

Abstract
Eukaryotic DNA methylation prevents genomic instability by regulating the expression of oncogenes and tumor-suppressor genes. The negative effects of dysregulated DNA methylation are highlighted by a strong correlation between mutations in the de novo DNA methyltransferase gene DNA methyltransferase 3α (DNMT3A) and poor prognoses among acute myeloid leukemia (AML) patients. We show here that clinically observed DNMT3A mutations dramatically alter enzymatic activity, including mutations that lead to 6-fold hypermethylation and 3-fold hypomethylation of the human cyclin-dependent kinase inhibitor 2B (CDKN2B or p15) gene promoter. Our results provide insights into the clinically observed heterogeneity of p15 methylation in AML. Cytogenetically normal AML (CN-AML) constitutes 40–50% of all AML cases and is the most epigenetically diverse AML subtype with pronounced changes in non-CpG DNA methylation. We identified a subset of DNMT3A mutations that enhance the enzyme's ability to perform non-CpG methylation by 2–8–fold. Many of these mutations mapped to DNMT3A regions known to interact with proteins that themselves contribute to AML, such as thymine DNA glycosylase (TDG). Using functional mapping of TDG–DNMT3A interactions, we provide evidence that TDG and DNMT3-like (DNMT3L) bind distinct regions of DNMT3A. Furthermore, DNMT3A mutations caused diverse changes in the ability of TDG and DNMT3L to affect DNMT3A function. Cell-based studies of one of these DNMT3A mutations (S714C) replicated the enzymatic studies and revealed that it causes dramatic losses of genome-wide methylation. In summary, mutations in DNMT3A lead to diverse levels of activity, interactions with epigenetic machinery components and cellular changes.
Keywords: enzyme catalysis, enzyme mutation, enzyme kinetics, epigenetics, DNA methyltransferase, DNA methylation, DNA binding protein
Introduction
5-Methylcytosine (5-MC)2 is a naturally occurring epigenetic modification of mammalian DNA associated with essential cellular processes such as transcriptional regulation and cellular differentiation (1, 2). Like many cancers, acute myeloid leukemia (AML) involves changes to gene expression arising from aberrant DNA methylation patterning (3, 4). Yet unlike most cancers, and compared with genetic abnormalities, epigenetic aberrations appear to be more prevalent in AML (5). In fact, AML is marked by recurring mutations in genes of epigenetic modifiers including the de novo DNA methyltransferase, DNMT3A, in which mutations throughout the DNMT3A gene are observed in 22% of all AML patients (6, 7). Furthermore, DNA methylation profiles of AML patients reveal that aberrant methylation is heterogenous and can occur as either hyper- or hypomethylation (4). In addition to changes in DNA methylation, specific mutations in DNMT3A sufficiently disrupt interactions with regulatory components, which can be restored pharmacologically (7). The ability to rationally direct such changes will require a fundamental biochemical understanding of mutations in DNMT3A.
DNMT3A forms homo- and heterotetrameric complexes, and prior structure–function studies highlighted the importance of residues in the DNMT3A interfaces for methylation activity, processive catalysis, and oligomerization (8). In addition, some mutations, like R882H, coincided with those observed in AML patients, thereby highlighting the contributions of specific residues to catalysis and regulation through interactions that stabilize the homotetramer as well as complexes involving partner proteins (8). Due to its striking prevalence in AML patients, R882H has been extensively studied and its biochemical characterization has provided possible mechanisms of how this substitution may manifest itself in AML (8–10). Therefore, establishing a fundamental biochemical understanding of additional AML mutations in DNMT3A may broaden our understanding of the role aberrant DNMT3A activity plays in AML.
AML patients harbor a wide-range of mutations dispersed throughout the DNMT3A gene at varying frequencies with distinct predicted consequences to enzymatic function (Fig. 1) (11). Here we combine a detailed functional analysis of DNMT3A with mutations identified in AML patients that remain largely unexplored at the level of activity and regulation through interactions with partner proteins. Some DNMT3A mutants show enhanced activity, whereas others show an attenuated ability to methylate the p15 promoter, a genomic target that is linked to AML (12–15). The methylation of non-CpG sites is altered in AML subtypes, and we show that this activity is enhanced in some DNMT3A mutants (16, 17). Several mutants show differential regulation by thymine-DNA glycosylase (TDG), a component of the base excision repair (BER) system (18, 19), and DNMT3-like (DNMT3L), another partner protein (20, 21). Overall, we show how clinically relevant DNMT3A mutations may contribute to the aberrant DNA methylation in these patients.
Figure 1.
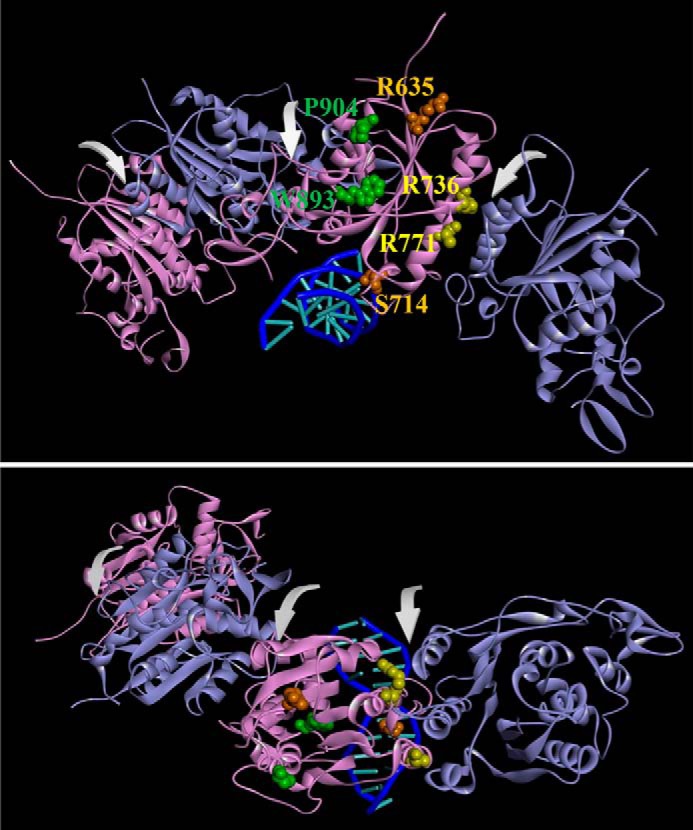
Mutations from AML patients in a DNMT3A homotetramer model. A model of the DNMT3A homotetramer (alternating purple and cyan monomers) bound to DNA was generated by aligning DNMT3A monomers to DNMT3L in a DNMT3A–DNMT3L heterotetramer crystal structure (PDB ID code 2QRV) followed by a subsequent alignment of a DNMT3A monomer to a M.HhaI-dsDNA co-crystal structure (PDB ID code 3EEO). Arrows in front view (A) and top view (B) indicate dimer and tetramer interfaces. Mutated residues are categorized based on location as follows: surface, orange; tetramer interface, yellow; and internal, green.
Results
Mutations from AML patients in the DNMT3A catalytic domain lead to varying degrees of DNA methylation
A common feature of AML patients carrying mutations in DNMT3A is the heterogeneity in both patterns and levels of DNA methylation, including the promoter region of the p15 tumor suppressor gene (12, 13). Furthermore, patients with mutations in DNMT3A display hypermethylation of the p15 promoter and reduced levels of this tumor suppressor (14, 15). Due to the diverse spatial distribution of the mutations throughout the DNMT3A catalytic domain, we sought to determine whether individual mutations vary to the extent and mechanism of DNMT3A functional changes, thereby contributing to the heterogeneity in DNA methylation observed within the AML population. We studied the ability of the WT and mutant DNMT3A (catalytic domain) to methylate the p15 promoter by inserting the promoter into a vector lacking any CpG sites (pCpGL) (21), referred to as p15-pCpGL. Due to the limited number of CpG sites, DNMT3A has reduced activity on the p15 human promoter (compared with poly(dI-dC) (Table 1) (21). Poly(dI-dC) represents the extreme of a high site-density substrate, whereas the p15-pCpGL represents a low-density substrate, but being derived from a CpG island, is still higher density than is typical of the human genome.
Table 1.
kcat values for wildtype and AML patient-derived DNMT3A mutants using poly(dI-dC) and p15-pCpGL DNA substrates
Mutations mapped to similar locations within the DNMT3A catalytic domain display different levels of activity, such as mutations at the tetramer interface, which display both loss- and gain-of-function. All enzymes were at 150 nm tetramer, corresponding to 27 nm active tetramer enzyme (24). DNA substrate concentrations were: 5 μm bp for poly(dI-dC) and 10 μm bp for p15-pCpGL plasmid. Product formation was measured after 1 h and the data for each substrate were fit to a linear regression and kcat values were obtained by dividing Vmax by the amount of active enzyme. Data reflect the results from at least 3 independent reactions. Mutations are categorized based on their respective location within the DNMT3A catalytic domain (red denotes hypomethylation and green denotes hypermethylation).
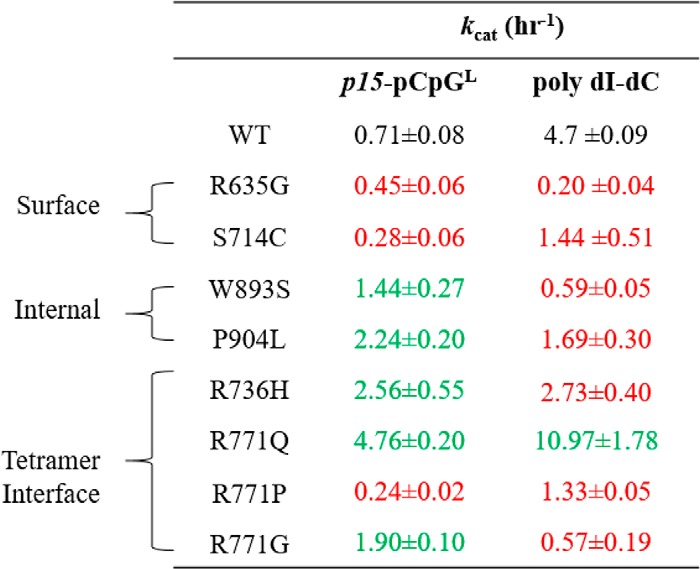
Relative to WT enzyme, R771P, S714C, and R635G, led to a 3-, 2.5-, and 1.5-fold decrease in activity, respectively, on the p15-pCpGL substrate (Table 1). Alternatively, we found that R736H, R771Q, R771G, W893S, and P904L displayed varying levels of p15-pCpGL hypermethylation compared with WT (Table 1). R771Q showed the most robust hypermethylation with a 6-fold increase in activity, followed by R736H and P904L with nearly a 4-fold increase, then R771G and W893S with roughly a 2-fold increase (Table 1). Our results suggest mutations in DNMT3A identified in AML patients significantly vary in their ability to methylate p15-pCpGL, which is in agreement with the heterogeneity in both patterns and levels of DNA methylation observed in these patients.
The kcat for WT DNMT3A using poly(dI-dC) is 4.7 h−1, and is comparable with previous findings using similar substrate and enzyme concentrations (Table 1) (21). Like that observed in p15-pCpGL, the impact on enzyme function observed in the various AML mutants varied extensively with poly(dI-dC) as a substrate. However, in contrast to our results using the p15-pCpGL substrate, most AML mutants displayed hypomethylation of poly(dI-dC) relative to WT. Although R635G, W893S, and R771G displayed minimal levels of DNA methylation, S714C, P904L, R771P, and R736H resulted in at least a 2-fold decrease in activity relative to WT (Table 1). Of the mutations studied here, including additional substitutions to the R771 codon, R771Q was the only enzyme that exhibited an elevated rate of poly(dI-dC) methylation with a 2-fold increase relative to WT (Table 1). However, for several mutants we observed enhanced activity on p15-pCpGL showing that the density of sites contributes to how these mutations impact function. Although R771Q, S714C, and R771P displayed a similar trend as WT, P904L, W893S, R771G, and R635G led to enhanced activity on p15-pCpGL (Fig. 2).
Figure 2.
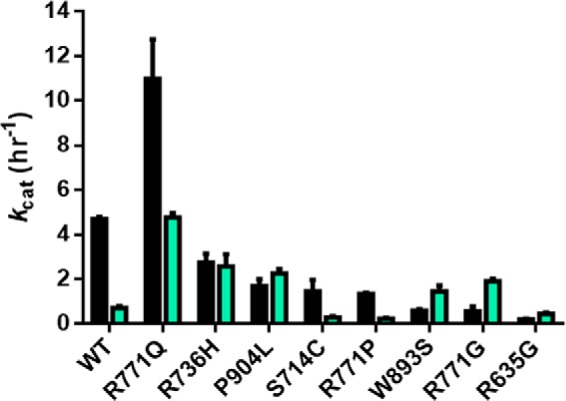
A subset of mutations display little change or enhanced activity for p15-pCpGL relative to the multiple CpG site substrate poly(dI-dC). WT DNMT3A has significantly reduced activity on p15-pCpGL (cyan, 20 μm bp) relative to poly(dI-dC) (black, 5 μm bp) due to the limited number of CpG sites on the human promoter substrate (∼10-fold less). R736H and P904L display minimal change in activity across DNA substrates, whereas W893S, R771G, and R635G lead to enhanced activity in p15-pCpGL. R771Q maintains significantly higher activity for both DNA substrates. Enzyme concentrations are 150 nm tetramer (27 nm active tetramer) (24) and kcat (h−1) values were determined as described under “Experimental procedures.”
In summary, compared with the WT DNMT3A, five of the eight mutants show differential changes when comparing the two substrates (poly(dI-dC) and p15-pCpGL). For example, R635G is significantly worse with the high-density substrate than p15-pCpGL, whereas R771Q shows the opposite trend. Even more surprising, some mutants actually reverse the trends compared with the WT enzyme; thus, W893S shows dramatically worse activity than the WT enzyme on poly(dI-dC) but shows improved activity with the p15-pCpGL substrate. R736H and R771G show a similar trend. Thus, the mutants may result in highly variable changes in their ability to methylate regions of high- and low-density CpG sites.
A subset of AML mutations display enhanced ability to perform non-CpG methylation (Fig. 4)
Figure 4.
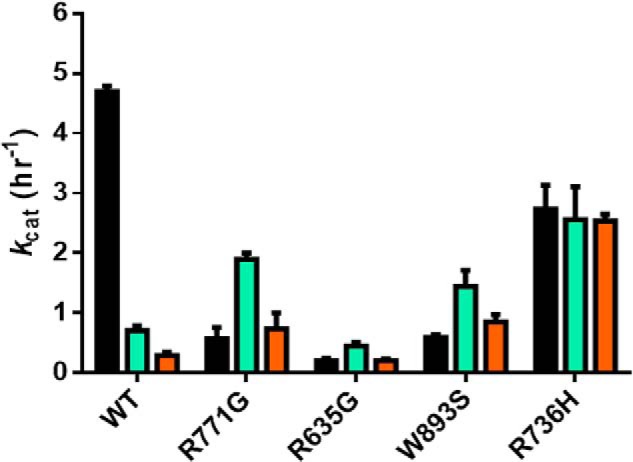
Some mutations result in enhanced activity at non-CpG sites. WT DNMT3A activity is affected by the availability of CpG sites on the DNA substrate as noted by the drastic activity loss from poly(dI-dC) (black, 5 μm bp poly(dI-dC)) to p15-pCpGL (cyan, 20 μm bp) and limited activity on a non-CpG substrate (orange, 20 μm bp pCpGL). In contrast to the WT enzyme, R771G, R635G, and W893S display a similar pattern of activity across substrates with enhancement on p15-pCpGL (cyan, 20 μm bp) and comparable levels on poly(dI-dC) (black, 5 μm bp poly(dI-dC)) and non-CpG substrate (orange, 20 μm bp pCpGL). R736H activity on the non-CpG substrate is significantly higher and virtually equal to substrates with multiple CpG sites. Enzyme concentrations are 150 nm tetramer (27 nm active tetramer) (24). Data reflect the results from at least three independent reactions.
In healthy cells, DNMT3A-mediated non-CpG methylation is associated with maintaining pluripotency (16, 22). Alternatively, compared with healthy bone marrow cells, non-CpG regions of cytogenetically normal AML (CN-AML) cells show the most pronounced changes in DNA methylation. Mutations in DNMT3A may contribute to this aberrant methylation and disease pathology (17). The pCpGL vector, lacking any CpG sites and the p15 insert, provide an opportunity to measure cytosine methylation at non-CpG sites (Fig. 3). Given that WT DNMT3A has minimal activity with pCpGL (kcat, 0.1–0.2 h−1) and the rate of product formation increases with the number of substrate CpG sites, the pattern observed for the rate of product formation by WT DNMT3A is as follows: poly(dI-dC) > p15-pCpGL > pCpGL (21). Unlike the WT enzyme, R771G, R635G, and W893S show comparable activity on poly(dI-dC) and pCpGL and the greatest activity with p15-pCpGL (Fig. 4). Alternatively, R736H displayed virtually equal levels of activity across poly(dI-dC), p15-pCpGL, and pCpGL. Along with distinct patterns of activity across the substrates tested, R771G, W893S, and R736H displayed a 3-, 4-, and 9-fold increase in non-CpG methylation, respectively, compared with the WT enzyme (Fig. 4).
Figure 3.

Substrate diagram and characteristics. Although poly(dI-dC) contains an extensive number of CpG sites with virtually no space between, p15-pCpGL consists of a limited number of sites available for methylation that are heterogeneously spread. The pCpGL vector, to which the p15 human promoter was inserted, is 3,872 bp in size and lacks any CpG sites for methylation.
Most AML mutations are unresponsive to modulation by TDG
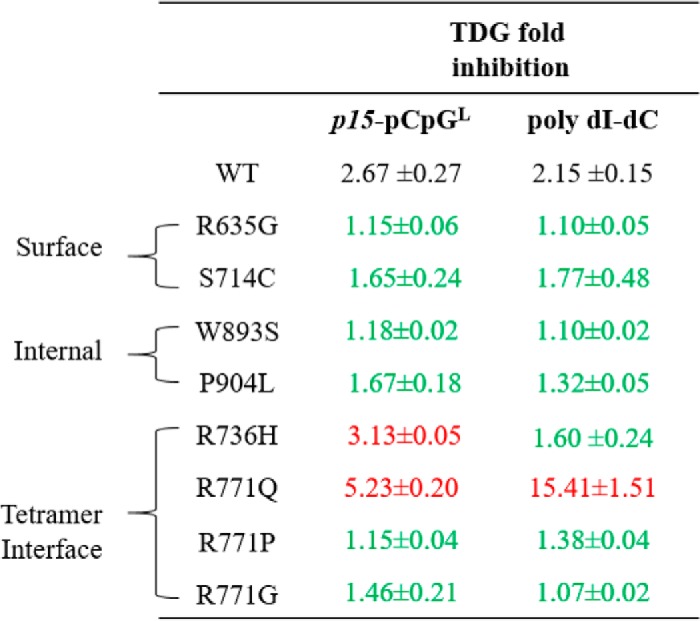
TDG, a component of the BER system, is responsible for removing single T bases in G–T mismatches that arise from spontaneous deamination of 5-methylcytosine (23). Furthermore, there is in vivo and in vitro evidence suggesting the direct DNMT3A–TDG interaction affects the activity of both enzymes in a reciprocal manner (18). Given that components of the BER system are affected in AML, the regulatory effect of TDG on the activity of AML-derived DNMT3A mutants was assessed (19). Li et al. (18) show that TDG inhibits DNMT3A activity in a dose-dependent manner with 2-fold excess TDG to DNMT3A having a greater inhibitory effect than equimolar concentrations of both proteins. In our hands, increasing concentrations of TDG relative to WT DNMT3A (1:1, 1:2, and 1:3, DNMT3A:TDG) did not further augment the 3-fold TDG-mediated inhibition (Fig. S1) (24). Therefore, the inhibitory effect of TDG on the various DNMT3A AML variants was tested at equimolar concentration of both enzymes. Given that DNMT3A and TDG contribute to the normal methylation and de-methylation of the p15 promoter, p15-pCpGL presents a biologically relevant platform to assess the interactions among the two enzymes (25). Co-incubation of TDG with WT DNMT3A and the p15-pCpGL human promoter substrate, led to approximately a 2.5-fold decrease in methylation, which we also observed in all mutants except for R771Q and R736H (Table 2). Interestingly, although the WT enzyme shows a similar effect when using the high site-density substrate (poly(dI-dC)), the TDG-mediated inhibition for the R771Q mutant is ∼15-fold with this substrate (Table 2).
Table 2.
Fold inhibition by TDG for wildtype and AML patient-derived DNMT3A mutants using poly(dI-dC) and p15-pCpGL DNA substrates
Fold inhibition was determined by product formed by DNMT3A alone divided by product formed by DNMT3A with TDG. Except for R771Q, all other surface, tetramer interface, and internal mutations lead to comparable levels of reduced sensitivity to inhibition by TDG. All enzymes were at 150 nm tetramer (27 nm active tetramer for DNMT3A) (24) and DNA substrate concentrations were: 5 μm bp for poly(dI-dC) and 10 μm bp for p15-pCpGL plasmid. Wildtype DNMT3A and mutant variants were preincubated with TDG for 1 h at 37 °C prior to initiating the reaction by the addition of substrate DNA. Mutations are categorized based on their respective location within the DNMT3A catalytic domain (red denotes greater inhibition, while green denotes lower inhibition, relative to wildtype). Data reflect the results from at least three independent DNMT3A-TDG co-incubation reactions.
AML mutations are variably responsive to stimulation by DNMT3-like (DNMT3L)
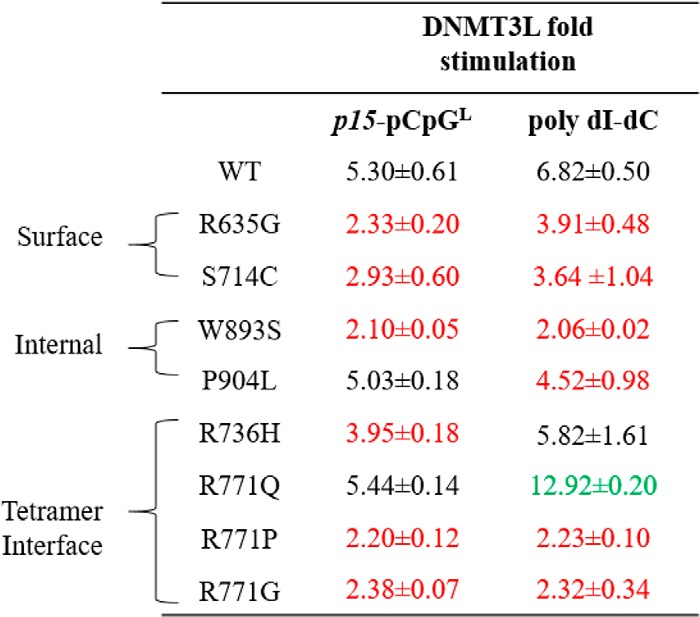
Ultimately our interest is to better understand how protein–protein interactions influence DNMT3A function, and how mutations might impact those interactions. Although the number of proteins shown or implicated in interacting with DNMT3A is extensive (26–28), we know very little about the regions on the DNMT3A surface involved in these complexes and the functional consequences of these altered interactions. The DNMT3A–DNMT3L co-crystal structure provides compelling insights into such interactions (29). Ley et al. (9) showed that although primary tissue samples from AML patients lack expression of the catalytically inactive member of the DNMT family, DNMT3L, spliced variants of DNMT3L transcripts were detected in various patients. Furthermore, DNMT3L provides an example of how an auxiliary protein interacts with and modulates DNMT3A function, and how mutations may impact on that function. A 1-h preincubation of DNMT3L with DNMT3A at a 1:1 ratio activates WT DNMT3A activity by ∼5-fold on p15-pCpGL, which we also observed in P904L and R771Q (Table 3). Alternatively, the rest of the mutants assessed in this study appeared to be desensitized to DNMT3L and displayed approximately half the extent of DNMT3L activation (Table 3). Under identical conditions and comparable with previous findings, DNMT3L activation was ∼7-fold for WT DNMT3A using poly(dI-dC) as a substrate (Table 3) (20). With the same high site-density substrate (poly(dI-dC)), R736H was the only substitution with comparable levels of DNMT3L stimulation as WT (Table 3). Although R771Q resulted in a 2-fold increase in DNMT3L activation, the remaining mutants assessed in this study displayed reduced modulation of DNA methylation activity by DNMT3L (Table 3). In summary, our results imply that in addition to direct changes in DNMT3A activity, AML patient-derived mutations in DNMT3A additionally disrupt modulation of DNMT3A by partner proteins.
Table 3.
Fold stimulation by DNMT3L for wildtype and AML patient-derived DNMT3A mutants using poly(dI-dC) and p15-pCpGL DNA substrates
Fold stimulation was determined by the sum of product formed by DNMT3A with DNMT3L divided by product formed by DNMT3A without DNMT3L. DNMT3L, known to interact at the tetramer interface of DNMT3A, leads to varying levels of responsiveness for tetramer interface and internal mutations, but comparable reduced stimulation in surface mutations. All DNMT3As were at 150 nm tetramer, corresponding to 27 nm active tetramer enzyme (24). DNA substrate concentrations were: 5 μm bp for poly(dI-dC) and 10 μm bp for p15-pCpGL plasmid. Wildtype and DNMT3A mutants were preincubated with DNMT3L (1:1) for 1 h at 37 °C prior to initiating the reaction by the addition of substrate DNA. Mutations are categorized based on their respective location within the DNMT3A catalytic domain (red corresponds to decreased stimulation and green corresponds to increased stimulation, compared to wildtype). Data reflect the results from at least three independent DNMT3A–DNMT3L co-incubation reactions.
P904L and R736H display distinct alterations to processive catalysis that are sensitive to substrate CpG density
WT DNMT3A is highly processive on both biological and synthetic DNA substrates (20, 21). Furthermore, mutational analyses suggest residues at the dimer interface are essential for DNMT3A to perform processive catalysis (8). The ability of WT DNMT3A, and the two mutants P904L and R736H to processively methylate poly(dI-dC) or p15-pCpGL was assessed using the chase processivity assay (21). Comparable with previous findings, WT DNMT3A continually methylates the initially bound substrate for multiple rounds of methylation as noted by the same degree of poly(dI-dC) methylation observed in both the substrate only and chase conditions for 100 min following the addition of chase DNA (pCpGL, Fig. 5A). Alternatively, addition of chase DNA led to a considerable decrease in the ability of P904L to methylate poly(dI-dC), indicating this mutant rapidly dissociates from the poly(dI-dC) substrate during catalysis (Fig. 5B). As a control, the addition of a mixture of both substrate and chase DNA at the start of the reaction virtually eliminates all methylation activity with the WT enzyme (Fig. 5A). However, R736H appears to treat chase DNA (which lacks CpG sites) as a substrate in the presence of poly(dI-dC) because we observe nearly identical levels of methylation under all conditions tested (Fig. 5C). As in the case of the high site-density substrate (poly(dI-dC)), addition of chase DNA (pCpGL) to the WT enzyme failed to disrupt processive catalysis in the presence of p15-pCpGL indicating a high degree of processivity. In contrast P904L displayed substantial disruptions of processivity on both substrates (Fig. 6B); the fact that the R736H mutant shows such strikingly different responses to the type of substrate being tested (Fig. 6B) suggests that the mutations have distinctive consequences on the ability of DNMT3A to carry out repeated catalysis on the same DNA molecule.
Figure 5.
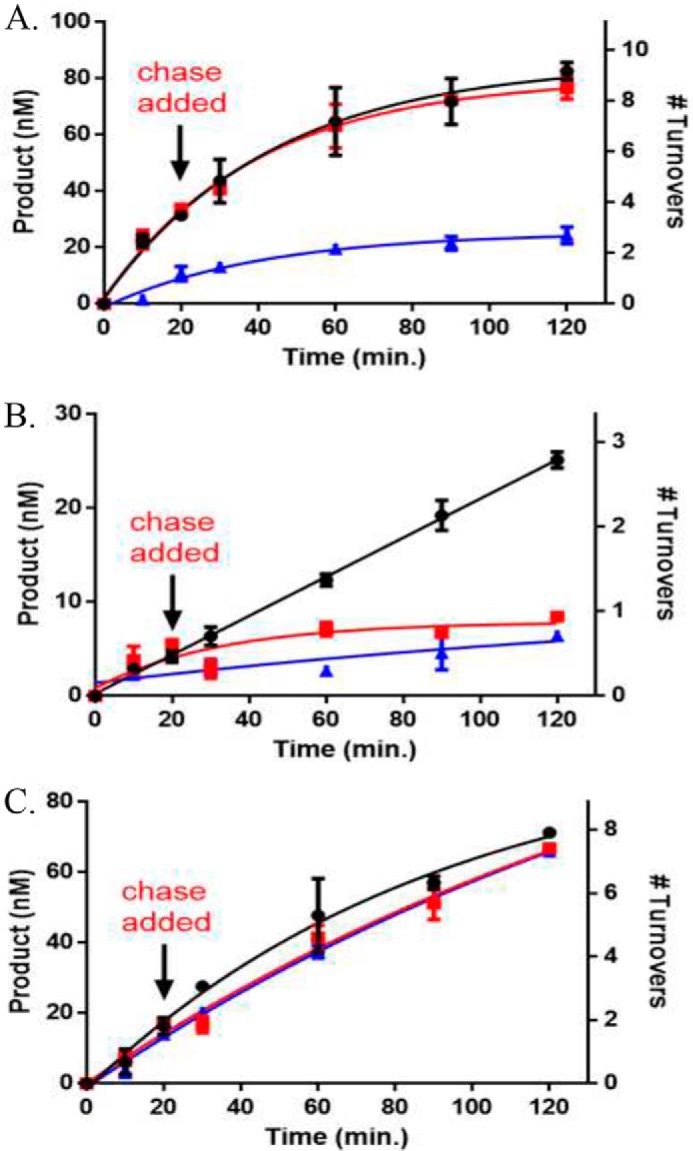
AML mutants display unique alterations to processive catalysis on the poly(dI-dC) substrate. A, WT DNMT3A; B, P904L; and C, R736H at 50 nm tetramer. Substrates were added at time 0 to start the reaction and chase assay conditions were as follows: ●, substrate only (2 μm bp poly(dI dC)); red square, substrate and then 20 min chase (40 μm bp pCpGL); blue triangle, substrate (2 μm bp poly(dI-dC)) and chase (40 μm bp pCpGL) at the start of the reaction. Data reflect the results from at least three independent experiments.
Figure 6.
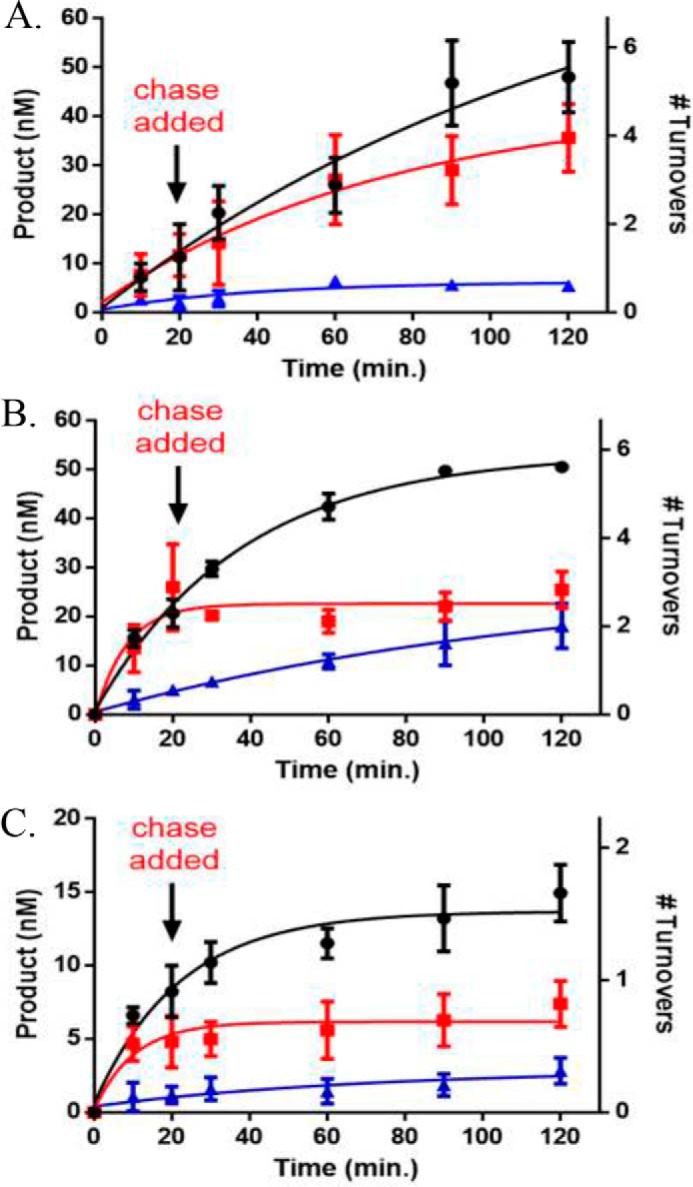
AML mutations display loss of processive catalysis on the p15-pCpGL human promoter substrate. A, WT DNMT3A retains the ability to perform processive catalysis when tested with the p15-pCpGL substrate, whereas B, R736H and C, P904L resulted in a loss of processivity. All enzyme concentrations were 50 nm tetramer. Substrates were added at time 0 to start the reaction and chase assay conditions were as follows: ●, substrate only (10 μm bp p15-pCpGL); red square, substrate and then 20 min chase (200 μm bp pCpGL); blue triangle, substrate (10 μm bp p15-pCpGL) and chase (200 μm bp pCpGL) at the start of the reaction. Data reflect the results from at least three independent experiments.
TDG does not compete with DNMT3L for binding on the DNMT3A tetramer interface
The DNMT3A–DNMT3L co-crystal structure implicates the DNMT3A tetramer interface as a region for binding by DNMT3L (29). Although DNMT3A interacts with a wide range of partner proteins, the specific region on the DNMT3A surface (e.g. tetramer interface or elsewhere) involved in formation of heterotetrameric complexes remains unknown (26–28). Functional assays can be used to define a protein–protein interface; for example, Doherty et al. (30) mapped regions on the Werner syndrome helicase important for replication protein A binding. Using a nonfunctional approach (size-exclusion chromatography), Chen et al. (31) showed SKI proto-oncogene (SKI) and CREB-binding protein compete for the same binding region on SMAD3. To elucidate the region on DNMT3A for TDG binding, we determined whether TDG and DNMT3L compete for binding to the DNMT3A tetramer interface. We first determined the relative binding affinities of each for DNMT3A. DNMT3A co-incubations with DNMT3L resulted in a KD,app of 80 ± 12 nm (Fig. 7A), whereas a KD,app of 223 ± 74 nm was determined from DNMT3A co-incubations with TDG (Fig. 7B). To assess whether DNMT3L or TDG binding to DNMT3A are mutually exclusive, all three proteins were preincubated for 1 h prior to starting the reaction by the addition of substrate DNA and DNMT3A activity measured at 30, 60, and 90 min. The combined results (Fig. 8, light grey) are distinct from the results when DNMT3A is incubated with DNMT3L (Fig. 8, medium grey) or TDG (Fig. 8, dark grey) alone. Instead, WT DNMT3A, DNMT3L, and TDG preparations (Fig. 8, light grey) reflected virtually the same level of activity as WT DNMT3A alone (Fig. 8, black). The results obtained from WT DNMT3A–DNMT3L–TDG co-incubations suggest: 1) TDG does not bind the DNMT3A tetramer interface and the activity of a WT DNMT3A–DNMT3L–TDG complex equals that of WT DNMT3A alone, or 2) co-incubation results in formation of a subset of DNMT3A–DNMT3L and DNMT3A–TDG complexes that yield the same level of activity as WT DNMT3A alone, or 3) DNMT3L and TDG interact with one another leaving WT DNMT3A alone.
Figure 7.
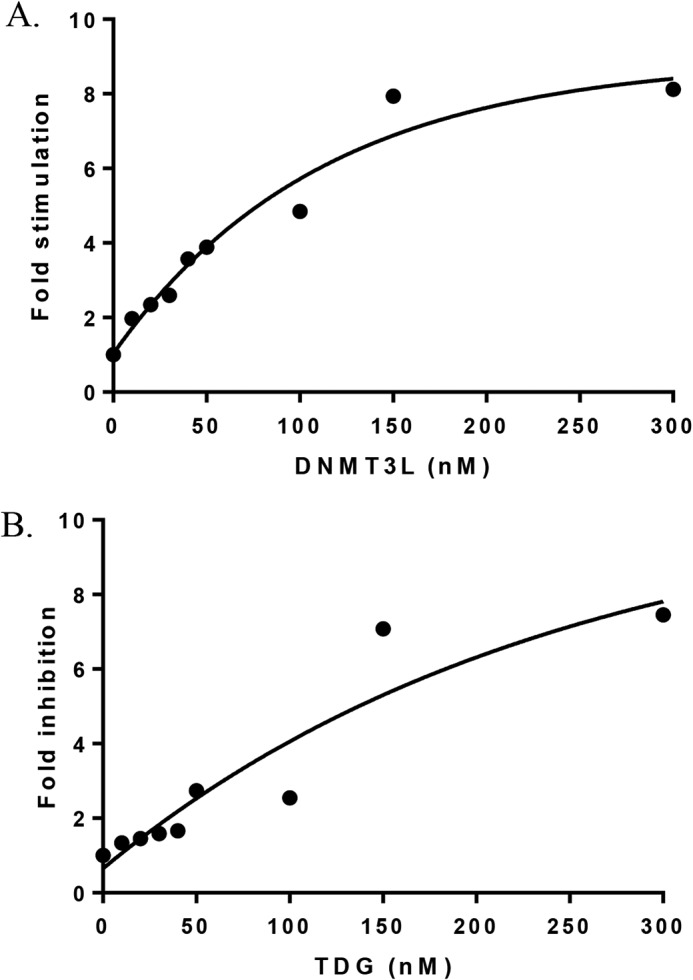
DNMT3L has a higher affinity for binding DNMT3A compared with TDG. In A and B, DNMT3A (10 nm) was preincubated in reaction buffer for 1 h at 37 °C with varying concentrations of TDG or DNMT3L (0–300 nm). Reactions were initiated by the addition of 5 μm bp poly(dI-dC) and run for 1 h. Fold stimulation was determined by the sum of product formed by DNMT3A with DNMT3L divided by product formed by DNMT3A without DNMT3L (A). Fold inhibition was determined by product formed by DNMT3A alone divided by product formed by DNMT3A with TDG (B). Data reflect the results from at least three independent co-incubation reactions.
Figure 8.
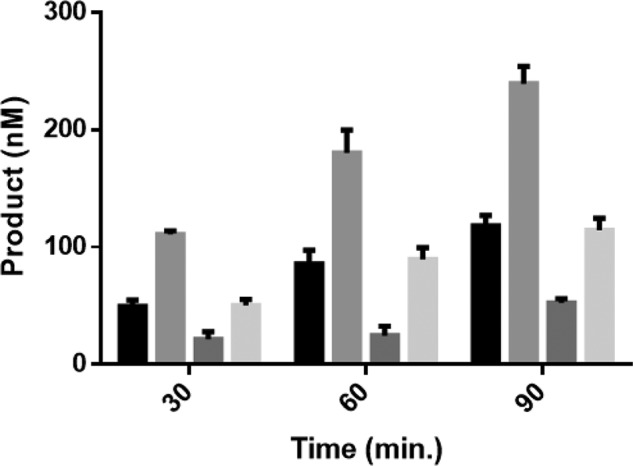
DNMT3L or TDG binding on DNMT3A tetramer interface are not mutually exclusive. WT DNMT3A, DNMT3L, and TDG co-incubations did not reflect the modulatory effect observed when either DNMT3L or TDG are preincubated with DNMT3A. Enzyme concentrations for all the reactions performed were 150 nm and at 1:1 for co-incubations. Prior to initiating the reaction by the addition of 5 μm bp poly(dI-dC); light grey, WT DNMT3A, DNMT3L, and TDG were preincubated for 1 h at 37 °C. Under similar conditions, the following reactions were performed as controls: black, WT DNMT3A; medium grey, WT DNMT3A and DNMT3L; dark grey, WT DNMT3A and TDG. Data reflect the results from at least three independent competitive co-incubation experiments.
To further assess whether DNMT3L or TDG binding to the DNMT3A tetramer interface are mutually exclusive events, active DNMT3A–DNMT3L (or DNMT3A–TDG) heterotetramers were challenged by the addition of TDG (or DNMT3L). In reactions where WT DNMT3A–DNMT3L co-incubations ran for 30 min, the addition of TDG did not disrupt active WT DNMT3A–DNMT3L heterotetramers (Fig. 9A, green) and displayed comparable activity as WT DNMT3A–DNMT3L co-incubations (Fig. 9A, black). However, the addition of DNMT3L to active WT DNMT3A–TDG heterotetramers (Fig. 9B, green) led to a rapid increase in product formation that was greater than WT DNMT3A–TDG co-incubations (Fig. 9B, black). Taken together, the results suggest: 1) TDG does not compete with DNMT3L for binding to the DNMT3A tetramer interface, 2) the DNMT3A tetramer interface is accessible for DNMT3L binding in the presence of TDG, and 3) the location for TDG binding on DNMT3A is inaccessible to TDG in the presence of DNMT3L.
Figure 9.
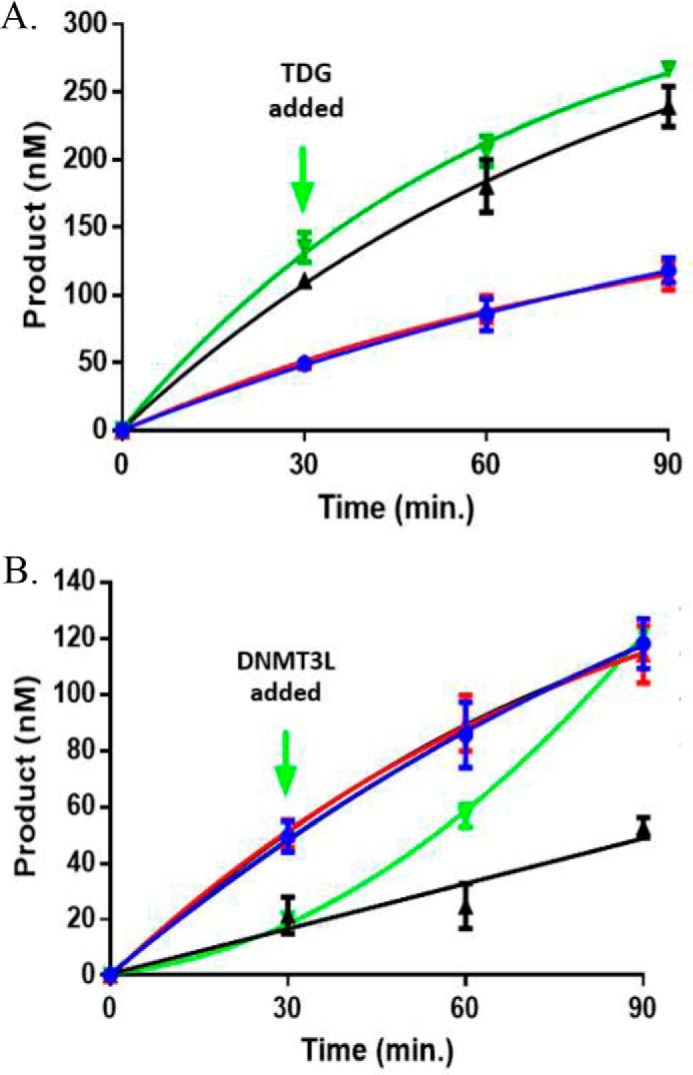
TDG does not compete with DNMT3L for binding to DNMT3A. The addition of TDG does not disrupt the activity of a functional DNMT3A–DNMT3L heterotetramer, but the addition of DNMT3L increases the activity of a functional DNMT3A–TDG heterotetramer. In all experiments performed, enzyme concentrations were 150 nm (1:1 for co-incubations or binding competitions) and reaction were initiated by the addition of 5 μm bp poly(dI-dC). A, green, WT DNMT3A was preincubated with DNMT3L for 1 h at 37 °C and the reaction run for 30 min prior to the addition of TDG. B, green, WT DNMT3A was preincubated with TDG for 1 h at 37 °C and the reaction run for 30 min prior to the addition of DNMT3L. The following reactions were also tested as controls: A, black, WT DNMT3A and DNMT3L; B, black, WT DNMT3A and TDG; A and B, blue, WT DNMT3A; A and B, red, WT DNMT3A, DNMT3L, and TDG preincubated at the start of the reaction (A and B). Data reflect the results from three independent experiments.
Whereas R771Q leads to a modest decrease in methylation, S714C appears to be catalytically inactive in murine embryonic stem cells
To determine the cell-based activity of the DNMT3A mutants, we utilized a system allowing the induction of full-length doxycycline-inducible DNMT3A–GFP fusion expression in methylation-deficient murine embryonic stem cells (DNMT3A/3B double knock-out mESCs; DKO mESCs). DNMT3A mutant-expressing cells were sorted by flow cytometry after doxycycline induction for 2 weeks (Fig. 2, A–D). The mean fluorescence intensity between mutant and control samples was similar (Fig. S2E), suggesting that protein expression level of DNMT3AWT, DNMT3AS714C, and DNMT3AR771Q was comparable. The methylation level of DKO mESCs was undetectable using a dot-blot assay to measure 5-mC (Fig. 10). Re-expressing DNMT3AWT significantly increases overall DNA methylation. In addition, DNMT3AR771Q showed a slight decrease of DNA methyltransferase activity compared with the cells expressing DNMT3AWT, whereas the DNMT3AS714C mutant seemed to be a catalytic inactive mutation with negligible DNA methylation detectable after introduction into the ES cells (Fig. 10).
Figure 10.

S714C reduces DNA methylation in mESCs. The images show the methylation level as determined by a dot-blot assay in doxycycline-inducible DNMT3A mutant-expressing DKO mESCs after doxycycline induction for 2 weeks. The upper blot represents serial dilution of DNA derived from the indicated cells in the dot-blot, probed with an antibody against 5-mC. The lower blot represents serial dilution of standard methylated DNA as a control.
Discussion
Our motivation for this study is based on several intertwined observations. Mutations in DNMT3A have emerged as very important drivers of disease in various hematological disorders including AML (9, 32–35). Our prior work suggests that the functional consequences from such mutations involve changes in methylation efficiency, the enzyme's oligomeric state, and ability to carry out processive catalysis (8, 20). Finally, DNMT3A directly and indirectly interacts with a large number of partner proteins; in many cases such interactions are known to directly influence the enzyme's specificity, subcellular localization, and overall activity (26, 27, 36, 37).
Our focus on the subset of clinically relevant DNMT3A mutants (Table 1) was determined by their relative frequency in AML patients, a lack of any previous biochemical characterization, and our prior work demonstrating that mutations at the dimer and tetramer DNMT3A–DNMT3A interfaces disrupt the oligomeric state of the tetramer (8). Multiple regions of DNMT3A form minor mutational hot spots, and the frequency and position of these hot spots vary significantly in a cancer-specific fashion; for example, in AML, Arg-882 is mutated in 58% of patients and mutations to Arg-736 are observed in 2%. The fact that patient-identified mutations appear across the entire DNMT3A protein suggests that they may result in distinctive molecular phenotypes. A working hypothesis is that some of these regions and mutations alter protein–protein interactions between DNMT3A and its partner proteins.
The most striking feature of our study is the range of functional consequences resulting from the different DNMT3A mutations, in some cases involving the same position (e.g. R771Q, -P, and -G), or spatially related residues like W893S and P904L. Thus, compared with the WT enzyme, we observe dramatic increases (∼7-fold, R771Q on p15-pCpGL) and decreases (24-fold, R635G on poly(dI-dC)) in activity (Table 1). This is also revealed in how the different mutations impact the ability of DNMT3A to act processively in carrying out methylation of spatially proximal sites. For example, P904L is dramatically decreased in this activity, whereas R736H is not impacted at all (Fig. 5). Changes in this activity are anticipated to impact the ability of the enzyme to efficiently methylate regions of the genome undergoing rapid methylation (38). Furthermore, R736H, unlike the WT enzyme, is fully capable of methylating cytosines at non-CpG positions (Figs. 3 and 5). This feature of R736H is not unique, as W893S and R771G both show enhanced non-CpG activity relative to the WT enzyme. Non-CpG methylation, associated with maintaining pluripotency, is accomplished by DNMT3A in mammalian stem cells (16, 28). CN-AML account for 40–50% of all AML cases observed (39). CN-AML cells show pronounced changes in non-CpG methylation when compared with healthy CD34+ bone marrow cells (17). Therefore, mutations in DNMT3A with altered non-CpG methylation activity may have detrimental consequences in vivo.
The functional consequences of methylating the p15-pCpGL substrate are particularly relevant because a hallmark of AML patients with mutations in DNMT3A is the heterogeneity in both patterns and levels of DNA methylation in the promoter region of the p15 tumor suppressor gene (12, 13). The range of changes relative to the WT enzyme, 3-fold reduction in R771P to 7-fold enhancement in R771Q (Table 1), suggest that these mutations will profoundly differ in their impact on DNA methylation in vivo. Studies have additionally shown that AML patients carrying mutations in DNMT3A display reduced p15 levels and hypermethylation of the p15 promoter (14, 15). Our results show that most mutations led to hypermethylation of the p15-pCpGL substrate, which is in agreement with that observed in AML patients.
Perhaps surprisingly, the positions of particular changes (e.g. Fig. 1, orange, surface, yellow, tetramer, or green, internal) are not uniformly correlated with any particular functional consequence. The four changes at the tetramer interface (three at Arg-771 and R736H) result in decreases (R771P) and increases (R736H, R771Q, and R771G) in activity with p15-pCpGL (Table 1). However, the two tetramer interface mutations R771G and R736H both result in dramatic enhancement of non-CpG methylation (Fig. 4). Also, the two internal substitutions (W893S and P904L) both result in decreased activity with p15-pCpGL, and the two surface substitutions (R635G and S714C) both show increases in this activity relative to the WT enzyme (Table 1). Although the effect of some mutants may be predicted by their location on DNMT3A, our results highlight the importance of functional characterization given that mutations on the same location on DNMT3A can lead to pronounced changes in activity.
Proteins that direct epigenetic changes in distinct pathways (e.g. DNA methylation, histone modification, RNA) are now understood to rely on extensive cross-talk, largely mediated through protein–protein interactions (40, 41). Because many proteins have been identified to interact with DNMT3A, we sought to determine how the mutations studied here impact some of these interactions, and how those interactions alter DNMT3A function. The DNMT3A–DNMT3L co-crystal structure implicates the DNMT3A tetramer interface as a region for binding and regulation of DNMT3A by a partner protein. Therefore, if the DNMT3A tetramer interface region is the only surface occupied by partner proteins, mutations at the tetramer interface would be predicted to have similar responses to TDG and DNMT3L modulation. However, tetramer interface mutants were differentially responsive to TDG inhibition (Table 2) and DNMT3L stimulation (Table 3), and overall, most mutations were desensitized to modulation by TDG or DNMT3L. Given that the region on DNMT3A for DNMT3L binding is well-defined, we then sought to explore whether TDG competes with DNMT3L for binding on the DNMT3A tetramer interface, or alternatively, TDG binds a different surface on DNMT3A. Our results from DNMT3L/TDG competitions for binding to DNMT3A suggest TDG does not compete with DNMT3L for binding to DNMT3A. Furthermore, the DNMT3A tetramer interface is accessible to DNMT3L in the presence of TDG and the surface for TDG binding on DNMT3A is inaccessible to TDG in the presence of DNMT3L (Figs. 8 and 9). DNMT3A has been shown to directly interact with a wide range of partner proteins that can impact enzymatic activity (25, 27, 28). The functional mapping used in this study by competitive binding using a well-defined regulatory protein, like DNMT3L, provides a rapid approach to explore the location of additional partner proteins of DNMT3A.
Our goal is to understand how mutations in DNMT3A derived from AML patients alter DNMT3A activity, either directly, or through interactions with other cellular components. Prior work has focused largely on R882H, which displays substantial hypomethylation in focal regions of the genome, although global methylation levels are comparable with WT DNMT3A (42, 43). In addition, the changes in methylation patterns observed in R882H appear to be dependent on the cell context and factors therein (42). Both enzyme- and cell-based studies show that R882H disrupts the oligomeric state of the protein and alters its ability to act on DNA (8, 10, 42). Here we show that S714C results in undetectable cellular levels of global methylation, whereas R771Q showed a slight decrease in methylation compared with WT (Fig. 10). For S714C, the enzyme and cell-based data are in accordance, suggesting that this substitution disrupts intrinsic enzymatic activity, which cannot be restored by additional regulatory components found in cells.
On the other hand, whereas the R771Q displayed increased DNA methylation activity in vitro, this hypermethylation activity was not observed in the ES cell-based assay; instead, a moderate decrease in activity was observed. These discordant results with R771Q could be explained by a number of factors. In the enzyme studies, we are using a specific substrate, poly(dI-dC), that is routinely used to measure DNMT3A activity (44), and the plasmid bearing the p15 promoter. In contrast, the cell-based assay measures the aggregate effect of methylation changes over the whole genome, including many different types of targets. Thus, the enhanced activity seen with specific substrates and R771Q may simply result from composition of the DNA substrates, and the enzyme activity across the whole genome is, on aggregate, largely unchanged from the WT DNMT3A.
Another likely possibility is that cellular factors modulate the activity of particular mutants directly or indirectly. Of note, the ex vivo methylation assays were performed with the catalytic domain of DNMT3A, whereas the cell-based assays were performed with the full-length version of DNMT3A. The N-terminal regulatory domains (45, 46) of DNMT3A, including the ATX–DNMT3A–DNMT3L (ADD) and PWWP domains, may modulate the enzyme's activities. These and other DNMT3A sequences are known to interact with numerous partner proteins. Thus, cellular factors may regulate the ultimate outcomes of particular DNMT3A mutations. Indirect support for this explanation comes from the observation that a Tatten-Brown-Rahman syndrome patient with the R882H mutation had modest changes in methylation patterns of blood cells (peripheral blood polymorphonuclear cells, monocytes, and T cells), whereas more substantial changes are observed in R882H expressing primary AML samples (42). Thus, implying cellular factors appear to ultimately dictate the cellular outcomes of a particular DNMT3A mutation throughout AML progression.
In conclusion, mutations in DNMT3A can directly alter DNMT3A function as well as indirectly through changing the enzyme's interaction with partner proteins. Diseases that show evidence of changes in DNA methylation, including cancer and in particular AML, are likely to result from a blend of these direct and indirect mechanisms. Interestingly, for those mutations studied here, the indirect mechanisms involving partner proteins do not restore the enzyme's WT activity. The latter changes may well be approachable therapeutically, because small molecules that interfere with protein–protein interactions, whereas challenging to design, have achieved some success.
Experimental procedures
Expression constructs
The catalytic domain of DNMT3A (residues 634–912) was used for all DNMT3A experiments, given the catalytic domain and full-length enzyme have comparable kinetic parameters (kcat, KmDNA, KmAdoMet, processivity, and DNMT3L stimulation) (47, 48). The codon-optimized plasmids used for recombinant protein expression include pET28a–hDNMT3A_CD (Δ1–611) for the DNMT3A catalytic domain (24), pTYB1–3L for full-length DNMT3L (49), and pET28a-hTDG (Δ1–175) for the TDG construct (50). The catalytic domain mutants in the pET28a–hDNMT3A_CD (Δ1–611) expression construct were generated using a QuikChange Lightning Site-directed Mutagenesis kit (Agilent).
Protein expression and purification
DNMT3A (WT and mutants), DNMT3L, and TDG were expressed in NiCo21 (DE3) competent Escherichia coli cells (New England Biolabs). Cell cultures were grown in LB medium at 37 °C to an A600 nm of 0.7 (WT and mutant DNMT3A), 0.5 (DNMT3L), and 0.8 (TDG). Expression was induced by the addition of 1 mm isopropyl β-d-thiogalactopyranoside (Gold Biotechnology) at 28 °C. Induction times were 6 h for DNMT3A (WT and mutants), overnight for DNMT3L, and 4 h for TDG. Cells were then collected by centrifugation and stored frozen at −80 °C. All proteins were purified from cells lysed by sonication in 50 mm HEPES, 350 mm NaCl, 50 mm imidazole, 10% glycerol, and 1% phenylmethylsulfonyl fluoride, pH 7.8, which were then clarified by centrifugation. Lysates were loaded onto a ÄKTA start FPLC system (GE Healthcare) for purification using a nickel-nitrilotriacetic acid column (GE Healthcare). Columns were equilibrated with 50 mm HEPES, 350 mm NaCl, 50 mm imidazole, 10% glycerol, and 1% phenylmethylsulfonyl fluoride, pH 7.8, and washed with identical buffer but 70 mm imidazole. WT DNMT3A, along with all mutant variants, were eluted with 162.5 mm imidazole, 200 mm imidazole for DNMT3L, and 250 mm imidazole for TDG, and stored at −80 °C in storage buffer (50 mm KH2PO4/K2HPO4, 20% glycerol, pH 7.8).
Methylation assays
Assays were carried out to measure total methyl groups transferred from the AdoMet cofactor to substrate DNA by DNMT3A. Reactions took place at 37 °C in a buffer composed of 50 mm KH2PO4/K2HPO4, 1 mm EDTA, 1 mm DTT, 0.2 mg/ml of BSA, 20 mm NaCl, and 5 μm AdoMet (from a 50 μm stock composed of 45 μm unlabeled and 5 μm 3H-methyl labeled) at pH 7.8. 15-μl Aliquots were taken from a larger reaction and quenched by mixing with 0.1% SDS (1:1). Samples were spotted onto Hybond-XL membranes (GE Healthcare), washed, and dried as previously established (51).
Catalysis (kcat)
WT DNMT3A and AML mutants (150 nm tetramer) correspond to 27 nm active tetramer, as previously determined (24). Reactions were initiated by the addition of substrate DNA (5 μm bp poly(dI-dC), 10 μm bp p15-pCpGL or 20 μm) at saturation, run for 1 h at 37 °C, and quenched as stated above. Values were determined as described in Holz-Schietinger et al. (20). In brief, data were fit to a linear regression and kcat values were obtained by dividing Vmax by the amount of active enzyme (Prism version 6.01). Data reflect the results from at least three independent reactions.
DNMT3L and TDG assays
DNMT3A was preincubated in reaction buffer with either DNMT3L or TDG for 1 h at 37 °C prior to initiating the reaction by the addition of DNA (5 μm bp poly(dI-dC) or 10 μm bp p15-pCpGL). Reactions were quenched as stated above after 1 h at 37 °C. DNMT3L yields maximum activation of DNMT3A when preincubated at a ratio of 1:1 (21); therefore, equal concentrations of 150 nm of both enzymes were used for all DNMT3L assays. TDG inhibition of DNMT3A was tested at 1:1, 1:2, and 1:3 ratios of DNMT3A to TDG concentration. Given that no significant changes were observed on DNMT3A inhibition with varying TDG concentrations, equal concentrations of 150 nm DNMT3A and TDG were used for TDG assays. DNMT3L stimulation was calculated by the sum of product formed by DNMT3A with DNMT3L divided by product formed by DNMT3A in the absence of DNMT3L as previously described in Holz-Schietinger et al. (21). TDG-fold inhibition was calculated by product formed by DNMT3A alone divided by product formed by DNMT3A with TDG. Data reflect the results from at least three independent co-incubation reactions.
Processivity assays
Processivity assays were performed to assess the fidelity of enzymes to methylate a multiple CG site DNA substrate when presented with an excess concentration of additional DNA lacking CG-methylation sites. Assays were performed as previously established (21) with enzyme concentrations of 50 nm tetramer, and substrate DNA concentrations of 2 μm bp poly(dI-dC) or 40 μm bp pCpGL. Following a 3-min preincubation at 37 °C, the reaction was initiated by the addition of 2 μm bp poly(dI-dC) and the enzyme was allowed to carry out 1–2 turnovers on poly(dI-dC) (20 min). 40 μm bp pCpGL (20-fold excess) was then added at 20 min to generate the chase condition. A reaction with a mixture of 2 μm bp poly(dI-dC) and 40 μm bp pCpGL at the start of the reaction was used as a control. The data were fit to a nonlinear regression (one phase decay) using Prism (version 6.01) as described in Holz-Schietinger et al. (8). Data reflect the results from at least three independent experiments.
Functional competitive binding assay
The DNMT3A–DNMT3L co-crystal structure provides a defined region on DNMT3A for DNMT3L binding and formation of a heterotetrameric complex. Although this tetramer interface region is well-defined, the surface on DNMT3A for TDG binding remains unknown. Therefore, a functional methylation assay was employed to assess whether TDG and DNMT3L compete for the same surface on DNMT3A. DNMT3A, DNMT3L, and TDG were preincubated at 1:1:1 (150 nm) in reaction buffer for 1 h at 37 °C prior to initiating the reaction by the addition of DNA (5 μm bp poly(dI-dC)). To further challenge whether TDG also binds DNMT3A at the tetramer interface, DNMT3A and DNMT3L (1:1 at 150 nm) were preincubated for 1 h at 37 °C, the reaction was initiated by the addition of 5 μm bp poly(dI-dC) and 150 nm TDG was added to the active heterotetramer at 30 min. Using similar conditions, a reciprocal reaction in which a preformed TDG–DNMT3A complex was challenged by the addition of DNMT3L was performed. As controls, reactions involving DNMT3A only, DNMT3A with either DNMT3L or TDG, and all three proteins together at the beginning of the reaction were performed. Data reflect the results from at least three independent co-incubation reactions.
Binding affinity of TDG or DNMT3L to DNMT3A
To assess the binding affinities of TDG or DNMT3L to DNMT3A, the effect of increasing concentrations of TDG or DNMT3L (10–300 nm) were tested on a fixed concentration of DNMT3A (10 nm). DNMT3A was preincubated in reaction buffer for 1 h at 37 °C with varying concentrations of TDG or DNMT3L, reactions were initiated by the addition of 5 μm bp poly(dI-dC) and run for 1 h. Apparent affinity (KD,app) were determined from a one-site specific binding model using Prism (version 6.01). Data reflect the results from at least three independent co-incubation reactions.
DNA sequences
DNA used for substrates include poly(dI-dC) (Sigma) and p15-pCpGL and pCpGL (non-CpG substrate) plasmids that were prepared as described in Holz-Schietinger et al. (21). Concentrations for DNA substrates are given in bp and were calculated from absorbance at 260 nm using the following extinction coefficients as previously determined: 6.9 mm−1 cm−1 for poly(dI-dC) and 6.8 mm−1 cm−1 for p15-pCpGL and pCpGL plasmids (21). The p15-pCpGL extinction coefficient considers both p15 promoter and pCpGL plasmid sequences to define the p15-pCpGL substrate concentration in number of base pairs.
Generation of doxycycline-inducible DNMT3A expressing cell line
Generation of doxycycline-inducible DNMT3A constructs was previously reported (52). In brief, full-length DNMT3A cDNA, which fused with a GFP sequence, was cloned into pDONR223 using a Gateway cloning BP clonase II enzyme mix. Two DNMT3A mutations, S714C and R771Q, were generated in pDONR–DNMT3A–GFP vectors using a QuikChange II site-directed mutagenesis kit. DNMT3AWT, DNMT3AS714C, and DNMT3AR771Q GFP fusion cDNA were then Gateway-cloned into a pinducer20-BSD vector using Gateway LR clonase II enzyme mix.
DNMT3A/3B DKO mESCs were previously described (53). Lentiviral particles of DNMT3A/S714C/R771Q fusion GFP were generated using a previous published protocol (54) and then infected into DKO mESCs for 48 h. Lentivirus-infected DKO mESCs were treated with 4 μg/ml of blasticidin for 72 h and then recovered after 1 week and treated with 4 μg/ml of blasticidin for 1 week. DNMT3A-GFP expressing cells were then sorted using a FACSArial II sorter after 2 weeks of doxycycline induction and DNA was then extracted using a Purelink DNA extraction kit.
Dot-blot assay
A 5-mC dot-blot assay was previously described (55). In short, 500 ng of DNA was serially diluted and then treated with 1 m NaOH, 25 mm EDTA at 95 °C for 10 min. Ice-cold 2 m ammonium acetate was added to NaOH-treated DNA and incubated on ice for 10 min. Membranes of the dot-blot apparatus (Bio-Rad) were washed with 200–400 μl of TE buffer, loaded with denatured DNA, and then washed with 200–400 μl of 2× SSC and air-dried for 20 min. Membranes were baked in 80 °C for 2 h, blocked using 5% nonfat milk TBST for 1 h, and incubated with 1:1000 anti–5-mC antibody at 4 °C overnight. The next day, membranes were washed with TBST for 10 min four times and then incubated with horseradish peroxidase-conjugated anti-rabbit secondary antibody at room temperature for 1 h. Membranes were washed with TBST for 10 min four times and reacted with ECL.
Author contributions
J. E. S., Y.-H. H., and M. A. G. data curation; J. E. S., Y.-H. H., M. A. G., and N. O. R. formal analysis; J. E. S., Y.-H. H., and M. A. G. investigation; J. E. S., Y.-H. H., and A. M. methodology; J. E. S., Y.-H. H., M. A. G., and N. O. R. writing-original draft; J. E. S., Y.-H. H., M. A. G., and N. O. R. writing-review and editing; M. A. G. conceptualization; M. A. G. and N. O. R. supervision; M. A. G. and N. O. R. funding acquisition; M. A. G. and N. O. R. project administration; N. O. R. resources.
Supplementary Material
This work was supported by National Science Foundation Grant 1808775 (to N. R.) and National Institutes of Health Grants CA183252, DK092883-01, and CA222736 (to M. G.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1 and S2.
- 5-mC
- 5-methylcytosine
- AML
- acute myeloid leukemia
- TDG
- thymine-DNA glycosylase
- BER
- base excision repair
- CN-AML
- cytogenetically normal AML
- GFP
- green fluorescent protein
- mESC
- murine embryonic stem cells
- DKO
- double knock-out
- AdoMet
- S-adenosylmethionine
- CREB
- cAMP-response element-binding protein.
References
- 1. Bird A. (2002) DNA methylation patterns and epigenetic memory. Genes Dev. 16, 6–21 10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- 2. Reik W., Dean W., and Walter J. (2001) Epigenetic reprogramming in mammalian development. Science 293, 1089–1093 10.1126/science.1063443 [DOI] [PubMed] [Google Scholar]
- 3. Robertson K. D. (2005) DNA methylation and human disease. Nat. Rev. Genet. 6, 597–610 10.1038/nrg1655 [DOI] [PubMed] [Google Scholar]
- 4. Figueroa M. E., Lugthart S., Li Y., Erpelinck-Verschueren C., Deng X., Christos P. J., Schifano E., Booth J., van Putten W., Skrabanek L., Campagne F., et al. (2010) DNA methylation signatures identify biologically distinct subtypes in acute myeloid leukemia. Cancer Cell 17, 13–27 10.1016/j.ccr.2009.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Melnick A. M. (2010) Epigenetics in AML. Best Pract. Res. Clin. Haematol. 23, 463–468 [DOI] [PubMed] [Google Scholar]
- 6. Hou H. A., and Tien H. F. (2016) Mutations in epigenetic modifiers in acute myeloid leukemia and their clinical utility. Expert Rev. Hematol. 9, 447–469 10.1586/17474086.2016.1144469 [DOI] [PubMed] [Google Scholar]
- 7. Rau R. E., Rodriguez B. A., Luo M., Jeong M., Rosen A., Rogers J. H., Campbell C. T., Daigle S. R., Deng L., Song Y., Sweet S., et al. (2016) DOT1L as a therapeutic target for the treatment of DNMT3A-mutant acute myeloid leukemia. Blood 128, 971–981 10.1182/blood-2015-11-684225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holz-Schietinger C., Matje D. M., and Reich N. O. (2012) Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J. Biol. Chem. 287, 30941–30951 10.1074/jbc.M112.366625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ley T. J., Ding L., Walter M. J., McLellan M. D., Lamprecht T., Larson D. E., Kandoth C., Payton J. E., Baty J., Welch J., Harris C. C., et al. (2010) DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 363, 2424–2433 10.1056/NEJMoa1005143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Russler-Germain D. A., Spencer D. H., Young M. A., Lamprecht T. L., Miller C. A., Fulton R., Meyer M. R., Erdmann-Gilmore P., Townsend R. R., Wilson R. K., and Ley T. J. (2014) The R882H DNMT3A mutation associated with AML dominantly inhibits wild-type DNMT3A by blocking its ability to form active tetramers. Cancer Cell 25, 442–454 10.1016/j.ccr.2014.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Forbes S. A., Beare D., Boutselakis H., Bamford S., Bindal N., Tate J., Cole C. G., Ward S., Dawson E., Ponting L., Stefancsik R., et al. (2017) COSMIC: somatic cancer genetics at high-resolution. Nucleic Acids Res. 45, D777–D783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aggerholm A., Guldberg P., Hokland M., and Hokland P. (1999) Extensive intra- and interindividual heterogeneity of p15INK4B methylation in acute myeloid leukemia. Cancer Res. 59, 436–441 [PubMed] [Google Scholar]
- 13. Cameron E. E., Baylin S. B., and Herman J. G. (1999) p15INK4B CpG island methylation in primary acute leukemia is heterogeneous and suggests density as a critical factor for transcriptional silencing. Blood 94, 2445–2451 [PubMed] [Google Scholar]
- 14. Esteller M. (2002) CpG island hypermethylation and tumor suppressor genes: a booming present, a brighter future. Oncogene 21, 5427–5440 10.1038/sj.onc.1205600 [DOI] [PubMed] [Google Scholar]
- 15. Santini V., Kantarjian H. M., and Issa J. P. (2001) Changes in DNA methylation in neoplasia: pathophysiology and therapeutic implications. Ann. Int. Med. 134, 573–586 10.7326/0003-4819-134-7-200104030-00011 [DOI] [PubMed] [Google Scholar]
- 16. Ramsahoye B. H., Biniszkiewicz D., Lyko F., Clark V., Bird A. P., and Jaenisch R. (2000) Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. U.S.A. 97, 5237–5242 10.1073/pnas.97.10.5237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Qu Y., Lennartsson A., Gaidzik V. I., Deneberg S., Karimi M., Bengtzén S., Höglund M., Bullinger L., Döhner K., and Lehmann S. (2014) Differential methylation in CN-AML preferentially targets non-CGI regions and is dictated by DNMT3A mutational status and associated with predominant hypomethylation of HOX genes. Epigenetics 9, 1108–1119 10.4161/epi.29315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li Y. Q., Zhou P. Z., Zheng X. D., Walsh C. P., and Xu G. L. (2006) Association of Dnmt3a and thymine DNA glycosylase links DNA methylation with base-excision repair. Nucleic Acids Res. 35, 390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Esposito M. T., and So C. W. E. (2014). DNA damage accumulation and repair defects in acute myeloid leukemia: implications for pathogenesis, disease progression, and chemotherapy resistance. Chromosoma. 123, 545–561 10.1007/s00412-014-0482-9 [DOI] [PubMed] [Google Scholar]
- 20. Holz-Schietinger C., Matje D. M., Harrison M. F., and Reich N. O. (2011) Oligomerization of DNMT3A controls the mechanism of de novo DNA methylation. J. Biol. Chem. 286, 41479–41488 10.1074/jbc.M111.284687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Holz-Schietinger C., and Reich N. O. (2010) The inherent processivity of the human de novo methyltransferase 3A (DNMT3A) is enhanced by DNMT3L. J. Biol. Chem. 285, 29091–29100 10.1074/jbc.M110.142513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Laurent L., Wong E., Li G., Huynh T., Tsirigos A., Ong C. T., Low H. M., Kin Sung K. W., Rigoutsos I., Loring J., and Wei C. L. (2010) Dynamic changes in the human methylome during differentiation. Genome Res. 20, 320–331 10.1101/gr.101907.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hardeland U., Bentele M., Lettieri T., Steinacher R., Jiricny J., and Schär P. (2001) Thymine DNA glycosylase. Prog. Nucleic Acid Res. Mol. Biol. 68, 235–253 10.1016/S0079-6603(01)68103-0 [DOI] [PubMed] [Google Scholar]
- 24. Purdy M. M., Holz-Schietinger C., and Reich N. O. (2010) Identification of a second DNA binding site in human DNA methyltransferase 3A by substrate inhibition and domain deletion. Arch. Biochem. Biophys. 498, 13–22 10.1016/j.abb.2010.03.007 [DOI] [PubMed] [Google Scholar]
- 25. Thillainadesan G., Chitilian J. M., Isovic M., Ablack J. N., Mymryk J. S., Tini M., and Torchia J. (2012) TGF-β-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol. Cell 46, 636–649 10.1016/j.molcel.2012.03.027 [DOI] [PubMed] [Google Scholar]
- 26. Hervouet E., Vallette F. M., and Cartron P. F. (2009) Dnmt3/transcription factor interactions as crucial players in targeted DNA methylation. Epigenetics 4, 487–499 10.4161/epi.4.7.9883 [DOI] [PubMed] [Google Scholar]
- 27. Fuks F., Burgers W. A., Godin N., Kasai M., and Kouzarides T. (2001) Dnmt3a binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 20, 2536–2544 10.1093/emboj/20.10.2536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Stirzaker C., Song J. Z., Ng W., Du Q., Armstrong N. J., Locke W. J., Statham A. L., French H., Pidsley R., Valdes-Mora F., Zotenko E., and Clark S. J. (2017) Methyl-CpG-binding protein MBD2 plays a key role in maintenance and spread of DNA methylation at CpG islands and shores in cancer. Oncogene 36, 1328–1338 10.1038/onc.2016.297 [DOI] [PubMed] [Google Scholar]
- 29. Jia D., Jurkowska R. Z., Zhang X., Jeltsch A., and Cheng X. (2007) Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 449, 248–251 10.1038/nature06146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Doherty K. M., Sommers J. A., Gray M. D., Lee J. W., von Kobbe C., Thoma N. H., Kureekattil R. P., Kenny M. K., and Brosh R. M. (2005) Physical and functional mapping of the replication protein A interaction domain of the werner and bloom syndrome helicases. J. Biol. Chem. 280, 29494–29505 10.1074/jbc.M500653200 [DOI] [PubMed] [Google Scholar]
- 31. Chen W., Lam S. S., Srinath H., Schiffer C. A., Royer W. E., and Lin K. (2007) Competition between Ski and CREB-binding protein for binding to Smad proteins in transforming growth factor-β signaling. J. Biol. Chem. 282, 11365–11376 10.1074/jbc.M700186200 [DOI] [PubMed] [Google Scholar]
- 32. Shiba N., Taki T., Park M. J., Shimada A., Sotomatsu M., Adachi S., Tawa A., Horibe K., Tsuchida M., Hanada R., Tsukimoto I., Arakawa H., and Hayshi Y. (2012) DNMT3A mutations are rare in childhood acute myeloid leukaemia, myelodysplastic syndromes and juvenile myelomonocytic leukaemia. Br. J. Haematol. 156, 413–414 10.1111/j.1365-2141.2011.08879.x [DOI] [PubMed] [Google Scholar]
- 33. Stegelmann F., Bullinger L., Schlenk R. F., Paschka P., Griesshammer M., Blersch C., Kuhn S., Schauer S., Döhner H., and Döhner K. (2011) DNMT3A mutations in myeloproliferative neoplasms. Leukemia 25, 1217–1219 10.1038/leu.2011.77 [DOI] [PubMed] [Google Scholar]
- 34. Couronné L., Bastard C., and Bernard O. A. (2012) TET2 and DNMT3A mutations in human T-cell lymphoma. N. Engl. J. Med. 366, 95–96 10.1056/NEJMc1111708 [DOI] [PubMed] [Google Scholar]
- 35. Grossmann V., Haferlach C., Weissmann S., Roller A., Schindela S., Poetzinger F., Stadler K., Bellos F., Kern W., Haferlach T., Schnittger S., and Kohlmann A. (2013) The molecular profile of adult T-cell acute lymphoblastic leukemia: mutations in RUNX1 and DNMT3A are associated with poor prognosis in T-ALL. Genes Chromosomes Cancer 52, 410–422 10.1002/gcc.22039 [DOI] [PubMed] [Google Scholar]
- 36. Suzuki M., Yamada T., Kihara-Negishi F., Sakurai T., Hara E., Tenen D. G., Hozumi N., and Oikawa T. (2006) Site-specific DNA methylation by a complex of PU.1 and Dnmt3a/b. Oncogene 25, 2477–2488 10.1038/sj.onc.1209272 [DOI] [PubMed] [Google Scholar]
- 37. Fuks F., Hurd P. J., Deplus R., and Kouzarides T. (2003) The DNA methyltransferases associate with HP1 and the SUV39H1 histone methyltransferase. Nucleic Acids Res. 31, 2305–2312 10.1093/nar/gkg332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Turker M. S. (2002) Gene silencing in mammalian cells and the spread of DNA methylation. Oncogene 21, 5388–5393 10.1038/sj.onc.1205599 [DOI] [PubMed] [Google Scholar]
- 39. Vardiman J. W., Thiele J., Arber D. A., Brunning R. D., Borowitz M. J., Porwit A., Harris N. L., Le Beau M. M., Hellström-Lindberg E., Tefferi A., and Bloomfield C. D. (2009) The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood 114, 937–951 10.1182/blood-2009-03-209262 [DOI] [PubMed] [Google Scholar]
- 40. Du J., Johnson L. M., Jacobsen S. E., and Patel D. J. (2015) DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Biol. 16, 519–532 10.1038/nrm4043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Matzke M. A., and Mosher R. A. (2014) RNA-directed DNA methylation: an epigenetic pathway of increasing complexity. Nat. Rev. Genet. 15, 394–408 10.1038/nrg3683 [DOI] [PubMed] [Google Scholar]
- 42. Spencer D. H., Russler-Germain D. A., Ketkar S., Helton N. M., Lamprecht T. L., Fulton R. S., Fronick C. C., O'Laughlin M., Heath S. E., Shinawi M., Westervelt P., et al. (2017) CpG island hypermethylation mediated by DNMT3A is a consequence of AML progression. Cell 168, 801–816.e13 10.1016/j.cell.2017.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chen D., Christopher M., Helton N. M., Ferguson I., Ley T. J., and Spencer D. H. (2018) DNMT3A R882-associated hypomethylation patterns are maintained in primary AML xenografts, but not in the DNMT3A R882C OCI-AML3 leukemia cell line. Blood Cancer J. 8, 38 10.1038/s41408-018-0072-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pedrali-Noy G., and Weissbach A. (1986) Mammalian DNA methyltransferases prefer poly(dI-dC) as substrate. J. Biol. Chem. 261, 7600–7602 [PubMed] [Google Scholar]
- 45. Galonska C., Charlton J., Mattei A. L., Donaghey J., Clement K., Gu H., Mohammad A. W., Stamenova E. K., Cacchiarelli D., Klages S., Timmermann B., Cantz T., Schöler H. R., Gnirke A., Ziller M. J., and Meissner A. (2018) Genome-wide tracking of dCas9-methyltransferase footprints. Nat. Commun. 9, 597 10.1038/s41467-017-02708-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lei Y., Huang Y. H., and Goodell M. A. (2018) DNA methylation and de-methylation using hybrid site-targeting proteins. Genome Biol. 19, 187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen T., Ueda Y., Dodge J. E., Wang Z., and Li E. (2003) Establishment and maintenance of genomic methylation patterns in mouse embryonic stem cells by Dnmt3a and Dnmt3b. Mol. Cell. Biol. 23, 5594–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hata K., Okano M., Lei H., and Li E. (2002) Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129, 1983–1993 [DOI] [PubMed] [Google Scholar]
- 49. Kareta M. S., Botello Z. M., Ennis J. J., Chou C., and Chédin F. (2006) Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J. Biol. Chem. 281, 25893–25902 10.1074/jbc.M603140200 [DOI] [PubMed] [Google Scholar]
- 50. Schuermann D., Scheidegger S. P., Weber A. R., Bjørås M., Leumann C. J., and Schär P. (2016) 3CAPS, a structural AP-site analogue as a tool to investigate DNA base excision repair. Nucleic Acids Res. 44, 2187–2198 10.1093/nar/gkv1520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peterson S. N., and Reich N. O. (2006) GATC flanking sequences regulate Dam activity: evidence for how Dam specificity may influence pap expression. J. Mol. Biol. 355, 459–472 10.1016/j.jmb.2005.11.003 [DOI] [PubMed] [Google Scholar]
- 52. Su J., Huang Y. H., Cui X., Wang X., Zhang X., Lei Y., Xu J., Lin X., Chen K., Lv J., Goodell M. A., and Li W. (2018) Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol. 19, 108 10.1186/s13059-018-1492-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Galonska C., Charlton J., Mattei A. L., Donaghey J., Clement K., Gu H., Mohammad A. W., Stamenova E. K., Cacchiarelli D., Klages S., Timmermann B., et al. (2018) Genome-wide tracking of dCas9-methyltransferase footprints. Nat. Commun. 9, 597 10.1038/s41467-017-02708-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huang Y. H., Su J., Lei Y., Brunetti L., Gundry M. C., Zhang X., Jeong M., Li W., and Goodell M. A. (2017) DNA epigenome editing using CRISPR-Cas SunTag-directed DNMT3A. Genome Biol. 18, 176 10.1186/s13059-017-1306-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gu T., Lin X., Cullen S. M., Luo M., Jeong M., Estecio M., Shen J., Hardikar S., Sun D., Su J., Rux D., et al. (2018) DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol. 19, 88 10.1186/s13059-018-1464-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.