal dogs (n = 7) and dogs with chronic intracoronary microembolization‐induced HF (n = 14). All dogs were fed the same diet throughout the study (Purina Pro Plan Sport, Nestle Purina PetCare Company, St. Louis, MO). The canine model of chronic HF used in this study was previously described in detail.23 In this study, 14 healthy purpose‐bred class A dealer mongrel male dogs, weighing between 20.8 and 25.7 kg, underwent serial intracoronary microembolizations, performed 1 to 2 weeks apart, to produce chronic HF. Embolizations were discontinued when left ventricular ejection fraction, determined angiographically, was ~30%. The study was approved by the Henry Ford Health System Institutional Animal Care and Use Committee and conformed to the National Institute of Health ‘Guide and Care for Use of Laboratory Animals’ (NIH Publication No. 85‐23). Six weeks after the last microembolizations, HF dogs were randomized to 3 months of therapy with subcutaneous injections of ELAM (0.5 mg/kg once daily, n = 7, HF + ELAM) or subcutaneous injections of saline as vehicle (once daily, n = 7). Vehicle‐treated HF dogs served as controls (HF‐CON). The haemodynamic, ventriculographic, and echocardiographic findings in these dogs, as well as the cardiac mitochondrial function measures, were previously reported.24
Measurements of skeletal muscle fibre‐type composition
At the end of 3 months of therapy, and after completing the final haemodynamic study (while the dog was under general anaesthesia), triceps skeletal muscle samples were obtained from all 14 HF dogs using an open biopsy procedure, as previously described.13 Triceps skeletal muscle samples from seven normal dogs were also obtained and used for comparisons. Skeletal muscle type 1 and type 2 fibres were differentiated histologically by myofibrillar adenosine triphosphatase staining.13 The proportion of type 1 and type 2 fibres and the average cross‐sectional area of each fibre type was assessed in five randomly selected skeletal muscle fields/dogs each containing ~100 fibres.
Measurements of skeletal muscle mitochondrial function
Skeletal muscle biopsies
Fresh skeletal muscle open biopsies (~6 g each) were obtained from the hind leg vastus lateralis muscle of six normal (NL) and six untreated HF anaesthetized dogs. Specimens were immersed in Solution A (consisting of 20 mM imidazole–HCl, 0.16 mM MES, 5 mM ATP, 7.5 mM phosphocreatine, 0.5 mM DTT, 20 mM taurine, 3 mM MgCl2, and 10 mM EGTA at pH = 7.4). Myofibre bundles were subsequently isolated as previously described.25 Briefly, tissue was cut into 100‐ to 200‐μm‐thin longitudinal sections and washed two times with Buffer A and subsequently washed once with Buffer B consisting of 10 mM EGTA, 3 mM MgCl2, 20 mM taurine, 0.5 mM DTT, 20 mM imidazole, 0.16 mM MES, and 10 mg/mL bovine serum albumin at pH = 7.4. Myofibre bundles were then divided into four equal portions and each resuspended in 20 mL of Solution B. One portion of each was incubated in 0 (vehicle–saline), 0.01, 0.10, and 1.0 μM concentration of ELAM, respectively, for 1 h at 37°C.
Isolation of mitochondria
After treatment, skeletal muscle fibres were washed and resuspended in 10 mL CP2 medium (100 mM KCl, 50 mM MOPS, 5 mM MgSO4·7H2O, 5 mM potassium phosphate, 5 mM ATP, and +200 mg bovine serum albumin, pH = 7.4) followed by homogenization by polytron at Setting 4 for 10 min and three strokes using a Potter Elvejhem homogenizer (Setting 60). The homogenate was then centrifuged at 584 g for 10 min. The pellet was discarded, and the supernatant was centrifuged at 3000 g for 10 min. The resulting pellet was washed once with 10 mL consisting of buffer 100 mM KCl, 5 mM MOPS, and 0.5 mM EGTA, pH = 7.4 and finally resuspended in a small volume (200 μL) of the same buffer.
Mitochondrial function
Isolated mitochondria were used to determine several measures of mitochondrial function as previously described.16 The measures included (i) ADP‐dependent mitochondrial respiration as previously described using a Strathklein respirometer; (ii) maximum rate of ATP synthesis (ATPsyn) using the bioluminescent ApoSENSOR assay kit (BioVision, Milpitas, CA)26; (iii) cytochrome c oxidase (COX‐IV) activity measured polarographically; (iv) mitochondrial membrane potential (Δψm) measured using the commercially available JC‐1 cationic fluorescent dye kit (Sigma, St. Louis, MO)26; and (v) mitochondrial permeability transition pore (mPTP) opening assessed using a MitoProbe Transition Pore assay kit (Molecular Probes, Eugene, OR, USA Cat# M34153).26
Western blotting
Skeletal muscle specimens were used to assess protein levels of iNOS and β‐actin (an internal control). Protein measurements of iNOS were normalized to β‐actin to adjust for loading conditions. Isolated mitochondria from skeletal muscle were also used to examine protein levels of indicators of cellular apoptosis, specifically protein levels of cytochrome c and levels of HtrA2, a proapoptotic mitochondrial serine protease involved in caspase‐dependent and caspase‐independent cell death. Levels of cytochrome c and HtrA2 were normalized to porin, a mitochondrial outer membrane protein that is unchanged in HF. Levels of all proteins were determined by western blotting and bands quantified in densitometric units.24 Primary antibodies for HtrA2 and cytochrome c were obtained from Santa Cruz, CA, antibody for porin from Abcam, Cambridge, MA, and antibodies for iNOS from LifeSpan Biosciences, Inc., Seattle, WA.
Statistical analysis
Within each group, comparisons of mitochondrial functional measures were made using repeated measures analysis of variance (ANOVA) with α set at 0.05. If significance was attained, pairwise comparisons between no treatment and each escalating ELAM treatment concentration were made using the Student–Newman–Keuls test with P < 0.05 considered significant. Comparisons of biochemical and histomorphometric measures between normal, HF‐CON, and HF + ELAM dogs were made using one‐way ANOVA with α set at 0.05. If significance was attained by ANOVA, pairwise comparisons were performed using the Student–Newman‐Keuls test with P < 0.05 considered significant. Comparisons of baseline levels (no ELAM exposure) between normal and HF dogs for all mitochondrial functional measures were made using a t‐statistic test for two means with P < 0.05 considered significant. The study was not blinded, and all data are reported as mean ± standard error of the mean.
Results
Histomorphometric findings
The proportion of skeletal muscle type 1 fibres was lower and type 2 fibres higher in HF‐CON dogs compared with normal dogs (Table 1 and Figure 1 ), leading to a significantly lower fibre‐type ratio. Treatment with ELAM restored a near‐normal fibre‐type composition. There were no differences in fibre cross‐sectional area among study groups (Table 1).
Table 1.
Skeletal muscle fibre‐type distribution and inducible nitric oxide synthase isoform level in normal, HF‐CON (n = 7), and HF + ELAM (n = 7) dogs
| Normal | HF‐CON | HF + ELAM | |
|---|---|---|---|
| Type 1 skeletal muscle fibres (%) | 32 ± 5 | 23 ± 4* | 31 ± 7** |
| Type 2 skeletal muscle fibres (%) | 68 ± 5 | 77 ± 4* | 69 ± 7** |
| Skeletal muscle type 1/type 2 ratio | 0.47 ± 0.04 | 0.30 ± 0.07* | 0.45 ± 0.13** |
| Average CSA type 1 fibres (μm2) | 2996 ± 176 | 2057 ± 415 | 3058 ± 354 |
| Average CSA type 2 fibres (μm2) | 3445 ± 240 | 3560 ± 378 | 3495 ± 258 |
| CSA type 1/CSA type 2 ratio | 0.88 ± 0.03 | 0.86 ± 0.05 | 0.87 ± 0.04 |
| β‐Actin (du) | 2.22 ± 0.11 | 2.36 ± 0.20 | 2.37 ± 0.10 |
| iNOS/β‐actin | 0.71 ± 0.08 | 2.01 ± 0.21* | 1.16 ± 0.08* , ** |
| Porin (du) | 0.62 ± 0.13 | 0.66 ± 0.05 | 0.64 ± 0.07 |
| Cytochrome c/porin | 2.17 ± 0.86 | 0.49 ± 0.06* | 1.20 ± 0.22* , ** |
| HtrA2/porin | 2.14 ± 0.49 | 0.34 ± 0.06* | 1.28 ± 0.15* , ** |
CSA, cross‐sectional area; HF‐CON, untreated heart failure control; HF + ELAM, elamipretide‐treated heart failure; iNOS, inducible nitric oxide synthase.
P < 0.05 vs. normal.
P < 0.05 vs. HF‐CON.
Figure 1.
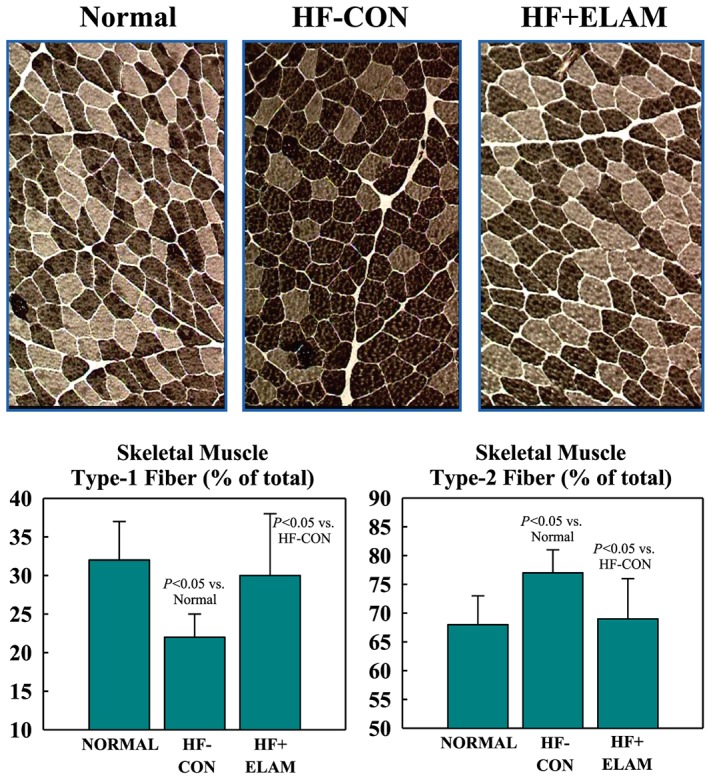
Top: ATPase‐stained histological panels (same magnification) depict skeletal muscle fibre‐type composition in triceps muscle. Type 1 fibres appear as lightly stained fibres (light brown) and type 2 fibres as dark‐stained fibres (dark brown). Left top panel: fibre‐type composition in a normal dog. Top middle panel: fibre‐type composition in an untreated heart failure control (HF‐CON) dog. Note that the number of type 1 fibres is reduced relative to normal dogs in the left panel. Top right panel: fibre‐type composition in an elamipretide‐treated heart failure (HF + ELAM) dog. Note that the number of type 1 fibres is increased relative to HF‐CON in middle panel. The bottom 2 graphs show the numerical distribution (mean ± standard error of the mean) of skeletal muscle type 1 and type 2 fibres in normal dogs and in both study groups.
Mitochondrial function measures and complex IV activity
At baseline (no exposure to ELAM), skeletal muscle mitochondria from dogs with HF showed significantly lower levels of ADP‐dependent mitochondrial respiration, Δψm, mPTP, maximum rate of ATPsyn, ATP/ADP ratio, and COX‐IV activity (Figures 2 and 3 ). Exposure of skeletal muscle myofibrillar mitochondria from normal dogs to ELAM for 1 h at concentration of 0.01 to 1.0 μM had no effect on any of the mitochondrial function parameters measured in this study (Figures 2 and 3 ). In contrast, exposure of skeletal muscle myofibrillar mitochondria from HF dogs to ELAM showed a dose‐dependent improvement/normalization of all measures of mitochondrial function. For all measures, statistical significance was attained at ELAM concentration of 0.1 and 1.0 μM.
Figure 2.
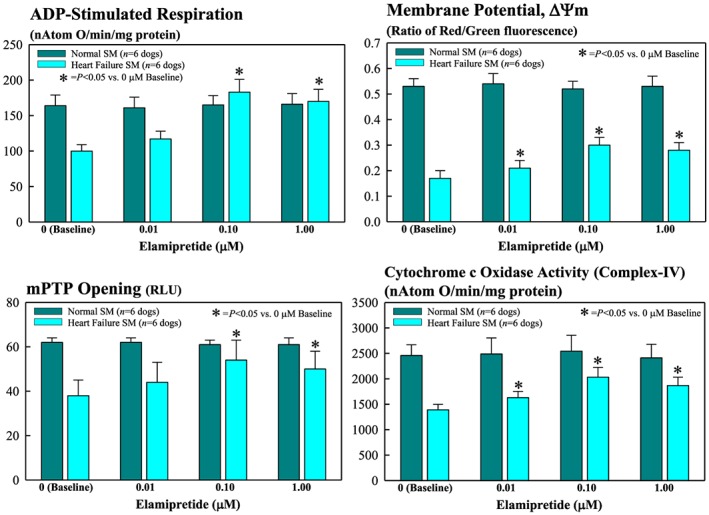
Bar graphs (mean ± standard error of the mean) depicting changes in mitochondrial functional measures of skeletal muscle (SM) myofibrillar mitochondria of normal and heart failure dogs. Depicted measures are ADP‐stimulated respiration (top left), membrane potential (top right), permeability transition pore (mPTP) opening (bottom left), and cytochrome c oxidase (complex IV) activity (bottom right) after exposure to 0.01, 0.10, and 1.0 μM elamipretide. RLU = relative light units.
Figure 3.
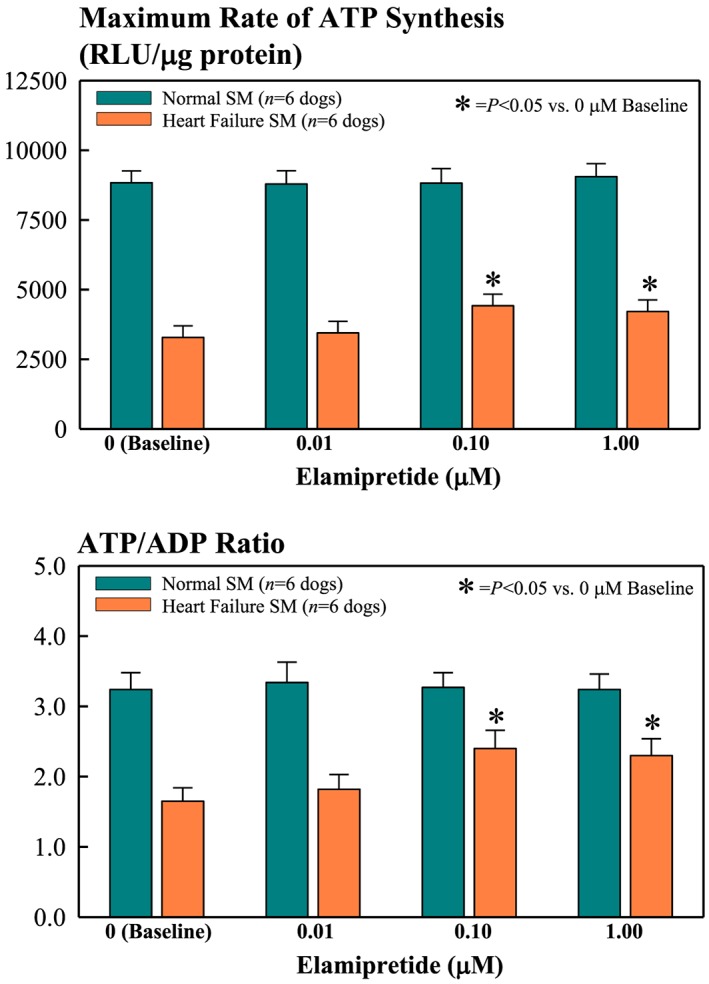
Bar graphs (mean ± standard error of the mean) depicting changes in mitochondrial functional measures of skeletal muscle (SM) myofibrillar mitochondria of normal and heart failure dogs. Depicted measures are maximum rate of ATP synthesis (top) and ATP/ADP ratio (bottom) after exposure to 0.01, 0.10, and 1.0 μM elamipretide. RLU = relative light units.
Protein levels of inducible nitric oxide synthase and indicators of apoptosis
There were no differences in protein levels of the internal control β‐actin between normal, HF‐CON, and HF + ELAM dogs (Table 1). Compared with the skeletal muscle of normal dogs, levels of iNOS were significantly increased in HF‐CON dogs. Treatment with ELAM reduced protein levels of iNOS (Table 1).
There were no differences in protein levels of the mitochondrial porin between normal, HF‐CON, and HF + ELAM dogs (Table 1). Compared with the skeletal muscle of normal dogs, levels of cytochrome c and HtrA2 normalized to porin were significantly reduced in HF‐CON dogs. Treatment with ELAM increased protein levels of both cytochrome c and HtrA2 normalized to porin (Table 1).
Discussion
In a previous study conducted in our laboratory on dogs with advanced HF, we showed that long‐term therapy with ELAM improved left ventricular systolic function, normalized plasma biomarkers, and reversed mitochondrial abnormalities in cardiomyocytes.24 Based on these results, we designed the current study to evaluate the effects of ELAM on the skeletal muscle of dogs with HF. Results of the present study showed that dogs with HF had a lower proportion of skeletal muscle type 1 fibres and a higher proportion of skeletal muscle type 2 fibres compared with normal dogs. This type of alteration of skeletal muscle fibre‐type composition is associated with exercise intolerance in patients with chronic HF.8, 16, 27 Patients with chronic HF had a reduced percentage of slow‐twitch type 1 fibres and a higher percentage of fast‐twitch type 2 fibres in vastus lateralis skeletal muscle biopsies.16 However, patients with HF who engaged in regular physical exercise had enhanced oxidative enzyme activity in skeletal muscle and a concomitant return to type 1 fibres.16 In addition, patients with HF also had a reduction in myosin heavy chain type 1,27 an isoform that is more abundant in skeletal muscle type 1 aerobic fibres. We previously showed a decrease in the relative composition of oxidative, slow‐twitch, fatigue‐resistant type 1 fibres and an increase in fast‐twitch, anaerobic, glycogen‐dependent type 2 fibres in dogs with coronary microembolization‐induced HF.13 This shift was associated with reduced exercise tolerance but not with atrophy of either fibre type.13 The shift in skeletal muscle fibre‐type composition in HF may be partly due to skeletal muscle mitochondrial abnormalities with associated reduction of ATP synthesis needed by aerobic type 1 fibres.28 Lack of ATP can lead to an adaptation of type 1 fibres to utilize glycogen as an energy source and thus shift towards a fast‐twitch phenotype. Direct support for this, however, remains to be elucidated. In the present study, long‐term treatment with ELAM restored skeletal muscle fibre‐type composition to a more normal distribution. Although not confirmed in the present study, one would speculate this correction or return to normal skeletal muscle fibre‐type distribution would lead to improved exercise tolerance.
The excess formation of ROS by dysfunctional mitochondria contributes to an increase of inner mitochondrial membrane permeability, which leads to a loss of transmembrane potential and ultimately tissue injury and cell death.29 Heart failure is associated with an opening of the mPTP, increased levels of cytosolic cytochrome c, and increased cardiomyocyte apoptosis.8, 30, 31 In the present study, measurements of cytochrome c and HtrA2 in isolated mitochondria showed a decrease of cytochrome c and HtrA2 in HF‐CON dogs compared with normal dogs, indicating translocation of cytochrome c and HtrA2 from the mitochondria to the cytosol, the latter a known trigger for activation of caspase‐3‐dependent and caspase‐3‐independent apoptotic pathways.24, 32 Treatment with ELAM increased mitochondrial levels of cytochrome c and HtrA2, signalling a potential reduction in the burden of programmed cell death. In this study, we also showed that exposure of skeletal muscle myofibrillar mitochondria from dogs with HF to ELAM resulted in a dose‐dependent improvement/normalization of all measures of mitochondrial function (ADP‐dependent mitochondrial respiration, Δψm, mPTP, maximum rate of ATPsyn, ATP/ADP ratio, and COX‐IV activity). These results support those from a previous study where we showed that chronic ELAM therapy improved the maximum rate of ATP synthesis and increased the ATP/ADP ratio in the failing myocardium of dogs.24 We also previously showed that in this same model of HF, chronic treatment with ELAM improved mitochondria state 3 respiration, Δψm, and mPTP and normalized the activity of complex I and complex IV in cardiomyocytes, thus limiting the formation of ROS. The skeletal muscle findings of the present study suggest that drugs that influence mitochondrial function of the failing heart can also positively impact mitochondrial function in peripheral skeletal muscle and potentially restore function of this muscle group with the potential for improved exercise tolerance.
Heart failure, regardless of aetiology, is associated with an enhanced inflammatory state evidenced by an increase in many proinflammatory cytokines. This proinflammatory state can lead to increased expression of iNOS, which, in turn, can result in suppression of mitochondrial respiration.33, 34, 35 Previous studies showed that increased expression of iNOS in skeletal muscle of patients with HF was inversely correlated with mitochondrial creatine kinase expression and exercise capacity.14 In the same study, cell experiments confirmed a causal relationship via nitric oxide.14 Results from the present study showed that long‐term therapy with ELAM in dogs with HF was also associated with normalization of iNOS along with normalization of mitochondrial state 3 respiration and overall mitochondrial function in skeletal muscle myofibres compared with untreated control dogs.
There are limitations to the study that merit considerations. No measures of overall muscle mass in the dogs were obtained as part of this study. Also, there were no measures obtained for the proportion of fat to lean tissue. These measures would have been helpful for future studies and for assessing the overall merits of restoration of skeletal muscle mitochondrial function. The present study also did not include studies with isolated mitochondria from HF dogs treated with ELAM. This would have been useful in determining whether changes in mitochondrial function are sustained over time. Finally, ultrastructural examination of muscle fibres and mitochondria in normal, HF, and HF dogs treated with ELAM would have been useful in supporting the underlying hypothesis of the role of mitochondria in skeletal muscle abnormalities in HF.
In conclusion, the results of this study indicate that long‐term therapy with ELAM, a mitochondria‐targeting peptide, normalized skeletal muscle fibre‐type composition and reversed the increased expression of iNOS in dogs with chronic HF. Furthermore, the study shows that treatment of skeletal muscle myofibres with ELAM normalizes mitochondrial function. These results, when viewed in concert, suggest that therapy with ELAM can improve skeletal muscle morphology and metabolism and, in doing so, potentially set the stage for an improvement of exercise tolerance, a key abnormality in HF.
Conflict of interest
H.N.S. has received research grants from the Stealth BioTherapeutics, Inc., and is a consultant of Stealth BioTherapeutics. R.C.G., V.S.‐G., and K.Z. have no conflicts of interest to disclose.
Funding
This study was supported, in part, by a research grant from the Stealth BioTherapeutics, Inc., and by the National Heart, Lung, and Blood Institute 1RO1HL132154‐01A1.
Sabbah H. N., Gupta R. C., Singh‐Gupta V., and Zhang K. (2019) Effects of elamipretide on skeletal muscle in dogs with experimentally induced heart failure, ESC Heart Failure, 6: 328–335. 10.1002/ehf2.12408.
References
- 1. Bayeva M, Gheorghiade M, Ardehali H. Mitochondria as a therapeutic target in heart failure. J Am Coll Cardiol 2013; 61: 599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mancuso M, Angelini C, Bertini E, Carelli V, Comi GP, Minetti C, Moggio M, Mongini T, Servidel S, Tonin P, Toscano A, Uziel G, Zeviani M, Siciliano G, Nation‐wide Italian Collaborative Network of Mitochondrial Diseases . Fatigue and exercise intolerance in mitochondrial diseases. Literature revision and experience of the Italian network of mitochondrial diseases. Neuromuscul Disord 2012; 22: S226–S229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rosca MG, Hoppel CL. Mitochondrial dysfunction in heart failure. Heart Fail Rev 2013; 18: 607–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 2012; 48: 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sorescu D, Griendling KK. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. CHF 2002; 8: 132–140. [DOI] [PubMed] [Google Scholar]
- 6. Drexler H, Riede U, Münzel T, König H, Funke E, Just H. Alterations of skeletal muscle in chronic heart failure. Circulation 1992; 85: 1751–1759. [DOI] [PubMed] [Google Scholar]
- 7. Wilson JR, Mancini DM, Dunkman WB. Exertional fatigue due to skeletal muscle dysfunction in patients with heart failure. Circulation 1993; 87: 470–475. [DOI] [PubMed] [Google Scholar]
- 8. Sullivan MJ, Green HJ, Cobb FR. Skeletal muscle biochemistry and histology in ambulatory patients with long‐term heart failure. Circulation 1990; 81: 518–527. [DOI] [PubMed] [Google Scholar]
- 9. Minotti JR, Christoph I, Oka R, Weiner MW, Wells I, Massie BM. Impaired skeletal muscle function in patients with congestive heart failure: relationship to systemic exercise performance. J Clin Invest 1991; 88: 2077–2082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mancini DM, Walter G, Reichek N, Lenkinski R, McCully KK, Mullen JL, Wilson JR. Contribution of skeletal muscle atrophy to exercise intolerance and altered muscle metabolism in heart failure. Circulation 1992; 85: 1364–1373. [DOI] [PubMed] [Google Scholar]
- 11. Mancini DM, Coyle E, Coggan A, Beltz J, Ferraro N, Montain S, Wilson JR. Contribution of intrinsic skeletal muscle changes to 31P NMR skeletal muscle metabolic abnormalities in patients with chronic heart failure. Circulation 1989; 80: 1338–1346. [DOI] [PubMed] [Google Scholar]
- 12. De Sousa E, Veksler V, Bigard X, Mateo P, Ventura‐Clapier R. Heart failure affects mitochondrial but not myofibrillar intrinsic properties of skeletal muscle. Circulation 2000; 102: 1847–1853. [DOI] [PubMed] [Google Scholar]
- 13. Sabbah HN, Hansen‐Smith F, Sharov VG, Kono T, Lesch M, Gengo PJ, Steffen RP, Levine TB, Goldstein S. Decreased proportion of type I myofibers in skeletal muscle of dogs with chronic heart failure. Circulation 1993; 87: 1729–1737. [DOI] [PubMed] [Google Scholar]
- 14. Hambrecht R, Volker A, Gielen S, Linke A, Mobius‐Winkler A, Yu J, Niebauer J, Jiang H, Fiehn E, Schuler G. Exercise intolerance in patients with chronic heart failure and increased expression of inducible nitric oxide synthase in skeletal muscle. J Am Coll Cardiol 1999; 33: 174–179. [DOI] [PubMed] [Google Scholar]
- 15. Hambrecht R, Niebauer J, Fiehn E, Kalberer B, Offner B, Hauer K, Riede U, Schlierf G, Kubler W, Schuler G. Physical training in patients with stable chronic heart failure: effects on cardiorespiratory fitness and ultrastructural abnormalities of leg muscle. J Am Coll Cardiol 1995; 25: 1239–1249. [DOI] [PubMed] [Google Scholar]
- 16. Hambrecht R, Fiehn E, Yu J, Niebauer J, Weigl C, Hilbrich L, Adams V, Riede U, Schuler G. Effects of endurance training on mitochondrial ultrastructure and fiber type distribution in skeletal muscle of patients with stable chronic heart failure. J Am Coll Cardiol 1997; 29: 1067–1073. [DOI] [PubMed] [Google Scholar]
- 17. Birk AV, Liu S, Soong Y, Mills W, Singh P, Warren JD, Seshan SV, Pardee JD, Szeto HH. The mitochondrial‐targeted compound SS‐31 re‐energizes ischemic mitochondria by interacting with cardiolipin. J Am Soc Nephrol 2013; 24: 1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Birk AV, Chao WM, Bracken C, Warren JD, Szeto HH. Targeting mitochondrial cardiolipin and the cytochrome c/cardiolipin complex to promote electron transport and optimize mitochondrial ATP synthesis. Br J Pharmacol 2014; 171: 2017–2028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Szeto HH, Liu S, Soong Y, Wu D, Darrah SF, Cheng FY, Zhao Z, Ganger M, Tow CY, Seshan SV. Mitochondria‐targeted peptide accelerates ATP recovery and reduces ischemic kidney injury. J Am Soc Nephrol 2011; 22: 1041–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Siegel MP, Kruse SE, Percival JM, Goh J, White CC, Hopkins HC, Kavanagh TJ, Szeto HH, Rabinovitch PS, Marcinek DJ. Mitochondrial‐targeted peptide rapidly improves mitochondrial energetics and skeletal muscle performance in aged mice. Aging Cell 2013; 12: 763–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Talbert EE, Smuder AJ, Min K, Kwon OS, Szeto HH, Powers SK. Immobilization‐induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria‐targeted antioxidant. J Appl Physiol 2013; 115: 529–538. [DOI] [PubMed] [Google Scholar]
- 22. Brown DA, Hale SL, Baines CP, Del Rio CL, Hamlin RL, Yueyama Y, Kijtawornrat A, Yeh ST, Frasier C, Stewart LM, Moukdar F, Shaikh SR, Fisher‐Wellman KH, Neufer PD, Kloner RA. Reduction of early reperfusion injury with the mitochondria‐targeting peptide bendavia. J Cardiovasc Pharmacol Ther 2014; 19: 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sabbah HN, Stein PD, Kono T, Gheorghiade M, Levine TB, Jafri S, Hawkins ET, Goldstein S. A canine model of chronic heart failure produced by multiple sequential coronary microembolizations. Am J Physiol 1991; 260: H1379–H1384. [DOI] [PubMed] [Google Scholar]
- 24. Sabbah HN, Gupta RC, Kohli S, Mengjun W, Hachem S, Zhang K. Chronic therapy with elamipretide (MTP‐131), a novel mitochondria‐targeting peptide, improves left ventricular and mitochondrial function in dogs with advanced heart failure. Circulation 2016; 9: 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pipinos II, Sharov VG, Shepard AD, Anagnostopoulos PV, Katsamouris A, Todor A, Filis KS, Sabbah HN. Abnormal mitochondrial respiration in skeletal muscle in patients with peripheral arterial disease. J Vasc Surg 2003; 38: 827–832. [DOI] [PubMed] [Google Scholar]
- 26. Sharov VG, Todor AV, Khanal S, Imai M, Sabbah HN. Cyclosporine A attenuates mitochondrial permeability transition and improves mitochondrial respiratory function in cardiomyocytes isolated from dogs with heart failure. J Mol Cell Cardiol 2007: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sullivan MJ, Duscha BD, Klitgaard H, Kraus WE, Cobb FR, Saltin B. Altered expression of myosin heavy chain in human skeletal muscle in chronic heart failure. Med Sci Sports Exerc 1997; 29: 860–866. [DOI] [PubMed] [Google Scholar]
- 28. Marcinek DJ, Schenkman KA, Ciesielski WA, Lee D, Conley KE. Reduced mitochondrial coupling in vivo alters cellular energetics in aged mouse skeletal muscle. J Physiol 2005; 569: 467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bernardi P, Di Lisa F. The mitochondrial permeability transition pore: molecular nature and role as a target in cardioprotection. J Mol Cell Cardiol 2015; 78: 100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sharov VG, Sabbah HN, Shimoyama H, Goussev AV, Lesch M, Goldstein S. Evidence of cardiocyte apoptosis in myocardium of dogs with chronic heart failure. Am J Pathol 1996; 148: 141–149. [PMC free article] [PubMed] [Google Scholar]
- 31. Sharov VG, Todor AV, Silverman N, Goldstein S, Sabbah HN. Abnormal mitochondrial respiration in failed human myocardium. J Mol Cell Cardiol 2000; 32: 2361–2367. [DOI] [PubMed] [Google Scholar]
- 32. Liu H‐R, Gao E, Hu A, Tao L, Qu Y, Most P, Koch WJ, Christopher TA, Lopez BL, Alnemri ES, Zervos AS, Ma XL. Role of Omi/HtrA2 in apoptotic cell death after myocardial ischemia and reperfusion. Circulation 2005; 111: 90–96. [DOI] [PubMed] [Google Scholar]
- 33. Brown GC, Borutaite V. Nitric oxide and mitochondrial respiration in the heart. Cardiovasc Res 2007; 75: 283–290. [DOI] [PubMed] [Google Scholar]
- 34. Brown GC. Nitric oxide and mitochondrial respiration. Biochemica et Biophysica Acta 1999; 1411: 351–369. [DOI] [PubMed] [Google Scholar]
- 35. Garcia JA, Ortiz F, Miana J, Doerrier C, Fernandez‐Ortiz M, Rusanova I, Escames G, Garcia JJ, Acuña‐Castroviejo D. Contribution of inducible and neuronal nitric oxide synthases to mitochondrial damage and melatonin rescue in LPS‐treated mice. J Physiol Biochem 2017; 73: 235–244. [DOI] [PubMed] [Google Scholar]