

Doravirine is a novel nonnucleoside reverse transcriptase inhibitor for the treatment of human immunodeficiency virus 1 (HIV-1) infection. A population pharmacokinetic (PK) model was developed for doravirine using pooled data from densely sampled phase 1 trials and from sparsely sampled phase 2b and phase 3 trials evaluating doravirine administered orally as a single entity or as part of a fixed-dose combination of doravirine-lamivudine-tenofovir disoproxil fumarate.
KEYWORDS: pharmacokinetics, doravirine, exposure-response modeling, population pharmacokinetics
ABSTRACT
Doravirine is a novel nonnucleoside reverse transcriptase inhibitor for the treatment of human immunodeficiency virus 1 (HIV-1) infection. A population pharmacokinetic (PK) model was developed for doravirine using pooled data from densely sampled phase 1 trials and from sparsely sampled phase 2b and phase 3 trials evaluating doravirine administered orally as a single entity or as part of a fixed-dose combination of doravirine-lamivudine-tenofovir disoproxil fumarate. A one-compartment model with linear clearance from the central compartment adequately described the clinical PK of doravirine. While weight, age, and healthy versus HIV-1 status were identified as statistically significant covariates affecting doravirine PK, the magnitude of their effects was not clinically meaningful. Other intrinsic factors (gender, body mass index, race, ethnicity, and renal function) did not have statistically significant or clinically meaningful effects on doravirine PK. Individual exposure estimates for individuals in the phase 2b and 3 trials obtained from the final model were used for subsequent exposure-response analyses for virologic response (proportion of individuals achieving <50 copies/ml) and virologic failure. The exposure-response relationships between these efficacy endpoints and doravirine PK were generally flat over the range of exposures achieved for the 100 mg once-daily regimen in the phase 3 trials, with a minimal decrease in efficacy in individuals in the lowest 10th percentile of steady-state doravirine concentration at 24 h values. These findings support 100 mg once daily as the selected dose of doravirine, with no dose adjustment warranted for the studied intrinsic factors.
INTRODUCTION
Doravirine is a novel nonnucleoside reverse transcriptase inhibitor (NNRTI) for the treatment of human immunodeficiency virus type 1 (HIV-1) infection (1). It is administered with other antiretroviral agents or as a three-drug single-tablet regimen with lamivudine (3TC) and tenofovir disoproxil fumarate (TDF) (2, 3). In a phase 2b trial in antiretroviral therapy-naive adults with HIV-1, doravirine at 100 mg once daily (QD) in combination with emtricitabine (FTC)-TDF demonstrated potent antiretroviral activity and an immunological effect at week 48 and was generally well tolerated (4). Phase 3 trials in treatment-naive adults with HIV-1 have also demonstrated the noninferior efficacy of doravirine-3TC-TDF versus efavirenz (EFV)-FTC-TDF (5) and of doravirine at 100 mg QD versus darunavir-ritonavir, both in combination with two nucleoside reverse transcriptase inhibitors (6).
The pharmacokinetics (PK) of doravirine have been extensively evaluated both as a single entity and as a fixed-dose combination of doravirine at 100 mg, 3TC at 300 mg, and TDF at 300 mg (equivalent to 245 mg of tenofovir disoproxil) in healthy individuals and in individuals with HIV-1 (7–11). Based on dedicated phase 1 trials, there were no clinically meaningful effects of food (11), age or gender (10), or renal impairment (12) on doravirine PK. Doravirine is primarily metabolized by CYP3A4 (13).
Here, we describe the development of a population PK model for doravirine in healthy individuals and individuals with HIV-1. The main aim was to identify and quantify the impact of individual covariates on doravirine PK in order to inform dose recommendations in special populations. The population PK model was also used to obtain individual PK parameter values to characterize the relationship between doravirine PK and efficacy for the proportion of individuals achieving undetectable HIV-1 RNA levels (<50 copies/ml) and for virologic failure.
RESULTS
Population PK.
In total, this analysis included 341 healthy individuals and 959 individuals with HIV-1 receiving oral doses of doravirine as a single entity or as doravirine-3TC-TDF. A summary of the baseline characteristics is shown in Table 1, for the total pooled phase 1 (n = 353), phase 2b (n = 217), and phase 3 (n = 730) populations. The analysis population was comprised predominantly of male (80.8%), white (65.2%), non-Hispanic (76.9%) individuals. For each component of the pooled analysis, the following PK observations were included: phase 1, 5,552 PK observations (52.2% of the total) at doses ranging from 6 to 200 mg (a single dose and multiple doses); phase 2, 1,132 PK observations (10.6% of the total) at once-daily doses of 25, 50, 100, and 200 mg; phase 3, 3,959 PK observations (37.2% of the total) at a once-daily dose of 100 mg.
TABLE 1.
Pooled demographic and disease characteristicsa
| Characteristic | No. (%) of subjects from: |
||||
|---|---|---|---|---|---|
| Population PK analysisb | Exposure-response analysisc | P007 trial | P018 trial | P021 trial | |
| Individuals with PK data | 1,300 | 947 | 217 | 375 | 355 |
| Gender | |||||
| Male | 1,051 (80.8) | 811 (85.6) | 200 (92.2) | 311 (82.9) | 300 (84.5) |
| Female | 249 (19.2) | 136 (14.4) | 17 (7.8) | 64 (17.1) | 55 (15.5) |
| Race | |||||
| American Indian/Alaska native | 15 (1.2) | 14 (1.5) | 3 (1.4) | 3 (0.8) | 8 (2.3) |
| Asian | 87 (6.7) | 78 (8.2) | 12 (5.5) | 7 (1.9) | 59 (16.6) |
| Black/African American | 281 (21.6) | 186 (19.6) | 40 (18.4) | 81 (21.6) | 65 (18.3) |
| Multiracial | 66 (5.1) | 56 (5.9) | 1 (0.5) | 6 (1.6) | 49 (13.8) |
| Native Hawaiian/other Pacific Islander | 2 (0.2) | 1 (0.1) | 0 | 1 (0.3) | 0 |
| White | 847 (65.2) | 611 (64.5) | 160 (73.7) | 277 (73.9) | 174 (49.0) |
| Not reported/unknown | 2 (0.2) | 1 (0.1) | 1 (0.5) | 0 | 0 |
| Ethnicity | |||||
| Hispanic | 286 (22.0) | 246 (26.0) | 34 (15.7) | 92 (24.5) | 120 (33.8) |
| Not Hispanic | 1,000 (76.9) | 687 (72.5) | 177 (81.6) | 277 (73.9) | 233 (65.6) |
| Not reported/unknown | 14 (1.1) | 14 (1.5) | 6 (2.8) | 6 (1.6) | 2 (0.6) |
| Formulation | |||||
| Doravirine tablets | 920 (70.8) | 592 (62.5) | 217 (100) | 375 (100) | 0 |
| Doravirine-3TC-TDF tablets | 380 (29.2) | 355 (37.5) | 0 | 0 | 355 (100) |
| Health status | |||||
| Healthy | 341 (26.2) | 0 | 0 | 0 | 0 |
| HIV-1 infected | 959 (73.8) | 947 (100) | 217 (100) | 375 (100) | 355 (100) |
3TC, lamivudine; HIV, human immunodeficiency virus; TDF, tenofovir disoproxil fumarate.
Data from the phase 1, 2b, and 3 trials were pooled for the population PK analysis.
Data from the phase 2b and 3 trials were pooled for the exposure-response analysis.
Doravirine PK were characterized by a one-compartment model with first-order absorption and linear clearance (CL/F) from the central compartment (Table 2). The less-than-dose-proportional increases in doravirine exposure were described by different relative bioavailabilities for the dose ranges of <30 mg and >120 mg in comparison with the reference dose range of 30 to 120 mg, which included the clinical dose of 100 mg. Interindividual variability random effects on CL/F and the apparent volume of distribution (V/F) were included.
TABLE 2.
Parameter estimates for final population PK model for doravirinea
| Parameter | Final estimate | % RSE | % CV | Lower 5% | Upper 95% | η shrinkage |
|---|---|---|---|---|---|---|
| Fixed effects | ||||||
| CL/F (liters/h) | 6.34 | 1.37 | 6.17 | 6.51 | ||
| V/F (liters) | 162 | 3.17 | 152 | 172 | ||
| Ka (1/h) | 1.40 | 4.57 | 1.28 | 1.53 | ||
| F1 < 30 mg | 1.20 | 5.87 | 1.06 | 1.34 | ||
| F1 30–120 mg (reference) | 1.00 | |||||
| F1 > 120 mg | 0.895 | 9.32 | 0.732 | 1.06 | ||
| Age on CL/F | −0.00508 | 16.6 | −0.00673 | −0.00343 | ||
| Healthy vs HIV-1 status on V/F | −0.220 | 12.3 | −0.273 | −0.167 | ||
| Wt on V/F | 0.00788 | 15.4 | 0.00551 | 0.0102 | ||
| Interindividual variability | ||||||
| CL/F | 0.117 | 7.93 | 35.2 | 0.0986 | 0.135 | 11.4 |
| V/F | 0.101 | 8.26 | 32.6 | 0.0848 | 0.118 | 38.7 |
| Residual variability (SD) | ||||||
| Phase 1 for ≤0.5 h | 0.224 | 3.77 | 22.4 | 0.207 | 0.241 | |
| Phase 1 for >0.5 h | 1.25 | 6.89 | 125 | 1.08 | 1.42 | 7.37 |
| Phase 2b/3 | 0.521 | 4.01 | 52.1 | 0.48 | 0.562 |
Clearance and the volume of distribution were calculated as follows: CLi = CLTV [1 + θ(agei − 34)] and Vi = VTV [1 + θweight(weighti − 75)]·(1 + θhealthy·flag), where CLi is the clearance for individual i, CLTV is the typical value for clearance, agei is the age of individual i, Vi is the volume of distribution for individual i, VTV is the typical value of the volume of distribution, θ is the estimated effect of age, θweight is the estimated effect of weight, θhealthy is the estimated effect of health status, weighti is the weight of individual i, and flag is equal to 1 for healthy volunteers and 0 for HIV-infected individuals (the most common population). CL/F, apparent clearance; CV, coefficient of variation; F1, oral bioavailability; Ka, absorption rate; RSE, relative standard error; V/F, apparent volume of distribution.
An additive residual error model was employed for the log-transformed concentration data. Separate residual errors were estimated for the phase 1 data versus the phase 2b/3 data and within the phase 1 data for the first 0.5 h postdose versus the remaining time points. A separate additive error term was included for phase 2b and 3 relative to phase 1 doravirine concentrations, as the residual variability in doravirine concentrations was generally higher in the phase 2b and 3 data than in the phase 1 data. The higher variability observed in the phase 2b and 3 data is likely related to PK sampling in the phase 2b/3 trials being subject to additional noise, as dose and sampling times were not as tightly controlled as they were in the phase 1 trials. With a single additive error term for the phase 1 trials, the variability in doravirine concentrations at 0.5 h postdose in the phase 1 trials was not well described. To address this, a separate additive error term was introduced for phase 1 doravirine concentrations after 0.5 h postdose. A similar term was not needed for the phase 2b and 3 data, as potential variability in the absorption patterns immediately following dosing would not be observed in the more sparsely sampled phase 2b and 3 data.
Based on stepwise covariate modeling (SCM) analysis, only body weight, age, and healthy versus HIV-1 status were identified as statistically significant covariates impacting doravirine PK. Hence, the final population PK model included the effects of body weight and healthy versus HIV-1 status on V/F and age on CL/F. Gender, race, ethnicity, renal function, and coadministration with 3TC and TDF were also evaluated and did not have significant effects on doravirine PK.
Goodness-of-fit diagnostic plots for the final population PK model are provided in Fig. S1 in the supplemental material. Visual predictive checks confirmed that the model accurately characterized the central tendency of the observed data and that an appropriate distribution of the observed data fell within the 5th and 95th percentiles of the model-simulated data, indicating that the model adequately describes doravirine plasma concentrations (Fig. S2).
Typical derived steady-state exposure estimates (geometric mean [percent coefficient of variation]) based on the post hoc PK parameters for individuals with HIV-1 in the phase 3 trials receiving 100 mg doravirine QD were as follows: area under the concentration-time curve (AUC) from 0 to 24 h (AUC0–24), 37.8 μM h (27.0%); concentration at 24 h (C24), 930 nM (41.6%); and maximum plasma concentration (Cmax), 2,260 nM (18.4%). Despite identification of healthy versus HIV-1 status as a significant covariate on the doravirine V/F, these values are consistent with those reported following intensive sampling in healthy individuals in a phase 1 trial (protocol 020 [P020], n = 19), where the steady-state geometric mean AUC0–24, C24, and Cmax values were 41.1 μM h, 902 nM, and 2.88 μM, respectively (9).
The effects of key intrinsic factors on doravirine AUC0–24, Cmax, and C24 at steady state, as estimated based on the population PK model, are shown in Fig. 1. Covariate effects in a representative population with HIV-1 were characterized by simulation of doravirine steady-state PK based on the data for individuals observed in the phase 2b and 3 trials and their covariates. For age, gender, and renal impairment, for which dedicated phase 1 trials have been conducted (7, 10, 12), the observed effects on doravirine PK were also included for comparison. All covariate effects were modest (largest increase, 69%; largest decrease, 34%). Moreover, the population PK model-estimated effects of age, gender, and renal impairment were generally consistent with the effects observed in the phase 1 trials.
FIG 1.

Effect of intrinsic factors on doravirine steady-state AUC0–24, Cmax, and C24 (the population PK [popPK] geometric mean ratio [GMR] and 90% confidence interval [CI] shown are based on a simulation of 1,000 individuals with HIV-1 per covariate subgroup for categorical covariates or a total of 1,000 individuals with HIV-1 for continuous covariates). AUC, area under the concentration-time curve; Cmax, maximum plasma concentration; C24, concentration at 24 h; RI, renal impairment; ref, reference value.
Age was identified as having a significant impact on doravirine CL/F. This effect corresponded with AUC and C24 estimates 15% and 22% higher, respectively, for a 59-year-old (the 95th percentile of age in the data set) than for a 34-year-old (the median age) when all other covariates were held constant. Similarly, AUC and C24 estimates were 6% and 9% lower, respectively, for a 21-year-old (the 5th percentile of age) than for a 34-year-old. To characterize the effect of age in the intended target population, doravirine steady-state AUC0–24, C24, and Cmax for individuals with HIV-1 aged ≥65 years or <65 years were simulated based on the covariate distributions observed in the phase 2b and 3 trials: the doravirine steady-state AUC0–24 was 30% higher, the steady-state C24 was 63% higher, and the steady-state Cmax was 12% higher in individuals with HIV-1 and ≥65 years of age than in individuals with HIV-1 and <65 years of age.
Body weight was also identified as a significant covariate impacting V/F. However, while significant, the magnitude of the effect of body weight on doravirine PK was minor: steady-state doravirine AUC0–24, C24, and Cmax differed by <10% for individuals weighing <64.5 kg or ≥83 kg compared with those weighing 64.5 to <83 kg. Given the correlation between weight and body mass index (BMI), BMI was not evaluated as a covariate in the SCM analysis. However, the effects of BMI were explored by simulation to better understand any differences in steady-state doravirine PK exposures in the intended target population. Doravirine steady-state AUC0–24 and Cmax differed by <10% for underweight (BMI < 18.5 kg/m2), preobese (BMI = 25.0 to 29.99 kg/m2), and obese (BMI ≥ 30 kg/m2) individuals compared with individuals within the normal weight range (BMI = 18.5 to 24.99 kg/m2). The doravirine steady-state C24 was 34% lower in underweight individuals and 13% and 20% higher in preobese and obese individuals, respectively, than in individuals within the normal weight range.
Exposure-response analysis.
The exposure-response data sets included virologic response (proportion of individuals with HIV-1 RNA levels of <50 copies/ml) and protocol-defined virologic failure (PDVF) data at week 48 of treatment from 217 individuals with HIV-1 who received oral doses of doravirine as a single entity in combination with FTC-TDF in the phase 2b trial (P007) and data from a total of 730 individuals with HIV-1 who received oral doses of doravirine as a single entity in combination with FTC-TDF or abacavir (ABC)-3TC (P018) or as doravirine-3TC-TDF (P021) in phase 3 trials.
In the phase 2b analysis, no statistically significant exposure-response relationships were identified between any of the doravirine PK parameters (AUC0–24, C24, or Cmax) and efficacy endpoints over the range of exposures achieved at the 25- to 200-mg QD doses. The exposure-response relationships for the proportion of individuals with HIV-1 RNA levels of <50 copies/ml (Fig. 2A) and PDVF (Fig. 2B) as a function of the doravirine steady-state C24 were flat, with no trends between response and doravirine exposure.
FIG 2.

Predicted and observed proportion of individuals achieving HIV-1 RNA levels of <50 copies/ml (A) and with protocol-defined virologic failure (B) as a function of doravirine steady-state C24 quartiles following administration of doravirine at 25 to 200 mg QD with FTC-TDF (n = 217). The heavy solid lines represent the mean predicted exposure-response relationship. The shaded areas represent the 95% CI of the prediction. Markers summarize the observed endpoint frequency (the median and 95% CI are shown) by C24 quartile. Horizontal box plots denote the distribution of individual C24 values at the 25-, 50-, 100-, and 200-mg QD doses in P007, where the point is the median, the box corresponds to the 25th and 75th percentiles, and the whiskers correspond to the 5th and 95th percentiles.
Statistically significant exposure-response relationships were identified between doravirine PK and the proportion of individuals achieving <50 copies/ml and PDVF in the phase 3 trials over the exposures achieved at the 100-mg QD dose. However, the relationships were generally flat, with a minimal decrease in efficacy in individuals with the lowest 10% of doravirine PK values. The exposure-response relationships were characterized by log-linear logistic regressions for each efficacy endpoint and doravirine steady-state AUC0–24, Cmax, and C24. The strongest exposure-response associations were observed for steady-state C24 rather than AUC0–24 or Cmax; therefore, the focus of this analysis was on the doravirine C24. The effect of the covariates age, gender, weight, race, ethnicity, and screening HIV-1 RNA level were also evaluated based on stepwise forward addition (P < 0.01) and backward elimination (P < 0.001); the only significant covariate identified was the screening HIV-1 RNA level (≤100,000 or >100,000 copies/ml) on the slope of the exposure-response relationship for the virologic response endpoints.
The relationship between doravirine C24 and the proportion of individuals achieving HIV-1 RNA levels of <50 copies/ml at week 48 stratified by screening HIV-1 RNA level is shown in Fig. 3. The relationship between doravirine C24 and the virologic response in individuals with a low screening HIV-1 RNA level appeared to be driven mainly by a slightly lower virologic response in individuals with the lowest 10% of doravirine C24 values, with a flat exposure-response relationship across the range of exposure above the 10th percentile. In contrast, the relationship between doravirine C24 and the virologic response was flat in individuals with a high screening HIV-1 RNA level.
FIG 3.

Predicted and observed proportions of individuals with HIV-1 achieving <50 copies/ml as a function of the doravirine steady-state C24 deciles stratified by screening HIV-1 RNA level following administration of doravirine at 100 mg QD with FTC-TDF or ABC-3TC or QD administration of doravirine-3TC-TDF (n = 730). The solid lines represent the mean predicted exposure-response relationship (by stratum). The shaded areas represent the 95% CI of the prediction. Markers summarize the observed endpoint frequency (the median and 95% CI are shown) by C24 quartile.
Of note, there was a disproportionate number of individuals in the first decile of doravirine C24 values who had at least one PK sample below the limit of quantitation (BLQ) during the 48-week treatment period (eight in the first decile, compared with four or less in each of the other deciles), possibly indicating missed doses. Individuals with at least one PK sample BLQ also achieved the endpoint of <50 copies/ml at a lower rate than the other individuals. Consequently, the disproportionate presence of these individuals in the lowest 10% of C24 values contributes to the lower virologic response observed in this group. In a sensitivity analysis excluding individuals with at least one PK sample with a C24 BLQ, the exposure-response relationship for the proportion of individuals achieving <50 copies/ml was weakened, with visibly flatter slopes (Fig. 4A).
FIG 4.

Predicted and observed proportions of individuals with HIV-1 achieving <50 copies/ml as a function of doravirine steady-state C24 deciles, excluding individuals with at least one sample with a C24 BLQ following administration of doravirine at 100 mg QD with FTC-TDF or ABC-3TC or QD administration of doravirine-3TC-TDF (n = 698), using a log-linear logistic regression model (A) and a linear logistic regression model (B). The solid lines represent the mean predicted exposure-response relationship. The shaded areas represent the 95% CI of the prediction. Markers summarize the observed endpoint frequency (the median and 95% CI are shown) by C24 quartile.
The final model for PDVF included only a relationship between log-transformed doravirine C24 and PDVF with no covariate effects. Similar to the virologic response analysis, the relationship between PDVF through week 48 and doravirine plasma PK was generally flat over the range of exposures achieved at the 100-mg dose in the phase 3 trials (Fig. 5), with a slightly higher virologic failure rate being seen in individuals with the lowest 10% of doravirine C24 values. No trends in exposure-response were identified for PDVF in the phase 2b analysis.
FIG 5.
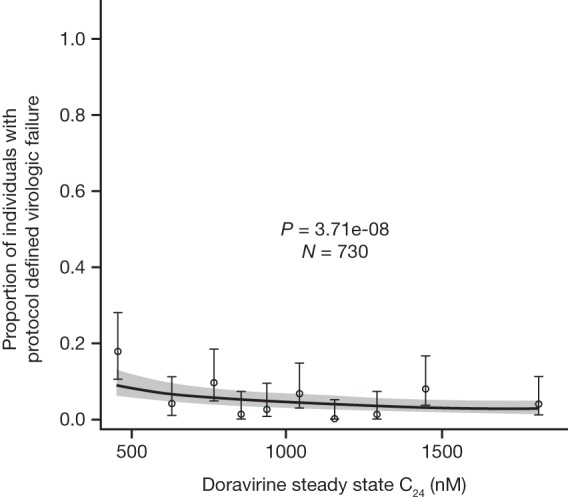
Predicted and observed proportions of individuals with HIV-1 with protocol-defined virologic failure as a function of doravirine steady-state C24 deciles following administration of doravirine at 100 mg QD with FTC-TDF or ABC-3TC or QD administration of doravirine-3TC-TDF (n = 730). The solid line represents the mean predicted exposure-response relationship. The shaded area represents the 95% CI of the prediction. Markers summarize the observed endpoint frequency (the median and 95% CI are shown) by C24 quartile.
DISCUSSION
In the current analysis, the pooled data set comprising data for individuals that participated in phase 1, phase 2, and phase 3 trials provided an adequate number of observations to support the characterization of doravirine PK, as well as determine any intrinsic factors that may influence doravirine PK in both healthy and HIV-1-infected participants. The pooled data set included data from individuals administered doravirine as a single entity and as a fixed-dose combination tablet with 3TC and TDF. Dedicated drug-drug interaction trials have demonstrated that there is no interaction between doravirine and 3TC or TDF (M. S. Anderson, J. Gilmartin, L. Fan, K. L. Yee, W. K. Kraft, I. Triantafyllou, C. Reitmann, Y. Guo, R. Liu, M. Iwamoto, M. O. Behm, submitted for publication). Coadministration of 3TC and TDF with doravirine was also evaluated as a covariate in the population PK analysis described herein, and no significant interaction was identified, which is consistent with the results of the drug-drug interaction trials and which supports the pooling of these data for the population PK and exposure-response analyses. Moreover, a data set consisting of data pooled from the dose-ranging study conducted in the phase 2b trial and the large number of participants in both phase 3 trials supported the exposure-response analysis.
A one-compartment PK model for doravirine with linear clearance from the central compartment was found to adequately describe the clinical PK of doravirine in the dose range of 6 to 200 mg and was suitable to provide individual exposure estimates for individuals with HIV-1 in the phase 2b and 3 trials for subsequent exposure-response analyses, despite a moderate level of shrinkage in V/F. As part of the stepwise covariate analysis, statistically significant effects were found for weight and healthy versus HIV-1 status on V/F and age on CL/F. While healthy versus HIV-1 status was identified as a significant covariate on doravirine V/F, the effect of this covariate was modest and steady-state doravirine PK parameter values estimated for the phase 3 population with HIV-1 were consistent with those observed in healthy individuals following intensive sampling in a phase 1 trial (9). Consequently, the PK data obtained in healthy individuals in the phase 1 trials are directly informative of doravirine PK and covariate effects in the population with HIV-1.
Similarly, the effects of age and body weight on doravirine exposure and C24 were also modest, corresponding to a less than 25% change in doravirine PK parameter values in a typical population with HIV-1. The effects of other covariates (race, ethnicity, gender, BMI, and renal function) were also evaluated and resulted in nonsignificant changes in doravirine exposure or C24 ranging from a 34% decrease to a 69% increase. Moreover, the covariate effects estimated in the population PK analysis were generally consistent with those observed in dedicated phase 1 trials that assessed the effects of age, gender, and renal impairment on doravirine PK (7, 10, 12). In the trial evaluating the effect of age and gender on doravirine PK in 24 elderly men and women, the doravirine AUC from 0 h to infinity (AUC0–inf), C24, and Cmax in elderly individuals were within 20% of those observed in nonelderly men and women (10). In the population PK analysis, elderly individuals were estimated to have a 30% higher AUC0–24 and a 63% higher C24, which may reflect decreased hepatic function or metabolism with increasing age (14, 15). Of note, elderly individuals comprised only 3% of the population PK analysis population, and the effect of age on doravirine CL/F was assessed continuously across the age range in the analysis population. Thus, changes in doravirine CL/F within the nonelderly population (age, 18 to 64 years) due to age were captured and the estimated effects on AUC0-24 and C24 reflect this relationship. In the same phase 1 trial, doravirine PK in women were demonstrated to be similar to those in men (10); in the population PK analysis, no significant difference in doravirine PK was identified between the genders. In a phase 1 trial to assess the effect of severe renal impairment on doravirine PK, doravirine AUC0–inf and C24 were increased by 43% and 38%, respectively (12). Correspondingly, in the population PK analysis, no relationship between renal function, as determined by the Modification of Diet in Renal Disease (MDRD) study-estimated glomerular filtration rate (eGFR), and doravirine PK was detected. However, the population PK analysis included only eight individuals with severe renal impairment from the phase 1 trial, so the ability to detect an effect of severe renal impairment with this model may be limited.
Analysis of the efficacy endpoints in the phase 3 data set indicated a statistically significant positive exposure-response relationship. However, the relationships were generally flat over the range of exposures achieved at the 100-mg QD dose, with a decrease in efficacy in individuals in the lowest 10th decile of steady-state doravirine PK values. Thus, the positive exposure-response relationship detected between doravirine PK and the proportion of individuals achieving HIV-1 RNA levels of <50 copies/ml appeared to be driven by low response rates in individuals in the first decile of doravirine PK values. The proportion of individuals achieving <50 copies/ml was flat, with no trend between response and C24 between the 2nd and 10th deciles. The HIV-1 RNA level at screening was a significant covariate affecting the slope of the exposure-response relationships but did not have a clinically relevant impact on the exposure-response relationships, and a robust antiviral response was observed across the range of exposures associated with the 100-mg QD dose regardless of the screening HIV-1 RNA level.
In this analysis, all missing data were treated as treatment failure regardless of the reason, including the early discontinuation of study therapy. Exposure-response analyses were also conducted using the observed failure approach, where individuals who discontinued treatment due to a lack of efficacy were considered to have treatment failure thereafter and data for individuals with data missing for other reasons and for the viral detection threshold of <40 copies/ml were excluded. Additional exposure-response analyses were conducted for the viral detection threshold of <40 copies/ml. Exposure-response relationships between these endpoints and doravirine C24 were similar to those already described for the proportion of individuals achieving <50 copies/ml. Virologic failure rates also followed the same trend, though the directionality was reversed; virologic failure was slightly higher in individuals in the lowest decile of C24, with no apparent trend between virologic failure and doravirine PK over the subsequent 2nd to 10th deciles.
To understand the factors that may influence the virologic response and failure rates in individuals with the lowest exposures, potential covariate imbalances between the lowest 10% of the doravirine C24 and subsequent deciles were explored. A screen of demographic covariates showed no imbalance of covariates across doravirine C24 deciles. However, there was a higher prevalence of individuals in the first decile with at least one reported sample with a C24 BLQ during the 48 weeks of sparse sampling, suggesting lower compliance. Given the apparent terminal half-life of doravirine of 12 to 21 h (7), the washout of doravirine plasma concentrations to levels BLQ likely reflects several days of consecutive missed doses. To assess the impact of individuals with potential missed doses on the virologic response exposure-response relationship, a sensitivity analysis was conducted where individuals with at least 1 PK sample BLQ were excluded. This had a clear impact on the statistical significance of the exposure-response relationships, with a marked increase in the P value associated with the exposure-response slope and visibly flatter slopes. The use of log-transformed exposures in the log-linear logistic regression model is intended to better characterize the response at the extremes of the exposure values. In particular, steady-state C24 values in the lowest decile were as low as 2.44 nM, with only a few individuals having C24 values below ∼40 nM. While individuals with these concentrations were not excluded in the sensitivity analysis, these plasma concentrations likely also reflect nonadherence to once-daily dosing. Consequently, the significant log-linear relationship identified in the sensitivity analysis is likely driven by a limited number of individuals with the lowest reported C24 values. Of note, no statistically significant exposure-response relationship for virologic response was identified after the exclusion of individuals with at least one sample with a C24 BLQ when a linear logistic regression model was assessed (Fig. 4B). These results suggest that the apparent trend in exposure-response observed in the phase 3 data set is a reflection of lower compliance and, therefore, reduced efficacy in a subset of individuals in the lowest 10% of exposures, rather than a true reduction in efficacy at lower exposures. Consistent with this, no significant relationships were established for the same endpoint in the phase 2b trial for exposures achieved over the 25- to 200-mg QD range, and the exposure-response relationship was also flat for the proportion of individuals achieving HIV-1 RNA levels of <50 copies/ml at week 48 over the exposures achieved in the 25-mg to 200-mg dose range in the phase 2b trial. These results are also consistent with the lack of a dose-response observed in the phase 2b trial, where doses of 25 to 200 mg demonstrated antiretroviral activity and an immunological effect that was similar to that of EFV at 600 mg through 24 weeks of treatment (4). The flat exposure-response relationships and efficacy observed in the phase 2b and phase 3 trials support the appropriateness of the 100-mg QD dose. Moreover, clinically observed doravirine concentrations associated with the 100-mg QD dose also conferred inhibitory activity against wild-type HIV-1 and common mutants in in vitro assays (16).
The 10th percentile of C24 values corresponds to approximately 60% of the geometric mean C24 value achieved at the 100-mg QD dose in the phase 3 data set. At C24 values higher than the 10th percentile, the differences in responses across the distribution of exposures associated with the proposed clinical dose of 100 mg QD are predicted to be small and of limited clinical relevance. As no covariate was associated with a decrease in the doravirine C24 of >40%, based on the population PK analysis, none of the covariates studied in this analysis had clinically meaningful effects on doravirine PK that may lead to a decrease in efficacy, and no dose adjustments to the 100-mg QD dose for special populations are recommended.
In summary, a one-compartment model with linear clearance from the central compartment adequately described the clinical PK of doravirine in the dose range of 6 to 200 mg and was suitable to provide individual exposure estimates for exposure-response analyses. The effects of age, gender, weight, BMI, race, ethnicity, and mild, moderate, or severe renal impairment on doravirine PK were not clinically meaningful, and no dose adjustment to the 100-mg QD dose is warranted. The exposure-response analyses indicate the existence of flat or, at most, shallow exposure-response relationships for the different efficacy endpoints for doravirine. A decrease in response in individuals with exposures below the 10th percentile observed for the 100-mg QD regimen may be associated with suboptimal compliance, indicating the importance of adherence to once-daily dosing.
MATERIALS AND METHODS
Population PK model.
Source data were obtained from 20 phase 1 trials (protocols MK-1439-001, MK-1439-002, MK-1439-003, MK-1439-005 [ClinicalTrials.gov registration number NCT01466985], MK-1439-009, MK-1439-010, MK-1439-011 [ClinicalTrials.gov registration number NCT03272347], MK-1439-016, MK-1439-019 [ClinicalTrials.gov registration number NCT02089659], MK-1439-020, MK-1439A-026, MK-1439A-029, MK-1439-034, MK-1439-035, MK-1439-037, MK-1439-038, MK-1439-039, MK-1439-042, MK-1439-043, and MK-1439-051 [ClinicalTrials.gov registration number NCT02641067]), a phase 2b trial (protocol MK-1439-007 [ClinicalTrials.gov registration number NCT01632345]), and two phase 3 trials (protocols MK-1439-018 [ClinicalTrials.gov registration number NCT02275780] and MK-1439A-021 [ClinicalTrials.gov registration number NCT02403674]).
The phase 1 PK data used in this analysis were obtained after fasted administration of the oral compressed tablet (OCT) or film-coated tablet of doravirine (Merck & Co., Inc., Kenilworth, NJ, USA), for which bioequivalence has been demonstrated. Additionally, doravirine data from two phase 1 trials where the fixed-dose combination doravirine-3TC-TDF was administered were included (P026, P029). The data from the phase 2b and phase 3 PK trials were obtained after the administration of the doravirine OCT as a single entity together with FTC-TDF (P007) and either FTC-TDF or ABC-3TC (P018) or a fixed-dose combination of doravirine-3TC-TDF (P021). Further to the lack of a food effect in the dedicated phase 1 trial (11), doravirine was administered without regard to food in the phase 2b and phase 3 trials. Due to slightly less than dose-proportional increases in exposure over the range of doses evaluated in clinical development (7), only data for doses of 200 mg or less (actual dose range, 6 to 200 mg) were included in the analysis.
In the phase 1 trials, PK data were densely sampled (as many as 12 samples over the first 24-h period and every 24 h thereafter), while a sparse sampling scheme was used in the phase 2b and phase 3 trials. In the phase 2b trial, PK sampling time points were predose and ∼2 h postdose at the week 4 and 12 visits, and, irrespective of the time of the dose, at the week 8 and 16 visits. In the phase 3 trials, PK sampling time points were predose on day 1, predose at the week 4 visit, pre- or postdose at the week 8 visit, and predose and within 0.5 to 2 h postdose at the week 24 and 48 visits.
The population PK analysis was performed on log-transformed doravirine plasma concentrations using a nonlinear mixed-effects modeling approach. First, a structural model was established and interindividual variability and residual random effects were included in the model to capture the observed data. The less-than-dose-proportional increases in doravirine exposure, which are due to absorption limitations, were described by estimation of different relative bioavailabilities across the dose range included in the analysis. A screening of different dose cutoffs was performed in a stepwise fashion to find the range of doses where the relative bioavailability significantly differed from that of the clinical dose of 100 mg. Subsequently, identification of significant covariates impacting doravirine PK was done using SCM. This procedure involved stepwise testing of covariate relationships in a forward inclusion (change in the objective function value [ΔOFV], 6.63; P < 0.01 for 1 degree of freedom [df]) and backward exclusion (ΔOFV, 10.8; P < 0.001 for 1 df) procedure. The intrinsic factors investigated were age, weight, gender, race, ethnicity, renal function (based on the MDRD study-estimated eGFR), and healthy versus HIV-1 status. The administration of doravirine as a single entity or as the fixed-dose combination of doravirine, 3TC, and TDF was also investigated as a covariate. The precision and stability of the parameter estimates and evidence for a reduction in unexplained variability were considered in the decision to include covariates meeting the predefined statistical criteria in the final model. The reliability of the final model was checked with diagnostic plots (individual predicted concentration versus observed concentration, population predicted concentration versus observed concentration, conditional weighted residuals versus time), visual predictive checks of concentration versus time stratified by dose and by phase 1 versus phase 2b/3, and nonparametric bootstrap analysis (n = 1,000).
Individual doravirine steady-state parameters (AUC0–24, Cmax, C24) were estimated for each participant in the phase 2b and phase 3 trials. Individual post hoc compartmental PK parameters from the final population PK model were used to generate, without residual error, individual 24-h plasma concentration-time profiles for calculation of the steady-state AUC0–24, Cmax, and C24 for each individual in the phase 2b and 3 trials. Summary statistics were calculated for each PK parameter.
Simulations were performed with the final population PK model to address the magnitude of the covariate effects on doravirine PK to enable an assessment of their clinical relevance. The following simulations were performed: (i) characterization of univariate covariate effects for significant covariates on doravirine PK identified from the SCM covariate analysis (simulation 1) and (ii) simulation of the effects of the covariates of interest on doravirine steady-state PK parameters in a typical population with HIV-1, considering covariate correlations (simulation 2). For simulation 1, for the covariates included in the final model, single individuals were simulated per covariate subgroup (with subgroups defined as the 5th and 95th percentiles for continuous covariates), with the values for the other covariates being fixed to the population median value (continuous covariates) or those for the most prevalent group (categorical covariates). Based on the final model including only fixed effects, 24-h concentration-time profiles were simulated and individual values of the steady-state AUC0–24, Cmax, and C24 were calculated. For simulation 2, 1,000 individuals per subgroup (categorical covariates) or 1,000 individuals overall (continuous covariates) from the phase 2b/3 trials were resampled with their covariate information. For renal impairment and HIV-1 status, the full analysis data set (including data from phase 1 trials) was used as a basis for the resampling to enable informative comparisons for these subgroups (e.g., individuals with severe renal impairment versus lesser degrees of renal impairment, healthy versus HIV-1-infected individuals), as there were no participants in the phase 2b or 3 trials with severe renal impairment or without HIV-1. Based on the final model with interindividual as well as residual random effects, individual 24-h concentration-time profiles at steady state were simulated for the subgroups of selected covariates, and individual values of the steady-state AUC0–24, Cmax, and C24 were calculated. The geometric mean ratios and 90% confidence intervals (CI) for comparisons between subgroups were calculated.
Exposure-response modeling.
Exposure-response analyses were conducted with data from the phase 2b trial (P007) and two phase 3 trials (P018 and P021), in which oral doses of doravirine were administered as a single entity with FTC-TDF or ABC-3TC or as doravirine-3TC-TDF. The efficacy endpoints evaluated included the proportion of individuals achieving HIV-1 RNA levels of <50 copies/ml at week 48 and the occurrence of PDVF. Separate analyses were conducted with data from the phase 2b and phase 3 trials due to different definitions of PDVF and potential differences in response due to study conduct and treatment of individuals with PDVF (e.g., discontinuation rates). In the phase 2b trial, PDVF was defined as a nonresponse in individuals who never achieved HIV-1 RNA levels of <40 copies/ml by week 24 or as a rebound in individuals who, after an initial response of HIV-1 RNA levels of <40 copies/ml, had, at or after week 24, two consecutive measurements of HIV-1 RNA levels of ≥40 copies/ml at least 1 week apart. In the phase 3 trials, PDVF was defined as either (i) two consecutive measurements at least 1 week apart of HIV-1 RNA levels of ≥200 copies/ml at week 24 or week 36 or two consecutive measurements at least 1 week apart of HIV-1 RNA levels of ≥50 copies/ml at week 48 or (ii) two consecutive measurements at least 1 week apart of HIV-1 RNA levels of ≥50 copies/ml after an initial response of HIV-1 RNA levels of <50 copies/ml at any time during the study. Data from the two phase 3 trials were pooled, as the doravirine PK were similar when doravirine was administered as a single-entity tablet (P018) or as doravirine-3TC-TDF (P021). As the PK target for efficacy has focused on maintenance of minimal trough plasma concentrations, the steady-state C24 for each participant was considered the primary exposure variable for the efficacy PK/pharmacodynamic analysis; AUC0–24 and Cmax were also evaluated for completeness.
Exposure-response evaluations were performed through logistic regression. As an initial model, a linear exposure-response relationship was investigated. In the case of a statistically significant exposure-response relationship (P < 0.05), models of increased complexity (specifically, a log-linear model utilizing log-transformed exposure parameters) were explored, and the impact of covariates (age, weight, gender, race, screening HIV-1 RNA status [≤100,000 or >100,000 copies/ml]) was explored to address potential confounding between the effects of doravirine exposure and these covariates. The goodness of fit of the final models was assessed by overlaying the observed responses with the model-predicted responses across quantiles of exposure and by numerical predictive check, where simulated proportions of events and observed proportions were compared across quantiles of exposure.
Data availability.
The data-sharing policy of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, including restrictions, is available at http://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this research was provided by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA. Medical writing assistance, under the direction of the authors, was provided by Annette Smith of CMC Affinity, a division of McCann Health Medical Communications Ltd, Macclesfield, UK, in accordance with Good Publication Practice (GPP3) guidelines. This medical writing assistance was funded by Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA.
K.L.Y. and L.W. are employees of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, and may own stock and/or stock options in Merck & Co., Inc., Kenilworth, NJ, USA. A.O., A.C., and R.D.G. are employees of Certara, through which they provided paid consultancy services to Merck & Co., Inc., Kenilworth, NJ, USA.
K.L.Y. substantially contributed to the conception, design, or planning of the study. K.L.Y., A.O., A.C., R.D.G., and L.W. all substantially contributed to analysis of the data, interpretation of the results, and critically reviewing or revising the manuscript for important intellectual content. K.L.Y. also substantially contributed to drafting of the manuscript. All authors reviewed and approved the version of the manuscript to be submitted.
We thank Pavan Vaddady for his help with the analysis of data from individuals with at least one sample below the limit of quantitation.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AAC.02502-18.
REFERENCES
- 1.Cote B, Burch JD, Asante-Appiah E, Bayly C, Bedard L, Blouin M, Campeau LC, Cauchon E, Chan M, Chefson A, Coulombe N, Cromlish W, Debnath S, Deschenes D, Dupont-Gaudet K, Falgueyret JP, Forget R, Gagne S, Gauvreau D, Girardin M, Guiral S, Langlois E, Li CS, Nguyen N, Papp R, Plamondon S, Roy A, Roy S, Seliniotakis R, St-Onge M, Ouellet S, Tawa P, Truchon JF, Vacca J, Wrona M, Yan Y, Ducharme Y. 2014. Discovery of MK-1439, an orally bioavailable non-nucleoside reverse transcriptase inhibitor potent against a wide range of resistant mutant HIV viruses. Bioorg Med Chem Lett 24:917–922. doi: 10.1016/j.bmcl.2013.12.070. [DOI] [PubMed] [Google Scholar]
- 2.Merck Sharp & Dohme Corp. 2018. Pifeltro (doravirine) prescribing information. Merck & Co., Inc, Whitehouse Station, NJ. [Google Scholar]
- 3.Merck Sharp & Dohme Corp. 2018. Delstrigo (doravirine, lamivudine, and tenofovir disoproxil fumarate) prescribing information. Merck & Co., Inc, Whitehouse Station, NJ. [Google Scholar]
- 4.Gatell JM, Morales-Ramirez JO, Hagins DP, Thompson M, Keikawus A, Hoffmann C, Rugina S, Osiyemi O, Escoriu S, Dretler R, Harvey C, Xu X, Teppler H. 2014. Forty-eight-week efficacy and safety and early CNS tolerability of doravirine (MK-1439), a novel NNRTI, with TDF/FTC in ART-naive HIV-positive patients. J Int AIDS Soc 17:19532. doi: 10.7448/IAS.17.4.19532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Orkin C, Squires KE, Molina JM, Sax PE, Wong WW, Sussmann O, Kaplan R, Lupinacci L, Rodgers A, Xu X, Lin G, Kumar S, Sklar P, Nguyen BY, Hanna GJ, Hwang C, Martin EA. DRIVE-AHEAD Study Group. 2019. Doravirine/lamivudine/tenofovir disoproxil fumarate is non-inferior to efavirenz/emtricitabine/tenofovir disoproxil fumarate in treatment-naive adults with human immunodeficiency virus-1 infection: week 48 results of the DRIVE-AHEAD trial. Clin Infect Dis 68:535–544. doi: 10.1093/cid/ciy540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Molina JM, Squires K, Sax PE, Cahn P, Lombaard J, DeJesus E, Lai MT, Xu X, Rodgers A, Lupinacci L, Kumar S, Sklar P, Nguyen BY, Hanna GJ, Hwang C, DRIVE-FORWARD Study Group. 2018. Doravirine versus ritonavir-boosted darunavir in antiretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 48-week results of a randomised, double-blind, phase 3, non-inferiority trial. Lancet HIV 5:e211–e220. doi: 10.1016/S2352-3018(18)30021-3. [DOI] [PubMed] [Google Scholar]
- 7.Anderson MS, Gilmartin J, Cilissen C, De Lepeleire I, van Bortel L, Dockendorf MF, Tetteh E, Ancona JK, Liu R, Guo Y, Wagner JA, Butterton JR. 2015. Safety, tolerability and pharmacokinetics of doravirine, a novel HIV non-nucleoside reverse transcriptase inhibitor, after single and multiple doses in healthy subjects. Antivir Ther 20:397–405. doi: 10.3851/IMP2920. [DOI] [PubMed] [Google Scholar]
- 8.Schürmann D, Sobotha C, Gilmartin J, Robberechts M, De Lepeleire I, Yee KL, Guo Y, Liu R, Wagner F, Wagner JA, Butterton JR, Anderson MS. 2016. A randomized, double-blind, placebo-controlled, short-term monotherapy study of doravirine in treatment-naive HIV-infected individuals. AIDS 30:57–63. doi: 10.1097/QAD.0000000000000876. [DOI] [PubMed] [Google Scholar]
- 9.Yee KL, Sanchez RI, Auger P, Liu R, Fan L, Triantafyllou I, Lai MT, Di Spirito M, Iwamoto M, Khalilieh SG. 2017. Evaluation of doravirine pharmacokinetics when switching from efavirenz to doravirine in healthy subjects. Antimicrob Agents Chemother 61:e01757-16. doi: 10.1128/AAC.01757-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Behm MO, Yee KL, Fan L, Fackler P. 2017. Effect of gender and age on the relative bioavailability of doravirine: results of a phase I trial in healthy subjects. Antivir Ther 22:337–344. doi: 10.3851/IMP3142. [DOI] [PubMed] [Google Scholar]
- 11.Behm MO, Yee KL, Liu R, Levine V, Panebianco D, Fackler P. 2017. The effect of food on doravirine bioavailability: results from two pharmacokinetic studies in healthy subjects. Clin Drug Investig 37:571–579. doi: 10.1007/s40261-017-0512-5. [DOI] [PubMed] [Google Scholar]
- 12.Ankrom W, Yee KL, Sanchez RI, Adedoyin A, Fan L, Marbury T, Preston RA, Iwamoto M, Khalilieh SG. 2018. Severe renal impairment has minimal impact on doravirine pharmacokinetics. Antimicrob Agents Chemother 62:e00326-18. doi: 10.1128/AAC.00326-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanchez RI, Fillgrove KL, Yee KL, Liang Y, Lu B, Tatavarti A, Liu R, Anderson MS, Behm MO, Fan L, Li Y, Butterton JR, Iwamoto M, Khalilieh SG. 28 March 2018. Characterisation of the absorption, distribution, metabolism, excretion and mass balance of doravirine, a non-nucleoside reverse transcriptase inhibitor in humans. Xenobiotica doi: 10.1080/00498254.2018.1451667. [DOI] [PubMed] [Google Scholar]
- 14.Sotaniemi EA, Arranto AJ, Pelkonen O, Pasanen M. 1997. Age and cytochrome P450-linked drug metabolism in humans: an analysis of 226 subjects with equal histopathologic conditions. Clin Pharmacol Ther 61:331–339. doi: 10.1016/S0009-9236(97)90166-1. [DOI] [PubMed] [Google Scholar]
- 15.McLachlan AJ, Pont LG. 2012. Drug metabolism in older people—a key consideration in achieving optimal outcomes with medicines. J Gerontol A Biol Sci Med Sci 67:175–180. doi: 10.1093/gerona/glr118. [DOI] [PubMed] [Google Scholar]
- 16.Feng M, Sachs NA, Xu M, Grobler J, Blair W, Hazuda DJ, Miller MD, Lai M-T. 2016. Doravirine suppresses common nonnucleoside reverse transcriptase inhibitor-associated mutants at clinically relevant concentrations. Antimicrob Agents Chemother 60:2241–2247. doi: 10.1128/AAC.02650-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data-sharing policy of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ, USA, including restrictions, is available at http://engagezone.msd.com/ds_documentation.php. Requests for access to the clinical study data can be submitted through the EngageZone site or via email to dataaccess@merck.com.