

Abstract
Heterogeneous Pd‐containing nanocatalysts, based on poly (propylene imine) dendrimers immobilized in silica pores and networks, obtained by co‐hydrolysis in situ, have been synthesized and examined in the hydrogenation of various unsaturated compounds. The catalyst activity and selectivity were found to strongly depend on the carrier structure as well as on the substrate electron and geometric features. Thus, mesoporous catalyst, synthesized in presence of both polymeric template and tetraethoxysilane, revealed the maximum activity in the hydrogenation of various styrenes, including bulky and rigid stilbene and its isomers, reaching TOF values of about 230000 h−1. Other mesoporous catalyst, synthesized in the presence of polymeric template, but without addition of Si(OEt)4, provided the trans‐cyclooctene formation with the selectivity of 90–95 %, appearing as similar to homogeneous dendrimer‐based catalysts. Microporous catalyst, obtained only on the presence of Si(OEt)4, while dendrimer molecules acting as both anchored ligands and template, demonstrated the maximum activity in the hydrogenation of terminal linear alkynes and conjugated dienes, reaching TOF values up to 400000 h−1. Herein the total selectivity on alkene in the case of terminal alkynes and conjugated dienes reached 95–99 % even at hydrogen pressure of 30 atm. The catalysts synthesized can be easily isolated from reaction products and recycled without significant loss of activity.
Keywords: palladium nanoparticles, hydrogenation, organic-inorganic hybrid composites, immobilized dendrimers
Introduction
Systems based on metal nanoparticles are the most prospective catalysts for hydrogenation, oxidation and cross‐coupling reactions.1 They may be applied both as homogeneous and heterogeneous catalysts. In the last case metal nanoparticles are supported on the various inorganic carriers, such as mesoporous silica and zeolites,2 Al2O3,3 carbon doped silica4 and carbon materials (activated carbon,5 carbon nanofibers and nanotubes,6 polyaromatic frameworks7) to prevent particles agglomeration and subsequent catalyst deactivation. In homogeneous catalysts various donor organic ligands, both low‐molecular (amines,8 ionic liquids,1a,8b–8d, 9 phosphines8b,8d,10) and supramolecular ligands (poly‐(N‐vinyl pyrrolidone),1a,8b,8d,11 poly(ethylene glycol),1a,12 poly(ethylene imine),11a,13 calix‐arenes,14 cyclodextrines15 and dendrimers1a,16), are used to stabilize metal nanoparticles.
Due to their physical and chemical properties, dendrimers, which are the spherically symmetrical globular macromolecules with branched regular structure,17 appeared as the most interesting for their application in catalysis. The primary merits of dendrimers are: 1) sorption of strictly defined metal quantity;17c 2) control of solubility and substrate selectivity due to the dendrimer end group modification;17a,18 3) recyclability due to the fractional precipitation of dendritic species from the solution.19
For the first time dendrimers were suggested as structure‐forming agents for metal nanoparticles‐based catalytic systems instead of block co‐polymers20 to prevent exchange by macromolecules and metal ions between micelles. Thus especial nanoreactors, in which metal nanoparticles were encapsulated within the dendrimer molecule, were proposed (Figure 1).21
Figure 1.
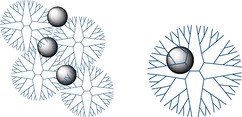
Dendrimer‐stabilized (left) and dendrimer‐encapsulated nanoparticles (right).21
This approach can be realized only under the following conditions:22 1) dendrimer peripheral groups must not be able to complex formation to prevent nanoparticle stabilization; 2) dendrimer end groups must be charged to provide repulsion between dendrimer molecules; 3) dendrimer generation must be high enough to enclose nanoparticle. In this connection the much more widespread is the dendrimer stabilization (Figure 1), that does not require especial synthesis conditions23 and includes simple complex formation of dendrimers with the ions of metal sorbed, followed by reduction with sodium borohydride.17b
Using this approach, metal nanoparticles of various size and nature, stabilized by carbosilane,24 poly(aryl ether),19a,19f–19g poly(amido amine) (PAMAM) or poly(propylene imine) (PPI) dendrimers with amino,25 hydroxyl,26 thiol27 or carboxyl end groups28 can be obtained. Dendrimer molecules here act as surfactants,29 adsorbed on the surface of growing nanoparticles, resulting in the larger size of the latters and wider particle size distribution in comparison with dendrimer‐encapsulated nanoparticles.21, 22
The main disadvantage of such systems is the gradual metal leaching from stabilizing micelles, leading to catalyst deactivation.19f–19g In this connection the promising may appear the heterogenisation of dendrimer‐based catalysts, allowing to combine the advantages of both conventional homogeneous and heterogeneous catalysts, for that several approach were developed.
The first one was the cross‐linking of amino‐terminated poly(propylene imine) (PPI) and (poly)amidoamine (PAMAM) dendrimers by various bifunctional agents (diepoxides, diisocyanates), followed by metal ions impregnation and subsequent reduction (Scheme 1).30 The driving force for the metal sorption here was the complex formation. Metal ions and nanoparticles were retained in the carrier due to large amount of donor coordination groups in the network and also by the multiple sterical hindrances, originated from the latter.
Scheme 1.
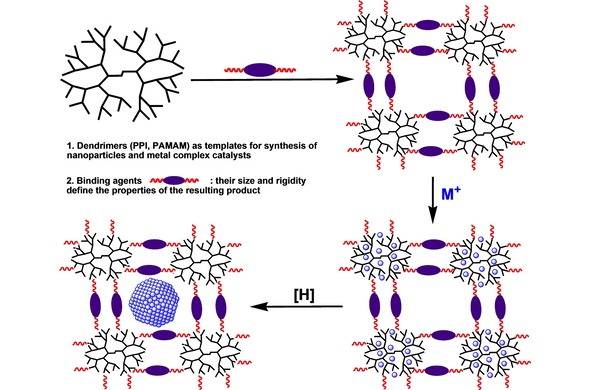
Approach to the synthesis of metal nanoparticles, encapsulated into the matrices of cross‐linked PPI or PAMAM dendrimers.30
Ru‐ and Rh containing catalysts, synthesized by this way, appeared as highly active catalysts for exhausting hydrogenation of aromatic compounds and phenols under two‐phase conditions, maintaining their efficacy at recycling.31 Analogously Pd nanoparticles, encapsulated into cross‐linked networks of PPI and PAMAM dendrimers, proved themselves as highly active, selective and stable catalysts for semi‐hydrogenation of phenyl acetylene and conjugated dienes.32 It was demonstrated, that the nature and generation of dendrimer used, the nature, size and rigidity of cross‐linking agent, as well as the synthesis conditions (temperature, solvent and the presence of polymeric template) had an essential influence on both physical chemical properties (particles size distribution, metal weight content, metal surface valency states etc.), activity and selectivity of the catalyst obtained.31c,32a
Similar approach was also developed in the works.33 Pd nanoparticles here were synthesized in situ, encapsulated in PAMAM dendrimers, co‐polymerized with ethylene glycol diacrylate.33a Herein nanoparticles growth occurred simultaneously with the meshy polymeric matrix formation, and the pore size was limited by the dendrimer diameter. Thus synthesized catalyst appeared as highly active in Suzuki‐Miyaura cross‐coupling in water, giving a yield exceeding 90 % within 2 hours, maintained during 8 cycles. Acid network‐like material, based on PPI dendrimers, cross‐linked with Sc(OTf)3, successfully catalyzed Mukaiyama aldol addition, Friedel‐Crafts acylation and Diels‐Alder coupling.33b
The main drawback of such materials, namely synthesized with the use of diisocyanates as cross‐linking agents, is the liability to metal‐catalyzed hydrolysis under two‐phase conditions, resulting in partial network destruction and, as a consequence, in particles agglomeration.31a
The alternative approach for heterogenization of dendrimer‐based catalysts is the grafting of dendrimers or dendrons on the surface of organic or inorganic insoluble carriers, such as SiO2 (both amorphous and mesoporous),34 cross‐linked poly(styrene),35 poly(thiophenes),36 poly(vinylpyridines)37 etc., by means of covalent or electrostatic interactions. Thus synthesized materials, impregnated with Pd, Ag, Au, Ru and Rh nanoparticles, proved their effectiveness in the selective hydrogenation of olefins,16,34a conjugated dienes,16,34b aromatic and heterocyclic compounds,16,36b cross‐coupling,16,35c,38 aldol condensation,35b carbonylation39 and oxidation reactions.40 Also immobilized dendrimers and dendrons can be impregnated by metal nanoparticles, already stabilized by simple organic ligands, e. g. ionic liquids.41
Multiwall carbon nanotubes (MWCNTs) were also proposed as prospective carriers for anchoring dendrimer‐based catalysts. Herein, as in the case of polystyrene or silica,34, 35 grafting the dendrons was performed by surface modification with amino groups followed by repeated Michael addition and subsequent end group activation.42 For this Pd‐PAMAM‐g‐MWCNTs catalysts not typical positive dendritic effect was observed in Mizoraki‐Heck cross‐coupling reaction: the reaction rate increased with the increase of dendron generation, that authors42 explained in terms of the enhanced stability due to sterical hindrances surrounding the catalytic centers.
These catalysts, based on the anchored dendrons, are stable at recycling and, as a rule, resistant to metal leaching.34, 35,42 Nonetheless, these materials are characterized by irregular dendrimer coating due to mutual sterical hindrances, arisen from bulky dendrons and resulting in irregular metal distribution through the carrier. To solve this problem, two different approaches were suggested.
The first one was the immobilization of ready dendrimers (not dendron grafting) on the silica polyamine composite via Mannich reaction (Scheme S1).43 Herein anchoring the dendrimers was performed, considering the space, occupied by the latters on surface of the carrier. Impregnated with Pd and PdAg nanoparticles, this material was characterized by regular and narrow particle size distribution and proved its high efficacy and stability in selective hydrogenation of phenyl acetylene, linear alkynes and conjugated dienes.43, 44 Similar approach was earlier applied for ready poly(ether imine) phosphine modified dendrimer, anchored to amorphous silica, preliminary functionalized with 3‐chloropropylsilane.45 Impregnated with Pd nanoparticles, this material was successfully applied in the hydrogenation of various alkenes.
Analogously, using aminomethylation reaction, PPI dendrimers can be anchored on the surface of ordered mesoporous phenol‐formaldehyde resins (OMR, MPF).46 Impregnated with Pd nanoparticles, such materials appeared as stable, very active and selective catalysts in semi‐hydrogenation of both terminal and internal liner alkynes, giving TOF values up 120000 h−1.46
An alternative approach was the co‐hydrolysis of Si(OEt)4 with PPI dendrimers, modified with (3‐glycidoxy)propyltri‐methoxysilane, in situ, resulting in regular porous material: microporous in the absence of the additional polymeric template (dendrimer appeared as both ligand for nanoparticles stabilization and template for pore formation) and mesoporous in the presence of polymeric PEG‐based template (Schemes 2 and 3).46 Impregnated with Ru nanoparticles, these catalysts were applied for the effective hydrogenation of phenols to corresponding cyclohexanols47 and for selective hydrogenation of levulinic acid and its esters to γ‐valerolactone.48
Scheme 2.
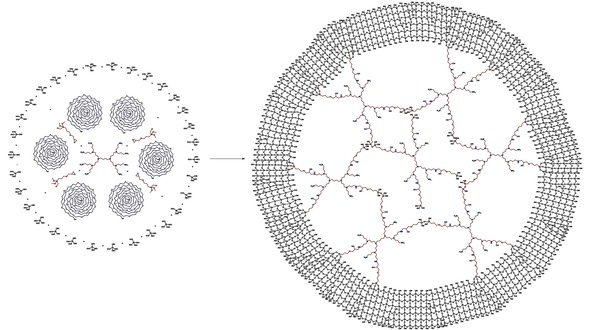
The synthesis of hybrid G2‐dendr‐meso‐SiO2 material.47
Scheme 3.
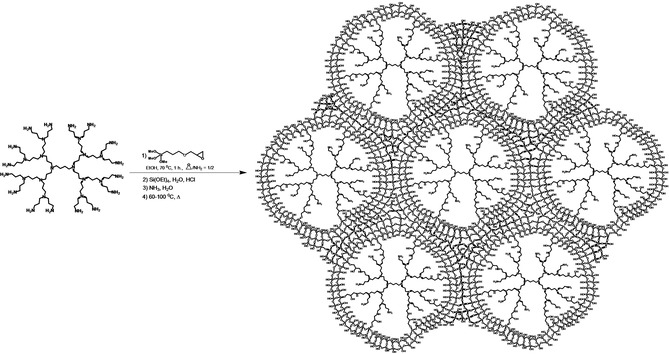
The synthesis of hybrid G3‐dendr‐SiO2 material.47
In the present work we propose the use of these hybrid dendrimer‐based organo‐silica materials as carriers for effective Pd nanocatalysts in the selective hydrogenation of unsaturated compounds.
Results and Discussion
The Synthesis of Bimetallic Dendrimer‐Based Heterogeneous Catalysts
In this work, in addition to earlier synthesized hybrid organo‐silica materials, denounced as G2‐dendr‐meso‐SiO2 and G3‐dendr‐SiO2, prepared by cohydrolysis tetra(ethoxy)silane with PPI dendrimers, modified with (3‐glycidoxy)propyl trimethoxysilane in the presence or absence of Pluronic template correspondingly (Schemes 2 and 3),46 recently we have developed a new organo‐silica carrier with low Si/N ratio. The latter have been achieved by exception of Si(OEt)4 from the synthetic procedure. As a consequence, PPI dendrimers of 2nd generation, partly modified with (3‐glycidoxy)propyl trimethoxysilane, underwent co‐hydrolysis with each other in the presence of polymeric template Pluronic P123 (Scheme S4).
Physical chemical properties of thus obtained meso‐G2‐dendr‐Si carrier, as well as of earlier synthesized G2‐dendr‐meso‐SiO2 and G3‐dendr‐SiO2, are presented in Tables S1 and S2. As seen from Figures S1–S3, in contrast to G2‐dendr‐meso‐SiO2 and G3‐dendr‐SiO2, meso‐G2‐dendr‐Si does not have well‐marked pores and channels in its structure and looks like as a typical dendrimer networks.31a One may suggest, that relatively bulk and flexible (in comparison with Si(OEt)4) PPI dendrimers are not able to form a rigid ensemble around the Pluronic micelle, resulting in amorphous structure.
XPS and solid‐state NMR spectroscopy of meso‐G2‐dendr‐Si sample (Table S1) confirmed the presence of fragments, typical for PPI dendrimers (NCH2CH2CH2N, NCH2CH2CH2NH2, etc.)49 and modifying groups, that link dendimers each other (SiCH2CH2CH2O, OCH2CH(OH)CH2NH).49, 50 Herein silicon was not detected, that probably might be due to its very low content in the sample. The another reason might be too deep location of Si groups under the surface, not detectable by XPS method, in contrast to dendrimer fragments, characterized by high surface C and N atomic concentrations.
13C NMR spectrum for meso‐G2‐dendr‐Si is relatively well resolved similar to G3‐dendr‐SiO2, while G2‐dendr‐meso‐SiO2 carrier is consisted of poorly solved, broadened signals, related to glycidyl (∼66–80, 80–96 ppm), dendrimer (∼21–66 ppm) and oxypropylsilane (∼12–21 ppm) fragments (Figure S4). Thus observed results may be explained by that dendrimers, anchored to the inner surface of mesopores in G2‐dendr‐meso‐SiO2, possess by partial mobility and, as a consequence, are subjected to much stronger vacillation in comparison to those in meso‐G2‐dendr‐Si, bounded into the network, or in G3‐dendr‐SiO2, where dendrimer molecules, being simultaneously a templates for pore formation, are anchored to their walls by all modified end groups (Schemes 3–4).35a,51
Scheme 4.
The synthesis of hybrid meso‐G2‐dendr‐Si material.
All three carriers: G2‐dendr‐meso‐SiO2, G3‐dendr‐SiO2 and meso‐G2‐dendr‐Si – were used as supports for Pd catalysts. Pd deposition was performed according to earlier published procedures for dendrimer‐based materials by wetness impregnation method, using Pd(OAc)2 in chloroform as a metal source and NaBH4 as a reducing agent (Scheme 5).43
Scheme 5.
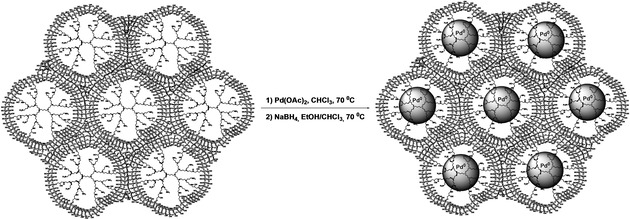
Encapsulation of Pd nanoparticles into dendrimer‐based organo‐silica carriers on the example.
The catalysts synthesized were characterized by means of atomic emission spectroscopy with inductively coupled plasma (ICP‐AES), transmission electron microscopy (TEM), X‐ray photoelectron (XPS) and solid‐state NMR spectroscopy. Physical chemical properties of the catalysts obtained are listed in Table 1 and illustrated in Figures 2 and 3 and S5–S13.
Table 1.
Physical chemical properties of the dendrimer‐based Pd‐containing catalysts.
| Entry | Catalyst | Pd content, wt. % | d, nm | XPS atomic concentrations, % | Pd 3d5/2 valency states, % (eV) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pd | C | N | O | Si | Pd0 | PdOx/Pd | PdO | Pd2+ | Pd(OAc)2, Pd (II) N‐bound | ||||
| 1 | G3‐dendr‐SiO2‐Pd | 6.70 | 2.81±0.71 | 3.1 | 28.3 | 4.2 | 48.5 | 15.9 | 54.5 (335.8) | – | 41.2 (337.4) | – | 4.3 (338.7) |
| 2 | G2‐dendr‐meso‐SiO2‐Pd | 7.58 | 3.29±0.34 | 2.7 | 32.1 | 3.6 | 48.2 | 13.1 | 63.9 (335.2) | 18.2 (336.5) | – | 16.3 (338.0) | 2.5 (339.0) |
| 3 | meso‐G2‐dendr‐Si‐Pd | 7.71 | 2.71±0.44 | 2.3 | 62.5 | 18.6 | 16.6 | – | 26.4 (335.3) | 45.0 (336.6) | – | 28.6 (337.8) | – |
Figure 2.
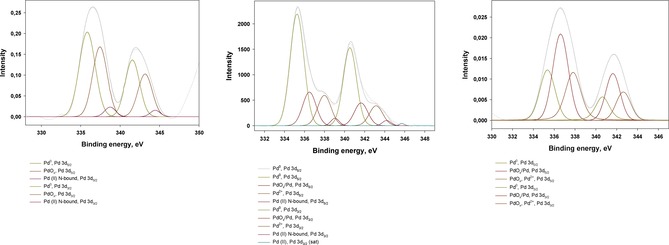
Pd 3d XPS spectra of the synthesized organo‐silica dendrimer‐based catalysts.
Figure 3.
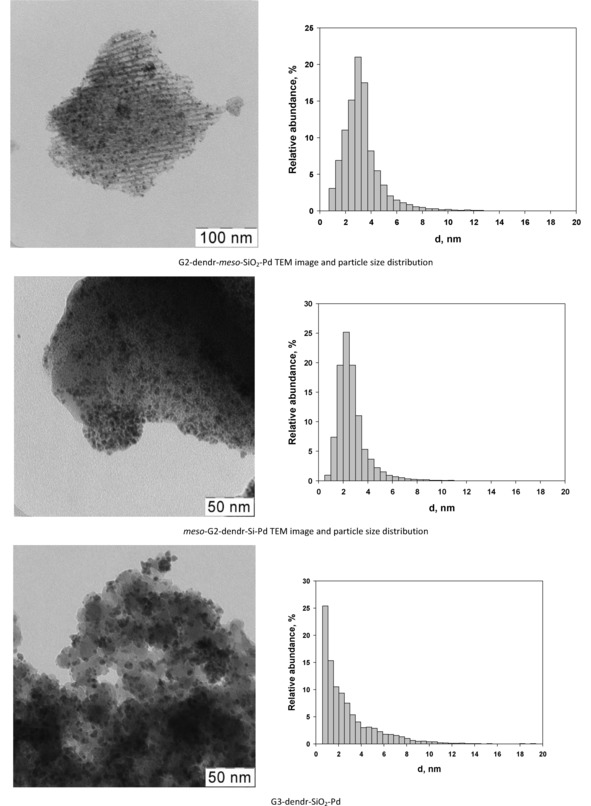
TEM images and particle size distributions of the synthesized Pd‐containing organo‐silica dendrimer‐based catalysts.
As one can see, both silica‐containing catalysts are characterized by Pd content, close to the theoretical one (Table 1). The latter can be calculated as the mass ratio of Pd, loaded in the synthesis, to the sum of Pd loaded and carrier mass and reached ∼8.7–9.3 % for G3‐dendr‐SiO2‐Pd and G2‐dendr‐meso‐SiO2‐Pd. In meso‐G2‐dendr‐Si‐Pd the found Pd content, vice versa, is inferior to theoretical (that is of 19.9 %) by ∼2.5 times. Similar phenomenon was earlier observed for meso‐G3‐PPI‐DMDPDI‐Pd catalyst, based on the PPI dendrimer network, also synthesized in the presence of polymeric template.32a
As in the case of dendrimer‐based hybrid organo‐silica Ru‐containing catalysts,46 the use of mesoporous carrier did not result in the significant particle size increase (Table 1). Nonetheless, the particle size distribution and morphology were different for each sample. G2‐dendr‐meso‐SiO2‐Pd was characterized by particle size distribution with a maximum at 3.0 nm and preferentially regular distribution of particles inside pores of the carrier (Figures 3 and S5). The electron diffraction, obtained from the larger nanoparticles by HRTEM method, also confirmed the presence of Pd crystallites in the sample (Table S3, Figures S6–S7). Herein Pd nanoparticles apparently ordered and limited vacillations of dendrimers, resulting in the better resolution for corresponding 13C NMR spectrum (Figure S8).
For meso‐G2‐dendr‐Si‐Pd particle size distribution was close to Gauss function with a maximum at 2.2 nm (Figures 3 and S9), often typical for the catalysts, based on the dendrimer networks.32 However particle distribution through the carrier surface was not regular, and sample contained areas both with high and low particle density (Figure S9).
Particle size distribution in G3‐dendr‐SiO2‐Pd is very similar to pore size distribution of the corresponding carrier with a maximum of ∼0.85 nm, a minimum diameter for Pd nanoparticles (Figures 3, S10–S11).2g,52 One may assume, that particles with diameter range of 0.85–2.5 nm are located predominantly in the micropores of the carrier, where the larger size corresponds to modified PPI dendrimer of 3rd generation in the expanded state. Larger crystallites (Figures S10, S12) are located respectively in the outer pore space, on the carrier surface, resulting in increase of the mean particle diameter. It can also be noted, that inclusion of Pd particles in the carrier structure had no a sufficient influence on the 13C NMR spectrum of the latter (Figure S13).
According to the XPS data (Table 1, Figure 2), Pd in all three samples was presented both in zero‐valent (Pd0, 335.0–335.7 eV at Pd 3d5/2)53 and oxidized forms (PdOx, 336.5 eV at Pd 3d5/2; PdO, 337.4 eV at Pd 3d5/2),54 that is typical for catalyst stored in the air55 and containing N‐ and O‐coordination groups in their structures.53a–53b,56 Nonetheless, the distribution of the latters was significantly dependent on the carrier structure for the certain catalyst. Thus, meso‐G2‐dendr‐Si‐Pd, in spite of the lowest oxygen content (∼17 %), was characterized by the lowest Pd0 portion (∼26.5 %) and maximum portion of the oxidized species in comparison with both G2‐dendr‐meso‐SiO2‐Pd and G3‐dendr‐SiO2‐Pd (Table 1, Figure 1). The ratio of surface Pd concentration, Cs (2.3 %), to the volume Pd content, Cv (7.7 %), was also the lowest for this catalyst an reached approximately 0.3. Herein it should be noted, that an oxygen portion, relating to silica knots, is extremely low in this sample; therefore Pd is appeared to bound with oxygen both from glycidyl groups of the carrier and adsorbed from the air.
G2‐dendr‐meso‐SiO2‐Pd, vice versa, was characterized by the highest portion of Pd0 state, reaching ∼64 % of total surface Pd content (Table 1). G3‐dendr‐SiO2‐Pd had the highest Cs/Cv ratio of ∼0.5 and Pd/O ratio close to those for G2‐dendr‐meso‐SiO2‐Pd, while Pd0 portion was ∼55 % (Table 1, Figure 1). The main difference for this sample is a relatively high binding energy for Pd0 state, as compared with G2‐dendr‐meso‐SiO2‐Pd and meso‐G2‐dendr‐Si‐Pd (Table 1). This phenomenon can be explained in terms of small particle sizes, predominating in the G3‐dendr‐SiO2‐Pd sample: the smaller particle size, the higher uncompensated surface partly positive charge, and, as a consequence, the higher binding energy for Pd0.57
Also G3‐dendr‐SiO2‐Pd sample contained a low, but noticeable portion of Pd2+ ions, strongly bounded with the dendrimer fragments (Table 1). Analogous tendency was earlier observed for Pd nanoparticles, encapsulated in PPI dendrimers of 3rd generation, immobilized on the silica‐polyamine composite surface,43 as well as for Pd nanoparticles, encapsulated in high densely cross‐linked networks of PPI and PAMAM dendrimers.32a
It appears, that hyper‐branched PPI dendrimers of 3rd generation or PAMAM dendrimers of 2nd generation are able not only to stabilize metal nanoparticles through electron donation,1a,16,17b–17c but also to interfere the metal ions reduction due to formation of stable chelate complexes.
Pd2+ ions, those may origin from the rest Pd(OAc)2 or weakly bounded Pd complexes with dendrimers, are absent in G3‐dendr‐SiO2‐Pd, as compared with G2‐dendr‐meso‐SiO2‐Pd and meso‐G2‐dendr‐Si‐Pd (Table 1). At the same time the remarkable portion of PdO in this sample can be referred not only to the oxidation in the air,55 but also to the strong interactions of Pd species with silica matrix,53a to those small particles are subjected extremely.57
Hydrogenation of Unsaturated Compounds in the Presence of Hybrid Dendrimer‐Based Pd Catalysts
Synthesized catalysts were tested in the hydrogenation of various unsaturated compounds, such as styrenes, alkynes and dienes. Catalyst activity, defined as the reaction turnover frequency (TOF), was calculated as amount of the substrate reacted (νsubstr) per mole of metal (νPd) per unit of time, according to formula:
where ω is the substrate conversion, expressed in the unit fractions and t is the minimal reaction time, for which the reaction progress is measured.
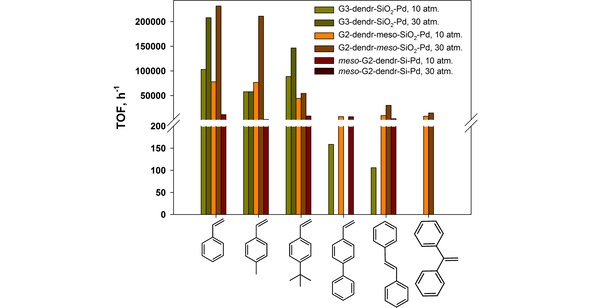
In hydrogenation of styrenes corresponding substituted alkylbenzenes were the only reaction products for all catalysts. It was established, that catalyst activity was simultaneously dependent on the carrier structure, substrate size and geometry, electron factors. Mesoporous G2‐dendr‐meso‐SiO2‐Pd catalyst appeared the highest activity for the most of substrates (Table 2, Figure 4). For styrene it exceeded 75000 h−1 at molar substrate/Pd ratio of ∼36760 and hydrogen pressure of 10 atm. and 230000 h−1 at molar substrate/Pd ratio of about 91900 and hydrogen pressure of 30 atm., that was extremely superior to Pd catalyst, based on PPI dendrimers, anchored on the silica polyamine composite (Pd‐G3‐dendr@BP‐1), earlier tested in the analogous reaction.43 So high efficacy for G2‐dendr‐meso‐SiO2‐Pd can be attributed to its regular mesoporous structure, providing the well access for substrate to the active Pd nanoparticles, while the ratio of Pd0/(PdOx+Pd2+) was approximately equal for both catalysts. Nonetheless, it should also be noted, that in the present work we have used higher temperatures (80 °C instead of 70 °C43) and carried out the experiments for liquid substrates in the absence of solvent, thus avoiding the competitive adsorption of the latter and the dilution effect.
Table 2.
Hydrogenation of styrenes in the presence of G2‐dendr‐meso‐SiO2‐Pd catalyst.[a]
| Entry | Substrate | Catalyst loading, mg | Substrate/Pd (mol/mol) | P (H2), atm. | Conv., % | TOF, h−1 |
|---|---|---|---|---|---|---|
| 1 | Styrene | 0.5 | 36760 | 10 | 53 | 77930 |
| 2 | 0.5 | 36760 | 30 | 100 | 147040 | |
| 3 | 0.5 | 91900 | 30 | 63 | 231590 | |
| 4 | 4‐methylstyrene | 0.5 | 38360 | 10 | 50 | 76725 |
| 5 | 0.5 | 38360 | 30 | 94 | 144240 | |
| 6 | 0.5 | 95905 | 30 | 55 | 210995 | |
| 7 | 4‐tert‐butylstyrene | 0.5 | 18395 | 10 | 60 | 44155 |
| 8 | 0.5 | 18395 | 30 | 74 | 54455 | |
| 9 | 4‐phenylstyrene | 1 | 2335 | 10 | 79 | 7385 |
| 10 | 0.5 | 4665 | 10 | 10.5 | 1965 | |
| 11 | trans‐stilbene | 1 | 2335 | 10 | 98.5 | 9205 |
| 12 | 0.5 | 4665 | 10 | 50.5 | 9440 | |
| 13 | 0.5 | 4665 | 30 | 100 | 18695 | |
| 14 | 0.5 | 9330 | 30 | 81 | 30285 | |
| 15 | 0.5 | 14000 | 30 | 46 | 25795 | |
| 16 | 1,1‐diphenylethylene | 1 | 2385 | 10 | 91 | 8685 |
| 17 | 0.5 | 4770 | 10 | 43 | 8205 | |
| 18 | 0.5 | 4770 | 30 | 77 | 14695 |
[a] Reaction conditions are: 80 °C, 15 min.
Figure 4.
Hydrogenation of styrenes in the presence of the dendrimer‐based organo‐silica palladium nanocatalysts. Reaction conditions are: 80 °C, 15 min.
With the substrate size increased, catalytic activity for G2‐dendr‐meso‐SiO2‐Pd decreased (Table 2, Figure 4), that was typical for the dendrimer‐based catalysts.17c,18a This downfall was especially remarkable for substrates, such as trans‐stilbene, 4‐phenylstyrene and 1,1‐diphenylethylene, whose substituents (phenyl rings) possessed the weak –M‐effect, along with bulk and rigid substrate geometry, hindering the adsorption of the latter. Even slightly increase of substrate/Pd ratio from ∼2300–2400 to ∼4700–4800 resulted in notable decrease in conversion: from 98 to 50 % for trans‐stilbene, from 79 to 10 % for 4‐phenylstyrene and from 91 to 43 % for 1,1‐diphenylethylene within 15 minutes at hydrogen pressure of 10 atm. (Table 2).
The lowest conversion for 4‐phenylstyrene in comparison with its isomers, 1,1‐diphenylethylene and trans‐stilbene, can be explained in terms of geometric factors: too closely located, phenyl rings in the 4‐phenylstyrene are perpendicularly oriented to each other, minimizing the steric energy and, as a consequence, sufficiently retarding the substrate adsorption. In 1,1‐diphenylethylene molecule phenyl rings are located farther from each other, facilitating the adsorption. In the trans‐stilbene molecule both C=C double bond and phenyl rings are located in the same plane, making the adsorption the most favourable. Increase in hydrogen pressure up to 30 atm. allowed to achieve conversions of 77 and 100 % for 1,1‐diphenylethylene and trans‐stilbene respectively (Table 2).
Activity of the microporous G3‐dendr‐SiO2‐Pd catalyst was strongly affected by both electron and geometry factors (Figure 4), that was originated form its hindered structure (Scheme 5). Thus, in the styrene hydrogenation turnover frequency for G3‐dendr‐GlycdSiO2‐Pd was comparable with that for G2‐dendr‐meso‐SiO2‐Pd, reaching 103140 h−1 at molar substrate/Pd ratio of ∼41560 and hydrogen pressure of 10 atm. and 207945 h−1 at molar substrate/Pd ratio of ∼103970 and hydrogen pressure of 30 atm. (Table 3). Then it dropped down for 4‐methylstyrene and rose again, when 4‐tert‐butylstyrene was used as s substrate (4‐tert‐butylstyrene). In the last case turnover frequency for G3‐dendr‐GlycdSiO2‐Pd reached 88805 h−1 at molar substrate/Pd ratio of ∼14465 and 10 atm. of H2 and 146525 h−1 at molar substrate/Pd ratio of ∼41625 and 30 atm. of H2 within 15 min. (Table 3), noticeably exceeding that for G2‐dendr‐meso‐SiO2‐Pd (Table 2).
Table 3.
Hydrogenation of styrenes in the presence of G3‐dendr‐SiO2‐Pd catalyst.[a]
| Entry | Substrate | Catalyst loading, mg | Substrate/Pd (mol/mol) | P (H2), atm. | Conv., % | TOF, h−1 |
|---|---|---|---|---|---|---|
| 1 | Styrene | 0.5 | 41590 | 10 | 62 | 103140 |
| 2 | 0.5 | 41590 | 30 | 100 | 166365 | |
| 3 | 0.5 | 103970 | 30 | 50 | 207945 | |
| 4 | 4‐methylstyrene | 0.5 | 14465 | 10 | 100 | 57870 |
| 5 | 0.5 | 21700 | 10 | 33 | 28645 | |
| 6 | 0.5 | 21700 | 30 | 64 | 55555 | |
| 7 | 4‐tert‐butylstyrene | 0.5 | 27750 | 10 | 80 | 88805 |
| 8 | 0.5 | 27750 | 30 | 95 | 105455 | |
| 9 | 0.5 | 41625 | 10 | 22.5 | 37465 | |
| 10 | 0.5 | 41625 | 30 | 88 | 146525 | |
| 11 | 4‐phenylstyrene | 1 | 2645 | 10 | 4 [b] | 105 |
| 12 | trans‐stilbene | 1 | 2645 | 10 | 6 [b] | 160 |
[a] Reaction conditions are: 80 0C, 15 min. [b] 1 h.
The phenomenon observed can be explained in terms of strong +I‐effect of the tert‐butyl group, located oppositely to C=C double bond in the side chain of 4‐tert‐butylstyrene molecule (the concerted action of substituents). G3‐dendr‐SiO2‐Pd is characterized by less portion of Pd0 on its surface (Table 1, Figure 1) and higher portion of small particles, having uncompensated partly positive charge on their surface (Figure 6). Therefore, the strong +I‐effect of 4‐tert‐butylstyrene can play a play key role in the preferential adsorption and the subsequent hydrogenation of the latter in comparison with 4‐methylstyrene and other bulky substrates.
Figure 6.
Hydrogenation of alkynes in the presence of the dendrimer‐based organo‐silica palladium catalysts. Reaction conditions are: 80 °C, 15 min.
Extremely low conversions for rigid 4‐phenylstyrene and trans‐stilbene (4 and 6 % within 1 hour correspondingly, Table 3) confirm our suggestion, as we think. Herein both sterical factors (hindered catalyst structure, bulky and rigid geometry of substrate) and electron factors (−M‐substituents) act together, resulting in the retarded hydrogenation.
G2‐dendr‐meso‐SiO2‐Pd and G3‐dendr‐SiO2‐Pd were tested on the possibility of recycling on the example of styrene (Figure 5). The recycling test was performed analogously to a standard hydrogenation procedure in the presence of heterogeneous dendrimer‐based palladium catalysts.32a,43 For the better sedimentation of the catalyst and, therefore, for the prevention of mechanical losses, the reaction products were additionally diluted with n‐hexane, which is a poor solvent for dendrimers.32a,43 The resulting solution was separated by decantation, and the remaining catalyst after sedimentation (∼30 min.) was used in the subsequent cycles without additional loading and regeneration.
Figure 5.
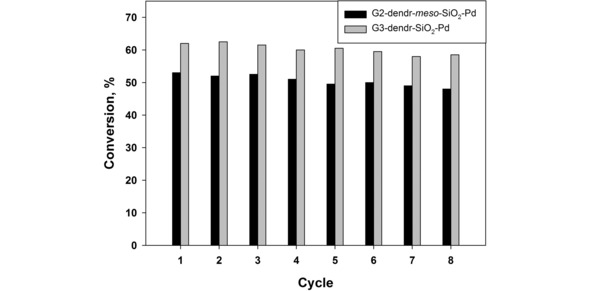
Recycling of G2‐dendr‐meso‐SiO2‐Pd and G3‐dendr‐SiO2‐Pd in the hydrogenation of styrene. Reaction conditions: 0.5 mg of cat., 1500 μL of the substrate (substrate/Pd ≈ 36760 for G2‐dendr‐meso‐SiO2‐Pd and 41590 for G3‐dendr‐SiO2‐Pd), 15 min., 80 °C, 10 atm. of H2.
This experiment revealed, that the catalysts said can be used six or more times without significant loss in activity. Total turnover number for eight cycles was 110280 for G2‐dendr‐meso‐SiO2‐Pd and 148885 for G3‐dendr‐SiO2‐Pd. Pd leaching during the test did not exceed 0.5 % according to ICP‐AES. Herein we suppose some mechanical losses of the catalysts as inevitable due to their fine dispersity and very low initial loading.
In contrast to G2‐dendr‐meso‐SiO2‐Pd and G3‐dendr‐SiO2‐Pd, meso‐G2‐dendr‐Si‐Pd revealed unexpectedly low activity in the hydrogenation of styrene (Table 4, Figure 4). An induction period was observed in the hydrogenation of all substrates (Figure S14); therefore the TOF values for meso‐G2‐dendr‐Si‐Pd were calculated with the use of time ranges, corresponding the maximum slopes on the kinetic curves (Figure S15).
Table 4.
Hydrogenation of styrenes in the presence of meso‐G2‐dendr‐Si−Pd.[a]
| Entry | Substrate | Catalyst loading, mg | Substrate/Pd (mol/mol) | t, min | Conv., % | TOF, h−1 |
|---|---|---|---|---|---|---|
| 1 | Styrene | 1 | 3010 | 60 | 100 | 8310 |
| 2 | 30 | 96 | ||||
| 3 | 15 | 27 | ||||
| 4 | 0.5 | 6025 | 60 | 96.5 | 11325 | |
| 5 | 30 | 69 | ||||
| 6 | 15 | 22 | ||||
| 7 | 4‐methylstyrene | 1 | 3145 | 60 | 18.5 | 1700 |
| 8 | 30 | 18 | ||||
| 9 | 15 | 4.5 | ||||
| 10 | 0.5 | 6285 | 60 | 10 | 880 | |
| 11 | 30 | 6.5 | ||||
| 12 | 15 | 3.5 | ||||
| 13 | 4‐tert‐butylstyrene | 1 | 3015 | 60 | 85.5 | 7595 |
| 14 | 30 | 81 | ||||
| 15 | 15 | 18 | ||||
| 16 | 0.5 | 6030 | 60 | 72 | 8555 | |
| 17 | 45 | 62 | ||||
| 18 | 30 | 20.5 | ||||
| 19 | 15 | 9 | ||||
| 20 | 4‐phenylstyrene | 1 | 2300 | 60 | 95.5 | 7030 |
| 21 | 30 | 80 | ||||
| 22 | 15 | 3.5 | ||||
| 23 | 0.5 | 4595 | 60 | 7 | 550 | |
| 24 | 30 | 4.5 | ||||
| 25 | 15 | 3 | ||||
| 26 | trans‐stilbene | 1 | 2300 | 60 | 63 | 3030 |
| 27 | 30 | 34.5 | ||||
| 28 | 15 | 1.5 |
[a] Reaction conditions are: 80 0C, 10 atm. of H2.
As seen from Figure S14 and Table 4, even slight increase in molar substrate/Pd ratio from ∼2300–3100 to 4600–6300 resulted in sharp decrease in the reaction rate for the most of the substrates. Prolongation of the induction period was also observed (Figure S14). We suppose, such behavior of the meso‐G2‐dendr‐Si‐Pd catalyst can be connected with the predominance of the oxidized Pd species on the surface (Table 1, Figure 1). This supposal may be confirmed by the results of 4‐tert‐butylstyrene hydrogenation, being significantly superior to those of 4‐methylstyrene. Herein 4‐tert‐butylstyrene by the strong +I‐effect was characterized.
The structure of meso‐G2‐dendr‐Si carrier is expanded and loose (Scheme 4). It resulted in elimination of the sterical factors in the hydrogenation of bulky and rigid 4‐phenylstyrene and trans‐stilbene (Figure 4, Table 4). Nonetheless, due to the −M‐effect of substituents, they are still inferior to 4‐tert‐butylstyrene in their propensity to hydrogenation, that was especially notable at increased substrate/Pd ratios (Table 4, Figure S14).
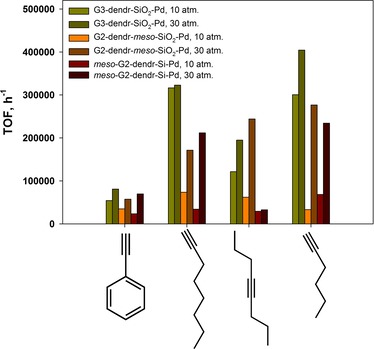
Selective hydrogenation of acetylene compounds is one of the most important problems in petrochemistry, since they poison polymerization catalysts.59 Therefore, the catalysts synthesized were tested in the hydrogenation of various alkynes, such as phenylacetylene, 1‐octyne, 4‐octyne and 1‐hexyne. As seen from Figure 6, phenylacetylene was subjected to hydrogenation in the significantly lesser degree in comparison with linear alkynes for all catalysts tested.
These results can be explained in terms of both sterical and electron factors. Thus, on the one hand, rigid phenylacetylene, with its planar geometry, requires much larger adsorption area for successful hydrogenation of the C≡C triple bond, than flexible linear alkynes. As a consequence, the same Pd nanoparticle can simultaneously adsorb linear alkyne molecules in rather large quantity, as compared with those of phenylacetylene. On the other hand, alkyl substituents in the 1‐octyne, 4‐octyne and 1‐hexyne molecules possess the weak +I‐effect, that facilitates substrate adsorption on Pd nanoparticles – in contrast to weak −M‐effect of phenyl ring in the phenylacetylene molecule.19b,32a
The presence of basic dendrimer amino groups strengthens polarization of terminal C−H bond near to the C≡C triple bond in phenylacetylene molecule and, as a consequence, the acidity of the terminal proton. On the one hand, it leads to increase in the electron density on the C≡C triple bond and, as a consequence, to the enhanced selectivity on styrene.32a On the other hand, it favours to the predominant perpendicular adsorption of phenylacetylene molecule, not suitable for the subsequent hydrogenation to styrene. Nonetheless, due to the dendrimer action, a portion of PhCH2−C≡Pd and PhCH2−CH=Pd adsorbed species,57a,58 leading to ethylbenzene formation decreases, replaced by surface Ph−C≡Cδ−−Pdδ+ complexes.60 As a result, the reaction turnover frequency decreases (Figure 6) along with the styrene selectivity increased (Table 5).
Table 5.
Hydrogenation of phenyl acetylene in the presence of the dendrimer‐based organo‐silica palladium catalysts.[a]
| Entry | Catalyst | d, nm | DM, %[b] |
|
Substrate/Pd, (mol/mol) | P (H2), atm. | Conv., % | TOF, h−1 | Selectivity on styrene, % | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | G3‐dendr‐SiO2‐Pd | 2.81±0.71 | 31.5 | 1.2 | 7230 | 10 | 99.5 | 28780 | 90.5 | |
| 2 | 14455 | 10 | 77 | 44545 | 98 | |||||
| 3 | 14455 | 30 | 97 | 56115 | 95 | |||||
| 4 | 18070 | 30 | 82 | 59300 | 97 | |||||
| 5 | 21680 | 10 | 57 | 49465 | 98 | |||||
| 6 | 21680 | 30 | 79 | 68555 | 98 | |||||
| 7 | 28910 | 10 | 47 | 54380 | 98 | |||||
| 8 | 28910 | 40 | 70 | 80995 | 96 | |||||
| 9 | G2‐dendr‐meso‐SiO2‐Pd | 3.29±0.34 | 26.9 % | 4.4 | 6390 | 10 | 98 | 25055 | 67 | |
| 10 | 12785 | 10 | 63 | 32215 | 95.5 | |||||
| 11 | 12785 | 30 | 98 | 50115 | 84 | |||||
| 12 | 15980 | 30 | 90 | 57530 | 90.5 | |||||
| 13 | 19175 | 10 | 45.5 | 34900 | 96.5 | |||||
| 14 | 19175 | 30 | 75 | 57530 | 92 | |||||
| 15 | meso‐G2‐dendr‐Si‐Pd | 2.71±0.44 | 32.6 % | 2.5 | 6285 | 10 | 73 | 18350 | 92 | |
| 16 | 12570 | 10 | 47 | 23630 | 98 | |||||
| 17 | 12570 | 30 | 98 | 49279 | 87.5 | |||||
| 18 | 18850 | 30 | 92.5 | 69755 | 91.5 | |||||
| 19 | 25135 | 10 | 20.5 | 20610 | 98 | |||||
| 20 | 25135 | 30 | 53.5 | 53795 | 93.5 |
[a] Reaction conditions are: 0.5 mg of catalyst, 80 °C, 15 min;
[b] Particle dispersity DM can be calculated according to the formula: ,2g where d is mean particle diameter, expressed in nm, and 0.885 is a Pd reduced factor, corresponding to the ratio of the atomic phase volume vM to the average atomic effective area aM on the particle surface.61 It can be calculated according to the formula: .
Catalyst efficiency in the phenyl acetylene hydrogenation was strongly influenced by the carrier structure and the reaction conditions (substrate/Pd ratio, hydrogen pressure etc.). G3‐dendr‐SiO2‐Pd appeared as the most selective and efficient catalyst among the others tested: the portion of styrene among the reaction products reached 95–98 % even at hydrogen pressures of 30 atm. and was just slightly dependent on the substrate/Pd ratio (Table 5). Herein the maximum TOF value might reach 80995 h−1 (Table 5, Entry 8), and the maximum styrene yield was of 92 % (conversion of 97 %, selectivity of 95 %) was achieved at substrate/Pd ratio of ∼14455 and hydrogen pressure of 30 atm. (Table 5, Entry 3).
Mesoporous catalysts G2‐dendr‐meso‐SiO2‐Pd and meso‐G2‐dendr‐Si‐Pd, based on PPI dendrimers of 2nd generation, were noticeably inferior to G3‐dendr‐SiO2‐Pd in both activity and selectivity (Figure 6, Table 5). The maximum styrene yield of 81.5 % (conversion of 90 %, selectivity of 90.5 %) for G2‐dendr‐meso‐SiO2‐Pd catalyst, as well as TOF values of 57530 h−1, can be achieved at molar substrate/Pd ratio of ∼15980 and hydrogen pressure of 30 atm. within 15 minutes (Table 5, Entry 12). In the case of meso‐G2‐dendr‐Si‐Pd the best conditions for the selective phenyl acetylene hydrogenation were the substrate/Pd ratio of ∼18850 and hydrogen pressure of 30 atm., allowing to achieve the styrene yield of 84.5 % within 15 minutes (conversion of 92.5 %, selectivity of 91.5 %) at TOF value of 69755 h−1 (Table 5, Entry 18).
As one can see from Table 5, there is no certain dependency between catalyst efficiency and mean particle diameter, typical for many both homogeneous62 and heterogeneous catalysts6c,63 and connected with the ratio of [111] (active in phenylacetylene hydrogenation) and [110] planes (active in phenylacetylene adsoption and complex formation).6c,57a–57b,58b,63, 64 Analogously catalyst activity does not seem to be influenced by the ratio of reduced and oxidized valency states on the surface of Pd nanoparticles (Table 5). Moreover, the catalyst G3‐dendr‐SiO2‐Pd, characterized by the lowest Pd0/(PdO+Pd2+) ratio, appeared as the most active and selective in the phenyl acetylene hydrogenation, whereas oxidized Pd forms are known decrease selectivity on styrene.43
Therefore, we assume the carrier structure and ligand microenvironment appear to be the main factors, influencing the activity and selectivity of the dendrimer‐based organo‐silica palladium catalysts. Indeed, in the microporous G3‐dendr‐SiO2‐Pd catalyst, prepared in the absence of polymeric template, PPI dendrimers of the 3rd and higher generation are presumably located closely to each other. It results in Pd nanoparticles, stabilized by the large amount of donor amino groups simultaneously.1a,16,32a As a consequence, Pd surface is partly filled with electrons and much stronger adsorbs phenylacetylene with its high electron density on the C≡C triple bond (additionally polarized by the interactions with the basic dendrimer amino groups) in comparison with styrene, resulting both in higher reaction rate and selectivity on styrene.32a,58b,65 Herein for organo‐silica dendrimer‐based catalysts a positive dendritic effect was observed: the more dendrimer's generation, the more donor amino groups it has and, as a consequence, the more stabilized and electron‐enriched nanoparticles appearing to be.
We think our supposal to be indirectly confirmed by the results of the selective 1‐hexyne hydrogenation in the presence of Pd nanoparticles, stabilized by imidazolium‐modified 2,2’‐bipyridyl.66 Herein alkenes formed were not subjected to the further hydrogenation even at quantitative conversions and long reaction times (4–8 hours). It was established, that adsorption constants decreased in the following order: K1 (alkyne) > K (nitrogen ligand)≫K2 (alkene).
Addition of piperidine allowed not only to maintain high selectivity on 1‐butene in 1‐butyne hydrogenation, but also to noticeably increase the reaction rate for small particles in comparison with the reference Pd/Al2O3 catalyst.65c PAMAM dendron‐stabiliized bimetallic PtPd nanoparticles effectively catalyzed methylphenylacetylene hydrogenation to β‐methylstyrene with the yield of 94 %.18b Analogously poly(ethylene imine) based Pd‐containing catalysts, both homogeneous and heterogeneous, allowed to obtain cis‐stilbene from diphenylacetylene in 1,4‐dioxane medium with the selectivity of 94–97 %, which did not decrease even at very long reaction times (10–40 hours) and quantitative conversions.13,67
Another important advantage for the use of N‐containing donor ligands is the suppression of alkyne polymerization, typical for small particles (< 4 nm), especially at high substrate/Pd ratios.63a,68 Herein ligands presumably occupy low coordination electron‐deficient adsorption sites, characterized by the lowest selectivity in alkyne hydrogenation.63a,66
All catalysts studied appeared to be inferior to the amorphous Pd‐G3‐dendr@BP‐1 catalyst, based on PPI dendrimers of the 3rd generation,43 but surpassed it in the selectivity on styrene. As we think, this difference in behavior for the catalysts with the similar composition may originate from the features of Pd nanoparticles localization in the carrier. In Pd‐G3‐dendr@BP‐1 both dendrimers and Pd nanoparticles are located only on the outer surface of the carrier:43 it results in the better substrate access to catalytic centers in comparison with catalysts, where Pd nanoparticles are predominantly located in pores or in the network cavities. On the other hand, due to its outer surface localization, Pd nanoparticles in the Pd‐G3‐dendr@BP‐1 catalyst are less coated with the dendrimer amino groups and, as a consequence, appeared less selective in the phenylacetylene hydrogenation, especially as compared with G3‐dendr‐SiO2‐Pd catalyst.
With respect to alkynes, the reaction turnover frequency as well as alkene selectivity was additionally influenced by the substrate size and C≡C triple bond position in the alkyne molecule. As seen from Figure 6, linear 1‐octyne and 1‐hexyne were hydrogenated with much better efficacy, than that for phenylacetylene, with 1‐hexyne hydrogenated much faster as less‐size substrate. The maximum TOF value appeared as 404415 h−1 for G3‐dendr‐SiO2‐Pd catalyst at molar substrate to Pd ratio of ∼103700 and hydrogen pressure of 30 atm. within 15 minutes (Table S4, Entry 5). Mesoporous catalysts G2‐dendr‐meso‐SiO2‐Pd and meso‐G2‐dendr‐Si‐Pd again revealed the similar activity, giving maximum TOF values for 1‐hexyne of about 234300–276805 h‐1 at substrate/Pd ratios of 90100–91600 and hydrogen pressure of 30 atm. within 15 minutes (Table S4, Entries 13 and 19).
As in the case of phenylacetylene hydrogenation, the selectivity on terminal alkene was strongly dependent on both reaction conditions (substrate/Pd ratio, hydrogen pressure) and catalyst structure. At low substrate/Pd ratios thermodynamic reaction control prevailed, resulting in high isomerization degrees for rest alkenes among the reaction products (Tables S4 and S5).
Isomerization of the C=C double bond position is the typical process for linear alkenes in the presence of Pd catalysts.19d It was especially noticeable for the products of less‐size 1‐hexyne hydrogenation (Table S4). Herein G3‐dendr‐SiO2‐Pd catalyst favoured for the maximum isomerization degree among the other catalysts (Table S4). This phenomenon can be explained in terms of highly branched moieties of PPI dendrimers of the 3rd generation, those retarded alkenes formed in close proximity of Pd nanoparticles, making the formers undergo the further isomerization.
Increase in substrate/Pd ratio led to the predominance of kinetic control, appearing in increase of the total alkene fraction among the reaction products and lowering the isomerization degree, while the rise of hydrogen pressure resulted in slight increase of alkane and isomer portions (Tables S4 and S5). Moreover, hydrogen pressure had a significant influence on the efficacy of the alkyne hydrogenation (Figure 6). The more noticeable it was for mesoporous G2‐dendr‐meso‐SiO2‐Pd and meso‐G2‐dendr‐Si‐Pd catalysts, allowing to increase the reaction turnover frequency by 2.5–8.5 times (Figure 6, Tables S4 and S5). This is typical for heterogeneous Pd catalysts6c,69 and may be explained by the fact, that in mesoporous catalysts, based on PPI dendrimers of the 2nd generation, have enough free space, that can be occupied by both substrate molecules and adsorbed hydrogen species, thus favouring to the hydrogenation process. The adsorption density here also increases.
For microporous G3‐dendr‐SiO2‐Pd catalyst increase in hydrogen pressure from 10 to 30 atm. gives increase in the reaction efficiency only by 1–1.5 times (Figure 6, Tables S4 and S5). This may be due to initial higher local ligand to Pd ratio, originated from PPI dendrimers of the 3rd generation, constituting the ligand basis of the G3‐dendr‐SiO2‐Pd catalyst, and the predominance of small particles (Figures 3). Moreover, G3‐dendr‐SiO2‐Pd allowed to obtain higher conversions and, as a consequence, higher TOF values at lower hydrogen pressures even at the highest substrate/Pd ratios (Figure 6, Tables S4 and S5). It was especially remarkable for 1‐octyne, for which conversion and TOF at 10 atm. of H2 reached 98 % and 316545 h−1 respectively (substrate/Pd ∼80750, 15 min.), therefore being remarkably superior to that for both G2‐dendr‐meso‐SiO2‐Pd and meso‐G2‐dendr‐Si‐Pd at 30 atm. of H2 (TOF ∼17130–211900 h−1 at substrate/Pd of ∼70170–71400, within 15 min.) (Figure 6, Table S5).
This phenomenon can be explained in terms of hindered catalyst structure, retarding the substrate molecules and, therefore, increasing their local density. As a consequence, the probability of substrate adsorption on Pd nanoparticles increases in comparison with more expanded mesoporous catalysts. By the careful adjusting the reaction conditions, it was possible to reach high terminal alkene yield (≥90 %) for all catalysts tested.
Similar to phenylacetylene, 1‐octyne and 1‐hexyne, at highest substrate/Pd ratios, when alkyne not consumed completely, caused slight Pd leaching from catalysts, that is typical for terminal alkynes due to the complex formation.60 At the same time, the reasonable selectivity on alkene (≥ 90 %) for 1‐octyne and 1‐hexyne can be achieved only at much higher substrate/Pd ratios, than for phenylacetylene (Tables S4 and S5). This may be due to the flexible structure of linear alkynes and their semi‐hydrogenation products, resulting in their retarding by dendrimer branches, whereas rigid and planar phenylacetylene moves apart the denrimers branches, thus providing the free space for the fast desorption of styrene formed.
The second possible reason is the less polarization of terminal C−H bond due to the +I‐effect of substituent near to C≡C triple bond in the linear alkyne molecule – as compared with phenylacetylene one with −M‐effect of phenyl ring. As a consequence, the difference between electron densities on C≡C and C=C bonds with alkyl substituents is diminished, resulting in facilitated readsorption of alkene formed. We think, this supposal is confirmed by the results of 4‐octyne hydrogenation, whose internal C=C triple bond has two +I‐ substituents from both sides and cannot be polarized.
As seen from Figure 6, when 4‐octyne is hydrogenated in the presence of G2‐dendr‐meso‐SiO2‐Pd catalyst, the reaction TOF reaches values as high as 243980 h−1 at substrate/Pd ratio of ∼71760 and 30 atm. of H2 within 15 minutes, that is just slightly less (Table S6, Entry 10), than for 1‐hexyne (Table S4, Entry 13). We assume, this phenomenon is originated both from less hindered mesoporous structure of G2‐dendr‐meso‐SiO2‐Pd, providing the well access for substrate molecules to Pd nanoparticles, and from internal position of C≡C triple bond, surrounded by two +I‐substituents. This assumption is confirmed by the fact, that for G3‐dendr‐SiO2‐Pd and meso‐G2‐dendr‐Si−Pd, with more hindered structures, activity for 4‐octyne, with the internal C≡C triple bond, sharply decreased, as compared with terminal 1‐octyne and 1‐hexyne, and did not exceed 194840 h−1 (Figure 6, Table S6, Entry 5).
Another remarkable difference in the 4‐octyne hydrogenation is the predominance of octane among the reaction products for all catalysts even at highest substrate/Pd ratios (Table S6). In the presence of dendrimer moieties on the carrier surface internal alkyne is retarded to adsorb and, as a consequence, its semi‐hydrogenation products with the internal C=C bonds are retarded to desorb away. It resulted in the subsequent hydrogenation of alkenes formed and corresponding high portion of n‐octane among the reaction products (Table S6) in comparison not only with bimetallic PdAg‐G3‐dendr@BP‐144 and monometallic MPF‐PPI‐G3‐Pd catalysts,46 which allowed to achieve near‐to‐quantitative yield of 4‐octene by carefully adjusting the reaction conditions under the thermodynamic reaction control,44 but also with monometallic supported Pd/SiO2 and Pd/Al2O3 catalysts, providing the selective hydrogenation of diphenylacetylene to cis‐stilbene3c,70.
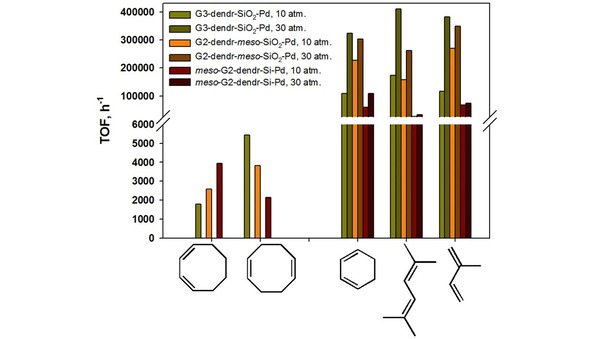
In the hydrogenation of dienes several main features may be marked. Flexible and tiny isoprene and 2,5‐dimethyl‐2,4‐hexadiene, as well as small 1,3‐cyclohexadiene (which is partly isomorphic to 2,5‐dimethyl‐2,4‐hexadiene in cis‐conformation) were hydrogenated with high efficacy, reaching the TOF values of 100000–400000 h−1 (Figure 7, Tables S7–S9), sharply exceeding those not only for traditional heterogeneous catalysts, but also for the materials, based on PPI or PAMAM dendrimers, grafted to amorphous silica surface34b,43 or dendritic networks.32a
Figure 7.
Hydrogenation of dienes in the presence of the dendrimer‐based organo‐silica palladium catalysts. Reaction conditions: 80 °C, 15 min.
In contrary, bulky 1,3‐ and 1,5‐cyclooctadienes were hydrogenated very slowly: their conversion after 1 hour does not reach 100 % for all catalysts at minimal substrate/Pd ratios (∼2800–3200). Herein 1,5‐cyclooctadiene very often underwent isomerization to conjugated 1,3‐cyclooctadiene, that was the appearance of the reaction thermodynamic control. The predominant reaction product for G2‐dendr‐meso‐SiO2‐Pd and G3‐dendr‐SiO2‐Pd catalysts was cyclooctane, whereas the literature reports, as a rule, about the cyclooctene formation in the presence of dendrimer‐based catalysts.18,21,34b,71
Trans‐cyclooctene with the selectivity of ∼5 % was detected for less active G3‐dendr‐SiO2‐Pd catalyst only in the beginning of the reaction, at conversion not exceeding 10 % (Figure S15). It should also be noted, that the literature data are referred mostly to the homogeneous catalysts, tested at the ambient temperature and pressure.18,21,34b,71
At the same time, unexpectedly high selectivity on cyclooctenes was revealed in the presence of meso‐G2‐dendr‐Si‐Pd catalyst. Herein 1,3‐cyclooctadiene, for which the induction period was observed (Figure S16), gave predominantly trans‐cyclooctene with the selectivity of 85–90 % at conversion of 44–46 % within 0.5–1 hour. Trans‐cyclooctene formation may originate from simultaneous adsorption of 1,3‐cyclooctadiene on both C=C double bonds.67 Vice versa, 1,5‐cyclooctadiene gave predominantly cis‐cyclooctene with the selectivity of 80–90 % in the beginning of the reaction (Figure S17, Table S9), that was the result of the adsorption only by one C=C bond. Nonetheless, this cis‐cyclooctene, due to its configuration, suitable for readsorption, easily underwent further hydrogenation to cyclooctane: the portion of the latter reached 92 % at 1,5‐cyclooctadiene conversion of 22 % within 1 hour.
The results, obtained for meso‐G2‐dendr‐Si‐Pd, may elucidate the main features of cyclooctadiene hydrogenation in the presence of other dendrimer‐based heterogeneous catalysts. Indeed, in the presence of severely hindered G3‐dendr‐SiO2‐Pd catalyst 1,3‐cyclooctadiene have not enough space for simultaneous adsorption on both two C=C double bonds; as a consequence, its hydrogenations proceeds similar to 1,5‐cyclooctadiene, with the formation of cis‐cyclooctene as an intermediate product. The latter, being as less stable, tense cycle, fast undergoes the subsequent hydrogenation to cyclooctane, possibly without desorption and readsorption act (maintaining as adsorbed species on Pd surface).
Hydrogenation in the presence of mesoporous, less hindered G2‐dendr‐meso‐SiO2‐Pd catalyst apparently proceeds in the same way. It may indicate, that Pd nanoparticles in this sample, though less intricated by dendrimer branches, nonetheless, have similar environment and shape, interfering the simultaneous double adsorption for 1,3‐cyclooctadiene. Thereafter, meso‐G2‐dendr‐Si‐Pd has respectively to consist of ensembles (Figure S18), similar to those in homogeneous catalysts, providing the selective formation of cyclooctene.
Hydrogenation of flexible linear or branched conjugated dienes, such as isoprene or 2,5‐dimethyl‐2,4‐hexadiene, proceeded according Scheme 6. Herein the product distribution was determined by the stability of corresponding π‐allyl intermediates (Figure S19), being formed at the first stage of hydrogenation, and was additionally affected by such factors, as mean particle size, local ligand/Pd ratio etc.68
Scheme 6.
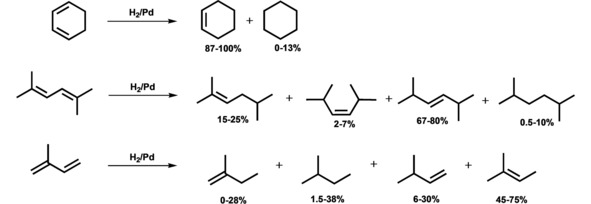
Product distribution in the hydrogenation of conjugated dienes in the presence of the dendrimer‐based organo‐silica palladium nanocatalysts.
The use of lower substrate/Pd ratios as well as increase in hydrogen pressures favoured to thermodynamic reaction control, resulting in increased portions for corresponding alkane and thermodynamically the most stable internal alkene, being the products of 1,4‐addition (2‐methyl‐2‐butene, 2,5‐dimethyl‐trans‐3‐hexene) (Tables S7–S9) – in contract to Pd nanocatalyst, based on arylcarbosilane triazol‐containing dendrimers21 – while the rise of substrate/Pd ratio furthered correspondingly to increased portions for 1,2‐addition products, being thermodynamically less stable terminal alkenes (2‐methyl‐1‐ and 3‐methyl‐1‐butenes) or cis‐isomers of internal alkenes (2,5‐dimethyl‐cis‐3‐hexene) (Tables S7–S9). In the last case the substrate, being in large excess, easily replaced olefin formed. As a result, the latter is desorbed without undergoing further hydrogenation or isomerization.43,71 On the other hand, thermodynamically less stable products of the kinetic control, as a rule, sterically less hindered (2‐methyl‐1‐ and 3‐methyl‐1‐butenes) and geometrically more suitable for readsorption (2,5‐dimethyl‐cis‐3‐hexene), were subjected to hydrogenation in the first instance. In the case of symmetrical and cyclic 1,3‐cyclohexadiene both 1,2‐ and 1,4‐addition of hydrogen resulted to the same product, cyclohexene (Figure S19). Similar phenomenon was earlier observed in the hydrogenation of isoprene in the presence of homogeneous Pd2(Ph2PCH2PPh2)3 catalyst at various substrate/Pd ratios.72
It should be noted, that the presence of PPI dendrimers as anchored ligands allowed not only to avoid the diene chemisorption and, as a consequence, polymerization and substrate inhibition, typical for small particles (< 4 nm), especially at high substrate/Pd ratios,63a,68 but also to maintain high total selectivity on olefins (90–99 %) (Tables S7–S9). PPI dendrimers, due to the electron‐donor properties of their amino groups, not only displace alkene formed from palladium surface, but also prevent readsorption of the latter (olefins are less electron rich in contrast to conjugated dienes), thus preventing the coke formation and polymerization of substrate and its semi‐hydrogenation products.73
The activity of the catalysts synthesized in the hydrogenation of conjugated dienes decreased in the following order: G3‐dendr‐SiO2‐Pd > G2‐dendr‐meso‐SiO2‐Pd ≫ meso‐G2‐dendr‐Si‐Pd (Figure 7). For mesoporous G2‐dendr‐meso‐SiO2‐Pd and meso‐G2‐dendr‐Si‐Pd catalysts size and geometry of the substrate appeared as main factors, determining their exposure to hydrogenation (Figure 7). Therefore, small 1,3‐cyclohexadiene with the rigid geometry (rotation of C=C double bonds is impossible) underwent hydrogenation faster, than others. Also small isoprene, but containing a substituent in central position at C=C double bond, was interfered to adsorb and, as a result, hydrogenated more slowly. Respectively, more hindered 2,5‐dimethyl‐2,4‐hexadiene was more inferior to isoprene in the tend to hydrogenation. At such size and structure dependency it is obvious the poor activity of heterogeneous dendrimer‐based catalysts in the hydrogenation of cyclooctadienes (Figure 7). For G2‐dendr‐meso‐SiO2‐Pd the thermodynamic control was predominant, resulting in higher portions for the products of 1,4‐addition even at highest substrate/Pd ratios (Table S7).
As earlier for styrenes and alkynes, the electron factors appeared as the crucial, when G3‐dendr‐SiO2‐Pd was used as the catalyst, even overbalancing the sterical factors, being expectable as more important for catalyst with such hindered structure. Thus, we can see, that the most substituted 2,5‐dimethyl‐2,4‐hexadiene, being able to form the most stable π‐allyl intermediates (Figure S19), was hydrogenated with the efficacy, just slightly superior to that for less substituted isoprene (Figure 7). The last, in its turn, was slightly superior to 1,3‐cyclohexadiene, which cannot form tertiary, the most stable π‐allyl intermediates (Figure S19). We attribute this tendency for G3‐dendr‐SiO2‐Pd catalyst to interact better with donor substrates, able to form more stable intermediates, to the predominance of small particles (Figure 3), characterized by the partial positive charge on the surface.63a,68 G3‐dendr‐SiO2‐Pd was influenced by the kinetic control, appearing in higher portions for the products of 1,2‐addition not only at highest substrate/Pd ratios (Table S8).
G3‐dendr‐SiO2‐Pd was tested on the possibility of diene recycling on the examples of 1,3‐cyclohexadiene and 2,5‐dimethyl‐2,4‐hexadiene (Figure 8). The recycling test revealed the slight, gradual decrease of activity, that might be caused mostly by the mechanical losses of the catalyst under the conditions, at which reaction becomes sensitive even to slight increase in substrate/Pd ratio. Leaching portion did not exceed 1 % according to ICP‐AES. Herein total selectivity on alkenes maintained at 96–99.5 %. Total turnover number for six cycles reached 125405 for 1,3‐cyclohexadiene and 169130 for 2,5‐dimethyl‐2,4‐hexadiene.
Figure 8.
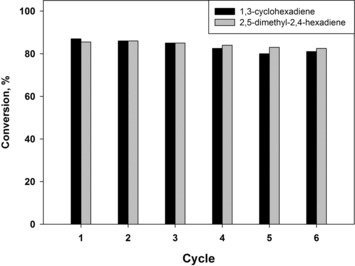
Recycling of G3‐dendr‐SiO2‐Pd in the hydrogenation of 1,3‐cyclohexadiene and 2,5‐dimethyl‐2,4‐hexadiene. Reaction conditions are: 0.5 mg of catalyst, 750 μL of 1,3‐cyclohexadiene (substrate/Pd ≈ 25005) or 1500 μL of 2,5‐dimethyl‐2,4‐hexadiene (substrate/Pd ≈ 33425), 15 min., 80 °C, 10 atm. of H2.
Increase in hydrogen pressure up to 30 atm. allowed to increase TOF values in diene hydrogenation by 1.3–1.7 times for G2‐dendr‐meso‐SiO2‐Pd and by 2.4–3 times for G3‐dendr‐SiO2‐Pd (Figure 7, Tables S7 and S8). Herein the total selectivity on olefin maintained still high, dropping only by 1–5 %. Analogous tendency was also the characteristic of alkyne hydrogenation in the presence of the dendrimer‐based organo‐silica Pd nanocatalysts, studied in this work (Tables S4–S6). This compares dendrimer‐based organo‐silica Pd nanocatalysts favourably with traditional heterogeneous Pd catalysts (including Lindlar catalyst).74 In spite of the fact, that the latters allow to achieve selectivities on alkene as high as 85–100 % and TOF values of 30000–100000 h−1 in some cases, reactions in their presence are carried out, as a rule, at ambient temperatures and pressures (Tables S10–S11) and have to be stopped at conversions of 70–90 % to avoid overhydrogenation, that becomes inevitable, when the most portion of alkyne or diene is consumed.58a,63b,74 Herein increase in temperature or pressure obviously results in accelerating the reaction rate and further downfall in the selectivity on alkene.3b,64c
In the case of meso‐G2‐dendr‐Si‐Pd, increase in hydrogen pressure did not result in the sufficient rise of efficacy (only by 1–1.2 times), when isoprene and 2,5‐dimethyl‐2,4‐hexadiene were used as substrates (Figure 7, Table S9). We assume, it may be connected with the adsorption features or nanoparticle local microenvironment, interfering the efficient hydrogenation (and simultaneously providing high selectivity on cyclooctene, when 1,3‐cyclooctadiene was used as the substrate). In general, meso‐G2‐dendr‐Si‐Pd appeared as poor catalyst for conjugated diene hydrogenation as compared with G2‐dendr‐meso‐SiO2‐Pd and G3‐dendr‐SiO2‐Pd. It tended to thermodynamic control in 2,5‐dimethyl‐2,4‐hexadiene hydrogenation, resulting in higher portions for the products of 1,4‐addition even at highest substrate/Pd ratios (Table S9), and to kinetic control in the isoprene hydrogenation, giving to portions of 2‐methyl‐1‐butene, 3‐methyl‐1‐butene and 2‐methyl‐2‐butene, close to the classical 1 : 1 : 2 (Table S9).68
Hydrogenation of unconjugated dicyclopentadiene proceeded stepwise (Scheme 7).32a The double bond, located opposite to the methylene bridge, primarily underwent hydrogenation due to its less sterical hindrance and geometrical strain (Figure 9).32a,75 Herein the configuration of initial dicyclopentadiene was retained. In contrast to the earlier published heterogeneous Pd catalysts, based on the dendrimer networks,32a organo‐silica Pd catalysts appeared much higher activity in the dicyclopentadiene hydrogenation along with the lowered selectivity on dihydrodicyclopentadiene (Tables 6 and 7).
Scheme 7.
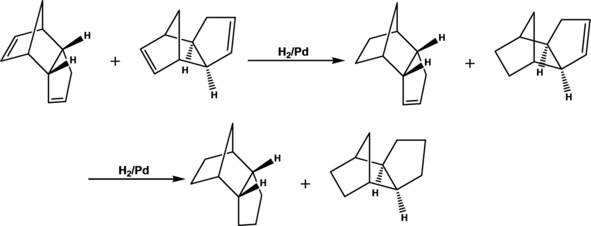
Stepwise hydrogenation of dicyclopentadiene in the presence of Pd catalysts.32a
Figure 9.
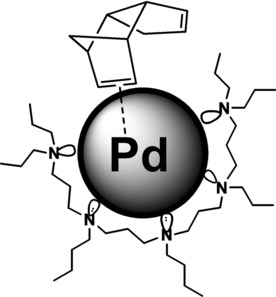
Adsorption of dicyclopentadiene molecule on the surface of palladium nanoparticle, stabilized by PPI dendrimers.[32a]
Table 6.
Hydrogenation of dicyclopentadiene in the presence of G2‐dendr‐meso‐SiO2‐Pd catalyst.[a]
| Entry | Catalyst loading, mg | Substrate/Pd (mol/mol) | Conv., % | TOF, h−1 | Product distribution, % |
|---|---|---|---|---|---|
| 1 | 1 | 2655 | 100 | 10620 | Dihydrodicyclopentadiene 5 % Tetrahydrodicyclopentadiene 95 % |
| 2 | 0.5 | 5310 | 100 | 21240 | Dihydrodicyclopentadiene 28.5 % Tetrahydrodicyclopentadiene 71.5 % |
| 3 | 0.5 | 10620 | 99.5 | 42265 | Dihydrodicyclopentadiene 57 % Tetrahydrodicyclopentadiene 43 % |
| 4 | 0.5 | 21240 | 96 | 81560 | Dihydrodicyclopentadiene 85.5 % Tetrahydrodicyclopentadiene 14.5 % |
| 5 | 0.5 | 42480 | 31.5 | 53525 | Dihydrodicyclopentadiene 93 % Tetrahydrodicyclopentadiene 7 % |
| 6 | 0.5 | 70800 | 18 | 50975 | Dihydrodicyclopentadiene 62 % |
| Tetrahydrodicyclopentadiene 6 % | |||||
| Cyclopentadiene 1 % | |||||
| Cyclopentene 23 % | |||||
| Cyclopentane 8 % |
[a] Reaction conditions are: 80 0C, 10 atm. of H2, 15 min.
Table 7.
Hydrogenation of dicyclopentadiene in the presence of G3‐dendr‐SiO2‐Pd catalyst.[a]
| Entry | Catalyst loading, mg | Substrate/Pd (mol/mol) | P (H2), atm. | Conv., % | TOF, h−1 | Product distribution, % |
|---|---|---|---|---|---|---|
| 1 | 1 | 3005 | 10 | 99.5 | 11955 | Dihydrodicyclopentadiene 42 % Tetrahydrodicyclopentadiene 55.5 % Retro‐diene decay 2.5 % |
| 2 | 0.5 | 6010 | 10 | 99.5 | 23910 | Dihydrodicyclopentadiene 69 % Tetrahydrodicyclopentadiene 17.5 % Retro‐diene decay 13.5 % |
| 3 | 0.5 | 18020 | 10 | 98 | 70645 | Dihydrodicyclopentadiene 66 % Tetrahydrodicyclopentadiene 4 % Retro‐diene decay 30 % |
| 4 | 0.5 | 24030 | 10 | 94 | 90350 | Dihydrodicyclopentadiene 94 % Tetrahydrodicyclopentadiene 4.5 % Retro‐diene decay 1.5 % |
| 5 | 0.5 | 48060 | 10 | 41 | 59115 | Dihydrodicyclopentadiene 95 % Tetrahydrodicyclopentadiene 3 % Retro‐diene decay 2 % |
| 6 | 0.5 | 48060 | 30 | 70 | 100925 | Dihydrodicyclopentadiene 90 % Tetrahydrodicyclopentadiene 9 % Retro‐diene decay 1 % |
| 7 | 0.5 | 64080 | 10 | 13 | 24990 | Dihydrodicyclopentadiene 90.5 % Tetrahydrodicyclopentadiene 2.5 % Retro‐diene decay 7 % |
| 8 | 0.5 | 64080 | 30 | 51 | 98040 | Dihydrodicyclopentadiene 93 % |
| Tetrahydrodicyclopentadiene 5 % | ||||||
| Retro‐diene decay 2 % |
[a] Reaction conditions are: 80 °C, 15 min.
Thus, the portion of tetrahydrodicyclopentadiene, the product of exhausting hydrogenation of dicyclopentadiene, in the presence of G2‐dendr‐meso‐SiO2‐Pd catalyst reached 95 % within 15 minutes at molar substrate/Pd ratio of ∼2655 and 10 atm. of hydrogen (Table 6), while in the presence of Pd nanoparticles, encapsulated into dendritic networks it was only 5–10 % even at less substrate/Pd ratios.32a Increase in the latters led to the corresponding increase in dihydrodicyclopentadiene portion; nonetheless, there also took place the abrupt downfall in conversion (Table 6).
The use of G3‐dendr‐SiO2‐Pd catalyst, containing in its structure more hindered dendrimers of the 3rd generation with larger amount of donor amino groups, allowed to achieve the higher selectivity on dihydrodicyclopentadiene (∼95 %) at near to quantitative conversions of dicyclopentadiene (∼95 %) within 15 minutes (Table 7). However, this dihydrodicyclopentadiene easily underwent the subsequent hydrogenation to tetrahydrodicyclopentadiene with the prolonged reaction time (Figure 10), that was typical for traditional supported Pd catalysts (Pd/C, Pd/Al2O3 and Pd/SiO2).76
Figure 10.
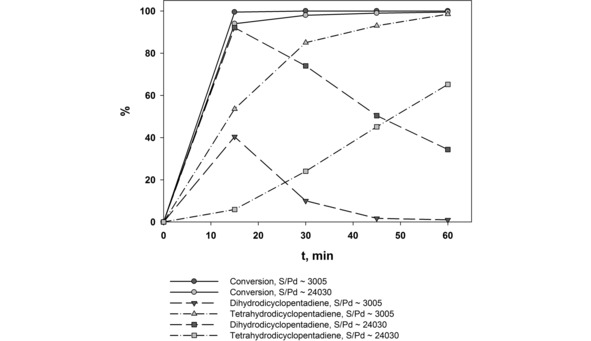
Hydrogenation kinetics of dicyclopentadiene in the presence of G3‐dendr‐SiO2‐Pd catalyst. Reaction conditions are: 80 °C, 10 atm. of H2.
Another feature of the dendrimer‐based organo‐silica Pd catalysts in the hydrogenation of dicyclopentadiene was the high portion of retro‐diene decay. In the case of G2‐dendr‐meso‐SiO2‐Pd it reached 32 % and appeared only at highest substrate/Pd ratios and the lowest dicyclopentadiene conversions (Table 6). When G3‐dendr‐SiO2‐Pd used as the catalyst, a small portion of the retro‐diene decay appeared even at quantitative dicyclopentadiene conversions and initially sharply increased with the increase in substrate/Pd ratio (the maximum of 30 % was observed at substrate/Pd ∼18020) and then abruptly downfall (Table 7). Increase in hydrogen pressure led to the decrease in the retro‐diene decay portion (Table 7).
Hence, one may assume, that retro‐diene decay occurs on the walls of silica pores, possessing a some of acidity. For its appearance the highest substrate/Pd ratio is required, when the mesoporous material G2‐dendr‐meso‐SiO2‐Pd is used as the catalyst. For much more hindered microporous G3‐dendr‐SiO2‐Pd it appears already at low and moderate substrate/Pd ratios, however the further increase in substrate/Pd ratio presumably results in displacement of the adsorbed species from the carrier surface before they succeed to undergo retro‐diene decay.
Conclusions
New hybrid organo‐silica Pd‐containing nanocatalysts, based on poly(propylene imine) dendrimers, have been synthesized using co‐hydrolysis of the silane‐modified dendrimer moieties in situ. Herein the synthesis can be performed either with or without addition of tetraethoxysilane, in the presence or in the absence of polymeric template.
Thus prepared catalysts were examined in the hydrogenation of various unsaturated compounds: styrenes, alkynes and dienes. The activity and selectivity of these catalysts were found to depend on the carrier structure, substrate size and geometry, as well as on the electron properties.
Mesoporous G2‐dendr‐meso‐SiO2‐Pd catalyst, synthesized in presence of template with the addition of tetraethoxysilane, revealed the maximum activity in the hydrogenation of styrenes, reaching TOF values of about 230000 h−1. Due to the larger particle size and much larger pore diameter, as well as due to the highest portion of Pd0 on the nanoparticle surface, it favoured to the quantitative conversions for bulky and rigid substrates with −M‐substituents, such as trans‐stilbene, 4‐phenylstyrene and 1,1‐diphenyl ethylene.
Microporous catalyst G3‐dendr‐SiO2‐Pd, based on PPI dendrimes of the 3rd generation and synthesized with the addition of tetraethoxysilane, but in the absence of template, appeared as the most active in terminal alkyne and flexible and small conjugated diene hydrogenation, reaching TOF values more than 400000 h−1 for 1‐hexyne and 2,5‐dimethyl‐2,4‐hexadiene. Herein the high selectivity on alkene (90–99 %) was maintained even at hydrogen pressure of 30 atm. The main feature of this catalyst was the predominance of small particles (∼0.9–2 nm) in the structure and, as a consequence, the strong influence by the electron factors, resulting in the unexpectedly high activity for the bulky, long and hindered substrates with +I‐substituents, such as 4‐tert‐butylstyrene, 1‐octyne and 2,5‐dimethyl‐2,4‐hexadiene. Herein the moieties of 3rd generation PPI dendrimers, containing the large amount of donor amino groups, provided the better Pd nanoparticle stabilization and, as a consequence, the highest selectivity on alkenes in the hydrogenation of terminal alkynes and tiny and flexible conjugated dienes.
The catalyst meso‐G2‐dendr‐Si‐Pd, prepared in presence of template, but without addition of tetraethoxysilane, appeared as significantly inferior both to G2‐dendr‐meso‐SiO2‐Pd and G3‐dendr‐SiO2‐Pd in its efficiency. At the same time it was the only catalyst, providing the trans‐cyclooctene formation with the selectivity of 90–95 %, having elucidated the mechanism of cyclooctadiene hydrogenation. This makes it similar to both homogeneous and heterogeneous dendrimer‐based catalysts, described in literature.
Pd‐containing dendrimer‐based organo‐silica nanocatalysts can be easily separated from the reaction products and reused several times without significant loss in activity. However their apparent stability depends on the reaction sensitivity to the substrate/Pd ratio.
Experimental Section
Chemicals
The following substances were used as substrates and reference compounds: phenylacetylene C6H5CH≡CH (Aldrich, 98 %); styrene C6H5CH=CH2 (Aldrich,≥99 %); ethylbenzene C6H5CH2CH3 (Reachim, Purum); p‐methylstyrene p‐CH3C6H4CH=CH2 (Aldrich, 96 %); p‐tert‐butylstyrene p‐(CH3)3CC6H4CH=CH2 (Aldrich, 94 %); 2,5‐dimethyl‐2,4‐hexadiene (CH3)2C=CH–CH=C(CH3)2 (Aldrich, 98 %); isoprene H2C=C(CH3)−CH=CH2 (Aldrich, 99 %); 1,3‐cyclohexadiene (Aldrich, 97 %); dicyclopentadiene C10H12 (Aldrich, 97 %).
Ethanol C2H5OH (IREA 2000, Analytical Grade), methanol (Acros Organics, 99.9 %), chloroform CHCl3 (Chimmed, Reagent Grade) and n‐hexane n‐C6H14 (Chimmed, Reagent Grade) were used as solvents.
For the synthesis of hybrid dendrimer‐based catalysts the following substances were used: Pd acetate (II) Pd(OAc)2 (Aldrich, 99.9 %); poly(propylene imine) (PPI) dendrimers of the 2nd generation with a diaminobutane core DAB(NH2)8, earlier prepared according to literature procedure;77 (3‐glycidoxypropyl)trimethoxysilane (Acros Organics,≥97 %); poly(ethylene glycol)‐block‐poly(propylene glycol)‐block‐poly(ethylene glycol) with the average number molecular weight Mn of 5800 HO(CH2CH2O)20(CH2CH(CH3)O)70(CH2CH2O)20H (Pluronic P123, Aldrich); microporous G3‐dendr‐SiO2 and mesoporous G2‐dendr‐meso‐SiO2 carriers, synthesized earlier according to the published procedure.46
Analyses and Instrumentations
Analysis by transmission electron microscopy (TEM) was conducted using a LEO912 AB OMEGA microscope with electron tube voltage of 100 kV. The count of particles and calculation of mean particle size was performed by processing the obtained micro‐images using the “Image J” program. High resolution transmission electron microscopy (HRTEM) was performed using a JEM‐2100 microscope with an electron tube voltage of 200 kV (LaB6 cathode). The d‐spacing of the metal nanoparticles was made by means of fast Fourier transforms of the micrographs obtained, using the “Image J” program.
X‐ray photoelectron spectroscopy (XPS) analysis was carried out on Kratos Axis Ultra DLD and LAS‐3000 instruments equipped with an OPX‐150 hemispherical retarding‐field electron‐energy analyzer. The X‐radiation of an aluminum anode (Al K=1486.6 eV) was used for the excitation of photoelectrons at a tube voltage of 12 kV and an emission current of 20 mA. Photoelectron peaks were calibrated on the C 1s line of carbon with a binding energy of 284.8 eV. Deconvolution of palladium peaks was processed with the PeakFit 4.11 program using the Gauss‐Lorentz‐Ampere approximation.
The quantitative determination of palladium in the samples was performed by atomic emission spectrometry with inductively coupled plasma (ICP AES) by means of IRIS Interpid II XPL instrument (Thermo Electron Corp., USA) with both radial and axial viewings at wavelengths of 310 and 95.5 nm.
Analysis by means of nitrogen low‐temperature adsorption and desorption was carried out on a Gemini VII 2390 instrument. Nitrogen adsorption‐desorption isotherms were obtained at 77 K. The samples being analyzed were previously outgassed at 150 °C for 12 h. Surface area measurements were performed according to the Brunauer‐Emmett‐Teller (BET) method from nitrogen adsorption points in the range P/P0=0.05–0.2. Pore size distribution was calculated using the Barrett‐Joyner‐Halenda model (BJH). The diameter corresponding to the maximum of the pore size distribution curve was suggested as the average pore size. Total pore volume was obtained at a relative pressure P/P0 of 0.985.
A ChromPack CP9001 gas chromatograph, equipped with a capillary column (SE‐30 grafted phase, 30 m×0.2 mm) and flame‐ionization detector, was used for analysis of substrates and reaction products. Chromatograms were recorded and analyzed on a computer with the use of the Maestro 1.4 software. Conversion was determined by changes in the relative peak areas (in %) of the substrates and products.
Synthetic Procedures for Porous Hybrid Dendrimer‐Based Materials
The synthesis of meso‐G2‐dendr‐Si network.78 The synthesis of mesoporous dendrimer‐based material was performed according to the following procedure Into a 25 mL single‐neck round‐bottom flask, equipped with a magnetic stirrer and a reflux condenser, 1.33 g (1.7 mmol) of DAB(NH2)8 dendrimer and 5 mL of ethanol were placed. Then 1.52 mL (6.9 mmol) of (3‐glycidoxypropyl)triethoxysilane were added dropwise at room temperature, while stirring. Reaction was carried out at 80 °C for 2 h while stirring and the flask contents gradually turned into an orange glass‐like mass, that was further evaporated on a rotary evaporator, giving an insoluble residue, weighing 3.3 g.
For the next stage of the synthesis, 2.5 g of Pluronic P123 were placed into a 100 mL glass beaker, containing a solution of 90 mL of deionized water and 0.55 mL of concentrated hydrochloric acid (Sigma‐Tech, Chemical Grade). The mixture obtained was subjected to stirring at 50 °C for 3 h, gradually growing turbid. After that the previously obtained and ground modified dendrimer was added to the resultant colloid solution of polymer, followed by stirring for 3 h. at 50 °C, resulting in red‐orange residue. This residue was further stored in a drying chamber for 3 h with a gradual temperature increase from 60 to 110 °C, and after that it was subjected to stirring with 25 mL of water and 100 mL of ethanol at 70 °C, to wash out the polymer. Then the residue obtained was subjected to centrifugation and dried in the air. The product was isolated as a fluffy yellow powder, weighing 3.25 g.
XPS: 285.8 (C 1s, 67.6 %); 399.4 (N 1s, 16.1 %); 532.7 (O 1s, 16.3 %).
13C NMR (δ, ppm): 74 (−OCH2 CH2OCH2); 68 (−(CH2)2 CHOH); 65 (SiCH2CH2 CH2O); 53 (NCH2CH2CH2 CH2N); 51 (NCH2CH2 CH2N, NHCH2CH2O); 47 (NCH2CH2 CH2NHCH2−); 38 (NCH2CH2 CH2NH2); 22 (NCH2 CH2CH2N, NCH2 CH2CH2CH2N, SiCH2 CH2CH2O); 9.6 (SiCH2CH2CH2O).
The synthesis of G3‐dendr‐SiO2‐Pd catalyst.43 Into a 25 mL single neck round‐bottom flask, equipped with magnetic stirrer and reflux condenser, 500 mg of the microporous hybrid G3‐dendr‐SiO2 carrier and 5 mL of chloroform were placed. Then 100 mg (0.45 mmol) of Pd(OAc)2 in 5 mL of chloroform were added to the resulting suspension while stirring. The reaction was carried out for 6 h at 70 °C. The color of the reaction mixture changed from red‐ocher to brown‐black. After the reaction, the suspension was evaporated in a rotary evaporator at 50 °C. The resulting material was a grey‐brown powder weighing 562 mg (yield of 93.5 %).
To reduce Pd (II) to Pd (0), 562 mg of the earlier obtained catalyst precursor, 7 mL of chloroform and 3 mL of methanol were placed to a 25 mL single neck round‐bottom flask, equipped with magnetic stirrer and reflux condenser. Then 171 mg (4.5 mmol) of sodium borohydride were portion‐wise added to the suspension obtained while stirring. The reaction mixture rapidly turned black and gas evolution was observed. The reaction was carried out for 6 h at 70 °C. After the reaction, the resulting solid mixture was washed with water and methanol to remove sodium tetraborate, and the residue isolated by centrifugation and dried in the air. The resulting material was a black powder weighing 494 mg (yield of 90 %).
XPS (eV): 102.9 (SiO2, Si 2p, 15.9 %); 283.6 (OCH2CH2 CH2Si, C 1s, 3.8 %), 284.7 (NCH2 CH2CH2N, NCH2 CH2CH2CH2N, OCH2 CH2CH2Si, C 1s, 6.2 %), 286.4 (NCH2CH2 CH2N, OCH2CH2CH2Si, OCH2CH2O/N, C 1s, 14.6 %), 288.2 (−O−C−O−, −CH2 CH2O→PdOx, C 1s, 2.9 %), 289.4 (orthoester, epoxide or NCH2CH2CH2N/OCH2CH2O (sat), C 1s, 0.9 %); 335.8 (Pd0, Pd 3d5/2, 1.7 %), 337.4 (PdO, Pd 3d5/2, 1.3 %), 338.7 (Pd (II) N‐bound, Pd 3d5/2, 0.1 %), 341.6 (Pd0, Pd 3d3/2), 343.2 (PdO, Pd 3d3/2), 344.4 (Pd (II) N‐bound, Pd 3d3/2); 398.7 (NCH2CH2CH2N, NCH2CH2CH2NH2, N 1s, 0.7 %), 399.9 (CH2NH2→Pd0, −CH2NH2→Pdδ+, N 1s, 1.5 %), 401.1 (NH3R+, −CH=NH+−, N 1s, 1.3 %), 402.5 (NHR3 +, R3N+→O−, N 1s, 0.5 %), 404.4 (R3N+→O−, N 1s, 0.05 %), 405.8 (N 1s (sat), 0.1 %); 531.1 (PdOx, O 1s, 3.9 %), 532.8 (OCH2CH2O, SiO2, O 1s, 17.7 %), 534.4 (SiO2, −CH2O→Pdδ+, N/O…H2O, O 1s, 15.4 %), 536.0 (ordered H2O, O 1s, 9.1 %), 537.8 (O 1s (sat), 2.4 %).
13C NMR (δ, ppm): 73 (−OCH2 CH2OCH2−); 66 (−(CH2)2 CHOH); 57 (NCH2CH2CH2 CH2N); 51 (NCH2CH2 CH2N, NHCH2CH2O); 46 (NCH2CH2 CH2NHCH2−); 33 (NCH2CH2 CH2NH2); 23 (NCH2 CH2CH2N, NCH2 CH2CH2CH2N); 16 (SiCH2 CH2CH2O); 9.6 (SiCH2CH2CH2O).
ICP‐AES: 6.70 % wt. of Pd.
The synthesis of G2‐dendr‐meso‐SiO2‐Pd catalyst.43 The synthesis was performed similar to the previously described procedure. 1000 mg of mesoporous hybrid G2‐dendr‐meso‐SiO2 carrier and 215 mg (0.96 mmol) of Pd(OAc)2 in 15 mL of chloroform were taken as initial substances. The reaction was carried out for 12 h at 70 °C. To reduce Pd (II) to Pd (0), the resulting intermediate product (847 mg), isolated as yellow‐brown powder, was introduced into reaction with 365 mg (9.6 mmol) of sodium borohydride in a mixture of 10 mL of chloroform and 5 mL of ethanol. The weight of the end product, isolated as a black powder, was 926 mg (71 %).
XPS (eV): 103.1 (SiO2, Si 2p, 13.1 %), 153.5 (SiO2, Si 2 s); 284.8 (NCH2 CH2CH2N, NCH2 CH2CH2CH2N, OCH2 CH2CH2Si, C 1s, 13.6 %), 286.3 (NCH2CH2 CH2N, OCH2CH2CH2Si, OCH2CH2O/N, C 1s, 15.1 %), 288.0 (−O−C−O−, −CH2 CH2O→PdOx, C 1s, 3.4 %); 335.2 (Pd0, Pd 3d5/2, 1.70 %), 336.5 (PdOx/Pd, Pd 3d5/2, 0.5 %), 338.0 (Pd2+, PdOx, Pd 3d5/2, 0.4 %), 339.0 (Pd (II) N‐bound, Pd 3d5/2, 0.1 %), 340.5 (Pd0, Pd 3d3/2), 341.6 (PdOx/Pd, Pd 3d3/2), 343.1 (Pd2+, PdOx, Pd 3d3/2), 344.2 (Pd (II) N‐bound, Pd 3d3/2), 345.7 (Pd2+, Pd 3d3/2 sat); 397.6 (−C=N−C−, −N≡Pdads, N 1s, 0.2 %), 399.9 (NCH2CH2CH2N, OCH2CH2NHCH2−, (−CH2NH2→Pd0, −CH2NH2→Pdδ+, N 1s, 2.9 %), 402.0 (NHR3 +, [−CH=NH−]+, N 1s, 0.5 %); 532.8 (OCH2CH2O, SiO2, O 1s, 48.2 %).
13C NMR (δ, ppm): 75 (−OCH2 CH2OCH2); 57 (NCH2CH2 CH2N, NCH2CH2CH2 CH2N, NHCH2CH2O, br); 51 (NCH2CH2 CH2NHCH2‐, br); 33 (NCH2CH2 CH2NH2, br); 25 (NCH2 CH2CH2N, NCH2 CH2CH2CH2N); 15 (SiCH2 CH2CH2O); 8 (SiCH2CH2CH2O).
ICP‐AES: 7.58 % wt. of Pd.
The synthesis of meso‐G2‐dendr‐Si‐Pd catalyst.43 The synthesis was carried out using a previously described procedure. 500 mg of meso‐G2‐dendr‐Si network and 262 mg (1.17 mmol) of Pd(OAc)2 in 15 mL of chloroform were taken as initial substances. The reaction was carried out for 6 h at 70 °C. To reduce Pd (II) to Pd (0), the resulting intermediate product (500 mg), isolated as dark‐brown powder, was introduced into reaction with 443 mg (11.66 mmol) of sodium borohydride in a mixture of 10 mL of chloroform and 5 mL of ethanol. The weight of the end product, isolated as a black powder, was 430 mg (69 %).
XPS (eV): 284.3 (NCH2 CH2CH2N, NCH2 CH2CH2CH2N, OCH2 CH2CH2Si, OCH2CH2 CH2Si, C 1s, 13.6 %), 286.3 (NCH2CH2 CH2N, OCH2CH2CH2Si, OCH2CH2O/N, C 1s, 32.4 %), 288.2 (−O−C−O−, −CH2 CH2O→PdOx, C 1s, 11.0 %), 290.1 (−C(OCH2−)3 or NCH2CH2CH2N/OCH2CH2O (sat), C 1s, 5.4 %); 335.3 (Pd0, Pd 3d5/2, 0.6 %), 336.6 (PdOx/Pd, Pd 3d5/2, 1.0 %), 337.8 (Pd2+, PdOx, Pd 3d5/2, 0.7 %), 340.6 (Pd0, Pd 3d3/2), 341.6 (PdOx/Pd, Pd 3d3/2), 342.6 (Pd2+, PdOx, Pd 3d3/2); 398.0 (−C=N−C−, −N≡Pdads, N 1s, 2.2 %), 399.3 (NCH2CH2CH2N, OCH2CH2NHCH2−, N 1s, 11.2 %), 400.4 (−CH2NH2→Pd0, −CH2NH2→Pdδ+, N 1s, 3.2 %), 401.4 (NH3R+, −CH=NH+−, N 1s, 0.4 %), 403.8 (R3N+→O−, N 1s, 1.2 %), 405.0 (N 1s (sat), 0.4 %); 530.1 (O 1s, PdOx/Pd, 0.9 %), 531.2 (PdOx, O 1s, 1.4 %), 532.5 (OCH2CH2O, SiO2, O 1s, 12.2 %), 533.7 (SiO2, −CH2O→Pdδ+, 1 s, 1.4 %), 534.7(N/O…H2O, O 1s, 0.7 %).
ICP‐AES: 7.71 % wt. of Pd.
Protocol for the Catalytic Experiments
A catalyst and a substrate in a required ratio were placed into a thermostatically controlled steel autoclave, equipped with an glass insertion tube and a magnetic stirrer. If the solid substrate was used (4‐phenylstyrene, trans‐stilbene), a toluene as a solvent was added with a concentration of 2 μL of the solvent per 1 mg of the substrate. The autoclave was tightly closed, filled with hydrogen to a required pressure, and connected to a thermostat. The reaction was carried out at 80 °C with intense stirring for a required time, after which the reactor was cooled below room temperature. The reaction products were analyzed by means of gas‐liquid chromatography.
Activity of the catalysts (TOF=turnover frequency) was calculated as the amount of reacted substrate (νsubstr) per mole of metal (νPd) per unit of time:
where ω is substrate conversion, expressed in the unit fractions. Herein t is the minimal reaction time, for which the reaction progress is measured.
The re‐use of dendrimer‐based Pd hybrid catalysts was conducted as follows. The desired amounts of catalysts and substrate were placed in a thermostatically controlled steel autoclave equipped with a glass insertion tube and a magnetic stirrer. The autoclave was tightly closed and filled with hydrogen up to a pressure of 10 or 30 atm. and then connected to the thermostat. The reaction was carried out at 80 °C with intense stirring for 15 min. After the reaction completed, the stirring was turned off and the autoclave was cooled below room temperature in a cold water bath and opened. The reaction mixture was diluted with ∼2 mL of hexane for the better sedimentation of the catalyst and allowed to stand for 20–30 min. The solution with reaction products was separated by decantation and analyzed by gas‐liquid chromatography. Catalyst, remained in the test tube, was repeatedly used without additional loading or purification.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by RFBR grant No 16–33‐60101. We also thank Natalia Tabachkova (National University of Science and Technology ‘MISIS’, Moscow, Russia) for HRTEM facilities.
E. A. Karakanov, A. V. Zolotukhina, A. O. Ivanov, A. L. Maximov, ChemistryOpen 2019, 8, 358.
References
- 1.
- 1a. Astruc D., Lu F., Aranzaes J. Ruiz, Angew. Chem. Int. Ed. 2005, 44, 7852–7872, [DOI] [PubMed] [Google Scholar]
- 1b. Stakheev A.Yu., Mashkovskii I. S., Baeva G. N., Telegina N. S., Russ. J. Gen. Chem. 2010, 80, 618–629; [Google Scholar]
- 1c. Pina C. D., Falletta E., Rossi M., Chem. Soc. Rev. 2012, 41, 350–369; [DOI] [PubMed] [Google Scholar]
- 1d. Favier I., Teuma E., Gómez M., C. R. Chim. 2009, 12, 533–545; [Google Scholar]
- 1e. Hagiwara H., Nakamura T., Okunaka N., Hoshi T., Suzuki T., Helv. Chim. Acta 2010, 93, 175–182; [Google Scholar]
- 1f. Karakhanov E. A., Maksimov A. L., Zolotukhina A. V., Kardasheva Yu. S., Russ. Chem. Bull. 2013, 62, 1465–1492; [Google Scholar]
- 1g. Widegren J. A., Finke R. G., J. Mol. Catal. A 2003, 191, 187–207; [Google Scholar]
- 1h. Polshettiwar V., Molnár Á., Tetrahedron 2007, 63, 6949–6976. [Google Scholar]
- 2.
- 2a. Thomas J. M., Johnson B. F. G., Raja R., Sankar G., Midgley P. A., Acc. Chem. Res. 2003, 26, 20–30; [DOI] [PubMed] [Google Scholar]
- 2b. Johnson B. F. G., Raynor S. A., Brown D. B., Shephard D. S., Mashmeyer T., Thomas J. M., Hermans S., Raja R., Sankar G., J. Mol. Catal. A: Chem. 2002, 182–183, 89–97; [Google Scholar]
- 2c. Liu J.-L., Zhu L.-J., Pei Ya., Zhuang J.-H., Li H., Li H.-X., Qiao M.-H., Fan K.-N., Appl. Catal. A. 2009, 353, 282–287; [Google Scholar]
- 2d. Miao Sh., Liu Zh., Han B., Huang J., Sun Zh., Zhang J., Jiang T., Angew. Chem. Int. Ed. 2006, 45, 266–269; [DOI] [PubMed] [Google Scholar]
- 2e. Liu J.-L., Zhu L.-J.n, Pei Ya., Zhuang J.-H., Li H., Li H.-X., Qiao M.-H., Fan K.-N., Appl. Catal. A 2009, 353, 282–287; [Google Scholar]
- 2f. Jacquin M., Jones D. J., Rozière J., Albertazzi S., Vaccari A., Lenarda M., Storaro L., Ganzerla R., Appl. Catal. A 2003, 251, 131–141; [Google Scholar]
- 2g. Mastalir Á., Király Z., Berger F., Appl. Catal. A 2004, 269, 161–168; [Google Scholar]
- 2h. Papp A., Molnár Á., Mastalir Á., Appl. Catal. A. 2005, 289, 256–266; [Google Scholar]
- 2i. Webb J. D., MacQuarrie S., McEleney K., Crudden C. M., J. Catal. 2007, 252, 97–109; [Google Scholar]
- 2j. Budroni G., Corma A., García H., Primo A., J. Catal. 2007, 251, 345–353; [Google Scholar]
- 2k. Das D. D., Sayari A., J. Catal. 2007, 246, 60–65; [Google Scholar]
- 2l. Ren N., Yang Y.-H., Zhang Y.-H., Wang Q.-R., Tang Y., J. Catal. 2007, 246, 215–222; [Google Scholar]
- 2m. Nie Yu., Jaenicke S., Bekkum H. van, Chuah G.-Kh., J. Catal. 2007, 246, 223–231. [Google Scholar]
- 3.
- 3a. Suppino R. Soeiro, Landers R., Cobo A. J. Gomez, Appl. Catal. A 2013, 452, 9–16; [Google Scholar]
- 3b. Piccolo L., Valcarcel A., Bausach M., Thomazeau C., Uzio D., Berhault G., Phys. Chem. Chem. Phys. 2008, 10, 5504–5506; [DOI] [PubMed] [Google Scholar]
- 3c. Markov P. V., Turova O. V., Mashkovsky I. S., Khudorozhkov A. K., Bukhtiyarov V. I., Stakheev A.Yu., Mendeleev Commun. 2015, 25, 367–369; [Google Scholar]
- 3d. Domínguez-Domínguez S., Berenguer-Murcia Á., Linares-Solano Á., Cazorla-Amorós D., J. Catal. 2008, 257, 87–95; [Google Scholar]
- 3e. Liu H., Jiang T., Han B., Liang Sh., Zhou Yi., Science 2009, 326, 1250–1252; [DOI] [PubMed] [Google Scholar]
- 3f. Pérez Y., Fajardo M., Corma A., Catal. Commun. 2011, 12, 1071–1074; [Google Scholar]
- 3g. Solladié-Cavallo A., Baram A., Choucair E., Norouzi-Arasi H., Schmitt M., Garin F., J. Mol. Catal. A 2007, 273, 92–98. [Google Scholar]
- 4.
- 4a. Su F., Lv L., Lee F. Y., Liu T., Cooper A. I., Zhao X. S., J. Am. Chem. Soc. 2007, 129, 14213–14223; [DOI] [PubMed] [Google Scholar]
- 4b. Wan Yi., Wang H., Zhao Q., Klingstedt M., Terasaki O., Zhao D., J. Am. Chem. Soc. 2009, 131, 4541–4550. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Maegawa T., Akashi A., Yaguchi K., Iwasaki Yo., Shigetsura M., Monguchi Ya., Sajiki H., Chem. Eur. J. 2009, 15, 6953–6963; [DOI] [PubMed] [Google Scholar]
- 5b. Güvenatam B., Kurşun O., Heeres E. H. J., Pidko E. A., Hensen E. J. M., Catal. Today, 2014, 233, 83–91; [Google Scholar]
- 5c. Park K. H., Jang K., Kim H. J., Son S. U., Angew. Chem. Int. Ed. 2007, 46, 1152–1155; [DOI] [PubMed] [Google Scholar]
- 5d. Chase Z. A., Fulton J. L., Camaioni D. M., Mei D., Balasubramanian M., Pham V.-T., Zhao Ch., Weber R. S., Wang Yo., Lercher J. A., J. Phys. Chem. C 2013, 117, 17603–17612; [Google Scholar]
- 5e. Hiyoshi N., Rode Ch., Sato O., Tetsuka H., Shirai M., J. Catal. 2007, 252, 57–68; [Google Scholar]
- 5f. Mastalir Á., Király Z., Patzkó Á., Dékány I., L'Argentiere P., Carbon 2008, 46, 1631–1637; [Google Scholar]
- 5g. Handa P., Wikander K., Holmberg K., Microporous Mesoporous Mater. 2009, 117, 126–135. [Google Scholar]
- 6.
- 6a. Wang H., Zhao F., Fujita S., Arai M., Catal. Commun. 2008, 9, 362–368; [Google Scholar]
- 6b. Domínguez-Domínguez S., Berenguer-Murcia Á., Pradhan B. K., Linares-Solano Á., Cazorla-Amorós Diego, J. Phys. Chem. C 2008, 112, 3827–3834; [Google Scholar]
- 6c. Karakhanov E. A., Aksenov I. A., Kardashev S. V., Maksimov A. L., Putilin F. N., Shatokhin A. N., Savilov S. V., Appl. Catal. A: Gen. 2013, 464–465, 253–260; [Google Scholar]
- 6d. Wang Y., Yao J., Li H., Su D., Antonietti M., J. Am. Chem. Soc. 2011, 133, 2362–2365; [DOI] [PubMed] [Google Scholar]
- 6e. Ma Y., Huang Ya., Cheng Yo., Wang L., Li X., Appl. Catal. A. Gen. 2014, 484, 154–160; [Google Scholar]
- 6f. Li L., Zhao Hu., Wang R., ACS Catal. 2015, 5, 948–955; [Google Scholar]
- 6g. Ruiz M. Pilar, Faria J., Shen M., Drexler S., Prasomsri T., Resasco D. E., ChemSusChem 2011, 4, 964–974; [DOI] [PubMed] [Google Scholar]
- 6h. Starodubtseva E. V., Vinogradov M. G., Turova O. V., Bumagin N. A., Rakov E. G., Sokolov V. I., Catal. Commun. 2009, 10, 1441–1442; [Google Scholar]
- 6i. Semagina N., Renken A., Kiwi-Minsker L., Chem. Eng. Sci. 2007, 62, 5344–5348. [Google Scholar]
- 7.
- 7a. Maximov A., Zolotukhina A., Kulikov L., Kardasheva Yu., Karakhanov E., React. Kinet. Mech. Cat. 2016, 117, 1–15; [Google Scholar]
- 7b.E. Karakhanov, Yu. Kardasheva, L. Kulikov, A. Maximov, A. Zolotukhina, M. Vinnikova, A. Ivanov, Catalysts 2016, 6, 122, doi:10.3390/catal6080122.
- 8.
- 8a. Gonzalez-Galvez D., Lara P., Rivada-Wheelaghan O., Conejero S., Chaudret B., Philippot K., Leeuwen P. W. N. M. van, Catal. Sci. Technol. 2013, 3, 99–105; [Google Scholar]
- 8b. Lara P., Philippot K., Chaudret B., ChemCatChem 2013, 5, 28–45; [Google Scholar]
- 8c. Nasar K., Fache F., Lemaire M., Béziat J.-C., Besson M., Gallezot P., J. Mol. Catal. 1994, 87, 107–115; [Google Scholar]
- 8d. Jansat S., Picurelli D., Pelzer K., Philippot K., Gómez M., Muller G., Lecante P., Chaudret B., New J. Chem. 2006, 30, 115–122; [Google Scholar]
- 8e. Gual A., Godard C., Castillon S., Claver Carmen, Dalton Trans. 2010, 39, 11499–11512; [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Bras J. Le, Mukherjee D. Kumar, González S., Tristany M., Ganchegui B., Moreno-Mañas M., Pleixats R., Hénin F., Muzart J., New J. Chem. 2004, 28, 1550–1553; [Google Scholar]
- 9b. Nowicki A., Boulaire V. Le, Roucoux A., Adv. Synth. Catal. 2007, 349, 2326–2330; [Google Scholar]
- 9c. Silveira E. T., Umpierre A. P., Rossi L. M., Machado G., Morais J., Soares G. V., Baumvol I. J. R., Teixeira S. R., Fichtner P. F. P., Dupont J., Chem. Eur. J. 2004, 10, 3734–3740; [DOI] [PubMed] [Google Scholar]
- 9d. Rossi L. M., Machado G., J. Mol. Catal. A: Chem. 2009, 298, 69–73; [Google Scholar]
- 9e. Maksimov A. L., Kuklin S. N., Kardasheva Yu.S., Karakhanov E. A., Petrol. Chem. 2013, 53, 157–163; [Google Scholar]
- 9f. Beller M., Fischer H., Kühlein K., Reisinger C.-P., Herrmann W. A., J.Organomet. Chem. 1996, 520, 257–259; [Google Scholar]
- 9g. Yang X., Fei Zh., Zhao D., Ang W. H., Li Yo., Dyson P. J., Inorg. Chem. 2008, 47, 3292–3297; [DOI] [PubMed] [Google Scholar]
- 9h. Calò V., Nacci A., Monopoli A., Montingelli F., J. Org. Chem. 2005, 70, 6040–6044; [DOI] [PubMed] [Google Scholar]
- 9i. Prechtl M. H. G., Scariot M., Scholten J. D., Machado G., Teixeira S. R., Dupont J., Inorg. Chem. 2008, 47, 8995–9001. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Guerrero M., Roucoux A., Denicourt-Nowicki A., Bricoute H., Monflier E., Collière V., Fajerwerg K., Philippot K., Catal. Today 2012, 183, 34–41; [Google Scholar]
- 10b. González-Gálvez D., Nolis P., Philippot K., Chaudret B., van Leeuwen P. W. N. M., ACS Catal. 2012, 2, 317–321; [Google Scholar]
- 10c. Gual A., Axet M. R., Philippot K., Chaudret B., Nowicky A. D., Roucoux A., Castillon S., Claver C., Chem. Commun. 2008, 2759–2761; [DOI] [PubMed] [Google Scholar]
- 10d. Bresó-Femenia E., Chaudret B., Castillón Sergio, Catal. Sci. Technol. 2015, 5, 2741–2751; [Google Scholar]
- 10e. Liu R., Zhao F., Fujita Sh., Arai M., Appl. Catal. A. Gen. 2007, 316, 127–133; [Google Scholar]
- 10f. Woo J. O., Park J.-E., Han J.-S., Kwon T. S., Kim Ye.-J., Son K.-S., Appl. Organometal. Chem. 2014, 28, 151–155; [Google Scholar]
- 10g. Tatumi R., Akita T., Fujihara H., Chem. Commun. 2006, 3349–3351. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Bayer E., Schumann W., J. Chem. Soc., Chem. Commun. 1986, 12, 949–952; [Google Scholar]
- 11b. Lu F., Liu J., Xu J., Mater. Chem. Phys. 2008, 108, 369–374; [Google Scholar]
- 11c. Lu F., Liu J., Xu J., J. Mol. Catal. A. Chem. 2007, 271, 6–13; [Google Scholar]
- 11d. Zhu J.-F., Tao G.-H., Liu H.-Yu, He L., Sun Q.-H., Liu H.-Ch., Green Chem. 2014, 16, 2664–2669; [Google Scholar]
- 11e. Telkar M. M., Rod C. V., Chaudhari R. V., Joshi S. S., Nalawade A. M., Appl. Catal. A: Gen. 2004, 273, 11–19; [Google Scholar]
- 11f. Lu F., Liu J., Xu J., Adv. Synth. Catal. 2006, 348, 857–861. [Google Scholar]
- 12.
- 12a. Sawoo S., Srimani D., Dutta P., Lahiri R., Sarkar A., Tetrahedron 2009, 65, 4367–4374; [Google Scholar]
- 12b. Srimani D., Sawoo S., Sarkar A., Org. Lett. 2007, 9, 3639–3642. [DOI] [PubMed] [Google Scholar]
- 13. Mori Sh., Ohkubo T., Ikawa T., Kume A., Maegawa T., Monguchi Ya., Sajiki H., J. Mol. Catal. A: Chem. 2009, 307, 77–87 [Google Scholar]
- 14. Huc V., Pelzer K., J. Colloid Interface Sci. 2008, 318, 1–4. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. Nowicky A., Zhang Y., Léger B., Rolland J.-P., Bricout H., Monflier E., Roucoux A., Chem. Commun. 2006, 296–298; [DOI] [PubMed] [Google Scholar]
- 15b. Kuklin S., Maximov A., Zolotukhina A., Karakhanov E., Catal. Commun. 2016, 73, 63–68; [Google Scholar]
- 15c. Noël S., Léger B., Herbois R., Ponchel A., Tilloy S., Wenz G., Monflier E., Dalton Trans. 2012, 41, 13359–13363; [DOI] [PubMed] [Google Scholar]
- 15d. Noël S., Léger B., Ponchel A., Philippot K., Denicourt-Nowicki A., Roucoux A., Monflier E., Catal. Today 2014, 235, 20–32. [Google Scholar]
- 16.
- 16a. Andrés R., Jesús E., Flores J. C., New J. Chem. 2007, 31, 1161–1191; [Google Scholar]
- 16b. Astruc D., Boisselier E., Ornelas C., Chem. Rev. 2010, 110, 1857–1959; [DOI] [PubMed] [Google Scholar]
- 16c. Mery D., Astruc D., Coord. Chem. Rev. 2006, 250, 1965–1979; [Google Scholar]
- 16d. Wang D., Astruc D., Coord. Chem. Rev. 2013, 257, 2317–2334. [Google Scholar]
- 17.
- 17a. Vögtle F., Gestermann S., Hesse R., Schwierz H., Windisch B., Prog. Polym. Sci. 2000, 25, 987–1041; [Google Scholar]
- 17b. Crooks R. M., Zhao M., Sun L., Chechik V., Yeung L. K., Acc. Chem. Res. 2001, 34, 181–190; [DOI] [PubMed] [Google Scholar]
- 17c. Niu Y., Crooks R. M., C. R. Chimie 2003, 6, 1049–1059; [Google Scholar]
- 17d. Newkome G. R., Shreiner C. D., Polym. 2008, 49, 1–173. [Google Scholar]
- 18.
- 18a. Ooe M., Murata M., Mizugaki T., Ebitani T., Kaneda K., Nanolett. 2002, 2, 999–1002; [Google Scholar]
- 18b. Murata M., Tanaka Yu., Mizugaki T., Ebitani K., Kaneda K., Chem. Lett. 2005, 34, 272–273. [Google Scholar]
- 19.
- 19a. Gopidas K. R., Whitesell J. K., Fox M. A., Nano Lett. 2003, 3, 1757–1760; [Google Scholar]
- 19b. Ooe M., Murata M., Mizugaki T., Ebitani K., Kaneda K., J. Am. Chem. Soc. 2004, 126, 1604–1605; [DOI] [PubMed] [Google Scholar]
- 19c. Yang Ya., Ogasawara Sh., Li G., Kato Sh., Polym. J. 2015, 47, 1–8; [Google Scholar]
- 19d. Karakhanov E. A., Maximov A. L., Tarasevich B. N., Skorkin V. A., J. Mol. Catal. A: Chem. 2009, 297, 73–79; [Google Scholar]
- 19e. Singh A., Chandler B. D., Langmuir 2003, 19, 7682–7684; [Google Scholar]
- 19f.Q.-H. Fan, Y.-M. Chen, X.-M. Chen, D.-Zh. Jiang, F. Xi, A. S. C. Chan, Chem. Commun 2000, 789–790;
- 19g.G.-J. Deng, Q.-H. Fan, X.-M. Chen, D.-Sh. Liu, A. S. C. Chan, Chem. Commun 2002, 1570–1571. [DOI] [PubMed]
- 20.
- 20a. Antonietti M., Wenz E., Bronstein L., Seregina M., Adv. Mater. 1995, 7, 1000–1005; [Google Scholar]
- 20b. Spatz J. P., Roescher A., Möller M., Adv. Mater. 1996, 8, 337–340. [Google Scholar]
- 21. Ornelas C., Ruiz J., Salmon L., Astruc D., Chem. Eur. J. 2008, 14, 50–64. [DOI] [PubMed] [Google Scholar]
- 22. Bronstein L. M., Shifrina Z. B. , Chem. Rev. 2011, 111, 5301–5344. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Esumi K., Suzuki A., Aihara N., Usui K., Torigoe K., Langmuir 1998, 14, 3157–3159; [Google Scholar]
- 23b. Garcia M. E., Baker L. A., Crooks R. M., Anal. Chem. 1999, 71, 256–258. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Chandra S., Lang H., Mater. Chem. Phys. 2009, 114, 926–932; [Google Scholar]
- 24b. Li C., Li D., Zhao Z.-S., Duan X.-M., Hou W., Colloid. Surf. A: Physicochem. Eng. Asp. 2010, 366, 45–49. [Google Scholar]
- 25.
- 25a. Lakowicz J. R., Gryczynski I., Gryczynski Z., Murphy C. J. J., Phys. Chem. B 1999, 103, 7613–7620; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Shi X. Y., Ganser T. R., Sun K., Balogh L. P., Baker J. R. Jr., Nanotechnol. 2006, 17, 1072–1078. [Google Scholar]
- 26. Niu Y., Yeung L. K., Crooks R. M., J. Am. Chem. Soc. 2001, 123, 6840–6846 [Google Scholar]
- 27. Wisher A. C., Bronstein I., Chechik V., Chem. Commun. 2006, 1637–1639 [DOI] [PubMed] [Google Scholar]
- 28. Sooklal K., Hanus L. H., Ploehn H. J., Murphy C. J., Adv. Mater. 1998, 10, 1083–1087 [Google Scholar]
- 29.
- 29a. Gröhn F., Bauer B. J., Akpalu Y. A., Jackson C. L., Amis E. J., Macromol. 2000, 33, 6042–6050; [Google Scholar]
- 29b. Boisselier E., Diallo A. K., Salmon L., Ornelas C., Ruiz J., Astruc D., J. Am. Chem. Soc. 2010, 132, 2729–2742; [DOI] [PubMed] [Google Scholar]
- 29c. Diallo A. K., Ornelas C., Salmon L., Ruiz J., Astruc D., Angew. Chem. Int. Ed. 2007, 46, 8644–8648; [DOI] [PubMed] [Google Scholar]
- 29d.C. Ornelas, L. Salmon, J. Ruiz, D. Astruc, Chem. Commun 2007, 4946–4948; [DOI] [PubMed]
- 29e. Ornelas C., Ruiz J., Salmon L., Astruc D., Adv. Synth. Catal. 2008, 350, 837–845. [Google Scholar]
- 30. Karakhanov E. A., Maximov A. L., Skorkin V. A., Zolotukhina A. V., Smerdov A. S., Tereshchenko A.Yu., Pure Appl. Chem. 2009, 81, 2013–2023 [Google Scholar]
- 31.
- 31a. Maximov A., Zolotukhina A., Murzin V., Karakhanov E., Rosenberg E., ChemCatChem 2015, 7, 1197–1210; [Google Scholar]
- 31b. Karakhanov E. A., Maximov A. L., Zolotukhina A. V., Terenina M. V., Vutolkina A. V., Petrol. Chem. 2016, 56, 491–502; [Google Scholar]
- 31c. Karakhanov E., Maximov A., Zolotukhina A., Kardasheva Yu., Talanova M., J. Inorg. Organomet. Polym. Mater. 2016, 26, 1264–1279; [Google Scholar]
- 31d. Zakharyan E. M., Ma Gouqiung, Maksimov A. L., Karakhanov E. A., Voronina Z. D., Petrol. Chem. 2014, 54, 412–419. [Google Scholar]
- 32.
- 32a. Karakhanov E. A., Maximov A. L., Zakharyan E. M., Zolotukhina A. V., Ivanov A. O., Mol. Catal. 2017, 440, 107–119; [Google Scholar]
- 32b. Karakhanov E. A., Maksimov A. L., Zolotukhina A. V., Kardashev S. V., Filippova T.Yu., Petrol. Chem. 2012, 52, 289–298; [Google Scholar]
- 32c. Karakhanov E. A., Maksimov A. L., Zakharian E. M., Kardasheva Yu.S., Savilov S. V., Truhmanova N. I., Ivanov A. O., Vinokurov V. A., J. Mol. Catal. A: Chem. 2015, 397, 1–18. [Google Scholar]
- 33.
- 33a. Ogasawara S., Kato S., J. Am. Chem. Soc. 2010, 132, 4608–4613; [DOI] [PubMed] [Google Scholar]
- 33b. Reetz M. T., Giebel D., Angew. Chem. Int. Ed. 2000, 39, 2948–2951 [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Jiang Yi., Gao Q., J. Am. Chem. Soc. 2006, 128, 716–717; [DOI] [PubMed] [Google Scholar]
- 34b. Zweni P. P., Alper H., Adv. Synth. Catal. 2006, 348, 725–731; [Google Scholar]
- 34c. Wu H., Liu Z., Wang X., Zhao B., Zhang J., Li C., J. Colloid Interface Sci. 2006, 30 2, 142–148; [DOI] [PubMed] [Google Scholar]
- 34d. Chung Y. M., Rhee H. K., Korean J. Chem. Eng. 2004, 21, 81–97; [Google Scholar]
- 34e. Li H., Lü J., Zheng Zh., Cao R., J. Colloid Interface Sci. 2011, 353, 149–155; [DOI] [PubMed] [Google Scholar]
- 34f. Xu Yi., Zhang Zh., Zheng J., Du Q., Li Yi., Appl. Organometal. Chem. 2013, 27, 13–18; [Google Scholar]
- 34g. Zheng Zh., Li H., Liu T., Cao R., J. Catal. 2010, 270, 268–274; [Google Scholar]
- 35.
- 35a. Krishnan G. R., Sreekumar K., Soft Mater. 2010, 8, 114–129; [Google Scholar]
- 35b. Suijkerbuijk B. M. J. M., Shu L. J., Gebbink R. J. M. Klein, Schlüter A. D., Koten G. van, Organometallics, 2003, 22, 4175–4177; [Google Scholar]
- 35c. Dahan A., Portnoy M., Org. Lett. 2003, 5, 1197–1200; [DOI] [PubMed] [Google Scholar]
- 36.
- 36a. Alvarez J., Sun L., Crooks R. M., Chem. Mater. 2002, 14, 3995–4001; [Google Scholar]
- 36b. Guo Z., Feng H., Ma H. C., Kang Q. X., Yang Z. W., Polym. Adv. Technol. 2004, 15, 100–104. [Google Scholar]
- 37. Murugan E., Rangasamy R., J. Polym. Sci A: Polym. Chem. 2010, 48, 2525–2532 [Google Scholar]
- 38.
- 38a. Alper H., Arya P., Bourque S. C., Jefferson G. R., Manzer L. E., Can. J. Chem. 2000, 78, 920–924; [Google Scholar]
- 38b. Chanthateyanonth R., Alper H., J. Mol. Catal. A: Chem. 2003, 201, 23–31 [Google Scholar]
- 39.
- 39a. Chanthateyanonth R., Alper H., Adv. Synth. Catal. 2004, 346, 1375–1384; [Google Scholar]
- 39b. Lu Sh.-M., Alper H., J. Am. Chem. Soc. 2005, 127, 14776–14784; [DOI] [PubMed] [Google Scholar]
- 39c. Touzani R., Alper H., J. Mol. Catal. A: Chem. 2005, 227, 197–207; [Google Scholar]
- 39d. Antebi Sh., Arya P., Manzer L. E., Alper H., J. Org. Chem. 2002, 67, 6623–6631; [DOI] [PubMed] [Google Scholar]
- 39e. Reynhardt J. P. K., Alper Howard, J. Org. Chem. 2003, 68, 8353–8360. [DOI] [PubMed] [Google Scholar]
- 40. Zweni P. P., Alper H., Adv. Synth. Catal. 2004, 346, 849–854 [Google Scholar]
- 41. Hagiwara H., Sasaki H., Tsubokawa N., Hoshi T., Suzuki T., Tsuda T., Kuwabata S., SynLett. 2010, 1990–1996 [Google Scholar]
- 42. Nabid M. Reza, Bide Ya., Rezaei S. J. Tabatabaei, Appl.Catal. A. Gen. 2011, 406, 124–132 [Google Scholar]
- 43. Karakhanov E., Maximov A., Kardasheva Yu., Semernina V., Zolotukhina A., Ivanov A., Abbott G., Rosenberg E., Vinokurov V., ACS Appl. Mater. Interfaces 2014, 6, 8807–8816 [DOI] [PubMed] [Google Scholar]
- 44. Karakhanov E. A., Maximov A. L., Zolotukhina A. V., Yatmanova N., Rosenberg E., Appl. Organomet. Chem. 2015, 29, 777–784 [Google Scholar]
- 45. Jayamurugan G., Umesh C. P., Jayaraman N., J. Mol. Catal. A. Chem. 2009, 307, 142–148 [Google Scholar]
- 46. Boronoev M. P., Zolotukhina A. V., Ignatyeva V I., Terenina M. V., Maximov A. L., Karakhanov E. A., Macromol. Symp. 2016, 363, 57–63 [Google Scholar]
- 47. Karakhanov E., Maximov A., Zolotukhina A., Mamadli E., Vutolkina A., Ivanov A., Catalysts 2017, 7, 86, doi:10.3390/catal7030086 [Google Scholar]
- 48. Maximov A. L., Zolotukhina A. V., Mamedli A. A., Kulikov L. A., Karakhanov E. A., ChemCatChem 2018, 10, 222–233 [Google Scholar]
- 49. Beamson G., Briggs D., Database High Resolution XPS of Organic Polymers: The Scienta ESCA300,Sons John Wiley &, Chichester, UK, 1992, p. 295. [Google Scholar]
- 50.
- 50a. Briggs D., Beamson G., Anal. Chem. 1992, 64, 1729–1736; [Google Scholar]
- 50b. Strohmeier B. R., Appl. Surf. Sci. 1991, 47, 225–234; [Google Scholar]
- 50c. Pertsin A. J., Gorelova M. M., Levin V. Y., Makarova L. I., J. Appl. Polym. Sci. 1992, 45, 1195–1202; [Google Scholar]
- 50d. Laoharojanaphand P., Lin T. J., Stoffer J. O., J. Appl. Polymer Sci. 1990, 40, 369–384. [Google Scholar]
- 51. González B., Colilla M., Laorden C. López de, Vallet-Regí M., J. Mater. Chem. 2009, 19, 9012–9024 [Google Scholar]
- 52.
- 52a. Anderson J. R., Structure of Metallic Catalysts, London, Academic Press, 1975; [Google Scholar]
- 52b. Aben P. C., J. Catal. 1968, 10, 224–229 [Google Scholar]
- 53.
- 53a.V. I. Nefedov, I. A. Zakharova, I. I. Moiseev, M. A. Porai-koshits, M. N. Vargoftik, A. P. Belov, Zh. Neorg. Khimii 1973, 18, 3264 (Russ. J. Inorg. Chem 1973, 18, 3264);
- 53b. Nefedov V. I., Salyn Y. V., Moiseev I. I., Sadovskii A. P., Berenbljum A. S., Knizhnik A. G., Mund S. L., Inorg. Chim. Acta 1979, 35, L343–L344; [Google Scholar]
- 53c. Powell C. J., J. Electron Spectrosc. Relat. Phenom. 2012, 185, 1–3; [Google Scholar]
- 53d. Nyholm R., Mårtensson N., J. Phys. C 1980, 13, L279–L284. [Google Scholar]
- 54.
- 54a. Militello M. C., Simko S. J., Surf. Sci. Spectra 1994, 3, 395–401; [Google Scholar]
- 54b. Shafeev G. A., Themlin J.-M., Bellard L., Marine W., Cros A., J. Vac. Sci. Technol. A 1996, 14, 319–326; [Google Scholar]
- 54c. Noack K., Zbinden H., Schlögl R., Catal. Lett. 1990, 4, 145–155; [Google Scholar]
- 54d. Fleisch T. H., Zaiac G. W., Schreiner J. O., Mains G. J., Appl. Surf. Sci. 1986, 26, 488–497 [Google Scholar]
- 55. Xiao Z. Yi, Zhao Yu., Wang A., Perumal J., Ki D.-P., Lab. Chip 2011, 11, 57–62 [DOI] [PubMed] [Google Scholar]
- 56.
- 56a. Masahiro Ya., Ichiro M., Tasuku I., Yoshiki W., Tadaoki M., Isao I., Bull. Chem. Soc. Jpn. 1985, 58, 2336–2339; [Google Scholar]
- 56b. Masahiro Ya., Ichiro M., Isao I., Bull. Chem. Soc. Jpn. 1985, 58, 2697–2698 [Google Scholar]
- 57.
- 57a. Hub S., Hilaire L., Touroude R., Appl. Catal. 1988, 36, 307–322; [Google Scholar]
- 57b. Ryndin Yu.A., Nosova L. V., Boronin A. I., Chuvilin A. L., Appl. Catal. 1988, 42, 131–141; [Google Scholar]
- 57c. Nosova L. V., Stenin M. V., Nogin Yu.N., Ryndin Yu.A., Appl. Surf. Sci. 1992, 55, 43–48; [Google Scholar]
- 57d. Mason M. G., Gerenser L. J., Lee S. -T., Phys. Rev. Lett. 1977, 39, 288–291; [Google Scholar]
- 57e. Takasu Yu., Unwin R., Tesche B., Grunze A. M. Bradshaw M., Surf. Sci. 1978, 77, 219–232; [Google Scholar]
- 57f. Fritsch A., Légaré P., Surf. Sci. 1985, 162, 742–746; [Google Scholar]
- 57g. Baetzold R. C., J. Catal. 1973, 29, 129–137 [Google Scholar]
- 58.
- 58a. Aramendía M. A., Borau V., Jiménez C., Marinas J. M., Sempere M. E., Urbano F. J., Appl. Catal. 1990, 63, 375–389; [Google Scholar]
- 58b. Molnár A., Sárkány A., Varga M., J. Mol. Catal. A. Chem. 2001, 173, 185–221 [Google Scholar]
- 59. Hub S., Touroude R., J. Catal. 1988, 114, 411–421 [Google Scholar]
- 60. He G., Song Ya., Kang X., Chen Sh., Electrochim. Acta 2013, 94, 98–103 [Google Scholar]
- 61. Anderson J. R., Structure of Metallic Catalysts, London, Academic Press, 1975. [Google Scholar]
- 62. Semagina N., Renken A., Kiwi-Minsker L., J. Phys. Chem. C 2007, 111, 13933–13937 [Google Scholar]
- 63.
- 63a. Crespo-Quesada M., Cardenas-Lizana F., Dessimoz A.-L., Kiwi-Minsker L., ACS Catal. 2012, 2, 1773–1786; [Google Scholar]
- 63b. Boitiaux J. P., Cosyns J., Vasudevan S., Appl. Catal. 1983, 6, 41–51 [Google Scholar]
- 64.
- 64a. Augustyn W. G., McCrindle R. I., Coville N. J., Appl. Catal. A: Gen. 2010, 388, 1–6; [Google Scholar]
- 64b. Gigola C. E., Aduriz H. R., Bodnariuk P., Appl. Catal. 1986, 27, 133–144; [Google Scholar]
- 64c. Silvestre-Albero J., Rupprechter G., Freund H.-J., Chem. Commun. 2006, 80–82; [DOI] [PubMed] [Google Scholar]
- 64d. Hardeveld R. Van, Hartog F., Surf. Sci. 1969, 15, 189–230; [Google Scholar]
- 64e. Uzio D., Berhault G., Catal. Rev. Sci. Eng. 2010, 52, 106–131. [Google Scholar]
- 65.
- 65a. Mallat T., Baiker A., Appl. Catal. A. Gen. 2000, 200, 3–22; [Google Scholar]
- 65b. Sulman E. M., Russ. Chem. Rev. 1994, 63, 923–936; [Google Scholar]
- 65c. Boitiaux J. P., Cosyns J., Vasudevan S., Appl. Catal. 1985, 15, 317–326 [Google Scholar]
- 66. Crespo-Quesada M., Dykeman R. R., Laurenczy G., Dyson P. J., Kiwi-Minsker L., J. Catal. 2011, 279, 66–74 [Google Scholar]
- 67. Long W., Brunelli N. A., Didas S. A., Ping E. W., Jones C. W, ACS Catal. 2013, 3, 1700–1708 [Google Scholar]
- 68.
- 68a. Bond G. C., Rawle A. F., J. Mol. Catal. A. Chem. 1996, 109, 261–271; [Google Scholar]
- 68b. Bond G. C., J. Mol. Catal. A. Chem. 1997, 11 8, 333–339; [Google Scholar]
- 68c. Joice B. J., Rooney J. J., Wells P. B., Wilson G. R., Discuss. Faraday Soc. 1966, 223–236 [Google Scholar]
- 69. Carturan G., Facchin G., Gottardi V., Guglielmi M., Navazio G., J. Non-Cryst. Solids 1982, 48, 219–226 [Google Scholar]
- 70. Stakheev A.Yu., Markov P. V., Taranenko A. S., Bragina G. O., Baeva G. N., Tkachenko O. P., Mashkovskii I. S., Kashin A. S., Kinet. Catal. 2015, 56, 733–740 [Google Scholar]
- 71. Chung Yo.-M., Rhee H.-K., J. Mol. Catal. A. Chem. 2003, 206, 291–298 [Google Scholar]
- 72. Stern E. W., Maples P. K., J. Catal. 1972, 27, 120–133 [Google Scholar]
- 73.
- 73a. Beisembaeva Z. T., Gudkov B. S., Kiperman S. L., Bull. Russ. Acad. Sci. Div. of Chem. Sci. 1984, 33, 481–485; [Google Scholar]
- 73b. Beisembaeva Z. T., Gudkov B. S., Kharson M. S., Popov N. I., Kiperman S. L., Bull. Russ. Acad. Sci. Div. of Chem. Sci. 1975, 24, 863–865 [Google Scholar]
- 74.
- 74a. Fujii H., Bailar J. C. Jr., J. Catal. 1978, 52, 342–345; [Google Scholar]
- 74b. Lin T.-B., Chou T.-C., Appl. Catal. A. Gen. 1994, 108, 7–19; [Google Scholar]
- 74c. Duca D., Liotta L. F., Deganello G., Catal.Today 1995, 24, 15–21; [Google Scholar]
- 74d. Carturan G., Facchin G., Cocco G., Enzo S., Navazio G., J. Catal. 1982, 76, 405–417; [Google Scholar]
- 74e. Carturan G., Cocco G., Facchin G., Navazio G., J. Mol. Catal. 1984, 26, 375–384; [Google Scholar]
- 74f. Anderson J. A., Mellor J., Wells R. P. K., J. Catal. 2009, 261, 208–216; [Google Scholar]
- 74g. Weerachawanasak P., Mekasuwandumrong O., c M. Arai, Fujita Sh.-I., Praserthdam P., Panpranot J., J. Catal. 2009, 262, 199–205 [Google Scholar]
- 75. Zou J.-J., Zhang X., Kong J., Wang L., Fuel 2008, 87, 3655–3659 [Google Scholar]
- 76.
- 76a. Sokolsky D. V., Ualikhanova A., Temirbulatova A.Ye., Dembitsky A. A., Mailyubaev B. T., Russ. J. Appl. Chem. 1988, 61, 2360–2363 (Russ.); [Google Scholar]
- 76b. Antonova T. N., Abramov I. A., Feldblyum V. S., Abramov I. G., Danilova A. S., Petrol. Chem. 2009, 49, 366–368; [Google Scholar]
- 76c. Behr A., Manz V., Lux A., Ernst A., Catal. Lett. 2013, 143, 241–245; [Google Scholar]
- 76d. Liu G., Mi Zh., Wang X. L., Zhang Ind., Eng. Chem. Res. 2005, 44, 3846–3851; [Google Scholar]
- 76e. Skála D., Hanika J., Petrol Coal 2003, 45, 105–108; [Google Scholar]
- 76f. Hao M., Yang B., Wang H., Liu G., Qi S., J. Phys. Chem. A 2010, 114, 3811–3817 [DOI] [PubMed] [Google Scholar]
- 77. Brabander-van den Berg E. M. M. de, Meijer E. W., Angew. Chem. Int. Ed. Engl. 1993, 32, 1308–1312 [Google Scholar]
- 78. Karaki M., Karout A., Toufaily J., Rataboul F., Essayem N., Lebeau B., J. Catal. 2013, 305, 204–216. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary