

Abstract
Innate immunity, the first line of defense against invading pathogens, is an ancient form of host defense found in all animals, from sponges to humans. During infection, innate immune receptors recognize conserved molecular patterns, such as microbial surface molecules, metabolites produces during infection, or nucleic acids of the microbe’s genome. When initiated, the innate immune response activates a host defense program that leads to the synthesis proteins capable of pathogen killing. In mammals, the induction of cytokines during the innate immune response leads to the recruitment of professional immune cells to the site of infection, leading to an adaptive immune response. While a fully functional innate immune response is crucial for a proper host response and curbing microbial infection, if the innate immune response is dysfunctional and is activated in the absence of infection, autoinflammation and autoimmune disorders can develop. Therefore, it follows that the innate immune response must be tightly controlled to avoid an autoimmune response from host-derived molecules, yet still unencumbered to respond to infection. In this review, we will focus on the innate immune response activated from cytosolic nucleic acids, derived from the microbe or host itself. We will depict how viruses and bacteria activate these nucleic acid sensing pathways and their mechanisms to inhibit the pathways. We will also describe the autoinflammatory and autoimmune disorders that develop when these pathways are hyperactive. Finally, we will discuss gaps in knowledge with regard to innate immune response failure and identify where further research is needed.
1. INTRODUCTION
Several nucleic sensing pathways have been identified over the last few decades that have increased our understanding of diseases that occur when these pathways are dysfunctional. The last decade has experienced an upsurge of new discoveries regarding nucleic acid sensing, many due to advances in technology. Nevertheless, intricacies of how the failure of nucleic acid-sensing mechanisms leads to autoinflammation and autoimmunity remain unsolved. In this review, we explore what is known about how the failure of nucleic acid-sensing mechanisms leads to autoimmunity and autoinflammation as well as highlight questions that remain to be solved.
Autoinflammation and autoimmunity are often mistakenly used interchangeably but refer to the origin of disease. Namely, while autoinflammatory diseases are driven primarily by dysregulation of the innate immune system and do not rely on T cells or B cells for disease to progress, autoimmune diseases are driven primarily by T-cell and B–cell mediation (Arakelyan et al., 2017). Several autoimmune diseases originate from the innate immune system but then require lymphocytes for progression, indicating that disease is often a result of more than one component of immunity. Details that differentiate autoimmunity and autoinflammation remain controversial, but our current understanding of these pathologies is based on our ample understanding of innate immune recognition.
Central to the autoinflammatory and autoimmune disorders discussed in this review are the sensors that detect cytosolic nucleic acids to stimulate an innate immune response. The primary role of the innate immune system is to be the first line of defense against foreign microbes. The recognition of microbes and nucleic acid depends on pattern recognition receptors (PRRs) that recognize a variety of pathogen-derived molecules called pathogen-associated molecular patterns (PAMPs). While many types of PAMPs are encountered only during microbial infection, nucleic acid-mediated PRR activation can be microbe-or host-derived. That is, the innate immune response can be activated by the presence of self-nucleic acids that escape the nucleus, a major etiological cause of the autoinflammatory and autoimmune disorders discussed here. As such, in this review, we will describe the receptors of RNA and DNA nucleic acids and the signaling pathways that each stimulate. Then, for both the RNA and DNA sensing pathways, we will identify the specific types of microbial infections that activate innate immune responses and how each microbe has evolved mechanisms to inhibit these pathways. Finally, we will discuss the autoinflammatory and autoimmune disorders that develop when these nucleic acid signaling pathways are dysfunctional.
2. RNA NUCLEIC ACID SENSING IN VIRAL IMMUNOLOGY AND AUTOIMMUNITY
2.1. Detection of Intracellular RNA
2.1.1. Protein Kinase R
Protein kinase R (PKR) is encoded by the EIF2AK2 gene, and it is an interferon (IFN)-induced, double-stranded RNA (dsRNA)-dependent protein kinase that phosphorylates the alpha subunit of eukaryotic initiation factor 2 (eIF2α), resulting in the inhibition of mRNA translation initiation (Kitajewski et al., 1986). PKR has also been shown to be a component of IFN activation to facilitate a robust innate immune response (Balachandran et al., 2000). PKR itself is induced by IFN, resulting in a positive feed-forward loop that further amplifies the innate immune response (Li et al., 2011). Double-stranded RNA activation of PKR results in its dimerization and autophosphorylation (Dever, 2002). Downstream IFN gene induction is induced primarily by the NF-κB transcription factor following PKR-mediated phosphorylation of IκB (Kumar et al., 1994), but not IRF3 activation (Smith et al., 2001). A number of viruses have encoded mechanisms to block PKR activation to allow for enhanced virus replication, such as adenovirus, reovirus, influenza virus, and hepatitis C virus (Gale et al., 1997; Katze et al., 1987; Lloyd and Shatkin, 1992; Lu et al., 1995).
The activation of PKR is also inhibited by other cellular factors, such as the gene encoded by DNAJC3, namely P58IPK (Lee et al., 1992). Specifically, P58IPK interacts with PKR at the site that promotes its dimerization and autophosphorylation (Gale et al., 1996). Importantly, P58IPK is activated during influenza virus infection and P58IPK dysfunction results in late onset type 1 diabetes, which will be discussed in a following section (Ladiges et al., 2005; Melville et al., 1999).
2.1.2. Toll-Like Receptors
Toll-like receptors (TLRs) are integral membrane glycoproteins, have a tri-modular structure, and contain 16–28 leucine-rich repeats (LRRs), which are necessary for interaction and recruitment of several adaptor proteins (Kawasaki et al., 2011; Matsushima et al., 2007). TLRs are a subset of PRRs expressed on the cell membrane of professional immune cells like monocytes, macrophages, dendritic cells, B cells, and non-immune cells like keratinocytes and epithelial cells (Kawasaki et al., 2011; Novak et al., 2010). TLRs are classified by their ectodomain for ligand binding (Kawai and Akira, 2009; Kumar et al., 2009). Thirteen TLRs have been identified in mammals but only TLR3, TLR7/8, and TLR9 recognize microbial nucleic acids in endolysosomal compartments while the others bind to bacterial or parasitic PAMPs such as triacyl lipopeptides, peptidoglycan, or lipopolysaccharide on the cell surface (Kawai and Akira, 2009; Kawasaki et al., 2011).
TLR3 recognizes dsRNA, a product of RNA virus replication lifecycles, in endolysosomal compartments. Recognition of dsRNA by TLR3 leads to signaling through NF−κB and subsequent activation of IFNs (Alexopoulou et al., 2001). This IFN–β promoter activation is uniquely mediated by the adaptor protein, Toll-interleukin 1 receptor domain (TIR)-containing adaptor inducing IFN-β (TRIF or TICAM-1) (Oshiumi et al., 2003). This recognition process by TLR3 must be tightly regulated to ensure IFN activation only in the presence of non-self nucleic acid. The structural composition of the dsRNA is important in efficient recognition by TLR3. For example, the 2′-OH group in cytidylic acid is necessary for the dsRNA to be recognized by TLR3 (Okahira et al., 2005). More recently, work has been done to examine the functional structure of TLR3 that has the ability to recognize dsRNA. Glycosylation and cathepsin cleavage of TLR3 occur as it is transported from the endoplasmic reticulum through the Golgi apparatus and into an endolysosome (Toscano et al., 2013). The TLR3 C terminal and N terminal cleavage product complex that results is necessary for the recognition of dsRNA (Garcia-Cattaneo et al., 2012; Murakami et al., 2014). This mature TLR3 is fully functional in the endolysosome and ready to accurately recognize non-self dsRNA (Toscano et al., 2013). An interesting exception has also been discovered whereby poliovirus-derived single-stranded RNA segments that have loop structures resembling dsRNA can also activate TLR3 when they come from damaged or inflamed cells (Tatematsu et al., 2013). As we will discuss below, TLR3 plays a key role in controlling viral infections that involve dsRNA structures and it is also involved in the manifestation of autoimmune disorders such as type 1 diabetes.
TLR7/8 resides in endolysosomal compartments and recognizes ssRNA as the ligand of activation. Signaling occurs through the adaptor molecule MyD88 and IRF7 (Diebold et al., 2004; Heil et al., 2004; Kawai et al., 2004). For example, TLR7 and TLR8 have been shown to aid in host defense against the paramyxovirus, Sendai virus (Melchjorsen et al., 2005). HIV is also an antagonist for TLR7/8 mediated antiviral responses robust sensing by these TLRs leads to recruitment of effector cells to the site of viral infection (Schlaepfer and Speck, 2008). Mechanistically, TLR7/8 sensing of ssRNA occurs in a sequence-independent manner. The uridine and ribose molecules of RNA are known antagonists of TLR7 (Diebold et al., 2006). TLR7/8 activation induces a robust IFN response alongside the production of other cytokines such as: interleukin (IL)-1β, IL-6 and IL-12. IL-1β in particular is produced when non-self ssRNA and TLR7/8 activation results in activation caspase-1 (Nicholas et al., 2011). More recently, researchers also determined that the RNA editing phenomenon of adenosine-to-inosine conversion enhances TLR7/8 activation. In this experiment, TLR7 sensing of ssRNA was enhanced in inosine-modified viral RNA (Sarvestani et al., 2014). The cooperative role between TLR7 and TLR8 is also an important part of the mechanism that results in an IFN response to ssRNA. Influenza virus infection was effectively controlled in a rat study where the dual administration of TLR7/8 was administered and effectively suppressed viral load (Hammerbeck et al., 2007). Additionally, during Japanese encephalitis virus infection, TLR8 can compensate for a lack of TLR7 by activating an effective response alone (Awais et al., 2017). TLR7/8 is a necessary component of the antiviral response that responds to foreign ssRNA in endolysosomal compartments and activates an IFN response.
2.1.3. RIG-I-Like Receptors
RIG-I-like receptors (RLRs) recognize viral RNA and initiate innate immune response signaling (Loo and Gale, 2011). RLRs are characterized by their central DExD/H box RNA helicase domain that senses cytosolic dsRNA (Gack, 2014). There are three known RLRs with unique functions: retinoic acid-inducible gene I (RIG-I) (Yoneyama et al., 2004), melanoma differentiation-associated gene 5 (MDA5) (Kang et al., 2004), and laboratory of genetics and physiology 2 (LGP2) (Cui et al., 2001). RLRs are expressed in most human tissues to allow for widespread type I IFN induction upon viral infection. RLRs are expressed at low levels in resting cells, and then the expression is increased in response to viral infection. RIG-I and MDA5 have similar functions such that they initiate antiviral signals to induce IFN gene activation (Kang et al., 2004; Yoneyama et al., 2004). LGP2 functions as a regulator of RIG-I and MDA5 (Yoneyama et al., 2005). Therefore, the antiviral response is a balanced system where the outcome of a viral infection is determined by the level of viral replication as compared to the level of antiviral response activation.
RIG-I and MDA5 are structurally similar with two caspase-recruitment domains (CARDs) at the N-terminus region, a central DExD/H domain, and C-terminal domain (CTD) (Kang et al., 2004, Yoneyama et al., 2004). Once the RIG-I and MDA5 central DExD/H domain and CTD bind viral RNA, the CARD domains interact with the mitochondrial antiviral signaling (MAVS) adaptor protein. MAVS is composed of an N-terminal CARD-like domain and C-terminal transmembrane domain, both of which are necessary for protein function and signaling (Seth et al., 2005). The activation signal is then transmitted through Fas-associated protein with the death domain (FADD)/receptor-interacting protein 1 (RIP1) that leads to the translocation of the NF-κB transcription factor into the nucleus (Honda et al., 2006). Signaling through MAVS can also activate IKKε and TANK-binding kinase 1 (TBK1) that phosphorylate IRF3 and allows for translocation to the nucleus (Sharma et al., 2003). NF-κB and IRF3 are transcription factors that lead to the production of proinflammatory cytokines, namely IFN-β for type I IFN response to viral infection (Yoneyama et al., 2005).
Mitochondrial antiviral-signaling protein (MAVS, also known as IPS-1, VISA, and Cardif ) is an adaptor molecule that also induces IFN from RIG-I and MDA5 signaling (Kawai et al., 2005). The MAVS molecule has a CARD domain that interacts with the CARD domains of RIG-I and MDA5. MAVS signaling requires TBK1 and IKKε protein kinases and activates transcription factors NF-κB and IRF3, leading to IFN induction (Kawai et al., 2005). Autoamplification of IFN signaling ensues because RIG-I and MDA5 are IFN-inducible (Honda et al., 2006). The abundance of IFNs induces up-regulation of IFN-stimulated genes (ISGs) (Pine et al., 1990). This can occur through the Jak/STAT (signal transducer and activator of transcription) upon binding by IFN-β (Darnell et al., 1994). The antiviral signals are then spread to surrounding infected and uninfected cells. Cells enter into antiviral states that control the infection by resisting viral replication. ISGs are responsible for amplifying the antiviral response and encoding proteins that have direct antiviral activity (Yoneyama et al., 1996). The production of innate immune cytokines and chemokines recruits professional immune cells to the site of infection and initiate the adaptive immune response (Kadowaki et al., 2000). Activation of RIG-I and MDA5 by dsRNA leads to activation of a signaling cascade and subsequent IFN induction during antiviral innate immune responses (Fig. 1).
Fig. 1.
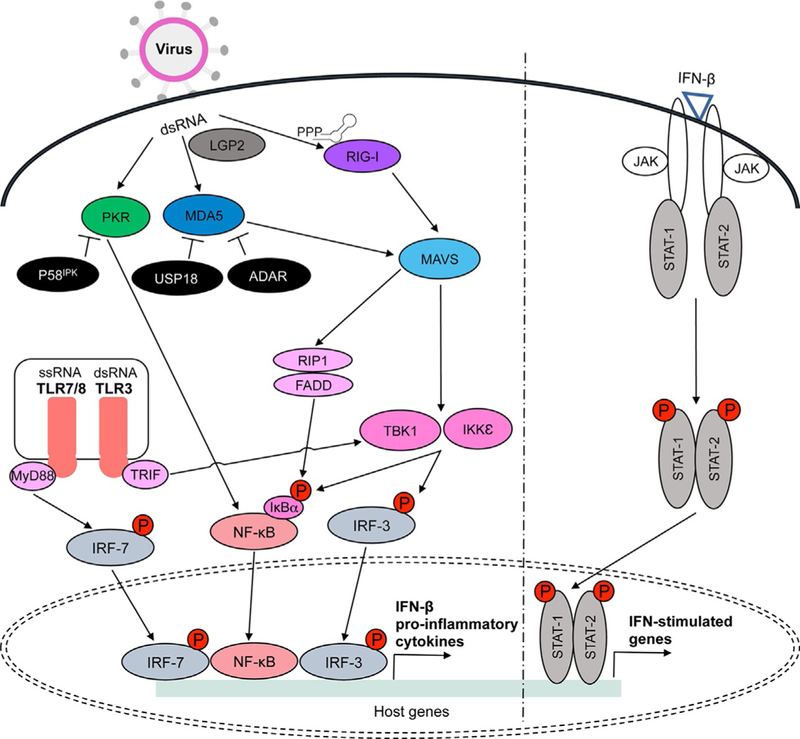
RIG-I, MDA5, LGP2, PKR, and TLRs3/7/8 are activated during viral infection in the presence of non-self dsRNA. RIG-I recognizes short dsRNA and non-self ssRNA with 5′-triphosphate or 5′-diphosphate, while MDA5 recognizes long dsRNA. LGP2 acts upstream to regulate RIG-I and MDA5 activation in the presence of dsRNA. RIG-I and MDA5 interact with MAVS adaptor protein located on the mitochondrial membrane. MAVS then activates FADD/RIP1 and TBK1/IKKε protein kinases that induce nuclear translocation of NF-κB and IRF3 following phosphorylation of IκBα or IRF3. PKR is also activated in the presence of dsRNA and it transmits activation signals through NF-κB. Endolysosomal TLRs3 and 7/8 lead to TBK1 and IRF7 activation via adaptors TRIF and MyD88, respectively. NF-κB, IRF3, and IRF7 are transcription factors that induce proinflammatory cytokine production, including IFN-β. Autoamplification of IFN signaling occurs through activation of the Jak/STAT pathway upon IFN-β binding. While IFN-stimulated genes spread antiviral signals to surrounding cells, viral proteins inhibit multiple steps of these pathways (see Table 1). Inhibitors of these signaling pathway also work to control activation signals and dysfunctions in these molecules can contribute to various autoimmune disorders. For example, P58IPK inhibits PKR while USP18 and ADAR suppress MDA5 activity (see Table 2).
Since an effective IFN response requires equilibrium within the system, regulators such as LGP2 are key in modulating activation signals. LGP2 has a different structure as compared to RIG-I and MDA5 because it lacks the two CARD domains (Murali et al., 2008). The ATPase domain of LGP2 functions upstream of RIG-I and MDA5 to facilitate recognition of cytosolic viral RNA (Satoh et al., 2010). Studies have shown that LGP2 binds dsRNA and negatively regulates RIG-I and MDA5 activation. These studies observed negative feedback regulation during Sendai virus and Newcastle disease virus infection (Rothenfusser et al., 2005; Yoneyama et al., 2005). However, a recent study has suggested that LGP2 might also have a positive enhancement regulation mechanism for MDA5 and RIG-I. Structural analysis of LGP2 revealed that RNA-dependent binding of dsRNA by LGP2 enhanced MDA5 antiviral signaling (Uchikawa et al., 2016). LGP2 has also been described as a necessary component of an effective IFN response during positive-sense picornavirus infection (Satoh et al., 2010). However, other studies have shown that LGP2 negatively regulates RIG-I function by sequestering viral RNA or competing with IKKε for interaction with MAVS (Komuro and Horvath, 2006; Rothenfusser et al., 2005; Saito et al., 2007). To clarify these seemingly disparate roles for LGP2, it was shown that in mice lacking LGP2, there was increased resistance during negative-sense vesicular stomatitis virus (VSV) infection while IFN signaling was defective during Cardiovirus A, a member of the Picornaviridae family, infection, causing the mice to be more susceptible to infection (Venkataraman et al., 2007). Taken together, LGP2 has an important regulatory role in IFN signaling, but the response is variable among different RNA viruses.
RIG-I and MDA5 have variable recognition abilities for foreign cytosolic RNA (Kato et al., 2006). The varying abilities for RIG-I and MDA5 to discriminate between self and non-self allows for specification in the antiviral innate immune response. RIG-I and MDA5 both have the ability to recognize dsRNA. However, RIG-I is known to recognize Orthomyxoviridae, Flaviviridae, and Paramyxoviridae family viruses while MDA5 specifically recognizes picornavirus infections (Kato et al., 2006; Loo et al., 2008). RIG-I and MDA5 activation together can also be essential to induce strong IFN responses during viral infections, such as West Nile virus (Errett et al., 2013). The length of the dsRNA plays a key role in activation of RIG-I or MDA5. RIG-I recognizes short dsRNA (up to 1 kb) while MDA5 recognizes long dsRNA molecules (more than 2 kb) (Kato et al., 2008). For example, the replicative form of the picornavirus genome is a 7.5 kb dsRNA intermediate that has been shown to robustly activate MDA5 in infected cells (Feng et al., 2012). RIG-I has additional abilities to discriminate between self and non-self by recognition of single-stranded RNA with a 5′-triphosphate (5′-PPP) or with a 5′-diphosphate (5′-PP) (Goubau et al., 2014; Hornung et al., 2006; Pichlmair et al., 2006). This was determined because influenza virus infection activates RIG-I in the absence of a dsRNA intermediate during the replication life-cycle (Pichlmair et al., 2006). Host ssRNA exists in the cytosol with a 5′-guanosine cap because it has undergone post-transcriptional modifications. Therefore, the 5′-PPP is an important feature in discrimination because self and non-self because it is unique to viral cytosolic ssRNA. The 5′-PPP is a component of the RNA molecule after viral polymerase replication has taken place (Hornung et al., 2006). It was also described that RIG-I can recognize ssRNA with 5′-PP, a special feature of mammalian reoviruses (Goubau et al., 2014). Recently, it was shown that a conserved residue (H830) of RIG-I is essential to prevent sensing of self RNA that bears a N1-2′-O-methyl group. Additionally, yellow fever virus encodes a methyl transferase to allow escape of viral RNA recognition (Schuberth-Wagner et al., 2015). Together, RIG-I and MDA5 recognize different features of foreign RNA molecules that lead to unique activation of these RLRs during different viral infections. The length of the viral dsRNA along with 5′-PPP, 5′-PP, or cap methylation features on ssRNA dictates the activation of either RIG-I or MDA5 in antiviral innate immune responses.
RNA binding to RIG-I and MDA5 requires specific molecular mechanisms to facilitate controlled and effective activation of the RLRs. Positive and negative regulatory mechanisms of RIG-I and MDA5 are necessary to tightly control IFN signaling. It has been shown that RIG-I retains an inactive configuration until RNA binding occurs and the ATPase activity increases to induce signal transduction of the antiviral response (Gee et al., 2008). Exposure of the RIG-I and MDA5 CARD domains leads to structural changes, such as ubiquitination and phosphorylation, that vary levels of activation. The RNF125 ubiquitin E3 ligase suppresses RIG-I and MDA5 activity by causing ubiquitination of their CARD domains and subsequent proteasomal degradation (Arimoto et al., 2007). Further, RNF125 enhancement by IFN creates a negative feedback loop that controls RIG-I and MDA5 activation during infection (Arimoto et al., 2007). In contrast, strong activation of RIG-I is facilitated by K63-polyubiquitin chains binding to RIG-I at the CARD domains (Zeng et al., 2010). RIG-I and MDA5 are also regulated by phosphorylation and dephosphorylation of its CARD domains through PP1α and PP1γ phosphatases. The function of PP1α and PP1γ to dephosphorylate the CARD domains of RIG-I and MDA5 is needed to facilitate strong induction of an IFN response (Wies et al., 2013). This positive and negative regulation of RIG-I and MDA5 is important in maintaining appropriate control of IFN induction and serving as a first line of defense against viral infections. There are many regulatory mechanisms for RIG-I-and MDA5-mediated IFN induction that help in effective viral control. However, dysfunctions in recognition and signaling can lead to increased viral susceptibility and even autoimmunity.
2.2. RNA Sensing During Viral Infections
Mutations within the genes coding for RNA sensors have been shown to increase susceptibility to a variety of viral infections. Additionally, viruses have mechanisms to antagonize specific aspects of the antiviral innate immune response. Decreasing IFN induction during viral infection is advantageous for the virus because it increases its infectivity potential. Several RNA viruses that exhibit these characteristics include: hepatitis C virus, Dengue virus, West Nile virus, Zika virus, respiratory syncytial virus, Nipah virus, Ebola virus, and Rotavirus (Table 1).
Table 1.
Summary of Host Proteins That Sense RNA Virus Infection, Viral Proteins That Inhibit Host Responses, and Polymorphisms That Affect Pathogenesis
| Virus | Family | RLR Detection | Innate Immune Inhibition by Viral Components | Genetic Polymorphisms That Increase Susceptibility |
|---|---|---|---|---|
| Hepatitis C virus (HCV) | Flaviviridae (+) ssRNA | • RIG-I (Sumpter et al., 2005) • TLR7 (Stone et al., 2014) |
• NS3/4A viral protease (Ding et al., 2012; Li et al., 2005) • NS3 (Kasama et al., 2012) • NS4B (Ding et al., 2013) |
• Polymorphisms in TLRs3/7/8 result in increased HCV susceptibility (El-Bendary et al., 2018) |
| Dengue virus (DENV) | Flaviviridae (+) ssRNA | • RIG-I and MDA5 (Nasirudeen et al., 2011) • TLR3 (Rodriguez-Madoz et al., 2010) |
• Virus induced double-membrane vesicles (Junjhon et al., 2014; Uchida et al., 2014) • NS4B, NS2A, NS4A (Muñoz-Jordán et al., 2003) • NS2B/NS3 (Angleró-Rodríguez et al., 2014; Rodriguez-Madoz et al., 2010) • NS2A/NS4B (Dalrymple et al., 2015) • NS5 (Ashour et al., 2009) |
• Type 2 diabetes increases susceptibility to severe viral infection (Lee et al., 2013) |
| West Nile virus (WNV) | Flaviviridae (+) ssRNA | • RIG-I and MDA5 (Fredericksen et al., 2008) • TLR7 (Xie et al., 2013) • TLR3 (Szretter et al., 2010) |
• NS1 (Zhang et al., 2017b) | • Lack of MAVS in myeloid cells (Pinto et al., 2014) • RFC1-DNA polymerase activator (Loeb et al., 2011) • Lack of MAVS (Suthar et al., 2010) • Type 2 diabetes (Kumar et al.,2012) |
| Zika virus (ZIKA) | Flaviviridae (+)ssRNA | • RIG-I, MDA5, and TLR3 (Hamel et al., 2015) • TLRs3/8 (Luo et al., 2018) |
• NS5 (Grant et al., 2016; Kumar et al., 2016) • NS1 (Xia et al., 2018) |
• MAVS (Piret et al., 2018) • IRF3, IRF5, IRF7 (Lazear et al., 2016) |
| Respiratory syncytial virus (RSV) | Paramyxoviridae (–)ssRNA | • RIG-I (Loo et al., 2008) • TLRs3/7 (Qi et al., 2015) |
• NS2 (Ramaswamy et al., 2004, 2006) • NS1 (Xu et al., 2014b) |
• FokI-vitamin D receptor (Hansdottir et al., 2010; Janssen et al., 2007; Stoppelenburg et al., 2014) • Plasmacytoid dendritic cells (Marr et al., 2014) |
| Nipah virus (NiV) | Paramyxoviridae (–)ssRNA | • RIG-I (Habjan et al., 2008) | • Phosphoprotein (Ciancanelli et al., 2009; Rodriguez et al., 2004; Shaw et al., 2004) •Nucleoprotein (Sugai et al., 2017) • Matrix protein (Bharaj et al., 2016) •Nonstructural C protein (Yamaguchi et al., 2014) |
|
| Ebola virus (EBOV) | Filoviridae (–) ssRNA | • RIG-I (Habjan et al., 2008) | • VP35 (Caballero et al., 2016; Hartman et al., 2008; Luthra et al., 2013; Yen et al., 2014) | |
| Rotavirus (RV) | Reoviridae dsRNA-RT | • RIG-I and MDA5 (Broquet et al., 2011) • TLRs3/7 (Yang et al., 2016) |
• NSP1 (Barro and Patton, 2005; Graff et al., 2009; Qin et al., 2011) | • Variable RNA transcripts (Uzri and Greenberg, 2013) |
2.2.1. Hepatitis C Virus
Hepatitis C virus (HCV) is a positive-sense ssRNA virus within the Flaviviridae family that activates RIG-I during infection (Sumpter et al., 2005). HCV uses viral proteases and viral proteins to target specific components of IFN signaling and decrease antiviral responses. The HCV-NS3/4A viral protease is involved in many mechanisms of antiviral suppression. HCV-NS3/4A is known to cleave MAVS and decrease IFN signaling. A point mutation in MAVS at Cys-508 renders it resistant to NS3/4A cleavage (Li et al., 2005). HCV-NS3/4A are also known to induce expression of the translocase of outer mitochondrial membrane 70 (TOM70) and induce IFN signaling in hepatocytes (Kasama et al., 2012). However, the HCV nonstructural (NS) protein 3 (HCV-NS3) then suppresses TOM70 induction of IRF3 mediated immunity by cleaving MAVS upstream of TOM70. Also, HCV-NS3/4A viral protease has also been shown to inhibit IL-28 induction. IL-28 contains NF-κB and IRF3 binding sites, meaning it can be induced by these transcription factors (Ding et al., 2012). Thus, the actions of HCV-NS3/4A aid in viral persistence within the host. The HCV-NS4B viral protein is also involved in antiviral suppression. HCV-NS4B interferes with TBK1 interactions, inhibiting these proteins from properly relaying IFN signaling (Ding et al., 2013). Finally, the HCV core protein was shown to inhibit TLR7-mediated IFN induction as well as IRF7 and STAT1 expression in plasmacytoid dendritic cells (Stone et al., 2014), and polymorphisms in genes encoding TLR3/7/8 result in increased susceptibility to HCV infection (El-Bendary et al., 2018). Together, HCV-NS3/4A, HCV-NS4B, and HCV-Core are viral components that suppress IFN induction and evade innate immune responses.
2.2.2. Dengue Virus and West Nile Virus
Dengue virus (DENV) and West Nile virus (WNV) are both mosquitoborne viruses within the Flaviviridae family (Ahlers and Goodman, 2018; Fredericksen et al., 2008; Loo et al., 2008; Nasirudeen et al., 2011). DENV and WNV both activate the IFN-mediated innate immune response through RIG-I and MDA5. Knockdown of RIG-I and MDA5 resulted in increased susceptibility to DENV infection (Nasirudeen et al., 2011). This highlights the importance of RIG-I and MDA5 in controlling DENV infection. Innate immune responses to WNV infection occur in two major phases, with RIG-I and MDA5 being important sensors at both phases. The initial response to WNV leads to IRF3 activation and ISG induction. Later stages in WNV infection are dominated by IFN-dependent antiviral gene expression. In response to WNV infection, RIG-I and MDA5 act through MAVS as they work together to stimulate a strong IFN response and subsequent signal amplification (Fredericksen et al., 2008). DENV and WNV both have antiviral evasion mechanisms that antagonize IFN induction after RIG-I and MDA5 sensing.
DENV has many mechanisms of innate immune suppression at different stages of infection. During early antiviral responses, before IFN induction, DENV induces the production of autophagic proteins, and autophagy activation suppresses the antiviral response (Huang et al., 2016). As the infection progresses, DENV also suppresses IFN induction by keeping viral dsRNA within intracellular membranes to hide the dsRNA from recognition by RLR receptors (Uchida et al., 2014). Virus induced double-membrane vesicles form early in infection and contain the dsRNA along with viral nonstructural proteins and replication machinery (Junjhon et al., 2014; Mackenzie et al., 1996). Therefore, viral replication can be occur within these vesicles in the first 48 h of infection with little dsRNA exposure in the cytosol (Uchida et al., 2014). However, other studies have shown that DENV NS4A is part of the membrane-bound replication complex that is also associated with viral RNA (Miller et al., 2007), also similar to the flavivirus Kunjin virus (Roosendaal et al., 2006). Additionally, DENV viral RNA was shown to associate with the rough ER (Grief et al., 1997). Therefore, it remains to be fully clarified if DENV viral RNA is fully incorporated within virus-induced membrane structures to avoid RLR recognition or if they are on the surface of cytosolic membrane structures. To evade host immune responses, DENV also uses nonstructural viral proteins direct suppression IFN signaling. Three nonstructural proteins of DENV are known to down-regulate IFN-β expression. The presence of DENV-NS4B caused the most significant decrease in IFN activity while DENV-NS2A and DENV-NS4A also antagonized IFN, but to a lesser extent than DENV-NS4B (Muñoz-Jordán et al., 2003). Additional research following this study described the mechanism by which the nonstructural proteins decreased innate immune responses during DENV infection. A viral nonstructural protein complex (DENV-NS2B/NS3) interacts with IKKε to inhibit kinase activity and ultimately decrease IFN induction (Angleró-Rodríguez et al., 2014). Also, DENV-NS2A/NS4B inhibits TBK1 phosphorylation in a dose dependent manner (Dalrymple et al., 2015). Experiments also indicated that DENV infection caused a reduction in STAT2 expression suggesting the virus antagonized this innate immune response gene (Ashour et al., 2009). DENV-NS5 is the viral polymerase and it has a role in inhibiting STAT2 function. Mature DENV-NS5 is the product of a polyprotein and this was important for STAT2 binding and subsequent inhibition. Precursor DENV-NS5 in the form of a polyprotein had a single role in inducing degradation of STAT2 while the proteolytically processed, mature DENV-NS5 protein had a single role in binding STAT2 (Ashour et al., 2009). DENV-NS5-mediated degradation of STAT2 is facilitated by the host E3 ubiquitin ligase, UBR4 (Morrison et al., 2013). Finally, the DENV protease complex (NS2B/NS3) has been shown to inhibit TLR3-mediated IFN induction (Rodriguez-Madoz et al., 2010). Together these studies highlight that many DENV nonstructural proteins function in suppressing IFN induction during innate immune antiviral responses.
WNV also encodes mechanisms of innate immune suppression. IFN responses in myeloid cells are necessary for control of WNV infection. Mice lacking MAVS and the type I IFN receptor had extremely low cytokine production coupled with high WNV replication. These findings highlight the importance of IFN signaling through MAVS to control WNV infection in myeloid cells (Pinto et al., 2014). Due to the neuroinvasive nature of WNV, IFN responses to control WNV infection in the central nervous system have been studied (Ramos et al., 2012). In vivo experiments showed a significant upregulation of IL-1β during acute WNV infection. IL-1β showed a synergistic role with IFN signaling to control WNV infection in neurons (Pinto et al., 2014). WNV susceptibility has also been linked to gene expression changes in peripheral blood cells. Individuals with resistance to WNV infection had higher IL-4 levels in serum as compared to individuals who developed severe symptoms. These IL-4 levels altered expression of many genes and correlated with disease outcome (Qian et al., 2014). Neuroinvasive human cases from WNV infection were also linked to mutations in RFC1 gene through a screen of common genetic polymorphisms. RFC1 aids in proper activation of DNA polymerase (Loeb et al., 2011). Additionally, experiments in mice revealed that during WNV infection, MAVS is essential for RLR signaling. Mice that lacked MAVS exhibited uncontrolled infection and a lack of regulatory T-cell expansions that is normally a characteristic of acute WNV infection. These findings highlight the importance of MAVS mediated RLR signaling to control WNV infection (Suthar et al., 2010). WNV NS1 inhibited K63-linked ubiquitination of RIG-I and blocked IRF3 signaling upon WNV recognition. It was specifically determined that WNV-NS1 interaction with MDA5 and RIG-I induced proteasome degradation of these intracellular receptors (Zhang et al., 2017b). Lastly, an attenuated WNV strain containing a mutant NS4B protein (P38G), exhibited increased T-cell priming via a TLR7-mediated mechanism (Xie et al., 2013), and TLR3 is important in blocking WNV replication and spread into the brain (Szretter et al., 2010; Wang et al., 2004). Together, there are a variety of antiviral mechanisms in different cell types employed by WNV to antagonize the IFN response.
Interestingly, type 2 diabetes has been shown to increase susceptibility of severe DENV and WNV infection. One study showed that DENV infected patients with type 2 diabetes were at a higher risk of developing dengue hemorrhagic fever as compared to DENV infected patients without type 2 diabetes. This was quantified by an increase in IL-4 and IL-10 cytokine production in patients with DENV and type 2 diabetes because these cytokines are an important immunopathogenesis marker for dengue hemorrhagic fever (Lee et al., 2013). Another study described an increase in WNV titer in serum, peripheral tissue, and the brain of the diabetic mouse model. Type 2 diabetes caused a non-specific WNV response that increased susceptibility of neuroinvasive WNV infection (Kumar et al., 2012). These studies highlight the important balance in innate immune signaling that must be present to efficiently clear a viral infection.
2.2.3. Zika Virus
Similar to DENV and WNV, Zika virus (ZIKV) is part of the Flaviviridae family and is transmitted by mosquitoes. A robust IFN response is also needed to control ZIKV infection. ZIKV infection induced RLR signaling in human skin fibroblasts to control infection. Human skin fibroblasts infected with ZIKV showed upregulation of TLR3, RIG-I, MDA5, and ISGs. There was also sequential activation observed where TLR3 was activated 6 h post-infection and RIG-I and MDA5 were activated later in infection (Hamel et al., 2015). Other mosquito-borne flaviviruses have been known to infect dendritic cells, and this was consistent with ZIKV. Researchers observed that RIG-I response to ZIKV in human dendritic cells was activated during infection. However, there was observable inhibition of IFN protein translation. Mechanistically, ZIKV was also able to control IFN signaling by blocking STAT1 and STAT2 phosphorylation (Bowen et al., 2017). These studies show that ZIKV infection leads to the activation of RIG-I and MDA5 but that ZIKV also has mechanisms to antagonize down-stream IFN signaling.
There are specific IFN adaptor molecules that increase ZIKV susceptibility and some nonstructural proteins of the virus antagonize IFN signaling, as shown for DENV and WNV. The MAVS adaptor protein within the IFN signaling cascade is important in the early IFN response because mice lacking MAVS had higher viremia than control mice (Piret et al., 2018). During the development of a ZIKV mouse model, experiments revealed that a triple knockout of IRF3, IRF5, and IRF7 increased susceptibility to ZIKV infection through decreased IFN signaling. Also, mice lacking the IFN receptor had higher viral load in the brain and spinal cord that could not be controlled, which correlates with the neuroinvasive nature of the virus (Lazear et al., 2016). On the other hand, increased IFN induction and placental inflammation can lead to brain damage in newborns, and inhibition of TLR3 and TLR8 led to decreased pro-inflammatory cytokine responses in trophoblasts (Luo et al., 2018). Many ZIKV nonstructural proteins have been shown to facilitate the inhibition of IFN signaling observed during infection. For example, ZIKV-NS5 binds STAT2 and its expression correlates with subsequent proteasome degradation of STAT2 (Kumar et al., 2016). The IFN inhibitory role of ZIKV-NS5 in other flaviviruses is consistent for that of ZIKV. DENV-NS5 acts on STAT2 but the mechanism is slightly different than that of ZIKV because it involves the E3 ubiquitin ligase UBR4 to induce degradation (Grant et al., 2016). ZIKV-NS1 has also been shown to inhibit IFN signaling induction through binding of TBK1. This inhibitory role of the ZIKV-NS1 was observed only in ZIKV strains that caused epidemics after 2012. This highlights the importance of a fixed mutation in viral evolution because the ZIKV-NS1 fixed mutation helped the virus increase infectivity by decreasing IFN-β induction (Xia et al., 2018). Therefore, adaptor molecules are important for regulating an efficient antiviral response and without them IFN signaling may be uncontrolled. Additionally, ZIKV-NS5 and ZIKV-NS1 have IFN inhibitory roles during viral infection.
2.2.4. Respiratory Syncytial Virus
Respiratory syncytial virus (RSV) disproportionately causes more severe symptoms in infants as compared to older populations. RSV has a negativesense non-segmented RNA genome and it is part of the Pneumovirus genus of the Paramyxoviridae family. RSV dsRNA is sensed by RLRs and TLRs (reviewed in Klein Klouwenberg et al., 2009; Mukherjee and Lukacs, 2013), and host immune responses play a major role in the differences in viral susceptibility (Loo et al., 2008; van Drunen Littel-van den Hurk and Watkiss, 2012). RSV infection is known to specifically cause severe lower respiratory tract infection in newborn infants (Marr et al., 2014). Differences in the innate immune response to RSV determine the disease severity in a child. One study found polymorphisms in immune-related genes of pre-term babies that correlated with increased disease susceptibility as compared to babies carried to term (Siezen et al., 2009). Another study found a consistent association in 22 single-nucleotide polymorphisms (SNPs) within 21 innate immune genes and the development of severe RSV bronchiolitis. These SNPs were identified in a cohort of children hospitalized for severe RSV bronchiolitis as compared to the control population. One SNP identified in this study with a highly significant association with bronchiolitis was the vitamin D receptor (Janssen et al., 2007). Vitamin D is known to mediate NF-κB and STAT1 expression (Stoppelenburg et al., 2014). Vitamin D deficiencies have been correlated with increased risk and severity of RSV (Hansdottir et al., 2010). Specifically, the FokI vitamin D receptor polymorphism abrogated vitamin D’s control of the STAT1-mediated antiviral response (Stoppelenburg et al., 2014). Together, these findings illustrate the importance of vitamin D in controlling the induction of STAT1 during antiviral responses and the importance of this regulation in RSV infection. Many SNPs have been linked to RSV susceptibility and loss-of-function SNPs in the vitamin D receptor have a strong association with the development of severe RSV bronchiolitis.
Increased susceptibility to RSV has also been linked to dysfunctions in IFN signaling from loss-of-function in plasmacytoid dendritic cells (pDCs) and direct inhibition of IFN signaling by RSV nonstructural proteins. Researchers have determined that pDCs are important in activating IFN responses in lungs during RSV infection. This epidemiological study found that newborn infants had increased susceptibility to RSV because they did not have fully functioning pDCs and therefore there was low IFN induction by RIG-I (Marr et al., 2014). Respiratory macrophages and pDCs are critical in combating RSV infection, not only through RLR signaling, but also through TLR3-and TLR7-mediated mechanisms (Qi et al., 2015). Using mouse pneumonia virus, the rodent-specific form of RSV, it was shown that TLR7 was critical for the host defense response and IFN induction during infection (Davidson et al., 2011). These results were corroborated through analysis of RSV-infected human pDCs (Schijf et al., 2013). RSV is also known to decrease IFN signaling in airway epithelial cells through degradation of STAT2 (Ramaswamy et al., 2004) and through an RSV-NS1-dependent but TLR3-independent mechanism (Xu et al., 2014b). Additionally, RSV-NS2 was identified as inducing these inhibitory effects on the IFN response (Ramaswamy et al., 2006). This innate immune response inhibition by RSV aids in viral replication specifically within airway epithelial cells. The control of RSV is complex, and it requires fully functioning proteins within the IFN pathway. As seen for other viruses, the RSV nonstructural proteins have individual roles in antagonizing the antiviral response.
2.2.5. Nipah Virus
Nipah virus (NiV) is another virus in the Paramyxoviridae family, but unlike RSV, it is part of the Henipavirus genus (Ciancanelli et al., 2009). NiV is a deadly zoonotic virus, and there is a 40–90% case mortality rate in human infected with NiV in Southeast Asia (Bharaj et al., 2016). NiV dsRNA is sensed by RIG-I, but not MDA5, to initiate IFN induction (Habjan et al., 2008). Research has shown that many of the NiV proteins contribute the deadly symptoms of NiV by antagonizing IFN stimulated antiviral responses (Bharaj et al., 2016). First, the NiV phosphoprotein (P) gene encodes the NiV-P, NiV-V, and NiV-W proteins that all antagonize IFN signaling (Ciancanelli et al., 2009). The NiV-V protein, mainly found in the cytoplasm, binds STAT1 and STAT2 proteins to prevent dimerization and nuclear transport (Rodriguez et al., 2004). The NiV-W protein has a very similar role to the NiV-V protein except it sequesters STAT1 in the nucleus to inhibit subsequent ISG activation. Therefore, the NiV-V and NiV-W proteins have dual roles but are located in either the nucleus or cytoplasm to block STAT1 and induce antiviral functions. The NiV-P protein can also bind STAT1 to sequester the inactive protein in the nucleus, but to a lesser extent than NiV-V and NiV-W (Ciancanelli et al., 2009; Shaw et al., 2004). In comparison, Cedar virus is a henipavirus that is not pathogenic to humans and the Cedar virus P gene products do not antagonize STAT proteins as seen in NiV infection. This suggests that the antagonizing properties of NiV-P gene protein products greatly contribute to the highly lethal nature of the virus (Lieu et al., 2015). Second, the NiV nucleoprotein also decreased IFN responses in a dose dependent manner. The specific mechanism of inhibition is through targeting of STAT1 and STAT2 complex formation in the cytoplasm. NiV nucleoprotein decreases STAT1/STAT2 nuclear transport and subsequently down-regulates ISG expression (Sugai et al., 2017). Third, the NiV matrix protein has been shown to inhibit IKKε kinase activity that is involved in IFN signaling. The NiV matrix protein acts by degrading TRIM6, an E3-ubiquitin ligase that generates unanchored polyubiquitin chains for IKKε activation. Therefore, this degradation by the NiV matrix protein results in decreased activity of IKKε activity and decreases IFN signaling (Bharaj et al., 2016). Finally, the nonstructural C protein of paramyxoviruses inhibits IFN signaling (Gotoh et al., 2003; Mathieu et al., 2012), via inhibition of TLR7 in pDCs (Yamaguchi et al., 2014). Together, the gene products from NiV phosphoprotein, nucleoprotein, and matrix protein have been shown to antagonize IFN induction at different steps in its signaling pathway. This likely contributes to the high virulence of NiV in humans.
2.2.6. Ebola Virus
Ebola virus is negative-sense RNA virus with a non-segmented genome similar to that of RSV and NiV. Ebola virus is a hemorrhagic fever virus in the Filoviridae family. Zaire ebolavirus (EBOV) has a fatality rate of up to 90% in humans and the viral dsRNA is sensed by RIG-I to activate innate immune responses (Habjan et al., 2008; Kash et al., 2006). Early studies on EBOV were performed in human liver cells. In this study, researchers determined that IFN signaling was suppressed during infection. Through genomic analysis of gene expression, many antiviral genes were suppressed during infection. Researchers hypothesized a global IFN suppression model and also identified specific antiviral genes, such as IRF3, as being suppressed during EBOV infection (Kash et al., 2006). Further research revealed that the EBOV viral protein 35 (EBOV-VP35) had a specific inhibitor mechanism for IRF3. This was a highly specific suppression model because a point mutation in EBOV-VP35 altered its inhibitor function. EBOV-VP35 also had a role in enhancing viral replication (Hartman et al., 2008). Furthermore, EBOV-VP35 binds dsRNA, inhibiting RIG-I-mediated detection of viral dsRNA (Cardenas et al., 2006), and EBOV-VP35 is an important cofactor in the viral polymerase complex (Prins et al., 2010). Together, EBOV-VP35 plays an important role in EBOV pathogenesis both for spread of the virus within the host as well as IFN specific suppression. Further research on EBOV-VP35 enhanced the understanding of its inhibition mechanism. PACT (PKR activator) is a dsRNA binding protein that is known to induce activation of RIG-I. Experiments showed that EBOV-VP35 was able to inhibit PACT from activating RIG-I through direct binding. This was observed in a dose-dependent manner where increased presence of EBOV-VP35 increased PACT inhibition. EBOV-VP35 can also bind viral dsRNA and decrease activation of RIG-I during EBOV infection (Luthra et al., 2013). Finally, EBOV-VP35 can inhibit TLR3-mediated signaling, but this occurs in a dsRNA-independent manner suggesting that the TLRs can circumvent VP35-mediated IFN inhibition (Leung et al., 2011; Yen et al., 2014). Taken together, EBOV-VP35 is a key antagonist of IFN induction during EBOV infection.
Further in vivo experiments have shown a more complex picture of EBOV infection than just the antagonizing properties of EBOV-VP35 (Caballero et al., 2016). Transcriptomics analysis of peripheral blood mononuclear cells from cynomolgus macaques infected with EBOV showed strong innate immune activation during viral infection. Many ISGs were upregulated in response to viral infection via an intramuscular injection. These findings were contrasting to previous studies that had shown the EBOV-VP35 protein targeted the IFN response for suppression. The authors of this study proposed a model of infection where EBOV-VP35 inhibits IRF3 activation in the single infected cell. Then, in neighboring cells, EBOV-VP35 might be inducing IRF3 nuclear translocation to facilitate induction of IFN signaling through an unknown mechanism. This hypothesis aims to explain why in isolated EBOV-VP35 experiments, the protein appears to decrease IFN signaling but then during in vivo EBOV infection, ISGs are upregulated (Caballero et al., 2016). This elevated pro-inflammatory response to EBOV has been described in other studies. Rhesus macaques were infected with EBOV and monitored daily throughout the infection (Ebihara et al., 2011). Blood samples described the cytokine and chemokine profile throughout the course of infection. IL-1β and IL-6 pro-inflammatory cytokines were upregulated during infection. Interestingly, anti-inflammatory cytokines such as IL-10 and IL-13 were also increased in fatal EBOV cases. This shows that the cytokine balance during infection might be an important indicator of disease outcome. This study also showed anti-coagulation induced by the virus and ultimately contributed to overall pathogenesis (Ebihara et al., 2011). These findings were also evident in samples for fatal human cases of Ebola virus disease, symptoms of which include hemorrhagic fever. Many cytokines and chemokines were upregulated in fatal human cases. When looking at the disease progression in vivo, uncontrolled pro-inflammatory responses are a characteristic of fatal EBOV cases. Biomarkers of infection were identified as IL-1β and IL-6 along with IL-8 at late stages of infection (McElroy et al., 2014). These EBOV studies highlight the important balance of the innate immune response in determining disease outcome. EBOV viral proteins have been identified to inhibit IFN signaling during infection.
2.2.7. Rotavirus
Rotavirus (RV) is a segmented dsRNA virus within the Reoviridae family. It causes severe diarrhea in young children and is known to infect epithelial cells of the small intestine (Barro and Patton, 2005). RV dsRNA has been shown to activate RIG-I like receptors within intestinal epithelial cells. This virus is unique in that it is sensed by both RIG-I and MDA5 either together or separately. When MAVS, RIG-I, or MDA5 were silenced, RV titer increased and IFN-β production decreased. These findings suggested that RV induced IFN-β production through MAVS signaling after RIG-I and MDA5 activation (Broquet et al., 2011). Researchers have looked further into the specific RV RNA transcripts that activate IFN signaling. Nascent single-stranded RNA transcripts produced during viral replication were a strong inducer of IFN signaling (Uzri and Greenberg, 2013). Also, large RNA transcripts produced 6 h after cells were infected with RV activated IFN signaling. This finding was only observed 6 h after infection but not 1 h after infection. Both the single-stranded RNA transcripts and large RNA produced 6 h after infection had uncapped RNA at the 5′ end. RNA lacking 2′-O-methylated 5′ cap was detected in the large RNA as well. The viral protein 3 enzyme of RV is known to encode guanylyltransferase and methyltransferase. Therefore, inaccuracy in this enzyme would result in a lack of a 5′ cap or 2′-O-methyl group on the 5′ cap structure of viral RNA. Then, RNA lacking these structural components would activate RIG-I-like receptors and subsequent IFN-β production (Uzri and Greenberg, 2013). In conclusion, RNA sensing by RIG-I or MDA5 is important for controlled IFN induction during RV infection. Variations in RNA transcripts and processing are also an important indicator of IFN-β production levels. Regarding TLR signaling and RV, interestingly, TLR3 and TLR7 were required to protect the gut from inflammatory-inducing conditions through proper recognition of the gut virome, which includes RV (Yang et al., 2016).
RV nonstructural proteins are also known to antagonize innate immune responses. Specifically, RV-NS1 (or NSP1) is an antagonist of IRF3 through a common mechanism found in other viruses where it induces proteasome-dependent degradation of IRF3 to decrease IFN signaling (Barro and Patton, 2005). RV-NS1 also has a novel mechanism of decreasing NF-κB activation. This viral protein induces proteasome-dependent degradation of β-transducin repeat containing protein, a cellular protein that functions within the multi-subunit complex responsible for NF-κB activation (Graff et al., 2009). Another study also demonstrated that RV-NS1 decreased IFN-β production through a mechanism independent of IRF3 degradation as previously reported. RV-NS1 directly bound to RIG-I to decrease activation and IFN-β production. This direct interaction was confirmed with immunoprecipitation and RV-NS1 interacted with RIG-I outside the IRF3 binding domain (Qin et al., 2011). Therefore, RV is sensed by RLRs and this sensing is enhanced with inefficiency of viral enzymes that make RNA products of replication more noticeable to the RLR sensing domain. However, the virus also has ways to combat this innate immune activation and RV-NS1 is a main driver that decreased IFN-β production through antagonizing of RIG-I, NF-κB, and IRF3 activation.
2.3. Defects in RNA Sensing and Autoimmunity
Hyperactive or non-functional mutations within the genes coding for RNA sensors or their interacting proteins have been show to lead to autoimmune-like phenotypes, such as type 1 diabetes, Aicardi–Goutières syndrome, Crohn’s disease, Singleton–Merten syndrome, and systemic lupus erythematosus. As described below, many of these phenotypes are the result of imbalanced IFN or insulin signaling, autophagy, or responses to pathogens (Table 2).
Table 2.
Summary of Polymorphisms in RNA Sensors and Their Associated Syndromes
| RNA Sensor | RNA Sensor Associated Protein | Genetic Polymorphisms That Causes Dysfunction | Associated Syndrome/Pathology |
|---|---|---|---|
| MDA5 | 1. Four loss-of-function variants IFIH1-gene that encodes MDA5 (Lincez et al., 2015; Liu et al., 2009; Nejentsev et al., 2009; Shigemoto et al., 2009) 2. Gain-of-function IFIH1 mutations (Crow et al., 2015) 3. Missense mutation in IFIH1 (Funabiki et al., 2014) 4. Missense mutation in IFIH1 leading to overactive MDA5 (Rutsch et al., 2015) |
1. Type 1 diabetes 2. Aicardi–Goutieres syndrome 3. Lupus-like symptoms 4. Singleton–Merten Syndrome |
|
| USP18 | 1. Loss-of-function in USP18 induces overactivation of MDA5 (Santin et al., 2012) | 1. Type 1 diabetes | |
| ADAR | 1. Loss-of-function in ADAR induces overactivation of MDA5 (Crow et al., 2015; Pestal et al., 2015) 2. Gain-of-function mutation in IFIH1 led to over expressed MDA5 (Crow et al., 2015; Schmelzer et al., 2018) |
1–2. Aicardi–Goutieres syndrome | |
| PKR | |||
| P58IPK interaction with PERK | 1. Knockout of P58IPK (Ladiges et al., 2005) 2. Loss-of-function mutations in Dnajc3 (Synofzik et al., 2014) 3. Homozygous stop mutation (Synofzik et al., 2014) |
1. Symptoms associated with type 1 and late stage type 2 diabetes 2. Juvenile-onset diabetes and multisystemicneurodegenerative disorders 3. Monogenic recessive diabetes mellitus |
|
| RIG-I | 1. Knockout of RIG-I (Wang et al., 2007) 2. Downregulation of G protein αi2 subunit (Hampe et al., 2001b) 3. Decreased expression of RIG-I (Funke et al., 2011) 4. Variants of DDX58 causing overactive RIG-I (Jang et al., 2015) |
1. Colitis-like phenotype associated with inflammatory bowel disease 2. Human inflammatory bowel disease 3. Crohn’s disease 4. Singleton–Merten Syndrome |
|
| NOD2 | 1. NOD2 mutations that imbalance IFN signaling through RIG-I (Morosky et al., 2011) | 1. Crohn’s disease | |
| TLR3 | 1. Polymorphisms in TLR3 (Assmann et al., 2014) | 1. Type 1 diabetes | |
| TLR7 | 1. Polymorphisms in TLR7 promoter (Skonieczna et al., 2018) | 1. Systemic lupus erythematosus | |
2.3.1. The Role of MDA5 in Type 1 Diabetes and Aicardi–Goutières Syndrome
Type 1 diabetes (T1D) is characterized by the development of autoantibodies that target beta cells of the human pancreas. These autoantibodies and auto-reactive immune cells then trigger the destruction of insulin-producing pancreatic beta cells and lead to T1D. Researchers have found that there is a genetic component to disease susceptibility along with viral infection and environmental variables. Environmental factors are known to be associated with disease manifestation, and one example is vitamin D deficiency. Also, Caucasians living in Europe have a high disease incidence that highlights the exogenous factors of T1D. Cohort studies linked enterovirus or coxsackie B virus infection with increased susceptibility to the development of T1D (Knip et al., 2005). Enterovirus is known to target beta cells in the human pancreas. Autopsied pancreases of T1D patients revealed a tropism of enterovirus for human pancreatic islet cells (Ylipaasto et al., 2004). A Finnish Diabetes Prediction and Prevention Study indicated a positive association between the development of autoantibodies for human pancreatic beta cells and enterovirus infection. Autoantibodies were more likely to be present in children infected with enterovirus as compared to the control groups (Lönnrot et al., 2000). In Europe there is a high prevalence of T1D and a low prevalence of enterovirus as compared to other regions of the world, such as Cuba. Therefore, another study observed the relationship between enterovirus and T1D in Cuba, where T1D incidence is low and enterovirus prevalence is high. The researchers found a direct correlation between enterovirus infection and the development of preclinical and clinical stages of T1D (Sarmiento et al., 2013). Molecular mimicry is a proposed mechanism for this correlation because antibodies made against enterovirus could also target beta cells (Jang et al., 2015). Acute cytolytic damage from enterovirus was also a proposed mechanism for the association of enterovirus infection with T1D (Sarmiento et al., 2013). A population-based cohort study in Taiwan of age-and sex-matched children also showed that T1D incidence was higher in the enterovirus-infected cohort as compared to non-enterovirus-infected cohort (Jang et al., 2015). A similar correlation is also evident for coxsackie B virus. Data collected throughout many countries in Europe concluded that diabetic children were more likely to have antibodies against coxsackie B virus 1 than the control group of children without diabetes (Oikarinen et al., 2014). Therefore, infections with enterovirus or coxsackie B virus increase susceptibility to T1D. This highlights the connection between innate immune signaling dysfunction in response to viral infection and the development of autoimmunity.
Dysfunctions in MDA5 have also been linked to the development of T1D. Genetic analysis through a genome-wide association study (GWAS) revealed genetic components of T1D susceptibility. SNPs have been associated with the development of T1D. Four SNPs with a strong association to disease susceptibility were located within the IFIH1 gene that codes for MDA5. High IFIH1 gene expression in peripheral blood mononuclear cells also correlated with susceptible genotypes. This study highlights an interesting genetic association and gene expression importance for IFIH1 in the development of T1D (Liu et al., 2009). Another GWAS further linked T1D and IFIH1. It identified four rare variants of IFIH1 that strongly correlated with a decreased risk of developing T1D (Nejentsev et al., 2009). Two of these IFIH1 variants were correlated with a loss-of-function to MDA5 (Shigemoto et al., 2009). This resistance to T1D was evident when mice heterozygous for IFIH1 (MDA5+/–) on a NOD/Ltj background, a known mouse model of T1D, had decreased levels of MDA5 protein as compared to wild-type mice. Mice heterozygous for MDA5 drove a regulatory T-cell response that was protective against T1D during coxsackievirus infection. Mice homozygous for MDA5 on the NOD/Ltj background had high MDA5 protein levels that resulted in a strong effector T-cells response and beta cell destruction during T1D during coxsackievirus infection (Lincez et al., 2015). These findings show how MDA5 function correlates with T1D susceptibility. Reduced MDA5 expression has also been linked to increased susceptibility to encephalomyocarditis virus strain D (EMCV-D) and diabetes. EMCV-D infects insulin-producing beta cells in the pancreas similar to enterovirus or coxsackie B virus. Therefore, a lack of viral infection control is likely to lead to the production of beta cell antigens that can cause destruction of the pancreas and increase susceptibility of T1D. Mouse experiments revealed that MDA5 along with TLR3 were essential in initiating a controlled IFN response against EMCV-D. Also, mice with knockouts in MDA5 and/or TLR3 had increased susceptibility to T1D (McCartney et al., 2011). Taken together, loss-of-function in MDA5 due to genetic polymorphisms or viral infection can lead to autoimmune disorders, namely T1D.
Over activation of MDA5 can also lead to autoimmune disorders. Researchers found that mice with a missense mutation in IFIH1 quickly developed lupus-like autoimmune symptoms without the presence of a viral infection (Funabiki et al., 2014). This means that MDA5 not only has a role in controlling viral infections, but can also control autoimmune disorders. The IFIH1 missense mutation leads to a gain-of-function in MDA5 and IFN induction spread throughout multiple organs. The cytokine production was directly correlative to MAVS activity, and without MAVS, the mice with the IFIH1 missense mutation did not develop autoimmune symptoms. This clarified the importance of MDA5 signaling through MAVS in the development of autoimmune disorders (Funabiki et al., 2014). A regulator of IFN signaling, ubiquitin-specific peptidase (USP18), has an important cross talk function with MDA5. USP18 controls IFN-stimulated gene 15 protease during IFN signaling through MAVS. A lack of USP18 resulted in over activation of MDA5 and an uncontrolled pro-inflammatory response in pancreatic beta cells. The authors suggested that USP18 was a major regulator of IFN responses and clarified a mechanism by which MDA5 expression might influence the development of T1D (Santin et al., 2012). Therefore, MDA5 gain-of-function also causes unregulated IFN signaling that can lead uncontrolled beta cell destruction and the development of T1D.
Dysfunctions in the tight control of MDA5-mediated signaling have also been implicated in another autoimmune disease, Aicardi–Goutiéres syndrome (AGS) (Crow et al., 2015). AGS is an autoinflammatory, neurodevelopmental disorder that affects the brain, skin, and immune system. Characteristics range from chilblains (skin lesions) to cerebral calcifications and atrophy that may be present before birth but ultimately result in slow neurological decline due to the excess production of IFN-α (Rice et al., 2007). Analysis of AGS patient genomes revealed seven gene mutations linked associated with the disease. With regards to RNA sensing pathways, candidate genes of particular interest were ADAR1, encoding for an RNA-editing enzyme, and IFIH1, encoding for MDA5. Mutations in these genes led to an increase in IFN signaling in cerebrospinal fluid and serum that correlated with disease symptoms (Crow et al., 2015). ADAR1 is a RNA-editing enzyme known to inhibit MDA5 signaling through MAVS (Pestal et al., 2015). Therefore, a loss-of-function mutation in ADAR1 contributed to uncontrolled MDA5 antiviral response (Mannion et al., 2014; Pestal et al., 2015). More specifically, mice with a knock-in mutation in ADAR1 that inhibited its RNA-editing ability exhibited embryonic lethality at embryonic day 13.5 due to MDA5 hyperactivity (Liddicoat et al., 2015). A study has also shown that the loss of ADAR1 in human cells results in PKR hyperactivation and translational shutdown (Chung et al., 2018). A gain-of-function mutation in IFIH1 led to over expression of MDA5 and contributed to an over-reactive IFN response (Crow et al., 2015). It is important to note that clinical studies have shown that these gene mutations do not always lead to the same clinical symptoms in patients. One clinical study observed two siblings both with the loss-of-function mutation in ADAR1. Even though they both had the same mutation, their symptoms presented at different ages in childhood and symptoms were also variable. This is important information because early diagnosis and treatment of AGS can help with early administration of IFN suppression therapy to minimize brain damage induced by inflammation (Schmelzer et al., 2018). These disease phenotypes in relationship to uncontrolled IFN signaling show the functional importance of MDA5 and other genes involved in facilitating a controlled IFN response (Crow et al., 2015). Moreover, mutations in IFIH1 in AGS patients reduce the tolerance of MDA5 to Alu retroelements, and that these retroelements can also activate MDA5 during the loss of ADAR1 function. A gain-of-function mutation in MDA5 (G495R) also retains the minimal Alu duplex length to 30–40 bp (Ahmad et al., 2018). In conclusion, mutations in genes related to MDA5 signaling have been linked to the development of AGS.
2.3.2. P58IPK, PERK, TLRs, and Type 1 Diabetes
The dynamic balance between the eIF2α kinases, such as PKR, with P58IPK is important in determining the outcome of viral infection and the development of diabetes. As discussed previously, PKR is involved in amplifying the IFN response, and P58IPK is a known inhibitor of PKR. P58IPK is important for innate immune responses to viral infection along with the development of autoimmune disorders. P58IPK was originally discovered and purified from influenza virus-infected cells (Lee et al., 1990). During influenza virus infection, P58IPK inhibits PKR’s ability to phosphorylate eIF2α, thus facilitating efficient translation of influenza virus mRNAs. In influenza virus infected cells lacking P58IPK, there was a notable decrease in influenza mRNA translation and increase in eIF2α phosphorylation. In cells lacking the inhibitory target of P58IPK, PKR, the results were reversed, and influenza virus mRNA translation increased while eIF2α phosphorylation decreased. This suggests efficient influenza virus replication if facilitated by P58IPK inhibition of PKR and this has downstream ramifications on eIF2α phosphorylation. These results were also observed during VSV infection (Goodman et al., 2007). In vivo experiments enhanced our understanding of P58IPK and identified it as a cellular inhibitor of the host defense (CIHD). Observation of P58IPK–/– mice infected with influenza virus showed the P58IPK helps in host survival. P58IPK–/– mice had increased mortality but similar viral load as compared to the wild-type control mice. Additionally, in P58IPK–/– mice, there was an observed increase in eIF2α and PKR phosphorylation during influenza virus infection. These findings suggested that P58IPK activation during viral infection actually enhanced host survival while also prolonging viral replication (Goodman et al., 2009). Through computational modeling, it was shown that during influenza virus infection there was a significant increase in P58IPK while PKR and eIF2α phosphorylation were decreased. The infectious dose of virus also influenced this relationship. These findings were confirmed with vaccinia virus infection where rapid activation of P58IPK was also observed (Goodman et al., 2011). Proviral roles of P58IPK were further demonstrated during coxsackievirus B3 infection where P58IPK suppressed virally induced apoptosis (Zhang Huifang et al., 2013). P58IPK is an important regulatory in influenza virus and vesicular stomatitis virus infection within the antiviral innate immune response signaling cascade. The tight regulation of PKR by P58IPK is an important indicator of viral infection outcome.
P58IPK also inhibits PERK, pancreatic ER-localized eIF2α kinase, which is encoded by EIF2AK3. Similar to that of PKR, PERK has a kinase binding domain very similar to that found in PKR where P58IPK interacts to inhibit function. During ER stress, defined as the continued presence of unfolded proteins, P58IPK is activated. P58IPK then interacts with PERK and inhibits its activity through decreased phosphorylation. PERK functions during ER stress to decrease protein production in the ER. However, it is important to have regulatory proteins, such as P58IPK, to prevent excess protein loss during stress. These findings show that P58IPK is an important regulator of PERK in maintaining a controlled ER-stress response (Yan et al., 2002). P58IPK and PERK are known to be expressed in pancreatic cells, hinting at an important connection between these proteins and the development of diabetes (Shi et al., 1999). In mice lacking PERK, progressive diabetes mellitus and exocrine pancreatic insufficiency develop (Harding et al., 2001). PERK has a high importance in controlling protein synthesis during the ER stress response. The important inhibitory role of P58IPK on PERK was confirmed in in vivo experiments where adult mice lacking P58IPK developed glucosuria, hyperglycemia, and hypoinsulinemia (Ladiges et al., 2005). These mice had greater levels of insulin producing beta cell destruction in the pancreas. The gene expression profiles in these mice were significantly altered to favor apoptosis of pancreatic islets. These findings suggest an important regulatory role of P58IPK in preventing uncontrolled cell destruction during stress. In the absence of P58IPK, these mice developed insulin deficiency phenotypes that are very similar to what is seen in type 1 and late stage type 2 diabetes (Ladiges et al., 2005). When PERK or P58IPK were dysfunctional, ER stress homeostasis was disrupted leading to uncontrolled pancreatic beta cell apoptosis and severe diabetic phenotypes. The importance of the DNAJC3 gene has also been shown recently in humans to be linked to autoimmunity. A large screen of individuals with diabetes revealed a loss-of-function mutation in DNAJC3 that correlated with juvenile-onset diabetes and multisystemic neurodegenerative disorders. The identified homozygous stop mutation in Dnajc3 in humans leads to a monogenic recessive form of diabetes mellitus (Synofzik et al., 2014). Taken together, P58IPK is a key regulator in viral infection and autoimmune diseases through its interactions with PKR and PERK.
In addition to the importance of translational control pathways, TLR signaling has also been implicated in regulating T1D. Pancreatic beta cells respond to dsRNA treatment via a TLR3 and TRIF dependent manner, leading to type I IFN induction. Additional treatment with IFN-γ leads to beta cell apoptosis (Rasschaert et al., 2005). Furthermore, beta cells that were knocked out for TLR3 or the type I IFN receptor were protected from apoptosis during dsRNA treatment (Dogusan et al., 2008). The presence of reactive oxygen species was also required for TLR3-mediated NF-κB activation and the induction of IFN-β and TNF-α (Seleme et al., 2012). In patients that died from fulminant T1D, a subtype of diabetes mellitus, TLR3 expression was detected in 85% of T cells that had infiltrated the pancreas and in 63% of infiltrated macrophages, leading to beta cell death (Shibasaki et al., 2010). Considering the important role of TLR3 in T1D pathogenesis, it follows that polymorphisms in TLR3 were shown to be associated with increased risk for T1D (Assmann et al., 2014). Finally, in insulitic islets isolated by laser capture microdissection from patients with recent onset T1D, there was increased ISG expression. Specifically, TLR3 and EIF2AK2 were of the significantly overexpressed ISGs, which further bolsters the link between translational control and innate immune signaling in T1D (Lundberg et al., 2016). In addition to TLR3, TLR7 also plays an important role in T1D autoimmune diabetes, since treating non-obese diabetic mice with TLR7 agonists accelerated the onset of autoimmune diabetes (Lee et al., 2011). In patients that are genetically susceptible to T1D who display increased levels of autoanti-bodies, there were increased levels of IL-1β, and treating peripheral blood mononuclear cells with TLR3 or TLR7 agonists led to increased percentages of IL-1β dendritic cells (Alkanani et al., 2012). As such, it has been shown that in TLR7 deficient mice, there was attenuated diabetic retinopathy (Liao et al., 2017). Taken together, targeting the TLR pathway may provide an opportunity for therapy in patients that may be susceptible to the onset of T1D.
2.3.3. Implication of RIG-I in Crohn’s Disease
RIG-I has also been linked to autoimmune diseases such as inflammatory bowel disease (IBD) that encompasses Crohn’s disease (CD) and ulcerative colitis (UC). Dysfunctions in innate immune response signaling, specifically through RIG-I, have been associated with the development of IBD symptoms. In RIG-I knockout mice, colitis-like phenotypes developed. In these mice, there was a noticeable decrease in the size of Peyer’s patches that are an essential component of immunity as they defend against pathogens in the intestine. Therefore, the observed cellular apoptosis and decreased size of Peyer’s patches in RIG-I deficient mice may increase susceptibility to the colitis-like phenotypes observed. These deficient mice also showed down-regulation of G protein αi2 subunit (Gαi2) in many tissues and negatively regulated T-cell responses (Wang et al., 2007). Gαi2 is necessary for many cellular processes and is a candidate gene associated with the development of human IBD (Hampe et al., 2001b). IBD developed in Gαi2–/– mice and increases in lymphocyte apoptosis lead to a decrease in the size of Peyer’s patches (Ohman et al., 2002). The importance of RIG-I in the development of CD was confirmed in a global gene expression analysis. Gut tissue samples of CD and UC diagnosed patients were obtained. A decrease in transcription of RIG-I in epithelial layer of the ileum was associated with CD patients specifically. These results show that RIG-I is not only important in controlling viral infection, but it is also important in controlling the development of CD (Funke et al., 2011). Lack of tight regulation in RIG-I can lead to changes in Peyer’s patches that increase susceptibility to autoimmune diseases, such as IBD.
An emerging correlation between autophagy regulation and RLR signaling in innate immune responses is important in autoimmune disease pathology of CD. Autophagy and IFN-mediated immunity are meticulously balanced in healthy individuals. Dysfunction in this balance can lead to auto-immune disorders, such as CD (reviewed in Deretic, 2016; Plantinga et al., 2012; Takahama et al., 2018). Autophagy is a degradation process that has been tied to antiviral innate immune responses. Atg5–Atg12 conjugation is a known regulator of autophagy and directly interacted with the CARD domain of RIG-I and MAVS individually. This binding decreases IFN pathway signaling and production. In this study, VSV replication was enhanced by Atg5–Atg12 activity because IFN production was suppressed. Therefore, autophagic regulation is an important indicator for infection outcome. Importantly, Atg5–Atg12 can interact with MAVS even in the absence of viral infection. This means that Atg5–Atg12 has an important role in cellular homeostasis that negatively regulates the IFN response through RIG-I and MAVS signaling in healthy individuals (Jounai et al., 2007). In autophagic deficient cells lacking Atg5, RLR signaling and IFN secretion increase to resist VSV replication. This deficiency also results in increased dysfunctional mitochondrial function and the mitochondrial associated protein, MAVS. Dysfunctional mitochondria enhance reactive oxygen species present in the cell and this further enhances RLR signaling to further amplify IFN signals. Therefore, autophagic signaling is an important regulator of RLR signaling and these data show the important balance between autophagy and IFN-mediated immunity (Tal et al., 2009). NOD2, which is a member of the NOD-like receptor (NLR) family, is a PRR for bacterial lipopolysaccharides (Inohara et al., 2001; Ogura et al., 2001b), and mutations in NOD2 have been associated with the onset of CD (Hampe et al., 2001a; Hugot et al., 2001; Ogura et al., 2001a). NOD2-mediated autophagy has also been proposed as a component of CD onset. NOD2 directly interacts with RIG-I to negatively regulate IFN induction. Three specific NOD2 mutations associated with CD enhance the capacity of NOD2 and RIG-I to negatively signal IFN as compared to wild-type NOD2, while mutations in NOD2 exhibit impaired autophagosome formation. Together, RIG-I signaling with NOD2 not only functions in response to pathogens, but it also influences the onset of CD (Morosky et al., 2011). Negative regulation of IFN via autophagy through RIG-I with MAVS or NOD2 protects healthy individuals from developing autoimmune disorders such as CD, highlighting the interactions among NLRs, RLRs, autophagy and CD (further reviewed in Coutermarsh-Ott et al., 2016; de Bruyn and Vermeire, 2017).
In CD individuals, the balance of autophagy negative regulation and IFN signaling is disrupted. The loss of autophagy regulation has been proposed as a key pathogenesis mechanism in CD. One study found that an important autophagic response pathway is suppressed in CD inflamed epithelial tissue. The EIF2AK4-EIF2A-ATF4 pathway was identified as being important in controlling intracellular replication of adherent-invasive Escherichia coli in leading to robust autophagic gene expression. EIF2AK4 encodes the GCN2 eIF2α kinase, which is activated during amino acid starvation. GCN2 then activates ATF4 and subsequently autophagy (B’Chir et al., 2013). This autophagic pathway is suppressed in inflamed CD tissue and there is not sufficient autophagic induction to control the intracellular E. coli replication. The adherent-invasive E. coli that colonize intestinal mucosa are in high abundance because autophagy is not properly activated thereby leading to increased inflammation in previously inflamed CD epithelial cells. This study highlights the relationships among translational control, autophagy, and inflammatory autoimmunity (Bretin et al., 2016). Autophagy can also be increased when the NF-κB transcription factor is inhibited. Porcine follicle development experiments showed that the follicle stimulating hormone inhibits NF-κB and subsequent IFN induction. This inhibition of NF-κB then led to enhanced autophagic activity through Jun N-terminal kinase signaling (Gao et al., 2016). Meta-analysis of individuals with and without CD linked two gene polymorphisms with disease susceptibility. Polymorphisms in ATG16L1 that limit interaction with Atg12p–Atg5p to induce autophagy signaling were correlated with an increase in an individual’s susceptibility to CD. Mutations in IRGM were also strongly associated with the development of CD and this gene encodes the GTP-binding protein that induces autophagy signaling through RIG-I Polymorphisms in ATG16L1 and IRGM were also documented as being risk factors for UC disease (Palomino-Morales et al., 2009). One of the CD-risk mutations in ATG16L1, T300A, was shown to improve overall survival in colorectal cancer patients. While this mutation was not associated with a change in autophagy, there was increased type I IFN production and sensitivity to dsRNA treatment via MAVS (Grimm et al., 2016). The mechanism behind the control that IRGM has on autophagy was shown to be through the interaction between IRGM and NOD2. In fact, IRGM, NOD2, and ATG16L1 form a complex that regulates the autophagic response to microbes (Chauhan et al., 2015). Together, IRGM confers antimicrobial and anti-inflammatory states that are important in regulating CD.
2.3.4. Singleton–Merten Syndrome, Systemic Lupus Erythematosus, and Prolidase Disorder
Dysfunction in IFN induction through MDA5 and RIG-I has also been linked to Singleton–Merten Syndrome (SMS) (Jang et al., 2015; Rutsch et al., 2015). SMS is a rare autoimmune disorder and classical SMS manifests in many ways such as dental dysplasmia, aortic calcification, glaucoma, and osteopenia. The disorder is known to have autosomal-dominant inheritance but clues about the genetic profile contributing to disease susceptibility have only recently been reported. Mutations in IFIH1 and DDX58, the gene that encodes RIG-I, led to increased induction of IFN-β production and contributed to disease symptoms. The missense mutation in IFIH1 enhanced MDA5 function and contributed to the enhanced IFN-β production in blood and dental tissue (Rutsch et al., 2015). Researchers also identified an atypical SMS phenotype in patients that exhibited variations in the clinical manifestations listed above. Two different variants of DDX58 led to constitutively active functionality of RIG-I that led to uncontrolled IFN induction. The Glu373Ala variant of DDX58 correlated with classical SMS symptoms except there were no dental abnormalities observed in these patients. The patients with the Cys268Phe variant of DDX58 had glaucoma and skeletal abnormalities without dental dysplasmia or aortic calcification (Jang et al., 2015). These genetic analyses show the importance of regulated RIG-I and MDA5 induction because overproduction of IFN-β can lead to the characteristic clinical manifestations of SMS. Another rare autosomal disorder, prolidase disorder (PD), is associated with dysfunction in regulated in IFN induction. Mutations in the gene that encodes prolidase, PEPD, cause PD in newborns that present with dermatological symptoms, that range from rashes to lower extremity ulcers, due to increased immunoglobulin levels and decreased complement factor C1q. (Falik-Zaccai et al., 2010; Kurien et al., 2013; Pandit et al., 2013). Prolidase deficiency is also associated with systemic lupus erythematosus (SLE), an autoinflammatory disorder characterized by the production of antibodies against self-DNA and RNA (Arbuckle et al., 2003; Dubois and Tuffanelli, 1964; Lisnevskaia et al., 2014; Tan et al., 1966), which helps explain the phenotypic similarities between PD and SLE (Butbul Aviel et al., 2012; Kurien et al., 2013). Interestingly, during flaviviral infection, viral NS5 binds PEPD, resulting in decreased expression of the type I IFN receptor. Additionally, PD patients also exhibit decreased type I IFN receptor expression, further linking PD with defects in innate immunity (Lubick et al., 2015).
TLR-mediated detection of nucleic acids has been shown to exacerbate pathogenesis of SLE (Deane et al., 2007). Furthermore, lupus-prone TLR7-deficient mice do not generate antibodies to RNA-containing antigens, thus displaying less severe symptoms and prolonged survival (Christensen et al., 2006). Since the production of autoantibodies against self-nucleic acids is a major driving force behind SLE pathogenesis, it follows that B cells play a major role in the development of SLE (Fan et al., 2018). Indeed, galectin-9, an s-type lectin that induces apoptosis in activated TH1 and TH17 cells while promoting Treg cell differentiation (Wu et al., 2014), inhibits maturation of pDCs and B cells, thereby decreasing cytokine and antibody production in response to TLR7 ligands (Panda et al., 2018). A recent study also showed that polymorphisms in the promoter of TLR7 were associated with SLE (Skonieczna et al., 2018). Finally, while pDCs secrete increased levels of type I IFN in a TLR7-mediated manner in SLE patients (Murayama et al., 2017), lupus nephritis was recently shown to be independent of type I IFN yet remained dependent on TLR7 signaling (Wolf et al., 2018).
Taken together, diabetes, Aicardi–Goutières syndrome, inflammatory bowel diseases, lupus, and Singleton–Merten syndrome have all been linked to dysfunctions in RLR-and TLR-mediated initiation of IFN signaling. These autoimmune disorders demonstrate the important balance of the innate immune response needed to prevent the development of disorders.
3. INTRACELLULAR RECOGNITION OF DNA
Since 2000, multiple research groups have identified at least 15 different DNA binding proteins that induce an innate immune response, referred to as DNA sensors. For example, the DNA-dependent activator of IFN-regulatory factors (DAI) was the first cytosolic DNA sensor identified to bind directly to the Z and B forms of DNA as well as detect herpes simplex virus infection (Furr et al., 2011; Takaoka et al., 2007). When activated, DAI, also known as Z-DNA binding protein 1 (ZBP1), induces receptor-interacting protein kinase 3 (RIPK3)-mediated necroptosis that is inhibited by RIPK1 (Lin et al., 2016; Newton et al., 2016). However, in addition to DAI signaling, two discoveries in the last 10 years have exposed the complexity of DNA-sensing mechanisms as well as highlight the conserved function of different sensors to protect the host from exogenous DNA. The first discovery was the identification of STING (also known as MYPS/ERIS/MITA), encoded by the TMEM173 gene, as a central adaptor protein for immunity to cytosolic nucleic acids and a PRR for cyclic dinucleotides (CDN) (Burdette et al., 2011; Ishikawa and Barber, 2008; Jin et al., 2008; Sun et al., 2009; Zhong et al., 2008). The second discovery came when cGAS was shown to be a direct DNA sensor that synthesizes cGAMP for STING activation (Gao et al., 2013b; Sun et al., 2013; Wu et al., 2013). Several other DNA sensors have been shown to interact with STING signaling such as DDX41, LSm14A, and MRE11 (Kondo et al., 2013; Li et al., 2012b; Zhang et al., 2011c). Other DNA sensors induce an innate immune response independent of STING, such as RNA polymerase III, LRRFIP1, Sox2, Rad50, DHX9/36, and AIM2 (Bürckstümmer et al., 2009; Chiu et al., 2009; Fernandes-Alnemri et al., 2009; Kim et al., 2010b; Roth et al., 2014; Xia et al., 2015; Yang et al., 2010). Additionally, the DNA sensors Ku70 and IFI16 have been shown to activate STING in cell-and pathogen-specific manners (Sui et al., 2017; Unterholzner et al., 2010). It is possible that other DNA sensors will be identified or that previously identified proteins will be shown to serve as DNA sensors whether dependent or independent of STING. Importantly, the existence of diverse DNA sensors, including synthases, DNA repair proteins, helicases, and inflammasomes, indicates that recognition of endogenous and exogenous DNA is a critical component of the host immune response. Below we describe the centrality of the cGAS/STING signaling pathway, the pathways induced by STING-dependent sensors, and lastly the pathways induced by STING-independent sensors (Fig. 2).
Fig. 2.

Sensing of cytosolic DNA or cyclic dinucleotides via multiple sensors leads through the induction of interferon and proinflammatory cytokines. Cytosolic DNA derived from viral or bacterial infection (see Table 3), as well as self-DNA, is sensed by receptors that signal through STING (pink), receptors that are independent of STING (red), and receptors that signal via STING-dependent or -independent mechanisms (blue). STING binds to cyclic dinucleotides, by-products of some bacterial infection and also metabolized by cGAS upon binding to dsDNA. While AIM2 leads to caspase and inflammasome activation, DNA-mediated activation of the other receptors leads to IRF or NF-κB transcription factor activation and the induction of IFN and proinflammatory cytokines. Dysfunction or hyperactivation of these DNA-sensing pathways can lead to autoimmunity (see Table 4).
3.1. A Central DNA-Sensing Pathway via cGAS and STING
The presence of host-or pathogen-derived dsDNA in the cytosol can be recognized by cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS), an enzyme that was recently identified as a critical DNA sensor (Gao et al., 2013b; Li et al., 2013). cGAS is encoded by the MB21D1 gene and single-molecule assays have shown that the N terminus of human cGAS plays an important role in its own activation upon binding nonspecific DNA inducing downstream signaling through STING (Tao et al., 2017). Unlike some DNA sensors that recognize dsDNA in a sequence-specific manner, cGAS activation is independent of the DNA sequence because binding depends on electrostatic and hydrogen bonding interactions between the negative sugar-phosphate backbone of DNA and positive-charged surfaces of cGAS (Civril et al., 2013). Indeed, the crystallization of human cGAS revealed its unique zinc-ribbon motif insertion which provides DNA-binding specificity (Kranzusch et al., 2013). In this manner, cGAS is a general DNA sensor for its unique ability to bind sequence-independent B form dsDNA. In addition, cGAS is unique for its synthase function which catalyzes the production of 2′−3′-cyclic GMP-AMP (cGAMP), the noncanonical CDN after binding DNA (Gao et al., 2013b). cGAMP has been shown to bind to STING to induce an innate immune response that results in the production of type I IFN (Ablasser et al., 2013; Diner et al., 2013; Shang et al., 2012; Sun et al., 2013). Several biochemical studies have also shown that cGAMP contains mixed phosphodiester linkages, a unique distinction from bacterial CDNs, and that the strength of IFN response depends on the phosphodiester link-age. That is, 2′−3′-cGAMP has the strongest affinity for human STING. Nonetheless, the versatility of STING in recognizing CDNs continues to be an area of intense investigation and suggests that some regulation of this pathway depends on binding affinity (Ablasser et al., 2013; Gao et al., 2013c; Opoku-Temeng et al., 2016; Sun et al., 2013; Wu et al., 2013).
Upon binding of cGAMP, STING dimerizes and translocates in autophagosomes to a perinuclear region which is necessary for downstream signaling (Barker et al., 2013; Diner et al., 2013; Ishikawa et al., 2009; Moretti et al., 2017; Sun et al., 2009, 2013). STING is an ER-resident host protein that contains four transmembrane domains and globular carboxy-terminal domain (CTD) that enable the binding of STING to TBK1 (Bhat and Fitzgerald, 2014; Burdette and Vance, 2013). The binding of STING and TBK1 causes autophosphorylation of TBK1 at Serine-172, which then facilitates the direct phosphorylation of STING at its Serine-366 and Leucine-374 residues by TBK1. Consequently, phosphorylation of STING by TBK1 recruits IRF3 to also be phosphorylated by TBK1 (Li et al., 2017c; Liu et al., 2015a; Tanaka and Chen, 2012). The association of TBK1 with STING facilitates the dsDNA-mediated activation of the NF-κB and IRF3 transcription factors. Specifically, TBK1 controls the activation of STING-mediated NF-κB signaling through its IKKαβ activation loop while the kinase domain of the ribosomal protein S6 kinase 1 (S6K1) binds to STING to facilitate the formation of a S6K1-STING-TBK1 complex that is necessary for the phosphorylation of IRF3 (Abe and Barber, 2014; Wang et al., 2016). Additionally, the activation of NF-κB by IKK depends on the phosphorylation of IκBα, which induces its degradation and activates NF-κB (Baeuerle and Baltimore, 1988; Beg and Baldwin, 1993; Israel, 2010; Mathes et al., 2008). Once activated in a cGAS/STING-dependent manner, IRF3 and NF-κB translocate to the nucleus and induce the expression of type I IFN, IL-6, IL-1β, and the production of proinflammatory cytokines like TNF-α (Paludan and Bowie, 2013). Additionally, STING translocation from the ER to an ER-Golgi intermediate compartment and the Golgi apparatus is an important rate-limiting event in signal transduction even in the absence of cGAMP (Chen et al., 2016; Dobbs et al., 2015). A recent study has demonstrated that STING senses bacterial c-di-AMP as a vita-PAMP, a sub-class of PAMPs derived only form living microbes, to induce ER stress and produce IFN (Moretti et al., 2017). This study and others indicate that ER-phagy and autophagy pathways play an important role in STING regulation during bacterial infections (Watson et al., 2015).
The cGAS/STING signaling pathway is induced by a variety of pathogens including DNA viruses, RNA viruses, retroviruses, and bacteria, all of which have evolved mechanisms to avoid detection by this pathway. Through a variety of mechanisms, several DNA viruses such as adenovirus, herpes simplex virus (HSV)-1, and human papilloma virus (HPV) have been shown to induce or actively inhibit a STING-dependent type I IFN response (Anghelina et al., 2016; Ishikawa et al., 2009; Lam and Falck-Pedersen, 2014; Lam et al., 2014; Liang et al., 2015; Sunthamala et al., 2014). For example, over a dozen proteins of HSV-1 have been found to actively suppress cytosolic-DNA recognition by the cGAS/STING pathway (Christensen et al., 2016; Horan et al., 2013; Ishikawa et al., 2009; Kalamvoki and Roizman, 2014; Su and Zheng, 2017; Xu et al., 2017; Ye et al., 2017; Zhang et al., 2016; Zheng, 2018). Similarly, the E2 proteins of HPV16 inhibit the transcription of different ISGs by targeting STING (Sunthamala et al., 2014). Another study has shown that both human and mouse cytomegalovirus (CMV) induce a cGAS/STING-dependent type I IFN response or actively inhibit STING through its UL82 tegument protein or US9 glycoprotein (Choi et al., 2018; Fu et al., 2017; Lio et al., 2016). Several studies have shown that some retroviruses including HIV, murine leukemia virus, and simian immunodeficiency virus can induce a type I IFN response due to recognition of reverse-transcribed DNA by cGAS and subsequent production of cGAMP (Gao et al., 2013a; Lahaye et al., 2013; Rasaiyaah et al., 2013). DENV can antagonize STING signaling by utilizing viral NS2B3 proteases to bind and cleave human STING although this cleavage does not occur in mouse or nonhuman primate STING (Aguirre and Fernandez-Sesma, 2017; Aguirre et al., 2012; Stabell et al., 2018; Yu et al., 2012). HCV has also been shown to antagonize STING signaling, highlighting the multifaceted role of STING in pathogen evasion beyond direct DNA sensing through cGAS (Ding et al., 2013; Maringer and Fernandez-Sesma, 2014; Moriyama et al., 2007; Nitta et al., 2013).
The fact that STING potentiates a type I IFN response in response to bacterial CDNs introduced a role for STING in regulating bacterial infection. First, it was found that synthetic c-di-GMP could induce a type I IFN response (McWhirter et al., 2009). Then, it was found that Listeria monocytogenes secretes c-di-AMP that also induced a type I IFN response (Woodward et al., 2010). Further, a mutant mouse strain called Gold-enticket (Gt) had a null mutation in its STING allele (StingGt/Gt) that rendered it unable to detect c-di-GMP and c-di-AMP from Listeria infection (Sauer et al., 2011). Finally, STING was shown to be a direct sensor of c-di-GMP (Burdette et al., 2011). Since then, studies have shown that STING mediates a type I IFN response specifically in response to CDNs such as those produced by Staphylococcus aureus and Chlamydia trachomatis (Barker et al., 2013; Gries et al., 2016; Zhang et al., 2014). Undoubtedly, the role of STING in detecting CDN-mediated type I IFN response was authenticated by the discovery that cGAS produces cGAMP in a DNA-dependent manner (Sun et al., 2013). Further studies since the discovery of cGAS and STING have further connected the CDN and DNA-mediated activation of this central pathway. In 2013, Chen’s group showed that cGAS–/– macrophages, fibroblasts, and dendritic cells as well as cGAS–/– mice could not produce type I IFNs or cytokines in response to DNA treatment or infection with HSV-1 or vaccinia virus, but infection with Sendai virus, an RNA virus, did induce type I IFN (Li et al., 2013). These infections were further tested in StingGt/Gt mice and cells. Notably, the production of IFN-β in cGAS–/– cells was rescued by the delivery of cGAMP, but not in StingGt/Gt cells, further supporting the role of STING in binding to CDNs. Importantly, this was the first study to use cGAMP as a potential adjuvant in the context of STING. They found that injection of the protein antigen ovalbumin (OVA) in the presence or absence of cGAMP in wild-type or StingGt/Gt mice could boost the development of OVA-specific antibodies in wild-type mice only (Desmet and Ishii, 2012; Li et al., 2013). Clinical trials using STING agonists have followed. For example, a current Phase I clinical study is using the synthetic, STING-activating CDN agonist MIW815 (ADU-S100) to study its safety and efficacy in treating patients with advanced/metastatic solid tumors or lymphomas via intratumoral injections (ClinicalTrials.gov Identifier: NCT02675439). Another study has shown that c-di-GMP can be complexed with simple cell-penetrating peptides that enhance cellular delivery and biological activity in murine splenocytes but also that the bacterial 3′−3′-cGAMP is a superior stimulator of IFN genes ligand than c-di-GMP in human peripheral blood mononuclear cells (Yildiz et al., 2015). A more recent study has shown that expression of an inducible c-di-GMP-producing diguanylate cyclase in Klebsiella pneumonia increased endogenous concentrations of c-di-GMP which attenuated its virulence in the lung of mice (Rosen et al., 2017). Importantly, the attenuation of virulence was independent of STING, supporting the idea that CDNs can be used to influence pathogen virulence (Karaolis et al., 2007a,b; Rosen et al., 2017). Indeed, the use of CDNs as adjuvants has increased in the last few years and will likely continue to expand as more pathogens and diseases are studied under this framework (Dubensky et al., 2013; Junkins et al., 2018; Karaolis et al., 2007a; Miyabe et al., 2014; Škrnjug et al., 2014).
As seen by the diversity of pathogens that cGAS and STING signaling are able to regulate, the centrality of this pathway is evident. Many DNA sensors had been identified before cGAS, but our understanding of host defense mechanisms has greatly increased due to the unique synthase function of cGAS in producing cGAMP in response to the presence of viral DNA, thus acting as an amplifier of cGAS-mediated DNA sensing. In addition, the centrality of STING is highlighted by its ability to use host-derived CDN, 2′−3′-cGAMP, or bacterial CDNs like 3′−3′-cGAMP, c-di-GMP, and c-di-AMP to an innate immune response. Indeed, several groups are investigating the potential use of CDNs as adjuvants in treating human diseases but also in controlling bacterial factors like virulence. Other groups are investigating novels ways, such as ER-phagy, that STING induces an innate immune response. All of these ongoing studies reflect the centrality of STING in the innate immune response to cytosolic nucleic acids (Table 3).
Table 3.
Summary of Host Proteins That Sense DNA Virus or Bacterial Infections
| DNA Sensor | DNA Sensor Associated Protein | Pathogens That Stimulate Sensor |
|---|---|---|
| cGAS | STING | • Adenovirus (AdV) (Anghelina et al., 2016; Ishikawa et al., 2009; Lam and Falck-Pedersen, 2014; Lam et al., 2014) • Herpes simplex virus 1 (HSV-1) (Christensen et al., 2016; Horan et al., 2013; Ishikawa et al., 2009; Kalamvoki and Roizman, 2014; Li et al., 2013; Su and Zheng, 2017; Xu et al., 2017; Ye et al., 2017; Zhang et al., 2016; Zheng, 2018) • Human papillomavirus (HPV) (Sunthamala et al., 2014) • Human and mouse cytomegalovirus (HCMV or MCMV) (Choi et al., 2018; Fu et al., 2017; Lio et al., 2016) • Human immunodeficiency (HIV) (Gao et al., 2013a; Lahaye et al., 2013; Rasaiyaah et al., 2013) • Dengue virus (DENV) (Aguirre and Fernandez-Sesma, 2017; Aguirre et al., 2012; Stabell et al., 2018; Yu et al., 2012) • Hepatitis C virus (HCV) (Ding et al., 2013; Maringer and Fernandez-Sesma, 2014; Moriyama et al., 2007; Nitta et al., 2013) • Listeria monocytogenes (Sauer et al., 2011; Woodward et al., 2010) • Staphylococcus aureus (Gries et al., 2016; Zhang et al., 2014) • Chlamydia trachomatis (Barker et al., 2013) |
| DDX41 | STING | • HSV-1 (Zhang et al., 2011c) • AdV (Zhang et al., 2011c) • Newcastle disease virus (NDV) (Cheng et al., 2017) • Listeria monocytogenes (Parvatiyar et al., 2012) |
| DAI | STING | • HSV-1 (Furr et al., 2011) |
| IFI16 | 1. STING 2. ASC |
• (1) Epstein-Barr virus (EBV) (Pisano et al., 2017) • (1) HSV-1 ( Johnson et al., 2014; Merkl et al., 2018; Orzalli et al., 2012; Unterholzner et al., 2010) • (2) Kaposi’s sarcoma-associated herpesvirus (KSHV) (Kerur et al., 2011) |
| Ku70 | STING-dependent or independent | • Hepatitis B virus (HBV) (Li et al., 2016) • Human T lymphotropic virus type 1 (HTLV-1) (Wang et al., 2017) • HSV-2 (Zhang et al., 2011a) |
| MRE11 | STING | • MRE11 is not required or induce a type I IFN response to HSV-1 or Listeria monocytogenes infection but plays a role in DNA damage recognition (Kondo et al., 2013) |
| LSm14A | STING | • HSV-1 (Li et al., 2012b) |
| RNA pol. III | RIG-I/MAVS | • Legionella pneumophila (Chiu et al., 2009) • EBV (Ablasser et al., 2009) • Invertebrate iridescent virus 6 (IIV-6) (Ahlers et al., 2016) • Varicella zoster virus (VZV) (Ogunjimi et al., 2017) |
| LRRFIP1 | β-Catenin | • Vesicular stomatitis virus (VSV) (Yang et al., 2010) • Listeria monocytogenes (Yang et al., 2010) |
| Sox2 | TAB2, TAK1 | • HPV (Martınez-Ramırez et al., 2017) |
| DHX9/36 | MyD88 | • HSV-1 (Kim et al., 2010a) |
| Rad50 | CARD9 | • Vaccinia virus (VV) (Alcamí and Smith, 1996; Roth et al., 2014) • AdV (Pancholi and Weitzman, 2018; Stracker et al., 2002) |
| AIM2 | ASC | • Francisella tularensis (Fernandes-Alnemri et al., 2010; Jones et al., 2010) • Listeria monocytogenes (Kim et al., 2010a,b) • Streptococcus pneumonia, Mycobacterium tuberculosis, Legionella pneumophila, Staphylococcus aureus (Fang et al., 2011; Ge et al., 2012; Hanamsagar et al., 2014; Saiga et al., 2012) • Francisella novicida (Meunier et al., 2015) • Chlamydia muridarum, Chlamydia trachomatis (Finethy et al., 2015) • MCMV (Shi et al., 2015) • HPV (Milutin Gasperov et al., 2014) • Influenza virus (Schattgen et al., 2016; Zhang et al., 2017a) |
| TLR9 | MyD88 | • HSV-1/2 (Krug et al., 2004; Lundberg et al., 2003) • Campylobacter jejuni, Klebsiella pneumonia, Staphylococcus aureus (Bhan et al., 2007; Dalpke et al., 2006) • VZV (Yu et al., 2011) • HCMV (Varani et al., 2007) • MCMV (Krug et al., 2004; Puttur et al., 2016) • EBV (Fiola et al., 2010; Lim et al., 2006) • KSHV (West et al., 2011) • VV and Ectromelia virus (Samuelsson et al., 2008) • AdV (Appledorn et al., 2008; Basner-Tschakarjan et al., 2006) |
3.2. STING-Dependent Sensors: DDX41, Ku70, MRE11, and LSm14A
3.2.1. DDX41
DDX41 is a member of the DEXDc helicase family that was recently identified as a DNA sensor in myeloid dendritic cells (mDCs). Knockdown of DDX41 by short-hairpin RNA prevents induction of type I IFN response via IRF3 and NF-κB activation after treatment with DNA or viral DNA infection but not RNA (Fullam and Schroöder, 2013; Jiang et al., 2017; Zhang et al., 2011c). DDX41 can also bind directly to bacterial CDNs after which downstream signaling occurs via the recruitment of STING and TBK1 to activate IRF3 (Omura et al., 2016; Parvatiyar et al., 2012). Two residues of DDX41 (Tyr364 and Tyr414) are necessary for its recognition of DNA, and activation of DDX41 depends on phosphorylation of its Tyr414 residue by BTK (Bruton’s tyrosine kinase), which then facilitates binding to STING (Lee et al., 2015). It is also known that the E3 ligase TRIM21 interacts with the DEAD domain of DDX41 and that DDX41 is subsequently degraded via ubiquitination of its Lys9 and Lys115 residues. These results were corroborated by the observation that knockdown of TRIM21 results in over-induction of type I IFN and overexpression of TRIM21 results in reduced IFN-β production (Zhang et al., 2012). One study showed that TBK1, NF-κB, and IRF3 activation are mediated by direct interaction of DDX41 with STING in dendritic cells, bone marrow-derived DCs, and human monocytes after infection with HSV-1 or adenovirus (Zhang et al., 2011c). This study found that knockdown of DDX41 or STING in THP-1 cells resulted in lower IFN-β production after DNA treatment or infection with HSV-1. Another study identified chicken DDX41 as a DNA sensor which results in the production of IFN-β in a STING-dependent manner after DNA treatment or infection with Newcastle disease virus (Cheng et al., 2017). This study used a ssRNA virus to show the role of DDX41 in mediating an innate immune response; however, it lacked experiments investigating the role of DDX41 in the context of DNA viruses and bacterial infection (Jiang et al., 2017). Nonetheless, it is important to note that DDX41 can bind to CDNs such as those produced by L. monocytogenes (Parvatiyar et al., 2012). Both DNA-and CDN-sensing roles of DDX41 indicate that it may detect a variety of intracellular viruses and bacteria.
3.2.2. Ku70
Ku70 is a DNA repair subunit protein that binds to DNA double-strand break ends and helps repair DNA via the non-homologous end-joining (NHEJ) pathway (Mimori et al., 1986). Recently, Ku70 was identified as a cytosolic DNA sensor that induces the production of IFN-λ1, a type III IFN, in primary human cell lines after viral infection after translocating from nucleus to cytosol to interact with STING (Sui et al., 2017; Zhang et al., 2011a). IRF1, IRF3, and IRF7 are implicated in this Ku70/STING pathway. One study has found that the Ku70/80 complex can directly sense hepatitis B virus (HBV) DNA which results in recruitment of PARP1, activation and translocation of IRF1 to the nucleus, and upregulation of chemokine secretion (Li et al., 2016). The relevance of this study is that it shows the role of Ku70 in mediating an innate immune response of a DNA virus. Similarly, another study has found that Ku70 can sense the DNA of human T lymphotropic virus type 1 (HTLV-1). Specifically, this study also found that knockdown of Ku70 led to decreased IRF3 phos-phorylation and induction of IFN-β, TNF-α, and IFN-stimulated gene 56 (ISG56) (Wang et al., 2017). Importantly, Ku70 was shown to associate directly with STING to produce IFN-β. While HTLV-1 is a retrovirus, the involvement of Ku70 in detecting viral DNA indicates that Ku70 is an important DNA sensor that may act as reverse transcription intermediate and its role in inducing an innate immune response awaits further investigation. Other studies have shown that Ku70 induces a type III IFN response after HSV-2 infection independently of STING (Zhang et al., 2011a). Nonetheless, the binding of Ku70 to STING as well as mediating an IFN-λ1 response to cytosolic DNA and DNA viruses highlights ability of Ku70 to function both independently and dependently of STING.
3.2.3. MRE11
Meiotic recombination 11 homolog A (MRE11) is an exonuclease better known for its role in microhomology-mediated end-joining (MMEJ). Over-expression of MRE11 has been shown to cause mutations that lead to breast cancer (Sharma et al., 2015; Spehalski et al., 2017; Yuan et al., 2012). One study has shown that MRE11 can physically interact with cytosolic DNA, trigger STING translocation, and subsequently activate IRF3 via interaction with Rad50, another DNA repair protein. However, this response was not observed during infection with Listeria or HSV-1, suggesting that MRE11 may induce a type I IFN response specifically to DNA damage instead of pathogen defense (Kondo et al., 2013). The MRE11/Rad50/NBS1 (MRN) complex is known for its conserved role in DNA repair (Maser et al., 1997), yet in Kondo et al., NBS1 was shown to be dispensable for inducing STING trafficking upon treatment with exogenous DNA. An important function of the MRN complex is to induce a DNA damage response (DDR) from stimuli that results in DNA damage (Chapman and Jackson, 2008; He et al., 2012; Paull and Deshpande, 2014). How viruses persist despite active DDR remains an unanswered question. For example, how is HPV able to establish infection despite inducing a DDR, presumably when it causes double-stranded breaks during its replication cycle (Bristol et al., 2017; Kadaja et al., 2009). Therefore, a survey of the MRE11-STING pathway with multiple pathogens would help clarify the role of MRE11 in inducing an innate immune response via STING.
3.2.4. LSm14A
LSm14A is a processing body-associated sensor of viral RNA and DNA that was recently shown to induce a specific type I IFN response via induction of IFN-α, IFN-β, and IL-6 through regulation of STING activation in mouse dendritic cells but not mouse embryonic fibroblasts or macrophages. This indicates that LSm14A is a cell-specific sensor of DNA that functions through nuclear mRNA processing. Further investigation into this DNA sensor will clarify its role in DNA-sensing and how it may function. Currently, only HSV-1 has been shown to induce an innate immune response through LSm14A and STING (Li et al., 2012b; Liu et al., 2016).
3.2. STING-Independent DNA Sensors: TLR9, RNA Polymerase III, DHX9/DHX36, AIM2, IFI16, Sox2, LRRFIP1, and Rad50
3.3.1. TLR9
TLR9 was first identified by its ability to recognize unmethylated 2′-deoxyribo(cytidine-phosphate-guanosine) (CpG) DNA from bacteria, and viral DNA is also recognized by TLR9 (Bauer et al., 2001; Hemmi et al., 2000; Hochrein et al., 2004; Tabeta et al., 2004). Additionally, TLR9 detects ssDNA of at least 21 nucleotides, and methylated ssDNA or dsDNA only weakly activate TLR9 (Pohar et al., 2015a,b). However, the addition of oligonucleotides as short as two nucleotides augments TLR activation (Pohar et al., 2017). In support of this, it has been shown that the endonuclease DNaseII is also required for a robust TLR9-mediated response (Chan et al., 2015). Preference for microbial DNA rather than self-DNA is due to TLR9’s location in the endolysosomes (Li et al., 2012a). TLR9 is localized in intracellular vesicles of the ER, lysosomes, and endosomes of resting cells. DNA recognition by TLR9 occurs after transport to endolysosomes, which is mediated by the multi-spanning protein UNC93B (Kawasaki et al., 2011; Latz et al., 2007; Tabeta et al., 2004, 2006). UNC93B physically interacts with TLR9 through its transmembrane domain 2 where the ectodomain of TLR9 is processed by cathepsins to become active (Ewald et al., 2011; Latz et al., 2007; Park et al., 2008; Sepulveda et al., 2009; Tabeta et al., 2006). TLR9 contains a large nonconserved Z-loop between LRR14 and LRR15 that is susceptible to cathepsin-mediated proteolysis. While processing of the Z-loop is required for subsequent TLR9 oligomerization, DNA binding to TLR9 is independent of the Z-loop (Ewald et al., 2011; Li et al., 2012a; Ohto et al., 2015). The chromatin protein HMGB1 (high mobility group box 1) enhances DNA recognition by TLR9 by bending it and bringing it closer to the RAGE (receptor for advanced glycation endproducts) receptor and delivering the HMGB1-DNA complex to early endosomes (Li et al., 2012a; Murugesapillai et al., 2017; Tian et al., 2007). Additionally, it has been recently shown that TLR9 has two DNA-binding sites, namely CpG and 5′-TCG binding sites, both of which contribute to the dimerization of TLR9 (Ohto et al., 2018). Additional activation of TLR9 is enhanced by endosome maturation and acidification (Latz et al., 2004; Wagner, 2004; Yasuda et al., 2005). After DNA binds to TLR9, there is subsequent induction of an innate immune response via two mechanisms. The first is through activation of NF-κB-dependent proinflammatory cytokines, and the second is through IRF7-dependent type I IFN induction. While both pathways are mediated by the adapter protein MyD88 (myeloid differentiation primary response 88) and the TNFR-associated factor TRAF6, IRF7-dependent IFN-α production requires TLR9 trafficking from endosomes to endolysosomes which is mediated by adaptor protein 3 (AP3) (Gohda et al., 2004; Medzhitov et al., 1998; Sasai et al., 2010). In contrast, TLR9 that is transported by UNC93B to the early endosome is cleaved and signals through NF-κB to induce proinflammatory cytokine genes that encode TNF-α, IL-6, and IL-12 (Sasai et al., 2010).
Pathogen-specific studies investigating the mechanism by which TLR9 induces an innate immune response were driven by the discovery that TLR9 recognizes unmethylated CpGs and that human CpGs are methylated and therefore immunologically inert. For example, the genome of HSV-1/2 is heavily unmethylated and CpG-rich, and thus induces a TLR9-dependent immune response in murine bone marrow-derived macrophages (BMDMs) (Lundberg et al., 2003). Bacteria are also capable of activating a TLR9-dependent innate immune response. For example, bacterial species such as Campylobacter jejuni, K. pneumonia, and S. aureus were shown to activate TLR9 based on the abundance of [CG] content. That is, higher CG base content resulted in increased activation of TLR9 and IL-8 production (Dalpke et al., 2006). One study has demonstrated that TLR9−/− mice infected with K. pneumonia had impaired activation and maturation of dendritic cells, reduced TNF-α induction, and lower production of TNF-α and IL-12. Interestingly, when dendritic cells from wild-type mice were intratracheally transferred into TLR9−/− mice, bacterial load was significantly reduced and TNF-α and IL-12 cytokine production increased (Bhan et al., 2007). Of note, human TLR9 is predominantly expressed in plasmacytoid dendritic cells and B cells, resulting in a strong type I IFN response via MyD88/IRF7 signaling. Conversely, murine TLR9 is expressed abundantly in myeloid immune cells and can also result in IFN-γ production and the recruitment of NK, αβ-, and γδ-T cells (Hartmann, 2017; Hornung et al., 2002; Krug et al., 2004; Walker et al., 2010). For example, it has been shown that CMV infection in mice activates a TLR9/MyD88 response that occurs selectively in CD11+ dendritic cells (Krug et al., 2004; Puttur et al., 2016). Other DNA viruses that have been implicated in activating an innate immune response through TLR9 are Varicella zoster virus (VZV), Epstein-Barr virus (EBV), Kaposi sarcoma-associated herpesvirus (KSHV), vaccinia virus (VV), adenovirus (AdV), and human CMV (Appledorn et al., 2008; Basner-Tschakarjan et al., 2006; Fiola et al., 2010; Lim et al., 2006; Samuelsson et al., 2008; Varani et al., 2007; West et al., 2011; Yu et al., 2011). However, the majority of viruses will result in the activation of lymphocytes that can then initiate adaptive responses that differ between mouse and human models of infection. These species-specific differences highlight the importance of understanding how evolution of host defense mechanisms relates to TLR9-mediated immune responses.
3.3.2. RNA Polymerase III
RNA polymerase III transcribes AT-rich dsDNA into an RNA-containing 5′-triosphate moiety which can then be recognized by RIG-I (retinoic acid-inducible gene I) and induce a type I IFN response through IRF3, IRF7, and NF-κB (Ablasser et al., 2009; Chiu et al., 2009). The observation that RNA polymerase III can sense cytosolic DNA has helped researchers investigate the complex ways that pathogens avoid recognition by the innate immune system (Bauernfeind et al., 2010). While Chen’s group showed that inhibition of RNA polymerase III resulted in abrogated IFN-β production from Legionella pneumophila infection, Hornung’s group found that inhibition of RNA polymerase III resulted in abrogated IFN-α production from EBV infection (Ablasser et al., 2009, Chiu et al., 2009). Another study demonstrated that during infection with invertebrate iridescent virus 6 (IIV-6), an insect DNA virus, the mammalian host requires RNA polymerase III to produce IFN-β (Ahlers et al., 2016). Through whole exome sequencing of 21 human patients, a recent study found that different mutations in the RNA polymerase III gene can explain why some patients suffer from severe acute VZV infection (Ogunjimi et al., 2017). In contrast, sequencing of 222 patients suffering from herpesviral encephalitis did not result in identification of RNA polymerase III mutations, which may be explained by the presence of high AT base content in several genome islands of VZV and overall lower cGAS expression in blood cells (Gram et al., 2017; Ogunjimi et al., 2017). Together, these studies indicate the important role of RNA polymerase III in mediating an innate immune response to DNA viruses.
3.3.3. DHX9/DHX36
DEAH box proteins 9 and 36 (DHX9 and DHX36) are cytosolic helicases that were found to bind to CpG viral DNAs but not RNAs in human primary dendritic cells (Kim et al., 2010b). Specifically, DHX9 and DHX36 sense CpG-oligodeoxynucleotides, CpG-B and CpG-A, respectively, through the adaptor protein MyD88. DHX9 and DHX36 both bind directly to the TIR domain of MyD88 independent of TLR9 signaling (Hochrein et al., 2004; Hokeness-Antonelli et al., 2007; Kim et al., 2010b; Ohnishi et al., 2009). Cytosolic binding of DHX9/DHX36 to CpG-B/A is corroborated by fractionation experiments that showed DHX9/36 are not present in any endosomal structures (Kim et al., 2010b). After binding to MyD88, DHX9 activates NF-κB, which then induces TNF-α and IL-6 production while DHX36 activates IRF7 and induces high IFN-α production. Interestingly, DHX9 and DHX36 were first identified as RNA helicases as they are part of the DExD/H box family, including proteins that have critical roles in RNA metabolism. However, DHX9 and DHX36 have also been shown to act as DNA helicases (Linder, 2006; Zhou et al., 2003). The role of DHX9 as an RNA or DNA helicase has been studied in the context of cancer such as colorectal and lung cancer, where DHX9 regulation is cancer-specific (He et al., 2017; Mi et al., 2016; Rahman et al., 2017; Sun et al., 2014). It is also known that DHX9 binds to viral dsRNA in myeloid dendritic cells, leading to the activation of NF-κB and IRF3, along with the production of IFN-α/β (Zhang et al., 2011d). Similarly, DHX36 has been shown to form a complex with DDX1 and DDX21 to function as a dsRNA sensor that uses the TRIF adaptor molecule to activate NF-κB and a type I IFN response in dendritic cells (Zhang et al., 2011b). As DNA sensors, DHX9/36 proteins have shown to mediate an innate immune response to HSV but not influenza virus (Kim et al., 2010b). These and other studies evidence suggests that the function of RNA and DNA sensing in DHX proteins depends both on cell type and viral genome that highlights the need for more in-depth investigation in this family of proteins.
3.3.4. AIM2 and IFI16
Cytosolic DNA can also bind directly to the AIM2 (absent in melanoma 2) inflammasome protein and activate ASC (apoptosis-associated speck-like protein containing a CARD), a critical component of the inflammasome complex (Fernandes-Alnemri et al., 2007, 2009; Muruve et al., 2008). AIM2 is member of an IFN-inducible HIN-200 family of proteins that contains an N-terminal pyrin domain and a C-terminal oligonucleotide/oligosaccharide-binding domain (Ludlow et al., 2005). Recruitment and activation of caspase-1 by AIM2 depends on the pyrin domain and direct interaction with the adaptor protein ASC. Direct binding of cytoplasmic DNA to AIM2 results in oligomerization which results in the formation of the oligomeric ASC pyroptosome that is required for caspase-1-dependent inflammatory cell death known as pyroptosis (Fernandes-Alnemri et al., 2007, 2009). While other inflammasome proteins such as NALP3 belong to the NLR protein family, AIM2 is not an NLR due to its unique pyrin domain needed for binding of host and pathogen-derived DNA in the cytosol which was first found to directly activate the inflammasome complex independent of NALP3 (Buürckstuümmer et al., 2009; Fernandes-Alnemri et al., 2009; Halle et al., 2008; Pétrilli et al., 2007). Nevertheless, AIM2-dependent DNA binding also results in the maturation of proinflammatory cytokines like pro-IL-1β and pro-IL-18 into their active and secreted forms, IL-1β and IL-18 (Muruve et al., 2008; Pétrilli et al., 2007). A recent study has shown that AIM2-like receptor knockout mice respond differently to endogenous retroviral DNA which suggests that AIM2-like receptors might have a more important role in sensing endogenous DNA (Nakaya et al., 2017).
Several bacterial species activate the AIM2 receptor to induce an inflammasome-mediated immune response, such as Francisella tularensis and L. monocytogenes (Fernandes-Alnemri et al., 2010; Jones et al., 2010; Kim et al., 2010a). Listeria is known to activate several innate immune pathways, but in the context of a inflammasome-dependent caspase-1 activation response, AIM2 compensates for an inflammasome response in NLRC4-and NLRP3-deficient mouse macrophages (Kim et al., 2010a). Other bacterial species that have been shown to activate AIM2 include Streptococcus pneumonia, some Mycobacterium species, L. pneumophila, and S. aureus (Fang et al., 2011; Ge et al., 2012; Hanamsagar et al., 2014; Saiga et al., 2012). The disruption of bacterial vacuoles allows bacterial DNA to be recognized by AIM2. For example, Francisella novicida infection increases the expression of IRF1 which then induces the expression and activation of IFN-inducible GTPases called guanylate-binding proteins (GBPs) to disrupt the bacterial vacuole (Meunier et al., 2015). GBPs colocalize with the IFN-inducible protein IRGB10 to cause bacteriolysis and ultimate exposure of DNA that can be recognized by AIM2 (Man et al., 2015). Recently, Chlamydia muridarum and trachomatis have also been shown to employ GBPs to induce caspase-1 and caspase-11-mediated inflammasome responses via NLRP3 and AIM2 (Finethy et al., 2015). In contrast, the role of AIM2 in detecting viral DNA is much less explored. For example, mouse CMV and HPV have been shown to activate an AIM2-mediated inflammasome assembly but the mechanism whereby viral DNA is exposed in the cytosol remains unclear (Milutin Gašperov et al., 2014; Shi et al., 2015). Interestingly, recent studies have showed that influenza virus can induce lung damage that results in the release of host DNA and subsequent AIM2-mediated activation (Schattgen et al., 2016; Zhang et al., 2017a). However, Zhang et al. showed that AIM2-deficient human and mouse cells are still able to induce caspase-1 activation, which may be due to difference in viral dose, virus propagation source, or difference between cell culture and in vivo models. It will be interesting to see how further research into different host cell types, in vivo models and accessory proteins that mediate AIM2-dependent activation of caspase-1 will clarify pathogen and host-specific differences.
Another DNA sensor closely related to AIM2 is IFI16 (gamma-IFN-inducible protein Ifi-16 or IFN-inducible myeloid differentiation transcriptional activator), which is part of the pyrin and HIN-200 domain-containing protein family. In contrast to AIM2, which induces the assembly of an inflammasome complex, IFI16 is known to induce a type I IFN response upon binding to intracellular dsDNA (Trapani et al., 1994; Unterholzner et al., 2010). Knockdown of IFI16 has shown that it is required to maintain EBV latency in Akata cells, an EBV-producing cell line (Pisano et al., 2017). A more recent study has shown that IFI16-depleted human foreskin fibroblasts display increased replication of HSV-1 as well as expression of HSV-1 immediate-early, early, and late proteins, which confirms previous studies using siRNA to knockdown IFI16 ( Johnson et al., 2014; Merkl et al., 2018). Although IFI16 is thought to induce a type I IFN response through STING, one study also links IFI16 to caspase-1 and ASC induction in response to KSHV, showing that the inflammasome also functions in the nucleus (Kerur et al., 2011). However, it has been shown that in primary fibroblasts, IFI16 is not required for an immune response to human CMV and that the AIM-like receptors are unnecessary for the response to immunostimulatory DNA (Gray et al., 2016). Further research into the specific pathway in which IFI16 mediates an innate immune response to different pathogens might indeed show more similarities between AIM2 and IFI16 as inflammasome assemblers and their specific roles in a cytosolic DNA-mediated immune response.
3.3.5. Sox2, LRRFIP1, and Rad50
Sex determining region Y-box 2 (Sox2) is a transcription factor that is expressed in the cytosol of neutrophils and that was recently discovered to bind bacterial DNA in a sequence-specific manner (Xia et al., 2015). Specifically, upon bacterial DNA stimulation, Sox2 interacts with TAB2, which causes the TAB2/TAK1 kinase complex to dimerize and lead to the activation of NF-κB and AP-1 signaling. A recent study analyzed the long control regions, which are important sites of viral replication regulation for HPV16, and found that overexpression that Sox2 can repress the E6 and E7 oncogene expression of HPV16 (Martínez-Ramírez et al., 2017). Although this study did not investigate innate immune induction by Sox2, this study highlights the role of Sox2 in mediating a DNA virus infection and will need further investigation to clarify its mechanism. While further research is needed to understand the DNA sensing pathway by Sox2 in humans, the discovery of Sox2 as a DNA sensor also highlights the conserved sequence-specific recognition system of foreign DNA used by eubacteria and archaea (Jinek et al., 2012; Xia et al., 2016).
Leucine-rich repeat flightless-interacting protein 1 (LRRFIP1) is a transcriptional repressor that binds to a GC-rich consensus sequence (5-AGCCCCCGGCG-3) and is thought to control smooth cell proliferation via platelet-derived growth factor repression, and positively regulate TLR signaling (Choe et al., 2013; Dai et al., 2009; Labbé et al., 2017). One study showed that LRRFIP1 can mediate a type I IFN response to VSV and Listeria infection via a β-catenin-dependent pathway (Yang et al., 2010). Specifically, this study showed that LRRFIP1 interacts and promotes β-catenin activation, resulting in the binding of β-catenin to IRF3. The LRRFIP1-β-catenin interaction results in the recruitment of the acetyltransferase p300 to the IFN-β-enhanceosome through IRF3. Importantly, Yang et al. showed that knockdown of LRRFIP1 decreased the expression of IFN-β mRNA induced by bacterial DNA in the cytosol but not by extracellular LPS. Similarly, the presence of LRRFIP1 increased production of IFN-β following Listeria and VSV infection. While further studies have examined the role of LRRFIP1 in inducing a type I IFN response during RNA virus infection, additional studies investigating the role of LRRFIP1 in sensing cytosolic DNA would be beneficial (Bagashev et al., 2010; Liu et al., 2015b). For example, while the crystal structure of LRRFIP1 shows that it remains highly extended while bound to DNA but aggregates at high concentrations of DNA (Nguyen and Modis, 2013), further studies examining the conformational changes of LRRFI1P1 from different DNA pathogens will help elucidate how DNA-binding mediates signaling through LRRFIP1.
As described previously, Rad50 is an important DNA repair protein in the MRN complex. Recently, a study has shown that Rad50 directly interacts with DNA to form a complex with the adaptor protein CARD9 and induce an immune response resulting in the production of IL-1β (Roth et al., 2014; Zhong et al., 2018). These groups observed that delivery of dsDNA or infection with vaccinia virus resulted in recruitment of Rad50 binds which recruited CARD9 and then the B-cell lymphoma/leukemia 10 protein (Bcl10) is recruited through its CARD-binding domain, the entire complex being necessary to activate NF-κB, increased transcription of pro-IL-1β, and IL-1β production. Importantly, this study also showed that activation of NF-κB and subsequent proinflammatory response was independent of an inflammasome-mediated immune response or type I IFN response via STING as seen by unaffected levels of caspase-1 and IRF3. The implications of this study highlight the importance of the MRN complex in sensing DNA and inducing an innate immune response through STING-dependent and independent mechanisms. Specifically, many viruses have developed strategies to avoid Rad50-mediated NF-κB activation and subsequent IL-1β production. For example, adenovirus is known to inhibit Rad50 signaling while several vaccinia virus strains produce a soluble IL-1 receptor that can bind the host-produce IL-1β and prevent fever (Alcamí and Smith, 1996; Stracker et al., 2002). A recent study has shown that even different serotypes of adenovirus have different effects in their mechanism of inhibiting MRN/ATM activation (Pancholi and Weitzman, 2018). Further investigation into the MRN proteins like MRE11 and Rad50 will help elucidate the pathogen-specific mechanisms by which host innate immune response is blocked by DNA viruses as well as create the possibility of viral targets.
In summary, a number of DNA-sensing proteins have been identified over the past decade: TLR9, DAI, AIM2, RNA polymerase III, LRRFIP1, DHX9/DHX36, IFI16, Ku70, DDX41, Sox2, and cGAS. Notably, DAI has recently been shown to recognize viral RNA in addition to its role in DNA sensing to induce necroptosis (Maelfait et al., 2017; Thapa et al., 2016). The ability of STING to potentiate signals from many of these DNA sensors and bind CDNs places it central to the DNA sensing pathway for viral and bacterial infections. Indeed, the wide diversity of proteins that can recognize DNA and induce an innate immune response shows that host defense mechanisms have a critical role in promoting protection and host survival. When these DNA-sensing mechanisms are dysfunctional, they can result in autoimmune and autoinflammatory disorders, a significant area of research driven by the need to discover new therapeutic targets. The next section will discuss autoimmune and autoinflammatory disorders in the context of DNA-sensing.
3.4. Autoimmune and Autoinflammatory Disorders Derived From Immune System Dysfunction
A critical application of innate and adaptive immunity in the context of human disease has been to dissect how autoimmune and autoinflammatory disorders occur as a result of immune system dysfunction. Autoinflammatory disorders are generally driven by innate immune components and do not actively rely on adaptive immune components. In contrast, autoimmunity generally refers to disorders that originate from defects in the innate immune system but require adaptive immune components such as lymphocyte influx or T and B-cell responses. A classic group of autoinflammatory disorders are the cryopyrin-associated periodic syndromes (CAPS), also known as familial cold autoinflammatory syndrome (FCAS), which result from gain-of-function mutations in the NLRP3/CIAS1 gene (Cordero et al., 2018; Hoffman et al., 2001; Kanneganti et al., 2006; Li et al., 2017a). On the other hand, classic autoimmune disorders include Aicardi–Goutières syndrome (AGS), and although several mutations in AGS patients are rooted in innate immune dysfunction, AGS has both autoimmune and autoinflammatory components. Differences in disease progression have been observed between human and mouse models, illustrating species-specific differences in these disease phenotypes. In characterizing autoimmune and autoinflammatory diseases, it is important to dissect the origins of disease. Our understanding of autoimmune disorders relies on our knowledge of innate immune recognition proteins such as the DNA sensors described above. While not all autoimmune/inflammatory disorders have a common DNA-sensing origin, this section will focus on disorders that originate in failures of DNA sensing pathways.
3.5. DNA-Sensing Molecular Mechanisms of Diseases and Nucleic Acid Accumulation
A group of disorders that result from errors in the innate immune system that result in constitutive activation of type I IFN signaling are collectively termed type I interferonopathies. While there are different causes of type I interferonopathies, two classes can summarize the majority of those derived from errors in DNA-sensing proteins with an end result of unregulated production of IFNs. The first derives from mutations in genes encoding enzymes that regulate accumulation of nucleic acids that ultimately feed into the cGAS/STING pathway. The second results from constitutive activation or hypersensitivity of the cGAS/STING and RLR pathways. In the next subsections we describe how nucleic acid accumulation through mutations or dysfunction of genes encoding the proteins TREX1, RNase H2, SAMHD1, and STING results in autoimmune/autoinflammatory diseases such as AGS, SLE, Sjögren’s syndrome, STING-associated vasculopathy with onset in infancy (SAVI), and familial chilblain lupus (Table 4).
Table 4.
Summary of Polymorphisms in DNA Sensors and Their Associated Syndromes
| Protein | Genetic Polymorphisms That Lead to Autoimmunity/Autoinflammation | Associated Syndrome/Pathology |
|---|---|---|
| TLR9 | 1. Loss of TLR9 leads to increased lymphocyte activation, type I IFN, and autoantibodies (Christensen et al., 2006) 2. Impaired TLR9 signaling in B cells with decreased CD19/21 (Gies et al., 2018) |
1–2. Systemic lupus erythematosus |
| TREX1 | 1. Loss-of-function mutations in TREX1 of AGS patients (Abe et al., 2014; Bailey et al., 2012; Crow, 2011; Crow et al., 2006a; Grieves et al., 2015; Lindahl et al., 2009; Namjou et al., 2011; Olivieri et al., 2013; Orebaugh et al., 2011; Uyur Yalçın et al., 2015) 2. TREX1-deficient mice have cGAMP accumulation and produce high amounts of proinflammatory cytokines (Ahn et al., 2012, 2014; Gao et al., 2015; Gray et al., 2015) 3. Missense TREX1 mutations present in systemic lupus erythematosus patients (Lee-Kirsch et al., 2007) |
1–3. Aicardi–Goutières syndrome, familial chilblain lupus, systemic lupus erythematosus |
| RNase H2 | 1. Mutations in RNASEH2A, RNASEH2B, and RNASH2C lead to nucleic acid accumulation (Chon et al., 2013; Coffin et al., 2011; Günther et al., 2015; Kind et al., 2014; Pizzi et al., 2015; Ramantani et al., 2010; Reijns et al., 2011) 2. RNase H2-deficient mice are embryonic lethal; all subunits are necessary (Hiller et al., 2012; Reijns et al., 2012) 3. Mice that are RNASEH2A-and RNASEH2B-null have increased nucleic acid accumulation and increased ISG expression (Mackenzie et al., 2016; Pokatayev et al., 2016) |
1–3. Aicardi–Goutières syndrome, lupus-like symptoms |
| SAMHD1 | 1. Over 16 missense, nonsense, and 12-nucleotide deletion mutations (e.g., AGS82, AGS92, and AGS91) have been identified in AGS patients and their families (Rice et al., 2009) 2. Mice deficient in Samhd1 lack AGS symptoms but have increased ISG expression (Maelfait et al., 2016) |
1. Aicardi–Goutières syndrome 2. Increased cGAS/STING signaling |
| STING | 1. De novo gain-of-function mutations in TMEM173 (Dobbs et al., 2015; Jeremiah et al., 2014; Konig et al., 2017; Konno et al., 2018; Liu et al., 2014, 2015a; Melki et al., 2017; Munoz et al., 2015; Picard et al., 2016; Warner et al., 2017) 2. Activation of STING via agonist leads to development of anti-STING antibodies, results in production of IFN-β in salivary gland cells (Papinska et al., 2018) |
1. SAVI, familial chilblain lupus 2. Sjögren’s syndrome |
3.5.1. TLR9, TREX1, RNase H2, and SAMHD1
As introduced in the previous section, AGS is characterized as a heritable inflammatory disease that can lead to severe neurological disorders. In addition to the roles that ADAR1 and MDA5 have in AGS, AGS has also been documented to be caused by genetic mutations encoding nucleic-acid-metabolizing proteins, such as TREX1, RNASEH2A-C (Ribonuclease H2), and SAMHD1 (Crow et al., 2006a,b; Rice et al., 2009, 2012, 2014). Mutations in three of these proteins, namely TREX1, RNase H2, and SAMHD1, result in the direct accumulation of nucleic acids and subsequent activation of the cGAS/STING signaling pathway. Dysfunctions in nucleic acid sensing are also found in SLE patients and include hyperactivity of TREX1 or TLR9 in B cells (Barrat et al., 2005; Christensen et al., 2005; Crispín et al., 2013; Lenert, 2010; Wu et al., 2009; Yu, 2006).
As described in the previous section focused on cytosolic RNA sensing, mice lacking TLR7 exhibited decreased SLE pathology, due to lower levels of autoantibodies and lymphocyte activation. In contrast, the loss of TLR9 results in increased SLE pathology, lymphocyte activation, type I IFN, and autoantibodies (Christensen et al., 2006). B cells from SLE patients display impaired TLR9 signaling and decreased levels of CD19 and CD21, while pDCs from SLE patients displayed normal TLR9 signaling. However, TLR7 signaling was normal in SLE B cells (Gies et al., 2018). Moreover, when lupus-prone Sle1 mice were crossed with TLR9−/− mice, SLE pathology was exacerbated, and TLR7 expression was increased, leading to increased antigen presentation on DCs (Celhar et al., 2018). Similar results were also observed in TLR9−/− mice treated epicutaneously with imiquimod, which induces local inflammation, as compared to wild-type mice (Liu et al., 2018). Recently, a mechanism behind TLR9-mediated tolerance of SLE was shown to function through the AhR transcription factor, which is activated in apoptotic cells via TLR9 and drives anti-inflammatory IL-10 production. The loss of AhR also resulted in a more severe SLE phenotype (Shinde et al., 2018). Together, these studies highlight the protective role TLR9 in TLR7-mediated SLE and the role of apoptotic-cell DNA in promoting B-cell tolerance.
An important detector of cytosolic DNA is TREX1 (three prime pair exonuclease 1), whose major function is to digest ssDNA and dsDNA in the cytosol and prevent autoimmune activation (de Silva et al., 2007; Hoss, 1999; Lehtinen et al., 2008; Mazur and Perrino, 1999; Yang et al., 2007). Unlike other proofreading DNases, TREX1 is anchored to the ER by its C-terminal region (Chowdhury et al., 2006; Mazur and Perrino, 2001; Richards et al., 2007; Stetson et al., 2008; Wolf et al., 2016). Indeed, the role of TREX1 in AGS has been documented extensively by several groups which have identified different loss-of-function mutations in the TREX1 gene in AGS (Abe et al., 2014; Bailey et al., 2012; Crow, 2011; Crow et al., 2006a; Grieves et al., 2015; Lindahl et al., 2009; Namjou et al., 2011; Olivieri et al., 2013; Orebaugh et al., 2011; Uyur Yalçın et al., 2015). Mouse models of TREX1 deficiency have demonstrated that TREX1−/− mice have increased type I IFN signaling, inflammation in multiple tissues, and increased mortality (Gall et al., 2012; Peschke et al., 2016; Stetson et al., 2008; Xu et al., 2014a). Importantly, disease progression in TREX1−/− mice depends significantly on cGAS, STING, and sub-sequent IRF3 activation, due in part by the accumulation of 2′−3′-cGAMP in TREX1−/− mice. Importantly, double knockout cGAS/TREX1 mice lack the autoinflammatory phenotype due to the loss of the cGAS DNA sensor (Ahn et al., 2012; Gao et al., 2015; Gray et al., 2015). The origin of cytosolic DNA that leads to overstimulation of cGAS and STING has also been studied extensively (Ablasser et al., 2014). Some groups have shown that TREX1 may function specifically to be a detector of viral retroelements which is supported by its role in metabolizing the of HIV-derived DNA and overall HIV pathogenesis (Booiman et al., 2014; Pontillo et al., 2013; Wheeler et al., 2016; Yan et al., 2010). Indeed, Beck-Engeser et al. suggest that the treatment of AGS patients with retroelement inhibitors may is a potential therapy, but Achleitner et al. has shown that these types of drugs do not improve disease (Achleitner et al., 2017; Beck-Engeser et al., 2011). Alternatively, it could be that TREX1 functions primarily to prevent DNA damage and serves a dual purpose in feeding into the cGAS/STING pathway when it detects cytosolic DNA. This hypothesis is supported in part from evidence showing that TREX1 metabolizes cytosolic DNA and contributes to the prevention of genome instability (Ahn et al., 2014; Domínguez-Sánchez et al., 2011; Yang et al., 2007). Indeed, a recent study has shown that TREX1 has a nuclease-independent function in preventing L1-mediated retrotransposon-induced DNA damage, thus maintaining genome integrity (Li et al., 2017b).
AGS is also caused by mutations in alleles in the trimeric protein RNase H2, whose two main functions are to degrade RNA/DNA hybrids resulting from misincorporation of ribonucleotides and to cleave dsDNA at a phosphodiester linkage adjacent to a single ribonucleotide (Crow et al., 2006b; Reijns et al., 2012). It is now well-documented that mutations in RNASEH2A, RNASEH2B, and RNASEH2C lead to the abnormal accumulation of nucleic acids which result in AGS (Chon et al., 2013; Coffin et al., 2011; Günther et al., 2015; Hiller et al., 2012; Kind et al., 2014; Pizzi et al., 2015; Pokatayev et al., 2016; Ramantani et al., 2010; Reijns et al., 2011). RNase H2-deficient mouse models show that mutations in this complex are embryonic lethal, indicating a critical function of all subunits (Reijns et al., 2012). Nonetheless, mice expressing mutant RNASEH2A and RNASEH2B alleles show that these mutations result in the direct accumulation of nucleic acids that induce IFN through cGAS/STING signaling (Mackenzie et al., 2016; Pokatayev et al., 2016). Evidence suggests that the specific origin of nucleic acids that leads to RNase H2-mediated inflammatory disease may occur through DNA damage responses as well as recognition of retroelements (Bartsch et al., 2017, 2018; Günther et al., 2015; Zhao et al., 2018). Furthermore, two recent studies have also shown that DNA damage leading to the formation of micronuclei induces an IFN response through cGAS and that this occurs in frequently in the absence of RNASEH2B (Harding et al., 2017; Mackenzie et al., 2017).
A third and recently characterized protein that plays a role in AGS is SAMHD1 (SAM domain and HD domain-containing protein 1), which encodes an enzyme with phosphohydrolase activity as well as antiviral protection from HIV (Beloglazova et al., 2013; Goldstone et al., 2011; Powell et al., 2011; Rice et al., 2009). Specifically, SAMHD1 detects and cleaves deoxyribose adenine triphosphates (dNTPs) to prevent the reverse transcription of the HIV genome (Goldstone et al., 2011; Lahouassa et al., 2012). A recent study using SAMHD1-deficent mice showed these mice lack autoimmune phenotypes but are hyperactive in cGAS/STING signaling and induce the expression of type I IFN genes Ifit1 and Ifi44 (Behrendt et al., 2013; Maelfait et al., 2016). Additionally, SAMHD1 has also been implicated in DNA damage responses through its ability to maintain genome stability, digest ssDNA fragments at stalling replication forks, and inhibiting a cGAS/STING inflammatory response (Coquel et al., 2018; Kretschmer et al., 2015; Medeiros et al., 2018). A recent study has shown that SAMHD1 can suppress innate immune induction independently of its dNTPase activity. Specifically, SAMHD1 can mediate suppression of NF-κB through its interaction with NF-κB1/2 and preventing phosphorylation of IκBα, and also by interacting with IKKε to prevent IKKε-dependent phosphorylation of IRF7 (Chen et al., 2018). Importantly, this study suggests that because SAMHD1 negatively regulates an innate immune response, it may serve as a therapeutic target. Indeed, all of these recent findings highlight the need to continue studying the multiple mechanisms that TREX1, RNase H2, and SAMHD1 have in mediating an inflammatory response that can lead to AGS and other inflammatory diseases.
3.5.2. Sjögren’s Syndrome
Sjögren’s syndrome (SS) is an autoimmune disorder characterized by dry mouth and eyes due to reduced lacrimal and salivary gland secretion first described in 1933 by Henrik Sjögren (Mutlu and Scully, 1993). The molecular mechanisms behind SS are unclear as symptoms are similar to rheumatoid arthritis and many groups have grappled with defining indicators and causes (Baldini et al., 2018; Daniels and Fox, 1992; Wang et al., 2018; Yang et al., 2018). Studies have documented that the DNA sensor IFI16 is upregulated in SS patients and leads to increased antibodies against IFI16 (Alunno et al., 2015; Baer et al., 2016; Mondini et al., 2006). A recent study has implicated STING hyperactivation as a possible cause of SS, shown by the increase in circulating levels of the IFN-α, IFN-β, IL-6, and TNF-α proinflammatory cytokines in primary salivary gland cells of mice after DMXAA-mediated activation of STING. Notably, these mice developed antibodies to STING, and STING/TBK1/IRF3 signaling resulted in increased IFN-β production in the salivary glands. In addition, transfection with cGAMP in salivary glands also resulted in robust production on IFN-β which suggests that a possible mechanisms for SS might be due to the presence of cytosolic DNA that leads to the activation of cGAS and subsequent STING signaling (Papinska et al., 2018). Importantly, this is the first study to associate SS with accumulation of nucleic acids that activate the cGAS/STING signaling pathway, but further dissection of the pathway in this disorder will be needed to clarify a mechanism.
In summary, it is evident that accumulation of nucleic acids leads to autoinflammatory and autoimmune diseases such as AGS, SLE, and SS. The proteins TREX1, RNase H2, and SAMHD1 all have important roles in protecting the cell from hyperactive cytosolic nucleic acid signaling via the cGAS/STING signaling pathway. Further studies connecting cytosolic DNA presence to AGS, SLE, and SS will clarify how DNA damage responses and innate immune responses like cGAS/STING lead to autoimmune and autoinflammatory disorders.
3.6. Hypersensitivity of cGAS/STING Signaling
In addition to accumulation of nucleic acids, another important cause of autoimmune and autoinflammatory disorders is hypersensitivity of the cGAS/STING pathway. This type of dysfunction is due to mutations that result in the constitutive activation of STING and type I IFN in the presence or absence of ligands, namely CDNs. Two examples of such disorders are SAVI and familial chilblain lupus where mutations in the TMEM173 gene encoding the STING protein result in constitutive production of IFN and proinflammatory cytokines. Lastly, we describe an example of a therapeutic option in a mouse model of MS that indirectly connects it to the cGAS/STING signaling pathway.
3.6.1. SAVI and Familial Chilblain Lupus
De novo, gain-of-function mutations in the TMEM173 gene were documented to cause SAVI (Liu et al., 2014). The disease begins in the first few months of life and autoinflammatory symptoms include rash, flares, nodules, blistering of fingers, toes, nose, cheeks, fever, and joint pain (de Jesus et al., 2015; Jeremiah et al., 2014; Munoz et al., 2015; Picard et al., 2016). Other characteristics of SAVI include ulcerating skin lesions that resemble chilblain lupus. Indeed, sequencing of TMEM173 in chilblain lupus patients has also revealed gain-of-function mutations in STING which lead to disease (König et al., 2017). Previously, the identified gain-of-function mutations in patient samples had involved one of four amino acids in STING at positions 147, 154, 155, or 166 which sequester STING in the ER, simulate ligand binding, resulting in increased production of IFNs (Dobbs et al., 2015; Liu et al., 2014; Zhang et al., 2013). However, substitutions in the amino acid residues 206, 281, and 284 of STING implicated a ligand-independent mechanism of STING activation (Melki et al., 2017). Additionally, a recent study in which whole exome sequencing was performed from a 9-month-old Ecuadorian boy displaying fever and a severe neck abscess revealed he had a missense heterozygous mutation resulting in the STING variant R284S which constitutively activates STING in the absence of CDNs (Konno et al., 2018). Interestingly, the chilblain lupus mutations that result in activation of STING and production of IFN-β are also induced in the absence of CDNs, indicating that other STING-trafficking mechanisms need to be investigated. For example, the constitutive production of IFN-β in SAVI has been thought to occur by activation of TBK1 and IRF3. However, a study generated STING N153S heterozygous knock-in mice and observed that these mutants develop disease symptoms independent of IRF3 which suggests that other type I IFN-independent mechanisms are involved causing diseased state or that species-specific differences exist molecularly (Warner et al., 2017).
As seen by the gain-of-function mutations in TMEM173, it is evident that overstimulation or activation of STING is enough to induce a diseased state. The use of JAK1/2 inhibitors is currently a growing therapeutic approach being used treat other inflammatory diseases such as rheumatoid arthritis and myelofibrosis that may also be used to treat SAVI. Indeed, a recent study showed that when the JAK inhibitor baricitinib was used to treat SAVI patients, resulting in amelioration of vasculitis (Sanchez et al., 2018). Nevertheless, many questions remain regarding JAK inhibitors such as their side effects which prevent FDA approval (Shreberk-Hassidim et al., 2017). Another therapeutic strategy lies in the development of STING-specific inhibitors to treat STING-mediated autoinflammatory diseases. In fact, covalent small-molecules have been developed that target a transmembrane domain of STING to block its palmitoylation and subsequent multimeric assembly at the Golgi (Haag et al., 2018). These molecules improve inflammatory disease in TREX1−/− mice and may soon be used for clinical trials in SAVI patients or other STING-mediated interferonopathies.
3.6.2. S6K1 and Multiple Sclerosis
One interesting therapeutic example for autoimmune disorders has been observed through the use of the mouse model of multiple sclerosis (MS), namely experimental autoimmune encephalitis (EAE). MS is an autoimmune disease that leaves scars, namely sclerosis, in the myelin sheath of multiple nerve fibers over time by the influx of T lymphocytes into the CNS (McFarland and Martin, 2007). Symptoms include muscle weakness, fatigue, difficulty balancing and walking, as well tremors, speech problems, and cognitive issues like cerebral and brainstem dysfunction (Landtblom et al., 2010; Minagar, 2014). There are varying degrees of MS ranging from subtle to severe that change over time as the disease progresses. Microarray analysis, large-and small-scale gene expression studies, and GWAS have been performed in patients and have revealed over 20 mutations that may lead to disease, some of which induce the production of cytokines by Th1 and Th17 cells (Munoz-Culla et al., 2013; Murugaiyan et al., 2011). There is no explicit evidence supporting DNA-sensing mechanisms involved in MS pathogenesis, however, there is one possible therapeutic option that indirectly connects to the cGAS/STING signaling pathway. Earlier, the ribosomal kinase S6K1 was described in the mechanism for cGAS/STING signaling where it forms a complex with STING and TBK1 to induce IRF3 activation in the context of cytosolic DNA sensing (Wang et al., 2016). Inhibition of S6K1 with a pan-ribosomal S6 kinase inhibitor BI-D1870 protected mice from EAE (Takada et al., 2016). Furthermore, when EAE mice were injected with DNA nanoparticles (DNPs) which led to selective activation of STING, subsequent IFN-α/β release, and overall suppression of EAE by reducing the recruitment of Th1 and Th17 cells into the CNS (Lemos et al., 2014). This therapeutic option mediated by DNPs suggests that other accessory proteins may indeed be able to serve as targets for other autoimmune or inflammatory diseases.
While type I interferonopathies are characterized by varying degrees of autoinflammation, autoimmunity, or immunodeficiency, only a few have been documented to be caused directly or indirectly by dysregulation of DNA-sensing mechanisms. Specifically, the molecular dissection of autoinflammatory and autoimmune disorders like AGS, SS, SAVI, and familial chilblain lupus has all revealed a commonality in DNA-sensing errors. Namely, mutations in the proteins TREX1, RNase H2, and SAMHD1 lead to nucleic acid accumulation while mutations in TMEM173 lead to overstimulation of STING and subsequent overproduction of IFN-β. On the other hand, SLE and SMS are indirectly connected to DNA-sensing mechanisms but nonetheless represent the complexity of the mechanisms behind these disorders.
4. PROSPECTIVE
Detailed mechanistic understanding of innate immunity is crucial for the development of novel therapies to combat microbial infection and provide relief to those suffering from certain autoimmune pathologies. Already, it has been shown that the STING pathway can be exploited to reduce infectious load of a number of viral (Guo et al., 2017; Skouboe et al., 2018) and bacterial (Barker et al., 2013; Karaolis et al., 2007b; Rosen et al., 2017) pathogens. Additionally, STING-specific adjuvants can boost vaccine efficiency ( Junkins et al., 2018; Sˇkrnjug et al., 2014) and be used for cancer immunotherapy (Miyabe et al., 2014). Similarly, RIG-I agonists can be used to reduce viral burden (Bedard et al., 2012; Coch et al., 2017; Green et al., 2016; Nielsen et al., 2017; Pattabhi et al., 2015) and induce anti-tumor activity (Dassler-Plenker et al., 2016). Given these advances in developing small molecules and methods to activate innate immunity to reduce microbial burden, methods to inhibit hyperactive innate immune signaling may aid in the development of therapies for autoimmune disorders. Already, small molecules have been developed that target cGAS and reduce its activity (An et al., 2015; Hall et al., 2017; Vincent et al., 2017). Given the evolutionary arms race between pathogens and host, especially with respect to the co-evolution of DNA-and retro-viruses with humans (Elde and Malik, 2009), might it be possible to develop virally based strategies or molecules to inhibit hyperactive innate immunity? As the saying goes, “…keep your enemies closer.”
ACKNOWLEDGMENTS
We thank the anonymous reviewers for their thoughtful comments and constructive criticism of our chapter contribution.
Research in the Goodman lab is funded by NIH Grant R21 AI128103 and funds from Washington State University. K.M.M. is supported by the Barry Goldwater Scholarship and Excellence in Education Foundation.
REFERENCES
- Abe T, Barber GN, 2014. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-kappaB activation through TBK1. J. Virol 88, 5328–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe J, Nakamura K, Nishikomori R, Kato M, Mitsuiki N, Izawa K, Awaya T, Kawai T, Yasumi T, Toyoshima I, Hasegawa K, Ohshima Y, Hiragi T, Sasahara Y, Suzuki Y, Kikuchi M, Osaka H, Ohya T, Ninomiya S, Fujikawa S, Akasaka M, Iwata N, Kawakita A, Funatsuka M, Shintaku H, Ohara O, Ichinose H, Heike T, 2014. A nationwide survey of Aicardi-Goutieres syndrome patients identifies a strong association between dominant TREX1 mutations and chilblain lesions: Japanese cohort study. Rheumatology 53, 448–458. [DOI] [PubMed] [Google Scholar]
- Ablasser A, Bauernfeind F, Hartmann G, Latz E, Fitzgerald KA, Hornung V, 2009. RIG-I-dependent sensing of poly(dA:dT) through the induction of an RNA polymerase III-transcribed RNA intermediate. Nat. Immunol 10, 1065–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner K-P, Ludwig J, Hornung V, 2013. cGAS produces a 2′−5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ablasser A, Hemmerling I, Schmid-Burgk JL, Behrendt R, Roers A, Hornung V, 2014. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol 192, 5993–5997. [DOI] [PubMed] [Google Scholar]
- Achleitner M, Kleefisch M, Hennig A, Peschke K, Polikarpova A, Oertel R, Gabriel B, Schulze L, Lindeman D, Gerbaulet A, Fiebig U, Lee-Kirsch MA, Roers A, Behrendt R, 2017. Lack of Trex1 causes systemic autoimmunity despite the presence of antiretroviral drugs. J. Immunol 199, 2261–2269. [DOI] [PubMed] [Google Scholar]
- Aguirre S, Fernandez-Sesma A, 2017. Collateral damage during dengue virus infection: making sense of DNA by cGAS. J. Virol 91, e01081–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre S, Maestre AM, Pagni S, Patel JR, Savage T, Gutman D, Maringer K, Bernal-Rubio D, Shabman RS, Simon V, Rodriguez-Madoz JR, Mulder LCF, Barber GN, Fernandez-Sesma A, 2012. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog 8, e1002934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlers LRH, Goodman AG, 2018. The immune responses of the animal hosts of west nile virus: a comparison of insects, birds, and mammals. Front. Cell. Infect. Microbiol 8, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlers LRH, Bastos RG, Hiroyasu A, Goodman AG, 2016. Invertebrate iridescent virus 6, a DNA virus, stimulates a mammalian innate immune response through RIG-I-like receptors. PLoS One 11, e0166088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, Zhang CZ, Hur S, 2018. Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation. Cell 172 (797–810), e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Gutman D, Saijo S, Barber GN, 2012. STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. U.S.A 109, 19386–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Ruiz P, Barber GN, 2014. Intrinsic self-DNA triggers inflammatory disease dependent on STING. J. Immunol 193, 4634–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcamí A, Smith GL, 1996. A mechanism for the inhibition of fever by a virus. Proc. Natl. Acad. Sci. U.S.A 93, 11029–11034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexopoulou L, Holt AC, Medzhitov R, Flavell RA, 2001. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature 413, 732. [DOI] [PubMed] [Google Scholar]
- Alkanani AK, Rewers M, Dong F, Waugh K, Gottlieb PA, Zipris D, 2012. Dys-regulated Toll-like receptor-induced interleukin-1beta and interleukin-6 responses in subjects at risk for the development of type 1 diabetes. Diabetes 61, 2525–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alunno A, Caneparo V, Carubbi F, Bistoni O, Caterbi S, Bartoloni E, Giacomelli R, Gariglio M, Landolfo S, Gerli R, 2015. Interferon gamma-inducible protein 16 in primary Sjögren’s syndrome: a novel player in disease pathogenesis? Arthritis Res. Ther 17, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An J, Woodward JJ, Sasaki T, Minie M, Elkon KB, 2015. Cutting edge: antimalarial drugs inhibit IFN-beta production through blockade of cyclic GMP-AMP synthase-DNA interaction. J. Immunol 194, 4089–4093. [DOI] [PubMed] [Google Scholar]
- Anghelina D, Lam E, Falck-Pedersen E, 2016. Diminished innate antiviral response to adenovirus vectors in cGAS/STING-deficient mice minimally impacts adaptive immunity. J. Virol 90, 5915–5927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anglero-Rodriguez YI, Pantoja P, Sariol CA, 2014. Dengue virus subverts the interferon induction pathway via NS2B/3 protease-IκB kinase epsilon interaction. Clin. Vaccine Immunol 21, 29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appledorn DM, Patial S, McBride A, Godbehere S, Van Rooijen N, Parameswaran N, Amalfitano A, 2008. Adenovirus vector-induced innate inflammatory mediators, MAPK signaling, as well as adaptive immune responses are dependent upon both TLR2 and TLR9 in vivo. J. Immunol 181, 2134–2144. [DOI] [PubMed] [Google Scholar]
- Arakelyan A, Nersisyan L, Poghosyan D, Khondkaryan L, Hakobyan A, Löffler-Wirth H, Melanitou E, Binder H, 2017. Autoimmunity and autoinflammation: a systems view on signaling pathway dysregulation profiles. PLoS One 12 (11), e0187572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arbuckle MR, McClain MT, Rubertone MV, Scofield RH, Dennis GJ, James JA, Harley JB, 2003. Development of autoantibodies before the clinical onset of systemic lupus erythematosus. N. Engl. J. Med 349, 1526–1533. [DOI] [PubMed] [Google Scholar]
- Arimoto K-I, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K, 2007. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc. Natl. Acad. Sci. U.S.A 104, 7500–7505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashour J, Laurent-Rolle M, Shi P-Y, Garcıa-Sastre A, 2009. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol 83, 5408–5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assmann TS, Brondani Lde A, Bauer AC, Canani LH, Crispim D, 2014. Polymor-phisms in the TLR3 gene are associated with risk for type 1 diabetes mellitus. Eur. J. Endocrinol 170, 519–527. [DOI] [PubMed] [Google Scholar]
- Awais M, Wang K, Lin X, Qian W, Zhang N, Wang C, Wang K, Zhao L, Fu ZF, Cui M, 2017. TLR7 deficiency leads to TLR8 compensative regulation of immune response against JEV in mice. Front. Immunol 8, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baer AN, Petri M, Sohn J, Rosen A, Casciola-Rosen L, 2016. Association of antibodies to interferon-inducible protein-16 with markers of more severe disease in primary Sjögren’s syndrome: anti-IFI16 antibodies and severe disease in Sjögren’s syndrome. Arthritis Care Res 68, 254–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeuerle P, Baltimore D, 1988. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science 242, 540–546. [DOI] [PubMed] [Google Scholar]
- Bagashev A, Fitzgerald MC, LaRosa DF, Rose PP, Cherry S, Johnson AC, Sullivan KE, 2010. Leucine-rich repeat (in flightless I) interacting protein-1 regulates a rapid type I interferon response. J. Interf. Cytokine Res 30, 843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey SL, Harvey S, Perrino FW, Hollis T, 2012. Defects in DNA degradation revealed in crystal structures of TREX1 exonuclease mutations linked to autoimmune disease. DNA Repair 11, 65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, Barber GN, 2000. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 13, 129–141. [DOI] [PubMed] [Google Scholar]
- Baldini C, Ferro F, Mosca M, Fallahi P, Antonelli A, 2018. The association of Sjögren syndrome and autoimmune thyroid disorders. Front. Endocrinol 9, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker JR, Koestler BJ, Carpenter VK, Burdette DL, Waters CM, Vance RE, Valdivia RH, 2013. STING-dependent recognition of cyclic di-AMP mediates type I interferon responses during Chlamydia trachomatis infection. mBio 4, e00018–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, Coffman RL, 2005. Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J. Exp. Med 202, 1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barro M, Patton JT, 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl. Acad. Sci. U.S.A 102, 4114–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartsch K, Knittler K, Borowski C, Rudnik S, Damme M, Aden K, Spehlmann ME, Frey N, Saftig P, Chalaris A, Rabe B, 2017. Absence of RNase H2 triggers generation of immunogenic micronuclei removed by autophagy. Hum. Mol. Genet 26, 3960–3972. [DOI] [PubMed] [Google Scholar]
- Bartsch K, Damme M, Regen T, Becker L, Garrett L, Ho€lter SM, Knittler K, Borowski C, Waisman A, Glatzel M, Fuchs H, Gailus-Durner V, Hrabe de Angelis M, Rabe B, 2018. RNase H2 loss in murine astrocytes results in cellular defects reminiscent of nucleic acid-mediated autoinflammation. Front. Immunol 9, 587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basner-Tschakarjan E, Gaffal E, O’Keeffe M, Tormo D, Limmer A, Wagner H, Hochrein H, Tüting T, 2006. Adenovirus efficiently transduces plasmacytoid dendritic cells resulting in TLR9-dependent maturation and IFN-α production. J. Gene Med 8, 1300–1306. [DOI] [PubMed] [Google Scholar]
- Bauer S, Kirschning CJ, Häcker H, Redecke V, Hausmann S, Akira S, Wagner H, Lipford GB, 2001. Human TLR9 confers responsiveness to bacterial DNA via species-specific CpG motif recognition. Proc. Natl. Acad. Sci. U.S.A 98, 9237–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind F, Ablasser A, Kim S, Bartok E, Hornung V, 2010. An unexpected role for RNA in the recognition of DNA by the innate immune system. RNA Biol 7, 151–157. [DOI] [PubMed] [Google Scholar]
- B’Chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P, Bruhat A, 2013. The eIF2alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res 41, 7683–7699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck-Engeser GB, Eilat D, Wabl M, 2011. An autoimmune disease prevented by antiretroviral drugs. Retrovirology 8, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard KM, Wang ML, Proll SC, Loo YM, Katze MG, Gale M Jr., Iadonato SP, 2012. Isoflavone agonists of IRF-3 dependent signaling have antiviral activity against RNA viruses. J. Virol 86, 7334–7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beg AA, Baldwin AS, 1993. The I kappa B proteins: multifunctional regulators of Rel/NF-kappa B transcription factors. Genes Dev 7, 2064–2070. [DOI] [PubMed] [Google Scholar]
- Behrendt R, Schumann T, Gerbaulet A, Nguyen LA, Schubert N, Alexopoulou D, Berka U, Lienenklaus S, Peschke K, Gibbert K, Wittmann S, Lindemann D, Weiss S, Dahl A, Naumann R, Dittmer U, Kim B, Mueller W, Gramberg T, Roers A, 2013. Mouse SAMHD1 has antiretroviral activity and suppresses a spontaneous cell-intrinsic antiviral response. Cell Rep 4, 689–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beloglazova N, Flick R, Tchigvintsev A, Brown G, Popovic A, Nocek B, Yakunin AF, 2013. Nuclease activity of the human SAMHD1 protein implicated in the aicardi-goutières syndrome and HIV-1 restriction. J. Biol. Chem 288, 8101–8110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhan U, Lukacs NW, Osterholzer JJ, Newstead MW, Zeng X, Moore TA, McMillan TR, Krieg AM, Akira S, Standiford TJ, 2007. TLR9 is required for protective innate immunity in gram-negative bacterial pneumonia: role of dendritic cells. J. Immunol 179, 3937–3946. [DOI] [PubMed] [Google Scholar]
- Bharaj P, Wang YE, Dawes BE, Yun TE, Park A, Yen B, Basler CF, Freiberg AN, Lee B, Rajsbaum R, 2016. The matrix protein of nipah virus targets the E3-ubiquitin ligase TRIM6 to inhibit the IKKε kinase-mediated type-I IFN antiviral response. PLoS Pathog 12, e1005880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhat N, Fitzgerald KA, 2014. Recognition of cytosolic DNA by cGAS and other STING-dependent sensors: HIGHLIGHTS. Eur. J. Immunol 44, 634–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booiman T, Setiawan LC, Kootstra NA, 2014. Genetic variation in Trex1 affects HIV-1 disease progression. AIDS 28, 2517–2521. [DOI] [PubMed] [Google Scholar]
- Bowen JR, Quicke KM, Maddur MS, O’Neal JT, McDonald CE, Fedorova NB, Puri V, Shabman RS, Pulendran B, Suthar MS, 2017. Zika virus antagonizes type I interferon responses during infection of human dendritic cells. PLoS Pathog 13, e1006164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretin A, Carriere J, Dalmasso G, Bergougnoux A, B’Chir W, Maurin A-C, Müller S, Seibold F, Barnich N, Bruhat A, Darfeuille-Michaud A, Nguyen HTT, 2016. Activation of the EIF2AK4-EIF2A/eIF2α-ATF4 pathway triggers autophagy response to Crohn disease-associated adherent-invasive Escherichia coli infection. Autophagy 12, 770–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bristol M, Das D, Morgan I, 2017. Why human papillomaviruses activate the DNA damage response (DDR) and how cellular and viral replication persists in the presence of DDR signaling. Viruses 9, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broquet AH, Hirata Y, McAllister CS, Kagnoff MF, 2011. RIG-I/MDA5/MAVS are required to signal a protective IFN response in rotavirus-infected intestinal epithelium. J. Immunol 186, 1618–1626. [DOI] [PubMed] [Google Scholar]
- Bürckstümmer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H, Planyavsky M, Bilban M, Colinge J, Bennett KL, Superti-Furga G, 2009. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat. Immunol 10, 266–272. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Vance RE, 2013. STING and the innate immune response to nucleic acids in the cytosol. Nat. Immunol 14, 19–26. [DOI] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, Vance RE, 2011. STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butbul Aviel Y, Mandel H, Avitan Hersh E, Bergman R, Adiv OE, Luder A, Brik R, 2012. Prolidase deficiency associated with systemic lupus erythematosus (SLE): single site experience and literature review. Pediatr. Rheumatol. Online J 10, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caballero IS, Honko AN, Gire SK, Winnicki SM, Mele M, Gerhardinger C, Lin AE, Rinn JL, Sabeti PC, Hensley LE, Connor JH, 2016. In vivo Ebola virus infection leads to a strong innate response in circulating immune cells. BMC Genomics 17, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardenas WB, Loo YM, Gale M Jr., Hartman AL, Kimberlin CR, Martinez-Sobrido L, Saphire EO, Basler CF, 2006. Ebola virus VP35 protein binds double-stranded RNA and inhibits alpha/beta interferon production induced by RIG-I signaling. J. Virol 80, 5168–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celhar T, Yasuga H, Lee HY, Zharkova O, Tripathi S, Thornhill SI, Lu HK, Au B, Lim LHK, Thamboo TP, Akira S, Wakeland EK, Connolly JE, Fairhurst AM, 2018. TLR9 deficiency breaks tolerance to RNA-associated antigens and upregulates TLR7 protein in Sle1 mice. Arthritis Rheumatol in press. [DOI] [PMC free article] [PubMed]
- Chan MP, Onji M, Fukui R, Kawane K, Shibata T, Saitoh S, Ohto U, Shimizu T, Barber GN, Miyake K, 2015. DNase II-dependent DNA digestion is required for DNA sensing by TLR9. Nat. Commun 6, 5853. [DOI] [PubMed] [Google Scholar]
- Chapman JR, Jackson SP, 2008. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO Rep 9, 795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan S, Mandell MA, Deretic V, 2015. IRGM governs the core autophagy machinery to conduct antimicrobial defense. Mol. Cell 58, 507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Q, Sun L, Chen ZJ, 2016. Regulation and function of the cGAS–STING pathway of cytosolic DNA sensing. Nat. Immunol 17, 1142–1149. [DOI] [PubMed] [Google Scholar]
- Chen S, Bonifati S, Qin Z, St. Gelais C, Kodigepalli KM, Barrett BS, Kim SH, Antonucci JM, Ladner KJ, Buzovetsky O, Knecht KM, Xiong Y, Yount JS, Guttridge DC, Santiago ML, Wu L, 2018. SAMHD1 suppresses innate immune responses to viral infections and inflammatory stimuli by inhibiting the NF-κB and interferon pathways. Proc. Natl. Acad. Sci. U.S.A 115, E3798–E3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Liu Y, Wang Y, Niu Q, Gao Q, Fu Q, Ma J, Wang H, Yan Y, Ding C, Sun J, 2017. Chicken DNA virus sensor DDX41 activates IFN-β signaling pathway dependent on STING. Dev. Comp. Immunol 76, 334–342. [DOI] [PubMed] [Google Scholar]
- Chiu Y-H, MacMillan JB, Chen ZJ, 2009. RNA polymerase III detects cytosolic DNA and induces type I interferons through the RIG-I pathway. Cell 138, 576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe N, Kwon J-S, Kim J-R, Eom GH, Kim Y, Nam K-I, Ahn Y, Kee HJ, Kook H, 2013. The microRNA miR-132 targets Lrrfip1 to block vascular smooth muscle cell proliferation and neointimal hyperplasia. Atherosclerosis 229, 348–355. [DOI] [PubMed] [Google Scholar]
- Choi HJ, Park A, Kang S, Lee E, Lee TA, Ra EA, Lee J, Lee S, Park B, 2018. Human cytomegalovirus-encoded US9 targets MAVS and STING signaling to evade type I interferon immune responses. Nat. Commun 9, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chon H, Sparks JL, Rychlik M, Nowotny M, Burgers PM, Crouch RJ, Cerritelli SM, 2013. RNase H2 roles in genome integrity revealed by unlinking its activities. Nucleic Acids Res 41, 3130–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung J-S, Demple B, Perrino FW, Lieberman J, 2006. The exonuclease TREX1 is in the SET complex and acts in concert with NM23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell 23, 133–142. [DOI] [PubMed] [Google Scholar]
- Christensen SR, Kashgarian M, Alexopoulou L, Flavell RA, Akira S, Shlomchik MJ, 2005. Toll-like receptor 9 controls anti-DNA autoantibody production in murine lupus. J. Exp. Med 202, 321–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ, 2006. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25, 417–428. [DOI] [PubMed] [Google Scholar]
- Christensen MH, Jensen SB, Miettinen JJ, Luecke S, Prabakaran T, Reinert LS, Mettenleiter T, Chen ZJ, Knipe DM, Sandri-Goldin RM, Enquist LW, Hartmann R, Mogensen TH, Rice SA, Nyman TA, Matikainen S, Paludan SR, 2016. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J 35, 1385–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, Rice CM, 2018. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172 (811–824), e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciancanelli MJ, Volchkova VA, Shaw ML, Volchkov VE, Basler CF, 2009. Nipah virus sequesters inactive STAT1 in the nucleus via a P gene-encoded mechanism. J. Virol 83, 7828–7841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civril F, Deimling T, de Oliveira Mann CC, Ablasser A, Moldt M, Witte G, Hornung V, Hopfner K-P, 2013. Structural mechanism of cytosolic DNA sensing by cGAS. Nature 498, 332–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coch C, Stumpel JP, Lilien-Waldau V, Wohlleber D, Kummerer BM, Bekeredjian-Ding I, Kochs G, Garbi N, Herberhold S, Schuberth-Wagner C, Ludwig J, Barchet W, Schlee M, Hoerauf A, Bootz F, Staeheli P, Hartmann G, Hartmann E, 2017. RIG-I activation protects and rescues from lethal influenza virus infection and bacterial superinfection. Mol. Ther 25, 2093–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffin SR, Hollis T, Perrino FW, 2011. Functional consequences of the RNase H2A subunit mutations that cause aicardi-goutières syndrome. J. Biol. Chem 286, 16984–16991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coquel F, Silva M-J, Techer H, Zadorozhny K, Sharma S, Nieminuszczy J, Mettling C, Dardillac E, Barthe A, Schmitz A-L, Promonet A, Cribier A, Sarrazin A, Niedzwiedz W, Lopez B, Costanzo V, Krejci L, Chabes A, Benkirane M, Lin Y-L, Pasero P, 2018. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 557, 57–61. [DOI] [PubMed] [Google Scholar]
- Cordero MD, Alcocer-Gomez E, Ryffel B, 2018. Gain of function mutation and inflammasome driven diseases in human and mouse models. J. Autoimmun 91, 13–22. [DOI] [PubMed] [Google Scholar]
- Coutermarsh-Ott S, Eden K, Allen IC, 2016. Beyond the inflammasome: regulatory NOD-like receptor modulation of the host immune response following virus exposure. J. Gen. Virol 97, 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crispín JC, Hedrich CM, Tsokos GC, 2013. Gene-function studies in systemic lupus erythematosus. Nat. Rev. Rheumatol 9, 476–484. [DOI] [PubMed] [Google Scholar]
- Crow YJ, 2011. Type I interferonopathies: a novel set of inborn errors of immunity: type I interferonopathies. Ann. N. Y. Acad. Sci 1238, 91–98. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, Corry PC, Cowan FM, Frints SG, Klepper J, Livingston JH, Lynch SA, Massey RF, Meritet JF, Michaud JL, Ponsot G, Voit T, Lebon P, Bonthron DT, Jackson AP, Barnes DE, Lindahl T, 2006a. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat. Genet 38, 917–920. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R, Baumann C, Baxter P, Bertini E, Chandler KE, Chitayat D, Cau D, Dery C, Fazzi E, Goizet C, King MD, Klepper J, Lacombe D, Lanzi G, Lyall H, Martınez-Frıas ML, Mathieu M, McKeown C, Monier A, Oade Y, Quarrell OW, Rittey CD, Rogers RC, Sanchis A, Stephenson JBP, Tacke U, Till M, Tolmie JL, Tomlin P, Voit T, Weschke B, Woods CG, Lebon P, Bonthron DT, Ponting CP, Jackson AP, 2006b. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat. Genet 38, 910–916. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GMA, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, Abdel-Hamid MS, Abdel-Salam GM, Ackroyd S, Aeby A, Agosta G, Albin C, Allon-Shalev S, Arellano M, Ariaudo G, Aswani V, Babul-Hirji R, Baildam EM, Bahi-Buisson N, Bailey KM, Barnerias C, Barth M, Battini R, Beresford MW, Bernard G, Bianchi M, Billette de Villemeur T, Blair EM, Bloom M, Burlina AB, Carpanelli ML, Carvalho DR, Castro-Gago M, Cavallini A, Cereda C, Chandler KE, Chitayat DA, Collins AE, Sierra Corcoles C, Cordeiro NJV, Crichiutti G, Dabydeen L, Dale RC, D’Arrigo S, De Goede CGEL, De Laet C, De Waele LMH, Denzler I, Desguerre I, Devriendt K, Di Rocco M, Fahey MC, Fazzi E, Ferrie CD, Figueiredo A, Gener B, Goizet C, Gowrinathan NR, Gowrishankar K, Hanrahan D, Isidor B, Kara B, Khan N, King MD, Kirk EP, Kumar R, Lagae L, Landrieu P, Lauffer H, Laugel V, La Piana R, Lim MJ, Lin J-PSM, Linnankivi T, Mackay MT, Marom DR, Marques Lourenco C, McKee SA, Moroni I, Morton JEV, Moutard M-L, Murray K, Nabbout R, Nampoothiri S, Nunez-Enamorado N, Oades PJ, Olivieri I, Ostergaard JR, Perez-Duenas B, Prendiville JS, Ramesh V, Rasmussen M, Regal L, Ricci F, Rio M, Rodriguez D, et al. , 2015. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 167A, 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Li M, Walton KD, Sun K, Hanover JA, Furth PA, Hennighausen L, 2001. The Stat3/5 locus encodes novel endoplasmic reticulum and helicase-like proteins that are preferentially expressed in normal and neoplastic mammary tissue. Genomics 78, 129–134. [DOI] [PubMed] [Google Scholar]
- Dai P, Jeong SY, Yu Y, Leng T, Wu W, Xie L, Chen X, 2009. Modulation of TLR signaling by multiple MyD88-interacting partners including leucine-rich repeat Fli-I-interacting proteins. J. Immunol 182, 3450–3460. [DOI] [PubMed] [Google Scholar]
- Dalpke A, Frank J, Peter M, Heeg K, 2006. Activation of toll-like receptor 9 by DNA from different bacterial species. Infect. Immun 74, 940–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalrymple NA, Cimica V, Mackow ER, 2015. Dengue virus NS proteins inhibit RIG-I/MAVS signaling by blocking TBK1/IRF3 phosphorylation: dengue virus serotype 1 NS4A is a unique interferon-regulating virulence determinant. mBio 6, e00553–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels TE, Fox PC, 1992. Salivary and oral components of Sjo€gren’s syndrome. Rheum. Dis. Clin. North Am 18, 571–589. [PubMed] [Google Scholar]
- Darnell JE, Kerr IM, Stark GR, 1994. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264, 1415–1421. [DOI] [PubMed] [Google Scholar]
- Dassler-Plenker J, Reiners KS, van den Boorn JG, Hansen HP, Putschli B, Barnert S, Schuberth-Wagner C, Schubert R, Tuting T, Hallek M, Schlee M, Hartmann G, Pogge von Strandmann E, Coch C, 2016. RIG-I activation induces the release of extracellular vesicles with antitumor activity. Oncoimmunology 5, e1219827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S, Kaiko G, Loh Z, Lalwani A, Zhang V, Spann K, Foo SY, Hansbro N, Uematsu S, Akira S, Matthaei KI, Rosenberg HF, Foster PS, Phipps S, 2011. Plasmacytoid dendritic cells promote host defense against acute pneumovirus infection via the TLR7-MyD88-dependent signaling pathway. J. Immunol 186, 5938–5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruyn M, Vermeire S, 2017. NOD2 and bacterial recognition as therapeutic targets for Crohn’s disease. Expert Opin. Ther. Targets 21, 1123–1139. [DOI] [PubMed] [Google Scholar]
- de Jesus AA, Canna SW, Liu Y, Goldbach-Mansky R, 2015. Molecular mechanisms in genetically defined autoinflammatory diseases: disorders of amplified danger signaling. Annu. Rev. Immunol 33, 823–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva U, Choudhury S, Bailey SL, Harvey S, Perrino FW, Hollis T, 2007. The crystal structure of TREX1 explains the 3′ nucleotide specificity and reveals a poly-proline II helix for protein partnering. J. Biol. Chem 282, 10537–10543. [DOI] [PubMed] [Google Scholar]
- Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S, 2007. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity 27, 801–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deretic V, 2016. Autophagy in leukocytes and other cells: mechanisms, subsystem organization, selectivity, and links to innate immunity. J. Leukoc. Biol 100, 969–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desmet CJ, Ishii KJ, 2012. Nucleic acid sensing at the interface between innate and adaptive immunity in vaccination. Nat. Rev. Immunol 12, 479–491. [DOI] [PubMed] [Google Scholar]
- Dever TE, 2002. Gene-specific regulation by general translation factors. Cell 108, 545–556. [DOI] [PubMed] [Google Scholar]
- Diebold SS, Kaisho T, Hemmi H, Akira S, Reise Sousa C, 2004. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 303, 1529–1531. [DOI] [PubMed] [Google Scholar]
- Diebold SS, Massacrier C, Akira S, Paturel C, Morel Y, Reis e Sousa C, 2006. Nucleic acid agonists for toll-like receptor 7 are defined by the presence of uridine ribonucleotides. Eur. J. Immunol 36, 3256–3267. [DOI] [PubMed] [Google Scholar]
- Diner EJ, Burdette DL, Wilson SC, Monroe KM, Kellenberger CA, Hyodo M, Hayakawa Y, Hammond MC, Vance RE, 2013. The innate immune DNA sensor cGAS produces a noncanonical cyclic dinucleotide that activates human STING. Cell Rep 3, 1355–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, Huang B, Lu J, Liu Y-J, Zhong J, 2012. Hepatitis C virus NS3/4A protease blocks IL-28 production. Eur. J. Immunol 42, 2374–2382. [DOI] [PubMed] [Google Scholar]
- Ding Q, Cao X, Lu J, Huang B, Liu Y-J, Kato N, Shu H-B, Zhong J, 2013. Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol 59, 52–58. [DOI] [PubMed] [Google Scholar]
- Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, Yan N, 2015. STING activation by translocation from the ER is associated with infection and autoinflammatory disease. Cell Host Microbe 18, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dogusan Z, Garcia M, Flamez D, Alexopoulou L, Goldman M, Gysemans C, Mathieu C, Libert C, Eizirik DL, Rasschaert J, 2008. Double-stranded RNA induces pancreatic beta-cell apoptosis by activation of the toll-like receptor 3 and interferon regulatory factor 3 pathways. Diabetes 57, 1236–1245. [DOI] [PubMed] [Google Scholar]
- Domínguez-Sánchez MS, Barroso S, Gómez-González B, Luna R, Aguilera A, 2011. Genome instability and transcription elongation impairment in human cells depleted of THO/TREX. PLoS Genet 7, e1002386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubensky TW, Kanne DB, Leong ML, 2013. Rationale, progress and development of vaccines utilizing STING-activating cyclic dinucleotide adjuvants. Ther. Adv. Vaccines 1, 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubois EL, Tuffanelli DL, 1964. Clinical manifestations of systemic lupus erythematosus: computer analysis of 520 cases. JAMA 190, 104–111. [DOI] [PubMed] [Google Scholar]
- Ebihara H, Rockx B, Marzi A, Feldmann F, Haddock E, Brining D, LaCasse RA, Gardner D, Feldmann H, 2011. Host response dynamics following lethal infection of rhesus macaques with Zaire ebolavirus. J. Infect. Dis 204, S991–S999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Bendary M, Neamatallah M, Elalfy H, Besheer T, Elkholy A, El-Diasty M, Elsareef M, Zahran M, El-Aarag B, Gomaa A, Elhammady D, El-Setouhy M, Hegazy A, Esmat G, 2018. The association of single nucleotide polymorphisms of Toll-like receptor 3, Toll-like receptor 7 and Toll-like receptor 8 genes with the susceptibility to HCV infection. Br. J. Biomed. Sci 1–7. [DOI] [PubMed]
- Elde NC, Malik HS, 2009. The evolutionary conundrum of pathogen mimicry. Nat. Rev. Microbiol 7, 787–797. [DOI] [PubMed] [Google Scholar]
- Errett JS, Suthar MS, McMillan A, Diamond MS, Gale M, 2013. The essential, non-redundant roles of RIG-I and MDA5 in detecting and controlling West Nile virus infection. J. Virol 87, 11416–11425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald SE, Engel A, Lee J, Wang M, Bogyo M, Barton GM, 2011. Nucleic acid recognition by Toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J. Exp. Med 208, 643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falik-Zaccai TC, Khayat M, Luder A, Frenkel P, Magen D, Brik R, Gershoni-Baruch R, Mandel H, 2010. A broad spectrum of developmental delay in a large cohort of prolidase deficiency patients demonstrates marked interfamilial and intrafamilial phenotypic variability. Am. J. Med. Genet. B Neuropsychiatr. Genet 153B, 46–56. [DOI] [PubMed] [Google Scholar]
- Fan H, Ren D, Hou Y, 2018. TLR7, a third signal for the robust generation of spontaneous germinal center B cells in systemic lupus erythematosus. Cell. Mol. Immunol 15, 286–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Tsuchiya K, Kawamura I, Shen Y, Hara H, Sakai S, Yamamoto T, Fernandes-Alnemri T, Yang R, Hernandez-Cuellar E, Dewamitta SR, Xu Y, Qu H, Alnemri ES, Mitsuyama M, 2011. Critical roles of ASC inflammasomes in caspase-1 activation and host innate resistance to Streptococcus pneumoniae infection. J. Immunol 187, 4890–4899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q, Hato SV, Langereis MA, Zoll J, Virgen-Slane R, Peisley A, Hur S, Semler BL, van Rij RP, van Kuppeveld FJM, 2012. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell Rep 2, 1187–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, Rosenberg S, Zhang J, Alnemri ES, 2007. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 14, 1590–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu J-W, Datta P, Wu J, Alnemri ES, 2009. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T, Yu J-W, Juliana C, Solorzano L, Kang S, Wu J, Datta P, McCormick M, Huang L, McDermott E, Eisenlohr L, Landel CP, Alnemri ES, 2010. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat. Immunol 11, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finethy R, Jorgensen I, Haldar AK, de Zoete MR, Strowig T, Flavell RA, Yamamoto M, Nagarajan UM, Miao EA, Coers J, 2015. Guanylate binding proteins enable rapid activation of canonical and noncanonical inflammasomes in chlamydia-infected macrophages. Infect. Immun 83, 4740–4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiola S, Gosselin D, Takada K, Gosselin J, 2010. TLR9 contributes to the recognition of EBV by primary monocytes and plasmacytoid dendritic cells. J. Immunol 185, 3620–3631. [DOI] [PubMed] [Google Scholar]
- Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M, 2008. Establishment and maintenance of the innate antiviral response to west nile virus involves both RIG-I and MDA5 signaling through IPS-1. J. Virol 82, 609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y-Z, Su S, Gao Y-Q, Wang P-P, Huang Z-F, Hu M-M, Luo W-W, Li S, Luo M-H, Wang Y-Y, Shu H-B, 2017. Human cytomegalovirus tegument protein UL82 inhibits STING-mediated signaling to evade antiviral immunity. Cell Host Microbe 21, 231–243. [DOI] [PubMed] [Google Scholar]
- Fullam A, Schröder M, 2013. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim. Biophys. Acta 1829, 854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funabiki M, Kato H, Miyachi Y, Toki H, Motegi H, Inoue M, Minowa O, Yoshida A, Deguchi K, Sato H, Ito S, Shiroishi T, Takeyasu K, Noda T, Fujita T, 2014. Autoimmune disorders associated with gain of function of the intracellular sensor MDA5. Immunity 40, 199–212. [DOI] [PubMed] [Google Scholar]
- Funke B, Lasitschka F, Roth W, Penzel R, Meuer S, Saile M, Gretz N, Sido B, Schirmacher P, Autschbach F, 2011. Selective downregulation of retinoic acidinducible gene I within the intestinal epithelial compartment in Crohn’s disease. Inflamm. Bowel Dis 17, 1943–1954. [DOI] [PubMed] [Google Scholar]
- Furr SR, Chauhan VS, Moerdyk-Schauwecker MJ, Marriott I, 2011. A role for DNA-dependent activator of interferon regulatory factor in the recognition of herpes simplex virus type 1 by glial cells. J. Neuroinflammation 8, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gack MU, 2014. Mechanisms of RIG-I-like receptor activation and manipulation by viral pathogens. J. Virol 88, 5213–5216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale MJ Jr., Korth MJ, Tang NM, Tan SL, Hopkins DA, Dever TE, Polyak SJ, Gretch DR, Katze MG, 1997. Evidence that hepatitis C virus resistance to interferon is mediated through repression of the PKR protein kinase by the nonstructural 5A protein. Virology 230, 217–227. [DOI] [PubMed] [Google Scholar]
- Gale M, Tan SL, Wambach M, Katze MG, 1996. Interaction of the interferon-induced PKR protein kinase with inhibitory proteins P58IPK and vaccinia virus K3L is mediated by unique domains: implications for kinase regulation. Mol. Cell. Biol 16, 4172–4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall A, Treuting P, Elkon KB, Loo Y-M, Gale M, Barber GN, Stetson DB, 2012. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Wu J, Wu YT, Du F, Aroh C, Yan N, Sun L, Chen ZJ, 2013a. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 341, 903–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Wu Y, Barchet W, Gaffney BL, Zillinger T, Serganov AA, Liu Y, Jones RA, Hartmann G, Tuschl T, Patel DJ, 2013b. Cyclic [G(2′,5′)pA (3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao P, Ascano M, Zillinger T, Wang W, Dai P, Serganov AA, Gaffney BL, Shuman S, Jones RA, Deng L, Hartmann G, Barchet W, Tuschl T, Patel DJ, 2013c. Structure-function analysis of STING activation by c[G(2′,5′)pA (3′,5′)p] and targeting by antiviral DMXAA. Cell 154, 748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao D, Li T, Li X-D, Chen X, Li Q-Z, Wight-Carter M, Chen ZJ, 2015. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc. Natl. Acad. Sci. U.S.A 112, E5699–E5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Lin L, Haq IU, Zeng S-M, 2016. Inhibition of NF-κB promotes autophagy via JNK signaling pathway in porcine granulosa cells. Biochem. Biophys. Res. Commun 473, 311–316. [DOI] [PubMed] [Google Scholar]
- Garcia-Cattaneo A, Gobert F-X, Müller M, Toscano F, Flores M, Lescure A, Del Nery E, Benaroch P, 2012. Cleavage of Toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc. Natl. Acad. Sci. U.S.A 109, 9053–9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge J, Gong YN, Xu Y, Shao F, 2012. Preventing bacterial DNA release and absent in melanoma 2 inflammasome activation by a Legionella effector functioning in membrane trafficking. Proc. Natl. Acad. Sci. U.S.A 109, 6193–6198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee P, Chua PK, Gevorkyan J, Klumpp K, Najera I, Swinney DC, Deval J, 2008. Essential role of the N-terminal domain in the regulation of RIG-I ATPase activity. J. Biol. Chem 283, 9488–9496. [DOI] [PubMed] [Google Scholar]
- Gies V, Schickel JN, Jung S, Joublin A, Glauzy S, Knapp AM, Soley A, Poindron V, Guffroy A, Choi JY, Gottenberg JE, Anolik JH, Martin T, Soulas-Sprauel P, Meffre E, Korganow AS, 2018. Impaired TLR9 responses in B cells from patients with systemic lupus erythematosus. JCI Insight 3, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohda J, Matsumura T, Inoue J. i., 2004. Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but Not Toll/IL-1 receptor domain-containing adaptor-inducing IFN-β (TRIF)-dependent pathway in TLR signaling. J. Immunol 173, 2913–2917. [DOI] [PubMed] [Google Scholar]
- Goldstone DC, Ennis-Adeniran V, Hedden JJ, Groom HCT, Rice GI, Christodoulou E, Walker PA, Kelly G, Haire LF, Yap MW, de Carvalho LPS, Stoye JP, Crow YJ, Taylor IA, Webb M, 2011. HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480, 379–382. [DOI] [PubMed] [Google Scholar]
- Goodman AG, Smith JA, Balachandran S, Perwitasari O, Proll SC, Thomas MJ, Korth MJ, Barber GN, Schiff LA, Katze MG, 2007. The cellular protein P58IPK regulates influenza virus mrna translation and replication through a PKR-mediated mechanism. J. Virol 81, 2221–2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AG, Fornek JL, Medigeshi GR, Perrone LA, Peng X, Dyer MD, Proll SC, Knoblaugh SE, Carter VS, Korth MJ, Nelson JA, Tumpey TM, Katze MG, 2009. P58IPK: A novel “CIHD” member of the host innate defense response against pathogenic virus infection. PLoS Pathog 5, e100438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AG, Tanner BCW, Chang ST, Esteban M, Katze MG, 2011. Virus infection rapidly activates the P58(IPK) pathway, delaying peak kinase activation to enhance viral replication. Virology 417, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotoh B, Takeuchi K, Komatsu T, Yokoo J, 2003. The STAT2 activation process is a crucial target of Sendai virus C protein for the blockade of alpha interferon signaling. J. Virol 77, 3360–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goubau D, Schlee M, Deddouche S, Pruijssers AJ, Zillinger T, Goldeck M, Schuberth C, Van der Veen AG, Fujimura T, Rehwinkel J, Iskarpatyoti JA, Barchet W, Ludwig J, Dermody TS, Hartmann G, Reis E, Sousa C, 2014. Ant-iviral immunity via RIG-I-mediated recognition of RNA bearing 5′-diphosphates. Nature 514, 372–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graff JW, Ettayebi K, Hardy ME, 2009. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog 5, e1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gram AM, Sun C, Landman SL, Oosenbrug T, Koppejan HJ, Kwakkenbos MJ, Hoeben RC, Paludan SR, Ressing ME, 2017. Human B cells fail to secrete type I interferons upon cytoplasmic DNA exposure. Mol. Immunol 91, 225–237. [DOI] [PubMed] [Google Scholar]
- Grant A, Ponia SS, Tripathi S, Balasubramaniam V, Miorin L, Sourisseau M, Schwarz MC, Sánchez-Seco MP, Evans MJ, Best SM, García-Sastre A, 2016. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 19, 882–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Treuting PM, Woodward JJ, Stetson DB, 2015. Cutting edge: cGAS is required for lethal autoimmune disease in the Trex1-deficient mouse model of Aicardi–Goutières syndrome. J. Immunol 195, 1939–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Winship D, Snyder JM, Child SJ, Geballe AP, Stetson DB, 2016. The AIM2-like receptors are dispensable for the interferon response to intracellular DNA. Immunity 45, 255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RR, Wilkins C, Pattabhi S, Dong R, Loo Y, Gale M Jr., 2016. Transcriptional analysis of antiviral small molecule therapeutics as agonists of the RLR pathway. Genom. Data 7, 290–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grief C, Galler R, Cortes LM, Barth OM, 1997. Intracellular localisation of dengue-2 RNA in mosquito cell culture using electron microscopic in situ hybridisation. Arch. Virol 142, 2347–2357. [DOI] [PubMed] [Google Scholar]
- Gries CM, Bruger EL, Moormeier DE, Scherr TD, Waters CM, Kielian T, 2016. Cyclic di-AMP released from Staphylococcus aureus biofilm induces a macrophage type I interferon response. Infect. Immun 84, 3564–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieves JL, Fye JM, Harvey S, Grayson JM, Hollis T, Perrino FW, 2015. Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proc. Natl. Acad. Sci. U.S.A 112, 5117–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm WA, Messer JS, Murphy SF, Nero T, Lodolce JP, Weber CR, Logsdon MF, Bartulis S, Sylvester BE, Springer A, Dougherty U, Niewold TB, Kupfer SS, Ellis N, Huo D, Bissonnette M, Boone DL, 2016. The Thr300Ala variant in ATG16L1 is associated with improved survival in human colorectal cancer and enhanced production of type I interferon. Gut 65, 456–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther C, Kind B, Reijns MAM, Berndt N, Martinez-Bueno M, Wolf C, Tüngler V, Chara O, Lee YA, Hübner N, Bicknell L, Blum S, Krug C, Schmidt F, Kretschmer S, Koss S, Astell KR, Ramantani G, Bauerfeind A, Morris DL, Cunninghame Graham DS, Bubeck D, Leitch A, Ralston SH, Blackburn EA, Gahr M, Witte T, Vyse TJ, Melchers I, Mangold E, No€then MM, Aringer M, Kuhn A, Lüthke K, Unger L, Bley A, Lorenzi A, Isaacs JD, Alexopoulou D, Conrad K, Dahl A, Roers A, Alarcon-Riquelme ME, Jackson AP, Lee-Kirsch MA, 2015. Defective removal of ribonucleotides from DNA promotes systemic autoimmunity. J. Clin. Investig 125, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo F, Tang L, Shu S, Sehgal M, Sheraz M, Liu B, Zhao Q, Cheng J, Zhao X, Zhou T, Chang J, Guo JT, 2017. Activation of stimulator of interferon genes in hepatocytes suppresses the replication of hepatitis B virus. Antimicrob. Agents Chemother 61, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haag SM, Gulen MF, Reymond L, Gibelin A, Abrami L, Decout A, Heymann M, Goot FGV, Turcatti G, Behrendt R, Ablasser A, 2018. Targeting STING with covalent small-molecule inhibitors. Nature 559, 269–273. [DOI] [PubMed] [Google Scholar]
- Habjan M, Andersson I, Klingström J, Schümann M, Martin A, Zimmermann P, Wagner V, Pichlmair A, Schneider U, Mühlberger E, Mirazimi A, Weber F, 2008. Processing of genome 5′ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS One 3, e2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J, Brault A, Vincent F, Weng S, Wang H, Dumlao D, Aulabaugh A, Aivazian D, Castro D, Chen M, Culp J, Dower K, Gardner J, Hawrylik S, Golenbock D, Hepworth D, Horn M, Jones L, Jones P, Latz E, Li J, Lin LL, Lin W, Lin D, Lovering F, Niljanskul N, Nistler R, Pierce B, Plotnikova O, Schmitt D, Shanker S, Smith J, Snyder W, Subashi T, Trujillo J, Tyminski E, Wang G, Wong J, Lefker B, Dakin L, Leach K, 2017. Discovery of PF-06928215 as a high affinity inhibitor of cGAS enabled by a novel fluorescence polarization assay. PLoS One 12, e0184843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT, 2008. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol 9, 857–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamel R, Dejarnac O, Wichit S, Ekchariyawat P, Neyret A, Luplertlop N, Perera-Lecoin M, Surasombatpattana P, Talignani L, Thomas F, Cao-Lormeau V-M, Choumet V, Briant L, Desprès P, Amara A, Yssel H, Missé D, 2015. Biology of Zika virus infection in human skin cells. J. Virol 89, 8880–8896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammerbeck DM, Burleson GR, Schuller CJ, Vasilakos JP, Tomai M, Egging E, Cochran FR, Woulfe S, Miller RL, 2007. Administration of a dual toll-like receptor 7 and toll-like receptor 8 agonist protects against influenza in rats. Antiviral Res 73, 1–11. [DOI] [PubMed] [Google Scholar]
- Hampe J, Cuthbert A, Croucher PJ, Mirza MM, Mascheretti S, Fisher S, Frenzel H, King K, Hasselmeyer A, MacPherson AJ, Bridger S, van Deventer S, Forbes A, Nikolaus S, Lennard-Jones JE, Foelsch UR, Krawczak M, Lewis C, Schreiber S, Mathew CG, 2001a. Association between insertion mutation in NOD2 gene and Crohn’s disease in German and British populations. Lancet 357, 1925–1928. [DOI] [PubMed] [Google Scholar]
- Hampe J, Lynch NJ, Daniels S, Bridger S, Macpherson AJ, Stokkers P, Forbes A, Lennard-Jones JE, Mathew CG, Curran ME, Schreiber S, 2001b. Fine mapping of the chromosome 3p susceptibility locus in inflammatory bowel disease. Gut 48, 191–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanamsagar R, Aldrich A, Kielian T, 2014. Critical role for the AIM2 inflammasome during acute CNS bacterial infection. J. Neurochem 129, 704–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansdottir S, Monick MM, Lovan N, Powers L, Gerke A, Hunninghake GW, 2010. Vitamin D decreases respiratory syncytial virus induction of NF-kappaB-linked chemokines and cytokines in airway epithelium while maintaining the antiviral state. J. Immunol 184, 965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, Jungries R, Chung P, Plesken H, Sabatini DD, Ron D, 2001. Diabetes mellitus and exocrine pancreatic dysfunction in perk mice reveals a role for translational control in secretory cell survival. Mol. Cell 7, 1153–1163. [DOI] [PubMed] [Google Scholar]
- Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA, 2017. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 548, 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartman AL, Ling L, Nichol ST, Hibberd ML, 2008. Whole-genome expression profiling reveals that inhibition of host innate immune response pathways by Ebola virus can be reversed by a single amino acid change in the VP35 protein. J. Virol 82, 5348–5358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann G, 2017. Nucleic acid immunity. In: Advances in Immunology Elsevier. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Shi LZ, Truong LN, Lu C-S, Razavian N, Li Y, Negrete A, Shiloach J, Berns MW, Wu X, 2012. Rad50 zinc hook is important for the Mre11 complex to bind chromosomal DNA double-stranded breaks and initiate various DNA damage responses. J. Biol. Chem 287, 31747–31756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Chen Y, Wu Y, Xu Y, Zhang Z, Liu Z, 2017. Nucleic acid sensing pattern recognition receptors in the development of colorectal cancer and colitis. Cell. Mol. Life Sci 74, 2395–2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S, 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303, 1526–1529. [DOI] [PubMed] [Google Scholar]
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S, 2000. A toll-like receptor recognizes bacterial DNA. Nature 408, 740–745. [DOI] [PubMed] [Google Scholar]
- Hiller B, Achleitner M, Glage S, Naumann R, Behrendt R, Roers A, 2012. Mammalian RNase H2 removes ribonucleotides from DNA to maintain genome integrity. J. Exp. Med 209, 1419–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochrein H, Schlatter B, O’Keeffe M, Wagner C, Schmitz F, Schiemann M, Bauer S, Suter M, Wagner H, 2004. Herpes simplex virus type-1 induces IFN-production via Toll-like receptor 9-dependent and -independent pathways. Proc. Natl. Acad. Sci. U.S.A 101, 11416–11421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD, 2001. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and muckle-wells syndrome. Nat. Genet 29, 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hokeness-Antonelli KL, Crane MJ, Dragoi AM, Chu W-M, Salazar-Mather TP, 2007. IFN-alphabeta-mediated inflammatory responses and antiviral defense in liver is TLR9-independent but MyD88-dependent during murine cytomegalovirus infection. J. Immunol 179, 6176–6183. [DOI] [PubMed] [Google Scholar]
- Honda K, Takaoka A, Taniguchi T, 2006. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity 25, 349–360. [DOI] [PubMed] [Google Scholar]
- Horan KA, Hansen K, Jakobsen MR, Holm CK, Soby S, Unterholzner L, Thompson M, West JA, Iversen MB, Rasmussen SB, Ellermann-Eriksen S, Kurt-Jones E, Landolfo S, Damania B, Melchjorsen J, Bowie AG, Fitzgerald KA, Paludan SR, 2013. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol 190, 2311–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdorfer B, Giese T, Endres S, Hartmann G, 2002. Quantitative expression of toll-like receptor 1–10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol 168, 4531–4537. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, Poeck H, Akira S, Conzelmann K-K, Schlee M, Endres S, Hartmann G, 2006. 5′-Triphosphate RNA is the ligand for RIG-I. Science 314, 994–997. [DOI] [PubMed] [Google Scholar]
- Hoss M, 1999. A human DNA editing enzyme homologous to the Escherichia coli DnaQ/MutD protein. EMBO J 18, 3868–3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Yue Y, Li D, Zhao Y, Qiu L, Chen J, Pan Y, Xi J, Wang X, Sun Q, Li Q, 2016. Antibody-dependent enhancement of dengue virus infection inhibits RLR-mediated Type-I IFN-independent signalling through upregulation of cellular autophagy. Sci. Rep 6, 22303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G, 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603. [DOI] [PubMed] [Google Scholar]
- Inohara N, Ogura Y, Chen FF, Muto A, Nunez G, 2001. Human Nod1 confers responsiveness to bacterial lipopolysaccharides. J. Biol. Chem 276, 2551–2554. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN, 2008. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, Barber GN, 2009. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israel A, 2010. The IKK complex, a central regulator of NF-κB activation. Cold Spring Harb. Perspect. Biol 2, a000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang M-A, Kim EK, Now H, Nguyen NTH, Kim W-J, Yoo J-Y, Lee J, Jeong Y-M, Kim C-H, Kim O-H, Sohn S, Nam S-H, Hong Y, Lee YS, Chang S-A, Jang SY, Kim J-W, Lee M-S, Lim SY, Sung K-S, Park K-T, Kim BJ, Lee J-H, Kim D-K, Kee C, Ki C-S, 2015. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am. J. Hum. Genet 96, 266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen R, Bont L, Siezen CLE, Hodemaekers HM, Ermers MJ, Doornbos G, van ‘t Slot R, Wijmenga C, Goeman JJ, Kimpen JLL, van Houwelingen HC, Kimman TG, Hoebee B, 2007. Genetic susceptibility to respiratory syncytial virus bronchiolitis is predominantly associated with innate immune genes. J. Infect. Dis 196, 826–834. [DOI] [PubMed] [Google Scholar]
- Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg M-C, Goudin N, Frémond M-L, Nitschke P, Molina TJ, Blanche S, Picard C, Rice GI, Crow YJ, Manel N, Fischer A, Bader-Meunier B, Rieux-Laucat F, 2014. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Investig 124, 5516–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Zhu Y, Liu Z-J, Ouyang S, 2017. The emerging roles of the DDX41 protein in immunity and diseases. Protein Cell 8, 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC, 2008. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol. Cell. Biol 28, 5014–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E, 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KE, Bottero V, Flaherty S, Dutta S, Singh VV, Chandran B, 2014. IFI16 restricts HSV-1 replication by accumulating on the HSV-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog 10, e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, Chan S, Dong J, Qu Y, Roose-Girma M, Dixit VM, Monack DM, 2010. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. U.S.A 107, 9771–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin K-Q, Ishii KJ, Kawai T, Akira S, Suzuki K, Okuda K, 2007. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc. Natl. Acad. Sci. U.S.A 104, 14050–14055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junjhon J, Pennington JG, Edwards TJ, Perera R, Lanman J, Kuhn RJ, 2014. Ultrastructural characterization and three-dimensional architecture of replication sites in dengue virus-infected mosquito cells. J. Virol 88, 4687–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junkins RD, Gallovic MD, Johnson BM, Collier MA, Watkins-Schulz R, Cheng N, David CN, McGee CE, Sempowski GD, Shterev I, McKinnon K, Bachelder EM, Ainslie KM, Ting JPY, 2018. A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J. Control. Release 270, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadaja M, Isok-Paas H, Laos T, Ustav E, Ustav M, 2009. Mechanism of genomic instability in cells infected with the high-risk human papillomaviruses. PLoS Pathog 5, e1000397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki N, Antonenko S, Lau JY-N, Liu Y-J, 2000. Natural interferon α/β-producing cells link innate and adaptive immunity. J. Exp. Med 192, 219–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalamvoki M, Roizman B, 2014. HSV-1 degrades, stabilizes, requires, or is stung by STING depending on ICP0, the US3 protein kinase, and cell derivation. Proc. Natl. Acad. Sci. U.S.A 111, E611–E617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang D-C, Gopalkrishnan RV, Lin L, Randolph A, Valerie K, Pestka S, Fisher PB, 2004. Expression analysis and genomic characterization of human melanoma differentiation associated gene-5, mda-5: a novel type I interferon-responsive apoptosis-inducing gene. Oncogene 23, 1789–1800. [DOI] [PubMed] [Google Scholar]
- Kanneganti T-D, Őzören N, Body-Malapel M, Amer A, Park J-H, Franchi L, Whitfield J, Barchet W, Colonna M, Vandenabeele P, Bertin J, Coyle A, Grant EP, Akira S, Núñez G, 2006. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature 440, 233–236. [DOI] [PubMed] [Google Scholar]
- Karaolis DKR, Means TK, Yang D, Takahashi M, Yoshimura T, Muraille E, Philpott D, Schroeder JT, Hyodo M, Hayakawa Y, Talbot BG, Brouillette E, Malouin F, 2007a. Bacterial c-di-GMP is an immunostimulatory molecule. J. Immunol 178, 2171–2181. [DOI] [PubMed] [Google Scholar]
- Karaolis DKR, Newstead MW, Zeng X, Hyodo M, Hayakawa Y, Bhan U, Liang H, Standiford TJ, 2007b. Cyclic di-GMP stimulates protective innate immunity in bacterial pneumonia. Infect. Immun 75, 4942–4950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasama Y, Saito M, Takano T, Nishimura T, Satoh M, Wang Z, Ali SNES, Harada S, Kohara M, Tsukiyama-Kohara K, 2012. Translocase of outer mitochondrial membrane 70 induces interferon response and is impaired by hepatitis C virus NS3. Virus Res 163, 405–409. [DOI] [PubMed] [Google Scholar]
- Kash JC, Mühlberger E, Carter V, Grosch M, Perwitasari O, Proll SC, Thomas MJ, Weber F, Klenk H-D, Katze MG, 2006. Global suppression of the host antiviral response by Ebola-and Marburgviruses: increased antagonism of the type I interferon response is associated with enhanced virulence. J. Virol 80, 3009–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh C-S, Reis e Sousa C, Matsuura Y, Fujita T, Akira S, 2006. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, Hiiragi A, Dermody TS, Fujita T, Akira S, 2008. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med 205, 1601–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katze MG, DeCorato D, Safer B, Galabru J, Hovanessian AG, 1987. Adenovirus VAI RNA complexes with the 68 000 Mr protein kinase to regulate its autophosphorylation and activity. EMBO J 6, 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Akira S, 2009. The roles of TLRs, RLRs and NLRs in pathogen recognition. Int. Immunol 21, 317–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, Takeuchi O, Akira S, 2004. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol 5, 1061–1068. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, Ishii KJ, Takeuchi O, Akira S, 2005. IPS-1, an adaptor triggering RIG-I-and Mda5-mediated type I interferon induction. Nat. Immunol 6, 981–988. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Kawai T, Akira S, 2011. Recognition of nucleic acids by pattern-recognition receptors and its relevance in autoimmunity: nucleic acid recognition and autoimmunity. Immunol. Rev 243, 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerur N, Veettil MV, Sharma-Walia N, Bottero V, Sadagopan S, Otageri P, Chandran B, 2011. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to kaposi sarcoma-associated herpesvirus infection. Cell Host Microbe 9, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Bauernfeind F, Ablasser A, Hartmann G, Fitzgerald KA, Latz E, Hornung V, 2010a. Listeria monocytogenes is sensed by the NLRP3 and AIM2 inflammasome. Eur. J. Immunol 40, 1545–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim T, Pazhoor S, Bao M, Zhang Z, Hanabuchi S, Facchinetti V, Bover L, Plumas J, Chaperot L, Qin J, Liu YJ, 2010b. Aspartate-glutamate-alanine-histidine box motif (DEAH)/RNA helicase A helicases sense microbial DNA in human plas-macytoid dendritic cells. Proc. Natl. Acad. Sci. U.S.A 107, 15181–15186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kind B, Muster B, Staroske W, Herce HD, Sachse R, Rapp A, Schmidt F, Koss S, Cardoso MC, Lee-Kirsch MA, 2014. Altered spatio-temporal dynamics of RNase H2 complex assembly at replication and repair sites in Aicardi–Goutières syndrome. Hum. Mol. Genet 23, 5950–5960. [DOI] [PubMed] [Google Scholar]
- Kitajewski J, Schneider RJ, Safer B, Munemitsu SM, Samuel CE, Thimmappaya B, Shenk T, 1986. Adenovirus VAI RNA antagonizes the antiviral action of interferon by preventing activation of the interferon-induced eIF-2 alpha kinase. Cell 45, 195–200. [DOI] [PubMed] [Google Scholar]
- Klein Klouwenberg P, Tan L, Werkman W, van Bleek GM, Coenjaerts F, 2009. The role of Toll-like receptors in regulating the immune response against respiratory syncytial virus. Crit. Rev. Immunol 29, 531–550. [DOI] [PubMed] [Google Scholar]
- Knip M, Veijola R, Virtanen SM, Hyo€ty H, Vaarala O, Akerblom HK, 2005. Environmental triggers and determinants of type 1 diabetes. Diabetes 54 (Suppl. 2), S125–S136. [DOI] [PubMed] [Google Scholar]
- Komuro A, Horvath CM, 2006. RNA-and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J. Virol 80, 12332–12342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo T, Kobayashi J, Saitoh T, Maruyama K, Ishii KJ, Barber GN, Komatsu K, Akira S, Kawai T, 2013. DNA damage sensor MRE11 recognizes cytosolic double-stranded DNA and induces type I interferon by regulating STING trafficking. Proc. Natl. Acad. Sci. U.S.A 110, 2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- König N, Fiehn C, Wolf C, Schuster M, Cura Costa E, Tüngler V, Alvarez HA, Chara O, Engel K, Goldbach-Mansky R, Günther C, Lee-Kirsch MA, 2017. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann. Rheum. Dis 76, 468–472. [DOI] [PubMed] [Google Scholar]
- Konno H, Chinn IK, Hong D, Orange JS, Lupski JR, Mendoza A, Pedroza LA, Barber GN, 2018. Pro-inflammation associated with a gain-of-function mutation (R284S) in the innate immune sensor STING. Cell Rep 23, 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranzusch PJ, Lee AS-Y, Berger JM, Doudna JA, 2013. Structure of human cGAS reveals a conserved family of second-messenger enzymes in innate immunity. Cell Rep 3, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer S, Wolf C, König N, Staroske W, Guck J, H€ausler M, Luksch H, Nguyen LA, Kim B, Alexopoulou D, Dahl A, Rapp A, Cardoso MC, Shevchenko A, Lee-Kirsch MA, 2015. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann. Rheum. Dis 74, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krug A, French AR, Barchet W, Fischer JAA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M, 2004. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity 21, 107–119. [DOI] [PubMed] [Google Scholar]
- Kumar A, Haque J, Lacoste J, Hiscott J, Williams BR, 1994. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc. Natl. Acad. Sci. U.S.A 91, 6288–6292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Akira S, 2009. Toll-like receptors and innate immunity. Biochem. Biophys. Res. Commun 388, 621–625. [DOI] [PubMed] [Google Scholar]
- Kumar M, Roe K, Nerurkar PV, Namekar M, Orillo B, Verma S, Nerurkar VR, 2012. Impaired virus clearance, compromised immune response and increased mortality in type 2 diabetic mice infected with west nile virus. PLoS One 7, e44682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A, Hou S, Airo AM, Limonta D, Mancinelli V, Branton W, Power C, Hobman TC, 2016. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep 17, 1766–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurien BT, D’Sousa A, Bruner BF, Gross T, James JA, Targoff IN, Maier-Moore JS, Harley IT, Wang H, Scofield RH, 2013. Prolidase deficiency breaks tolerance to lupus-associated antigens. Int. J. Rheum. Dis 16, 674–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labbe P, Faure E, Lecointe S, Le Scouarnec S, Kyndt F, Marrec M, Le Tourneau T, Offmann B, Duplaa C, Zaffran S, Schott JJ, Merot J, 2017. The alternatively spliced LRRFIP1 isoform-1 is a key regulator of the Wnt/β-catenin transcription pathway. Biochim. Biophys Acta 1864, 1142–1152. [DOI] [PubMed] [Google Scholar]
- Ladiges WC, Knoblaugh SE, Morton JF, Korth MJ, Sopher BL, Baskin CR, MacAuley A, Goodman AG, LeBoeuf RC, Katze MG, 2005. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 54, 1074–1081. [DOI] [PubMed] [Google Scholar]
- Lahaye X, Satoh T, Gentili M, Cerboni S, Conrad C, Hurbain I, El Marjou A, Lacabaratz C, Lelièvre J-D, Manel N, 2013. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity 39, 1132–1142. [DOI] [PubMed] [Google Scholar]
- Lahouassa H, Daddacha W, Hofmann H, Ayinde D, Logue EC, Dragin L, Bloch N, Maudet C, Bertrand M, Gramberg T, Pancino G, Priet S, Canard B, Laguette N, Benkirane M, Transy C, Landau NR, Kim B, Margottin-Goguet F, 2012. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol 13, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam E, Falck-Pedersen E, 2014. Unabated adenovirus replication following activation of the cGAS/STING-dependent antiviral response in human cells. J. Virol 88, 14426–14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam E, Stein S, Falck-Pedersen E, 2014. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J. Virol 88, 974–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landtblom A-M, Fazio P, Fredrikson S, Granieri E, 2010. The first case history of multiple sclerosis: Augustus d’Esté (1794–1848). Neurol. Sci 31, 29–33. [DOI] [PubMed] [Google Scholar]
- Latz E, Schoenemeyer A, Visintin A, Fitzgerald KA, Monks BG, Knetter CF, Lien E, Nilsen NJ, Espevik T, Golenbock DT, 2004. TLR9 signals after translocating from the ER to CpG DNA in the lysosome. Nat. Immunol 5, 190–198. [DOI] [PubMed] [Google Scholar]
- Latz E, Verma A, Visintin A, Gong M, Sirois CM, Klein DCG, Monks BG, McKnight CJ, Lamphier MS, Duprex WP, Espevik T, Golenbock DT, 2007. Ligand-induced conformational changes allosterically activate Toll-like receptor 9. Nat. Immunol 8, 772–779. [DOI] [PubMed] [Google Scholar]
- Lazear HM, Govero J, Smith AM, Platt DJ, Fernandez E, Miner JJ, Diamond MS, 2016. A mouse model of zika virus pathogenesis. Cell Host Microbe 19, 720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TG, Tomita J, Hovanessian AG, Katze MG, 1990. Purification and partial characterization of a cellular inhibitor of the interferon-induced protein kinase of Mr 68,000 from influenza virus-infected cells. Proc. Natl. Acad. Sci. U.S.A 87, 6208–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TG, Tomita J, Hovanessian AG, Katze MG, 1992. Characterization and regulation of the 58,000-dalton cellular inhibitor of the interferon-induced, dsRNA-activated protein kinase. J. Biol. Chem 267, 14238–14243. [PubMed] [Google Scholar]
- Lee AS, Ghoreishi M, Cheng WK, Chang TY, Zhang YQ, Dutz JP, 2011. Toll-like receptor 7 stimulation promotes autoimmune diabetes in the NOD mouse. Diabetologia 54, 1407–1416. [DOI] [PubMed] [Google Scholar]
- Lee I-K, Hsieh C-J, Chen R-F, Yang Z-S, Wang L, Chen C-M, Liu C-F, Huang C-H, Lin C-Y, Chen Y-H, Yang KD, Liu J-W, 2013. Increased production of interleukin-4, interleukin-10, and granulocyte-macrophage colony-stimulating factor by type 2 diabetes’ mononuclear cells infected with dengue virus, but not increased intracellular viral multiplication. Biomed. Res. Int 2013, 965853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K-G, Kim SS-Y, Kui L, Voon DC-C, Mauduit M, Bist P, Bi X, Pereira NA, Liu C, Sukumaran B, Rénia L, Ito Y, Lam K-P, 2015. Bruton’s tyrosine kinase phosphorylates DDX41 and activates its binding of dsDNA and STING to initiate type 1 interferon response. Cell Rep 10, 1055–1065. [DOI] [PubMed] [Google Scholar]
- Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, de Silva U, Bailey SL, Witte T, Vyse TJ, Kere J, Pfeiffer C, Harvey S, Wong A, Koskenmies S, Hummel O, Rohde K, Schmidt RE, Dominiczak AF, Gahr M, Hollis T, Perrino FW, Lieberman J, Hübner N, 2007. Mutations in the gene encoding the 3’−5’ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat. Genet 39 (9), 1065–1077. [DOI] [PubMed] [Google Scholar]
- Lehtinen DA, Harvey S, Mulcahy MJ, Hollis T, Perrino FW, 2008. The TREX1 double-stranded DNA degradation activity is defective in dominant mutations associated with autoimmune disease. J. Biol. Chem 283, 31649–31656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos H, Huang L, Chandler PR, Mohamed E, Souza GR, Li L, Pacholczyk G, Barber GN, Hayakawa Y, Munn DH, Mellor AL, 2014. Activation of the STING adaptor attenuates experimental autoimmune encephalitis. J. Immunol 192, 5571–5578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenert P, 2010. Nucleic acid sensing receptors in systemic lupus erythematosus: development of novel DNA-and/or RNA-like analogues for treating lupus. Clin. Exp. Immunol 161 (2), 208–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung LW, Park MS, Martinez O, Valmas C, Lopez CB, Basler CF, 2011. Ebolavirus VP35 suppresses IFN production from conventional but not plasmacytoid dendritic cells. Immunol. Cell Biol 89, 792–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-D, Sun L, Seth RB, Pineda G, Chen ZJ, 2005. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. U.S.A 102, 17717–17722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X-L, Ezelle HJ, Hsi TY, Hassel BA, 2011. A central role for RNA in the induction and biological activities of type 1 interferons. Wiley Interdiscip. Rev. RNA 2, 58–78. [DOI] [PubMed] [Google Scholar]
- Li Y, Berke IC, Modis Y, 2012a. DNA binding to proteolytically activated TLR9 is sequence-independent and enhanced by DNA curvature: ligand-binding study of cleaved TLR9. EMBO J 31, 919–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chen R, Zhou Q, Xu Z, Li C, Wang S, Mao A, Zhang X, He W, Shu HB, 2012b. LSm14A is a processing body-associated sensor of viral nucleic acids that initiates cellular antiviral response in the early phase of viral infection. Proc. Natl. Acad. Sci. U.S.A 109, 11770–11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ, 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wu Y, Zheng X, Cong J, Liu Y, Li J, Sun R, Tian ZG, Wei HM, 2016. Cytoplasm-translocated Ku70/80 complex sensing of HBV DNA induces hepatitis-associated chemokine secretion. Front. Immunol 7, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Tan X, Zhang J, Li S, Mo W, Han T, Kuang W, Zhou Y, Deng J, 2017a. Gene mutations and clinical phenotypes in 15 Chinese children with cryopyrin-associated periodic syndrome (CAPS). Sci. China Life Sci 60, 1436–1444. [DOI] [PubMed] [Google Scholar]
- Li P, Du J, Goodier JL, Hou J, Kang J, Kazazian HH Jr., Zhao K, Yu XF, 2017b. Aicardi-Goutieres syndrome protein TREX1 suppresses L1 and maintains genome integrity through exonuclease-independent ORF1p depletion. Nucleic Acids Res 45, 4619–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wilson HL, Kiss-Toth E, 2017c. Regulating STING in health and disease. J. Inflamm 14, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang D, Xiao-Feng H, Guan-Jun D, Er-Ling H, Sheng C, Ting-Ting W, Qin-Gang H, Yan-Hong N, Ya-Yi H, 2015. Activated STING enhances Tregs infiltration in the HPV-related carcinogenesis of tongue squamous cells via the c-jun/CCL22 signal. Biochim. Biophys. Acta 1852, 2494–2503. [DOI] [PubMed] [Google Scholar]
- Liao YR, Li ZJ, Zeng P, Lan YQ, 2017. TLR7 deficiency contributes to attenuated diabetic retinopathy via inhibition of inflammatory response. Biochem. Biophys. Res. Commun 493, 1136–1142. [DOI] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR, 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349, 1115–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieu KG, Marsh GA, Wang L-F, Netter HJ, 2015. The non-pathogenic Henipavirus Cedar paramyxovirus phosphoprotein has a compromised ability to target STAT1 and STAT2. Antiviral Res 124, 69–76. [DOI] [PubMed] [Google Scholar]
- Lim WH, Kireta S, Russ GR, Coates PTH, 2006. Human plasmacytoid dendritic cells regulate immune responses to Epstein-Barr virus (EBV) infection and delay EBV-related mortality in humanized NOD-SCID mice. Blood 109, 1043–1050. [DOI] [PubMed] [Google Scholar]
- Lin J, Kumari S, Kim C, Van TM, Wachsmuth L, Polykratis A, Pasparakis M, 2016. RIPK1 counteracts ZBP1-mediated necroptosis to inhibit inflammation. Nature 540, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lincez PJ, Shanina I, Horwitz MS, 2015. Reduced expression of the MDA5 Gene IFIH1 prevents autoimmune diabetes. Diabetes 64, 2184–2193. [DOI] [PubMed] [Google Scholar]
- Lindahl T, Barnes DE, Yang Y-G, Robins P, 2009. Biochemical properties of mam-malian TREX1 and its association with DNA replication and inherited inflammatory disease. Biochem. Soc. Trans 37, 535–538. [DOI] [PubMed] [Google Scholar]
- Linder P, 2006. Dead-box proteins: a family affair—active and passive players in RNP-remodeling. Nucleic Acids Res 34, 4168–4180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lio C-WJ, McDonald B, Takahashi M, Dhanwani R, Sharma N, Huang J, Pham E, Benedict CA, Sharma S, 2016. cGAS-STING signaling regulates initial innate control of cytomegalovirus infection. J. Virol 90, 7789–7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisnevskaia L, Murphy G, Isenberg D, 2014. Systemic lupus erythematosus. Lancet 384, 1878–1888. [DOI] [PubMed] [Google Scholar]
- Liu S, Wang H, Jin Y, Podolsky R, Reddy MVPL, Pedersen J, Bode B, Reed J, Steed D, Anderson S, Yang P, Muir A, Steed L, Hopkins D, Huang Y, Purohit S, Wang C-Y, Steck AK, Montemari A, Eisenbarth G, Rewers M, She J-X, 2009. IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum. Mol. Genet 18, 358–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, Lee C-CR, DiMattia MA, Cowen EW, Gonzalez B, Palmer I, DiGiovanna JJ, Biancotto A, Kim H, Tsai WL, Trier AM, Huang Y, Stone DL, Hill S, Kim HJ, St. Hilaire C, Gurprasad S, Plass N, Chapelle D, Horkayne-Szakaly I, Foell D, Barysenka A, Candotti F, Holland SM, Hughes JD, Mehmet H, Issekutz AC, Raffeld M, McElwee J, Fontana JR, Minniti CP, Moir S, Kastner DL, Gadina M, Steven AC, Wingfield PT, Brooks SR, Rosenzweig SD, Fleisher TA, Deng Z, Boehm M, Paller AS, Goldbach-Mansky R, 2014. Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med 371, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Cai X, Wu J, Cong Q, Chen X, Li T, Du F, Ren J, Wu YT, Grishin NV, Chen ZJ, 2015a. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zou Z, Zhu B, Hu Z, Zeng P, Wu L, 2015b. LRRFIP1 inhibits hepatitis C virus replication by inducing type I interferon in hepatocytes. Hepat. Mon 15, e28473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T-T, Yang Q, Li M, Zhong B, Ran Y, Liu L-L, Yang Y, Wang Y-Y, Shu H-B, 2016. LSm14A plays a critical role in antiviral immune responses by regulating MITA level in a cell-specific manner. J. Immunol 196, 5101–5111. [DOI] [PubMed] [Google Scholar]
- Liu Y, Seto NL, Carmona-Rivera C, Kaplan MJ, 2018. Accelerated model of lupus autoimmunity and vasculopathy driven by toll-like receptor 7/9 imbalance. Lupus Sci. Med 5, e000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd RM, Shatkin AJ, 1992. Translational stimulation by reovirus polypeptide sigma 3: substitution for VAI RNA and inhibition of phosphorylation of the alpha subunit of eukaryotic initiation factor 2. J. Virol 66, 6878–6884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb M, Eskandarian S, Rupp M, Fishman N, Gasink L, Patterson J, Bramson J, Hudson TJ, Lemire M, 2011. Genetic variants and susceptibility to neurological complications following West Nile virus infection. J. Infect. Dis 204, 1031–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loönnrot M, Korpela K, Knip M, Ilonen J, Simell O, Korhonen S, Savola K, Muona P, Simell T, Koskela P, Hyöty H, 2000. Enterovirus infection as a risk factor for beta-cell autoimmunity in a prospectively observed birth cohort: the Finnish diabetes prediction and prevention study. Diabetes 49, 1314–1318. [DOI] [PubMed] [Google Scholar]
- Loo Y-M, Gale M, 2011. Immune signaling by RIG-I-like receptors. Immunity 34, 680–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo Y-M, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, Akira S, Gill MA, García-Sastre A, Katze MG, Gale M, 2008. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J. Virol 82, 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Wambach M, Katze MG, Krug RM, 1995. Binding of the influenza virus NS1 protein to double-stranded RNA inhibits the activation of the protein kinase that phos-phorylates the elF-2 translation initiation factor. Virology 214, 222–228. [DOI] [PubMed] [Google Scholar]
- Lubick KJ, Robertson SJ, McNally KL, Freedman BA, Rasmussen AL, Taylor RT, Walts AD, Tsuruda S, Sakai M, Ishizuka M, Boer EF, Foster EC, Chiramel AI, Addison CB, Green R, Kastner DL, Katze MG, Holland SM, Forlino A, Freeman AF, Boehm M, Yoshii K, Best SM, 2015. Flavivirus antagonism of type I interferon signaling reveals prolidase as a regulator of IFNAR1 surface expression. Cell Host Microbe 18, 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludlow LEA, Johnstone RW, Clarke CJP, 2005. The HIN-200 family: more than interferon-inducible genes? Exp. Cell Res 308, 1–17. [DOI] [PubMed] [Google Scholar]
- Lundberg P, Welander P, Han X, Cantin E, 2003. Herpes simplex virus type 1 DNA is immunostimulatory in vitro and in vivo. J. Virol 77, 11158–11169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg M, Krogvold L, Kuric E, Dahl-Jorgensen K, Skog O, 2016. Expression of interferon-stimulated genes in insulitic pancreatic islets of patients recently diagnosed with type 1 diabetes. Diabetes 65, 3104–3110. [DOI] [PubMed] [Google Scholar]
- Luo H, Winkelmann ER, Fernandez-Salas I, Li L, Mayer SV, Danis-Lozano R, Sanchez-Casas RM, Vasilakis N, Tesh R, Barrett AD, Weaver SC, Wang T, 2018. Zika, dengue and yellow fever viruses induce differential anti-viral immune responses in human monocytic and first trimester trophoblast cells. Antiviral Res 151, 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthra P, Ramanan P, Mire CE, Weisend C, Tsuda Y, Yen B, Liu G, Leung DW, Geisbert TW, Ebihara H, Amarasinghe GK, Basler CF, 2013. Mutual antagonism between the Ebola virus VP35 protein and the RIG-I activator PACT determines infection outcome. Cell Host Microbe 14, 74–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie JM, Jones MK, Young PR, 1996. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 220, 232–240. [DOI] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B, Kilanowski F, Grimes G, Fluteau A, Devenney PS, Hill RE, Reijns MAM, Jackson AP, 2016. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J 35, 831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, Osborn RT, Wheeler AP, Nowotny M, Gilbert N, Chandra T, Reijns MAM, Jackson AP, 2017. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature 548, 461–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait J, Bridgeman A, Benlahrech A, Cursi C, Rehwinkel J, 2016. Restriction by SAMHD1 limits cGAS/STING-dependent innate and adaptive immune responses to HIV-1. Cell Rep 16, 1492–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maelfait J, Liverpool L, Bridgeman A, Ragan KB, Upton JW, Rehwinkel J, 2017. Sensing of viral and endogenous RNA by ZBP1/DAI induces necroptosis. EMBO J 36, 2529–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man SM, Karki R, Malireddi RKS, Neale G, Vogel P, Yamamoto M, Lamkanfi M, Kanneganti T-D, 2015. The transcription factor IRF1 and guanylate-binding proteins target activation of the AIM2 inflammasome by Francisella infection. Nat. Immunol 16, 467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellaker C, Vesely C, Ponting CP, McLaughlin PJ, Jantsch MF, Dorin J, Adams IR, Scadden AD, Ohman M, Keegan LP, O’Connell MA, 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9, 1482–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maringer K, Fernandez-Sesma A, 2014. Message in a bottle: lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor Rev 25, 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marr N, Wang T-I, Kam SHY, Hu YS, Sharma AA, Lam A, Markowski J, Solimano A, Lavoie PM, Turvey SE, 2014. Attenuation of respiratory syncytial virus-induced and RIG-I-dependent type I IFN responses in human neonates and very young children. J. Immunol 192, 948–957. [DOI] [PubMed] [Google Scholar]
- Martínez-Ramírez I, del-Castillo-Falconi V, Mitre-Aguilar I, Amador-Molina A, Carrillo-García A, Langley E, Zentella-Dehesa A, Soto-Reyes E, García-Carrancá A, Herrera L, Lizano M, 2017. SOX2 as a new regulator of HPV16 transcription. Viruses 9, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser RS, Monsen KJ, Nelms BE, Petrini JH, 1997. hMre11 and hRad50 nuclear foci are induced during the normal cellular response to DNA double-strand breaks. Mol. Cell. Biol 17, 6087–6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathes E, O’Dea EL, Hoffmann A, Ghosh G, 2008. NF-κB dictates the degradation pathway of IκBα. EMBO J 27, 1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathieu C, Guillaume V, Volchkova VA, Pohl C, Jacquot F, Looi RY, Wong KT, Legras-Lachuer C, Volchkov VE, Lachuer J, Horvat B, 2012. Non-structural Nipah virus C protein regulates both the early host proinflammatory response and viral virulence. J. Virol 86, 10766–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushima N, Tanaka T, Enkhbayar P, Mikami T, Taga M, Yamada K, Kuroki Y, 2007. Comparative sequence analysis of leucine-rich repeats (LRRs) within vertebrate toll-like receptors. BMC Genomics 8, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazur DJ, Perrino FW, 1999. Identification and expression of the TREX1 and TREX2 cDNA sequences encoding mammalian 3′ 5′ exonucleases. J. Biol. Chem 274, 19655–19660. [DOI] [PubMed] [Google Scholar]
- Mazur DJ, Perrino FW, 2001. Excision of 3′ termini by the Trex1 and TREX2 3′ 5′ exo-nucleases: characterization of the recombinant proteins. J. Biol. Chem 276, 17022–17029. [DOI] [PubMed] [Google Scholar]
- McCartney SA, Vermi W, Lonardi S, Rossini C, Otero K, Calderon B, Gilfillan S, Diamond MS, Unanue ER, Colonna M, 2011. RNA sensor-induced type I IFN prevents diabetes caused by a β cell–tropic virus in mice. J. Clin. Invest 121, 1497–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy AK, Erickson BR, Flietstra TD, Rollin PE, Nichol ST, Towner JS, Spiropoulou CF, 2014. Ebola hemorrhagic fever: novel biomarker correlates of clinical outcome. J. Infect. Dis 210, 558–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland HF, Martin R, 2007. Multiple sclerosis: a complicated picture of autoimmunity. Nat. Immunol 8, 913–919. [DOI] [PubMed] [Google Scholar]
- McWhirter SM, Barbalat R, Monroe KM, Fontana MF, Hyodo M, Joncker NT, Ishii KJ, Akira S, Colonna M, Chen ZJ, Fitzgerald KA, Hayakawa Y, Vance RE, 2009. A host type I interferon response is induced by cytosolic sensing of the bacterial second messenger cyclic-di-GMP. J. Exp. Med 206, 1899–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros AC, Soares CS, Coelho PO, Vieira NA, Baqui MMA, Teixeira FR, Gomes MD, 2018. DNA damage response signaling does not trigger redistribution of SAMHD1 to nuclear foci. Biochem. Biophys. Res. Commun 499, 790–796. [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA, 1998. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell 2, 253–258. [DOI] [PubMed] [Google Scholar]
- Melchjorsen J, Jensen SB, Malmgaard L, Rasmussen SB, Weber F, Bowie AG, Matikainen S, Paludan SR, 2005. Activation of innate defense against a paramyxo-virus is mediated by RIG-I and TLR7 and TLR8 in a cell-type-specific manner. J. Virol 79, 12944–12951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki I, Rose Y, Uggenti C, Van Eyck L, Fremond M-L, Kitabayashi N, Rice GI, Jenkinson EM, Boulai A, Jeremiah N, Gattorno M, Volpi S, Sacco O, Terheggen-Lagro SWJ, Tiddens HAWM, Meyts I, Morren M-A, De Haes P, Wouters C, Legius E, Corveleyn A, Rieux-Laucat F, Bodemer C, Callebaut I, Rodero MP, Crow YJ, 2017. Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J. Allergy Clin. Immunol 140 543–552.e5. [DOI] [PubMed] [Google Scholar]
- Melville MW, Tan SL, Wambach M, Song J, Morimoto RI, Katze MG, 1999. The cellular inhibitor of the PKR protein kinase, P58(IPK), is an influenza virus-activated co-chaperone that modulates heat shock protein 70 activity. J. Biol. Chem 274, 3797–3803. [DOI] [PubMed] [Google Scholar]
- Merkl PE, Orzalli M, Knipe DM, 2018. Mechanisms of host IFI16, PML and Daxx protein restriction of herpes simplex virus 1 replication. J. Virol 92, e00057–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier E, Wallet P, Dreier RF, Costanzo S, Anton L, Rühl S, Dussurgey S, Dick MS, Kistner A, Rigard M, Degrandi D, Pfeffer K, Yamamoto M, Henry T, Broz P, 2015. Guanylate-binding proteins promote activation of the AIM2 inflammasome during infection with Francisella novicida. Nat. Immunol 16, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi J, Ray P, Liu J, Kuan C-T, Xu J, Hsu D, Sullenger BA, White RR, Clary BM, 2016. In vivo selection against human colorectal cancer xenografts identifies an aptamer that targets RNA helicase protein DHX9. Mol. Ther. Nucleic Acids 5, e315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller S, Kastner S, Krijnse-Locker J, Buhler S, Bartenschlager R, 2007. The non-structural protein 4A of dengue virus is an integral membrane protein inducing membrane alterations in a 2K-regulated manner. J. Biol. Chem 282, 8873–8882. [DOI] [PubMed] [Google Scholar]
- Milutin Gašperov N, Farkas SA, Nilsson TK, Grce M, 2014. Epigenetic activation of immune genes in cervical cancer. Immunol. Lett 162, 256–257. [DOI] [PubMed] [Google Scholar]
- Mimori T, Hardin JA, Steitz JA, 1986. Characterization of the DNA-binding protein antigen Ku recognized by autoantibodies from patients with rheumatic disorders. J. Biol. Chem 261, 2274–2278. [PubMed] [Google Scholar]
- Minagar A, 2014. Multiple sclerosis: an overview of clinical features, pathophysiology, neuroimaging, and treatment options. Colloq. Ser. Integr. Syst. Physiol. Mol. Funct 6, 1–117. [Google Scholar]
- Miyabe H, Hyodo M, Nakamura T, Sato Y, Hayakawa Y, Harashima H, 2014. A new adjuvant delivery system ‘cyclic di-GMP/YSK05 liposome’ for cancer immunotherapy. J. Control. Release 184, 20–27. [DOI] [PubMed] [Google Scholar]
- Mondini M, Vidali M, Andrea MD, Azzimonti B, Airo P, D’Ambrosio R, Riboldi P, Meroni PL, Albano E, Shoenfeld Y, Gariglio M, Landolfo S, 2006. A novel autoantigen to differentiate limited cutaneous systemic sclerosis from diffuse cutaneous systemic sclerosis: the interferon-inducible gene IFI16. Arthritis Rheum 54, 3939–3944. [DOI] [PubMed] [Google Scholar]
- Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, Lamming DW, Chen ZJ, Horng T, Yeretssian G, Green DR, Blander JM, 2017. STING senses microbial viability to orchestrate stress-mediated autophagy of the endoplasmic reticulum. Cell 171 809–823.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyama M, Kato N, Otsuka M, Shao R-X, Taniguchi H, Kawabe T, Omata M, 2007. Interferon-beta is activated by hepatitis C virus NS5B and inhibited by NS4A, NS4B, and NS5A. Hepatol. Int 1, 302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morosky SA, Zhu J, Mukherjee A, Sarkar SN, Coyne CB, 2011. Retinoic acid-induced gene-I (RIG-I) associates with nucleotide-binding oligomerization domain-2 (NOD2) to negatively regulate inflammatory signaling. J. Biol. Chem 286, 28574–28583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison J, Laurent-Rolle M, Maestre AM, Rajsbaum R, Pisanelli G, Simon V, Mulder LCF, Fernandez-Sesma A, García-Sastre A, 2013. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog 9, e1003265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S, Lukacs NW, 2013. Innate immune responses to respiratory syncytial virus infection. Curr. Top. Microbiol. Immunol 372, 139–154. [DOI] [PubMed] [Google Scholar]
- Munoz J, Rodiere M, Jeremiah N, Rieux-Laucat F, Oojageer A, Rice GI, Rozenberg F, Crow YJ, Bessis D, 2015. Stimulator of interferon genes-associated vasculopathy with onset in infancy: a mimic of childhood granulomatosis with polyangiitis. JAMA Dermatol 151, 872. [DOI] [PubMed] [Google Scholar]
- Munoz-Culla M, Irizar H, Otaegui D, 2013. The genetics of multiple sclerosis: review of current and emerging candidates. Appl. Clin. Genet 6, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Jordan JL, Sánchez-Burgo GG Laurent-Rolle M, García-Sastre A, 2003. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. U.S.A 100, 14333–14338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami Y, Fukui R, Motoi Y, Kanno A, Shibata T, Tanimura N, Saitoh S, Miyake K, 2014. Roles of the cleaved N-terminal TLR3 fragment and cell surface TLR3 in double-stranded RNA sensing. J. Immunol 193, 5208–5217. [DOI] [PubMed] [Google Scholar]
- Murali A, Li X, Ranjith-Kumar CT, Bhardwaj K, Holzenburg A, Li P, Kao CC, 2008. Structure and function of LGP2, a DEX(D/H) helicase that regulates the innate immunity response. J. Biol. Chem 283, 15825–15833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama G, Furusawa N, Chiba A, Yamaji K, Tamura N, Miyake S, 2017. Enhanced IFN-alpha production is associated with increased TLR7 retention in the lyso-somes of palasmacytoid dendritic cells in systemic lupus erythematosus. Arthritis Res. Ther 19, 234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL, 2011. Silencing MicroRNA-155 ameliorates experimental autoimmune encephalomyelitis. J. Immunol 187, 2213–2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murugesapillai D, McCauley MJ, Maher LJ, Williams MC, 2017. Single-molecule studies of high-mobility group B architectural DNA bending proteins. Biophys. Rev 9, 17–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muruve DA, Petrilli V, Zaiss AK, White LR, Clark SA, Ross PJ, Parks RJ, Tschopp J, 2008. The inflammasome recognizes cytosolic microbial and host DNA and triggers an innate immune response. Nature 452, 103–107. [DOI] [PubMed] [Google Scholar]
- Mutlu S, Scully C, 1993. The person behind the eponym: Henrik Sjogren (1899–1986). J. Oral Pathol. Med 22, 439. [DOI] [PubMed] [Google Scholar]
- Nakaya Y, Lilue J, Stavrou S, Moran EA, Ross SR, 2017. AIM2-like receptors positively and negatively regulate the interferon response induced by cytosolic DNA. mBio 8, e00944–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namjou B, Kothari PH, Kelly JA, Glenn SB, Ojwang JO, Adler A, Alarcon-Riquelme ME, Gallant CJ, Boackle SA, Criswell LA, Kimberly RP, Brown E, Edberg J, Stevens AM, Jacob CO, Tsao BP, Gilkeson GS, Kamen DL, Merrill JT, Petri M, Goldman RR, Vila LM, Anaya JM, Niewold TB, Martin J, Pons-Estel BA, Sabio JM, Callejas JL, Vyse TJ, Bae SC, Perrino FW, Freedman BI, Scofield RH, Moser KL, Gaffney PM, James JA, Langefeld CD, Kaufman KM, Harley JB, Atkinson JP, 2011. Evaluation of the TREX1 gene in a large multi-ancestral lupus cohort. Genes Immun 12, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasirudeen AMA, Wong HH, Thien P, Xu S, Lam K-P, Liu DX, 2011. RIG-I, MDA5 and TLR3 synergistically play an important role in restriction of dengue virus infection. PLoS Negl. Trop. Dis 5, e926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, Todd JA, 2009. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324, 387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Wickliffe KE, Maltzman A, Dugger DL, Strasser A, Pham VC, Lill JR, Roose-Girma M, Warming S, Solon M, Ngu H, Webster JD, Dixit VM, 2016. RIPK1 inhibits ZBP1-driven necroptosis during development. Nature 540, 129–133. [DOI] [PubMed] [Google Scholar]
- Nguyen JB, Modis Y, 2013. Crystal structure of the dimeric coiled-coil domain of the cytosolic nucleic acid sensor LRRFIP1. J. Struct. Biol 181, 82–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas SA, Bubnov VV, Yasinska IM, Sumbayev VV, 2011. Involvement of xan-thine oxidase and hypoxia-inducible factor 1 in Toll-like receptor 7/8-mediated activation of caspase 1 and interleukin-1β. Cell. Mol. Life Sci 68, 151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen AE, Hantho JD, Mancini RJ, 2017. Synthetic agonists of NOD-like, RIG-I-like, and C-type lectin receptors for probing the inflammatory immune response. Future Med. Chem 9, 1345–1360. [DOI] [PubMed] [Google Scholar]
- Nitta S, Sakamoto N, Nakagawa M, Kakinuma S, Mishima K, Kusano-Kitazume A, Kiyohashi K, Murakawa M, Nishimura-Sakurai Y, Azuma S, Tasaka-Fujita M, Asahina Y, Yoneyama M, Fujita T, Watanabe M, 2013. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology 57, 46–58. [DOI] [PubMed] [Google Scholar]
- Novak N, Koch S, Allam JP, Bieber T, 2010. Dendritic cells: bridging innate and adaptive immunity in atopic dermatitis. J. Allergy Clin. Immunol 125, 50–59. [DOI] [PubMed] [Google Scholar]
- Ogunjimi B, Zhang S-Y, Sørensen KB, Skipper KA, Carter-Timofte M, Kerner G, Luecke S, Prabakaran T, Cai Y, Meester J, Bartholomeus E, Bolar NA, Vandeweyer G, Claes C, Sillis Y, Lorenzo L, Fiorenza RA, Boucherit S, Dielman C, Heynderickx S, Elias G, Kurotova A, Auwera AV, Verstraete L, Lagae L, Verhelst H, Jansen A, Ramet J, Suls A, Smits E, Ceulemans B, Van Laer L, Plat Wilson G, Kreth J, Picard C, Von Bernuth H, Fluss J, Chabrier S, Abel L, Mortier G, Fribourg S, Mikkelsen JG, Casanova J-L, Paludan SR, Mogensen TH, 2017. Inborn errors in RNA polymerase III underlie severe varicella zoster virus infections. J. Clin. Investig 127, 3543–3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH, 2001a. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606. [DOI] [PubMed] [Google Scholar]
- Ogura Y, Inohara N, Benito A, Chen FF, Yamaoka S, Nunez G, 2001b. Nod2, a Nod1/Apaf-1 family member that is restricted to monocytes and activates NF-kappaB. J. Biol. Chem 276, 4812–4818. [DOI] [PubMed] [Google Scholar]
- Ohman L, Franzén L, Rudolph U, Birnbaumer L, Hoörnquist EH, 2002. Regression of Peyer’s patches in G alpha i2 deficient mice prior to colitis is associated with reduced expression of Bcl-2 and increased apoptosis. Gut 51, 392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi H, Tochio H, Kato Z, Orii KE, Li A, Kimura T, Hiroaki H, Kondo N, Shirakawa M, 2009. Structural basis for the multiple interactions of the MyD88 TIR domain in TLR4 signaling. Proc. Natl. Acad. Sci. U.S.A 106, 10260–10265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohto U, Shibata T, Tanji H, Ishida H, Krayukhina E, Uchiyama S, Miyake K, Shimizu T, 2015. Structural basis of CpG and inhibitory DNA recognition by Toll-like receptor 9. Nature 520, 702–705. [DOI] [PubMed] [Google Scholar]
- Ohto U, Ishida H, Shibata T, Sato R, Miyake K, Shimizu T, 2018. Toll-like receptor 9 contains two DNA binding sites that function cooperatively to promote receptor dimerization and activation. Immunity 48 649–658.e4. [DOI] [PubMed] [Google Scholar]
- Oikarinen S, Tauriainen S, Hober D, Lucas B, Vazeou A, Sioofy-Khojine A, Bozas E, Muir P, Honkanen H, Ilonen J, Knip M, Keskinen P, Saha M-T, Huhtala H, Stanway G, Bartsocas C, Ludvigsson J, Taylor K, Hyöty H, VirDiab Study G, 2014. Virus antibody survey in different European populations indicates risk association between coxsackievirus B1 and type 1 diabetes. Diabetes 63, 655–662. [DOI] [PubMed] [Google Scholar]
- Okahira S, Nishikawa F, Nishikawa S, Akazawa T, Seya T, Matsumoto M, 2005. Interferon-β induction through toll-like receptor 3 depends on double-stranded RNA structure. DNA Cell Biol 24, 614–623. [DOI] [PubMed] [Google Scholar]
- Olivieri I, Cattalini M, Tonduti D, Piana RL, Uggetti C, Galli J, Meini A, Tincani A, Moratto D, Fazzi E, Balottin U, Orcesi S, 2013. Dysregulation of the immune system in Aicardi-Goutières syndrome: another example in a TREX1-mutated patient. Lupus 22, 1064–1069. [DOI] [PubMed] [Google Scholar]
- Omura H, Oikawa D, Nakane T, Kato M, Ishii R, Ishitani R, Tokunaga F, Nureki O, 2016. Structural and functional analysis of DDX41: a bispecific immune receptor for DNA and cyclic dinucleotide. Sci. Rep 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opoku-Temeng C, Zhou J, Zheng Y, Su J, Sintim HO, 2016. Cyclic dinucleotide (c-di-GMP, c-di-AMP, and cGAMP) signalings have come of age to be inhibited by small molecules. Chem. Commun 52, 9327–9342. [DOI] [PubMed] [Google Scholar]
- Orebaugh CD, Fye JM, Harvey S, Hollis T, Perrino FW, 2011. The TREX1 exo-nuclease R114H mutation in Aicardi-Goutières syndrome and lupus reveals dimeric structure requirements for DNA degradation activity. J. Biol. Chem 286, 40246–40254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orzalli MH, DeLuca NA, Knipe DM, 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U.S.A 109 (44), E3008–E3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshiumi H, Matsumoto M, Funami K, Akazawa T, Seya T, 2003. TICAM-1, an adaptor molecule that participates in toll-like receptor 3-mediated interferon-β induction. Nat. Immunol 4, 161–167. [DOI] [PubMed] [Google Scholar]
- Palomino-Morales RJ, Oliver J, Gómez-García M, López-Nevot MA, Rodrigo L, Nieto A, Alizadeh BZ, Martín J, 2009. Association of ATG16L1 and IRGM genes polymorphisms with inflammatory bowel disease: a meta-analysis approach. Genes Immun 10, 356–364. [DOI] [PubMed] [Google Scholar]
- Paludan SR, Bowie AG, 2013. Immune sensing of DNA. Immunity 38, 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancholi NJ, Weitzman MD, 2018. Serotype-specific restriction of wild-type adenovi-ruses by the cellular Mre11-Rad50-Nbs1 complex. Virology 518, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda SK, Facchinetti V, Voynova E, Hanabuchi S, Karnell JL, Hanna RN, Kolbeck R, Sanjuan MA, Ettinger R, Liu YJ, 2018. Galectin-9 inhibits TLR7-mediated autoimmunity in murine lupus models. J. Clin. Invest 128, 1873–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandit RA, Chen C-J, Butt TA, Islam N, 2013. Identification and analysis of a novel mutation in PEPD gene in two Kashmiri siblings with prolidase enzyme deficiency. Gene 516, 316–319. [DOI] [PubMed] [Google Scholar]
- Papinska J, Bagavant H, Gmyrek GB, Sroka M, Tummala S, Fitzgerald KA, Deshmukh US, 2018. Activation of stimulator of interferon genes (STING) and Sjögren syndrome. J. Dent. Res 97, 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B, Brinkmann MM, Spooner E, Lee CC, Kim Y-M, Ploegh HL, 2008. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat. Immunol 9, 1407–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvatiyar K, Zhang Z, Teles RM, Ouyang S, Jiang Y, Iyer SS, Zaver SA, Schenk M, Zeng S, Zhong W, Liu Z-J, Modlin RL, Liu Y-J, Cheng G, 2012. The helicase DDX41 recognizes the bacterial secondary messengers cyclic di-GMP and cyclic di-AMP to activate a type I interferon immune response. Nat. Immunol 13, 1155–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabhi S, Wilkins CR, Dong R, Knoll ML, Posakony J, Kaiser S, Mire CE, Wang ML, Ireton RC, Geisbert TW, Bedard KM, Iadonato SP, Loo YM, Gale M Jr., 2015. Targeting innate immunity for antiviral therapy through small molecule agonists of the RLR pathway. J. Virol 90, 2372–2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paull TT, Deshpande RA, 2014. The Mre11/Rad50/Nbs1 complex: recent insights into catalytic activities and ATP-driven conformational changes. Exp. Cell Res 329, 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peschke K, Achleitner M, Frenzel K, Gerbaulet A, Ada SR, Zeller N, Lienenklaus S, Lesche M, Poulet C, Naumann R, Dahl A, Ravens U, Günther C, Müller W, Knobeloch K-P, Prinz M, Roers A, Behrendt R, 2016. Loss of Trex1 in dendritic cells is sufficient to trigger systemic autoimmunity. J. Immunol 197, 2157–2166. [DOI] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB, 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pétrilli V, Dostert C Muruve DA, Tschopp J, 2007. The inflammasome: a danger sensing complex triggering innate immunity. Curr. Opin. Immunol 19, 615–622. [DOI] [PubMed] [Google Scholar]
- Picard C, Thouvenin G, Kannengiesser C, Dubus J-C, Jeremiah N, Rieux-Laucat F, Crestani B, Belot A, Thivolet-Béjui F, Secq V, Ménard C, Reynaud-Gaubert M, Reix P, 2016. Severe pulmonary fibrosis as the first manifestation of interferonopathy (TMEM173 mutation). Chest 150, e65–e71. [DOI] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Näslund TI, Liljeström P, Weber F, Reis e Sousa C, 2006. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phos-phates. Science 314, 997–1001. [DOI] [PubMed] [Google Scholar]
- Pine R, Decker T, Kessler DS, Levy DE, Darnell JE, 1990. Purification and cloning of interferon-stimulated gene factor 2 (ISGF2): ISGF2 (IRF-1) can bind to the promoters of both beta interferon-and interferon-stimulated genes but is not a primary transcriptional activator of either. Mol. Cell. Biol 10, 2448–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto AK, Ramos HJ, Wu X, Aggarwal S, Shrestha B, Gorman M, Kim KY, Suthar MS, Atkinson JP, Gale M Jr., Diamond MS, 2014. Deficient IFN signaling by myeloid cells leads to MAVS-dependent virus-induced sepsis. PLoS Pathog 10, e1004086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piret J, Carbonneau J, Rhéaume C, Baz M, Boivin G, 2018. Predominant role of IPS-1 over TRIF adaptor proteins in early innate immune response against Zika virus in mice. J. Gen. Virol 99, 209–218. [DOI] [PubMed] [Google Scholar]
- Pisano G, Roy A, Ahmed Ansari M, Kumar B, Chikoti L, Chandran B, 2017. Interferon-γ-inducible protein 16 (IFI16) is required for the maintenance of Epstein-Barr virus latency. Virol. J 14, 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzi S, Sertic S, Orcesi S, Cereda C, Bianchi M, Jackson AP, Lazzaro F, Plevani P, Muzi-Falconi M, 2015. Reduction of hRNase H2 activity in Aicardi–Goutières syndrome cells leads to replication stress and genome instability. Hum. Mol. Genet 24, 649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantinga TS, Joosten LA, van der Meer JW, Netea MG, 2012. Modulation of inflammation by autophagy: consequences for Crohn’s disease. Curr. Opin. Pharmacol 12, 497–502. [DOI] [PubMed] [Google Scholar]
- Pohar J, Kuznik Krajnik A, Jerala R, Bencina M, 2015a. Minimal sequence requirements for oligodeoxyribonucleotides activating human TLR9. J. Immunol 194, 3901–3908. [DOI] [PubMed] [Google Scholar]
- Pohar J, Lainscek D, Fukui R, Yamamoto C, Miyake K, Jerala R, Bencina M, 2015b. Species-specific minimal sequence motif for oligodeoxyribonucleotides activating mouse TLR9. J. Immunol 195, 4396–4405. [DOI] [PubMed] [Google Scholar]
- Pohar J, Lainscek D, Ivicak-Kocjan K, Cajnko MM, Jerala R, Bencina M, 2017. Short single-stranded DNA degradation products augment the activation of Toll-like receptor 9. Nat. Commun 8, 15363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, Morris HD, Yan N, Crouch RJ, 2016. RNase H2 catalytic core Aicardi-Goutières syndromerelated mutant invokes cGAS–STING innate immune-sensing pathway in mice. J. Exp. Med 213, 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontillo A, Girardelli M, Catamo E, Duarte AJ, Crovella S, 2013. Polymorphisms in TREX1 and susceptibility to HIV-1 infection. Int. J. Immunogenet 40, 492–494. [DOI] [PubMed] [Google Scholar]
- Powell RD, Holland PJ, Hollis T, Perrino FW, 2011. Aicardi-Goutières syndrome gene and HIV-1 restriction factor SAMHD1 is a dGTP-regulated deoxynucleotide triphosphohydrolase. J. Biol. Chem 286, 43596–43600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prins KC, Binning JM, Shabman RS, Leung DW, Amarasinghe GK, Basler CF, 2010. Basic residues within the ebolavirus VP35 protein are required for its viral poly-merase cofactor function. J. Virol 84, 10581–10591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttur F, Francozo M, Solmaz G, Bueno C, Lindenberg M, Gohmert M, Swallow M, Tufa D, Jacobs R, Lienenklaus S, Kuöhl AA, Borkner L, Cicin-Sain L, Holzmann B, Wagner H, Berod L, Sparwasser T, 2016. Conventional dendritic cells confer protection against mouse cytomegalovirus infection via TLR9 and MyD88 signaling. Cell Rep 17, 1113–1127. [DOI] [PubMed] [Google Scholar]
- Qi F, Wang D, Liu J, Zeng S, Xu L, Hu H, Liu B, 2015. Respiratory macrophages and dendritic cells mediate respiratory syncytial virus-induced IL-33 production in TLR3-or TLR7-dependent manner. Int. Immunopharmacol 29, 408–415. [DOI] [PubMed] [Google Scholar]
- Qian F, Thakar J, Yuan X, Nolan M, Murray KO, Lee WT, Wong SJ, Meng H, Fikrig E, Kleinstein SH, Montgomery RR, 2014. Immune markers associated with host susceptibility to infection with West Nile virus. Viral Immunol 27, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin L, Ren L, Zhou Z, Lei X, Chen L, Xue Q, Liu X, Wang J, Hung T, 2011. Rotavirus nonstructural protein 1 antagonizes innate immune response by interacting with retinoic acid inducible gene I. Virol. J 8, 526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MM, Bagdassarian E, Ali MAM, McFadden G, 2017. Identification of host DEAD-box RNA helicases that regulate cellular tropism of oncolytic Myxoma virus in human cancer cells. Sci. Rep 7, 15710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramantani G, Kohlhase J, Hertzberg C, Innes AM, Engel K, Hunger S, Borozdin W, Mah JK, Ungerath K, Walkenhorst H, Richardt H-H, Buckard J, Bevot A, Siegel C, von Stuölpnagel C, Ikonomidou C, Thomas K, Proud V, Niemann F, Wieczorek D, Höausler M, Niggemann P, Baltaci V, Conrad K, Lebon P, Lee-Kirsch MA, 2010. Expanding the phenotypic spectrum of lupus erythematosus in Aicardi-Goutières syndrome. Arthritis Rheum 62, 1469–1477. [DOI] [PubMed] [Google Scholar]
- Ramaswamy M, Shi L, Monick MM, Hunninghake GW, Look DC, 2004. Specific inhibition of type I interferon signal transduction by respiratory syncytial virus. Am. J. Respir. Cell Mol. Biol 30, 893–900. [DOI] [PubMed] [Google Scholar]
- Ramaswamy M, Shi L, Varga SM, Barik S, Behlke MA, Look DC, 2006. Respiratory syncytial virus nonstructural protein 2 specifically inhibits type I interferon signal transduction. Virology 344, 328–339. [DOI] [PubMed] [Google Scholar]
- Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, Sodhi K, Treuting PM, Busch MP, Norris PJ, Gale MG Jr., 2012. IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog 8, e1003039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ, 2013. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503, 402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasschaert J, Ladriere L, Urbain M, Dogusan Z, Katabua B, Sato S, Akira S, Gysemans C, Mathieu C, Eizirik DL, 2005. Toll-like receptor 3 and STAT-1 contribute to double-stranded RNA+ interferon-gamma-induced apoptosis in primary pancreatic beta-cells. J. Biol. Chem 280, 33984–33991. [DOI] [PubMed] [Google Scholar]
- Reijns MAM, Bubeck D, Gibson LCD, Graham SC, Baillie GS, Jones EY, Jackson AP, 2011. The structure of the human RNase H2 complex defines key interaction interfaces relevant to enzyme function and human disease. J. Biol. Chem 286, 10530–10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijns MAM, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, Devenney PS, Sexton D, Grimes G, Holt IJ, Hill RE, Taylor MS, Lawson KA, Dorin JR, Jackson AP, 2012. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell 149, 1008–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, Brunner HG, Burke CJ, Carr IM, Carvalho DR, Chandler KE, Christen H-J, Corry PC, Cowan FM, Cox H, D’Arrigo S, Dean J, De Laet C, De Praeter C, Dery C, Ferrie CD, Flintoff K, Frints SGM, Garcia-Cazorla A, Gener B, Goizet C, Goutieres F, Green AJ, Guet A, Hamel BCJ, Hayward BE, Heiberg A, Hennekam RC, Husson M, Jackson AP, Jayatunga R, Jiang Y-H, Kant SG, Kao A, King MD, Kingston HM, Klepper J, van der Knaap MS, Kornberg AJ, Kotzot D, Kratzer W, Lacombe D, Lagae L, Landrieu PG, Lanzi G, Leitch A, Lim MJ, Livingston JH, Lourenco CM, Lyall EGH, Lynch SA, Lyons MJ, Marom D, McClure JP, McWilliam R, Melancon SB, Mewasingh LD, Moutard M-L, Nischal KK, Østergaard JR, Prendiville J, Rasmussen M, Rogers RC, Roland D, Rosser EM, Rostasy K, Roubertie A, Sanchis A, Schiffmann R, Scholl-Buörgi S, Seal S, Shalev SA, Corcoles CS, Sinha GP, Soler D, Spiegel R, Stephenson JBP, Tacke U, Tan TY, Till M, Tolmie JL, et al. , 2007. Clinical and molecular phenotype of Aicardi-Goutières syndrome. Am. J. Hum. Genet 81, 713–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, Fuller JC, Jackson RM, Lamb T, Briggs TA, Ali M, Gornall H, Couthard LR, Aeby A, Attard-Montalto SP, Bertini E, Bodemer C, Brockmann K, Brueton LA, Corry PC, Desguerre I, Fazzi E, Cazorla AG, Gener B, Hamel BCJ, Heiberg A, Hunter M, van der Knaap MS, Kumar R, Lagae L, Landrieu PG, Lourenco CM, Marom D, McDermott MF, van der Merwe W, Orcesi S, Prendiville JS, Rasmussen M, Shalev SA, Soler DM, Shinawi M, Spiegel R, Tan TY, Vanderver A, Wakeling EL, Wassmer E, Whittaker E, Lebon P, Stetson DB, Bonthron DT, Crow YJ, 2009. Mutations involved in Aicardi-Goutières syndrome implicate SAMHD1 as regulator of the innate immune response. Nat. Genet 41, 829–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GMA, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, Jenkinson EM, Bacino CA, Battini R, Bertini E, Brogan PA, Brueton LA, Carpanelli M, De Laet C, de Lonlay P, del Toro M, Desguerre I, Fazzi E, Garcia-Cazorla A, Heiberg A, Kawaguchi M, Kumar R, Lin J-PSM, Lourenco CM, Male AM, Marques W, Mignot C, Olivieri I, Orcesi S, Prabhakar P, Rasmussen M, Robinson RA, Rozenberg F, Schmidt JL, Steindl K, Tan TY, van der Merwe WG, Vanderver A, Vassallo G, Wakeling EL, Wassmer E, Whittaker E, Livingston JH, Lebon P, Suzuki T, McLaughlin PJ, Keegan LP, O’Connell MA, Lovell SC, Crow YJ, 2012. Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature. Nat. Genet 44, 1243–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, del Toro Duany Y, Jenkinson EM, Forte GMA, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, Casarano M, Chouchane M, Cimaz R, Collins AE, Cordeiro NJV, Dale RC, Davidson JE, De Waele L, Desguerre I, Faivre L, Fazzi E, Isidor B, Lagae L, Latchman AR, Lebon P, Li C, Livingston JH, Lourenco CM, Mancardi MM, Masurel-Paulet A, McInnes IB, Menezes MP, Mignot C, O’Sullivan J, Orcesi S, Picco PP, Riva E, Robinson RA, Rodriguez D, Salvatici E, Scott C, Szybowska M, Tolmie JL, Vanderver A, Vanhulle C, Vieira JP, Webb K, Whitney RN, Williams SG, Wolfe LA, Zuberi SM, Hur S, Crow YJ, 2014. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet 46, 503–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards A, van den Maagdenberg AMJM, Jen JC, Kavanagh D, Bertram P, Spitzer D, Liszewski MK, Barilla-LaBarca M-L, Terwindt GM, Kasai Y, McLellan M, Grand MG, Vanmolkot KRJ, de Vries B, Wan J, Kane MJ, Mamsa H, Schäfer R, Stam AH, Haan J, de Jong PTVM, Storimans CW, van Schooneveld MJ, Oosterhuis JA, Gschwendter A, Dichgans M, Kotschet KE, Hodgkinson S, Hardy TA, Delatycki MB, Hajj-Ali RA, Kothari PH, Nelson SF, Frants RR, Baloh RW, Ferrari MD, Atkinson JP, 2007. C-terminal truncations in human 3′-5′ DNA exonuclease TREX1 cause autosomal dominant retinal vasculopathy with cerebral leukodystrophy. Nat. Genet 39, 1068–1070. [DOI] [PubMed] [Google Scholar]
- Rodriguez JJ, Cruz CD, Horvath CM, 2004. Identification of the nuclear export signal and STAT-binding domains of the Nipah virus V protein reveals mechanisms underlying interferon evasion. J. Virol 78, 5358–5367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Madoz JR, Belicha-Villanueva A, Bernal-Rubio D, Ashour J, Ayllon J, Fernandez-Sesma A, 2010. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. J. Virol 84, 9760–9774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roosendaal J, Westaway EG, Khromykh A, Mackenzie JM, 2006. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol 80, 4623–4632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen DA, Twentyman J, Hunstad DA, 2017. High levels of cyclic Di-GMP in Kleb-siella pneumoniae attenuate virulence in the lung. Infect. Immun 86, e00647–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth S, Rottach A, Lotz-Havla AS, Laux V, Muschaweckh A, Gersting SW, Muntau AC, Hopfner K-P, Jin L, Vanness K, Petrini JHJ, Drexler I, Leonhardt H, Ruland J, 2014. Rad50-CARD9 interactions link cytosolic DNA sensing to IL-1β production. Nat. Immunol 15, 538–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, Yamamoto M, Akira S, Fitzgerald KA, 2005. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J. Immunol 175, 5260–5268. [DOI] [PubMed] [Google Scholar]
- Rutsch F, MacDougall M, Lu C, Buers I, Mamaeva O, Nitschke Y, Rice GI, Erlandsen H, Kehl HG, Thiele H, Nuörnberg P, Höhne W, Crow YJ, Feigenbaum A, Hennekam RC, 2015. A specific IFIH1 gain-of-function mutation causes Singleton-Merten syndrome. Am. J. Hum. Genet 96, 275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiga H, Kitada S, Shimada Y, Kamiyama N, Okuyama M, Makino M, Yamamoto M, Takeda K, 2012. Critical role of AIM2 in mycobacterium tuberculosis infection. Int. Immunol 24, 637–644. [DOI] [PubMed] [Google Scholar]
- Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, Akira S, Fujita T, Gale M Jr., 2007. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. U.S.A 104, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuelsson C, Hausmann J, Lauterbach H, Schmidt M, Akira S, Wagner H, Chaplin P, Suter M, O’Keeffe M, Hochrein H, 2008. Survival of lethal poxvirus infection in mice depends on TLR9, and therapeutic vaccination provides protection. J. Clin. Investig 118, 1776–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, Schalm S, Murias S, Dare JA, Brown D, Stone DL, Gao L, Klausmeier T, Foell D, de Jesus AA, Chapelle DC, Kim H, Dill S, Colbert RA, Failla L, Kost B, O’Brien M, Reynolds JC, Folio LR, Calvo KR, Paul SM, Weir N, Brofferio A, Soldatos A, Biancotto A, Cowen EW, Digiovanna JJ, Gadina M, Lipton AJ, Hadigan C, Holland SM, Fontana J, Alawad AS, Brown RJ, Rother KI, Heller T, Brooks KM, Kumar P, Brooks SR, Waldman M, Singh HK, Nickeleit V, Silk M, Prakash A, Janes JM, Ozen S, Wakim PG, Brogan PA, Macias WL, Goldbach-Mansky R, 2018. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J. Clin. Invest 128, 3041–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santin I, Moore F, Grieco FA, Marchetti P, Brancolini C, Eizirik DL, 2012. USP18 is a key regulator of the interferon-driven gene network modulating pancreatic beta cell inflammation and apoptosis. Cell Death Dis 3, e419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarmiento L, Cubas-Dueñas I, Cabrera-Rode E, 2013. Evidence of association between type 1 diabetes and exposure to enterovirus in cuban children and adolescents. MEDICC Rev 15, 29–32. [DOI] [PubMed] [Google Scholar]
- Sarvestani ST, Tate MD, Moffat JM, Jacobi AM, Behlke MA, Miller AR, Beckham SA, McCoy CE, Chen W, Mintern JD, O’Keeffe M, John M, Williams BRG, Gantier MP, 2014. Inosine-mediated modulation of RNA sensing by toll-like receptor 7 (TLR7) and TLR8. J. Virol 88, 799–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasai M, Linehan MM, Iwasaki A, 2010. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science 329, 1530–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh T, Kato H, Kumagai Y, Yoneyama M, Sato S, Matsushita K, Tsujimura T, Fujita T, Akira S, Takeuchi O, 2010. LGP2 is a positive regulator of RIG-I-and MDA5-mediated antiviral responses. Proc. Natl. Acad. Sci. U.S.A 107, 1512–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauer J-D, Sotelo-Troha K, von Moltke J, Monroe KM, Rae CS, Brubaker SW, Hyodo M, Hayakawa Y, Woodward JJ, Portnoy DA, Vance RE, 2011. The N-ethyl-N-nitrosourea-induced Goldenticket mouse mutant reveals an essential function of STING in the in vivo interferon response to Listeria monocytogenes and cyclic dinucleotides. Infect. Immun 79, 688–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattgen SA, Gao G, Kurt-Jones EA, Fitzgerald KA, 2016. Cutting edge: DNA in the lung microenvironment during influenza virus infection tempers inflammation by engaging the DNA sensor AIM2. J. Immunol 196, 29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schijf MA, Lukens MV, Kruijsen D, van Uden NO, Garssen J, Coenjaerts FE, Van’t Land B, van Bleek GM, 2013. Respiratory syncytial virus induced type I IFN production by pDC is regulated by RSV-infected airway epithelial cells, RSV-exposed monocytes and virus specific antibodies. PLoS One 8, e81695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer E, Speck RF, 2008. Anti-HIV activity mediated by natural killer and CD8 + cells after toll-like receptor 7/8 triggering. PLoS One 3, e1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmelzer L, Smitka M, Wolf C, Lucas N, Tüngler V, Hahn G, Tzschach A, Di Donato N, Lee-Kirsch MA, von der Hagen M, 2018. Variable clinical phenotype in two siblings with Aicardi-Goutières syndrome type 6 and a novel mutation in the ADAR gene. Eur. J. Paediatr. Neurol 22, 186–189. [DOI] [PubMed] [Google Scholar]
- Schuberth-Wagner C, Ludwig J, Bruder AK, Herzner AM, Zillinger T, Goldeck M, Schmidt T, Schmid-Burgk JL, Kerber R, Wolter S, Stumpel JP, Roth A, Bartok E, Drosten C, Coch C, Hornung V, Barchet W, Kummerer BM, Hartmann G, Schlee M, 2015. A conserved histidine in the RNA sensor RIG-I controls immune tolerance to N1–2′O-methylated self RNA. Immunity 43, 41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seleme MC, Lei W, Burg AR, Goh KY, Metz A, Steele C, Tse HM, 2012. Dys-regulated TLR3-dependent signaling and innate immune activation in superoxide-deficient macrophages from nonobese diabetic mice. Free Radic. Biol. Med 52, 2047–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulveda FE, Maschalidi S, Colisson R, Heslop L, Ghirelli C, Sakka E, Lennon-Duménil A-M, Amigorena S, Cabanie L Manoury B, 2009. Critical role for aspar-agine endopeptidase in endocytic toll-like receptor signaling in dendritic cells. Immunity 31, 737–748. [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea C-K, Chen ZJ, 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 122, 669–682. [DOI] [PubMed] [Google Scholar]
- Shang G, Zhu D, Li N, Zhang J, Zhu C, Lu D, Liu C, Yu Q, Zhao Y, Xu S, Gu L, 2012. Crystal structures of STING protein reveal basis for recognition of cyclic di-GMP. Nat. Struct. Mol. Biol 19, 725–727. [DOI] [PubMed] [Google Scholar]
- Sharma S, tenOever BR, Grandvaux N, Zhou G-P, Lin R, Hiscott J, 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300, 1148–1151. [DOI] [PubMed] [Google Scholar]
- Sharma S, Javadekar SM, Pandey M, Srivastava M, Kumari R, Raghavan SC, 2015. Homology and enzymatic requirements of microhomology-dependent alternative end joining. Cell Death Dis 6, e1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw ML, García-Sastre A, Palese P, Basler CF, 2004. Nipah virus V and W proteins have a common STAT1-binding domain yet inhibit STAT1 activation from the cytoplasmic and nuclear compartments, respectively. J. Virol 78, 5633–5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, An J, Liang J, Hayes SE, Sandusky GE, Stramm LE, Yang NN, 1999. Characterization of a mutant pancreatic eIF-2α kinase, PEK, and co-localization with somatostatin in islet delta cells. J. Biol. Chem 274, 5723–5730. [DOI] [PubMed] [Google Scholar]
- Shi X, Dong Y, Li Y, Zhao Z, Li H, Qiu S, Li Y, Guo W, Qiao Y, 2015. Inflammasome activation in mouse inner ear in response to MCMV induced hearing loss. J. Otol 10, 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki S, Imagawa A, Tauriainen S, Iino M, Oikarinen M, Abiru H, Tamaki K, Seino H, Nishi K, Takase I, Okada Y, Uno S, Murase-Mishiba Y, Terasaki J, Makino H, Shimomura I, Hyoty H, Hanafusa T, 2010. Expression of toll-like receptors in the pancreas of recent-onset fulminant type 1 diabetes. Endocr. J 57, 211–219. [DOI] [PubMed] [Google Scholar]
- Shigemoto T, Kageyama M, Hirai R, Zheng J, Yoneyama M, Fujita T, 2009. Identification of loss of function mutations in human genes encoding RIG-I and MDA5. J. Biol. Chem 284, 13348–13354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinde R, Hezaveh K, Halaby MJ, Kloetgen A, Chakravarthy A, da Silva Medina T, Deol R, Manion KP, Baglaenko Y, Eldh M, Lamorte S, Wallace D, Chodisetti SB, Ravishankar B, Liu H, Chaudhary K, Munn DH, Tsirigos A, Madaio M, Gabrielsson S, Touma Z, Wither J, De Carvalho DD, McGaha TL, 2018. Apoptotic cell-induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat. Immunol 19, 571–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shreberk-Hassidim R, Ramot Y, Zlotogorski A, 2017. Janus kinase inhibitors in dermatology: a systematic review. J. Am. Acad. Dermatol 76 745–753.e19. [DOI] [PubMed] [Google Scholar]
- Siezen CLE, Bont L, Hodemaekers HM, Ermers MJ, Doornbos G, van’t Slot R, Wijmenga C, Houwelingen HCV, Kimpen JLL, Kimman TG, Hoebee B, Janssen R, 2009. Genetic susceptibility to respiratory syncytial virus bronchiolitis in preterm children is associated with airway remodeling genes and innate immune genes. Pediatr. Infect. Dis. J 28, 333. [DOI] [PubMed] [Google Scholar]
- Skonieczna K, Wozniacka A, Czajkowski R, Styczynski J, Krenska A, Robak E, Gawrych M, Kaszewski S, Wysocki M, Grzybowski T, 2018. X-linked TLR7 gene polymorphisms are associated with diverse immunological conditions but not with discoid lupus erythematosus in polish patients. Postepy Dermatol. Alergol 35, 26–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skouboe MK, Knudsen A, Reinert LS, Boularan C, Lioux T, Perouzel E, Thomsen MK, Paludan SR, 2018. STING agonists enable antiviral cross-talk between human cells and confer protection against genital herpes in mice. PLoS Pathog 14, e1006976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Škrnjug I, Guzmán CA, Ruecker C, 2014. Cyclic GMP-AMP displays mucosal adju-vant activity in mice. PLoS One 9, e110150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EJ, Marie I, Prakash A, Garcia-Sastre A, Levy DE, 2001. IRF3 and IRF7 phos-phorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or Ikappa B kinase but is blocked by Vaccinia virus E3L protein. J. Biol. Chem 276, 8951–8957. [DOI] [PubMed] [Google Scholar]
- Spehalski E, Capper KM, Smith CJ, Morgan MJ, Dinkelmann M, Buis J, Sekiguchi JM, Ferguson DO, 2017. MRE11 promotes tumorigenesis by facilitating resistance to oncogene-induced replication stress. Cancer Res 77, 5327–5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stabell AC, Meyerson NR, Gullberg RC, Gilchrist AR, Webb KJ, Old WM, Perera R, Sawyer SL, 2018. Dengue viruses cleave STING in humans but not in non-human primates, their presumed natural reservoir. eLife 7, e31919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Ko JS, Heidmann T, Medzhitov R, 2008. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell 134, 587–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone AE, Mitchell A, Brownell J, Miklin DJ, Golden-Mason L, Polyak SJ, Gale MJ Jr., Rosen HR, 2014. Hepatitis C virus core protein inhibits interferon production by a human plasmacytoid dendritic cell line and dysregulates interferon regulatory factor-7 and signal transducer and activator of transcription (STAT) 1 protein expression. PLoS One 9, e95627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppelenburg AJ, von Hegedus JH, Huis in’t Veld R, Bont L, Boes M, 2014. Defective control of vitamin D receptor-mediated epithelial STAT1 signalling predisposes to severe respiratory syncytial virus bronchiolitis. J. Pathol 232, 57–64. [DOI] [PubMed] [Google Scholar]
- Stracker TH, Carson CT, Weitzman MD, 2002. Adenovirus oncoproteins inactivate the Mre11-Rad50-NBS1 DNA repair complex. Nature 418, 348–352. [DOI] [PubMed] [Google Scholar]
- Su C, Zheng C, 2017. Herpes simplex virus 1 abrogates the cGAS/STING-mediated cytosolic DNA-sensing pathway via its virion host shutoff protein, UL41. J. Virol 91, e02414–e02416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugai A, Sato H, Takayama I, Yoneda M, Kai C, 2017. Nipah and Hendra virus nucle-oproteins inhibit nuclear accumulation of signal transducer and activator of transcription 1 (STAT1) and STAT2 by interfering with their complex formation. J. Virol 91, e01136–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui H, Zhou M, Imamichi H, Jiao X, Sherman BT, Lane HC, Imamichi T, 2017. STING is an essential mediator of the Ku70-mediated production of IFN-λ1 in response to exogenous DNA. Sci. Signal 10, eaah5054. [DOI] [PubMed] [Google Scholar]
- Sumpter R, Loo Y-M, Foy E, Li K, Yoneyama M, Fujita T, Lemon SM, Gale M, 2005. Regulating intracellular antiviral defense and permissiveness to Hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J. Virol 79, 2689–2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z, 2009. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proc. Natl. Acad. Sci. U.S.A 106, 8653–8658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, Chen ZJ, 2013. Cyclic GMP-AMP synthase is a cyto-solic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z, Wang L, Eckloff BW, Deng B, Wang Y, Wampfler JA, Jang J, Wieben ED, Jen J, You M, Yang P, 2014. Conserved recurrent gene mutations correlate with pathway deregulation and clinical outcomes of lung adenocarcinoma in never-smokers. BMC Med. Genomics 7, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunthamala N, Thierry F, Teissier S, Pientong C, Kongyingyoes B, Tangsiriwatthana T, Sangkomkamhang U, Ekalaksananan T, 2014. E2 Proteins of high risk human papillomaviruses down-modulate STING and IFN-κ transcription in keratinocytes. PLoS One 9, e91473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suthar MS, Ma DY, Thomas S, Lund JM, Zhang N, Daffis S, Rudensky AY, Bevan MJ, Clark EA, Kaja M-K, Diamond MS, Gale M, 2010. IPS-1 is essential for the control of West Nile virus infection and immunity. PLoS Pathog 6, e1000757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Synofzik M, Haack TB, Kopajtich R, Gorza M, Rapaport D, Greiner M, Schönfeld C, Freiberg C, Schorr S, Holl RW, Gonzalez MA, Fritsche A, Fallier-Becker P, Zimmermann R, Strom TM, Meitinger T, Züchner S, Schüle R, Schöls L, Prokisch H, 2014. Absence of BiP co-chaperone DNAJC3 causes diabetes mellitus and multisystemic neurodegeneration. Am. J. Hum. Genet 95, 689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szretter KJ, Daffis S, Patel J, Suthar MS, Klein RS, Gale M Jr., Diamond MS, 2010. The innate immune adaptor molecule MyD88 restricts West Nile virus replication and spread in neurons of the central nervous system. J. Virol 84, 12125–12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, Alexopoulou L, Flavell RA, Beutler B, 2004. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cyto-megalovirus infection. Proc. Natl. Acad. Sci. U.S.A 101, 3516–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, Mudd S, Mann N, Sovath S, Goode J, Shamel L, Herskovits AA, Portnoy DA, Cooke M, Tarantino LM, Wiltshire T, Steinberg BE, Grinstein S, Beutler B, 2006. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat. Immunol 7, 156–164. [DOI] [PubMed] [Google Scholar]
- Takada I, Yogiashi Y, Makishima M, 2016. The ribosomal S6 kinase inhibitor BI-D1870 ameliorated experimental autoimmune encephalomyelitis in mice. Immunobiology 221, 188–192. [DOI] [PubMed] [Google Scholar]
- Takahama M, Akira S, Saitoh T, 2018. Autophagy limits activation of the inflamma-somes. Immunol. Rev 281, 62–73. [DOI] [PubMed] [Google Scholar]
- Takaoka A, Wang Z, Choi MK, Yanai H, Negishi H, Ban T, Lu Y, Miyagishi M, Kodama T, Honda K, Ohba Y, Taniguchi T, 2007. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 448, 501–505. [DOI] [PubMed] [Google Scholar]
- Tal MC, Sasai M, Lee HK, Yordy B, Shadel GS, Iwasaki A, 2009. Absence of autophagy results in reactive oxygen species-dependent amplification of RLR signaling. Proc. Natl. Acad. Sci. U.S.A 106, 2770–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan EM, Schur PH, Carr RI, Kunkel HG, 1966. Deoxybonucleic acid (DNA) and antibodies to DNA in the serum of patients with systemic lupus erythematosus. J. Clin. Investig 45, 1732–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Chen ZJ, 2012. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci. Signal 5, ra20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao J, Zhang X-W, Jin J, Du X-X, Lian T, Yang J, Zhou X, Jiang Z, Su X-D, 2017. Nonspecific DNA binding of cGAS N terminus promotes cGAS activation. J. Immunol 198, 3627–3636. [DOI] [PubMed] [Google Scholar]
- Tatematsu M, Nishikawa F, Seya T, Matsumoto M, 2013. Toll-like receptor 3 recognizes incomplete stem structures in single-stranded viral RNA. Nat. Commun 4, 1833. [DOI] [PubMed] [Google Scholar]
- Thapa RJ, Ingram JP, Ragan KB, Nogusa S, Boyd DF, Benitez AA, Sridharan H, Kosoff R, Shubina M, Landsteiner VJ, Andrake M, Vogel P, Sigal LJ, tenOever BR, Thomas PG, Upton JW, Balachandran S, 2016. DAI senses influenza a virus genomic RNA and activates RIPK3-dependent cell death. Cell Host Microbe 20, 674–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Avalos AM, Mao S-Y, Chen B, Senthil K, Wu H, Parroche P, Drabic S, Golenbock D, Sirois C, Hua J, An LL, Audoly L, La Rosa G, Bierhaus A, Naworth P, Marshak-Rothstein A, Crow MK, Fitzgerald KA, Latz E, Kiener PA, Coyle AJ, 2007. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol 8, 487–496. [DOI] [PubMed] [Google Scholar]
- Toscano F, Estornes Y, Virard F, Garcia-Cattaneo A, Pierrot A, Vanbervliet B, Bonnin M, Ciancanelli MJ, Zhang S-Y, Funami K, Seya T, Matsumoto M, Pin J-J, Casanova J-L, Renno T, Lebecque S, 2013. Cleaved/associated TLR3 represents the primary form of the signaling receptor. J. Immunol 190, 764–773. [DOI] [PubMed] [Google Scholar]
- Trapani J, Dawson M, Apostolidis V, Browne K, 1994. Genomic organization of IFI16, an interferon-inducible gene whose expression is associated with human myeloid cell differentiation: correlation of predicted protein domains with exon organization. Immunogenetics 40, 415–424. [DOI] [PubMed] [Google Scholar]
- Uchida L, Espada-Murao LA, Takamatsu Y, Okamoto K, Hayasaka D, Yu F, Nabeshima T, Buerano CC, Morita K, 2014. The dengue virus conceals double-stranded RNA in the intracellular membrane to escape from an interferon response. Sci. Rep 4, 7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchikawa E, Lethier M, Malet H, Brunel J, Gerlier D, Cusack S, 2016. Structural analysis of dsRNA binding to anti-viral pattern recognition receptors LGP2 and MDA5. Mol. Cell 62, 586–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unterholzner L, Keating SE, Baran M, Horan KA, Jensen SB, Sharma S, Sirois CM, Jin T, Latz E, Xiao TS, Fitzgerald KA, Paludan SR, Bowie AG, 2010. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol 11, 997–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uyur Yalçın E, Maraş Genç H, Kara B, 2015. Clinical and neuroradiologic variability of Aicardi-Goutiéres syndrome: two siblings with RNASEH2C mutation and a boy with TREX1 mutation. Turk. J. Pediatr 57, 504–508. [PubMed] [Google Scholar]
- Uzri D, Greenberg HB, 2013. Characterization of rotavirus RNAs that activate innate immune signaling through the RIG-I-like receptors. PLoS One 8, e69825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Drunen Littel-van den Hurk S, Watkiss ER, 2012. Pathogenesis of respiratory syncytial virus. Curr. Opin. Virol 2, 300–305. [DOI] [PubMed] [Google Scholar]
- Varani S, Cederarv M, Feld S, Tammik C, Frascaroli G, Landini MP, Soderberg-Naucler C, 2007. Human cytomegalovirus differentially controls B Cell and T Cell responses through effects on plasmacytoid dendritic cells. J. Immunol 179, 7767–7776. [DOI] [PubMed] [Google Scholar]
- Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, Iwakura Y, Barber GN, 2007. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol 178, 6444–6455. [DOI] [PubMed] [Google Scholar]
- Vincent J, Adura C, Gao P, Luz A, Lama L, Asano Y, Okamoto R, Imaeda T, Aida J, Rothamel K, Gogakos T, Steinberg J, Reasoner S, Aso K, Tuschl T, Patel DJ, Glickman JF, Ascano M, 2017. Small molecule inhibition of cGAS reduces interferon expression in primary macrophages from autoimmune mice. Nat. Commun 8, 750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner H, 2004. The immunobiology of the TLR9 subfamily. Trends Immunol 25, 381–386. [DOI] [PubMed] [Google Scholar]
- Walker WE, Booth CJ, Goldstein DR, 2010. TLR9 and IRF3 cooperate to induce a systemic inflammatory response in mice injected with liposome:DNA. Mol. Ther 18, 775–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA, 2004. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat. Med 10, 1366–1373. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang H-X, Sun Y-P, Liu Z-X, Liu X-S, Wang L, Lu S-Y, Kong H, Liu Q-L, Li X-H, Lu Z-Y, Chen S-J, Chen Z, Bao S-S, Dai W, Wang Z-G, 2007. Rig-I−/−mice develop colitis associated with down-regulation of G alpha i2. Cell Res 17, 858–868. [DOI] [PubMed] [Google Scholar]
- Wang F, Alain T, Szretter KJ, Stephenson K, Pol JG, Atherton MJ, Hoang H-D, Fonseca BD, Zakaria C, Chen L, Rangwala Z, Hesch A, Chan ESY, Tuinman C, Suthar MS, Jiang Z, Ashkar AA, Thomas G, Kozma SC, Gale M, Fitzgerald KA, Diamond MS, Mossman K, Sonenberg N, Wan Y, Lichty BD, 2016. S6K-STING interaction regulates cytosolic DNA-mediated activation of the transcription factor IRF3. Nat. Immunol 17, 514–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Kang L, Song D, Liu L, Yang S, Ma L, Guo Z, Ding H, Wang H, Yang B, 2017. Ku70 senses HTLV-1 DNA and modulates HTLV-1 replication. J. Immunol 199, 2475–2482. [DOI] [PubMed] [Google Scholar]
- Wang JJ, Reed JH, Colella AD, Russell AJ, Murray-Brown W, Chataway TK, Jackson KJL, Goodnow CC, Gordon TP, 2018. Molecular profiling and clonal tracking of secreted rheumatoid factors in primary Sjögren’s syndrome. Arthritis Rheumatol 1–9. in press. [DOI] [PubMed]
- Warner JD, Irizarry-Caro RA, Bennion BG, Ai TL, Smith AM, Miner CA, Sakai T, Gonugunta VK, Wu J, Platt DJ, Yan N, Miner JJ, 2017. STING-associated vasculopathy develops independently of IRF3 in mice. J. Exp. Med 214, 3279–3292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RO, Bell SL, MacDuff DA, Kimmey JM, Diner EJ, Olivas J, Vance RE, Stallings CL, Virgin HW, Cox JS, 2015. The cytosolic sensor cGAS detects mycobacterium tuberculosis DNA to induce type I interferons and activate autophagy. Cell Host Microbe 17, 811–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JA, Gregory SM, Sivaraman V, Su L, Damania B, 2011. Activation of plasmacytoid dendritic cells by Kaposi’s sarcoma-associated herpesvirus. J. Virol 85, 895–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheeler LA, Trifonova RT, Vrbanac V, Barteneva NS, Liu X, Bollman B, Onofrey L, Mulik S, Ranjbar S, Luster AD, Tager AM, Lieberman J, 2016. TREX1 knockdown induces an interferon response to HIV that delays viral infection in humanized mice. Cell Rep 15, 1715–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wies E, Wang MK, Maharaj NP, Chen K, Zhou S, Finberg RW, Gack MU, 2013. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 38, 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf C, Rapp A, Berndt N, Staroske W, Schuster M, Dobrick-Mattheuer M, Kretschmer S, König N, Kurth T, Wieczorek D, Kast K, Cardoso MC, Günther C, Lee-Kirsch MA, 2016. RPA and Rad51 constitute a cell intrinsic mechanism to protect the cytosol from self DNA. Nat. Commun 7, 11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf SJ, Theros J, Reed TJ, Liu J, Grigorova IL, Martinez-Colon G, Jacob CO, Hodgin JB, Kahlenberg JM, 2018. TLR7-mediated lupus nephritis is independent of type I IFN signaling. J. Immunol 201, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodward JJ, Iavarone AT, Portnoy DA, 2010. c-di-AMP secreted by intracellular Listeria monocytogenes activates a host type I interferon response. Science 328, 1703–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu O, Chen GP, Chen H, Li XP, Xu JH, Zhao SS, Sheng J, Feng JB, Cai J, Fang XH, Zhang WH, Li LH, Zhang N, Li J, Li JJ, Pan FM, Wang CZ, Ye DQ, 2009. The expressions of toll-like receptor 9 and T-bet in circulating B and T cells in newly diagnosed, untreated systemic lupus erythematosus and correlations with disease activity and laboratory data in a Chinese population. Immunobiology 214, 392–402. [DOI] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, Chen ZJ, 2013. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Thalhamer T, Franca RF, Xiao S, Wang C, Hotta C, Zhu C, Hirashima M, Anderson AC, Kuchroo VK, 2014. Galectin-9-CD44 interaction enhances stability and function of adaptive regulatory T cells. Immunity 41, 270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P, Wang S, Ye B, Du Y, Huang G, Zhu P, Fan Z, 2015. Sox2 functions as a sequence-specific DNA sensor in neutrophils to initiate innate immunity against microbial infection. Nat. Immunol 16, 366–375. [DOI] [PubMed] [Google Scholar]
- Xia P, Wang S, Gao P, Gao G, Fan Z, 2016. DNA sensor cGAS-mediated immune recognition. Protein Cell 7, 777–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia H, Luo H, Shan C, Muruato AE, Nunes BTD, Medeiros DBA, Zou J, Xie X, Giraldo MI, Vasconcelos PFC, Weaver SC, Wang T, Rajsbaum R, Shi P-Y, 2018. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun 9, 414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie G, Welte T, Wang J, Whiteman MC, Wicker JA, Saxena V, Cong Y, Barrett AD, Wang T, 2013. A West Nile virus NS4B-P38G mutant strain induces adaptive immunity via TLR7-MyD88-dependent and independent signaling pathways. Vaccine 31, 4143–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zoltick PW, Gamero AM, Gallucci S, 2014a. TLR ligands up-regulate Trex1 expression in murine conventional dendritic cells through type I interferon and NF-κB-dependent signaling pathways. J. Leukoc. Biol 96, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Zheng J, Zheng K, Hou Y, Zhao F, Zhao D, 2014b. Respiratory syncytial virus NS1 protein degrades STAT2 by inducing SOCS1 expression. Intervirology 57, 65–73. [DOI] [PubMed] [Google Scholar]
- Xu H, Su C, Pearson A, Mody CH, Zheng C, 2017. Herpes simplex virus 1 UL24 abrogates the DNA sensing signal pathway by inhibiting NF-κB activation. J. Virol 91, e00025–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Kitagawa Y, Zhou M, Itoh M, Gotoh B, 2014. An anti-interferon activity shared by paramyxovirus C proteins: inhibition of toll-like receptor 7/9-dependent alpha interferon induction. FEBS Lett 588, 28–34. [DOI] [PubMed] [Google Scholar]
- Yan W, Frank CL, Korth MJ, Sopher BL, Novoa I, Ron D, Katze MG, 2002. Control of PERK eIF2α kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Nat. Acad. Sci. U.S.A 99, 15920–15925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan N, Regalado-Magdos AD, Stiggelbout B, Lee-Kirsch MA, Lieberman J, 2010. The cytosolic exonuclease TREX1 inhibits the innate immune response to human immunodeficiency virus type 1. Nat. Immunol 11, 1005–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y-G, Lindahl T, Barnes DE, 2007. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 131, 873–886. [DOI] [PubMed] [Google Scholar]
- Yang P, An H, Liu X, Wen M, Zheng Y, Rui Y, Cao X, 2010. The cytosolic nucleic acid sensor LRRFIP1 mediates the production of type I interferon via a β-catenin-dependent pathway. Nat. Immunol 11, 487–494. [DOI] [PubMed] [Google Scholar]
- Yang JY, Kim MS, Kim E, Cheon JH, Lee YS, Kim Y, Lee SH, Seo SU, Shin SH, Choi SS, Kim B, Chang SY, Ko HJ, Bae JW, Kweon MN, 2016. Enteric viruses ameliorate gut inflammation via toll-like receptor 3 and toll-like receptor 7-mediated interferon-beta production. Immunity 44, 889–900. [DOI] [PubMed] [Google Scholar]
- Yang H, Bian S, Chen H, Wang L, Zhao L, Zhang X, Zhao Y, Zeng X, Zhang F, 2018. Clinical characteristics and risk factors for overlapping rheumatoid arthritis and Sjögren’s syndrome. Sci. Rep 8, 6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda K, Ogawa Y, Yamane I, Nishikawa M, Takakura Y, 2005. Macrophage activation by a DNA/cationic liposome complex requires endosomal acidification and TLR9-dependent and -independent pathways. J. Leukoc. Biol 77, 71–79. [DOI] [PubMed] [Google Scholar]
- Ye R, Su C, Xu H, Zheng C, 2017. Herpes simplex virus 1 ubiquitin-specific protease UL36 abrogates NF-κB activation in DNA sensing signal pathway. J. Virol 91, e02417–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen B, Mulder LC, Martinez O, Basler CF, 2014. Molecular basis for ebolavirus VP35 suppression of human dendritic cell maturation. J. Virol 88, 12500–12510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz S, Alpdundar E, Gungor B, Kahraman T, Bayyurt B, Gursel I, Gursel M, 2015. Enhanced immunostimulatory activity of cyclic dinucleotides on mouse cells when complexed with a cell-penetrating peptide or combined with CpG: innate immunity. Eur. J. Immunol 45, 1170–1179. [DOI] [PubMed] [Google Scholar]
- Ylipaasto P, Klingel K, Lindberg AM, Otonkoski T, Kandolf R, Hovi T, Roivainen M, 2004. Enterovirus infection in human pancreatic islet cells, islet tropism in vivo and receptor involvement in cultured islet beta cells. Diabetologia 47, 225–239. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Suhara W, Fukuhara Y, Sato M, Ozato K, Fujita T, 1996. Autocrine amplification of type I interferon gene expression mediated by interferon stimulated gene factor 3 (ISGF3). J. Biochem 120, 160–169. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T, 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol 5, 730–737. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, Foy E, Loo Y-M, Gale M, Akira S, Yonehara S, Kato A, Fujita T, 2005. Shared and unique functions of the DExD/H-Box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol 175, 2851–2858. [DOI] [PubMed] [Google Scholar]
- Yu P, 2006. Toll-like receptor 9-independent aggravation of glomerulonephritis in a novel model of SLE. Int. Immunol 18, 1211–1219. [DOI] [PubMed] [Google Scholar]
- Yu H-R, Huang H-C, Kuo H-C, Sheen J-M, Ou C-Y, Hsu T-Y, Yang KD, 2011. IFN-α production by human mononuclear cells infected with varicella-zoster virus through TLR9-dependent and -independent pathways. Cell. Mol. Immunol 8, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu C-Y, Chang T-H, Liang J-J, Chiang R-L, Lee Y-L, Liao C-L, Lin Y-L, 2012. Dengue virus targets the adaptor protein MITA to subvert host innate immunity. PLoS Pathog 8, e1002780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S-SF, Hou M-F, Hsieh Y-C, Huang C-Y, Lee Y-C, Chen Y-J, Lo S, 2012. Role of MRE11 in cell proliferation, tumor invasion, and DNA repair in breast cancer. J. Natl. Cancer Inst 104, 1485–1502. [DOI] [PubMed] [Google Scholar]
- Zeng W, Sun L, Jiang X, Chen X, Hou F, Adhikari A, Xu M, Chen ZJ, 2010. Reconstitution of the RIG-I pathway reveals a signaling role of unanchored polyubiquitin chains in innate immunity. Cell 141, 315–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Huifang M, Qiu Y, Ye X, Hemida Maged G, Hanson P, Yang D, 2013. P58IPK inhibits coxsackievirus-induced apoptosis via the PI3K/Akt pathway requiring activation of ATF6a and subsequent upregulation of mitofusin 2. Cell. Microbiol 16, 411–424. [DOI] [PubMed] [Google Scholar]
- Zhang X, Brann TW, Zhou M, Yang J, Oguariri RM, Lidie KB, Imamichi H, Huang DW, Lempicki RA, Baseler MW, Veenstra TD, Young HA, Lane HC, Imamichi T, 2011a. Cutting edge: Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J. Immunol 186, 4541–4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Kim T, Bao M, Facchinetti V, Jung SY, Ghaffari AA, Qin J, Cheng G, Liu Y-J, 2011b. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity 34, 866–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu Y-J, 2011c. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nat. Immunol 12, 959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Yuan B, Lu N, Facchinetti V, Liu YJ, 2011d. DHX9 pairs with IPS-1 to sense double-stranded RNA in myeloid dendritic cells. J. Immunol 187, 4501–4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Bao M, Lu N, Weng L, Yuan B, Liu Y-J, 2012. The E3 ubiquitin ligase TRIM21 negatively regulates the innate immune response to intracellular double-stranded DNA. Nat. Immunol 14, 172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, Chen ZJ, 2013. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Yeruva L, Marinov A, Prantner D, Wyrick PB, Lupashin V, Nagarajan UM, 2014. The DNA sensor, cyclic GMP–AMP synthase, is essential for induction of IFN-β during Chlamydia trachomatis infection. J. Immunol 193, 2394–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Su C, Zheng C, 2016. Herpes simplex virus 1 serine protease VP24 blocks the DNA-sensing signal pathway by abrogating activation of interferon regulatory factor 3. J. Virol 90, 5824–5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Luo J, Alcorn JF, Chen K, Fan S, Pilewski J, Liu A, Chen W, Kolls JK, Wang J, 2017a. AIM2 inflammasome is critical for influenza-induced lung injury and mortality. J. Immunol 198, 4383–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H-L, Ye H-Q, Liu S-Q, Deng C-L, Li X-D, Shi P-Y, Zhang B, 2017b. West Nile virus NS1 antagonizes interferon beta production by targeting RIG-I and MDA5. J. Virol 91, e02396–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Zhu M, Limbo O, Russell P, 2018. RNase H eliminates R-loops that disrupt DNA replication but is nonessential for efficient DSB repair. EMBO Rep 19, e45335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, 2018. Evasion of cytosolic DNA-stimulated innate immune responses by herpes simplex virus 1. J. Virol 92, e00099–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB, 2008. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 29, 538–550. [DOI] [PubMed] [Google Scholar]
- Zhong X, Chen B, Yang L, Yang Z, 2018. Molecular and physiological roles of the adaptor protein CARD9 in immunity. Cell Death Dis 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou K, Choe K-T, Zaidi Z, Wang Q, Mathews MB, Lee C-G, 2003. RNA helicase A interacts with dsDNA and topoisomerase IIalpha. Nucleic Acids Res 31, 2253–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]