

Summary
We identified ∼30-mer amyloid-β protein (Aβ) assemblies, termed amylospheroids, from brains of patients with Alzheimer disease (AD) as toxic entities responsible for neurodegeneration and showed that Na+,K+-ATPase α3 (NAKα3) is the sole target of amylospheroid-mediated neurodegeneration. However, it remains unclear where in neurons amylospheroids form and how they reach their targets to induce neurodegeneration. Here, we present an in vitro culture system designed to chronologically follow amylospheroid formation in mature neurons expressing amyloid precursor protein bearing early-onset AD mutations. Amylospheroids were found to accumulate mainly in the trans-Golgi network of excitatory neurons and were initially transported in axons. Proteasome inhibition dramatically increased amylospheroid amounts in trans-Golgi by increasing Aβ levels and induced dendritic transport. Amylospheroids were secreted and caused the degeneration of adjacent NAKα3-expressing neurons. Interestingly, the ASPD-producing neurons later died non-apoptotically. Our findings demonstrate a link between ASPD levels and proteasome function, which may have important implications for AD pathophysiology.
Subject Areas: Pathophysiology, Neuroscience, Cellular Neuroscience
Graphical Abstract
Highlights
-
•
Proteasome inhibition increases ASPD in excitatory neurons by increasing APP and Aβ
-
•
Proteasome inhibition shifts the Aβ and ASPD distribution from axons to dendrites
-
•
ASPDs are secreted and kill surrounding ATPase α3 neurons by cell surface binding
-
•
Following apoptosis in α3 neurons, ASPD-accumulating neurons die non-apoptotically
Pathophysiology; Neuroscience; Cellular Neuroscience
Introduction
Alzheimer's disease (AD) impairs neural networks involved in cognitive function by affecting synaptic connections and causing neuronal degeneration (Kouchi et al., 1998, Parthasarathy et al., 2015, Serrano-Pozo et al., 2011). Amyloid-β protein (Aβ), a small protein produced physiologically by proteolytic cleavages of the amyloid precursor protein (APP) (Haass et al., 2012), has been long considered to play a primary role in this process (Hardy and Selkoe, 2002). Aβ has the ability to form a range of structurally distinct assemblies, which exert different toxic functions via different targets (Benilova et al., 2012, Chiti and Dobson, 2009, Glabe, 2008, Hardy and Selkoe, 2002, Hoshi et al., 2003, Jarosz-Griffiths et al., 2016, Klein et al., 2001, Matsumura et al., 2011, Noguchi et al., 2009, Ohnishi et al., 2015, Roychaudhuri et al., 2009, Walsh and Selkoe, 2007). However, in neurons, the subcellular site and the regulation mechanism of Aβ production still remain to be determined (Haass et al., 2012), as do the mechanisms of Aβ assembly into neurotoxic structures and the manner in which these structures encounter their target neurons.
We previously identified a population of neurotoxic Aβ assemblies, amylospheroids (ASPD), stable assemblies containing approximately 30 monomers (Hoshi et al., 2003, Matsumura et al., 2011, Noguchi et al., 2009, Parthasarathy et al., 2015). ASPD bind to the neuron-specific α3 subunit of the Na+, K+-ATPase pump (NAKα3), with a Kd in the low nanomolar range, and induce neurodegeneration by impairing the pump activity (Ohnishi et al., 2015). Importantly, ASPD concentration correlated with disease severity and progression in patients (Ohnishi et al., 2015). Interestingly, ASPD levels were high in the cerebral cortex, where NAKα3-expressing neurons were lost, whereas their levels were low in the cerebellum, where NAKα3-expressing neurons were preserved. These findings showed that NAKα3 was a neurotoxic target of ASPD and also suggested a difference in the ability to form ASPD between brain regions (Ohnishi et al., 2015).
In vitro studies have shown that the pathway of ASPD assembly from Aβ monomers is distinct from those of other Aβ oligomers (Matsumura et al., 2011). A key question remaining was how ASPD formation occurred in vivo. We reasoned that because the primary component of patient-derived ASPD appears to be Aβ (Ohnishi et al., 2015), ASPD formation should be linked to Aβ production from APP. Full-length APP is synthesized in the endoplasmic reticulum (ER) and transported through the Golgi apparatus to the trans-Golgi network (TGN), where the highest concentration of APP is found in neurons in steady state (Greenfield et al., 1999, Hartmann et al., 1997, Xu et al., 1997). From the TGN, a population of APP is further transported in the TGN-derived secretory vesicles to the plasma membrane (PM) where it is either cleaved by α-secretase to produce a soluble molecule, sAPPα, or re-internalized via an endosomal/lysosomal degradation pathway (Haass et al., 2012). Aβ has been reported to be generated from mature APP through β- and γ-secretase cleavages in the above sorting pathways that interconnect the TGN, the cell surface, and the endosomes (Haass et al., 2012). Therefore we hypothesized that ASPD might also be generated in these pathways.
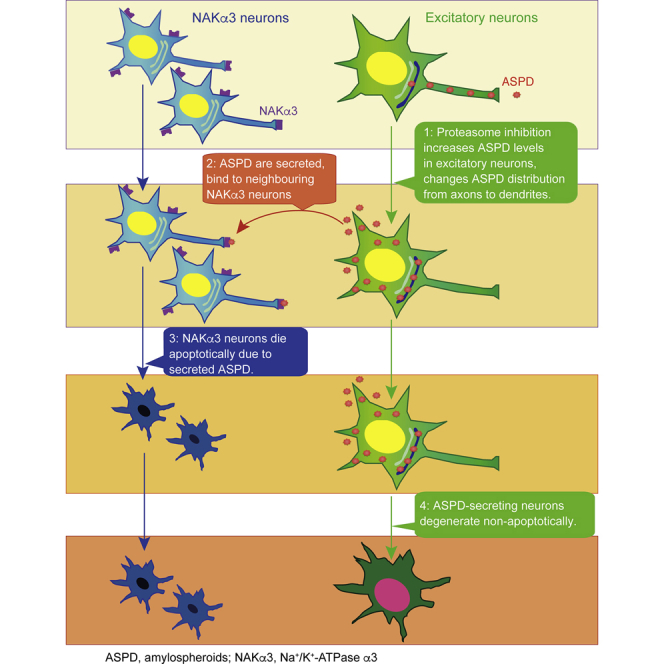
Proteasomes may also be involved in APP/Aβ degradation (Wang and Saunders, 2014). Proteasomes are present mainly in the cytoplasm in mammalian cells, but immune electron microscopy has shown that they can also be associated with the cytoplasmic surface of the ER in mammalian cells (Palmer et al., 1996, Rivett et al., 1992). Membrane proteins like APP are retro-translocated from the ER to the cytoplasm and degraded by the ER-associated proteasomes. This process is termed ER-associated degradation (ERAD) (Vembar and Brodsky, 2008). In general, upward of 30% of newly synthesized proteins are degraded by proteasomes (Schubert et al., 2000), including APP and Aβ (Wang and Saunders, 2014). Proteasome activities are lower in AD brains than in age-matched control brains (Keck et al., 2003, Keller et al., 2000), and proteomic studies have suggested that proteasome impairment might be a risk factor for AD (Manavalan et al., 2013, Tonoki et al., 2009). We report here studies to determine the role of proteasomes in ASPD formation. Our approach was to inhibit the proteasome-mediated degradation of APP and Aβ and determine its effect on ASPD formation in mature neurons. For this purpose, we established an in vitro cellular system to monitor ASPD formation in neurons expressing APP-bearing mutations linked to familial early-onset AD. As summarized in the Graphical Abstract, we found that proteasome inhibition dramatically increased intra-neuronal ASPD levels and changed ASPD distribution from the axon to dendrites. ASPD were then secreted and killed neighboring NAKα3 neurons. These findings deepen our understanding of the formation and delivery of toxic Aβ oligomers in AD brains, which in the future may open up the possibility of developing anti-assembly drugs for AD by modifying APP/Aβ degradation.
Results
Introduction of Human APP770 Gene Bearing the Early-Onset Mutations into Mature Hippocampal Neurons by Using an AAV Vector
To establish a mature neuron-based system, we introduced human APP770 gene with a familial AD mutation into rat hippocampal neuronal cultures at 10 days in vitro (DIV) using an adeno-associated virus 1-derived (AAV) vector (Li et al., 2006) (Transparent Methods, Figure 1A). Two types of mutations were selected. One was the Swedish mutation (APPswe), which results in the substitution of Lys670 and Met671, two amino acids adjacent to the β-secretase cleavage site, into Asn670 and Leu671, respectively (Mullan et al., 1992). The other was the Osaka mutation (APPosk), which involves deletion of the entire codon 693 encoding glutamate (corresponding to glutamate at position 22 of Aβ; accordingly designated as E22Δ) (Tomiyama et al., 2008). Western blot and immunocytochemistry confirmed that mature human APP was expressed in neurons transduced with either APPswe or APPosk gene (Figures 1B and 1C). The level of expressed human APP was on average 2.7 times (as to APPswe) or 5.1 times (as to APPosk) as much as that of endogenous rodent APP, based on quantification in western blots (Figure 1B). As reported previously (Powell et al., 2016), the AAV vector showed tropism for neurons over astrocytes. In our study, transduction efficiency of rat hippocampal neurons with the AAV vector was usually >85%. Consistently, in the AAV-infected cultures, the human APP770-specific antibody detected human APP770 protein in almost all the neurons (Figure 1C and enlarged views in insets) and minimal expression in astrocytes (Figure 1C).
Figure 1.

Expression of Human APP in Mature Neurons
(A) Experiments were performed as shown here except for the staining in the upper panels of Figure 5A (performed at 30 DIV).
(B) Representative western blot of whole lysates (10 μg/lane) of primary rat hippocampal neuronal cultures with or without AAV-APP transduction, detected by anti-APP or anti-actin antibody (see Transparent Methods). Arrows show O-glycosylated or N-glycosylated human APP, above which rat APP was also detected.
(C) Primary rat hippocampal neuronal cultures, with or without AAV-APP transduction, were stained with antibodies against MAP2, GFAP, and human APP at 22 DIV (see Transparent Methods). Representative images are shown. Scale bar, 50 μm. An enlarged view of the field is shown in the insets.
Proteasome Inhibition Affects Aβ Metabolism
We first examined the effect of proteasome inhibition on Aβ production by treating the AAV-transduced mature neurons with a proteasome inhibitor, MG132 (IC50 of 100 nM; Tsubuki et al., 1996), at 75 nM for 24 h. Because the rate-limiting step in Aβ production is cleavage of APP by BACE-1 (Sinha and Lieberburg, 1999), we monitored cleavage levels using an Aβ N-terminal-end-specific antibody that does not cross-react with full-length APP. As shown in Figure 2A, proteasome inhibition markedly increased Aβ N-terminal-end-specific (N-terAβ) staining in human APP-transduced mature neurons. Although the degree of increase varied widely from neuron to neuron, N-terAβ staining appeared to increase in almost all the neurons (Figures 2A and S1), consistent with human APP expression in almost all the neurons (Figure 1C). Similarly, we found that human APP770-specific staining increased markedly in the neuronal cell body after 24-h MG132 treatment (Figure 2B). At this time, Aβ assemblies were trimeric, as assessed by SDS-PAGE (red arrow in Figure 3 left panel). Western blot showed that Aβ and human APP levels increased approximately 3- and 10-fold, respectively (Figure 3). We confirmed that proteasome inhibition did not accelerate Aβ secretion as the level of secreted Aβ, either Aβ1-40 or Aβ1-42, was not changed or even slightly decreased (*p = 0.04 by Scheffé post hoc test; n = 4) after 24-h MG132 treatment (Figure S2). Collectively, these data showed that proteasome inhibition led to an increase in the steady-state level of Aβ in our system, either directly by inhibiting ERAD-mediated Aβ degradation or indirectly by increasing the APP available for Aβ production.
Figure 2.

Effect of Proteasome Inhibition on Aβ and APP
(A and B) Primary rat hippocampal neuronal cultures, with or without AAV-APP transduction, were treated with or without 75 nM MG132 for 24 h. The cultures were triple stained with DAPI, anti-MAP2, and anti-Aβ N-terminal end-specific (N-terAβ) antibodies in (A) or stained with DAPI, anti-MAP2, and anti-human APP770-specific antibodies in (B) (Transparent Methods). Representative images are shown. For each condition, left columns show the triple staining, whereas right columns show the corresponding DAPI/N-terAβ staining in (A) or DAPI/human APP770 staining in (B). An enlarged view is shown in the insets. Scale bar, 50 μm.
Figure 3.

Effect of Proteasome Inhibition on Aβ and APP
Representative western blot of whole lysates (8 μg/lane) of primary rat hippocampal neuronal cultures with or without AAV-APP transduction, treated with or without 75 nM MG132 for 24 h, detected by anti-Aβ N-terminal, anti-APP, or anti-actin antibody (see Transparent Methods). Left panel, Aβ appeared at the position of the trimer in SDS-PAGE (red arrowhead). The right lane represents western blot of synthetic Αβ1-42 (4 pmol/lane). Black arrows show monomer, dimer, and trimer positions. The asterisk indicates non-specific bands detected by anti-Aβ N-terminal antibodies. Middle panel, arrows show O-glycosylated or N-glycosylated human APP, above which rat APP was also detected. Right panel, green asterisk shows non-specific bands detected by an anti-actin antibody. See Figure S2.
Inhibition of Proteasome Activities Increases ASPD Accumulation in Mature Neurons Expressing Human APP Bearing the Early-Onset Swedish Mutation
We next examined the effect of proteasome inhibition on ASPD formation using the APPswe transgene, which increases Aβ production by facilitating APP processing mediated by β-secretase without any change in the Aβ sequence (Cai et al., 1993, Citron et al., 1992). As shown in Figures 4A and S3A, after 24-h treatment with MG132, APPswe-transduced neurons showed a marked increase in ASPD staining. Consistent with AAV tropism for neurons (Figure 1C), ASPD staining was increased essentially in microtubule-associated protein 2 (MAP2)-positive neurons, but rarely in glial fibroblast acidic protein (GFAP)-positive astrocytes, after 24-h MG132 treatment (Figure 4A left). However, whereas N-terAβ antibody staining was observed in some degree in almost all the neurons, only a limited population of the APPswe-transduced neurons showed an increased ASPD staining (arrowheads in Figure S1). The level of ASPD staining thus does not simply correlate with N-terAβ staining. In contrast with APPswe-transduced neurons, untransduced neurons (without human APP) that produce only endogenous rodent Aβ rarely accumulate ASPD. This is reasonable because, owing to the three amino acid substitutions, Arg5Gly, His13Arg, and Tyr10Phe, rodent Aβ is less prone to form toxic, smaller aggregates when compared with human Aβ, resulting in higher resistance to the development of AD-like neuropathology (Roychaudhuri et al., 2015).
Figure 4.

Proteasome Inhibition Increases Intra-neuronal ASPD Accumulation
(A) Primary rat hippocampal neuronal cultures, with or without AAV-APPswe transduction, were treated with 75 nM MG132 for 24 h. The cultures were triple stained with antibodies against N-terAβ, MAP2, and DAPI on the right side, or against MAP2, GFAP, and ASPD (rpASD1) on the left side. Representative images are shown. Scale bar, 50 μm. In this work, ASPD were detected by immunohistochemical staining with ASPD-specific rpASD1 antibody or mASD3 antibody (Noguchi et al., 2009), both of which have been used to detect ASPD in immunopathological studies of human brains (Noguchi et al., 2009, Ohnishi et al., 2015).
(B) Primary rat hippocampal neuronal cultures, with or without AAV-APPswe transduction, were treated with 75 nM MG132 for the indicated time. The cultures were double stained with anti-ASPD rpASD1 antibody and DAPI. An enlarged view of the field enclosed by a hatched line is shown immediately on the right. Representative images are shown. Scale bar, 50 μm. The number of ASPD and MAP2 double-positive cells was counted by using a GE Healthcare Life Sciences “IN Cell Analyzer” system and is shown as “ASPD + neuron” in the inset upper panel. Then the total amount of rpASD1-immunostained ASPD that was detected in MAP2-positive cells was determined by using an IN Cell Analyzer and was divided by the number of ASPD and MAP2 double-positive cells, which is shown as “ASPD level/neuron” in the inset lower panel (mean ± SD, n = 3; Scheffé post hoc test; ***p < 0.0001, **p = 0.0006, *p = 0.0176).
To confirm that proteasomal dysfunction caused the ASPD formation, we examined the concentration-dependent effects of various pharmacologically unrelated inhibitors on ASPD formation. In addition to a reversible inhibitor, MG132 (Figure S3A), we examined an irreversible inhibitor, epoxomicin, (IC50 of ∼4 nM; Kim et al., 1999; Figure S3B), and an irreversible inhibitor, lactacystin, (IC50 of 4.8 μM; Csizmadia et al., 2010; Figure S3C). All these proteasome inhibitors increased the neuronal ASPD staining at 24 h in a dose-dependent fashion (Figures 4A and S3). Based on these observations, further experiments were performed using MG132 at 75 nM.
We next examined spatiotemporal change in ASPD staining after 75 nM MG132 treatment. As shown in Figure 4B, left columns, baseline levels of ASPD staining were detected in untransduced control cultures, which did not change after 24-h MG132 treatment (see quantification in Figure 4B right). In contrast with control cultures, MG132 treatment increased the ASPD levels substantially and significantly with time in APPswe-transduced neurons (see quantification in Figure 4B right). Initially, very diffuse ASPD staining was consistently detected in the APPswe-transduced neurons before proteasome inhibition (see images of AAV-APPswe at 0 h in Figures 4B and S3A–S3C). This diffuse ASPD staining in neuronal cell bodies was usually accompanied with axonal ASPD staining that was co-stained by an axonal marker, an antibody against neurofilament heavy chain (arrows at 0 h in Figure 4B right and orange arrows in upper panels of Figure 5A). MG132 treatment for 6 h markedly increased the ASPD staining in neuronal cell bodies (representative image of the 6-h MG132 treatment: Figure 4B middle and right). At this time point, in addition to the axonal ASPD staining, dendritic ASPD staining appeared (arrowheads at 6 h in Figure 4B right). MG132 treatment for 24 h dramatically increased both the number of neurons that contained ASPD and the ASPD level in each neuron (an image of the 24-h MG132 treatment is shown in Figure 4B, middle and right; quantification on the right). Interestingly, at this stage, the axonal ASPD staining disappeared (orange arrows in lower panels of Figure 5A). Conversely, the dendritic ASPD staining became stronger, and its distribution changed (double-lined white arrows in lower panels of Figure 5A). At 6-h MG132 treatment, the dendritic ASPD staining was distributed along the entire length of the dendrite, all the way to its tip (arrowhead at 6 h in Figure 4B right). However, at 24-h treatment, the dendritic ASPD staining reached the dendritic branch points, but the staining at the tip had weakened and become patchy (double-lined arrows at 24 h in Figure 4B rightmost panel and double-lined white arrows in lower panels of Figure 5A). High-power images showed that ASPD mostly appeared to stay inside of the dendrites (orange arrows in Figure 5B), but some dendritic ASPD signals on MAP2-labeled dendrites (39.6% ± 4.7%; n = 7 and total count = 1,614) appeared on the surface proximal to the Bassoon staining (double-lined white arrows in Figure 5B). The spatiotemporal change in ASPD staining is schematically illustrated in Figure 5C. Thus proteasome inhibition markedly increased the intra-neuronal ASPD level in steady state and led to changes in its intra-neuronal distribution.
Figure 5.

Proteasome Inhibition Changes ASPD Distribution from Axon to Dendrites
(A) Upper panels: primary rat hippocampal neuronal cultures with AAV-APPswe transduction were quadruple stained at 30 DIV with the anti-ASPD rpASD1 antibody, anti-NFH antibody, anti-MAP2 antibody, and DAPI (see Transparent Methods). Lower panels: primary rat hippocampal neuronal cultures with AAV-APPswe transduction were treated with 75 nM MG132 for 24 h and quadruple stained at 22 DIV as in the upper panels. A representative image, along with the corresponding single red, green, or magenta image, is shown with 50-μm scale bars. Orange arrows mark the axon, whereas white double-lined arrows mark dendrites. ASPD were co-localized with NFH (orange arrows) in the upper panels, but co-localized with MAP2 (white double-lined arrows) in the lower panels.
(B) APPswe-transduced primary rat hippocampal neuronal cultures were treated with 75 nM MG132 for 24 h. The cultures were triple stained with antibodies against MAP2, Bassoon, and ASPD (rpASD1) on the left side. Representative images are shown. Scale bar, 1 μm.
(C) Spatiotemporal changes in the ASPD distribution observed in Figures 3B and 4A are schematically shown in red.
ASPD Accumulate in the Trans-Golgi Network
Next, we examined the subcellular location of ASPD in the APPswe-transduced neurons by double labeling neurons with an ASPD-specific rpASD1 antibody and organelle-specific antibodies. Representative images and their single-channel images are shown in Figures 6 and S4, respectively. Before MG132 treatment, the number of ASPD signals was small, but they were present mainly where the trans-Golgi network-38 (TGN38) signal (Humphrey et al., 1993) was found (see arrowheads in enlarged views of TGN38/rpASD1 staining of Figure 6, and their line scans in Figure 7). With longer MG132 treatment, more ASPD accumulated at the TGN (compare 0-, 6-, and 24-h MG132 treatment in enlarged views of Figures 6 and 7). In contrast, ASPD staining was not co-localized with the ER marker, protein disulfide isomerase (PDI) (Wilkinson and Gilbert, 2004), or a cis-Golgi marker, GM130 (Nakamura et al., 1995), without proteasome inhibition and only rarely co-stained with them at 6-h MG132 treatment (see arrowhead in lower part of Figure 6; their line scans in Figure 7). After 24-h MG132 treatment, more ASPD signals were found together with PDI and GM130 signals, but the coexistence rate in TGN38/ASPD appeared to be higher than that in PDI/ASPD or GM130/ASPD (compare staining of TGN38/ASPD with that of PDI or GM130/ASPD in Figures 6 and S4). ASPD staining in mature neurons was also rarely co-localized with early endosome antigen 1 (EEA1) (Barysch et al., 2009) or lysosomal-associated membrane protein-1 (LAMP1) (Carlsson and Fukuda, 1989) at 6-h MG132 treatment (Figures 6, single channel images in S4, and line scans in Figure 7). This basically remained the case at 24-h MG132 treatment, at which time the vast majority of ASPD staining did not overlap with EEA1 or LAMP1 staining, although some overlap of the ASPD signal with them was seen (see arrowheads in lower part of Figure 6). ASPD staining was rarely observed in untransduced neurons with or without MG132 treatment (Figure S5).
Figure 6.

ASPD Accumulate in the Trans-Golgi of Mature Neurons
Primary rat hippocampal neuronal cultures, with or without AAV-APPswe transduction, were treated with 75 nM MG132 for the indicated time. The cultures were double-stained with anti-ASPD rpASD1 antibody and antibody against organelle marker protein (see Transparent Methods). A highly sensitive, quantitative single-photon-counting system employing avalanche photodiodes on a Zeiss LSM710 microscope was used to perform detailed analyses along the z axis in high-power views of each neuronal cell body doubly-stained with ASPD-specific rpASD1 antibody and an antibody against an organelle marker. Representative images obtained with a 100X oil objective lens are shown. An enlarged view of the field enclosed by a hatched line is shown below. Solid and hatched scale bars, 10 and 1 μm, respectively. Single red and green images for each time point are shown in Figure S4. See also representative images of control cultures without AAV transduction in Figure S5. See Figures 7, S4, and S5.
Figure 7.

Line scans of ASPD Accumulate in the Trans-Golgi of Mature Neurons
Primary rat hippocampal neuronal cultures, with or without AAV-APPswe transduction, were treated with 75 nM MG132 for the indicated time. The cultures were double stained with anti-ASPD rpASD1 antibody and antibody against organelle marker protein as in Figure 6 (see Transparent Methods). Representative line scans of enlarged views shown in Figure 6 are included. See Figure 6
Although the TGN has been presumed to be a sorting site for newly synthesized membrane and secretory proteins, recent studies suggested that the recycling endosomes (REs), detected by an antibody either against the transferrin receptor (TfR) or Rab11 (Kobayashi and Fukuda, 2013), may serve as a sorting hub to direct proteins to the PM (Lasiecka and Winckler, 2011). Interestingly, we found that ASPD were present in the REs of APPswe-transduced neurons at 24-h MG132 treatment (Figures 8 and S6), which is not surprising, given the close and dynamic cross-talk between the TGN and the REs in neurons (Schmidt and Haucke, 2007). However, whereas ASPD always appears to co-localize with the TGN, ASPD co-localization with the REs was rarely detected up to 6-h MG132 treatment. The results strongly support the idea that the TGN is the major and initial site of ASPD localization in mature neurons.
Figure 8.

ASPD Are Also Present in the Recycling Endosomes of Mature Neurons
Primary rat hippocampal neuronal cultures, with or without AAV-APPswe transduction, were treated with 75 nM MG132 for the indicated time. The cultures were double-stained with anti-ASPD rpASD1 antibody and anti-transferrin receptor (TfR) antibody (see Transparent Methods). Representative images obtained with a highly sensitive, direct photon-counting system with a 100X oil objective lens are shown. An enlarged view of the field enclosed by a hatched line is shown below. Solid and hatched scale bars, 10 and 1 μm, respectively. Single red and green images for each time point are shown in Figure S6. See Figure S6.
To further elucidate ASPD localization in the TGN, we used brefeldin A (BFA), a lactone antibiotic and ATPase inhibitor for protein transport (IC50 of 0.06 μg/mL in HCT116 cells; Zhu et al., 2000), which rapidly collapses the Golgi apparatus into the ER by inhibiting protein transport from the ER to the Golgi apparatus (Nebenfuhr et al., 2002, Sciaky et al., 1997). In our system, BFA treatment at 0.6 μg/mL was enough to destroy the Golgi structure without affecting the cell viability in untransduced neurons (compare neurons with the preserved Golgi structure [orange arrow] and those with BFA-destroyed Golgi structure [green arrow] in Figure S7). When MG132-treated APPswe-transduced mature neurons were treated with 0.6 μg/mL BFA for 24 h, the level of ASPD staining was markedly reduced to 36% ± 13% (n = 3) of that without BFA treatment (see high-power images in the right, quantification results below in Figure 9). We also found that BFA treatment of MG132-treated APPswe-transduced neurons substantially decreased the ASPD and TGN38 signals in a time-dependent manner (Figure S8). These results further support the idea that ASPD are mainly present in the TGN under steady-state conditions.
Figure 9.

BFA Markedly Reduced ASPD Accumulation
Primary rat hippocampal neuronal cultures, with or without AAV-APPswe transduction, were treated with 75 nM MG132 for 24 h in the absence or presence of 0.6 μg/mL BFA. We determined this concentration by examining BFA concentrations from 0.6 to 10 μg/mL in our system and found that 0.6 μg/mL BFA treatment was enough to destroy the Golgi structure without affecting the cell viability. The cultures were triple-stained with rpASD1, anti-TGN38 antibody (TGN marker), and DAPI. Representative images are shown. High-power images on the right were obtained with a highly sensitive, direct photon-counting system with a 100X oil objective lens. Solid and hatched scale bars, 10 and 1 μm, respectively. See also representative images of control cultures without AAV transduction in Figure S7. The total amount of rpASD1-immunostained ASPD detected in MAP2-positive neurons was determined by using an IN Cell Analyzer as in Figure 4B, right, and is shown as “ASPD level” below (mean ± SD, n = 3; Scheffé post hoc test, ***p < 0.0001, **p = 0.001).
See Figures S7 and S8.
We then examined the steady-state distribution of human APP770 and N-terAβ in APPswe-transduced mature neurons treated with MG132 for 24 h. As shown in Figure S9, human APP770-specific signals increased most intensely in the TGN and also in the ER in APPswe-transduced neurons with MG132 treatment. In the case of N-terAβ staining, the signal was increased mainly in the cis-Golgi, and also on the TGN, the REs, and the ER surface, but was rarely observed in the early endosomes (EEs) or the Lys in our system (Figures S10 and S11). In contrast, without MG132 treatment, the signal intensity of either human APP770 or N-terAβ was too weak to determine its subcellular distribution (representative images in Figures S9–S11).
Finally, we performed a quantitative co-localization analysis of N-terAβ staining and ASPD staining with organelle markers in MG132-treated APPswe-transduced cells. As shown in Figure 10, although N-terAβ and ASPD co-localized to some extent with EEA1 and LAMP1-stained areas, the levels of co-localization in PDI-, GM130-, TGN38-, and TfR-stained areas were always substantially and significantly (p < 0.0001 by Scheffé post hoc test, see legend in Figure 10) higher than those in EEA1- and LAMP1-stained areas. Interestingly, among PDI-, GM130-, TGN38-, and TfR-stained areas, the co-localization coefficients for N-terAβ were consistently significantly higher in GM130-stained areas, whereas the coefficients for ASPD were always significantly higher in TGN38-stained areas (p < 0.0001 by Scheffé post hoc test, see the legend in Figure 10) in steady state.
Figure 10.

Quantitative Co-localization Analyses
The weighted colocalization coefficients were obtained using ZEN 2009 software (Zeiss) exactly according to the manufacturer's instructions (see Transparent Methods). The weighted colocalization coefficients represent the number of red pixels (rpASD1 or N-terAβ) that co-localize with green pixels (organelle markers) divided by the total number of red pixels. Analyses were performed using different sets of MG132-treated APP-expressing neurons. A representative scattergram is shown below. Statistical significance was calculated using Scheffé post hoc test (green asterisks: ***p < 0.0001, **p = 0.0008 [for ASPD] and 0.0019 [for N-ter Aβ], *p = 0.021 [for ASPD] and 0.0316 [for N-ter Aβ], compared with negative control using DAPI staining; pink asterisks and black asterisks ***p < 0.0001). See Figures S9–S11.
ASPD Accumulate in the Trans-Golgi Network and the Recycling Endosomes of Mature Neurons Expressing APP Bearing the Osaka Mutation
The Osaka mutation causes the deletion of a single amino acid, residue E693 (corresponding to E22 of Aβ). Because Aβ fibrils appeared to be absent in the brains of patients and mice carrying this mutation (Tomiyama et al., 2008, Tomiyama et al., 2010), the mutation was believed to accelerate only the formation of Aβ oligomers. However, precise conformational and assembly kinetic studies of the mutant peptides in vitro revealed that the primary biophysical effect of this mutant is to accelerate conformational changes in the monomer that facilitate oligomerization and fibril formation (Inayathullah and Teplow, 2011). To address whether this mutation facilitates ASPD formation in mature neurons, we first examined whether E22Δ-Aβ1-42 (Aβ1-42-osk) formed neurotoxic ASPD by using a toxicity assay, transmission electron microscopic analysis, and dot blotting with anti-ASPD antibody rpASD1 (Figure 11A). Interestingly, ASPD derived from Aβ1-42-osk were more toxic to mature hippocampal neurons than ASPD from wild-type Aβ1-42 (compare viability data at 18 nM in Figure 11A). Treatment of the APPosk-transduced neurons with 75 nM MG132 for 24 h led to a marked increase in both the number of the ASPD-containing neurons and the ASPD levels in each neuron (see Figure 11B), as observed in the case of the APPswe transduction, except that the ASPD level in each neuron was significantly lower in the case of APPosk transduction, compared with APPswe transduction (n = 3, p < 0.0001 by Scheffé post hoc test, Figure 11B below). As observed in APPswe-transduced neurons, proteasome inhibition increased N-terAβ and human APP770 staining in almost all APPosk-expressing neurons (Figures 2), whereas ASPD accumulation was detected only in some of the N-terAβ-labeled neurons (Figure S1). High-power images of these ASPD-containing APPosk-expressing neurons showed that ASPD staining was co-localized mainly with TGN38 and also with the cis-Golgi GM130 and the RE marker, TfR, as observed in the APPswe-expressing neurons, at 24-h MG132 treatment (Figure 11C). Finally, the Golgi-destroying BFA treatment confirmed that ASPD accumulated in the Golgi apparatus of the APPosk-transduced neurons (Figure S12). Thus based on the findings in neurons expressing the two different APP transgenes, we conclude that the TGN is the primary site of ASPD accumulation in steady state in APP-expressing mature neurons.
Figure 11.

ASPD Form from Aβ1-42-osk
(A) Primary rat hippocampal neuronal cultures at 21 DIV were treated with synthetic ASPD produced either from Aβ1-42 or Aβ1-42-osk (E22Δ) at the indicated concentration for 24 h. Viability of each culture was determined by using highly water-soluble tetrazolium salt, WST-8, (Transparent Methods; mean ± SD, n = 3; Scheffé post hoc test, ***p < 0.0001 and **p = 0.0007 compared with ASPD-untreated neurons; *p = 0.0048 compared with Aβ1-42-osk-derived ASPD). Representative dot blotting (using anti-Aβ N-terminal 82E1 and ASPD-specific rpASD1 antibodies) and a representative transmission electron microscopic image of the negatively stained Aβ1-42-osk-derived ASPD (arrows) are shown on the right.
(B) Primary rat hippocampal neuronal cultures with AAV-APPosk transduction were treated with 75 nM MG132 for the indicated time, stained, and photographed as in Figure 4B (left column). Representative images are shown. Scale bar, 50 μm. The ASPD staining of each culture was quantified using the In Cell Analyzer (GE Healthcare Lifesciences) as in Figure 4B (mean ± SD, n = 3; Scheffé post hoc test; ***p < 0.0001, **p < 0.0005, *p = 0.01).
(C) Primary rat hippocampal neuronal cultures with AAV-APPosk transduction were treated with 75 nM MG132 for the indicated time, stained, and photographed as in Figures 6 and 8. High-power images were obtained with a highly sensitive, direct photon-counting system with a 100X oil objective lens. For the 24-h MG132 treatment, the single red or green image is also shown. Representative images are shown. Scale bar, 10 μm.
Intra-neuronal ASPD Are Secreted and Are Toxic to NAKα3-Expressing Neurons
Our previous studies have shown that NAKα3 is the only ASPD-binding protein that is directly linked to ASPD neurotoxicity (Ohnishi et al., 2015). ASPD bind to the extracellular region of NAKα3 located in the L7/8 extracellular loop encompassing Asn879 and Trp880 and induce neurodegeneration by inhibiting NAKα3 pump activity (Ohnishi et al., 2015). Accordingly, ASPD that form inside neurons should be secreted to be neurotoxic. We therefore examined whether intra-neuronal ASPD were secreted and exerted neurotoxicity against NAKα3-expressing neurons. To this end, we developed a chemiluminescent enzyme immunoassay (CLEIA) that selectively detects ASPD with two ASPD-specific antibodies (see scheme in Figure 12A). This ASPD-specific CLEIA system enabled us to detect ASPD in the picomolar range. It did not detect low-molecular-weight Aβ, mostly monomers and dimers, which passed through a 50-kDa molecular weight cutoff filter (Xiao et al., 2015) (Figure 12A). Note that in Figures 12A, 12B, and S13, to enable easier comparison between ASPD and Aβ, the concentration of ASPD (128 kDa average mass; Matsumura et al., 2011) is expressed in terms of Aβ1-42 monomer (4.53 kDa mass). We found that ASPD were recovered in the 10× concentrated 100-kDa retentate fraction of the culture supernate of the MG132-treated APPswe-transduced neurons at a concentration of 12.8 ± 4.8 pM (n = 3) (Figure 12B). ASPD were not detected in the 100-kDa flow-through fraction (Figure S13 left). The culture supernates of the MG132-treated APPswe-expressing neurons also contained Aβ1-40 and Aβ1-42 at 237.5 ± 53.7 pM and 39.9 ± 9.4 pM (n = 3), respectively, which were mostly recovered in the 100-kDa flow-through fraction (Figure S13 right). The ASPD level was below the detection limit in the 10× concentrated 100-kDa retentate fraction of the culture supernate of untransduced neurons (Figure 12B), which is in line with the quantification showing only a trace amount of ASPD formed in untransduced neurons (Figure 4B right). Before MG132 treatment, the ASPD levels were approximately 5.7 pM in the 10× concentrated 100-kDa retentates, suggesting that ASPD secretion increased after MG132 treatment.
Figure 12.

Intra-neuronal ASPD Were Secreted and Kill NAKα3 Neurons
(A) The CLEIA system was developed based on our previous enzyme-linked immunosorbent assay (ELISA) that selectively detected ASPD with two ASPD-specific antibodies (Noguchi et al., 2009). The newly established ASPD-specific CLEIA (see a schematic image in upper panel and details in Transparent Methods) detects picomolar levels of ASPD (expressed as Aβ monomer concentration) but does not respond to low-molecular-weight (LMW)-Aβ in the 50-kDa flow-through fraction. R2 = 0.99891 for ASPD and 0.01162 for LMW-Aβ.
(B) ASPD level in culture supernates detected by ASPD-specific CLEIA in (A). A 10× concentrated culture supernate was obtained using 100-kDa filters from the neuronal cultures (with or without AAV-APPswe) treated with 75 nM MG132 for 24 h (see Methods). Aβ1-40 and Aβ1-42 were also quantified (see Figure S13). Mean ± SD, n = 3.
(C) APPswe-transduced or untransduced control neuronal cultures were treated with 75 nM MG132 for 24 h in the presence or absence of mASD3 antibody (1 ng/mL) that can block ASPD neurotoxicity (Noguchi et al., 2009) and stained with anti-NAKα3 antibody and DAPI (Transparent Methods). The NAKα3 level was quantified using a Yokogawa CQ1 (see Transparent Methods; mean ± SD, n = 28 for MG132-treated control, 19 for mASD3-treated MG132-treated control, 52 for MG132-treated APPswe, or 31 for mASD3-treated MG132-treated APPswe; Scheffé post hoc test; ***p < 0.0001). Representative images, used for the quantification, are shown with 100-μm scale bars. Note that NAKα3 was detected by an antibody that specifically recognizes an internal region of NAKα3 (SANTA CRUZ sc-16051-R). Therefore, ASPD binding to the extracellular region of NAKα3 does not interfere with the binding of the anti-NAKα3 antibody to NAKα3 (Ohnishi et al., 2015).
(D) Culture supernates were collected from 24-h MG132-treated APPswe-transduced or untransduced control neuronal cultures. From the collected culture supernates, ASPD were immunodepleted by mASD3-covered magnetic beads, and another 22 DIV primary rat hippocampal neuronal culture was treated with the resulting immunodepleted culture supernates. As a control, the collected culture supernates were also incubated with normal mouse IgG-covered magnetic beads (see Transparent Methods). The NAKα3 level was quantified as in (C) after 24 h. Mean ± SD, n = 8, Scheffé post hoc test; *p = 0.0172 compared with IgG-treated control supernate. Representative images, used for the quantification, are shown with 100-μm scale bars.
See Figure S13
We next examined whether NAKα3-expressing neurons degenerated in the MG132-treated AAV-APPswe-transduced cells. The level of NAKα3-expressing neurons in MG132-treated AAV-APPswe-transduced cultures significantly decreased to 72.9% ± 23.9% (n = 52) of that in MG132-treated untransduced cultures (***p < 0.0001 by Scheffé post hoc test), which correlated with the ASPD amounts in culture supernates. Previously, we showed that pretreatment of mASD3 antibody (with a Kd of 0.003 nM for ASPD) specifically blocked ASPD neurotoxicity against mature neurons (Noguchi et al., 2009). Consistent with this observation, the addition of an excess amount of mASD3 antibody in culture media of MG132-treated AAV-APPswe-transduced mature neurons completely blocked the decrease in NAKα3-expressing neurons (Figure 12C). We confirmed that incubation of MG132-treated untransduced mature neurons with mASD3 antibody did not affect the level of NAKα3-expressing neurons (Figure 12C). Given that antibodies usually do not cross the cell membrane, these results demonstrate that secreted ASPD caused the neurodegeneration observed in the MG132-treated AAV-APPswe-transduced mature neurons.
To further test our claim, we collected culture media from MG132-treated APPswe-transduced or untransduced cultures and applied them to untransduced mature neurons. Before application, ASPD were immunodepleted from the collected media by 1-h incubation with mASD3-antibody-covered magnetic beads. As a control, the collected media were separately incubated with normal mouse-IgG-covered magnetic beads. As clearly shown in Figure 12D, the collected media that were incubated with normal IgG induced degeneration in NAKα3-expressing neurons. This neurodegeneration was not observed when ASPD in the media were immunodepleted by prior incubation with mASD3-antibody-covered magnetic beads (Figure 12D). These results collectively demonstrate that ASPD were secreted and were toxic to NAKα3-expressing neurons.
We next performed detailed viability analyses of the ASPD-producing neurons. WST-8 assay (Figure 13A) showed that the viability of the APPswe-transduced mature hippocampal neurons at 21 DIV was lower than that of the untransduced neurons before the MG132 treatment (p = 0.089 by Scheffé post hoc test; n = 3). The 24-h MG132 treatment further reduced the viability of the APPswe-transduced mature hippocampal neurons (Figure 13A). Similar results were obtained with APPosk-transduced neurons (Figure 13A). The viability of the APPosk-transduced mature hippocampal neurons was significantly lower than that of the untransduced neurons before the MG132 treatment (**p = 0.0268 by Scheffé post hoc test; n = 3), and the 24-h MG132 treatment further reduced the viability of the APPosk-transduced neurons (Figure 13A). We confirmed that neither 24-h MG132 treatment nor AAV transduction of GFP itself affected cell viability (Figure 13A). Taken together, the viability of the neurons declined concomitantly with the accumulation of ASPD.
Figure 13.

Secreted ASPD Induced Apoptotic Degeneration of Surrounding Neurons
(A) APP- or GFP-transduced or untransduced control neuronal cultures were treated with 75 nM MG132 for the indicated time. Viability was determined with a Cell Counting Kit-8 (see Transparent Methods; mean ± SD, n = 3; Scheffé post hoc test, ***p < 0.0001, **p = 0.0268). The viability of GFP-transduced cultures was 93.5% ± 7.0% of that of the untransduced control cultures (n = 3, p = 0.1854 by Scheffé posy hoc test), indicating that AAV transduction itself does not affect cell viability.
(B) APP-transduced or untransduced control cultures were treated with 75 nM MG132 for the indicated time and stained with the anti-ASPD rpASD1 antibody, DAPI, and TUNEL (arrowhead) or with the anti-MAP2 antibody (Transparent Methods). Solid and hatched scale bars, 50 and 100 μm, respectively.
(C) APP-transduced or untransduced control cultures were treated with or without 75 nM MG132 for 24 h and quadruple-stained with rpASD1, DAPI, TUNEL, and MAP2/GFAP. The number of MAP2-labeled neurons or GFAP-labeled astrocytes in APPswe-transduced cultures was counted and is shown (n = 7, Scheffé post hoc test, ***p < 0.0001). Representative images are shown in Figure S14.
(D) APPswe-transduced primary rat hippocampal neuronal cultures were treated with 75 nM MG132 for the indicated time; triple-stained with propidium iodide (PI), calcein-AM, and anti-ASPD mASD3 antibody (see Transparent Methods); and photographed (see Figure S15). Representative images of the corresponding PI/mASD3 staining are shown with 50-μm scale bars. PI-detectable non-apoptotic death was detected in ASPD-producing neurons at 48-h MG132 treatment.
See Figures S14 and S15.
We next examined whether apoptotic cell death occurred in these neurons by detecting DNA fragmentation using TUNEL staining. To our surprise, TUNEL-positive apoptosis did not occur in the ASPD-producing neurons but did occur in the neurons surrounding these ASPD-positive neurons (arrowhead in Figure 13B, left and middle columns, and S14). MAP2 immunostaining showed a marked reduction of neurons in the APPswe- and APPosk-transduced mature neuronal cultures treated with 75 nM MG132 for 24 h (Figures 13B right and S14). We counted the number of MAP2- or GFAP-positive cells in MG132-treated AAV-APPswe-transduced cultures that were double stained with TUNEL and MAP2/GFAP and found that, after 24-h MG132 treatment, on an average 54% of MAP2-positive neurons was lost in APPswe-transduced cells (n = 7), whereas the number of GFAP-positive astrocytes was not changed (Figures 13C and S14). Following apoptotic cell death in the surrounding neurons, the ASPD-producing neurons themselves degenerated after 48 h of 75 nM MG132 treatment, as indicated by propidium iodide (PI) staining (Figure 13D, see also arrowheads in Figure S15). PI-detectable, non-apoptotic neurodegeneration was rarely observed until 24-h MG132 treatment (Figures 13D and S15). This suggests that the ASPD-producing neurons either die very slowly or degenerate due to the lack of synaptic inputs from the surrounding neurons.
ASPD Appear to Primarily Form in Excitatory Neurons
In Figures 4A and S1, we showed that ASPD increased in only a limited population of neurons. Therefore we examined in which type of neurons the intra-neuronal ASPD increase. We first showed that the AAV transduction ratio did not vary according to the neuronal subtype by introducing non-toxic GFP (as shown in Figure 13A) by AAV. As shown in Figure S16, GFP was transduced to almost all neurons, irrespective of Math2-positive excitatory neurons (Schwab et al., 2000) (99.1% ± 1.4%, n = 30) or parvalbumin (PV)-positive GABAergic interneurons (97.0% ± 3.7%, n = 30; see representative images of Figure S16). Although APPswe transduction itself does not seem to affect the ratio between Math2-positive excitatory neurons and PV-labeled interneurons (Figure S17), interestingly, most of the Math2-positive excitatory neurons accumulated ASPD (84% ± 14%, n = 6), whereas most of the PV-labeled interneurons did not accumulate ASPD (8.7% ± 10.0%, n = 24) (Figure 14). We also examined calbindin-labeled neurons, which are mostly inhibitory neurons (McDonald and Mascagni, 2001) and found that most of the calbindin-positive inhibitory neurons did not accumulate ASPD (9.0% ± 3.2%, n = 5) (Figure S18). Thus it appears that ASPD primarily form in the excitatory neurons and are secreted from these neurons, exhibiting neurotoxicity toward the surrounding NAKα3-expressing neurons (Graphical Abstract; see Discussion).
Figure 14.

ASPD Accumulate Only in Excitatory Neurons
APPswe-transduced neuronal cultures were treated with 75 nM MG132 for 24 h and stained with the indicated antibodies. The ratio of the ASPD-containing neurons among either MAP2/PV or MAP2/Math2 double-positive neurons was quantified using a Yokogawa CQ1 (see Transparent Methods; inset, mean ± SD, n = 6 for MAP2/Math, 24 for MAP2/PV). ASPD were detected by anti-ASPD rpASD1 or mASD3 antibody. GABAergic interneurons are composed of a highly diverse population of inhibitory neurons, which represent about 20% of cortical/hippocampal neurons (Amaral, 1978). For quantification purposes, we used an antibody against PV, because PV-labeled interneurons represent ∼40% of the total number of GABAergic interneurons in the CA1 and CA3 of hippocampus (Ribak et al., 1990) and the PV antibody clearly stains the neuronal cell body. Representative images, used for the quantification, are shown with 100 μm scale bars. See control data in Figures S16 and S17 and calbindin data in Figure S18. See Figures S16–18.
Discussion
In AD, Aβ oligomers accumulate in the brain. This accumulation might arise because of defects in APP processing or Aβ degradation, a shift in Aβ profiles to longer Aβs, or a change in Aβ assembly propensity caused by biological or chemical factors (Haass et al., 2012). Here we studied how impairment of the proteasome function affected APP/Aβ turnover and oligomer formation. We found that the inhibition of proteasomes by a variety of inhibitors led to higher levels of ASPD in steady state by increasing Aβ levels in APP-expressing neurons and triggered changes in the subcellular distribution of ASPD from the axon to dendrites. Interestingly, ASPD appeared to form primarily in excitatory neurons and were secreted into the medium, leading to apoptotic death of surrounding NAKα3-expressing neurons (Figure 12). This is consistent with our previous finding that ASPD bind to NAKα3 on synapses and cause neuronal degeneration by inactivating NAKα3 pump activity (Ohnishi et al., 2015). NAKα3 neurons died apoptotically due to secreted ASPD, whereas ASPD-producing neurons that killed NAKα3 neurons died slowly and non-apoptotically (Figures 13, S14, and S15).
In using APP-overexpressing neurons as a model, we are aware that some phenotypes of APP-overexpressing mice models, such as sudden death, may be the result of APP overexpression per se and thus may not be reflective of the disease process in humans (Sasaguri et al., 2017). Nevertheless, these mouse APP overexpression model systems have proved to be useful and relevant in numerous studies of the production or deposition of Aβ (Sasaguri et al., 2017). In our system, as shown in Figure 11B, MG132 treatment substantially and significantly (p < 0.0001 by Scheffé post hoc test) increased the levels of ASPD and the number of ASPD-containing neurons, relative to control neurons, suggesting that the effects we observed were not merely due to APP overexpression. More importantly, intra-neuronal accumulation of ASPD is indeed present in the brains of patients with AD (Noguchi et al., 2009). The AAV-APP system used in this work thus appears capable of revealing how ASPD may form in neurons in the human brain and affect disease pathology.
We showed that proteasome inhibition led to an increase in Aβ in steady state, by inhibiting ERAD-mediated degradation of Aβ or APP, without affecting its secretion (Figures 2, 3, and S2). The rate-limiting step in ERAD is ubiquitination performed by ER-membrane-anchored E3 ubiquitin ligases, which seem to target only a small number of proteins. Recent studies have identified several E3 ubiquitin ligases for APP that directly interact with APP and mediate APP degradation by proteasomes (Kaneko et al., 2010, Kumar et al., 2007, Wang and Saunders, 2014, Watanabe et al., 2012). Notably, gene suppression of one of the E3 ubiquitin ligases for APP, hydroxymethylglutaryl-coenzyme A reductase degradation protein 1 (HRD1), leads to APP accumulation and an increase in Aβ production, and vice versa (Kaneko et al., 2010). MG132 inhibits APP degradation induced by increased HRD1 expression (Kaneko et al., 2010). In the case of Aβ, another E3 ubiquitin ligase, parkin, promotes proteasome-mediated clearance of Aβ1-42 in neurons (Burns et al., 2009). This is consistent with findings in a parkin-null mouse model, where the accumulation of Aβ deposits was observed in the brain even without modification of APP, presenilins, or secretases (Rodriguez-Navarro et al., 2008). These findings support the notion that proteasomes may play a role in regulating APP/Aβ metabolism. Our findings demonstrate a link between ASPD levels and proteasome function, which may have important implications for AD pathophysiology. It will be interesting in the future to determine which E3 ubiquitin ligases or other regulators of APP/Aβ degradation are indeed involved in increasing ASPD formation.
Previous reports have shown that Aβ can be generated in all possible compartments of the secretory pathways, including the ER (Cook et al., 1997, Greenfield et al., 1999, Hartmann et al., 1997), the TGN (Greenfield et al., 1999, Xu et al., 1997), the PM (Chyung and Selkoe, 2003, Tarassishin et al., 2004), the endosomes (Grbovic et al., 2003, Koo and Squazzo, 1994, Perez et al., 1999), and the lysosomes (Yu et al., 2004). We consistently detected the N-terminal end of Aβ (N-terAβ) in steady state in compartments ranging from the ER to the lysosomes (Figure 10). Among the possible compartments, the subcellular localization of BACE suggested that either the TGN or endosomes would serve as the locus of Aβ production. Recent studies have emphasized the role of EEs in Aβ production, because blocking endocytosis reduces Aβ production (Carey et al., 2005, Perez et al., 1999), whereas enhancing endocytosis increases it (Grbovic et al., 2003). Proteins related to endocytosis have been reported to be involved in APP processing (Sannerud et al., 2011, Schneider et al., 2008, Ubelmann et al., 2017) (see also review Rajendran and Annaert, 2012). However, these studies do not rule out a role for the TGN in β-cleavage. For example, fluorescence resonance energy transfer analyses have shown that APP and BACE interact at least in the GM130-detected cis-Golgi, in addition to at the cell surface and in the EEs (Kinoshita et al., 2003). Aβ40 has been reported to be generated in the TGN depending on retromer-mediated APP recycling from the EEs to the TGN (Choy et al., 2012). Accumulating data support the importance of the sorting mechanisms that transport APP and BACE from the cell surface to the endosomes and from the endosomes to the TGN. The exact location of APP processing will depend on the balance of traffic of APP and secretases. Therefore some discrepancies in existing data may derive from the use of different cell types and conditions. In particular, to establish and maintain functionally distinct domains in membranes, neurons have developed trafficking systems to deliver transmembrane proteins to the correct compartments (Lasiecka and Winckler, 2011). Accordingly, the trafficking and processing of APP in highly polarized neurons might well be different from the processes in non-neuronal cells. For example, the sorting signal(s) mediating transport of APP to the axon or dendrites in neurons is completely different from that mediating the basolateral transport in MDCK cells (Back et al., 2007, Szodorai et al., 2009). In our system, ASPD signals were broadly distributed in steady state, but the TGN was shown to be the initial and highest localization site of ASPD in APP-expressing neurons (Figures 6 and 10). This suggests the possibility that ASPD may form mainly in the TGN. However, other possibilities cannot be excluded given the close and dynamic cross talk between TGN and endosomes (Schmidt and Haucke, 2007). Real-time imaging that enables detection of ASPD separately from Aβ monomers in mature neurons is needed to give a more definitive answer as to the exact location of ASPD formation in mature neurons. Interestingly, ASPD are present not only in the TGN, but also in the REs, which lie between the ER-TGN system and the endosome system. Because the RE serves as a hub to direct proteins to the PM, it is possible that the REs serve as a port for ASPD to be secreted. The REs, which are clustered tightly near the nucleus in close proximity to the TGN in non-neuronal cells, are spread almost evenly throughout the cell body, dendrites, and the axons in neurons (Wang et al., 2008). One may speculate that axon-directed REs or dendrite-directed REs separately serve as hubs to direct ASPD to the axons or dendrites. We showed in Figure 5B that some population of dendritic ASPD co-localizes with Bassoon, suggesting that ASPD could be released at synapses. This observation is consistent with our previous study showing that ASPD bind to the pre-synaptic side of the neuron to induce neurodegeneration (Noguchi et al., 2009). Scheme 1 integrates our results with those extant.
Scheme 1.

Steady-State Distribution of APP, N-terminal end of Aβ, and ASPD, Detected in This Work under Physiological Conditions without Proteasome Inhibition and with Proteasome Inhibition, Integrated with Previous Findings as to APP Degradation and Trafficking
In mature neurons, APP was widely detected in the ER-TGN as reported previously (Greenfield et al., 1999, Hartmann et al., 1997, Xu et al., 1997). Proteasome inhibition leads to a higher steady-state level of APP and N-terAβ in APP-expressing neurons, possibly by inhibiting ERAD (see text). Under physiological conditions without proteasome inhibition, N-terAβ staining is too weak to allow its subcellular distribution to be determined (representative images in Figures S10 and S11). From the red enhanced images, it appears that the weak N-terAβ signal is co-localized with the ER or the cis-Golgi, but not with the TGN, the EE, the RE, or the Lys (Figures S10 and S11). After 24-h MG132 treatment, N-terAβ staining was detected mainly in the cis-Golgi, and also on the TGN, the RE, and the ER surface, but was rarely observed in the EE or the Lys in our system (Figures 10, S10, and S11). In the case of ASPD, a limited amount of ASPD was detected mainly in the TGN under physiological conditions (Figures 6 and 7). With proteasome inhibition, the steady-state level of ASPD increased markedly, in particular, in the TGN, and also in the cis-Golgi and the RE. ASPD were also detected to some extent in the ER, and a very few were present in the EE and the Lys. Proteasome inhibition changes both N-terAβ and ASPD distribution from axons to dendrites (Figures 5 and S19). Blue arrows, APP; green arrows, N-terAβ; pink arrows, ASPD. Higher levels of APP and N-terAβ were detected after inhibiting ERAD (see text). Hatched brown arrows show possible cross-talk reported previously. N, nucleus; ER, endoplasmic reticulum; TGN, trans-Golgi network; PM, plasma membrane; EE, early endosome; RE, recycling endosome; LE/Lys, late endosome and lysosome.
Previously, we observed substantial accumulation of ASPD in the cerebral cortex and little accumulation in the cerebellum (Ohnishi et al., 2015). These findings strongly suggest that there exist regional or cell-type differences in ASPD accumulation. Consistently, we found that only excitatory neurons appear to form ASPD (Figure 14). This may be due to factors intrinsic to these cells that affect assembly propensity or it may be that only inhibitory neurons have a degradation system for ASPD, about which we do not know anything. Interestingly, proteasome inhibition led to a distribution change of both ASPD and Aβ from axons to dendrites (Figures 5 and S19), suggesting the presence of a common mechanism underlying this distribution change. The reason why such a change in ASPD delivery occurs remains to be established, as do the molecular mechanisms of ASPD secretion. Neurons have specialized Golgi compartments called “Golgi outposts” within dendritic arbors, which are essential for the proper elaboration of dendrites and neuronal polarity (Lasiecka and Winckler, 2011). Golgi outposts are discontinuous from the somatic Golgi and are frequently localized at dendritic branch points (Lasiecka and Winckler, 2011). Aβ and ASPD might use this system to be delivered to dendrites. We found that after 24 h of MG132 treatment the Golgi structure appeared to be fragmented in most of the APPswe-transduced neurons. Because the Golgi did not appear to be fragmented and ASPD had already accumulated at the TGN at 6-h MG132 treatment (Figure 6), it is unlikely that Golgi fragmentation causes ASPD accumulation at the TGN. Golgi fragmentation enhances vesicle budding and accelerates protein trafficking (Wang et al., 2008), and reversible fragmentation has been detected in hyperactive neurons (Thayer et al., 2013). We therefore speculate that Golgi fragmentation at 24-h MG132 treatment might be a compensatory reaction to allow faster protein trafficking when the cells are under stress.
In this work, we found that Aβ1-42-osk (E22Δ-Aβ1-42) forms ASPD. As shown in quantification in Figure 11, the formation ratio of ASPD from Aβ1-42-osk was lower than that of ASPD from wild-type Aβ1-42 in APPswe-transduced neurons. Nevertheless, the total cell death level of the APPosk-transduced cultures was equivalent to that of the APPswe-transduced cultures, as shown by viability assay, MAP2 staining, and TUNEL staining (Figures 13A, 13B, and S14). This could be due to increased toxicity of the ASPD derived from Aβ1-42-osk compared with that of ASPD derived from wild-type Aβ1-42. Our previous solution nuclear magnetic resonance (NMR) analysis of ASPD showed that the E22 residue resides in a core region of ASPD (Ohnishi et al., 2015). Therefore it is unlikely that E22 directly affects ASPD binding to NAKα3. It is more likely that the E22Δ mutation alters the conformation of the Aβ monomer (Inayathullah and Teplow, 2011), which leads to differences in the structure of ASPD. Further solution NMR studies will be needed to determine at the atomic level precisely how the deletion of E22 changes the structure of ASPD and thus their toxicity.
Biomarker studies on humans indicate that accumulation of Aβ precedes the clinical onset of AD by more than a decade (Weiner et al., 2012). Notably, western blot analyses of human brains obtained from the Nun study showed distinct trends in levels of dimers, trimers, and dodecamers, according to the disease stage, suggesting that dodecamers might form earlier than trimers or dimers (Lesné et al., 2013). However, the lack of specific antibodies makes it difficult to monitor their actual formation. We have identified ASPD from brains of patients with AD as the key species responsible for neurodegeneration and also obtained ASPD-specific antibodies that could specifically detect the native tertiary structure of ASPD (Noguchi et al., 2009, Ohnishi et al., 2015). Building on that work, we established a mature neuron-derived system that was usable to monitor where ASPD form. As described above, the sorting and processing mechanisms of APP in neurons are expected to be more complicated than those in non-neuronal cells owing to dynamic cross talk between the TGN and the endosome system. Disturbances in these complex sorting and processing systems in neurons could affect Aβ production and have been linked to AD pathogenesis. Thus the mature neuron-based system established in the present work, and the findings resulting from its use, should further deepen our understanding of how Aβ assemblies may form in the human brain.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Manami Akioka, Masafumi Inoue, Yoshitaka Matsumura, and Takashi Nishiyama for technical help; Masatoshi Takeichi and Shigenobu Yonemoto for help in using the TEM instrument; and David B. Teplow, Koshi Murata, Yugo Fukazawa, and Masatoshi Hagiwara for helpful discussion. M.H. thanks the Ministry of Education, Culture, Sports, Science and Technology (Grant-in-Aid for Scientific Research B), Niigata University (the Collaborative Research Project of the Brain Research Institute), and Takeda Science Foundation (Life Science Research Grant) for support.
Author Contributions
Conceptualization, M.H.; Methodology, H.K., S.K., T.S., Y.A., N.T., Y.-I.N., S.-I.M., I.K., and M.H.; Investigation, H.K., S.K., T.S., Y.A., K.S., N.T., M.S., T.O., and M.H.; Writing – Original Draft, H.K., S.K., T.S., S.-I.M., I.K., and M.H.; Writing – Review & Editing, all authors; Visualization, H.K., S.K., T.S., and M.H.; Supervision, M.H.; Funding Acquisition, M.H.
Declaration of Interests
M.H. has served as a technical advisor to TAO Health Life Pharma Co. Ltd., a Kyoto University-derived bioventure, with the permission of the Conflict of Interest Committee of Kyoto University and the Foundation for Biomedical Research and Innovation at Kobe; H.K., Y.A., S.K., T.S., K.S., and T.O. are employees of TAO Health Life Pharma Co. Ltd.
Published: February 28, 2019
Footnotes
Supplemental Information includes Transparent Methods and 19 figures and can be found with this article online at https://doi.org/10.1016/j.isci.2019.01.018.
Supplemental Information
References
- Amaral D.G. A Golgi study of cell types in the hilar region of the hippocampus in the rat. J. Comp. Neurol. 1978;182:851–914. doi: 10.1002/cne.901820508. [DOI] [PubMed] [Google Scholar]
- Back S., Haas P., Tschape J.A., Gruebl T., Kirsch J., Muller U., Beyreuther K., Kins S. β-amyloid precursor protein can be transported independent of any sorting signal to the axonal and dendritic compartment. J. Neurosci. Res. 2007;85:2580–2590. doi: 10.1002/jnr.21239. [DOI] [PubMed] [Google Scholar]
- Barysch S.V., Aggarwal S., Jahn R., Rizzoli S.O. Sorting in early endosomes reveals connections to docking- and fusion-associated factors. Proc. Natl. Acad. Sci. U S A. 2009;106:9697–9702. doi: 10.1073/pnas.0901444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benilova I., Karran E., De Strooper B. The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes. Nat. Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- Burns M.P., Zhang L., Rebeck G.W., Querfurth H.W., Moussa C.E. Parkin promotes intracellular Aβ1-42 clearance. Hum. Mol. Genet. 2009;18:3206–3216. doi: 10.1093/hmg/ddp258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai X.D., Golde T.E., Younkin S.G. Release of excess amyloid beta protein from a mutant amyloid beta protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- Carey R.M., Balcz B.A., Lopez-Coviella I., Slack B.E. Inhibition of dynamin-dependent endocytosis increases shedding of the amyloid precursor protein ectodomain and reduces generation of amyloid β protein. BMC Cell Biol. 2005;6:30. doi: 10.1186/1471-2121-6-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson S.R., Fukuda M. Structure of human lysosomal membrane glycoprotein 1. assignment of disulfide bonds and visualization of its domain arrangement. J. Biol. Chem. 1989;264:20526–20531. [PubMed] [Google Scholar]
- Chiti F., Dobson C.M. Amyloid formation by globular proteins under native conditions. Nat. Chem. Biol. 2009;5:15–22. doi: 10.1038/nchembio.131. [DOI] [PubMed] [Google Scholar]
- Choy R.W.Y., Cheng Z.L., Schekman R. Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc. Natl. Acad. Sci. U S A. 2012;109:E2077–E2082. doi: 10.1073/pnas.1208635109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chyung J.H., Selkoe D.J. Inhibition of receptor-mediated endocytosis demonstrates generation of amyloid β-protein at the cell surface. J. Biol. Chem. 2003;278:51035–51043. doi: 10.1074/jbc.M304989200. [DOI] [PubMed] [Google Scholar]
- Citron M., Oltersdorf T., Haass C., McConlogue L., Hung A.Y., Seubert P., Vigo-Pelfrey C., Lieberburg I., Selkoe D.J. Mutation of the β-amyloid precursor protein in familial Alzheimer's disease increases β-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Cook D.G., Forman M.S., Sung J.C., Leight S., Kolson D.L., Iwatsubo T., Lee V.M.Y., Doms R.W. Alzheimer's Aβ(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nat. Med. 1997;3:1021–1023. doi: 10.1038/nm0997-1021. [DOI] [PubMed] [Google Scholar]
- Csizmadia V., Csizmadia E., Silverman L., Simpson C., Raczynski A., O'Brien L., Gallacher M., Cardoza K., Kadambi V.J., Fedyk E.R. Effect of proteasome inhibitors with different chemical structures on the ubiquitin-proteasome system in vitro. Vet. Pathol. 2010;47:358–367. doi: 10.1177/0300985809358423. [DOI] [PubMed] [Google Scholar]
- Glabe C.G. Structural classification of toxic amyloid oligomers. J. Biol. Chem. 2008;283:29639–29643. doi: 10.1074/jbc.R800016200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grbovic O.M., Mathews P.M., Jiang Y., Schmidt S.D., Dinakar R., Summers-Terio N.B., Ceresa B.P., Nixon R.A., Cataldo A.M. Rab5-stimulated Up-regulation of the endocytic pathway increases intracellular β-cleaved amyloid precursor protein carboxyl-terminal fragment levels and Aβ production. J. Biol. Chem. 2003;278:31261–31268. doi: 10.1074/jbc.M304122200. [DOI] [PubMed] [Google Scholar]
- Greenfield J.P., Tsai J., Gouras G.K., Hai B., Thinakaran G., Checler F., Sisodia S.S., Greengard P., Xu H. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer β-amyloid peptides. Proc. Natl. Acad. Sci. U S A. 1999;96:742–747. doi: 10.1073/pnas.96.2.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C., Kaether C., Thinakaran G., Sisodia S. Trafficking and proteolytic processing of APP. Cold Spring Harb. Perspect. Med. 2012;2:a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J., Selkoe D.J. The Amyloid hypothesis of alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hartmann T., Bieger S.C., Bruhl B., Tienari P.J., Ida N., Allsop D., Roberts G.W., Masters C.L., Dotti C.G., Unsicker K. Distinct sites of intracellular production for Alzheimer's disease Aβ40/42 amyloid peptides. Nat. Med. 1997;3:1016–1020. doi: 10.1038/nm0997-1016. [DOI] [PubMed] [Google Scholar]
- Hoshi M., Sato M., Matsumoto S., Noguchi A., Yasutake K., Yoshida N., Sato K. Spherical aggregates of β-amyloid (amylospheroid) show high neurotoxicity and activate tau protein kinase I/glycogen synthase kinase-3β. Proc. Natl. Acad. Sci. U S A. 2003;100:6370–6375. doi: 10.1073/pnas.1237107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey J.S., Peters P.J., Yuan L.C., Bonifacino J.S. Localization of TGN38 to the trans-Golgi network: involvement of a cytoplasmic tyrosine-containing sequence. J. Cell. Biol. 1993;120:1123–1135. doi: 10.1083/jcb.120.5.1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inayathullah M., Teplow D.B. Structural dynamics of the ΔE22 (Osaka) familial Alzheimer's disease-linked amyloid β-protein. Amyloid. 2011;18:98–107. doi: 10.3109/13506129.2011.580399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarosz-Griffiths H.H., Noble E., Rushworth J.V., Hooper N.M. Amyloid-β receptors: the good, the bad, and the prion protein. J. Biol. Chem. 2016;291:3174–3183. doi: 10.1074/jbc.R115.702704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko M., Koike H., Saito R., Kitamura Y., Okuma Y., Nomura Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-β generation. J. Neurosci. 2010;30:3924–3932. doi: 10.1523/JNEUROSCI.2422-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keck S., Nitsch R., Grune T., Ullrich O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer's disease. J. Neurochem. 2003;85:115–122. doi: 10.1046/j.1471-4159.2003.01642.x. [DOI] [PubMed] [Google Scholar]
- Keller J.N., Hanni K.B., Markesbery W.R. Impaired proteasome function in alzheimer's disease. J. Neurochem. 2000;75:436–439. doi: 10.1046/j.1471-4159.2000.0750436.x. [DOI] [PubMed] [Google Scholar]
- Kim K.B., Myung J., Sin N., Crews C.M. Proteasome inhibition by the natural products epoxomicin and dihydroeponemycin: insights into specificity and potency. Bioorg. Med. Chem. Lett. 1999;9:3335–3340. doi: 10.1016/s0960-894x(99)00612-5. [DOI] [PubMed] [Google Scholar]
- Kinoshita A., Fukumoto H., Shah T., Whelan C.M., Irizarry M.C., Hyman B.T. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J. Cell. Sci. 2003;116:3339–3346. doi: 10.1242/jcs.00643. [DOI] [PubMed] [Google Scholar]
- Klein W.L., Krafft G.A., Finch C.E. Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- Kobayashi H., Fukuda M. Arf6, Rab11 and transferrin receptor define distinct populations of recycling endosomes. Commun. Integr. Biol. 2013;6:e25036. doi: 10.4161/cib.25036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo E.H., Squazzo S.L. Evidence that production and release of amyloid β-protein involves the endocytic pathway. J. Biol. Chem. 1994;269:17386–17389. [PubMed] [Google Scholar]
- Kouchi Z., Kinouchi T., Sorimachi H., Ishiura S., Suzuki K. The deletion of the C-terminal tail and addition of an endoplasmic reticulum targeting signal to Alzheimer's amyloid precursor protein change its localization, secretion, and intracellular proteolysis. Eur. J. Biochem. 1998;258:291–300. doi: 10.1046/j.1432-1327.1998.2580291.x. [DOI] [PubMed] [Google Scholar]
- Kumar P., Ambasta R.K., Veereshwarayya V., Rosen K.M., Kosik K.S., Band H., Mestril R., Patterson C., Querfurth H.W. CHIP and HSPs interact with β-APP in a proteasome-dependent manner and influence Aβ metabolism. Hum. Mol. Genet. 2007;16:848–864. doi: 10.1093/hmg/ddm030. [DOI] [PubMed] [Google Scholar]
- Lasiecka Z.M., Winckler B. Mechanisms of polarized membrane trafficking in neurons – focusing in on endosomes. Mol. Cell. Neurosci. 2011;48:278–287. doi: 10.1016/j.mcn.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesné S.E., Sherman M.A., Grant M., Kuskowski M., Schneider J.A., Bennett D.A., Ashe K.H. Brain amyloid-β oligomers in ageing and Alzheimer's disease. Brain. 2013;136:1383–1398. doi: 10.1093/brain/awt062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.G., Okada T., Kodera M., Nara Y., Takino N., Muramatsu C., Ikeguchi K., Urano F., Ichinose H., Metzger D. Viral-mediated temporally controlled dopamine production in a rat model of Parkinson disease. Mol. Ther. 2006;13:160–166. doi: 10.1016/j.ymthe.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Manavalan A., Mishra M., Feng L., Sze S.K., Akatsu H., Heese K. Brain site-specific proteome changes in aging-related dementia. Exp. Mol. Med. 2013;45:e39. doi: 10.1038/emm.2013.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura S., Shinoda K., Yamada M., Yokojima S., Inoue M., Ohnishi T., Shimada T., Kikuchi K., Masui D., Hashimoto S. Two distinct amyloid β-protein (Aβ) assembly pathways leading to oligomers and fibrils identified by combined fluorescence correlation spectroscopy, morphology, and toxicity analyses. J. Biol. Chem. 2011;286:11555–11562. doi: 10.1074/jbc.M110.181313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald A.J., Mascagni F. Colocalization of calcium-binding proteins and GABA in neurons of the rat basolateral amygdala. Neuroscience. 2001;105:681–693. doi: 10.1016/s0306-4522(01)00214-7. [DOI] [PubMed] [Google Scholar]
- Mullan M., Crawford F., Axelman K., Houlden H., Lilius L., Winblad B., Lannfelt L. A pathogenic mutation for probable Alzheimer's disease in the APP gene at the N-terminus of β-amyloid. Nat. Genet. 1992;1:345–347. doi: 10.1038/ng0892-345. [DOI] [PubMed] [Google Scholar]
- Nakamura N., Rabouille C., Watson R., Nilsson T., Hui N., Slusarewicz P., Kreis T.E., Warren G. Characterization of a cis-golgi matrix protein, GM130. J. Cell Biol. 1995;131:1715–1726. doi: 10.1083/jcb.131.6.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nebenfuhr A., Ritzenthaler C., Robinson D.G. Brefeldin a: deciphering an enigmatic inhibitor of secretion. Plant Physiol. 2002;130:1102–1108. doi: 10.1104/pp.011569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noguchi A., Matsumura S., Dezawa M., Tada M., Yanazawa M., Ito A., Akioka M., Kikuchi S., Sato M., Ideno S. Isolation and characterization of patient-derived, toxic, high mass amyloid β-protein (Aβ) assembly from alzheimer disease brains. J. Biol. Chem. 2009;284:32895–32905. doi: 10.1074/jbc.M109.000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi T., Yanazawa M., Sasahara T., Kitamura Y., Hiroaki H., Fukazawa Y., Kii I., Nishiyama T., Kakita A., Takeda H. Na, K-ATPase α3 is a death target of Alzheimer patient amyloid-β assembly. Proc. Natl. Acad. Sci. U S A. 2015;112:E4465–E4474. doi: 10.1073/pnas.1421182112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A., Rivett A.J., Thomson S., Hendil K.B., Butcher G.W., Fuertes G., Knecht E. Subpopulations of proteasomes in rat liver nuclei, microsomes and cytosol. Biochem. J. 1996;316:401–407. doi: 10.1042/bj3160401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parthasarathy S., Inoue M., Xiao Y., Matsumura Y., Nabeshima Y., Hoshi M., Ishii Y. Structural insight into an alzheimer's brain-derived spherical assembly of amyloid β by solid-state NMR. J. Am. Chem. Soc. 2015;137:6480–6483. doi: 10.1021/jacs.5b03373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez R.G., Soriano S., Hayes J.D., Ostaszewski B., Xia W.M., Selkoe D.J., Chen X.H., Stokin G.B., Koo E.H. Mutagenesis identifies new signals for β-amyloid precursor protein endocytosis, turnover, and the generation of secreted fragments, including Aβ42. J. Biol. Chem. 1999;274:18851–18856. doi: 10.1074/jbc.274.27.18851. [DOI] [PubMed] [Google Scholar]
- Powell S.K., Khan N., Parker C.L., Samulski R.J., Matsushima G., Gray S.J., McCown T.J. Characterization of a novel adeno-associated viral vector with preferential oligodendrocyte tropism. Gene Ther. 2016;23:807–814. doi: 10.1038/gt.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L., Annaert W. Membrane trafficking pathways in Alzheimer's disease. Traffic. 2012;13:759–770. doi: 10.1111/j.1600-0854.2012.01332.x. [DOI] [PubMed] [Google Scholar]
- Ribak C.E., Nitsch R., Seress L. Proportion of parvalbumin-positive basket cells in the GABAergic innervation of pyramidal and granule cells of the rat hippocampal formation. J. Comp. Neurol. 1990;300:449–461. doi: 10.1002/cne.903000402. [DOI] [PubMed] [Google Scholar]
- Rivett A.J., Palmer A., Knecht E. Electron microscopic localization of the multicatalytic proteinase complex in rat liver and in cultured cells. J. Histochem. Cytochem. 1992;40:1165–1172. doi: 10.1177/40.8.1619280. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Navarro J.A., Gomez A., Rodal I., Perucho J., Martinez A., Furio V., Ampuero I., Casarejos M.J., Solano R.M., de Yebenes J.G. Parkin deletion causes cerebral and systemic amyloidosis in human mutated tau over-expressing mice. Hum. Mol. Genet. 2008;17:3128–3143. doi: 10.1093/hmg/ddn210. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R., Yang M., Hoshi M.M., Teplow D.B. Amyloid β-protein assembly and alzheimer disease. J. Biol. Chem. 2009;284:4749–4753. doi: 10.1074/jbc.R800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roychaudhuri R., Zheng X., Lomakin A., Maiti P., Condron M.M., Benedek G.B., Bitan G., Bowers M.T., Teplow D.B. Role of species-specific primary structure differences in Aβ42 assembly and neurotoxicity. ACS Chem. Neurosci. 2015;6:1941–1955. doi: 10.1021/acschemneuro.5b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sannerud R., Declerck I., Peric A., Raemaekers T., Menendez G., Zhou L.J., Veerle B., Coen K., Munck S., De Strooper B. ADP ribosylation factor 6 (ARF6) controls amyloid precursor protein (APP) processing by mediating the endosomal sorting of BACE1. Proc. Natl. Acad. Sci. U S A. 2011;108:E559–E568. doi: 10.1073/pnas.1100745108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaguri H., Nilsson P., Hashimoto S., Nagata K., Saito T., De Strooper B., Hardy J., Vassar R., Winblad B., Saido T.C. APP mouse models for Alzheimer's disease preclinical studies. EMBO J. 2017;36:2473–2487. doi: 10.15252/embj.201797397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt M.R., Haucke V. Recycling endosomes in neuronal membrane traffic. Biol. Cell. 2007;99:333–342. doi: 10.1042/BC20070007. [DOI] [PubMed] [Google Scholar]
- Schneider A., Rajendran L., Honsho M., Gralle M., Donnert G., Wouters F., Hell S.W., Simons M. Flotillin-dependent clustering of the amyloid precursor protein regulates its endocytosis and amyloidogenic processing in neurons. J. Neurosci. 2008;28:2874–2882. doi: 10.1523/JNEUROSCI.5345-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U., Anton L.C., Gibbs J., Norbury C.C., Yewdell J.W., Bennink J.R. Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature. 2000;404:770–774. doi: 10.1038/35008096. [DOI] [PubMed] [Google Scholar]
- Schwab M.H., Bartholomae A., Heimrich B., Feldmeyer D., Druffel-Augustin S., Goebbels S., Naya F.J., Zhao S., Frotscher M., Tsai M.J. Neuronal basic helix-loop-helix proteins (NEX and BETA2/Neuro D) regulate terminal granule cell differentiation in the hippocampus. J. Neurosci. 2000;20:3714–3724. doi: 10.1523/JNEUROSCI.20-10-03714.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciaky N., Presley J., Smith C., Zaal K.J., Cole N., Moreira J.E., Terasaki M., Siggia E., Lippincott-Schwartz J. Golgi tubule traffic and the effects of brefeldin a visualized in living cells. J. Cell Biol. 1997;139:1137–1155. doi: 10.1083/jcb.139.5.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano-Pozo A., Frosch M.P., Masliah E., Hyman B.T. Neuropathological alterations in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2011;1:a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S., Lieberburg I. Cellular mechanisms of β-amyloid production and secretion. Proc. Natl. Acad. Sci. U S A. 1999;96:11049–11053. doi: 10.1073/pnas.96.20.11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szodorai A., Kuan Y.H., Hunzelmann S., Engel U., Sakane A., Sasaki T., Takai Y., Kirsch J., Muller U., Beyreuther K. APP anterograde transport requires Rab3A GTPase activity for assembly of the transport vesicle. J. Neurosci. 2009;29:14534–14544. doi: 10.1523/JNEUROSCI.1546-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarassishin L., Yin Y.I., Bassit L., Li Y.M. Processing of Notch and amyloid precursor protein by γ-secretase is spatially distinct. Proc. Natl. Acad. Sci. U S A. 2004;101:17050–17055. doi: 10.1073/pnas.0408007101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thayer D.A., Jan Y.N., Jan L.Y. Increased neuronal activity fragments the Golgi complex. Proc. Natl. Acad. Sci. U S A. 2013;110:1482–1487. doi: 10.1073/pnas.1220978110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiyama T., Matsuyama S., Iso H., Umeda T., Takuma H., Ohnishi K., Ishibashi K., Teraoka R., Sakama N., Yamashita T. A mouse model of amyloid β oligomers: their contribution to synaptic alteration, abnormal tau phosphorylation, glial activation, and neuronal loss in vivo. J. Neurosci. 2010;30:4845–4856. doi: 10.1523/JNEUROSCI.5825-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiyama T., Nagata T., Shimada H., Teraoka R., Fukushima A., Kanemitsu H., Takuma H., Kuwano R., Imagawa M., Ataka S. A new amyloid β variant favoring oligomerization in alzheimer's-type dementia. Ann. Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- Tonoki A., Kuranaga E., Tomioka T., Hamazaki J., Murata S., Tanaka K., Miura M. Genetic evidence linking age-dependent attenuation of the 26S proteasome with the aging process. Mol. Cell. Biol. 2009;29:1095–1106. doi: 10.1128/MCB.01227-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubuki S., Saito Y., Tomioka M., Ito H., Kawashima S. Differential inhibition of calpain and proteasome activities by peptidyl aldehydes of Di-leucine and Tri-leucine. J. Biochem. 1996;119:572–576. doi: 10.1093/oxfordjournals.jbchem.a021280. [DOI] [PubMed] [Google Scholar]
- Ubelmann F., Burrinha T., Salavessa L., Gomes R., Ferreira C., Moreno N., Almeida C.G. Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep. 2017;18:102–122. doi: 10.15252/embr.201642738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar S.S., Brodsky J.L. One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 2008;9:944–957. doi: 10.1038/nrm2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D.M., Selkoe D.J. Aβ Oligomers - a decade of discovery. J. Neurochem. 2007;101:1172–1184. doi: 10.1111/j.1471-4159.2006.04426.x. [DOI] [PubMed] [Google Scholar]
- Wang H., Saunders A.J. The role of ubiquitin-proteasome in the metabolism of amyloid precursor protein (APP): implications for novel therapeutic strategies for alzheimer's disease. Discov. Med. 2014;18:41–50. [PubMed] [Google Scholar]
- Wang Y., Wei J.H., Bisel B., Tang D., Seemann J. Golgi cisternal unstacking stimulates COPI vesicle budding and protein transport. PLoS One. 2008;3:e1647. doi: 10.1371/journal.pone.0001647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T., Hikichi Y., Willuweit A., Shintani Y., Horiguchi T. FBL2 regulates amyloid precursor protein (APP) metabolism by promoting ubiquitination-dependent APP degradation and inhibition of APP endocytosis. J. Neurosci. 2012;32:3352–3365. doi: 10.1523/JNEUROSCI.5659-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner M.W., Veitch D.P., Aisen P.S., Beckett L.A., Cairns N.J., Green R.C., Harvey D., Jack C.R., Jagust W., Liu E. The alzheimer's disease neuroimaging initiative: a review of papers published since its inception. Alzheimers Dement. 2012;8:S1–S68. doi: 10.1016/j.jalz.2011.09.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson B., Gilbert H.F. Protein disulfide isomerase. Biochim. Biophys. Acta. 2004;1699:35–44. doi: 10.1016/j.bbapap.2004.02.017. [DOI] [PubMed] [Google Scholar]
- Xiao Y., Ma B., McElheny D., Parthasarathy S., Long F., Hoshi M., Nussinov R., Ishii Y. Aβ(1-42) fibril structure illuminates self-recognition and replication of amyloid in Alzheimer's disease. Nat. Struct. Mol. Biol. 2015;22:499–505. doi: 10.1038/nsmb.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Sweeney D., Wang R., Thinakaran G., Lo A.C., Sisodia S.S., Greengard P., Gandy S. Generation of Alzheimer β-amyloid protein in the trans-Golgi network in the apparent absence of vesicle formation. Proc. Natl. Acad. Sci. U S A. 1997;94:3748–3752. doi: 10.1073/pnas.94.8.3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu W.H., Kumar A., Peterhoff C., Kulnane L.S., Uchiyama Y., Lamb B.T., Cuervo A.M., Nixon R.A. Autophagic vacuoles are enriched in amyloid precursor protein-secretase activities: implications for β-amyloid peptide over-production and localization in Alzheimer's disease. Int. J. Biochem. Cell. Biol. 2004;36:2531–2540. doi: 10.1016/j.biocel.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Zhu J.W., Nagasawa H., Nagura F., Mohamad S.B., Uto Y., Ohkura K., Hori H. Elucidation of strict structural requirements of brefeldin a as an inducer of differentiation and apoptosis. Bioorg. Med. Chem. 2000;8:455–463. doi: 10.1016/s0968-0896(99)00297-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.