

Abstract
Hypoxia-inducible factor-2α (HIF2α) is a nuclear transcription factor that plays a critical role in cell survival including metabolic adaptation under hypoxia as well as normoxia, but whether HIF2α contributes to the control of whole-body metabolic balance is unclear. In this study, we found that the hypothalamic HIF2α protein level rapidly increases in young mice that are centrally stimulated with insulin. However, this insulin-induced HIF2α up-regulation is substantially attenuated in mice of advanced age. This attenuation is comparable with the effect of high-calorie feeding in young mice. Of note, unlike high-calorie feeding conditions, age-dependent HIF2α attenuation occurs without impaired activation of the hypothalamic IR/IRS-2/AKT/FOXO1 pathway in response to insulin. Molecular and physiological analyses revealed that hypothalamic HIF2α contributes to the action of central insulin in regulation of proopiomelanocortin (Pomc) gene expression and food intake. HIF2α knockout in POMC neurons led to age-dependent excess weight gain and fat increase, a phenotype that was associated with a mild degree of glucose intolerance and insulin resistance. In conclusion, hypothalamic HIF2α responds to insulin, and the up-regulation is involved in adaptive metabolic regulation as age increases, whereas impairment of HIF2α in the hypothalamus contributes to weight gain and glucose disorders in age-dependent manners.
Keywords: hypoxia-inducible factor (HIF), insulin resistance, obesity, aging, hypothalamus, cell signaling, diabetes
Introduction
The hypoxia-inducible factor-α (HIFα)2 family, including HIF1α, HIF2α, and the less well-characterized HIF3α, are oxygen-sensitive transcription factors capable of regulating anaerobic metabolism, vascular growth, proliferation, and cell survival (1, 2). The physiological role of HIF in the brain is unclear, despite studies that were conducted in stroke models (3–5). Although HIF is important for reducing the intracellular metabolic rate in ischemic conditions, this paradigm does not apply to metabolic regulation in normal physiology, in which oxygen levels in the brain only fluctuate to a small degree. On the other hand, studies have demonstrated that the HIF signaling pathway can be regulated by a wide variety of signals, not necessarily related to oxygen levels. For instance, glucose has been shown to regulate HIF through multiple mechanisms. It has been reported that glucose metabolites such as pyruvate can inhibit prolyl 4-hydroxylases to stabilize HIFα (6). The mechanism of glucose-induced HIF up-regulation can be mediated by the action of carbohydrate response element-binding protein (7). Furthermore, fumarate and succinate were also found to increase HIF2α protein levels partially through inhibiting prolyl 4-hydroxylases (8). Notably, this HIF2α up-regulation through TCA-cycle intermediates is part of glucose sensing in the hypothalamus (8), pointing to the direction that the HIF2α isoform might be physiologically relevant in neural mechanisms governing metabolic balance.
In addition to glucose, insulin has been shown to increase HIF1α protein levels (9), suggesting that HIF could be a transcriptional component in insulin signaling. Also, it is well appreciated that insulin resistance and impaired glucose tolerance are key factors in the development of type 2 diabetes, and both are commonly seen in elderly populations. For example, the postprandial glucose excursion is substantially greater and remains elevated longer in nondiabetic elderly adults than in nondiabetic younger adults (10), indicating an age-related decline in insulin sensitivity and glucose tolerance. Furthermore, aging is associated with body composition changes including excess fat mass (11). However, it is unclear if metabolic disorders in advanced age are mediated through similar or different mechanisms compared with the dietary etiologies of diabetes such as chronic high-calorie feeding. Recently, hypothalamic dysfunction has been implicated in the development of not only dietary metabolic syndromes (12–14) but also aging (15, 16). Insulin signaling in the hypothalamus is important in metabolic regulation (17), but how it contributes to age-dependent metabolic changes is unclear. In this context, this study investigates whether hypothalamic HIF2α responds to insulin and how this reaction is affected under different age and dietary conditions. Also, because HIF can induce Pomc gene transcription (8), we further study if HIF2α contributes to insulin-dependent Pomc gene expression, and how loss of HIF2α in Pomc neurons affects metabolic physiology at different ages.
Results
Hypothalamic HIF2α up-regulation by insulin and its impairment at advanced age
Although glucose infusion has been shown to increase HIF2α protein levels (8), it is unknown whether insulin influences hypothalamic HIF2α, despite evidence that HIF1α mRNA and protein were shown to increase after insulin stimulation in human retinal epithelial cells (9). To investigate the effect of insulin on hypothalamic HIF2α, we assessed if HIF2α expression in the hypothalamus is altered after hypothalamic third ventricle insulin injection in chow-fed 3-month-old mice. We found hypothalamic HIF2α protein levels increased dramatically and rapidly post-insulin injection (Fig. 1, A and B). Chow-fed 15-month-old mice were comparatively studied, showing that up-regulation of hypothalamic HIF2α protein by insulin was substantially attenuated (Fig. 1, A and B). Apart from the aged mice, we also included a group of 3-month-old mice fed a high-fat diet (HFD) for 6 weeks, which was enough time to cause body weight gain. Despite the young age of these animals, HFD feeding greatly diminished the ability of insulin to up-regulate the hypothalamic HIF2α protein, to a similar degree as seen in advanced age (Fig. 1, A and B). Because the IR/IRS-2/AKT/FOXO1 cascade in insulin signaling is known to be inactive in diet-induced obesity (DIO) conditions (18), we further analyzed IR/IRS-2/AKT/FOXO1 signaling in the same cohorts of 3-month-old chow-fed, 3-month-old HFD, and 15-month-old chow-fed mice, as described above. We found that insulin injection increased activation of this pathway, as evidenced by increased phosphorylation of IRS-1, IRS-2, AKT (Ser-473), and FOXO1 in young mice. However, insulin-stimulated activation of this pathway was blunted in HFD-fed mice compared with age-matched chow-fed mice (Fig. 1, A and C–F). Interestingly, phosphorylation of these proteins was similar in 3-month-old and 15-month-old chow-fed mice (Fig. 1, A and C–F). Thus, we identified a time window in middle-aged mice in which the core components of insulin signaling are still not affected, given the evidence that this pathway is inhibited in older mice (19). Taken together, impairment in insulin-dependent HIF2α induction in the hypothalamus is a common change in advanced age and HFD feeding, unlike the IR/IRS-2/AKT/FOXO1 core pathway activation in response to insulin, which differs between these two etiological conditions.
Figure 1.

Insulin-induced HIF2α versus other signaling components in middle-aged versus DIO mice. Following overnight fasting, 3-month-old chow-fed, 3-month-old HFD-fed, and 15-month-old chow-fed mice were infused in the hypothalamic third-ventricle with vehicle (Veh) or insulin (INS). At 90 min post-infusion, the hypothalamic samples were harvested for Western blot analysis of HIF2α, tyrosine phosphorylation of insulin receptor substrate 1 (p-IRS–1), insulin receptor substrate-1 (IRS–1), tyrosine phosphorylation of insulin receptor substrate 2 (p-IRS-2), insulin receptor substrate-2 (IRS-2), Akt phosphorylation, and Foxo1 phosphorylation. A, representative Western blots. B–F, Western blotting quantification. HIF2α was normalized by β-actin, and p-IRS-1, p-IRS-2, p-AKT, and p-FOXO1 were normalized by the respective total protein levels. **, p < 0.01; ***, p < 0.001; n = 4 mice per group (biological replicates). Bars represent mean ± S.E. AU, arbitrary unit. NS, not significant.
Decline in insulin-induced Pomc mRNA up-regulation at advanced age
We then examined whether middle age could affect insulin-induced regulation of Pomc gene expression in the hypothalamus. For comparison, we analyzed gene expression of neuropeptides neuropeptide Y (Npy) and agouti-related transcript (Agrp), because they are produced by AgRP neurons, which are located in the same hypothalamic subregion as POMC neurons but are known to functionally oppose them (20). A less relevant neuropeptide neurotensin (NT) was further included in the assay as an additional reference. Indeed, central injection of insulin led to a great increase in hypothalamic Pomc mRNA levels as compared with vehicle injection in 3-month-old chow-fed mice. In contrast, this effect of insulin dropped substantially in 15-month-old chow-fed mice, although the remaining effect was still statistically significant (Fig. 2A). Insulin-induced hypothalamic Pomc gene expression was more strongly inhibited in HFD-fed mice, which was statistically nonsignificant compared with young controls (Fig. 2A). We did not observe any obvious differences in Pomc mRNA levels between 3-month-old chow-fed, 3-month-old HFD, and 15-month-old chow-fed mice with vehicle injection. On the other hand, insulin did not significantly affect the expression levels of other neuropeptides at this time point (Fig. 2A). These results agree with previous work showing that hypothalamic insulin injection did not acutely affect Agrp and Npy gene expression (21, 22). Hence, whereas hypothalamic POMC is a neuropeptide that is sensitive to insulin, this sensitivity dramatically declines in both conditions of DIO and middle age. Considering that these two conditions have different influences on the IR/IRS-2/Akt/FOXO1 signaling cascade, it prompted us to question if HIF could be particularly important for the effect of aging on POMC.
Figure 2.

Insulin-induced Pomc gene expression in middle age versus DIO conditions. Following overnight fasting, 3-month-old chow-fed, 3-month-old HFD-fed, and 15-month-old chow-fed mice were infused in the hypothalamic third ventricle with vehicle (Veh) or insulin (INS). At 90 min post-infusion, the hypothalamic samples were harvested for measurements of Pomc mRNA (A), Agrp mRNA (B), Npy mRNA (C), and Nt mRNA (D). *, p < 0.05; ***, p < 0.001, n = 8–10 per group (biological replicates). Error bars reflect mean ± S.E. AU, arbitrary unit. NS, not significant.
Loss of HIF2α in POMC neurons reduces the up-regulation of Pomc mRNA by insulin
To experimentally address the question raised above, we investigated whether loss of HIF2α in POMC neurons could affect insulin signaling and further regulate hypothalamic Pomc gene transcription. To do so, we ablated HIF2α in hypothalamic Pomc neurons by crossing POMC-Cre mice with HIF2αlox/lox mice, leading to compound offspring termed POMC/HIF2αlox/lox mice. Immunofluorescence staining was used to detect HIF2α in POMC neurons of this knockout mouse model compared with littermate controls. As shown in Fig. 3, A–C, HIF2α protein was present in the majority of POMC neurons in control mice but not detectable in the majority of POMC neurons in POMC/HIF2αlox/lox mice. It is also worth mentioning that HIF2α ablation did not reduce the number of hypothalamic POMC neurons, and consistent with this profile, HIF2α knockout mice were metabolically indistinguishable from littermate controls when young. To profile insulin-induced hypothalamic Pomc expression, knockout mice and controls received a single infusion of insulin through a cannula implanted in the hypothalamic third ventricle. As shown in Fig. 3, D and E, insulin increased hypothalamic Pomc mRNA in chow-fed controls at a young age but did less so in chow-fed controls of middle age. Relatively, HIF2α knockout in POMC neurons blunted insulin's ability to increase Pomc mRNA more so in middle-aged mice than young mice (Fig. 3, D and E), suggesting that loss of HIF2α might affect additional age-dependent pathway(s), which can stimulate Pomc gene expression. Under HFD conditions, insulin's ability to increase Pomc mRNA was abolished similarly in control and POMC/HIF2αlox/lox mice (Fig. 3F), indicating that HFD feeding and HIF2α loss might share an overlapping mechanism to inhibit Pomc gene expression. Thus, HIF2α is an appreciable factor contributing to insulin-induced Pomc gene expression, which is sensitive to age and dietary conditions.
Figure 3.

Role of HIF2α in Pomc neurons in insulin-dependent regulation of Pomc mRNA. A, hypothalamic sections from POMC/HIF2αlox/lox mice and control HIF2αlox/lox mice were stained for HIF2α (red) and POMC (green) in the region containing the arcuate nucleus (Arc). DAPI staining shows the nuclei of cells. Arrows point to POMC neurons that are strongly positive for HIF2α. Scale bar: 25 μm. B and C, POMC neurons (B) and HIF2α-positive population (C) in the Arc sections were counted according to immunostaining. ***, p < 0.001, n = 4 mice per group (biological replicates). Error bars reflect mean ± S.E. D–F, POMC/HIF2αlox/lox mice and HIF2αlox/lox mice at the indicated ages and diet conditions were fasted 24 h and received a third-ventricle injection of insulin (INS) versus vehicle (Veh), and 2 h later the hypothalamus was harvested for the measurement of Pomc mRNA. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n = 8 mice per group (biological replicates). Error bars reflect mean ± S.E. NS, not significant.
Loss of HIF2α in POMC neurons desensitizes insulin-dependent regulation of feeding
Using physiological approaches, we further examined whether HIF2α knockout in POMC neurons could compromise the feeding regulation of hypothalamic insulin. This study was based on the experimental condition that prolonged fasting (24 h) induces a feeding response that is inhibited by hypothalamic insulin administration. Indeed, insulin was able to suppress food intake by 55–60% in chow-fed control young mice and middle-aged mice (Fig. 4, A–D). In contrast, this inhibitory effect of insulin was blunted in POMC/HIF2αlox/lox mice, as evidenced by insulin suppressing food intake by 28% in young mice and 16% in middle-aged mice, compared with their respective controls (Fig. 4, A–D). These data may also implicate that certain factors during middle age can work to potentiate the metabolic consequences of HIF2α loss. In HFD-fed mice, insulin barely inhibited food intake in control and POMC/HIF2αlox/lox mice even when young (Fig. 4, E and F). Thus, feeding dysregulation under HFD feeding is strong enough to mask the metabolic effects of HIF2α loss. Altogether, impairment of HIF2α in POMC neurons is sufficient to reduce insulin sensitivity in the regulation of feeding.
Figure 4.
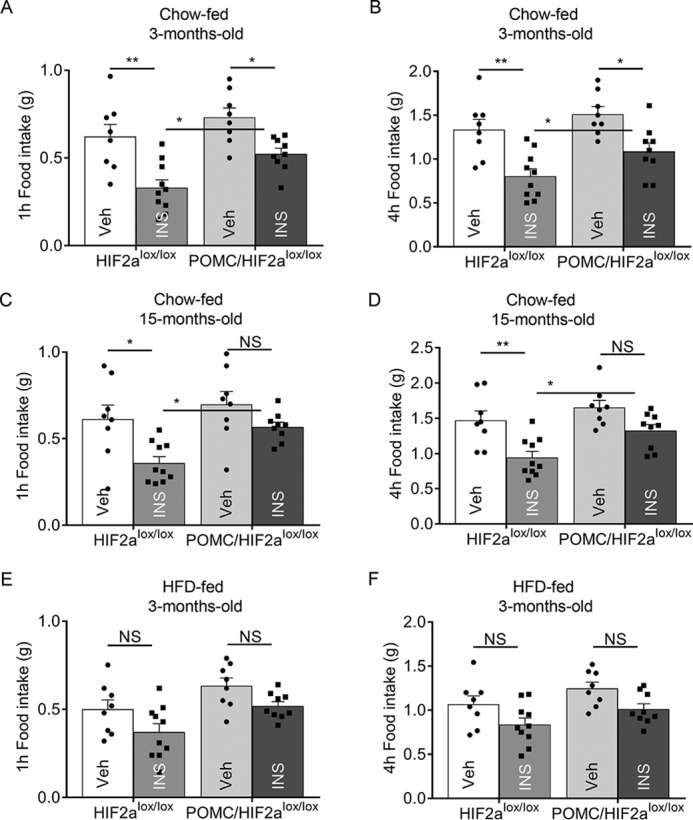
Role of HIF2α in POMC neurons in insulin-dependent regulation of feeding. POMC/HIF2αlox/lox mice versus littermate control HIF2αlox/lox mice at the age of 3 versus 15 months under chow feeding (A–D) and at the age of 3 months under HFD feeding (E and F) were fasted for 24 h and received a third-ventricle injection of insulin or the vehicle. Food was placed in each cage and measured for 1 and 4 h. *, p < 0.05; **, p < 0.01; n = 8–10 per group (biological replicates). Error bars reflect mean ± S.E. NS, not significant.
Loss of HIF2α in POMC neurons led to age-dependent weight excess and fat gain
To completely profile the metabolic outcomes of POMC/HIF2αlox/lox mice, we set up a group of these animals for longitudinal follow-up of body weight and feeding, compared with the littermate HIF2αlox/lox controls and age-matched POMC-Cre controls. To single out the metabolic effects of HIF2α inactivation, the experiment was performed using standard normal chow feeding. As shown in Fig. 5A, these three groups of mice were similar in terms of weight gain until middle age. Roughly starting ∼6 months of age, POMC/HIF2αlox/lox mice showed a gradual increase in body weight compared with both controls (Fig. 5A). At around 8–12 months old, weight gain levels in the knockout mice was quite apparent. Magnetic resonance imaging scanning was performed for these mice 3 times during the experimental follow-up, showing that the excess weight gain in knockout mice was attributed to fat mass instead of lean mass (Fig. 5, B and C). Food intake profiling revealed that knockout mice were often hyperphagic; although modest when young, it became increasingly obvious with advanced age (Fig. 5D). Water intake was similar between POMC/HIF2αlox/lox and control mice during the follow-up. We also subjected subgroups of POMC/HIF2αlox/lox mice and the littermate HIF2αlox/lox controls to metabolic cages to measure oxygen consumption levels. As shown in Fig. 5, E and F, knockout mice had reduced oxygen consumption during the day and night, despite no significant difference in locomotive activities (Fig. 5G). Thus, apart from food intake, reduced energy expenditure is likely another contributor to the body weight phenotype of these knockout mice. Taken together, HIF2α loss in POMC neurons can mimic the effect of advanced age leading to excess weight and body fat.
Figure 5.

Long-term effects of HIF2α ablation in POMC neurons on body weight and energy balance. A, body weight of chow-fed POMC/HIF2αlox/lox and controls (POMC-Cre and HIF2αlox/lox mice) were longitudinally measured. B and C, lean mass (B) and fat mass (C) of chow-fed POMC/HIF2αlox/lox and controls (POMC-Cre and HIF2αlox/lox mice) were analyzed at the age of 12, 30, and 48 weeks. D, chow-fed POMC/HIF2αlox/lox and controls (POMC-Cre and HIF2αlox/lox mice) were longitudinally measured for food intake. E–G, chow-fed POMC/HIF2αlox/lox and littermate control HIF2αlox/lox mice at the age of 12 months were analyzed for O2 consumption and locomotive activities via metabolic cages. Data show 24-h tracings (E, left panels), correlation of O2 consumption with lean mass (E, right panels), average values according to day and night time (F), and 24-h locomotive activities (G). *, p < 0.05; **, p < 0.01, compared with each control group, n = 8 per group (biological replicates). Error bars reflect mean ± S.E. NS, not significant.
Loss of HIF2α in POMC neurons increases glucose intolerance at advanced age
Impaired glucose tolerance is characteristic of a pre-diabetic state as well as aging. Therefore, to determine whether HIF2α knockout in POMC neurons plays a role in the development of glucose intolerance, we performed glucose tolerance tests (GTT) in 3-month-old versus 12-month-old POMC/HIF2αlox/lox mice, compared with age-matched controls. These mice were all maintained on a chow diet as described above. We observed that POMC/HIF2αlox/lox mice had normal glucose tolerance at 3 months, but by 12 months old, they displayed a mild to moderate degree of glucose intolerance when compared with either HIF2αlox/lox controls or POMC-Cre controls (Fig. 6, A–D). Because insulin resistance is an important factor for the development of glucose intolerance, we further examined insulin tolerance in these mice. In agreement with glucose intolerance, knockout mice showed a normal level of glucose responsiveness to insulin when young, but this response was diminished in 12-month-old mice (Fig. 6, E–H). Although the magnitudes of glucose intolerance and insulin insensitivity were not dramatic, these changes agreed with the moderate overweight phenotype in these mice at these ages. To summarize, loss of HIF2α in POMC neurons leads to weight increase and glucose disorders in an age-dependent manner. These findings support the hypothesis that reduction of HIF2α in the hypothalamus has a mechanistic contribution to the development and progression of age-related metabolic disorders.
Figure 6.

Long-term effects of HIF2α ablation in POMC neurons on blood glucose homeostasis. POMC/HIF2αlox/lox mice and matched control groups (POMC-Cre and HIF2αlox/lox mice) were maintained under chow feeding and were examined for glucose and insulin tolerance via GTT (A–D) and ITT (E–H) at 3 months of age (A, B, E, and F) versus 12 months (C, D, G, and H). AUC, area under curve. *, p < 0.05; **, p < 0.01, compared with each control group, n = 8 for each group (biological replicates). Data are presented as mean ± S.E. NS, not significant.
Discussion
Previous research has studied insulin signaling in the hypothalamus and the central nervous system. Although deletion of IRs in POMC neurons has minimal impact on body weight and blood glucose (23), selective decrease of insulin receptor protein within cells in the medial portion of the arcuate nucleus in the hypothalamus causes hyperphagia and increased fat mass (24, 25). Central injection of insulin clearly suppresses food intake (26), and this anorexigenic action has been shown to be mediated by phosphoinositide 3-kinase activation via IRS2 leading to the inactivation of Foxo1 (27). However, the relationship between hypothalamic insulin signaling and HIF has not been studied thus far, although insulin was shown to activate HIF1α in certain peripheral cells such as retinal epithelial cells (9). In this study, we found that central insulin injection greatly increases hypothalamic HIF2α protein. This effect of central insulin is rapid, suggesting that HIF2α might contribute to the transcriptional program of hypothalamic insulin signaling.
In research, hypothalamic insulin resistance has been implicated in obesity. For example, activation of hypothalamic IR/IRS-2/AKT/FOXO1 by insulin is impaired in DIO animals (18). Compared with DIO, which is substantive for diabetic development, little is known for the condition of age advancement. Although previous research has shown that insulin-stimulated hypothalamic IR, GSK3, AKT, and p70S6K phosphorylation decreased in older than 2-year aged rats (28), it is entirely unclear how hypothalamic signaling is changed at middle age (for example, at 12–15 months), representing a time window that may set up the mechanisms for aging pathology. We evaluated middle-aged mice in this study and found that IR/IRS-2/AKT/FOXO1 pathway activation in these mice is comparable with young mice. In contrast, insulin-induced hypothalamic HIF2α up-regulation dramatically dropped in these middle-aged mice. This drop at advanced age is comparable with the change seen in short-term DIO. Thus, hypothalamic HIF2α up-regulation by insulin is impaired in both DIO and advanced age. Compared with hypothalamic IR/IRS-2/AKT/FOXO1 in insulin response, HIF2α is selectively affected at middle age, indicating that this HIF2α decline might be highly sensitive and essential for the initiation of metabolic disorders during the progression of aging.
Hypothalamic POMC neurons are fundamentally important for the central regulation of feeding and metabolic homeostasis via the secretion of several bioactive neuropeptides that are cleaved from the POMC precursor (29–31). POMC-derived peptides, including α melanocyte-stimulating hormone and β-endorphin (32), can each play particular roles in the control of the metabolic physiology. Indeed, both Pomc-deficient rodent models and human patients with POMC mutations invariably exhibit hyperphagia and marked obesity (33–35). Because of the strong anorexigenic action of POMC neurons, these cells are counterbalanced by NPY/AgRP neurons (36). We observed that, in contrast to up-regulating Pomc gene expression, single injection of insulin did not acutely affect Npy or Agrp mRNA levels. These data are consistent with several prior studies (21, 22), and further indicates that the Pomc gene is sensitive to insulin regulation. In this context, we found that insulin-induced Pomc gene up-regulation is partially lost at an advanced age. In agreement, the ability of central insulin to inhibit food intake is also impaired in middle-aged mice. In the context of previous work showing that Pomc is a transcriptional target of HIF (8), we generated mice with HIF2α knockout in POMC neurons so that studies were more specifically focused on this neuronal subtype. Our findings from this model confirmed that HIF2α significantly but partially accounts for hypothalamic insulin signaling in the regulation of the Pomc gene and feeding. Besides, with increasing age in these knockout mice, impaired insulin regulation of Pomc and feeding appeared slightly worse, implicating that HIF2α loss may work with some age-dependent changes to enhance these impairments.
To obtain a more complete picture, we then longitudinally studied this POMC–HIF2α knockout model for metabolic physiology until middle age. Lack of HIF2α in POMC neurons led to late-onset overweight phenotypes compared with age-matched controls. Body weight and metabolic profiling of these knockout mice were rather normal during the first 5 months of life, but thereafter, gradually increased more than controls, and by 12 months, these mice were considerably heavier due to increased fat mass. Furthermore, these POMC/HIF2αlox/lox mice at middle age had classical metabolic syndrome, including impaired glucose tolerance and reduced glucose responsiveness to insulin. Overall, metabolic problems due to HIF2α loss in POMC neurons are rather less striking, compared with the amplitude of these disorders induced by strong factors such as HFD feeding. The extent of these disorders, however, is rather comparable with that seen in age-associated weight gain and glucose intolerance. Also, it should be pointed out that, because this study just focused on POMC neurons, the genetic model might only represent a fraction of the mechanism and phenotype. If so, some other hypothalamic neurons may play additional roles, directly and indirectly, for the development of age-related metabolic disorders. Hence, future research is still needed to address these questions.
To summarize, our study demonstrated that HIF2α in the hypothalamus is sensitively up-regulated by local insulin signaling, whereas this effect remarkably declines at advanced age. This hypothalamic change under pre-aging conditions does not rely on decline in IR/IRS-2/AKT/FOXO1 activation by insulin. Genetic deletion of HIF2α in POMC neurons results in a late-onset overweight phenotype together with mild but appreciable levels of impaired glucose tolerance and impaired blood glucose response to insulin. According to these findings, we can speculate that reduced response of hypothalamic HIF2α to insulin could potentially and at least partially be responsible for the development of the age-related metabolic syndrome. In conclusion, HIF2α in POMC neurons contributes to the central control of the Pomc gene and feeding by insulin, whereas loss of this function is sufficient to cause body weight and blood glucose imbalance at advance age.
Materials and methods
Animal models and metabolic phenotyping
C57BL/6 mice were purchased from Jackson Laboratories and housed under standard conditions in a temperature- and humidity-controlled facility with a 12-h light–dark cycle at Albert Einstein College of Medicine. HIF2αlox/lox mice were originally obtained from Jackson Laboratories. HIF2αlox/lox mice and POMC-Cre mice described previously (8) were maintained on the C57BL/6 strain background. Male mice were used for all experiments. To induce DIO and middle age, mice were maintained on high-fat diet (HFD, 60 kcal % fat, 20 kcal % protein, and 20 kcal % carbohydrate) or normal chow (10 kcal % fat, 20 kcal % protein, and 70 kcal % carbohydrate) for the indicated periods. Magnetic resonance imaging was used to determine body composition of mice. O2 consumption and physical activity of mice were measured using metabolic chambers (Columbus Instrument, Inc.) at the core at Albert Einstein College of Medicine. The Institutional Animal Care and Use Committee at Albert Einstein College of Medicine approved all the procedures.
Third ventricle cannulation and infusion
As previously described (8), an ultra-precise small animal stereotactic apparatus (David Kopf Instrument) was used to implant a guide cannula into the third ventricle of anesthetized mice at the midline coordinates of 1.8 mm posterior to the bregma and 5.0 mm below the bregma. The mice were allowed at least 1 week for surgical recovery. Individual mice were restrained in a mouse restrainer, and infused with an indicated reagent over the indicated time period using a 26-gauge guide cannula and 33-gauge injector (Plastics One) connected to a Hamilton Syringe and infusion pump (Harvard Apparatus).
Test of insulin-suppressed food intake
Individually-housed mice were deprived of food for 24 h but with free access to water and were intracerebroventricular injected via pre-implanted cannula with insulin versus vehicle in a 2-μl volume over 5 min. Afterward, food was returned to the cages and food intake was determined by measuring the difference between the weight initially given and the weight left at 1 and 4 h post-injection. Food spilling was carefully checked to ensure the accuracy of actual consuming by each animal.
GTT and ITT
GTT was performed for overnight fasted mice that received i.p. injection of glucose (Sigma) (2 g/kg body weight). At the indicated times, blood was collected from the tail vein for measuring glucose levels with an Accu-Chek glucose meter (Roche Applied Science, Mannheim, Germany). For the insulin tolerance test, mice were fasted 6 h before i.p. injection of insulin (Lilly, Bad Homburg, Germany) (0.75 units/kg of body weight) and blood glucose levels were measured at the indicated times.
Immunofluorescence staining
Animals under anesthesia were perfused with 4% paraformaldehyde, then the brains were harvested, post-fixed in 4% paraformaldehyde, and infiltrated in 20–30% sucrose. Brain sections were made at 20-μm thickness using a cryostat at −20 °C. Fixed tissues were blocked with serum of appropriate species, penetrated with 0.3% Triton X-100, treated with primary antibodies including rabbit anti-HIF2α, goat anti-POMC (Novus Biologicals), and subsequently followed by a fluorescent reaction with Alexa Fluor 488 or 555 secondary antibody (Invitrogen). Naive IgGs of the appropriate species were included to provide negative controls. DAPI nuclear staining was employed to reveal the cells in sections. A confocal microscope was used to capture images.
Western blotting
Mice were sacrificed 90 min after a single injection of through pre-implanted cannula in the hypothalamic third ventricle. The hypothalamus was cut along the anterior border of the optic chiasm, the posterior border of the mammillary body, the upper border of anterior commissure, and lateral border halfway from the lateral sulcus in the ventral side of the brain. Tissues were homogenized, proteins were dissolved in a lysis buffer, and Western blotting was performed as previously described (8). Protein extracts were separated by SDS-PAGE and detected by immunoblotting using antibodies against anti-HIF2α, phospho-IRS-1, IRS-1, phospho-IRS-2, IRS-2, phospho-AKT, AKT, phospho-Foxo1, Foxo1, and β-actin (Cell Signaling), and then reacted with horseradish peroxidase-conjugated anti-rabbit or anti-mouse secondary antibodies (Pierce). Intensity of specific blots was analyzed using Image Lab 3.0 software (Bio-Rad).
Quantitative PCR
Total RNA isolation and quantitative PCR were performed as described previously (8). Briefly, mice were sacrificed 90 min after insulin injection via pre-implanted cannula in the hypothalamic third ventricle. Total RNA from tissues was isolated using TRIzol reagent (Life Technologies), treated with RNase-free DNase (Promega), and re-extracted with TRIzol. Then, cDNA was synthesized from 2 μg of RNA using the High Capacity cDNA Reverse Transcription Kit (Life Technologies). Quantitative PCR were performed in duplicate with 100 ng of cDNA using SYBR Green PCR master mix (Life Technologies) and specific primers. The primer sequences were: Pomc, 5′-atgccgagattctgctacagtcg-3′, and 5′-ttcatctccgttgccaggaaacac-3′; Nt, 5′-aatgtttgcagcctcataaataac-3′, and 5′-tgccaacaaggtcgtcatc-3′; Npy, 5′-cagaaaacgcccccagaa-3′, and 5′-aaaagtcgggagaacaagtttcatt-3′; Agrp, 5′-cggaggtgctagatccacaga-3′, and 5′-aggactcgtgcagccttacac-3′; cyclophilin, 5′-aaggtgaaagaaggcatgaac-3′; and 5′-agctgtccacagtcggaaatg-3′. The relative quantification of mRNA levels normalized to cyclophilin was calculated using the ΔΔCT method from CT values obtained from ABI SDS software.
Statistical analyses
All measured data were presented as mean ± S.E. Analysis of variance and Turkey' post hoc analyses were used for comparisons when experiments had more than two groups. Student's t test was used for comparisons when experiments involved only two groups. Software for performing statistics included Excel and GraphPad Prism. Statistical significance was set at p < 0.05.
Author contributions
Z. W. data curation; Z. W. formal analysis; Z. W., S. K., and D. C. investigation; Z. W. methodology; Z. W. and S. K. writing-original draft; D. C. conceptualization; D. C. supervision; D. C. funding acquisition; D. C. project administration; D. C. writing-review and editing.
Acknowledgments
We sincerely thank all Cai laboratory members and the Einstein Physiology Core for technical support.
This work was supported by National Institutes of Health Grants RO1 DK 099136 and AG 031774. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- HIF2α
- hypoxia-inducible factor-2α
- IR
- insulin receptor
- IRS-1
- insulin receptor substrate-1
- IRS-2
- insulin receptor substrate-2
- Akt
- protein kinase B
- Foxo1
- Forkhead box protein O-1
- POMC
- proopiomelanocortin
- HFD
- high-fat diet
- DIO
- diet-induced obesity
- NPY
- neuropeptide Y
- AgRP
- agouti-related protein
- NT
- neurotrophin
- GTT
- glucose tolerance test
- ITT
- insulin tolerance test
- GSK3
- glycogen synthase kinase 3
- DAPI
- 4′,6-diamidino-2-phenylindole.
References
- 1. Semenza G. L. (2012) Hypoxia-inducible factors in physiology and medicine. Cell 148, 399–408 10.1016/j.cell.2012.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keith B., and Simon M. C. (2007) Hypoxia-inducible factors, stem cells, and cancer. Cell 129, 465–472 10.1016/j.cell.2007.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Shi H. (2009) Hypoxia Inducible Factor 1 as a Therapeutic Target in Ischemic Stroke. Curr. Med. Chem. 16, 4593–4600 10.2174/092986709789760779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lai T. W., Lin S. Z., Lee H. T., Fan J. R., Hsu Y. H., Wang H. J., Yu Y. L., and Shyu W. C. (2012) HIF-1α binding to the Epac1 promoter recruits hematopoietic stem cells to the ischemic brain following stroke. J. Mol. Cell Biol. 4, 184–187 10.1093/jmcb/mjs009 [DOI] [PubMed] [Google Scholar]
- 5. Sharp F. R., Bergeron M., and Bernaudin M. (2001) Hypoxia-inducible factor in brain. Adv. Exp. Med. Biol. 502, 273–291 10.1007/978-1-4757-3401-0_18 [DOI] [PubMed] [Google Scholar]
- 6. Bento C. F., and Pereira P. (2011) Regulation of hypoxia-inducible factor 1 and the loss of the cellular response to hypoxia in diabetes. Diabetologia 54, 1946–1956 10.1007/s00125-011-2191-8 [DOI] [PubMed] [Google Scholar]
- 7. Isoe T., Makino Y., Mizumoto K., Sakagami H., Fujita Y., Honjo J., Takiyama Y., Itoh H., and Haneda M. (2010) High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Int. 78, 48–59 10.1038/ki.2010.99 [DOI] [PubMed] [Google Scholar]
- 8. Zhang H., Zhang G., Gonzalez F. J., Park S. M., and Cai D. (2011) Hypoxia-inducible factor directs POMC gene to mediate hypothalamic glucose sensing and energy balance regulation. Plos Biol. 9, e1001112 10.1371/journal.pbio.1001112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Treins C., Giorgetti-Peraldi S., Murdaca J., Semenza G. L., and Van Obberghen E. (2002) Insulin stimulates hypoxia-inducible factor 1 through a phosphatidylinositol 3-kinase/target of rapamycin-dependent signaling pathway. J. Biol. Chem. 277, 27975–27981 10.1074/jbc.M204152200 [DOI] [PubMed] [Google Scholar]
- 10. Short K. R., Bigelow M. L., Kahl J., Singh R., Coenen-Schimke J., Raghavakaimal S., and Nair K. S. (2005) Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. U.S.A. 102, 5618–5623 10.1073/pnas.0501559102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He X., Li Z., Tang X., Zhang L., Wang L., He Y., Jin T., and Yuan D. (2018) Age- and sex-related differences in body composition in healthy subjects aged 18 to 82 years. Medicine (Baltimore) 97, e11152 10.1097/MD.0000000000011152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang X., Zhang G., Zhang H., Karin M., Bai H., and Cai D. (2008) Hypothalamic IKKβ/NF-κB and ER stress link overnutrition to energy imbalance and obesity. Cell 135, 61–73 10.1016/j.cell.2008.07.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yan J., Zhang H., Yin Y., Li J., Tang Y., Purkayastha S., Li L., and Cai D. (2014) Obesity- and aging-induced excess of central transforming growth factor-β potentiates diabetic development via an RNA stress response. Nat. Med. 20, 1001–1008 10.1038/nm.3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang Y., Reichel J. M., Han C., Zuniga-Hertz J. P., and Cai D. (2017) Astrocytic process plasticity and IKKβ/NF-κB in central control of blood glucose: blood pressure, and body weight. Cell Metab. 25, 1091–1102.e1094 10.1016/j.cmet.2017.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Y., Kim M. S., Jia B., Yan J., Zuniga-Hertz J. P., Han C., and Cai D. (2017) Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 548, 52–57 10.1038/nature23282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang G., Li J., Purkayastha S., Tang Y., Zhang H., Yin Y., Li B., Liu G., and Cai D. (2013) Hypothalamic programming of systemic ageing involving IKK-β, NF-κB and GnRH. Nature 497, 211–216 10.1038/nature12143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Coll A. P., and Yeo G. S. (2013) The hypothalamus and metabolism: integrating signals to control energy and glucose homeostasis. Curr. Opin. Pharmacol. 13, 970–976 10.1016/j.coph.2013.09.010 [DOI] [PubMed] [Google Scholar]
- 18. Ropelle E. R., Pauli J. R., Prada P., Cintra D. E., Rocha G. Z., Moraes J. C., Frederico M. J., da Luz G., Pinho R. A., Carvalheira J. B. C., Velloso L. A., Saad M. A., and De Souza C. T. (2009) Inhibition of hypothalamic Foxo1 expression reduced food intake in diet-induced obesity rats. J. Physiol. (London) 587, 2341–2351 10.1113/jphysiol.2009.170050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sartorius T., Peter A., Heni M., Maetzler W., Fritsche A., Häring H. U., and Hennige A. M. (2015) The brain response to peripheral insulin declines with age: a contribution of the blood-brain barrier? Plos One 10, e0126804 10.1371/journal.pone.0126804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dietrich M. O., and Horvath T. L. (2013) Hypothalamic control of energy balance: insights into the role of synaptic plasticity. Trends Neurosci. 36, 65–73 10.1016/j.tins.2012.12.005 [DOI] [PubMed] [Google Scholar]
- 21. Brown L. M., Clegg D. J., Benoit S. C., and Woods S. C. (2006) Intraventricular insulin and leptin reduce food intake and body weight in C57BL/6J mice. Physiol. Behav. 89, 687–691 10.1016/j.physbeh.2006.08.008 [DOI] [PubMed] [Google Scholar]
- 22. Clegg D. J., Gotoh K., Kemp C., Wortman M. D., Benoit S. C., Brown L. M., D'Alessio D., Tso P., Seeley R. J., and Woods S. C. (2011) Consumption of a high-fat diet induces central insulin resistance independent of adiposity. Physiol. Behav. 103, 10–16 10.1016/j.physbeh.2011.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hill J. W., Elias C. F., Fukuda M., Williams K. W., Berglund E. D., Holland W. L., Cho Y. R., Chuang J. C., Xu Y., Choi M., Lauzon D., Lee C. E., Coppari R., Richardson J. A., Zigman J. M., et al. (2010) Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 11, 286–297 10.1016/j.cmet.2010.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Obici S., Feng Z., Karkanias G., Baskin D. G., and Rossetti L. (2002) Decreasing hypothalamic insulin receptors causes hyperphagia and insulin resistance in rats. Nat. Neurosci. 5, 566–572 10.1038/nn0602-861 [DOI] [PubMed] [Google Scholar]
- 25. Grillo C. A., Tamashiro K. L., Piroli G. G., Melhorn S., Gass J. T., Newsom R. J., Reznikov L. R., Smith A., Wilson S. P., Sakai R. R., and Reagan L. P. (2007) Lentivirus-mediated downregulation of hypothalamic insulin receptor expression. Physiol. Behav. 92, 691–701 10.1016/j.physbeh.2007.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Woods S. C., Lotter E. C., McKay L. D., and Porte D. Jr. (1979) Chronic intracerebroventricular infusion of insulin reduces food intake and body weight of baboons. Nature 282, 503–505 10.1038/282503a0 [DOI] [PubMed] [Google Scholar]
- 27. Kim M. S., Pak Y. K., Jang P. G., Namkoong C., Choi Y. S., Won J. C., Kim K. S., Kim S. W., Kim H. S., Park J. Y., Kim Y. B., and Lee K. U. (2006) Role of hypothalamic Foxo1 in the regulation of food intake and energy homeostasis. Nat. Neurosci. 9, 901–906 10.1038/nn1731 [DOI] [PubMed] [Google Scholar]
- 28. Garcia-San Frutos M., Fernández-Agulló T., De Solís A. J., Andrés A., Arribas C., Carrascosa J. M., and Ros M. (2007) Impaired central insulin response in aged Wistar rats: role of adiposity. Endocrinology 148, 5238–5247 10.1210/en.2007-0543 [DOI] [PubMed] [Google Scholar]
- 29. Mercer A. J., Hentges S. T., Meshul C. K., and Low M. J. (2013) Unraveling the central proopiomelanocortin neural circuits. Front. Neurosci. 7, 19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Morton G. J., Cummings D. E., Baskin D. G., Barsh G. S., and Schwartz M. W. (2006) Central nervous system control of food intake and body weight. Nature 443, 289–295 10.1038/nature05026 [DOI] [PubMed] [Google Scholar]
- 31. Millington G. W. (2007) The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr. Metab. 4, 18 10.1186/1743-7075-4-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cone R. D. (2005) Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 8, 571–578 10.1038/nn1455 [DOI] [PubMed] [Google Scholar]
- 33. Krude H., Biebermann H., Schnabel D., Tansek M. Z., Theunissen P., Mullis P. E., and Grüters A. (2003) Obesity due to proopiomelanocortin deficiency: three new cases and treatment trials with thyroid hormone and ACTH4–10. J. Clin. Endocrinol. Metab. 88, 4633–4640 10.1210/jc.2003-030502 [DOI] [PubMed] [Google Scholar]
- 34. Yaswen L., Diehl N., Brennan M. B., and Hochgeschwender U. (1999) Obesity in the mouse model of proopiomelanocortin deficiency responds to peripheral melanocortin. Nat. Med. 5, 1066–1070 10.1038/12506 [DOI] [PubMed] [Google Scholar]
- 35. Krude H., Biebermann H., Luck W., Horn R., Brabant G., and Grüters A. (1998) Severe early-onset obesity, adrenal insufficiency and red hair pigmentation caused by POMC mutations in humans. Nat. Genet. 19, 155–157 10.1038/509 [DOI] [PubMed] [Google Scholar]
- 36. Woods S. C., Schwartz M. W., Baskin D. G., and Seeley R. J. (2000) Food intake and the regulation of body weight. Annu. Rev. Psychol. 51, 255–277 10.1146/annurev.psych.51.1.255 [DOI] [PubMed] [Google Scholar]