

Abstract
A major pharmacological barrier to peptide therapeutics is their susceptibility to proteolytic degradation and poor membrane permeability, which, in principle, can be overcome by nanoparticle-based delivery technologies. Proteins, by definition, are nano materials and have been clinically proven as an efficient delivery vehicle for small molecule drugs. Here we describe the design of a protein-based peptide drug carrier derived from the tetramerization domain of the chimeric oncogenic protein Bcr/Abl of chronic myeloid leukemia. A dodecameric peptide inhibitor of the p53-MDM2/MDMX interaction, termed PMI, was grafted to the N-terminal helical region of Bcr/Abl tetramer. To antagonize intracellular MDM2/MDMX for p53 activation, we extended this protein, PMIBcr/Abl, by a C-terminal Arg-repeating hexapeptide to facilitate its cellular uptake. The resultant tetrameric protein PMIBcr/Abl-R6 adopted an alpha-helical conformation in solution and bound to MDM2 at an affinity of 32 nM.PMIBcr/Abl-R6 effectively induced apoptosis of HCT116 p53+/+ cells in vitro in a p53-dependent manner and potently inhibited tumor growth in a nude mouse xenograft model by stabilizing p53 in vivo. Our protein-based delivery strategy thus provides a clinically viable solution to p53-inspired anticancer therapy and is likely applicable to the development of many other peptide therapeutics to target a great variety of intracellular protein-protein interactions responsible for disease initiation and progression.
Introduction
Intracellular protein-protein interactions (PPIs) control many essential cellular pathways implicated in human diseases [1, 2], representing an important class of therapeutic targets that are considered to be The Holy Grail in drug discovery and development [3, 4]. Among various PPI inhibitors with therapeutic potential, small peptides, compared with low molecular weight compounds, often excel due to their high potency and selectivity and low toxicity [5, 6]. However, major pharmacological disadvantages of peptide inhibitors exist. For example, peptides are susceptible to enzymatic degradation because they generally do not possess a stable tertiary structure to confer resistance to proteolysis; peptides also lack the ability to actively traverse the cell membrane, thus failing to reach intracellular drug targets. Poor proteolytic stability and membrane permeability severely limit peptide bioavailability and therapeutic efficacy [5, 7]. Various elaborate medicinal chemistry approaches and peptide delivery techniques have been developed to overcome these pharmacological barriers [8–16]. While considerable success has been achieved in using peptides to target intracellular PPIs [17–21], much still remains to be done to fulfill their full therapeutic potential.
Nanotechnology has been widely used in the development of new strategies for drug delivery and cancer therapy [22, 23]. Nanoparticle-based traditional delivery tools include, but are not limited to, micelle, liposome, dendrimer, gold nanoshell, and polymer [24, 25]. As unique biopolymers in the nanoscale, proteins are superior in many aspects as a drug carrier to synthetic polymers [26, 27]. Protein-based drug carriers are attractive also because they are amenable to both biological and chemical modifications so that their properties such as molecular size, site of conjugation, and loading capacity can be controlled [28]. In addition, novel functionalities can be engineered into proteins to facilitate cellular uptake and improve targeting specificity. Albumin, a natural transport protein with multiple ligand binding sites, cellular receptor engagement, and a long circulatory half-life, represents a clinically proven platform for the delivery of various drug molecules [29, 30]. Despite the obvious advantages of protein-based drug delivery of low molecular weight compounds, it remains challenging to efficiently deliver peptide therapeutics to target intracellular PPIs. We aim to alleviate this problem by using molecular grafting approaches to design a stable protein scaffold with multiple functionalities.
Most protein scaffolds used for peptide grafting are stabilized by disulfide bonds [31–33], thus unsuitable for targeting PPIs in the cytoplasmic space, where the reducing environment can structurally destabilize disulfide-bridged proteins, prompting their proteolytic degradation. To circumvent this severe limitation, we have identified the disulfide-devoid tetramerization domain of the chimeric oncoprotein Bcr/Abl of chronic myeloid leukemia (CML) [34], which forms a highly stable tetramer in solution, as a novel protein scaffold for molecular grafting of therapeutic peptides of an α-helical nature. In this proof of concept study, we introduced into the N-terminus of Bcr/Abl a potent dodecameric peptide antagonist, termed PMI, of both MDM2 and MDMX – the two oncogenic proteins that functionally inhibit the tumor suppressor protein p53 in many tumor types [35, 36]. C-terminal extension of PMIBcr/Abl by an Arg-repeating hexapeptide (R6) endowed the protein with the ability to traverse the cell membrane. PMIBcr/Abl-R6 potently inhibited tumor growth in vitro and in vivo by antagonizing MDM2/MDMX to reactivate the p53 pathway. This protein scaffold, Bcr/Abl-R6, has the potential to be used as an efficient delivery tool for α-helical peptides to target a great variety of intracellular PPIs for disease intervention.
Results and Discussion
Design strategy.
In many tumor cells harboring wild type p53, the E3 ubiquitin ligase MDM2 and/or its homolog MDMX (also known as MDM4) block the transcriptional activity of p53 and target the tumor suppressor protein for proteasomal degradation, conferring tumor development and progression [35–37]. MDM2/MDMX antagonism has been validated as an effective therapeutic strategy for cancer treatment. Since MDMX potentiates MDM2 function in p53 inhibition, dual-specificity antagonists of both MDM2 and MDMX are particularly attractive as therapeutic agents for robust and sustained p53 activation [37]. We have previously identified PMI, a series of high-affinity and dual-specificity dodecameric peptide antagonists of MDM2 and MDMX, through combinatorial library screening and structure-based rational design approaches [38, 39]. Although PMI peptides tightly bind, in an α-helical conformation, to the p53-binding pocket of MDM2 and MDMX at affinities ranging from high pM to low nM, they are not inhibitory against tumor growth due mainly to their inability to traverse the cell membrane [38, 39].
To carry therapeutic peptides of an α-helical nature for cancer therapy, we surmise that the protein must meet the following five criteria: (1) structurally amenable to peptide grafting with a pre-existing short α-helix, (2) sufficiently large in size (via oligomerization, for example) to alleviate renal excretion, (3) resistant to proteolytic degradation by adopting a stable structure with few flexible loops and disordered regions, (4) devoid of disulfide bonds, and (5) efficient in membrane permeabilization. Bcr/Abl tetramerization domain comprises 72 amino acid residues and forms a coiled-coil tetramer, with each monomer consisting of a short N-terminal α-helix, a connecting loop, and a long C-terminal α-helix [34]. This protein is thus ideally suited as a nano-carrier of PMI for cancer therapy because it readily meets the first four criteria defined. To enable its membrane permeability, however, additional modifications such as introduction of a cationic penetrating peptide sequence to Bcr/Abl tetramerization domain is warranted. Our design strategy is schematically illustrated in Fig. 1.
Fig. 1. Strategy for the design of a protein-based nano-carrier of PMI for cancer therapy.
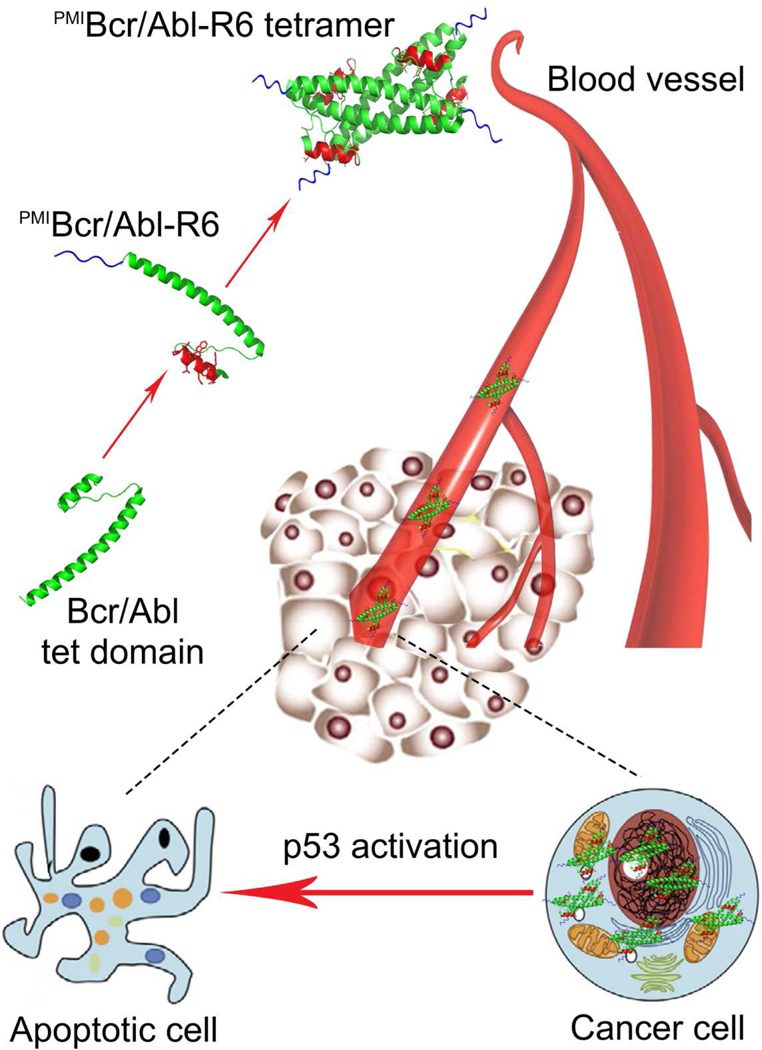
The tetramerization domain of 72 amino acid residues of Bcr/Abl (green) comprises an N-terminal α-helix linked via a flexible loop to an elongated C-terminal α-helix that mediates tetramer formation. PMI in red is grafted to the short α-helical region in place of residues 5–16 of Bcr/Abl, resulting in PMIBcr/Abl. To facilitate membrane permeabilization, PMIBcr/Abl is C-terminally extended by an Arg-repeating hexapeptide (R6) in blue, yielding PMIBcr/Abl-R6. PMIBcr/Abl-R6 forms a stable tetramer, circulates in the blood, accumulates in the tumor, traverse the cell membrane, and activates p53 by antagonizing MDM2/MDMX, leading to inhibition of tumor growth in experimental animals.
Synthesis and biochemical and biophysical characterization of PMIBcr/Abl-R6.
Structural studies indicate that the N-terminal α-helix of Bcr/Abl (residues 5–15) does not contribute to protein tetramerization, which is mediated predominantly by the elongated C-terminal α-helix (residues 28–67) [34] (Fig. 2A). Since PMI (TSFAEYWALLSP) [38, 39] and residues 5–16 of Bcr/Abl (VGFAEAWKAQFP) share some degrees of sequence identity and structural similarity (Fig. 2A), we simply replaced the latter with the former in the amino acid sequence. Further, we extended the C-terminus of Bcr/Abl by an Arg-repeating hexapeptide (R6) to enhance cellular uptake, ultimately yielding PMIBcr/Abl-R6 (Fig. 2B). PMIBcr/Abl-R6 of 78 amino acid residues was chemically synthesized via native chemical ligation [40, 41] of two peptide fragments as illustrated in Figs. 2B and S1. Ala38 was mutated to Cys to enable the ligation reaction, which was reverted to Ala, after ligation, though desulfurization as described [42]. The final product was purified by reversed phase HPLC and its molecular mass ascertained by electrospray ionization mass spectrometry (Fig. 2C). We folded the synthetic proteins by dissolving the polypeptides at 1 mg/ml in 6 M GuHCl, followed by a six-fold dilution with and dialysis against PBS containing 0.5 mM TCEP, pH 7.4. As shown in Fig. 2D, both Bcr/Abl-R6 and PMIBcr/Abl-R6 adopted similar α-helical conformations in solution as evidenced by their similar circular dichroism spectra showing double minima at 208 and 222 nm and a positive peak at 195 nm, consistent with the known structural features of Bcr/Abl [34]. As negative controls, Bcr/Abl-R6 and PMIBcr/Abl were also chemically synthesized essentially as described for PMIBcr/Abl-R6 (Fig. 2B).
Fig. 2. Chemical synthesis of Bcr/Abl proteins via native chemical ligation.
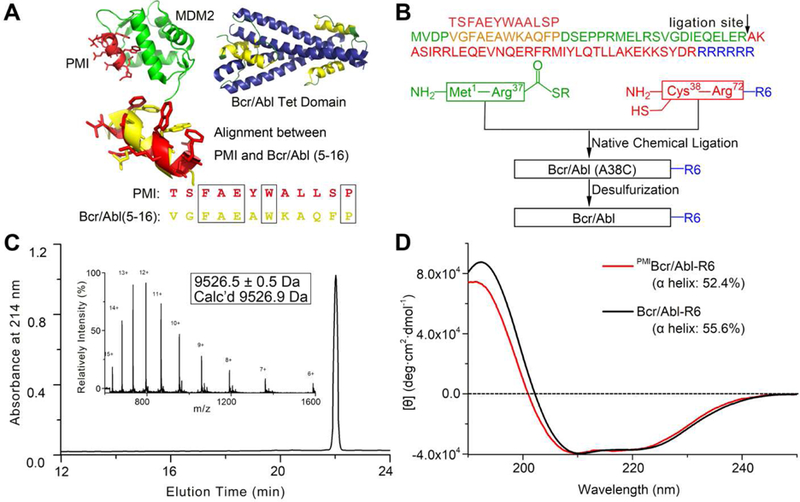
(A) Structure-based rational design of PMIBcr/Abl-R6. The crystal structures of PMI (red) in complex with MDM2 (green) [39] and of the tetramerization domain of Bcr/Abl (blue/yellow) [34] are shown in ribbons. The dodecameric peptide PMI (TSFAEYWALLSP) and residues 5–16 of Bcr/Abl (VGFAEAWKAQFP) share certain degrees of sequence and structural similarity. (B) Total chemical synthesis of PMIBcr/Abl, Bcr/Abl-R6 and PMIBcr/Abl-R6 via native chemical ligation [40, 41]. All peptides were synthesized on appropriate resin using Boc-chemistry solid phase peptide synthesis [66]. Ligation reactions were carried out in 0.1 M phosphate buffer containing 6 M GuHCl, 100 mM MPAA and 40 mM TCEP, pH 7.4. Desulfurization of the ligation product was achieved by dissolving the peptide at 1 mg/mL in 0.1 M phosphate buffer containing 6 M GuHCl, 0.01 M VA-044, 0.5 M TCEP, 20% t-BuSH. (C) PMIBcr/Abl-R6 analyzed by HPLC and electrospray ionization mass spectrometry (ESI-MS). Analytical HPLC was performed on a reversed-phase C18 column (Waters XBridge™ 3.5 µm, 4.6×150 mm) at 40 °C. (D) Circular dichroism (CD) spectra of Bcr/Abl-R6 (black) and PMIBcr/Abl-R6 (red) at 20 µM in 20 mM phosphate buffer, pH 7.4, obtained on a Jasco spectrometer at 25 °C. Proteins were quantified spectroscopically by UV measurements at 280 nm using a molar extinction coefficient of 9970 calculated as described [67]. Percent helicity was calculated from the ratio of [θ]222 to [θ]max, where [θ]max = −39500 × [1−(2.57/n)] [68].
We also characterized Bcr/Abl-R6 and PMIBcr/Abl-R6 using size exclusion chromatography (Figs. 3A and S2–S3), dynamic light scattering (Figs. 3B and S3–S4), fluorescence polarization (Fig. 3C), and small angle X-ray scattering (Fig. 3D-E). All data unambiguously demonstrated that the synthetic proteins existed in aqueous buffer as tetramers at concentrations above 100 nM (Fig. 3C). Of note, measurements of the zeta potential of both PMIBcr/Abl and PMIBcr/Abl-R6 confirmed that the Arg-repeating hexapeptide R6 substantially increased protein surface charges, as expected (Fig. S4). Importantly, PMIBcr/Abl-R6 bound to the p53-binding domain of MDM2 with an affinity of 32 nM as measured by isothermal titration calorimetry (ITC) (Fig. 3F). By contrast, Bcr/Abl-R6 showed no binding to MDM2 under identical conditions (Fig. S5). PMI, with a binding affinity of 0.52 nM for MDM2 (Fig. 3F) (KD=0.5 nM, determined by surface plasmon resonance [38]), was significantly more potent than PMIBcr/Abl-R6. ITC data analysis indicates that despite a small net gain in entropy for PMIBcr/Abl-R6 as opposed to a large loss for PMI, an expected outcome instigated by molecular grafting, PMIBcr/Abl-R6 lost a substantial amount of enthalpy for binding (Table S1), suggesting that structurally rigidified PMI in the context of Bcr/Abl was energetically suboptimal for MDM2 binding. Nevertheless, ITC-based binding assays clearly validated the molecular design at the functional level of a protein antagonist of MDM2.
Fig 3. Biochemical and biophysical characterization of PMIBcr/Abl-R6.
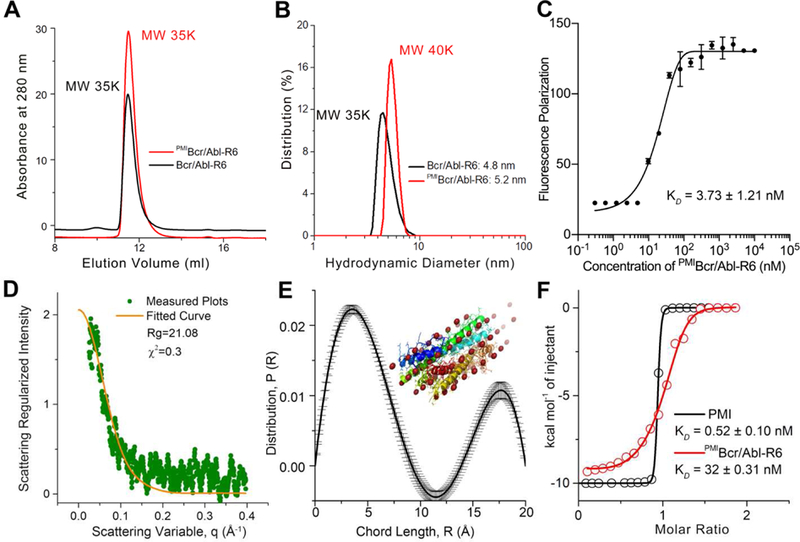
(A) Size exclusion chromatography of Bcr/Abl-R6 (black) and PMIBcr/Abl-R6 (red) performed on a GE Superdex 75 column (10/300 GL) running PSB at a flow rate of 0.5 ml/min at room temperature. The apparent molecular weights of Bcr/Abl-R6 and PMIBcr/ABL-R6 were calculated according to the standard calibration curve shown in Fig. S2, indicating that they exist in aqueous buffer as tetramers. (B) Dynamic light scattering analysis of Bcr/Abl-R6 (black) and PMIBcr/Abl-R6 (red) at 20 µM in PBS performed on a Malvin Zetasizer Nano system. The apparent molecular weights were calculated using manufacturer-supplied software. (C) Monomer-tetramer equilibrium of serially diluted PMIBcr/Abl-R6 (from 10 µM to 0.3 nM in 20 mM Tris/HCl, pH 7.4) measured in 386-well black plates by fluorescence polarization, yielding a KD value of 3.73±1.21 nM (KD=(monomer)4/(tetramer), where the concentrations of monomeric and tetrameric PMIBcr/Abl-R6 were derived from fluorescence polarization values). PMIBcr/Abl-R6 was N-terminally labeled with a fluorophore, BDP TR (Excitation 589 nm, Emission 616 nm). (D) Small angle X-ray scattering (SAXS) diffractograms of PMIBcr/Abl-R6 measured at 20 µ M in PBS. The orange line is the least squares fit to the data (green points) using a rod model. (E) SAXS analysis of PMIBcr/Abl-R6 at 10 µM in PBS at room temperature. The chord length distribution that describes the size, shape and spatial arrangement of PMIBcr/Abl-R6 was obtained from SAXS data. The simulated structure of PMIBcr/Abl-R6 is largely in agreement with the crystal structure of tetrameric Bcr/Abl (PDB code: 1K1F [34]) (inset). (F) Measurements of the binding affinity of PMI and PMIBcr/Abl-R6 for MDM2 in 20 mM Tris/HCl, pH 7.4, by isothermal titration calorimetry on a MicroCal ITC 200 instrument at 25 °C. Titrations were carried out by 20 stepwise injections, 2 µL at a time, of 80 µM PMIBcr/Abl-R6 in the syringe to 8 µM MDM2 in the cell. For the PMI-MDM2 interaction, the concentrations were 100 µM and 10 µM, respectively. Data were analyzed using the MicroCal Origin program. The KD value of 0.52 nM, measured as described [39], is nearly identical to the published value of PMI determined by surface plasmon resonance [38]. Values of enthalpy change, ∆H, entropy change, ∆S, and binding stoichiometry, n are listed in Table S1.
PMIBcr/Abl-R6 with enhanced proteolytic stability efficiently permeabilizes HCT116 p53+/+ tumor cells via an endocytosis-independent pathway.
As was demonstrated previously [39], PMI was inactive in killing HCT116 p53+/+ cells due to its poor proteolytic stability and inability to traverse the cell membrane. We compared proteolytic stability of free PMI and PMIBcr/Abl-R6 in the presence of human serum (mainly serine proteases) or the intracellular cysteine protease cathepsin B. Intact peptide or protein was identified by mass spectrometry and quantified by RP-HPLC. As shown in Fig. 4A-B, the half-life of PMIBcr/Abl-R6, compared with that of PMI, increased by 5-fold in human serum and 12-fold in cathepsin B. These data demonstrate that PMIBcr/Abl-R6 is significantly more stable than free PMI in the presence of proteases.
Fig. 4. Proteolytic stability and membrane permeability of PMIBcr/Abl-R6.
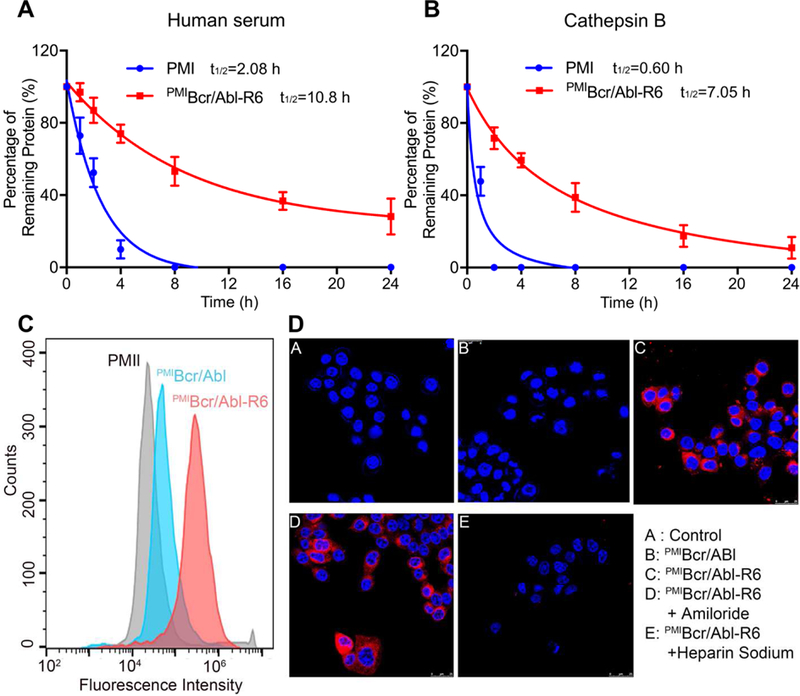
(A) Degradation kinetics of PMI and PMIBcr/Abl-R6 at 1 mg/ml in 20 mM Tris-HCl, pH 7.4, containing 10% human serum from a healthy donator. Intact peptide and protein were verified by ESI-MS and quantified by analytical C18 HPLC. (B) Degradation kinetics of PMI and PMIBcr/Abl-R6 at 1 mg/ml in 20 mM sodium acetate buffer, pH 5.0, containing cathepsin B at 10 units/ml. (C) Cellular uptake of PMI, PMIBcr/Abl and PMIBcr/Abl-R6 analyzed by flow cytometry. PMI, PMIBcr/Abl and PMIBcr/Abl-R6 were N-terminally labeled with BDP TR (excitation 589 nm, emission 616 nm). HCT 116 p53+/+ cells were seeded in a 12-well plate at a density of 30,000 cells/well, cultured for 24 h, and treated with peptide or protein at 10 µM for 4 h before flow cytometric analysis. (D) Cellular uptake of BDP TR-labeled PMI, PMIBcr/Abl and PMIBcr/Abl-R6 by HCT 116 p53+/+ cells, treated by peptide or protein at 10 µM each for 4 h, and visualized by a confocal laser scanning microscope (panels A-C). Hoechst 33342 blue dye was used for nuclei staining. For the experiments presented in panels D-E, amiloride (3 mM) or heparin sodium (5 mM) was incubated with cells for 12 h before the addition of PMIBcr/Abl-R6.
Next, we examined peptide/protein internalization and cytosolic release using both confocal microscopy and flow cytometry. As shown in Fig. 4C, flow cytometric analysis indicated that PMIBcr/Abl-R6 N-terminally labeled with a BODIPY (borondipyrromethene) dye, BDP TR (589/616 nm), traversed HCT116 p53+/+ cell membranes much more efficiently than PMIBcr/Abl or free PMI labeled with the same fluorophore. Confocal microscopic analysis confirmed this finding by showing the cytosolic distribution of PMIBcr/Abl-R6, but not of PMIBcr/Abl (Fig. 4D, panels A-C).
Cationic cell penetrating peptides as a carrier are known to promote cellular uptake of cargos primarily through the non-endocytic uptake pathway, or direct membrane translocation, which is inhibited by heparin but not by amiloride [43, 44]. To better understand the mechanism of cellular uptake of PMIBcr/Abl-R6, we performed confocal microscopic analysis on cells treated with heparin or amiloride. As shown in Fig. 4D (panels D-E), while amiloride had little effect on PMIBcr/Abl-R6 internalization, heparin almost completely blocked it, suggesting that the cellular uptake pathway for PMIBcr/Abl-R6 is indeed endocytosis-independent.
PMIBcr/Abl-R6 kills HCT116 p53+/+ tumor cells in vitro by reactivating the p53 pathway.
To evaluate the tumor-killing activity of PMIBcr/Abl-R6 in vitro, we treated isogenic HCT116 p53+/+ and HCT116 p53−/− cell lines expressing abundant MDM2 [45, 46] with the protein at concentrations from 1.56 µM to 50 µM. PMI and Bcr/Abl-R6 were used as a negative control and Nutlin-3, an extensively studied small molecule antagonist of MDM2 [47], as a positive control. As expected, while neither PMI nor Bcr/Abl-R6 had any effect on the viability of HCT116 cells 48 h after treatment (Figs. 5A and S6), a dose-dependent growth inhibition of HCT116 p53+/+ cells was observed with PMIBcr/Abl-R6 and Nutlin-3 that were similarly active (Fig. 5A). Unlike PMIBcr/Abl-R6, Nutlin-3 was toxic at 50 µM against HCT116 p53−/− cells, indicative of its smaller therapeutic window than PMIBcr/Abl-R6.
Fig. 5. PMIBcr/Abl-R6 kills HCT116 p53+/+ tumor cells in vitro by reactivating the p53 pathway.
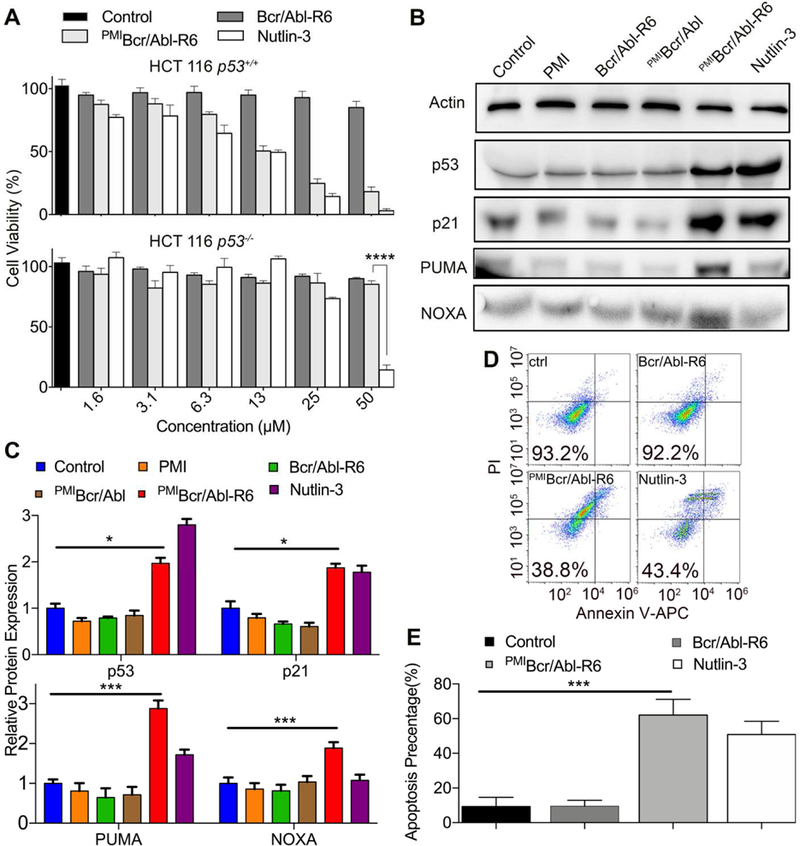
(A) Cell viability of HCT116 p53+/+ and HCT116 p53−/− cells (3×103 cells/well in McCoys’s 5A medium with 10% FBS) 48 h after treatment with varying concentrations of Bcr/Abl-R6, PMIBcr/Abl-R6 and Nutlin-3. Following a 2-h incubation with CCK-8 reagents, absorbance values at 450 nm were measured on a microplate reader, and percent cell viability was calculated as (Atreatment−Ablank)/(Acontrol−Ablank) × 100%. The data are the means of three independent assays. Except for HCT116 p53−/− cells treated by PMIBcr/Abl-R6 and Nutlin-3 at 50 µM (****, p<0.0001), no statistically significant difference in activity between PMIBcr/Abl-R6 and Nutlin-3 was found. (B) Representative Western blotting analysis of p53, p21, PUMA, NOXA in HCT116 p53+/+ cells (2×104 cells/well) 48 h after treatment with PMI, PMIBcr/Abl, PMIBcr/Abl-R6, Bcr/Abl-R6 and Nutlin-3 at 12.5 µM each, normalized to β-actin. The primary antibodies were from Santa Cruz Biotechnology (p53), Calbiochem (p21, PUMA and NOXA) and Sigma-Aldrich (β-actin), and secondary antibodies conjugated with horseradish peroxidase from Calbiochem. (C) Quantitative Western blotting analysis (via Image J software) of HCT116 p53+/+ cells treated with PMI, Bcr/Abl-R6, PMIBcr/Abl, PMIBcr/Abl-R6 and Nutlin-3 at 12.5 µ M for 48 h. T-test was performed for statistical analysis, * standing for p<0.05, *** for p<0.001. The data are the means ± SD of three independent Western blotting assays. (D) Representative data on apoptosis of HCT116 p53+/+ cells 48 h after treatment with Bcr/Abl-R6, PMIBcr/Abl-R6 and Nutlin-3 as analyzed by flow cytometry. Cells were seeded in a 12-well plate with a density of 20,000/well and treated with 12.5 µM PMIBcr/Abl-R6, Bcr/Abl-R6 or 10 µ M Nutlin-3. Apoptosis was detected using a standard apoptotic kit from Biolegend, including APC labeled anti-annexin V antibody and a propidium iodide solution. (E) Statistical analysis of apoptosis of HCT116 p53+/+ cells quantified by flow cytometry. Three independent FACS assays were performed, and data are shown as the means ± SD (n=3). p values were calculated by t-test (***, p<0.001).
To investigate into the mechanisms of action of PMIBcr/Abl-R6, we analyzed by Western blotting the expression of p53, p21, PUMA and NOXA in HCT116 p53+/+ cells 48 h after treatment. As shown in Fig. 5B-C, compared with mock-treated and PMI-, PMIBcr/Abl- or Bcr/Abl-R6-treated cells, PMIBcr/Abl-R6 or Nutlin-3 treatment significantly stabilized p53 in HCT116 p53+/+ cells, leading to upregulation of the p53-responsive genes p21, PUMA and NOXA important for cell cycle arrest and apoptosis [48, 49]. Consistent with these results, FACS analysis confirmed that HCT116 p53+/+ cells underwent similar degrees of apoptosis when treated with PMIBcr/Abl-R6 or Nutlin-3, whereas mock-treated or Bcr/Abl-R6-treated cells were largely unaffected (Fig. 5D-E). Collectively, these data demonstrate that PMIBcr/Abl-R6 induces apoptosis of wild type p53-harboring tumor cells by antagonizing MDM2 to activate the p53 signaling pathway.
PMIBcr/Abl-R6 accumulates and retains extendedly in solid tumors in vivo.
Nanoparticles can actively accumulate in solid tumors through leaky blood vessels in diseased tissues – a phenomenon known as the enhanced permeability and retention (EPR) effect [50, 51]. To examine the biodistribution of PMIBcr/Abl-R6, we fluorescently labeled it with BDP TR and subcutaneously injected the protein into BALB/c nude mice with palpable tumors grown from subcutaneously inoculated HCT116 p53+/+ cells. The biodistribution of PMIBcr/Abl-R6 in the heart, lung, spleen, kidney, liver and tumor at three different time points (12, 24, and 48 h) was semi-quantitatively evaluated on an in vivo optical imaging system. As shown in Fig. 6, the protein reached a maximum level at 24 h and accumulated predominantly in the kidney, liver and tumor. However, only the liver and tumor were found to harbor significant amounts of PMIBcr/Abl-R6 at 48 h. These data suggest that PMIBcr/Abl-R6 is capable of accumulating and retaining extendedly in solid tumors likely via the EPR effect.
Fig. 6. Biodistribution of PMIBcr/Abl-R6 in vivo.
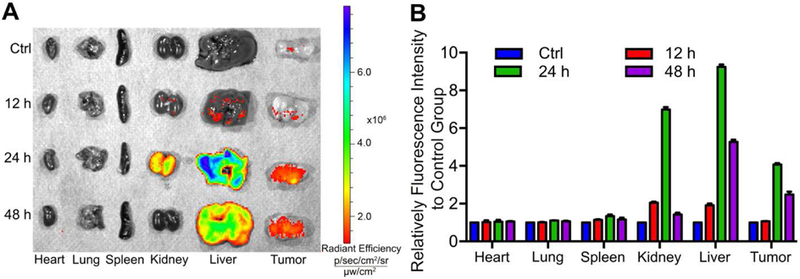
(A) Representative ex vivo fluorescence images of major organs and tumors 12 h, 24 h, 48 h after subcutaneous injection of BDP TR-labeled PMIBcr/Abl-R6. HCT116 p53+/+ cells (4×106 cells/site) were injected subcutaneously into BALB/c nude mice of four weeks old. Three weeks after tumor cell inoculation, tumor-bearing mice were each injected with 100 µL BDP TR-labeled PMIBcr/Abl-R6 at a dose of 5 mg/Kg, and sacrificed for imaging at indicated time points. (B) Semi-quantitative ex vivo analysis of biodistribution of BDP TR-labeled PMIBcr/Abl-R6 in the organs and tumor. Fluorescence intensity in each organ was determined using Living Image 3.0. software from IVIS fluorescence data expressed as radiant efficiency (mean ± SD, n=3).
PMIBcr/Abl-R6 potently inhibits tumor growth in xenograft mice by inducing p53-dependent apoptotic responses in vivo.
To evaluate the therapeutic efficacy of PMIBcr/Abl-R6 in vivo, we established a nude mouse xenograft model where animals were subcutaneously inoculated with HCT116 p53+/+ cells (3×106). Thirty-six tumor-bearing mice were randomly divided into 6 groups (n = 6) and received a 3-week subcutaneous treatment with medium, Nutlin-3, free PMI, Bcr/Abl-R6, PMIBcr/Abl and PMIBcr/Abl-R6 at the same dose of 5 mg/Kg every other day. As shown in Fig. 7A-D, while free PMI and Bcr/Abl-R6 had no effect on tumor growth, both Nutlin-3 and PMIBcr/Abl-R6 significantly inhibited it. Interestingly, PMIBcr/Abl was marginally active, suggesting that this protein, even without R6, was probably still able to partially traverse the cell membrane at high concentrations, albeit at a greatly reduced efficiency (Fig. 4C). It is important to point out that the molecular mass of PMIBcr/Abl-R6 is 16-fold higher than that of Nutlin-3. The fact that PMIBcr/Abl-R6 was even more effective than Nutlin-3 in inhibiting tumor growth suggests that PMIBcr/Abl-R6, on the basis of molar concentration, is at least 16-fold more active as a monomer and 64-fold more active as a tetramer than Nutlin-3 in vivo.
Fig. 7. PMIBcr/Abl-R6 inhibits tumor growth in vivo.
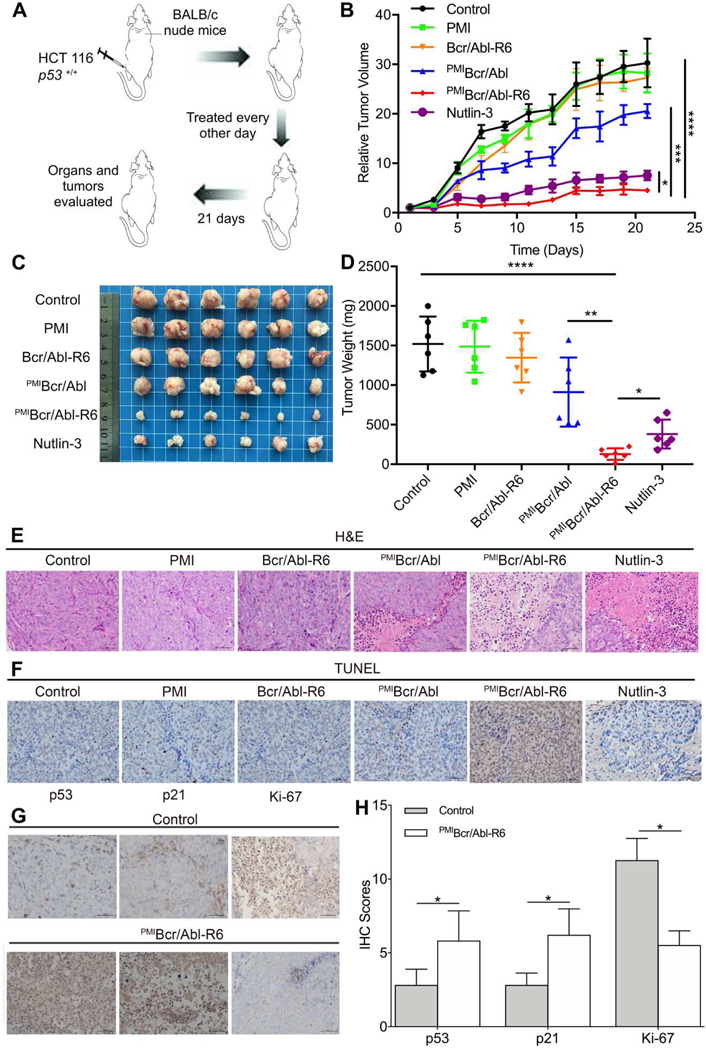
(A) Schematic diagram of therapy. Thirty six athymic nude mice (BALB/c) bearing HCT116 p53+/+ xenograft tumors, subcutaneously established in two weeks as a palpable mass (50–100 mm3 in size), were randomly divided into six groups (n=6/group), and treated every other day for three weeks via subcutaneous injection of 20 mM Tris-HCl (mock treatment) and PMI, Bcr/Abl-R6, PMIBcr/Abl, PMIBcr/Abl-R6 or Nutlin-3 at a dose of 5 mg/Kg. (B) Curves of inhibition of tumor growth during the 21-day treatment. Tumor length (L) and width (W) were measured with a caliper, and tumor volume (V) was calculated using the following equation: V=LxW2/2. The data represent the mean ± SD (n=6). Statistical analysis was performed using T-test, * standing for p<0.05, *** for p<0.001, and **** for p<0.0001. (C) Images of tumors collected upon conclusion of the three-week treatment. (D) The average weight of tumors excised from each group of mice at the end of treatment. Statistical analysis was performed using T-test, * standing for p<0.05, ** for p<0.01, and **** for p<0.0001. (E) Histopathological analysis using hematoxylin and eosin (H&E) staining. Representative tumors from each treatment group were fixed with formaldehyde, dehydrated and sliced into 5 µm-thick sections, and subjected to H&E staining according to standard protocols (scale bar: 50 µm). (F) Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay to stain fragmented DNA in apoptotic cells (scale bar: 50 µm). (G) Immunohistochemical (IHC) staining of tumor tissues using commercially available antibodies against p53, p21 and Ki-67 (scale bar: 50 µ m). Prepared tissue sections at 5 µm thickness were incubated with various antibodies at 4°C overnight, and subsequently stained using the Labeled Streptavidin-Biotin (LSAB) staining method. Each stained section was evaluated by a minimum of 10 randomly selected x20 high-power fields for further statistical analysis. (H) Statistical analysis of IHC scores. We used a numeric score ranging from 0 to 3 to evaluate immunostaining intensity (I): 0, no staining; 1, weak staining; 2, moderate staining; 3, strong staining. To evaluate immunostaining area (A), we used a numeric score ranging from 1 to 4: 1, positive area<10%; 2, 10%<positive area<50%; 3, 50%<positive area<90%; 4, positive area>90%. The total score, IxA, was calculated accordingly, and statistical analysis was performed using T-test, * standing for p<0.05.
Consistent with the above findings from the in vivo efficacy study, histopathological analysis using hematoxylin and eosin (H&E) (Fig. 7E) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) (Fig. 7F) staining techniques revealed massive necrotic and apoptotic tumor cells in the tissues from the PMIBcr/Abl-R6-treated group and, to a lesser extent, in the tissues from the Nutlin-3-treated group. H&E and TUNEL staining also confirmed the partial activity of PMIBcr/Abl and the lack of activity of PMI and Bcr/Abl-R6, as expected. Immunohistochemistry analysis demonstrated that PMIBcr/Abl-R6 treatment significantly increased the expression of p53 and p21 in tumor tissues but decreased the expression of the tumor progression marker Ki-67 (Fig. 7G-H). Taken together, our in vivo data unequivocally validate the design of PMIBcr/Abl-R6 as a potent antitumor agent that inhibits tumor growth in a p53-dependent fashion.
PMIBcr/Abl-R6 is minimally immunogenic and non-toxic to blood cells and kidney and liver tissues.
Immunogenicity of peptide/protein therapeutics often impedes their clinical use. We evaluated immunogenicity of PMI and PMIBcr/Abl-R6 in immune-competent C57BL/6 mice by measuring the level of the cytokines IL-2, TNF-α and erythropoietin (EPO) in the blood in response to subcutaneous treatments with PMI and PMIBcr/Abl-R6 for three weeks, every other day, at a dose of 5 mg/Kg. IL-2 and TNF-α were used as markers because T cell responses are known to play a critical role in the development of immunogenic responses to therapeutic peptides and proteins [52, 53]. Since biotherapeutics can potentially generate cross-reactive neutralizing antibodies that inhibit endogenous proteins such as EPO, leading to anemia known as antibody-mediated pure red-cell aplasia, EPO was also used as a marker for immunogenicity in our study. As shown in Fig. 8A-C, while free PMI noticeably increased IL-2 and TNF-α levels and decreased the level of EPO, only slight changes in the amount of IL-2, TNF-α and EPO were observed with PMIBcr/Abl-R6, suggesting that grafting PMI to the Bcr/Abl protein scaffold significantly dampened immunogenicity of the peptide drug.
Fig. 8. Safety evaluation of PMIBcr/Abl-R6 in vivo.
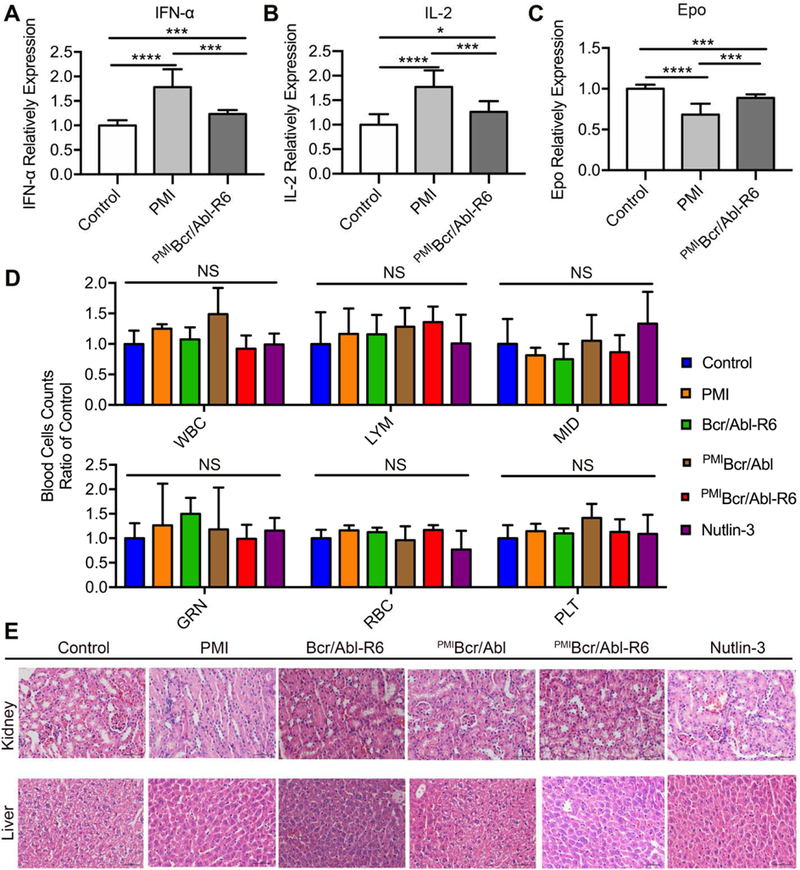
(A-C) Immunogenicity of PMI and PMIBcr/Abl-R6 in immune-competent C57BL/6 mice (n=6/group) as measured by the level of IL-2 (A), TNF-α (B) and erythropoietin (EPO) (C) in the blood in response to subcutaneous treatments with PMI and PMIBcr/Abl-R6 for three weeks, every other day, at a dose of 5 mg/Kg. PBS was used as a negative control for mock treatment; IL-2, TNF-α and EPO in the blood collected at the end of the treatment were quantified by ELISA kits (R&D Systems) using protein standards from Sigma-Aldrich. The data from each group are presented as the mean ± SD (n=6), and statistical analysis was performed using T-test, * standing for p<0.05, *** for p<0.001, and **** for p<0.0001. (D) Counts of different types of blood cells from a complete blood cell analysis after the 21-day treatment with PMI, Bcr/Abl-R6, PMIBcr/Abl, PMIBcr/Abl-R6 and Nutlin-3. WBC, white blood cell; LYM, lymphocyte; MID, monocyte; GRN, granulocytes; RBC, red blood cell; PLT, platelet. Statistical analysis was performed using T-test, NS standing for no significant difference. (E) Representative H&E staining of liver and kidney tissues from mice treated with with PMI, Bcr/Abl-R6, PMIBcr/Abl, PMIBcr/Abl-R6 and Nutlin-3 for three weeks (scale bar: 50 µm).
Some small molecule antagonists of MDM2 have showed cytotoxicity against B lymphocytes and hematopoietic stem cells in clinical trials, resulting in side effects such as thrombocytopenia, leukopenia and neutropenia [54]. We established the cytotoxicity profile of PMI, Bcr/Abl-R6, PMIBcr/Abl, PMIBcr/Abl-R6 and Nutlin-3 at the end of the three-week treatment by counting white blood cells, lymphocytes, monocytes, granulocytes, red blood cells, and platelets in a complete blood cell analysis. As shown in Fig. 8D, no statistically significant difference in the number of each cell type was observed for all five treatment groups compared with the mock-treated control group.
Since PMIBcr/Abl-R6 accumulates in the liver and kidney in addition to the solid tumor (Fig. 6), the two principal organs for drug metabolism and elimination, we also examined in vivo toxicity of PMI, Bcr/Abl-R6, PMIBcr/Abl, PMIBcr/Abl-R6 and Nutlin-3 to liver and kidney tissues by H&E staining at the end of the three-week treatment. As shown in Fig. 8E, no overt toxicity was observed at the doses used in our study. Taken together, the in vivo immunogenicity and toxicity data validate the safety of PMIBcr/Abl-R6.
Small molecule and peptide versus protein activators of p53.
The p53-MDM2/MDMX interaction has garnered much attention as an important intracellular drug target for the development of MDM2/MDMX antagonists or p53-activating agents for anticancer therapy [37, 55–57]. Small molecule antagonists are generally mono-specific for MDM2, and several are in clinical trials with promising early results [54, 58]. By contrast, peptide antagonists are often dual-specific for both MDM2 and MDMX, potentially affording more robust and sustained p53 activation. One notable example is ALRN-6924, a hydrocarbon-stapled peptide antagonist of MDM2 and MDMX kills tumor cells harboring wild-type p53 in phase 2 clinical trials for advanced solid tumors and lymphomas [59]. More recently, ALRN-6924 has been reported to be effective against acute myeloid leukemia in vitro and in vivo [60]. The hydrocarbon-stapling technique pioneered by Verdine and colleagues enables side-chain cross-linked and conformationally stabilized helical peptides to traverse the cell membrane with improved proteolytic stability and enhanced biological activity [61, 62]. Of note, a hydrocarbon- or dithiocarbamate-stapled PMI has been shown to be a potent p53 activator in vitro and in vivo [63–65]. Despite these successes, it is worth noting that small peptides do not have a sufficiently long circulation half-life in vivo due to renal excretion (<20 KDa), thus adversely affecting their therapeutic efficacy. By contrast, our protein construct PMIBcr/Abl-R6, a stable tetramer of 35 KDa that can be readily prepared in large quantity via recombinant expression, is expected to have excellent bioavailability compared with small peptide therapeutics.
Conclusion
We have demonstrated the tetrameric Bcr/Abl scaffold as an ideal protein-based nanocarrier of p53-activating peptides to target the p53-MDM2/MDMX interaction for cancer therapy. MDM2 and MDMX cooperate to persistently inhibit p53 function and target the tumor suppressor protein for proteasomal degradation, contributing to tumor development and progression. PMIBcr/Abl-R6 as a dual-specificity antagonist of MDM2 and MDMX and a powerful p53 activator in vitro and in vivo is superior in many aspects to mono-specific small molecule inhibitors of MDM2 as well as stapled peptide antagonists currently in clinical trials, promising a novel class of antitumor agents with significant therapeutic potential. Importantly, this protein-based nanocarrier is also suitable for the design of different classes of peptide therapeutics of an α-helical nature to target intracellular PPIs involved in many other human diseases.
Supplementary Material
Acknowledgement
We thank Prof. Bert Vogelstein of Johns Hopkins University for providing isogenic HCT116 cell lines. We also thank the staff from BL19U2 beamline of the National Center for Protein Sciences at Shanghai Synchrotron Radiation Facility, for assistance during data collection. This work was partially supported by the US National Institutes of Health Grants CA167296 and CA219150 (to WL), China Scholarship Council, and the Natural Science Foundation of China (31770146 to DX).
Footnotes
Conflicts of Interest
There are no conflicts of interest to declare.
Data Availability
The raw/processed data required to reproduce these findings cannot be shared at this time due to legal or ethical reasons.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Ideker T, Sharan R, Protein networks in disease, Genome research 18(4) (2008) 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Stelzl U, Worm U, Lalowski M, Haenig C, Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A, Koeppen S, A human protein-protein interaction network: a resource for annotating the proteome, Cell 122(6) (2005) 957–968. [DOI] [PubMed] [Google Scholar]
- [3].Milroy L-G, Grossmann TN, Hennig S, Brunsveld L, Ottmann C, Modulators of protein–protein interactions, Chemical reviews 114(9) (2014) 4695–4748. [DOI] [PubMed] [Google Scholar]
- [4].Barabási A-L, Gulbahce N, Loscalzo J, Network medicine: a network-based approach to human disease, Nature reviews genetics 12(1) (2011) 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Craik DJ, Fairlie DP, Liras S, Price D, The future of peptide-based drugs, Chemical biology & drug design 81(1) (2013) 136–147. [DOI] [PubMed] [Google Scholar]
- [6].Scott DE, Bayly AR, Abell C, Skidmore J, Small molecules, big targets: drug discovery faces the protein–protein interaction challenge, Nature Reviews Drug Discovery 15(8) (2016) 533. [DOI] [PubMed] [Google Scholar]
- [7].Vlieghe P, Lisowski V, Martinez J, Khrestchatisky M, Synthetic therapeutic peptides: science and market, Drug discovery today 15(1–2) (2010) 40–56. [DOI] [PubMed] [Google Scholar]
- [8].Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ, Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix, Science 305(5689) (2004) 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Robinson JA, β-Hairpin peptidomimetics: design, structures and biological activities, Accounts of chemical research 41(10) (2008) 1278–1288. [DOI] [PubMed] [Google Scholar]
- [10].Zuckermann RN, Kodadek T, Peptoids as potential therapeutics, Curr. Opin. Mol. Ther 11(3) (2009) 299–307. [PubMed] [Google Scholar]
- [11].Simon RJ, Kania RS, Zuckermann RN, Huebner VD, Jewell DA, Banville S, Ng S, Wang L, Rosenberg S, Marlowe CK, Peptoids: a modular approach to drug discovery, Proceedings of the National Academy of Sciences 89(20) (1992) 9367–9371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Daly NL, Craik DJ, Bioactive cystine knot proteins, Current opinion in chemical biology 15(3) (2011) 362–368. [DOI] [PubMed] [Google Scholar]
- [13].Gradauer K, Barthelmes J, Vonach C, Almer G, Mangge H, Teubl B, Roblegg E, Dünnhaupt S, Fröhlich E, Bernkop-Schnürch A, Liposomes coated with thiolated chitosan enhance oral peptide delivery to rats, Journal of controlled release 172(3) (2013) 872–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Niu Z, Conejos-Sánchez I, Griffin BT, O’Driscoll CM, Alonso MJ, Lipid-based nanocarriers for oral peptide delivery, Advanced drug delivery reviews 106 (2016) 337–354. [DOI] [PubMed] [Google Scholar]
- [15].Liu M, Pazgier M, Li C, Yuan W, Li C, Lu W, A Left-Handed Solution to Peptide Inhibition of the p53–MDM2 Interaction, Angewandte Chemie International Edition 49(21) (2010) 3649–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Shi Y, Teng P, Sang P, She F, Wei L, Cai J, gamma-AApeptides: Design, Structure, and Applications, Acc Chem Res 49(3) (2016) 428–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mitragotri S, Burke PA, Langer R, Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies, Nature reviews Drug discovery 13(9) (2014) 655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tan ML, Choong PF, Dass CR, Recent developments in liposomes, microparticles and nanoparticles for protein and peptide drug delivery, Peptides 31(1) (2010) 184–193. [DOI] [PubMed] [Google Scholar]
- [19].Mitra S, Montgomery JE, Kolar MJ, Li G, Jeong KJ, Peng B, Verdine GL, Mills GB, Moellering RE, Stapled peptide inhibitors of RAB25 target context-specific phenotypes in cancer, Nature communications 8(1) (2017) 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bernal F, Wade M, Godes M, Davis TN, Whitehead DG, Kung AL, Wahl GM, Walensky LD, A stapled p53 helix overcomes HDMX-mediated suppression of p53, Cancer cell 18(5) (2010) 411–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu M, Li C, Pazgier M, Li C, Mao Y, Lv Y, Gu B, Wei G, Yuan W, Zhan C, D-peptide inhibitors of the p53–MDM2 interaction for targeted molecular therapy of malignant neoplasms, Proceedings of the National Academy of Sciences 107(32) (2010) 14321–14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Shi J, Votruba AR, Farokhzad OC, Langer R, Nanotechnology in drug delivery and tissue engineering: from discovery to applications, Nano letters 10(9) (2010) 3223–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wang C, Cheng L, Liu Z, Drug delivery with upconversion nanoparticles for multi-functional targeted cancer cell imaging and therapy, Biomaterials 32(4) (2011) 1110–1120. [DOI] [PubMed] [Google Scholar]
- [24].Ma Y, Liang X, Tong S, Bao G, Ren Q, Dai Z, Gold Nanoshell Nanomicelles for Potential Magnetic Resonance Imaging, Light-Triggered Drug Release, and Photothermal Therapy, Advanced functional materials 23(7) (2013) 815–822. [Google Scholar]
- [25].Al-Jamal WT, Kostarelos K, Liposomes: from a clinically established drug delivery system to a nanoparticle platform for theranostic nanomedicine, Accounts of chemical research 44(10) (2011) 1094–1104. [DOI] [PubMed] [Google Scholar]
- [26].Altunbas A, Lee SJ, Rajasekaran SA, Schneider JP, Pochan DJ, Encapsulation of curcumin in self-assembling peptide hydrogels as injectable drug delivery vehicles, Biomaterials 32(25) (2011) 5906–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Elzoghby AO, Samy WM, Elgindy NA, Protein-based nanocarriers as promising drug and gene delivery systems, Journal of controlled release 161(1) (2012) 38–49. [DOI] [PubMed] [Google Scholar]
- [28].Lohcharoenkal W, Wang L, Chen YC, Rojanasakul Y, Protein nanoparticles as drug delivery carriers for cancer therapy, BioMed research international 2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Elzoghby AO, Samy WM, Elgindy NA, Albumin-based nanoparticles as potential controlled release drug delivery systems, Journal of controlled release 157(2) (2012) 168–182. [DOI] [PubMed] [Google Scholar]
- [30].Peng Q, Zhang S, Yang Q, Zhang T, Wei X-Q, Jiang L, Zhang C-L, Chen Q-M, Zhang Z-R, Lin Y-F, Preformed albumin corona, a protective coating for nanoparticles based drug delivery system, Biomaterials 34(33) (2013) 8521–8530. [DOI] [PubMed] [Google Scholar]
- [31].Chan LY, Gunasekera S, Henriques ST, Worth NF, Le S-J, Clark RJ, Campbell JH, Craik DJ, Daly NL, Engineering pro-angiogenic peptides using stable, disulfide-rich cyclic scaffolds, Blood 118(25) (2011) 6709–6717. [DOI] [PubMed] [Google Scholar]
- [32].Li C, Pazgier M, Liu M, Lu WY, Lu W, Apamin as a Template for Structure-Based Rational Design of Potent Peptide Activators of p53, Angewandte Chemie International Edition 48(46) (2009) 8712–8715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Li C, Liu M, Monbo J, Zou G, Li C, Yuan W, Zella D, Lu W-Y, Lu W, Turning a scorpion toxin into an antitumor miniprotein, Journal of the American Chemical Society 130(41) (2008) 13546–13548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhao X, Ghaffari S, Lodish H, Malashkevich VN, Kim PS, Structure of the Bcr-Abl oncoprotein oligomerization domain, Nature Structural and Molecular Biology 9(2) (2002) 117. [DOI] [PubMed] [Google Scholar]
- [35].Khoo KH, Verma CS, Lane DP, Drugging the p53 pathway: understanding the route to clinical efficacy, Nature reviews Drug discovery 13(3) (2014) 217–236. [DOI] [PubMed] [Google Scholar]
- [36].Zhang Q, Zeng SX, Lu H, Targeting p53-MDM2-MDMX loop for cancer therapy, Mutant p53 and MDM2 in Cancer, Springer; 2014, pp. 281–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wade M, Li YC, Wahl GM, MDM2, MDMX and p53 in oncogenesis and cancer therapy, Nat Rev Cancer 13(2) (2013) 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Li C, Pazgier M, Li C, Yuan W, Liu M, Wei G, Lu W-Y, Lu W, Systematic mutational analysis of peptide inhibition of the p53–MDM2/MDMX interactions, Journal of molecular biology 398(2) (2010) 200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Pazgier M, Liu M, Zou G, Yuan W, Li C, Li C, Li J, Monbo J, Zella D, Tarasov SG, Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX, Proceedings of the National Academy of Sciences 106(12) (2009) 4665–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Dawson PE, Muir TW, Clark-Lewis I, Kent S, Synthesis of proteins by native chemical ligation, Science 266(5186) (1994) 776–779. [DOI] [PubMed] [Google Scholar]
- [41].Dawson PE, Kent SB, Synthesis of native proteins by chemical ligation, Annual review of biochemistry 69(1) (2000) 923–960. [DOI] [PubMed] [Google Scholar]
- [42].Yan LZ, Dawson PE, Synthesis of peptides and proteins without cysteine residues by native chemical ligation combined with desulfurization, Journal of the American Chemical Society 123(4) (2001) 526–533. [DOI] [PubMed] [Google Scholar]
- [43].Patel LN, Zaro JL, Shen W-C, Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives, Pharmaceutical research 24(11) (2007) 1977–1992. [DOI] [PubMed] [Google Scholar]
- [44].Åmand HL, Rydberg HA, Fornander LH, Lincoln P, Nordén B, Esbjörner EK, Cell surface binding and uptake of arginine-and lysine-rich penetratin peptides in absence and presence of proteoglycans, Biochimica et Biophysica Acta (BBA)-Biomembranes 1818(11) (2012) 2669–2678. [DOI] [PubMed] [Google Scholar]
- [45].Geyer RK, Zhong KY, Maki CG, The MDM2 RING-finger domain is required to promote p53 nuclear export, Nature cell biology 2(9) (2000) 569. [DOI] [PubMed] [Google Scholar]
- [46].Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA, Kinzler KW, Vogelstein B, Papadopoulos N, A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53, Proceedings of the National Academy of Sciences 106(10) (2009) 3964–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, In vivo activation of the p53 pathway by small-molecule antagonists of MDM2, Science 303(5659) (2004) 844–848. [DOI] [PubMed] [Google Scholar]
- [48].Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D, p21 is a universal inhibitor of cyclin kinases, nature 366(6456) (1993) 701. [DOI] [PubMed] [Google Scholar]
- [49].Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ, Adams JM, Strasser A, p53-and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa, Science 302(5647) (2003) 1036–1038. [DOI] [PubMed] [Google Scholar]
- [50].Maeda H, Nakamura H, Fang J, The EPR effect for macromolecular drug delivery to solid tumors: Improvement of tumor uptake, lowering of systemic toxicity, and distinct tumor imaging in vivo, Advanced drug delivery reviews 65(1) (2013) 71–79. [DOI] [PubMed] [Google Scholar]
- [51].Torchilin V, Tumor delivery of macromolecular drugs based on the EPR effect, Advanced drug delivery reviews 63(3) (2011) 131–135. [DOI] [PubMed] [Google Scholar]
- [52].Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS, T-cell dependent immunogenicity of protein therapeutics: preclinical assessment and mitigation, Clinical immunology 149(3) (2013) 534–555. [DOI] [PubMed] [Google Scholar]
- [53].De Groot AS, Scott DW, Immunogenicity of protein therapeutics, Trends in immunology 28(11) (2007) 482–490. [DOI] [PubMed] [Google Scholar]
- [54].Burgess A, Chia KM, Haupt S, Thomas D, Haupt Y, Lim E, Clinical Overview of MDM2/X-Targeted Therapies, Front Oncol 6 (2016) 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Khoo KH, Verma CS, Lane DP, Drugging the p53 pathway: understanding the route to clinical efficacy, Nature reviews. Drug discovery 13(3) (2014) 217–36. [DOI] [PubMed] [Google Scholar]
- [56].Zhang Q, Zeng SX, Lu H, Targeting p53-MDM2-MDMX loop for cancer therapy, Subcell Biochem 85 (2014) 281–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Estrada-Ortiz N, Neochoritis CG, Domling A, How To Design a Successful p53-MDM2/X Interaction Inhibitor: A Thorough Overview Based on Crystal Structures, ChemMedChem 11(8) (2016) 757–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Zhao Y, Aguilar A, Bernard D, Wang S, Small-molecule inhibitors of the MDM2-p53 protein-protein interaction (MDM2 Inhibitors) in clinical trials for cancer treatment, J Med Chem 58(3) (2015) 1038–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Meric-Bernstam F, Saleh MN, Infante JR, Goel S, Falchook GS, Shapiro G, Chung KY, Conry RM, Hong DS, Wang JS-Z, Steidl U, Walensky LD, Guerlavais V, Payton M, Annis DA, Aivado M, Patel MR, Phase I trial of a novel stapled peptide ALRN-6924 disrupting MDMX- and MDM2-mediated inhibition of WT p53 in patients with solid tumors and lymphomas, Journal of Clinical Oncology 35(15_suppl) (2017) 2505–2505. [Google Scholar]
- [60].Carvajal LA, Neriah DB, Senecal A, Benard L, Thiruthuvanathan V, Yatsenko T, Narayanagari SR, Wheat JC, Todorova TI, Mitchell K, Kenworthy C, Guerlavais V, Annis DA, Bartholdy B, Will B, Anampa JD, Mantzaris I, Aivado M, Singer RH, Coleman RA, Verma A, Steidl U, Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia, Sci Transl Med 10(436) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Bernal F, Tyler AF, Korsmeyer SJ, Walensky LD, Verdine GL, Reactivation of the p53 tumor suppressor pathway by a stapled p53 peptide, J Am Chem Soc 129(9) (2007) 2456–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Verdine GL, Hilinski GJ, Stapled peptides for intracellular drug targets, Methods Enzymol 503 (2012) 3–33. [DOI] [PubMed] [Google Scholar]
- [63].Brown CJ, Quah ST, Jong J, Goh AM, Chiam PC, Khoo KH, Choong ML, Lee MA, Yurlova L, Zolghadr K, Joseph TL, Verma CS, Lane DP, Stapled peptides with improved potency and specificity that activate p53, ACS Chem Biol 8(3) (2013) 506–12.23214419 [Google Scholar]
- [64].Chen X, Tai L, Gao J, Qian J, Zhang M, Li B, Xie C, Lu L, Lu W, Lu W, A stapled peptide antagonist of MDM2 carried by polymeric micelles sensitizes glioblastoma to temozolomide treatment through p53 activation, J Control Release 218 (2015) 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Li X, Tolbert WD, Hu H-G, Gohain N, Zou Y, Niu F, He W-X, Yuan W, Su J-C, Pazgier M, Lu W, Dithiocarbamate-inspired side chain stapling chemistry for peptide drug design Chemical science (2019). [DOI] [PMC free article] [PubMed]
- [66].Schnolzer M, Alewood P, Jones A, Alewood D, Kent SB, In situ neutralization in Boc-chemistry solid phase peptide synthesis. Rapid, high yield assembly of difficult sequences, Int J Pept Protein Res 40(3–4) (1992) 180–93. [DOI] [PubMed] [Google Scholar]
- [67].Pace CN, Vajdos F, Fee L, Grimsley G, Gray T, How to measure and predict the molar absorption coefficient of a protein, Protein Sci 4(11) (1995) 2411–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Chen Y-H, Yang JT, Martinez HM, Determination of the secondary structures of proteins by circular dichroism and optical rotatory dispersion, Biochemistry 11(22) (1972) 4120–4131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.