

Abstract
Background:
Hypertrophic cardiomyopathy (HCM) is frequently caused by mutations in myosin binding protein C3 (MYBPC3) resulting in a premature termination codon (PTC). The underlying mechanisms of how PTC mutations in MYBPC3 lead to the onset and progression of HCM are poorly understood. This study’s aim was to investigate the molecular mechanisms underlying the pathogenesis of HCM associated with MYBPC3 PTC mutations by utilizing human isogenic induced pluripotent stem cell-derived cardiomyocytes (iPSC-CMs).
Methods:
Isogenic iPSC lines were generated from patients harboring MYBPC3 PTC mutations (p.R943x; p.R1073P_Fsx4) using genome editing and then differentiated into cardiomyocytes. Comprehensive phenotypical and transcriptome analyses were performed.
Results:
We observed aberrant calcium handling properties with prolonged decay kinetics and elevated diastolic calcium levels in HCM iPSC-CMs compared to isogenic controls without structural abnormalities or contractile dysfunction. The mRNA expression levels of MYBPC3 were significantly reduced in mutant iPSC-CMs, but the protein levels were comparable among isogenic iPSC-CMs, suggesting that haploinsufficiency of MYBPC3 does not contribute to the pathogenesis of HCM in vitro. Furthermore, truncated MYBPC3 peptides were not detected. At the molecular level, the nonsense-mediated decay (NMD) pathway was activated, and a set of genes involved in major cardiac signaling pathways was dysregulated in HCM iPSC-CMs, indicating an HCM gene signature in vitro. Specific inhibition of the NMD pathway in mutant iPSC-CMs resulted in reversal of the molecular phenotype and normalization of calcium handling abnormalities.
Conclusions:
iPSC-CMs carrying MYBPC3 PTC mutations displayed aberrant calcium signaling and molecular dysregulations in the absence of significant haploinsufficiency of MYBPC3 protein. Here we provided the first evidence of the direct connection between the chronically activated NMD pathway and HCM disease development.
Keywords: hypertrophic cardiomyopathy, MYBPC3, iPSC, cardiomyocyte, nonsense-mediated decay
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is the most common inherited heart disease with a prevalence of 1 in 500 in the population.1 Generally, HCM is characterized by various degrees of left ventricular wall thickening, diastolic dysfunction, progressive heart failure, and sudden cardiac death. HCM is predominantly caused by mutations in myosin heavy chain 7 (MYH7) and myosin binding protein C3 (MYBPC3); the majority of mutations in MYBPC3 result in a premature termination codon (PTC).2 Previous studies suggest that MYBPC3 PTC mutations are associated with low mRNA and absent truncated peptides derived from the diseased allele due to the activation of the nonsense-mediated decay (NMD) pathway and the ubiquitin-proteasome system (UPS), respectively, resulting in haploinsufficiency of the healthy allele.3,4 Despite the progress in understanding the pathogenesis of HCM, the underlying molecular mechanisms that link PTC mutations in MYBPC3 to HCM onset and progression are incompletely understood.
In recent years, the emergence of human induced pluripotent stem cell (iPSC) and genome editing technologies has dramatically advanced disease modeling of cardiomyopathies in vitro.5 Combining iPSC technology and genome editing now provides new opportunities for a better understanding of the molecular mechanisms underlying the pathogenesis of genetic HCM in vitro.6–8 To study the molecular mechanisms of HCM associated with MYBPC3 PTC mutations, we generated isogenic iPSC-derived cardiomyocytes (iPSC-CMs) by either correcting PTC mutations (p. 943x; p.R1073) in iPSCs derived from HCM patients or by introducing a PTC mutation (p.R943x) into a healthy iPSC line. We performed structural and functional analyses to investigate the HCM phenotypes in vitro and observed aberrant calcium handling abnormalities in HCM iPSC-CMs compared to their respective isogenic controls in vitro. Although MYBPC3 mRNA expression levels were significantly reduced in HCM iPSC-CMs, we observed no haploinsufficiency of MYBPC3 at the protein levels when compared to isogenic controls. Transcriptomic analysis revealed a molecular HCM signature characterized by activation of pathways associated with cardiac hypertrophy, calcium signaling, translation, RNA metabolism, and the NMD. Molecular inhibition of the NMD pathway in HCM iPSC-CMs carrying the MYBPC3 p.R943x mutation resulted in the reversal of the molecular phenotype and normalization of calcium handling kinetics, suggesting a direct connection between the constitutively activated NMD pathway and the molecular mechanisms associated with the pathogenesis of HCM.
METHODS
A full description of the methods is presented in the Supplemental Methods section. The data, analytic methods, and study materials are available to other researchers for purposes of reproducing the results or replicating the procedures upon request. The iPSC lines have been deposited to the Stanford’s Cardiovascular Institute iPSC Biobank and are available to other researchers upon request.
iPSC Generation and Cardiac Differentiation.
Patients and healthy controls (Supplemental Figure S1a) were enrolled in the study with an informed consent. The study was approved by the Stanford Institutional Review Board (IRB) and Stem Cell Research Oversight (SCRO) Committee. Peripheral blood mononuclear cells (PBMCs) were collected and subsequently reprogrammed using the non-integrating Sendai virus (CytoTune™-iPS 2.0 Sendai Reprogramming Kit, Thermo Fisher Scientific). A single-nucleotide polymorphism (SNP) array (OmniExpress24, Illumina) was used to assess the karyotype of the iPSC clones, and one clone per iPSC line was chosen for subsequent studies. All experiments were performed using iPSC-CMs generated from iPSCs with passage numbers between 18 and 35. Differentiation into iPSC-CMs was performed using a monolayer small molecule Wnt-activation/inhibition protocol.9 Beating iPSC-CMs were maintained in RPMI 1640 medium (Thermo Fisher Scientific) supplemented with B27 supplements (Thermo Fisher Scientific). All experiments were performed between day 35 and day 50 after initiation of differentiation. Dissociation of iPSC-CMs was performed using pre-warmed TrypLE select 10x (Thermo Fisher Scientific) at 37°C. After detaching, cells were collected using RPMI 1640 with B27 supplement media and centrifuged. The cells were resuspended in RPMI 1640 with B27 supplement media and plated in Matrigel-coated dishes.
Genome Editing.
Genome editing was performed using the engineered nucleases TALEN (Transcription Activator-like Effector Nucleases) or CRISPR/Cas9 (Clustered Regularly Interspaced Short Palindromic Repeats/Cas9) as previously described.6,10,11 For genome editing, the respective iPSC lines were used at passage numbers between 20 and 25. Expanded methods can be found in the Online Data Supplement.
Droplet Digital PCR.
Droplet digital PCR (ddPCR) was performed based on the ddPCR Applications Guide (BioRad). To establish the specificity of the primer and probe combinations, and the optimal annealing temperature, gBlocks (IDT) were used with either the healthy or the mutant cDNA sequence. Total RNA was extracted from iPSC-CMs and reverse transcribed, and 1 ng of RNA-equivalent cDNA was mixed with primers, probes, and ddPCR Supermix reaction (BiodRad; total volume 20 μl). Droplet formation was carried out using a BioRad QX100 droplet generator. PCR amplification was performed and the samples were analyzed using a BioRad QX100 reader. The following probes and primers were used:
MYBPC3_943: FW: CACACATCGATACTGGTGAAGG; REV: GCTCCGTGGTGGTAA CAG; wild type probe: FAM-TTTCCGAGTGCGGGCACACAATAT-black-hole; mutant probe: hex-TTTCTGAGTGCGGGCACACAATAT- black-hole.
MYBPC3_1073: FW: TTGTTGACAAGCCAAGTCCT; REV: CTCCATGGTCTTCTTGT CGG; wild type probe: FAM-TCCGGGTGACTGACGCCTGGGGT-black-hole; mutant probe HEX-TCCCGGGTGACTGACGCCTGGGGT-black-hole.
RNA-Sequencing.
Sequencing libraries were generated using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina. Clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina). Sequencing was carried out on an Illumina HiSeq platform. For each sample in the whole transcriptome sequencing library, 30–40 million 150-base pair paired-end reads were acquired from the sequencer. Raw sequence data were checked for quality with FastQC (Version 0.11.5) and results were aggregated with MultiQC. Sequence data were aligned to the human genome (hg19) using STAR (Version 2.5.1b) with ENCODE options for long RNA-seq pipeline. Alignment results were assessed using Samtools and aggregated with MultiQC (Version 0.9). Uniquely mapped reads were quantified using featureCounts (Version 1.28.0) and differentially expressed genes were identified using DESeq2 (Version 1.18.1). The DESeq2 Bioconductor package was used to identify genes that were differentially expressed between groups.12 Pairwise comparisons between groups were conducted by applying the Wald test of the negative binomial distribution to the log2 gene counts and then Benjamini–Hochberg correction was applied. A significant regulation was suggested with a fold-change of ±1.4 and a q value < 0.05. The expression of wild type vs. mutant MYBPC3 mRNA was assessed using the Integrative Genomics Viewer (IGV; Broad institute).
Contractility Assays.
Throughout the study, contraction analysis was performed using a high-speed video microscopy followed by motion vector analysis to investigate the contractile characteristics of iPSC-CM monolayers. Depending on the design of the experiment, iPSC-CMs were seeded at 2×105 cells/cm2 into 384-well or 96-well plates. Contractility was measured using the Sony SI8000 cell motion imaging system. Video imaging of beating iPSC-CMs was recorded for 10 sec at a frame rate of 75 fps, a resolution of 1024 × 24 pixels, and a depth of 8 bits using a 10× objective on a fully automated Nikon microscope (Eclipse Ti, Nikon). Motion detection and analysis were performed using the Sony Cardio-analysis software (based on a block matching algorithm).13 For stress experiments, iPSC-CMs were treated with the following options: 30 nM triiodothyronine (T3), 1 µM dexamethasone (dex), 20 ng/µl insulin-like growth factor 1 (IGF1) for 72 hr (all Sigma Aldrich); 25 nM digoxin (Sigma Aldrich) for 3 days,1 nM digoxin for 7 days; 10 µM phenylephrine (PE), 25 pg/ml endothelin-1 (ET1), and 100 nM isoproterenol (Iso) for 7 days (all from Sigma Aldrich).
Preparation and Culture of EHTs.
Engineered heart tissues (EHTs) were prepared using a fibrin gel as previously described.14 Briefly, EHTs were produced around silicone posts in agarose casting molds in a 24-well plate. Each EHT contained 1×106 iPSC-CMs in a fibrin matrix (100 µL total) composed of 10 µL Matrigel (Corning), 5 mg/ml bovine fibrinogen (2.53 µL of 200 mg/ml fibrinogen reconstituted in 0.9% NaCl), and supplemented with 0.1 mg/mL aprotinin Sigma Aldrich. 5.57 µL of 2x high glucose DMEM with 10% horse serum and 1% penicillin/streptomycin (Thermo Fischer Scientific), 78.8 µL of high glucose DMEM (Thermo Fischer Scientific) with 10% FBS and 1% L-glutamine (Invitrogen 25030081), 10 µM Y-27632, and 3 U/ml thrombin (Sigma T7513). EHTs were incubated for 2 hr at 37o C and then transferred to a new plate containing RPMI supplemented with B27 and 10% KnockOut serum replacement (Thermo Fischer Scientific). The medium was replaced every two days with RPMI+B27 with 33 μg/ml aprotinin.
Contractile Analysis of EHTs.
EHT contractile motion was recorded using the SI8000 Cell Motion Imaging System (Sony). The SI8000R Analyzer Software (Sony) was used to detect motion vectors and track the movement of each EHT post. The distance of maximum post deflection from rest was extracted from each contraction cycle in the tracking data using a custom MATLAB script. Deflection was converted to force using the equation for deflection of an end-loaded cantilever beam with elastic modulus of 1.7 MPa, radius of 0.5 mm, and length of 10 mm.
Ca2+ Imaging
Ratiometric calcium imaging was performed on iPSC-CMs loaded with Fura-2 under continuous electrical stimulation with excitation wavelengths at 340 nm and 380 nm. Raw data exported from Nikon NIS-Elements were analyzed using a custom Python script (accessible at: https://github.com/GeorgeMcMullen/CalciPy)15 built specifically to automate processing of ratiometric calcium imaging data for high throughput analysis. An expanded description can be found in the Online Data Supplement.
Nanofluidics Immuno Assay (NIA).
NIA was performed on a PeggySue® system (ProteinSimple; Bio-Techne). The primary antibodies anti-MYBPC3 (Abgent) and anti-aSA (Abcam) were diluted to be used in a final dilution of 1:50. The determination of the sizes, areas, heights, and signal-to-noise (S/N) ratios of MYBPC3 and aSA proteins were automatically determined using on Compass for Simple Western® software, version 3.1.7. A detailed protocol is outlined in the expanded methods in the Online Data Supplement.
Mass Spectrometry.
Protein lysates from isogenic lines (943cor and 943het iPSC-CMs, as well as ctrl and ctrl943 iPSC-CMs), were obtained from four independent differentiation batches. After protein digestion and labeling with 10-plex tandem mass tags (Thermo), mass spectrometry was performed using a Thermo Orbitrap Fusion high-resolution mass spectrometer. A detailed protocol is outlined in the expanded methods in the Online Data Supplement.
cDNA Overexpression of Healthy and Mutated MYBPC3.
Healthy MYBPC3 cDNA in a pCMV-SPORT6 vector was obtained as glycerol stock from Dharmacon (MHS6278–202800272). The plasmid was transformed into competent cells (Zymo Research) and plated overnight. After colony picking, plasmid extraction was performed (Plasmid Miniprep Kit, Qiagen) and the vector was sequence validated by Sanger sequencing. Before final use, the clone was expanded and plasmid extraction was performed (Plasmid Midiprep Kit, Qiagen). Mutagenesis of the wild type MYBPC3 cDNA vector to introduce the p.R943x mutation was performed using the QuikChange Lightning Site-Directed Mutagenesis Kit according to the manufacturer’s protocol (Agilent). Wild type or mutated vectors (1 μg) were transfected into HEK293T cells using Lipofectamine 3000 according to the manufacturer’s protocol (Thermo Fisher Scientific). Protein expression was evaluated 72 hr post-transfection.
siRNA-Mediated Knockdown.
Gene knockdown experiments were performed using Lipofectamine RNAiMax (Life Technologies) according to the manufacturer’s instructions. Cells were transfected with either scrambled or siRNA against UPF1 (siGENOME siRNA, GE Healthcare Dharmacon, 25 nM) for 48 hr before being subjected to subsequent downstream analyses.
Statistical Analysis.
Data analysis was performed using Microsoft Excel and Graphpad Prism5 software. Heatmaps were generated using the web-based tool found at https://software.broadinstitute.org/morpheus/.16 For statistical analysis, the Student’s t-test was used to compare differences between two data sets. For comparisons among multiple groups, one-way ANOVA was used, and Bonferroni post-hoc test was used for comparisons of all groups, depending on the properties of the data sets. For data sets with two factors, 2-way ANOVA was used to calculate the significance of the source of variation. Once a significant variation by treatment and interaction of the two factors was confirmed, the positive differences in increase between treatments were analyzed by one-way ANOVA. Correlation analysis was performed using the Spearman’s rank test. Overall, a p-value <0.05 was considered statistically significant. All data in bar graphs are presented as mean ± standard deviation unless indicated otherwise.
RESULTS
Generation of isogenic iPSC lines with MYBPC3 truncation mutations
To study PTC mutations in MYBPC3 in the pathogenesis of HCM, we enrolled a patient diagnosed with a severe hypertrophic phenotype of HCM as well as with a positive family history of HCM, carrying an heterozygous mutation in MYBPC3 resulting in a PTC in exon 27 (c. 2827 C>T; p.R943x; hereafter referred to as “943het”) (Figure 1a, Supplemental Figures S1a, b). To generate isogenic iPSC lines, we corrected the mutation in the patient’s iPSCs (hereafter referred to as “943cor”), and also introduced the mutation in the healthy allele to obtain an iPSC line homozygous for the p.R943x mutation (hereafter referred to as “943hom”) (Figure 1b). We also introduced a heterozygous p.R943x mutation into a healthy control iPSC line (hereafter referred to as “ctrl943” and “ctrl”, respectively; Supplemental Figures S1a, e).
Figure 1: Genome editing and structural phenotype of MYBPC3 HCM iPSC-CMs.
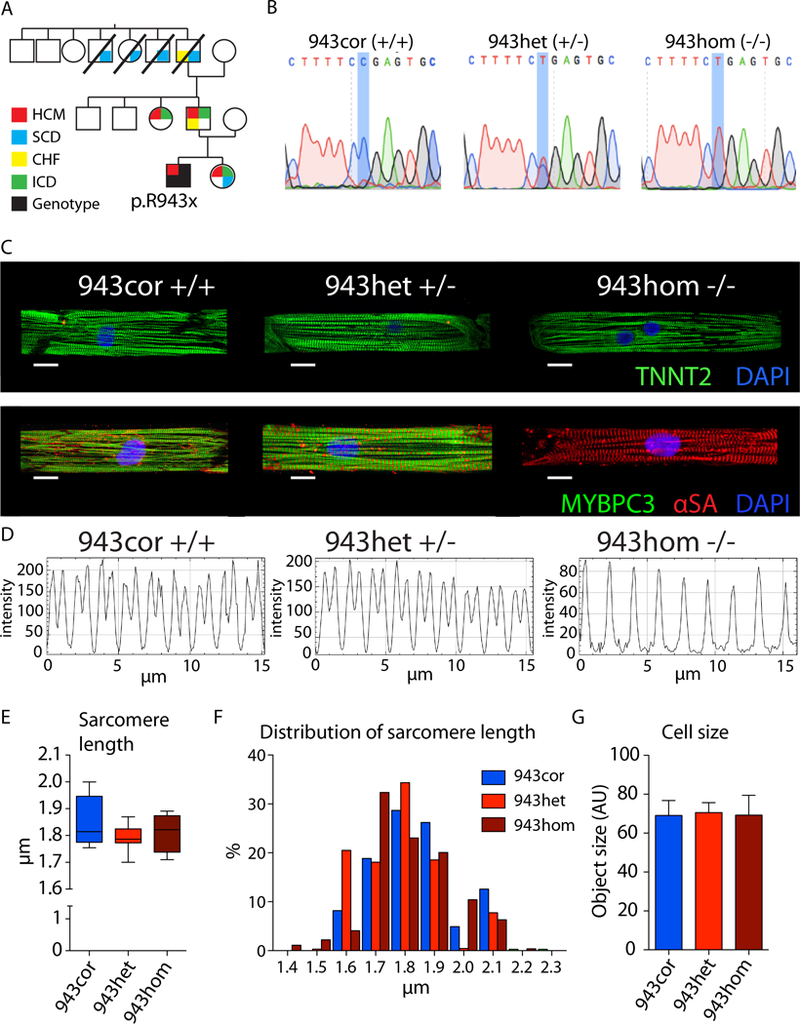
(A) Pedigree of the affected family. The proband was diagnosed with HCM and a MYBPC3 truncation mutation (p.R943x) was identified by genetic screening. (B) Sanger sequencing of patient corrected (943cor), patient (943het), and patient homozygous (943hom) iPSCs. (C) Micropatterned iPSC-CMs stained with Troponin (green) and DAPI (top row), and MYBPC3 (green), alpha sarcomeric actin (αSA; red), and DAPI (bottom row; scale bar 10 µm). (D) Representative plots of fluorescence intensity longitudinally throughout myofibers analyzed using plot profile (ImageJ), showing MYBPC3 expression in micro-patterned iPSC-CMs (943cor, 943het) and αSA (943hom). (E) Quantification of sarcomeric length based on αSA stained micro-patterned iPSC-CMs (n=10, 11, and 4 cells, respectively). (F) Distribution of the obtained sarcomere length measurements in 943cor (blue), 943het (red), and 943hom (dark). (G) Cell size measurement of unpatterned iPSC-CMs (n=4–6 differentiation batches with 20–130 cells each, respectively). HCM - hypertrophic cardiomyopathy; SCD - sudden cardiac death; CHF - congestive heart failure; ICD - implantable cardioverter defibrillator; AU - arbitrary units.
Two additional unrelated patients were enrolled, one carrying the same MYBPC3 mutation (hereafter referred to as “943het2”) and one with a p.R1073P_Fsx4 mutation in MYBPC3 (hereafter referred to as “1073het”; Supplemental Figures S1a, b). These iPSC lines were also successfully corrected using CRISPR/Cas9-mediated genome editing (“943cor2” and “1073cor”; Supplemental Figures S1a, f).
Structural and functional phenotype of HCM iPSC-CMs
The pathological hallmarks of HCM at the histological level are myocyte hypertrophy, myocardial disarray, and interstitial fibrosis.17 To analyze the sarcomeric integrity, single cells were micropatterned on a soft polyacrylamide hydrogel (12 kPa) and immunostained for cardiac troponin T (TNNT2), MYBPC3, and alpha sarcomeric actin (αSA; Figure 1c and Supplemental Figure 2a). We observed that 943cor, 943het and 943hom iPSC-CMs as well as ctrl and ctrl943 iPSC-CMs formed regular sarcomeres, and there were no significant differences in sarcomere length (Figure 1d-f and Supplemental Figure S2a-c). This is in agreement with results from transgenic mouse models showing that MYBPC3+/− and the MYBPC3−/− mice did not exhibit obvious structural and ultrastructural differences in tissue morphology or in isolated cardiomyocytes.18,19 Furthermore, we observed no significant differences in cell size of iPSC-CMs among all isogenic comparisons (Figure 1g; Supplemental Figure S2d and e), corroborating the findings of a recent study.20 Taken together, these data suggest that the investigated MYBPC3 PTC mutations do not cause any structural abnormalities at the sarcomeric level and do not significantly increase the cell size in iPSC-CMs in vitro.
Recent studies have provided evidence for impaired contractile force generation in cardiomyocytes from HCM patients harboring MYBPC3 mutations.21 To evaluate the contractile function in our study, monolayers of isogenic iPSC-CMs were analyzed using vector based-motion detection.13 We observed no significant differences in contractility between 943cor and 943het iPSC-CMs (Figure 2a), and between ctrl and ctrl943 iPSC-CMs (Supplemental Figure S3a). A recent report suggests that HCM iPSC-CMs harboring a MYBPC3 PTC mutation (c.2373_2374insG) treated with dexamethasone, triiodothyronine, and insulin-like growth factor 1 (Dex; T3; IGF1) showed significantly lower response in contractile force generation than non-isogenic controls.20 Treatment with Dex/T3/IGF1 resulted in a uniform contractile response in both healthy and heterozygous iPSC-CMs (Figure 2b, Supplemental Figure S3b). However, the contractile response to treatment was blunted in 943hom iPSC-CMs (Figure 2b). Furthermore, we observed similar contractile responses in isogenic iPSC-CMs exposed to inotropic agents, including isoproterenol (Iso), phenylephrine (PE), endothelin-1/IGF1 and digoxin (Supplemental Figure S3c, d). To measure absolute forces, we next generated 3D engineered heart tissue (EHT) constructs from all isogenic lines (Supplemental Figures 3e).14 We observed no significant differences in force generation within isogenic comparisons of EHTs between heterozygous and healthy iPSC-CMs (Supplemental Figures 3f). However, EHTs generated from homozygous iPSC-CMs (943hom) showed significantly impaired force generation (Supplemental Figure 3f). Taken together, these data suggest the absence of a significant contractile phenotype in iPSC-CMs carrying a heterozygous MYBPC3 PTC (p.R943x or p.R1073P_Fsx4) mutation in vitro.
Figure 2: Functional evaluation of HCM iPSC-CMs.
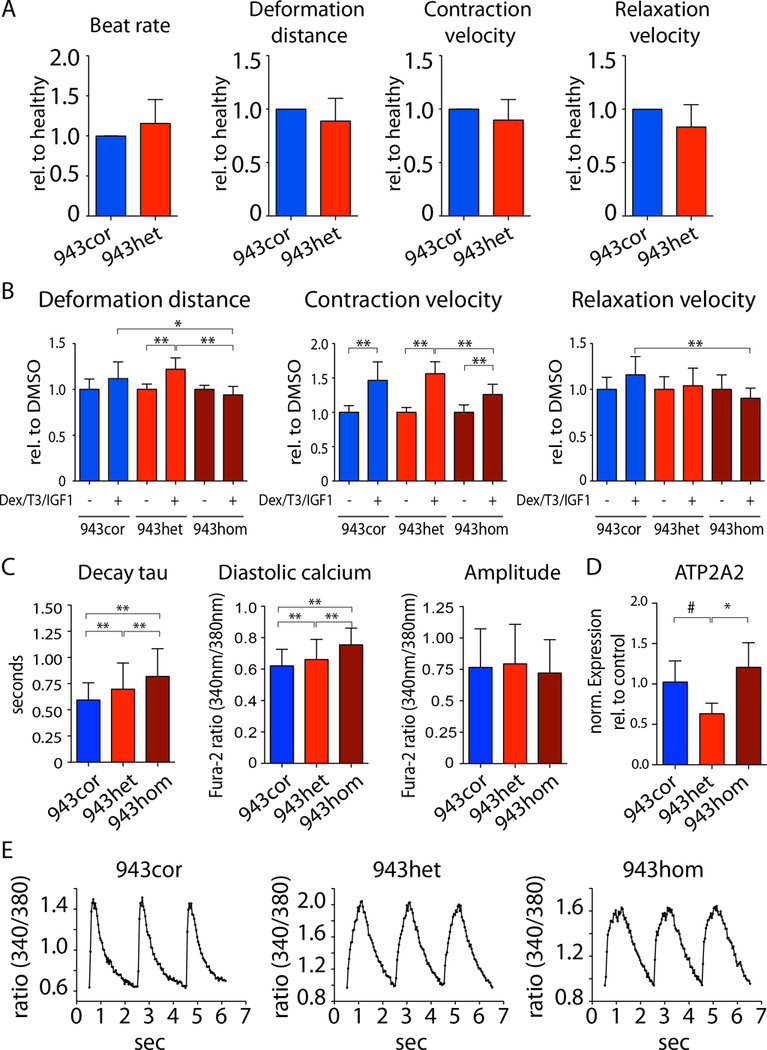
(A) Contraction analysis of iPSC-CM monolayers using a vector-based imaging approach. The relative differences of disease (943het) vs. healthy (943cor) were calculated for each respective experiment (n=5). (B) iPSC-CMs from 943cor, 943het, and 943hom were treated with dexamethasone (Dex), triiodothyronine (T3), and insulin like growth factor 1 (IGF1) for 72 hours prior to contractility analysis (n=3 batches). Analysis was performed using vector-based imaging approach. (C) Analysis of calcium handling properties in isogenic iPSC-CMs (943cor, 943het, 943hom) using ratiometric measurements of fluorescence after Fura-2 staining under continuous field potential stimulation (n=4/4/6 differentiation batches each, 40 cells per batch). (D) Relative expression of ATP2A2 in iPSC-CMs (943cor, 943het, 943hom; n=3–6 differentiation batches each). (E) Representative raw traces of intracellular calcium signaling. # p<0.1, * p<0.05; ** p<0.01.
As alterations in calcium (Ca2+) cycling are a hallmark of HCM,22,23 we next compared the calcium handling properties of isogenic iPSC-CMs. We observed both elevated diastolic Ca2+ levels and prolonged relaxation kinetics in 943het compared to 943cor iPSC-CMs that were exacerbated in 943hom iPSC-CMs (Figure 2c). Similarly, the relaxation kinetics were consistently impaired in all other HCM iPSC-CMs when compared to their respective isogenic controls (ctrl943 vs. ctrl, 943het2 vs. 943cor2, and 1073het vs. 1073cor; Supplemental Figures S4a, c, e). In accordance, the expression of ATP2A2, which encodes for sarco/endoplasmic reticulum calcium ATPase cardiac isoform 2a (SERCA2a), was significantly downregulated in HCM iPSC-CMs when compared to the respective isogenic controls (Figure 2d, Supplemental Figures S4b, d, f). Taken together, these results suggest that both p.R943x and p.R1073P_Fsx4 MYBPC3 PTC mutations induce calcium cycling abnormalities in HCM iPSC-CMs in vitro.
PTC mutations do not result in MYBPC3 protein haploinsufficiency in isogenic iPSC-CMs
The prevailing hypothesis is that MYBPC3 PTC mutations induce MYBPC3 protein haploinsufficiency,3 and recent studies suggest that the MYBPC3 mutant mRNA is degraded via the NMD pathway.4 We observed that the total MYBPC3 mRNA expression was reduced by 45% and 76% in 943het and 943hom iPSC-CMs, respectively, when compared to 943cor iPSC-CMs (Figure 3a). Similarly, we observed a significantly reduced expression of MYBPC3 mRNA in the other isogenic comparisons (ctrl943 vs. ctrl, 943het2 vs. 943cor2, and 1073het vs. 1073cor; Supplemental Figures S5a, c). In addition, the mRNA expression levels of other sarcomeric genes, such as TNNT2 and MYH7, were comparable in 943cor and 943het iPSC-CMs, but were significantly increased in 943hom iPSC-CMs (Figure 3b). As the fraction of MYBPC3 mRNA carrying a PTC mutation has been shown to be reduced in patient-derived samples,3 we next quantified the expression of healthy and mutated alleles in HCM iPSC-CMs. In HCM iPSC-CMs, 27.7 ± 4.4% of total MYBPC3 mRNA was generated from the mutated allele (943het, Figure 3c; 943het2, 1073het; Supplemental Figure S5d), whereas this fraction was 13.5 ± 4.7% in ctrl943 iPSC-CMs (Supplemental Figure S5b), indicating the degradation of the mutant mRNA. At the protein level, we observed comparable expression of MYBPC3 among isogenic iPSC-CMs by western blot analysis using an antibody detecting the N-terminus of MYBPC3 in all isogenic comparisons, while MYBPC3 protein expression was undetectable in 943hom iPSC-CMs (Figure 3d, Supplemental Figures S5e-f, and Supplemental Figures S6a-c). We corroborated these findings with mass spectrometry analysis of isogenic iPSC-CMs (Figure 3e). Furthermore, we performed western blot analysis on protein lysates from EHTs of all isogenic lines to evaluate the protein expression of MYBPC3 in 3D constructs and observed comparable levels of MYBPC3 in EHTs derived from HCM iPSC-CMs when compared to the respective isogenic controls (Supplemental Figure S6d). We next evaluated a possible expression of truncated MYBPC3 peptides in HCM iPSC-CMs. We did not detect truncated MYBPC3 peptides by western blot analysis using a validated antibody or by mass spectrometry analysis (Figure 3d, Supplemental Figure S5e-f, Supplemental Figure S6a-d and Supplemental Figure S7a, b). Given the involvement of the UPS in the clearance of the truncated MYBPC3 peptides,24 we investigated the potential role of the UPS in HCM iPSC-CMs harboring PTC mutations. After treating 943hom iPSC-CMs with various proteasome inhibitors (Supplemental Figures S7c), we were still unable to detect truncated MYBPC3 protein in 943hom iPSC-CMs by western blot analysis (Supplemental Figures S7d). In contrast, low levels of putative truncated MYBPC3 peptide were detected at the predicted molecular weight of 125 kDa with high sensitivity nanofluidic-immunoassay after proteasome inhibition (Supplemental Figures S7e, f). Taken together, these data do not support either a MYBPC3 haploinsufficiency or a truncated MYBPC3 ‘poison’ peptide hypothesis as the underlying cause of HCM.
Figure 3: MYBPC3 expression in HCM iPSC-CMs.
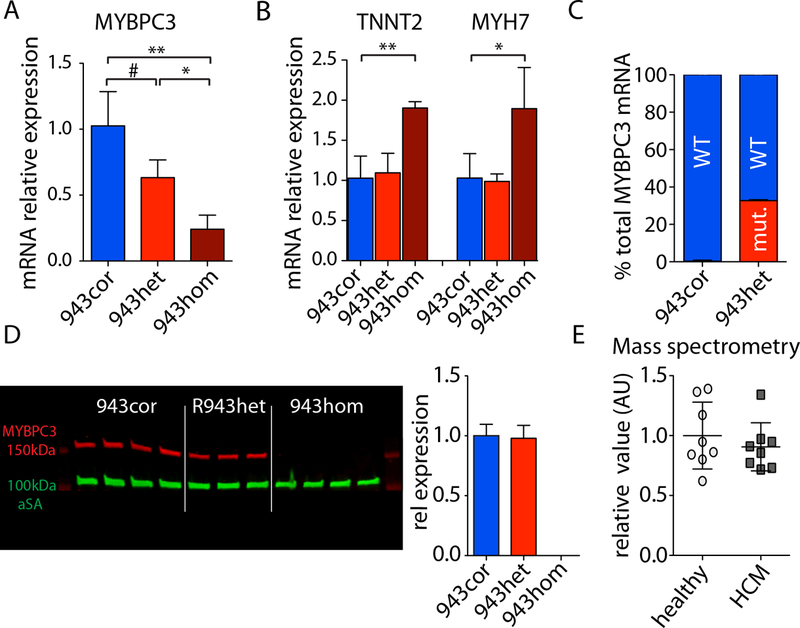
(A,B) Total MYBPC3, troponin T (TNNT2), and myosin heavy chain 7 (MYH7) mRNA expression in isogenic iPSC-CMs measured by qPCR (943cor, 943het, 943hom; n=3–4, respectively). (C) Fraction of mutant mRNA (mut.) and wild type (WT) within total MYBPC3 mRNA accounts for 32.7% ± 0.6% in 943het iPSC-CMs (quantification using RNA-seq data; 943cor and 943het; n=3, respectively). (D) Western blot analysis of MYBPC3 protein levels and quantification of MYBPC3 protein expression normalized to alpha sarcomeric actin (αSA) in isogenic iPSC-CMs (using Santa Cruz E7 anti-MYBPC3; 943cor and 943het). (E) Mass spectrometry analysis of MYBPC3 protein expression in healthy (943cor, ctrl) and HCM (943het, ctrl943) iPSC-CMs (n=4 differentiation batches, respectively). # p<0.1; * p<0.05; ** p<0.01.
RNA-sequencing of MYBPC3 PTC iPSC-CMs reveals a gene signature of NMD activation
To further elucidate the molecular mechanism underlying the pathogenesis of the HCM phenotype associated with MYBPC3 PTC mutation, we next performed RNA-sequencing in isogenic iPSC-CMs (943cor vs. 943het; ctrl vs. ctrl943; Supplemental Figure S8a). We detected 125 upregulated and 173 downregulated genes in HCM compared to isogenic control iPSC-CMs (Figure 4a and Supplemental Table S4). Within the most significantly regulated genes, we observed a gene signature with a significant representation of genes involved in calcium handling (ATP2A2, ATP2B2, and CASQ1), cardiac hypertrophy (GP130, JAK2, RRAS, MEK1, TWEAKR, and NPPB), stress response (HSPB1, HSPB6, HSPB7, IGF1, and IGF2), and structural organization of sarcomeres and mechanosensors (CSRP3 and TCAP; Figure 4b and Supplemental Figure S9a). These genes were uniformly dysregulated in iPSC-CMs carrying the p.R1073P_Fsx4 mutation (Supplemental Figure S9b, c). Furthermore, gene-set enrichment analysis (GSEA) of the RNA-sequencing data revealed a significant enrichment of pathways involved in protein trafficking, translation, RNA metabolism, and NMD in HCM compared to isogenic control iPSC-CMs (Figure 4c). Taken together, these data suggest a common molecular HCM phenotype in iPSC-CMs carrying MYBPC3 PTC mutations characterized by the activation of the NMD pathway and a HCM gene signature in comparison to isogenic healthy iPSC-CMs.
Figure 4: RNA-sequencing reveals a molecular HCM phenotype.
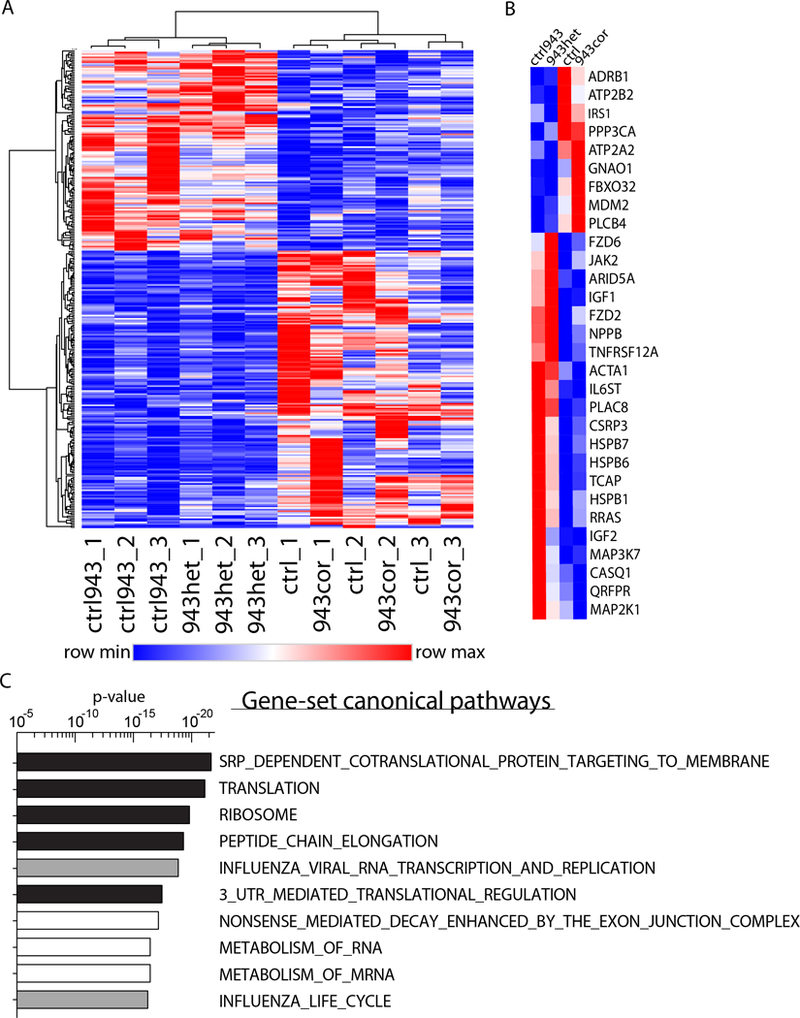
(A) Heatmap of the 298 significantly differentially regulated genes comparing HCM (943het, ctrl943) and healthy (943cor, ctrl) iPSC-CMs (fold-change ± 1.4; q-value <0.05; n=3 differentiation batches each iPSC line). 125 genes were upregulated and 173 genes were downregulated in HCM compared to healthy iPSC-CMs. (B) Within the top significant genes, a set of 30 genes were identified to be involved in cardiac hypertrophy, anabolic processes, and calcium signaling. (C) Top 10 canonical pathways activated in HCM iPSC-CMs identified by gene-set enrichment analysis (GSEA) of the genes. Pathways involved in translation are highlighted in black and pathways involved in RNA metabolism are highlighted in white.
NMD inhibition ameliorates MYBPC3 PTC-associated functional and molecular phenotypes
To evaluate whether there is a direct interaction between the upregulation of NMD and the observed molecular phenotype in iPSC-CMs, we next used a siRNA approach to inhibit UPF1, a key component of the NMD pathway.25 UPF1 is one of the crucial regulators for initiation of NMD in response to mRNA transcripts with a PTC mutation. The recruitment of the SURF complex (SMG1, UPF1, ERF1, and ERF3 factors) due to a PTC mutation distinguishes the PTC from the authentic termination site.26 SMG1-mediated phosphorylation of UPF1 leads to initiation of mRNA and peptide degradation.26 In iPSC-CMs carrying the p.R943x mutation, UPF1 inhibition resulted in the normalization of the expression of the majority of genes included in the HCM gene signature (Figure 5a, b and Supplemental Figure S10a). In contrast, knockdown of UPF1 in isogenic control iPSC-CMs showed no specific regulation of the HCM gene signature (Supplemental Figure S10b). We next investigated whether manipulating the NMD pathway would affect the functional phenotype of HCM iPSC-CMs. Inhibition of UPF1 ameliorated the aberrant calcium handling properties of 943het iPSC-CMs, characterized by a significant increase in Ca2+ reuptake kinetics (Figure 5c). On the contrary, the disease specific response of increased Ca2+ reuptake kinetics upon UPF1 inhibition was not observed in the 943cor isogenic control iPSC-CMs (Supplemental Figure S10c). These findings suggest a direct link between MYBPC3 PTC mutation and sustained NMD activation at the molecular level that precedes the emergence of the HCM phenotype (Figure 5d). At this stage, these mechanisms are independent of haploinsufficiency on MYBPC3 protein level or an accumulation of truncated peptides.
Figure 5: Modulation of nonsense-mediated decay pathway reverses HCM phenotype.
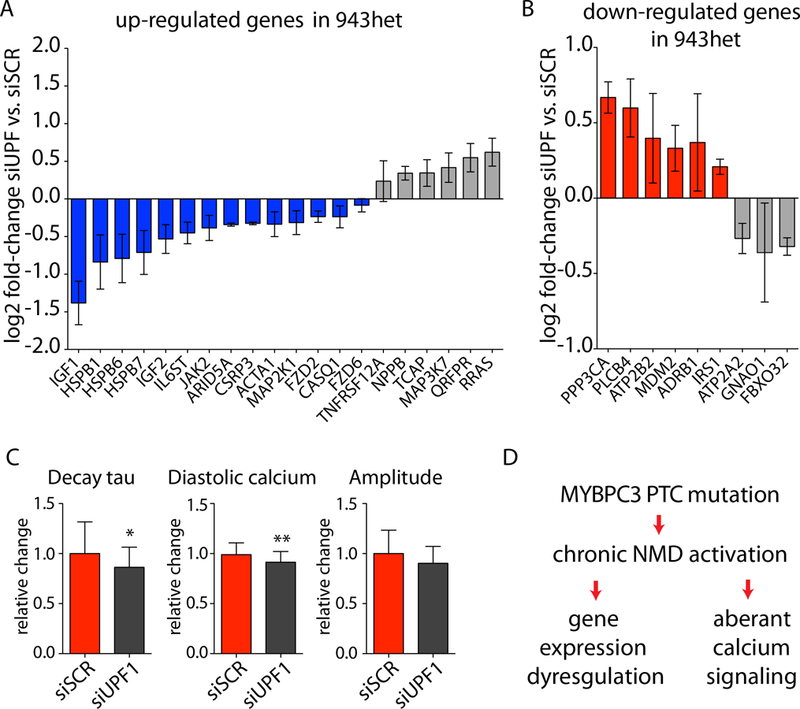
Regulation of genes contributing to the molecular HCM phenotype by siUPF1 treatment compared to scramble (siSCR) control in 943het iPSC-CMs. (A) Genes upregulated in HCM (943het, ctrl) being reversed by NMD inhibition are highlighted in blue and (B) genes downregulated in HCM being reversed by NMD inhibition are highlighted in red (943het iPSC-CMs; n=3 differentiation batches; data are presented as mean log2 fold-change of siUPF1 vs. siSCR control ± SEM). (C) NMD modulation by UPF1 inhibition resulted in a reversion of calcium handling abnormalities in 943het iPSC-CMs. (D) Proposed mechanism of NMD activation by PTC carrying MYBPC3 mRNA resulting in a molecular HCM phenotype and abnormal calcium signaling. Short-term inhibition of UPF1 results in a partial reversion of the HCM phenotypes supporting a direct link between NMD activation and HCM. * p<0.05; ** p<0.01.
DISCUSSION
MYBPC3 PTC mutations are the most common cause of HCM.2 Although recent studies have shown that iPSC-CMs recapitulate certain aspects of HCM phenotypes,8,20,27 the mechanistic link between MYBPC3 PTC mutations and the pathogenesis of HCM is still lacking. Several studies have investigated the underlying mechanism of how MYBPC3 PTC mutations lead to HCM and two separate hypotheses have been proposed: i) a deleterious truncated peptide, and ii) MYBPC3 haploinsufficiency. Studies using cardiac tissue from HCM patients harboring MYBPC3 PTC mutations3,28 and transgenic animal models29,30 suggest that the truncated MYBPC3 peptides are cleared by the UPS.24,31 Haploinsufficiency of MYBPC protein has been shown in cardiac samples derived from patients with in PTC mutations of MYBPC3.32 Previous studies comparing HCM to unrelated control iPSC-CMs have yielded conflicting results in regards to MYBPC3 protein haploinsufficiency,20,33 most likely reflecting the influence of the genetic background on the iPSC-CM phenotype34 when comparing non-isogenic iPSCs.
In this study, we combined human iPSCs and genome editing technologies to interrogate the mechanisms underlying the pathogenesis of HCM associated with MYBPC3 PTC mutations in vitro. We demonstrated that HCM iPSC-CMs display aberrant Ca2+ handling properties without evidence for haploinsufficiency of MYBPC3 at the protein level and absence of any detectable truncated peptides in isogenic iPSC-CMs carrying two separate MYBPC3 PTC mutations (p.R943x and p.R1073P_Fsx4) in both 2D monolayers and 3D EHT preparations. These results challenge the concept of haploinsufficiency or poison peptides as the underlying mechanisms for HCM caused by MYBPC3 PTC mutations, suggesting that early pathophysiological processes at the molecular level may precede the development of disease. Indeed, we identified a molecular HCM signature characterized by the activation of NMD pathway and the dysregulation of a set of genes involved in major cardiac signaling pathways. The NMD has been shown to be the major pathway involved in clearance of MYBPC3 mRNA with PTC mutations in mice.4 However, besides its role as a cellular surveillance pathway, NMD also interacts with other major pathways involved in RNA and protein homeostasis35 that functionally tunes both the transcriptomes and proteomes in mammalian cells.25 We demonstrated that inhibition of the NMD pathway in HCM iPSC-CMs ameliorates the HCM phenotype at both the functional and molecular level, supporting a role of NMD in the pathogenesis of HCM associated with MYBPC3 PTC mutations.
In conclusion, we have provided the first evidence directly connecting the sustained activation of the NMD pathway and HCM disease development associated MYBPC3 PTC mutations. Further elucidation of the specific molecular mechanisms linking NMD to HCM holds promise for uncovering valuable new therapeutic targets in HCM.
Supplementary Material
Clinical Perspective.
What is new?
Combining iPSC technology and genome editing technologies, we have shown that MYBPC3 PTC mutations chronically activate the NMD pathway.
The study challenges the current concept of haploinsufficiency or poison peptides as the underlying mechanisms for HCM caused by MYBPC3 PTC mutations.
The results suggest that early pathophysiological processes at the molecular level may precede the development of disease.
Short-term inhibition of NMD resulted in a reversal of the HCM phenotype in vitro.
What are the clinical implications?
The study offers a novel perspective on potential therapies for genetic HCM.
Strategies targeting the NMD pathway might prove successful in halting the inevitable progression of HCM.
ACKNOWLEDGMENTS
We gratefully acknowledge the Stanford Translational Applications Service Center (J.E. Liliental, S. Patel) for performing the nanofluidics immunoassays and the Stanford Functional Genomics Facility (J. Coller, D. Wagh) for assistance performing with the ddPCR analysis. Furthermore, we acknowledge the Stanford Neuroscience Microscopy Service, supported by NIH NS069375.
T.S. conceived, performed, and interpreted the experiments and wrote the manuscript. R.S. and C.C. performed and interpreted the experiments. L.C.K performed the calcium handling experiments. W.L.M. performed contractility assays. E.L performed the proteomics analysis and data analysis. A.W. generated the EHTs and performed the contractility analysis. G.M. wrote and optimized automated calcium analysis script. M.G. performed the RNA-sequencing data analysis. J.L. and A.O. provided experimental advice and contributed to manuscript writing. S.L., F.Y., and H.Y. helped establish the micro-pattering experiments. M.M. provided experimental advice and contributed to manuscript writing. M.W. and E.A.A. referred the patients included in the study. J.C.W and I.K. conceived the experimental design, and provided experimental advice, manuscript writing, and provided funding support.
FUNDING SOURCES
Funding support was provided by the German Funding Foundation (DFG) (T.S.); California Institute of Regenerative Medicine (CIRM) (R.S); NIH R01 HL139679, R00 HL104002 and American Heart Association (AHA) 17IRG33410532 (I.K.); NIH R01 HL141851, NIH R01 HL128170, NIH R01 HL130020, NIH R01 HL126527, American Heart Association (AHA) Merit 33610009, and Burroughs Wellcome Fund 1015009 (J.C.W).
Footnotes
DISCLOSURES
None
REFERENCES
- 1.Maron BJ, Ommen SR, Semsarian C, Spirito P, Olivotto I, Maron MS. Hypertrophic Cardiomyopathy: Present and future, with translation into contemporary cardiovascular medicine. JACC 2014;64:83–99. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy After 20 Years. JACC 2012;60:705–715. [DOI] [PubMed] [Google Scholar]
- 3.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 2009;105:219–222. [DOI] [PubMed] [Google Scholar]
- 4.Vignier N, Schlossarek S, Fraysse B, Mearini G, Kramer E, Pointu H, Mougenot N, Guiard J, Reimer R, Hohenberg H, Schwartz K, Vernet M, Eschenhagen T, Carrier L. Nonsense-mediated mRNA decay and ubiquitin-proteasome system regulate cardiac myosin-binding protein C mutant levels in cardiomyopathic Mice. Circ Res 2009;105:239–248. [DOI] [PubMed] [Google Scholar]
- 5.Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circ Res 2015;117:80–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Karakikes I, Termglinchan V, Cepeda DA, Lee J, Diecke S, Hendel A, Itzhaki I, Ameen M, Shrestha R, Wu H, Ma N, Shao N-Y, Seeger T, Woo NA, Wilson KD, Matsa E, Porteus MH, Sebastiano V, Wu JC. A comprehensive TALEN-based knockout library for generating human induced pluripotent stem cell-based models for cardiovascular diseases. Circ Res 2017;120:1561–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Garg P, Oikonomopoulos A, Chen H, Li Y, Lam CK, Sallam K, Perez M, Lux RL, Sanguinetti MC, Wu JC. Genome editing of induced pluripotent stem cells to decipher cardiac channelopathy variant. JACC 2018;72:62–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma N, Zhang J, Itzhaki I, Zhang SL, Chen H, Haddad F, Kitani T, Wilson KD, Tian L, Shrestha R, Wu H, Lam CK, Sayed N, Wu JC. Determining the pathogenicity of a genomic variant of uncertain significance using CRISPR/Cas9 and human-induced pluripotent stem cells. Circulation 2018; epub ahead of print; DOI: 10.1161/CIRCULATIONAHA.117.032273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, Hsiao C, Kamp TJ, Palecek SP. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/β-catenin signaling under fully defined conditions. Nat Protoc 2013;8:162–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Termglinchan V, Seeger T, Chen C, Wu JC, Karakikes I. Efficient genome editing in induced pluripotent stem cells with engineered nucleases in vitro. Methods Mol Biol 2017;1521:55–68. [DOI] [PubMed] [Google Scholar]
- 11.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8:2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayakawa T, Kunihiro T, Ando T, Kobayashi S, Matsui E, Yada H, Kanda Y, Kurokawa J, Furukawa T. Image-based evaluation of contraction–relaxation kinetics of human-induced pluripotent stem cell-derived cardiomyocytes: Correlation and complementarity with extracellular electrophysiology. J Mol Cell Cardiol 2014;77:178–191. [DOI] [PubMed] [Google Scholar]
- 14.Breckwoldt K, Letuffe-Brenière D, Mannhardt I, Schulze T, Ulmer B, Werner T, Benzin A, Klampe B, Reinsch MC, Laufer S, Shibamiya A, Prondzynski M, Mearini G, Schade D, Fuchs S, Neuber C, Krämer E, Saleem U, Schulze ML, Rodriguez ML, Eschenhagen T, Hansen A. Differentiation of cardiomyocytes and generation of human engineered heart tissue. Nat Protoc 2017;12:1177–1197. [DOI] [PubMed] [Google Scholar]
- 15.McMullen G. CalciPy: A Python script for processing ratiometric calcium fluorescence data. Github https://github.com/GeorgeMcMullen/CalciPy. Accessed Sep 21, 2018.
- 16.Morpheus: Versatile matrix visualization and analysis software Broad Instutute. https://software.broadinstitute.org/morpheus/. Accessed Sep 21, 2018.
- 17.Hughes SE. The pathology of hypertrophic cardiomyopathy. Histopathology 2004;44:412–427. [DOI] [PubMed] [Google Scholar]
- 18.Harris SP, Bartley CR, Hacker TA, McDonald KS, Douglas PS, Greaser ML, Powers PA, Moss RL. Hypertrophic cardiomyopathy in cardiac myosin binding protein-C knockout mice. Circ Res 2002;90:594–601. [DOI] [PubMed] [Google Scholar]
- 19.Pohlmann L, Kroger I, Vignier N, Schlossarek S, Kramer E, Coirault C, Sultan KR, El-Armouche A, Winegrad S, Eschenhagen T, Carrier L. Cardiac myosin-binding protein C is required for complete relaxation in intact myocytes. Circ Res 2007;101:928–938. [DOI] [PubMed] [Google Scholar]
- 20.Birket MJ, Ribeiro MC, Kosmidis G, Ward D, Leitoguinho AR, van de Pol V, Dambrot C, Devalla HD, Davis RP, Mastroberardino PG, Atsma DE, Passier R, Mummery CL. Contractile defect caused by mutation in MYBPC3 revealed under conditions optimized for human PSC-cardiomyocyte function. Cell Rep 2015;13:733–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Dijk SJ, Boontje NM, Heymans MW, ten Cate FJ, Michels M, dos Remedios C, Dooijes D, van Slegtenhorst MA, van der Velden J, Stienen GJM. Preserved cross-bridge kinetics in human hypertrophic cardiomyopathy patients with MYBPC3 mutations. Pflugers Arch 2014;466:1619–1633. [DOI] [PubMed] [Google Scholar]
- 22.Frey N, Luedde M, Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol 2011;9:91–100. [DOI] [PubMed] [Google Scholar]
- 23.Helms AS, Alvarado FJ, Yob J, Tang VT, Pagani F, Russell MW, Valdivia HH, Day SM. Genotype-dependent and -independent calcium signaling dysregulation in human hypertrophic cardiomyopathy. Circulation 2016;134:1738–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sarikas A, Carrier L, Schenke C, Doll D, Flavigny J, Lindenberg K, Eschenhagen T, Zolk O. Impairment of the ubiquitin proteasome system by truncated cardiac myosin binding protein C mutants. Cardiovasc Res 2005;66:33–44. [DOI] [PubMed] [Google Scholar]
- 25.Lykke-Andersen S, Jensen TH. Nonsense-mediated mRNA decay: an intricate machinery that shapes transcriptomes. Nat Rev Mol Cell Biol 2015;16:665–677. [DOI] [PubMed] [Google Scholar]
- 26.Kashima I, Yamashita A, Izumi N, Kataoka N, Morishita R, Hoshino S, Ohno M, Dreyfuss G, Ohno S. Binding of a novel SMG-1-Upf1-eRF1-eRF3 complex (SURF) to the exon junction complex triggers Upf1 phosphorylation and nonsense-mediated mRNA decay. Genes Dev 2006;20:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lan F, Lee AS, Liang P, Sanchez-Freire V, Nguyen PK, Wang L, Han L, Yen M, Wang Y, Sun N, Abilez OJ, Hu S, Ebert AD, Navarrete EG, Simmons CS, Wheeler M, Pruitt B, Lewis R, Yamaguchi Y, Ashley EA, Bers DM, Robbins RC, Longaker MT, Wu JC. Abnormal calcium handling properties underlie familial hypertrophic cardiomyopathy pathology in patient-specific induced pluripotent stem cells. Cell Stem Cell 2013;12:101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Dijk SJ, Dooijes D, dos Remedios C, Michels M, Lamers JMJ, Winegrad S, Schlossarek S, Carrier L, ten Cate FJ, Stienen GJM, van der Velden J. Cardiac myosin-binding protein C mutations and hypertrophic cardiomyopathy: haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation 2009;119:1473–83. [DOI] [PubMed] [Google Scholar]
- 29.Carrier L, Knöll R, Vignier N, Keller DI, Bausero P, Prudhon B, Isnard R, Ambroisine M- L, Fiszman M, Ross J, Schwartz K, Chien KR. Asymmetric septal hypertrophy in heterozygous cMyBP-C null mice. Cardiovas Res 2004;63:293–304. [DOI] [PubMed] [Google Scholar]
- 30.Barefield D, Kumar M, Gorham J, Seidman JG, Seidman CE, de Tombe PP, Sadayappan S. Haploinsufficiency of MYBPC3 exacerbates the development of hypertrophic cardiomyopathy in heterozygous mice. J Mol Cell Cardiol 2015;79:234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schlossarek S, Singh SR, Geertz B, Schulz H, Reischmann S, Hübner N, Carrier L. Proteasome inhibition slightly improves cardiac function in mice with hypertrophic cardiomyopathy. Front Physiol 2014;5:484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marston S, Copeland O, Jacques A, Livesey K, Tsang V, McKenna WJ, Jalilzadeh S, Carballo S, Redwood C, Watkins H. Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circ Res 2009;105:219–222. [DOI] [PubMed] [Google Scholar]
- 33.Prondzynski M, Krämer E, Laufer SD, Shibamiya A, Pless O, Flenner F, Müller OJ, Münch J, Redwood C, Hansen A, Patten M, Eschenhagen T, Mearini G, Carrier L. Evaluation of MYBPC3 trans-splicing and gene replacement as therapeutic options in human iPSC-derived cardiomyocytes. Mol Ther Nucleic Acids 2017;7:475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsa E, Burridge PW, Yu K-H, Ahrens JH, Termglinchan V, Wu H, Liu C, Shukla P, Sayed N, Churko JM, Shao N, Woo NA, Chao AS, Gold JD, Karakikes I, Snyder MP, Wu JC. Transcriptome profiling of patient-specific human iPSC-cardiomyocytes predicts individual drug safety and efficacy responses in vitro. Cell Stem Cell 2016;19:311–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Karam R, Lou C-H, Kroeger H, Huang L, Lin JH, Wilkinson MF. The unfolded protein response is shaped by the NMD pathway. EMBO rep 2015;16:599–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.