

Abstract
The structure of neuronal circuits that subserve cognitive functions in the brain is shaped and refined throughout development and into adulthood. Evidence from human and animal studies suggests that the cellular and synaptic substrates of these circuits are atypical in neuropsychiatric disorders, indicating that altered structural plasticity may be an important part of the disease biology. Advances in genetics have redefined our understanding of neuropsychiatric disorders and have revealed a spectrum of risk factors that impact pathways known to influence structural plasticity. In this Review, we discuss the importance of recent genetic findings on the different mechanisms of structural plasticity and propose that these converge on shared pathways.
TOC blurb
The morphology of dendrites and dendritic spines changes with development and as a result of activity-dependent plasticity mechanisms. Penzes and colleagues describe the altered dendritic structural plasticity that is associated with some neuropsychiatric disorders and consider the underlying molecular mechanisms, based on recent genetic discoveries.
The highly specialized morphology of neurons provides the substrate to build the circuits that underlie human cognition and behaviour. The shaping of neuronal circuits occurs in sequential developmental phases and is refined by activity-dependent mechanisms and environmental cues1. Many neuronal subcompartments have been shown to change in response to activity; these include dendritic arbors and spines, the axon initial segment and presynaptic boutons2. The different sites of structural plasticity that work in concert to ensure precise brain wiring are therefore sensitive to dysregulation at multiple levels and on different timescales. In particular, morphological changes in dendritic spines have been the focus of intense investigation owing to their proposed role in several neuropsychiatric disorders3.
Neuropsychiatric disorders are a heterogeneous group of mental disorders that manifest psychiatric symptoms and demonstrable brain pathology and are thought to arise from disruptions of the synaptic circuits that subserve cognitive, social and emotional processing4. Since the advent of genomic technologies, genetic risk factors for neuropsychiatric disorders are being identified at an unprecedented rate, providing a window into the molecular basis of disease. Remarkably, the pathways that are being uncovered intersect with mechanisms regulating the growth and structural plasticity of synapses, which may be important for understanding the nature of these disorders. Structural plasticity of circuits involves changes in both excitatory and inhibitory neurons and in both presynaptic and postsynaptic termini (reviewed in REF 2); however, genetic studies indicate that excitatory postsynaptic compartments may be particularly important in the aetiology of a wide range of neuropsychiatric disorders5–8.
In this Review, we focus on the structural plasticity that occurs at postsynaptic sites, the dendrites and spines of excitatory neurons, which are atypical in several neuropsychiatric disorders and in which synaptic plasticity mechanisms have been extensively studied. We emphasize the integration of biological insights from large-scale genomic studies of idiopathic neuropsychiatric disorders of neurodevelopmental origin, including intellectual disability, epilepsy, autism spectrum disorder (ASD), schizophrenia and bipolar disorder. Although epilepsy is typically classified as a neurological condition, we include it in this Review because it shares many genetic risk factors with ASD and intellectual disability9. In addition, epilepsy is often comorbid with intellectual disability and ASD, and individuals with epilepsy often present with various neuropsychiatric symptoms10. Each neuropsychiatric disorder that we discuss is associated with a distinct pattern of dendritic spine pathology, and mounting evidence implicates the altered structural plasticity of spines as a central disease mechanism in these disorders.
Dendritic structural plasticity
Although multiple brain regions are likely to have a role in the development of neuropsychiatric disorders, the human neocortex has an important role in higher-order brain functions (such as cognition) that are dysfunctional in many neuropsychiatric disorders. Thus, we here consider the postsynaptic elements of cortical circuits — the dendrites and spines of pyramidal neurons — and describe their developmental timeline and the mechanisms regulating their activity-dependent remodelling.
Dendritic spines and brain development.
Cortical pyramidal neurons are the main excitatory cells of the neocortex11. During development, pyramidal neurons elaborate complex dendritic arbors, which increase their surface area and connectivity (FIG. 1a). Maturing dendrites become studded with small protrusions called dendritic spines, which host the majority of the excitatory synapses in the brain. The total excitatory input that a neuron can receive is dependent on the complexity of the dendritic arbor and the density and size of spines, which together form the neuron’s synaptic input field. The growth of dendrites and spines is therefore fundamental to the development of neuronal circuits.
Figure 1|. Spine and dendrite development in health and disease.
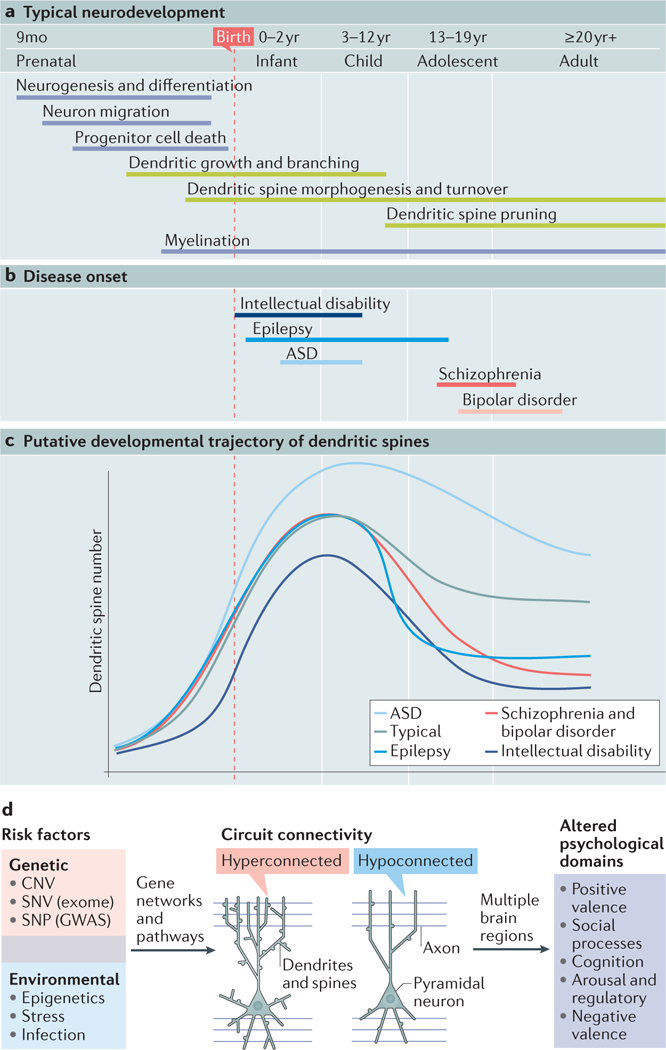
a,b| Timeline of the principal cellular events occurring during human brain development1,265 and their coincidence with neuropsychiatric diseases of varying onset. Morphological events regulating dendritic structure that are discussed in this Review are highlighted in green. Intellectual disability usually presents in early infancy or early childhood, although in some cases, it cannot be formally diagnosed until later (when standardized tests can be implemented)266. Epilepsy encompasses a broad range of seizure disorders, each with their own typical age of onset. Here, we focus on genetic forms of epilepsy that have their onset largely in the age range depicted (see REF. 267 for review). The symptoms of autism spectrum disorder (ASD) are recognized early in development and are noticeable in the first few years of life268. These early-onset disorders overlap with the developmental processes of dendritic growth and spine morphogenesis. Bipolar disorder269 and schizophrenia270 are late-onset neurodevelopmental disorders that appear in late adolescence or adulthood, coinciding with the biological process of spine pruning. c | Putative developmental trajectories of spine development during typical development or in neuropsychiatric disorders, predicted from post-mortem studies. Studies have found that spine density is reduced in intellectual disability39 and increased in ASD41, which may suggest a developmental alteration in spine morphogenesis. Spine and dendrite loss in epilepsy is thought to be caused by the onset of seizures45 and is depicted as a sharp decline in spine density (however, a primary defect in spine density before seizure onset cannot be excluded). Schizophrenia and bipolar disorder are associated with fewer dendritic spines post-mortem48,53, possibly caused by excessive spine pruning. d | Genetic and environmental risk factors for neuropsychiatric disorders are hypothesized to converge on a subset of genetic networks and pathways that regulate dendritic structure and alter the synaptic input field. Both hypoconnected and hyperconnected cells disrupt local and distal connectivity in the brain. Depending on the brain regions affected, this will impact different psychological domains (here, we use the National Institutes of Health Research Domain Criteria (RDoC) relevant to neuropsychiatric disorders; see ‘Related links’). CNV, copy number variant; GWAS, genome-wide association study; mo, month; SNP, single-nucleotide polymorphism; SNV, single-nucleotide variation.
Dendrites provide the support on which dendritic spines grow, and during early rodent development, the growth of both structures is intimately linked12. Later in development, dendritic branches become stabilized and uncoupled from the morphological changes to dendritic spines. Dendrites are therefore much less dynamic than dendritic spines in mature neurons. Dendritic spines compartmentalize the postsynaptic machinery and assist in the independent regulation of synaptic inputs13. Spines are typically less than 1 (μm3 in size and exist on a morphological continuum from long, thin filopodia to large, mature spines with a defined spine head14. Filopodia are thought of as immature spines because they arise early in development, are highly dynamic and often lack functional synapses. By contrast, mature (or ‘mushroom’) spines are typically more stable and have an enlarged spine head containing neurotransmitter receptors and a postsynaptic density (PSD). Given that certain neuronal subtypes do not have spines, the reason for the existence of such a broad array of spine morphologies in pyramidal neurons remains somewhat enigmatic; however, their precise shape and plasticity are likely to influence the regulation and compartmentalization of biochemical and electrical signalling that are required to form complex and adaptable circuits15.
In the human neocortex, dendrites can be recognized approximately halfway through gestation (17–25 weeks), whereas spines appear late in the second trimester (26–34 weeks) (FIG. 1a). The appearance of spines coincides with a period of intensive dendritic growth and cortical thickening16. The number of spines increases rapidly in the perinatal and postnatal periods and reaches its peak early in infancy (1–2 years) before progressively declining in a period known as ‘pruning’, which continues into adolescence and adulthood17. The elimination of spines indicates the start of a fine structural reorganization of the cortex and is concurrent with the observation of cortical thinning (measured by MRI) in late childhood to adolescence1,18. The developmental changes in this period are consistent with the emergence of higher cognitive functions such as attention, working memory, cognitive control and response inhibition1. In humans, childhood is a period of massive structural refinement at the level of dendritic arbors and spines that is critically important for attaining properly functioning and adaptable synaptic circuits. Importantly, early life witnesses the intersection of a waning developmental and a growing experience-dependent influence on plasticity, each affecting spines on their own timescales.
Activity-dependent structural plasticity.
Spines are highly dynamic in nature and have a remarkable capacity to undergo structural changes in the young and adult brain19,20. Spines can grow, shrink, form de novo and be maintained or be eliminated, all of which contribute to the formation and experience-dependent optimization of neuronal circuits20. In addition, the morphological modifications of spines are widely regarded as the structural basis for learning and encoding memories21,22. Spine plasticity is affected by multiple factors, including developmental stage, brain region, sensory experience and synaptic activity20. Recent super-resolution imaging techniques have been instrumental in shaping our knowledge of the type and extent of the structural changes that take place (BOX 1).
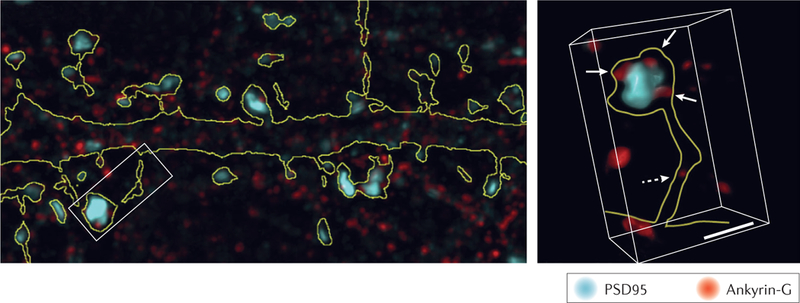
Box 1 |. Super-resolution imaging of dendritic spines.
In recent years, super-resolution techniques that are able to break the diffraction limit of light microscopy (~200 nm) have routinely allowed fluorescent imaging with resolutions down to 50 nm (reviewed in REF 245). This ability has led to characterization of the dendritic spine in unprecedented detail and revealed novel mechanisms of structural regulation. Structured illumination microscopy108 and novel image deconvolution protocols246 allow resolving power down to ~100–200 nm. This increased resolution allowed for the characterization of ankyrin-G nanodomains within spine necks and their regulatory role in dendritic spine morphology (see the figure)108. However, techniques that rely on single-molecule localization (stochastic optical reconstruction microscopy (STORM) and photoactivated localization microscopy) or stimulated excitation emission depletion (STED) have garnered great interest owing to their ability to observe filament formation and protein complex distributions within spines. For example, STORM imaging was pivotal in the observation of previously uncharacterized presynaptic mitochondria that contribute to long-term plasticity247 and endogenous brain-derived neurotrophic factor (BDNF) granules that were enriched in the presynaptic but not postsynaptic areas248. In addition, the resolution afforded by STED microscopy revealed that the spine neck is vital in compartmentalizing synaptic signals249 and undergoes actin-dependent changes in size250 in response to long-term potentiation251.
A major advantage of super-resolution techniques over electron microscopy is the ability to observe changes within live cells. For example, dynamic imaging of dendritic spine extension and retraction was achieved at nanoscale resolution using membrane-selective photo-switchable probes252. In addition, super-resolution imaging has been applied to actin dynamics within the spine over extended periods of structural change with lateral resolutions exceeding 50 nm (REFS 250,253,254). Such studies revealed the role of actin in driving vesicle–membrane fusion255, a distinct AMPA receptor (AMPAR) trafficking mechanism within subsets of spines256 and the formation of postsynaptic density–AMPAR scaffolds that contribute to receptor clustering and responsiveness257,258.
Thus, the rapid development of fluorescent imaging techniques has provided previously unachievable insight into dendritic spine structure and regulation. Increasing access to these imaging techniques will undoubtedly result in an even greater understanding of the structural mechanisms underlying neuronal function. PSD95, postsynaptic density protein 95 (also known as DLG4). Figure adapted with permission from REF. 108, Elsevier.
Activity-dependent changes in spines are known to occur via at least two distinct cellular mechanisms: Hebbian and homeostatic plasticity. Hebbian synaptic plasticity is a mechanism through which correlated pre-synaptic and postsynaptic activity leads to durable changes in synaptic function23. Long-term potentiation (LTP) and long-term depression (LTD) are two forms of Hebbian plasticity that can induce long-lasting increases or decreases in spine size, respectively24 (FIG. 2a). LTP is also associated with an increase in the number and stability of spines. LTP involves actin polymerization, an enlargement of the PSD and an increase in AMPA receptor (AMPAR) surface trafficking25. Opposing structural rearrangements occur during LTD, including spine shrinkage, a decrease in actin polymerization and a reduction of surface AMPARs.
Figure 2|. Mechanisms of structural plasticity.
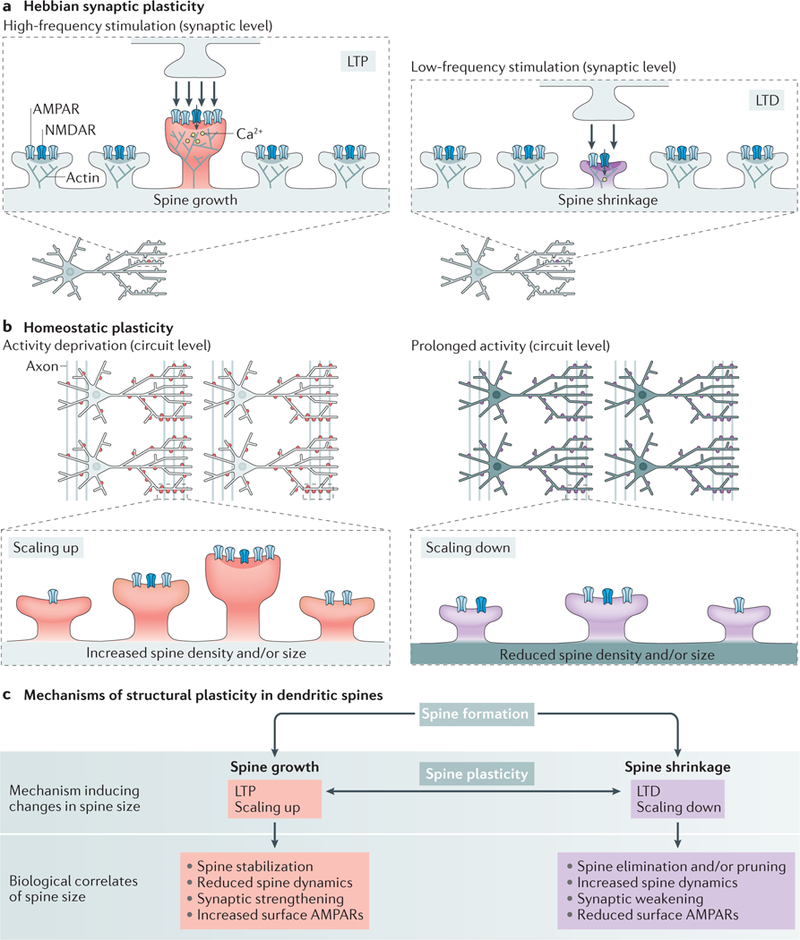
a | Hebbian synaptic plasticity mechanisms25,94. High-frequency synaptic activity associated with high calcium entry causes long-term potentiation (LTP), which induces spine growth, an enlargement of the postsynaptic density (PSD) and actin polymerization and promotes the surface expression of AMPA receptors (AMPARs). Low-frequency synaptic activity associated with modest calcium entry through NMDA receptors (NMDARs) causes long-term depression (LTD), which induces shrinking of the spine and PSD, actin depolymerization and a reduction of surface AMPAR expression. b | Proposed mechanisms of homeostatic synaptic plasticity2. Synaptic scaling is a form of homeostatic plasticity that allows neurons to modify their overall synaptic input (excitability) in response to changes in circuit activity26. Activity deprivation causes neurons to scale up, proportionally strengthening synapses by increasing surface AMPAR expression to increase overall synaptic input. Studies have shown that activity blockade in vitro and activity deprivation in vivo can also cause an increase in spine size (or density)29,31,32. Prolonged circuit activity causes neurons to scale down, proportionally reducing synaptic strength by removal of surface AMPARs. Prolonged activity during seizures also reduces the size and number of spines33,34, opposing the morphological effects of activity deprivation. c | Summary of mechanisms that can regulate spine development and plasticity. Spine growth can be induced by LTP or scaling up and is associated with spine stability, reduced spine dynamics, increased synaptic strength and increased surface AMPAR expression14. Spine shrinkage can be induced by LTD or scaling down and is associated with spine destabilization, possibly leading to elimination or pruning of spines, synaptic weakening and a reduction of surface AMPAR expression14.
Homeostatic plasticity is an overarching form of plasticity that restrains cellular and circuit excitability within physiological limits2. Synaptic scaling is a form of homeostatic plasticity through which neurons can adjust the strength of their synaptic connections, by regulating the surface expression of AMPARs, in response to global changes in circuit activity26. Morphological correlates of synaptic scaling have been demonstrated27, which is perhaps not surprising given the strong correlation between spine size and AMPAR content28. Activity deprivation, a model for studying homeostatic plasticity, has been shown to increase the density or size of spines in vitro29 and in vivo30–32, presumably to compensate for the loss of synaptic input (FIG. 2b). By contrast, prolonged activity (such as that experienced during seizures) reduces spine number and size33,34. However, the exact morphological changes in spines that occur when modelling homeostatic plasticity in vivo appear to be variable and highly dependent on the experimental model used35,36, highlighting our incomplete knowledge of this process.
Although neuronal activity plays a critical part in shaping the growth and structure of spines, recent studies have also shown that spines can form in the absence of glutamate release37. Thus, activity-dependent and activity-independent forces influence structural plasticity and, together, these dynamically regulate the overall number and morphology of spines (FIG. 2c).
Atypical spine and dendrite development
Spine pathology is a common feature of many neuropsychiatric disorders and refers to any shift in the size, shape or density of dendritic spines from that expected in a given brain region and developmental time point (FIG. 1b,c). Changes in spine morphology are often reported in human and animal models because they are easily observable by light microscopy. Given the tight relationship between spine structure and function, even small alterations in average spine size may reveal a profound dysfunction at the cellular or circuit level. Furthermore, imbalances in the processes governing spine formation and elimination can lead to a net change in spine density that, when summated with changes in dendritic complexity, can dramatically alter a neuron’s synaptic input field (FIG. 1d). However, detailed reports of atypical spine development in human neuropsychiatric disorders remain scarce, presumably owing to the limited availability of post-mortem brain tissue. This limitation also makes individual findings difficult to generalize to wider populations. In addition, such studies are subject to many confounding factors38 that can impede the discovery of robust disease pathophysiology. These factors have led to variations in findings across studies; however, certain trends can be distilled.
Spine pathology in intellectual disability.
Post-mortem studies of unclassified intellectual disability (previously termed mental retardation) were among the first to suggest a connection between dendritic defects and neuropsychiatric disorders. Spines on cortical neurons from individuals with intellectual disability have been described as long and thin, indicative of an immature morphology, and display fewer spines per dendritic branch39. Several studies have noted reduced complexity of both the apical and the basal dendrites of cortical pyramidal neurons in the brain tissue of individuals with intellectual disability compared with controls40.
Spine pathology in autism spectrum disorder.
Studies of individuals with ASD have found that they exhibit higher spine densities than control subjects on the apical dendrites of layer II pyramidal neurons in the frontal, parietal and temporal lobes and the layer V projection neurons of the temporal lobe41,42. These observations suggest a disruption of the balance between spine formation and elimination. Coincident with these results, longitudinal MRI studies have demonstrated an enlargement of brain volume and cortical grey matter in ASD that begins before 2 years of age43,44. This period parallels the rapid expansion of spine and dendrite formation in the human brain that peaks in the first 2 years of life16,17, suggesting that spine development during this period leads to an overabundance of spines in ASD.
Spine pathology in epilepsy.
In patients with temporal lobe epilepsy, layer III cortical pyramidal neurons have lower spine densities and fewer dendritic branch points than controls; however, it is thought that these changes may be caused by excitotoxicity45. The reductions in grey matter observed by MRI in temporal lobe epilepsy patients appear not to be due to cell loss but rather to reflect the occupation of a smaller volume by neurons with sparser dendritic trees46. Nonetheless, it is not yet clear whether the dendritic changes observed in epilepsy are the cause or consequence of epileptic seizures34.
Spine pathology in schizophrenia and bipolar disorder.
In schizophrenia, aberrant spine density has been reported in multiple brain regions (reviewed in REF. 47). The most reproducible observation is a reduction of spine density on layer III pyramidal neurons of the neocortex compared with controls48. More recently, a study found that pyramidal neurons in the same layer of the auditory cortex had a specific reduction in the number of small spines49. Post-mortem studies of schizophrenia have also found evidence for reduced complexity of basal dendrites in layer III and layer V pyramidal neurons of the prefrontal cortex (PFC) compared with controls50,51. Atypical dendritic development was again reported in a more recent study that found reduced dendrite length in layer III pyramidal neurons in the PFC of individuals with schizophrenia and, for the first time, bipolar disorder; however, this study did not replicate the lower spine density reported previously for schizophrenia52. A separate study reported lower dendrite length and spine density in the PFC of individuals with bipolar disorder but no changes in spine density in individuals treated with lithium, suggesting that lithium ameliorates spine pathology in bipolar disorder53. Despite these important findings, it is not known when the structural deficits occur in these disorders, as brain samples are typically collected from deceased adults. The favoured hypothesis in schizophrenia is that the lower spine density is the result of accelerated pruning of synapses during adolescence54. This view is supported by longitudinal MRI studies reporting higher rates of grey matter reductions in individuals who go on to develop schizophrenia55, which may in part be due to spine and dendrite loss. Furthermore, studies show that the onset of cognitive symptoms preceding psychosis coincides with the biological process of spine pruning in late childhood to adolescence56.
Spine pathology in syndromic disorders.
In addition to idiopathic disorders, there has been a prominent focus on dendritic structure in Mendelian forms of disease with known genetic aetiology, such as Fragile X syndrome, Rett syndrome and Down syndrome, each of which has a characteristic spine pathology (reviewed in REF. 57). In these cases, human post-mortem data have been supported by findings from animal and stem cell models (BOX 2), which together provide a more persuasive assessment of spine pathology. These models also open up avenues to investigate the underlying mechanisms in syndromic disorders58. Studies of syndromic intellectual disability demonstrate that stratification of individuals on the basis of genetic aetiology can help to identify distinct spine pathologies and that different spine pathologies can converge on cognitive dysfunction40. However, animal models often fail to recapitulate the inherent polygenic nature of many idiopathic neuropsychiatric disorders, which is why the analysis of human brain tissue and stem cells remains essential. Models of syndromic disease also have limited utility in modelling idiopathic neuropsychiatric disorders as intellectual disability, ASD and epilepsy are often comorbid in the same individual (as observed in Fragile X syndrome).
Box 2 |. Stem cell models of atypical dendrite and spine development.
Progress in stem cell reprogramming technologies has revolutionized the way we can study neuropsychiatric conditions. Analysis of the structural and activity-dependent development of human induced pluripotent stem cell (iPSC)-derived neurons is still nascent and promises to complement human post-mortem and MRI studies, enabling research into the early molecular and cellular events that lead to neuropsychiatric disorders. To date, there have been several key insights from studies looking at dendritic and spine dysmorphogenesis in genetically defined human stem cells:
iPSC-derived human neurons in which SHANK3 is conditionally deleted display neurodevelopmental deficits with decreased dendritic complexity and synapse density103.
iPSC-derived human neurons generated from individuals with Timothy syndrome (in which there are mutations in CACNA1C) display defects in activity-dependent dendritic retraction121.
A single-nucleotide polymorphism near microRNA locus MIR137 that was identified in a genome-wide association study of schizophrenia is associated with increased dendritic complexity (see the figure) and an increased proportion of spines containing the AMPAR subunit GluA1 in patient iPSC-derived neurons259.
Deletion of chromosome band 7q11.23 causes Williams–Beuren syndrome, and the same copy number variant is also associated with autism spectrum disorder (ASD). Neurons derived from iPSCs from individuals with the typical 7q11.23 deletion have a larger number of dendrites and dendritic spines and increased total dendrite length compared with controls260.
Deletion of MECP2 causes Rett syndrome, whereas duplication of MECP2 causes a severe neurodevelopmental disorder with similar phenotypes. IPSC-derived neurons from individuals with Rett syndrome have a decreased spine density. Conversely, iPSC-derived neurons from individuals with MECP2 duplications have increased dendritic complexity concomitant with an increased number of immature spines and a reduction of mature spines but no change in total spine number261,262.
iPSC-derived neurons from individuals with atypical Rett syndrome, caused by mutations in CDKL5, have an increased number of dendritic protrusions263.
A heterozygous NRXN1 deletion akin to those found in individuals with schizophrenia and ASD does not impair synapse number or dendrite development in iPSC-derived human neurons264.
Figure adapted with permission from REF. 257, Elsevier.

Patterns of spine pathology.
As described above, defective cortical pyramidal neurons have been observed in a range of different neuropsychiatric disorders, mostly in the frontal and temporal lobes. Early-onset developmental disorders such as intellectual disability and ASD are associated with too few or too many dendritic connections, respectively. Epilepsy, which is often comorbid with intellectual disability and ASD, is mainly characterized by fewer dendrites and spines. By contrast, schizophrenia and bipolar disorder, the later-onset psychiatric disorders, are both reported to have fewer dendritic spines and simplified arbors compared with controls. Further studies at different time points of illness are required to know exactly how and when these dendritic changes occur.
Genetic risk and structural plasticity
Neuropsychiatric genetics.
Neuropsychiatric disorders are known to have a strong genetic component, but their genetic architecture is complex59. Genetic studies of common variants and rare variants in neuropsychiatric disorders have discovered a range of genes and chromosomal regions involved in disease risk. Genome-wide association studies (GWAS) of common risk variants in schizophrenia and bipolar disorder have been successful in identifying a growing number of synaptic genes that are related to disease60,61 (TABLE 1; Supplementary Table S1). In addition, a wide range of rare and highly penetrant copy number variants (CNVs), consisting of large chromosomal deletions or duplications, have been implicated in the aetiology of multiple neuropsychiatric disorders62, several of which alter the structural plasticity of neurons (TABLE 1).
Table 1|.
Neuropsychiatric risk loci with a reported effect on structural plasticity
| Gene or copy number variant | Gene name or description | Type of effect on spines and/or dendrites | Disease associations |
|---|---|---|---|
| Cell adhesion | |||
| NRXN1 | Neurexin 1 | Spine stability79 | SZ281 and ASD6 |
| NLGN3 | Neuroligin 3 | Spine density80 | ASD64 |
| CNTNAP2 | Contactin-associated protein-like 2 (also known as CASPR2) | Spine and dendrite stability86–88 | ID and ASD83 |
| DSCAM | DS cell adhesion molecule | Dendrite and spine development282 | ASD64 |
| PCDH10 | Protocadherin 10 | Spine density283 | ASD283 and SZ63 |
| Glutamate receptors | |||
| GRIA1 | Glutamate ionotropic receptor AMPA type subunit 1 | Spine size93 | SZ60, ID65 and ASD64 |
| PRRT2 | Proline-rich transmembrane protein 2 | Spine density284 | Epilepsy285 |
| CACNG2 | Calcium voltage-gated channel auxiliary subunit-γ2 (also known as TARP-γ2) | Dendritic arborization246 | ID7 |
| GRIN2A | Glutamate ionotropic receptor NMDA type subunit 2A | Dendritic arborization99 | SZ60 and epilepsy286 |
| GRIN2B | Glutamate ionotropic receptor NMDA type subunit 2B | Spine density97 | ASD64 |
| Scaffold proteins | |||
| SHANK3 | SH3 and multiple ankyrin repeat domains 3 | Dendrite and spine development103,106,107 | ASD64,287 |
| ANK3 | Ankyrin 3 (also known as ankyrin-G) | Spine size and density and activity-dependent spine enlargement108 | BD61 |
| DLG4 | Discs large MAGUK scaffold protein 4 (also known as PSD95) | Spine density and size; activity-dependent structural plasticity288,289 | ASD and/or ID290 |
| DLGAP1 | DLG associated protein 1 (also known as GKAP and SAPAP1) | Spine size291 | ASD and/or ID290 |
| CASK | Calcium/calmodulin-dependent serine protein kinase | Spine density and size292 | ASD and/or ID290 and DD67 |
| Calcium signalling | |||
| CACNA1C | Calcium voltage-gated channel subunit-α1C | Spine density120 | BD61 and SZ60 |
| CACNB4 | Calcium voltage-gated channel auxiliary subunit-β4 | Spine density | SZ115,116 |
| CAMK2B | Calcium/calmodulin-dependent protein kinase ||β | Activity-dependent spine formation293 and dendritic arborization294 | ID295 |
| CAMK2A | Calcium/calmodulin-dependent protein kinase ||α | Activity-dependent spine formation293 | ASD64 and ID295 |
| GTPase signalling | |||
| SYNGAP1 | Synaptic RAS GTPase-activating protein 1 | Spine formation, development and activity-dependent structural plasticity131,132,296 | ID297, DD67 and ASD6 |
| TRIO | Trio Rho guanine nucleotide exchange factor | Dendrite development and activity-dependent structural plasticity146 | ASD and/or ID290 |
| KALRN | Kalirin Rho-GEF kinase | Spine morphogenesis and activity-dependent structural plasticity139 | DD67 |
| NF1 | Neurofibromin 1 | Spine development and activity-dependent structural plasticity136 | ID297 |
| ARHGEF6 | Rho guanine nucleotide exchange factor 6 | Dendrite development and spine density156 | ID297 |
| OPHN1 | Oligophrenin 1 | Dendrite and spine development155 | ID297 and DD67 |
| Copy number variant models | |||
| 7q11.23 deletion | Williams–Beuren syndrome | Increased dendrite length and spine number260 | ASD6 |
| 15q11–13 duplication | Dup15q syndrome | Increased spine turnover and deceased spine density81,298 | ASD6 and SZ281 |
| 16p11.2 duplication | None | Dendrite arborization299 | ASD6 and SZ281 |
| 22q11.2 deletion | DiGeorge/velocardiofacial syndrome | Decreased spine density and stability and increased spine turnover248,300 | SZ281 |
As different neuropsychiatric disorders often share the same risk factors, other disease associations exist that are not referenced herein. ASD, autism spectrum disorder; BD, bipolar disorder; DD, developmental disorder; GEF, guanine nucleotide exchange factor; GWAS, genome-wide association study; ID, intellectual disability; SZ, schizophrenia.
The most recent success in revealing the genetic landscape of psychiatric disorders has come from studies using whole-genome sequencing and exome sequencing to identify protein-coding variants63–67. To aid in the interpretation of these genetic risk factors, enrichment analyses have been an invaluable and unbiased way to identify functionally relevant gene sets and biological pathways relevant to neuropsychiatric disorders8. Through such analyses, processes including cytoskeletal organization68,69, transcriptional regulation and chromatin modification6,70,71 have all been suggested to be altered in different neuropsychiatric disorders; however, postsynaptic signalling at glutamatergic synapses appears to be the most prominent shared biological process across multiple disorders5–8,63,68,72, and many of the genes identified are likely to impact spine plasticity. This observation has prompted the need for a detailed examination of the relationship between neuropsychiatric risk factors and the mechanisms of structural plasticity.
Exome sequencing studies have been particularly helpful for biologists as they implicate specific genes rather than the multigene loci often associated with GWAS and CNV studies. Therefore, exome data provide a rich source of information that can assist in disentangling the biological pathways involved in neuropsychiatric disorders and may be critical in unravelling the disease pathways that could underlie spine deficits in neuropsychiatric disorders (FIG. 3).
Figure 3|. Neuropsychiatric risk factors and biological pathways regulating structural plasticity.
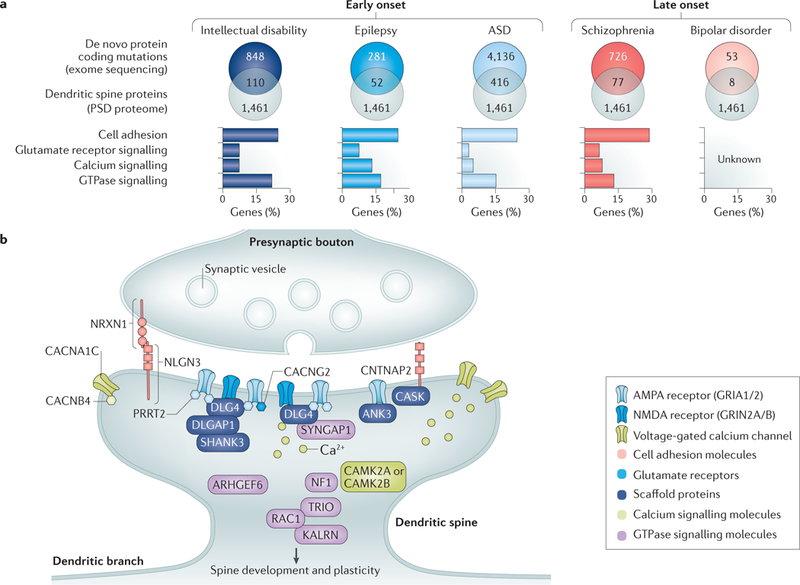
a |Schematic illustration of a systems biology approach for identifying novel genes likely to affect spine plasticity in neuropsychiatric disorders. Exome sequencing studies of de novo variants are particularly useful for uncovering genetic risk networks involved in different disorders. Although many genes discovered in exome sequencing studies are individually nonsignificant, as a whole, they can reveal a mutational spectrum for each disorder and provide clues for the underlying pathways and networks involved in pathogenesis. In particular, genes with de novo mutations for which the protein products localize to the postsynaptic density (PSD) are excellent candidates for future studies on the dysfunctional pathways altering structural plasticity in neuropsychiatric disorders. To highlight such genes, de novo mutations affecting protein coding for each disorder were intersected with proteins present in the human PSD proteome271 (a proxy for the dendritic spine). The number of genes in each overlap is indicated (top panels). All of the de novo data were obtained from denovo-db (see ‘Related links’)272 accessed on 21 September 2017. Additional studies for schizophrenia273–275 that were not in the denovo-db were obtained from supplementary data compiled in REF276. Only de novo variants affecting protein-coding regions or splice sites were included (that is, missense, frameshift and splice site mutations, altered stop codons, altered start codons, insertions and deletions). Human PSD proteome data were obtained from REF. 271. Gene ontologies (GOs) were then used to classify PSD-associated mutations into biological processes relevant to spine structure and function. To do so, GO categories from each data set were defined in DAVID (see ‘Related links’) v6.8, accessed on 11 November 2017, using the functional annotation chart. Homo sapiens was used as a background, and the ‘biological process’ ontology (GOTERM_BP_FAT) was used to annotate gene lists. The following ontologies were used to cluster genes into functional pathways: cell adhesion (GO:0007155-cell adhesion), GluR signalling (GO:0007215-glutamate signalling pathway), calcium (GO:0006816-calcium ion transport) and GTPase (G00043087-regulation of GTPase activity). This analysis shows that the de novo mutations affecting PSD proteins cluster into distinct biological pathways to varying degrees, providing an overview of the contribution of each pathway to each disorder (bottom panels). The percentage of genes in each GO category is depicted on the y-axis. Exome sequencing data for bipolar disorder are currently insufficient to classify mutations into categories. b | Illustration of a dendritic spine containing different functional groups of neuropsychiatric-disorder-associated risk factors (TABLE 1) that regulate spine structure. Functional groups are colour coded. ANK3, ankyrin 3; ARHGEF9, Rho guanine nucleotide exchange factor 9; ASD, autism spectrum disorder; CACNA1C, voltage-gated calcium channel subunit-α1Cav1.2; CACNG2, voltage-dependent calcium channel-γ2 subunit; CACNB4, calcium channel voltage-dependent subunit-β4; CAMK2A/B, calcium/calmodulin-dependent protein kinase type II subunit-α/β; CASK, calcium/calmodulin-dependent serine protein kinase; CNTNAP2, contactin-associated protein-like 2; DLG4, disks large homologue 4; DLGAP1, disks large-associated protein 1; GRIA1/2, glutamate receptor ionotropic, AMPA1/2; GRIN2A/B, glutamate receptor ionotropic NMDA2A/B; KALRN, kalirin; NF1, neurofibromin 1; NLGN3, neuroligin 3; NRXN1, neurexin 1; PRRT2, proline-rich transmembrane protein 2; RAC1, RAS-related C3 botulinum toxin substrate 1; SHANK3, SH3 and multiple ankyrin repeat domains protein 3; SYNGAP1, RAS/RAP GTPase-activating protein SynGAP 1; TRIO, triple functional domain protein.
The complex polygenic nature of neuropsychiatric disorders suggests that many different mutations of varying severity are expected to hit a range of synaptic pathways that converge on spine plasticity. Each pathway, or functional group of proteins, could represent a different dimension that may contribute to deficits in structural plasticity (FIG. 3). Below, we discuss prominent classes of proteins and pathways that are at the convergence point of dendritic structural plasticity and neuropsychiatric genetics, exemplifying each class with well-characterized representatives. These examples may help the functional interpretation of other potential candidate genes that are identified in genetic studies of neuropsychiatric disorders.
Cell adhesion.
Cell adhesion molecules (CAMs) comprise a diverse set of proteins with large extracellular domains that serve as trans-synaptic anchors, ligands, receptors and mediators of activity-dependent signalling73. Neurexins and neuroligins are among the best studied CAMs and have been strongly implicated in several neuropsychiatric disorders. Deletions in NRXN1 were some of the first CNVs to be associated with schizophrenia74, whereas a range of loss-of-function and de novo mutations in neuroligin genes (NLGN1–3 and NLGN4X) are detected in individuals with ASD64,75. Alterations in neurexin family members have also been reported in epilepsy and in neuropsychiatric disorders that are comorbid with epilepsy76,77.
The presynaptic neurexins (neurexin 1 (NRXN1)–NRXN3) participate in heterophilic trans-synaptic interactions with the postsynaptic neuroligins, which triggers the recruitment of the scaffolding molecules and receptors necessary for synapse formation in co-culture models78. In vitro, disruption of all neurexins in hippocampal neurons reduces synaptic stability and increases synapse elimination, implicating neurexins in the stabilization of nascent synapses79. In cultured rat hippocampal neurons, neuroligin 1 (NLGN1) overexpression increases spine density, whereas knockdown of NLGN1, NLGN2 or NLGN3 reduces spine number80. NLGN3-R451C mice, which model a human ASD mutation, have a marked increase in spine dynamics due to spine destabilization in the anterior frontal cortex81. The same mutation increases the number of dendritic branch points in the stratum radiatum region of the hippocampus82.
Contactin-associated protein-like 2 (CNTNAP2; also known as CASPR2) is a more distantly related member of the neurexin family, with a role in structural neurodevelopment. Homozygous loss of CNTNAP2 in humans causes a severe neurodevelopmental syndrome, whereas heterozygous mutations have been found in individuals with schizophrenia, epilepsy and autism83–85. In vitro, mature mouse cortical neurons lacking Cntnap2 have a reduced number of spines, which exhibit an altered morphology and reduced levels of the AMPAR subunit GluA1 (also known as GRIA1)86. Live imaging of layer V pyramidal cells in vivo shows that spine deficits may be caused by increased spine elimination, as spine formation is unaffected in Cntnap2-knockout mice87. In vitro knockdown of CNTNAP2 in developing mouse neurons causes the collapse of dendritic branches and reduction of spine head size without affecting spine or synapse density88. Together, these experiments support a role for CNTNAP2 in the structural integrity of mature dendritic branches and the stability of newly formed spines.
Mutations in a wide variety of other synaptic CAMs, including protocadherins, contactins, Down syndrome cell adhesion molecule (DSCAM) and cadherins, are linked to both early-onset and late-onset neuropsychiatric disorders and have marked effects on structural plasticity (TABLE 1; Supplementary Table S1) These functions at multiple stages of dendritic development may explain why variations in CAMs are associated with a wide range of disorders with varying onset. A prominent role of CAM networks in neuropsychiatric disorder aetiology is consistent with their well-documented roles in multiple steps of circuit formation and remodelling, including synapse formation, specification and stabilization. As these adhesion events are largely activity-independent, CAMs may underlie activity-independent spine pathology processes.
Glutamate receptors.
Excitatory neurotransmission in the brain is mediated primarily by AMPARs and NMDA receptors (NMDARs). Each glutamate receptor consists of four subunits, encoded by multiple genes (GRIA1–GRIA4 (AMPAR) and GRIN1, GRIN2A–GRIN2D and GRIN3 (NMDAR)), which are expressed with distinct spatio-temporal patterns during brain development. Glutamate receptors and their associated auxiliary subunits have been implicated in neuropsychiatric disorders by a variety of different genetic studies (TABLE 1). GRIA1 and GRIN2A were identified as genome-wide significant susceptibility genes in the largest GWAS of schizophrenia to date60. Exome sequencing studies have revealed de novo variants affecting GRIN2A in individuals with schizophrenia, and recurrent de novo variants in GRIN2B have been identified in individuals with ASD, making it one of the strongest candidate ASD susceptibility genes63,64.
AMPARs have a central role in the structure and function of spines. Spine size is robustly correlated with AMPAR content, with large spines containing more AMPARs, enhancing their synaptic strength28. Glutamate signalling through AMPARs is sufficient to induce the formation of new spines and maintain existing spines in hippocampal slice cultures, suggesting that appropriate levels of AMPARs are required for activity-dependent sculpting of neuronal circuits89,90. The GluA1 subunit is particularly important in activity-dependent AMPAR insertion into spines during LTP and experience-dependent plasticity in vivo91,92. The carboxy-terminal tail of GluA1 may be directly required to generate stable increases in spine size in vitro during chemically induced LTP, providing a link between structural and functional plasticity93.
NMDARs are well recognized for their role in the induction of LTP and LTD, which cause long-lasting changes in spine size94 (FIG. 2a). However, blockade of NMDAR in postnatal hippocampal slice cultures results in increased dendritic arborization and synapse number, demonstrating that NMDARs can also influence neuronal development95. Knockdown of the obligatory NMDAR subunit GluN1 (encoded by GRIN1), in hippocampal organotypic slice cultures causes increased spine motility and destabilization of spine structure, which is dependent on the GluN1 intracellular carboxyl terminus, implicating NMDARs in the control of spine dynamics96. An ASD-linked rare mutation in the GluN2B (encoded by GRIN2B)carboxy-terminal tail results in decreased surface trafficking and a reduction in the spine density of cultured hippocampal neurons97. In vivo, loss of GRIN2B causes reductions in spine density in CA1 hippocampal neurons, whereas deletion of GRIN2A causes decreases in dendritic complexity in the hippocampal dentate gyrus98,99. Interestingly, the predominant subunit used in the assembly of NMDARs changes during development from GluN2B (ASD-associated) to GluN2A (schizophrenia-associated)100. This developmental switch may reflect the importance of NMDA subunits at different phases of synaptic maturation and may implicate distinct NMDA-dependent mechanisms in governing the neurobiology and onset of ASD and schizophrenia.
AMPARs and NMDARs are the centrepiece of the activity-dependent mechanisms shaping dendritic spines. Hence, alterations in glutamate receptor expression, trafficking or function, caused by genetic variation, are likely to have important structural implications, especially in postnatal structural plasticity and activity-dependent development.
Scaffold proteins.
One of the most recognizable features of dendritic spines is the presence of a PSD, constructed from a complex network of scaffold proteins. Scaffold proteins have a highly modular domain structure and contain protein interaction sites enabling them to tether receptors, ion channels and cytoskeletal elements together to regulate synaptic structure101. SH3 and multiple ankyrin repeat domains (SHANK) and ankyrin (ANK) proteins are two types of scaffold proteins from the ankyrin repeat domain-containing protein (ANKRD) family. SHANK2 and SHANK3 mutations are two of the most prominent risk factors for ASD102. In addition, exonic mutations in SHANK1–SHANK3 have been identified in a wide range of neurodevelopmental disorders63,64,67. Large GWAS have implicated ANK3 as one of the very few genome-wide significant susceptibility genes for bipolar disorder61. These studies and others strongly implicate postsynaptic scaffolds in the pathogenesis of neuropsychiatric disorders (TABLE 1).
In Shank3-knockout mice, hippocampal neurons have decreased dendritic complexity and synapse density in vitro103. In addition to hippocampal dysfunction, SHANK3-deficient mice have reduced total spine density in medium spiny neurons expressing the dopamine D2 receptor but not the D1 receptor in vivo104. These observations implicate SHANK3 in the structural development of specific striatal circuits important for social behaviour. Interestingly, re-expression of SHANK3 in adulthood can rescue dendritic spine deficits and behavioural abnormalities105, indicating that neuronal circuits can be readjusted even after the major neurodevelopmental events shaping brain architecture have passed. The schizophrenia-linked R1117X mutation in SHANK3 reduces spine density in cortical neurons in vivo, consistent with post-mortem studies in schizophrenia106. The ASD-linked InsG3680 mutation also causes a significant decrease in spine density in homozygous mice. Another set of SHANK3 mutations identified in individuals with ASD causes deficits in hippocampal spine morphology and actin polymerization in vitro107.
The scaffold protein ANK3 (also known as ankyrin-G) is another ANKRD protein with important functions in spine plasticity108. The 190 kDa ANK3 isoform regulates spine head size and neck diameter. ANK3 accumulates in spine heads after chemical LTP, and knockdown of ANK3 disrupts LTP-induced increases in spine head size and density in cortical cultures. Notably, ANK3–190 is driven into spines upon lithium treatment in rats109, suggesting that this treatment induces similar mechanisms to LTP. Lithium has pleiotropic effects on cellular signalling; however, part of its effect on ANK3 transport may result from its role as a regulator of microtubule-related trafficking110. Thus, reduced ANK3 expression in spines may compromise the structure of mature spines and their ability to undergo activity-dependent structural changes.
Scaffold proteins are central hubs of the synapse, bringing upstream CAMs and receptors together with downstream signalling proteins and cytoskeletal elements. These are all classes of proteins that are individually relevant for neuropsychiatric disorders. Genetic insults to these structures would be expected to have diverse effects on both activity-dependent and activity-independent pathways regulating spine structure, possibly contributing to the wide spectrum of disorders with genetic variants in protein scaffolds (TABLE 1).
Calcium signalling.
The regulation of calcium influx into spines is crucial for activity-dependent signalling in neurons. Calcium channels are also important for activity-dependent postsynaptic structural refinement111,112. Calcium entry into spines is primarily controlled by synaptic activity and the gating properties of NMDARs and voltage-gated calcium channels (VGCCs), which trigger calcium-dependent signalling cascades113. Calcium channel genes CACNA1C and CACNB2 were found to be genome-wide significant in a GWAS combining data from five major psychiatric disorders114. In addition, rare coding variants in calcium-channel-related genes have been discovered in a number of different neuropsychiatric disorders and are enriched in individuals with schizophrenia115,116.
CACNA1C encodes the pore-forming a-subunit of the L-type VGCC Cav1.2 and is a consistently replicated genome-wide-significant risk factor for bipolar disorder and schizophrenia60,61. Heterozygous mutations in CACNA1C cause Timothy syndrome, a rare developmental disorder associated with cognitive dysfunction and autism117. Cav1.2 channels are widely expressed in dendrites and spines, where they regulate calcium entry118. Brain imaging studies have found that the CACNA1C risk allele is associated with an age-related prefrontal cortical thinning in individuals with bipolar disorder119. Activity-dependent pruning of the excitatory synapses of medium spiny neurons is dependent on Ca2+ entry through Cav1.2 in co-culture models, indicating that pruning may play a part in this age-related thinning120. Transgenic mice engineered with a Timothy-syndrome-related mutation in CACNA1C have decreased basal dendrite complexity in layer II/III, reminiscent of post-mortem analyses in bipolar disorder121. Disruption of downstream calcium-dependent signalling can also impair neuronal structure. A de novo ASD mutation in the catalytic domain of calcium/calmodulin-dependent kinase type II (CAMKII) decreases binding to other ASD-related proteins including SHANK3, the NMDAR subunit GluN2B, and the VGCC β2a subunit, causing reduced targeting to spines and reduced spine density in hippocampal cultures122.
Thus, elements of the calcium signalling pathway are directly involved in the structural rearrangement of neurons and may be particularly important for circuit remodelling after synaptic activity. Calcium triggers multiple downstream signalling pathways and may therefore affect an array of different cellular events linked to structural plasticity. Disruption to calcium signalling caused by genetic mutation would conceivably contribute to pathogenesis in early and late neuro-developmental events (TABLE 1).
Small GTPases.
Small GTPases are a large superfamily of regulatory proteins that induce activation of downstream signalling upon binding of GTP, which they then hydrolyse to GDP. RAS-related C3 botulinum toxin substrate 1 (RAC1) and RAS activation have been extensively characterized in vitro as causative factors in spine formation, enlargement, maturation and stabilization as well as synapse strengthening123,124, whereas transforming protein RhoA (RHOA)125 and RAP126 activation causes spine shrinkage and elimination as well as synapse weakening. GTPase activation is provided by the action of guanine nucleotide exchange factors (GEFs) and inactivation by GTPase-activating proteins (GAPs).
RAS has a well-documented role in regulating synaptic plasticity127 and has been implicated in intellectual disability128. RAS is classically activated in response to stimulation of receptor tyrosine kinases by neuro-trophins, inducing phosphoinositide 3-kinase (PI3K) and extracellular-signal-regulated kinase (ERK) signalling that promotes AMPAR membrane insertion and spine enlargement129 (FIG. 4). RAS/RAP GTPase-activating protein SynGAP (SYNGAP1) is a brain-specific RAS-GAP that is highly abundant in the PSD. Mutations within SYNGAP1 cause intellectual disability, epilepsy and ASD130. Defects in spine and synapse formation were observed in cultured Syngap1-knockout mouse neurons131,132, whereas Syngap1−/+ mice accumulated glutamate receptors in spines earlier than wild-type mice, resulting in significantly higher spine density and impaired synaptic plasticity133. Although heterozygous Syngap1 mutants display few synaptic defects in adulthood, conditional knockout during an early developmental stage was sufficient to induce learning deficits in adult mice, suggesting that SYNGAP1 and RAS regulation are critical during early neuronal development134.
Figure 4|. Pharmacological targets and associated structural pathways within the dendritic spine.
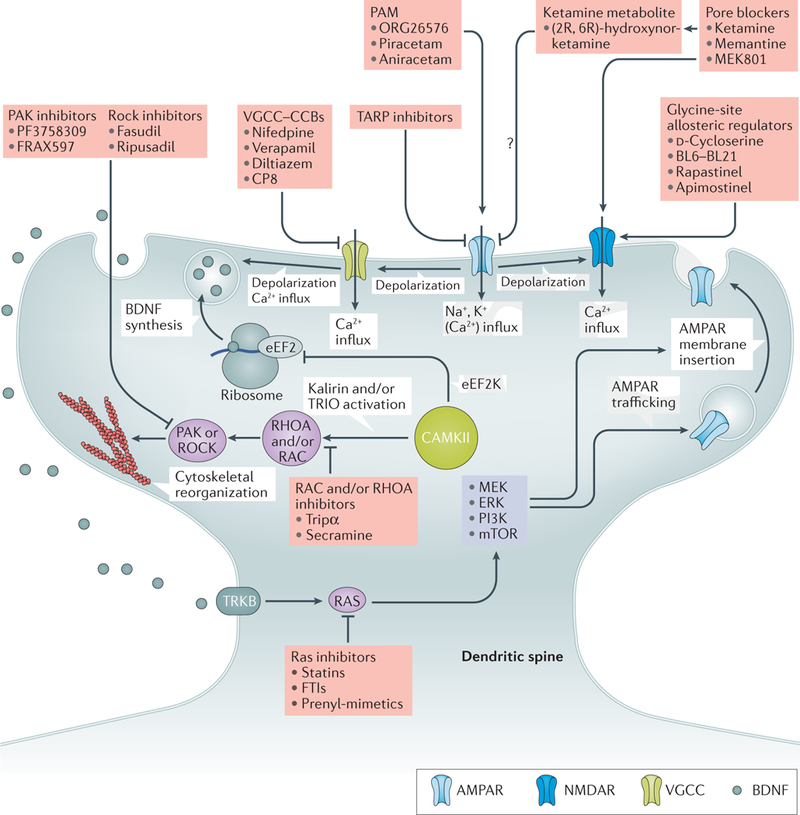
Within the dendritic spine, AMPA receptors (AMPARs) govern fast synaptic transmission in response to glutamatergic signalling. Glutamate binds to, and activates, AMPARs in response to synaptic activity. The resulting influx of sodium and potassium ions through the open channel results in depolarization277. NMDA receptor (NMDAR) activation occurs in response to glycine and glutamate binding concurrently with depolarization. In addition, depolarization results in the opening of voltage-gated calcium channels (VGCCs; including L-type, Cav1.2/1.3-containing channels). Depolarization and VGCC activation result in brain-derived neurotrophic factor (BDNF) exocytosis278. Calcium influx activates calcium/calmodulin-dependent kinase type II (CAMKII)279, resulting in activation of GTPase signalling by phosphorylation of kalirin139 and/ or triple functional domain protein (TRIO)140. These guanine nucleotide exchange factors regulate RAS-related C3 botulinum toxin substrate 1 (RAC)/transforming protein RhoA (RHOA) to alter cytoskeletal dynamics. GTPase activation can either increase (RAC pathway) or decrease (RHOA pathway) actin polymerization through serine/threonine protein kinase PAK (PAK) or Rho-associated protein kinase (ROCK) pathways to promote spine growth or retraction, respectively124,125. In addition, calcium results in the activation of eukaryotic elongation factor 2 kinase (eEF2K), phosphorylation of elongation factor 2 (EF2) at Thr56 (REF. 192) and inhibition of BDNF translation at ribosomes. BDNF release results in the activation of the BDNF/NT3 growth factor receptor (TRKB; also known as NTRK2) and induction of the RAS pathway through growth factor receptor-bound protein 2 (GRB2) and son of sevenless homologue 1 (SOS1; not shown), which in turn activates the phosphatidylinositol 3-kinase (PI3K) and MEK (MAPK/ERK kinase) and/ or extracellular-signal-regulated kinase (ERK) pathways, contributing to long-term potentiation via AMPAR trafficking and membrane insertion within the spine280. Many regulatory compounds targeting key synaptic signalling proteins have been isolated (indicated in the pink boxes). These, or other related small molecules, may have utility in neuropsychiatric disorders by regulating synaptic function and spine dynamics. CCBs, calcium channel blockers; FTIs, farnesyl transferase inhibitors; mTOR, mammalian target of rapamycin; PAM, positive allosteric modulator; TARP, transmembrane AMPAR regulator protein. TRIPα, TRIO inhibitory aptamer; Question mark indicates unknown mechanism.
Mutations within NF1, which encodes neuro-fibromin (another RAS-GAP present within spines), cause neurofibromatosis and intellectual disability135. Neurofibromin is required for normal spine density136. In addition, conditional knockout of Tsc1 and Tsc2, two genes encoding GAPs for the RAS-like GTP-binding protein Rheb (RHEB), in mice revealed the central importance of these molecules in maintaining normal spine size and density137. In humans, mutations in TSC1 and TSC2 cause tuberous sclerosis complex, a disorder associated with intellectual disability, ASD and epilepsy138. Therefore, RAS and/or RHEB regulation by GAPs is integral to the formation and development of spines, and its dysregulation is intimately linked to neuropsychiatric disorders.
RHOA and RAC1 are the archetypal RHO family members and control separate and opposing rearrangements of the actin cytoskeleton124,125 (FIG. 4). Several RHO-GEFs and RHO-GAPs have been associated with neuropsychiatric disorders and regulate spines. Kalirin and triple functional domain protein (TRIO) are highly homologous, multifunctional proteins that contain both RAC-GEF and RHO-GEF domains. Both proteins are abundant in spines and regulate spine maintenance139. A dual Trio and Kalrn knockout in mice resulted in an 80% loss of spines within CA1 hippocampal neurons and profound effects on LTP induction140. A rare mutation in the RAC-GEF domain of KALRN, detected in individuals with schizophrenia and major depressive disorder (MDD), induces spine loss in vitro and is associated with reduced cortical thickness in individuals with schizophrenia carrying the mutation141. Another mutation in KALRN, P2255T, is the only exonic mutation with an association with schizophrenia142 and recent studies suggest that it causes dendrite simplification143. Furthermore, a homozygous missense variant in KALRN causes intellectual disability144. Mutations in TRIO and/or KALRN have been detected in intellectual disability145,146 and in whole-exome sequencing studies in ASD70 and developmental disorders144. Notably, alterations in the kalirin–RAC–serine/threonine protein kinase PAK (PAK1) pathway have been reported in post-mortem studies in schizophrenia. These changes include reduced expression of kalirin 7 (REFS 147,148), increased expression of kalirin 9 (REF 149), reduced PAK1 phosphorylation and increased myosin light chain phosphorylation148. [Au:OK?] Similarly, levels of the closely related RAC1 homologue, CDC42, were reduced in human schizophrenia cortex148,150. Interestingly, despite their overlapping function within spines, mutations within TRIO and altered KALRN expression are linked to different disease states — KALRN with schizophrenia128,129 and TRIO with ASD and/or intellectual disability70,151. This difference may be a direct result of disrupted GTPase activity at key stages of spine formation and maturation, given that TRIO is expressed during early development152 and the main neuronal kalirin isoform, kalirin 7, is expressed at later stages153.
Several other RAC1 and/or RHO regulators and targets have been linked to intellectual disability. Of these, OPHN1 (encoding oligophrenin 1)154,155, ARHGEF6 (REF. 156) and PAK3 regulate spine plasticity157,158. Interestingly, mutations in GIT1, encoding a protein that regulates synapse formation159 and spine density160 and alters RAC and/or PAK3 signalling, has been linked to schizophrenia161. The related family members, PAK2 and PAK7, have also been found to play a key role in regulating neuroplasticity and psychoses, suggesting a functional link to schizophrenia162,163. In addition, the intellectual-disability-linked gene ARHGEF2, encoding the GEF H1, has been implicated in regulating spine density and length as well as AMPAR mediated structural plasticity164. The gene encoding WAVE-associated RAC GTPase-activating protein (WRP), SRGAP3, may be associated with intellectual disability165 and has been functionally linked to impaired spine precursor formation on dendrites, resulting in reduced spine density and mushroom spine number. Indeed, loss of WRP impairs learning and memory in heterozygous and null mouse models, highlighting the importance of this GAP in spine development and neuropsychiatric disorders166.
A prominent role for small GTPase regulatory pathways in neuropsychiatric disorders (TABLE 1 ) is consistent with their regulation of multiple spine and/or dendrite remodelling processes. Most severe mutations in these genes seem to be associated with early-onset neuropsychiatric disorders, such as intellectual disability and ASD, suggesting an important role in the early formation of synaptic circuits.
Novel therapeutic avenues
Traditional pharmacological treatments for neuropsychiatric disorders have typically targeted serotonin reuptake (tricyclic or selective serotonin reuptake inhibitor antidepressants167) and dopamine receptors (antipsychotics for schizophrenia and bipolar disorder168), whereas epilepsy treatments employ a range of sodium, calcium and GABAergic channel regulators depending on seizure type169. Although efficacious for many patients, these treatments suffer from various serious drawbacks. For example, antidepressants and anti-psychotics can cause serious side effects that result in high rates of noncompliance. Furthermore, antipsychotic use ameliorates the positive symptoms of schizophrenia and bipolar disorder with limited efficacy in treating negative and cognitive defects, whereas many sufferers of depression are resistant to treatment170. These limitations have prompted the search for novel drug targets, informed by molecular and genetic studies, which can circumvent the negative side effects of traditional therapeutics while improving patient outcomes (FIG. 4). A greater understanding of the molecular substrates of dendritic and spine alterations in neuropsychiatric disorders may facilitate the development of novel therapeutic approaches to reverse or delay neuronal defects and, therefore, the course of disease.
Despite the prevalence of adhesion and scaffold molecules identified in genetic studies of neuropsychiatric disorders, difficulty in targeting such classes has precluded the development of any clinically relevant drugs. Of the risk genes associated with neuropsychiatric disorders, membrane receptors have received the greatest attention in drug development, although recent clinical studies suggest that GTPase signalling pathways represent a novel therapeutic avenue. Below, we review recent developments in drug development targeting glutamatergic, calcium and GTPase signalling pathways within the synapse.
Targeting glutamate receptors.
Owing to their central importance in synaptic transmission and plasticity and their extensive genetic links to neuropsychiatric disorders, NMDARs171 and AMPARs172 have been the focus of many drug development attempts. Emerging data suggest that structural modulation of neurons through pharmacological intervention underlies some of the beneficial effects of NMDAR and AMPAR regulators in disease173,174. Indeed, as described above, it is hypothesized that reducing excessive glutamatergic activity and/or spine formation in ASD may be beneficial, whereas enhancing spine formation, stabilization and plasticity may be the goal in schizophrenia and bipolar disorder.
Interest in the NMDARs as a pharmacological target was spurred by the striking effect of the pore blocker ketamine on individuals with depression175. In contrast to typical antidepressants, a single intravenous injection of ketamine can produce a rapid, long-lasting reduction in the symptoms of depression (reviewed in REF. 176). Atypical spiny synapse connectivity has been reported in MDD177 and bipolar disorder52, and stress, a risk factor in MDD and bipolar disorder, causes spine alterations in animal models178,179. Accordingly, evidence shows that ketamine acts, at least in part, by modulating spine plasticity180. For example, ketamine rapidly increased the number and function of new spiny synapses in the PFC of rats through mechanisms involving mammalian target of rapamycin (mTOR) signalling, reversing stress-induced synaptic deficits181. In another study, ketamine enhanced spine formation and induced a larger-scale remodelling of the dendritic arbor at different rates173.
Despite its remarkable antidepressant effects, ketamine can induce psychotic behaviour and cognitive defects in healthy individuals and exacerbate schizophrenia psychoses182–184. Furthermore, controversy exists over its mechanism of action. Indeed, mechanisms involving AMPAR regulation185, reduction of presynaptic inhibitory GABAergic input186, alterations in NMDA-mediated miniature excitatory postsynaptic currents 187 and metabolism of ketamine to (2R,6R)-hydroxynorketamine188 have been proposed (reviewed in REFS 189,190). The underlying effects have been extensively linked to the altered translation of brain-derived neurotrophic factor (BDNF), which has been shown to have a crucial role in mediating the sustained antidepressant effects of ketamine191,192. Therefore, a greater understanding of ketamine’s modulation of spine plasticity is crucial to engineer drugs developed for maximal efficacy in neuropsychiatric disorders with minimal psychomimetic and cognitive side effects.
NMDAR partial agonists, such as glycine/D-serine and D-cycloserine, have been shown to be efficacious in reducing depression in schizophrenia193–195 and ASD196 and lack the psychomimetic side effects of ketamine. The success of NMDAR partial agonism is exemplified by rapastinel, a tetrapeptide derivative of the B6-B21 monoclonal antibody, which targets the glycine-binding site197. This brain-permeable compound has shown promise in both preclinical and clinical trials, including fast-acting antidepressant effects similar to ketamine in the absence of psychomimetic side effects198, which has warranted ‘fast-tracking’ to phase III clinical trials (NCT02951988 (REF. 199) and NCT02943564 (REF. 200)). Of particular interest is the ability of rapastinel to reverse social phenotypes in preclinical animal models, suggesting that NMDAR regulation is a potential treatment for ASD201,202. Several studies show that rapastinel acts, at least in part, by modulating spine plasticity; treatment rapidly increased the number and function of spine synapses in the apical dendritic tuft of layer V pyramidal neurons in the medial PFC180, enhanced the recovery of spines after insults203 and increased sensitivity to LTP and metaplasticity204. Apimostinel, a rapastinel derivative that is suitable for oral dosage, has recently been developed, and early clinical trials for depression have recently been completed, although results are pending (NCT02067793 (REF. 205)).
Increased AMPAR ligand binding has been observed in the striatum of individuals with schizophrenia and suicide victims206, suggesting deregulation of AMPAR activity in MDD and schizophrenia. Indeed, preclinical study of the role of positive allosteric modulators (PAMs; including ampakines) revealed a striking antidepressant effect in a variety of mouse models (reviewed in REF. 207). Furthermore, PAMs increased synaptic plasticity and showed promise in cognitive enhancement208. Thus, AMPARs may represent a promising therapeutic target in regulating cognitive impairment and mood disorders, such as bipolar disorder, schizophrenia and MDD. Ampakines have been shown to modulate spine plasticity, which may underlie part of their action. They facilitate dendritic recovery and improve synaptic plasticity and memory in middle-aged rats209 and promote spine actin polymerization in a model of Angelman syndrome210. The ampakines CX546 and aniracetam reduced prepulse inhibition in a mouse model of schizophrenia211. This effect may be linked to the ability of CX546 to prime spines for structural plasticity, promote spine head enlargement and (in presynaptic terminals) increase the probability of neurotransmitter release212.
Clinical trials assessing the efficacy of ampakines in disease yielded mixed results. Phase I clinical trials assessing the suitable dosage of the AMPAR–PAM, Org 26576, revealed the drug was well tolerated, and a second study suggested both an improvement in depressive symptomology and signs of cognitive enhancement213,214. However, the PAM CX516 showed varying efficacy, failing to produce significant improvement in cognitive function in individuals with fragile X syndrome or schizophrenia215. Despite the potential identified in preclinical studies, these results suggest that clinical efficacy is dependent on the drug mechanism or molecular and genetic context of disease. Further studies are required to confirm the potential of ampakines in AMPAR-related neuropsychiatric disorders.
Inhibition of AMPAR may be beneficial in neuropsychiatric disorders associated with an increased excitatory-to-inhibitory ratio216, such as epilepsy, ASD and some forms of intellectual disability. Indeed, the clinically approved AMPAR inhibitor perampanel has efficacy in reducing tonic-clonic seizures217. However, general AMPAR inhibition leads to an array of side effects and, to date, no selective subunit inhibitors have been isolated218. To address this lack of specificity and avoid unwanted side effects, novel brain-region-specific protein modulators of AMPAR activity have been developed that inhibit the interaction of AMPARs with transmembrane AMPAR regulatory proteins (TARPs; encoded by genes CACNG1–CACNG8 (REF. 191)). Of particular interest is TARPλ8, which is expressed within the forebrain where it regulates AMPAR protein levels, surface expression and function215. As a result, disrupting the TARPλ8–AMPAR interaction yields brain-region-selective AMPAR regulation. Recently, compounds that selectively inhibit the AMPAR–TARPλ8 interaction have been shown to prevent seizures in animal models without the motor side effects associated with general AMPAR inhibition219,220. As TARPλ8 is underexpressed in schizophrenia221 and CACNG2 and CACNG8 have been implicated as risk factors in schizophrenia and bipolar disorder (TABLE 1 ), compounds able to stabilize AMPAR–TARP complexes may provide a therapeutic strategy. TARPs target AMPAR to the PSD222 and are required for LTP222, suggesting that TARP regulators may be of substantial benefit to disorders stemming from abnormal structural plasticity.
Targeting calcium signalling.
Calcium signalling is central to dendritic development and spine plasticity, with the L-type VGCC mediating synaptic calcium influx in response to membrane depolarization223. Within the brain, Cav1.2 and Cav1.3 (encoded by CACNA1C and CACNA1D, respectively) are the predominant forms of the pore-forming calcium channel subunits and are major risk genes in a range of neuropsychiatric disorders (TABLE 1). They have been the target of effective VGCC channel blockers (CCBs; including nifedipine, verapamil and diltiazem223) developed for hypertension and arrhythmia. The ability to repurpose VGCC–CBCs for neuropsychiatric disorders prompted studies in the 1980s and 1990s, with several positive outcomes as adjunctive therapies to antipsychotics224. However, critical evaluation of multiple studies found that these drugs were as effective as placebo or produced inconsistent results225,226. Identification of CACNA1C in GWAS as a risk factor in schizophrenia227 and bipolar disorder61 has prompted assessment of isradipine in schizophrenia (currently recruiting, NCT01658150 (REF 228)), and early trials suggest potential in bipolar disorder229. The observation that Cav1.2 is the major target of cardiac-protective and pulmonary-protective drugs prompted attempts to target Cav1.3 (REF 230). Such a strategy may avoid non-neural activation of Cav1.2 and thus facilitate Cav1.3-selective brain targeting and a higher tolerable dosage. A recent screen identified Cp8 as a compound exhibiting 600-fold selectivity for Cav1.3 over Cav1.2 (REF 230). Such Cav1.3 selective compounds could provide a novel route to regulate CACNA1D-dependent neuropsychiatric disorders. Future work aimed at revealing the modulation of dendritic and spine plasticity by such compounds may support VGCCs as a valuable therapeutic target in neurodevelopmental disorders.
Targeting small GTPases.
Small GTPases have been the target of extensive drug discovery efforts, largely owing to their role in cancers, providing a bounty of therapeutic agents with already well-characterized tolerability. Thus, reversing the RAS dysregulation caused by NF1 and SYNGAP1 mutations (see above) may be a valuable target for repurposing efforts in neuropsychiatric disorders.
RAS requires post-translation prenylation for its membrane insertion and full activity. The observation that limiting the addition of a farnesyl group to oncogenic RAS abrogated its transforming activity spurred the development of compounds to inhibit this process (reviewed in REF 231). Most of these agents inhibit farnesyltransferase (FT), but others (including statins) target farnesyl diphosphate synthesis to limit RAS activity. The potential of FT inhibitors and statins to treat neurological disease has been shown preclinically in mouse models of neurofibromatosis type 1 (REFS 232,233), prompting clinical trials in neurofibromatosis type 1 patients. The FT inhibitor tipifarnib produced significant increases in emotional health and suggestive evidence of improved cognitive function234. Similarly, lovastatin improved synaptic plasticity235 and learning and memory in neurofibromatosis type 1 patients during early-phase clinical trials236,237. However, a recent, larger trial of lovastatin failed to show benefit in cognition to children with neurofibromatosis type 1 (REF 238). Thus, these studies suggest that RAS inhibitors are useful in neuropsychiatric disorders stemming from overactive RAS. Despite preclinical promise, conclusive evidence of the effects of RAS regulation in human neurodevelopmental disease is lacking.
Conclusions and future directions
Spine pathology is a convergence point of many neuropsychiatric disorders, as supported by mounting evidence from human post-mortem, cellular, animal and genetic studies. The most important advance in the past decade has come from translating human genetic information into bona fide risk factors and meaningful disease pathways in order to successfully model structural deficits under experimental conditions. Although this Review has covered many of the critical pathways involved in spine plasticity and disease, there are clearly other important pathways and genes that we have not been able to cover, including those affecting Wnt signalling, the actin cytoskeleton and chromatin remodelling71,239,240. As the genetic studies of neuropsychiatric disorders progress, we can expect more disease-relevant biological processes to be revealed as well as a more complete understanding of the role of structural plasticity in other psychiatric conditions where genetic and experimental data are still emerging, such as attention-deficit hyperactivity disorder (ADHD), anxiety and depression. Functional analysis of these newly discovered genetic risk factors will be paramount to uncover novel mechanisms and pathways leading to atypical spine and dendrite development. In this endeavour, omic technologies and systems biology-level investigations will be essential to deconstruct the global alterations taking place in model systems. Drug discovery efforts may then be designed with genetic and pathway disruptions as a guide to better target underlying disease mechanisms.
In addition to genetics, new technological advancements in stem cell reprogramming and imaging (BOXES 1,2) have substantially improved our understanding of the mechanisms regulating spine plasticity in health and disease. To further our understanding of structural deficits in human brain, future neuropathological studies should aim to stratify individuals on the basis of genetics and disease severity to address the considerable heterogeneity in neuropsychiatric disorders. The challenge for the future will be to integrate our expanding knowledge of human disorders into more comprehensive models of disease. Human neurons derived from genetically defined individuals will likely play a critical role (BOX 2), overcoming the limited genetic complexity and species differences inherent in animal models. Stem cell models provide a unique opportunity to examine the effects of new drugs that target structural plasticity pathways directly in dysmorphic patient neurons and in a high-throughput manner. The fact that structural pathology can be observed before cognitive symptoms in certain disorders suggests that there are critical time windows for treatment strategies to prevent disease onset241. Furthermore, several mouse models have also shown that certain structural and behavioural deficits can be reversed in adult animals105,242–244, offering hope for treating human conditions.
Supplementary Material
Key points
The development of dendritic branches and spines, which host the excitatory postsynaptic machinery, is atypical in neuropsychiatric disorders.
Genetic risk factors for neuropsychiatric disease converge on genes that encode proteins present in excitatory postsynaptic termini with known roles in dendritic structural plasticity.
Risk variants in genes regulating structural plasticity may explain the atypical dendrite and spine development observed in individuals with neuropsychiatric disorders.
Drug discovery efforts targeting the genetic risk factors affecting the structural plasticity of dendritic spines have been successful in improving patient outcomes. Genetics may facilitate the prioritization, or elucidation, of drug targets within neuropsychiatric disorders.
Stem cell models can be used to model the structural deficits in individuals with neuropsychiatric disease, offering an avenue for screening personalized treatments.
Subject categories
Biological sciences / Neuroscience / Spine regulation and structure / Spine plasticity [URI /631/378/2597/2599]
Biological sciences / Neuroscience / Diseases of the nervous system / Autism spectrum disorders [URI /631/378/1689/1373]
Biological sciences / Neuroscience / Diseases of the nervous system / Schizophrenia [URI /631/378/1689/1799]
Biological sciences / Neuroscience / Diseases of the nervous system / Bipolar disorder [URI /631/378/1689/1333]
Techniques terms
Life sciences techniques, Genomic analysis [Genome-wide association studies]
Life sciences techniques, Genomic analysis [DNA sequencing]
Life sciences techniques [Cellular imaging]
RELATED LINKS
NIH Research Domain Criteria (RDoC): https://www.nimh.nih.gov/research-priorities/rdoc/index.shtml
denovo-db (collection of human de novo variants): http://denovo-db.gs.washington.edu/denovo-db/
DAVID (Database for annotation, visualization and integrated discovery): https://david.ncifcrf.gov/
ALL LINKS ARE ACTIVE IN THE ONLINE PDF
Dendritic spines
Micrometre protrusions on dendritic branches of neurons that host the majority of excitatory synapses in the brain.
Genetic risk factors
DNA sequence variants that are associated with increased disease susceptibility.
Synaptic input field
The total number and spatial arrangement of a neuron’s postsynaptic receptors. This postsynaptic field is determined by the architecture of the dendritic tree and the number and size of receptor domains contained within dendritic spines.
Postsynaptic density(PSD).
A specialization on excitatory dendritic spines, originally identified by electron microscopy, which contains glutamate receptors and many associated scaffolding and trafficking proteins that are crucial for excitatory synaptic transmission.
Long-term potentiation(LTP).
Long-lasting increase in synaptic strength between neurons, usually resulting from synchronous or temporally coordinated presynaptic and postsynaptic activity,
Long-term depression(LTD).
Long-lasting weakening of synaptic strength between neurons, often resulting from asynchronous presynaptic and postsynaptic activity.
Cell reprogramming technologies
Techniques in biotechnology used to convert one cell type into another using defined biological factors; for example transcription factors can be introduced into adult somatic cells to reprogramme them into induced pluripotent stem cells or induced neurons.
Common variants
Alterations in a DNA sequence, often with small effect on disease risk, which are present in large proportion of the general population (>1%). These are typically single-nucleotide polymorphisms that are identified in genome-wide association study (GWAS).
Rare variants
Mutations in a DNA sequence, often with a large effect on disease risk, which occur only in a small fraction of the general population (<1%). These types of variants are usually identified in family, exome sequencing or copy number variant (CNV) studies.
Genome-wide association studies(GWAS).
Genetic study designed to identify variants in the genome that associate with a disease or trait. Large-scale GWAS have been especially successful in identifying many common risk variants in schizophrenia and bipolar disorder.
Exome sequencing
Genomic sequencing technique used to sequence all of the protein-coding regions (exons) of the genome.
Enrichment analyses
Bioinformatic technique used to identify over-represented gene sets or biological processes from large omic data sets. This method is typically used to understand the concerted function of hundreds of genes and/or proteins from genomic and proteomic data.
Acknowledgements
The authors thank M. D. Martin-de-Saavedra and K. Myczek for lively discussions and critical input on preparing the manuscript.
Footnotes
Competing interests
The authors declare no competing interests.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer Information
Nature Reviews Neuroscience thanks L. Pozzo-Miller, H. Zhang and the other anonymous reviewer(s), for their contribution to the peer review of this work.
References
- 1.Tau GZ & Peterson BS Normal development of brain circuits. Neuropsychopharmacology 35, 147–168 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wefelmeyer W, Puhl CJ & Burrone J Homeostatic plasticity of subcellular neuronal structures: from inputs to outputs. Trends Neurosci. 39, 656–667 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Penzes P, Cahill ME, Jones KA, VanLeeuwen JE & Woolfrey KM Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 14, 285–293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taber KH, Hurley RA & Yudofsky SC Diagnosis and treatment of neuropsychiatric disorders. Annu. Rev. Med. 61, 121–133 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Kirov G et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry 17, 142–153 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanders SJ et al. Insights into autism spectrum disorder genomic architecture and biology from 71 risk loci. Neuron 87, 1215–1233 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hamdan FF et al. Excess of de novo deleterious mutations in genes associated with glutamatergic systems in nonsyndromic intellectual disability. Am. J. Hum. Genet. 88, 306–316 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall J, Trent S, Thomas KL, O’Donovan MC & Owen MJ Genetic risk for schizophrenia: convergence on synaptic pathways involved in plasticity. Biol. Psychiatry 77, 52–58 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Epi4K Consortium et al. De novo mutations in epileptic encephalopathies. Nature 501, 217–221 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanner AM Management of psychiatric and neurological comorbidities in epilepsy. Nat. Rev. Neurol. 12, 106–116 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Spruston N Pyramidal neurons: dendritic structure and synaptic integration. Nat. Rev. Neurosci. 9, 206–221 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Koleske AJ Molecular mechanisms of dendrite stability. Nat. Rev. Neurosci. 14, 536–550 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y & Sabatini BL Signaling in dendritic spines and spine microdomains. Curr. Opin. Neurobiol. 22, 389–396 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berry KP & Nedivi E Spine dynamics: are they all the same? Neuron 96, 43–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tonnesen J & Nagerl UV Dendritic spines as tunable regulators of synaptic signals. Front. Psychiatry 7, 101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mrzljak L & Uylings HB, Kostovic I & Van Eden CG. Prenatal development of neurons in the human prefrontal cortex: I. A qualitative Golgi study. J. Comp. Neurol. 271, 355–386 (1988). [DOI] [PubMed] [Google Scholar]
- 17.Petanjek Z et al. Extraordinary neoteny of synaptic spines in the human prefrontal cortex. Proc. Natl Acad. Sci. USA 108, 13281–13286 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tamnes CK et al. Development of the cerebral cortex across adolescence: a multisample study of inter-related longitudinal changes in cortical volume, surface area, and thickness. J. Neurosci. 37, 3402–3412 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zuo Y, Lin A, Chang P & Gan WB Development of long-term dendritic spine stability in diverse regions of cerebral cortex. Neuron 46, 181–189 (2005). [DOI] [PubMed] [Google Scholar]
- 20.Caroni P, Donato F & Muller D Structural plasticity upon learning: regulation and functions. Nat. Rev. Neurosci. 13, 478–490 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Yang G, Pan F & Gan WB Stably maintained dendritic spines are associated with lifelong memories. Nature 462, 920–924 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bourne J & Harris KM Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol. 17, 381–386 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Zenke F, Gerstner W & Ganguli S The temporal paradox of Hebbian learning and homeostatic plasticity. Curr. Opin. Neurobiol. 43, 166–176 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Bosch M & Hayashi Y Structural plasticity of dendritic spines. Curr. Opin. Neurobiol. 22, 383–388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Herring BE & Nicoll RA Long-term potentiation: from CaMKII to AMPA receptor trafficking. Annu. Rev. Physiol. 78, 351–365 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Turrigiano G Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb. Perspect. Biol. 4, a005736 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wallace W & Bear MF A morphological correlate of synaptic scaling in visual cortex. J. Neurosci. 24, 6928–6938 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsuzaki M et al. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 4, 1086–1092 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kirov SA, Goddard CA & Harris KM Age-dependence in the homeostatic upregulation of hippocampal dendritic spine number during blocked synaptic transmission. Neuropharmacology 47, 640–648 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Hofer SB, Mrsic-Flogel TD, Bonhoeffer T & Hubener M Experience leaves a lasting structural trace in cortical circuits. Nature 457, 313–317 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keck T et al. Synaptic scaling and homeostatic plasticity in the mouse visual cortex in vivo. Neuron 80, 327–334 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Barnes SJ et al. Deprivation-induced homeostatic spine scaling in vivo is localized to dendritic branches that have undergone recent spine loss. Neuron 96, 871–882.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jiang M, Lee CL, Smith KL & Swann JW Spine loss and other persistent alterations of hippocampal pyramidal cell dendrites in a model of early-onset epilepsy. J. Neurosci. 18, 8356–8368 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wong M & Guo D Dendritic spine pathology in epilepsy: cause or consequence? Neuroscience 251, 141–150 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Chen CC, Bajnath A & Brumberg JC The impact of development and sensory deprivation on dendritic protrusions in the mouse barrel cortex. Cereb. Cortex 25, 1638–1653 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou Y, Lai B & Gan WB Monocular deprivation induces dendritic spine elimination in the developing mouse visual cortex. Sci. Rep. 7, 4977 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sigler A et al. Formation and maintenance of functional spines in the absence of presynaptic glutamate release. Neuron 94, 304–311.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McCullumsmith RE, Hammond JH, Shan D & Meador-Woodruff JH Postmortem brain: an underutilized substrate for studying severe mental illness. Neuropsychopharmacology 40, 1307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Purpura DP Dendritic spine “dysgenesis” and mental retardation. Science 186, 1126–1128 (1974). [DOI] [PubMed] [Google Scholar]
- 40.Kaufmann WE & Moser HW Dendritic anomalies in disorders associated with mental retardation. Cereb. Cortex 10, 981–991 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Hutsler JJ & Zhang H Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309, 83–94 (2010). [DOI] [PubMed] [Google Scholar]
- 42.Tang G et al. Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hazlett HC et al. Early brain development in infants at high risk for autism spectrum disorder. Nature 542, 348–351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schumann CM et al. Longitudinal magnetic resonance imaging study of cortical development through early childhood in autism. J. Neurosci. 30, 4419–4427 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Multani P, Myers RH, Blume HW, Schomer DL & Sotrel A Neocortical dendritic pathology in human partial epilepsy: a quantitative Golgi study. Epilepsia 35, 728–736 (1994). [DOI] [PubMed] [Google Scholar]
- 46.Bothwell S et al. Neuronal hypertrophy in the neocortex of patients with temporal lobe epilepsy. J. Neurosci. 21, 4789–4800 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glausier JR & Lewis DA Dendritic spine pathology in schizophrenia. Neuroscience 251, 90–107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Glantz LA & Lewis DA Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch. Gen. Psychiatry 57, 65–73 (2000). [DOI] [PubMed] [Google Scholar]
- 49.MacDonald ML et al. Selective loss of smaller spines in schizophrenia. Am. J. Psychiatry 174, 586–594 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kalus P, Muller TJ, Zuschratter W & Senitz D The dendritic architecture of prefrontal pyramidal neurons in schizophrenic patients. Neuroreport 11, 3621–3625 (2000). [DOI] [PubMed] [Google Scholar]
- 51.Broadbelt K, Byne W & Jones LB Evidence for a decrease in basilar dendrites of pyramidal cells in schizophrenic medial prefrontal cortex. Schizophr. Res. 58, 75–81 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Konopaske GT, Lange N, Coyle JT & Benes FM Prefrontal cortical dendritic spine pathology in schizophrenia and bipolar disorder. JAMA Psychiatry 71, 1323–1331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tobe BTD et al. Probing the lithium-response pathway in hiPSCs implicates the phosphoregulatory set-point for a cytoskeletal modulator in bipolar pathogenesis. Proc. Natl Acad. Sci. USA 114, E4462–E4471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moyer CE, Shelton MA & Sweet RA Dendritic spine alterations in schizophrenia. Neurosci. Lett. 601, 46–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dietsche B, Kircher T & Falkenberg I Structural brain changes in schizophrenia at different stages of the illness: a selective review of longitudinal magnetic resonance imaging studies. Aust. N. Z. J. Psychiatry 51, 500–508 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Mollon J & Reichenberg A Cognitive development prior to onset of psychosis. Psychol. Med. 48, 392–403 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Phillips M & Pozzo-Miller L Dendritic spine dysgenesis in autism related disorders. Neurosci. Lett. 601, 30–40 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Levenga J & Willemsen R Perturbation of dendritic protrusions in intellectual disability. Prog. Brain Res. 197, 153–168 (2012). [DOI] [PubMed] [Google Scholar]
- 59.Sullivan PF, Daly MJ & O’Donovan M Genetic architectures of psychiatric disorders: the emerging picture and its implications. Nat. Rev. Genet. 13, 537–551 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 511, 421–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Psychiatric GWAS Consortium Bipolar Disorder Working Group. Large-scale genome-wide association analysis of bipolar disorder identifies a new susceptibility locus near ODZ4. Nat. Genet. 43, 977–983 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kirov G CNVs in neuropsychiatric disorders. Hum. Mol. Genet. 24, R45–R49 (2015). [DOI] [PubMed] [Google Scholar]
- 63.Fromer M et al. De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yuen RKC et al. Whole genome sequencing resource identifies 18 new candidate genes for autism spectrum disorder. Nat. Neurosci. 20, 602–611 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lelieveld SH et al. Meta-analysis of 2,104 trios provides support for 10 new genes for intellectual disability. Nat. Neurosci. 19, 1194–1196 (2016). [DOI] [PubMed] [Google Scholar]
- 66.Epi4K Consortium. De novo mutations in SLC1A2 and CACNA1A are important causes of epileptic encephalopathies. Am. J. Hum. Genet. 99, 287–298 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Deciphering Developmental Disorders Study. Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chang J, Gilman SR, Chiang AH, Sanders SJ & Vitkup D Genotype to phenotype relationships in autism spectrum disorders. Nat. Neurosci. 18, 191–198 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gilman SR et al. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 70, 898–907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.De Rubeis S et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 515, 209–215 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McCarthy SE et al. De novo mutations in schizophrenia implicate chromatin remodeling and support a genetic overlap with autism and intellectual disability. Mol. Psychiatry 19, 652–658 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kochinke K et al. Systematic phenomics analysis deconvolutes genes mutated in intellectual disability into biologically coherent modules. Am. J. Hum. Genet. 98, 149–164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Dalva MB, McClelland AC & Kayser MS Cell adhesion molecules: signalling functions at the synapse. Nat. Rev. Neurosci. 8, 206–220 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rujescu D et al. Disruption of the neurexin 1 gene is associated with schizophrenia. Hum. Mol. Genet. 18, 988–996 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Jamain S et al. Mutations of the X-linked genes encoding neuroligins NLGN3 and NLGN4 are associated with autism. Nat. Genet. 34, 27–29 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zweier C et al. CNTNAP2 and NRXN1 are mutated in autosomal-recessive Pitt-Hopkins-like mental retardation and determine the level of a common synaptic protein in Drosophila. Am. J. Hum. Genet. 85, 655–666 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Moller RS et al. Exon-disrupting deletions of NRXN1 in idiopathic generalized epilepsy. Epilepsia 54, 256–264 (2013). [DOI] [PubMed] [Google Scholar]
- 78.Graf ER, Zhang X, Jin SX, Linhoff MW & Craig AM Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119, 1013–1026 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Quinn DP et al. Pan-neurexin perturbation results in compromised synapse stability and a reduction in readily releasable synaptic vesicle pool size. Sci. Rep. 7, 42920 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Chih B, Engelman H & Scheiffele P Control of excitatory and inhibitory synapse formation by neuroligins. Science 307, 1324–1328 (2005). [DOI] [PubMed] [Google Scholar]
- 81.Isshiki M et al. Enhanced synapse remodelling as a common phenotype in mouse models of autism. Nat. Commun. 5, 4742 (2014). [DOI] [PubMed] [Google Scholar]
- 82.Etherton M et al. Autism-linked neuroligin-3 R451C mutation differentially alters hippocampal and cortical synaptic function. Proc. Natl Acad. Sci. USA 108, 13764–13769 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rodenas-Cuadrado P et al. Characterisation of CASPR2 deficiency disorder — a syndrome involving autism, epilepsy and language impairment. BMC Med. Genet. 17, 8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Bakkaloglu B et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am. J. Hum. Genet. 82, 165–173 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Friedman JI et al. CNTNAP2 gene dosage variation is associated with schizophrenia and epilepsy. Mol. Psychiatry 13, 261–266 (2008). [DOI] [PubMed] [Google Scholar]
- 86.Varea O et al. Synaptic abnormalities and cytoplasmic glutamate receptor aggregates in contactin associated protein-like 2/Caspr2 knockout neurons. Proc. Natl Acad. Sci. USA 112, 6176–6181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gdalyahu A et al. The autism related protein contactin-associated protein-like 2 (CNTNAP2) stabilizes new spines: an in vivo mouse study. PLoS ONE 10, e0125633 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Anderson GR et al. Candidate autism gene screen identifies critical role for cell-adhesion molecule CASPR2 in dendritic arborization and spine development. Proc. Natl Acad. Sci. USA 109, 18120–18125 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McKinney RA, Capogna M, Durr R, Gahwiler BH & Thompson SM Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat. Neurosci. 2, 44–49 (1999). [DOI] [PubMed] [Google Scholar]
- 90.Richards DA et al. Glutamate induces the rapid formation of spine head protrusions in hippocampal slice cultures. Proc. Natl Acad. Sci. USA 102, 6166–6171 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hayashi Y et al. Driving AMPA receptors into synapses by LTP and CaMKII: requirement for GluR1 and PDZ domain interaction. Science 287, 2262–2267 (2000). [DOI] [PubMed] [Google Scholar]
- 92.Zhang Y, Cudmore RH, Lin DT, Linden DJ & Huganir RL Visualization of NMDA receptor-dependent AMPA receptor synaptic plasticity in vivo. Nat. Neurosci. 18, 402–407 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kopec CD, Real E, Kessels HW & Malinow R GluR1 links structural and functional plasticity at excitatory synapses. J. Neurosci. 27, 13706–13718 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luscher C & Malenka RC NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. 4, a005710 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Luthi A, Schwyzer L, Mateos JM, Gahwiler BH & McKinney RA NMDA receptor activation limits the number of synaptic connections during hippocampal development. Nat. Neurosci. 4, 1102–1107 (2001). [DOI] [PubMed] [Google Scholar]
- 96.Alvarez VA, Ridenour DA & Sabatini BL Distinct structural and ionotropic roles of NMDA receptors in controlling spine and synapse stability. J. Neurosci. 27, 7365–7376 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Liu S et al. A rare variant identified within the GluN2B C-terminus in a patient with autism affects NMDA receptor surface expression and spine density. J. Neurosci. 37, 4093–4102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brigman JL et al. Loss of GluN2B-containing NMDA receptors in CA1 hippocampus and cortex impairs long-term depression, reduces dendritic spine density, and disrupts learning. J. Neurosci. 30, 4590–4600 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kannangara TS et al. Deletion of the NMDA receptor GluN2A subunit significantly decreases dendritic growth in maturing dentate granule neurons. PLoS ONE 9, e103155 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sanz-Clemente A, Nicoll RA & Roche KW Diversity in NMDA receptor composition: many regulators, many consequences. Neuroscientist 19, 62–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhu J, Shang Y & Zhang M Mechanistic basis of MAGUK-organized complexes in synaptic development and signalling. Nat. Rev. Neurosci. 17, 209–223 (2016). [DOI] [PubMed] [Google Scholar]
- 102.Monteiro P & Feng G SHANK proteins: roles at the synapse and in autism spectrum disorder. Nat. Rev. Neurosci. 18, 147–157 (2017). [DOI] [PubMed] [Google Scholar]
- 103.Yi F et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science 352, aaf2669 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wang W et al. Striatopallidal dysfunction underlies repetitive behavior in Shank3-deficient model of autism. J. Clin. Invest. 127, 1978–1990 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mei Y et al. Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhou Y et al. Mice with Shank3 mutations associated with ASD and schizophrenia display both shared and distinct defects. Neuron 89, 147–162 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Durand CM et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol. Psychiatry 17, 71–84 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Smith KR et al. Psychiatric risk factor ANK3/ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron 84, 399–415 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nanavati D et al. The effects of chronic treatment with mood stabilizers on the rat hippocampal postsynaptic density proteome. J. Neurochem. 119, 617–629 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gottschalk MG et al. Lithium reverses behavioral and axonal transport-related changes associated with ANK3 bipolar disorder gene disruption. Eur. Neuropsychopharmacol 27, 274–288 (2017). [DOI] [PubMed] [Google Scholar]
- 111.Kang MG et al. A functional AMPA receptor-calcium channel complex in the postsynaptic membrane. Proc. Natl Acad. Sci. USA 103, 5561–5566 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wong RO & Ghosh A Activity-dependent regulation of dendritic growth and patterning. Nat. Rev. Neurosci. 3, 803–812 (2002). [DOI] [PubMed] [Google Scholar]
- 113.Higley MJ & Sabatini BL Calcium signaling in dendritic spines. Cold Spring Harb. Perspect. Biol. 4, a005686 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet 381, 1371–1379 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Purcell SM et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 506, 185–190 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Heyes S et al. Genetic disruption of voltage-gated calcium channels in psychiatric and neurological disorders. Prog. Neurobiol. 134, 36–54 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Splawski I et al. Cay1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31 (2004). [DOI] [PubMed] [Google Scholar]
- 118.Obermair GJ, Szabo Z, Bourinet E & Flucher BE Differential targeting of the L-type Ca2+ channel alpha 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur. J. Neurosci. 19, 2109–2122 (2004). [DOI] [PubMed] [Google Scholar]
- 119.Soeiro-de-Souza MG et al. The CACNA1C risk allele rs1006737 is associated with age-related prefrontal cortical thinning in bipolar I disorder. Transl Psychiatry 7, e1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tian X, Kai L, Hockberger PE, Wokosin DL & Surmeier DJ MEF-2 regulates activity-dependent spine loss in striatopallidal medium spiny neurons. Mol. Cell Neurosci. 44, 94–108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Krey JF et al. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 16, 201–209 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Stephenson JR et al. A novel human CAMK2A mutation disrupts dendritic morphology and synaptic transmission, and causes ASD-related behaviors. J. Neurosci. 37, 2216–223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Penzes P et al. Rapid induction of dendritic spine morphogenesis by trans-synaptic ephrinB-EphB receptor activation of the Rho-GEF kalirin. Neuron 37, 263–274 (2003). [DOI] [PubMed] [Google Scholar]
- 124.Tashiro A & Yuste R Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol. Cell Neurosci. 26, 429–440 (2004). [DOI] [PubMed] [Google Scholar]
- 125.Tashiro A, Minden A & Yuste R Regulation of dendritic spine morphology by the rho family of small GTPases: antagonistic roles of Rac and Rho. Cereb. Cortex 10, 927–938 (2000). [DOI] [PubMed] [Google Scholar]
- 126.Woolfrey KM et al. Epac2 induces synapse remodeling and depression and its disease-associated forms alter spines. Nat. Neurosci. 12, 1275–1284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Stornetta RL & Zhu JJ Ras and Rap signaling in synaptic plasticity and mental disorders. Neuroscientist 17, 54–78 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.San Martin A & Pagani MR Understanding intellectual disability through RASopathies. J. Physiol. 108, 232–239 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kim MJ, Dunah AW, Wang YT & Sheng M Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron 46, 745–760 (2005). [DOI] [PubMed] [Google Scholar]
- 130.Berryer MH et al. Mutations in SYNGAP1 cause intellectual disability, autism, and a specific form of epilepsy by inducing haploinsufficiency. Hum. Mutat. 34, 385–394 (2013). [DOI] [PubMed] [Google Scholar]
- 131.Clement JP et al. Pathogenic SYNGAP1 mutations impair cognitive development by disrupting maturation of dendritic spine synapses. Cell 151, 709–723 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Vazquez LE, Chen HJ, Sokolova I, Knuesel I & Kennedy MB SynGAP regulates spine formation. J. Neurosci. 24, 8862–8872 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Komiyama NH et al. SynGAP regulates ERK/MAPK signaling, synaptic plasticity, and learning in the complex with postsynaptic density 95 and NMDA receptor. J. Neurosci. 22, 9721–9732 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Aceti M et al. Syngapl haploinsufficiency damages a postnatal critical period of pyramidal cell structural maturation linked to cortical circuit assembly. Biol. Psychiatry 77, 805–815 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gutmann DH et al. Neurofibromatosis type 1. Nat. Rev. Dis. Primers 3, 17004 (2017). [DOI] [PubMed] [Google Scholar]
- 136.Wang HF et al. Valosin-containing protein and neurofibromin interact to regulate dendritic spine density. J. Clin. Invest. 121, 4820–4837 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tavazoie SF, Alvarez VA, Ridenour DA, Kwiatkowski DJ & Sabatini BL Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat. Neurosci. 8, 1727–1734 (2005). [DOI] [PubMed] [Google Scholar]
- 138.Jones AC et al. Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am. J. Hum. Genet. 64, 1305–1315 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Xie Z et al. Kalirin-7 controls activity-dependent structural and functional plasticity of dendritic spines. Neuron 56, 640–656 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Herring BE & Nicoll RA Kalirin and Trio proteins serve critical roles in excitatory synaptic transmission and LTP. Proc. Natl Acad. Sci. USA 113, 2264–2269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Russell TA et al. A sequence variant in human KALRN impairs protein function and coincides with reduced cortical thickness. Nat. Commun. 5, 4858 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kushima I et al. Resequencing and association analysis of the KALRN and EPHB1 genes and their contribution to schizophrenia susceptibility. Schizophr. Bull. 38, 552–560 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Russell TA et al. A schizophrenia-linked KALRN coding variant alters neuron morphology, protein function, and transcript stability. Biol. Psichiatry 83, 499–508 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Makrythanasis P et al. Exome sequencing discloses KALRN homozygous variant as likely cause of intellectual disability and short stature in a consanguineous pedigree. Hum. Genom. 10, 26 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Pengelly RJ et al. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J. Med. Genet. 53, 735–742 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ba W et al. TRIO loss of function is associated with mild intellectual disability and affects dendritic branching and synapse function. Hum. Mol. Genet. 25, 892–902 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Hill JJ, Hashimoto T & Lewis DA Molecular mechanisms contributing to dendritic spine alterations in the prefrontal cortex of subjects with schizophrenia. Mol. Psychiatry 11, 557–566 (2006). [DOI] [PubMed] [Google Scholar]
- 148.Rubio MD, Haroutunian V & Meador- Woodruff JH Abnormalities of the Duo/Ras-related C3 botulinum toxin substrate 1/p21-activated kinase 1 pathway drive myosin light chain phosphorylation in frontal cortex in schizophrenia. Biol. Psychiatry 71, 906–914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Deo AJ et al. Increased expression of Kalirin-9 in the auditory cortex of schizophrenia subjects: its role in dendritic pathology. Neurobiol. Dis. 45, 796–803 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Datta D, Arion D, Corradi JP & Lewis DA Altered expression of CDC42 signaling pathway components in cortical layer 3 pyramidal cells in schizophrenia. Biol. Psychiatry 78, 775–785 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Sadybekov A, Tian C, Arnesano C, Katritch V & Herring BE An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun. 8, 601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ma XM, Huang JP, Eipper BA & Mains RE Expression of Trio, a member of the Dbl family of Rho GEFs in the developing rat brain. J. Comp. Neurol. 482, 333–348 (2005). [DOI] [PubMed] [Google Scholar]
- 153.McPherson CE, Eipper BA & Mains RE Genomic organization and differential expression of Kalirin isoforms. Gene 284, 41–51 (2002). [DOI] [PubMed] [Google Scholar]
- 154.Billuart P et al. Oligophrenin-1 encodes a rhoGAP protein involved in X-linked mental retardation. Nature 392, 923–926 (1998). [DOI] [PubMed] [Google Scholar]
- 155.Govek EE et al. The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat. Neurosci. 7, 364–372 (2004). [DOI] [PubMed] [Google Scholar]
- 156.Ramakers GJ et al. Dysregulation of Rho GTPases in the alphaPix/Arhgef6 mouse model of X-linked intellectual disability is paralleled by impaired structural and synaptic plasticity and cognitive deficits. Hum. Mol. Genet. 21, 268–286 (2012). [DOI] [PubMed] [Google Scholar]
- 157.Allen KM et al. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 20, 25–30 (1998). [DOI] [PubMed] [Google Scholar]
- 158.Boda B et al. The mental retardation protein PAK3 contributes to synapse formation and plasticity in hippocampus. J. Neurosci. 24, 10816–10825 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang H, Webb DJ, Asmussen H & Horwitz AF Synapse formation is regulated by the signaling adaptor GIT1. J. Cell Biol. 161, 131–142 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Menon P et al. Impaired spine formation and learning in GPCR kinase 2 interacting protein-1 (GIT1) knockout mice. Brain Res. 1317, 218–226 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kim MJ et al. Functional analysis of rare variants found in schizophrenia implicates a critical role for GIT1-PAK3 signaling in neuroplasticity. Mol. Psychiatry 22, 417–429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Morris DW et al. An inherited duplication at the gene p21 protein-activated kinase 7 (PAK7) is a risk factor for psychosis. Hum. Mol. Genet. 23, 3316–3326 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Mulle JG et al. Microdeletions of 3q29 confer high risk for schizophrenia. Am. J. Hum. Genet. 87, 229–236 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Kang MG, Guo Y & Huganir RL AMPA receptor and GEF-H1/Lfc complex regulates dendritic spine development through RhoA signaling cascade. Proc. Natl Acad. Sci. USA 106, 3549–3554 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Endris V et al. The novel Rho-GTPase activating gene MEGAP/ srGAP3 has a putative role in severe mental retardation. Proc. Natl Acad. Sci. USA 99, 11754–11759 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Carlson BR et al. WRP/srGAP3 facilitates the initiation of spine development by an inverse F-BAR domain, and its loss impairs long-term memory. J. Neurosci. 31, 2447–2460 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Vaswani M, Linda FK & Ramesh S Role of selective serotonin reuptake inhibitors in psychiatric disorders: a comprehensive review. Prog. Neuropsychopharmacol. Biol. Psychiatry 27, 85–102 (2003). [DOI] [PubMed] [Google Scholar]
- 168.Forray C & Buller R Challenges and opportunities for the development of new antipsychotic drugs. Biochem. Pharmacol. 143, 10–24 (2017). [DOI] [PubMed] [Google Scholar]
- 169.Yamatogi Y Principles of antiepileptic drug treatment of epilepsy. Psychiatry Clin. Neurosci. 58, S3–S6 (2004). [DOI] [PubMed] [Google Scholar]
- 170.Al-Harbi KS Treatment-resistant depression: therapeutic trends, challenges, and future directions. Patient Prefer. Adherence 6, 369–388 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Gonda X Basic pharmacology of NMDA receptors. Curr. Pharm. Des. 18, 1558–1567 (2012). [DOI] [PubMed] [Google Scholar]
- 172.Pirotte B, Francotte P, Goffin E & de Tullio P AMPA receptor positive allosteric modulators: a patent review. Expert Opin. Ther. Pat. 23, 615–628 (2013). [DOI] [PubMed] [Google Scholar]
- 173.Phoumthipphavong V, Barthas F, Hassett S & Kwan AC Longitudinal effects of ketamine on dendritic architecture in vivo in the mouse medial frontal cortex. eNeuro 3, ENEUR0.0133–15.2016 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Li N et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 329, 959–964 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Berman RM et al. Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 47, 351–354 (2000). [DOI] [PubMed] [Google Scholar]
- 176. Kavalali ET & Monteggia LM How does ketamine elicit a rapid antidepressant response? Curr. Opin. Pharmacol. 20, 35–39 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Duman CH & Duman RS Spine synapse remodeling in the pathophysiology and treatment of depression. Neurosci. Lett. 601, 20–29 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Magarinos AM et al. Effect of brain-derived neurotrophic factor haploinsufficiency on stress-induced remodeling of hippocampal neurons. Hippocampus 21, 253–264 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Conrad CD, McLaughlin KJ, Huynh TN, El-Ashmawy M & Sparks M Chronic stress and a cyclic regimen of estradiol administration separately facilitate spatial memory: relationship with hippocampal CA1 spine density and dendritic complexity. Behav. Neurosci. 126, 142–156 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Liu RJ et al. GLYX-13 produces rapid antidepressant responses with key synaptic and behavioral effects distinct from ketamine. Neuropsychopharmacology 42, 1231–1242 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Abelaira HM et al. Effects of ketamine administration on mTOR and reticulum stress signaling pathways in the brain after the infusion of rapamycin into prefrontal cortex. J. Psychiatr. Res. 87, 81–87 (2017). [DOI] [PubMed] [Google Scholar]
- 182.Breier A, Malhotra AK, Pinals DA, Weisenfeld NI & Pickar D Association of ketamine- induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am. J. Psychiatry 154, 805–811 (1997). [DOI] [PubMed] [Google Scholar]
- 183.Malhotra AK et al. Ketamine-induced exacerbation of psychotic symptoms and cognitive impairment in neuroleptic-free schizophrenics. Neuropsychopharmacology 17, 141–150 (1997). [DOI] [PubMed] [Google Scholar]
- 184.Honey GD et al. Individual differences in psychotic effects of ketamine are predicted by brain function measured under placebo. J. Neurosci. 28, 6295–6303 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Maeng S et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amin o-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol. Psychiatry 63, 349–352 (2008). [DOI] [PubMed] [Google Scholar]
- 186.Aligny C et al. Ketamine alters cortical integration of GABAergic interneurons and induces long-term sex-dependent impairments in transgenic Gad67-GFP mice. Cell Death Dis. 5, e1311 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Suzuki K, Nosyreva E, Hunt KW, Kavalali ET & Monteggia LM Effects of a ketamine metabolite on synaptic NMDAR function. Nature 546, E1–E3 (2017). [DOI] [PubMed] [Google Scholar]
- 188.Zanos P et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 533, 481–486 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Abdallah CG, Sanacora G, Duman RS & Krystal JH Ketamine and rapid-acting antidepressants: a window into a new neurobiology for mood disorder therapeutics. Annu. Rev. Med. 66, 509–523 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Aleksandrova LR, Phillips AG & Wang YT Antidepressant effects of ketamine and the roles of AMPA glutamate receptors and other mechanisms beyond NMDA receptor antagonism. J. Psychiatry Neurosci. 42, 222–229 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Song M, Martinowich K & Lee FS BDNF at the synapse: why location matters. Mol. Psychiatry 22, 1370–1375 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Bjorkholm C & Monteggia LM BDNF — a key transducer of antidepressant effects. Neuropharmacology 102, 72–79 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Goff DC & Evins AE Negative symptoms in schizophrenia: neurobiological models and treatment response. Harv. Rev. Psychiatry 6, 59–77 (1998). [DOI] [PubMed] [Google Scholar]
- 194.Goff DC et al. Once-weekly D-cycloserine effects on negative symptoms and cognition in schizophrenia: an exploratory study. Schizophr. Res. 106, 320–327 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Tuominen HJ, Tiihonen J & Wahlbeck K Glutamatergic drugs for schizophrenia: a systematic review and meta-analysis. Schizophr. Res. 72, 225–234 (2005). [DOI] [PubMed] [Google Scholar]
- 196.Wink LK et al. d-Cycloserine enhances durability of social skills training in autism spectrum disorder. Mol. Autism 8, 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Moskal JR et al. GLYX-13: a monoclonal antibody- derived peptide that acts as an N-methyl- D-aspartate receptor modulator. Neuropharmacology 49, 1077–1087 (2005). [DOI] [PubMed] [Google Scholar]
- 198.Preskorn S et al. Randomized proof of concept trial of GLYX-13, an N-methyl-D-aspartate receptor glycine site partial agonist, in major depressive disorder nonresponsive to a previous antidepressant agent. J. Psychiatr. Pract. 21, 140–149 (2015). [DOI] [PubMed] [Google Scholar]
- 199.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02951988 (2018).
- 200.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02943564 (2018).
- 201.Moskal JR, Burgdorf J, Kroes RA, Brudzynski SM & Panksepp J A novel NMDA receptor glycine-site partial agonist, GLYX-13, has therapeutic potential for the treatment of autism. Neurosci. BiobehavRev. 35, 1982–1988 (2011). [DOI] [PubMed] [Google Scholar]
- 202.Santini AC et al. Glix 13, a new drug acting on glutamatergic pathways in children and animal models of autism spectrum disorders. Biomed. Res. Int. 2014, 234295 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Zhang XL, Sullivan JA, Moskal JR & Stanton PKA NMDA receptor glycine site partial agonist, GLYX-13, simultaneously enhances LTP and reduces LTD at Schaffer collateral-CA1 synapses in hippocampus. Neuropharmacology 55, 1238–1250 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Burgdorf J et al. The long-lasting antidepressant effects of rapastinel (GLYX-13) are associated with a metaplasticity process in the medial prefrontal cortex and hippocampus. Neuroscience 308, 202–211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT02067793 (2006).
- 206.Gupta DS et al. Metabotropic glutamate receptor protein expression in the prefrontal cortex and striatum in schizophrenia. Synapse 57, 123–131 (2005). [DOI] [PubMed] [Google Scholar]
- 207.Nisenbaum, E. S. & Witkin, J. M. in Glutamate-Based Therapies for Psychiatric Disorders (ed. Skolnick, P.] 39–56 (2010).
- 208.O’Neill MJ & Dix S AMPA receptor potentiators as cognitive enhancers. IDrugs 10, 185–192 (2007). [PubMed] [Google Scholar]
- 209.Lauterborn JC et al. Chronic ampakine treatments stimulate dendritic growth and promote learning in middle-aged rats. J. Neurosci. 36, 1636–1646 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Baudry M et al. Ampakines promote spine actin polymerization, long-term potentiation, and learning in a mouse model of Angelman syndrome. Neurobiol. Dis. 47, 210–215 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Lipina T, Weiss K & Roder J The ampakine CX546 restores the prepulse inhibition and latent inhibition deficits in mGluR5-deficient mice. Neuropsychopharmacology 32, 745–756 (2007). [DOI] [PubMed] [Google Scholar]
- 212.Chang PK, Prenosil GA, Verbich D, Gill R & McKinney RA Prolonged ampakine exposure prunes dendritic spines and increases presynaptic release probability for enhanced long-term potentiation in the hippocampus. Eur. J. Neurosci. 40, 2766–2776 (2014). [DOI] [PubMed] [Google Scholar]
- 213.Nations KR et al. Examination of Org 26576, an AMPA receptor positive allosteric modulator, in patients diagnosed with major depressive disorder: an exploratory, randomized, double-blind, placebocontrolled trial. J. Psychopharmacol 26, 1525–1539 (2012). [DOI] [PubMed] [Google Scholar]
- 214.Nations KR et al. Maximum tolerated dose evaluation of the AMPA modulator Org 26576 in healthy volunteers and depressed patients: a summary and method analysis of bridging research in support of phase II dose selection. Drugs R. D. 12, 127–139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Goff DC et al. A placebo-controlled add-on trial of the Ampakine, CX516, for cognitive deficits in schizophrenia. Neuropsychopharmacology 33, 465–472 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gao R & Penzes P Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr. Mol. Med. 15, 146–167 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Krauss GL Perampanel: a selective AMPA antagonist for treating seizures. Epilepsy Curr. 13, 269–272 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Ko D, Yang H, Williams B, Xing D & Laurenza A Perampanel in the treatment of partial seizures: time to onset and duration of most common adverse events from pooled Phase III and extension studies. Epilepsy Behav. 48, 45–52 (2015). [DOI] [PubMed] [Google Scholar]
- 219.Kato AS et al. Forebrain-selective AMPA-receptor antagonism guided by TARP gamma-8 as an antiepileptic mechanism. Nat. Med. 22, 1496–1501 (2016). [DOI] [PubMed] [Google Scholar]
- 220.Maher MP et al. Discovery and characterization of AMPA receptor modulators selective for TARP-gamma8. J. Pharmacol. Exp. Ther. 357, 394–414 (2016). [DOI] [PubMed] [Google Scholar]
- 221.Drummond JB, Tucholski J, Haroutunian V & Meador-Woodruff JH Transmembrane AMPA receptor regulatory protein (TARP) dysregulation in anterior cingulate cortex in schizophrenia. Schizophr. Res. 147, 32–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Sumioka A, Yan D & Tomita S TARP phosphorylation regulates synaptic AMPA receptors through lipid bilayers. Neuron 66, 755–767 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Ortner NJ & Striessnig J L-Type calcium channels as drug targets in CNS disorders. Channels 10, 7–13 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kabir ZD, Martinez-Rivera A & Rajadhyaksha AM From gene to behavior: L-type calcium channel mechanisms underlying neuropsychiatric symptoms. Neurotherapeutics 14, 588–613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Hollister LE & Trevino ES Calcium channel blockers in psychiatric disorders: a review of the literature. Can. J. Psychiatry 44, 658–664 (1999). [DOI] [PubMed] [Google Scholar]
- 226.Cipriani A et al. A systematic review of calcium channel antagonists in bipolar disorder and some considerations for their future development. Mol. Psychiatry 21, 1324–1332 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Lencz T & Malhotra AK Targeting the schizophrenia genome: a fast track strategy from GWAS to clinic. Mol. Psychiatry 20, 820–826 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.US National Library of Medicine. ClinicalTrials.gov https://clinicaltrials.gov/ct2/show/NCT01658150 (2017).
- 229.Ostacher MJ et al. Pilot investigation of isradipine in the treatment of bipolar depression motivated by genome-wide association. Bipolar Disord. 16, 199–203 (2014). [DOI] [PubMed] [Google Scholar]
- 230.Kang S et al. CaV1.3-selective L-type calcium channel antagonists as potential new therapeutics for Parkinson’s disease. Nat. Commun. 3, 1146 (2012). [DOI] [PubMed] [Google Scholar]
- 231.Basso AD, Kirschmeier P & Bishop WR Lipid posttranslational modifications. Farnesyl transferase inhibitors. J. Lipid Res. 47, 15–31 (2006). [DOI] [PubMed] [Google Scholar]
- 232. Costa RM et al. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 415, 526–530 (2002). [DOI] [PubMed] [Google Scholar]
- 233. Li W et al. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr. Biol. 15, 1961–1967 (2005). [DOI] [PubMed] [Google Scholar]
- 234.Widemann BC et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro Oncol. 16, 707–718 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Mainberger F et al. Lovastatin improves impaired synaptic plasticity and phasic alertness in patients with neurofibromatosis type 1. BMC Neurol. 13, 131 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Acosta MT et al. Lovastatin as treatment for neurocognitive deficits in neurofibromatosis type 1: phase I study. Pediatr. Neurol. 45, 241–245 (2011). [DOI] [PubMed] [Google Scholar]
- 237.Bearden CE et al. A randomized placebocontrolled lovastatin trial for neurobehavioral function in neurofibromatosis I. Ann. Clin. Transl Neurol. 3, 266–279 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Payne JM et al. Randomized placebo-controlled study of lovastatin in children with neurofibromatosis type 1. Neurology 87, 2575–2584 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Yan Z, Kim E, Datta D, Lewis DA & Soderling SH Synaptic actin dysregulation, a convergent mechanism of mental disorders? J. Neurosci. 36, 11411–11417 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Packer A Enrichment of factors regulating canonical Wnt signaling among autism risk genes. Mol. Psychiatry 22, 492–493 (2018). [DOI] [PubMed] [Google Scholar]
- 241.Marin O Developmental timing and critical windows for the treatment of psychiatric disorders. Nat. Med. 22, 1229–1238 (2016). [DOI] [PubMed] [Google Scholar]
- 242.Robinson L et al. Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain 135, 2699–2710 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Boda B, Mendez P, Boury-Jamot B, Magara F & Muller D Reversal of activity-mediated spine dynamics and learning impairment in a mouse model of Fragile X syndrome. Eur. J. Neurosci. 39, 1130–1137 (2014). [DOI] [PubMed] [Google Scholar]
- 244.Ehninger D, Li W, Fox K, Stryker MP & Silva AJ Reversing neurodevelopmental disorders in adults. Neuron 60, 950–960 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Wegel E et al. Imaging cellular structures in superresolution with SIM, STED and localisation microscopy: a practical comparison. Sci. Rep. 6, 27290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Lam F, Cladiere D, Guillaume C, Wassmann K & Bolte S Super-resolution for everybody: an image processing workflow to obtain high-resolution images with a standard confocal microscope. Methods 115, 17–27 (2017). [DOI] [PubMed] [Google Scholar]
- 247.Younts TJ et al. Presynaptic protein synthesis is required for long-term plasticity of GABA release. Neuron 92, 479–492 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Andreska T, Aufmkolk S, Sauer M & Blum R High abundance of BDNF within glutamatergic presynapses of cultured hippocampal neurons. Front. Cell. Neurosci. 8, 107 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Tønnenson J, Katona G, Rózsa B & Näggerl UV Spine neck plasticity regulates compartmentalization of synapses. Nat. Neurosci. 17, 678–685 (2014). [DOI] [PubMed] [Google Scholar]
- 250.Bar J, Kobler O, van Bommel B & Mikhaylova M Periodic F-actin structures shape the neck of dendritic spines. Sci. Rep. 6, 37136 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Urban NT, Willig KI, Hell SW & Nagerl UV STED nanoscopy of actin dynamics in synapses deep inside living brain slices. Biophys. J. 101, 1277–1284 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Shim SH et al. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. Proc. Natl Acad. Sci. USA 109, 13978–13983 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Frost NA, Kerr JM, Lu HE & Blanpied TA A network of networks: cytoskeletal control of compartmentalized function within dendritic spines. Curr. Opin. Neurobiol. 20, 578–587 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Izeddin I et al. Super-resolution dynamic imaging of dendritic spines using a low-affinity photoconvertible actin probe. PLOS ONE 6, e15611 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Wen PJ et al. Actin dynamics provides membrane tension to merge fusing vesicles into the plasma membrane. Nat. Commun. 7, 12604 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hoze N et al. Heterogeneity of AMPA receptor trafficking and molecular interactions revealed by superresolution analysis of live cell imaging. Proc. Natl Acad. Sci. USA 109, 17052–17057 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.MacGillavry HD, Song Y, Raghavachari S & Blanpied TA Nanoscale scaffolding domains within the postsynaptic density concentrate synaptic AMPA receptors. Neuron 78, 615–622 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Nair D et al. Super-resolution imaging reveals that AMPA receptors inside synapses are dynamically organized in nanodomains regulated by PSD95. J. Neurosci. 33, 13204–13224 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Forrest MP et al. Open chromatin profiling in hiPSC- derived neurons prioritizes functional noncoding psychiatric risk variants and highlights neurodevelopmental loci. Cell Stem Cell 21, 305–318. e8 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Chailangkarn T et al. A human neurodevelopmental model for Williams syndrome. Nature 536, 338–343 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Marchetto MC et al. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 143, 527–539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Nageshappa S et al. Altered neuronal network and rescue in a human MECP2 duplication model. Mol. Psychiatry 21, 178–188 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Ricciardi S et al. CDKL5 ensures excitatory synapse stability by reinforcing NGL-1-PSD95 interaction in the postsynaptic compartment and is impaired in patient iPSC-derived neurons. Nat. Cell Biol. 14, 911–923 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Pak C et al. Human neuropsychiatric disease modeling using conditional deletion reveals synaptic transmission defects caused by heterozygous mutations in NRXN1. Cell Stem Cell 17, 316–328 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Silbereis JC, Pochareddy S, Zhu Y, Li M & Sestan N The cellular and molecular landscapes of the developing human central nervous system. Neuron 89, 248–268 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Moeschler JB, Shevell M & Committee on Genetics. Comprehensive evaluation of the child with intellectual disability or global developmental delays. Pediatrics 134, e903–e918 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Neubauer BA, Gross S & Hahn A Epilepsy in childhood and adolescence. Dtsch. Arztebl Int. 105, 319–328 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Jones EJ, Gliga T, Bedford R, Charman T & Johnson MH Developmental pathways to autism: a review of prospective studies of infants at risk. Neurosci. Biobehav. Rev. 39, 1–33 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Leboyer M, Henry C, Paillere-Martinot ML & Bellivier F Age at onset in bipolar affective disorders: a review. Bipolar Disord. 7, 111–118 (2005). [DOI] [PubMed] [Google Scholar]
- 270.Jones PB Adult mental health disorders and their age at onset. Br. J. Psychiatry 202, S5–S10 (2013). [DOI] [PubMed] [Google Scholar]
- 271.Bayes A et al. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 14, 19–21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Turner TN et al. denovo-db: a compendium of human de novo variants. Nucleic Acids Res. 45, D804–D811 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Xu B et al. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat. Genet. 44, 1365–1369 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Xu B et al. Exome sequencing supports a de novo mutational paradigm for schizophrenia. Nat. Genet. 43, 864–868 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Girard SL et al. Increased exonic de novo mutation rate in individuals with schizophrenia. Nat. Genet. 43, 860–863 (2011). [DOI] [PubMed] [Google Scholar]
- 276.Genovese G et al. Increased burden of ultra-rare protein-altering variants among 4,877 individuals with schizophrenia. Nat. Neurosci. 19, 1433–1441 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Huganir RL & Nicoll RA AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Lepack AE, Fuchikami M, Dwyer JM, Banasr M & Duman RS BDNF release is required for the behavioral actions of ketamine. Int. J. Neuropsychopharmacol. 18, pyu033 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Lee SJ, Escobedo-Lozoya Y, Szatmari EM & Yasuda R Activation of CaMKII in single dendritic spines during long-term potentiation. Nature 458, 299–304 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Yoshii A & Constantine-Paton M Postsynaptic BDNF-TrkB signaling in synapse maturation, plasticity, and disease. Dev. Neurobiol. 70, 304–322 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Marshall CR et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat. Genet. 49, 27–35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Maynard KR & Stein E DSCAM contributes to dendrite arborization and spine formation in the developing cerebral cortex. J. Neurosci. 32, 16637–16650 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Schoch H et al. Sociability deficits and altered amygdala circuits in mice lacking Pcdh10, an autism associated gene. Biol. Psychiatry 81, 193–202 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Liu YT et al. PRRT2 mutations lead to neuronal dysfunction and neurodevelopmental defects. Oncotarget 7, 39184–39196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Heron SE et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am. J. Hum. Genet. 90, 152–160 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Lemke JR et al. Mutations in GRIN2A cause idiopathic focal epilepsy with rolandic spikes. Nat. Genet. 45, 1067–1072 (2013). [DOI] [PubMed] [Google Scholar]
- 287.Durand CM et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39, 25–27 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.El-Husseini AE, Schnell E, Chetkovich DM, Nicoll RA & Bredt DS PSD-95 involvement in maturation of excitatory synapses. Science 290, 1364–1368 (2000). [PubMed] [Google Scholar]
- 289.Wu Q, Sun M, Bernard LP & Zhang H Postsynaptic density 95 (PSD-95) serine 561 phosphorylation regulates a conformational switch and bidirectional dendritic spine structural plasticity. J. Biol. Chem. 292, 16150–16160 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Stessman HA et al. Targeted sequencing identifies 91 neurodevelopmental-disorder risk genes with autism and developmental-disability biases. Nat. Genet. 49, 515–526 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Hung AY, Sung CC, Brito IL & Sheng M Degradation of postsynaptic scaffold GKAP and regulation of dendritic spine morphology by the TRIM3 ubiquitin ligase in rat hippocampal neurons. PLOS ONE 5, e9842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Chao HW, Hong CJ, Huang TN, Lin YL & Hsueh YP SUMOylation of the MAGUK protein CASK regulates dendritic spinogenesis. J. Cell Biol. 182, 141–155 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Jourdain P, Fukunaga K & Muller D Calcium/ calmodulin-dependent protein kinase II contributes to activity-dependent filopodia growth and spine formation. J. Neurosci. 23, 10645–10649 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Fink CC et al. Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 39, 283–297 (2003). [DOI] [PubMed] [Google Scholar]
- 295.Kury S et al. De novo mutations in protein kinase genes CAMK2A and CAMK2B cause intellectual disability. Am. J. Hum. Genet. 101, 768–788 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Araki Y, Zeng M, Zhang M & Huganir RL Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron 85, 173–189 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Johns Hopkins University. OMIM, online mendelian inheritance in man. OMIM; https://omim.org/(2018). [Google Scholar]
- 298.Wang M et al. Distinct defects in spine formation or pruning in two gene duplication mouse models of autism. Neurosci. Bull. 33, 143–152 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Blizinsky KD et al. Reversal of dendritic phenotypes in 16p11.2 microduplication mouse model neurons by pharmacological targeting of a network hub. Proc. Natl Acad. Sci. USA 113, 8520–8525 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Moutin E et al. Palmitoylation of cdc42 promotes spine stabilization and rescues spine density deficit in a mouse model of 22q11.2 deletion syndrome. Cereb. Cortex 27, 3618–3629 (2017). [DOI] [PubMed] [Google Scholar]
- 301. Fenelon K et al. The pattern of cortical dysfunction in a mouse model of a schizophrenia-related microdeletion. J. Neurosci. 33, 14825–14839 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.