

Abstract
Sirtuin 3 (Sirt3)-modified mitochondrial fission participates in the progression of several types of cancers. However, its role in tongue cancer requires investigation. The aim of our study is to determine whether Sirt3 knockdown regulates the viability of tongue cancer cells via modulating mitochondrial fission. Two types of tongue cancer cells were used in the present study, and siRNA was transfected into the cells to suppress Sirt3 expression. Mitochondrial function and cell apoptosis were determined via immunofluorescence, Western blotting, ELISA, and qPCR assays. A pathway blocker was applied to verify the role of the JNK-Fis1 signaling pathway in regulation of mitochondrial fission. The present study showed that loss of Sirt3 promoted tongue cancer cell death in a manner dependent on mitochondrial apoptosis. Mitochondrial oxidative stress, energy metabolism disorder, mitochondrial cyt-c liberation, and mitochondrial apoptosis activation were observed after Sirt3 silencing. Furthermore, we demonstrated that Sirt3 knockdown activated mitochondrial stress via triggering Fis1-related mitochondrial fission and that inhibition of Fis1-related mitochondrial fission abrogated the pro-apoptotic effect of Sirt3 knockdown on tongue cancer cells. To this end, we found that Sirt3 modulated Fis1 expression via the c-Jun N-terminal kinases (JNK) signaling pathway and that blockade of the JNK pathway attenuated mitochondrial stress and repressed apoptosis in Sirt3 knockdown cells. Altogether, our results identified a tumor-suppressive role for Sirt3 deficiency in tongue cancer via activation of the JNK-Fis1 axis and subsequent initiation of fatal mitochondrial fission. Given these findings, strategies to repress Sirt3 activity and enhance the JNK-Fis1-mitochondrial fission cascade have clinical benefits for patients with tongue cancer.
Keywords: Tongue cancer, Mitochondrial fission, Sirt3, JNK-Fis1 signaling pathway, Mitochondrial dysfunction
Introduction
Oral cancer is the sixth most common type of cancer in the global human population. Tongue cancer that occurs on the anterior part of the tongue is also termed oral tongue cancer (Zhou et al. 2017f). The symptoms of tongue cancer include a red or white patch on the tongue, pain while swallowing, and a constant sore throat (Xu et al. 2017). Although advances have been made in the diagnosis and treatment of tongue cancer over the last decade, the morbidity and mortality in young patients (< 40 years old) have significantly increased (Zhong et al. 2017). Moreover, the tumorigenesis of the tongue has not been fully elucidated. Accordingly, achieving a better understanding of the molecular mechanism underlying the development of tongue cancer is essential for the design of effective treatment approaches.
Sirtuin 3 (Sirt3) is associated with cancer survival, including ovarian cancer cells, renal carcinoma, and colorectal cancer (Wang et al. 2018b). At the molecular level, lower Sirt3 expression has been associated with mitochondrial oxidative stress and ATP depletion (Li et al. 2018). Moreover, decreased Sirt3 induces mitochondrial dysfunction and cell apoptosis, and Sirt3 inhibition can repress cancer migration via modifying the Src/Fak signaling pathway (van Beijnum et al. 2017). Besides, Sirt3 knockout also leads to c-Jun N-terminal kinases (JNK) pathway activation and ROS overproduction (Jing et al. 2011). These data confirm the tumor-suppressive property of Sirt3 inhibition on cancer survival, metabolism, and invasion (Schluter et al. 2017). Interestingly, no study has explored the detailed role of Sirt3 in tongue cancer.
Based on previous findings, mitochondria are the primary downstream target of Sirt3 (Kalyanaraman 2017). Structurally, Sirt3 is a type of a mitochondrial NAD+-dependent deacetylase that is preferentially localized to mitochondria and closely modulates mitochondrial function and structure (Pryds et al. 2017). Decreased Sirt3 in cervical cancer HeLa cells triggers significant mitochondrial ROS overproduction. In colorectal cancer, knockdown of Sirt3 activates mitochondrial fission that initiates mitochondrial apoptosis in a manner dependent on caspase-9 (Wang et al. 2018b). Sirt3 repression also impairs the amplification of mitochondrial DNA and consequently inhibits mitochondrial biogenesis (Turner et al. 2017). In addition to the well-documented role of Sirt3 inhibition in mediating mitochondrial damage, exploring whether Sirt3 inhibition contributes to tongue cancer cell apoptosis via inducing mitochondrial stress is worthwhile.
Recently, mitochondrial fission has been connected to cancer apoptosis via multiple mechanisms (Zhou et al. 2018e). Several mitochondrial pathological processes are closely handled by mitochondrial fission, including mitochondrial redox balance (Zhou et al. 2018b), mitochondrial autophagy (Jin et al. 2018), mitochondrial calcium management (Zhu et al. 2018a), mitochondrial energy synthesis (Yan et al. 2018a), and mitochondrial pro-apoptotic factor liberation (Li et al. 2017). Abnormal mitochondrial fission generates massive mitochondrial debris with lower mitochondrial potential that cannot produce ATP to ensure cancer metabolism. Moreover, due to mitochondrial membrane damage, fragmented mitochondria release pro-apoptotic factors and/or calcium into the cytoplasm/nucleus and activate the mitochondria-dependent apoptotic pathway (Zhao et al. 2018). Mechanistically, mitochondrial fission is primarily regulated by mitochondrial fission factors, such as Fis1 and its ligand Drp1. Interestingly, Fis1-related mitochondrial fission has been associated with tongue cancer apoptosis through a poorly understood mechanism. In other cancer types, Fis1-related mitochondrial fission is also associated with cancer mitochondrial damage and cell apoptosis. However, whether Fis1-related mitochondrial fission modifies tongue cancer apoptosis via regulating mitochondrial function is unknown. Therefore, the aim of our study is to explore the roles of Sirt3 and Fis1-related mitochondrial fission in tongue cancer apoptosis with a particular focus on mitochondrial stress.
Materials and methods
Cell culture and treatment
Two types of tongue cancer cell lines [SCC-15 (ATCC® CRL-1623™) and SCC-9 (ATCC® CRL-1629™) cells] were used in the present study. These cells were cultured in L-DMEM (Thermo Fisher Scientific, Inc., Waltham, MA, USA) with 10% fetal bovine serum (HyClone; GE Healthcare Life Sciences, Logan, UT, USA) under 37 °C/5% CO2. After the cells reached 70–80% confluence, two independent siRNAs against Sirt3 were transfected into the cells. To inhibit JNK activation, SP600125 (25 μM, Selleck Chemicals, Houston, TX, USA) was added to the medium for 2 h.
TUNEL staining and MTT assay
Cellular death was measured via TUNEL assay. TUNEL staining was performed using a One Step TUNEL Apoptosis Assay Kit (Beyotime, China, Cat. No.: C1086) according to the manufacturer’s instructions. The MTT assay was performed according to the methods used in a previous study. Cells were plated onto a 96-well plate. MTT solution (Beyotime, China, Cat. No.: C0009) was then added into the medium, and the cells were incubated for approximately 2 h at 37 °C/5% CO2. The optical density (OD) of the MTT solution was recorded using a microplate reader (490 nm absorbance; Epoch 2; BioTek Instruments, Inc., Winooski, VT, USA) 24 h after siRNA transfection (Tenreiro et al. 2017). An LDH release assay was conducted using a commercial kit (Beyotime, China, Cat. No.: C0016) according to the manufacturer’s instructions (Gadicherla et al. 2017).
Western blotting
Cells were lysed in RIPA Lysis Buffer (Beyotime, China, Cat. No.: P0013C). After high-speed centrifugation, the proteins were collected and quantified with the Enhanced BCA Protein Assay Kit (Beyotime, China, Cat. No.: P0009) (Ackermann et al. 2017). Subsequently, 40–60 μg of protein was loaded onto 10% SDS-PAGE gels and transferred to PVDF membranes. The membranes were washed with TBST and then blocked with 5% non-fat milk for 45 min at room temperature. The primary antibodies used in the present study were as follows (Zhai et al. 2017): Bcl2 (1:1000, Cell Signaling Technology, #3498), Bax (1:1000, Cell Signaling Technology, #2772), caspase9 (1:1000, Cell Signaling Technology, #9504), pro-caspase3 (1:1000, Abcam, #ab13847), cleaved caspase3 (1:1000, Abcam, #ab49822), t-JNK (1:1000; Cell Signaling Technology, #4672), p-JNK (1:1000; Cell Signaling Technology, #9251), Drp1 (1:1000, Abcam, #ab56788), Fis1 (1:1000, Abcam, #ab71498), Mff (1:1000, Cell Signaling Technology, #86668), PARP (1:1000, Abcam, #ab32064), and Sirt3 (1:1000, Abcam, no. ab86671). The experiments were performed in triplicate and repeated three times with similar results.
Immunofluorescence
Cells were washed with PBS at room temperature to remove the DMEM. Then, the cells were fixed in 3.7% paraformaldehyde for 30 min at room temperature and permeabilized with 0.1% Triton X-100 for 10 min at 4 °C. The cells were then washed with PBS, and 10% goat serum albumin was used to block the samples for 45 min at room temperature. The samples were again washed with PBS and then incubated overnight with the following primary antibodies: cyt-c (1:500; Abcam; #ab90529), Fis1 (1:1000, Abcam, #ab71498), and Tom-20 (1:1000, Abcam, #ab186735) (Zhou et al. 2018c). The average length of the mitochondria was measured on an inverted microscope to quantify mitochondrial fission (BX51; Olympus Corporation, Tokyo, Japan) as described in the previous study (Zhou et al. 2018f). The experiments were performed in triplicate and repeated three times with similar results.
Mitochondrial potential observation
The mitochondrial membrane potential was determined by JC-1 staining. Live cells were washed with PBS, and a JC-1 solution was then added to the medium (Lee et al. 2017). The cells were incubated at 37 °C/5% CO2 for 30 min, washed with PBS, loaded with DAPI, and then observed under a fluorescence microscope.
Caspase activity detection
The caspase-3 and caspase-9 activities were determined using commercial kits (Beyotime Institute of Biotechnology). The levels of antioxidant factors, including GPX, SOD, and GSH, were measured with ELISA kits purchased from the Beyotime Institute of Biotechnology. The experiments were performed in triplicate and repeated three times with similar results (Blackburn et al. 2017).
Flow cytometry for mROS
Mitochondrial ROS production was measured via flow cytometry as described by a previous study. Cells were washed three times in PBS and incubated with MitoSOX Red Mitochondrial Superoxide Indicator (Molecular Probes, USA) for 30 min at 37 °C/5% CO2 in the dark (Zhang et al. 2016). After incubation, the cells were washed three times in PBS at room temperature, and the mitochondrial ROS production was measured via flow cytometry.
Transfection
siRNA transfection was used to suppress Sirt3 and Fis1 expression. Two independent siRNAs against Sirt3 (siRNA1-Sirt3 and siRNA2-Sirt3, Yangzhou Ruibo Biotech Co., Ltd., Yangzhou, China) and siRNA against Fis1 (siRNA-Fis1, Yangzhou Ruibo Biotech Co., Ltd., Yangzhou, China) were transfected into the tongue cancer cell lines using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol (Das et al. 2017). The negative control group was transfected with a negative control siRNA. The transfection was performed for approximately 48 h. Then, Western blotting was performed to verify the knockdown efficiency after harvesting the transfected cells. The experiments were performed in triplicate and repeated three times with similar results (Zhou et al. 2018e).
Statistical analysis
All results presented in this study were acquired from at least three independent experiments. The statistical analyses were performed using SPSS 16.0 (SPSS, Inc., Chicago, IL, USA). All results in the present study were analyzed by one-way analysis of variance, followed by Tukey’s test. p < 0.05 was considered statistically significant.
Results
Sirt3 silencing promotes apoptosis in tongue cancer cells
To investigate the role of Sirt3 in tongue cancer apoptosis, two independent siRNAs were transfected into the SCC-15 and SCC-9 cells. The knockdown efficiency was confirmed via Western blotting (Fig. 1a–d). Then, cell viability was measured using the MTT assay. Compared to that of the control group, Sirt3 siRNA transfection significantly reduced the cell viability of both the SCC-15 and SCC-9 cells (Fig. 1e, f). Notably, Z-VAD-FMK treatment could abolish the pro-apoptotic effects of Sirt3 siRNAs on tongue cancer, indicating that Sirt3 mediates cell apoptosis in tongue cancer. To evaluate whether the reduction in cell viability was attributable to increased apoptosis, TUNEL staining was performed. As shown in Fig. 1g–i, compared to that of the control group, loss of Sirt3 elevated the ratio of TUNEL-positive cells, suggesting that Sirt3 knockdown promoted cell apoptosis in both the SCC-15 and SCC-9 cells. This result was confirmed via evaluating the caspase-3 activity. As shown in Fig. 1j, k, Sirt3 deficiency increased caspase-3 activity in both the SCC-15 and SCC-9 cells. In addition to caspase-3 activation, cleaved caspase-3 and PARP (the substrate of caspase-3 activation) expression were upregulated in response to Sirt3 siRNA transfection (Fig. 1l–q). Altogether, these data indicated that Sirt3 knockdown promoted apoptosis in tongue cancer cells.
Fig. 1.

Sirt3 knockdown induces apoptosis in tongue cancer cells in vitro. a–d Two independent Sirt3 siRNAs were transfected into SCC-15 and SCC-9 tongue cancer cells. Western blotting was used to observe the knockdown efficiency. e–f The MTT assay was used for cell viability detection. Two independent Sirt3 siRNAs were transfected into SCC-15 and SCC-9 tongue cancer cells. g–i TUNEL staining for apoptotic cells. Green dots were recorded, and the ratio of TUNEL-positive cells was evaluated to reflect cell apoptosis. j and k Caspase-3 activity was measured via ELISA. Two independent siRNAs were transfected into SCC-15 and SCC-9 tongue cancer cells. l–q Western blotting was performed to analyze the expression of pro-apoptotic proteins, such as cleaved caspase-3 and PARP. *p < 0.05 vs. si-ctrl (control siRNA); #p < 0.05 vs. si2-sirt3 (Sirt3 siRNA1)
Sirt3 silencing activates mitochondrial apoptosis
Subsequently, we explored the mechanism by which Sirt3 knockdown induced apoptosis in tongue cancer cells. Previous studies have suggested that Sirt3 is a mitochondrial NAD+-dependent deacetylase that modulates mitochondrial homeostasis (Anamika et al. 2017; Parodi-Rullan et al. 2018). Based on this information, we investigated whether Sirt3 knockdown activated mitochondria-dependent apoptosis in tongue cancer. The early feature of mitochondrial apoptosis is a reduction in the mitochondrial membrane potential that reflects mitochondrial function, such as ATP production and oxidative stress (Fuhrmann and Brune 2017). Using JC-1 staining, which is a probe of the mitochondrial membrane potential, we found that Sirt3 deficiency reduced the mitochondrial potential compared to that of the control group (Fig. 2a–d). In light of the central role of the mitochondrial potential in ATP production, we measured total ATP production in the Sirt3-deleted cells. Compared to that of the control group, Sirt3 knockdown significantly alleviated ATP production in both the SCC-15 and SCC-9 cells (Fig. 2e, f). We also found that mitochondrial ROS production was significantly increased in response to Sirt3 knockdown as assessed via flow cytometry (Fig. 2g–j), indicating that Sirt3 deficiency induced mitochondrial oxidative injury.
Fig. 2.

Mitochondrial apoptosis is activated by Sirt3 knockdown. a–d Mitochondrial membrane potential was observed using JC-1 staining. e and f ATP production was measured in SCC-15 and SCC-9 cells. Two independent siRNAs were transfected into SCC-15 and SCC-9 tongue cancer cells. g–j Mitochondrial ROS production was analyzed using flow cytometry. The relative ROS content was recorded as a ratio to that of the control group. k–r Western blotting was performed to analyze the expression of mitochondrial apoptotic proteins. Two independent siRNAs were transfected into SCC-15 and SCC-9 tongue cancer cells. *p < 0.05 vs. si-ctrl (control siRNA)
The late feature of mitochondrial apoptosis is the activation of mitochondrial apoptotic proteins (Zhou et al. 2017a). With the help of Western blotting, we found that the pro-apoptotic factors related to mitochondrial apoptosis, such as caspase-9 and Bax, were significantly upregulated in the Sirt3-deleted cells (Fig. 2k–r). Conversely, the anti-apoptotic protein related to mitochondrial apoptosis, such as Bcl-2, was obviously downregulated in response to Sirt3 silencing (Fig. 2k–r). Altogether, our data illustrated that Sirt3 knockdown was accompanied by activation of mitochondrial apoptosis in tongue cancer.
Loss of Sirt3 triggers Fis1-related mitochondrial fission
Next, experiments were conducted to analyze the mechanism by which Sirt3 knockdown activated mitochondrial apoptosis. Previous studies have found a causal role of mitochondrial fission in initiating the caspase-9-related mitochondrial apoptosis pathways in several cancer types (Wang et al. 2018b). Based on this information, we examined whether Sirt3 knockdown induced mitochondrial apoptosis in a manner dependent on mitochondrial fission. To answer this question, the mitochondrial morphology was observed in response to Sirt3 knockdown in both the SCC-15 and SCC-9 cells. As shown in Fig. 3a–d, compared to that of the control group, loss of Sirt3 generated significant massive fragmentation of the mitochondria. Subsequently, the mitochondrial length was measured to quantify mitochondrial fission according to the method described in previous reports (Zhou et al. 2017e). As shown in Fig. 3A–D, compared to that in the control group, the length of the mitochondria decreased to ~ 2.3 μm after Sirt3 was silenced, reconfirming activation of mitochondrial fission under Sirt3 knockdown.
Fig. 3.

Sirt3 knockdown activates mitochondrial fission in a manner dependent on Fis1. a–d Immunofluorescence for mitochondria using a mitochondrial-specific antibody Tom-20. The average length of the mitochondria was measured, and this parameter was used to quantify mitochondrial fission. e–l Western blotting was applied to evaluate the expression of pro-fission proteins, including Drp1, Mff, and Fis1. Relative Fis1 expression was recorded in response to Sirt3 deletion. *p < 0.05 vs. si-ctrl (control siRNA)
Based on previous findings (Zhou et al. 2018a; Zhou et al. 2019), Fis1 is an indispensable factor that regulates mitochondrial division. In the present study, using the Western blotting assay, we found that Fis1 expression was significantly increased in response to Sirt3 knockdown in both the SCC-9 and SCC-15 cells (Fig. 3e–l). As a consequence of Fis1 upregulation, the expression of pro-fission proteins, such as Drp1 and Mff, were mostly increased in the Sirt3-silenced cells compared to those in the control group (Fig. 3e–l). This information highlighted the promotive effects of Sirt3 knockdown on mitochondrial fission.
Inhibition of Fis1-related mitochondrial fission reduces mitochondrial stress and promotes cell survival in tongue cancer cells
The above data validated the regulatory effects of Sirt3 on Fis1-related mitochondrial fission (Sigala et al. 2017). However, whether Fis1-related mitochondrial fission is required for the mitochondrial damage and cell apoptosis induced by Sirt3 silencing is unknown. To answer this question, Fis1 was knocked down using siRNA. The knockdown efficiency was confirmed via Western blotting (Fig. 4a–d). Then, mitochondrial stress was determined via analyzing ROS production and cyt-c liberation. The feature of mitochondrial stress is cyt-c liberation into the cytoplasm/nucleus due to mitochondrial membrane damage (Zhou et al. 2017a). Once translocated from the mitochondria into the nucleus, cyt-c would initiate mitochondrial apoptosis. Using an immunofluorescence assay, we found that nuclear cyt-c expression was increased in response to Sirt3 knockdown in both the SCC-9 and SCC-15 cells (Fig. 4e–h), confirming that Sirt3 knockdown contributed to cyt-c migration to the nucleus. However, Sirt3 deficiency-mediated cyt-c liberation was negated by Fis1 silencing. This result was also confirmed via Western blotting (Fig. 4j, k). Subsequently, mitochondrial ROS was analyzed via flow cytometry. As shown in Fig. 4m–o, compared with that of the control group, Sirt3 knockdown promoted ROS overloading, and this effect was abolished by the Fis1 siRNA. This information verified the necessary role played by Fis1-related mitochondrial fission in aggravating Sirt3-mediated mitochondrial stress.
Fig. 4.

Loss of Fis1-related mitochondrial fission attenuates mitochondrial damage and cell apoptosis. a–d Fis1 siRNA was transfected into SCC-15 and SCC-9 tongue cancer cells. Western blotting was used to analyze the knockdown efficiency. e–h Immunofluorescence for cyt-c liberation. Relative nuclear cyt-c expression was recorded as a ratio to that of the control group. Fis1 siRNA was transfected into SCC-15 and SCC-9 tongue cancer cells. i–l Western blotting was used to observe the translocation of cyt-c from mitochondria into cytoplasm. m–o Flow cytometry was performed to analyze the mitochondrial ROS levels in response to Fis1 deletion. p and q Caspase-9 activity was measured using ELISA. *p < 0.05 vs. si-ctrl (control siRNA); #p < 0.05 vs. si1-sirt3 (Sirt3 siRNA1)
To assess cell apoptosis, a caspase-9 activity assay was performed. As shown in Fig. 4p, q, compared to that of the control group, caspase-9 activity was significantly increased in response to Sirt3 knockdown. However, loss of Fis1 abrogated the pro-apoptotic property of Sirt3 knockdown in tongue cancer cells. Altogether, these results supported the functional importance of Fis1-related mitochondrial fission in triggering Sirt3-related mitochondrial stress and cancer cell apoptosis.
Sirt3 inhibits Fis1 via activating the MAPK-JNK signaling pathway
To this end, we investigated the molecular mechanisms by which Sirt3 modulated Fis1 in tongue cancer cells. According to the previous study (Blackburn et al. 2017; Gao et al. 2017), the MAPK-JNK signaling pathway was involved in mitochondrial fission management via modifying the expression of pro-fission factors, such as Mff (Jin et al. 2018). In the present study, we evaluated whether Sirt3 affected Fis1 expression via the MAPK-JNK pathway. Western blotting demonstrated that phosphorylated JNK (p-JNK) was significantly increased in response to Sirt3 knockdown that was indicative of JNK activation under Sirt3 silencing (Fig. 5a–f). Subsequently, SP600125, which is a JNK pathway blocker, was added to the Sirt3-knocked down cells. Then, the p-JNK and Fis1 expression levels were observed. As shown in Fig. 5a–f, after blockade of the JNK pathway, p-JNK expression was obviously downregulated and was accompanied by a decrease in Fis1 expression. This result was further verified via immunofluorescence. Compared to that of the control group, abundant Fis1 expression was observed in the Sirt3-deleted group (Fig. 5g–j). However, the Fis1 levels were rapidly downregulated in response to SP600125 (Fig. 5g–j), reconfirming that the MAPK-JNK signaling pathway was involved in Fis1 expression.
Fig. 5.

Sirt3 regulates Fis1 via the JNK signaling pathway. a–f Western blotting was used to analyze p-JNK and Fis1 expression. The JNK blocker SP600125 was used to prevent JNK activation in Sirt3-deleted cells. g–j Immunofluorescence assay for Fis1. The relative fluorescence intensity of Fis1 was measured. *p < 0.05 vs. si-ctrl (control siRNA); #p < 0.05 vs. si1-sirt3 (Sirt3 siRNA1)
The MAPK-JNK signaling pathway also affects mitochondrial function and cell survival
Lastly, we explored whether the MAPK-JNK signaling pathway participated in mitochondrial stress and cancer cell apoptosis (Conradi et al. 2017). To address this question, a pathway blocker was used. As shown in Fig. 6a, b, compared to that of the control group, Sirt3 knockdown elevated caspase-9 activity, and this effect was nullified by SP600125. Total ATP production was also repressed by Sirt3 deletion, and this effect was reversed by SP600125 (Fig. 6c, d). These results indicated that inhibition of the JNK pathway alleviated Sirt3-mediated mitochondrial stress. To assess cell survival, the MTT and LDH release assays were performed. As shown in Fig. 6e, f, compared to that of the control group, cell viability as assessed by MTT was significantly reduced in response to Sirt3 knockdown and was reversed to near-normal levels by SP600125 treatment. Moreover, cell death as evaluated by the LDH release assay was enhanced by Sirt3. However, SP600125 treatment attenuated Sirt3-mediated cell death (Fig. 6g, h). Similarly, the number of TUNEL-positive cells was significantly increased in the Sirt3-deleted cells, and this effect was abolished by SP600125 (Fig. 6i–l). Altogether, these results illustrated that the MAPK-JNK pathway was involved in Sirt3-related mitochondrial stress and cell apoptosis in tongue cancer cells.
Fig. 6.

The JNK signaling pathway participates in mitochondrial stress and cell apoptosis. a and b Caspase-9 activity was measured by ELISA. SP600125 was used to inhibit JNK activation. c and d ATP production was determined in response to JNK inhibition using SP600125. e and f Cell viability was measured via the MTT assay, and SP600125 was used to inhibit JNK activation. g and h The LDH release assay was performed to analyze cell death via ELISA. i–l TUNEL staining for apoptotic cell and the number of apoptosis was recorded in response to SP600125. *p < 0.05 vs. si-ctrl (control siRNA); #p < 0.05 vs. si1-sirt3 (Sirt3 siRNA1)
Discussion
In the present study, our results identified the anti-tumor effects and mechanisms of Sirt3 knockdown in tongue cancer. Two types of tongue cancer cell lines were used in the present study, and two independent siRNAs targeting Sirt3 were transfected into these cells. Using an array of functional investigations, we demonstrated that Sirt3 knockdown promoted cell apoptosis via inducing mitochondrial stress. Our results further highlighted the necessary role of Fis1-related mitochondrial fission in mediating mitochondrial stress, because inhibition of Fis1-related mitochondrial fission sustained mitochondrial function and repressed cancer cell apoptosis. To this end, the molecular analysis illustrated that Sirt3 modulated Fis1 via the MAPK-JNK pathway that was also involved in Sirt3-mediated mitochondrial stress and cancer death. Collectively, this study is the first to explore the actions of Sirt3 and Fis1-related mitochondrial fission in tongue cancer cell viability (Fig. 7). Based on our results, the Sirt3-JNK-Fis1 signaling pathway should be considered as a potential target to treat tongue cancer via inducing cell apoptosis and mitochondrial stress (Fukumoto et al. 2018). Notably, more investigations in animal research and clinical practice are required to obtain more complete elucidation of the properties of Sirt3-modified Fis1 mitochondrial fission in tongue cancer death.
Fig. 7.
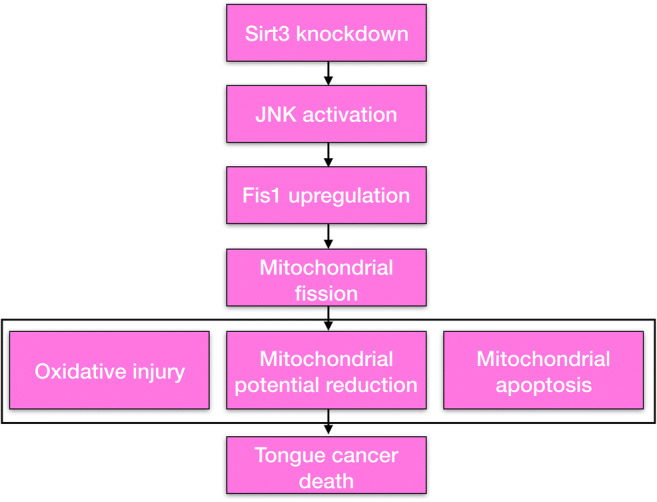
Loss of Sirt3 promoted tongue cancer cell death in a manner dependent on mitochondrial apoptosis. Sirt3 knockdown activated mitochondrial stress via triggering Fis1-related mitochondrial fission. Mitochondrial oxidative stress, energy metabolism disorder, mitochondrial cyt-c liberation, and mitochondrial apoptosis activation were observed after Sirt3 silencing. At the molecular levels, Sirt3 modulated Fis1 expression via the JNK signaling pathway
Strong evidence supports the pro-apoptotic role of mitochondrial fission in initiating the mitochondrial apoptosis pathway in various cancer types (Zhou et al. 2017b; Zhou et al. 2017c). For example, in lung cancer, ovarian cancer, breast cancer, liver cancer, pancreatic cancer, and colorectal cancer (Wang et al. 2018b), mitochondrial fission has been well recognized as a major pro-apoptotic factor via regulation of mitochondrial homeostasis, ER stress, and the inflammatory response (Zhou et al. 2017e). Mechanistically, excessive mitochondrial fission produces massive mitochondrial debris that contains a lower mitochondrial membrane potential and fragmentary DNA (Zhou et al. 2017a; Zhou et al. 2018b). These damaged mitochondria cannot generate sufficient ATP to fuel cell metabolism but instead liberate pro-apoptotic factors into the cytoplasm/nucleus to initiate the mitochondria-related apoptosis pathway (Yan et al. 2018b; Zhao et al. 2018). For example, in endothelial oxidative injury, mitochondrial fission impairs mitochondrial DNA transcription and replication and consequently represses mitochondrial respiratory complex expression (Zhou et al. 2017a), leading to ATP depletion. Additionally, in cardiac ischemia reperfusion injury (Zhou et al. 2018d), abnormal mitochondrial fission interrupts mitophagy and promotes mitochondrial calcium overloading, which obligates cardiomyocytes to undergo death. In rectal cancer (Li et al. 2017), aberrant mitochondrial fission mediates cellular oxidative stress that blunts cancer migration. In the present study, our results indicated that mitochondrial fission was associated with mitochondrial apoptosis and mitochondrial ROS overproduction (Koopman et al. 2017; Zhou et al. 2017d); inhibition of mitochondrial fission sustained mitochondrial metabolism and attenuated mitochondrial damage, finally promoting cancer cell survival (Souza et al. 2018). Therefore, our results combined with those of previous studies lay the foundation for understanding the molecular features of mitochondrial fission in mitochondrial damage and substantiate the sufficiency and necessity of mitochondrial fission in inducing cancer death (Koentges et al. 2017; Reddy et al. 2018).
In the present study, we provided a piece of evidence to support the regulatory effects of Fis1 in inducing mitochondrial fission in tongue cancer. This finding was similar to those of the previous studies (Shi et al. 2018; Zhu et al. 2018b). For example, lipopolysaccharide-induced oxidative injury in alveolar macrophages was associated with Fis1 activation and subsequent mitochondrial fission (Kiel et al. 2017; Lagerweij et al. 2017). Moreover, Fis1-related mitochondrial fission was also involved in the self-renewal of acute myeloid leukemia stem cells (Lin et al. 2017; Zhou et al. 2017b). In bladder cancer cells, activation of Fis1-related mitochondrial fission impairs cancer invasion and attenuates tumor chemoresistance (Hooshdaran et al. 2017; Jokinen et al. 2017). Importantly, a recent study has reported that the sensitivity of tongue cancer to cisplatin is closely regulated by miR-483-5p via modulating Fis1 expression (Hambright et al. 2017; Korbel et al. 2018). In the present study, we observed a direct role of Fis1-related mitochondrial fission in initiating mitochondrial apoptosis in tongue cells (Gonzalez et al. 2018; Morell et al. 2017). This finding helps further understanding of the causal action of Fis1 in tongue cancer cell viability. From a therapeutic perspective, clinicians should bear in mind that activation of Fis1-related mitochondrial fission is of utmost importance when designing anti-tumor therapies for tongue cancer cells (Fuhrmann and Brune 2017; Iggena et al. 2017).
Herein, we reported that Sirt3 modulated Fis1-related mitochondrial fission via the MAPK-JNK pathway. The relationship between the JNK pathway and Fis1-related mitochondrial fission has been extensively explored. In acute myocardial ischemia reperfusion injury and chronic heart fibrosis, activated JNK promotes mitochondrial fission and cardiomyocyte death. Similarly, in liver cancer, rectal cancer, gastric cancer, endometriosis, and cervical cancer, the JNK pathway has been identified as the upstream factor for mitochondrial fission activation. In agreement with previous studies, we also found that inhibition of the JNK pathway repressed Fis1 expression and attenuated mitochondrial fission. These results define the JNK pathway as a tumor suppressor that acts by triggering mitochondrial fission with potential implications for new approaches to tongue cancer therapy.
Altogether, the present study identified the Sirt3-JNK-Fis1-mitochondrial fission axis as a potential candidate target for new therapies against tongue cancer. Loss of Sirt3 activated the JNK pathway and then upregulated Fis1 expression. Increased Fis1 expression triggered mitochondrial fission that exacerbated mitochondrial damage, ultimately initiating the mitochondria-dependent apoptosis pathway in tongue cancer. Notably, a previous study has found that Sirt3 activation promotes Fis1 upregulation in the development of osteoarthritis (Wang et al. 2018a). This conclusion seems to oppose our observations. Thereby, our data, combined with the previous studies (Wang et al. 2018a), support Sirt3 having multiple effects on mitochondrial stress and its potential functions depend on different cell types and a cell-specific context. Although our present study presents a new signaling pathway responsible for tongue cancer cell death, additional investigations using animal studies or human samples are needed to validate our concept and help transform basic research findings into clinical application.
Funding
This research was supported by the Scientific Research Common Program of Beijing Municipal Commission of Education (KM201710025020).
Compliance with ethical standards
Competing interests
The authors declare that they have no competing interests.
Footnotes
This article has been retracted. Please see the retraction notice for more detail:https://doi.org/10.1007/s12192-021-01227-z
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
8/25/2021
A Correction to this paper has been published: 10.1007/s12192-021-01227-z
References
- Ackermann M, Kim YO, Wagner WL, Schuppan D, Valenzuela CD, Mentzer SJ, Kreuz S, Stiller D, Wollin L, Konerding MA. Effects of nintedanib on the microvascular architecture in a lung fibrosis model. Angiogenesis. 2017;20:359–372. doi: 10.1007/s10456-017-9543-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anamika KA, Acharjee P, Acharjee A, Trigun SK. Mitochondrial SIRT3 and neurodegenerative brain disorders. J Chem Neuroanat. 2017;95:43–53. doi: 10.1016/j.jchemneu.2017.11.009. [DOI] [PubMed] [Google Scholar]
- Blackburn NJR, Vulesevic B, McNeill B, Cimenci CE, Ahmadi A, Gonzalez-Gomez M, Ostojic A, Zhong Z, Brownlee M, Beisswenger PJ, Milne RW, Suuronen EJ. Methylglyoxal-derived advanced glycation end products contribute to negative cardiac remodeling and dysfunction post-myocardial infarction. Basic Res Cardiol. 2017;112:57. doi: 10.1007/s00395-017-0646-x. [DOI] [PubMed] [Google Scholar]
- Conradi LC, Brajic A, Cantelmo AR, Bouché A, Kalucka J, Pircher A, Brüning U, Teuwen LA, Vinckier S, Ghesquière B, Dewerchin M, Carmeliet P. Tumor vessel disintegration by maximum tolerable PFKFB3 blockade. Angiogenesis. 2017;20:599–613. doi: 10.1007/s10456-017-9573-6. [DOI] [PubMed] [Google Scholar]
- Das N, Mandala A, Naaz S, Giri S, Jain M, Bandyopadhyay D, Reiter RJ, Roy SS (2017) Melatonin protects against lipid-induced mitochondrial dysfunction in hepatocytes and inhibits stellate cell activation during hepatic fibrosis in mice. J Pineal Res 62. 10.1111/jpi.12404 [DOI] [PubMed]
- Fuhrmann DC, Brune B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. 2017;12:208–215. doi: 10.1016/j.redox.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto M, Kondo K, Uni K, Ishiguro T, Hayashi M, Ueda S, Mori I, Niimi K, Tashiro F, Miyazaki S, Miyazaki JI, Inagaki S, Furuyama T. Tip-cell behavior is regulated by transcription factor FoxO1 under hypoxic conditions in developing mouse retinas. Angiogenesis. 2018;21:203–214. doi: 10.1007/s10456-017-9588-z. [DOI] [PubMed] [Google Scholar]
- Gadicherla AK, Wang N, Bulic M, Agullo-Pascual E, Lissoni A, de Smet M, Delmar M, Bultynck G, Krysko DV, Camara A, Schlüter KD, Schulz R, Kwok WM, Leybaert L. Mitochondrial Cx43 hemichannels contribute to mitochondrial calcium entry and cell death in the heart. Basic Res Cardiol. 2017;112:27. doi: 10.1007/s00395-017-0618-1. [DOI] [PubMed] [Google Scholar]
- Gao Y, Xiao X, Zhang C, Yu W, Guo W, Zhang Z, Li Z, Feng X, Hao J, Zhang K, Xiao B, Chen M, Huang W, Xiong S, Wu X, Deng W (2017) Melatonin synergizes the chemotherapeutic effect of 5-fluorouracil in colon cancer by suppressing PI3K/AKT and NF-kappaB/iNOS signaling pathways. J Pineal Res 62. 10.1111/jpi.12380 [DOI] [PubMed]
- Gonzalez NR, Liou R, Kurth F, Jiang H, Saver J. Antiangiogenesis and medical therapy failure in intracranial atherosclerosis. Angiogenesis. 2018;21:23–35. doi: 10.1007/s10456-017-9578-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambright WS, Fonseca RS, Chen L, Na R, Ran Q. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol. 2017;12:8–17. doi: 10.1016/j.redox.2017.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooshdaran B, Kolpakov MA, Guo X, Miller SA, Wang T, Tilley DG, Rafiq K, Sabri A. Dual inhibition of cathepsin G and chymase reduces myocyte death and improves cardiac remodeling after myocardial ischemia reperfusion injury. Basic Res Cardiol. 2017;112:62. doi: 10.1007/s00395-017-0652-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iggena D, Winter Y, Steiner B (2017) Melatonin restores hippocampal neural precursor cell proliferation and prevents cognitive deficits induced by jet lag simulation in adult mice. J Pineal Res 62. 10.1111/jpi.12397 [DOI] [PubMed]
- Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma S, Zhu H, Ren J, Zhou H. DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol. 2018;14:576–587. doi: 10.1016/j.redox.2017.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, Kahn CR. Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc Natl Acad Sci U S A. 2011;108:14608–14613. doi: 10.1073/pnas.1111308108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokinen R, Pirnes-Karhu S, Pietilainen KH, Pirinen E. Adipose tissue NAD(+)-homeostasis, sirtuins and poly (ADP-ribose) polymerases -important players in mitochondrial metabolism and metabolic health. Redox Biol. 2017;12:246–263. doi: 10.1016/j.redox.2017.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyanaraman B. Teaching the basics of cancer metabolism: developing antitumor strategies by exploiting the differences between normal and cancer cell metabolism. Redox Biol. 2017;12:833–842. doi: 10.1016/j.redox.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel AM, Goodwill AG, Noblet JN, Barnard AL, Sassoon DJ, Tune JD. Regulation of myocardial oxygen delivery in response to graded reductions in hematocrit: role of K(+) channels. Basic Res Cardiol. 2017;112:65. doi: 10.1007/s00395-017-0654-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koentges C, Pepin ME, Müsse C, Pfeil K, Alvarez SVV, Hoppe N, Hoffmann MM, Odening KE, Sossalla S, Zirlik A, Hein L, Bode C, Wende AR, Bugger H. Gene expression analysis to identify mechanisms underlying heart failure susceptibility in mice and humans. Basic Res Cardiol. 2017;113:8. doi: 10.1007/s00395-017-0666-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman CD, Zimmermann WH, Knopfel T, de Boer TP. Cardiac optogenetics: using light to monitor cardiac physiology. Basic Res Cardiol. 2017;112:56. doi: 10.1007/s00395-017-0645-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel C, Gerstner MD, Menger MD, Laschke MW. Notch signaling controls sprouting angiogenesis of endometriotic lesions. Angiogenesis. 2018;21:37–46. doi: 10.1007/s10456-017-9580-7. [DOI] [PubMed] [Google Scholar]
- Lagerweij T, Dusoswa SA, Negrean A, Hendrikx EML, de Vries HE, Kole J, Garcia-Vallejo JJ, Mansvelder HD, Vandertop WP, Noske DP, Tannous BA, Musters RJP, van Kooyk Y, Wesseling P, Zhao XW, Wurdinger T. Optical clearing and fluorescence deep-tissue imaging for 3D quantitative analysis of the brain tumor microenvironment. Angiogenesis. 2017;20:533–546. doi: 10.1007/s10456-017-9565-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Jung YH, Choi GE, Ko SH, Lee SJ, Lee SH, Han HJ. BNIP3 induction by hypoxia stimulates FASN-dependent free fatty acid production enhancing therapeutic potential of umbilical cord blood-derived human mesenchymal stem cells. Redox Biol. 2017;13:426–443. doi: 10.1016/j.redox.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, He F, Zhao X, Zhang Y, Chu X, Hua C, Qu Y, Duan Y, Ming L. YAP inhibits the apoptosis and migration of human rectal cancer cells via suppression of JNK-Drp1-mitochondrial fission-HtrA2/Omi pathways. Cell Physiol Biochem. 2017;44:2073–2089. doi: 10.1159/000485946. [DOI] [PubMed] [Google Scholar]
- Li R, Xin T, Li D, Wang C, Zhu H, Zhou H. Therapeutic effect of sirtuin 3 on ameliorating nonalcoholic fatty liver disease: the role of the ERK-CREB pathway and Bnip3-mediated mitophagy. Redox Biol. 2018;18:229–243. doi: 10.1016/j.redox.2018.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Hoffmann K, Gao C, Petrulionis M, Herr I, Schemmer P (2017) Melatonin promotes sorafenib-induced apoptosis through synergistic activation of JNK/c-jun pathway in human hepatocellular carcinoma. J Pineal Res 62. 10.1111/jpi.12398 [DOI] [PubMed]
- Morell M, Burgos JI, Gonano LA, Vila Petroff M. AMPK-dependent nitric oxide release provides contractile support during hyperosmotic stress. Basic Res Cardiol. 2017;113:7. doi: 10.1007/s00395-017-0665-7. [DOI] [PubMed] [Google Scholar]
- Parodi-Rullan RM, Chapa-Dubocq XR, Javadov S. Acetylation of mitochondrial proteins in the heart: the role of SIRT3. Front Physiol. 2018;9:1094. doi: 10.3389/fphys.2018.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryds K, Nielsen RR, Jorsal A, Hansen MS, Ringgaard S, Refsgaard J, Kim WY, Petersen AK, Bøtker HE, Schmidt MR. Effect of long-term remote ischemic conditioning in patients with chronic ischemic heart failure. Basic Res Cardiol. 2017;112:67. doi: 10.1007/s00395-017-0658-6. [DOI] [PubMed] [Google Scholar]
- Reddy KRK, Dasari C, Duscharla D, Supriya B, Ram NS, Surekha MV, Kumar JM, Ummanni R. Dimethylarginine dimethylaminohydrolase-1 (DDAH1) is frequently upregulated in prostate cancer, and its overexpression conveys tumor growth and angiogenesis by metabolizing asymmetric dimethylarginine (ADMA) Angiogenesis. 2018;21:79–94. doi: 10.1007/s10456-017-9587-0. [DOI] [PubMed] [Google Scholar]
- Schluter KD, Wolf A, Weber M, Schreckenberg R, Schulz R. Oxidized low-density lipoprotein (oxLDL) affects load-free cell shortening of cardiomyocytes in a proprotein convertase subtilisin/kexin 9 (PCSK9)-dependent way. Basic Res Cardiol. 2017;112:63. doi: 10.1007/s00395-017-0650-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu S, Zhu P, Wang W, Zhou H. Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 2018;14:59–71. doi: 10.1016/j.redox.2017.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigala F, Efentakis P, Karageorgiadi D, Filis K, Zampas P, Iliodromitis EK, Zografos G, Papapetropoulos A, Andreadou I. Reciprocal regulation of eNOS, H2S and CO-synthesizing enzymes in human atheroma: correlation with plaque stability and effects of simvastatin. Redox Biol. 2017;12:70–81. doi: 10.1016/j.redox.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza LEB, Beckenkamp LR, Sobral LM, Fantacini DMC, Melo FUF, Borges JS, Leopoldino AM, Kashima S, Covas DT. Pre-culture in endothelial growth medium enhances the angiogenic properties of adipose-derived stem/stromal cells. Angiogenesis. 2018;21:15–22. doi: 10.1007/s10456-017-9579-0. [DOI] [PubMed] [Google Scholar]
- Tenreiro MM, Correia ML, Brito MA. Endothelial progenitor cells in multiple myeloma neovascularization: a brick to the wall. Angiogenesis. 2017;20:443–462. doi: 10.1007/s10456-017-9571-8. [DOI] [PubMed] [Google Scholar]
- Turner CJ, Badu-Nkansah K, Hynes RO. Endothelium-derived fibronectin regulates neonatal vascular morphogenesis in an autocrine fashion. Angiogenesis. 2017;20:519–531. doi: 10.1007/s10456-017-9563-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Beijnum JR, Nowak-Sliwinska P, van Berkel M, Wong TJ, Griffioen AW. A genomic screen for angiosuppressor genes in the tumor endothelium identifies a multifaceted angiostatic role for bromodomain containing 7 (BRD7) Angiogenesis. 2017;20:641–654. doi: 10.1007/s10456-017-9576-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wang K, Huang C, Lin D, Zhou Y, Wu Y, Tian N, Fan P, Pan X, Xu D, Hu J, Zhou Y, Wang X, Zhang X. SIRT3 activation by dihydromyricetin suppresses chondrocytes degeneration via maintaining mitochondrial homeostasis. Int J Biol Sci. 2018;14:1873–1882. doi: 10.7150/ijbs.27746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Sun X, Ji K, du L, Xu C, He N, Wang J, Liu Y, Liu Q. Sirt3-mediated mitochondrial fission regulates the colorectal cancer stress response by modulating the Akt/PTEN signalling pathway. Biomed Pharmacother. 2018;105:1172–1182. doi: 10.1016/j.biopha.2018.06.071. [DOI] [PubMed] [Google Scholar]
- Xu X, Zhang P, Kwak D, Fassett J, Yue W, Atzler D, Hu X, Liu X, Wang H, Lu Z, Guo H, Schwedhelm E, Böger RH, Chen P, Chen Y. Cardiomyocyte dimethylarginine dimethylaminohydrolase-1 (DDAH1) plays an important role in attenuating ventricular hypertrophy and dysfunction. Basic Res Cardiol. 2017;112:55. doi: 10.1007/s00395-017-0644-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan H, Qiu C, Sun W, Gu M, Xiao F, Zou J, Zhang L. Yap regulates gastric cancer survival and migration via SIRT1/Mfn2/mitophagy. Oncol Rep. 2018;39:1671–1681. doi: 10.3892/or.2018.6252. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Yan H, Xiao F, Zou J, Qiu C, Sun W, Gu M, Zhang L. NR4A1-induced increase in the sensitivity of a human gastric cancer line to TNFalpha-mediated apoptosis is associated with the inhibition of JNK/Parkin-dependent mitophagy. Int J Oncol. 2018;52:367–378. doi: 10.3892/ijo.2017.4216. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Zhai M, Li B, Duan W, Jing L, Zhang B, Zhang M, Yu L, Liu Z, Yu B, Ren K, Gao E, Yang Y, Liang H, Jin Z, Yu S (2017) Melatonin ameliorates myocardial ischemia reperfusion injury through SIRT3-dependent regulation of oxidative stress and apoptosis. J Pineal Res 63. 10.1111/jpi.12419 [DOI] [PubMed]
- Zhang Y, Zhou H, Wu W, Shi C, Hu S, Yin T, Ma Q, Han T, Zhang Y, Tian F, Chen Y. Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca(2+)-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic Biol Med. 2016;95:278–292. doi: 10.1016/j.freeradbiomed.2016.03.035. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Ye M, Yang W, Wang M, Li M, Gu C, Zhao L, Zhang Z, Han W, Fan W, Meng Y. Effect of Mst1 on endometriosis apoptosis and migration: role of Drp1-related mitochondrial fission and Parkin-required mitophagy. Cell Physiol Biochem. 2018;45:1172–1190. doi: 10.1159/000487450. [DOI] [PubMed] [Google Scholar]
- Zhong W, Gao X, Wang S, Han K, Ema M, Adams S, Adams RH, Rosenblatt MI, Chang JH, Azar DT. Prox1-GFP/Flt1-DsRed transgenic mice: an animal model for simultaneous live imaging of angiogenesis and lymphangiogenesis. Angiogenesis. 2017;20:581–598. doi: 10.1007/s10456-017-9572-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, du W, Li Y, Shi C, Hu N, Ma S, Wang W, Ren J (2018a) Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res 64. 10.1111/jpi.12450 [DOI] [PubMed]
- Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P, Ma Q, Tian F, Chen Y (2017a) Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc 6. 10.1161/JAHA.116.005328 [DOI] [PMC free article] [PubMed]
- Zhou H, Li D, Zhu P, Hu S, Hu N, Ma S, Zhang Y, Han T, Ren J, Cao F, Chen Y (2017b) Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J Pineal Res 63. 10.1111/jpi.12438 [DOI] [PubMed]
- Zhou H, Shi C, Hu S, Zhu H, Ren J, Chen Y. BI1 is associated with microvascular protection in cardiac ischemia reperfusion injury via repressing Syk-Nox2-Drp1-mitochondrial fission pathways. Angiogenesis. 2018;21:599–615. doi: 10.1007/s10456-018-9611-z. [DOI] [PubMed] [Google Scholar]
- Zhou H, Wang J, Hu S, Zhu H, Toanc S, Ren J. BI1 alleviates cardiac microvascular ischemia-reperfusion injury via modifying mitochondrial fission and inhibiting XO/ROS/F-actin pathways. J Cell Physiol. 2019;234:5056–5069. doi: 10.1002/jcp.27308. [DOI] [PubMed] [Google Scholar]
- Zhou H, Wang J, Zhu P, Hu S, Ren J. Ripk3 regulates cardiac microvascular reperfusion injury: the role of IP3R-dependent calcium overload, XO-mediated oxidative stress and F-action/filopodia-based cellular migration. Cell Signal. 2018;45:12–22. doi: 10.1016/j.cellsig.2018.01.020. [DOI] [PubMed] [Google Scholar]
- Zhou H, Wang J, Zhu P, Zhu H, Toan S, Hu S, Ren J, Chen Y (2018d) NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2alpha. Basic Res Cardiol 113(23). 10.1007/s00395-018-0682-1 [DOI] [PubMed]
- Zhou H, Wang S, Hu S, Chen Y, Ren J. ER-mitochondria microdomains in cardiac ischemia-reperfusion injury: a fresh perspective. Front Physiol. 2018;9:755. doi: 10.3389/fphys.2018.00755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Wang S, Zhu P, Hu S, Chen Y, Ren J. Empagliflozin rescues diabetic myocardial microvascular injury via AMPK-mediated inhibition of mitochondrial fission. Redox Biol. 2017;15:335–346. doi: 10.1016/j.redox.2017.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q, Jin Q, Cao F, Tian F, Chen Y (2017d) Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res 63. 10.1111/jpi.12413 [DOI] [PMC free article] [PubMed]
- Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D, Hu S, Ren J, Cao F, Chen Y. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507. doi: 10.1016/j.redox.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2alpha-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018;25:1080–1093. doi: 10.1038/s41418-018-0086-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Zhang H, Wang H, Lutz AM, el Kaffas A, Tian L, Hristov D, Willmann JK. Early prediction of tumor response to bevacizumab treatment in murine colon cancer models using three-dimensional dynamic contrast-enhanced ultrasound imaging. Angiogenesis. 2017;20:547–555. doi: 10.1007/s10456-017-9566-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D, Zhou H, Chen Y. Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca(2+)]c/VDAC-[Ca(2+)]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones. 2018;23:101–113. doi: 10.1007/s12192-017-0827-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Hu S, Jin Q, Li D, Tian F, Toan S, Li Y, Zhou H, Chen Y. Ripk3 promotes ER stress-induced necroptosis in cardiac IR injury: a mechanism involving calcium overload/XO/ROS/mPTP pathway. Redox Biol. 2018;16:157–168. doi: 10.1016/j.redox.2018.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]