

Abstract
The hormone relaxin is considered a potential therapy for idiopathic pulmonary fibrosis (IPF). We have previously shown that a potential limitation to relaxin-based IPF therapy is decreased expression of a relaxin receptor, relaxin/insulin-like family peptide receptor 1 (RXFP1), in IPF fibroblasts. The mechanism that down-regulates RXFP1 in IPF remains unclear. To determine whether microRNAs (miRs) regulate RXFP1 gene expression, here we employed a bioinformatics approach to identify miRs predicted to target RXFP1 and identified a putative miR-144-3p target site in the RXFP1 mRNA. In situ hybridization of IPF lung biopsies revealed that miR-144-3p is expressed in fibroblastic foci. Furthermore, we found that miR-144-3p is up-regulated in IPF fibroblasts compared with lung fibroblasts from healthy donors. Transforming growth factor β increased miR-144-3p expression in both healthy and IPF lung fibroblasts in a SMAD family 2/3 (SMAD2/3)-dependent manner, and Jun proto-oncogene AP-1 transcription factor subunit (AP-1) was required for constitutive miR-144-3p expression. Overexpression of an miR-144-3p mimic significantly reduced RXFP1 mRNA and protein levels and increased expression of the myofibroblast marker α-smooth muscle actin (α-SMA) in healthy lung fibroblasts. IPF lung fibroblasts transfected with anti-miR-144-3p had increased RXFP1 expression and reduced α-SMA expression. Of note, a lentiviral luciferase reporter carrying the WT 3′ UTR of RXFP1 was significantly repressed in IPF lung fibroblasts, whereas a reporter carrying a mutated miR-144-3p–binding site exhibited less sensitivity toward endogenous miR-144-3p expression, indicating that miR-144-3p down-regulates RXFP1 in IPF lung fibroblasts by targeting its 3′ UTR. We conclude that miR-144-3p directly represses RXFP1 mRNA and protein expression.
Keywords: relaxin, fibroblast, myofibroblast, microRNA (miRNA), pulmonary fibrosis, transforming growth factor β (TGF-B), post-transcriptional regulation, epigenetics, idiopathic pulmonary fibrosis, lung disease, lung fibroblasts, miR-144-3p, relaxin/insulin-like family peptide receptor 1 (RXFP1)
Introduction
Idiopathic pulmonary fibrosis (IPF)3 is a chronic and progressive scarring of the lung parenchyma that ultimately compromises gas exchange and progresses to respiratory failure and death in many by four years after the diagnosis (1, 2). The hallmark of fibrosis, which is shared by all organs, is the dysregulated, and seemingly unending, architectural destruction caused by the accumulation of activated fibroblasts and the deposition of extracellular matrix. Multiple lines of evidence have suggested that the hormone relaxin is a potentially powerful inhibitor of fibrosis (3, 4). These include the seminal observations that mice genetically engineered to lose expression of the hormone relaxin (encoded in humans by RLN2) developed a progressive, age-related multiorgan fibrosis that was reversible with exogenous relaxin (4–6). In further support of the importance of this pathway, we have shown that gene expression levels for the relaxin receptor, RXFP1, in IPF is directly associated with pulmonary function (7). In IPF, RXFP1 protein expression is decreased in both diseased lungs and in fibroblasts isolated from diseased lungs compared with donor controls. Because the loss of RXFP1 expression in IPF may desensitize fibroblasts from the positive effects of relaxin-like agonists (7), a strategy to increase RXFP1 expression in IPF might prove to be an effective therapeutic approach.
Little is known, however, about the transcriptional regulation of RXFP1 expression. Many endocrine signals have been associated with RXFP1 expression (8, 9). We and others have shown that transforming growth factor (TGF) β stimulation can lead to decreased RXFP1 expression (7, 10). Further highlighting the heterogeneity of fibrosis in different organs, increased RXFP1 expression has been observed in liver fibrosis (11, 12). To our knowledge, there are no data on the regulation of RXFP1 by particular transcription factors.
One relatively unexplored mechanism to regulate RXFP1 expression is the role of microRNAs. MicroRNAs are noncoding small RNAs, about 22 nucleotides in length, that can bind to the 3′ UTR of target genes to repress their translation and/or induce degradation of target gene mRNA by incomplete base pairing. The role of mesenchymal cell (fibroblast) microRNA expression and function in pulmonary fibrosis has been demonstrated in several recent studies (13–18). Only synthetic microRNAs have been associated with regulation of RXFP1 expression (19). In this study, we tested the hypothesis that dysregulation of microRNA expression in IPF fibroblasts regulates RXFP1 gene expression.
Results
Lowest human RXFP1 mRNA expression is present in the IPF patients with the highest predicted mortality
Previously, we found that RXFP1 gene expression in IPF lungs was negatively correlated with pulmonary function (7). These data, however, were not analyzed for predicted mortality. In this new analysis, we calculated GAP (genomic-age-pulmonary function) stages (1 to 3) for the IPF patients in the Lung Tissue Research Consortium (LTRC) dataset (Fig. 1, A–C, n = 134). This three-stage system has been validated and is associated with increasing 1-year mortality in IPF (7, 20). Here, we plotted RXFP1 (and RXFP2 and RXFP3) gene expression data as a function of GAP stage. For all three genes, missing pulmonary function data (N = 13/134) were imputed to the severest pulmonary function category. We have found that patients in the highest GAP stage had the lowest RXFP1 gene expression. This was not observed for RXFP2 and -3. These data indicate that patients with the lowest RXFP1 gene expression were in the group of patients predicted to have the highest mortality.
Figure 1.

TGFβ is associated with decreased half-life of RXFP1 in human lung fibroblasts. GAP scores were calculated on the IPF patients in the LTRC dataset, and the patients were assigned a GAP stage from 1 to 3. Gene expression by microarray for RXFP1 (A), RXFP2 (B), and RXFP3 (C) were plotted as a function of increasing GAP stage. For GAP 1, n = 45; GAP 2, n = 75; and GAP 3, n = 14. Data were analyzed by Kruskal-Wallis, and p values are indicated in the panel. D, donor (n = 3), and E, IPF (n = 3) lung fibroblasts were processed for quantitative RT-PCR for RXFP1 at several time points following incubation with TGFβ and actinomycin D. Data represent the percentage of relative abundance of RXFP1 mRNA remaining compared with the 0-h time point, and the best fit curves for RXFP1 decay were plotted. F, dark line indicates data representing relative abundance of RXFP1 expression of RXFP1 mRNA remaining compared with the 0-h time point for donor fibroblasts. Dotted line indicates the percentage of relative abundance of RXFP1 expression in IPF fibroblasts at the indicated time points normalized to the levels of RXFP1 mRNA of donor fibroblasts at the 0-h time point. Data represent the relative abundance of miR-144-3p (ΔΔCt) remaining compared with the 0-h time point, and the best fit curves for miR-144-3p decay were plotted.
TGFβ is associated with decreased half-life of RXFP1 in human lung fibroblasts
Because a reduction in RXFP1 gene expression in the lung predicts reduced pulmonary function (7) and increased mortality, we next sought to understand the kinetics of RXFP1 mRNA stability, donor and IPF lung fibroblasts were incubated simultaneously with the transcriptional inhibitor actinomycin D and TGFβ. Cells were processed for quantitative RT-PCR at several time points. We determined that the half-life of RXFP1 from donor lung fibroblasts is ∼19.5 h, whereas that of TGFβ-treated donor lung fibroblasts is ∼13.6 h (Fig. 1D). Interestingly, we found that the half-life of RXFP1 from IPF lung fibroblasts is ∼33 h, whereas that of TGFβ-treated IPF lung fibroblasts is ∼11 h (Fig. 1E). The rate of degradation of RXFP1 in response to TGFβ is slightly faster in IPF lung fibroblasts compared with donor lung fibroblasts. When we compare IPF and donor lung fibroblasts for RXFP1 at baseline, we noted that the expression level of RXFP1 expression at baseline is much lower in IPF lung fibroblasts compared with donor lung fibroblasts (Fig. 1F). Taken together, this led us to speculate that post-transcriptional mechanisms may regulate RXFP1 expression in IPF fibroblasts and in response to TGFβ.
Human RXFP1 mRNA is a direct target of miR-144-3p
To understand potential post-transcriptional mechanisms that may regulate RXFP1 transcription in lung fibroblasts, we employed several public database prediction programs to identify potential microRNA species that may regulate RXFP1 gene expression. MiRanda (microrna.org)4 and Targetscan 7.0 predict that RXFP1 is a potential target of miR-144-3p (Fig. 2A). miR-144-3p is highly conserved in primates and lower vertebrates. Although the RXFP1 target site for miR-144-3p is conserved among primates (human, chimpanzee, rhesus, and bovine genomes), it is not conserved in mouse. Because hsa–miR-144-3p is predicted to have strong binding to 3′ UTR of hRXFP1 mRNA, we prioritized hsa–miR-144-3p for further testing. First, to determine whether miR-144-3p can regulate RXFP1 mRNA and protein levels, we transfected donor and IPF lung fibroblasts with miR-144-3p mimic and processed these cells for quantitative RT-PCR and immunoblotting of RXFP1 (Fig. 2, B and C). Quantitative RT-PCR results showed that miR-144-3p overexpression resulted in a significant down-regulation of RXFP1 mRNA levels in donor lung fibroblasts (Fig. 2B). We also show by qRT-PCR that IPF lung fibroblasts have significantly lower basal levels of RXFP1 mRNA compared with donor lung fibroblasts, confirming our previous findings (Fig. 2B) (7). These data support the hypothesis that hsa–miR-144-3p regulates RXFP1 expression. To verify miR-144-3p specificity on expression of RXFP1, we determined whether the effect manifested with another relaxin receptor gene, RXFP2. In silico, miR-144-3p is not predicted to target either RXFP2 or RXFP3. Expression of human RXFP2 was unaffected by miR-144-3p overexpression compared with control mimic based on qRT-PCR (Fig. 2C).
Figure 2.
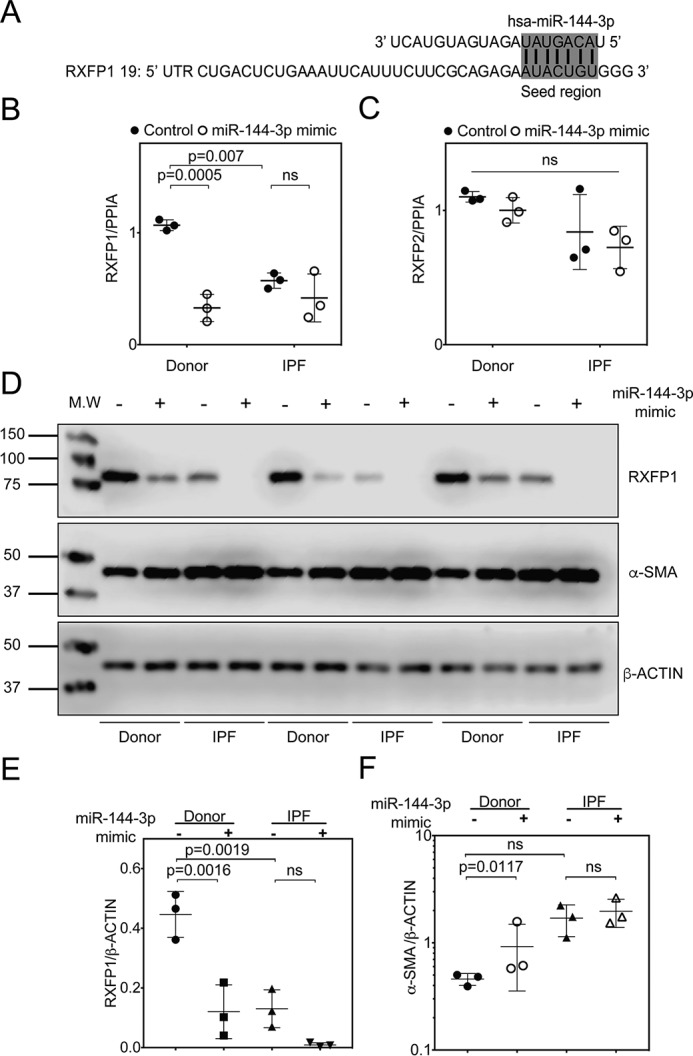
miR-144-3p targets RXFP1 in human lung fibroblasts. A, the seed region of hsa–miR-144-3p predicted to target the 3′ UTR of human RXFP1. B and C, donor and IPF lung fibroblasts were treated with miR-144-3p mimic or scrambled control. RNA was isolated and processed for qPCR for RXFP1 (B) or RXFP2 (C). Significantly higher basal levels of RXFP1 mRNA were detected in donor lung fibroblasts compared with IPF lung fibroblasts (p = 0.007, n = 3). Treatment of donor lung fibroblasts with the miR-144-3p significantly decreased RXFP1 expression (p = 0.0005, by two-way ANOVA and Tukey post hoc testing, n = 3). D, donor and IPF lung fibroblasts were transfected with 10 nm miR-144-3p mimic or control, and cells were lysed for immunoblotting for RXFP1, α-SMA, and β-actin. E, densitometry of immunoblotting presented in F. Data are expressed as mean ± S.D., normalized to β-actin. Less RXFP1 protein was detected in donor lung fibroblasts following transfection of miR-144-3p compared with scrambled control (p = 0.0016, n = 3). Significantly more RXFP1 was present in donor lung fibroblasts compared with IPF lung fibroblasts following transfection with the scrambled control (p = 0.0019, n = 3). Data were analyzed by two-way ANOVA and Tukey's post-hoc test. F, densitometry of α-SMA were normalized to β-actin. Transfection of miR-144-3p into donor lung fibroblasts significantly increased expression of α-SMA (p = 0.0117 for log-transformed data, n = 3, by two-way ANOVA followed by Tukey's post hoc testing).
We further analyzed if the effects of miR-144-3p on RXFP1 were detectable at the protein level. Using Western blotting and densitometry analyses, we found that forced expression of miR-144-3p mimic in donor and IPF lung fibroblasts resulted in a significant repression of RXFP1 protein levels (Fig. 2, D–F). We previously found that the loss of RXFP1 is also associated with increased expression of the myofibroblast differentiation marker, α-SMA (7). To determine whether miR-144-3p–mediated loss of RXFP1 is associated with α-SMA expression, we further analyzed its role in the response to miR-144-3p overexpression. We transfected donor and IPF fibroblasts with miR-144-3p mimic or negative control mimic. In Western blotting and densitometry analysis for α-SMA levels for the experiment from Fig. 2E and Fig. S2A; we found that miR-144-3p overexpression resulted in increased levels of α-SMA in donor lung fibroblasts; however, α-SMA levels increased significantly in IPF lung fibroblasts over baseline (Fig. 2F). Next, we determined if RXFP1 protein levels can be inhibited in a dose-dependent manner in donor lung fibroblasts using a range of mimic concentrations (0–10 nm). Western blotting and densitometry analysis show that miR-144-3p mimic decreased RXFP1 with increasing concentrations of miR-144-3p mimic, whereas completely inhibiting expression of RXFP1 at a concentration of 10 nm in donor lung fibroblasts (Fig. 3, A–C and Fig. S2B). Although RXFP1 levels decreased, α-SMA levels increased in a dose-dependent manner as well (Fig. 3C).
Figure 3.

miR-144-3p targets RXFP1 in a dose-dependent manner. A, donor lung fibroblasts were transfected with increasing concentrations of miR-144-3p mimic (0.1, 1, 5, and 10 nm). miR-144-3p mimic decreased the expression of RXFP1 and up-regulated the expression of α-SMA in a dose-dependent manner. Densitometry of RXFP1 (B) and α-SMA (C) from A (data analyzed by one-way ANOVA, for RXFP1, p value for trend 0.0006, R2 = 0.640) and for α-SMA (p value of log-transformed data for trend <0.02, R2 = 0.51). D, top panel, prediction of the seed region of hsa–miR-144-3p WT and mismatch mutant targeting the 3′ UTR of human RXFP1. Bottom panel, representative image of gel contraction experiments of donor lung fibroblasts following infection with WT miR-144-3p, mismatch miR-144-3p, or GFP-expressing particles or incubation with TGFβ (n = 3). E, quantification of collagen gel areas was performed using ImageJ and plotted (p < 0.001, vehicle + miR-144-3p versus vehicle + GFP; p < 0.05, TGFβ + miR-144-3p versus TGFβ + GFP; ns, untreated GFP versus untreated mismatch group). Values are presented as mean ± S.D.
To determine the effects of miR-144-3p mimic on the contractility of lung fibroblasts, we treated lung fibroblasts from healthy donors with lentiviral particles carrying a miR-144-3p mimic vector, a miR-144-3p mismatch mutant vector, or a GFP control vector (Fig. 3, D and E). Forty-eight hours after lentiviral infection, we evaluated the contractility using gel contraction assays, donor fibroblasts infected with lentiviral miR-144-3p mimic vector demonstrated greater gel contraction (∼55% reduction in area) compared with lenti-GFP control vector. The mismatch mutant did not have any significant effect on gel contraction. TGFβ increased the gel contraction of donor fibroblasts with lenti-GFP, but this TGFβ effect was not enhanced with lenti-miR-144-3p mimic vector. These data indicate that miR-144-3p expression globally increases myofibroblast contractility.
miR-144-3p is up-regulated in IPF lung fibroblasts
Lung biopsies were obtained from IPF subjects (n = 5) and processed for in situ hybridization (Fig. 4A) employing probes against the small nuclear RNA U6, miR-144-3p, and a scrambled control. Staining for the U6 positive control was present in nearly all cells observed in a nuclear pattern. No detectable staining was present with the scrambled control. The miR-144-3p probe showed staining in many cell types including fibroblasts in fibroblastic foci, the hallmark lesion of IPF.
Figure 4.

Regulation of miR-144-3p expression. A, lung biopsies were obtained from IPF subjects (n = 5). In situ hybridization was performed as described employing probes directed against U6 (left), scrambled control (center), or miR-144-3p (right). Low power images are shown on the top. Yellow inset squares are magnified at the bottom. Green inset bar = 200 μm and red inset bar = 50 μm. B, donor and IPF lung fibroblasts were stimulated with and without TGFβ, and RNA was isolated for qPCR. Significantly higher levels (>70-fold) of miR-144-3p were detected in IPF lung fibroblasts compared with donor controls (p < 0.0001, n = 6). TGFβ stimulation increased expression of miR-144-3p in donor lung fibroblasts by ∼5-fold (p < 0.0001), whereas it increased by ∼1.4-fold (p < 0.0001) in IPF lung fibroblasts. Data were log-transformed and analyzed by two-way ANOVA followed by Tukey's post hoc test. C, donor lung fibroblasts were incubated with Smad2 and/or Smad3 siRNA and stimulated with TGFβ. TGFβ significantly increased miR-144-3p expression in the presence of the scrambled control (p < 0.0001, n = 6). Smad2 and/or Smad3 siRNA significantly decreased the effect of TGFβ on miR-144-3p expression (p < 0.005, n = 6). Data were analyzed by two-way ANOVA, followed by Tukey's post hoc test. D, IPF lung fibroblasts were incubated with Smad2 and/or Smad3 siRNA and stimulated with TGFβ. TGFβ significantly increased miR-144-3p expression in the presence of the scrambled control (p < 0.0001, n = 6). Smad2 and/or Smad3 siRNA significantly decreased the effect of TGFβ on miR-144-3p expression (p < 0.0001, n = 6). Data were analyzed by two-way ANOVA, followed by Tukey's post hoc test. E and F, quantitative RT-PCR for Smad2 (E) and Smad3 (F) for experiments in C and D. Data were analyzed by two-way ANOVA followed by Tukey's post hoc test.
If miR-144-3p regulates RXFP1, we reasoned that miR-144-3p levels should be higher in response to TGFβ and in IPF. First, we determined the levels of miR-144-3p in IPF and donor fibroblasts using quantitative RT-PCR with and without TGFβ stimulation. We found that IPF fibroblasts showed >70-fold higher levels of mature miR-144-3p (Fig. 4B) compared with donor lung fibroblast controls. In donor lung fibroblasts, TGFβ increased miR-144-3p levels ∼5-fold. The already elevated levels of miR-144-3p in IPF fibroblasts increased further with TGFβ stimulation (∼1.4-fold). These data indicate that expression of miR-144-3p is downstream of TGFβ stimulation and is significant in IPF compared with donor controls. To determine the effect on miR-144-3p following actinomycin-D, we re-analyzed the experiments performed in Fig. 1C, and we found the half-life of miR-144-3p in IPF lung fibroblasts to be ∼18 h, whereas it is ∼12 h in response to TGFβ (Fig. S2C).
miR-144-3p is regulated by TGFβ/Smad2/Smad3 signaling in donor and IPF lung fibroblasts
We have previously observed that chemical inhibition of Smad signaling could block the effects of TGFβ on RXFP1 expression. In this experiment, we determined if silencing of Smad2/3 would decrease miR-144-3p expression. We silenced Smad2 and Smad3 individually as well as in combination by siRNA. Although vehicle treatment had no effect, TGFβ significantly increased the expression levels of mature miR-144-3p both in donor and IPF fibroblasts. Silencing of Smad2 and/or Smad3 reversed the effects of TGFβ on donor and IPF fibroblasts (Fig. 4, C and D). We confirmed that we achieved significant silencing of Smad2/3 gene expression in vehicle and TGFβ-treated cells. We did observe that silencing of Smad2/3 was not sufficient to bring the levels of miR-144-3p down compared with the levels of expression in vehicle-treated controls for donor and IPF lung fibroblasts (Fig. 4, E and F). This led us to speculate that there must be an additional mechanism of transcriptional regulation for maintaining basal (constitutive) expression of miR-144-3p in lung fibroblasts.
Innate immune signaling and inflammation have recently been associated with myofibroblast differentiation in IPF (21, 22). IL-1β is a principal cytokine of the innate immune system and mediates downstream inflammatory pathways. Because IL-1β is shown to up-regulate miR-144-3p (23) and IL1β is upstream of TGFβ (24), we determined the effects of IL1β on miR-144-3p. However, in our hands IL-1β showed no significant effect on the levels of miR-144-3p or RXFP1 both in donor and IPF lung fibroblasts (Fig. S6). By qPCR analysis, there were no significant differences in miR-144-3p or RXFP1 expression.
miR-144-3p is regulated by the AP-1 transcription factor (c-Jun/c-Fos) in donor and IPF lung fibroblasts
Previously, it has been shown that the AP-1 complex is one of the key regulators of miR-144 locus (25, 26). Furthermore, to determine whether AP-1 may be involved in regulation of miR-144-3p in lung fibroblasts, we silenced c-Jun or c-Fos by siRNA (Fig. 5, A and B). Individual and combined silencing of c-Jun and/or c-Fos resulted in a significant down-regulation of miR-144-3p levels in both donor and IPF fibroblasts. Successful silencing of c-Jun and c-Fos for this experiment are shown in Fig. 5, C and D. In gain-of-function experiments, overexpression of c-Jun using a lentiviral vector carrying c-Jun gene resulted in ∼6.4-fold increase in miR-144-3p levels (Fig. 6B) compared with miR-186-5p (Fig. 6C), an endogenous control microRNA that is not known to be affected by c-Jun. Furthermore, we showed that phorbol 12-myristate 13-acetate (PMA), which is known to activate the AP-1 complex, significantly augmented the levels of mature miR-144-3p by ∼3.8-fold (Fig. 6A). We found that IPF fibroblasts showed slightly higher expression of c-Jun and c-Fos (∼2.3- and ∼2.9-fold, respectively) when compared with that of donor fibroblasts (Fig. 5, E and F). This indicates that the AP-1 complex in IPF is responsible for constitutive expression of miR-144-3p.
Figure 5.
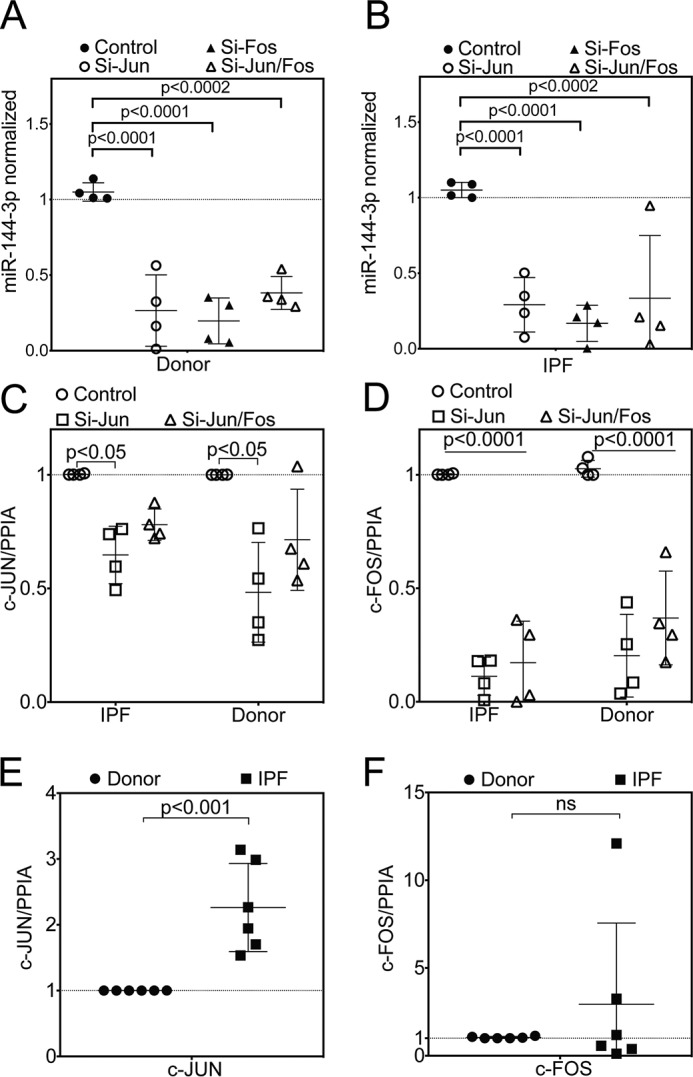
AP-1 complex is one of the key regulators of miR-144 locus. Quantitative RT-PCR for miR-144-3p following silencing of c-Jun and/or c-Fos in (A) donor or (B) IPF lung fibroblasts (data were analyzed by two-way ANOVA, followed by Tukey's post hoc test, n = 4). Experiments with donor and IPF lung fibroblasts were performed in parallel, and data from A and B were analyzed independently. C and D, quantitative RT-PCR for silencing of c-Jun (C) and c-Fos (D) from A and B, respectively. Data were analyzed by two-way ANOVA, followed by Tukey's post-hoc test, n = 4. E and F, quantitative RT-PCR for c-Jun (E) and c-Fos (F) between unstimulated donor and IPF lung fibroblasts. Data were analyzed by unpaired t test, n = 6.
Figure 6.

PMA and lentiviral overexpression of c-Jun induced expression of miR-144-3p. A, quantitative RT-PCR for miR-144-3p following incubation of donor lung fibroblasts with PMA, an activator of the AP-1 transcription factor (p < 0. 05, paired two-tailed t test, n = 6). B and C, quantitative RT-PCR for miR-144-3p (B) and miR-186-5p (C) following infection of donor lung fibroblasts with a lentiviral vector expressing GFP (control) or c-Jun (data were analyzed by paired t test, n = 5).
Anti-miR-144-3p reverses the suppression of RXFP1 in IPF lung fibroblasts
Having shown that miR-144-3p inhibits RXFP1 expression, we next questioned if inhibition of miR-144-3p would augment RXFP1 expression. As shown in Fig. 7A, RT-PCR analyses showed that transfection of donor lung fibroblasts with anti-miR-144-3p (antagomir) resulted in a 9-fold depletion of miR-144-3p levels in vehicle control. Although TGFβ treatment resulted in an increase in the levels of mature miR-144-3p by ∼40-fold, this effect was partially reversed by transfection of anti-miR-144-3p. We observed a ∼6.5-fold reduction in levels of mature miR-144-3p levels. We further showed, as a proof-of-principle, that the effect of suppression of RXFP1 expression by TGFβ treatment can be reversed by transfection of anti-miR-144-3p. This indicated that miR-144-3p is crucial in the suppression of RXFP1 expression and that anti-miR-144-3p can be used as a tool to reverse the loss of RXFP1 (Fig. 7B).
Figure 7.
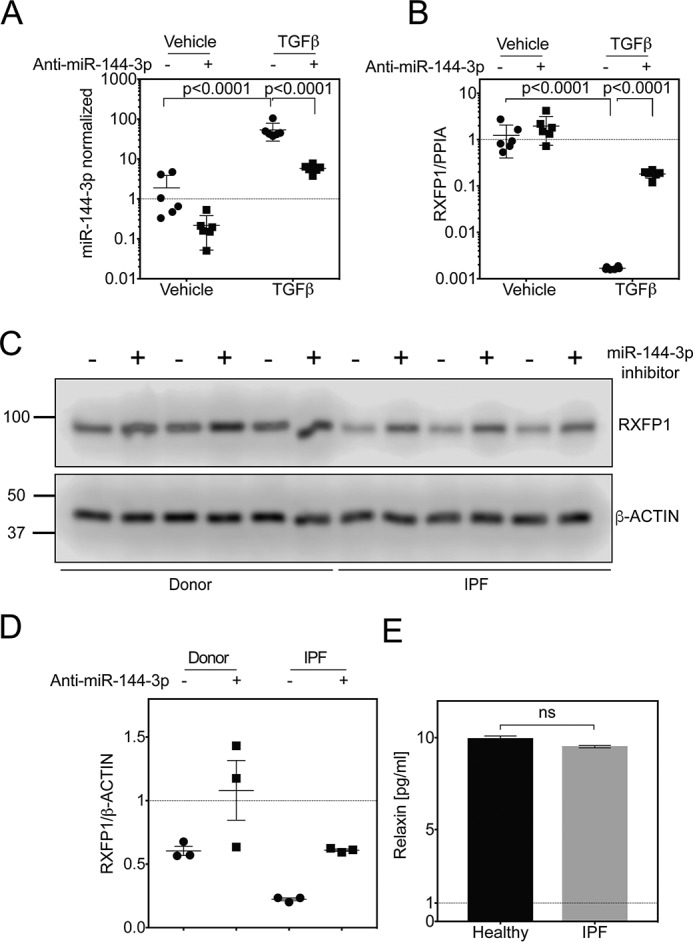
Anti-miR-144-3p reversed the suppression of RXFP1 in IPF lung fibroblasts. A and B, quantitative RT-PCR for miR-144-3p (A) or RXFP1 (B) from donor lung fibroblasts after transfection with a miR-144-3p antagonist (anti-miR-144-3p) and stimulation with TGFβ (p = 0.0001 for log-transformed data for miR-144-3p and RXFP1, by two-way ANOVA followed by Tukey's multiple comparisons test, n = 6). C, immunoblots for RXFP1 from donor and IPF lung fibroblasts following transfection with anti-miR-144-3p or scrambled controls (n = 3). D, densitometry of band intensity of RXFP1 normalized to β-actin from donor (E) and IPF (F) lung fibroblasts (p = ns for donor; p < 0.0001 for IPF, n = 3, data were analyzed by paired t test). E, Bio-Plex 200 multiplex analysis of bronchoalveolar lavage for Relaxin (n = 18 healthy controls and n = 126 IPF patients, p = not significant, Kolmogorov-Smirnov test).
Having shown that RXFP1 is a targeted gene by miR-144-3p, we next examined whether forced expression of anti-miR-144-3p in donor and IPF fibroblasts would augment RXFP1 protein expression (Fig. 7, C and D). As shown in Fig. 7, C and D, and Fig. S3 Western blotting and densitometry analyses show that the expression of RXFP1 protein was significantly increased by forced expression of the anti-miR-144-3p in both donor lung fibroblasts and IPF lung fibroblasts (p = not significant for donor; p < 0.0001 for IPF, n = 3).
We have previously observed that silencing of RXFP1 increased levels of phosphorylated myosin light chains in fibroblasts as a measure of myofibroblast-like contractility (7). We also showed that treatment of cells with a relaxin agonist could decrease phosphorylation of myosin light chains (7, 27). To estimate an in vivo concentration of relaxin in the lung, we assayed by bronchoalveolar lavage (BAL) patients with IPF or normal controls for relaxin. No significant differences in relaxin was detected by BAL between IPF and control. In BAL, the mean Relaxin concentration was 9.9 pg/ml (∼1.7 pm) in unaffected controls compared with 9.5 pg/ml in IPF patients (n = 18 healthy controls and n = 126 IPF patients, p = not significant, Kolmogorov-Smirnov test) (Fig. 7E, Table S1). Next, we performed in vitro experiments to alter expression of RXFP1 to determine the sensitivity at physiologic levels of relaxin compared with enhanced levels of relaxin, as a model for relaxin therapy (Fig. 8A). To decrease RXFP1 expression, donor fibroblasts were transfected with miR-144-3p mimic. To increase RXFP1 expression, IPF fibroblasts were transfected with anti-miR-144-3p. Transfection of miR-144-3p significantly decreased levels of RXFP1 and increased α-SMA in donor fibroblasts. pMLC20 levels were elevated significantly upon treatment with miR-144-3p mimic. Loss of RXFP1 was associated with insensitivity to low doses of recombinant relaxin as measured by phosphorylation of MLC20 and by expression of α-SMA. We found that the effects of miR-144-3p mimic on α-SMA can partially be reversed by pre-treatment with 10 nm Relaxin (high dose) but insensitive at physiological concentration of Relaxin at 10 pm (Fig. 8A, Fig. S4 and Fig. S8). We found that pMLC20 levels decreased with pre-treatment by Relaxin in a dose-dependent manner. Additionally, pre-treatment with Relaxin also inhibited pMLC20, and α-SMA levels in a dose-dependent manner (Fig. 8, B–E). In summary, these data show that transfection of anti-miR-144-3p in IPF fibroblasts resulted in a significant recovery of RXFP1.
Figure 8.

Relaxin reverses contractile phenotypes associated with miR-144-3p. A, transfection of donor lung fibroblasts with miR-144-3p mimic to decrease RXFP1 levels followed by incubation with low and high concentrations of relaxin. Right panel, transfection of IPF lung fibroblasts with anti-miR-144-3p followed by incubation with low and high concentrations of relaxin. Cells were lysed for immunoblotting for RXFP1, phospho-MLC20, total MLC20, α-SMA, and β-Actin (n = 3). B-E, densitometry of immunoblotting presented in A. Data were expressed as mean ± S.D., normalized to β-Actin or total MLC20. B, between mimic and no mimic, significant effects (p < 0.05) were observed for vehicle, relaxin low, and relaxin high in donor lung fibroblasts. More α-SMA protein was detected in donor lung fibroblasts following transfection of miR-144-3p compared with scrambled control. C, in IPF lung fibroblasts, no significant effects were observed with either low or high dose relaxin between mimic or no mimic groups. D, significant effects between mimic and no mimic were observed only with high dose of relaxin (p < 0.05). Significantly more phospho-MLC20 was present in IPF lung fibroblasts compared with donor lung fibroblasts at baseline. E, significant effects were observed only for relaxin high dose in IPF lung fibroblasts following transfection with antagomir (p < 0.05; n = 3). Data were analyzed by two-way ANOVA followed by the Holm-Sidak test.
miR-144-3p directly targets the 3′ UTR of RXFP1 mRNA
To determine whether there is a direct interaction between miR-144-3p and the 3′ UTR of RXFP1 mRNA, we subcloned both the WT and mutant RXFP1 3′ UTRs (containing the putative miR-144-3p–binding sequence) distal to luciferase ORF in pmirGLO luciferase reporter (Fig. 9A) and determined if the miR-144-3p mimic can suppress luciferase activity in 293T cells. We found that the WT RXFP1 3′ UTR construct was associated with a nearly 50% decrease in luciferase activity when compared with empty control vector. This loss of luciferase activity was abrogated by the scrambled control mimic (Fig. 9B). The luciferase activity of mutant RXFP1 3′ UTR was unaffected by miR-144-3p mimic (Fig. 9C). These results indicate the specificity of miR-144-3p binding to RXFP1 3′ UTR affects mRNA stability.
Figure 9.

miR-144-3p directly targets the 3′ UTR of RXFP1 mRNA. A, depiction of pmirGLO dual-luciferase reporter construct for the WT 3′ UTR of the RXFP1 seed region and mutations. B and C, 293T cells were plated 24 h prior to transfection. Cells were then co-transfected with 50 ng of either pmirGLO vector carrying WT (B) 3′ UTR of RXFP1 or mutated (C) 3′ UTR of RXFP1 with and without 100 nm miR-144-3p mimic or control mimic. miR-144-3p significantly repressed the luciferase activity of the reporter containing the WT 3′ UTR of RXFP1 (p < 0.005, unpaired t test, mean ± S.D., n = 3), whereas the mutated construct was insensitive in 293T cells (I, unpaired t test, mean ± S.D., n = 3). Luciferase activity was measured using Renilla luciferase as an internal control. D, lentiviral luciferase reporter construct carrying WT 3′ UTR of RXFP1 was used to transduce both donor and IPF lung fibroblasts. There was a reduction in luciferase activity with WT 3′ UTR of RXFP1 compared with mutated 3′ UTR of RXFP1. Also, the luciferase activity of WT 3′ UTR RXFP1 was highly suppressed in IPF lung fibroblasts compared with donor lung fibroblasts indicating higher levels of endogenous miR-144-3p in IPF lung fibroblasts compared with that of donor. Lentiviral Renilla luciferase vector was used as an internal control. Data shown are from a representative experiment (n = 3). E, depiction of mmu–miR-144-3p mimic and its modified version to enhance base pairing with mouse RXFP1 3′ UTR. There is a single nucleotide mismatch in mmu–miR-144-3p with mouse RXFP1 3′ UTR target region. E, primary MLF were isolated from C57/B6 mice (n = 2; repeated twice). MLF were transfected either with a mmu–miR-144-3p mimic or a modified mmus–miR-144-3p mimic (modified to match mouse RXFP1 target region) along with a negative control mimic. MLF were harvested to isolate total RNA and qRT-PCR was performed to determine the levels of mRXFP1, mPPIA, and mNFE2L2 (mNrf2).
To further verify direct targeting in vivo, donor and IPF lung fibroblasts were co-transduced with lentiviral particles with vectors of WT or mutant 3′ UTR RXFP1 sequences that are subcloned distal to luciferase ORF with Renilla luciferase lentivector. We determined that the luciferase activity of WT 3′ UTR RXFP1 was highly suppressed in IPF lung fibroblasts compared with donor lung fibroblasts. A mutant RXFP1 3′ UTR construct was less effective at suppressing luciferase activity (Fig. 9D) in both. The WT 3′ UTR lentivector titrated the endogenous levels of miR-144-3p, whereas the mutant construct failed. Altogether, these data collectively indicate that endogenous hsa–miR-144-3p directly targets human RXFP1 and regulates its expression in IPF patient lung fibroblasts. The RXFP1 target site for miR-144-3p is poorly conserved between the human and mouse genomes. Therefore, we asked if the mechanism of targeting by miR-144-3p is conserved despite a single nucleotide change in mouse RXFP1 3′ UTR target region (Fig. 9, E and F). Primary mouse lung fibroblasts (MLF) were isolated from C57/Bl6 mice and cultured as described (28). MLF were transfected either with mmu–miR-144-3p mimic or a modified mmu–miR-144-3p mimic to enhance base pairing with the 3′ UTR of RXFP1 along with a negative control mimic. Cells were harvested to isolate total RNA and qRT-PCR was performed to determine the levels of mRXFP1 and mNFE2L2 (mNrf2), a positive control, because it has at least two predicted target sites for miR-144-3p (29). We postulated that transfection of a modified miR-144-3p mimic that exhibits stronger bp matching should result in a suppression of mRXFP1 expression. We found that the modified mimic failed to suppress levels of mouse RXFP1 mRNA. As predicted, mmu–miR-144-3p mimic inhibited mNrf2 expression. The modified mimic had no effect on mNrf2. This indicates that the mechanism of miR-144-3p–based RXFP1 targeting is present in humans but not in mice. We also show that hsa–miR-144-3p can target the 3′ UTR of human Nrf2 at two sites and by qPCR, transfection of miR-144-3p significantly decreased Nrf2 expression in donor lung fibroblasts (Fig. S5).
Genes associated with fibrosis are expressed downstream of miR-144-3p
IPF fibroblasts express higher levels of collagens and extracellular matrix proteins (13, 14). Gene expression profiling of IPF lung tissues exhibited a unique gene signature with up-regulated genes that included a significant increase in expression of Collagen1a2 (COL1A2), Collagen3a1 (COL3A1), and Versican (VCAN) levels (30). TGFβ induced an increase in expression levels of fibronectin (FN1) and α-SMA (ACTA2) in MRC5 fibroblasts (14). Because relaxin signaling is associated with lower expression of matrix proteins (5, 6, 31), we hypothesized that loss of RXFP1 expression associated with miR-144-3p would be associated with increases in fibrosis-associated genes and that restoration of RXFP1 gene expression would decrease expression of fibrosis-associated genes. To determine whether miR-144-3p in associated with fibroblast activation phenotype, we transfected miR-144-3p in donor lung fibroblasts. In parallel experiments, we transfected anti-miR-144-3p into IPF fibroblasts (n = 4). Transfections were matched with scrambled controls. RNA was isolated and processed for qPCR for a set of target profibrotic genes (Fig. 10A) COL1A2, (Fig. 10B) COL3A1, (Fig. 10C) ACTA2, (Fig. 10D) VCAN, and (Fig. 10E) FN1. Significant increases in levels of both collagens COL1A2, and COL3A1, VCAN, and FN1 mRNA were observed with transfection of miR-144-3p in donor lung fibroblasts compared with scrambled controls. A trend toward increased ACTA2 (Fig. 10C) was observed. Transfection of antagomir significantly lowered levels of COL1A2, and COL3A1, VCAN, and FN1 mRNA in IPF lung fibroblasts compared with control inhibitor (paired, two-tailed Student's t test, n = 4). A trend decrease in ACTA2 expression was observed with the transfection of antagomir (Fig. 10C′). These data support the hypothesis that miR-144-3p drives expression of several pathologic fibrosis genes downstream of its effects on RXFP1.
Figure 10.
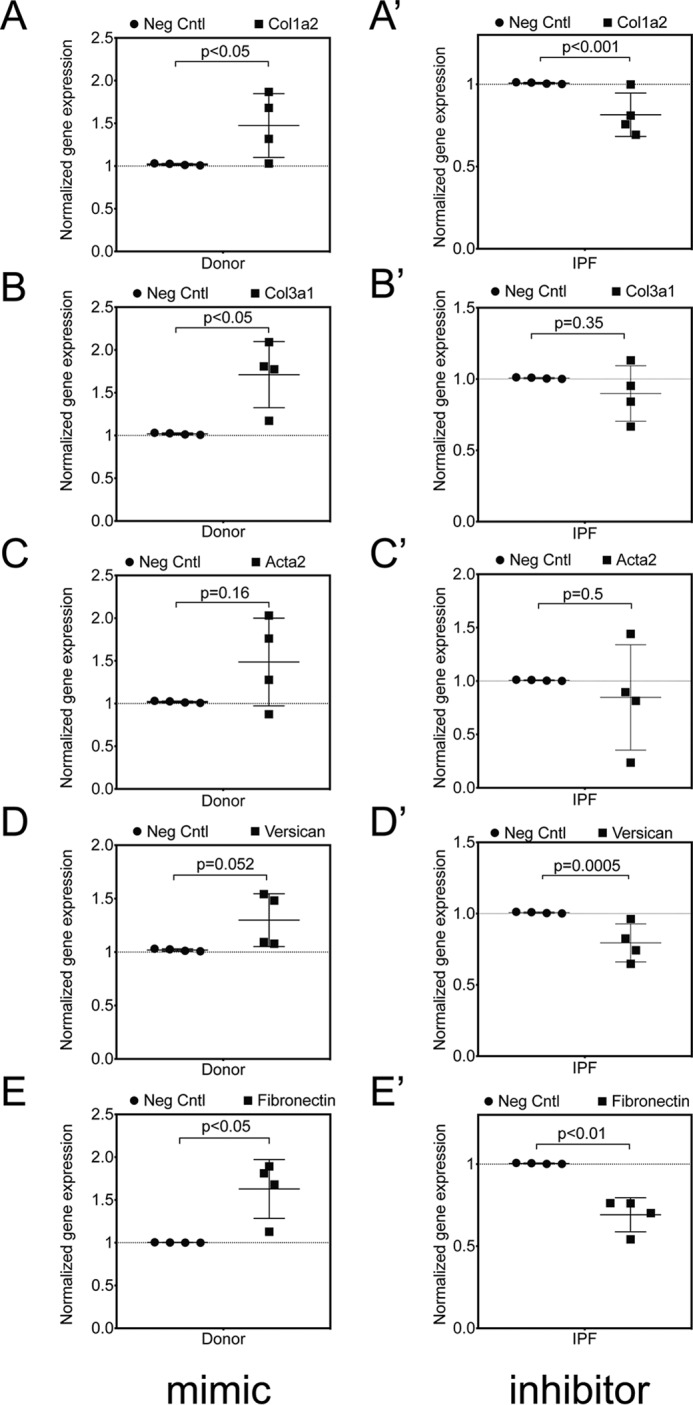
miR-144-3p controls fibrotic gene expression in lung fibroblasts. Donor and IPF lung fibroblasts (n = 4) were treated with miR-144-3p mimic or scrambled control and anti-miR-144-3p mimic or negative control inhibitor, respectively. RNA was isolated and processed for qPCR for (A, A′), COL1A2 (B, B′), COL3A1 (C, C′), ACTA2 (D, D′), and VCAN (E, E′) FN1. Transfection of mimic resulted in significantly higher basal levels of COL1A2, COL3A1, ACTA2, VCAN, and FN1 mRNA in donor lung fibroblasts compared with scrambled controls, whereas transfection of antagomir resulted in significantly lower basal levels of COL1A2, COL3A1, ACTA2, VCAN, and FN1 in IPF lung fibroblasts compared with a negative control inhibitor (paired, two-tailed Student's t test, n = 4).
Discussion
We have previously found that regulation of RXFP1 at the level of mRNA, and consequently at the protein level, may have important implications for the prognosis of IPF patients. In this study, we have observed that there is differential decay of RXFP1 mRNA following actinomycin D treatment, suggesting that RNA-destabilizing mechanisms regulate RXFP1 mRNA levels. Based on in silico predictions, we identified that miR-144-3p may inhibit RXFP1 expression. We found that miR-144-3p significantly reduced RXFP1 mRNA and protein in both donor and IPF lung fibroblasts, in a dose-dependent manner. miR-144-3p also increased expression of the myofibroblast marker α-SMA, also in a dose-dependent manner. In addition, silencing of RXFP1 by miR-144-3p was associated with increased expression of several matrix genes including COL1A2, COL3A1, ACTA2, VCAN, and FN1. Conversely, miR-144-3p blockade increased RXFP1 mRNA and protein, depleted endogenous miR-144-3p expression, and decreased expression of COL1A2, COL3A1, ACTA2, VCAN, and FN1 in IPF lung fibroblasts. These data suggest that blockade of miR-144-3p could be employed to enhance expression of RXFP1 therapeutically and could be a critical adjunct to relaxin-based therapies.
It seems rather unlikely that enhanced expression of miR-144-3p in IPF fibroblasts is the sole mechanism regulating RXFP1 mRNA stability. Certainly other microRNAs are predicted to target RXFP1 but were not tested in this study. Moreover, our study has not addressed transcription factors that are predicted to target sites 5′ or intronic promoter or enhancer sites. Differential gene, and microRNA expression, between IPF and donor lung fibroblasts likely explains the differences in the half-life of RXFP1 following actinomycin D treatment. Testing other microRNAs or silencing of certain transcription factors might be employed in the future to address some of these unanswered questions.
We have observed that silencing of Fos/Jun and Smad2/3 both decreased miR-144-3p expression. Based on our experiments, we found the Smad-mediated effects in the context of TGFβ. The Fos and Jun effects were observed in otherwise unstimulated cells. From these experiments, it is difficult to estimate the relative biologic importance of these two fundamental pathways. Future studies would address the upstream signaling mechanisms that lead to both Smad2/3 and AP-1 activation. These could be tested with the FDA-approved drugs for IPF, pirfenidone and nintedanib, both of which may be associated with fibrotic signaling (32). In addition, several current clinical trials in IPF are testing cell-signaling inhibitors that may be relevant here including the c-Jun-NH2 terminal kinase 2 (JNK2) inhibitor (33) or a PI3K/mTOR inhibitor (34). Both pathways are known to converge on Smad and AP-1 signaling (35–39). Although such drug studies are admittedly nonspecific and will likely have many overlapping effects on several microRNAs, the rationale for pursuing these studies is to explore how agents that are either approved for or under consideration as treatments for pulmonary fibrosis may impact Smad and Fos/Jun signaling and consequently miR-144-3p, RXFP1, and other potentially important downstream targets such as Nrf2.
So where does miR-144-3p fit in the larger context of fibrosis? We speculate that expression of miR-144-3p in lung fibroblasts may be part of a larger pathway regulating myofibroblast differentiation, an effect that may be partially related to RXFP1 expression. The precursor microRNA has been associated with reduced expression of the Smad signaling repressor TGFβ-induced factor homeobox (TGIF1) (40). A previous report has shown that the levels of miR-144-3p are elevated in the biopsy specimens from patients with the bronchiolitis obliterans syndrome (BOS), a form of airway fibrosis associated with chronic rejection following lung transplant. In this study, forced expression of miR-144-3p mimic increased levels of TGFβ and vice versa, which increased α-SMA and F-actin levels in MRC5 fibroblasts (40). Our observations that myofibroblast gene expression can be modulated in response to miR-144-3p–mediated silencing of RXFP1 and rescued by anti-miR-144-3p–mediated overexpression of RXFP1 does suggest that relative reduction of RXFP1 induced by TGFβ is likely a pathologic event rather than simply a marker of TGFβ activity.
So this question of miR-144-3p's place in the pathogenesis of pulmonary fibrosis also highlights a critical limitation of our studies, namely, the lack of biological activity of miR-144-3p on RXFP1 expression in mice and therefore the potential application of miR-144-3p to mouse models of pulmonary fibrosis. Although hsa–miR-144-3p decreased RXFP1 mRNA in human lung fibroblasts, we found that a mmu–miR-144-3p construct, designed to enhance base pairing and address the evolutionary mismatch, did not decrease mouse RXFP1. These data indicate that miR-144-3p is not a regulator of mouse RXFP1 expression, even when base pairing is enhanced to target murine RXFP1. We do, however, suggest that miR-144-3p blockade could be employed in mouse models of pulmonary fibrosis to increase the expression of Nrf2 (encoded by Nfe2l2), an important regulator of the antioxidant response, which may represent a parallel, anti-fibrotic pathway (41, 42).
In conclusion, we have identified a potential mechanism regulating expression of RXFP1 in pulmonary fibrosis. Because of the importance of relaxin signaling to fibrosis across many organs, we speculate that molecular strategies to increase RXFP1 expression may potentially sensitize fibroblasts, as the principal effector cells of fibrosis, to the beneficial effects of relaxin. Further study is needed to consider miR-144-3p inhibitors in animal models of fibrosis.
Experimental procedures
Ethical review
Collection of human lung tissue was approved by the Institutional Review Board and the Committee for Oversight of Research and Clinical Training Involved Decedents of the University of Pittsburgh and in accordance with the Declaration of Helsinki. This study was conducted in accordance with University of Pittsburgh IRB protocol numbers PRO14120072 and IRB0411036. Informed written consent to the internal ethics review board-approved clinical study protocol (Hannover Medical School IRB protocol number 2923-2015) was obtained from each individual before participation in this study, in accordance with the Declaration of Helsinki.
Isolation of human lung tissue
Human lung tissue from patients with IPF were obtained from the recipients undergoing lung transplantation and pieces cut from the upper and lower lobes and placed in 2% paraformaldehyde for fixation. Sections were then embedded in paraffin for sectioning.
Bioinformatics
Prediction of miRNA targets was conducted using miRanda (43) and Targetscan 7.0 (44). The putative sequences of primary microRNAs and the 3′ UTR of RXFP1, -2, and -3 were retrieved from the National Center for Biotechnology Information and ENSEMBLE server (45) and miRBase (46).
Primary cell culture
Primary lung fibroblasts were obtained as previously described (47). Donor human fibroblasts were isolated from lungs that appeared to have no injury by histology but were deemed unacceptable for lung transplant. IPF lung fibroblasts were obtained from patients either at explant or at autopsy (28). Briefly, lung tissue from explanted IPF lungs and age-matched normal donors was collected from the lower lobes to find age and sex of the lines included in the experiments (mean age for IPF 58 years ± 6 and 59 years ± 4 for controls, p = 0.76). Enzymatic digestion (0.05% trypsin; GIBCO) was used to isolate human lung fibroblasts, and later fibroblasts were grown with Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (Corning) containing 10% fetal bovine serum (Atlanta Biologicals) and 1% antimycotic–antibiotic (GIBCO). Cells were cultured at 37 °C, 5% CO2 and then expanded to select a homogeneous fibroblast population (experiments were performed with lines between passages 5 and 9). Cells were used for expression analysis of both RXFP1 and miR-144-3p. Anti-hsa–miR-144-3p and hsa–miR-144-3p mimic (Qiagen) were used to modulate miR144-3p levels. miScript mouse miRNA mimic miR-144-3p (Syn–mmu–miR-144-3p: 5 nmol, 5′-UACAGUAUAGAUGAUGUACU; catalog number MSY0000156) was purchased from Qiagen. Modified mouse miR-144-3p mimic (miRIDIAN mimic: 20 nmol, 5′-UACAGCAUAGAUGAUGUACU; mismatch highlighted) was custom-made to order from Dharmacon. ON-TARGETplus smartpool siRNAs for Smad2, Smad3, c-Fos, c-Jun, RXFP1, and Negative control scramble siRNA were purchased from Dharmacon. Fibroblasts were transfected with miRNA mimics or miRNA inhibitors using HiperFect transfection reagent (Qiagen). Briefly, transfection complexes were generated by mixing 0–10 nm of miRNA mimic/control mimic or 50 nm inhibitor/control inhibitor according to the manufacturer's protocol. After a 10-min incubation at room temperature, the transfection complexes were added dropwise onto 2 × 105 cells per well of a 6-well plate in 2000 μl of culture medium with 1% fetal bovine serum but without antibiotics. Cells were transfected on the day of plating (day 0) and again on day 1. To determine the transfection efficiency of antagomir, 0–50 nm 5′ FAM-labeled anti-miR-144-3p or 5′ FAM-labeled negative control were used to transfect donor lung fibroblasts, which are plated on coverslips, on days 0 and 1. 24 h after transfection, cells were washed in PBS and fixed in 4% paraformaldehyde for 30 min. Coverslips were washed in PBS and mounted in medium containing DAPI (4′,6-diamidino-2-phenylindole dihydrochloride). Fluorescent images of FAM and DAPI were taken at 488 and 358 nm, respectively, on a Nikon Eclipse TE200 microscope (Nikon, Tokyo, Japan) equipped with epifluorescence. Fields were selected at random and FITC+ cells were enumerated and scored as a percentage of all cells present/high-power field (hpf). About 60% cells were identified with fluorescent-labeled miRNA complexes. Representative images are shown in Fig. S7.
RNA extraction
Briefly, total RNAs were extracted from the donor and IPF lung fibroblasts using RNeasy (Qiagen). The RNA samples were reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). The product from each reverse transcription reaction was preamplified and then the mRNA expression analysis was performed by qRT-PCR using a ABI TaqMan system (Life Technologies) following the manufacturer's recommended protocol. qRT-PCR primers were purchased from Qiagen (Hsa-PPIA, catalog number QT01669542; Hsa-RXFP1, catalog number QT00041720; Hsa-RXFP2, catalog number 00095725; Hsa-PPIA (peptidylprolyl isomerase A). PPIA was used as the housekeeping gene for normalization. The global normalization process included the subtraction of the mean CT value of the reference set from the CT value of each gene of the same sample. Quantification of each sample is shown as 2−ΔΔCt value.
TaqMan real-time RT-PCR for miRNAs
Small RNAs were isolated from cells following the protocol of the miRNeasy kit (Qiagen). 100–300 ng of RNA sample was reverse transcribed into cDNA using the TaqManTM Advanced miRNA cDNA Synthesis Kit (Applied Biosystems) according to manufacturer's protocol. TaqMan probes (TaqManTM microRNA Control Assay for RNU43 and TaqManTM microRNA Advanced Assay for hsa–miR-144-3p) were purchased from Applied Biosystems. Real-time quantitative PCR was performed using the powerUP SYBR Green master mix and the TaqMan Fast Advanced Master Mix (Applied Biosystems). Quantification of each sample is shown as 2−ΔΔCt value. Error bars indicate the mean ± S.E. from biological replicates, each consisting of technical triplicates.
Analysis of bronchoalveolar lavage
Patients with presumed IPF at Hannover Medical School underwent bronchoscopy and BAL during the routine diagnostic work-up (Table S1). After routine clinical testing was performed, an aliquot of BAL was saved. Cells were spun down, the cell-free supernatant, BAL fluid, was subjected to multiplex Luminex analysis (Bio-Plex 200, Bio-Rad).
Western blotting
RXFP1 protein expression was analyzed using Western blotting. Western blotting was performed as described previously (7). Anti-RXFP1 (ab72159) and anti-αSMA antibodies (ab7817) were purchased from Abcam. pMLC20 and MLC20 antibodies were purchased from Cell Signaling. Mouse anti-actin (sc-47778), and mouse anti-human RXFP1 (sc-293228) antibodies were from Santa Cruz Biotechnology. Densitometry analyses were performed with the C-DIGIT imager.
Design of reporter constructs and Luciferase assays
Duplexed oligonucleotide pairs (IDT) were designed to contain the predicted miR-144-3p–binding region in the RXFP1 3′ UTR and when annealed and ligated into the pmirGLO vector, resulted in the miR-144-3p target region in the correct 5′ to 3′ orientation. The sequences of the duplexes used were as follows: RXFP1–Mir144–WT–duplex (sense strand, 5′-AAACTAGCGGCCGCTAGTTCTTCGCAGAGAATACTGTGGGGGTGT; antisense, 5′-CTAGACACCCCCACAGTATTCTCTGCGAAGAACTAGCGGCCGCTAGTTT) and RXFP1–Mir144–Mismatch duplex (sense strand: 5′-AAACTAGCGGCCGCTAGTTCTTCGCAGAGAATAAATGGGGGTGT; antisense strand, 5′-CTAGACACCCCCATTTATTCTCTGCGAAGAACTAGCGGCCGCTAGTTT). Overhangs in the designed duplex were complementary to those generated by PmeI and XbaI double digestion of the pmirGLO Vector (Promega). A NotI site was designed into the oligo duplex for clone confirmation. pmirGLO Vector was linearized with PmeI and XbaI to generate overhangs that are complementary to the duplex overhangs. Duplexed oligonucleotide-linearized vectors were ligated using a standard ligation protocol. Ligated pmirGLO was transformed using high-efficiency TOP10-competent cells (New England Biolabs). Clones were selected on carbenicillin-containing plates, and screened for those containing the duplex by digesting miniprep-purified DNA using the internal NotI site. All constructs and mutants were verified by DNA sequencing at the University of Pittsburgh Genomics Research Core. The purified plasmid DNA was used directly in transfections. HEK-293T cells in a 24-well plate were transfected with 50 ng of pmirGLO-RXFP1-WT in triplicates and 100 nm control or miR-144-3p mimic (Ambion) Lipofectamine 2000 (Invitrogen). 24 h post-transfection, luciferase assays were performed on the lysates from the cells using a Dual Luciferase Assay System (Promega) on a Spectramax L instrument. Data were normalized and presented as the ratio of firefly to Renilla luciferase activities.
Lentiviral vector cloning and transduction in lung fibroblasts
Lentiviral expression vector pLenti-GIII-CMV-GFP-2A-Puro carrying human c-JUN (LV194093), and pLenti-UTR-GFP-Blank (m014), lentiluciferase vectors pLenti-UTR-Luc-Blank (m012), and pLenti-III-Renilla (LV010079) were purchased from abmgood (Applied Biological Materials Inc., Richmond, BC, Canada). pLenti-RXFP1-UTR-Luc and its mutant were generated as follows: cassettes for lentiviral 3′ UTR vectors carrying RXFP1 target region were generated using the duplex oligonucleotide pairs that were synthesized at IDT (RXFP1-miR144-WT duplex-pLenti: sense strand, 5′-AATTCTCTTCGCAGAGAATACTGTGGGGGTGC; antisense strand, 5′-TCGAGCACCCCCACAGTATTCTCTGCGAAGAG; RXFP1-miR144-MsM duplex-pLenti: sense strand, 5′-AATTCTCTTCGCAGAGAATAAATGGGGGTGC, antisense strand, 5′-TCGAGCACCCCCATTTATTCTCTGCGAAGAG). Duplexes were designed to include unique restriction sites EcoRI and XhoI at the ends of the sequences. Annealed duplexes were ligated into pLenti-3′UTR-Luc vector that is cleaved by EcoRI and XhoI, and transformed into TOP10 cells. Lentiviral packaging was performed using viral packaging plasmids from the “ViraPower” kit (Invitrogen) according to the manufacturer's instructions. Filtered viral supernatants were obtained from transfection of 293FT cells according to the manufacturer's protocol. qPCR Lentivirus Titration Kit (abmgood) was used to measure the lentiviral titers according to the manufacturer's instructions. Viral medium was added to complete medium in the presence of Polybrene at a multiplicity of infection of 5 to infect donor and IPF lung fibroblasts for 72 h. Cells were lysed in passive lysis buffer and dual luciferase assays were performed as described above.
In situ hybridization of miRNAs
The protocol for in situ hybridization for miRNA detection was based on a previous report (48). Specifically, 4-μm lung sections were deparaffinized and rehydrated in Xylene and a decreasing ethanol gradient. Slides were further probed with digoxigenin (3′ DIG)-labeled miRCURY LNA hsa–miR-144-3p detection probe (hsa–miR-144-3p/DigN/AGTACATCATCTATACTGTA, 5 nmol/liter; Qiagen), miRCURY LNA scramble-miR probe (/DigN/GTGTAACACGTCTATACGCCCA, 1 nmol/liter; Negative control) and miRCURY LNA U6 probe (U6, hsammurno/DigN/CACGAATTTGCGTGTCATCCTT, 5 nmol/liter; positive control).
Gel contraction assays
Lentiviral overexpression vector carrying GFP–hsa–miR-144-3p was purchased from abmgood (catalog number mh11106). Mismatch mutant was generated using QuikChange site-specific mutagenesis with the following oligonucleotide primers (forward, 5′-GTTGACAGTGAGCGACTTCTGAAAAGATGATGTACTAATA-3′; reverse, 5′-TATTAGTACATCATCTTTTCAGAAGTCGCTCACTGTCAAC-3′) and the sequence was confirmed by Sanger sequencing. Lentiviruses were packaged as described above and used to infect donor and IPF lung fibroblasts at a multiplicity of infection of 5. 72 h after infection, cells were trypsinized, rinsed in 1× PBS without Ca2+ and Mg2+, and mixed with 3 mg/ml of neutralized bovine collagen I diluted in DMEM as 2 parts cells and 8 parts collagen matrix. 250 μl of cell and collagen mixture was seeded into a 48-well cell contraction plate (Cell Biolabs, No. 50211B) at 5 × 105 cells/well. The collagen gels were polymerized at 37 °C and 5% CO2 for 1 h. After polymerization, 0.5 ml of DMEM with 0.5% fetal bovine serum was added with and without 10 ng/ml of TGFβ on top of the collagen matrix. Wells were monitored at 37 °C and 5% CO2 for 48 h. Media was changed every 24 h. ImageJ suite was used to measure the diameters of the gels at 0 and 48 h from the image taken using a Bio-Rad imager.
Decay kinetics
Actinomycin D (0.5 μg/ml) and TGFβ (5 ng/ml) were added concurrently to serum-starved IPF and donor lung fibroblast lines. Total RNA was extracted as shown at 24-, 48-, and 72-h time points, and mRNA levels were measured with qRT-PCR. RXFP1 transcript or miR-144-3p concentrations were expressed as fractions of abundance in the samples prior to the addition of actinomycin D or TGFβ (n = 3). Nonlinear fit one-phase exponential decay equation curve from GraphPad Prism was used to calculate the transcript turnover rates and expressed as half-life for each experiment.
Statistical analysis
All data are shown as mean ± S.D. Statistical significance was assessed by performing t tests, one-way and two-way analysis of variance (ANOVA), where appropriate, followed by the Tukey's post hoc analysis for multiple comparisons using p < 0.05 in GraphPad Prism software (San Diego, CA) unless otherwise stated. Statistical analysis is indicated in the figure legends.
Author contributions
H. B., J. T., J. A. D., Y. Z., and D. J. K. conceptualization; H. B., J. T., J. A. D., and D. J. K. formal analysis; H. B., Y. Z., and D. J. K. supervision; H. B. and D. J. K. funding acquisition; H. B., J. T., J. A. D., S. B. S., J. S., A. P., Y. Z., and D. J. K. investigation; H. B., J. T., and D. J. K. writing-original draft; H. B. and D. J. K. project administration; H. B., J. T., J. A. D., S. B. S., J. S., D. A., M. R., Y. Z., and D. J. K. writing-review and editing; J. A. D., S. B. S., J. S., D. A., M. R., B. J., A. P., and D. J. K. resources; B. J. methodology.
Supplementary Material
This work was supported in whole or part by National Institutes of Health Grants R01 HL126990 and P50AR060780 (to D. J. K.) and the Dorothy P. and Richard P. Simmons Center for Interstitial Lung Disease. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1–S8.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- IPF
- idiopathic pulmonary fibrosis
- miR
- microRNA
- RXFP1
- relaxin/insulin-like family peptide receptor 1
- TGF
- transforming growth factor
- ANOVA
- analysis of variance
- GAP
- genomic-age-pulmonary function
- PMA
- phorbol 12-myristate 13-acetate
- BAL
- bronchoalveolar lavage
- MLF
- mouse lung fibroblast
- PPIA
- peptidylprolyl isomerase A
- qRT
- quantitative RT
- GFP
- green fluorescent protein
- IL
- interleukin
- α-SMA
- α-smooth muscle actin
- DIG
- digoxigenin.
References
- 1. Navaratnam V., Fleming K. M., West J., Smith C. J., Jenkins R. G., Fogarty A., and Hubbard R. B. (2011) The rising incidence of idiopathic pulmonary fibrosis in the U.K. Thorax 66, 462–467 10.1136/thx.2010.148031 [DOI] [PubMed] [Google Scholar]
- 2. Raghu G., Lynch D., Godwin J. D., Webb R., Colby T. V., Leslie K. O., Behr J., Brown K. K., Egan J. J., Flaherty K. R., Martinez F. J., Wells A. U., Shao L., Zhou H., Pedersen P. S., et al. (2014) Diagnosis of idiopathic pulmonary fibrosis with high-resolution CT in patients with little or no radiological evidence of honeycombing: secondary analysis of a randomised, controlled trial. Lancet Respir. Med. 2, 277–284 10.1016/S2213-2600(14)70011-6 [DOI] [PubMed] [Google Scholar]
- 3. Chihal H. J., and Espey L. L. (1973) Utilization of the releaxed symphysis pubis of guinea pigs for clues to the mechanism of ovulation. Endocrinology 93, 1441–1445 10.1210/endo-93-6-1441 [DOI] [PubMed] [Google Scholar]
- 4. Bennett R. G. (2009) Relaxin and its role in the development and treatment of fibrosis. Transl. Res. 154, 1–6 10.1016/j.trsl.2009.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Samuel C. S., Zhao C., Bathgate R. A., Bond C. P., Burton M. D., Parry L. J., Summers R. J., Tang M. L., Amento E. P., and Tregear G. W. (2003) Relaxin deficiency in mice is associated with an age-related progression of pulmonary fibrosis. FASEB J. 17, 121–123 10.1096/fj.02-0449fje [DOI] [PubMed] [Google Scholar]
- 6. Samuel C. S., Mookerjee I., Masterson R., Tregear G. W., and Hewitson T. D. (2005) Relaxin regulates collagen overproduction associated with experimental progressive renal fibrosis. Ann. N.Y. Acad. Sci. 1041, 182–184 10.1196/annals.1282.026 [DOI] [PubMed] [Google Scholar]
- 7. Tan J., Tedrow J. R., Dutta J. A., Juan-Guardela B., Nouraie M., Chu Y., Trejo Bittar H., Ramani K., Biswas P. S., Veraldi K. L., Kaminski N., Zhang Y., and Kass D. J. (2016) Expression of RXFP1 is decreased in idiopathic pulmonary fibrosis: implications for relaxin-based therapies. Am. J. Respir. Crit. Care Med. 194, 1392–1402 10.1164/rccm.201509-1865OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dehghan F., Muniandy S., Yusof A., and Salleh N. (2014) Sex-steroid regulation of relaxin receptor isoforms (RXFP1 & RXFP2) expression in the patellar tendon and lateral collateral ligament of female WKY rats. Int. J. Med. Sci. 11, 180–191 10.7150/ijms.6283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moore X. L., Su Y., Fan Y., Zhang Y. Y., Woodcock E. A., Dart A. M., and Du X. J. (2014) Diverse regulation of cardiac expression of relaxin receptor by α1- and β1-adrenoceptors. Cardiovasc. Drugs Ther. 28, 221–228 10.1007/s10557-014-6525-x [DOI] [PubMed] [Google Scholar]
- 10. Mazella J., Tang M., and Tseng L. (2004) Disparate effects of relaxin and TGFβ1: relaxin increases, but TGFβ1 inhibits, the relaxin receptor and the production of IGFBP-1 in human endometrial stromal/decidual cells. Hum. Reprod. 19, 1513–1518 10.1093/humrep/deh274 [DOI] [PubMed] [Google Scholar]
- 11. Fallowfield J. A., Hayden A. L., Snowdon V. K., Aucott R. L., Stutchfield B. M., Mole D. J., Pellicoro A., Gordon-Walker T. T., Henke A., Schrader J., Trivedi P. J., Princivalle M., Forbes S. J., Collins J. E., and Iredale J. P. (2014) Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology 59, 1492–1504 10.1002/hep.26627 [DOI] [PubMed] [Google Scholar]
- 12. Snowdon V. K., Lachlan N. J., Hoy A. M., Hadoke P. W., Semple S. I., Patel D., Mungall W., Kendall T. J., Thomson A., Lennen R. J., Jansen M. A., Moran C. M., Pellicoro A., Ramachandran P., Shaw I., et al. (2017) Serelaxin as a potential treatment for renal dysfunction in cirrhosis: preclinical evaluation and results of a randomized phase 2 trial. PLoS Med. 14, e1002248 10.1371/journal.pmed.1002248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Montgomery R. L., Yu G., Latimer P. A., Stack C., Robinson K., Dalby C. M., Kaminski N., and van Rooij E. (2014) MicroRNA mimicry blocks pulmonary fibrosis. EMBO Mol. Med. 6, 1347–1356 10.15252/emmm.201303604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu G., Friggeri A., Yang Y., Milosevic J., Ding Q., Thannickal V. J., Kaminski N., and Abraham E. (2010) miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J. Exp. Med. 207, 1589–1597 10.1084/jem.20100035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Christmann R. B., Wooten A., Sampaio-Barros P., Borges C. L., Carvalho C. R., Kairalla R. A., Feghali-Bostwick C., Ziemek J., Mei Y., Goummih S., Tan J., Alvarez D., Kass D. J., Rojas M., de Mattos T. L., et al. (2016) miR-155 in the progression of lung fibrosis in systemic sclerosis. Arthritis Res. Ther. 18, 155 10.1186/s13075-016-1054-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pandit K. V., Corcoran D., Yousef H., Yarlagadda M., Tzouvelekis A., Gibson K. F., Konishi K., Yousem S. A., Singh M., Handley D., Richards T., Selman M., Watkins S. C., Pardo A., Ben-Yehudah A., et al. (2010) Inhibition and role of let-7d in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 182, 220–229 10.1164/rccm.200911-1698OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mizuno K., Mataki H., Seki N., Kumamoto T., Kamikawaji K., and Inoue H. (2017) MicroRNAs in non-small cell lung cancer and idiopathic pulmonary fibrosis. J. Hum. Genet. 62, 57–65 10.1038/jhg.2016.98 [DOI] [PubMed] [Google Scholar]
- 18. Chen S., Puthanveetil P., Feng B., Matkovich S. J., Dorn G. W. 2nd, and Chakrabarti S. (2014) Cardiac miR-133a overexpression prevents early cardiac fibrosis in diabetes. J. Cell Mol. Med. 18, 415–421 10.1111/jcmm.12218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yong K. L., Callander G. E., Bergin R., Samuel C. S., and Bathgate R. A. (2013) Development of human cells with RXFP1 knockdown using retroviral delivery of microRNA against human RXFP1. Ital. J. Anat. Embryol. 118, 10–12 [PubMed] [Google Scholar]
- 20. Ley B., Ryerson C. J., Vittinghoff E., Ryu J. H., Tomassetti S., Lee J. S., Poletti V., Buccioli M., Elicker B. M., Jones K. D., King T. E. Jr., and Collard H. R. (2012) A multidimensional index and staging system for idiopathic pulmonary fibrosis. Ann. Intern. Med. 156, 684–691 10.7326/0003-4819-156-10-201205150-00004 [DOI] [PubMed] [Google Scholar]
- 21. O'Dwyer D. N., Ashley S. L., and Moore B. B. (2016) Influences of innate immunity, autophagy, and fibroblast activation in the pathogenesis of lung fibrosis. Am. J. Physiol. Lung Cell Mol. Physiol. 311, L590–L601 10.1152/ajplung.00221.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bringardner B. D., Baran C. P., Eubank T. D., and Marsh C. B. (2008) The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid. Redox. Signal 10, 287–301 10.1089/ars.2007.1897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu C., Li X., Zhang D., Xu B., Hu W., Zheng X., Zhu D., Zhou Q., Jiang J., and Wu C. (2018) IL-1β-mediated up-regulation of WT1D via miR-144-3p and their synergistic effect with NF-κB/COX-2/HIF-1α pathway on cell proliferation in LUAD. Cell Physiol. Biochem. 48, 2493–2502 10.1159/000492687 [DOI] [PubMed] [Google Scholar]
- 24. Gasse P., Mary C., Guenon I., Noulin N., Charron S., Schnyder-Candrian S., Schnyder B., Akira S., Quesniaux V. F., Lagente V., Ryffel B., and Couillin I. (2007) IL-1R1/MyD88 signaling and the inflammasome are essential in pulmonary inflammation and fibrosis in mice. J. Clin. Invest. 117, 3786–3799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li H., Zhou J., Wei X., Chen R., Geng J., Zheng R., Chai J., Li F., and Jiang S. (2016) miR-144 and targets, c-fos and cyclooxygenase-2 (COX2), modulate synthesis of PGE2 in the amnion during pregnancy and labor. Sci. Rep. 6, 27914 10.1038/srep27914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cheng C., Li W., Zhang Z., Yoshimura S., Hao Q., Zhang C., and Wang Z. (2013) MicroRNA-144 is regulated by activator protein-1 (AP-1) and decreases expression of Alzheimer disease-related a disintegrin and metalloprotease 10 (ADAM10). J. Biol. Chem. 288, 13748–13761 10.1074/jbc.M112.381392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huang X., Gai Y., Yang N., Lu B., Samuel C. S., Thannickal V. J., and Zhou Y. (2011) Relaxin regulates myofibroblast contractility and protects against lung fibrosis. Am. J. Pathol. 179, 2751–2765 10.1016/j.ajpath.2011.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tan J., Tedrow J. R., Nouraie M., Dutta J. A., Miller D. T., Li X., Yu S., Chu Y., Juan-Guardela B., Kaminski N., Ramani K., Biswas P. S., Zhang Y., and Kass D. J. (2017) Loss of Twist1 in the mesenchymal compartment promotes increased fibrosis in experimental lung injury by enhanced expression of CXCL12. J. Immunol. 198, 2269–2285 10.4049/jimmunol.1600610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sangokoya C., Telen M. J., and Chi J. T. (2010) microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 116, 4338–4348 10.1182/blood-2009-04-214817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bridges R. S., Kass D., Loh K., Glackin C., Borczuk A. C., and Greenberg S. (2009) Gene expression profiling of pulmonary fibrosis identifies Twist1 as an antiapoptotic molecular “rectifier” of growth factor signaling. Am. J. Pathol. 175, 2351–2361 10.2353/ajpath.2009.080954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Unemori E. N., Bauer E. A., and Amento E. P. (1992) Relaxin alone and in conjunction with interferon-γ decreases collagen synthesis by cultured human scleroderma fibroblasts. J. Invest. Dermatol. 99, 337–342 10.1111/1523-1747.ep12616665 [DOI] [PubMed] [Google Scholar]
- 32. Bahudhanapati H., and Kass D. J. (2017) Unwinding the collagen fibrils: elucidating the mechanism of pirfenidone and nintedanib in pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 57, 10–11 10.1165/rcmb.2017-0079ED [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Baig S., Veeranna V., Bolton S., Edwards N., Tomlinson J. W., Manolopoulos K., Moran J., Steeds R. P., and Geberhiwot T. (2018) Treatment with PBI-4050 in patients with Alstrom syndrome: study protocol for a phase 2, single-centre, single-arm, open-label trial. BMC Endocr. Disord. 18, 88 10.1186/s12902-018-0315-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mercer P. F., Woodcock H. V., Eley J. D., Platé M., Sulikowski M. G., Durrenberger P. F., Franklin L., Nanthakumar C. B., Man Y., Genovese F., McAnulty R. J., Yang S., Maher T. M., Nicholson A. G., Blanchard A. D., Marshall R. P., Lukey P. T., and Chambers R. C. (2016) Exploration of a potent PI3 kinase/mTOR inhibitor as a novel anti-fibrotic agent in IPF. Thorax 71, 701–711 10.1136/thoraxjnl-2015-207429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Conery A. R., Cao Y., Thompson E. A., Townsend C. M. Jr., Ko T. C., and Luo K. (2004) Akt interacts directly with Smad3 to regulate the sensitivity to TGF-β induced apoptosis. Nat. Cell Biol. 6, 366–372 10.1038/ncb1117 [DOI] [PubMed] [Google Scholar]
- 36. Remy I., Montmarquette A., and Michnick S. W. (2004) PKB/Akt modulates TGF-β signalling through a direct interaction with Smad3. Nat. Cell Biol. 6, 358–365 10.1038/ncb1113 [DOI] [PubMed] [Google Scholar]
- 37. Lamouille S., and Derynck R. (2007) Cell size and invasion in TGF-β-induced epithelial to mesenchymal transition is regulated by activation of the mTOR pathway. J. Cell Biol. 178, 437–451 10.1083/jcb.200611146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Angel P., and Karin M. (1991) The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim. Biophys. Acta 1072, 129–157 [DOI] [PubMed] [Google Scholar]
- 39. Kim S. J., Angel P., Lafyatis R., Hattori K., Kim K. Y., Sporn M. B., Karin M., and Roberts A. B. (1990) Autoinduction of transforming growth factor beta 1 is mediated by the AP-1 complex. Mol. Cell Biol. 10, 1492–1497 10.1128/MCB.10.4.1492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xu Z. P., Ramachandran S., Gunasekaran M., Zhou F. Y., Trulock E., Kreisel D., Hachem R., and Mohanakumar T. (2015) MicroRNA-144 dysregulates the transforming growth factor-β signaling cascade and contributes to the development of bronchiolitis obliterans syndrome after human lung transplantation. J. Heart Lung Transpl. 34, 1154–1162 10.1016/j.healun.2015.03.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y., Zhao Y., Cheng M., Qiao Y., Wang Y., Xiong W., and Yue W. (2018) Suppression of microRNA-144–3p attenuates oxygen-glucose deprivation/reoxygenation-induced neuronal injury by promoting Brg1/Nrf2/ARE signaling. J. Biochem. Mol. Toxicol. 32, e22044 10.1002/jbt.22044 [DOI] [PubMed] [Google Scholar]
- 42. Hecker L., Logsdon N. J., Kurundkar D., Kurundkar A., Bernard K., Hock T., Meldrum E., Sanders Y. Y., and Thannickal V. J. (2014) Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 6, 231ra247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Betel D., Koppal A., Agius P., Sander C., and Leslie C. (2010) Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome Biol. 11, R90 10.1186/gb-2010-11-8-r90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Agarwal V., Bell G. W., Nam J. W., and Bartel D. P. (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Aken B. L., Ayling S., Barrell D., Clarke L., Curwen V., Fairley S., Fernandez Banet J., Billis K., Garcia Giron C., Hourlier T., Howe K., Kahari A., Kokocinski F., Martin F. J., et al. (2016) The Ensembl gene annotation system. Database (Oxford) 2016, pii: baw093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kozomara A., and Griffiths-Jones S. (2014) miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 42, D68–73 10.1093/nar/gkt1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Álvarez D., Cardenes N., Sellarés J., Bueno M., Corey C., Hanumanthu V. S., Peng Y., D'Cunha H., Sembrat J., Nouraie M., Shanker S., Caufield C., Shiva S., Armanios M., Mora A. L., and Rojas M. (2017) IPF lung fibroblasts have a senescent phenotype. Am. J. Physiol. Lung Cell Mol. Physiol. 313, L1164–L1173 10.1152/ajplung.00220.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Parikh V. N., and Chan S. Y. (2013) Analysis of microRNA niches: techniques to measure extracellular microRNA and intracellular microRNA in situ. Methods Mol. Biol. 1024, 157–172 10.1007/978-1-62703-453-1_12 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.