

Abstract
DNA methylation represents a fundamental epigenetic modification that regulates chromatin architecture and gene transcription. Many diseases, including cancer, show aberrant methylation patterns that contribute to the disease phenotype. DNA methylation inhibitors have been used to block methylation dependent gene silencing to treat hematopoietic neoplasms and to restore expression of developmentally silenced genes. However, these inhibitors disrupt methylation globally and show significant off- target toxicities. As an alternative approach, we have been studying readers of DNA methylation, the 5-methylcytosine binding domain family of proteins, as potential therapeutic targets to restore expression of aberrantly and developmentally methylated and silenced genes. In this review, we discuss the role of DNA methylation in gene regulation and cancer development, the structure and function of the 5-methylcytosine binding domain family of proteins, and the possibility of targeting the complexes these proteins form to treat human disease.
Keywords: DNA methylation, 5-methylcytosine binding domain, chromatin, NuRD, gene regulation
1. Introduction
DNA methylation, a fundamental epigenetic mediator of gene regulation in animals, primarily involves the addition of a methyl group to carbon-5 of a cytosine base, creating 5-methylcytosine, first identified in biological material in 1925 (Johnson & Coghill, 1925). However, it was not until the late 1940s that Hotchkiss identified a unique base in mammalian tissue, which he referred to as epicytosine for its migration relative to cytosine in paper chromatography, and suggested it may represent 5-methylcytosine (Hotchkiss, 1948). Then in the early 1950s Wyatt isolated and quantified 5- methylcytosine from animal and plant tissues (Wyatt, 1950, 1951). The functional significance of this DNA modification in gene regulation was hypothesized by Holliday and Pugh (Holliday & Pugh, 1975) and Riggs (Riggs, 1975). Subsequent seminal work by Jones and Taylor in the late 1970s showed that the pyrimidine analog 5-azacytidine induced cellular differentiation by blocking enzymatic DNA methylation (Constantinides, Taylor, & Jones, 1978; Jones & Taylor, 1980; Taylor & Jones, 1979). Important early studies showing an inverse correlation between cytosine methylation and gene expression offered an explanation for how DNA methylation might control gene expression (McGhee & Ginder, 1979; Razin & Riggs, 1980; Shen & Maniatis, 1980; van der Ploeg & Flavell, 1980). 5-azacytidine eventually became the first epigenetic modifier to be used clinically (Charache, et al., 1983; Ley, et al., 1982), and remains one of the few, to be FDA approved for therapy (Itzykson & Fenaux, 2014; Scott, 2016). At the same time, Bird demonstrated that DNA methylation occurred almost exclusively on cytosine bases in a cytosine-guanosine dinucleotide (CpG) to generate a symmetrically methylated dinucleotide (mCpG) (Bird, 1978; Bird & Southern, 1978). Hence, cell division generates two daughter cells with hemimethylated dinucleotides, that can be converted into fully methylated sites to maintain the symmetric modification.
These foundational studies established a mechanism by which epigenetic information could be transmitted across cell division, and led to the exciting and active field of epigenetics. Despite extensive research over the past forty years and clinical experience with hypomethylating agents, the precise mechanistic role of DNA methylation in gene regulation is not fully understood, and only a few drugs that abrogate the effects of DNA methylation for therapeutic benefit have been developed. In this review, we will discuss how the methylation mark is interpreted by the 5- methylcytosine binding domain (MBD) family of proteins and explore the possibility of selectively targeting these proteins and protein complexes for therapy.
2. DNA methylation
2.1. The role of DNA methylation in gene regulation
DNA methylation is present across all three branches of life, yet the function and distribution vary. Prokaryotes methylate both adenosine (6-methyladenosine) and cytosine bases (4- and 5-methylcytosines) to regulate restriction endonuclease activity, gene expression, and transposons (Blow, et al., 2016; Casadesus & Low, 2006). In animals, the DNA methyltransferase (DNMT) enzymes, add methyl groups to the carbon-5 position of cytosine bases (5-methylcytosine), predominantly but not exclusively those found in CpGs (Fig. 1a). Invertebrate animals have relatively low levels of total 5-methylcytosine, and a few species apparently completely lack cytosine methylation (Suzuki & Bird, 2008). When present in invertebrates, it tends to be localized to gene bodies and transposable elements (Zemach, McDaniel, Silva, & Zilberman, 2010; Zemach & Zilberman, 2010). In contrast, vertebrate animals methylate the majority of CpGs throughout their genome, sparing only those that occur in regions with relatively increased numbers of CpGs, known as CpG islands (CGIs). These CGIs are largely unmethylated and overlap with promoters of most genes. When a promotor- associated CGI is methylated, the downstream gene tends to be silenced. Hence, the pattern of CpG methylation in vertebrates and association with gene silencing suggests that DNA methylation can directly regulate gene expression in a heritable fashion.
Fig. 1.
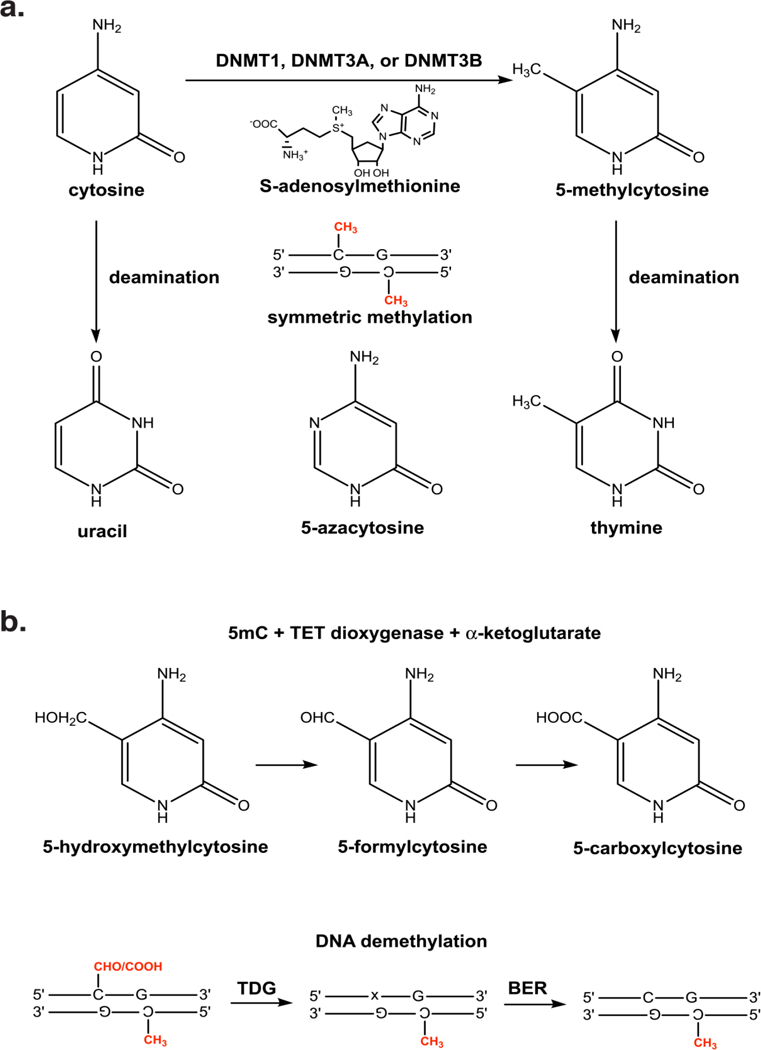
a) The DNMT enzymes add a methyl group to carbon-5 of symmetrically related cytosine bases in a CpG. Spontaneous deamination of cytosine and 5-methylcytosine generate uracil and thymine, respectively. Hence, deamination of 5-methylcytosine is a common cause of C → T and G → A transition mutations. 5-azacytosine contains a nitrogen at position 5 of the base which, when incorporated into DNA, inhibits methylation by the DNMT enzymes, b) The TET dioxygenase enzymes oxidize the methyl group of 5-methylcytosine to generate 5-hydroxymethylcytosine, 5- formylcytosine, and 5-carboxylcytosine. The latter two oxidation products can be excised by TDG, possibly in conjunction with other components of the base excision repair (BER) pathway, to form an abasic site (x) and demethylate the DNA.
Spontaneous deamination of 5-methylcytosines generates C → T and G → A transition mutations (Fig. 1a) (Cooper, Mort, Stenson, Ball, & Chuzhanova, 2010; Duncan & Miller, 1980). Therefore, CpG dinucleotides are a common source of mutation in vertebrate genomes. Over evolutionary timescales, CpG dinucleotides have been lost, presumably through spontaneous deamination, leading to an apparent deficiency of CpG (1.0%) and excess of TpG (7.4%) dinucleotides in the human genome (Jabbari & Bernardi, 2004; Josse, Kaiser, & Kornberg, 1961; Swartz, Trautner, & Kornberg, 1962). Hence, CGIs are defined by a relative increase in CpG density as compared to the rest of the genome, but this density often only approaches the level one would expect by random chance. A common definition of a CGI requires the incidence of CpGs in a 200 base-pair or more stretch of DNA to be greater than 60% of that expected for a random sequence containing a comparable GC content (Gardiner- Garden & Frommer, 1987; Illingworth & Bird, 2009). This observation raises a “chicken or egg” question, does active inhibition of methylation in promoters reduce spontaneous deamination thereby increasing the CpG density, or is the CpG density selectively retained for functional purposes that require a lack of methylation (Bird, 1986)?
This conundrum was recognized from the earliest studies of methylation dependent gene regulation. Bird proposed that active transcription involves binding of factors that block DNMT access to the DNA, such that promoter associated CGIs reflect passive demethylation of actively transcribed genes (Bird, 1986). In contrast, studies of 5- azacytidine and globin regulation demonstrated that demethylation of developmentally regulated genes could activate transcription (Busslinger, Hurst, & Flavell, 1983; Charache, et al., 1983; DeSimone, Heller, Hall, & Zwiers, 1982; Ginder, Whitters, Kelley, & Chase, 1983; Ginder, Whitters, & Pohlman, 1984; Ley, et al., 1982). This gene activation requires additional epigenetic modifiers or transcription factors, leading to the concept that an unmethylated CGI reflects a permissive but not prescriptive state for transcription. Most DNA methylation occurs at non-regulatory sites, such that changes in global methylation do not necessarily reflect changes in transcription. Therefore, large-scale changes in DNA methylation can obscure differential methylation that contributes to development gene regulation and carcinogenesis (Edwards, Yarychkivska, Boulard, & Bestor, 2017; Spencer, et al., 2017). Furthermore, DNA methylation contributes to X-inactivation, genetic imprinting, and inhibition of transposons, whereas enforced DNA methylation of specific genes is strongly associated with transcriptional silencing. Hence, while disagreement continues, the general consensus is that methylation of a promoter-associated CGI most often reflects long-term silencing, methylation of gene bodies is often associated with active transcription, and demethylation of promoter associated CGIs is necessary, but not sufficient, for transcription (Tirado-Magallanes, Rebbani, Lim, Pradhan, & Benoukraf, 2017; van der Ploeg & Flavell, 1980).
2.2. The ten-eleven translocation enzymes and DNA demethylation
The metazoan DNMT enzymes utilize S-adenosyl methionine to add a methyl group to carbon-5 of the cytosine base. The de novo methyltransferases, DNMT3A and DNMT3B, modify symmetrically related cytosine bases in a CpG dinucleotide whereas the maintenance methyltransferase, DNMT1, recognizes hemimethylated sites and restores symmetric methylation during replication (Jeltsch, 2006; Jurkowska & Jeltsch, 2016). Thus, the enzymatic mechanism by which new methylated sites are introduced and maintained across cell division has been well established. Until relatively recently, however, it remained unclear if or how methylation was actively removed from DNA. In the absence of an enzymatic demethylase, 5-methylcytosines can be lost through a passive mechanism in which DNMT1 fails to restore symmetric methylation after cell division. This mechanism, though, cannot explain the rapid wave of demethylation that occurs during early embryogenesis and in the absence of cell division (Mayer, Niveleau, Walter, Fundele, & Haaf, 2000).
One of the more exciting discoveries over the past decade was the identification of the ten-eleven translocation dioxygenase (TET) enzymes that specifically oxidize 5- methylcytosines to sequentially generate 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine bases (Fig. 1b) (He, et al., 2011; Tahiliani, et al., 2009). The latter two oxidative derivatives, 5-formylcytosine and 5-carboxylcytosine, can be recognized by thymine DNA-glycosylase (TDG) of the base-excision repair pathway, possibly with participation of other factors in this pathway, followed by enzymatic removal from DNA (Bochtler, Kolano, & Xu, 2017; Hashimoto, Hong, Bhagwat, Zhang, & Cheng, 2012; Hashimoto, Liu, et al., 2012; Hashimoto, Zhang, Vertino, & Cheng, 2015; Schomacher & Niehrs, 2017). The identification of TET oxidation provided a mechanistic answer to the long-standing question in the field of how 5-methylcytosine is actively removed. Although the oxidative modifications are present at much lower levels than 5-methylcytosine, some tissues and genomic loci show persistent hydroxymethylation, suggesting that this modification may represent an independent epigenetic mark (Hahn, Szabo, & Pfeifer, 2014). Nonetheless, a growing consensus views these oxidative derivatives as functioning primarily, but not exclusively, as a pathway for active demethylation.
2.3. The role of DNA methylation in cancer
Changes in DNA methylation patterns have been associated with carcinogenesis and, as such, extensively documented and investigated for the past forty years (Baylin, et al., 1986; Baylin & Jones, 2011; Burdon, 1966; Christman, 1984; Jones, 1986). Unlike loss-of-function tumor suppressor gene mutations or gain-of-function oncogene activating mutations, DNA methylation dependent changes in transcription are potentially reversible without genome editing. Therefore, as tumor suppressor genes frequently contain CpG rich promoters that are aberrantly silenced in tumors, DNA methylation inhibitors have the unique potential to restore the function of tumor suppressor genes. Vogelstein first demonstrated that changes in DNA methylation precede the development of invasive colorectal carcinoma (Feinberg & Vogelstein, 1983; Goelz, Vogelstein, Hamilton, & Feinberg, 1985). Dysplastic adenomas show global hypomethylation with selective hypermethylation of tumor suppressor genes. This observation leads to the hypothesis that epigenetic silencing of tumor suppressor genes contributes to carcinogenesis and suggests that reversing this silencing could inhibit cancer growth and possibly prevent its development. Subsequent work revealed that many of the methylated CpG islands in colorectal cancer are also methylated in normal colon in an age-dependent manner (Toyota, et al., 1999). Yet, in a subset of colorectal cancers, there is methylation of a distinct set of genes that are unmethylated in normal colonic epithelium, leading to the concept of a CpG island methylator phenotype (CIMP). The CIMP colorectal cancers often harbor methylated tumor suppressor genes, such as CDKN2A, and frequently display microsatellite instability with associated methylation and silencing of the DNA mismatch repair gene MLH1. More robust analyses of promoter methylation have supported the concept of CIMP within tumors showing microsatellite instability and found correlations with specific mutations such as SRAFV600E (Ang, Li, Soong, & lacopetta, 2009; Ogino, et al., 2007). However, no clear consensus has emerged for how to identify CIMP, nor for the overall prognostic significance of this category (Jia, Gao, Zhang, Hoffmeister, & Brenner, 2016; Jia, Jansen, et al., 2016; Kim, Huh, Kim, & Kim, 2017).
The CIMP concept has been extended to other solid tumors. For example, a subset of low-grade gliomas contains driver mutations in isocitrate dehydrogenase (IDH) and harbor hypermethylation across their genome. Specific mutations in IDH1 or IDH2 generate neomorphic enzymatic activity with the production of the oncometabolite D-2- hydroxyglutarate as opposed to the normal metabolite α-ketoglutarate (Dang, et al., 2009). Increased levels of D-2-hydroxyglutarate inhibit the TET enzymes, as well as other α-ketoglutarate dependent enzymes, thereby blocking active DNA demethylation (Gross, et al., 2010; Xu, et al., 2011; Zhao, et al., 2009). Consequently, mutations in the IDH proteins are associated with global increases in DNA methylation (Figueroa, et al., 2010; Noushmehr, et al., 2010; Turcan, et al., 2012). In brain tumors, the resulting change in methylation pattern inhibits DNA binding by the cohesion and CCCTC-binding factor (Hashimoto, et al., 2017; Liu, et al., 2016), which disrupts topologically associating domains and deregulates key oncogenes (Flavahan, et al., 2016). This model of methylation dependent formation of topologically associating domains represents one of the more exciting developments in the field, providing new insight into how DNA methylation can impact gene regulation. Additional studies have suggested that the link between DNA methylation, nucleosome occupancy, and cohesion and CCCTC-binding factor binding extends both to normal development and to other cancers (Ghirlando & Felsenfeld, 2016; Kang, et al., 2015; Teif, et al., 2014).
Changes in DNA methylation play a critical role in hematopoietic neoplasms (Issa, Baylin, & Herman, 1997; Jiang & Melnick, 2015; Toyota & Issa, 2005; Yang, Rau, & Goodell, 2015). Several of the most common mutations found in myelodysplasia, acute myeloid leukemia, and lymphoma involve the TET, DNMT3A, and IDH enzymes (Guillamot, Cimmino, & Aifantis, 2016). These mutations either lead to decreased (DNMT3A) or increased (TET and IDH) levels of DNA methylation, although these changes are not universally observed. Importantly, clones harboring mutations in these enzymes, along with 10–20 other genes, can be found in an age dependent manner in otherwise normal bone marrows (Buscarlet, et al., 2017; Zink, et al., 2017). These mutations have been associated with increased risk of subsequent hematopoietic neoplasm and often persist even after remission (Busque, et al., 2012; Genovese, et al., 2014; Jaiswal, et al., 2014). The observation that clones harboring these mutations do not necessarily lead to hematological malignancies inspired the concept of clonal hematopoiesis of indeterminate potential (CHIP) (Steensma, et al., 2015). The CHIP mutations are not sufficient for the development of a hematopoietic neoplasm but may set the stage for leukemia by promoting stem cell self-renewal.
The observation that CHIP mutations are found in normal bone marrow cells and are associated with both increased and decreased global levels of DNA methylation raise questions about the mechanistic role DNA methylation plays in driving aberrant hematopoiesis. In fact, a recent study by the Ley group suggests that increased DNA methylation in DNMT3A mutant leukemia may be a consequence of stem cell proliferation instead of a driver of leukemogenesis (Spencer, et al., 2017). Nonetheless, 5-azacytidine has shown significant efficacy in retrospective analyses of patients with myelodysplasia and TET mutations (Bejar, et al., 2014; Itzykson, et al., 2011) and in mouse models of neoplasms harboring TET and IDH mutations (Shih, et al., 2017), consistent the clinical utility of hypomethylating agents in myelodysplasia and subsets of acute myeloid leukemia (Plimack, Kantarjian, & Issa, 2007; Schuh, et al., 2017). A very recent study (Cimmino, et al., 2017) confirms that inducible knockdown of TET2 in bone marrow progenitors increases DNA methylation, augments bone marrow stem cell selfrenewal, and leads to a chronic myelomonocytic leukemia-like proliferation of white blood cells. Restoring TET2 expression reverses these changes, suggesting that augmenting TET2 function could be a viable therapy for leukemia. Furthermore, supplementation with vitamin C, a co-factor for a-ketoglutarate-dependent dioxygenases, increases DNA demethyation, sensitizes to PARP inhibition, and simulates the effects of restoring TET2 expression. This work raises the exciting possibility that vitamin C could selectively sensitize acute myeloid leukemia blasts to chemotherapy, but has yet to be tested in patients. Finally, work by the Issa group (Kelly, et al., 2017) has identified an acute myeloid leukemia CGI methylator phenotype that is independent of CHIP mutations and is associated with a better prognosis. Taken together, these findings suggest that regardless of its role in initiating abnormal hematopoiesis, aberrant DNA methylation may be critical for maintenance of tumor suppressor gene silencing in keeping with its apparent role in gene silencing in general (i.e. necessary but not sufficient). Hence, DNA methylation clearly plays an expanding, albeit complicated, role in hematopoietic neoplasms.
Finally, similar changes in DNA methylation patterns have been demonstrated in at least a large subset of breast (Davalos, Martinez-Cardus, & Esteller, 2017; Fang, et al., 2011; Holm, et al., 2016; Stefansson, et al., 2015), prostate (Kirby, et al., 2017; Massie, Mills, & Lynch, 2017), melanoma (Micevic, Theodosakis, & Bosenberg, 2017), and other common cancers (Hao, et al., 2017). Despite the frequent association between DNA methylation and cancer, however, hypomethylating agents have not demonstrated clinical utility except in hematopoietic neoplasms and few solid tumors (Gnyszka, Jastrzebski, & Flis, 2013). Therefore, research efforts are currently directed towards developing new hypomethylating agents and combinations (Gnyszka, et al., 2013; Shih, et al., 2017; Singh, Sharma, & Capalash, 2013) as well as alternative methods of inhibiting the transcriptional effects of DNA methylation.
3. The 5-methylcytosine binding domain
The discovery that DNA methylation modifies cellular differentiation (Jones & Taylor, 1980) inspired a search for factors that selectively recognize the 5-methylcytosine base. Early biochemical approaches generated two nuclear fractions, MeCP1 and MeCP2, that preferentially bound to methylated DNA (Lewis, et al., 1992; Meehan, Lewis, McKay, Kleiner, & Bird, 1989). The second of these, MeCP2, comprised a single protein that contained a small, approximately seventy amino-acid domain that preferentially bound methylated CpGs. Subsequently, four additional proteins were identified that contain a similar 5-methylcytosine binding domain (MBD1–4) that show varying degrees of binding selectivity for methylated CpGs (Hendrich, Abbott, et al., 1999; Hendrich & Bird, 1998; Nan, Meehan, & Bird, 1993). Shortly thereafter, Wade and others in the Wolffe laboratory isolated a large protein complex from Xenopus laevis containing histone deacetylase, chromatin remodeling, and methylated DNA binding activities (Wade, Jones, Vermaak, & Wolffe, 1998). The complex they identified comprised either MBD2 or MBD3 along with five additional components, each of which have multiple paralogs, that has become known as the NuRD complex (Alqarni, et al., 2014; Le Guezennec, et al., 2006; Ng, et al., 1999; Smits, Jansen, Poser, Hyman, & Vermeulen, 2013; Spruijt, et al., 2010; Y. Zhang, et al., 1999).
Of the remaining mammalian MBDs, MBD1 contains three additional CXXC DNA interacting domains that modify methylation selectivity (Ng, Jeppesen, & Bird, 2000). MBD1 recruits chromatin modifying enzymes to both methylated and unmethylated CGIs and largely silences transcription. In contrast, MBD4 contains a glycosylase domain that selectively recognizes G X (where X=T, U, or 5-hydroxymethyl-U) mismatches arising from spontaneous deamination of 5-methylcytosine, cytosine, and 5-hydroxymethylcytosine bases, respectively. MBD4 excises the mismatch base thereby initiating base-excision repair (Hendrich, Hardeland, Ng, Jiricny, & Bird, 1999; Morera, et al., 2012; Petronzelli, Riccio, Markham, Seeholzer, Genuardi, et al., 2000; Petronzelli, Riccio, Markham, Seeholzer, Stoerker, et al., 2000). The functional role of the MBD4 MBD remains unclear but likely contributes to localization of the protein to regions of increased mCpG content where G X mismatches are most likely to occur (Walavalkar, Cramer, Buchwald, Scarsdale, & Williams, 2014). More recently, two additional MBD proteins have been identified, MBD5 and MBD6; however, the MBDs of these paralogs do not contain critical residues for DNA binding and, instead, function as protein-protein interaction domains in a de-ubiquitinating polycomb repressive complex (Baymaz, et al., 2014; Laget, et al., 2010).
3.1. Structure of the 5-methylcytosine binding domain
The structure of the MBD free in solution (MBD1 and MeCP2) and bound to DNA (MBD1, MBD2, MBD3, MBD4, and MeCP2) has been solved by both NMR and x-ray crystallography techniques (Cramer, et al., 2014; Heitmann, et al., 2003; Ho, et al., 2008; Manvilla, Maiti, Begley, Toth, & Drohat, 2012; Ohki, et al., 2001; Ohki, Shimotake, Fujita, Nakao, & Shirakawa, 1999; Otani, et al., 2013; Rauch, et al., 2005; Scarsdale, Webb, Ginder, & Williams, 2011; Wakefield, et al., 1999; Walavalkar, et al., 2014). The domain fold consists of a 3–4 strand antiparallel β-sheet forming a curved base with an N-terminal loop and C-terminal α-helix packing against one face of this sheet (Fig. 2a). The middle two strands of the β-sheet form a finger-like projection that extends down the major groove of DNA with a short, dynamic loop connecting these strands. Three residues extend from the exposed surface of the β-sheet to make key, methylation- specific interactions with the DNA: two arginine residues that form bidentate hydrogen bonds with symmetrically related guanosine bases of the CpG dinucleotide and pack against the methyl groups of the two 5-methylcytosines, and a tyrosine residue that directly or indirectly interacts with a methyl groups from one of the two 5- methylcytosines. In addition to these base-specific and methylation specific interactions, the MBD interacts with only a few bases that surround the CpG as well as the phosphate backbone of DNA. Although the MBDs can show selectivity for surrounding bases, these are largely secondary effects with most of the base-specific favorable binding energy derived from interacting with the CpG (Clouaire, de Las Heras, Merusi, & Stancheva, 2010; Klose, etal., 2005; Scarsdale, etal., 2011).
Fig. 2.
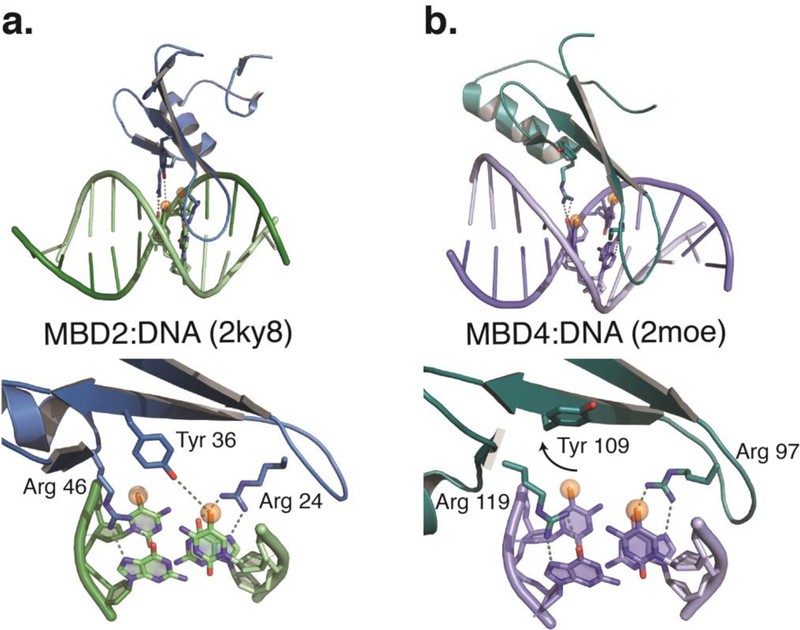
a) A cartoon diagram depicts the solution structure of the MBD from MBD2 bound to methylated DNA (Scarsdale, et al., 2011). The domain contains a 3–4 strand β-sheet with a long finger like projection that extends down the major groove and a single α-helix. An expanded view shows how a tyrosine and two arginine residues from critical interactions with the 5-methylcytosine and two guanosine bases, respectively, of the mCpG. b) For comparison, cartoon diagram of the solution structure of MBD4 bound to methylated DNA (Walavalkar, et al., 2014) shows that the α-helix contains one additional turn, but otherwise has a similar structure. However, an expanded view reveals that the critical tyrosine residue rotates away from the DNA and does not interact with 5-methylcytosine. This structural rearrangement likely contributes to reduced selectivity for methylated DNA.
3.2. Structural and functional differences between 5-methylcytosine binding domain proteins
MBD proteins have been identified from across metazoans (Cramer, et al., 2017), with invertebrates having a single MBD2/3 orthologue and vertebrates duplicating (MBD2 and MBD3) and expanding the family (MBD1, MeCP2, and MBD4) (Hendrich & Bird, 1998; Hendrich & Tweedie, 2003). Although each of the MBDs shares the same overall fold, detailed structural analyses have revealed differences that impact binding affinity and selectivity.
Perhaps the most substantive change, a phenylalanine replaces the critical tyrosine residue in MBD3, thereby modifying a key interaction with DNA. This change greatly reduces methylation selectivity (from ~100-fold to ~5-fold selectivity for mCpG over CpG) and contributes to a marked reduction in overall binding affinity for DNA (from low nanomolar to high micromolar dissociation constants) (Cramer, et al., 2014; Fraga, et al., 2003; Hashimoto, Liu, et al., 2012). Both MBD2 and MBD3 recruit the NuRD complex in a mutually exclusive manner (Le Guezennec, et al., 2006; W. Zhang, et al., 2016) with apparently distinct functions (Gunther, et al., 2013; Menafra & Stunnenberg, 2014). Genetic knockout of MBD3 is embryonic lethal while knockout of MBD2 causes only mild phenotypic effects (Hendrich, Guy, Ramsahoye, Wilson, & Bird, 2001). Concordantly, MBD3 plays an important, although debated, role in embryonic stem cells while MBD2 appears to play a less critical role at this stage of differentiation (dos Santos, et al., 2014; Rais, et al., 2013). These observations have led to the conclusion that MBD3-NuRD plays a vital role in cell differentiation independent of DNA binding. Yet, the MBD3 MBD itself has been retained, which implies that either this domain has gained alternative roles in protein-protein interactions or provides weak DNA binding that is critical for function. In support of the former, a few studies have mapped interaction between specific transcription factors and NuRD to the MBD3 MBD (Aguilera, et al., 2011). In support of the latter, we recently demonstrated that MBD3 retains a weak ability to recognize mCpGs and CpGs, and unlike MBD2, rapidly exchanges between mCpG specific and non-specific binding modes (Cramer, et al., 2014). This biophysical difference between MBD2 and MBD3 correlates with whole genome localization studies that found both MBD2 and MBD3 can localize to unmethylated CGIs while MBD2 more exclusively binds to methylated CGIs associated with silenced genes (Gunther, et al., 2013; Le Guezennec, et al., 2006; Menafra, et al., 2014; Shimbo, et al., 2013).
More recent work by the Williams lab suggests that differences in dynamic sliding along the DNA reflect the functional role of MBD2 in compacting and silencing genes containing methylated promoter-associated CGIs (Pan, et al., 2017). Perhaps as expected, we found that the MBD2 bound statically to methylated CGIs and could induce marked DNA bending. Surprisingly, though, we found that MBD2 showed rapid 1D-sliding along unmethylated CpG-rich DNA while demonstrating more restricted dynamic motion on CpG-poor DNA. Hence, we propose that the MBD2 primarily targets NuRD to methylated CGIs where it binds statically to stabilize nucleosome positioning and compacts chromatin, but can also target NuRD to unmethylated CGIs where it can mobilize nucleosomes and promote active remodeling. In contrast, MBD3 targets the NuRD complex to unmethylated CGIs and may permit active chromatin remodeling. This model helps explain several unresolved issues, such as why MBD2- and MBD3- NuRD complexes can be associated with unmethylated and actively transcribed genes and that knockdown of either leads to both activation or inhibition of transcription of differing gene sets. While gene inactivation upon MBD2 or MBD3 knockdown likely reflects indirect effects for many genes, our observations raise the possibility that when MBD2 and MBD3 localize to unmethylated CGIs they could directly contribute to transcription activation by helping to maintain open chromatin. Furthermore, it suggests that unmethylated CGIs play a functional role themselves, allowing for rapid mobility and remodeling by the NuRD complex. Nonetheless, this model remains speculative without additional data to determine how the intact proteins and associated NuRD complexes remodel chromatin on methylated and unmethylated CGIs.
The largest structural difference between MBDs involves an insertion of four amino acids in the α-helix of MeCP2 and MBD4, adding one turn and increasing the hydrophobic core of the domain (Ho, et al., 2008; Otani, et al., 2013; Walavalkar, et al., 2014). This insertion appears to stabilize the isolated domain and perhaps helps MeCP2 bind non-CpG 5-methylcytosines (mCpH, where H is A, T, or C) (M. J. Sperlazza, Bilinovich, Sinanan, Javier, & Williams, 2017). While most mammalian DNA methylation occurs on CpG dinucleotides, recent research has uncovered a small but stable fraction of mCpH in oocytes, embryonic stem cells, and brain. While mCpG can be maintained across cell division, mCpH will be lost since only one daughter will inherit the methylated site. Concordantly, the highest levels of mCpH are found in mitotically arrested cells, most notably in neurons. In addition, the overall incidence of mCpH in these cells correlates with the relative rates at which DNMT3a modifies different CpH dinucleotide sequences (mCpA > mCpT, mCpC) (Aoki, et al., 2001; Gowher & Jeltsch, 2001; Pinney, 2014; Ramsahoye, et al., 2000). Even though these observations indicate that mCpH may simply reflect the accumulation of incidentally methylated sites over time, recent work has suggested that mCpH accumulates in specific regions of the genome and plays a definitive role in neuron development. Both mCpH and hydroxymethylation accumulate in the gene bodies of long transcripts expressed in neurons (Gabel, et al., 2015; Kinde, Gabel, Gilbert, Griffith, & Greenberg, 2015). In addition, MeCP2 expression is highest in the brain where it appears to localize to these same genes. Importantly, in vitro binding analyses indicate that MeCP2 can bind to mCpH with comparable affinity to that of mCpG. Bird recently found that MeCP2 preferentially binds the mCAC trinucleotide particularly abundant in the brain (Lagger, et al., 2017), while we found a similar binding preference unique to MeCP2 as compared to MBD2 and attributed this preference to strand-specific interactions (M. J. Sperlazza, et al., 2017). Hence, MeCP2 shows a unique ability to bind mCpH and hydroxym ethyl - CpH sites that correlate with its functional role in the developing brain.
Finally, a structural change involving the critical tyrosine residue was revealed by the crystal and NMR structures of the MBD4 MBD bound to methylated DNA (Otani, et al., 2013; Walavalkar, et al., 2014). Instead of pointing towards the DNA and 5- methylcytosine, the tyrosine rotates away opening the protein-DNA interface (Fig. 2b). This structural change leads to a reduction in methylation specificity (approximately 5- fold selectivity for mCpG as compared to CpG), comparable to that measured for MBD3 (Walavalkar, et al., 2014). Otani (Otani, et al., 2013) propose that this structural rearrangement allows for binding to the bulkier oxidative derivatives of 5-methylcytosine (5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxylcytosine). In addition, we found that this rearrangement allows the MBD4 MBD to exchange more rapidly between methylated sites (Walavalkar, et al., 2014). Hence, we propose that the reorientation of this tyrosine promotes rapid scanning through methylated CGIs, where spontaneous deamination of 5-methylcytosines and resulting G X mismatches are most likely to occur. This possibility awaits further validation but does help explain why the MBD4 MBD does not modify the glycosylase reaction rates when using small substrates in vitro (Petronzelli, Riccio, Markham, Seeholzer, Genuardi, et al., 2000; Petronzelli, Riccio, Markham, Seeholzer, Stoerker, et al., 2000).
4. The nucleosome remodeling and deacetylase (NuRD) complex
The NuRD complex comprises at least six core proteins in addition to multiple sub- stoichiometric components (Kloet, et al., 2015; Smits, et al., 2013; Spruijt, et al., 2010; Vermeulen, Hubner, & Mann, 2008; Y. Zhang, et al., 1999). The core proteins include an MBD (MBD2 or 3), histone deacetylase protein (HDAC1 or 2), the chromodomain helicase DNA binding protein (CHD3, 4, or 5), as well as architectural proteins including retinoblastoma-binding protein (RBBP4 or 7), metastasis tumor associated (MTA1, 2, or 3), and GATA zinc-finger domain containing 2 (GATAD2A or B). One of the longstanding goals in the field has been to determine the structural details driving the formation of this macromolecular complex. However, its large size, heterogeneity in component paralogs, and intrinsic disorder have proven prohibitive to structural analyses of the full complex. Nonetheless, several key structures of NuRD subcomplexes have been reported in recent years, significantly improving our understanding of complex formation (Fig. 3).
Fig. 3.
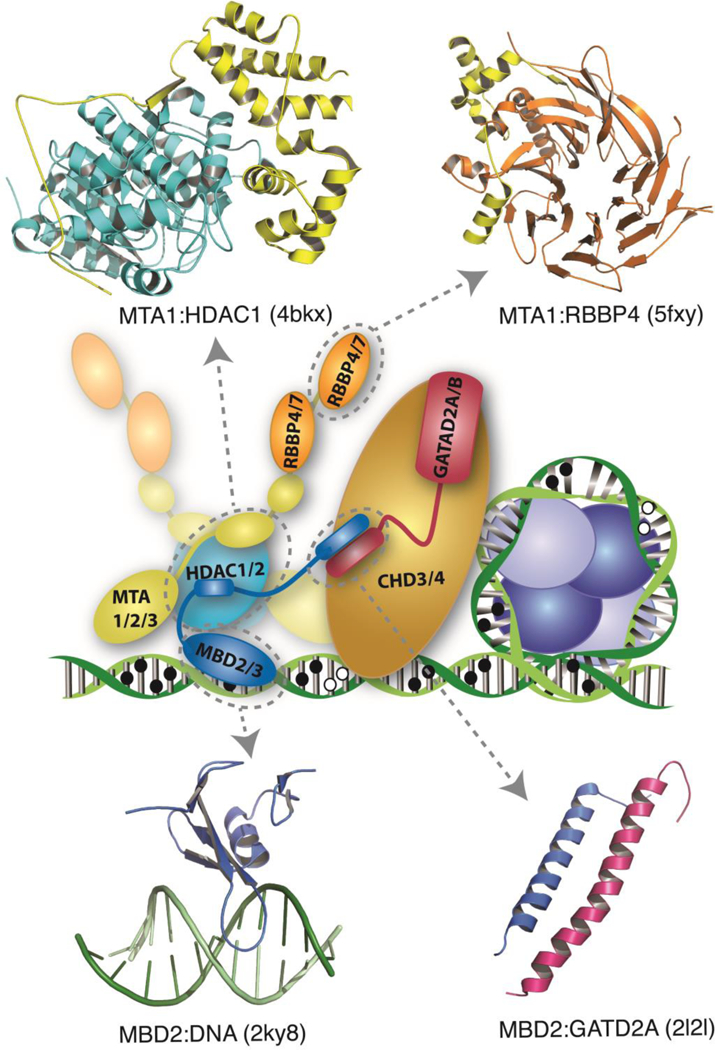
A schematic diagram shows the NuRD complex bound to methylated DNA next to a nucleosome. The complex comprises at least six core proteins, each of which have multiple paralogues (MBD2 or 3, in blue; MTA1,2, or 3, in yellow; HDAC1 or 2 in cyan; RBBP4 or 7, in orange; GATAD2A or B, in red; CHD3 or 4, in gold). While the structure of the full complex has not been determined to date, structures of several key interactions have been reported. Cartoon diagrams, generated with PyMOL (Schrodinger), are shown for structures that have been solved of the MTA1:HDAC1 (PDB ID - 4bkx) (Millard, et al., 2013), MTA1:RBBP4 (PDB ID - 5fxy) (Millard, et al., 2016), MBD2:DNA (PDB ID - 2ky8) (Scarsdale, et al., 2011), and MBD2:GATAD2A (PDB ID - 2l2l) (Gnanapragasam, et al., 2011) sub-complexes. Both stoichiometric (Acuna-Hidalgo, et al., 2017) and structural analyses indicate that the MTA1/2/3:HDAC1/2 sub-complex forms a dimer (Millard, et al., 2013), at least two RBBP4/7 bind to each MTA1/2/3 (Alqarni, et al., 2014; Schmidberger, et al., 2016), one to two copies of MBD2/3 and GATAD2A/B, and only copy of CHD3/4 are present in the complex.
One of the largest sub-complex structures to be determined comprises the ELM2- SANT domains of MTA1 and HDAC1. The crystal structure of this complex shows that the MTA protein wraps around the HDAC, such that the SANT domain interacts near the enzymatic cleft. Interestingly, D-myo-inositol-1,4,5,6-tetraphosphate (IP4) bridges the interaction between SANT and HDAC, raising the possibility that IP4 concentration could regulate the histone deacetylase activity of NuRD in a cell-cycle dependent manner (Millard, et al., 2013; Watson, Fairall, Santos, & Schwabe, 2012). The structure indicates that the BAH domain of MTA would be appropriately situated to deliver a histone tail to the enzymatic binding site of the HDAC. However, further work is necessary to confirm this model of MTA function.
Additional crystal structures have shown how two relatively short peptides within the C-terminal portion of MTA bind to separate RBBP proteins using a common interface (Alqarni, et al., 2014; Brasen, et al., 2017; Kloet, et al., 2015; Millard, et al., 2016; Schmidberger, et al., 2016). These structures help explain stoichiometric analyses that indicate multiple (up to eight) copies of the RBBP proteins are present in a full NuRD. Interestingly, the small RBBP binding motif was first identified within the N-terminal a- helix of histone H4 and two crystal structures were solved for that complex (Murzina, et al., 2008; Song, Garlick, & Kingston, 2008). However, formation of this complex between RBBP and H4 requires unfolding of the nucleosome core particle, raising questions about the functional context in which RBBP-H4 interaction takes place in cells.
We recently found that an intrinsically disordered region (IDR) in MBD2 is both necessary and sufficient for binding to the NuRD sub-complex containing RBBP, HDAC, and MTA proteins (Desai, et al., 2015). Mutation of two neighboring residues within the MBD2 IDR disrupts the interaction and can abrogate methylation dependent gene silencing by MBD2-NuRD. Therefore, a stable histone deacetylase core complex, as first described by Zhang et al (Y. Zhang, et al., 1999), forms between the MBD, HDAC, MTA, and RBBP proteins. Consequently, NuRD can be separated into histone deacetylase and chromatin remodeling sub-complexes.
Much less is known about recruitment of the chromatin remodeling component, CHD3, CHD4, or CHD5, into NuRD, perhaps reflecting a comparatively weak association (Low, et al., 2016). We previously determined the solution structure of a small coiled-coil complex formed between MBD2 and GATAD2A (Gnanapragasam, et al., 2011). This interaction involves a highly conserved peptide from GATAD2A (CR1) and the C-terminal coiled-coil domain from MBD2. Immunoprecipitation of the GATAD2A peptide brought down all the histone deacetylase core components (MBD, HDAC, MTA, and RBBP proteins), but not the GATAD2 or CHD proteins. This observation leads to a model in which the GATAD2 protein bridges between the chromatin remodeling and histone deacetylase functional halves of the complex. More recent work has supported this interpretation and shown that the CHD protein is a peripheral component of NuRD (Low, et al., 2016). The natural hypothesis arising from these observations is that the second conserved domain in the GATAD2 proteins binds to the CHD proteins; however, this relationship has yet to be confirmed experimentally.
5. The MBD proteins as therapeutic targets
The DNA hypomethylating agents, 5-azacytidine and 5-aza-2’-deoxycytidine (decitabine), have been approved for therapy of myelodysplasia and acute myeloid leukemia since 2004 and 2006, respectively. The clinical utility of these drugs has been limited by several factors including chemical instability, a lack of specificity leading to a global reduction in DNA methylation with concomitant increased genomic instability, DNA damage and cytotoxicity, and significant off-target effects (Brocks, et al., 2017; Gnyszka, et al., 2013; Gravina, et al., 2010; Gros, et al., 2012; Pechalrieu, Etievant, & Arimondo, 2017). Both 5-azacytidine and decitabine have been investigated for the treatment of hemoglobinopathies through relief of silencing of the fetal gamma globin gene resulting in therapeutic increases in HbF (Charache, et al., 1983; Ley, et al., 1982; Saunthararajah & DeSimone, 2004). However, the potential toxicities and off target effects have limited their application in this setting as well, especially in children for whom the greatest potential for major lifelong benefit could accrue. As an alternative, directly inhibiting individual readers of DNA methylation should provide more biological specificity and less off-target toxicity, as evidenced by the lack of major phenotypic changes associated with complete knockout of MBD2 (Hendrich, et al., 2001; Loughran, et al., 2017; Rupon, Wang, Gaensler, Lloyd, & Ginder, 2006; Wood, et al., 2016). While systemic toxicity is more manageable for short term chemotherapy of malignant neoplasms, long-term therapy for inherited disorders requires a very low-level of systemic toxicities. Hence, directly targeting the MBDs represents a potentially groundbreaking approach for restoring expression of epigenetically silenced genes to treat disease (Ballestar & Esteller, 2005; Ginder, 2015; Gnanapragasam, et al., 2011; Lopez-Serra & Esteller, 2008; Mian, et al., 2011; Sansom, et al., 2003). However, the MBDs lack enzymatic activities (with the notable exception of MBD4), which necessitates strategies to disrupt protein-protein and protein-DNA interactions specific to the individual MBD. In this section, we discuss the rationale and different approaches being considered for inhibiting the function of MBD proteins, focusing on MBD2 and MeCP2.
5.1. MBD2-NuRD and globin regulation
The connection between DNA methylation and gene regulation raises the possibility that the MBD proteins could be valuable therapeutic targets for restoring expression of epigenetically silenced genes. Shortly after the discovery that 5-azacytidine could block DNA methylation, this information was applied to the study of globin regulation. During primate development, the predominant hemoglobin expressed in erythrocytes switches from embryonic (HbE) and fetal hemoglobin (HbF) to adult hemoglobin (HbA). This transcriptional switch has been extensively studied both as a well-defined example of developmental gene regulation and as a potential therapeutic target for treating diseases associated with deficiencies in adult hemoglobin. In humans, fetal HbF contains two α-globin and two γ-globin chains and HbA contains two α-globin and two β- globin chains. Therefore, restoring expression of HbF can effectively ameliorate the pathophysiology associated with reduced expression of β-globin (β-thalassemia) or abnormal β-globin (sickle cell anemia). Early studies showed that 5-azacytidine could restore expression of fetal and embryonic globin genes in animal models and increase HbF in adult baboons and humans, implicating DNA methylation in globin gene silencing (Charache, etal., 1983; DeSimone, etal., 1982; Ginder, etal., 1984; Ley, et al., 1982). Both 5 azacytidine and decitabine have been used in the treatment of β-type hemoglobinopathies through relief of silencing of the fetal γ-globin gene resulting in therapeutic increases in HbF (Charache, etal., 1983; Ley, et al., 1982 Saunthararajah & DeSimone, 2004). However, the potential toxicities and off target effects of these agents have limited their application in this setting, especially in children for whom the greatest potential for major lifelong benefit could accrue.
A little over a decade ago, work in the Ginder laboratory demonstrated that the MBD2-NuRD complex bound directly to the methylated and silenced embryonic o-globin promoter in adult avian primary erythrocytes (Kransdorf, et al., 2006). Knockdown of MBD2 in mouse erythroleukemia cells carrying an avian p-globin mini-locus (MEL-p) and mouse erythroid cells carrying human β-globin locus on a yeast artificial chromosome (β-YAC) reversed methylation-dependent silencing of both the avian embryonic p- and human γ-globin genes, respectively. Complete knockout of MBD2 in β-YAC bearing mice likewise resulted in substantial augmentation of fetal γ-globin gene expression in erythrocytes of adult mice but with otherwise minimal phenotypic effects (Rupon, et al., 2006). Our laboratories subsequently showed that knockdown of MBD2 in human primary adult erythroid cells results in a major increase in fetal γ-globin gene expression (Amaya, et al., 2013). These studies strongly suggest that molecular disruption of MBD2 function could restore HbF expression for therapy of β- hemoglobinopathies without significant side effects. There remains the challenge of how to selectively disrupt MBD2 function given that it lacks enzymatic activity but instead functions by providing key protein-protein and protein-DNA interactions in recruiting the NuRD co-repressor complex.
To pursue this goal, we closely collaborated to study the molecular interactions between MBD2 and NuRD as potential therapeutic targets. Based on work by the Renkawitz group (Brackertz, Boeke, Zhang, & Renkawitz, 2002; Brackertz, Gong, Leers, & Renkawitz, 2006), we identified and determined the structure of a critical coiled-coil interaction formed between MBD2 and GATAD2A (Fig. 2) (Gnanapragasam, et al., 2011). The coiled-coil domains from each protein largely remain monomeric in isolation, yet form a tight (Kd ~ 42 nM) heterodimeric anti-parallel coiled-coil complex together (Walavalkar, Gordon, & Williams, 2013). Given the small size of the coiled-coil domains, we postulated that the GATAD2A coiled-coil peptide could inhibit the formation of a full MBD2-NuRD complex. Enforced expression of the peptide in both MEL-p cells and mouse cells carrying a β-YAC augmented expression of avian p-globin and human y-globin, respectively (Gnanapragasam, et al., 2011). Immunoprecipitation of the peptide pulled down all components of the NuRD complex except native GATAD2A and CHD4. Hence, the peptide appears to block recruitment of the chromatin remodeling portion of the complex to abrogate methylation dependent gene silencing (Fig 4a). To our knowledge, these experiments were the first to demonstrate that a small peptide can abrogate methylation dependent gene silencing by MBD2-NuRD and have motivated our continued efforts to identify key interaction interfaces within MBD2- NuRD as potential therapeutic targets.
Fig. 4.
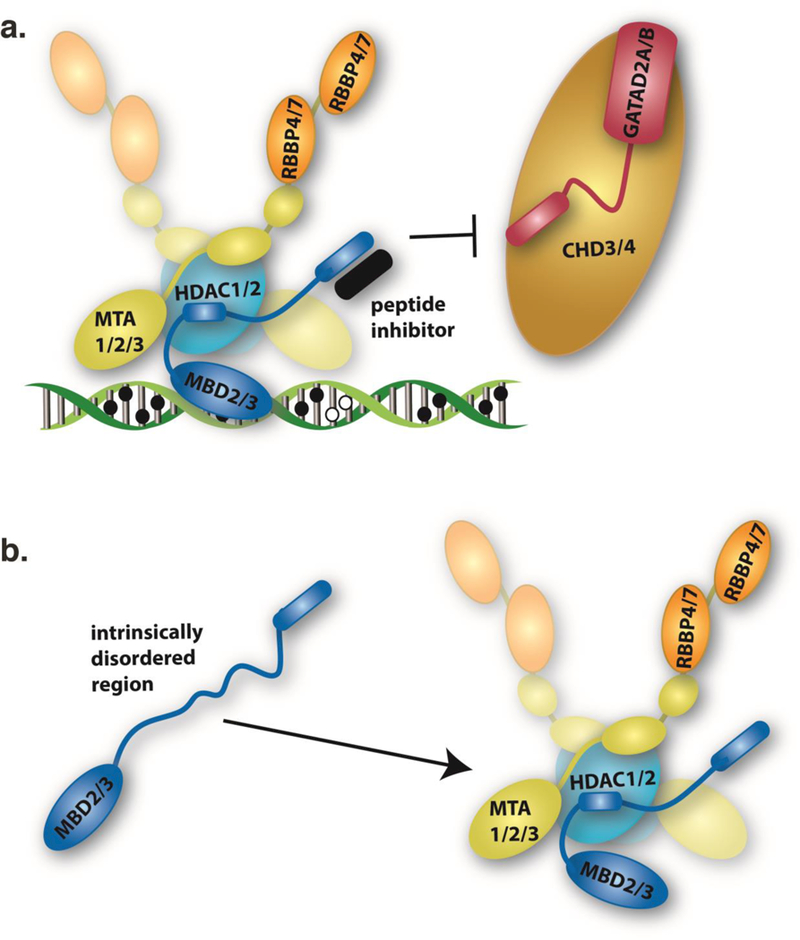
a) A schematic cartoon depicts a peptide inhibitor (black) of complex NuRD complex formation. We previously demonstrated that a coiled-coil peptide, derived from the GATAD2A coiled-coil domain, restores expression of fetal/embryonic hemoglobin in tissue culture models of globin regulation (Gnanapragasam, et al., 2011). Immunoprecipitation of this peptide indicates that it disrupts function by blocking recruitment of CHD3/4 to the complex. b) Recently, we found that an intrinsically disordered region (IDR) of MBD2 is necessary and sufficient to bind the histone deacetylase core complex of NuRD comprising MTA1/2/3, HDAC1/2, and RBBP4/7 (Desai, et al., 2015). Secondary structure propensity of the isolated IDR suggests that it forms a helix upon interacting with the complex. Mutation of two consecutive residues within the helical region of the IDR disrupts interaction with the NuRD complex and abrogates methylation dependent silencing by MBD2-NuRD. Importantly, IDR-protein interactions have proven amenable to small molecule inhibition (Vela & Marzo, 2015). Hence, the MBD2 IDR represents a potential target for small molecule inhibition of NuRD complex formation.
5.2. MBD2-NuRD and cancer
The association between hypermethylation of tumor suppressor genes and cancer suggests that MBD proteins would mediate aberrant silencing to promote carcinogenesis. In early studies, MBD2 was found at the methylated and silenced CDKN2A locus in colon cancer and HeLa cell lines, and occupancy inversely correlated with gene expression (Magdinier & Wolffe, 2001). Likewise, MBD2 was found to bind and silence the methylated promoter of the GSTP1 tumor suppressor in hepatocellular, breast, and prostate cancer cell lines (Bakker, Lin, & Nelson, 2002; Lin & Nelson, 2003; Singal, van Wert, & Bashambu, 2001). Subsequently, the Esteller group measured occupancy of MBDs at methylated CGIs for select tumor suppressors in cancer cell lines and found that different MBDs preferentially bound to the methylated promoters of distinct tumor suppressors (Lopez-Serra, et al., 2006; Lopez-Serra & Esteller, 2008). Furthermore, knockdown of MBDs restores expression of silenced tumor suppressor genes, with targeting of MBD2 the most effective at doing so (Lopez-Serra, et al., 2008).
In perhaps one of the more exciting studies, genetic knockout of MBD2 in a mouse model of adenomatous polyposis coli led to a significant increase in lifespan and approximately a ten-fold reduction in adenomas at death (Sansom, et al., 2003). The MBD2 knockout mice showed only mild phenotypic changes but were otherwise viable and fertile. Work in the Ginder laboratory has shown that knockout of MBD2 in triple negative breast cancer cell lines reduced cell proliferation in tissue culture and mouse xenografts (Mian, et al., 2011). In support of this observation, knockdown of MBD2 synergizes with 5-aza-2’-deoxycytidine to inhibit the growth of MCF7 breast cancer cells in tissue culture and xenografts while preventing increased invasiveness associated with 5-aza-2’-deoxycytidine treatment alone (Cheishvili, et al., 2014). More recently, the Ginder group found that knockdown of CHD4 in acute myeloid leukemia cell lines reduced colony formation in soft agar and sensitized both acute myeloid leukemia cell lines and primary leukemia cells to standard chemotherapy agents while sparing any effect on CD34 positive hematopoietic progenitor cells (J. Sperlazza, et al., 2015). These studies are in keeping with recent findings from the Baylin laboratory on the effects of CHD4 depletion in colon cancer (Xia, et al., 2017). Finally, a recent study reported that MBD2 is consistently overexpressed in glioblastoma multiforme (GBM) (Zhu, Flunter, Vertino, & Van Meir, 2011) and that MBD2 binds to methylated CGIs and silences gene expression. Knockdown of MBD2 restores expression of silenced genes, including the anti-angiogenic tumor suppressor BAI1. Hence, reducing MBD2 expression blocked migration of endothelial cells in a scratch-wound assay, raising the possibility that inhibiting MBD2 could inhibit the growth of GBM in patients. This possibility was further substantiated by a recent study showing that microRNA-520b inhibited the growth of glioma cells by reducing expression of MBD2 (Cui, et al., 2017).
In summary, work over the past decade has built a strong case for inhibiting MBD2 function as a potential therapy for a wide variety of cancers. The challenge that remains, though is how to selectively disrupt the MBD2-NuRD complex.
5.3. Selectively targeting MBD2-NuRD
Using a combination of structural biology and cell studies, we have characterized three interaction interfaces in MBD2. These comprise the N-terminal MBD which binds selectively to methylated DNA (Pan, et al., 2017; Scarsdale, et al., 2011), the C-terminal coiled-coil domain that binds to GATAD2A (Gnanapragasam, et al., 2011; Walavalkar, et al., 2013), and an intervening unstructured region that binds the histone deacetylase core complex of NuRD (Desai, et al., 2015). Each of these represents potential targets for disrupting co-repressor complex formation, and offer differing advantages and challenges.
The MBD2 MBD is responsible for the unique function of MBD2-NuRD as compared to MBD3-NuRD, and, as such, represents the most logical target for selectively inhibiting MBD2. However, apart from nuclear receptors (i.e. retinoic acid receptor and estrogen receptor), to our knowledge no DNA binding domains have been directly inhibited for therapy. This limitation likely reflects the challenge of competitively disrupting binding to DNA, which is present in large quantities at high concentration in the nucleus. Nonetheless, a recent study by the Nelson group reports using time resolved fluorescence resonance energy transfer to identify four compounds that inhibit DNA binding by MBD2 (Wyhs, Walker, Giovinazzo, Yegnasubramanian, & Nelson, 2014). Although most of the identified compounds inhibit multiple protein-DNA interactions and are not readily soluble in aqueous buffers, the results suggest that a selective inhibitor of MBD2 may be identified through additional optimization.
As discussed previously, we have successfully disrupted the MBD2-GATAD2A coiled-coil complex to block methylation dependent gene silencing of the fetal y-globin by MBD2-NuRD (Gnanapragasam, et al., 2011). This interaction entails unique features that encourage further development of a peptide therapeutic. First, the coiled-coil peptides are well-behaved, remain largely monomeric in isolation, and utilize a relatively small surface to selectively bind one another with high-affinity in a heterodimeric antiparallel complex (Walavalkar, et al., 2013). These features suggest that the isolated peptides should effectively compete with the native interaction without many off-target interactions that might otherwise be expected of coiled-coil domain proteins. Indeed, most successful peptide-based inhibitors of protein-protein interactions reported to date are stapled helices, making the MBD2-GATAD2A complex an appealing target. However, this type of peptide-peptide involves a relatively flat and small interface without an obvious binding pocket for developing a small molecule competitive inhibitor (Goncearenco, Li, Simonetti, Shoemaker, & Panchenko, 2017; Wells & McClendon, 2007). Hence, we are pursuing methods to stabilize the purified peptides against proteolytic degradation and to enhance cell penetration, both of which are challenging goals for a coiled-coil domain.
The intrinsically disordered region from MBD2 represents perhaps the most promising target for inhibiting complex formation. We found that this region is both necessary and sufficient for binding the histone deacetylase core and that disrupting this interaction by point mutations relieved methylation-dependent tumor suppressor gene silencing (Desai, et al., 2015). Importantly, interactions between IDRs and structured domains typically involve a disorder-to-order transition of the IDR (Fig. 4b), which then binds a relatively small, often groove-like, interface on the structured domain (Uversky, 2016). In addition, binding typically requires a relatively short and continuous stretch from the IDR, more contacts per residue, and extensive burial of hydrophobic residues (Meszaros, Tompa, Simon, & Dosztanyi, 2007). Therefore, small molecules have been developed that selectively disrupt an IDR-protein interaction critical to the function of the Bcl-2 family (Vela & Marzo, 2015). However, the structural details of the complex formed with the MBD2 IDR have yet to be determined; without this information, it remains unclear whether this interaction will be amenable to targeted inhibition.
5.4. MeCP2 and Rett syndrome
Rett syndrome (RTT) is the most common genetic cause of severe mental disability in females worldwide. The majority of RTT patients have pathological mutations in MeCP2 which lead to pervasive behavioral and cognitive deficits beginning between 618 months of life (Amir, et al., 1999). Restoring function of MeCP2 even after brain development, though, can abrogate the neurological deficits in mouse models (Gadalla, Bailey, & Cobb, 2011; Lu, et al., 2016). This finding has raised the possibility that ameliorating the functional effects of mutations in MeCP2 would reverse the clinical manifestations.
Over half of RTT associated missense mutations fall within the MBD highlighting the functional importance of this domain in neurological development. The functional significance of these mutations has been studied extensively over the past decade, including both in vitro and whole animal studies (reviewed in (Lyst & Bird, 2015)). When we mapped some of the most common MeCP2 missense mutations associated with RTT onto the MBD2 structure, we found that many involve interactions that bridge between dynamic and more stable regions of the protein (Scarsdale, et al., 2011). Based on these results, we proposed that these missense mutations disrupt the function of MeCP2 by destabilizing an inherently dynamic region of the protein without disrupting methylation specificity or eliminating DNA binding. Biophysical analyses of RTT associated missense mutations in the MBD confirm that many disrupt the structural stability of the domain (Ghosh, Horowitz-Scherer, Nikitina, Gierasch, & Woodcock, 2008). More recently, studies have demonstrated that MeCP2 can selectively bind to non-CpG 5-methylcytosines and hydroxymethylcytosines, which are found in relative abundance in brain tissue (Kinde, et al., 2015; Lagger, et al., 2017). We have found that MeCP2 can uniquely (as compared to other MBDs) accommodate and bind with high affinity to non-CpG methylated DNA (M. J. Sperlazza, et al., 2017). Importantly, we found that RTT associated missense mutations in the MBD did not inhibit methylation specific recognition of both CpG and non-CpG methylated sites.
Together, these studies raise the possibility that stabilizing mutant MeCP2 proteins could restore high-affinity methylation specific binding and ameliorate symptoms of the disease. This goal, however, requires stabilizing a poorly structured domain for therapeutic effect, an approach that has been explored for p53 mutants (Boeckler, et al., 2008; Bromley, Bauer, Fersht, & Daggett, 2016) but has not generated clinically useful drugs. In the case of RTT syndrome, the difficulties are further exacerbated by the need to cross the blood-brain barrier. Nonetheless, a small molecule MeCP2 stabilizing agent has the potential to help a large portion of RTT patients and would open a new therapeutic paradigm if successful.
5.5. Other 5-methylcytosine binding domain proteins as therapeutic targets
This review has focused on MBD2 and MeCP2 as potential therapeutic targets for several reasons. There is strong evidence that MBD2 targets many tumor suppressor genes (Lopez-Serra, et al., 2006), and its depletion results in more robust gene increases in expression of methylated and silenced genes than other MBD proteins (Lopez-Serra, et al., 2008). In addition, MeCP2 has a reversible and driving role in the pathophysiology of RTT (Lyst & Bird, 2015). In contrast, both MBD1 and MBD3 localize to unmethylated CGIs (Bianchi & Zangi, 2015), suggesting that neither will be good targets for blocking methylation dependent gene silencing. As discussed previously, MBD4 repairs G X mismatches arising from spontaneous deamination of 5- methylcytosine. Thus, inhibiting MBD4 would likely increase DNA mutations without necessarily sensitizing cancer cells to chemotherapy. Nonetheless, as research continues to delineate the functional specificity of each MBD protein, there may be currently unrecognized benefits to selectively targeting these other MBDs.
6. Concluding remarks
Since the studies by several groups in the late 1970s and early 1980s (Constantinides, et al., 1978; Jones & Taylor, 1980; Taylor & Jones, 1979), extensive research has been directed towards understanding the role of DNA methylation in development and disease. These efforts have uncovered new molecular pathways leading to pervasive changes in methylation across the spectrum of cancer types and other diseases and provided very exciting insight into gene regulation and cellular differentiation. Current sequencing technologies have rapidly expanded the database of DNA methylation, yet, this information has led to many new questions and challenges. As seems to be true across the field of epigenetics, a simple on-off binary model does not capture the complexity of gene regulation by DNA methylation. Instead, interpretation of this epigenetic mark depends both on genomic context, cell type, and methylation density. Nonetheless, inhibiting DNA methylation has shown clear therapeutic benefit for hematopoietic neoplasms and has the potential to restore developmentally silenced globin genes. Therefore, we anticipate that alternative approaches for manipulating methylation-dependent gene regulation will lead to new therapeutics with the potential to treat a wide-range of human disease.
Acknowledgement of our funding source:
NIH R01 GM098624 [to D.C.W.]; NIH R01 DK029902 and R56 DK029902 [to G.D.G]
Abbreviations:
- NuRD
nucleosome remodeling and deacetylase
- CpG
cytosine-guanosine dinucleotide
- mCpG
5-methylcytosine-guanosine dinucleotide
- MBD
5-methylcytosine binding domain
- DNMT
DNA methyltransferase
- CGI
CpG island
- TET
ten-eleven translocation dioxygenase
- TDG
thymine DNA-glycosylase
- CIMP
CpG island methylator phenotype
- IDH
isocitrate dehydrogenase
- CHIP
clonal hematopoiesis of indeterminate potential
- RTT
Rett syndrome
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest Statement: The authors declare that there are no conflicts of interest.
Reference List:
- Acuna-Hidalgo R, Sengul H, Steehouwer M, van de Vorst M, Vermeulen SH, Kiemeney L, Veltman JA, Gilissen C, & Hoischen A (2017). Ultra-sensitive Sequencing Identifies High Prevalence of Clonal Hematopoiesis-Associated Mutations throughout Adult Life. Am J Hum Genet, 101, 50–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguilera C, Nakagawa K, Sancho R, Chakraborty A, Hendrich B, & Behrens A (2011). c-Jun N-terminal phosphorylation antagonises recruitment of the Mbd3/NuRD repressor complex. Nature, 469, 231–235. [DOI] [PubMed] [Google Scholar]
- Alqarni SS, Murthy A, Zhang W, Przewloka MR, Silva AP, Watson AA, Lejon S, Pei XY, Smits AH, Kloet SL, Wang H, Shepherd NE, Stokes PH, Blobel GA, Vermeulen M, Glover DM, Mackay JP, & Laue ED (2014). Insight into the architecture of the NuRD complex: structure of the RbAp48-MTA1 subcomplex. J Biol Chem, 289, 21844–21855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya M, Desai M, Gnanapragasam MN, Wang SZ, Zu Zhu S, Williams DC Jr., & Ginder GD (2013). Mi2beta-mediated silencing of the fetal gamma-globin gene in adult erythroid cells. Blood, 121, 3493–3501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, & Zoghbi HY (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet, 23, 185–188. [DOI] [PubMed] [Google Scholar]
- Ang PW, Li WQ, Soong R, & lacopetta B (2009). BRAF mutation is associated with the CpG island methylator phenotype in colorectal cancer from young patients. Cancer Lett,273, 221–224. [DOI] [PubMed] [Google Scholar]
- Aoki A, Suetake I, Miyagawa J, Fujio T, Chijiwa T, Sasaki H, & Tajima S (2001). Enzymatic properties of de novo-type mouse DNA (cytosine-5) methyltransferases. Nucleic Acids Res, 29, 3506–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker J, Lin X, & Nelson WG (2002). Methyl-CpG binding domain protein 2 represses transcription from hypermethylated pi-class glutathione S-transferase gene promoters in hepatocellular carcinoma cells. J Biol Chem, 277, 22573–22580. [DOI] [PubMed] [Google Scholar]
- Ballestar E, & Esteller M (2005). Methyl-CpG-binding proteins in cancer: blaming the DNA methylation messenger. Biochem Cell Biol, 83, 374–384. [DOI] [PubMed] [Google Scholar]
- Baylin SB, Hoppener JW, de Bustros A, Steenbergh PH, Lips CJ, & Nelkin BD (1986). DNA methylation patterns of the calcitonin gene in human lung cancers and lymphomas. Cancer Res, 46, 2917–2922. [PubMed] [Google Scholar]
- Baylin SB, & Jones PA (2011). A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer, 11, 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baymaz HI, Fournier A, Laget S, Ji Z, Jansen PW, Smits AH, Ferry L, Mensinga A, Poser I, Sharrocks A, Defossez PA, & Vermeulen M (2014). MBD5 and MBD6 interact with the human PR-DUB complex through their methyl-CpG-binding domain. PROTEOMICS, 14, 2179–2189. [DOI] [PubMed] [Google Scholar]
- Bejar R, Lord A, Stevenson K, Bar-Natan M, Perez-Ladaga A, Zaneveld J, Wang H, Caughey B, Stojanov P, Getz G, Garcia-Manero G, Kantarjian H, Chen R, Stone RM, Neuberg D, Steensma DP, & Ebert BL (2014). TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood, 124, 2705–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi C, & Zangi R (2015). Molecular dynamics study of the recognition of dimethylated CpG sites by MBD1 protein. J Chem Inf Model, 55, 636–644. [DOI] [PubMed] [Google Scholar]
- Bird AP (1978). Use of restriction enzymes to study eukaryotic DNA methylation: II. The symmetry of methylated sites supports semi-conservative copying of the methylation pattern. J Mol Biol, 118, 49–60. [DOI] [PubMed] [Google Scholar]
- Bird AP (1986). CpG-rich islands and the function of DNA methylation. Nature, 321, 209–213. [DOI] [PubMed] [Google Scholar]
- Bird AP, & Southern EM (1978). Use of restriction enzymes to study eukaryotic DNA methylation: I. The methylation pattern in ribosomal DNA from Xenopus laevis. J Mol Biol, 118, 27–47. [DOI] [PubMed] [Google Scholar]
- Blow MJ, Clark TA, Daum CG, Deutschbauer AM, Fomenkov A, Fries R, Froula J, Kang DD, Malmstrom RR, Morgan RD, Posfai J, Singh K, Visel A, Wetmore K, Zhao Z, Rubin EM, Korlach J, Pennacchio LA, & Roberts RJ (2016). The Epigenomic Landscape of Prokaryotes. PLoS Genet, 12, e1005854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochtler M, Kolano A, & Xu GL (2017). DNA demethylation pathways: Additional players and regulators. BioEssays, 39, 1–13. [DOI] [PubMed] [Google Scholar]
- Boeckler FM, Joerger AC, Jaggi G, Rutherford TJ, Veprintsev DB, & Fersht AR (2008). Targeted rescue of a destabilized mutant of p53 by an in silico screened drug. Proc Natl Acad Sci U S A, 105, 10360–10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brackertz M, Boeke J, Zhang R, & Renkawitz R (2002). Two highly related p66 proteins comprise a new family of potent transcriptional repressors interacting with MBD2 and MBD3. J Biol Chem, 277, 40958–40966. [DOI] [PubMed] [Google Scholar]
- Brackertz M, Gong Z, Leers J, & Renkawitz R (2006). p66alpha and p66beta of the Mi-2/NuRD complex mediate MBD2 and histone interaction. Nucleic Acids Res, 34, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brasen C, Dorosz J, Wiuf A, Boesen T, Mirza O, & Gajhede M (2017). Expression, purification and characterization of the human MTA2-RBBP7 complex. Biochim Biophys Acta, 1865, 531–538. [DOI] [PubMed] [Google Scholar]
- Brocks D, Schmidt CR, Daskalakis M, Jang HS, Shah NM, Li D, Li J, Zhang B, Hou Y, Laudato S, Lipka DB, Schott J, Bierhoff H, Assenov Y, Helf M, Ressnerova A, Islam MS, Lindroth AM, Haas S, Essers M, Imbusch CD, Brors B, Oehme I, Witt O, Lubbert M, Mallm JP, Rippe K, Will R, Weichenhan D, Stoecklin G, Gerhauser C, Oakes CC, Wang T, & Plass C (2017). DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat Genet, 49, 1052–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromley D, Bauer MR, Fersht AR, & Daggett V (2016). An in silico algorithm for identifying stabilizing pockets in proteins: test case, the Y220C mutant of the p53 tumor suppressor protein. Protein Eng Des Sel, 29, 377–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdon RH (1966). Methylation of nucleic acids in Krebs II ascites tumour cells. Nature, 210, 797–799. [DOI] [PubMed] [Google Scholar]
- Buscarlet M, Provost S, Feroz Zada Y, Barhdadi A, Bourgoin V, Lepine G, Mollica L, Szuber N, Dube MP, & Busque L (2017). DNMT3A and TET2 dominate clonal hematopoiesis, demonstrate benign phenotypes and different genetic predisposition. Blood. [DOI] [PubMed] [Google Scholar]
- Busque L, Patel JP, Figueroa ME, Vasanthakumar A, Provost S, Hamilou Z, Mollica L, Li J, Viale A, Heguy A, Hassimi M, Socci N, Bhatt PK, Gonen M, Mason CE, Melnick A, Godley LA, Brennan CW, Abdel-Wahab O, & Levine RL (2012). Recurrent somatic TET2 mutations in normal elderly individuals with clonal hematopoiesis. Nat Genet, 44, 1179–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busslinger M, Hurst J, & Flavell RA (1983). DNA methylation and the regulation of globin gene expression. Cell, 34, 197–206. [DOI] [PubMed] [Google Scholar]
- Casadesus J, & Low D (2006). Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev, 70, 830–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charache S, Dover G, Smith K, Talbot CC Jr., Moyer M, & Boyer S (1983). Treatment of sickle cell anemia with 5-azacytidine results in increased fetal hemoglobin production and is associated with nonrandom hypomethylation of DNA around the gamma-delta- beta-globin gene complex. Proc Natl Acad Sci U S A, 80, 4842–4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheishvili D, Chik F, Li CC, Bhattacharya B, Suderman M, Arakelian A, Hallett M, Rabbani SA, & Szyf M (2014). Synergistic effects of combined DNA methyltransferase inhibition and MBD2 depletion on breast cancer cells; MBD2 depletion blocks 5-aza-2’- deoxycytidine-triggered invasiveness. Carcinogenesis, 35, 2436–2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christman JK (1984). DNA methylation in friend erythroleukemia cells: the effects of chemically induced differentiation and of treatment with inhibitors of DNA methylation. Curr Top Microbiol Immunol, 108, 49–78. [DOI] [PubMed] [Google Scholar]
- Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, Grillo I, Bakogianni S, Ndiaye-Lobry D, Martin MT, Guillamot M, Banh RS, Xu M, Figueroa ME, Dickins RA, Abdel-Wahab O, Park CY, Tsirigos A, Neel BG, & Aifantis I (2017). Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouaire T, de Las Heras JI, Merusi C, & Stancheva I (2010). Recruitment of MBD1 to target genes requires sequence-specific interaction of the MBD domain with methylated DNA. Nucleic Acids Res, 38, 4620–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Constantinides PG, Taylor SM, & Jones PA (1978). Phenotypic conversion of cultured mouse embryo cells by aza pyrimidine nucleosides. Dev Biol, 66, 57–71. [DOI] [PubMed] [Google Scholar]
- Cooper DN, Mort M, Stenson PD, Ball EV, & Chuzhanova NA (2010). Methylation- mediated deamination of 5-methylcytosine appears to give rise to mutations causing human inherited disease in CpNpG trinucleotides, as well as in CpG dinucleotides. Hum Genomics, 4, 406–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer JM, Pohlmann D, Gomez F, Mark L, Kornegay B, Hall C, Siraliev-Perez E, Walavalkar NM, Sperlazza MJ, Bilinovich S, Prokop JW, Hill AL, & Williams DC Jr. (2017). Methylation specific targeting of a chromatin remodeling complex from sponges to humans. Sci Rep, 7, 40674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cramer JM, Scarsdale JN, Walavalkar NM, Buchwald WA, Ginder GD, & Williams DC Jr. (2014). Probing the dynamic distribution of bound states for methylcytosinebinding domains on DNA. J Biol Chem, 289, 1294–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui S, Liu L, Wan T, Jiang L, Shi Y, & Luo L (2017). MiR-520b inhibits the development of glioma by directly targeting MBD2. Am J Cancer Res, 7, 1528–1539. [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, Marks KM, Prins RM, Ward PS, Yen KE, Liau M, Rabinowitz JD, Cantley LC, Thompson CB, Vander Heiden MG, & Su SM (2009). Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature, 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos V, Martinez-Cardus A, & Esteller M (2017). The Epigenomic Revolution in Breast Cancer: From Single-Gene to Genome-Wide Next-Generation Approaches. Am J Pathol. [DOI] [PubMed] [Google Scholar]
- Desai MA, Webb HD, Sinanan LM, Scarsdale JN, Walavalkar NM, Ginder GD, & Williams DC Jr. (2015). An intrinsically disordered region of methyl-CpG binding domain protein 2 (MBD2) recruits the histone deacetylase core of the NuRD complex. Nucleic Acids Res, 43, 3100–3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSimone J, Heller P, Hall L, & Zwiers D (1982). 5-Azacytidine stimulates fetal hemoglobin synthesis in anemic baboons. Proc Natl Acad Sci U S A, 79, 4428–4431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- dos Santos RL, Tosti L, Radzisheuskaya A, Caballero IM, Kaji K, Hendrich B, & Silva JC (2014). MBD3/NuRD facilitates induction of pluripotency in a context-dependent manner. Cell stem cell, 15, 102–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan BK, & Miller JH (1980). Mutagenic deamination of cytosine residues in DNA. Nature, 287, 560–561. [DOI] [PubMed] [Google Scholar]
- Edwards JR, Yarychkivska O, Boulard M, & Bestor TH (2017). DNA methylation and DNA methyltransferases. Epigenetics Chromatin, 10, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Morris LG, Shen R, Seshan V, Mo Q, Heguy A, Baylin SB, Ahuja N, Viale A, Massague J, Norton L, Vahdat LT, Moynahan ME, & Chan TA (2011). Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med, 3, 75ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg AP, & Vogelstein B (1983). Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature, 301, 89–92. [DOI] [PubMed] [Google Scholar]
- Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, Peeters JK, Liu W, Choe SE, Fantin VR, Paietta E, Lowenberg B, Licht JD, Godley LA, Delwel R, Valk PJ, Thompson CB, Levine RL, & Melnick A (2010). Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer ce!!, 18, 553–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flavahan WA, Drier Y, Liau BB, Gillespie SM, Venteicher AS, Stemmer-Rachamimov AO, Suva ML, & Bernstein BE (2016). Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature, 529, 110–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, & Esteller M (2003). The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucleic Acids Res, 31, 1765–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel HW, Kinde B, Stroud H, Gilbert CS, Harmin DA, Kastan NR, Hemberg M, Ebert DH, & Greenberg ME (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature, 522, 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadalla KK, Bailey ME, & Cobb SR (2011). MeCP2 and Rett syndrome: reversibility and potential avenues for therapy. Biochem J, 439, 1–14. [DOI] [PubMed] [Google Scholar]
- Gardiner-Garden M, & Frommer M (1987). CpG islands in vertebrate genomes. J Mol Biol, 196, 261–282. [DOI] [PubMed] [Google Scholar]
- Genovese G, Kahler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, Chambert K, Mick E, Neale BM, Fromer M, Purcell SM, Svantesson O, Landen M, Hoglund M, Lehmann S, Gabriel SB, Moran JL, Lander ES, Sullivan PF, Sklar P, Gronberg H, Hultman CM, & McCarroll SA (2014). Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N Engl J Med, 371, 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirlando R, & Felsenfeld G (2016). CTCF: making the right connections. Genes Dev, 30, 881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh RP, Horowitz-Scherer RA, Nikitina T, Gierasch LM, & Woodcock CL (2008). Rett syndrome-causing mutations in human MeCP2 result in diverse structural changes that impact folding and DNA interactions. J Biol Chem, 283, 20523–20534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginder GD (2015). Epigenetic regulation of fetal globin gene expression in adult erythroid cells. Transl Res, 165, 115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginder GD, Whitters M, Kelley K, & Chase RA (1983). In vivo demethylation of chicken embryonic beta-type globin genes with 5-azacytidine. Prog Clin Biol Res, 134, 501–510. [PubMed] [Google Scholar]
- Ginder GD, Whitters MJ, & Pohlman JK (1984). Activation of a chicken embryonic globin gene in adult erythroid cells by 5-azacytidine and sodium butyrate. Proc Natl Acad Sci U S A, 81, 3954–3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnanapragasam MN, Scarsdale JN, Amaya ML, Webb HD, Desai MA, Walavalkar NM, Wang SZ, Zu Zhu S, Ginder GD, & Williams DC Jr. (2011). p66Alpha-MBD2 coiled-coil interaction and recruitment of Mi-2 are critical for globin gene silencing by the MBD2-NuRD complex. Proc Natl Acad Sci U S A, 108, 7487–7492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnyszka A, Jastrzebski Z, & Flis S (2013). DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res, 33, 2989–2996. [PubMed] [Google Scholar]
- Goelz SE, Vogelstein B, Hamilton SR, & Feinberg AP (1985). Hypomethylation of DNA from benign and malignant human colon neoplasms. Science, 228, 187–190. [DOI] [PubMed] [Google Scholar]
- Goncearenco A, Li M, Simonetti FL, Shoemaker BA, & Panchenko AR (2017). Exploring Protein-Protein Interactions as Drug Targets for Anti-cancer Therapy with In Silico Workflows. Methods Mol Biol, 1647, 221–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowher H, & Jeltsch A (2001). Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: the enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J Mol Biol, 309, 1201–1208. [DOI] [PubMed] [Google Scholar]
- Gravina GL, Festuccia C, Marampon F, Popov VM, Pestell RG, Zani BM, & Tombolini V (2010). Biological rationale for the use of DNA methyltransferase inhibitors as new strategy for modulation of tumor response to chemotherapy and radiation. Mol Cancer, 9, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros C, Fahy J, Halby L, Dufau I, Erdmann A, Gregoire JM, Ausseil F, Vispe S, & Arimondo PB (2012). DNA methylation inhibitors in cancer: recent and future approaches. Biochimie, 94, 2280–2296. [DOI] [PubMed] [Google Scholar]
- Gross S, Cairns RA, Minden MD, Driggers EM, Bittinger MA, Jang HG, Sasaki M, Jin S, Schenkein DP, Su SM, Dang L, Fantin VR, & Mak TW (2010). Cancer-associated metabolite 2-hydroxyglutarate accumulates in acute myelogenous leukemia with isocitrate dehydrogenase 1 and 2 mutations. J Exp Med, 207, 339–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillamot M, Cimmino L, & Aifantis I (2016). The Impact of DNA Methylation in Hematopoietic Malignancies. Trends Cancer, 2, 70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther K, Rust M, Leers J, Boettger T, Scharfe M, Jarek M, Bartkuhn M, & Renkawitz R (2013). Differential roles for MBD2 and MBD3 at methylated CpG islands, active promoters and binding to exon sequences. Nucleic Acids Res, 41, 3010–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn MA, Szabo PE, & Pfeifer GP (2014). 5-Hydroxymethylcytosine: a stable or transient DNA modification? Genomics, 104, 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao X, Luo H, Krawczyk M, Wei W, Wang W, Wang J, Flagg K, Hou J, Zhang H, Yi S, Jafari M, Lin D, Chung C, Caughey BA, Li G, Dhar D, Shi W, Zheng L, Hou R, Zhu J, Zhao L, Fu X, Zhang E, Zhang C, Zhu JK, Karin M, Xu RH, & Zhang K (2017). DNA methylation markers for diagnosis and prognosis of common cancers. Proc Natl Acad Sci U S A, 114, 7414–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Hong S, Bhagwat AS, Zhang X, & Cheng X (2012). Excision of 5- hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation. Nucleic Acids Res, 40, 10203–10214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM, Zhang X, & Cheng X (2012). Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res, 40, 4841–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Wang D, Horton JR, Zhang X, Corces VG, & Cheng X (2017). Structural Basis for the Versatile and Methylation-Dependent Binding of CTCF to DNA. Mol Cell, 66, 711–720 e713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto H, Zhang X, Vertino PM, & Cheng X (2015). The Mechanisms of Generation, Recognition, and Erasure of DNA 5-Methylcytosine and Thymine Oxidations. J Biol Chem, 290, 20723–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q, Ding J, Jia Y, Chen Z, Li L, Sun Y, Li X, Dai Q, Song CX, Zhang K, He C, & Xu GL (2011). Tet-mediated formation of 5- carboxylcytosine and its excision by TDG in mammalian DNA. Science, 333, 1303–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitmann B, Maurer T, Weitzel JM, Stratling WH, Kalbitzer HR, & Brunner E (2003). Solution structure of the matrix attachment region-binding domain of chicken MeCP2. Eur J Biochem, 270, 3263–3270. [DOI] [PubMed] [Google Scholar]
- Hendrich B, Abbott C, McQueen H, Chambers D, Cross S, & Bird A (1999). Genomic structure and chromosomal mapping of the murine and human Mbd1, Mbd2, Mbd3, and Mbd4 genes. Mamm Genome, 10, 906–912. [DOI] [PubMed] [Google Scholar]
- Hendrich B, & Bird A (1998). Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol, 18, 6538–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B, Guy J, Ramsahoye B, Wilson VA, & Bird A (2001). Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev, 15, 710–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B, Hardeland U, Ng HH, Jiricny J, & Bird A (1999). The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature, 401, 301–304. [DOI] [PubMed] [Google Scholar]
- Hendrich B, & Tweedie S (2003). The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet, 19, 269–277. [DOI] [PubMed] [Google Scholar]
- Ho KL, McNae IW, Schmiedeberg L, Klose RJ, Bird AP, & Walkinshaw MD (2008). MeCP2 binding to DNA depends upon hydration at methyl-CpG. Mol Cell, 29, 525–531. [DOI] [PubMed] [Google Scholar]
- Holliday R, & Pugh JE (1975). DNA modification mechanisms and gene activity during development. Science, 187, 226–232. [PubMed] [Google Scholar]
- Holm K, Staaf J, Lauss M, Aine M, Lindgren D, Bendahl PO, Vallon-Christersson J, Barkardottir RB, Hoglund M, Borg A, Jonsson G, & Ringner M (2016). An integrated genomics analysis of epigenetic subtypes in human breast tumors links DNA methylation patterns to chromatin states in normal mammary cells. Breast Cancer Res, 18, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotchkiss RD (1948). The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem, 175, 315–332. [PubMed] [Google Scholar]
- Illingworth RS, & Bird AP (2009). CpG islands--’a rough guide’. FEBS Lett, 583, 1713–1720. [DOI] [PubMed] [Google Scholar]
- Issa JP, Baylin SB, & Herman JG (1997). DNA methylation changes in hematologic malignancies: biologic and clinical implications. Leukemia, 11 Suppl 1, S7–11. [PubMed] [Google Scholar]
- Itzykson R, & Fenaux P (2014). Epigenetics of myelodysplastic syndromes. Leukemia, 28, 497–506. [DOI] [PubMed] [Google Scholar]
- Itzykson R, Kosmider O, Cluzeau T, Mansat-De Mas V, Dreyfus F, Beyne-Rauzy O, Quesnel B, Vey N, Gelsi-Boyer V, Raynaud S, Preudhomme C, Ades L, Fenaux P, Fontenay M, & Groupe Francophone des M (2011). Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia, 25, 1147–1152. [DOI] [PubMed] [Google Scholar]
- Jabbari K, & Bernardi G (2004). Cytosine methylation and CpG, TpG (CpA) and TpA frequencies. Gene, 333, 143–149. [DOI] [PubMed] [Google Scholar]
- Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, Lindsley RC, Mermel CH, Burtt N, Chavez A, Higgins JM, Moltchanov V, Kuo FC, Kluk MJ, Henderson B, Kinnunen L, Koistinen HA, Ladenvall C, Getz G, Correa A, Banahan F, Gabriel S, Kathiresan S, Stringham HM, McCarthy MI, Boehnke M, Tuomilehto J, Haiman C, Groop L, Atzmon G, Wilson JG, Neuberg D, Altshuler D, & Ebert BL (2014). Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med, 371, 2488–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeltsch A (2006). Molecular enzymology of mammalian DNA methyltransferases. Curr Top Microbiol Immunol, 301, 203–225. [DOI] [PubMed] [Google Scholar]
- Jia M, Gao X, Zhang Y, Hoffmeister M, & Brenner H (2016). Different definitions of CpG island methylator phenotype and outcomes of colorectal cancer: a systematic review. Clin Epigenetics, 8, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia M, Jansen L, Walter V, Tagscherer K, Roth W, Herpel E, Kloor M, Blaker H, Chang- Claude J, Brenner H, & Hoffmeister M (2016). No association of CpG island methylator phenotype and colorectal cancer survival: population-based study. BrJ Cancer, 115, 1359–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, & Melnick A (2015). The epigenetic basis of diffuse large B-cell lymphoma. Semin Hematol, 52, 86–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TB, & Coghill RD (1925). Researches on pyrimidines. C111. The discovery of 5- methyl-cytosine in tuberculinic acid, the nucleic acid of the tubercle bacillusl. Journal of the American Chemical…. [Google Scholar]
- Jones PA (1986). DNA methylation and cancer. Cancer Res, 46, 461–466. [PubMed] [Google Scholar]
- Jones PA, & Taylor SM (1980). Cellular differentiation, cytidine analogs and DNA methylation. Cell, 20, 85–93. [DOI] [PubMed] [Google Scholar]
- Josse J, Kaiser AD, & Kornberg A (1961). Enzymatic synthesis of deoxyribonucleic acid. VIII. Frequencies of nearest neighbor base sequences in deoxyribonucleic acid. J Biol Chem, 236, 864–875. [PubMed] [Google Scholar]
- Jurkowska RZ, & Jeltsch A (2016). Enzymology of Mammalian DNA Methyltransferases. Adv Exp Med Biol, 945, 87–122. [DOI] [PubMed] [Google Scholar]
- Kang JY, Song SH, Yun J, Jeon MS, Kim HP, Han SW, & Kim TY (2015). Disruption of CTCF/cohesin-mediated high-order chromatin structures by DNA methylation downregulates PTGS2 expression. Oncogene, 34, 5677–5684. [DOI] [PubMed] [Google Scholar]
- Kelly AD, Kroeger H, Yamazaki J, Taby R, Neumann F, Yu S, Lee JT, Patel B, Li Y, He R, Liang S, Lu Y, Cesaroni M, Pierce SA, Kornblau SM, Bueso-Ramos CE, Ravandi F, Kantarjian HM, Jelinek J, & Issa JP (2017). A CpG island methylator phenotype in acute myeloid leukemia independent of IDH mutations and associated with a favorable outcome. Leukemia. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Huh JW, Kim HR, & Kim YJ (2017). CpG island methylator phenotype is an independent predictor of survival after curative resection for colorectal cancer: A prospective cohort study. J Gastroenterol Hepatol. [DOI] [PubMed] [Google Scholar]
- Kinde B, Gabel HW, Gilbert CS, Griffith EC, & Greenberg ME (2015). Reading the unique DNA methylation landscape of the brain: Non-CpG methylation, hydroxymethylation, and MeCP2. Proc Natl Acad Sci U S A, 112, 6800–6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby MK, Ramaker RC, Roberts BS, Lasseigne BN, Gunther DS, Burwell TC, Davis NS, Gulzar ZG, Absher DM, Cooper SJ, Brooks JD, & Myers RM (2017). Genome-wide DNA methylation measurements in prostate tissues uncovers novel prostate cancer diagnostic biomarkers and transcription factor binding patterns. BMC cancer, 17, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloet SL, Baymaz HI, Makowski M, Groenewold V, Jansen PW, Berendsen M, Niazi H, Kops GJ, & Vermeulen M (2015). Towards elucidating the stability, dynamics and architecture of the nucleosome remodeling and deacetylase complex by using quantitative interaction proteomics. FEBS J, 282, 1774–1785. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, & Bird AP (2005). DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell, 19, 667–678. [DOI] [PubMed] [Google Scholar]
- Kransdorf EP, Wang SZ, Zhu SZ, Langston TB, Rupon JW, & Ginder GD (2006). MBD2 is a critical component of a methyl cytosine-binding protein complex isolated from primary erythroid cells. Blood, 108, 2836–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laget S, Joulie M, Le Masson F, Sasai N, Christians E, Pradhan S, Roberts RJ, & Defossez PA (2010). The human proteins MBD5 and MBD6 associate with heterochromatin but they do not bind methylated DNA. PLoS ONE, 5, e11982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagger S, Connelly JC, Schweikert G, Webb S, Selfridge J, Ramsahoye BH, Yu M, He, Sanguinetti G, Sowers LC, Walkinshaw MD, & Bird A, (2017). MeCP2 recognizes cytosine methylated tri-nucleotide and di-nucleotide sequences to tune transcription in the mammalian brain. PLoS Genet, 13, e1006793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Guezennec X, Vermeulen M, Brinkman AB, Hoeijmakers WA, Cohen A, Lasonder E, & Stunnenberg HG (2006). MBD2/NuRD and MBD3/NuRD, two distinct complexes with different biochemical and functional properties. Mol Cell Biol, 26, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Meehan RR, Henzel WJ, Maurer-Fogy I, Jeppesen P, Klein F, & Bird A (1992). Purification, sequence, and cellular localization of a novel chromosomal protein that binds to methylated DNA. Cell, 69, 905–914. [DOI] [PubMed] [Google Scholar]
- Ley TJ, DeSimone J, Anagnou NP, Keller GH, Humphries RK, Turner PH, Young NS, Keller P, & Nienhuis AW (1982). 5-azacytidine selectively increases gamma-globin synthesis in a patient with beta+ thalassemia. N Engl J Med, 307, 1469–1475. [DOI] [PubMed] [Google Scholar]
- Lin X, & Nelson WG (2003). Methyl-CpG-binding domain protein-2 mediates transcriptional repression associated with hypermethylated GSTP1 CpG islands in MCF-7 breast cancer cells. Cancer Res, 63, 498–504. [PubMed] [Google Scholar]
- Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, & Jaenisch R (2016). Editing DNA Methylation in the Mammalian Genome. Cell, 167, 233–247 e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Serra L, Ballestar E, Fraga MF, Alaminos M, Setien F, & Esteller M (2006). A profile of methyl-CpG binding domain protein occupancy of hypermethylated promoter CpG islands of tumor suppressor genes in human cancer. Cancer Res, 66, 8342–8346. [DOI] [PubMed] [Google Scholar]
- Lopez-Serra L, Ballestar E, Ropero S, Setien F, Billard LM, Fraga MF, Lopez-Nieva P, Alaminos M, Guerrero D, Dante R, & Esteller M (2008). Unmasking of epigenetically silenced candidate tumor suppressor genes by removal of methyl-CpG-binding domain proteins. Oncogene, 27, 3556–3566. [DOI] [PubMed] [Google Scholar]
- Lopez-Serra L, & Esteller M (2008). Proteins that bind methylated DNA and human cancer: reading the wrong words. Br J Cancer, 98, 1881–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran SJ, Comoglio F, Hamey FK, Giustacchini A, Errami Y, Earp E, Gottgens B, Jacobsen SEW, Mead AJ, Hendrich B, & Green AR (2017). Mbd3/NuRD controls lymphoid cell fate and inhibits tumorigenesis by repressing a B cell transcriptional program. J Exp Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low JK, Webb SR, Silva AP, Saathoff H, Ryan DP, Torrado M, Brofelth M, Parker BL, Shepherd NE, & Mackay JP (2016). CHD4 Is a Peripheral Component of the Nucleosome Remodeling and Deacetylase Complex. J Biol Chem, 291, 15853–15866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Ash RT, He L, Kee SE, Wang W, Yu D, Hao S, Meng X, Ure K, Ito-Ishida A, Tang B, Sun Y, Ji D, Tang J, Arenkiel BR, Smirnakis SM, & Zoghbi HY (2016). Loss and Gain of MeCP2 Cause Similar Hippocampal Circuit Dysfunction that Is Rescued by Deep Brain Stimulation in a Rett Syndrome Mouse Model. Neuron, 91, 739–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst MJ, & Bird A (2015). Rett syndrome: a complex disorder with simple roots. Nat Rev Genet, 16, 261–275. [DOI] [PubMed] [Google Scholar]
- Magdinier F, & Wolffe AP (2001). Selective association of the methyl-CpG binding protein MBD2 with the silent p14/p16 locus in human neoplasia. Proc Natl Acad Sci U S A, 98, 4990–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manvilla BA, Maiti A, Begley MC, Toth EA, & Drohat AC (2012). Crystal structure of human methyl-binding domain IV glycosylase bound to abasic DNA. J Mol Biol, 420, 164–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massie CE, Mills IG, & Lynch AG (2017). The importance of DNA methylation in prostate cancer development. J Steroid Biochem Mol Biol, 166, 1–15. [DOI] [PubMed] [Google Scholar]
- Mayer W, Niveleau A, Walter J, Fundele R, & Haaf T (2000). Demethylation of the zygotic paternal genome. Nature, 403, 501–502. [DOI] [PubMed] [Google Scholar]
- McGhee JD, & Ginder GD (1979). Specific DNA methylation sites in the vicinity of the chicken beta-globin genes. Nature, 280, 419–420. [DOI] [PubMed] [Google Scholar]
- Meehan RR, Lewis JD, McKay S, Kleiner EL, & Bird AP (1989). Identification of a mammalian protein that binds specifically to DNA containing methylated CpGs. Cell, 58, 499–507. [DOI] [PubMed] [Google Scholar]
- Menafra R, Brinkman AB, Matarese F, Franci G, Bartels SJ, Nguyen L, Shimbo T, Wade PA, Hubner NC, & Stunnenberg HG (2014). Genome-wide binding of MBD2 reveals strong preference for highly methylated loci. PLoS ONE, 9, e99603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menafra R, & Stunnenberg HG (2014). MBD2 and MBD3: elusive functions and mechanisms. Front Genet, 5, 428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meszaros B, Tompa P, Simon I, & Dosztanyi Z (2007). Molecular principles of the interactions of disordered proteins. J Mol Biol, 372, 549–561. [DOI] [PubMed] [Google Scholar]
- Mian OY, Wang SZ, Zhu SZ, Gnanapragasam MN, Graham L, Bear HD, & Ginder G (2011). Methyl-binding domain protein 2-dependent proliferation and survival of breast cancer cells. Mol Cancer Res, 9, 1152–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micevic G, Theodosakis N, & Bosenberg M (2017). Aberrant DNA methylation in melanoma: biomarker and therapeutic opportunities. Clin Epigenetics, 9, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard CJ, Varma N, Saleh A, Morris K, Watson PJ, Bottrill AR, Fairall L, Smith CJ, & Schwabe JW (2016). The structure of the core NuRD repression complex provides insights into its interaction with chromatin. Elife, 5, e13941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millard CJ, Watson PJ, Celardo I, Gordiyenko Y, Cowley SM, Robinson CV, Fairall L, & Schwabe JW (2013). Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol Cell, 51, 57–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morera S, Grin I, Vigouroux A, Couve S, Henriot V, Saparbaev M, & Ishchenko AA (2012). Biochemical and structural characterization of the glycosylase domain of MBD4 bound to thymine and 5-hydroxymethyuracil-containing DNA. Nucleic Acids Res, 40, 9917–9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murzina NV, Pei XY, Zhang W, Sparkes M, Vicente-Garcia J, Pratap JV, McLaughlin SH, Ben-Shahar TR, Verreault A, Luisi BF, & Laue ED (2008). Structural basis for the recognition of histone H4 by the histone-chaperone RbAp46. Structure, 16, 1077–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nan X, Meehan RR, & Bird A (1993). Dissection of the methyl-CpG binding domain from the chromosomal protein MeCP2. Nucleic Acids Res, 21, 4886–4892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Jeppesen P, & Bird A (2000). Active repression of methylated genes by the chromosomal protein MBD1. Mol Cell Biol, 20, 1394–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng HH, Zhang Y, Hendrich B, Johnson CA, Turner BM, Erdjument-Bromage H, Tempst P, Reinberg D, & Bird A (1999). MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat Genet, 23, 58–61. [DOI] [PubMed] [Google Scholar]
- Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, Pan F, Pelloski CE, Sulman EP, Bhat KP, Verhaak RG, Hoadley KA, Hayes DN, Perou CM, Schmidt HK, Ding L, Wilson RK, Van Den Berg D, Shen H, Bengtsson H, Neuvial P, Cope LM, Buckley J, Herman JG, Baylin SB, Laird PW, Aldape K, & Cancer Genome Atlas Research, N. (2010). Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer cell, 17, 510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino S, Kawasaki T, Kirkner GJ, Kraft P, Loda M, & Fuchs CS (2007). Evaluation of markers for CpG island methylator phenotype (CIMP) in colorectal cancer by a large population-based sample. J Mol Diagn, 9, 305–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohki I, Shimotake N, Fujita N, Jee J, Ikegami T, Nakao M, & Shirakawa M (2001). Solution structure of the methyl-CpG binding domain of human MBD1 in complex with methylated DNA. Cell, 105, 487–497. [DOI] [PubMed] [Google Scholar]
- Ohki I, Shimotake N, Fujita N, Nakao M, & Shirakawa M (1999). Solution structure of the methyl-CpG-binding domain of the methylation-dependent transcriptional repressor MBD1. EMBO J, 18, 6653–6661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otani J, Arita K, Kato T, Kinoshita M, Kimura H, Suetake I, Tajima S, Ariyoshi M, & Shirakawa M (2013). Structural basis of the versatile DNA recognition ability of the methyl-CpG binding domain of methyl-CpG binding domain protein 4. J Biol Chem, 288, 6351–6362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Bilinovich SM, Kaur P, Riehn R, Wang H, & Williams DC Jr. (2017). CpG and methylation-dependent DNA binding and dynamics of the methylcytosine binding domain 2 protein at the single-molecule level. Nucleic Acids Res, 45, 9164–9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pechalrieu D, Etievant C, & Arimondo PB (2017). DNA methyltransferase inhibitors in cancer: From pharmacology to translational studies. Biochem Pharmacol, 129, 1–13. [DOI] [PubMed] [Google Scholar]
- Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Genuardi M, Karbowski M, Yeung AT, Matsumoto Y, & Bellacosa A (2000). Investigation of the substrate spectrum of the human mismatch-specific DNA N-glycosylase MED1 (MBD4): fundamental role of the catalytic domain. J Cell Physiol, 185, 473–480. [DOI] [PubMed] [Google Scholar]
- Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Stoerker J, Genuardi M, Yeung AT, Matsumoto Y, & Bellacosa A (2000). Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J Biol Chem, 275, 32422–32429. [DOI] [PubMed] [Google Scholar]
- Pinney SE (2014). Mammalian Non-CpG Methylation: Stem Cells and Beyond. Biology (Basel), 3, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plimack ER, Kantarjian HM, & Issa JP (2007). Decitabine and its role in the treatment of hematopoietic malignancies. Leuk Lymphoma, 48, 1472–1481. [DOI] [PubMed] [Google Scholar]
- Rais Y, Zviran A, Geula S, Gafni O, Chomsky E, Viukov S, Mansour AA, Caspi I, Krupalnik V, Zerbib M, Maza I, Mor N, Baran D, Weinberger L, Jaitin DA, Lara- Astiaso D, Blecher-Gonen R, Shipony Z, Mukamel Z, Hagai T, Gilad S, Amann- Zalcenstein D, Tanay A, Amit I, Novershtern N, & Hanna JH (2013). Deterministic direct reprogramming of somatic cells to pluripotency. Nature, 502, 65–70. [DOI] [PubMed] [Google Scholar]
- Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, & Jaenisch R (2000). Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A, 97, 5237–5242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch C, Trieb M, Wibowo FR, Wellenzohn B, Mayer E, & Liedl KR (2005). Towards an understanding of DNA recognition by the methyl-CpG binding domain 1. J BiomolStruct Dyn, 22, 695–706. [DOI] [PubMed] [Google Scholar]
- Razin A, & Riggs AD (1980). DNA methylation and gene function. Science, 210, 604–610. [DOI] [PubMed] [Google Scholar]
- Riggs AD (1975). X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet, 14, 9–25. [DOI] [PubMed] [Google Scholar]
- Rupon JW, Wang SZ, Gaensler K, Lloyd J, & Ginder GD (2006). Methyl binding domain protein 2 mediates gamma-globin gene silencing in adult human betaYAC transgenic mice. Proc Natl Acad Sci U S A, 103, 6617–6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom OJ, Berger J, Bishop SM, Hendrich B, Bird A, & Clarke AR (2003). Deficiency of Mbd2 suppresses intestinal tumorigenesis. Nat Genet, 34, 145–147. [DOI] [PubMed] [Google Scholar]
- Saunthararajah Y, & DeSimone J (2004). Clinical studies with fetal hemoglobin-enhancing agents in sickle cell disease. Semin Hematol, 41, 11–16. [DOI] [PubMed] [Google Scholar]
- Scarsdale JN, Webb HD, Ginder GD, & Williams DC Jr. (2011). Solution structure and dynamic analysis of chicken MBD2 methyl binding domain bound to a target-methylated DNA sequence. Nucleic Acids Res, 39, 6741–6752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidberger JW, Sharifi Tabar M, Torrado M, Silva AP, Landsberg MJ, Brillault L, AlQarni S, Zeng YC, Parker BL, Low JK, & Mackay JP (2016). The MTA1 subunit of the nucleosome remodeling and deacetylase complex can recruit two copies of RBBP4/7. Protein Sci, 25, 1472–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomacher L, & Niehrs C (2017). DNA repair and erasure of 5-methylcytosine in vertebrates. BioEssays, 39. [DOI] [PubMed] [Google Scholar]
- Schuh AC, Dohner H, Pleyer L, Seymour JF, Fenaux P, & Dombret H (2017). Azacitidine in adult patients with acute myeloid leukemia. Crit Rev Oncol Hematol, 116, 159–177. [DOI] [PubMed] [Google Scholar]
- Scott LJ (2016). Azacitidine: A Review in Myelodysplastic Syndromes and Acute Myeloid Leukaemia. Drugs, 76, 889–900. [DOI] [PubMed] [Google Scholar]
- Shen CK, & Maniatis T (1980). Tissue-specific DNA methylation in a cluster of rabbit beta-like globin genes. Proc Natl Acad Sci U S A, 77, 6634–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih AH, Meydan C, Shank K, Garrett-Bakelman FE, Ward PS, Intlekofer AM, Nazir A, Stein EM, Knapp K, Glass J, Travins J, Straley K, Gliser C, Mason CE, Yen K, Thompson CB, Melnick A, & Levine RL (2017). Combination Targeted Therapy to Disrupt Aberrant Oncogenic Signaling and Reverse Epigenetic Dysfunction in IDH2- and TET2-Mutant Acute Myeloid Leukemia. Cancer Discov, 7, 494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimbo T, Du Y, Grimm SA, Dhasarathy A, Mav D, Shah RR, Shi H, & Wade PA (2013). MBD3 localizes at promoters, gene bodies and enhancers of active genes. PLoS Genet, 9, e1004028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singal R, van Wert J, & Bashambu M (2001). Cytosine methylation represses glutathione S- transferase P1 (GSTP1) gene expression in human prostate cancer cells. Cancer Res, 61, 4820–4826. [PubMed] [Google Scholar]
- Singh V, Sharma P, & Capalash N (2013). DNA methyltransferase-1 inhibitors as epigenetic therapy for cancer. Curr Cancer Drug Targets, 13, 379–399. [DOI] [PubMed] [Google Scholar]
- Smits AH, Jansen PW, Poser I, Hyman AA, & Vermeulen M (2013). Stoichiometry of chromatin-associated protein complexes revealed by label-free quantitative mass spectrometry-based proteomics. Nucleic Acids Res, 41, e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JJ, Garlick JD, & Kingston RE (2008). Structural basis of histone H4 recognition by p55. Genes Dev, 22, 1313–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer DH, Russler-Germain DA, Ketkar S, Helton NM, Lamprecht TL, Fulton RS, Fronick CC, O’Laughlin M, Heath SE, Shinawi M, Westervelt P, Payton JE, Wartman LD, Welch JS, Wilson RK, Walter MJ, Link DC, DiPersio JF, & Ley TJ (2017). CpG Island Hypermethylation Mediated by DNMT3A Is a Consequence of AML Progression. Cell, 168, 801–816 e813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperlazza J, Rahmani M, Beckta J, Aust M, Hawkins E, Wang SZ, Zu Zhu S, Podder S, Dumur C, Archer K, Grant S, & Ginder GD (2015). Depletion of the chromatin remodeler CHD4 sensitizes AML blasts to genotoxic agents and reduces tumor formation. Blood, 126, 1462–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperlazza MJ, Bilinovich SM, Sinanan LM, Javier FR, & Williams DC Jr. (2017). Structural Basis of MeCP2 Distribution on Non-CpG Methylated and Hydroxymethylated DNA. J Mol Biol, 429, 1581–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt CG, Bartels SJ, Brinkman AB, Tjeertes JV, Poser I, Stunnenberg HG, & Vermeulen M (2010). CDK2AP1/DOC-1 is a bona fide subunit of the Mi-2/NuRD complex. Mol Biosyst, 6, 1700–1706. [DOI] [PubMed] [Google Scholar]
- Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, & Ebert BL (2015). Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood, 126, 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefansson OA, Moran S, Gomez A, Sayols S, Arribas-Jorba C, Sandoval J, Hilmarsdottir H, Olafsdottir E, Tryggvadottir L, Jonasson JG, Eyfjord J, & Esteller M (2015). A DNA methylation-based definition of biologically distinct breast cancer subtypes. Mol Oncol, 9, 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, & Bird A (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet, 9, 465–476. [DOI] [PubMed] [Google Scholar]
- Swartz MN, Trautner TA, & Kornberg A (1962). Enzymatic synthesis of deoxyribonucleic acid. XI. Further studies on nearest neighbor base sequences in deoxyribonucleic acids. J Biol Chem, 237, 1961–1967. [PubMed] [Google Scholar]
- Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, & Rao A (2009). Conversion of 5-methylcytosine to 5- hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science, 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SM, & Jones PA (1979). Multiple new phenotypes induced in 10T1/2 and 3T3 cells treated with 5-azacytidine. Cell, 17, 771–779. [DOI] [PubMed] [Google Scholar]
- Teif VB, Beshnova DA, Vainshtein Y, Marth C, Mallm JP, Hofer T, & Rippe K (2014). Nucleosome repositioning links DNA (de)methylation and differential CTCF binding during stem cell development. Genome Res, 24, 1285–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirado-Magallanes R, Rebbani K, Lim R, Pradhan S, & Benoukraf T (2017). Whole genome DNA methylation: beyond genes silencing. Oncotarget, 8, 5629–5637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, & Issa JP (1999). CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA, 96, 8681–8686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyota M, & Issa JP (2005). Epigenetic changes in solid and hematopoietic tumors. Semin Oncol, 32, 521–530. [DOI] [PubMed] [Google Scholar]
- Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, Levine R, Heguy A, Viale A, Morris LG, Huse JT, Mellinghoff IK, & Chan TA (2012). IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature, 483, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky VN (2016). Dancing Protein Clouds: The Strange Biology and Chaotic Physics of Intrinsically Disordered Proteins. J Biol Chem, 291, 6681–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ploeg LHT, & Flavell RA (1980). DNA methylation in the human y63-globin locus in erythroid and nonerythroid tissues. Cell, 19, 947–958. [DOI] [PubMed] [Google Scholar]
- Vela L, & Marzo I (2015). Bcl-2 family of proteins as drug targets for cancer chemotherapy: the long way of BH3 mimetics from bench to bedside. Curr Opin Pharmacol, 23, 74–81. [DOI] [PubMed] [Google Scholar]
- Vermeulen M, Hubner NC, & Mann M (2008). High confidence determination of specific protein-protein interactions using quantitative mass spectrometry. Curr Opin Biotechnol, 19, 331–337. [DOI] [PubMed] [Google Scholar]
- Wade PA, Jones PL, Vermaak D, & Wolffe AP (1998). A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr Biol, 8, 843–846. [DOI] [PubMed] [Google Scholar]
- Wakefield RI, Smith BO, Nan X, Free A, Soteriou A, Uhrin D, Bird AP, & Barlow PN (1999). The solution structure of the domain from MeCP2 that binds to methylated DNA. J Mol Biol, 291, 1055–1065. [DOI] [PubMed] [Google Scholar]
- Walavalkar NM, Cramer JM, Buchwald WA, Scarsdale JN, & Williams DC Jr. (2014). Solution structure and intramolecular exchange of methyl-cytosine binding domain protein 4 (MBD4) on DNA suggests a mechanism to scan for mCpG/TpG mismatches. Nucleic Acids Res, 42, 11218–11232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walavalkar NM, Gordon N, & Williams DC Jr. (2013). Unique features of the anti-parallel, heterodimeric coiled-coil interaction between methyl-cytosine binding domain 2 (MBD2) homologues and GATA zinc finger domain containing 2A (GATAD2A/p66alpha). J Biol Chem, 288, 3419–3427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PJ, Fairall L, Santos GM, & Schwabe JW (2012). Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature, 481, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells JA, & McClendon CL (2007). Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature, 450, 1001–1009. [DOI] [PubMed] [Google Scholar]
- Wood KH, Johnson BS, Welsh SA, Lee JY, Cui Y, Krizman E, Brodkin ES, Blendy JA, Robinson MB, Bartolomei MS, & Zhou Z (2016). Tagging methyl-CpG-binding domain proteins reveals different spatiotemporal expression and supports distinct functions. Epigenomics, 8, 455–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt GR (1950). Occurrence of 5-Methyl-Cytosine in Nucleic Acids. Nature, 166, 237–238. [DOI] [PubMed] [Google Scholar]
- Wyatt GR (1951). Recognition and estimation of 5-methylcytosine in nucleic acids. Biochem J, 48, 581–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyhs N, Walker D, Giovinazzo H, Yegnasubramanian S, & Nelson WG (2014). Time- Resolved Fluorescence Resonance Energy Transfer Assay for Discovery of Small- Molecule Inhibitors of Methyl-CpG Binding Domain Protein 2. J Biomol Screen, 19, 1060–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia L, Huang W, Bellani M, Seidman MM, Wu K, Fan D, Nie Y, Cai Y, Zhang YW, Yu LR, Li H, Zahnow CA, Xie W, Chiu Yen RW, Rassool FV, & Baylin SB (2017). CHD4 Has Oncogenic Functions in Initiating and Maintaining Epigenetic Suppression of Multiple Tumor Suppressor Genes. Cancer cell, 31, 653–668 e657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, & Xiong Y (2011). Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer cell, 19, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Rau R, & Goodell MA (2015). DNMT3A in haematological malignancies. Nat Rev Cancer, 15, 152–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, McDaniel IE, Silva P, & Zilberman D (2010). Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science, 328, 916–919. [DOI] [PubMed] [Google Scholar]
- Zemach A, & Zilberman D (2010). Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr Biol, 20, R780–785. [DOI] [PubMed] [Google Scholar]
- Zhang W, Aubert A, Gomez de Segura JM, Karuppasamy M, Basu S, Murthy AS, Diamante A, Drury TA, Balmer J, Cramard J, Watson AA, Lando D, Lee SF, Palayret M, Kloet SL, Smits AH, Deery MJ, Vermeulen M, Hendrich B, Klenerman D, Schaffitzel C, Berger I, & Laue ED (2016). The Nucleosome Remodeling and Deacetylase Complex NuRD Is Built from Preformed Catalytically Active Sub-modules. J Mol Biol, 428, 2931–2942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird A, & Reinberg D (1999). Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev, 13, 1924–1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, & Xiong Y (2009). Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science, 324, 261–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu D, Hunter SB, Vertino PM, & Van Meir EG (2011). Overexpression of MBD2 in glioblastoma maintains epigenetic silencing and inhibits the antiangiogenic function of the tumor suppressor gene BAI1. Cancer Res, 71, 5859–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, Thorgeirsson TE, Sigurdsson A, Gudjonsson SA, Gudmundsson J, Jonasson JG, Tryggvadottir L, Jonsson T, Helgason A, Gylfason A, Sulem P, Rafnar T, Thorsteinsdottir U, Gudbjartsson DF, Masson G, Kong A, & Stefansson K (2017). Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. [DOI] [PMC free article] [PubMed] [Google Scholar]