

Abstract
Compared to enhancers, silencers are notably difficult to identify and validate experimentally. In search for human silencers, we utilized H3K27me3-DNase I hypersensitive site (DHS) peaks with tissue specificity negatively correlated with the expression of nearby genes across 25 diverse cell lines. These regions are predicted to be silencers since they are physically linked, using Hi-C loops, or associated, using expression quantitative trait loci (eQTL) results, with a decrease in gene expression much more frequently than general H3K27me3-DHSs. Also, these regions are enriched for the binding sites of transcriptional repressors (such as CTCF, MECOM, SMAD4, and SNAI3) and depleted of the binding sites of transcriptional activators. Using sequence signatures of these regions, we constructed a computational model and predicted approximately 10,000 additional silencers per cell line and demonstrated that the majority of genes linked to these silencers are expressed at a decreased level. Furthermore, single nucleotide polymorphisms (SNPs) in predicted silencers are significantly associated with disease phenotypes. Finally, our results show that silencers commonly interact with enhancers to affect the transcriptional dynamics of tissue-specific genes and to facilitate fine-tuning of transcription in the human genome.
Regulatory elements (REs) are generally categorized into two major classes based on their impact on the transcription of their target genes: positive regulators, such as promoters and enhancers; and negative regulators, such as silencers and insulators. Precisely unveiling the maps of these REs is an essential step toward understanding gene regulatory mechanisms (Maston et al. 2006).
High-throughput sequencing techniques have provided a wealth of genome-wide landscapes of various epigenetic modifications and transcription factor (TF) binding sites. These data, together with the increasing knowledge of histone modification patterns of functional REs, facilitates the identification of positive and negative REs (Ernst and Kellis 2012; Hoffman et al. 2013). Despite these advances, the studies of positive REs largely outnumber those of negative REs (Petrykowska et al. 2008). The investigative studies of negative REs, especially silencers, have lagged far behind those of positive REs partially due to the experimental difficulty in distinguishing negative regulators from either repressed positive regulators or neutral DNA (Gaszner and Felsenfeld 2006; Maston et al. 2006; Qi et al. 2015).
The methylation of H3K9, H3K27, or H4K20 is commonly associated with transcriptional down-regulation (Mozzetta et al. 2015). In particular, H3K9me3 features heterochromatin formation of chromosomal regions with tandem repeat structures (Kim and Kim 2012). H3K27me3, which is catalyzed and maintained by Polycomb repressive complexes (PRCs), directly inhibits gene transcription by impeding or poising the occupancy of RNA polymerase II (van Kruijsbergen et al. 2015). Transcriptional dynamics during development and disease initiation have been attributed to H3K27me3 (Simon and Kingston 2009; Sexton et al. 2012; Jiang et al. 2013; Pinello et al. 2014; Laprell et al. 2017). Notwithstanding the fact that H3K27me3 is well-known as a modification of silencers, the regulatory contribution of H3K27me3 has been asserted to be context-dependent (Young et al. 2011; Lv et al. 2016). H3K27me3 and H3K4me1, simultaneously recruited by Polycomb repressive complex 2 (PRC2), collaborate to precisely fine-tune the expression of targeting genes in pluripotent cells (Mikkelsen et al. 2007; Schwartz and Pirrotta 2008). We thus argued that H3K27me3 is a descriptive feature of silencers, but it is not a determinant factor of silencers on its own.
Results
H3K27me3 is not a specific mark and frequently co-occurs with other histone marks
In this study, we focused on open chromatin regions that are accessible for transcriptional activity and can be characterized by deoxyribonuclease I (DNase I) hypersensitive sites (DHSs). We overlapped DNase-seq peaks and H3K27me3 chromatin immunoprecipitation-high throughput sequencing (ChIP-seq) peaks reported by the Roadmap Epigenomics Mapping Consortium (Roadmap Epigenomics Consortium et al. 2015) and built genome-wide maps of H3K27me3-DHSs in a cohort of 25 distinct human cell lines, including stem cells (H1), normal human lung fibroblast primary cells (NHLF), and cancer cells, such as hepatocellular carcinoma cells (HepG2) and myelogenous leukemia cells (K562) (Supplemental Fig. S1). After excluding promoter regions, we obtained a total of 481,101 distal H3K27me3-DHSs, with an average of 46,457 per cell line.
Approximately 75% of the H3K27me3-DHSs coincided with the well-known activator-associated histone modifications H3K4me1, H3K4me3, or H3K27ac. Similarly, 15% of the regulatory activators, identified using DHS peaks with H3K4me1, H3K4me3, or H3K27ac signals, displayed H3K27me3 signals, while only 7% of these regions have been annotated as bivalent enhancers by the Chromatin Hidden Markov Model (ChromHMM) (Roadmap Epigenomics Consortium et al. 2015). Overall, the H3K27me3 mark appears to be characteristic of different groups of REs, such as silencers and poised, bivalent, and often active enhancers. The intensive co-occurrence of H3K27me3 with activator-associated histone marks suggests that H3K27me3 alone is inadequate to accurately identify silencers.
Silencer identification using the correlation between H3K27me3-DHS activity and gene expression
We herein propose a computational framework for building genome-wide silencer maps. In this framework, focusing on the DHSs overlapping H3K27me3 peaks (H3K27me3-DHSs), we explore their correlation with the expression of the nearby genes across tissues and then the sequence patterns of H3K27me3-DHSs to build genome-wide maps of silencers (Supplemental Fig. S2).
We focused on the H3K27me3-DHSs present in at least three cell lines, which account for about 30% of all H3K27me3-DHS peaks (Supplemental Fig. S3), as cross-tissue profiles of these marks and gene expression levels are required to evaluate the tissue-specificity correlation between these H3K27me3-DHSs and their neighboring genes. Based on these correlation estimates, we categorized these H3K27me3-DHSs into three groups: significantly negatively correlated (negCOR); significantly positively correlated (posCOR); and uncorrelated (unCOR) with the expression of neighboring genes (see Methods; Fig. 1A). In total, we identified 4574 negCORs and 4451 posCORs, with an average of 1334 negCORs and 1364 posCORs per cell line (Supplemental Fig. S4). For example, myocyte enhancer factor 2A (MEF2A), a landmark gene in muscle cells (Narlikar et al. 2010; Liu et al. 2014), hosts three negCORs in its neighborhood that are present in a broad spectrum of 13 nonmuscle cell lines, including stem, progenitor, pancreas, skin, and liver cells (Fig. 1B). Similarly, the negCOR close to PPARG coactivator 1 alpha (PPARGC1A) is not present in the tissues where PPARGC1 is highly expressed, such as muscle fiber and liver and pancreas (Supplemental Fig. S5; Lin et al. 2005).
Figure 1.

Silencer identification based on correlation of cross-tissue profiles of gene expression and H3K27me3-DHS activity. (A) Multi-tissue H3K27me3-DHSs were categorized based on their correlation with the expression of proximal genes into three groups: positively correlated (posCOR); negatively correlated (negCOR); and uncorrelated (unCOR). negCORs were verified as silencers. (B) Three negCOR silencers in the locus of MEF2A.
The negative correlation between negCORs and their neighboring genes does not necessarily indicate that negCORs function as silencers. An alternative hypothesis would characterize negCORs as enhancers acting distantly on genes other than the flanking ones. To examine this hypothesis, we identified target genes of negCORs using Hi-C data available for seven cell lines (see Supplemental “Hi-C data”; Rao et al. 2014). We analyzed the normalized expression levels of genes linked to different classes of REs by Hi-C data. As compared to all genes and those linked to DHSs, the genes linked only to unCOR H3K27me3-DHSs displayed an insignificant tendency toward a decrease of gene expression (Wilcoxon rank-sum test P = 0.08). The genes linked to negCORs were significantly lowly expressed compared to unCOR genes, which was independent of whether genes harbor negCORs in their neighborhood (P = 4 × 10−12) (shown as “proximal negCORs” in Fig. 2A) or genes are targeted by negCORs only through Hi-C loops while skipping intermediate gene(s) (P = 3 × 10−24) (termed as “distal negCORs” in Fig. 2A). While the expressional decrease of proximal negCOR genes is expected due to the negatively correlated tissue specificity between negCORs and their neighboring genes, the expressional decrease of distal negCOR genes is an independent verification of the silencing activity of negCORs. Also, the genes targeted by the ChromHMM “bivalent elements” (see Supplemental “Bivalent elements defined by ChromHMM”), but not by either negCORs or posCORs, displayed no significant expression differences from the unCOR genes (P = 0.5), indicating that the identified negCORs yield transcriptional impact different from bivalent elements. Together, these observations demonstrate that a large fraction of the identified negCORs represent silencers. Therefore, we will refer to negCORs as negCOR silencers or simply silencers, although we note that a fraction of the predicted silencers might not act as silencers in vivo.
Figure 2.

Expression of genes linked to different classes of DNA sequences by Hi-C data (A) and eQTL associations (B). Background consists of all genes assayed in RNA-seq experiments. Proximal negCOR genes host negCORs in their loci, while distal negCOR genes are linked by Hi-C/eQTL to the negCOR(s) separated from them by at least one intermediate gene. Expression of the distal negCOR genes represents an independent test on the effect of negCORs. Bivalent represents the genes associated with bivalent elements reported by ChromHMM but not with either negCOR silencers or posCORs. The number below each x-axis label is the number of the associated genes. (C) DNase-seq signal intensity of negCOR silencers (red) and posCORs (gray) across cell lines. (D) Motif-based TFBS enrichment in negCOR silencers. PosCORs were used as controls for enrichment analyses. The averages and standard deviations of the enrichment values are represented by the red diamonds and the gray lines flanking the red diamonds, respectively. The blue bar identifies TFBSs ubiquitously enriched in negCOR silencers across cell lines. (E) ChIP-seq TFBS density (gray bars) and enrichment (diamonds) in K562 and GM12878 cell lines.
We further validated the silencers using eQTL associations. Through measuring the correlation between genotypes and transcriptional levels, eQTL studies pair genomic loci, the majority of which are noncoding, with their highly correlated genes, predicting that these genomic loci regulate the transcription of the paired genes in a way of either cis-acting or trans-acting (Grundberg et al. 2012; Albert and Kruglyak 2015). We used eQTL associations reported by the Genotype-Tissue Expression (GTEx) project (see Supplemental “GTEx eQTLs”; The GTEx Consortium 2015). The eQTL-associated genes of silencers had significantly lower expression (P < 10−6) (Fig. 2B), while the genes associated with unCORs or ChromHMM bivalent elements showed no significant trend toward either increase or decrease of gene expression (P = 0.57 and 0.33, respectively) (Fig. 2B). Consistent with the results based on Hi-C connections, these findings again advocate for the effectiveness of the proposed framework in silencer identification.
In addition, we observed that the genes associated with H3K27me3-DHS posCORs were highly expressed (Hi-C: P = 2 × 10−5; eQTL: P = 2 × 10−19) (Fig. 2A; Fig. 2B, respectively), indicating that posCORs represent either active or, more likely, poised enhancers, as they contain H3K27me3 marks but not silencers. These findings further support the functional complexity of the H3K27me3-DHSs and the competency of our approach to distinguish silencers from general H3K27me3-DHSs. Despite opposite transcriptional influence, silencers and posCORs feature multiple similarities. For example, silencers and posCORs exhibit comparable signal intensity of DHS and H3K27me3 (Fig. 2C; Supplemental Fig. S6), similar GC content, repeat density, distance to the nearest transcriptional start sites, and similar sequence length across all cell lines (Supplemental Figs. S7, S8; Supplemental Table S1). PosCORs were thus used as controls in further analysis.
Silencers feature abundant binding sites of transcriptional repressors
TF occupancy and histone modifications along DNA sequences constitute two layers of transcriptional regulation machinery. Since H3K27me3 alone is not able to discern silencers from other H3K27me3-DHSs (Supplemental Fig. S6), we speculated that TF binding plays a substantial role in establishing the biological function of different H3K27me3-DHS peaks. To identify TFs specific to silencers, we mapped putative TF binding sites (TFBSs) using 1388 distinct TF binding motifs collected from published resources and evaluated enrichment of these putative TFBSs in silencers from different cell lines using posCORs as background (see Methods). The binding sites of well-known ubiquitous repressors were enriched in silencers across most cell lines, as highlighted by the blue bar in Figure 2D. These repressors included MECOM (Cattaneo et al. 2007) and its partners from SMAD family of proteins (Bard-Chapeau et al. 2012), FOXG1 (Roth et al. 2010), CTCF (Holwerda and de Laat 2013; Qi et al. 2015), TCF (Bienz 1998), and SOX21 (Ohba et al. 2004). In addition to the cross-tissue enrichment of several repressor TFBSs, we observed tissue-specific TFBS enrichment in silencers (Fig. 2D). TFs, such as the STAT family of proteins (Schroder et al. 2002), EHF (Tugores et al. 2001), TFAP2A (Scibetta et al. 2010), NKX6-1 (Iype et al. 2004), and E2F4 (Lee et al. 2011), display binding site enrichment in selected cell lines only and have dual transcription roles in different contexts.
We also explored TFBSs detected in ChIP-seq experiments in which the binding events are captured in vivo. In K562, silencers displayed an elevated TFBS density of CTCF and its working partners SMC3 and RAD21 (fold enrichment > 1.2, binomial test P < 10−100) (Fig. 2E), which is in line with the binding motif analysis (Supplemental Fig. S9). Also, the enrichment of the ChIP-seq peaks of RCOR1 (a REST corepressor) further supports the suppressive influence of silencers. In GM12878, silencers were enriched for the ChIP-seq TFBSs of well-known repressors, such as PAX5 (McManus et al. 2011; Somasundaram et al. 2015), YY1 (Kleiman et al. 2016), and RUNX3 (Spender et al. 2005), not to mention CTCF and its interacting partner RAD21 (fold enrichment > 1.2, binomial test P < 10−100) (Fig. 2E). In HepG2 and the cervical carcinoma cell line HeLa S3, elevated AP-1 binding was observed in silencers (Supplemental Figs. S10, S11). AP-1 enrichment in the tested cancer cells, namely K562, HepG2, and HeLa S3, is in accordance with the suppressive function of activator protein 1 (AP-1) during carcinogenesis (Shaulian and Karin 2002; Fan et al. 2016).
In addition, the depletion of EP300 ChIP-seq peaks in silencers was seen across all tested cells. Other well-known tissue-specific activators also displayed notable binding depletion in the silencers, suggesting that negCORs perform a function potentially opposite to activators. For example, BCL3 and NFATC1 were depleted on the silencer sequences in GM12878, HNF4A and HNF4G in HepG2, ELK4 and ELK1 in HeLa S3, and IRF1 in K562. The TFBS analysis of silencers identifies putative regulatory mechanisms at the foundation of silencer activity across different cell lines and provides directions for follow-up biochemical analyses of these sequences.
A machine learning model can accurately identify silencer sequences de novo
For the identification of silencers, we had to focus on 30% of H3K27me3-DHSs peaks only (Supplemental Fig. S3). To expand the reach of our silencer detection framework, we decided to capitalize on specific TFBS patterns of silencers (Fig. 2C,D). We trained a linear support vector machine (SVM) classifier for each cell line to distinguish silencers from posCORs using TFBS maps coupled with epigenetic and gene expression profiles, and then applied these SVM models to all H3K27me3-DHSs (excluding negCOR silencers and posCOR elements) to identify silencers beyond negCOR silencers (see Methods).
The classification performance of our SVM models varied across different cell lines, with the area under the curve (AUC) ranging from 0.62 to 0.89 in precision-recall analysis (PR) (Fig. 3A) and from 0.71 to 0.82 in receiver operating characteristic measurement (ROC) (Fig. 3A; see Supplemental “Performance evaluation of SVM classification”). For instance, in K562, the SVM model performed with AUC PR = 0.89 and AUC ROC = 0.80 on average (Fig. 3B). In each cell line, we applied the SVM classifiers to score H3K27me3-DHSs and marked the top-scoring ones as potential silencers by keeping the false positive rate of CV under 0.1 (see Supplemental “Performance evaluation of SVM classification”). These predicted silencers were named SVM silencers to distinguish them from negCOR silencers. We predicted, on average, 9194 SVM silencers per cell line, which are eight times more than negCOR counterparts (Supplemental Table S2). The identified SVM silencers show high tissue specificity, with 70% of them being present in a single cell line only (Supplemental Fig. S12).
Figure 3.

SVM classification performance. (A) AUC-PR and AUC-ROC of the classifiers built to discriminate negCOR silencers from posCORs, which were measured under a fivefold cross-validation. (B) PR and ROC curves in K562 cell line. Expression of genes linked to SVM silencers by Hi-C data (C) and eQTL associations (D).
To evaluate SVM silencers, we studied their transcriptional impact. Similar to negCOR silencers, SVM silencers were used to categorize genes associated with SVM silencers by Hi-C links into two groups—genes connected by Hi-C links only to an SVM silencer separated from them by one or more intermediate genes (termed “distal SVM” genes in Fig. 3C), and genes connected by Hi-C links to at least one of the SVM silencers residing in their locus (represented by “proximal SVM” genes in Fig. 3C). Genes targeted by SVM silencers according to Hi-C connections are significantly lowly expressed, regardless of whether they were “proximal SVM” or “distal SVM” genes (Wilcoxon rank-sum test, proximal: P = 6 × 10−61; distal: P = 9 × 10−35) (Fig. 3C). Distal SVM genes provided an independent validation of SVM silencers as the expression of these genes has no contribution to silencer prediction. Sixty-five percent of distal SVM genes are lowly expressed, with a normalized expressional level of <0, which is significantly higher than expected (65% of distal SVM genes versus 50% of all genes, binomial test P < 10−100) and thus suggests a significant repressive effect exerted by SVM silencers. Similarly, the eQTL-associated genes of SVM silencers were strongly lowly expressed in both proximal (P = 2 × 10−22) and distal cases (P = 4 × 10−5) (Fig. 3D). These findings, reminiscent of those of negCOR silencers, confirm the reliability of the newly identified silencers, which significantly expand the landscape of silencer elements.
In support of our model's ability to predict silencers, a large fraction of the predictions features the classical polycomb signature of repressive activity. In particular, in addition to the overlap with the H3K27me3 regions used as a foundation for the SVM scan, 54% of the predicted SVM silencers contain a binding site of enhancer of zeste homolog II (EZH2) and 13% of them contain a SUZ12 binding site, two enzyme components of the Polycomb repressive complex 2 (Supplemental Table S3). In addition, histone modifications H3K9me3 and H4K20me1 are well-known to be associated with the repression of gene regulation (Mozzetta et al. 2015). Predicted SVM silencers more frequently contain these histone modifications than background DHSs (Supplemental Fig. S13), further advocating for the repressive function of SVM silencers. On the other hand, SVM silencers and general H3K27me3-DHSs show similar enrichment for H3K9me3 and H4K20me1 modifications (Supplemental Fig. S13), suggesting that the well-known repressive histone marks (including H3K27me3, H3K9me3, and H4K20me1) are incapable of accurately identifying silencers on their own due to a complicated interaction among transcriptional regulators (including histone modifications and TFs). Additional analysis (such as the proposed model) is necessary to distinguish potential silencers from general DHSs carrying repressive histone marks.
It is important to note that SVM silencers are predicted learning from negCOR silencers. Thus, negCOR silencers represent an essential step toward building a larger map of silencers in the human genome. Also, as SVM classifiers are not perfect, negCOR silencers are more likely to accurately predict in vivo silencers than their more abundant SVM silencer counterparts.
Experimental validation of silencer predictions
To address the impact of the predicted silencers on the activity of a nearby gene experimentally, we placed 10 negCOR silencers (S1…S10) and four H3K27me3-DHS regions displaying no significant correlation to the flanking gene expression profiles (H1…H4) upstream of an enhancer + SV40 promoter + luciferase reporter gene construct in K562 cells (see Methods). All 14 tested sequences (S1…S10 and H1…H4) showed a significant decrease in the reporter gene activity (Fig. 4; Supplemental Table S4). We observed a decrease in the reporter gene activity for the H1…H4 sequences, which were not classified as silencers. Although this decrease is significant as compared to the reporter gene activity driven by the naked enhancer, it is at the level of other previously reported controls (Petrykowska et al. 2008). While our method is not rejecting a possible silencer function of these sequences and some of them might be acting as silencers, we used H1…H4 sequences as a strict control while screening for predictions, which would result in reporter gene activity lower than the activity associated with H1…H4 sequences. Five out of 10 predicted silencers (S1, S3, S7, S9, and S10) resulted in the decrease of reporter gene activity significantly stronger than at least three out four of the H1…H4 sequences (Student's t-test, P < 0.05) (Fig. 4). As a group, the S1…S10 predicted silencers drove the reporter gene expression 3.5-fold lower than the enhancer + promoter construct and 1.4-fold lower than the H1…H4 set. As in vitro enhancer validation is known to result in ∼50% detection of enhancer activity in a selected cell line (The ENCODE Project Consortium 2012; Sanyal et al. 2012), our 40%–50% validation of silencer activity in vitro using very strict controls is a strong indicator of the true silencing function of the predicted silencers.
Figure 4.
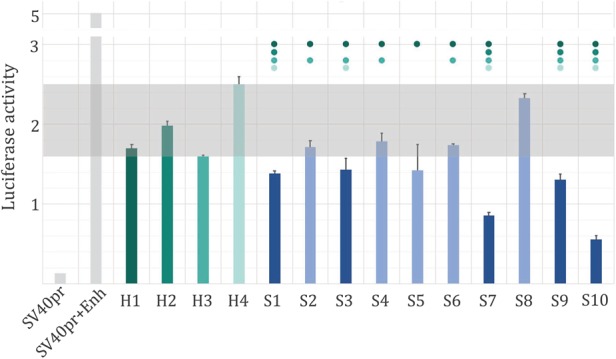
Predicted silencer activity in K562 cells. Ten predicted silencers (S1…S10) and four H3K27me3-DHS regions (H1…H4) were placed upstream of an SV40pr promoter and enhancer construct (SV40pr + Enh), and the activity of the luciferase reporter gene has been measured in K562 cells (see Methods). All predicted silencers resulted in a significant decrease of the enhancer-driven gene expression (blue bars), while five predicted silencers (represented by dark blue bars) reduced the activity of the reporter gene to levels lower than 3/4 H3K27me3-DHS regions. The dots above bars indicate that the reporter gene activity of a silencer was significantly lower (Student's t-test, P < 0.05) than that of the corresponding control with the matching color bar. The gray rectangle indicates the luciferase activity range of H1…H4.
Also, the results of massively parallel report assays (MPRAs), in which the impact of REs was measured using the expression of reporter genes (Melnikov et al. 2012), were used to test the function of predicted silencers. Based on the results of Sharpr-MPRA (Ernst et al. 2016), the predicted negCORs and SVM silencers are associated with negative Sharpr-MPRA scores, suggesting a repressive impact of these elements (Ernst et al. 2016). Also, the predicted silencers display a stronger decrease in the level of gene expression than the H3K27me3-DHSs regions not predicted as silencers (Supplemental Fig. S15). This independent test further supports the predicted silencers having a repressive function on the activity of nearby genes.
To counterbalance the sparsity of chromatin interaction data, we assumed that H3K27me3-DHSs act on their neighboring genes, rather than on distal genes, as a part of our model. This assumption resulted in an underprediction of silencers and can partially explain why some of the H1…H4 sequences might have been acting as silencers and thus resulting in a lower reporter gene expression (Fig. 4). Those H3K27me3-DHSs that only target distal genes would not be picked up by our model, while using additional information (such as chromatin interaction data across cell lines) in future studies could reduce the number of false negative predictions and result in a larger set of predicted silencers.
Silencers form clusters in tissue-specific gene loci
We next examined the genomic distribution of silencers. Following the widely used proximity rule, we assigned silencers and enhancers (marked by the DHSs overlapping both H3K27ac and H3K4me1) to their most proximal genes (see Methods). The genes next to the silencers displayed a higher value of tau (τ) than their enhancer counterparts (Wilcoxon rank-sum test, P = 2 × 10−186) (Fig. 5A), suggesting that the predicted silencers tend to be associated with tissue-specific genes, since a large τ indicates high tissue specificity (Yanai et al. 2005). To summarize, the tissue-specific genes (τ > 0.9) hosted 13.8% of silencers but only 10.7% of enhancers (binomial test, P < 10−10) (Fig. 5B). On the other hand, the housekeeping genes (τ < 0.6) hosted 15.9% of the silencers and 19.2% of the enhancers (P < 10−10). This suggests that silencers determine the identity of a cell through suppressing genes highly expressed in other cell lines. Also, we examined the tissue specificity of the genes targeted by enhancers and silencers via Hi-C loops (Rao et al. 2014). As compared with enhancers, silencers more frequently link to tissue-specific genes (13.8% of silencers versus 11.2% of enhancers, P = 1.2 × 10−6) and avert from the housekeeping genes (14.3% of silencers versus 17.2% of enhancers, P = 1.7 × 10−4) (Supplemental Fig. S15). All these observations advocate for the essential role of silencers in establishing a unique transcriptional profile of a cell.
Figure 5.

Distribution of silencers. τ of genes flanking silencers and enhancers (A) and fraction of tissue-specific (τ > 0.9) and housekeeping (τ < 0.6) gene loci harboring silencers or enhancers (B). (C) Clustering of silencers. Background clustering was established using randomly selected H3K27me3-DHSs peaks matching the number of silencers. The dashed lines indicate the average fraction of clustered elements across all cell lines (red = silencers, gray = background). (D) Association of silencers and silencer clusters with tissue-specific genes. (E) Enrichment of clustered silencers in the loci of superenhancers (SEs). Enhancers were used as a background reference.
Since regulatory cluster formation is well-known for enhancers (Parker et al. 2013; Zhou et al. 2014), we investigated the clustering tendency of silencers by detecting significant silencer cluster formation in gene loci with more than one silencer (binomial test P < 10−6) (see Methods). On average, these silencer clusters accounted for 28% of all silencers across tested cell lines (Fig. 5C), which is twofold higher than expected if silencers were randomly distributed among all H3K27me3-DHSs (P < 10−10). Silencer cluster formation might be beneficial for functional backup or battering (Spitz and Furlong 2012). This cluster formation might also provide evolutionary stability to the repression of gene expression in a specific cell where the activity of particular genes might be detrimental to the fitness of the species.
Relative to all H3K27me3-DHSs, as reported above, silencers are more likely to reside in the loci of tissue-specific genes in the tested tissues (binomial test, P < 10−20) (Fig. 5D). This effect greatly increased only when clustered silencers were considered (14.6% of silencer genes versus 25.8% of clustered-silencer genes, P < 10−20) (Fig. 5D). This trend is significant in almost all differentiated tissues. In HepG2, for instance, 28.6% of the loci hosting silencer clusters are tissue-specific, which is significantly higher than the background expectation (29% of clustered silencers versus 14.5% of all silencers versus 13.6% of all H3K27me3-DHSs, P < 10−20) (Fig. 5D). The enrichment of silencer clusters in the loci of tissue-specific genes is lower in the case of stem and progenitor cells. For instance, in H1-derived neuronal progenitor cells, 16% of the clustered silencer loci were tissue-specific. To address a possible bias caused by the locus length discrepancy (on average, the loci of tissue-specific genes are longer than those of housekeeping genes) (Supplemental Fig. S16), we compared silencers with the background, which was generated through randomly selecting H3K27me3-DHSs matching the corresponding silencers, and consistently observed a significant association of silencers from silencer clusters with tissue-specific genes, especially in differentiated cells (Supplemental Fig. S17).
Silencer clusters act alongside superenhancers
Superenhancers, i.e., groups of multiple enhancers, have been reported to frequently occur in the loci of tissue-specific genes (Lovén et al. 2013; Pott and Lieb 2015). The strong preference of clustered silencers toward tissue-specific genes, as reported above (Fig. 6C), prompted us to examine co-occurrence of silencers with superenhancers. We observed a depletion of silencers in the loci hosting active superenhancers. Such depletion became even more pronounced when only the clustered silencers were considered (binomial test, P < 10−5) (Fig. 5E). At first, this antagonistic presence of clustered silencers and superenhancers appears counterintuitive given the individual enrichment of each of these two groups of REs in the loci of tissue-specific genes. However, if we assume that the activity of silencers/silencer clusters and superenhancers is compartmentalized to different tissue specificities, the silencer clusters and superenhancers might be acting on the same genes but in a mutually exclusive way, i.e., the activity of one group determines the inactivity of the other group. Indeed, silencer clusters were significantly enriched in the genomic regions hosting superenhancers active in a cell line(s) other than the inquiry one (binomial test, P < 10−5) (Fig. 5E). This suggests a regulatory model for the genes controlled by superenhancers, which requires strong transcriptional deactivation in tissues or cell types where superenhancers are absent. We can hypothesize that this deactivation might be taking place by silencer clusters acting directly on superenhancers to deactivate key gene regulators instead of acting directly on promoters of these genes, as a direct competition with superenhancers might not be feasible. However, we would like to note that either a validation or rejection of this hypothesis would have to be performed by a follow-up biochemical study.
Figure 6.

Silencers in TF gene loci. Gene loci are stratified into three groups: silencer-rich loci; enhancer-rich loci; and silencer-enhancer loci (see Methods). The fraction of loci (A) and the fraction of TF genes (B) are binned by the level of gene expression (C). Green asterisks depict significant differences between silencer-enhancer loci and silencer-rich loci, while red asterisks indicate significant differences of TF fraction among silencer-enhancer loci across gene groups. Association of silencers and enhancers with eQTL SNPs (D) and GWAS SNPs (E).
Regulation of TF genes is fine-tuned by silencers and enhancers
To gain further insight into the cooperation between silencers and enhancers, we focused on gene loci hosting a mixture of enhancers and silencers (named silencer-enhancer loci) (see Methods). As gene expression increases, the fraction of silencer-enhancer loci among all the loci hosting at least one silencer or enhancer decreases, while the enhancer-rich loci gain prominence (Fig. 6A,C). About 33% of all loci are silencer-enhancer loci, and this silencer-enhancer loci fraction diminishes to 13.1% when the genes having normalized expression >2 are considered (binomial test, P < 10−100). Nevertheless, there is a notable fraction of highly expressed genes that require silencers for their proper regulation. Highly expressed silencer-enhancer genes were significantly enriched for TFs, including canonical TFs and transcriptional cofactors (Schmeier et al. 2017; see Supplemental “Human TFs”). Among the silencer-enhancer genes with normalized expression of >1, 18% are TFs, which is 1.4 times enhancer-rich counterparts (binomial test, P < 10−5) (Fig. 6B). The TF enrichment among silencer-enhancer genes increases with the increase of gene expression (P < 0.05) (Fig. 6B), highlighting the role of silencers collaborating with enhancers in the regulation of TFs. Collectively, highly expressed TFs, which are the backbone of the transcriptional regulation system (Vaquerizas et al. 2009), tend to be regulated simultaneously by enhancers and silencers. The opposite effect of enhancers and silencers enables a fine-tuning of these TFs and provides great sensitivity and robustness to the entire regulatory circuitry (Daniel et al. 2014).
Finally, we explored Hi-C connections to examine silencer-enhancer genes, although Hi-C connections are available only for a part of the enhancers and silencers. TFs are overrepresented among the highly expressed silencer-enhancer genes (binomial test, P < 0.05) (Supplemental Fig. S18), which is in line with the results presented in Figure 6B.
Silencer mutations likely have phenotypic consequences
We superimposed silencers onto the SNPs associated with phenotypic effects. We collected eQTL SNPs from seven tissues matching our studied cell lines from the GTEx project (The GTEx Consortium 2015; see Supplemental “GTEx eQTLs”). Silencers, as well as enhancers, were enriched for eQTL SNPs compared to general H3K27me3-DHSs (binomial test, P < 10−100) (Fig. 6D). Fifteen percent of the identified silencers carry at least one eQTL SNP, which is significantly higher than their background counterparts (P < 10−12, H3K27me3-DHSs and DHSs). Next, we turned to GWAS SNPs significantly associated with phenotypic traits or disorders. In this study, we used 324,454 GWAS SNPs that are associated with 1106 phenotypes, from body mass index to glioblastoma (see Supplemental “GWAS SNPs”). Silencers, as well as enhancers, are more likely to carry GWAS SNPs than the H3K27me3 background (P < 10−12) (Fig. 6E). Approximately 6.7% of silencers harbor at least one GWAS SNP, which is significantly higher than the background (P < 10−12) (Fig. 6E), although less than that of enhancers (P = 8 × 10−8). The enrichment of GTEx and GWAS SNPs in silencers, which is comparable to that of enhancers, suggests a role of silencers in human diseases and disorders that should not be overshadowed by enhancer studies.
Discussion
Histone modifications have been an extremely powerful approach for investigating the cell-type–specific regulatory landscape of the human and other genomes. Hundreds of thousands of tissue-specific enhancers have been successfully identified (Roadmap Epigenomics Consortium et al. 2015). However, it appears to be much more difficult to predict silencers. One of the challenges is the dual nature of the repressive H3K27me3 mark (i.e., being associated with both enhancers and silencers). To address this issue, we correlated H3K27me3 activity with gene expression across a set of distinct tissues to identify negCORs, i.e., the H3K27me3-DHSs peaks anticorrelating with the expression of nearby genes. We validated negCORs as silencers by demonstrating that 63% of genes linked to negCORs by Hi-C connections or eQTL associations show decreased expression, which is significantly higher than that of background genes.
NegCOR silencer sequences are enriched for the binding sites of repressors across different cell lines and therefore were exploited to build SVM models for identifying additional silencers. With these models, we expanded the silencer landscape of the human genome from 1334 negCOR silencers to 9194 SVM silencers per cell line. Identified silencers (negCOR and SVM silencers together) are significantly enriched for SNPs associated with human phenotypes and diseases as compared with general H3K27me3-DHSs. Their enrichment levels are comparable to enhancers, highlighting the biological importance of silencers.
The regulatory model suggesting an active interplay of different classes of REs is one of the dominant topics of the postsequencing era (Kolovos et al. 2012; Markenscoff-Papadimitriou et al. 2014; Long et al. 2016). We demonstrated that silencers form clusters of activity and intimately collaborate with enhancers and superenhancers to accurately orchestrate the transcription of genes, especially tissue-specific genes and TF genes. The interaction between silencers and enhancers seems to be bimodal: (1) Silencers and/or silencer clusters are present in a mutually exclusive way to enhancers, probably acting on enhancers or, more likely, superenhancers to deactivate their activity and down-regulate the target gene expression through this indirect activity; and, (2) silencers co-occur with enhancers to fine-tune highly expressed genes. Our results show that the former model mainly governs tissue-specific genes and is important for setting up a distinct identity of a cell. The latter greatly contributes to the regulation of highly expressed TFs and is essential for a stable and sensitive regulatory system.
Understanding the molecular mechanisms by which silencers function remains an outstanding topic in the field of transcriptional regulation (Blackledge et al. 2015). Establishing reliable genome-wide silencer maps is the crucial step in promoting these studies. Through exploring DNA sequence signatures, epigenetic as well as transcriptional profiles, we identified a multi-tissue map of silencers in the human genome with improved accuracy and demonstrated that both TF occupancy and epigenetic modification contribute to the activation of silencers.
Methods
A framework for predicting silencers
Using the DNase-seq peaks overlapping with H3K27me3 ChIP-seq peaks (defined as H3K27me3-DHSs) as input across different human cell lines, our method is composed of two sequential modules: a correlation-based module and a classification-based module (Supplemental Fig. S2). In the first module, we evaluated the correlation between the activity of H3K27me3-DHSs and the expression of their proximal genes by focusing on the H3K27me3-DHSs active in at least three tissues. For an intronic H3K27me3-DHS, the proximal gene is its host gene. An intergenic H3K27me3-DHS has two proximal genes, i.e., the nearest gene in the downstream direction and the nearest one in the upstream direction. A correlation between an H3K27me3-DHS region (h, h = 1 and 0 represents the presence and absence of a region, respectively) and its proximal gene is measured as
Gh = 1 is the collection of the expression levels of the tested gene when h = 1. avg(*) is the average of *, and p(a,b) is the Wilcoxon rank-sum test P-value measuring the significance of the difference between a and b. As such, the correlation corr(G,h) is the weighted significance of the change of gene expression due to presence/absence of a H3K27me3-DHS region (h = 1/0, respectively). A low absolute value of corr(G,h) indicates a strong association between G and h. A negative/positive corr(G,h) shows that the gene expression G decreases/increases as the element h appears in a locus. We marked the correlation of (G,h) as significantly negative when −0.05 < corr(G,h) < 0. An H3K27me3-DHS was regarded as negCOR only when at least one of its corr(G,h) values is significantly negative and none of its corr(G,h) values is significantly positive. Similarly, an H3K27me3-DHS was regarded as significantly positive correlated (posCOR) only when at least one of its corr(G,h) values is significantly positive and none of its corr(G,h) values is significantly negative.
In the second module, i.e., the classification-based module, SVMs were built by contrasting negCORs against posCORs. An H3K27me3-DHS is encoded by using the occurrence of putative TFBSs along its DNA sequence (as detailed below), the signal intensity of several histone modifications (including H3K4me1, H3K4me2, H3K4me3, H3K27ac, H3K79me1, H3K9ac, H3K9me3, H2A.Z, and H3K36me3), as well as the maximum and minimum of the expressions of proximal genes. The values of each feature were linearly normalized to be in the range of [0, 1]. SVMs were implemented in MATLAB with the function “svmtrain” having kernel_function = “linear” and method = “LS”. To test classification performance, a fivefold cross-validation scheme was used. In this scheme, a training data set was equally divided into five subsets. After using every subset for validating the SVM built on the other four subsets, we obtained validation results on all training samples and evaluated these results in terms of false positive rate, precision, and recall. Also, we applied all the built SVMs to score each of the H3K27me3-DHSs other than negCORs and posCORs and used the average of the SVM scores as a final estimate of the inquiry H3K27me3-DHSs. H3K27me3-DHSs having scores greater than a threshold were then marked as potential silencers. The threshold was determined in such a way that the false positive rate on the validation results was 0.1. After concatenating negCOR and SVM silencers, we delivered a silencer map for each tested cell line.
Analysis of gene expression data
We downloaded RNA-seq data for 25 cell lines (Supplemental Fig. S2) and discarded uninformative genes, i.e., those with expression levels of less than 0.01. We normalized gene expression (RPKM values) so that the normalized expression level of each gene had a median of 0 and a standard deviation of 1 across the tested cell lines. A positive/negative normalized level of expression suggests a lowly/highly expressed gene in the corresponding cell line.
With these RNA-seq data, we measured tissue specificity of genes using tau (τ), which has been shown to provide strong robustness to change of data and high sensitivity of detecting tissue-specific genes (Kryuchkova-Mostacci and Robinson-Rechavi 2017). τ is defined as
where xi is the expression of a tested gene in the cell line i. N is the number of the cell lines under consideration (N = 25 here). A high value of τ indicates a large variation in gene expression across tissues (i.e., high tissue specificity) of the corresponding gene. Genes having τ > 0.9 and τ < 0.6 are considered tissue-specific and housekeeping genes, respectively (Kryuchkova-Mostacci and Robinson-Rechavi 2017). In total, we identified 3419 tissue-specific and 2503 housekeeping genes.
Experimental characterization of silencers
Candidate silencer regions were synthesized by Thermo Fisher Scientific. Synthesized regions were cloned into plasmid DNA, upstream of an enhancer, SV40 promoter, and luciferase reporter gene using the Invitrogen Gateway Cloning System. Transfection of cells and measurement of expression were the same as previously published (Petrykowska et al. 2008).
Identification of silencer clusters
To identify silencer clusters, we used the distribution of enhancers (detected as the DHSs overlapping both H3K27ac and H3K4me1 peaks in this study) as a reference. For a cell line, we thereby established a genome-wide map of silencers and enhancers (consisting of nS silencers and nE enhancers) and measured the genome-wide fraction of silencers as P = nS/(nE + nS). Given a gene locus harboring s silencers and e enhancers, the enrichment of silencers in that locus was estimated as the probability of obtaining more than s silencers in (s + e) attempts of randomly selecting one element (silencer or enhancer) in the built genome-wide map. This probability can be calculated under a binomial distribution as
| (1) |
A given locus was regarded as being enriched for silencers when and s ≥ 2, in which all silencers were considered to form a silencer cluster. With Equation 1, we estimated silencer enrichment in each gene locus, then sorted out the loci enriched for silencers, and finally evaluated the fraction of the clustered silencers over all tested silencers. To estimate the significance of this fraction, we generated a background distribution through randomly shuffling silencer labels among all H3K27me3-DHSs and then calculating the fraction of clustered silencers. After 50 independent runs, the background distribution of the fraction of clustered silencers was empirically built to assess the significance of the predicted silencers being clustered.
The above analytics was also applied to estimate the enhancer enrichment in a gene locus. That is, the enrichment of enhancers in a locus (hosting e silencers and s silencers) was evaluated as
| (2) |
where q, the fraction of the enhancers on a genome-wide scale, was measured as q = 1 − p = nE/(nE + nS).
Silencer-rich, silencer-enhancer, and enhancer-rich loci
The estimates of and render us three groups of gene loci—“silencer-rich” representing the loci with a significant abundance of silencers, i.e., and s ≥ 2; “enhancer-rich” labeling the loci enriched for enhancers (i.e., and e ≥ 2; and finally, “silencer-enhancer loci” denoting the loci harboring both silencers and enhancers but enriched for neither silencers nor enhancers.
Binding sites of TFs
Transcription factor binding sites were identified through mapping of TF motifs, represented by position weight matrices (PWMs). We used 4004 human and mouse PWMs provided by MEME (http://meme-suite.org/db/motifs). To exclude PWM redundancy, we analyzed PWM mapping in random DNA sequences and excluded a PWM if its TFBSs were at least 90% similar to TFBSs of another PWM. The reduction in redundancy resulted in 1388 distinct PWMs, which correspond to 1119 TFs. To predict TFBSs, we applied FIMO (Grant et al. 2011) with default settings to the DNA sequences of H3K27me3-DHSs. We also used the TF ChIP-seq peaks to build TFBS profiles (see Supplemental “TFBS profiles based on TF ChIP-seq data”).
We used posCORs as a background to evaluate the TFBS signature of silencers. To ensure reliability of analysis, we focused only on the TFs for which TFBSs occurred in at least 3% of either enhancer sequences or silencer sequences. For example, in GM12878, EP300 was excluded from our study since <3% of H3K27me3-DHSs overlap with EP300 ChIP-seq peaks.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Program of the National Library of Medicine and the National Human Genome Research Institute, National Institutes of Health. We thank Dorothy L. Buchhagen for critical reading of the manuscript.
Author contributions: D.H. performed computational analysis and analyzed the data. H.M.P. and B.F.M. performed experimental validation of silencers and analyzed experimental results. I.O. supervised computational work, and L.E. supervised the experimental component of the study. D.H. prepared figures and tables. D.H., L.E., and I.O. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.247007.118.
Freely available online through the Genome Research Open Access option.
References
- Albert FW, Kruglyak L. 2015. The role of regulatory variation in complex traits and disease. Nat Rev Genet 16: 197–212. 10.1038/nrg3891 [DOI] [PubMed] [Google Scholar]
- Bard-Chapeau EA, Jeyakani J, Kok CH, Muller J, Chua BQ, Gunaratne J, Batagov A, Jenjaroenpun P, Kuznetsov VA, Wei C-L, et al. 2012. Ecotopic viral integration site 1 (EVI1) regulates multiple cellular processes important for cancer and is a synergistic partner for FOS protein in invasive tumors. Proc Natl Acad Sci 109: 2168–2173. 10.1073/pnas.1119229109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bienz M. 1998. TCF: transcriptional activator or repressor? Curr Opin Cell Biol 10: 366–372. 10.1016/S0955-0674(98)80013-6 [DOI] [PubMed] [Google Scholar]
- Blackledge NP, Rose NR, Klose RJ. 2015. Targeting Polycomb systems to regulate gene expression: modifications to a complex story. Nat Rev Mol Cell Biol 16: 643–649. 10.1038/nrm4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo F, Zakharov A, Nucifora G. 2007. EVI1 is a transcriptional repressor able to recruit SUV39H1 histone methylation activity. Blood 110: 1235. [Google Scholar]
- Daniel B, Nagy G, Nagy L. 2014. The intriguing complexities of mammalian gene regulation: how to link enhancers to regulated genes. Are we there yet? FEBS Lett 588: 2379–2391. 10.1016/j.febslet.2014.05.041 [DOI] [PubMed] [Google Scholar]
- The ENCODE Project Consortium. 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Kellis M. 2012. ChromHMM: automating chromatin-state discovery and characterization. Nat Methods 9: 215–216. 10.1038/nmeth.1906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst J, Melnikov A, Zhang X, Wang L, Rogov P, Mikkelsen TS, Kellis M. 2016. Genome-scale high-resolution mapping of activating and repressive nucleotides in regulatory regions. Nat Biotechnol 34: 1180 10.1038/nbt.3678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Bashari MH, Morelli E, Tonon G, Malvestiti S, Vallet S, Jarahian M, Seckinger A, Hose D, Bakiri L, et al. 2016. The AP-1 transcription factor JunB is essential for multiple myeloma cell proliferation and drug resistance in the bone marrow microenvironment. Leukemia 31: 1570–1581. 10.1038/leu.2016.358. [DOI] [PubMed] [Google Scholar]
- Gaszner M, Felsenfeld G. 2006. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet 7: 703–713. 10.1038/nrg1925 [DOI] [PubMed] [Google Scholar]
- Grant CE, Bailey TL, Noble WS. 2011. FIMO: scanning for occurrences of a given motif. Bioinformatics 27: 1017–1018. 10.1093/bioinformatics/btr064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundberg E, Small KS, Hedman AK, Nica AC, Buil A, Keildson S, Bell JT, Yang T-P, Meduri E, Barrett A, et al. 2012. Mapping cis- and trans-regulatory effects across multiple tissues in twins. Nat Genet 44: 1084–1089. 10.1038/ng.2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The GTEx Consortium. 2015. The genotype-tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 348: 648–660. 10.1126/science.1262110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman MM, Ernst J, Wilder SP, Kundaje A, Harris RS, Libbrecht M, Giardine B, Ellenbogen PM, Bilmes JA, Birney E, et al. 2013. Integrative annotation of chromatin elements from ENCODE data. Nucleic Acids Res 41: 827–841. 10.1093/nar/gks1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holwerda SJB, de Laat W. 2013. CTCF: the protein, the binding partners, the binding sites and their chromatin loops. Philos Trans R Soc B Biol Sci 368: 20120369 10.1098/rstb.2012.0369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iype T, Taylor DG, Ziesmann SM, Garmey JC, Watada H, Mirmira RG. 2004. The transcriptional repressor Nkx6.1 also functions as a deoxyribonucleic acid context-dependent transcriptional activator during pancreatic β-cell differentiation: evidence for feedback activation of the nkx6.1 gene by Nkx6.1. Mol Endocrinol 18: 1363–1375. 10.1210/me.2004-0006 [DOI] [PubMed] [Google Scholar]
- Jiang W, Wang J, Zhang Y. 2013. Histone H3K27me3 demethylases KDM6A and KDM6B modulate definitive endoderm differentiation from human ESCs by regulating WNT signaling pathway. Cell Res 23: 122–130. 10.1038/cr.2012.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim H. 2012. Recruitment and biological consequences of histone modification of H3K27me3 and H3K9me3. ILAR J 53: 232–239. 10.1093/ilar.53.3-4.232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiman E, Jia H, Loguercio S, Su AI, Feeney AJ. 2016. YY1 plays an essential role at all stages of B-cell differentiation. Proc Natl Acad Sci 113: E3911–E3920. 10.1073/pnas.1606297113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolovos P, Knoch TA, Grosveld FG, Cook PR, Papantonis A. 2012. Enhancers and silencers: an integrated and simple model for their function. Epigenetics Chromatin 5: 1 10.1186/1756-8935-5-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryuchkova-Mostacci N, Robinson-Rechavi M. 2017. A benchmark of gene expression tissue-specificity metrics. Brief Bioinformatics 18: 205–214. 10.1093/bib/bbw008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprell F, Finkl K, Müller J. 2017. Propagation of Polycomb-repressed chromatin requires sequence-specific recruitment to DNA. Science 356: 85–88. 10.1126/science.aai8266 [DOI] [PubMed] [Google Scholar]
- Lee B-K, Bhinge AA, Iyer VR. 2011. Wide-ranging functions of E2F4 in transcriptional activation and repression revealed by genome-wide analysis. Nucleic Acids Res 39: 3558–3573. 10.1093/nar/gkq1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. 2005. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab 1: 361–370. 10.1016/j.cmet.2005.05.004 [DOI] [PubMed] [Google Scholar]
- Liu N, Nelson BR, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel-Duby R, Olson EN. 2014. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proc Natl Acad Sci 111: 4109–4114. 10.1073/pnas.1401732111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Prescott SL, Wysocka J. 2016. Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167: 1170–1187. 10.1016/j.cell.2016.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovén J, Hoke Heather A, Lin Charles Y, Lau A, Orlando David A, Vakoc Christopher R, Bradner James E, Lee Tong I, Young Richard A. 2013. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153: 320–334. 10.1016/j.cell.2013.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv X, Han Z, Chen H, Yang B, Yang X, Xia Y, Pan C, Fu L, Zhang S, Han H, et al. 2016. A positive role for polycomb in transcriptional regulation via H4K20me1. Cell Res 26: 529–542. 10.1038/cr.2016.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markenscoff-Papadimitriou E, Allen WE, Colquitt BM, Goh T, Murphy KK, Monahan K, Mosley CP, Ahituv N, Lomvardas S. 2014. Enhancer interaction networks as a means for singular olfactory receptor expression. Cell 159: 543–557. 10.1016/j.cell.2014.09.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maston GA, Evans SK, Green MR. 2006. Transcriptional regulatory elements in the human genome. Annu Rev Genomics Hum Genet 7: 29–59. 10.1146/annurev.genom.7.080505.115623 [DOI] [PubMed] [Google Scholar]
- McManus S, Ebert A, Salvagiotto G, Medvedovic J, Sun Q, Tamir I, Jaritz M, Tagoh H, Busslinger M. 2011. The transcription factor Pax5 regulates its target genes by recruiting chromatin-modifying proteins in committed B cells. EMBO J 30: 2388–2404. 10.1038/emboj.2011.140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnikov A, Murugan A, Zhang X, Tesileanu T, Wang L, Rogov P, Feizi S, Gnirke A, Callan CG Jr., Kinney JB, et al. 2012. Systematic dissection and optimization of inducible enhancers in human cells using a massively parallel reporter assay. Nat Biotechnol 30: 271 10.1038/nbt.2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim T-K, Koche RP, et al. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448: 553–560. 10.1038/nature06008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mozzetta C, Boyarchuk E, Pontis J, Ait-Si-Ali S. 2015. Sound of silence: the properties and functions of repressive Lys methyltransferases. Nat Rev Mol Cell Biol 16: 499–513. 10.1038/nrm4029 [DOI] [PubMed] [Google Scholar]
- Narlikar L, Sakabe NJ, Blanski AA, Arimura FE, Westlund JM, Nobrega MA, Ovcharenko I. 2010. Genome-wide discovery of human heart enhancers. Genome Res 20: 381–392. 10.1101/gr.098657.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohba H, Chiyoda T, Endo E, Yano M, Hayakawa Y, Sakaguchi M, Darnell RB, Okano HJ, Okano H. 2004. Sox21 is a repressor of neuronal differentiation and is antagonized by YB-1. Neurosci Lett 358: 157–160. 10.1016/j.neulet.2004.01.026 [DOI] [PubMed] [Google Scholar]
- Parker SCJ, Stitzel ML, Taylor DL, Orozco JM, Erdos MR, Akiyama JA, van Bueren KL, Chines PS, Narisu N, NISC Comparative Sequencing Program, et al. 2013. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. Proc Natl Acad Sci 110: 17921–17926. 10.1073/pnas.1317023110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrykowska HM, Vockley CM, Elnitski L. 2008. Detection and characterization of silencers and enhancer-blockers in the greater CFTR locus. Genome Res 18: 1238–1246. 10.1101/gr.073817.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinello L, Xu J, Orkin SH, Yuan G-C. 2014. Analysis of chromatin-state plasticity identifies cell-type–specific regulators of H3K27me3 patterns. Proc Natl Acad Sci 111: E344–E353. 10.1073/pnas.1322570111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pott S, Lieb JD. 2015. What are super-enhancers? Nat Genet 47: 8–12. 10.1038/ng.3167 [DOI] [PubMed] [Google Scholar]
- Qi H, Liu M, Emery DW, Stamatoyannopoulos G. 2015. Functional validation of a constitutive autonomous silencer element. PLoS One 10: e0124588 10.1371/journal.pone.0124588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. 2014. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159: 1665–1680. 10.1016/j.cell.2014.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. 10.1038/nature14248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth M, Bonev B, Lindsay J, Lea R, Panagiotaki N, Houart C, Papalopulu N. 2010. FoxG1 and TLE2 act cooperatively to regulate ventral telencephalon formation. Development 137: 1553–1562. 10.1242/dev.044909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, Lajoie B, Jain G, Dekker J. 2012. The long-range interaction landscape of gene promoters. Nature 489: 109–113. 10.1038/nature11279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeier S, Alam T, Essack M, Bajic VB. 2017. TcoF-DB v2: update of the database of human and mouse transcription co-factors and transcription factor interactions. Nucleic Acids Res 45: D145–D150. 10.1093/nar/gkw1007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder AJ, Pavlidis P, Arimura A, Capece D, Rothman PB. 2002. Cutting edge: STAT6 serves as a positive and negative regulator of gene expression in IL-4-stimulated B lymphocytes. J Immunol 168: 996–1000. 10.4049/jimmunol.168.3.996 [DOI] [PubMed] [Google Scholar]
- Schwartz YB, Pirrotta V. 2008. Polycomb complexes and epigenetic states. Curr Opin Cell Biol 20: 266–273. 10.1016/j.ceb.2008.03.002 [DOI] [PubMed] [Google Scholar]
- Scibetta AG, Wong P-P, Chan KV, Canosa M, Hurst HC. 2010. Dual association by TFAP2A during activation of the p21cip/CDKN1A promoter. Cell Cycle (Georgetown, Tex) 9: 4525–4532. 10.4161/cc.9.22.13746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. 2012. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148: 458–472. 10.1016/j.cell.2012.01.010 [DOI] [PubMed] [Google Scholar]
- Shaulian E, Karin M. 2002. AP-1 as a regulator of cell life and death. Nat Cell Biol 4: E131–E136. 10.1038/ncb0502-e131 [DOI] [PubMed] [Google Scholar]
- Simon JA, Kingston RE. 2009. Mechanisms of Polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 10: 697–708. 10.1038/nrm2763 [DOI] [PubMed] [Google Scholar]
- Somasundaram R, Prasad MAJ, Ungerbäck J, Sigvardsson M. 2015. Transcription factor networks in B-cell differentiation link development to acute lymphoid leukemia. Blood 126: 144–152. 10.1182/blood-2014-12-575688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spender LC, Whiteman HJ, Karstegl CE, Farrell PJ. 2005. Transcriptional cross-regulation of RUNX1 by RUNX3 in human B cells. Oncogene 24: 1873–1881. 10.1038/sj.onc.1208404 [DOI] [PubMed] [Google Scholar]
- Spitz F, Furlong EEM. 2012. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13: 613–626. 10.1038/nrg3207 [DOI] [PubMed] [Google Scholar]
- Tugores A, Le J, Sorokina I, Snijders AJ, Duyao M, Reddy PS, Carlée L, Ronshaugen M, Mushegian A, Watanaskul T, et al. 2001. The epithelium-specific ETS protein EHF/ESE-3 is a context-dependent transcriptional repressor downstream of MAPK signaling cascades. J Biol Chem 276: 20397–20406. 10.1074/jbc.M010930200 [DOI] [PubMed] [Google Scholar]
- van Kruijsbergen I, Hontelez S, Veenstra GJC. 2015. Recruiting polycomb to chromatin. Int J Biochem Cell Biol 67: 177–187. 10.1016/j.biocel.2015.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM. 2009. A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10: 252–263. 10.1038/nrg2538 [DOI] [PubMed] [Google Scholar]
- Yanai I, Benjamin H, Shmoish M, Chalifa-Caspi V, Shklar M, Ophir R, Bar-Even A, Horn-Saban S, Safran M, Domany E, et al. 2005. Genome-wide midrange transcription profiles reveal expression level relationships in human tissue specification. Bioinformatics 21: 650–659. 10.1093/bioinformatics/bti042 [DOI] [PubMed] [Google Scholar]
- Young MD, Willson TA, Wakefield MJ, Trounson E, Hilton DJ, Blewitt ME, Oshlack A, Majewski IJ. 2011. ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res 39: 7415–7427. 10.1093/nar/gkr416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HY, Katsman Y, Dhaliwal NK, Davidson S, Macpherson NN, Sakthidevi M, Collura F, Mitchell JA. 2014. A Sox2 distal enhancer cluster regulates embryonic stem cell differentiation potential. Genes Dev 28: 2699–2711. 10.1101/gad.248526.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.