

Abstract
Social organisms combat pathogens through individual innate immune responses or through social immunity—behaviors among individuals that limit pathogen transmission within groups. Although we have a relatively detailed understanding of the genetics and evolution of the innate immune system of animals, we know little about social immunity. Addressing this knowledge gap is crucial for understanding how life-history traits influence immunity, and identifying if trade-offs exist between innate and social immunity. Hygienic behavior in the Western honey bee, Apis mellifera, provides an excellent model for investigating the genetics and evolution of social immunity in animals. This heritable, colony-level behavior is performed by nurse bees when they detect and remove infected or dead brood from the colony. We sequenced 125 haploid genomes from two artificially selected highly hygienic populations and a baseline unselected population. Genomic contrasts allowed us to identify a minimum of 73 genes tentatively associated with hygienic behavior. Many genes were within previously discovered QTLs associated with hygienic behavior and were predictive of hygienic behavior within the unselected population. These genes were often involved in neuronal development and sensory perception in solitary insects. We found that genes associated with hygienic behavior have evidence of positive selection within honey bees (Apis), supporting the hypothesis that social immunity contributes to fitness. Our results indicate that genes influencing developmental neurobiology and behavior in solitary insects may have been co-opted to give rise to a novel and adaptive social immune phenotype in honey bees.
Keywords: social immunity, selection, sociality, eusocial
Introduction
Living at high densities with close relatives increases the risk of epizootic outbreaks, yet these are the exact conditions in which social insects successfully live (Schmid-Hempel 1994; Zasloff 2002; Lawniczak et al. 2007; Nunn et al. 2015). Their success is due in part to their ability to mitigate epizootic risk through two forms of immunity. The first is the innate immune system ( e.g. Evans et al. 2006), composed of sets of genes that are conserved and well characterized across social and solitary taxa. This system is activated by a set of generally acting recognition proteins that detect pathogens and, through downstream signaling pathways, elicit the expression of proteins that eliminate or reduce the pathogenic threat. We have a deep understanding of the genetics and evolution of the innate immune system in social insects, in part because the genes underpinning innate immunity are taxonomically ancient and are largely conserved across insects (Evans et al. 2006; Harpur and Zayed 2013; Barribeau et al. 2015).
The second form of immunity is social immunity, an evolutionarily derived system of prophylactic or curative, occasionally altruistic, responses that limit the spread of pathogens (Cremer et al. 2007). Social immunity includes secretions that act to limit bacterial and fungal growth (Poulsen et al. 2003), self- or social-exclusion from all or part of the colony (Heinze and Walter 2010; Lecocq et al. 2016), removal or cannibalism of infected or deceased workers (Sun and Zhou 2013), grooming (Rosengaus et al. 1998), and/or the removal of dead or infected larvae (fig. 1A; Rothenbuhler 1964a, 1964b). These responses are very effective at eliminating the risk of epizootics. For example, in honey bees, some workers are able to detect and remove infected brood—a trait referred to as hygienic behavior (fig. 1A). Forms of Social immunity, such as hygiene, can be very effective at eliminating the risk of disease. Field trials demonstrate that hygienic behavior eliminates the risk of developing clinical symptoms of Chalkbrood disease and reduces the risk of developing symptoms of American Foul Brood disease by 61% (Spivak and Reuter 2001). Because of its evolutionary novelty in social insects, we do not yet know the genetic mechanisms underpinning social immunity. This hampers efforts to understand how social immunity evolves and to quantify potential genetic or evolutionary trade-offs between innate and social immunity (e.g., Sackton et al. 2007; Harpur and Zayed 2013; Barribeau et al. 2015).
Fig. 1.
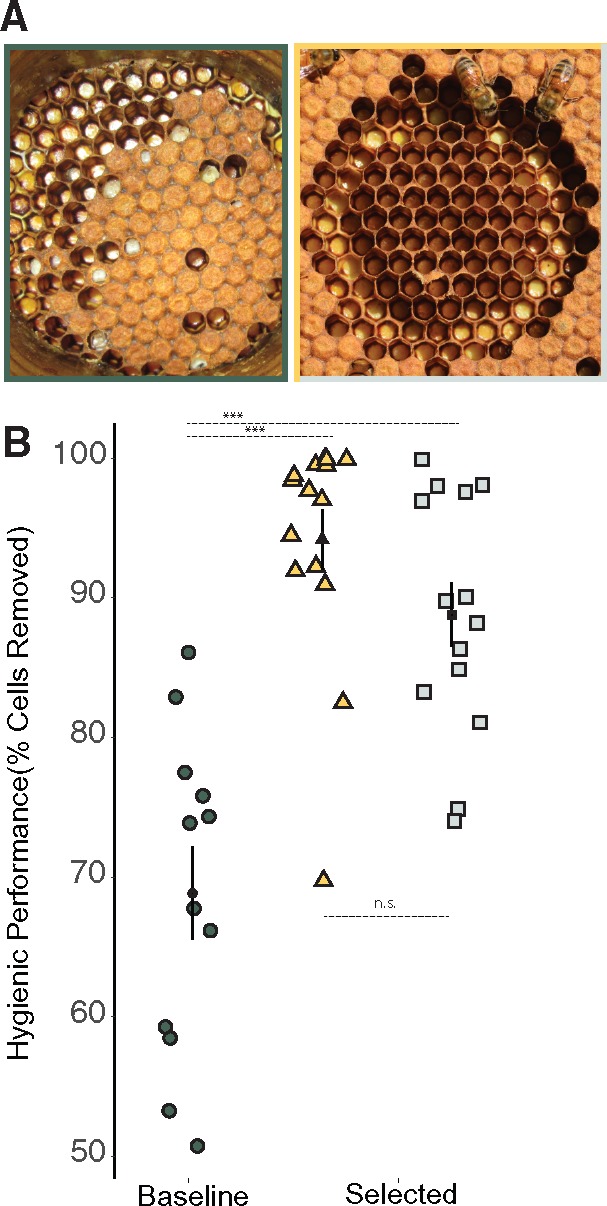
.—(A) Result of a freeze-killed brood (FKB) assay for two colonies showing (left panel) low uncapping and removal rates after 24 h and (right panel) high uncapping and removal after 24 h. The FKB assay is performed by freezing a section of capped honey bee brood (see left image) with liquid nitrogen. Once thawed, the frozen section is placed back inside the colony. After 24 h the section is removed once more and the number of uncapped and removed cells is counted. Hygienic behavior is scored as the percentage of cells uncapped and/or removed divided by the number initially frozen. (B) Hygienic response of independently selected populations and a baseline population after three generations of selection (baseline population was not artificially selected). Black points and whiskers represent mean and Standard Error for each sampled population while individual points represent individual colony measurements. Baseline population expressed hygiene significantly less than the two selected populations (68%; ANOVA; F2,38 = 25.8; P < 0.000001; Tukey HSD P < 0.00001, ***for selected vs. baseline; Tukey HSD selected vs. selected = 0.29. n.s.).
Here, we take an integrative genomic approach to study the genetics and evolution of loci associated with social immunity in honey bees. Hygienic behavior provides an ideal model for this study. It varies in expression within and among honey bee species and populations (Spivak and Gilliam 1998; Woyke et al. 2004; Woyke et al. 2012; Uzunov et al. 2014), has been the target of several breeding programs around the world (e.g., Spivak and Reuter 2001; Buchler et al. 2010; Pernal et al. 2012; Guarna et al. 2015), and has decades of research effort demonstrating that it has high narrow-sense heritability (Rothenbuhler 1964a, 1964b; Lapidge et al. 2002; Oxley et al. 2010. In this study, we created two artificially selected populations that highly express hygienic behavior and made use of high-depth full-genome sequencing to identify loci contributing to the variation in the expression of hygiene. We then integrated multiple independent genomic data sets to quantify patterns of natural and artificial selection at loci associated with hygienic behavior in honey bees.
Materials and Methods
Beekeeping and Breeding
Honey bee sampling, field testing, and breeding was performed at four locations in Western Canada: Selective breeding for hygienic behavior was conducted near Grand Forks, BC, whereas unselected colonies were maintained at the Research Farm of Agriculture and Agri-Food Canada in Beaverlodge, AB, and at the University of Manitoba in Winnipeg and propagated near Abbotsford, BC (for extensive details on breeding scheme see Guarna et al. 2015). Colonies were assessed for hygienic behavior using the freeze-killed brood (FKB) method (Spivak and Gilliam 1998), where the proportion of sealed cells that nurse bees fully uncap and remove dead pupae from is counted at 24 h using two separate tests performed 1 week apart on each colony. From an initial survey of 635 colonies, we created two selected populations (of 100 colonies each) and maintained them for three generations. Each generation we selectively bred for either high hygienic behavior or a combination of hygienic behavior and expression of protein markers associated with hygiene (for extensive sampling details see: Guarna et al. 2015, 2017). Along with these two selected populations, we also maintained a “baseline” population of 100 colonies that were randomly chosen from the survey population. To prevent migration among baseline and selected populations during selection, we maintained the baseline populations at separate apiaries. The baseline population was maintained throughout the experiment but allowed to openly mate. Our artificial selection procedures involved the following. For the first two generations, selected colonies were crossed using instrumental insemination: selected virgins were crossed with pooled semen collected from drones from 8 to 12 breeder colonies per site. Virgin queens from the third generation of selection were naturally closed mated, with mating apiaries located in an isolated mountain valley near Grand Forks and Christina Lake, Canada, respectively, where there were no other known feral or domestic sources honey bees. We sampled colonies from these two selected populations and from the baseline population at the third generation (see below).
We also sampled eight diploid adult workers from a random set of colonies within Ontario, Canada (supplementary table S1, Supplementary Material online). We included these samples to look for evidence of nonrandom introgression at regions tentatively associated with hygienic behavior.
Genome Alignment and Single Nucleotide Polymorphism Calling
The McGill University and Génome Québec Innovation Centre sequenced high molecular weight DNA from a total of 125 haploid male honey bees (drones) using Illumina HiSeq 2500 Rapid with 150-bp paired-ended reads to a mean depth of 33.07 reads. Drones were collected as larvae from randomly selected colonies of the control and selected lines with an average of 3.1 drones collected per colony. All samples were aligned, processed, and had single nucleotide polymorphisms (SNPs) called following a similar pipeline used previously by our group (Harpur, Kent, et al. 2014) and all sequencing data have been deposited with NCBI SRA (supplementary table S1, Supplementary Material online). Raw reads were trimmed of leading and tailing sequence with Trimmomatic v0.32, aligned to the honey bee reference genome (AMEL v4.5) using NextGenMapaligner v 0.4.12 (Sedlazeck et al. 2013), and removed of duplicate reads with Picard v1.8. For each colony, we created Variant Call Files (VCF) with GATK v 3.5 first by realigning around indels with RealignerTargetCreator followed by IndelRealigner to reduce any potential erroneous alignments (McKenna et al. 2010) then using UnifiedGenotyper (with options -stand_call_conf 60.0 -stand_emit_conf 40.0 –dcov 200 –min_base_quality_score 20) to call SNPs and then indels. We hard-filtered SNPs using VariantFiltration (QD < 5.0, FS > 40.0, MQ < 25.0, DP < 100.0) and excluded sequence from all unmapped scaffolds (AMEL v4.5; Groups 17 or Groups Un) because of low sequencing coverage in these small and gene-sparse scaffolds. Several genomic features can result erroneous variant calls (McKenna et al. 2010; Hodgkinson and Eyre-Walker 2011; Leffler et al. 2013). To account for these problems, we applied three additional filters to our data set prior to scanning for selection. First, we removed all SNPs within 10 nt of an indel using GATK’s VariantFiltration. Second, we eliminated 1.5xIQR outliers for depth within any alignment. Third, we aligned all drones individually to the honey bee reference genome; however, when calling SNPs with GATK (as above) we allowed the calls to be made as diploid with the expectation that heterozygotic calls would indicate areas of low complexity that may lead to subsequent sequencing error (Wallberg et al. 2014). We excluded any SNP within 5 bp of these low-complexity sites. This alignment procedure was followed for each drone as well as for pooled alignments of drones from the same colony. The later allowed us to infer the queen’s genotype for each colony, the data set we proceeded with for all analyses. SNPs were identified as nonsynonymous or synonymous using SNPEff v3.6 (Cingolani et al. 2012).
Quantifying the Effects of Genotyping Error
Inferring each queen’s genotype from a sample of at least three of her haploid sons may lead to genotyping error at heterozygotic sites due to the chance of not observing one of her two alleles. In cases where both alleles are not observed, we would falsely infer that a heterozygous genotype is homozygous, with the probability of assignment to either homozygotic genotype being the population-level allele frequency. This random error is not expected to influence our selection analysis because it affects both control and selected populations and is not expected to lead to consistent differences in allele frequency between the two populations. Nevertheless, we explored the impact of this random effect, by using the R package HardyWeinberg v1.6.1 to sample genotypes at 1,000,000 sites from a multinomial distribution with allele frequencies of p = 0.1, 0.2, 0.3, 0.4, and 0.5. We did this independently from three populations to represent queens in each of control and two selected populations; supplementary table S1, Supplementary Material online). For each heterozygotic genotype, we applied a binomial probability to determine the number of heterozygotes missed due to sampling error, assuming each queen’s genotype was inferred from three haploid sons. In the case where the sampled sons of a heterozygous queen all had the same allele, we probabilistically assigned the queen’s genotype to either of the two homozygous genotypes based on the underlying allele frequency. We then estimated Fst between selected and control populations for both the real data (i.e. without genotyping error and called ‘real’ hereafter) and for the data following genotyping error.
We found that genotyping error had minimal effect on allele frequency–based estimates of selection at heterozygotic sites. The real allele frequency and the allele frequency estimated with genotyping error were highly correlated (r = 0.99; P < 0.000001) and expectedly, so too were real Fst and Fst with genotyping error (r = 0.81; P < 0.000001). Fst with genotyping error (mean = 0.012) was slightly but significantly higher than real Fst (mean = 0.017; P < 0.00001). Genotyping error did not greatly impact our False Positive rate: there was only a 0.53% ± 0.0053 Standard Error chance across all allele frequencies of a single real ‘low’ Fst site (Fst < 95% of data) becoming a ‘high’ Fst site due to genotyping error alone (Fst > 95% of data). It is therefore very unlikely that a 1Kbp window (containing an average of 11 SNPs in our dataset) will falsely be called an outlier due to genotyping error alone.
Neutral Simulations
We created a neutral simulation of our sampling design using ms (Hudson 2002). We simulated sampling 100 mated queens from within a much larger Canadian honey bee population. Because the effective population size of Canadian honey bees is unknown, we varied the initial population size from which the three experimental populations were selected over a wide range (N0 = 50,000, 100,000, 150,000, 250,000, 500,000, and 1,000,000). We note that British Columbia and Alberta have a census population size of at least 350,000 honey bee colonies (Mukezangango and Page 2017) so our simulations likely cover the range of expected effective population size for honeybees in our sampling range and Canada more broadly. Honeybees in Canada have little genetic differentiation between provinces (Harpur et al. 2015) and have high levels of genetic diversity (Harpur et al. 2012); two properties that suggest high effective population sizes. Importantly, our conclusions where robust over the range of N0 simulated herein.
From each N0 simulated, we instantaneously sampled 100 mated queens each of which was assumed to be storing sperm from 18 haploid males (Tarpy et al. 2015). This equates to sampling from each N0 to an effective population size of 219 (Wright 1933; Zayed 2004). We created three such samples of 219 individuals to represent our three experimental populations and we allowed these three populations to evolve via drift with no migration for three generations. For each simulation, we assumed a recombination rate of 22 cM/Mb (Beye et al. 2006; Solignac et al. 2007) and a mutation rate of 3 × 10−9 (Liu et al. 2017). For each N0, we simulated 100,000 1-kbp windows and sampled 84 total chromosomes across the three populations, representing our experimental sampling scheme. From these samples, we then estimated pairwise Fst (Weir and Cockerham 1984) between each of the two populations representing the experimental selected populations against the single population representing the experimental control population. We also estimated Tajima’s D (Tajima 1989) within the selected populations independently. Therefore, our simulation allows us to quantify the expected distributions of Fst and Tajima’s D for our breeding trial without the effects of selection.
Identifying Positively Selected Loci
Our simulations demonstrated that the demographic effects on allele frequency differentiation among populations are minimal and so we sought to identify regions of the genome acted on by positive selection using several robust frequency-based approaches. Artificial positive selection shifts the allele frequency spectrum around selected loci by driving causal mutations and those linked to them to fixation (Nijhout and Paulsen 1997; Nielsen 2005). Alleles that are associated with a given trait will be among the first to fix and be detectable by differences in allele frequency between populations (Nijhout and Paulsen 1997; Akey et al. 2002; Nielsen 2005; De Kovel 2006). By sequencing the genomes of selected and unselected lines, we were able to look for these differences in allele frequency between lines using scans of pairwise Fst (Weir and Cockerham 1984) with the understanding that regions of high Fst relative to the rest of the genome are likely to be those acted on by selection (Akey et al. 2002). We used hapFLK analysis (Bonhomme et al. 2010; Fariello et al. 2013) to identify local haplotype clusters acted on by positive selection. We first ran hapFLK on each of the 16 chromosomes individually across all populations to create a pairwise Reynolds’ distances between populations. Using this kinship matrix, we used 20 haplotype clusters and scanned across each chromosome for 20 expectation maximization iterations with hapFLK using our baseline population as the outgroup. We estimated significance using chi-squared density and we corrected for False Discovery Rate by using Storey’s method (Storey and Tibshirani 2003) and taking only P values with Q < 0.01 (corresponding to P < 0.000001). We estimated the integrated haplotype score (Voight et al. 2006) using the R package rehh (Gautier et al. 2017). We estimated the shift in the allele frequency spectrum within selected populations using Tajima’s D (Tajima 1989) within 1-kb windows as estimated through VCFTOOLS v1.11 (Danecek et al. 2011). We compiled each of these three measures of selection into a single statistic, the single composite selection statistic (CSS) (Randhawa et al. 2014). We scanned each chromosome using a running median of 101 SNPs and extracted all regions with a −log10[CSS P value] > 1.3. Any region that was within 5 kb of any other significant region was pooled. For these methods, and all other methods requiring phased data, we phased all queen genotypes together for each chromosome individually using SHAPEIT v2.2 (O’Connell et al. 2014) with the additional options –rho 0.39 –window 0.5.
Comparisons to Previous Hygienic Behavior Association Studies and Evidence of Natural Selection
Broad Quantitative Trait Loci (QTLs) have been previously identified for hygienic behavior Lapidge et al. 2002; Oxley et al. 2010; Spotter et al. 2012; Tsuruda et al. 2012. We tested if genes acted on by artificial selection in our analysis localized to these broader regions. We remapped QTL regions based on microsatellites by using BlastN to identify the homologous regions within the Amelv4.5 genome. We have included the previously associated regions supplementary material (Data Set S1), Supplementary Material online. To quantify natural selection acting since the split of Apismellifera from its sister species Apiscerana, we used previous estimates of the selection coefficient (γ = 2Nes) on most genes within the honey bee genome (Harpur, Kent, et al. 2014); a selection coefficient >1 is indicative of positive selection driving the fixation of beneficial alleles. This analysis was performed with samples independent of those used in our artificial selection experiment.
Phenotype Association Analysis
We targeted our association analyses to quantify the relationship between haplotypes and the quantitative expression of hygiene within the 132 artificially selected regions. Haplotype analysis was performed within the baseline population only for a moving three SNP window using PLINK v 1.07 (–hap –hap-window 3) (Purcell et al. 2007; Chang et al. 2015). We extracted all 1,443 haplotypes that were significantly associated (P < 0.05) with hygienic behavior.
Admixture Analyses
We scanned the genome for evidence of differential admixture between selected and baseline populations and within North American populations using ELAI v 1.0 (Guan 2014). For each chromosome, we estimated local ancestry using the recommended default parameters of ELAI and assuming 200 generations since the initial admixture of source populations. Each run included both selected and baseline populations together.
Gene Ontology Analyses
We used DAVID v 6.7 (Huang et al. 2009) to identify if the list of candidate genes associated with hygienic behavior was enriched for Gene Ontology (GO) terms, focusing specifically on BP_4, MF_4, and CC_4. All tests we performed using Drosophila homologs identified with BlastP match (E-value threshold 1e-10) and because of our small gene list, we accepted any GO term with P < 0.1.
Statistical Analyses
All statistical analyses were performed with R v3.30 (R Core Team 2010). Statistical tests are reported within text and we performed parametric tests where data permitted, otherwise we report nonparametric results.
Results and Discussion
Sampling and Genome Sequencing
After three generations of artificial selection, our two selected populations expressed hygienic behavior significantly more (mean = 92% of dead brood and caps completely removed 24 h postfreezing) than the baseline population (mean = 68%; Analysis of variance [ANOVA]; F2,38 = 25.8; P < 0.000001; Tukey Honestly Significant Difference [HSD] P < 0.00001 for selected vs. baseline; Tukey HSD selected 1 vs. selected 2 = 0.29; fig. 1B). For all subsequent analyses, we have pooled the two selected populations unless otherwise stated. We sampled a total of 125 haploid drone larvae for colonies from each of the three populations. The queen genotypes of each colony were inferred given the genomes of their haploid drone sons, each sequenced to the same average mean site depth (supplementary table S1, Supplementary Material online; mean = 34.3×; ANOVA F1,37 = 2.15; P = 0.15; see Materials and Methods). Following alignment and variant calling, we were able to identify 2,340,950 segregating SNPs.
Selection Mapping
Strong selective events are expected to 1) increase the degree of differentiation between selected and unselected populations at loci contributing variation to the selected trait (Nijhout and Paulsen 1997), 2) increase differentiation of allele frequencies at loci that are nearby causal loci due to hitchhiking effects—called selective sweeps (Nielsen 2005), and 3) cause a shift in the allele frequency spectrum away from neutral expectations at and nearby causal loci in selected populations (Nielsen 2005). We used these expectations to identify regions of the genome that are associated with hygienic behavior. To that end, we made use of three tests for selection. The first was the haplotype-based outlier approach hapFLK (Qanbari et al. 2012; Fariello et al. 2013) applied on the selected populations using the baseline population as an outgroup. The hapFLK statistic is a measure of haplotype frequency differentiation scaled by relatedness between populations: A high hapFLK value is indicative of positive selection (i.e., artificial selection in our study) (Qanbari et al. 2012; Fariello et al. 2013). Second, we estimated the shift in the allele frequency spectrum within the selected populations using Tajima’s D (Tajima 1989)—lower Tajima’s D relative to the genomic average is indicative of positive artificial selection. Finally, we estimated the integrated haplotype score (Voight et al. 2006) for selected populations at each SNP within the genome. This statistic detects evidence of recent positive selection at a locus by comparing levels of linkage disequilibrium around alleles. These three statistics were combined into a single composite selection statistic (CSS) (Randhawa et al. 2014) that allowed us to find regions of the genome with robust signatures of artificial selection. This approach yielded 132 candidate regions across the genome that had significant evidence of positive selection within the selected populations (fig. 2). Combined, these regions account for at least 1,255 kb and 10,140 SNPs across the genome.
Fig. 2.

—Selection map highlighting regions associated with hygienic behavior. Each plot presents the significance of the Composite Selection Statistic (CSS) for a single chromosome. The horizontal, dotted line represents significance cut-off. Red boxes are regions (±1 Mb) that both have significant evidence of positive selection and have evidence of having haplotypes associated with hygiene within baseline populations. Horizontal bars are QTL regions for hygienic behavior (high bars: Oxley et al. 2010; Tsuruda et al. 2012) and QTLs for hygiene-associated behaviors of uncapping and brood removal (low bars: Oxley et al. 2010). Dots are the location of SNPs tentatively associated with hygiene from a previous association study (Spotter et al. 2012, 2016).
Observed Patterns of Diversity and Divergence in Selected Regions Are Not Expected by Drift and Sampling Alone
We developed neutral simulations to examine the distribution of population genetic statistics in three populations similar to the ones studied herein under a model of random sampling and genetic drift. Three lines of evidence suggest that the patterns used to identify artificially selected regions those having high divergence between selected populations and the control population and allele frequency shifts within selected populations—are not caused by drift alone. First, simulated Fst values among the three modeled populations were never as high as those observed within our experimental population (table 1). The maximum observed Fst between either selected population and the control population in 1,000 bp windows was 0.726, whereas the maximum simulated Fst was 0.334. Second, there was a significant excess of Fst windows that were high (Fst > 99% of the data) in both of the selected populations relative to the control population within our experimental population when compared with our neutral simulations (table 1; Fisher test P < 0.001). Finally, in the experimental data set, we found that windows with high Fst in both selected populations were enriched for having lower Tajima’s D (D < 99% of the data) relative to all windows in the genome (Fisher test; OR = 1.32; P < 0.013). We never observed this pattern within our simulations (Fisher test; P > 0.62 for all comparisons). In fact, such high Fst windows within the simulations had significantly higher Tajima's D relative to all windows within a simulation (AOV; P < 0.01). This evidence suggests that our selection scan adequately captured regions which were acted on by artificial selection: regions with high Fst between selected and control populations and low Tajima's D in selected populations (Nielsen, et al. 2007).
Table 1.
Patterns of Diversity and Divergence within Our Selected Populations Were Rarely or Never Observed within Simulated Neutral Data Sets
| N0 ( ×1,000) | Max Fst | OR(Fst) | ΔD | OR(D) |
|---|---|---|---|---|
| 50 | 0.334 | 1.51 | 0.52 | — |
| 100 | 0.242 | 1.64 | 0.64 | 0.80 |
| 150 | 0.195 | 2.12 | 0.58 | — |
| 250 | 0.164 | 3.27 | 0.77 | — |
| 500 | 0.089 | 7.94 | 0.66 | — |
| 1,000 | 0.063 | 51.60 | 1.04 | — |
| Observed | 0.726 | −0.03 | 1.32 | |
Note.—Max Fst: the maximum observed Fst between any selected and control comparisons. OR(Fst): the ratio of observed high Fst windows in both selected populations to all windows over the same ratio in simulations (significantly >1 indicates proportionally more jointly high outlier Fst windows in the experimental data). ΔD: the difference in mean D between windows with Fst in two populations and the genomic average mean D. OR(D): The ratio of low D windows overlapping with high Fst windows in two populations to all windows over the same ratio genome-wide (significantly greater than one indicates more low D windows overlapping with jointly-high Fst windows). Significant comparisons are in bold (P < 0.05). “—” indicates never observed.
Overlap with Previous QTL Studies
The regions we found to be significantly associated with hygienic behavior often overlapped with, or were near to, previous QTLs for the trait (fig. 2; Lapidge et al. 2002; Oxley et al. 2010; Spotter et al. 2012; Tsuruda et al. 2012). Previous studies found several broad QTLs (totaling ∼12 Mb) that explained variation in the expression of hygiene among colonies. Our regions fell directly inside the most informative QTL identified to date: hyg2 on chromosome 5 (fig. 2) that accounted for 13% of the phenotypic variation in the expression of hygienic behavior in an independent study (Oxley et al. 2010). Our regions also overlapped with two QTLs on chromosomes 1 and 9 that explain 3.9% and 6%, respectively, of the phenotypic variance of Varroa-Specific Hygiene—a form of hygienic behavior specific to brood parasitized by Varroa mites (Harbo and Harris 1999, 2005; Tsuruda et al. 2012). Two selected regions on chromosomes 10 and 9 that also overlapped with QTLs that explain 7% of the variation in brood removal and 7% of the variation in brood uncapping behavior, respectively (Oxley et al. 2010). Finally, we supported evidence of loci on chromosomes 3 and 6 that were found to be associated with hygienic behavior from a low resolution genome association study (fig. 2) (Spotter et al. 2012, 2016). The overlap between our work and previous genetic studies of hygienic behavior strongly supports our approach for identifying loci underpinning hygienic behavior in honey bees. However, our approach has higher resolution: hygienic-associated regions span 1,255 kb in our study, relative to a total of ∼12 Mb previously implicated in hygienic or associated behaviors from in QTL studies.
Candidate Regions Explain Variation in Hygienic Behavior in the Baseline Population
Overlap with known QTLs provides evidence that the regions we identified are associated with hygiene. We were able to provide additional support by using a targeted haplotype association approach (Purcell et al. 2007). We asked if hygiene-associated loci inferred from our population genomic contrasts between baseline and selected populations contained SNPs or haplotypes that explained phenotypic variation in hygiene in our baseline population (see Materials and Methods). We found 1,443 haplotypes (2,058 SNPs) within 99 of the 132 candidate loci inferred from population genomic contrasts that were significantly associated (P < 0.05) with differences in hygienic behavior in the baseline (unselected) population. For the proceeding functional and evolutionary analysis, we only included the 99 genomic regions (977 kb; supplementary table S2, Supplementary Material online) that had significant evidence of selection in our genomic contrast between selected and baseline populations and contained haplotypes that were significantly associated with hygienic behavior in our baseline population.
Candidate Genes for Hygienic Behavior
By integrating both selection and association mapping, we have narrowed the candidate loci underpinning variation in hygiene from the ∼12 Mb of bee’s genome previously implicated in QTL studies to ∼977 kb, representing an order of magnitude improvement in mapping resolution.
The reduction in sequence space is promising and provides a useful list of candidate genes for future functional investigation. However, we sought to narrow our search further by extracting from our list of candidate regions those genes with the greatest evidence of differentiation among selected and control populations. We extracted those genes with significant evidence of differentiation in and around the 99 associated windows (−log10[hapFLK P] > 2.5). In doing so, we narrowed the putative candidates to a set of 73 protein-coding genes (49 of which are within or near to QTL regions; supplementary table S3, Supplementary Material online). We studied the taxonomic origin, molecular and biological function, and evolution of these 73 genes to better understand the genetics, molecular biology and evolution of hygienic behavior.
After classifying these 73 candidate genes associated with hygienic behavior based on their phylogenetic origins (see supplemental in Harpur, Kent, et al. 2014; Jasper et al. 2015), we found that 85–98.7% of them are shared among Hymenopterans and Insects, respectively (supplementary table S3, Supplementary Material online). It has been hypothesized that novel social traits arise either via novel genes (i.e., evolutionary recent) or by reusing and remodeling existing genes and gene networks regulating analogous traits found in solitary ancestors (i.e., evo-devo/tool kit hypothesis) (Johnson and Linksvayer 2010; Rehan and Toth 2015). Our study supports the latter model for social immunity, given that most of our top candidate genes are taxonomically ancient.
Using enrichment analysis based on GO, we found that candidate genes were enriched for terms associated with neuronal development and early axon guidance (GO Analysis; supplementary table S4, Supplementary Material online). The most highly significant SNPs (−log10[hapFLK P] > 2.5) within the 73 genes were predominately found within introns (94% of these SNPs), a pattern that suggests the genes underpinning hygiene play a role in regulating gene expression. Taken together, our results suggest that mutations in our candidate genes play a role in regulating the expression of key genes involved in neuronal development.
Reducing our search further and focusing solely on the genes within only the most significant peaks and those within or near to previous QTLs alone, we recapitulate the broader results reported above. The significant CSS peak on chromosome 6 contains three genes (abscam, goosecoid, and tropomysin-2-like), all of which are critical to early neuronal development (Hahn and Jäckle 1996; Li and Gao 2003; Funada et al. 2007; Posnien et al. 2011). The most significantly differentiated of the candidates is abscam (GB45774) an ortholog of the Drosophila gene dscam2. Abscam is among the few honey bee genes that has been functionally characterized and is known to play a role in axon guidance (Funada et al. 2007). Isoforms of abscam are expressed during early development within the lamina, medulla, and lobula of the optic lobes, the glomeruli of the antennal lobes, the central body, and the mushroom bodies where expression promotes neural outgrowth, particularly of olfactory neural axons (Funada et al. 2007). It is the many isoforms of abscam that are involved in neuronal outgrowth and patterning, isoforms created by including or excluding immuno-globin domains through alternative splicing (Funada et al. 2007). The most significantly differentiated of the SNPs within this gene are intronic and are within or flank splice-site recognition regions surrounding immuno-globin domains.
The peaks at chromosomes 11 and 9 contain the ortholog to the Drosophila gene dyschronic (chromosome 11; GB45054) and Insulin-like receptor (Chr. 9; GB53353). Dyschronic is expressed during development and encodes several splice forms whose expression can affect axon guidance, overall neuroanatomy and locomotion (Jepson et al. 2012). In adult Drosophila, dyschronic protein is expressed in the mushroom bodies, ellipsoid body and antennal lobes where it interacts with Big Potassium (BK) channels and regulates neuronal excitability (Jepson et al. 2012). Variants of dyschronic, may act to alter the response thresholds of hygienic bees through its association with BK channels. BK channels are known to limit the action potential duration (Bean 2007) and their interaction with dyschronic can change the shape of response thresholds (Jepson et al. 2014). Highly differentiated mutations within dyschronic include one mutation within a splice-site region and two nonsynonymous variants. Insulin-like receptor on chromosome 9 shares similar functions with abscam and dyschronic: It is involved in neuronal pruning and axon guidance (Song et al. 2003; Wong et al. 2013). The CSS peak, on chromosome 5, contains GB44550 (similar to Drosophila sidestep), again known to be involved in axon guidance during development (Sink et al. 2001).
Though we are only able to explore candidate genes at this time, our results are consistent with mechanistic studies of hygienic behavior in honey bees. Previous experimental work revealed that variation in hygienic behavior is the result of variance in the response threshold of nurse bees to “dead-brood” signals (Masterman et al. 2001; McAfee et al. 2018) potentially caused by overactive octopaminergenic neurons in the antennal lobes or mushroom bodies of the brain (Spivak et al. 2003). Dead-brood signals are detected at olfactory chemo-sensory neurons of the antennae which are then transmitted to the antennal lobes and processed by the mushroom bodies. Hygienic bees are more receptive to these signals as a result of structural variation in the brain and have distinct patterns of gene and protein expression in brain and antennal regions (Parker et al. 2012; Boutin et al. 2015; Guarna et al. 2015). Our data suggest that differences in the expression of hygienic behavior between bees may be the result of differences in developmental trajectory during adult behavioral maturation or larval development.
Evidence of Positive Selection on Social Immune Loci
Social immunity is argued to be effective at reducing the risk of infection to such an extent that it relaxes constraint on the innate immune system (Evans et al. 2006; Cotter and Kilner 2010; Harpur, Chernyshova, et al. 2014; Lopez-Uribe et al. 2016). If the genes underpinning social immunity contribute to fitness in social lineages, we would expect those genes to be acted on by natural selection. To date, no study has explored the genetic evolution of social immunity in honey bees because the underlying genes were not known. Here, we examined patterns of adaptive evolution at our 73 top candidate genes relative to the rest of the honey bee’s protein-coding genome over the past ∼5–25 Ma (Harpur, Kent, et al. 2014). We achieved this by directly estimating selection coefficients at the 73 genes strongly associated with hygienic behavior and comparing them to other genes in the genome using a variant of the MK test applied to sequence data from A. mellifera and its sister species Apiscerana (as performed in Harpur, Kent, et al. [2014]).
We found that 13.6% of the 73 candidate genes had evidence of strong positive selection and that these genes had significantly higher selection coefficients than all other similar sized sets of genes in the genome (permutation test N = 10,000; P = 0.005). If we restrict our analysis to only the 49 candidate genes within QTL regions, we again find that hygiene candidates are more highly enriched for evidence of selection with 23.2% of those genes having evidence of selection (P = 0.01). If we estimate the average selection coefficient for all GO Biological Process sets in the honey bee database (Huang et al. 2009), we find that the hygienic candidates had higher selection coefficients than 90% of all Biological Process GO terms, with levels of selection similar to the biological processes of regulation of neurotransmitter levels (GO:0001505), learning or memory (GO:0007611), and detection of external stimulus (GO:0009581). Our analysis supports the hypothesis that hygienic behavior is important for fitness in honey bees. Further, fitness benefit is not strictly a result of management though beekeeping as our estimates of selection were derived from the African honey bee a population that is not typically used in commercial beekeeping.
C-Lineage Alleles Associated with Hygienic Behavior in Managed Bees
Comparisons within and across multiple studies suggest that subspecies of the honey bee’s C-lineage (e.g., A. m. ligustica or A. m. carnica) are more hygienic than subspecies of the M-lineage (e.g., A. m. mellifera) in Europe (Flores et al. 2001; Perez-Sato et al. 2009; Bak et al. 2010; Balhareth et al. 2012; Uzunov et al. 2014; Gerula et al. 2015) (supplementary table S5, Supplementary Material online). Managed North America honey bees are highly admixed, originating mostly from both the C- and M-lineage bees of Europe (Harpur et al. 2015). If the differences in hygienic behavior between the C- and M-lineages are genetically influenced, then we may expect to find a higher frequency of C-lineage alleles in managed North American populations that have been artificially selected for hygienic behavior.
In our artificially selected populations, we found that hygienic loci have significantly more C-lineage ancestry (median 87% C) relative to the baseline population (79% C) and relative to the genome as a whole (fig. 3A; Wilcoxon test, P < 0.0001). We found this same pattern of differential admixture at hygienic loci within an independent population of Canadian honey bees—colonies from the Province of Ontario that have been subjected to artificial selection for hygienic behavior for more than a decade (Harpur et al. 2012, 2015) (fig. 3B). Within the candidate genes above, at the most extreme, SNPs within Insulin-like receptor (chr 9; GB53353) are almost entirely fixed for C-lineage variants within selected and North American hygienic populations (median 95% C in selected and 91% within North America) but not within the baseline population (53% C).
Fig. 3.

—(A) Proportion of C-lineage ancestry at hygienic loci within selected populations compared with baseline populations. y axis represents the proportion of C-lineage ancestry in selected populations minus that of the baseline population; increasing values are indicative of more C-lineage ancestry in the selected populations. (B) This is a pattern that we also found within highly hygienic North American populations not included within our artificially selected populations. (***P < 0.01).
Conclusions
We used an integrative genomic approach to identify regions of the honey bee genome associated with hygienic behavior and to study the molecular function and evolutionary history of the top candidate genes within those regions. Our work provides new opportunities to explore how candidate genes contribute variation to the expression of hygienic behavior. We show that candidate genes are highly conserved, have evidence of positive selection, and are enriched for regulatory mutations that likely act to influence brain and neuronal development of worker bees. Our work now allows for future functional experiments to identify and confirm the mechanistic roles of the candidate genes identified herein.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank the staff at McGill University/Génome Québec Innovation Centre’s for their continued sequencing support. Appreciation is also owed to Charlie Whitfield, Nadia Tsvetkov, Clement Kent, Alivia Dey, and Kathleen Dogantzis, for comments on an early manuscript. We would further like to recognize Susan Cobey for assisting with instrumental insemination used in our breeding efforts. We also thank the reviewers and editorial team for their suggested improvements to this manuscript. Ethicsapproval andconsent toparticipate: No human or regulated animal subjects were part of this study. Funding: The project was funded by a seed grant from Ontario Genomics, a Discovery grant from the Natural Sciences and Engineering Council (NSERC) of Canada, and a York University Research Chair in Genomics to A.Z. The selected honey bee populations were generated by the BeeIPM project (107BEE) funded by Genome Canada, Genome British Columbia, Genome Alberta, the British Columbia Honey Producers Association’s Boone-Hodgson-Wilkinson Fund, the British Columbia Blueberry Council, the University of British Columbia, the University of Manitoba, the US Department of Agriculture, and Agriculture and Agri-Food Canada. B.A.H. was supported by an Elia Scholarship from York University, a NSERC Alexander Graham Bell Canadian Graduate Scholarship, and a NSERC Postdoctoral Fellowship. The funding bodies had no role in the design, analysis, nor interpretation of the study.
Author Contributions
The study was designed by L.J.F., R.W.C., S.F.P., A.Z., and B.A.H. Colonies were maintained, phenotyped, bred, and collected by M.M.G., E.H., H.H., K.-M.M., S.E.H., A.I., A.P.M., S.D., R.W.C., and S.F.P. Genomic data were created and analyzed by B.A.H. B.A.H. and A.Z. wrote the manuscript. All authors read and approved the manuscript.
Data deposition: This project has been deposited at NCBI under the BioProject accession PRJNA363032.
Literature Cited
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD.. 2002. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 12(12):1805–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arias MC, Sheppard WS.. 2005. Phylogenetic relationships of honey bees (Hymenoptera:Apinae:Apini) inferred from nuclear and mitochondrial DNA sequence data. Mol Phylogenet Evol. 37(1):25–35. [DOI] [PubMed] [Google Scholar]
- Bak B, Wilde J, Siuda M.. 2010. Comparison of hygienic behaviour between five honey bee breeding lines. J Apic Sci. 54:17–24. [Google Scholar]
- Balhareth HM, Alqarni AS, Owayss AA.. 2012. Comparison of hygienic and grooming behaviors of indigenous and exotic honeybee (Apis mellifera) races in Central Saudi Arabia. Int J Agric Biol. 14:1005–1008. [Google Scholar]
- Barribeau SM, et al. 2015. A depauperate immune repertoire precedes evolution of sociality in bees. Genome Biol. 16:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. 2007. The action potential in mammalian central neurons. Nat Rev Neurosci. 8(6):451–465. [DOI] [PubMed] [Google Scholar]
- Beye M, et al. 2006. Exceptionally high levels of recombination across the honey bee genome. Genome Res. 16(11):1339–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhomme M, et al. 2010. Detecting selection in population trees: the Lewontin and Krakauer test extended. Genetics 186(1):241–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutin S, Alburaki M, Mercier PL, Giovenazzo P, Derome N.. 2015. Differential gene expression between hygienic and non-hygienic honeybee (Apis mellifera L.) hives. BMC Genomics. 16:500.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchler R, Berg S, Le Conte Y.. 2010. Breeding for resistance to Varroa destructor in Europe. Apidologie 41:393–408. [Google Scholar]
- Chang CC, et al. 2015. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4:7.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P, et al. 2012. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso-2; iso-3. Fly (Austin) 6(2):80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotter SC, Kilner RM.. 2010. Personal immunity versus social immunity. Behav Ecol. 21(4):663–668. [Google Scholar]
- Cremer S, Armitage SAO, Schmid-Hempel P.. 2007. Social immunity. Curr Biol. 17(16):R693–R702. [DOI] [PubMed] [Google Scholar]
- Danecek P, et al. 2011. The variant call format and VCFtools. Bioinformatics 27(15):2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Kovel CGF. 2006. The power of allele frequency comparisons to detect the footprint of selection in natural and experimental situations. Genet Sel Evol. 38(1):3–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans JD, et al. 2006. Immune pathways and defence mechanisms in honey bees Apis mellifera. Insect Mol Biol. 15(5):645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fariello MI, Boitard S, Naya H, SanCristobal M, Servin B.. 2013. Detecting signatures of selection through haplotype differentiation among hierarchically structured populations. Genetics 193(3):929–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores JM, Ruiz JA, Ruz JM, Puerta F, Bustos M.. 2001. Hygienic behaviour of Apis mellifera iberica against brood cells artificially infested with varroa. J Apic Res. 40(1):29–34. [Google Scholar]
- Funada M, Hara H, Sasagawa H, Kitagawa Y, Kadowaki T.. 2007. A honey bee Dscam family member, AbsCAM, is a brain-specific cell adhesion molecule with the neurite outgrowth activity which influences neuronal wiring during development. Eur J Neurosci. 25(1):168–180. [DOI] [PubMed] [Google Scholar]
- Gautier M, Klassmann A, Vitalis R.. 2017. rehh 2.0: a reimplementation of the r package rehh to detect positive selection from haplotype structure. Mol Ecol Resour. 17(1):78–90. [DOI] [PubMed] [Google Scholar]
- Gerula D, Węgrzynowicz P, Panasiuk B, Bieńkowska M, Skowronek W.. 2015. Hygienic behaviour of honeybee colonies with different levels of polyandry and genotypic composition. J Apic Sci. 59(2):107–113. [Google Scholar]
- Guan Y. 2014. Detecting structure of haplotypes and local ancestry. Genetics 196(3):625–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarna MM, et al. 2015. A search for protein biomarkers links olfactory signal transduction to social immunity. BMC Genomics. 16:63.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guarna MM, et al. 2017. Peptide biomarkers used for the selective breeding of a complex polygenic trait in honey bees. Sci Rep. 7(1):8381.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M, Jäckle H.. 1996. Drosophila goosecoid participates in neural development but not in body axis formation. EMBO J. 15(12):3077.. [PMC free article] [PubMed] [Google Scholar]
- Harbo JR, Harris JW.. 1999. Heritability in honey bees (Hymenoptera: Apidae) of characteristics associated with resistance to Varroa jacobsoni (Mesostigmata: Varroidae). J Econ Entomol. 92(2):261–265. [Google Scholar]
- Harbo JR, Harris JW.. 2005. Suppressed mite reproduction explained by the behaviour of adult bees. J Apic Res. 44(1):21–23. [Google Scholar]
- Harpur BA, et al. 2015. Assessing patterns of admixture and ancestry in Canadian honey bees. Insectes Soc. 62(4):479–489. [Google Scholar]
- Harpur BA, Chernyshova A, et al. 2014. No genetic tradeoffs between hygienic behaviour and individual innate immunity in the honey bee, Apis mellifera. PLoS One 9(8):e104214.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpur BA, Kent CF, et al. 2014. Population genomics of the honey bee reveals strong signatures of positive selection on worker traits. Proc Natl Acad Sci U S A. 111(7):2614–2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpur BA, Minaei S, Kent CF, Zayed A.. 2012. Management increases genetic diversity of honey bees via admixture. Mol Ecol. 21(18):4414–4421. [DOI] [PubMed] [Google Scholar]
- Harpur BA, Zayed A.. 2013. Accelerated evolution of innate immunity proteins in social insects: adaptive evolution or relaxed constraint? Mol Biol Evol. 30(7):1665–1674. [DOI] [PubMed] [Google Scholar]
- Heinze J, Walter B.. 2010. Moribund ants leave their nests to die in social isolation. Curr Biol. 20(3):249–252. [DOI] [PubMed] [Google Scholar]
- Hodgkinson A, Eyre-Walker A.. 2011. Variation in the mutation rate across mammalian genomes. Nat Rev Genet. 12(11):756–766. [DOI] [PubMed] [Google Scholar]
- Huang DW, Sherman BT, Lempicki RA.. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 4(1):44–57. [DOI] [PubMed] [Google Scholar]
- Hudson RR. 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics. 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Jasper WC, et al. 2015. Large-scale coding sequence change underlies the evolution of postdevelopmental novelty in honey bees. Mol Biol Evol. 32(2):334–346. [DOI] [PubMed] [Google Scholar]
- Jepson JEC, et al. 2012. dyschronic, a Drosophila homolog of a deaf-blindness gene, regulates circadian output and slowpoke channels. PLoS Genet. 8:549–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jepson JEC, et al. 2014. Regulation of synaptic development and function by the Drosophila PDZ protein dyschronic. Development 141(23):4548–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BR, Linksvayer TA.. 2010. Deconstructing the superorganism: social physiology, groundplans, and sociogenomics. Q Rev Biol. 85(1):57–79. [DOI] [PubMed] [Google Scholar]
- Kotthoff U, Wappler T, Engel MS.. 2013. Greater past disparity and diversity hints at ancient migrations of European honey bee lineages into Africa and Asia. J Biogeogr. 40(10):1832–1838. [Google Scholar]
- Lapidge KL, Oldroyd BP, Spivak M.. 2002. Seven suggestive quantitative trait loci influence hygienic behavior of honey bees. Naturwissenschaften 89(12):565–568. [DOI] [PubMed] [Google Scholar]
- Lawniczak MKN, et al. 2007. Mating and immunity in invertebrates. Trends Ecol Evol. 22:48–55. [DOI] [PubMed] [Google Scholar]
- Lecocq A, Jensen AB, Kryger P, Nieh JC.. 2016. Parasite infection accelerates age polyethism in young honey bees. Sci Rep. 6:22042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler EM, et al. 2013. Multiple instances of ancient balancing selection shared between humans and chimpanzees. Science 339(6127):1578–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li WJ, Gao FB.. 2003. Actin filament-stabilizing protein tropomyosin regulates the size of dendritic fields. J Neurosci. 23(15):6171–6175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, et al. 2017. Direct determination of the mutation rate in the bumblebee reveals evidence for weak recombination-associated mutation and an approximate rate constancy in insects. Mol Biol Evol. 34(1):119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Uribe MM, Sconiers WB, Frank SD, Dunn RR, Tarpy DR.. 2016. Reduced cellular immune response in social insect lineages. Biol Lett. 12:20150984.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masterman R, Ross R, Mesce K, Spivak M.. 2001. Olfactory and behavioral response thresholds to odors of diseased brood differ between hygienic and non-hygienic honey bees (Apis mellifera L.). J Comp Physiol A Sens Neural Behav Physiol. 187:441–452. [DOI] [PubMed] [Google Scholar]
- McAfee A, et al. 2018. A death pheromone, oleic acid, triggers hygienic behavior in honey bees (Apis mellifera L.). Sci Rep. 8(1):5719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20(9):1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukezangango J, Page S.. 2017. Statistical overview of the Canadian honey and bee industry and the economic contribution of honey bee pollination. In: Canada HaCSDAaA-F, editor. Government of Canada, p. 3.
- Nielsen R. 2005. Molecular signatures of natural selection. Annu Rev Genet. 39(1):197–218. [DOI] [PubMed] [Google Scholar]
- Nielsen R, Hellmann I, Hubisz M, Bustamante C, Clark AG.. 2007. Recent and ongoing selection in the human genome. Nat Rev Genet. 8(11):857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhout HF, Paulsen SM.. 1997. Developmental models and polygenic characters. Am Nat. 149(2):394–405. [Google Scholar]
- Nunn CL, Jordan F, McCabe CM, Verdolin JL, Fewell JH.. 2015. Infectious disease and group size: more than just a numbers game. Philos Trans R Soc B Biol Sci. 370(1669): 20140111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell J, et al. 2014. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 10:e1004234.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxley PR, Spivak M, Oldroyd BP.. 2010. Six quantitative trait loci influence task thresholds for hygienic behaviour in honeybees (Apis mellifera). Mol Ecol. 19(7):1452–1461. [DOI] [PubMed] [Google Scholar]
- Parker R, et al. 2012. Correlation of proteome-wide changes with social immunity behaviors provides insight into resistance to the parasitic mite, Varroa destructor, in the honey bee (Apis mellifera). Genome Biol. 13(9):R81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Sato JA, Chaline N, Martin SJ, Hughes WOH, Ratnieks FLW.. 2009. Multi-level selection for hygienic behaviour in honeybees. Heredity 102:609–615. [DOI] [PubMed] [Google Scholar]
- Pernal SF, Sewalem A, Melathopoulos AP.. 2012. Breeding for hygienic behaviour in honeybees (Apis mellifera) using free-mated nucleus colonies. Apidologie 43(4):403–416. [Google Scholar]
- Posnien N, Koniszewski NDB, Hein HJ, Bucher G.. 2011. Candidate gene screen in the red flour beetle Tribolium reveals Six3 as ancient regulator of anterior median head and central complex development. PLoS Genet. 7(12):e1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulsen M, Bot ANM, Boomsma JJ.. 2003. The effect of metapleural gland secretion on the growth of a mutualistic bacterium on the cuticle of leaf-cutting ants. Naturwissenschaften 90(9):406–409. [DOI] [PubMed] [Google Scholar]
- Purcell S, et al. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 81(3):559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanbari S, et al. 2012. A high resolution genome-wide scan for significant selective sweeps: an application to pooled sequence data in laying chickens. PLoS One 7(11):e49525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2010. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing. [Google Scholar]
- Randhawa IAS, Khatkar MS, Thomson PC, Raadsma HW.. 2014. Composite selection signals can localize the trait specific genomic regions in multi-breed populations of cattle and sheep. BMC Genet. 15(1):34.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehan SM, Toth AL.. 2015. Climbing the social ladder: the molecular evolution of sociality. Trends Ecol Evol. 30:426–433. [DOI] [PubMed] [Google Scholar]
- Rosengaus RB, Maxmen AB, Coates LE, Traniello JFA.. 1998. Disease resistance: a benefit of sociality in the dampwood termite Zootermopsis angusticollis (Isoptera: Termopsidae). Behav Ecol Sociobiol. 44(2):125–134. [Google Scholar]
- Rothenbuhler WC. 1964a. Behavior genetics of nest cleaning in honey bees. 4. Responses of F1 and backcross generations to disease-killed brood. Am Zool. 4(2):111–123. [DOI] [PubMed] [Google Scholar]
- Rothenbuhler WC. 1964b. Behaviour genetics of nest cleaning in honey bees. I. Responses of 4 inbred lines to disease-killed brood. Anim Behav. 12(4):578.. [DOI] [PubMed] [Google Scholar]
- Sackton TB, et al. 2007. Dynamic evolution of the innate immune system in Drosophila. Nat Genet. 39(12):1461–1468. [DOI] [PubMed] [Google Scholar]
- Schmid-Hempel P. 1994. Infection and colony variability in social insects. Philos Trans R Soc Lond Ser B Biol Sci. 346:313–321. [Google Scholar]
- Sedlazeck FJ, Rescheneder P, von Haeseler A.. 2013. NextGenMap: fast and accurate read mapping in highly polymorphic genomes. Bioinformatics 29(21):2790–2791. [DOI] [PubMed] [Google Scholar]
- Sink H, Rehm EJ, Richstone L, Bulls YM, Goodman CS.. 2001. sidestep encodes a target-derived attractant essential for motor axon guidance in Drosophila. Cell 105(1):57–67. [DOI] [PubMed] [Google Scholar]
- Solignac M, Mougel F, Vautrin D, Monnerot M, Cornuet JM.. 2007. A third-generation microsatellite-based linkage map of the honey bee, Apis mellifera, and its comparison with the sequence-based physical map. Genome Biol. 8(4):R66.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JB, Wu LL, Chen Z, Kohanski RA, Pick L.. 2003. Axons guided by insulin receptor in Drosophila visual system. Science 300(5618):502–505. [DOI] [PubMed] [Google Scholar]
- Spivak M, Gilliam M.. 1998. Hygienic behaviour of honey bees and its application for control of brood diseases and varroa. Part I. Hygienic behaviour and resistance to American foulbrood. Bee World 79(3):124–134. [Google Scholar]
- Spivak M, Masterman R, Ross R, Mesce KA.. 2003. Hygienic behavior in the honey bee (Apis mellifera L.) and the modulatory role of octopamine. J Neurobiol. 55(3):341–354. [DOI] [PubMed] [Google Scholar]
- Spivak M, Reuter GS.. 2001. Resistance to American foulbrood disease by honey bee colonies Apis mellifera bred for hygienic behavior. Apidologie 32(6):555–565. [Google Scholar]
- Spotter A, Gupta P, Mayer M, Reinsch N, Bienefeld K.. 2016. Genome-wide association study of a varroa-specific defense behavior in honeybees (Apis mellifera). J Hered. 107:220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spotter A, Gupta P, Nurnberg G, Reinsch N, Bienefeld K.. 2012. Development of a 44K SNP assay focussing on the analysis of a Varroa-specific defence behaviour in honey bees (Apis mellifera carnica). Mol Ecol Resour. 12:323–332. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R.. 2003. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 100(16):9440–9445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Zhou XG.. 2013. Corpse management in social insects. Int J Biol Sci. 9(3):313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical-method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123(3):585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpy DR, Delaney DA, Seeley TD.. 2015. Mating frequencies of honey bee queens (Apis mellifera L.) in a population of feral colonies in the Northeastern United States. PLoS One 10(3):e0118734.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth AL, Robinson GE.. 2007. Evo-devo and the evolution of social behavior. Trends Genet. 23(7):334–341. [DOI] [PubMed] [Google Scholar]
- Tsuruda JM, Harris JW, Bourgeois L, Danka RG, Hunt GJ.. 2012. High-resolution linkage analyses to identify genes that influence varroa sensitive hygiene behavior in honey bees. PLoS One 7(11):e48276.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uzunov A, et al. 2014. Swarming, defensive and hygienic behaviour in honey bee colonies of different genetic origin in a pan-European experiment. J Apic Res. 53(2):248–260. [Google Scholar]
- Voight BF, Kudaravalli S, Wen XQ, Pritchard JK.. 2006. A map of recent positive selection in the human genome. PLoS Biol. 4:446–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallberg A, et al. 2014. A worldwide survey of genome sequence variation provides insight into the evolutionary history of the honeybee Apis mellifera. Nat Genet. 46(10):1081–1088. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC.. 1984. Estimating F-statistics for the analysis of population structure. Evolution 38(6):1358–1370. [DOI] [PubMed] [Google Scholar]
- Wong JJL, et al. 2013. A Cullin1-based SCF E3 ubiquitin ligase targets the InR/PI3K/TOR pathway to regulate neuronal pruning. PLoS Biol. 11:e1001657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woyke J, Wilde J, Phaincharoen M.. 2012. First evidence of hygienic behaviour in the dwarf honey bee Apis florea. J Apic Res. 51(4):359–361. [Google Scholar]
- Woyke J, Wilde J, Reddy CC.. 2004. Open-air-nesting honey bees Apis dorsata and Apis laboriosa differ from the cavity-nesting Apis mellifera and Apis cerana in brood hygiene behaviour. J Invertebr Pathol. 86(1–2):1–6. [DOI] [PubMed] [Google Scholar]
- Wright S. 1933. Inbreeding and homozygosis. Proc Natl Acad Sci U S A. 19(4):411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasloff M. 2002. Antimicrobial peptides of multicellular organisms. Nature 415(6870):389–395. [DOI] [PubMed] [Google Scholar]
- Zayed A. 2004. Effective population size in Hymenoptera with complementary sex determination. Heredity 93(6):627.. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.