

Abstract
Objective
To assess the contribution of education to cognitive reserve.
Methods
Analyses are based on older participants in a longitudinal clinical-pathologic cohort study who had annual cognitive testing (n = 2,899) and subgroups that developed incident dementia (n = 696), died, and underwent a neuropathologic examination from which 10 neurodegenerative and cerebrovascular markers were derived (n = 752), or both (n = 405). Cognitive test scores were converted to a standard scale and averaged to yield composite measures of cognition.
Results
Participants had a mean of 16.3 years of education (SD = 3.7, range 0–30). In all participants, education was associated with initial level of global cognition but not rate of cognitive change. In those who developed dementia, rate of global cognitive decline accelerated a mean of 1.8 years before the diagnosis, but education was not related to the onset or rate of accelerated decline. In the deceased, rate of global cognitive decline accelerated a mean of 3.4 years before death, but higher educational attainment was related to earlier (not later) onset of accelerated decline and unrelated to rate of acceleration. Higher education was associated with lower likelihood of gross and microscopic cerebral infarcts but not with other neuropathologic markers. Education was not related to global cognitive change not attributable to neuropathologic burden and did not decrease the association of higher neuropathologic burden with more rapid cognitive decline.
Conclusion
The results suggest that the contribution of education to cognitive reserve is limited to its association with level of cognitive function before old age.
Level of education is widely used as an indicator of cognitive reserve. That higher level of education is associated with lower risk of dementia1–5 supports this idea. However, prospective studies suggest that this association is mostly attributable to the association of education with level of cognitive function rather than rate of cognitive change.6–10 High cognitive reserve is also thought to alter the shape of cognitive trajectories by delaying the onset of cognitive symptoms. Evidence that education contributes to this aspect of reserve is mixed. Higher level of education has been associated with delayed onset of accelerated cognitive decline linked to incipient dementia, including analyses of data from the Bronx Aging Study11 and the Religious Orders Study/Memory and Aging Project (ROSMAP),12 and impending death.13 However, other studies have not observed these associations.14–17 Clinical-pathologic studies allow a more direct assessment of the role of education in cognitive reserve but results have been inconclusive. In prior analyses of Religious Orders Study data, the negative association of Alzheimer disease (AD) pathology with level of cognition proximate to death was reduced at higher levels of education.18,19 In another clinical-pathologic study, education was related to dementia proximate to death but did not modify the relation of postmortem neuropathologic markers to dementia.20 Thus, it remains uncertain whether education moderates pathologic influences on cognitive change.
The present analyses examine the role of education in cognitive reserve. Participants are older persons from 2 longitudinal clinical-pathologic cohort studies who had annual clinical evaluations that included cognitive testing. Those who died underwent a neuropathologic examination to quantify multiple neurodegenerative and cerebrovascular pathologies. In analyses, we test hypotheses based on the assumption that education is an indicator of cognitive reserve. Specifically, we test the hypotheses that higher education is associated with more positive cognitive trajectories in general and with later onset of accelerated cognitive decline associated with incident dementia and impending death in particular; education is unrelated to common neurodegenerative and cerebrovascular conditions; education has an association with cognitive change that is independent of neuropathologic burden; and education reduces the association of higher neuropathologic burden with more rapid cognitive decline.
Methods
Participants
Analyses are based on individuals who participated in 2 ongoing longitudinal clinical-pathologic cohort studies. The Religious Orders Study, which began in 1994, comprises older Catholic clergy members from across the United States.21,22 The Memory and Aging Project, which began in 1997, comprises older lay persons from the Chicago metropolitan region.23,24 Eligibility in both studies requires age of 50 years or older at enrollment, absence of a prior dementia diagnosis, and agreement to annual clinical evaluations and brain autopsy upon death.
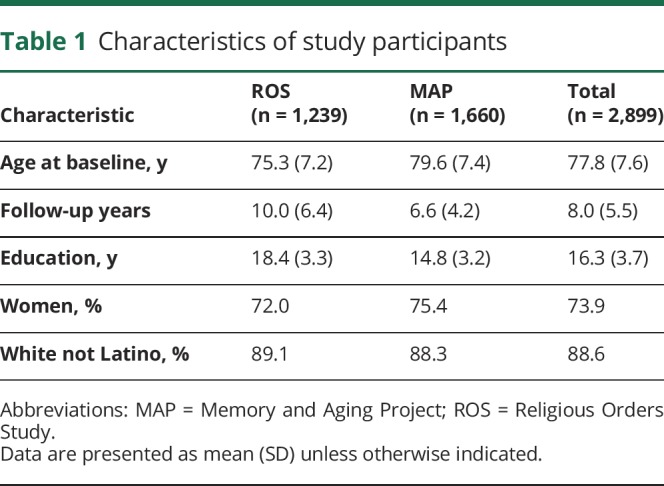
These analyses are based on data collected from 1994 through 2018. Eligibility required the absence of dementia at baseline and a valid cognitive score at baseline plus at least one follow-up evaluation. Of the 2,899 individuals who met these criteria (main group), 1,239 came from the Religious Orders Study and 1,660 came from the Memory and Aging Project (table 1). They had a mean age of 77.8 years at baseline, a mean of 8.0 years of follow-up, and a mean of 16.3 years of education; 2,143 (73.9%) were women and 2,569 (88.6%) were white and not Latino.
Table 1.
Characteristics of study participants
To allow for nonlinear change in cognitive function, most analyses were confined to subsets of the 2,005 individuals in the main group with a minimum of 4 years of annual follow-up (81% of the 2,478 survivors who had reached the fifth evaluation date). One subgroup (incident dementia subgroup) consisted of 696 individuals who developed incident dementia (mean age at diagnosis = 87.5 [SD = 6.5], mean of 10.5 years of follow-up [SD = 4.8], with a mean of 7.8 years [SD = 5.2] before the diagnosis and 2.7 years [SD = 2.9] after the diagnosis, mean education = 16.4 [SD = 3.7], 75.3% women, 90.2% white and not Latino).
There were 1,044 individuals who died with at least 4 years of follow-up. A brain autopsy was done on 958 (92%); at the time of these analyses, the neuropathologic examination had been completed in 752 persons who made up the neuropathologically examined subgroup. They had a mean age at death of 90.2 (SD = 6.3), mean of 10.1 years of follow-up (SD = 4.4), mean education of 16.5 (SD = 3.6); 67.8% were women and 95.2% were white and not Latino. A final subgroup (incident dementia neuropathologically examined subgroup) consisted of 405 individuals with incident dementia who died and underwent a neuropathologic examination (mean age at baseline = 80.5 [SD = 6.5], mean age at diagnosis = 87.7 [SD = 6.4], mean age at death = 91.3 [SD = 5.8], mean of 10.8 years of follow-up [SD = 4.5] with a mean of 7.2 years before the diagnosis and 3.7 years [SD = 3.0] after diagnosis, mean education = 16.7 [SD = 3.6], 70.4% women, 95.1% white and not Latino).
Standard protocol approvals, registrations, and patient consents
Following a presentation, interested persons had a detailed discussion with study personnel and signed informed consent forms and an anatomical gift act. Both studies were approved by the institutional review board of Rush University Medical Center.
Clinical evaluation
Participants had annual clinical evaluations that included a medical history, neurologic examination, and cognitive testing. On the basis of this evaluation, an experienced clinician, blinded to previously collected data, diagnosed dementia using the criteria of the National Institute of Neurological and Communicative Disorders and Stroke/Alzheimer's Disease and Related Disorders Association (NINCDS/ADRDA).25 These require a history of cognitive decline and impairment in at least 2 cognitive domains. Further information on the implementation of these criteria is published elsewhere.26
Assessment of cognitive function
A battery of cognitive tests was administered annually. One testing aim was to support clinical classification of dementia and mild cognitive impairment. As previously described,27–29 the tests were used to determine impairment in different cognitive domains in support of clinical classification of dementia. In addition, raw scores on 7 measures of episodic memory (immediate and delayed recall of Logical Memory Story A and the East Boston Story; Word List Memory, Recall, and Recognition), 3 measures of semantic memory (Boston Naming Test, Verbal Fluency, Reading Test), 3 measures of working memory (Digit Span Forward, Digit Span Backward, Digit Ordering), 2 measures of perceptual speed (Symbol Digit Modalities Test, Number Comparison), and 2 measures of visuospatial ability (Standard Progressive Matrices, Judgment of Line Orientation) were converted to z scores using the baseline mean and SD from all participants in both parent studies. In analyses, we used composite measures of global cognition (based on 17 tests), episodic memory (7 tests), and perceptual speed (2 tests) formed by averaging the z scores of component tests. Composites of individual tests can accommodate a wide range of cognitive ability thereby minimizing floor and ceiling artifacts in longitudinal analyses. Further information on the individual tests and these composite measures is published elsewhere.27–29
Neuropathologic examination
The central aim of the neuropathologic examination was to assess common conditions linked with cognitive impairment and dementia in old age. Because regional measures of a given pathologic condition are robustly intercorrelated in these cohorts,30,31 we combined measures across brain regions to minimize the number of pathologic markers in analyses while maximizing their metric properties (by basing pathologic scores on multiple tissue samples).
There was a standard protocol for brain removal and sectioning and preservation of the tissue.32,33 The cerebral hemispheres were cut coronally into 1-cm slabs that were examined for gross infarcts. We used hematoxylin & eosin staining to identify microinfarcts in 9 regions (6 cortical, 2 subcortical, 1 midbrain). Cerebral β-amyloid angiopathy was assessed in 4 regions (midfrontal cortex, inferior temporal cortex, calcarine cortex, angular cortex) with β-amyloid deposition in meningeal and parenchymal vessels in each region rated on a 5-point scale and averaged to yield a summary score.34 Arteriosclerosis ratings were based on visual inspection of the vessels of the circle of Willis, and arteriolar sclerosis was rated on hematoxylin & eosin sections of the basal ganglia.35 We assessed β-immunoreactive plaques in 8 brain regions (dorsolateral prefrontal cortex, superior frontal cortex, inferior parietal cortex, inferior temporal cortex, primary visual cortex, anterior cingulate cortex, entorhinal cortex, CA1/subiculum) with a monoclonal antibody (1:50, β-amyloid, clone 6F/3D; Dako, Carpinteria, CA). An antipaired helical filaments-tau antibody clone AT8 (1:2000; Thermo Scientific, Waltham, MA) was used to assess tau-immunoreactive neurofibrillary tangles in the same 8 brain regions. Regional scores were averaged to yield composite measures of β-amyloid burden and tau-tangle density, as previously described.30 TAR DNA-binding protein 43 (TDP-43) cytoplasmic inclusions were identified using monoclonal antibodies to phosphorylated TDP-43 (pS409/410; 1:10,000)36 in 6 brain regions (midfrontal cortex, middle temporal cortex, entorhinal cortex, dentate gyrus, CA1/subiculum, amygdala), with density of inclusions rated on a 4-point scale.37,38 Hippocampal sclerosis was defined as severe neuronal loss and gliosis in any hippocampal subfield or the subiculum.39 We assessed 6 brain regions (midfrontal cortex, superior or middle temporal cortex, inferior parietal cortex, anterior cingulate cortex, entorhinal cortex, substantia nigra) for Lewy bodies using a monoclonal antibody to α-synuclein (Zymed LB 509; 1:50).40 Hippocampal sclerosis and Lewy bodies were treated as present or absent in analyses.
Statistical analysis
We used mixed-effects models to assess change in cognitive function. This approach has 3 main strengths. First, in assessing linear cognitive change, it separates initial level from rate of change and includes random effects that allow the initial level to be higher or lower and rate of change to be faster or slower. Second, to accommodate nonlinear cognitive change, the model let rate of cognitive decline accelerate at some point and included random effects to allow for individual differences in the onset of the change point and the rate of cognitive decline following it. Third, by including random effects to capture individual differences in cognitive trajectory components (i.e., intercept, slope[s], change point), the mixed-effects models help account for within-person correlations.
In the main group, we assessed change in global cognition in a mixed-effects model with time treated as years since baseline and terms for education, age at baseline, and sex. We included terms for age and sex in this and subsequent models because of their associations with education and cognition. In the incident dementia subgroup, we constructed a mixed-effects model with time treated as years before (and after) diagnosis, allowing rate of decline to accelerate at some variable point, and including terms for education, age at diagnosis, and sex. We repeated these 2 analyses, first treating education as a categorical rather than continuous variable with a low education group contrasted with medium and high education groups, and then substituting specific cognitive outcomes for the global measure.
We conducted a series of analyses in the neuropathologically examined subgroup. First, we constructed a mixed-effects model with time treated as years before death, allowing rate of global cognitive decline to accelerate at some variable point before death, and including terms for education, age at death, and sex. To assess the relation of education to pathology, we separately regressed each neuropathologic marker on education, age at death, and sex. To test whether education had an association with cognitive change independent of neuropathologic burden, we constructed mixed-effects change point models (with time treated as years before death) that had terms for a neuropathologic marker, education, age at death, and sex (with separate analyses for each neuropathologic marker). To test whether education modified the relation of neuropathologic burden to global cognitive trajectories, we repeated each model with a term added for the interaction of education with the neuropathologic marker. To assess time metric effects, we constructed 3 mixed-effects change point models in the incident dementia neuropathologically examined subgroup with time anchored at baseline, dementia diagnosis, or death.
Data availability
All data included in these analyses are available through the Rush Alzheimer's Disease Center Research Resource Sharing Hub at radc.rush.edu. It includes descriptions of the studies and key variables. After logging in, qualified users can request deidentified data.
Results
Education and global cognitive function
Participants had completed a mean of 16.3 years of education (SD = 3.7, skewness = −0.1, range 0–30). Higher level of education was associated with younger age at baseline (r = −0.17, p < 0.001). Women had less schooling than men (mean of 16.0 vs 17.1, t1,170.4 = 6.1, p < 0.001).
At baseline, the composite measure of global cognition had an approximately normal distribution with a mean of 0.096 (SD = 0.531, skewness = −0.5). Higher level of education (r = 0.37, p < 0.001) and younger age at baseline (r = −0.35, p < 0.001) were each associated with higher baseline level of global cognition. Women had a higher baseline level of cognition than men (mean of 0.113 vs 0.047, t2,897 = 3.0, p = 0.003).
To assess the relation of education to late-life cognitive function, we constructed a mixed-effects model with terms for age (at baseline), sex, and education. In this analysis, the composite measure of global cognition declined a mean of 0.073 unit per year (SE = 0.002, p < 0.001). Level of global cognition at baseline was 0.052-unit higher for each additional year of educational attainment (SE = 0.003, p < 0.001). However, education was not related to linear rate of change in global cognition (estimate = −0.0004, SE = 0.001, p = 0.438). The model results are illustrated in figure 1A for typical participants with high (75th percentile, 18 years, solid line) vs low (25th percentile, 14 years, dashed line) levels of education.
Figure 1. Relation of education to trajectories of global cognitive change.
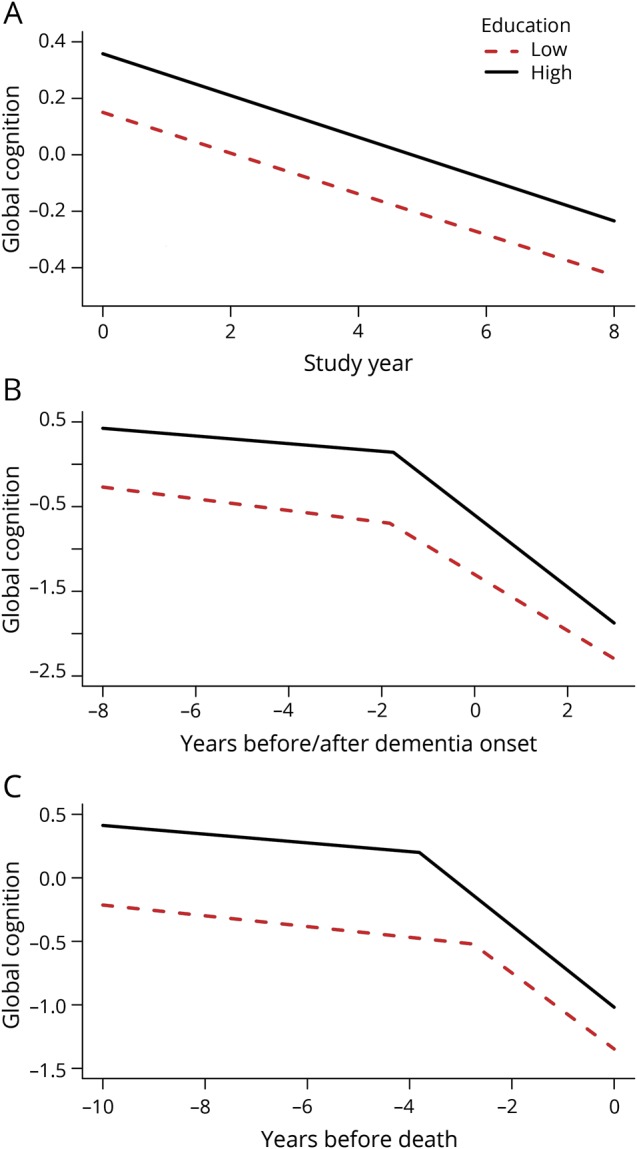
Predicted paths of global cognitive change in typical participants with high (75th percentile, 18 years, solid line) vs low (25th percentile, 14 years, dashed line) levels of education: (A) from the main group using a mixed-effects model adjusted for age at baseline and sex; (B) from the incident dementia group using a mixed-effects change point model adjusted for age at diagnosis and sex; (C) from the neuropathologically examined group using a mixed-effects change point model adjusted for age at death and sex.
To assess possible nonlinear education effects, we divided participants into low (12 years or less; n = 546), medium (13–16 years; n = 1,029), and high (17 years or more; n = 1,324) education groups. We then repeated the analysis contrasting the low education group with each of the other 2 education groups. Both medium (estimate = 0.313, SE = 0.026, p < 0.001) and high (estimate = 0.502, SE = 0.025, p < 0.001) education were associated with higher baseline level of global cognition than low education, but neither education group was related to global cognitive change.
Although education was not related to linear cognitive change, it has been reported to modify components of nonlinear cognitive trajectories, such as the onset of accelerated decline associated with incident dementia.11,12 Therefore, we constructed a mixed-effects model for the 696 participants who had at least 4 years of follow-up and developed incident dementia (24.0% of 2,899). We treated time as years before and after the dementia diagnosis (which was made at a mean age of 87.4, SD = 6.5) and allowed rate of cognitive decline to accelerate at some variable point. In this analysis (table 2), global cognition declined a mean of 0.059 unit annually until a mean of 1.8 years before dementia was diagnosed after which the mean rate of decline accelerated to a loss of 0.373 unit per year, a more than 6-fold increase. Although the statistical change point (mean age = 85.6, SD = 6.3) occurred slightly before the diagnosis of dementia, the correlation between the statistical and diagnostic measures approached unity (r = 0.98, p < 0.001). As illustrated in figure 1B, higher level of education was associated with higher initial cognitive level (intercept term in table 2) but was not related to the onset of accelerated decline (change point term in table 2) or the rate at which cognition declined before (first slope term in table 2) or after (second slope term in table 2) the change point. When the analysis was repeated treating education as a categorical rather than continuous variable, results were comparable. Thus, both the medium (n = 238; intercept estimate = 0.288, 95% credible interval [CI] 0.143–0.427) and high (n = 323; intercept estimate = 0.493, 95% CI 0.360–0.624) education groups had higher initial levels of global cognition than the low education group (n = 135), but education group was not related to the slope terms or change point.
Table 2.
Relation of demographic variables to trajectories of change in global cognition in incident dementia (n = 696)a

Of the 696 individuals with incident dementia, 584 were diagnosed with AD and no other conditions (NINCDS/ADRDA probable AD), 44 had AD plus at least one other cognition impairing condition (NINCDS/ADRDA possible AD), and 68 had conditions other than AD. To determine whether this clinical heterogeneity affected results, we conducted a second analysis restricted to the 584 individuals with probable AD. Results were essentially unchanged, with education related to initial level of global cognition but not to other cognitive trajectory components.
Because education has also been reported to modify the onset of terminal cognitive decline,13 we analyzed global cognitive change in deceased participants with at least 4 years of follow-up (n = 752) with a mixed-effects model that allowed rate of cognitive decline to accelerate at some variable point. In this analysis (table 3), the global cognitive score declined a mean of 0.038 unit per year until a mean of 3.4 years before death when the mean rate of decline accelerated to 0.312 unit per year, a more than 8-fold increase. As shown figure 1C, education was only related to one trajectory component, the change point, with the onset of terminal decline occurring a mean of 0.201 year earlier for each additional year of education (above the mean of the group), contrary to the cognitive reserve hypothesis.
Table 3.
Relation of demographic variables to trajectories of change in global cognition in decedents (n = 752)a

Education and cognitive domains
To determine whether the contribution of education to cognitive reserve differed across cognitive domains, we repeated the 3 core models separately with composite measures of episodic memory and perceptual speed as outcomes. These analyses provided little evidence that education enhances reserve. In the full group (n = 2,899), the only change from the analyses of global cognition was that higher education had a marginal association with more rapid decline in perceptual speed (estimate = −0.001, SE = 0.001, p = 0.028). In the incident dementia group (n = 696), the only change from the analysis of global cognition was that higher education was associated with more rapid accelerated decline in episodic memory (estimate = 0.174, 95% CI 0.113–0.237) and perceptual speed (estimate = 0.210, 95% CI 0.136–0.283).
Education and neuropathologic burden
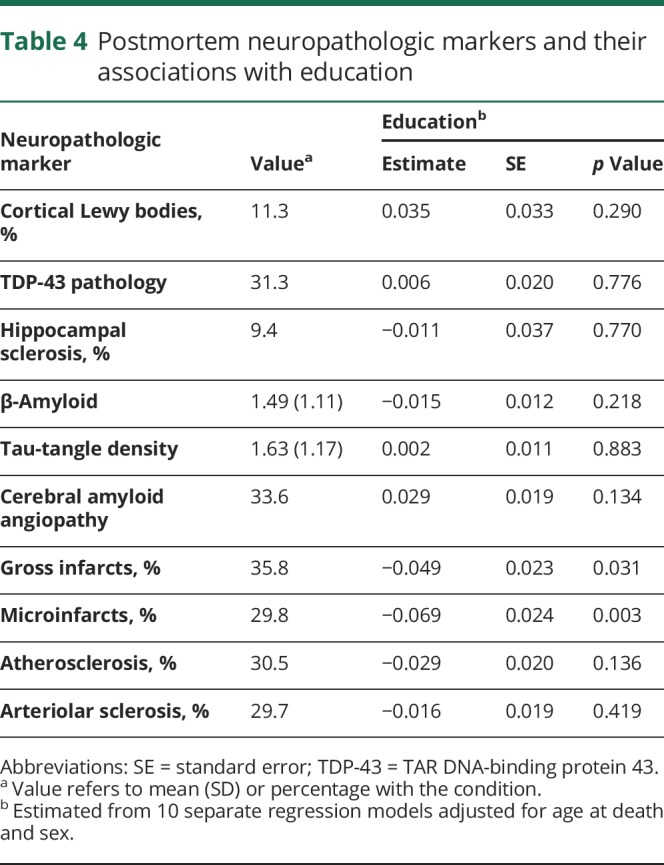
The cognitive reserve hypothesis assumes that education is not related to the pathologic drivers of dementia. Uniform neuropathologic examinations of study participants showed substantial evidence of neurodegenerative and cerebrovascular conditions (table 4, value column). In separate regression models adjusted for age at death and sex, higher education was associated with lower likelihood of gross and microscopic cerebral infarcts but not with other postmortem markers (table 4, education column).
Table 4.
Postmortem neuropathologic markers and their associations with education
Education, neuropathologic burden, and global cognition
Education could confer cognitive reserve in 2 ways: by an association with cognitive trajectories that is independent of neuropathologic burden (additive effect) or by reducing the negative association of neuropathologic burden with cognitive function (modifying effect). To address the possibility of an additive effect, we constructed change point models adjusted for age (at death), sex, and a given neuropathologic marker (with separate models for each marker) to test whether education had an association with residual cognitive change not related to neuropathologic burden. The only association of education with these residual cognitive trajectories was with the onset of terminal cognitive decline (regardless of the neuropathologic marker included in the model). Contrary to the cognitive reserve hypothesis, higher level of education was associated with earlier onset of residual terminal cognitive decline in all models.
The model results did show the expected negative associations of each neuropathologic marker except microscopic infarcts with the cognitive trajectories. To test the hypothesis that education modifies the relation of pathology to cognition, we repeated each analysis with terms added to assess the interaction of education with each postmortem neuropathologic measure. The interaction terms provide no support for education as a measure of cognitive reserve (table 5). The association of 3 neurodegenerative markers (TDP-43 pathology, hippocampal sclerosis, tau-tangle density) with the onset of accelerated terminal cognitive decline was modified by education but in the opposite direction predicted by the cognitive reserve hypothesis. As shown in figure 2, the association of pathology with earlier onset of accelerated decline was stronger at higher (solid lines) than lower (dashed lines) levels of education. In addition, the only interaction involving rate of terminal cognitive decline (stronger association of cortical Lewy bodies with more rapid cognitive decline at higher than lower levels of education) provides little support for the cognitive reserve hypothesis.
Table 5.
Interaction of education with neuropathologic markers on trajectories of change in global cognitiona

Figure 2. Relation of education and neurodegeneration to trajectories of global cognitive change.
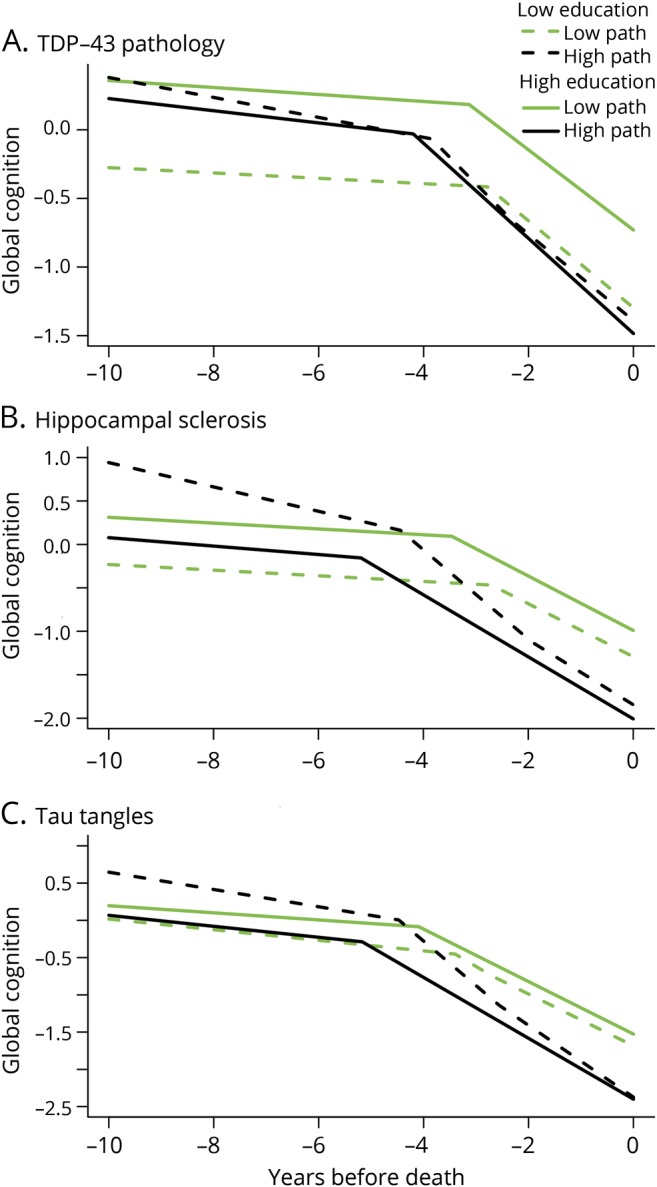
Predicted paths of global cognitive change in typical participants who died with high (75th percentile, 18 years, solid lines) vs low (25th percentile, 14 years, dashed lines) levels of education and high (black color) vs low (green color) postmortem levels of TDP-43 pathology (A), hippocampal sclerosis (B), and tau tangles (C), from mixed-effects change point models adjusted for age at death and sex. path = pathology; TDP-43 = TAR DNA-binding protein 43.
Treatment of time
In analyses, we treated time as years after baseline, years before and after diagnosis of dementia, and years before death. To determine whether the different time metrics affected results, we used all 3 metrics on the 405 individuals in the incident dementia neuropathologically examined subgroup. Higher education was not associated with a later change point in any model but was associated with an earlier change point when time was treated as years before death, consistent with the same analysis in the larger neuropathologically examined subgroup (table 2). Education was not associated with rate of global cognitive decline before or after the change point in any model, consistent with all previous analyses.
Discussion
The current analyses are based on older persons who had annual cognitive testing, died, and underwent a uniform neuropathologic examination. Higher education was associated with higher level of cognition at study entry. However, education was not associated with slower rate of linear cognitive decline, later onset of accelerated cognitive decline associated with incident dementia or impending death, or residual cognitive change not attributable to neuropathologic burden, and it did not lessen the deleterious association of neurodegenerative and cerebrovascular conditions with cognitive trajectories. The results suggest that the contribution of education to cognitive reserve is limited to its association with premorbid cognitive level and does not involve an association with cognitive aging trajectories or moderation of the relation of neuropathologic conditions to those trajectories.
The lack of association of education with linear change in cognitive function in these analyses is consistent with prior longitudinal studies with 3 or more measurement occasions.6–10 However, the relation of education to nonlinear cognitive trajectories has been difficult to establish. In analyses based on 117 cases of incident dementia, higher education was associated with later onset and faster rate of accelerated cognitive decline,11 consistent with the cognitive reserve hypothesis. Analyses based on 399 incident cases from ROSMAP yielded similar results.12 By contrast, higher education was associated with earlier onset of accelerated decline in analyses of 442 incident cases,14 and in the present analyses based on nearly 700 ROSMAP participants with incident dementia, education was not associated with either the onset or rate of accelerated cognitive decline. The discrepancy between the present and previous12 ROSMAP findings was unexpected. The present analyses are based on more participants and a longer observation period than previous analyses, thereby not only enhancing statistical power but also perhaps capturing a higher proportion of incident dementia cases with less rapid cognitive decline.
Higher educational attainment has also been associated with later onset of terminal cognitive decline,13 consistent with the cognitive reserve hypothesis. However, the long interval between cognitive assessments may have limited the ability to estimate terminal decline in this study, and higher education has been unrelated to terminal cognitive decline in other studies15,16 or related to earlier onset of terminal cognitive decline on some measures17 as in the present study, inconsistent with the cognitive reserve hypothesis.
A key component of the cognitive reserve hypothesis is that individuals with higher reserve are able to tolerate a higher neuropathologic burden than those with lower reserve. Aging research on this issue has primarily relied on cross-sectional cognitive outcomes (i.e., level of cognitive function or dementia diagnosis proximate to death). We found no evidence that education modified the relation of neuropathologies to level of cognition proximate to death (intercept column of table 4). This is consistent with a large study (n = 872) of education, neuropathology, and dementia,20 but it contradicts previous analyses of Religious Orders Study data in which the association of β-amyloid plaques with cognitive level proximate to death was weaker in those with higher vs lower education.18,19 In comparison to the previous Religious Orders Study analyses, the present analyses are based on more than 5 times as many participants, a broader array of postmortem neuropathologic markers, and a cognitive outcome with longitudinal as well as cross-sectional components, providing a stronger platform for assessing the role of education in cognitive reserve. Contrary to the cognitive reserve hypothesis, there was no evidence that the association of pathology with earlier onset of terminal cognitive decline was reduced in those with more education. In fact, the opposite pattern was observed for tau-tangle density, TDP-43 pathology, and hippocampal sclerosis. The only evidence that education modified the rate of terminal cognitive decline was also inconsistent with the cognitive reserve hypotheses: the association of cortical Lewy bodies with more rapid terminal decline was stronger at higher than lower levels of education. In addition, education did not have an association with change in cognition that was independent of neuropathologic burden.
That education apparently contributes little to cognitive reserve is surprising given its association with cognitive growth41 and changes in brain structure.42 However, formal education typically ends decades before old age begins whereas late-life level of cognitive activity (roughly analogous to schooling) has been associated with rate of cognitive change,43,44 as have other aspects of experience in adulthood and old age such as social activity,45 conscientiousness,46 cognitively demanding work,47 and purpose in life.48 This implies that influences on cognitive reserve vary over time, with recent experiences more influential than remote experiences such as schooling.
The concept of cognitive reserve was introduced to account for nonpathologic influences on cognition, but we found a direct association of higher education with lower likelihood of gross and microscopic cerebral infarcts. These associations were not particularly strong, which may explain why they have not been identified in some previous clinical-pathologic research.20 However, there have been previous reports linking higher level of education with lower risk of stroke,49,50 consistent with the present findings. That education is associated with cerebral infarction further complicates its use as a measure of cognitive reserve.
Limitations and strengths of this study should be noted. An important limitation is that participants were selected and so the generalizability of the findings is uncertain. A related issue is the relatively high education level in the cohort, with a mean of 16.3 years and few participants with little or no schooling. Although education showed the expected association with level of cognitive function in this cohort, it is possible that the other reserve effects attributable to education are primarily driven by variation at the lower end of the spectrum of educational attainment. Further cognitive-pathologic research in less educated groups is needed. In addition, these analyses do not rule out the possibility that the contribution of education to cognitive reserve depends on experiential or biologic factors. The availability of psychometrically sound composite cognitive outcomes, a mean of 8.0 annual assessments per individual, plus information on dementia onset and time of death allowed us to apply mixed-effects change point models to capture nonlinear trajectories of change in cognitive function. The availability of a broad spectrum of postmortem neurodegenerative and cerebrovascular markers allowed us to test whether education was directly related to specific neuropathologic processes or modified the relation of these neuropathologic processes to cognitive outcomes.
Acknowledgment
The authors thank the many Catholic nuns, priests, and monks who participated in the Religious Orders Study and the many Illinois residents who have participated in the Rush Memory and Aging Project; Traci Colvin, MPH, for coordination of the clinical data collection; Karen Skish, MS, for coordination of the pathologic data collection; and John Gibbons, MS, and Greg Klein, MS, for data management.
Glossary
- AD
Alzheimer disease
- ADRDA
Alzheimer's Disease and Related Disorders Association
- CI
credible interval
- NINCDS
National Institute of Neurological and Communicative Disorders and Stroke
- ROSMAP
Religious Orders Study/Memory and Aging Project
- TDP-43
TAR DNA-binding protein 43
Appendix. Authors
Footnotes
CME Course: NPub.org/cmelist
Study funding
Supported by NIH (R01AG17917, P30AG010161, R01AG15819, R01AG34374) and the Illinois Department of Public Health. The funding organizations had no role in the design or conduct of the study; the collection, analysis, or interpretation of the data; or the writing of the report or the decision to submit it for publication.
Disclosure
R. Wilson receives research support from NIH grants RF1AG022018, P30AG010161, RF1AG015819, R01AG017917, U01AG046152-02S1, R01AG054058, R01AG034374, R01AG033678, R01AG020048, U24AG056270, R01AG051635, RF1AG057532, R01NS093870, and U54TR002056. L. Yu receives research support from NIH grants R01AG053446, R01AG017917, RF1AG015819, RF1AG036042, U01AG046152, R01AG054058, R01AG033678, R01AG034374, R01DK099269, U01AG046161, R01AG052488, R01AG050631, and RF1AG048056. M. Lamar receives research support from NIH grants R21NS095723, P30AG010161, R01AG033678, R01AG029672, R01HL129153, and UH2NS100599. J. Schneider receives research support from NIH grants UH2NS100599, R01NS084965, RF1AG022018, P30AG010161, RF1AG015819, R01AG017917, U01AG046152, R01AG054058, R01AG034374, R01AG033678, R01NS089674, R01AG047976, R01AG056352, R01AG057911, R01AG048108, R01AG033570, U01AG046161, RF1AG054057, R01AG054476, P01AG014449, and U54TR002056. P. Boyle receives research support from NIH grants R01AG034374, R01AG033678, P30AG010161, R01AG055430, R01AG033570, UH2NS100599, and R01AG056533. D. Bennett receives research support from NIH grants P30AG010161, RF1AG015819, R01AG017917, RF1AG036042, U01AG046152, U01AG046152-02S1, R01AG054058, RF1AG052476, RF1AG057473, RF1AG057471, R01AG053446, RF1AG051641, R01AG057911, R01AG055430, R01AG048108, R01AG057914, R01AG033570, U01AG046161, R01AG057912, R01AG052488, R01NS086736, U24AG056270, RF1AG054057, R01AG054476, P01AG014449, R01AG050431, R01NS093870, R01AG050631, R01AG056533, UM1HG009443, RF1AG048056, R01AG046174, and R01AG057907. Go to Neurology.org/N for full disclosures.
References
- 1.Stern Y, Gurland B, Tatemichi TK, Tang MX, Wilder D, Mayeux R. Influence of education and occupation on the incidence of Alzheimer's disease. JAMA 1994;271:1004–1010. [PubMed] [Google Scholar]
- 2.Evans DA, Hebert LE, Beckett LA, et al. Education and other measures of socioeconomic status and risk of incident Alzheimer's disease in a defined population of older persons. Arch Neurol 1997;54:1399–1405. [DOI] [PubMed] [Google Scholar]
- 3.Karp A, Kåreholt I, Qiu C, Bellander T, Winblad B, Fratiglioni L. Relation of education and occupation-based socioeconomic status to incident Alzheimer's disease. Am J Epidemiol 2004;159:175–183. [DOI] [PubMed] [Google Scholar]
- 4.Caamano-Isoma F, Corral M, Montes-Martinez A, Takkouche B. Education and dementia: a meta-analytic study. Neuroepidemiology 2006;26:226–232. [DOI] [PubMed] [Google Scholar]
- 5.Meng X, D'Arcy C. Education and dementia in the context of the cognitive reserve hypothesis: a systematic review with meta-analyses and qualitative analyses. PLoS One 2012;7:e38268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chistensen H, Hofer SM, Mackinnon AJ, Korten AE, Jorm AF, Henderson AS. Age is no kinder to the better educated: absence of an association investigated using latent growth techniques in a community sample. Psychol Med 2001;31:15–28. [DOI] [PubMed] [Google Scholar]
- 7.Seeman TE, Huang MH, Bretsky P, Crimmins E, Launer L, Guralnik JM. Education and APOE e4 in longitudinal cognitive decline: MacArthur Studies of Successful Aging. J Gerontol B Psychol Sci Soc Sci 2005;60B:P74–P83. [DOI] [PubMed] [Google Scholar]
- 8.Van Dijk KRA, Van Gerven PWM, Van Boxtel MPJ, Van der Elst W, Jolles J. No protective effects of education during normal cognitive aging: results from the 6-year follow-up of the Maastricht Aging Study. Psychol Aging 2008;23:119–130. [DOI] [PubMed] [Google Scholar]
- 9.Winnock M, Lelenneur L, Jacqmin-Gadda H, Dallongeville J, Amouyd P, Dartigues JF. Longitudinal analysis of the effect of apolipoprotein E ε4 and education on cognitive performance in elderly subjects: the PAQUID study. J Neurol Neurosurg Psychiatry 2002;72:794–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson RS, Hebert LE, Scherr PA, Barnes LL, Mendes de Leon CF, Evans DA. Educational attainment and cognitive decline in old age. Neurology 2009;72:460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hall CB, Derby C, LeValley A, Katz MJ, Verghese J, Lipton RB. Education delays accelerated decline on a memory test in persons who develop dementia. Neurology 2007;69:1657–1664. [DOI] [PubMed] [Google Scholar]
- 12.Yu L, Boyle PA, Wilson RS, et al. A random change point model for cognitive decline in Alzheimer's disease and mild cognitive impairment. Neuroepidemiology 2012;39:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Muniz Terrera G, Minett T, Brayne C, Matthews FE. Education associated with a delayed onset of terminal decline. Age Ageing 2014;43:26–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Amieva H, Mokri H, Le Goff M, et al. Compensatory mechanisms in higher-educated subjects with Alzheimer's disease: a study of 20 years of cognitive decline. Brain 2014;137:1167–1175. [DOI] [PubMed] [Google Scholar]
- 15.Piccinin AM, Muniz G, Matthews FE, Johansson B. Terminal decline from within- and between-person perspectives, accounting for incident dementia. J Gerontol B Psychol Sci Soc Sci 2011;66:391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cadar D, Stephan BC, Jagger C, et al. The role of cognitive reserve on terminal decline: a cross-cohort analysis from two European studies: OCTO-Twin, Sweden, and Newcastle 85+, UK. Int J Geriatr Psychiatry 2016;31:601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Batterham PJ, Mackinnon AJ, Christensen H. The effect of education on the onset and rate of terminal decline. Psychol Aging 2011;26:339–350. [DOI] [PubMed] [Google Scholar]
- 18.Bennett DA, Wilson RS, Schneider JA, et al. Education modifies the relation of AD pathology to level of cognitive function in older persons. Neurology 2003;60:1909–1915. [DOI] [PubMed] [Google Scholar]
- 19.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Education modifies the association of amyloid but not tangles with cognitive function. Neurology 2005;65:953–955. [DOI] [PubMed] [Google Scholar]
- 20.Brayne C, Ince PG, Keage HA, et al. Education, the brain and dementia: neuroprotection or compensation? Brain 2010;133:2210–2216. [DOI] [PubMed] [Google Scholar]
- 21.Wilson RS, Bienias JL, Evans DA, Bennett DA. Religious Orders Study: overview and change in cognitive and motor speed. Aging Neuropsychol Cogn 2004;11:280–303. [Google Scholar]
- 22.Bennett DA, Schneider JA, Arvanitakis Z, Wilson RS. Overview and findings from the religious orders study. Curr Alzheimer Res 2012;9:628–645. PMCID: PMC3409291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bennett DA, Schneider JA, Buchman AS, Mendes de Leon CF, Wilson RS. The Rush Memory and Aging Project: study design and baseline characteristics of the study cohort. Neuroepidemiology 2005;25:163–175. [DOI] [PubMed] [Google Scholar]
- 24.Bennett DA, Schneider JA, Buchman AS, Barnes LL, Boyle PA, Wilson RS. Overview and findings from the Rush Memory and Aging Project. Curr Alzheimer Res 2012;9:646–663. PMCID: PMC3439198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Health and Human Services Task Force on Alzheimer's disease. Neurology 1984;34:939–944. [DOI] [PubMed] [Google Scholar]
- 26.Bennett DA, Schneider JA, Aggarwal NT, et al. Decision rules guiding the clinical diagnosis of Alzheimer's disease in two community-based cohort studies compared to standard practice in a clinic-based cohort study. Neuroepidemiology 2006;27:169–176. [DOI] [PubMed] [Google Scholar]
- 27.Wilson RS, Beckett LA, Barnes LL, et al. Individual differences in rates of change in cognitive abilities of older persons. Psychol Aging 2002;17:179–193. [PubMed] [Google Scholar]
- 28.Wilson RS, Barnes LL, Bennett DA. Assessment of lifetime participation in cognitively stimulating activities. J Clin Exp Neuropsychol 2003;25:634–642. [DOI] [PubMed] [Google Scholar]
- 29.Wilson RS, Barnes LL, Krueger KR, Hoganson G, Bienias JL, Bennett DA. Early and late life cognitive activity and cognitive systems in old age. J Int Neuropsychol Soc 2005;11:400–407. [PubMed] [Google Scholar]
- 30.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Neurofibrillary tangles mediate the association of amyloid with clinical AD and level of cognitive function. Arch Neurol 2004;61:348–384. [DOI] [PubMed] [Google Scholar]
- 31.Wilson RS, Nag S, Boyle PA, et al. Neural reserve, neuronal density in the locus coeruleus, and cognitive decline. Neurology 2013;80:1202–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett DA, Schneider JA, Arvanitakis Z, et al. Neuropathology of older persons without cognitive impairment from two community-based studies. Neurology 2006;66:1837–1844. [DOI] [PubMed] [Google Scholar]
- 33.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 2009;66:200–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boyle PA, Yu L, Nag S, et al. Cerebral amyloid angiopathy and cognitive outcomes in community-based older person. Neurology 2015;85:1930–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arvanitakis Z, Capuano AW, Leurgans SE, Bennett DA, Schneider JA. Relation of cerebral blood vessel disease to Alzheimer's disease dementia and cognitive function in elderly people: a cross-sectional study. Lancet Neurol 2016;15:934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neumann M, Kwong LK, Lee EB, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol 2009;117:137–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson RS, Yu L, Trojanowski JQ, et al. TDP-43 pathology, cognitive decline, and dementia in old age. JAMA Neurol 2013;70:1418–1424. PMCID: PMC3830649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nag S, Yu L, Wilson RS, Chen EY, Bennett DA, Schneider JA. TDP-43 pathology and memory impairment in elders without the pathological diagnoses of AD or FTLD. Neurology 2017;88:653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nag S, Yu L, Capuano AW, et al. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer's disease. Ann Neurol 2015;77:942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider JA, Li JL, Li Y, Wilson RS, Kordower JH, Bennett DA. Neurofibrillary tangles in the substantia nigra are related to gait impairment in older persons. Ann Neurol 2006;59:166–173. [DOI] [PubMed] [Google Scholar]
- 41.Ceci SJ. How much does schooling influence general intelligence and its cognitive components? A reassessment of the evidence. Dev Psychol 1991;27:703–722. [Google Scholar]
- 42.Draganski B, Gaser C, Kempermann G, et al. Temporal and spatial dynamics of brain structure changes during extensive learning. J Neurosci 2006;26:6314–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hertzog C, Kramer AF, Wilson RS, Lindenberger U. Enrichment effects on adult cognitive development: can the functional capacity of older adults be preserved or enhanced? Psychol Sci Public Interest 2009:9:1–65. [DOI] [PubMed] [Google Scholar]
- 44.Wilson RS, Boyle PA, Yu L, Barnes LL, Schenider JA, Bennett DA. Life-span cognitive activity, neuropathologic burden, and cognitive aging. Neurology 2013;81:314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bassuk SS, Glass TA, Berkman LF. Social disengagement and incident cognitive decline in community-dwelling elderly persons. Ann Intern Med 1999;131:165–173. [DOI] [PubMed] [Google Scholar]
- 46.Curtis RG, Windsor TD, Soubelet A. The relationship between Big-5 personality traits and cognitive decline ability in older adults: a review. Neuropsychol Dev Cogn B Aging Neuropsychol Cogn 2015;22:42–71. [DOI] [PubMed] [Google Scholar]
- 47.Pool LR, Weuve J, Wilson RS, Bültmann U, Evans DA, Mendes de Leon CF. Occupational cognitive requirements and late-life cognitive aging. Neurology 2016;86:1386–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boyle PA, Buchman AS, Wilson RS, Yu L, Schneider JA, Bennett DA. Effect of purpose in life on the relation between Alzheimer disease pathologic changes on cognitive function in advanced age. Arch Gen Psychiatry 2012;69:499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qureshi AI, Suri MF, Saad M, Hopkins LN. Educational attainment and risk of stroke and myocardial infarction. Med Sci Monit 2003;9:CR466–473. [PubMed] [Google Scholar]
- 50.McHutchinson CA, Backhouse EV, Cvoro V, Shenkin SD, Wardlaw JM. Education, socioeconomic status, and intelligence in childhood and stroke risk in later life: a meta-analysis. Epidemiology 2017;28:608–618. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data included in these analyses are available through the Rush Alzheimer's Disease Center Research Resource Sharing Hub at radc.rush.edu. It includes descriptions of the studies and key variables. After logging in, qualified users can request deidentified data.