

Abstract
Triple negative breast cancer (TNBC) represents an aggressive phenotype with poor prognosis compared with ER, PR, and HER2‐positive tumors. TNBC is a heterogeneous disease, and gene expression analysis has identified seven molecular subtypes. Accumulating evidence demonstrates that long non‐coding RNA (lncRNA) are involved in regulation of gene expression and cancer biology, contributing to essential cancer cell functions. In this study, we analyzed the expression profile of lncRNA in TNBC subtypes from 156 TNBC samples, and then characterized the functional role of LncKLHDC7B (ENSG00000226738). A total of 710 lncRNA were found to be differentially expressed between TNBC subtypes, and a subset of these altered lncRNA were independently validated. We discovered that LncKLHDC7B (ENSG00000226738) acts as a transcriptional modulator of its neighboring coding gene KLHDC7B in the immunomodulatory subtype. Furthermore, LncKLHDC7B knockdown enhanced migration and invasion, and promoted resistance to cellular death. Our findings confirmed the contribution of LncKLHDC7B to induction of apoptosis and inhibition of cell migration and invasion, suggesting that TNBC tumors with enrichment of LncKLHDC7B may exhibit distinct regulatory activity, or that this may be a generalized process in breast cancer. Additionally, in silico analysis confirmed for the first time that the low expression of KLHDC7B and LncKLHDC7B is associated with poor prognosis in patients with breast cancer.
Keywords: ENSG00000226738, invasion, LncKLHDC7B, long non‐coding RNA, migration, triple‐negative breast cancer
Abbreviations
- ADAM9
ADAM metallopeptidase domain 9
- AMOT
angiomotin
- APP
amyloid beta precursor protein
- BL1
basal‐like 1
- BL2
basal‐like 2
- CAV1
caveolin 1
- CCAR2
cell cycle and apoptosis regulator 2
- CDH3
cadherin 3
- CI
confidence interval
- ECM
extracellular matrix
- EPS8
epidermal growth factor receptor pathway substrate 8
- ER
estrogen receptor
- FDR
false discovery rate
- FFPE
formalin‐fixedparaffin‐embedded
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GBA
guilt‐by‐association analysis
- GEO
gene expression omnibus
- HER2
human epidermal growth factor receptor 2
- HR
hazard ratio
- HTA
human transcriptome array
- IM
immunomodulatory
- IPA
ingenuity pathway analysis
- KLHDC7B
Kelch domain containing 7B
- KLHL39
Kelch‐like protein 39
- LAR
luminal androgen receptor
- LncKLHDC7
long non‐coding KLHDC7B
- lncMAP
LncRNA modulator atlas in pan‐cancer
- lncRNA
long non‐coding RNA
- MHC
major histocompatibility complex
- M
mesenchymal
- MMP2
matrix metallopeptidase 2
- MSL
mesenchymal stem‐like
- PMAIP1
phorbol‐12‐myristate‐13‐acetate‐induced protein 1
- PR
progesterone receptor
- QBS
2‐amino‐N‐quinolin‐8‐yl‐benzenesulfonamide
- RAD21
RAD21 cohesin complex component
- shRNA
short hairpin RNA
- TCGA
The Cancer Genome Atlas
- TGF
transforming growth factor
- TN
triple‐negative
- TNBC
triple‐negative breast cancer
- TNC
tenascin C
- TNF
tumor necrosis factor
- U87
small nucleolar RNA U87
- UNS
unstable
1. Introduction
Triple negative breast cancer (TNBC), defined in the clinic by the negative expression of estrogen receptor (ER), progesterone receptor (PR), and lack of overexpression of human epidermal growth factor receptor 2 (HER2), is a heterogeneous tumor that represents ~ 14–23% of all breast cancers (BC) (Lara‐Medina et al., 2011; Martinez et al., 2013; Perez‐Rodriguez, 2015). TNBC currently lacks targeted treatment, is more aggressive, has worse global survival in metastatic disease, affects young women, and a higher proportion of African American and Hispanic women (Bauer et al., 2007; Dent et al., 2007; Foulkes et al., 2010; Rastelli et al., 2010). With the development and advancement of high throughput technologies, TNBC has been classified into intrinsic subtypes based on coding gene expression [messenger (m)RNA] and ontology analysis into the following groups: two basal‐like (BL1 and BL2), immunomodulatory (IM), mesenchymal (M), mesenchymal stem‐like (MSL), luminal androgen receptor (LAR), and unstable (UNS) subtypes (Lehmann et al., 2011). Subtyping of TNBC based on non‐coding RNA (ncRNA) expression and research on its therapeutic implications are still being refined (Lehmann et al., 2016).
During the development of cancer, major transcriptional alterations can be induced by ncRNA, leading to changes in global gene expression patterns and genomic instability (Wapinski and Chang, 2011). A large proportion of RNAs with no coding capacity (Consortium, 2012), such as long non‐coding RNA (lncRNA) (Martin and Chang, 2012), have emerged as key players in several biological processes. LncRNA have > 200 nucleotides, transcribed by RNA polymerase II, mainly located within the nucleus but also found in the cytosolic compartment, lack protein‐coding potential, and show lower expression compared with mRNA (Derrien et al., 2012). The lncRNA species are also considered to be new protagonists in the development of cancer, with potential roles in oncogenic and tumor suppressor pathways (Gibb et al., 2011; Li and Chen, 2013; Wapinski and Chang, 2011).
To analyze the differences in lncRNA expression landscapes in TNBC subtypes, we evaluated lncRNA expression profiles of 156 TNBC samples. TNBC subtypes were first defined by mRNA microarray expression profiling, and differentially expressed lncRNA were identified between subtypes. A significant number of differentially expressed lncRNA were identified between all subtypes, and some of them were validated in an independent cohort of TNBC samples. LncKLHDC7B, an lncRNA that was over‐expressed in the immunomodulatory subtype, was selected for further investigation of its functional role in TNBC through modulation of its expression in cell lines. Down‐modulation of LncKLHDC7B resulted in down‐regulation of its coding gene (KLHDC7B) and in the alteration of the expression of several other genes, as well as in an increase in migration and invasion, in addition to resistance to cell death not only in TNBC cell lines, but also in other breast tumor subtypes, suggesting its potential role in breast oncogenesis.
2. Materials and methods
2.1. Collection and processing of samples
A total of 156 formalin‐fixed, paraffin‐embedded (FFPE) tissues were collected from the Instituto de Enfermedades de la Mama FUCAM (Mexico) and the Istituto Nazionale dei Tumori (Italy) by retrospective collection from 2007 to 2015. Samples were defined as triple‐negative using the current international histopathological criteria, less than 1% of positive cells for ER and PR, and HER2. Immunohistochemical detection of the three markers was carried out using the following antibodies: ER (clone 1D5, Dako, Denmark, Carpinteria, CA, USA), PR (clone PgR636, Dako, Denmark) and HER2 (K5204, Dako). Cytokeratin 5/6 and Ki67 (clone D5/16B4, Dako, Denmark) were also evaluated to rule out potential false‐negative immunohistochemical reactions. All procedures were performed according to the Declaration of Helsinki, and were reviewed and approved by the Ethics and Research Committees of the National Institute of Genomic Medicine (approval number CEI2016/13).
A pathologist identified regions of the sample with the highest amount of cancer tissue, and tissue cores were obtained with a 2‐mm‐wide needle with a tissue arrayer. Tissue cores were used for RNA extraction using the AllPrep® nucleic acid purification FFPE kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Subsequently, RNA was quantified by spectrophotometry (Nanodrop, Thermo Fisher Scientific, Waltham, MA, USA) and stored at −80 °C until further processing.
2.2. Microarray processing and data analysis
Transcriptional profiles were analyzed using the Affymetrix Human Transcriptome Array V2.0, following the manufacturer's instructions. Briefly, ~ 200 ng of total RNA was converted into complementary (c)DNA and labeled with the SensationPlus™ FFPE Amplification and WT Labeling® kit (Affymetrix, Santa Clara, CA, USA) and hybridized on the array, which detects both mRNA and lncRNA. Arrays were washed, stained, and scanned using a Genechip Scanner 3000 7G (Affymetrix). The data were analyzed with the Robust Multichip Analysis (RMA) algorithm using Affymetrix default analysis settings and global scaling as normalization method on a Affymetrix Transcriptome Analysis Console (tac) V3.0 [Data accessibility gene expression omnibus (GEO): GSE86948]. The ComBat function was used to remove batch effects using the SVA package. The boxplots, heat maps, and correlation analyses were conducted in r software (http://cran.r-project.org; http://www.bioconductor.org; ggplots cran.r‐project.org) using tools from the Bioconductor project (http://www.bioconductor.org) (Wickham, 2009).
2.3. Messenger RNA subtyping of triple‐negative breast cancer samples and lncRNA expression profiles
The Web‐based TNBCtype algorithm (http://cbc.mc.vanderbilt.edu/tnbc/) was used to identify the mRNA‐based subtypes of TNBC in our cases (Chen et al., 2012). Four of 160 TNBC samples were excluded from lncRNA expression profile analysis, according to the bimodal filter of the TNBCtype algorithm (Chen et al., 2012). After mRNA subtyping, we analyzed the lncRNA expression profiles between subgroups using the Affymetrix Transcriptome Analysis Console (tac) v.3.0, through comparison of the samples belonging to a specific subtype against to the rest of the samples (e.g. IM vs other subtypes). A ≥ 1.5‐fold change, ANOVA P‐value less than or equal to 0.05, and false discovery rate (FDR) < 0.05 were considered as significant to detect expression changes between the TNBC subtypes, except for BL2 where FDR was < 0.5. A heatmap with the differentially expressed lncRNA is shown in Fig. 1. TNBC samples from the GEO database (Data accessibility GEO: GSE76250) were used for validation of differential gene expression in an independent cohort (Liu et al., 2016). Gene ontology and cellular pathway analysis were carried out using david (Frederick, MD, USA) (Huang da et al., 2009). Venn diagrams were used to show the relationship between groups of differentially expressed lncRNA (Heberle et al., 2015).
Figure 1.
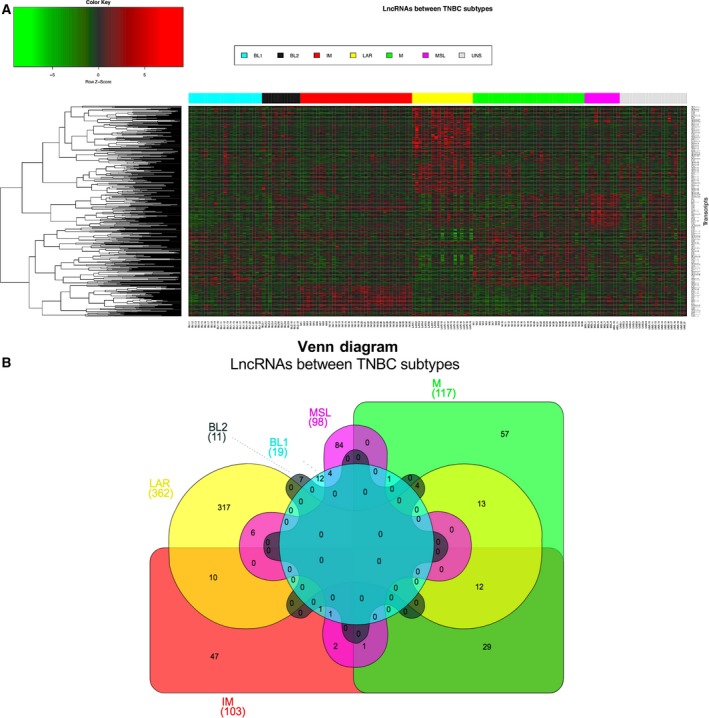
Differential expression of lncRNA across TNBC subtypes. (A) Supervised hierarchical clustering of the differentially expressed lncRNA in TNBC subtypes. Red represents up‐regulation and green down‐regulation. Rows correspond to transcripts and columns to samples, color bar represents each TNBC subtype. (B) Venn diagram shows the common and differential lncRNA between TNBC subtypes.
2.4. Guilt‐by‐association analysis
Enrichment pathway analysis was carried out by a guilt‐by‐association analysis (Guttman et al., 2009). Briefly, normalized gene expression of mRNA and lncRNA of the 156 profiled tumors were used to build a correlation matrix including only the differentially expressed lncRNA and mRNA for each subtype. For each lncRNA, mRNA were ranked according to their Spearman correlation coefficient and P value. A significant correlation between lncRNA and mRNA was considered as follows: Spearman correlation > 30% and P value < 0.05. The lncRNA‐mRNA co‐expression was computed to define candidate lncRNA prioritization by identifying which transcripts show a coordinated expression pattern across a group of samples. The significant correlated mRNA were then evaluated in an enrichment pathway analysis to define their impact on the signaling process, using enrichr (New York , NY, USA) (Kuleshov et al., 2016) to associate the altered lncRNA with pathways and signatures that were significantly enriched. The co‐expression correlations were the basis for constructing an enrichment pathway analysis in which each term represents a set of transcripts with a significant strength co‐expression relationship. To compute this weighted correlation analysis, some functions of the wgcna package were applied (Los Angeles, CA, USA) (Langfelder and Horvath, 2008).
2.5. Immunoscore prediction
Gene expression profile of four immune‐related classes (effector cells, immunosuppressive cells, MHC molecules, and selected immunomodulators) were evaluated to determine the Immunophenoscore* using the available r‐script deposited on GitHub (https://github.com/mui-icbi/Immunophenogram).
2.6. Analysis of LncKLHDC7B expression in breast cancer cell lines
MCF10A, BT20, MDA‐MB‐468, MDA‐MB‐231, Hs578‐T, MCF‐7, and HCC1187 cell lines were grown under standard conditions (DMEM or RPMI‐1640 medium, ATCC®, Manassas, VA, USA) according to each line, supplemented with 10% FBS (ATCC®) and incubated at ~ 37 °C with 5% CO2. Total RNA was isolated using the Trizol reagent (Invitrogen, Carlsbad, CA, USA) according the manufacturer's recommendation. Subsequently, RNA was quantified by spectrophotometry (Nanodrop) and stored at −80 °C until further processing.
2.7. Reverse transcription polymerase chain reaction
Complementary DNA was synthesized using SuperScript III RT‐PCR (Invitrogen) following the manufacturer's recommendations. Briefly, 100 ng of total RNA was used to synthesize cDNA in a final reaction volume of 20 μL. The real time PCR mixture contained 1 μL of cDNA, 5 μL 2× TaqMan Universal Master Mix (Applied Biosystems), 0.5 μL TaqMan® probes (Hs00536653_s1, custom from tcon_00029630), and 3.5 μL of nuclease‐free water. GAPDH (Hs99999905_m1) and U87 (Hs03298717_s1) were used as endogenous controls for coding and non‐coding genes, respectively. The fold change for each gene (KLHDC7B or LncKLHDC7B) in treated cells relative to control cells was calculated using the Ct (2^‐ΔΔCT) method, where ΔΔC t = ΔC t treated cell (shRNA‐1 or ‐2) – ΔC t control cell (NC); ΔC t = C t coding gene – C t GAPDH or ΔC t = Ct non‐coding gene – C t U87.
2.8. Long non‐coding RNA down‐regulation with shRNA
Short hairpin RNA were generated using the BLOCK‐iT™ U6 RNAi Entry Vector Kit (Invitrogen) following the manufacturer's instructions. Briefly, pairs of cDNA oligos were designed containing four nucleotide overhangs necessary for directional cloning. We generated the double‐stranded oligo (ds‐oligo) and subsequently performed the ligation reaction of ds‐oligo into the pENTR™/U6 vector, which was used to transform competent Escherichia coli One Shot® TOP10 cells. Sanger sequencing was used to corroborate the presence and correct orientations of the ds‐oligo insert. Two shRNA were used to silence LncKLHDC7B (shRNA‐1, 5′‐CACCGCCTCAGCCCAAGTCTTAACTCGAAAGTTAAGACTTGGGCTGAGGC‐3′ and shRNA‐2, 5′‐CACCGCCCAAGTCTTAACTTCAGCTCGAAAGCTGAAGTTAAGACTTGGGC‐3′) and negative control (NC, 5′‐CACCGGAATTACGGAGTCTTCTTCGCGAACGAAGAAGACTCCGTAATTCC‐3′), with a sequence that does not target any mRNA in the human genome GFP.
Silencing of LncKLHDC7B was performed in three cell lines: HCC1187 (TNBC), MCF‐7 (Luminal A) and BT‐20 (TNBC) using two shRNA (shRNA‐1 and shRNA‐2), which were transfected using Xfect™ Transfection Reagent Protocol (Clontech, Palo Alto, CA, USA). Briefly, HCC1187 cells (6.5 × 105 cells per well), BT‐20 (5 × 105 cells per well) and MCF‐7 (5 × 105 cells per well) were seeded in 6‐well plates and 3 μg of plasmid was transfected during 24 h. Nanoparticle complexes were removed and replaced with complete growth medium. After 48 h post‐transfection, expression of LncKLHDC7B was evaluated by RT‐PCR. The RNA was extracted using Trizol according the manufacturer's recommendations and stored at −80 °C until processing. A > 70% decrease of lncRNA expression was achieved after 48 h post‐transfection using 2–3.5 μg of plasmid. The experiments were carried out in triplicate. The effect of silencing LncKLHDC7B on the whole‐genome transcriptional landscape in HCC1187 was analyzed with the HTA 2.0 microarray (Affymetrix; Data accessibility GEO: GSE114468), as described above. Genes with a fold change of > 2 and < −2, and a P value <0.05 were considered significant and selected for biological pathway analysis using Ingenuity Pathway Analysis (ipa) software and genetrail2 (Stockel et al., 2016).
2.9. Migration and invasion assay
Cell migration and invasion assays were performed using Transwell migration chambers with 8‐μm pores (Corning, NY, USA). Transfected cells were harvested, re‐suspended in RPMI without FBS at a concentration of 5 × 104 cells in 100 μL, and seeded into the upper chamber of the 24‐well plate. The lower chamber was filled with 600 μL RPMI containing 10% FBS. For the invasion assay, 1 × 105 cells were plated on chambers precoated with 1.6 mg Matrigel (Corning). Cells were incubated (HCC1187 and MCF‐7 for 24 h, BT‐20 for 6 h) for migration assay and (HCC1187 for 36 h, BT‐20 and MCF‐7 for 48 h) for invasion assay at 37 °C, in 5% CO2. Each experiment was done in triplicate. At the end of the experiments, cells that migrated to the reverse side of the Transwell membrane were fixed with paraformaldehyde (3.7%), stained with crystal violet, and counted using imagej software (Madison, WI, USA) (Schneider et al., 2012).
2.10. Apoptosis assay
Apoptosis was induced with ~ 50 μm of 2‐amino‐N‐quinolin‐8‐yl‐benzenesulfonamide (QBS; A3105, Sigma‐Aldrich, St. Louis, MO, USA) for 24 h before the end of 48 h post‐transfection. The cells were then immediately harvested by trypsinization and washed with PBS. The cells were re‐suspended in binding buffer and stained with Annexin V and PI (FITC Annexin V/ Dead cell Apoptosis kit, Invitrogen) for 15 min in the dark at room temperature. The stained cells were examined by cytometry. The cells were categorized into early apoptotic cells and late apoptotic cells (bounded in red lines).
2.11. Statistical analysis
Statistical significance was analyzed using graphpad prism (version 6, San Diego, CA, USA) and stata (Version 12, College Station, TX, USA) software. Variances in Kruskal–Wallis tests, ANOVA test, Student's t test, and chi‐square tests were performed for all comparisons involving categorical variables. Correlation between variables was determined by Spearman's correlation coefficient. The Kaplan–Meier method and survival differences among groups were assessed by log‐rank test. A P value < 0.05 was considered significant (*P < 0.05, **P < 0.01, ***P < 0.001).
3. Results
3.1. Population characteristics and TNBC subtypes distribution
A total of 156 samples derived from patients with TNBC were included in this study. The mean age of patients was 53 years old (range 26–88) and infiltrating ductal carcinoma was the most frequent type of tumor. Clinical and pathological information on TNBC patients included in this study is shown in Table 1. TNBC subtypes were identified using the TNBCtype algorithm, which assigned samples to the six triple‐negative breast cancer subtypes. Analyzing our TNBC cohort, the majority of tumors were clustered as IM and M (22.4%, n = 35/156), followed by BL1 (14.7%, n = 23/156) and LAR subtypes (12.2%, n = 19/156), BL2 (7.7%, n = 12/156), MSL subtype (7%, n = 11/156), and UNS (13.5%, n = 21/156) (Fig. S1a). The distribution of the TNBC subtypes in our cohort was similar to previous studies (Lehmann et al., 2011; Liu et al., 2016; Masuda et al., 2013) (Fig. S1b). david enrichment analysis (Huang da et al., 2009) was applied to each of the TNBC subtypes to corroborate previously reported signaling pathways enriched in each TNBC subtype (Table S1, Fig. S2).
Table 1.
Clinical‐pathological characteristics of the population
| Characteristics | Number | BL1 | BL2 | IM | M | MSL | LAR | UNS | P‐valuea |
|---|---|---|---|---|---|---|---|---|---|
| (Total = 156) | (n = 23) | (n = 12) | (n = 35) | (n = 35) | (n = 11) | (n = 19) | (n = 21) | ||
| Age (Mean ± SD) | 145 | 47.7±10.4 | 59±14.6 | 50.1±12.6 | 55.2±14.2 | 53.3±10.4 | 59.1±15.6 | 51.9±14.6 | 0.0835b |
| Tumor grade | |||||||||
| ≤II | 26 | 1 (3.8) | 1 (2.1) | 3 (5.5) | 7 (5.9) | 1 (1.9) | 8 (3.1) | 5 (3.6) | |
| >II | 110 | 19 (16.2) | 10 (8.9) | 26 (23.5) | 24 (25.1) | 9 (8.1) | 8 (12.9) | 14 (15.4) | 0.0148 c |
| Unknown | 20 | ||||||||
| Tumor size (cm) | |||||||||
| ≤ 2 cm | 64 | 6 (9.3) | 6 (5.1) | 12 (14.4) | 15 (13.9) | 7 (4.6) | 9 (7.4) | 9 (9.3) | |
| > 2 cm | 74 | 14 (10.7) | 5 (5.9) | 19 (16.6) | 15 (16.1) | 3 (5.4) | 7 (8.6) | 11 (10.7) | 0.398c |
| Unknown | 18 | ||||||||
| Type histology | |||||||||
| IDC | 117 | 17 (15.9) | 6 (9.2) | 29 (26.7) | 28 (26.7) | 8 (8.4) | 13 (13.4) | 16 (16.7) | |
| Other | 23 | 2 (3.1) | 5 (1.8) | 3 (5.3) | 4 (5.3) | 2 (1.6) | 3 (2.6) | 4 (3.3) | 0.168c |
| Unknown | 16 | ||||||||
| Follow up, month (mean) | 55.8 | 50.5 | 56.6 | 55 | 48 | 65.8 | 59.8 | 62.1 | 0.562d |
| CI | 50.7–60.9 | 34.8–66.2 | 37.3–75.8 | 43.9–66.1 | 37.6–58.4 | 41.6–90.1 | 42–77.6 | 49.2–75.1 | |
| Dead event | |||||||||
| Yes | 30 | 8 (4.2) | 4 (2.3) | 9 (6.8) | 5 (6.8) | 1 (2.3) | 1 (3.4) | 2 (4.2) | 0.064c |
| No | 112 | 12 (15.8) | 7 (8.7) | 23 (25.2) | 27 (25.2) | 10 (8.7) | 15 (12.6) | 18 (15.8) | |
| Unknown | 14 | ||||||||
BL1, basal‐like 1; BL2, basal‐like 2; CI, 95% confidence interval; IDC, infiltrating ductal carcinoma; IM, immunomodulatory; LAR, luminal androgen receptor; M, mesenchymal; MSL, mesenchymal stem‐like; SD, standard deviation; UNS, undetermined.
Unknown data were not included for the statistical significance.
ANOVA test.
Chi‐square test P < 0.05 (in bold).
Kruskal–Wallis test.
3.2. Unique lncRNA expression profiles in TNBC subtypes
Once our tumor cohort was classified into TNBC subtypes, we identified expression patterns of lncRNA among the tumor subclasses (Fig. 1A). In particular, 710 lncRNA showed differential expression: 84 were altered in at least two of the TNBC subtypes, whereas 524 were only altered in a particular tumor subtype (Fig. 1B, Tables 2 and S2). Comparison between the LAR subtype and all other subtypes yielded the highest number of differentially expressed lncRNA (50.9%, n = 362/710) followed by M subtype (16.5%, n = 117/710) and IM subtype (14.5%, n = 130/710). The MLS subtype showed 98 altered lncRNA, whereas the basal subtypes (BL1 and BL2) had a lower number of deregulated lncRNA (19 and 11 transcripts, respectively) (Fig. 1B, Tables 2 and S2).
Table 2.
Up‐regulated and down‐regulated lncRNA with higher rate of change between TNBC subtypes
| lncRNA name | Fold changea | ANOVA P‐value | FDR P‐value | TNBC subtype |
|---|---|---|---|---|
| RP11‐532E4.2 | 16.98 | 0.000133 | 0.015499 | BL1 |
| LINC01956 | 3.26 | 6.72E‐13 | 4.54E‐08 | BL1 |
| LINC01123 | 1.88 | 7.11E‐07 | 0.000572 | BL1 |
| RP11‐619J20.1 | −1.65 | 0.000054 | 0.009449 | BL1 |
| RP11‐815J21.4 | −1.65 | 0.000489 | 0.031038 | BL1 |
| RP1‐142L7.5 | −1.82 | 0.000059 | 0.00983 | BL1 |
| RP11‐116G8.5 | 2.73 | 0.000015 | 0.060613 | BL2 |
| RP11‐3K16.2 | 2.22 | 0.000036 | 0.089263 | BL2 |
| LINC01133 | 1.69 | 0.000655 | 0.307124 | BL2 |
| LINC02095 | −7.91 | 0.000565 | 0.300484 | BL2 |
| RP4‐620F22.2 | 9.87 | 0.00E+00 | 0.00E+00 | IM |
| RP5‐1171I10.5 | 1.9 | 1.71E‐14 | 2.95E‐12 | IM |
| ELMO1‐AS1 | 1.71 | 7.48E‐10 | 5.31E‐08 | IM |
| RP13‐455A7.1 | −1.79 | 0.000545 | 0.014289 | IM |
| CTD‐2033D15.1 | −2.98 | 0.000041 | 0.001466 | IM |
| IFNG‐AS1 | 1.82 | 6.93E‐12 | 6.63E‐10 | IM |
| RP11‐206M11.7 | 80.48 | 1.78E‐15 | 1.08E‐12 | LAR |
| LINC00993 | 4.24 | 8.76E‐07 | 0.000048 | LAR |
| PCAT18 | 2.56 | 1.11E‐16 | 8.24E‐14 | LAR |
| AF178030.2 | −7.68 | 9.13E‐10 | 1.41E‐07 | LAR |
| LINC01152 | −2.27 | 0.000181 | 0.002271 | LAR |
| VIM‐AS1 | −1.83 | 3.63E‐07 | 0.000024 | LAR |
| GAS5 | −1.63 | 0.000023 | 0.000538 | LAR |
| LINC02095 | 20.31 | 1.11E‐16 | 1.50E‐12 | M |
| RP11‐84E24.2 | 7.5 | 0.00E+00 | 0.00E+00 | M |
| SOX9‐AS1 | 13.71 | 0.00E+00 | 0.00E+00 | M |
| H19 | 2.99 | 0.000066 | 0.002324 | M |
| ANO1‐AS1 | 2.13 | 0.000059 | 0.00212 | M |
| CASC15 | 1.82 | 9.47E‐08 | 0.00001 | M |
| RP11‐553K8.5 | −3.39 | 6.12E‐10 | 2.27E‐07 | M |
| RP11‐989E6.8 | −2.97 | 4.34E‐07 | 0.000037 | M |
| ANKRD44‐IT1 | −1.86 | 8.62E‐08 | 0.00001 | M |
| MEG3 | 4.21 | 5.29E‐11 | 4.35E‐08 | MSL |
| AC133106.2 | 3.97 | 1.11E‐09 | 6.08E‐07 | MSL |
| DNM3OS | 3.73 | 0.000039 | 0.003966 | MSL |
| VCAN‐AS1 | 3.22 | 0.000019 | 0.002127 | MSL |
| CARMN | 2.44 | 2.25E‐11 | 2.03E‐08 | MSL |
| TCONS_l2_00019097 | −1.59 | 0.000161 | 0.0125 | MSL |
| LINC00302 | −1.64 | 0.000782 | 0.042797 | MSL |
| TCONS_l2_00003601 | −1.89 | 0.000845 | 0.045414 | MSL |
Results from tac software (Affymetrix).
To validate the lncRNA expression patterns in the TNBC subtypes, we corroborated the expression of some candidates using an independent cohort containing lncRNA profiles. Consistent with our data, a set of lncRNA was differentially expressed in the comparisons between BL1, BL2, M, MSL, IM, and LAR tumors in our in silico analysis (Fig. S3). Validation of the expression of this set of lncRNA in an independent cohort suggests that they might play important biological roles in the establishment of TNBC subtypes.
3.3. Identifying biological pathways associated with lncRNA in TNBC subtypes
To gain insight into the biological relevance of the altered lncRNA, we used a guilt‐by‐association approach to investigate their relationship to dysregulated mRNA and their possible impact on different pathways. This analysis revealed a significant association between some of the differentially expressed lncRNA and mRNA, as well as the enrichment in key breast cancer‐related pathways of associated coding genes for each TNBC subtype. lncRNA altered in BL1 correlated most strongly with extracellular matrix organization, cell cycle and transforming growth factor‐beta (TGF‐β) signaling. IM lncRNA are most strongly related to antigen processing, interferon signaling and immune system. LAR contains lncRNA that are potentially involved in translation regulation. Altered lncRNA in M tumors are associated with toll‐like receptor and TNF signaling. Finally, the co‐expression correlation network of altered lncRNA‐mRNA in the MSL subtype was enriched in focal adhesion, ECM receptor interaction, and PI3K‐Akt signaling pathway (Fig. 2, Table S3). With the exception of the BL2 subtype, where we did not find any significant association, guilt‐by‐association analyses suggested functions for a set of altered lncRNA by their weighted co‐expression with related mRNA that defined a group of pathways with relevant correlations with lncRNA expression portraits in human tumors.
Figure 2.
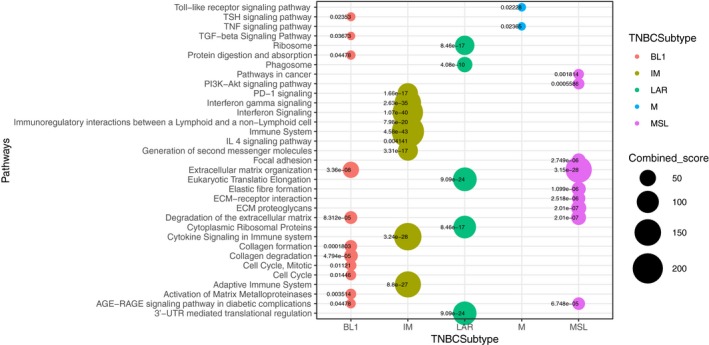
Overview of the biological pathways by guilt‐by‐association analysis across TNBC subtypes by lncRNA‐mRNA co‐expression. Guilt‐by‐association analysis showing the significant enriched pathways of the resulted co‐expression mRNA‐lncRNA. Pathway enrichment analysis resulted in significant association with cancer‐related signaling. The y‐axis label represents pathways and the x‐axis label represents TNBC subtypes. Bubble chart shows biological pathways enrichment by differential expression of lncRNA for each TNBC subtype. Size and color of the bubble represent the score each pathway and TNBC subtypes, respectively.
3.4. Co‐expression of lncRNA and coding genes in IM subtypes
Long non‐coding RNAs can function as enhancers of the expression of both adjacent and distant genes. To prioritize the most biologically relevant lncRNA in the IM subtypes (one of the most frequent subtypes), as a first approach we identified lncRNA (intergenic or intragenic) with possible regulatory functions over their neighboring genes; thus we analyzed lncRNA–mRNA pairs positively co‐expressed (Fig. S4, Table S4). This approach defined the positive correlation of KLHDC7B and ENSG00000226738.
3.5. LncKLHDC7B (ENSG00000226738) and KLHDC7B show a correlated expression pattern in the immunomodulatory subtype
The lncRNA ENSG00000226738 (which we call LncKLHDC7B) and its coding gene KLHDC7B were among one the most correlated pairs evaluated (r 2 = 0.9, P < 0.0001; Figs S4 and 3A–D). The overexpression of the pair LncKLHDC7B/KLHDC7B as well as their correlation in the IM subtype was also validated in TCGA data (Fig. 3C,D, Table S4). We evaluated the expression of both transcripts in a panel of breast cancer cell lines, which showed increased expression of both genes in many tumor cell lines, including IM cell line (HCC1187), in contrast to a non‐tumorigenic epithelial cell line MCF10A (Fig. 4A). Interestingly, these observations were reproducible in breast cancer tissues compared with normal tissue using databases from TCGA (Fig. S6). We noticed a distinct expression pattern of KLHDC7B and LncKLHDC7B among the different triple‐negative subgroups, both in tumors and cell line models (Figs 3A‐D, 4A and S6). To further validate the relationship of LncKLHDC7B with the immunophenotype, we calculated the immunophenoscore of our profiled cohort. Our findings showed distinct expression patterns across the scores, where the highest scores (9 and 10) present a significant enrichment of LncKLHDC7B, particularly in the IM subtype (Fig. 3E). Given these results, we selected the HCC1187 cell line to investigate further the functional role of LncKLHDC7B in the immunomodulatory subtype.
Figure 3.
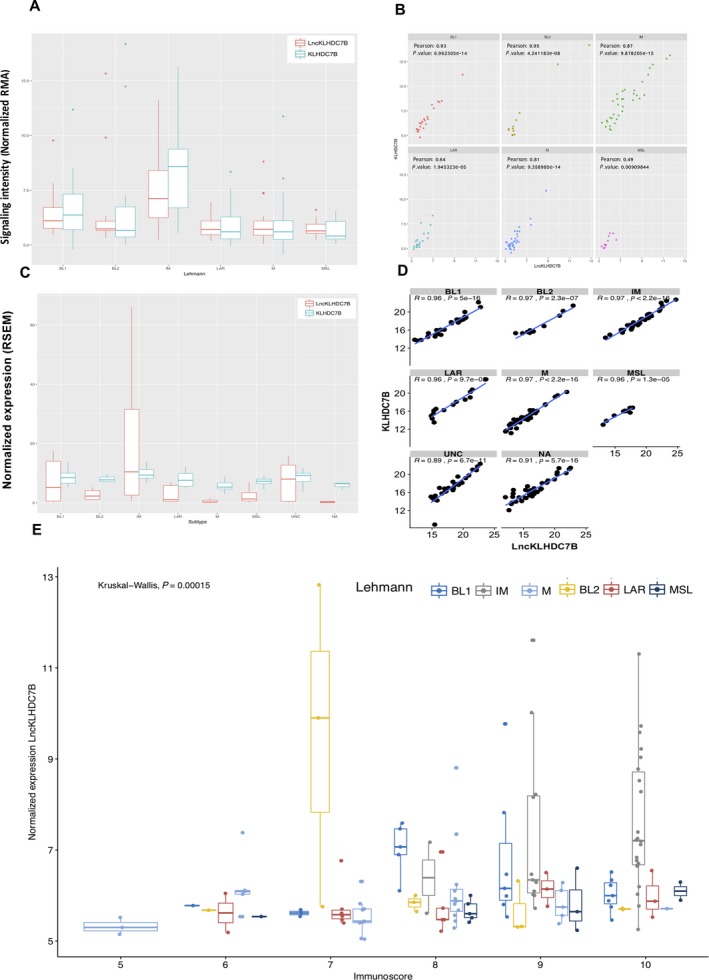
Evaluation of LncKLHDC7B and KLHDC7B expression in TNBC subtypes. (A) LncKLHDC7B and KLHDC7B expression levels in our cohort and (C) TCGA database. (B) Correlation between gene expression level of LncKLHDC7B and KLHDC7B across TNBC subtypes in our dataset, and TCGA database (D). (E) Relation across the immunoscore with the expression of LncKLHDC7B in our cohort. Kruskal–Wallis test was performed to determine significance. The y‐axis label represents the normalized expression of the LncKLHDC7B and the x‐axis label represents the immunoscore.
Figure 4.
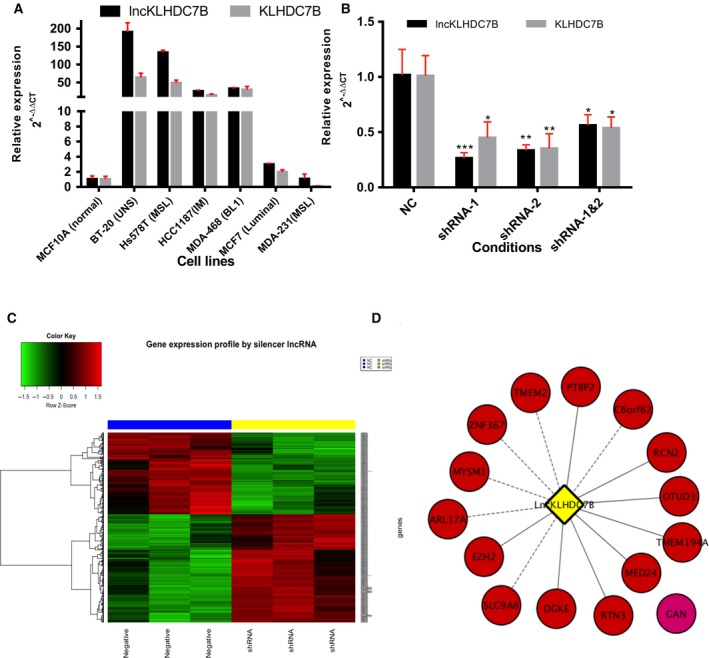
Impact of LncKLHDC7B silencing on the TNBC immunomodulatory phenotype. (A) Validation of LncKLHDC7B and KLHDC7B expression in a panel of breast cancer cell lines. (B) Expression levels of LncKLHDC7B in immunomodulatory phenotype (HCC1187) of breast cancer cell line after silencing by shRNA system. KLHDC7B coding gen expression is affected by LncKLHDC7B silencing. All data are shown as the mean ± SD of at least three independent experiments. Student's t test was performed to determine significance *P < 0.05, **P < 0.01, ***P < 0.001 of NC vs shRNA. (C) Heatmap from transcripts altered by LncKLHDC7B silenced. The microarray was performed in triplicate for the NC condition (blue bar) and shRNA‐1 (yellow bar). Red represents elevated and green down‐regulated expression. (D) Prediction of interactions between lncRNA and mRNA targets sub‐expressed by the silencing of the LncKLHDC7B. Continuous and dotted lines represent a medium and high interaction, respectively.
We then chose the LncKLHDC7B/KLHDC7B pair for further analysis for the following reasons: (1) LncKLHDC7B may be a subtype tumor marker of the IM subtype, but its function is not well described in the literature; (2) LncKLHDC7B presents a significant association with its coding gene (localized in the same locus), which indicates a possible regulatory activity; (3) our guilt‐by‐association analysis highlighted correlations between the lncRNA and key cancer‐related pathways such as antigen presentation, natural killer‐mediated cytotoxicity, interferon signaling, and cell adhesion (Fig. S5). All these data suggest a potential role for LncKLHDC7B in the TNBC subtype establishment and biological regulatory programs.
3.6. Silencing of LncKLHDC7B down‐regulates KLHDC7B and impacts the expression of a large number of genes
We evaluated the role of LncKLHDC7B on oncogenic phenotypes in the IM breast cancer HCC1187 cell line model. The knockdown of LncKLHDC7B was achieved with ~ 75% silencing efficiency (Fig. 4B) and a 50% inhibition of the expression of KLHDC7B gene (Fig. 4B). Furthermore, a complete genomic analysis of the transcriptomic state after silencing of LncKLHDC7B revealed an impact on the global expression of 1265 transcripts (Fig. 4C, Table S5), including several transcripts from the Kelch family. These data demonstrated that LncKLHDC7B might regulate gene expression both in cis, mediating the transcriptional activation of the KLHDC7B gene, and in trans, by regulating distant genes located throughout the genome.
Long non‐coding RNAs show a diversity of functions through diverse mechanisms, interactions with other RNA molecules being some of the most representative. We applied an mRNA‐lncRNA interaction prediction with a nearest‐neighbor method based on thermodynamic parameters using the LncTar tool (Li et al., 2015). Genes most significantly down‐regulated upon LncKLHDC7B silencing were evaluated. Our results indicate a high and medium direct interaction between the mRNA and LncKLHDC7B (ndG value ≤ −0.1 indicates a true predicted interaction that could not be considered a chance event). Thus, their down‐modulation might be a direct consequence of LncKLHDC7B knockdown. The regulator‐signaling network mediated by LncKLHDC7B in the IM cell line model is described in Fig. 4D and Table S6. This approach provided relevant information to identify valuable lncRNA–mRNA interactions for predicting RNA targets and figuring out the possible biological function of the lncRNA.
3.7. LncKLHDC7B modulates the expression of genes associated with relevant hallmarks of cancer
To investigate the global impact of LncKLHDC7B inhibition in the IM cell line model, we performed an Ingenuity Pathway Analysis on up‐ or down‐modulated targets after LncKLHDC7B reduction, which revealed a significant over‐representation of cancer‐relevant pathways related to cell migration, cellular death, apoptosis, and invasion (Fig. 5A).
Figure 5.
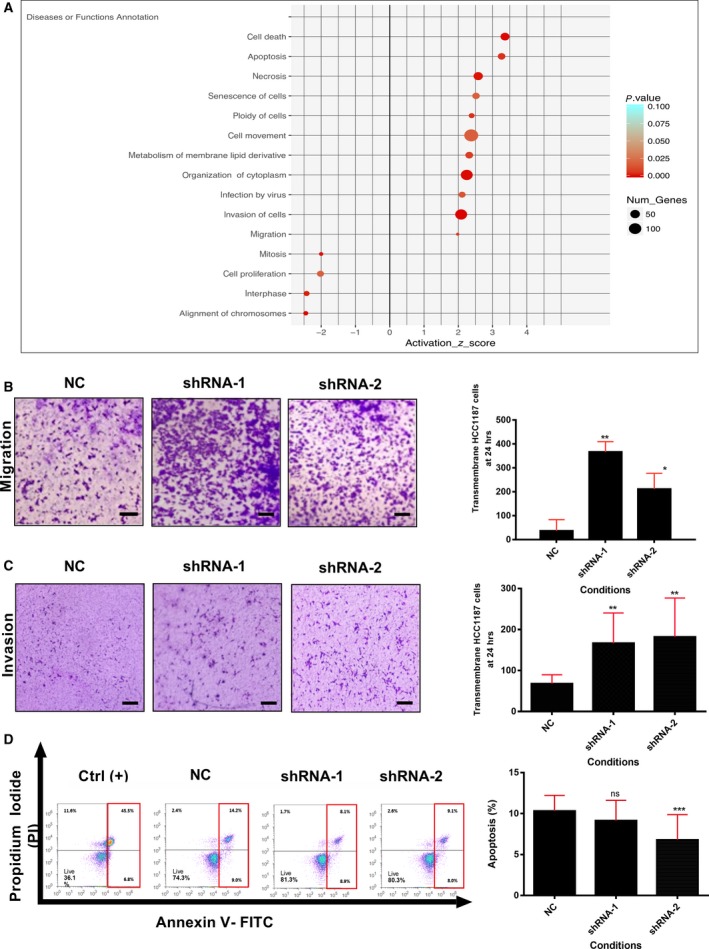
Enrichment and functional analysis for the silencing of LncKLHDC7B in HCC1187 cell line. (A) Enrichment of biological process from transcriptional alteration by LncKLHDC7B silencing in HCC1187 cell line (IPA analysis). The size of the circle represents the number of genes involved in the biological process; the color indicates the statistical significance. (B) Transwell migration and (C) invasion assay showed that LncKLHDC7B silencing increases the migration and invasion of HCC1187. Representative images are shown on the left and quantification on the right. Scale bar: 50 μm. (D) Flow cytometric analysis of apoptosis (early and late) in HCC1187 cell transfected with control and shRNA‐1 and ‐2 after Annexin V/PI staining. All data are shown as the mean ± SD of at least three independent experiments. Student's t test was performed to determine significance of NC vs shRNA: *P < 0.05, **P < 0.01, ***P < 0.001.
3.8. Silencing of LncKLHDC7B increases cellular migration and invasion, and decreases cell death
LncKLHDC7B knockdown significantly increased the ability of cells to migrate in comparison with control HCC1187 cells (Fig. 5B,C). This oncogenic phenotype could be achieved by the observed up‐modulation of TNC, CDH3, APP, EPS8, and CAV1, as previously reported (Baek et al., 2010; Chen et al., 2015a; Diaz et al., 2014; Lim et al., 2014; Zhang et al., 2016). As shown in Fig. 5C, the invasion ability of HCC1187 cells transfected with shRNA was significantly enhanced compared with the control group. This phenotype might be related to the up‐modulation (Table S5) of genes that have been previously involved in the invasion process, such as MMP2 (Fagan‐Solis et al., 2013; Kim and Rhee, 2016), ADAM9 (Micocci et al., 2013), and AMOT (Zhang and Fan, 2015). Additionally, the depletion of LncKLHDC7B decreased the number of apoptotic cells, reaching ~ 23% of reduction in HCC1187 cells going through apoptotic death (Fig. 5D). This effect might be explained by the down‐regulation of genes related to the induction of apoptosis, such as PMAIP1 (Zhao et al., 2014), RAD21 (Chen et al., 2002; Pati et al., 2002), and CCAR2 (known as DBC1) (Kim et al., 2008), that we observed to be under‐expressed after LncKLHDC7B silencing (Table S5).
To determine whether the silencing effect of LncKLHDC7B on cellular migration, invasion, and apoptosis events are exclusive for triple‐negative breast cancer, we silenced lncRNA in two additional cell lines (BT‐20 and MCF‐7). Interestingly, the down‐regulation of LncKLHDC7B in the MCF‐7 luminal cell line affected KLHDC7B mRNA expression (Fig. 6A). Moreover, silencing promoted migration and invasion (Fig. 6B,C) of MCF‐7 cells, as well as a resistance to apoptosis (18–24%), compared with the control group (Fig. 6D).
Figure 6.
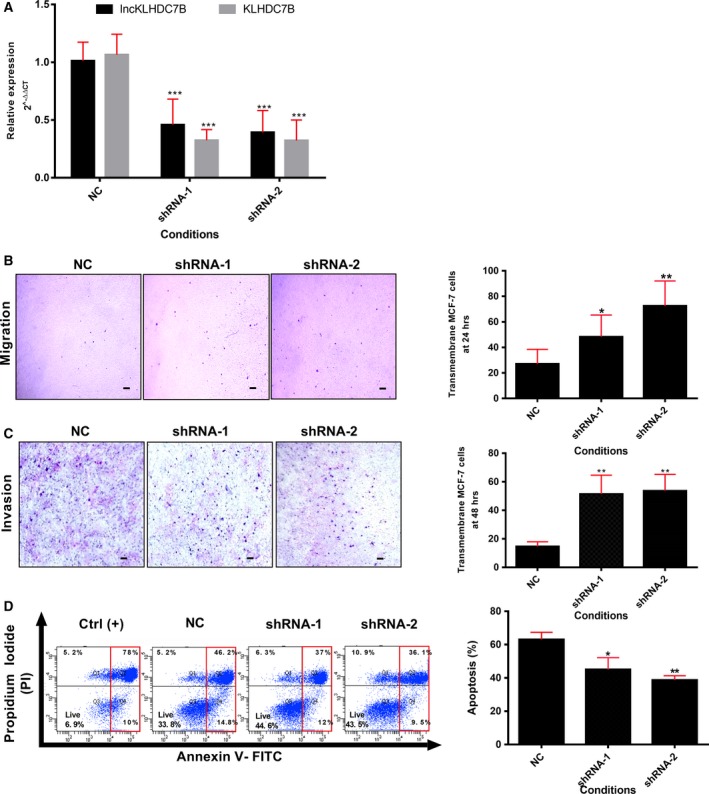
The silencing of LncKLHDC7B modulates breast cancer cell migration and invasion as well as resistance to apoptosis in MCF‐7. (A) The expression of LncKLHDC7B and KLHDC7B in MCF‐7 cells transfected with NC or shRNA‐1 or ‐2 was determined by qRT‐PCR. (B) Transwell migration and (C) invasion assay showed that LncKLHDC7B silencing increases the migration and invasion of MCF‐7. Representative images are shown on the left and quantification on the right. Scale bar: 25 μm. (D) Flow cytometric analysis of apoptosis (early and late) in MCF‐7 cell transfected with control and shRNA‐1 and ‐2 after Annexin V/PI staining. All data are shown as the mean ± SD of at least three independent experiments. Student's t test was performed to determine significance of NC vs shRNA: *P < 0.05, **P < 0.01, ***P < 0.001.
However, in the triple‐negative cell line BT‐20, we were only able to obtain a 35% of inhibition of LncKLHDC7B, and we only observed a 34% down‐regulation of KLHDC7B (Fig. S7a,b). In addition, the migration, invasion, and resistance to apoptosis were enhanced, albeit non‐significantly, by an shRNA (shRNA‐1) that had the greatest effect on the silencing the lncRNA in this particular cell line (Fig. S7b‐d).
3.9. Clinical implications of LncKLHDC7B and KLHDC7B expression in triple‐negative breast cancer
To investigate the potential clinical implication of down‐regulating LncKLHDC7B and KLHDC7B in triple‐negative breast cancer, we identified and analyzed public data (Anaya, 2016; Curtis et al., 2012; Lanczky et al., 2016) with available mRNA or lncRNA expression profiles and clinical information (disease‐free survival or overall survival). The down‐modulation of KLHDC7B was associated with an increased probability of a recurrent or metastatic event (n = 161, P = 0.0053, Fig. 7A), but not with risk of death (n = 133, P = 0.212; Fig. 7B) compared with samples showing a higher expression of the gene. We did not detect a significant probability of death in tumors with low expression of the lncRNA in triple‐negative breast cancer samples (Fig. 7C, but a generalized analysis in breast cancer showed that the low KLHDC7B and LncKLHDC7B expression was significantly associated with lower survival (HR 1.910, 95% CI 1.147–3.233, log‐rank test, P = 0.0137 and HR 1.933, 95% CI 1.161–3.273, log‐rank test, P = 0.0120, respectively) compared with patients who presented higher expression of both transcripts (Fig. S8).
Figure 7.
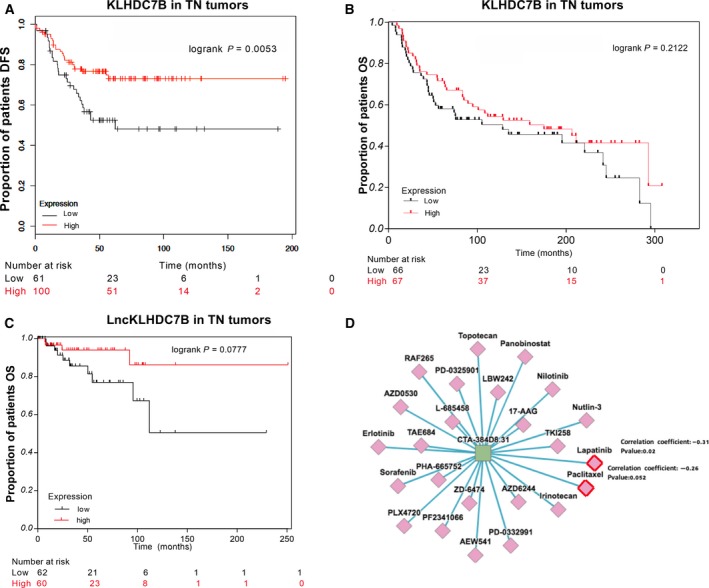
Clinical implications of the sub‐expression of LncKLHDC7B and KLHDC7B in TNBC. (A) Kaplan–Meier analysis curve of the disease‐free survival (DFS) of TNBC according to KLHDC7B expression. (B) Kaplan–Meier analysis of overall survival (OS) according to KLHDC7B expression. (C) Kaplan–Meier analysis of prognostic relevance of LncKLHDC7B expression from public data. (D) In silico prediction of susceptibility to USFDA‐approved treatment drugs related with LncLHDC7B expression in triple‐negative breast cancer cell line.
Previous studies and our results shown above indicated that lncRNA is differentially expressed between TN breast cancer subtypes, thus lncRNA may be explored as a new class of clinical biomarkers for therapy response or targets for drug discovery (Arun et al., 2018; Prabhakar et al., 2017; Wu and Du, 2017). To identify drug‐related lncRNA in breast cancer, we explored the association of the LncKLHDC7B levels and drug activity with the lncMAP tool (Li et al., 2018) (http://bio-bigdata.hrbmu.edu.cn/LncMAP/index.jsp), which computes the Spearman correlation coefficient between lncRNA expression and the half maximal inhibitory concentration (IC50) values of 24 drugs approved for oncology purposes in TNBC cell lines. We observed a significant negative correlation between the expression of LncKLHDC7B and concentrations of lapatinib and paclitaxel (Fig. 7D), which are being evaluated in various clinical trials (https://clinicaltrials.gov/ct2/home).
4. Discussion
Triple‐negative breast cancer is a heterogeneous disease at the cellular, molecular, and clinical levels (Lehmann et al., 2011). Long non‐coding RNA have recently emerged as important players implicated in relevant biological processes (Martin and Chang, 2012) as well as potential cancer biomarkers or therapeutic targets (Arun et al., 2018; Chandra Gupta and Nandan Tripathi, 2017; Prabhakar et al., 2017; Wan et al., 2016; Wu and Du, 2017). In the present study, we classified TNBC using mRNA expression profiles (Chen et al., 2012) and identified several lncRNA expressed in specific tumor subtypes. The most frequent subtypes in our sample collection were mesenchymal‐like and immunomodulatory, consistent with previous reports (Lehmann et al., 2011; Liu et al., 2016; Masuda et al., 2013). Regarding tumor subgrouping and clinical characteristics, we did not detect any significant association between TNBC subtypes and tumor size, histology or survival data, except for tumor grade.
A class comparison analysis identified a profile of 710 differentially expressed lncRNA among the 156 samples belonging to different TNBC subtypes. A guilt‐by‐association analysis revealed the possible biological pathways associated with the lncRNA expressed for each of the TNBC subtypes except for BL2, possibly due to lack of correlation between the expression of mRNA and lncRNA.
Overall, our findings indicate that lncRNA expression patterns can distinguish TNBC subtypes and that lncRNA might be involved in important pathways related to tumor biology, contributing to the regulatory circuits between lncRNA and mRNA. There was a quite interesting relationship between lncRNA and the immunomodulatory subtype, one of the altered tumor groups in the context of lncRNA transcriptional landscape, in which lncRNA activity seems to impact an important number of pathways involved in the establishment of the immune infiltrated phenotype. Given these results, we decided to further characterize the IM subtype.
In the IM subtype, LncKLHDC7B and KLHDC7B were up‐regulated with positive correlated expression (r 2 = 0.9, P < 0.0001; Fig. S4), suggesting that the antisense lncRNA might regulate the expression of the KLHDC7B coding gene.
Several lines of evidence indicated the importance of the LncKLHDC7B/KLHDC7B pair of transcripts, including that LncKLHDC7B expression is specific for the IM subtype (or, based on our analyses, on breast cancer in a generalized way). LncKLHDC7B expression levels are significantly correlated with its coding gene (localized in the same locus) and our guilt‐by‐association analysis highlighted a correlation between lncRNA and key cancer‐related pathways.
KLHDC7B (Kelch Domain Containing 7B) is a protein member of the Kelch superfamily, proteins related to cellular processes such as cytoskeletal rearrangement and protein degradation (Adams et al., 2000). Alterations in this protein superfamily have been associated with various types of cancer, including leukemia, lung, prostate, brain, and Hodgkin's disease (Gupta and Beggs, 2014). To the best of our knowledge, the only report that associates this gene with breast cancer, reported the hypermethylation and over‐regulation of this gene in mammary tumors, suggesting a possible role as an epigenetic marker (Kim et al., 2010). However, the role of its non‐coding antisense LncKLHDC7B was unknown.
LncKLHDC7B is a 260‐base transcript and has two exons. At the genomic level LncKLHDC7B is an antisense lncRNA of ~ 1.26 Kb, located on the long arm of chromosome 22q13.33, sharing a locus with the KLHDC7B gene (Kent et al., 2002; Volders et al., 2015). Different approaches have demonstrated the potential role of lncRNA as regulators of the expression of genes in cis or in trans (Engreitz et al., 2016; Gupta et al., 2010). We first corroborated the positive correlation in the expression of LncKLHDC7B and KLHDC7B in independent tumor datasets and in cell lines. This over‐expression was corroborated in the immunomodulatory HCC1187 cell line. Subsequently, we silenced the LncKLHDC7B using shRNA, which resulted in the repression of the KLHDC7B coding gene. Thus, LncKLHDC7B might be acting as a cis transcriptional regulator of its coding gene KLHDC7B.
We also analyzed the effects of LncKLHDC7B knockdown on the global transcriptional landscape in HCC11887 cells. We found that expression of several genes was modified after silencing the lncRNA, suggesting its potential role as a transcriptional regulator in trans. Enrichment pathways analysis identified a significant up‐modulation of cell death, necrosis, apoptosis, cell movement, migration, invasion, and organization of cytoplasm upon lncRNA silencing. Interestingly, we observed the deregulation of several genes of the Kelch family both upwards and downwards, some of them related to processes such as cell migration and invasion, as well as apoptosis (Lian et al., 2016; Ohta et al., 2010).
Previous studies showed that silencing KLHL39, another member of the Kelch family, increased cell migration and invasion, and decreased cell death by anoikis (Chen et al., 2015b). In accordance with these data, we showed that silencing LncKLHDC7B significantly increased the cellular migration and invasion, as well as the resistance to apoptosis in HCC1187 and MCF‐7 cells. These observations are also supported by our analysis of the BT‐20 cell line (which showed higher expression of LncKLHDC7B and KLHDC7B), where a ~ 30% silencing was obtained by one of the shRNA, although in a non‐significant way, of migration and cellular invasion with a tendency toward resistance to apoptosis. In general, these observations suggest that the effect of LncKLHDC7B and KLHDC7B inhibition might be applicable beyond the triple‐negative subtype.
Previous studies suggest that the overexpression of protein members of the Kelch family, such as KLHL2, increases apoptosis (Tseng and Bixby, 2011) or, in the case of silencing KLHDC7B, favors death resistance, as previously published (Jeong et al., 2018). Here, we show that the silencing of LncKLHDC7B led to resistance to cell death. Similarly, it has been reported that silencing of members of the Kelch family leads to death resistance by anoikis and is associated with lower survival in colorectal cancer (Chen et al., 2015b).
These results support the potential role of LncKLHDC7B as a gene expression regulator of KLHDC7B, and its association with cytoskeletal rearrangement, cell migration, and invasion. Further evidence of the potential role of the LncKLHDC7B–KLHDC7B correlated expression in cancer comes from the recent description of a novel 5′ alternative splicing site in the KLHDC7B gene in cervical squamous cell carcinoma. This novel splice site coincides with the location of a lncRNA (Tcons_00029745) and was related to cellular differentiation and tumor size in this disease (Guo et al., 2015).
Our data indicate that LncKLHDC7B is required for cell migration and invasion inhibition, and contributes to apoptosis induction, suggesting its important regulatory activity in TNBC tumors with enrichment of LncKLHDC7B, or maybe a generalized process in breast cancer.
To explore further the altered expression of LncKLHDC7B and KLHDC7B and their clinical implication in breast cancer, we evaluated the correlation between their expression and survival probability in a public database. Down‐regulation of KLHDC7B was associated with lower disease‐free survival and LncKLHDC7B was closely associated with poor clinical outcome in triple‐negative tumors and breast cancer in general. As far as we know, this is the first report of the association of KLHDC7B or LncKLHDC7B expression with poor prognosis in breast cancer. In addition, we analyzed the clinical implication of LncKLHDC7B expression and oncological drugs. Our results suggest a higher sensitivity to lapatinib and paclitaxel in cells over‐expressing the LncKLHDC7B; however, the limitation of this approach was the lack of in vitro validation.
5. Conclusions
We explored the lncRNA expression landscape of the very complex and heterogeneous disease represented by triple‐negative breast cancer, and identified several lncRNA with subtype‐specific expression. Through this analysis we unveiled a correlation in the expression of diverse lncRNA with their corresponding coding genes in the IM subtype, including LncKLHDC7B‐KLHDC7B. Silencing of LncKLHDC7B led to altered expression of KLHDC7B, a member of the Kelch family, and increased cell migration and invasion with decreased apoptosis, indicating that LncKLHDC7B may modulate the aggressiveness of breast cancer IM subtype at least in part by regulating the expression of its host gene KLHDC7B. We observed a consistent trend toward shorter survival for patients with low expression levels of LncKLHDC7B and KLHDC7B. Although the clinical relevance of LncKLHDC7B expression remains incompletely understood, our results reaffirm the down‐modulation of these transcripts in patients with aggressive clinical TN tumors and suggest their association with poor outcomes. These data must be further characterized to elucidate the exact mechanism involved in these observations.
Author contributions
B.A.: performed most of the experimental work and data analysis. R.C., C.T., R.R.: data analysis and discussion. R.V., A.R., J.M., R.G.: sample collection and processing. A.O.: data analysis. B.P.: histopathological review of the cases. D.R., V.C., T.T.: patient identification and clinical follow up. T.E., I.M., B.A., H.M.: project coordination and leadership, data analysis, and study design. All authors contributed to the writing of the manuscript.
Conflict of interest
A.H.M. has received grants from Astra Zeneca, but these are not related to this particular project. All other authors declare no conflict of interest.
Supporting information
Table S1. Gene ontology and KEGG pathway enrichment analysis from differentially expressed genes from TNBC subtypes.
Table S2. Differentially expressed lncRNA in the TNBC subtypes.
Table S3. In silico prediction of biological pathways by guilt‐by‐association analysis across lncRNA‐mRNA co‐expressed in the TNBC subtypes.
Table S4. Correlation analysis of coding and non‐coding genes co‐expressed positively in the immunomodulatory phenotype.
Table S5. Differentially expressed genes that are significantly modulated after lncKLHDC7B silencing in HCC1187 cells.
Table S6. Prediction lncRNA‐mRNA interactions from down‐regulated genes after silencing lncKLHDC7B.
Fig. S1. (a) Frequency of TNBC subtypes in this study and (b) other studies (Cancer Genome Atlas, 2012; Curtis et al., 2012; Lehmann et al., 2011; Liu et al., 2016; Masuda et al., 2013).
Fig. S2. Gene ontology from FFPE TNBC samples in this study.
Fig. S3. Independent validation of some LncRNA in TNBC subtypes.
Fig. S4. Correlation analysis of coding and non‐coding genes co‐expressed positively in the immunomodulatory phenotype.
Fig. S5. Guilt‐by‐association analysis.
Fig. S6. Up‐modulation of LncKLHDC7B and KLHDC7B in tumor samples of breast cancer.
Fig. S7. Functional analysis by the silencing of LncKLHDC7B in BT‐20 cell line.
Fig. S8. Kaplan–Meier curve of overall survival (OS).
Acknowledgements
Fredy Omar Beltrán Anaya is a doctoral student from Programa de Doctorado en Ciencias Biomédicas, Universidad Nacional Autónoma de México (UNAM), and received fellowship (CVU 416163) from CONACYT.
We would like to thank all the patients who participated in this study, and all the medical and nursing staff of FUCAM for their support. We appreciate the support of M.Sc. Raul Mojica from the INMEGEN microarray core lab, M.Sc. L. Nelly Patiño of the cytometry unit, and Dra. Diana I. Aparicio‐Baustista and Marco González for their help. This work was funded by the Mexican National Council of Science and Technology Basic Science grant (grant number 258936) and Frontiers in Science grant (number 1285).
References
- Adams J, Kelso R and Cooley L (2000) The Kelch repeat superfamily of proteins: propellers of cell function. Trends Cell Biol 10, 17–24. [DOI] [PubMed] [Google Scholar]
- Anaya J (2016) OncoLnc: linking TCGA survival data to mRNA, miRNAs, and lncRNAs. PeerJ Comput Sci 2:e67, 2e:67. [Google Scholar]
- Arun G, Diermeier SD and Spector DL (2018) Therapeutic targeting of long non‐coding RNAs in cancer. Trends Mol Med 24, 257–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek S, Lee YW, Yoon S, Baek SY, Kim BS and Oh SO (2010) CDH3/P‐Cadherin regulates migration of HuCCT1 cholangiocarcinoma cells. Anat Cell Biol 43, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer KR, Brown M, Cress RD, Parise CA and Caggiano V (2007) Descriptive analysis of estrogen receptor (ER)‐negative, progesterone receptor (PR)‐negative, and HER2‐negative invasive breast cancer, the so‐called triple‐negative phenotype: a population‐based study from the California cancer Registry. Cancer 109, 1721–1728. [DOI] [PubMed] [Google Scholar]
- Chandra Gupta S, Nandan Tripathi Y (2017) Potential of long non‐coding RNAs in cancer patients: from biomarkers to therapeutic targets. Int J Cancer 140, 1955–1967. [DOI] [PubMed] [Google Scholar]
- Chen HY, Hu JY, Chen TH, Lin YC, Liu X, Lin MY, Lang YD, Yen Y and Chen RH (2015b) KLHL39 suppresses colon cancer metastasis by blocking KLHL20‐mediated PML and DAPK ubiquitination. Oncogene 34, 5141–5151. [DOI] [PubMed] [Google Scholar]
- Chen F, Kamradt M, Mulcahy M, Byun Y, Xu H, McKay MJ and Cryns VL (2002) Caspase proteolysis of the cohesin component RAD21 promotes apoptosis. J Biol Chem 277, 16775–16781. [DOI] [PubMed] [Google Scholar]
- Chen X, Li J, Gray WH, Lehmann BD, Bauer JA, Shyr Y and Pietenpol JA (2012) TNBCtype: a subtyping tool for triple‐negative breast cancer. Cancer Informat 11, 147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liang Z, Huang W, Li X, Zhou F, Hu X, Han M, Ding X and Xiang S (2015a) Eps8 regulates cellular proliferation and migration of breast cancer. Int J Oncol 46, 205–214. [DOI] [PubMed] [Google Scholar]
- Consortium, E.P. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis C, Shah SP, Chin SF, Turashvili G, Rueda OM, Dunning MJ, Speed D, Lynch AG, Samarajiwa S, Yuan Y et al (2012) The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 486, 346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P and Narod SA (2007) Triple‐negative breast cancer: clinical features and patterns of recurrence. Clin Cancer Res 13, 4429–4434. [DOI] [PubMed] [Google Scholar]
- Derrien T, Johnson R, Bussotti G, Tanzer A, Djebali S, Tilgner H, Guernec G, Martin D, Merkel A, Knowles DG et al (2012) The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res 22, 1775–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz J, Mendoza P, Ortiz R, Diaz N, Leyton L, Stupack D, Quest AF and Torres VA (2014) Rab5 is required in metastatic cancer cells for Caveolin‐1‐enhanced Rac1 activation, migration and invasion. J Cell Sci 127, 2401–2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engreitz JM, Haines JE, Perez EM, Munson G, Chen J, Kane M, McDonel PE, Guttman M and Lander ES (2016) Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 539, 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagan‐Solis KD, Schneider SS, Pentecost BT, Bentley BA, Otis CN, Gierthy JF and Arcaro KF (2013) The RhoA pathway mediates MMP‐2 and MMP‐9‐independent invasive behavior in a triple‐negative breast cancer cell line. J Cell Biochem 114, 1385–1394. [DOI] [PubMed] [Google Scholar]
- Foulkes WD, Smith IE and Reis‐Filho JS (2010) Triple‐negative breast cancer. N Engl J Med 363, 1938–1948. [DOI] [PubMed] [Google Scholar]
- Gibb EA, Brown CJ and Lam WL (2011) The functional role of long non‐coding RNA in human carcinomas. Mol Cancer 10, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, Wang D, Wu J, Yang J, Ren T, Zhu B and Xiang Y (2015) The landscape of alternative splicing in cervical squamous cell carcinoma. Onco Targets Ther 8, 73–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta VA and Beggs AH (2014) Kelch proteins: emerging roles in skeletal muscle development and diseases. Skelet Muscle 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta RA, Shah N, Wang KC, Kim J, Horlings HM, Wong DJ, Tsai MC, Hung T, Argani P, Rinn JL et al (2010) Long non‐coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guttman M, Amit I, Garber M, French C, Lin MF, Feldser D, Huarte M, Zuk O, Carey BW, Cassady JP et al (2009) Chromatin signature reveals over a thousand highly conserved large non‐coding RNAs in mammals. Nature 458, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heberle H, Meirelles GV, da Silva FR, Telles GP and Minghim R (2015) InteractiVenn: a web‐based tool for the analysis of sets through Venn diagrams. BMC Bioinformat 16, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Huang W, Sherman BT and Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protocol 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Jeong G, Bae H, Jeong D, Ham J, Park S, Kim HW, Kang HS and Kim SJ (2018) A Kelch domain‐containing KLHDC7B and a long non‐coding RNA ST8SIA6‐AS1 act oppositely on breast cancer cell proliferation via the interferon signaling pathway. Sci Rep 8, 12922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM and Haussler D (2002) The human genome browser at UCSC. Genome Res 12, 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D and Rhee S (2016) Matrix metalloproteinase2 regulates MDAMB231 breast cancer cell invasion induced by active mammalian diaphanous‐related formin 1. Mol Med Rep 14, 277–282. [DOI] [PubMed] [Google Scholar]
- Kim JE, Chen J and Lou Z (2008) DBC1 is a negative regulator of SIRT1. Nature 451, 583–586. [DOI] [PubMed] [Google Scholar]
- Kim TW, Kim YJ, Lee HJ, Min SY, Kang HS and Kim SJ (2010) Hs.137007 is a novel epigenetic marker hypermethylated and up‐regulated in breast cancer. Int J Oncol 36, 1105–1111. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A et al (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanczky A, Nagy A, Bottai G, Munkacsy G, Szabo A, Santarpia L and Gyorffy B (2016) miRpower: a web‐tool to validate survival‐associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res Treat 160, 439–446. [DOI] [PubMed] [Google Scholar]
- Langfelder P and Horvath S (2008) WGCNA: an R package for weighted correlation network analysis. BMC Bioinformat 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara‐Medina F, Perez‐Sanchez V, Saavedra‐Perez D, Blake‐Cerda M, Arce C, Motola‐Kuba D, Villarreal‐Garza C, Gonzalez‐Angulo AM, Bargallo E, Aguilar JL et al (2011) Triple‐negative breast cancer in Hispanic patients: high prevalence, poor prognosis, and association with menopausal status, body mass index, and parity. Cancer 117, 3658–3669. [DOI] [PubMed] [Google Scholar]
- Lehmann BD, Bauer JA, Chen X, Sanders ME, Chakravarthy AB, Shyr Y and Pietenpol JA (2011) Identification of human triple‐negative breast cancer subtypes and preclinical models for selection of targeted therapies. J Clin Invest 121, 2750–2767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann BD, Jovanovic B, Chen X, Estrada MV, Johnson KN, Shyr Y, Moses HL, Sanders ME and Pietenpol JA (2016) Refinement of triple‐negative breast cancer molecular subtypes: implications for neoadjuvant chemotherapy selection. PLoS ONE 11, e0157368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li CH and Chen Y (2013) Targeting long non‐coding RNAs in cancers: progress and prospects. Int J Biochem Cell Biol 45, 1895–1910. [DOI] [PubMed] [Google Scholar]
- Li J, Ma W, Zeng P, Wang J, Geng B, Yang J and Cui Q (2015) LncTar: a tool for predicting the RNA targets of long noncoding RNAs. Brief Bioinform 16, 806–812. [DOI] [PubMed] [Google Scholar]
- Li Y, Li L, Wang Z, Pan T, Sahni N, Jin X, Wang G, Li J, Zheng X, Zhang Y et al (2018) LncMAP: Pan‐cancer atlas of long noncoding RNA‐mediated transcriptional network perturbations. Nucleic Acids Res 46, 1113–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian YF, Yuan J, Cui Q, Feng QS, Xu M, Bei JX, Zeng YX and Feng L (2016) Upregulation of KLHDC4 predicts a poor prognosis in human nasopharyngeal carcinoma. PLoS ONE 11, e0152820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S, Yoo BK, Kim HS, Gilmore HL, Lee Y, Lee HP, Kim SJ, Letterio J and Lee HG (2014) Amyloid‐beta precursor protein promotes cell proliferation and motility of advanced breast cancer. BMC Cancer 14, 928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YR, Jiang YZ, Xu XE, Yu KD, Jin X, Hu X, Zuo WJ, Hao S, Wu J, Liu GY et al (2016) Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype‐specific RNAs of triple‐negative breast cancer. Breast Cancer Res 18, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin L and Chang HY (2012) Uncovering the role of genomic ‘dark matter’ in human disease. J Clin Invest 122, 1589–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez ME, Wertheim BC, Natarajan L, Schwab R, Bondy M, Daneri‐Navarro A, Meza‐Montenegro MM, Gutierrez‐Millan LE, Brewster A, Komenaka IK et al (2013) Reproductive factors, heterogeneity, and breast tumor subtypes in women of mexican descent. Cancer Epidemiol Biomarkers Prev 22, 1853–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda H, Baggerly KA, Wang Y, Zhang Y, Gonzalez‐Angulo AM, Meric‐Bernstam F, Valero V, Lehmann BD, Pietenpol JA, Hortobagyi GN et al (2013) Differential response to neoadjuvant chemotherapy among 7 triple‐negative breast cancer molecular subtypes. Clin Cancer Res 19, 5533–5540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micocci KC, Martin AC, Montenegro Cde F, Durante AC, Pouliot N, Cominetti MR and Selistre‐de‐Araujo HS (2013) ADAM9 silencing inhibits breast tumor cell invasion in vitro. Biochimie 95, 1371–1378. [DOI] [PubMed] [Google Scholar]
- Ohta Y, Fujimura L, Nishio S, Arima M, Sakamoto A, Shimada H, Ochiai T, Tokuhisa T and Hatano M (2010) A Kelch family protein Nd1‐L functions as a metastasis suppressor in cancer cells via Rho family proteins mediated mechanism. Int J Oncol 36, 427–434. [PubMed] [Google Scholar]
- Pati D, Zhang N and Plon SE (2002) Linking sister chromatid cohesion and apoptosis: role of Rad21. Mol Cell Biol 22, 8267–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Rodriguez G (2015) Prevalence of breast cancer sub‐types by immunohistochemistry in patients in the Regional General Hospital 72, Instituto Mexicano del Seguro Social. Cir Cir 83, 193–198. [DOI] [PubMed] [Google Scholar]
- Prabhakar B, Zhong XB and Rasmussen TP (2017) Exploiting long noncoding RNAs as pharmacological targets to modulate epigenetic diseases. Yale J Biol Med 90, 73–86. [PMC free article] [PubMed] [Google Scholar]
- Rastelli F, Biancanelli S, Falzetta A, Martignetti A, Casi C, Bascioni R, Giustini L and Crispino S (2010) Triple‐negative breast cancer: current state of the art. Tumori 96, 875–888. [PubMed] [Google Scholar]
- Schneider CA, Rasband WS and Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stockel D, Kehl T, Trampert P, Schneider L, Backes C, Ludwig N, Gerasch A, Kaufmann M, Gessler M, Graf N et al (2016) Multi‐omics enrichment analysis using the GeneTrail2 web service. Bioinformatics 32, 1502–1508. [DOI] [PubMed] [Google Scholar]
- Tseng LA and Bixby JL (2011) Interaction of an intracellular pentraxin with a BTB‐Kelch protein is associated with ubiquitylation, aggregation and neuronal apoptosis. Mol Cell Neurosci 47, 254–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volders PJ, Verheggen K, Menschaert G, Vandepoele K, Martens L, Vandesompele J and Mestdagh P (2015) An update on LNCipedia: a database for annotated human lncRNA sequences. Nucleic Acids Res 43, D174–D180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan X, Huang W, Yang S, Zhang Y, Pu H, Fu F, Huang Y, Wu H, Li T and Li Y (2016) Identification of androgen‐responsive lncRNAs as diagnostic and prognostic markers for prostate cancer. Oncotarget 7, 60503–60518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O and Chang HY (2011) Long noncoding RNAs and human disease. Trends Cell Biol 21, 354–361. [DOI] [PubMed] [Google Scholar]
- Wickham H (2009) Ggplot2 Elegant Graphics for Data Analysis, Use R! Dordrecht: Springer, pp. 1 online resource (viii, 212 pages) illustrations (some color). [Google Scholar]
- Wu T and Du Y (2017) LncRNAs: From basic research to medical application. Int J Biol Sci 13, 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H and Fan Q (2015) MicroRNA‐205 inhibits the proliferation and invasion of breast cancer by regulating AMOT expression. Oncol Rep 34, 2163–2170. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yu B, Gu Y, Zhou S, Qian T, Wang Y, Ding G, Ding F and Gu X (2016) Fibroblast‐derived tenascin‐C promotes Schwann cell migration through beta1‐integrin dependent pathway during peripheral nerve regeneration. Glia 64, 374–385. [DOI] [PubMed] [Google Scholar]
- Zhao X, Liu X and Su L (2014) Parthenolide induces apoptosis via TNFRSF10B and PMAIP1 pathways in human lung cancer cells. J Exp Clin Cancer Res 33, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Gene ontology and KEGG pathway enrichment analysis from differentially expressed genes from TNBC subtypes.
Table S2. Differentially expressed lncRNA in the TNBC subtypes.
Table S3. In silico prediction of biological pathways by guilt‐by‐association analysis across lncRNA‐mRNA co‐expressed in the TNBC subtypes.
Table S4. Correlation analysis of coding and non‐coding genes co‐expressed positively in the immunomodulatory phenotype.
Table S5. Differentially expressed genes that are significantly modulated after lncKLHDC7B silencing in HCC1187 cells.
Table S6. Prediction lncRNA‐mRNA interactions from down‐regulated genes after silencing lncKLHDC7B.
Fig. S1. (a) Frequency of TNBC subtypes in this study and (b) other studies (Cancer Genome Atlas, 2012; Curtis et al., 2012; Lehmann et al., 2011; Liu et al., 2016; Masuda et al., 2013).
Fig. S2. Gene ontology from FFPE TNBC samples in this study.
Fig. S3. Independent validation of some LncRNA in TNBC subtypes.
Fig. S4. Correlation analysis of coding and non‐coding genes co‐expressed positively in the immunomodulatory phenotype.
Fig. S5. Guilt‐by‐association analysis.
Fig. S6. Up‐modulation of LncKLHDC7B and KLHDC7B in tumor samples of breast cancer.
Fig. S7. Functional analysis by the silencing of LncKLHDC7B in BT‐20 cell line.
Fig. S8. Kaplan–Meier curve of overall survival (OS).