

Abstract
Protein kinases are a superfamily of enzymes that control a wide range of cellular functions. These enzymes share a highly conserved catalytic core that folds into a similar bilobar three-dimensional structure. One highly conserved region in the protein kinase core is the glycine-rich loop (or G-loop), a highly flexible loop that is characterized by a consensus GxGxxG sequence. The G-loop points toward the catalytic cleft and functions to bind and position ATP for phosphotransfer. Of note, in many protein kinases, the second and third glycine residues in the G-loop triad flank residues that can be targets for phosphorylation (Ser, Thr, or Tyr) or other post-translational modifications (ubiquitination, acetylation, O-GlcNAcylation, oxidation). There is considerable evidence that cyclin-dependent kinases are held inactive through inhibitory phosphorylation of the conserved Thr/Tyr residues in this position of the G-loop and that dephosphorylation by cellular phosphatases is required for CDK activation and progression through the cell cycle. This review summarizes literature that identifies residues in or adjacent to the G-loop in other protein kinases that are targets for functionally important post-translational modifications.
Keywords: protein kinase, phosphorylation, ATP-positioning G-loop, post-translational modifications, oxidative stress
1. INTRODUCTION
Protein kinases play a central role in a large number of physiologic processes and they are implicated in the pathogenesis of many diseases. These factors have fueled interest in (and considerable investment by) both academia and the pharmaceutical industry toward solving the three-dimensional structures of many of these enzymes 1. We now recognize that eukaryotic protein kinases fold into a highly conserved bilobar structure, with a smaller mainly β-stranded N-lobe connected by a short flexible hinge region to a larger mainly α-helical C-lobe (Figure 1A). ATP is sandwiched in a deep cleft between the N- and C-lobes of this kinase domain structure; the substrate-binding site also is located in the cleft between the two lobes (Figure 1B).
Figure 1: Ser/Thr Kinase Catalytic Domain Structure.
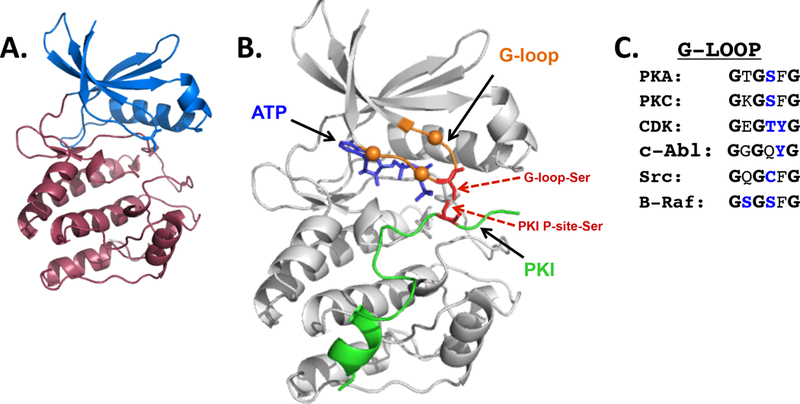
Figures are based upon the crystal structure of the mouse PKA catalytic domain complexed with ATP and the inhibitor peptide PKI (Protein Data Bank [PDB] accession number 1ATP). Panel A depicts a ribbon structure representation of the PKA N-lobe (residues 40–125, in blue) and C-lobe (residues 126–280, in red). The N- and C-terminal tails of PKA - that wrap around the catalytic core and serve as tethers to structure determinant in the catalytic core - are not depicted in this structure. Panel B: ATP (in blue) and the PKI inhibitor protein (in green, with an Ala-Ser substitution at the phosphoacceptor site [P-site] in red sticks) are introduced into the catalytic domain structure (in grey). The catalytic domain G-loop is highlighted in orange; G-loop glycine residues are depicted as orange spheres and the G-loop serine residue that is a target for phosphorylation is depicted as red sticks. The figure serves to emphasize the close proximity of the ATP-γ-P, the G-loop serine phosphorylation site, and the P-site on peptide substrate in the catalytic pocket. Panel C: An alignment of G-loop sequences that are targets for PTMs in PKA, PKC, CDK, c-Abl, Src, and B-Raf.
The structural and functional properties of many protein kinases are fine-tuned by phosphorylations at key sites within the kinase domain 2. For example, members of the AGC superfamily of eukaryotic protein kinases undergo a series of ordered ‘priming’ or regulatory phosphorylations at conserved motifs in the activation loop and C-terminus. Phosphorylations at these sites play a critical role to stabilize the catalytically competent conformation of the enzyme and regulate kinase activity. The notion that post-translational modifications at other positions strategically located in the structure can influence protein kinase function is not generally considered. This review focuses on recent studies indicating that the Gly-rich loop (or G-loop) constitutes an additional target for regulatory phosphorylation (or other regulatory post-translational modifications) on certain protein kinase enzymes.
The G-loop motif (characterized by the consensus GxGxxG sequence) connects the β1 and β2 strands of the N-lobe. The G-loop functions as a nucleotide-positioning motif to anchor ATP in an orientation that is optimal for catalysis and to shield bound nucleotide from solvent 3. The three glycine residues in the G-loop (residues that impose minimal steric interference) make the G-loop one of the most flexible elements in the catalytic core. These glycine residues are highly conserved across protein kinases, with the first glycine present in ~95% of kinases, the second in more than 99% of kinases, and the third conserved in ~85% of kinases (with replacement of this least conserved third position glycine largely restricted to small amino acids such as alanine or serine 3). Mutations localized to glycine residues in this triad typically disrupt G-loop conformation and/or sterically interfere with ATP binding and are not – or are only poorly – tolerated 4. Mutations at these sites that disrupt kinase activity have been implicated in certain human diseases. For example, a G75V substitution at the first position in the triad of RSK2 has been implicated in the pathogenesis of Coffin-Lowry syndrome (an X-linked disorder characterized by severe psychomotor retardation, facial and digital dysmorphisms, and progressive skeletal deformations 5) and Gly-Glu or Gly-Arg substitutions at this position in tropomyosin receptor kinase A (TRKA, the receptor tyrosine kinase for nerve growth factor) is implicated in congenital insensitivity to pain with anhidrosis (CIPA) syndrome 6, 7. A Gly-Arg substitution in protein kinase D1 has been implicated in syndromic-congenital heart disease (a disorder that presents with atrioventricular septal defects, developmental delay and limb abnormalities 8). Finally, a Gly-Val substitution at the third glycine in the triad in InsR disrupts activity and causes diabetes 9, a Gly-Arg substitution at this position in c-Kit inhibits activity and causes piebaldism (an autosomal dominant disorder caused by defective proliferation or migration of melanocytes from the neural crest during early development 10), and a Gly-Arg substitution at this position in the VEGFR3 (FLT4) gene that encodes vascular endothelial growth factor receptor 3 (VEGFR3) is implicated in Milroy disease (an autosomal dominant inherited form of primary lymphedema 11).
Residues between the second and third glycines in the GxGxxG triad extend outward toward the catalytic cleft, sit in close proximity to the substrate peptide recognition regions of the protein kinase, and also can be critical (Figure 1B). There is growing evidence that these G-loop residues (which have been characterized as hot spots for cancer-driving or drug resistance mutations) are targets for post-translational modifications (PTMs) that control the binding, positioning and/or recognition of substrate (Figure 1C). This review summarizes recent literature that identifies PTMs at the G loop as an underappreciated mechanism to regulate many protein kinase activities.
2. Cyclin-dependent kinases (CDKs)
CDKs are a family of serine/threonine kinases whose activity is influenced by a cyclin – a protein regulatory subunit named for its oscillatory pattern during the cell cycle. CDKs play a critical role in eukaryotic cell division and transcription. Detailed descriptions of the classification and function of the many CDK family members as regulators of cell-cycle progression have been published 12 and go beyond the scope of this review, which focuses on one specific aspect of CDK regulation involving G-loop phosphorylation.
CDKs were the first enzymes shown to be regulated through G-loop phosphorylation. CDKs are held inactive by phosphorylation at Tyr15 and to a lesser extent the adjacent Thr14 in the G-loop GEGTYG motif 13, 14. Thr14/Tyr15 phosphorylation (by the related Wee1 and Myt1 kinases) does not lead to major changes in CDK structure, but rather reduces the affinity of CDK for substrates and produces an unproductive binding mode for ATP 15. Progression through the cell cycle requires activation of dual specificity cell division cycle 25 (Cdc25) phosphatases that dephosphorylate these two residues and thereby activate CDK-cyclin complexes.
3. Protein kinase C (PKC)
3.1. PKCδ
PKCδ is a serine/threonine kinase that plays a key role in signal transduction pathways that control a wide range of normal cellular responses and also contribute to the pathogenesis of ischemia reperfusion injury 16, 17.
Like other PKC isoforms, PKCδ contains a highly conserved C-terminal catalytic domain and an N-terminal regulatory domain consisting of a lipid-binding C1 domain and a C2 domain. While most C2 domains function as membrane-targeting modules (being calcium-sensitive in the case of conventional PKCs, or calcium-insensitive in the case of novel PKCs), the PKCδ C2 domain is a topological variant that does not bind lipids, but rather functions as a phosphotyrosine (pY) binding motif that binds proteins with a consensus sequence (Y/F)-(S/A)-(V/I)-pY-(Q/R)-X-(Y/F) 18.
The conventional model for PKCδ activation by growth factor receptor pathways involves the generation of diacylglycerol, a second messenger that interacts with the lipid-binding C1 domain and anchors FL-PKCδ in an active conformation to membranes. This PKCδ activation mechanism accounts for the enzyme’s membrane-delimited actions, but it does not adequately explain the full repertoire of PKCδ’s actions in other cellular compartments. Recent studies address this dilemma by showing that PKCδ is activated via a distinct lipid-independent mechanism during oxidative stress 19. Oxidative stress leads to the activation of Src and Src-dependent phosphorylation of PKCδ at Tyr313 a residue in the V3 hinge region of the enzyme that is flanked by sequence that conforms to a C2 domain consensus-binding motif (VGI-Y313-QGF) 20. This results in an intramolecular interaction between the Tyr313-phosphorylated hinge region and the phosphotyrosine-binding C2 domain that controls PKCδ’s enzymology indirectly by inducing a long-range change in the phosphorylation status of Ser359, a site at the tip of the ATP-positioning G-loop in the kinase domain (GKGS359FG) 21. Further studies show that PKCδ is recovered from resting cells as a Ser359-phosphorylated enzyme that (when activated by lipid-cofactors) translocates to lipid membranes and acts as a serine kinase (i.e., shows a strong preference for substrates with a serine residue at the phosphoacceptor site). Oxidative stress triggers a redox-induced C2 domain-pTyr313 docking interaction that results in a long-range conformational change that facilitates PKCδ-Ser359 dephosphorylation and converts PKCδ into a lipid-independent Ser/Thr kinase; the redox-activated PKCδ enzyme is poised to phosphorylate substrates with either serine or threonine residues at the phosphoacceptor site throughout the cell – not just on lipid membranes. These results implicate G-loop phosphorylation at Ser359 as a dynamically-regulated mechanism that regulates PKCδ’s lipid-requirement for activation, changes its phosphoacceptor site specificity, and calibrates its activation of signaling pathways that contribute to cellular responses. This alternate mode for PKCδ activation during oxidative stress (that is presumed to contribute to the pathogenesis of ischemic injury) could be specifically targeted for therapeutic advantage.
3.2. Other PKCs and PKA.
The G-loop Ser phosphorylation site in PKCδ is highly conserved in other PKCs and in PKA; there is evidence that this site is phosphorylated in PKA 22 and tentative evidence that this site is O-GlycNAcylated (serves as an acceptor for a single O-linked β-N-acetyl glucosamine – or O-GlcNAc - sugar molecule) in PKCα, PKCβ, and/or PKCε 23 (Table 1). Studies to date, that examined the in vitro kinase activity of enzymes bearing single residue substitutions at the G-loop serine residue in PKA and PKCα, provide rather consistent evidence that G-loop phosphorylation plays a general role to inhibit PKA and PKCα activity 21, 24. Effects on substrate specificity remain uncertain, since the early evidence that a G-loop modification can also function to alter the substrate specificity of PKA was not subsequently substantiated 21, 24. The physiologic controls and in vivo functional consequences of G loop phosphorylation on these other enzymes have not been examined.
TABLE 1. KINASES WITH KNOWN G-LOOP POST-TRANSLATIONAL MODIFICATIONS.
| PROTEIN KINASE |
PTM at or adjacent to the G- loop |
Frequency with which the PTM has been identified |
References | Comments | |
|---|---|---|---|---|---|
| AGC Kinases | |||||
| PKA | TL GTGSFG | 6 | 22, 48–51 | ||
| TL GTGSFG | 2 | 52 | |||
| TL GTGSFG | 5 | 22, 48, 49, 53, 54 | |||
| PKCs | |||||
| PKCα | GKGSFG K | 2 | 55 | ||
| GKGSFG K | 1 | O-GlcNAcylation | |||
| GKGSFG K | 1 | 56 | Ubiquitination | ||
| PKCβ | GKGSFG K | 1 | 23 | O-GlcNAcylation | |
| PKCγ | GKGSFG K | 1 | Acetylation | ||
| GKGSFG K | 1 | 57 | ubiquitination | ||
| PKCδ | GKGSFG K | 3 | 21, 58,55 | ||
| GKGSFG K | 1 | 57 | ubiquitination | ||
| PKCε | GKGSFG K | 1 | 23 | O-GlcNAcylation | |
| GKGSFG K | 1 | Acetylation | |||
| PKCθ | GKGSFG K | 1 | 56 | ubiquitination | |
| PKCη | GKGSFG K | 2 | 55 | ||
| RSK Subfamily | |||||
| MSK1 | GTGAYG | 1 | |||
| MSK2 | KVL GTGAYG | 3 | 56, 59, 60 | ubiquitination | |
| KVL GTGAYG | 1 | 61 | |||
| KVL GTGAYG | 2 | 61 | |||
| p70S6K | GKGGYG K | 2 | 56, 62 | ubiquitination | |
| GKGGYG K | 2 | 56, 57 | ubiquitination | ||
| RSK1 | GQGSFG K | 3 | 56, 59 | ubiquitination | All RSK PTMs are located in the N-terminal kinase domain. PTMs have not been identified in RSK3. |
| GQGSFG K | 1 | Acetylation | |||
| RSK2 | GQGSFG K | 3 | 48, 63, 64 | ||
| KVL GQGSFG | 2 | 59 | ubiquitination | ||
| RSK4 | GQGSFG K | 1 | Acetylation | ||
| GQGSFG K | 3 | 56, 59 | ubiquitination | ||
| CAMK Group | |||||
| CaMK2 | |||||
| CaMK2α | GKGAFS | 11 | 57 | ubiquitination | |
| GKGAFS | 3 | 65–67 | |||
| CaMK2β | GKGAFS | 8 | 57 | ubiquitination | |
| GKGAFS | 2 | 66, 67 | |||
| CAMK2δ | GKGAFS | 1 | 68 | Acetylation | |
| GKGAFS | 2 | 66, 67 | |||
| CAMK2γ | GKGAFS | 1 | 68 | Acetylation | |
| GKGAFS | 6 | ubiquitination | |||
| GKGAFS | 2 | 66, 67 | |||
| AMPKα1 | TL GVGTFG | 3 | 54, 64, 69 | ||
| PIM1 | GSGGFG | 1 | |||
| PIM2 | GKGGFG | 25 | 56, 60, 70 | ubiquitination | |
| CMGC Group | |||||
| CDK1 | GEGTYG | 1846 | 13, 71–73 | ||
| GEGTYG | 4125 | 13, 71–77 | |||
| ERK2 | SYI GEGAYG | 1 | 47 | Not in ERK1 where S is replaced by Q | |
| SYI GEGAYG | 3 | Sequence conserved in ERK1, but phosphorylation not detected | |||
| GSK3β | GNGSFG | 2 | 48, 55 | not in GSK3α | |
| STE Group | |||||
| MEK1 | SEL GAGNGG | 4 | 78 | ||
| MEK2 | SEL GAGNGG | 1 | 79 | ||
| Tyrosine Kinases | |||||
| EGFRs | |||||
| EGFR | GSGAFG TVY | 8 | 48, 50, 51, 78, 80–82 | ||
| GSGAFG TVY | 150 | 78, 80, 83–95 | |||
| ErbB2 | GSGAFG TVY | 9 | 48, 50, 51, 78, 80, 81 | ||
| GSGAFG TVY | 148 | 78, 80, 83–88, 90, 92, 93, 95, 96 | |||
| ErbB3 | GSGVFG | 2 | 48, 53 | ||
| ErbB4 | GSGAFG TVY | 7 | 48, 50, 51, 78, 80, 81 | ||
| GSGAFG TVY | 147 | 78, 80, 83–88, 90, 92, 93, 95, 96 | |||
| InsR | GQGSFG | 5 | 43, 44, 46, 97, 98 | ||
| IGF1R | GQGSFG MVY | 1 | 48 | ||
| GQGSFG MVY | 3 | 83 | |||
| c-Abl | GGGQYG EVY | 288 | 33, 39, 40, 86, 99 | ||
| GGGQYG EVY | 339 | 32, 33, 39, 40, 54, 83, 86, 88, 92 | |||
| Alk | GHGAFG EVY | 22 | 100, 101 | ||
The data summarizes records curated by PhosphoSitePlus, a comprehensive online resource provided by Cell Signaling Technology. PhosphoSitePlus provides comprehensive information on post-translational modifications identified in studies that use both traditional low-throughput methods (i.e., studies that focus on few modification sites that are experimentally validated using robust techniques such as amino acid sequencing, phospho-specific antibodies, site-directed mutagenesis, dominant-negative constructs, etc.) as well as studies that use high throughput discovery mass spectrometry methods 102. Since the significance of a protein kinase G-loop PTM identified in only a single study using high-throughput MS methods is uncertain, these have generally not been included in the table, unless PTMs (phosphorylation, ubiquitination, acetylation, O-GlcNAcylation) are identified at multiple sites in or adjacent to the G-loop (or on the G-loops of multiple members of a single protein kinase subfamily).
4. RAF
Oncogenic mutations in B-RAF are identified in ~60% of malignant melanomas and they occur with moderate to high frequency in certain other cancers (papillary thyroid carcinomas, colorectal cancer, non-small cell lung cancer, and hairy cell leukemia 25). The V600E substitution in the activation loop (the 15–30 amino acid sequence flanked by the almost invariant DFG and APE motifs – a flexible loop that contributes to substrate recognition and functions to structure the enzyme for catalysis) accounts for ~80% of the cancer-associated B-Raf mutations, with other B-Raf mutations clustering in or adjacent to the activation loop or in the G-loop 26, 27. The V600E substitution increases B-Raf kinase activity and leads to constitutive activation of the mitogen-activated protein kinase (MAPK) pathway in cells 27. Structural studies suggest a mechanism for B-Raf activation by the V600E substitution. These studies identify a hydrophobic interaction between the DFG motif in the activation loop and the G-loop that stabilizes B-Raf in an inactive conformation. This model predicts that a V600E substitution (or other V600D, V600K, or V600R charge substitutions commonly found in tumors) disrupts the activation loop-G-loop interaction and lead to B-Raf activation – in a manner that mimics the effect of growth factor stimuli that activate B-Raf at least in part by promoting B-Raf autophosphorylation at adjacent residues in the highly conserved activation loop T599VKS602 motif 28.
Certain mutations in the G-loop also increase B-Raf activity, but the mechanism appears to be less straightforward, since Ala, Val or Ser substitutions at the 3rd glycine residue in the G-loop increase activity, but a Glu substitution at the identical site decreases catalytic activity 25, 27, 29. Hints that these phenotypes reflect an additional independent role of the G-loop to control B-Raf activity come from studies by Holderfield et al. showing that the two serine residues (GSGSFG) in the B-Raf G-loop are targets for inhibitory autophosphorylation. These authors proposed that under basal conditions B-Raf exists in an autoinhibited state as a result of constitutive G-loop autophosphorylation and that phosphatases reverse this phosphorylation during B-Raf activation 30. This model predicts that certain oncogenic mutations in the G-loop (adjacent to the autophosphorylation sites) activate B-Raf by preventing inhibitory G-loop autophosphorylations – effectively bypassing the built-in autoinhibitory brake on enzyme activity 30.
Finally, while B-Raf G-loop substitutions have been studied most intensively in models of clinically important cancers, it is worth noting that B-Raf G-loop mutations (S467A, F468S, and G469E) also have been identified in cardio-facio-cutaneous syndrome (a developmental disorder that has features similar to other Rasopathies – genetic syndromes such as Noonan’s syndrome or Costello syndrome that are due to mutations in genes that alter signaling through the Ras pathway 31).
5. Abelson murine leukemia virus (Abl) proto-oncogene
The c-Abl proto-oncogene is a ubiquitous non-receptor tyrosine kinase that is activated by extrinsic ligands such as growth factor receptors or intrinsic signals such as DNA damage or oxidative stress. c-Abl shuttles between the cytosolic and nuclear compartments, phosphorylates a diverse set of cellular substrates (including adaptor proteins, other kinases, cytoskeletal proteins, transcription factors, and chromatin modifiers) and controls signaling pathways that influence actin polymerization and cytoskeletal remodeling, cell adhesion and cell motility, transcriptional regulation, the DNA damage response, and cellular apoptosis.
The c-Abl G-loop contains a tyrosine residue (Tyr253) that can be phosphorylated 32, 33. While a single early study used a mutagenesis approach to show that Tyr253 phosphorylation functions to limit c-Abl activity (i.e., a Y253F substitution is sufficient to increase c-Abl activity 34), subsequent literature has focused primarily on the role of this tyrosine residue - and a second tyrosine residue adjacent to the G-loop (Tyr257) - in the context of the BCR-Abl oncoprotein, a fusion protein that results from a translocation between the BCR (breakpoint cluster region) on chromosome 22 and the ABL1 gene on chromosome 9. c-Abl tyrosine kinase activity is tightly regulated in normal cells, but the BCR-Abl fusion protein is a constitutively active kinase that plays a central role in the pathogenesis of essentially all cases of Philadelphia (Ph) chromosome-positive (Ph+) chronic myeloid leukemia (CML) as well as ~3–5% of pediatric acute lymphoblastic leukemia and 25% of adult acute lymphoblastic leukemias. The observation that the transforming ability of BCR-Abl is closely tied to its tyrosine kinase activity lead to the development of imatinib mesylate (also known as STI571 or Gleevec), a 2-phenylamino pyrimidine that targets the ATP binding site and stabilizes the Abl kinase-domain in an inactive conformation. Imatinib typically produces durable remissions in patients in the early more chronic phase of CML 35. However, advanced phase CML or blast crisis typically is characterized by the appearance of additional mutations in the BCR-Abl kinase domain that impair imatinib binding (i.e., mutations that result in escape from effective inhibition of BCR-Abl kinase activity 36). G-loop Y253F or Y253H mutations have been identified in a subset of patients with advanced CML and acquired imatinib resistance; these G-loop mutations (which eliminate the inhibitory break on enzyme activity) carry a dire prognosis as they typically are associated with a more aggressive phenotype and a shortened survival compared with other mutations 37, 38. A T315I substitution (at the gatekeeper residue in the kinase domain) is the more common mutation that confers resistance to imatinib as well as other 2nd generation Abl inhibitor compounds such as disatinib 36. Of note, the T315I drug-resistant mutation has been detected in imantinib-naïve CML patients in blast crisis, suggesting that this mutation confers an oncogenic fitness advantage over the wild-type BCR-Abl allele. The precise underlying mechanism remains somewhat uncertain. While there is evidence that the T315I mutant displays very high transformation potency but little-to-no kinase activity when tested against a panel of typical Abl substrates, other studies suggest that a T315I substitution may alter the enzyme’s substrate specificity, raising questions regarding the interpretation of studies that sample kinase activity with only a limited set of substrates 39. Of relevance to this review on G-loop modifications, there is evidence that the highly oncogenic T315I substitution leads to a high level of kinase domain autophosphorylation at Tyr257 (a site just N-terminal to the G-loop); c-Abl-T315I phosphorylation at Tyr253 is not detected, a finding that has been taken as tentative evidence that these phosphorylation events are mutually exclusive. These results have fueled speculation that an increase in Tyr257 phosphorylation forces an unfavorable G-loop conformation that limits access to Tyr253 kinases and prevents G-loop phosphorylation at Tyr253 (a PTM that inhibits kinase activity) This model predicts that strategies to enhance Tyr253 phosphorylation (by inhibiting the relevant phosphatase) or abrogate Tyr257 phosphorylation might be used to therapeutic advantage, particularly in patients harboring the drug-resistant T315I allele 39.
Finally, there is evidence that BCR-Abl contains several nuclear-localization signals but nevertheless is localized exclusively to the cytosol - in fact its oncogenicity requires its exclusion from the nucleus. Studies of mechanism indicate that BCR-Abl is retained in the cytosolic compartment through a mechanism that requires its kinase activity and specifically G-loop phosphorylation at Tyr253 and Tyr257 (i.e., that these post-translational modifications inhibit nuclear-localization signal function 40).
6. Redox inactivation of Src and other protein tyrosine kinases with G-loop cysteine residues.
Oxidative stress leads to changes in the activity of a large number of signaling responses. Most studies have focused on redox regulation of protein tyrosine phosphatases, enzymes that contain redox-sensitive catalytic cysteine residues in their activation site; oxidation of the free sulfhydryl groups on these cysteine residues disrupts protein tyrosine phosphatase activity. However, there also is evidence that reactive oxygen species (ROS) can alter protein tyrosine phosphorylation by directly regulating Src and certain other protein tyrosine kinase activities. Specifically, Src contains a cysteine residue at the tip of its G-loop (Cys277) that is prone to reversible oxidation. Kemble et al. used mutagenesis and biochemical approaches to show that this site within the G-loop is architecturally positioned to act as a redox-sensor and that oxidation of this strategically placed cysteine residue leads to a loss of Src activity 41. While the precise mechanism for oxidative inactivation of Src remains uncertain, there is evidence that Cys277 oxidation leads to formation of inactive Src homodimers or oxidized Src heterodimerization with Csk. This observation resonates with structural studies that identify a disulfide bond between Cys277 in c-Src and Cys290 in Csk 42, but the biological consequence of this disulfide linkage (between a Src molecule from one Csk-Src kinase substrate complex to a Csk in a different Csk-Src substrate complex) remains uncertain. It is worth noting that this redox-sensing mechanism is built into the G-loops of two other Src family kinase members (Yes and Fgr) and four members of the FGFR family of receptor tyrosine kinases, whereas it is not conserved in other protein tyrosine kinases (including other Src family kinases –that contain a Gln at the cognate position - or Csk).
7. Insulin Receptor (IR).
The insulin receptor (IR) is a tetrameric protein comprised of two extracellular insulin-binding α-subunits linked by disulfide bonds to two transmembrane β-subunits that display tyrosine kinase activity. Insulin binding to the IR-α-subunit leads to a conformational change that increases β-subunit tyrosine kinase activity and leads to β-subunit tyrosine transphosphorylation; this serves to further increase kinase activity toward exogenous substrates and it also provides docking sites for receptor binding-partners and substrates.
While tyrosine phosphorylation is essential for IR activation, serine/threonine phosphorylation of the IR in response to certain stimuli (including phorbol 12-myristate 13-acetate, cAMP, and insulin itself) provides an inhibitory brake that serves to prevent sustained/uncontrolled activation of downstream signaling pathways, severe perturbations of cellular metabolism, and excessive cellular growth responses that can contribute to tumorigenesis. Of note, Ser/Thr phosphorylation of the IR can become excessively increased, and lead to impaired IR activation, in various animal models of insulin resistance as well as certain insulin-resistant states in humans. While the IR β-subunit contains multiple serine phosphorylation sites, Ser994 in the G-loop has emerged as a functionally important inhibitory PTM. There is evidence that IR-Ser994 phosphorylation is altered in various models of insulin resistance 43 and mutagenesis studies support the conclusion that Ser994 phosphorylation is sufficient to disrupt basal and insulin-stimulated IR tyrosine kinase activity 44, 45. Efforts to identify the pathophysiologically relevant IR-Ser994 kinases have generally focused on PKC isoforms, which are increased in many insulin-resistant states and display in vivo IR-Ser994 kinase activity 43. However, there is limited evidence that TANK-binding kinase 1 (TBK1, an IκB kinase-related serine/threonine kinase that plays a role in certain inflammatory/immune responses) also can function as a IR-Ser994 kinase and thereby link inflammation to the pathogenesis of insulin resistance 46.
8. PTMs near the G-loop.
While the primary focus of this review is on PTMs within the G-loop, the notion that a PTM adjacent to the G-loop can force an unfavorable G-loop conformation and thereby impact on catalytic function has been evoked to explain phosphorylation-dependent regulation of BCR-Abl (see section 5). Of note, a review of the PTM data curated by PhosphositePlus provides many examples of PTMs (phosphorylation as well as lysine ubiquination or acetylation) at residues adjacent to the G-loop of many protein kinases (Table 1). In fact, there is evidence that ERK2 phosphorylation by serum and glucocorticoid-inducible protein kinase1 (SGK1) at Ser29 (a residue that sits just C-terminal to the G-loop) leads to increased MEK/ERK2 complex formation and enhanced ERK2 signaling; this mechanism is specific for ERK2, since this serine residue is replaced by a non-phosphorylatable Q in ERK1 47. The functional role of PTMs adjacent to the G-loop in other protein kinases remains to be determined.
9. CONCLUSION
This review summarizes literature that identifies an important role for the G-loop as a target of PTMs that influence the catalytic properties of many protein kinase enzymes. It is worth noting that with the exception of CDKs, the kinases and phosphatases that dynamically regulate G-loop phosphorylation – and could be targeted to prevent the development of clinical phenotypes - have not unambiguously been identified. This is relevant, since with the more widespread application of phosphoproteomic techniques to studies designed to identify molecular signatures for various cancer and other clinical disorders, G-loop phosphorylation is increasingly identified on many protein kinases. This suggests that PTMs localized to the G-loop may play a more general role in the regulation of protein kinase function and that strategies to manipulate G-loop phosphorylation might be harnessed for therapeutic advantage.
Acknowledgments:
This work is supported by the National Institutes of Health, National Heart, Blood, and Lung Institute grants HL112388 and HL123061.
ABBREVIATIONS:
- cAbl
Abelson murine leukemia virus proto-oncogene
- BCR
breakpoint cluster region
- CDK
cyclin-dependent kinase
- CML
chronic myeloid leukemia
- IR
Insulin Receptor
- O-GlcNAc
O-linked β-N-acetyl glucosamine
- PKA
protein kinase A
- PKC
protein kinase C
- PTM
post-translational modification
REFERENCES
- 1.Taylor SS, Zhang P, Steichen JM, Keshwani MM, Kornev AP. PKA: Lessons learned after twenty years. Biochim Biophys Acta. 2013;1834:1271–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Newton AC. Regulation of the ABC kinases by phosphorylation: Protein kinase C as a paradigm. Biochem J. 2003;370:361–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hemmer W, McGlone M, Tsigelny I, Taylor SS. Role of the glycine triad in the ATP-binding site of cAMP-dependent protein kinase. J Biol Chem. 1997;272:16946–16954 [DOI] [PubMed] [Google Scholar]
- 4.Torkamani A, Kannan N, Taylor SS, Schork NJ. Congenital disease SNPs target lineage specific structural elements in protein kinases. Proc Natl Acad Sci U S A. 2008;105:9011–9016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trivier E, De Cesare D, Jacquot S, Pannetier S, Zackai E, Young I, et al. Mutations in the kinase RSK-2 associated with Coffin-Lowry syndrome. Nature. 1996;384:567–570 [DOI] [PubMed] [Google Scholar]
- 6.Shaikh SS, Chen YC, Halsall SA, Nahorski MS, Omoto K, Young GT, et al. A comprehensive functional analysis of NTRK1 missense mutations causing hereditary sensory and autonomic neuropathy type IV (HSAN IV). Human mutation. 2017;38:55–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mardy S, Miura Y, Endo F, Matsuda I, Indo Y. Congenital insensitivity to pain with anhidrosis (CIPA): Effect of TRKA (NTRK1) missense mutations on autophosphorylation of the receptor tyrosine kinase for nerve growth factor. Hum Mol Genet. 2001;10:179–188 [DOI] [PubMed] [Google Scholar]
- 8.Sifrim A, Hitz MP, Wilsdon A, Breckpot J, Turki SH, Thienpont B, et al. Deciphering Developmental Disorders S, Hurles ME. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48:1060–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Odawara M, Kadowaki T, Yamamoto R, Shibasaki Y, Tobe K, Accili D, et al. Human diabetes associated with a mutation in the tyrosine kinase domain of the insulin receptor. Science. 1989;245:66–68 [DOI] [PubMed] [Google Scholar]
- 10.Syrris P, Malik NM, Murday VA, Patton MA, Carter ND, et al. Three novel mutations of the proto-oncogene KIT cause human piebaldism. Am J Med Genet. 2000;95:79–81 [DOI] [PubMed] [Google Scholar]
- 11.Gordon K, Spiden SL, Connell FC, Brice G, Cottrell S, Short J, et al. Flt4/VEGFR3 and Milroy disease: Novel mutations, a review of published variants and database update. Hum Mutat. 2013;34:23–31 [DOI] [PubMed] [Google Scholar]
- 12.Malumbres M Cyclin-dependent kinases. Genome Biol. 2014;15:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norbury C, Blow J, Nurse P. Regulatory phosphorylation of the p34cdc2 protein kinase in vertebrates. EMBO J. 1991;10:3321–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gu Y, Rosenblatt J, Morgan DO. Cell cycle regulation of CDK2 activity by phosphorylation of Thr160 and Tyr15. EMBO J. 1992;11:3995–4005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Welburn JP, Tucker JA, Johnson T, Lindert L, Morgan M, Willis A, et al. How tyrosine 15 phosphorylation inhibits the activity of cyclin-dependent kinase 2-cyclin A. J Biol Chem. 2007;282:3173–3181 [DOI] [PubMed] [Google Scholar]
- 16.Steinberg SF. Structural basis of protein kinase C isoform function. Physiol Rev. 2008;88:1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steinberg SF. Cardiac actions of protein kinase C isoforms. Physiology. 2012;27:130–139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benes CH, Wu N, Elia AE, Dharia T, Cantley LC, Soltoff SP. The C2 domain of PKCδ is a phosphotyrosine binding domain. Cell. 2005;121:271–280 [DOI] [PubMed] [Google Scholar]
- 19.Sumandea MP, Rybin VO, Hinken AC, Wang C, Kobayashi T, Harleton E, et al. Tyrosine phosphorylation modifies PKCδ dependent phosphorylation of cardiac troponin I. J Biol Chem. 2008;283:22680–22689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rybin VO, Guo J, Sabri A, Elouardighi H, Schaefer E, Steinberg SF. Stimulus-specific differences in protein kinase C-δ localization and activation mechanisms in cardiomyocytes. J Biol Chem. 2004;279:19350–19361 [DOI] [PubMed] [Google Scholar]
- 21.Gong J, Yao Y, Zhang P, Udayasuryan B, Komissarova EV, Chen J, et al. The C2 domain and altered ATP-binding loop phosphorylation at Ser359 mediate the redox-dependent increase in protein kinase C-δ activity. Mol Cell Biol. 2015;35:1727–1740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seidler J, Adal M, Kubler D, Bossemeyer D, Lehmann WD. Analysis of autophosphorylation sites in the recombinant catalytic subunit alpha of cAMP-dependent kinase by nano-UPLC-ESI-MS/MS. Anal Bioanal Chem. 2009;395:1713–1720 [DOI] [PubMed] [Google Scholar]
- 23.Robles-Flores M, Melendez L, Garcia W, Mendoza-Hernandez G, Lam TT, Castaneda-Patlan C, Gonzalez-Aguilar H. Posttranslational modifications on protein kinase C isozymes. Effects of epinephrine and phorbol esters. Biochim Biophys Acta. 2008;1783:695–712 [DOI] [PubMed] [Google Scholar]
- 24.Aimes RT, Hemmer W, Taylor SS. Serine-53 at the tip of the glycine-rich loop of cAMP-dependent protein kinase: Role in catalysis, P-site specificity, and interaction with inhibitors. Biochemistry. 2000;39:8325–8332 [DOI] [PubMed] [Google Scholar]
- 25.Dankner M, Rose AAN, Rajkumar S, Siegel PM, Watson IR. Classifying BRAF alterations in cancer: New rational therapeutic strategies for actionable mutations. Oncogene. 2018;37:3183–3199 [DOI] [PubMed] [Google Scholar]
- 26.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Mechanism of activation of the Raf-ERK signaling pathway by oncogenic mutations of B-Raf. Cell. 2004;116:855–867 [DOI] [PubMed] [Google Scholar]
- 27.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the B-Raf gene in human cancer. Nature. 2002;417:949–954 [DOI] [PubMed] [Google Scholar]
- 28.Thevakumaran N, Lavoie H, Critton DA, Tebben A, Marinier A, et al. Crystal structure of a BRAF kinase domain monomer explains basis for allosteric regulation. Nat Struct Mol Biol. 2015;22:37–43 [DOI] [PubMed] [Google Scholar]
- 29.Ikenoue T, Hikiba Y, Kanai F, Aragaki J, Tanaka Y, Imamura J, et al. Different effects of point mutations within the B-Raf glycine-rich loop in colorectal tumors on mitogen-activated protein/extracellular signal-regulated kinase kinase/extracellular signal-regulated kinase and nuclear factor κB pathway and cellular transformation. Cancer Res. 2004;64:3428–3435 [DOI] [PubMed] [Google Scholar]
- 30.Holderfield M, Merritt H, Chan J, Wallroth M, Tandeske L, Zhai H, et al. Raf inhibitors activate the MAPK pathway by relieving inhibitory autophosphorylation. Cancer Cell. 2013;23:594–602 [DOI] [PubMed] [Google Scholar]
- 31.Rodriguez-Viciana P, Tetsu O, Tidyman WE, Estep AL, Conger BA, Cruz MS, et al. Germline mutations in genes within the MAPK pathway cause cardio-facio-cutaneous syndrome. Science. 2006;311:1287–1290 [DOI] [PubMed] [Google Scholar]
- 32.Salomon AR, Ficarro SB, Brill LM, Brinker A, Phung QT, Ericson C, et al. Profiling of tyrosine phosphorylation pathways in human cells using mass spectrometry. Proc Natl Acad Sci U S A. 2003;100:443–448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steen H, Fernandez M, Ghaffari S, Pandey A, Mann M. Phosphotyrosine mapping in Bcr/Abl oncoprotein using phosphotyrosine-specific immonium ion scanning. Mol Cell Proteomics. 2003;2:138–145 [DOI] [PubMed] [Google Scholar]
- 34.Allen PB, Wiedemann LM. An activating mutation in the ATP binding site of the Abl kinase domain. J Biol Chem. 1996;271:19585–19591 [DOI] [PubMed] [Google Scholar]
- 35.Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, et al. Activity of a specific inhibitor of the BCR-Abl tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the philadelphia chromosome. N Engl J Med. 2001;344:1038–1042 [DOI] [PubMed] [Google Scholar]
- 36.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-Abl gene mutation or amplification. Science. 2001;293:876–880 [DOI] [PubMed] [Google Scholar]
- 37.Roumiantsev S, Shah NP, Gorre ME, Nicoll J, Brasher BB, et al. Clinical resistance to the kinase inhibitor STI-571 in chronic myeloid leukemia by mutation of Tyr253 in the Abl kinase domain P-loop. Proc.Natl.Acad.Sci.U.S.A. 2002;99:10700–10705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Branford S, Rudzki Z, Walsh S, Grigg A, Arthur C, Taylor K, et al. High frequency of point mutations clustered within the adenosine triphosphate-binding region of Bcr/Abl in patients with chronic myeloid leukemia or ph-positive acute lymphoblastic leukemia who develop imatinib (STI571) resistance. Blood. 2002;99:3472–3475 [DOI] [PubMed] [Google Scholar]
- 39.Skaggs BJ, Gorre ME, Ryvkin A, Burgess MR, Xie Y, Han Y, et al. Phosphorylation of the ATP-binding loop directs oncogenicity of drug-resistant Bcr-Abl mutants. Proc.Natl.Acad.Sci.U.S.A. 2006;103:19466–19471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Preyer M, Vigneri P, Wang JY. Interplay between kinase domain autophosphorylation and F-actin binding domain in regulating imatinib sensitivity and nuclear import of BCR-Abl. PloS one. 2011;6:e17020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kemble DJ, Sun G. Direct and specific inactivation of protein tyrosine kinases in the Src and FGFR families by reversible cysteine oxidation. Proc.Natl.Acad.Sci.U.S.A. 2009;106:5070–5075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Levinson NM, Seeliger MA, Cole PA, Kuriyan J. Structural basis for the recognition of c-Src by its inactivator CSK. Cell. 2008;134:124–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coba MP, Munoz MC, Dominici FP, Toblli JE, Pena C, et al. Increased in vivo phosphorylation of insulin receptor at serine 994 in the liver of obese insulin-resistant zucker rats. J Endocrinol. 2004;182:433–444 [DOI] [PubMed] [Google Scholar]
- 44.Strack V, Hennige AM, Krutzfeldt J, Bossenmaier B, Klein HH, Kellerer M, et al. Serine residues 994 and 1023/25 are important for insulin receptor kinase inhibition by protein kinase C isoforms β2 and θ. Diabetologia. 2000;43:443–449 [DOI] [PubMed] [Google Scholar]
- 45.Strack V, Stoyanov B, Bossenmaier B, Mosthaf L, Kellerer M, Haring HU. Impact of mutations at different serine residues on the tyrosine kinase activity of the insulin receptor. Biochem Biophys Res Commun. 1997;239:235–239 [DOI] [PubMed] [Google Scholar]
- 46.Munoz MC, Giani JF, Mayer MA, Toblli JE, Turyn D, Dominici FP. Tank-binding kinase 1 mediates phosphorylation of insulin receptor at serine residue 994: A potential link between inflammation and insulin resistance. J Endocrinol. 2009;201:185–197 [DOI] [PubMed] [Google Scholar]
- 47.Won M, Park KA, Byun HS, Kim YR, Choi BL, Hong JH, et al. Protein kinase SGK1 enhances MEK/ERK complex formation through the phosphorylation of ERK2: Implication for the positive regulatory role of SGK1 on the ERK function during liver regeneration. J Hepatol. 2009;51:67–76 [DOI] [PubMed] [Google Scholar]
- 48.Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Humphrey SJ, Yang G, Yang P, Fazakerley DJ, Stockli J, et al. Dynamic adipocyte phosphoproteome reveals that AKT directly regulates mTORC2. Cell Metab. 2013;17:1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daub H, Olsen JV, Bairlein M, Gnad F, Oppermann FS, Korner R, et al. Kinase-selective enrichment enables quantitative phosphoproteomics of the kinome across the cell cycle. Mol Cell. 2008;31:438–448 [DOI] [PubMed] [Google Scholar]
- 51.Dulla K, Daub H, Hornberger R, Nigg EA, Korner R. Quantitative site-specific phosphorylation dynamics of human protein kinases during mitotic progression. Mol Cel proteomics. 2010;9:1167–1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu X, Tian L, Li J, Zhang Y, Han V, Li Y, et al. Investigation of receptor interacting protein (RIP3)-dependent protein phosphorylation by quantitative phosphoproteomics. Mol Cell Proteomics. 2012;11:1640–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics. 2014;13:1690–1704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, et al. Toward a comprehensive characterization of a human cancer cell phosphoproteome. J Proteome Res. 2013;12:260–271 [DOI] [PubMed] [Google Scholar]
- 55.Mertins P, Qiao JW, Patel J, Udeshi ND, Clauser KR, Mani DR, et al. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat Methods. 2013;10:634–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Udeshi ND, Svinkina T, Mertins P, Kuhn E, Mani DR, et al. Refined preparation and use of anti-diglycine remnant (K-ε-GG) antibody enables routine quantification of 10,000s of ubiquitination sites in single proteomics experiments. Mol Cell Proteomics. 2013;12:825–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wagner SA, Beli P, Weinert BT, Scholz C, Kelstrup CD, Young C, et al. Proteomic analyses reveal divergent ubiquitylation site patterns in murine tissues. Mol Cell Proteomics. 2012;11:1578–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gong J, Park M, Steinberg SF. Cleavage alters the molecular determinants of protein kinase C-δ catalytic activity. Mol Cell Biol. 2017;37:e00324–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, et al. A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics. 2011;10:M111 013284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011;44:325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li H, Ren Z, Kang X, Zhang L, Li X, Wang Y, et al. Identification of tyrosine-phosphorylated proteins associated with metastasis and functional analysis of FER in human hepatocellular carcinoma cells. BMC cancer. 2009;9:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boeing S, Williamson L, Encheva V, Gori I, Saunders RE, Instrell R, et al. Multiomic analysis of the UV-induced DNA damage response. Cell Reports. 2016;15:1597–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3. [DOI] [PubMed] [Google Scholar]
- 64.Oppermann FS, Gnad F, Olsen JV, Hornberger R, Greff Z, Keri G, et al. Large-scale proteomics analysis of the human kinome. Mol Cell Proteomics. 2009;8:1751–1764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tweedie-Cullen RY, Reck JM, Mansuy IM. Comprehensive mapping of post-translational modifications on synaptic, nuclear, and histone proteins in the adult mouse brain. J Proteome Res. 2009;8:4966–4982 [DOI] [PubMed] [Google Scholar]
- 66.Trinidad JC, Thalhammer A, Specht CG, Lynn AJ, Baker PR, et al. Quantitative analysis of synaptic phosphorylation and protein expression. Mol Cell Proteomics. 2008;7:684–696 [DOI] [PubMed] [Google Scholar]
- 67.Vosseller K, Hansen KC, Chalkley RJ, Trinidad JC, Wells L, et al. Quantitative analysis of both protein expression and serine/threonine post-translational modifications through stable isotope labeling with dithiothreitol. Proteomics. 2005;5:388–398 [DOI] [PubMed] [Google Scholar]
- 68.Lundby A, Lage K, Weinert BT, Bekker-Jensen DB, Secher A, Skovgaard T, et al. Proteomic analysis of lysine acetylation sites in rat tissues reveals organ specificity and subcellular patterns. Cell Rep. 2012;2:419–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, et al. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics. 2012;11:215–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lumpkin RJ, Gu H, Zhu Y, Leonard M, Ahmad AS, Clauser KR, et al. Site-specific identification and quantitation of endogenous sumo modifications under native conditions. Nat Comm. 2017;8:1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Poon RY, Jiang W, Toyoshima H, Hunter T. Cyclin-dependent kinases are inactivated by a combination of p21 and Thr-14/Tyr-15 phosphorylation after UV-induced DNA damage. J Biol Chem. 1996;271:13283–13291 [DOI] [PubMed] [Google Scholar]
- 72.Atherton-Fessler S, Liu F, Gabrielli B, Lee MS, Peng CY, Piwnica-Worms H. Cell cycle regulation of the p34cdc2 inhibitory kinases. Mol Biol Cell. 1994;5:989–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Solomon MJ, Lee T, Kirschner MW. Role of phosphorylation in p34cdc2 activation: Identification of an activating kinase. Mol Biol Cell. 1992;3:13–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Krek W, Nigg EA. Differential phosphorylation of vertebrate p34cdc2 kinase at the G1/s and G2/m transitions of the cell cycle: Identification of major phosphorylation sites. EMBO J. 1991;10:305–316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parker LL, Atherton-Fessler S, Lee MS, Ogg S, Falk JL, et al. Cyclin promotes the tyrosine phosphorylation of p34cdc2 in a wee1+ dependent manner. EMBO J. 1991;10:1255–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Piwnica-Worms H, Atherton-Fessler S, Lee MS, Ogg S, Swenson KI, Parker LL. P107wee1 is a serine/threonine and tyrosine kinase that promotes the tyrosine phosphorylation of the cyclin/p34cdc2 complex. Cold Spring Harb Symp Quant Biol. 1991;56:567–576 [DOI] [PubMed] [Google Scholar]
- 77.McGowan CH, Russell P. Human wee1 kinase inhibits cell division by phosphorylating p34cdc2 exclusively on Tyr15. EMBO J. 1993;12:75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sharma K, D’Souza RC, Tyanova S, Schaab C, Wisniewski JR, et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of Tyr and Ser/Thr-based signaling. Cell Rep. 2014;8:1583–1594 [DOI] [PubMed] [Google Scholar]
- 79.Choudhary C, Olsen JV, Brandts C, Cox J, Reddy PN, Bohmer FD, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009;36:326–339 [DOI] [PubMed] [Google Scholar]
- 80.Zhang G, Fang B, Liu RZ, Lin H, Kinose F, Bai Y, et al. Mass spectrometry mapping of epidermal growth factor receptor phosphorylation related to oncogenic mutations and tyrosine kinase inhibitor sensitivity. J Proteome Res. 2011;10:305–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Schreiber TB, Mausbacher N, Keri G, Cox J, Daub H. An integrated phosphoproteomics work flow reveals extensive network regulation in early lysophosphatidic acid signaling. Mol Cell Proteomics. 2010;9:1047–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tong J, Taylor P, Moran MF. Proteomic analysis of the epidermal growth factor receptor (EGFR) interactome and post-translational modifications associated with receptor endocytosis in response to EGF and stress. Mol Cell Proteomics. 2014;13:1644–1658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Palacios-Moreno J, Foltz L, Guo A, Stokes MP, Kuehn ED, George L, et al. Neuroblastoma tyrosine kinase signaling networks involve FYN and LYN in endosomes and lipid rafts. PLoS Comput Biol. 2015;11:e1004130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tzouros M, Golling S, Avila D, Lamerz J, Berrera M, Ebeling M, et al. Development of a 5-plex SILAC method tuned for the quantitation of tyrosine phosphorylation dynamics. Mol Cell Proteomics. 2013;12:3339–3349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Klammer M, Kaminski M, Zedler A, Oppermann F, Blencke S, Marx S, et al. Phosphosignature predicts dasatinib response in non-small cell lung cancer. Mol Cell Proteomics. 2012;11:651–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bai Y, Li J, Fang B, Edwards A, Zhang G, Bui M, et al. Phosphoproteomics identifies driver tyrosine kinases in sarcoma cell lines and tumors. Cancer Res. 2012;72:2501–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chumbalkar V, Latha K, Hwang Y, Maywald R, Hawley L, Sawaya R, et al. Analysis of phosphotyrosine signaling in glioblastoma identifies Stat5 as a novel downstream target of ΔEGFR. J Proteome Res. 2011;10:1343–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ding VM, Boersema PJ, Foong LY, Preisinger C, Koh G, Natarajan S, et al. Tyrosine phosphorylation profiling in FGF-2 stimulated human embryonic stem cells. PloS one. 2011;6:e17538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li J, Rix U, Fang B, Bai Y, Edwards A, Colinge J, et al. A chemical and phosphoproteomic characterization of dasatinib action in lung cancer. Nat Chem Biol. 2010;6:291–299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moritz A, Li Y, Guo A, Villen J, Wang Y, MacNeill J, et al. Akt-Rsk-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci Signal. 2010;3:ra64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lu KV, Zhu S, Cvrljevic A, Huang TT, Sarkaria S, Ahkavan D, et al. Fyn and Src are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 2009;69:6889–6898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Guo A, Villen J, Kornhauser J, Lee KA, Stokes MP, Rikova K, et al. Signaling networks assembled by oncogenic EGFR and c-Met. Proc Natl Acad Sci U S A. 2008;105:692–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203 [DOI] [PubMed] [Google Scholar]
- 94.Thelemann A, Petti F, Griffin G, Iwata K, Hunt T, Settinari T, et al. Phosphotyrosine signaling networks in epidermal growth factor receptor overexpressing squamous carcinoma cells. Mol Cell Proteomics. 2005;4:356–376 [DOI] [PubMed] [Google Scholar]
- 95.Schulze WX, Deng L, Mann M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol Syst Biol. 2005;1:2005 0008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Guha U, Chaerkady R, Marimuthu A, Patterson AS, Kashyap MK, Harsha HC, et al. Comparisons of tyrosine phosphorylated proteins in cells expressing lung cancer-specific alleles of EGFR and K-Ras. Proc Natl Acad Sci U S A. 2008;105:14112–14117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Munoz MC, Argentino DP, Dominici FP, Turyn D, Toblli JE. Irbesartan restores the in-vivo insulin signaling pathway leading to AKT activation in obese Zucker rats. J Hypertens. 2006;24:1607–1617 [DOI] [PubMed] [Google Scholar]
- 98.Coba MP, Turyn D, Pena C. Synthesis and immunogenic properties of phosphopeptides related to the human insulin receptor. J Pept Res. 2003;61:17–23 [DOI] [PubMed] [Google Scholar]
- 99.Stokes MP, Farnsworth CL, Moritz A, Silva JC, Jia X, Lee KA, et al. PTMscan direct: Identification and quantification of peptides from critical signaling proteins by immunoaffinity enrichment coupled with LC-MS/MS. Mol Cell Proteomics. 2012;11:187–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu F, Wang P, Zhang J, Young LC, Lai R, Li L. Studies of phosphoproteomic changes induced by nucleophosmin-anaplastic lymphoma kinase (ALK) highlight deregulation of tumor necrosis factor (TNF)/Fas/TNF-related apoptosis-induced ligand signaling pathway in ALK-positive anaplastic large cell lymphoma. Mol Cell Proteomics. 2010;9:1616–1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005;23:94–101 [DOI] [PubMed] [Google Scholar]
- 102.Hornbeck PV, Zhang B, Murray B, Kornhauser JM, Latham V, Skrzypek E. PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic acids Res. 2015;43:D512–520 [DOI] [PMC free article] [PubMed] [Google Scholar]