

Summary
The mammalian brain is composed of diverse, specialized cell populations. To systematically ascertain and learn from these cellular specializations, we used Drop-seq to profile RNA expression in 690,000 individual cells, sampled from nine regions of the adult mouse brain. We identified 565 transcriptionally distinct groups of cells using computational approaches developed to distinguish biological from technical signals. Cross-region analysis of these 565 cell populations revealed features of brain organization, including a gene-expression module for synthesizing axonal and presynaptic components, patterns in the co-deployment of voltage-gated ion channels, functional distinctions among the cells of the vasculature and specialization of glutamatergic neurons across cortical regions. Systematic neuronal classifications for two complex basal ganglia nuclei and the striatum revealed a rare population of spiny projection neurons. This adult mouse brain cell atlas, accessible through interactive online software (DropViz), serves as a reference for development, disease, and evolution.
Keywords: Single-cell, brain, striatum, basal ganglia, transcriptional programs
Introduction
Cellular specialization is central to the function of the mammalian brain. At the coarsest level, cells of different classes (for example, neurons, astrocytes, and endothelial cells) interact to maintain homeostasis and enable electrochemical communication. At finer levels, subtle specializations – such as those that distinguish neuron subtypes in the same region – can control behaviors such as appetite (Andermann and Lowell, 2017; Sternson, 2013), sex drive (Anderson, 2012), habit formation (O’Hare et al., 2016; Wang et al., 2011), spatial mapping (Moser et al., 2008), and associative learning (Krabbe et al., 2017). Some cell populations have been characterized in detail; many remain uncharacterized or have yet to be discovered.
Systematic efforts – to identify cell populations, reveal the RNA repertoires of every cell type and state, and identify molecular markers for each population – would help to understand the functions and interactions of cells in the brain, including the roles of distinct cell types in disease. High-throughput single-cell RNA-seq (scRNA-seq) now enables RNA profiling in thousands of individual cells in complex tissue (Han et al., 2018; Klein et al., 2015; Macosko et al., 2015; Rosenberg et al., 2018; Zheng et al., 2017). To date, single-cell gene expression studies have yielded cell-type classifications in the mouse cerebral cortex (Tasic et al., 2016; Zeisel et al., 2015), retina (Shekhar et al., 2016), hypothalamic arcuate nucleus (Campbell et al., 2017), entopeduncular nucleus (Wallace et al., 2017) and amygdala (Wu et al., 2017).
In this work, we sought to analyze cellular diversity across many brain regions in order to investigate shared and region-specific patterns in cellular composition and gene expression. We overcame several challenges. First, dissociating adult brain into healthy, representative cell suspensions is difficult; many scRNA-seq studies have thus used younger mice in which developmental programs are comingled with the expression differences that underlie functional specializations. Here we developed techniques, borrowing ideas from preparations for electrophysiological recordings, that allowed adult brain tissue to be dissociated into intact cell bodies while representing all major cell classes. Second, scRNA-seq data are simultaneously shaped by cellular categories, continuously varying gradients, and technical artifacts; cell clusters derived from scRNA-seq often reflect unknown combinations of these effects. We developed analytical methods to separate biological and technical influences on single-cell data, enabling a more transparent understanding of the relationships driving cellular classifications.
Here, we describe a draft mouse brain cell atlas that we created by profiling (using Drop-seq) 690,000 individual cells from nine major regions of the adult mouse brain. By comparing single-cell transcriptional patterns within and across neuron types, we identified and validated a pervasive transcriptional program supporting axon and presynaptic function and discovered large-scale patterns in ion channel gene expression that may enact neurons’ electrophysiological properties. We found that, in the cortex, glutamatergic neurons tend to be specialized by region, while non-neuron cell classes, such as those that make up the vasculature, can be variably specialized across cortical and subcortical areas. We also highlight the neuronal diversity of individual brain regions through the classification of neuron types using examples from the basal ganglia. In the globus pallidus externus (GPe) and substantia nigra reticulata (SNr), where neuron types are not well-understood, we propose complete neuron-type classifications and identify selective markers for each population. In the striatum, where neuronal diversity is well charted, we nonetheless identify a novel group of principal neurons that had been overlooked in decades of research.
We hope that these data will advance a wide variety of efforts and nominate many unforeseen research questions for further study. To facilitate the exploration and utilization of these data, we developed an interactive analysis platform (“DropViz”, http://dropviz.org/) for comparing cell types, identifying cell populations that express genes of interest, and performing many other kinds of analyses.
Results
Isolation and Molecular Analysis of Cells for an Adult Brain Cell Atlas
To build an atlas of cell populations and cell-type-specific gene expression patterns across the adult (P60–70) mouse brain, we prepared single-cell suspensions (STAR Methods) from nine brain regions (Table S1 and Data S1) and used Drop-seq (Macosko et al., 2015) to profile the RNA expression of 690,207 individual cells (Figure 1A). The resulting cell suspensions, which recovered intact 40–50% of cells from most tissues (cortex: 0.46 ± 19 mean ± sem; striatum: 0.39 ± 20, Figure S4A–C), had cells with morphologies characteristic of neurons, astrocytes, and oligodendrocytes (Figure S1A). We generated and analyzed 13 billion sequencing reads from the resulting Drop-seq libraries, detecting 1.45 billion distinct mRNA transcripts (UMIs), which arose from 31,767 distinct genes. We ascertained an average of 17,480 reads (median=10,824), 2,218 mRNA transcripts (median=1,450 UMIs), and 1,169 genes per cell (median=900).
Figure 1. Single-cell transcriptional profiling of the adult mouse brain using Drop-seq and identification of transcriptional programs with independent component analysis.

(A) Sagittal schematic illustrating profiled brain regions and numbers of cells sampled (anatomical detail in Data S1). (B) Workflow for semi-supervised Independent Components Analysis (ICA)-based signal extraction and clustering (STAR Methods). In stage 1, the DGE matrix is clustered into cell classes (Figure S1) using ICA (“global clustering”). In stage 2 (“subclustering”), the process is repeated for each individual cluster from stage 1. In stage 2, however, the resulting ICs are curated as “technical” or “biological” with only “biological” ICs used as input for subclustering. (Figure S2) (C) tSNE plots for frontal cortex global clustering (left) and two representative subclusterings, GABAergic interneurons (cluster 1) and glutamatergic layer 2/3 and a subset of layer 5 neurons (cluster 6). (D) Examples of heterogeneous “Biological” ICs from frontal cortex cluster 6, representing a cell state (top, IC 16), cell type (middle, IC 22), and spatial anatomical signal (bottom, IC 29). For each example, a cell-loading tSNE plot, gene loading plot, and in situ hybridization experiment (Allen Mouse Brain Atlas, “Allen”) for a top-loading gene are shown from left to right. IC 16 corresponds to the immediate early gene signal. The IC 22 signal originates from layer 5a glutamatergic neurons, as suggested by Deptor expression. IC 29 represents a spatial signal, evidenced by a medial to lateral gradient of Lypd1. (E) Correspondence between heterogeneous transcriptional signals (biological ICs) and subclusters identified by modularity-based clustering (STAR Methods). Cell loadings for Biological ICs from frontal cortex cluster 6 and the resulting n=5 subclusters identified. Alternative subcluster solutions are shown in Figure S2K.
Cell-class Composition of Nine Adult Brain Regions
We separately analyzed data for each region using a two-stage procedure (Figure 1B and STAR Methods). The first stage of analysis robustly grouped cells into 8–11 broad classes (Data S2), including neurons, astrocytes, microglia/macrophages, oligodendrocytes, polydendrocytes (oligodendrocyte progenitor cells), and components of the vasculature – endothelial cells, fibroblast-like cells, and mural cells (Abbott et al., 2006; Marques et al., 2016; Vanlandewijck et al., 2018). The hippocampus, for example, yielded cells from all 11 cell classes (Figure S1B–D), including local cell classes native to the ventricle – the choroid plexus and ependymal cells - and a class undergoing adult neurogenesis (Habib et al., 2016; Hochgerner et al., 2018; Ming and Song, 2011). Distinct brain regions yielded cell classes in different proportions (Figure S1E).
Inference of Cell Types and States Using Independent Components Analysis
The recognition of subtle variation among cells of the same class presents a formidable analytical challenge for unsupervised analysis (Mayer et al., 2015; Satija et al., 2015; Shekhar et al., 2016; Tanay and Regev, 2017; Tasic et al., 2016). The size, diversity, and replicates for each atlas region highlighted limitations in current methods, exemplified by clusters specific to experimental replicates or driven by tissue digestion artifacts (see below). We sought a strategy that would (i) dissect biological from technical contributions to expression data and (ii) generate intermediate outputs (upstream of clustering) that could be critically evaluated and analyzed.
We therefore developed an analysis method based on independent components analysis (ICA)(Figure 1B–E). ICA reduces large datasets to a smaller number of dimensions in which entities (here cells) have score distributions that are maximally structured – as measured by deviation from a normal distribution (generally due to a spiky or clustered distribution of the cells in that dimension) – and statistically independent (Hyvärinen, 1999). Each of the inferred independent components (ICs) is a weighted combination of many genes (the weight of each gene’s contribution to an IC is the gene “loading”) and each cell is given a score for each IC (cell loading). This score reflects the degree to which the constellation of genes encoded by the IC is more or less expressed in that cell as compared to the average cell in the analysis. Each cell’s gene-expression profile is a weighted sum of ICs.
We found that individual ICs corresponded to recognizable biological phenomena (Figure 1D and Figure S2C–H)(Adamson et al., 2016), in contrast to results from principal components analysis (Figure S2A). For example, among glutamatergic neurons from frontal cortex cluster 6, we identified ICs whose strongly loading genes marked specific cell types, cell states or spatial gradients across anatomical axes (Figure 1D). Other ICs captured technical effects such as 1) cells from different replicate preparations; 2) RNA libraries of different sizes; 3) experimentally identified effects of tissue preparation; or 4) cell-cell “doublets” (Figure S2C-H). We found that the interpretability of individual ICs allowed us to distinguish presumed endogenous signals (called “biological ICs”) from ICs related to the technical signals described above. Removing technical ICs reduced spurious distinctions among cells (Figure S2I).
We analyzed the data from each class and region (109 analyses total) using semi-supervised ICA, in which we excluded 1,157 ICs as technical and classified the remaining 601 ICs as “biological ICs” (STAR Methods). We then grouped cells based on combinations of “biological IC” cell loadings into 565 subclusters using network-based clustering (Shekhar et al., 2016; Waltman and van Eck, 2013) (Figure 1E and Figures S2J,K). Of these subclusters, 323 were neuronal, derived from 368 biological ICs (Data S3). Across subclusters, patterns of similar RNA expression generally corresponded to cell class rather than brain region (Figure S1F,G).
Characteristics of the Cells of the Blood-Brain Barrier
Non-neuronal cells exhibited broadly consistent expression signals across brain regions. To better appreciate diversity among non-neuronal classes, we grouped single-cell libraries across regions by cell class and performed semi-supervised ICA on each of the seven non-neuronal datasets independently, identifying a total of 53 biological ICs (Figure 2A and Data S4A–G). We focus here on cell classes that form the blood-brain barrier – mural, fibroblast-like and endothelial cells – because they are disease-relevant (Sweeney et al., 2016) and remain incompletely characterized (Figure 2B).
Figure 2. Comprehensive description of transcriptional diversity within non-neurons as illustrated by cell classes of the vasculature.

(A) Number of Biological ICs identified during curation for each non-neuronal cell class. All non-neuronal ICs are shown in Data S4. (B) Vasculature cell classes. (C-E) Subcluster assignments and examples of two biological ICs for each vasculature cell class. Subclusters (color-coded), IC cell-loadings, and gene expression values displayed on tSNE plots. Left, subcluster assignments. Middle, IC cell- and gene-loadings. For each IC, the top ten loading genes are listed. Right, expression plots for individual genes. For Mural Cell IC 19, the bottom loading gene Acta2 is shown in purple. (F) Dot plots illustrating fractional representation of cells from each region contributing to fibroblast-like and endothelial subclusters. Other non-neuronal cell classes are shown Data S4H.
Mural cells are intrinsic to the endothelium and control vascular development, stability, and homeostasis (Sweeney et al., 2016; Trost et al., 2016). We identified 7 mural subclusters from 7 biological ICs (n=4,713 cells, Figure 2C and Data S4E). Mural cells have two subtypes: pericytes, which associate with capillaries, and smooth muscle alpha actin (SMA) cells, which associate with larger-bore vasculature and control blood flow (Hill et al., 2015; Hughes and Chan-Ling, 2004; Nehls and Drenckhahn, 1991). A single IC (IC 13) appeared to encode this distinction, with pericyte marker Vtn as the strongest loading gene (Figure 2C)(Vanlandewijck et al., 2018). Other enriched genes suggest specialized pericyte function. For example, expression of a potassium channel activated by diphosphate levels (encoded by Kcnj8 and Abcc9) and an ADP-ribosyltransferase (Art3) suggest signaling machinery that couples dinucleotide metabolites to membrane potential and post-translational modification. Among SMA cells, Acta2 expression correlates with a veinous versus arterial distribution (Vanlandewijck et al., 2018). IC 19 represented this difference in a graded rather than categorical way, as Rgs5/Acta2 expression and IC cell scores were continuously, rather than bimodally, distributed across these cells (Figure 2C)(Vanlandewijck et al., 2018). Our data also identify new mural cell diversity. For example, cluster 1 expressed pericyte (Rgs5) and SMA (Acta2) markers. Unique markers (e.g. Aldh1a1) will help determine the anatomical identify of this population (Data S4E).
While endothelial cells are known as the constituent cell class of blood vessels, fibroblast-like cells are a recently-described population with unknown function that inhabit the perivascular space in the brain (Vanlandewijck et al., 2018). We found 7 subclusters each of endothelial (n=16,248) and fibroblast-like (n=1,587) cells (Figure 2D,E). Among the fibroblast-like cells, two subclusters (4 and 5) selectively expressed many genes encoding membrane transporters (e.g., Slc38a2, Slc4a10, Slc26a2, and Slc47a1) and pumps (e.g., Fxyd5 and Atp1b1) (Figure 2D). To varying extents, subclusters 1, 2, and 3 expressed genes involved with extracellular matrix (ECM) secretion, angiogenesis, and contraction (Data S4F), such as the basement membrane collagen genes (Col4a1, Col4a2, and Col15a1) (Figure 2D). Interestingly, cluster 3 expressed higher levels of different collagen genes (Col1a1 and Col3a1) (Data S4F). These examples suggest that fibroblast-like cells are transcriptionally specialized to support membrane transport and ECM production; ECM secretion may involve different sets of co-regulated collagen genes.
We identified endothelial ICs (Figure 2E) with strong contributions from genes with arterial (IC 5 and 20) and veinous (IC 3 and 12) expression, suggesting heterogeneity related to vessel type (Data S4G) (Vanlandewijck et al., 2018). These genes (e.g. Tm4sf1 and Slc38a5) showed continuous, reciprocal expression (Data S4G), consistent with the smooth molecular transitions described for endothelial cells associated with arteries or veins (Vanlandewijck et al., 2018). Other ICs identified genes with more bimodal expression, indicating specializations within this artery/vein gradient. Subcluster 6 exhibited exclusive expression of Cytl1 and enrichment for arterial marker Bmx, along with other genes implicated in growth-factor dependent vascular remodeling (Mgp, Fbln5, Eln, Igfbp4, and Clu) (Figure 2E and Data S4G)(Boström et al., 2004; Contois et al., 2012; Fu et al., 2013; Guadall et al., 2011; Karnik et al.; Vanlandewijck et al., 2018). Other subpopulations could represent specializations shared across vessel types. For example, IC 10 loads onto cells with both artery and vein markers and strongly contributing genes suggest a signal related to host immunity, including genes encoding interferons (Ifit3, Ifit1, Ifit2, and Ifitm3), GTPases induced by interferons (Iigp1, Irgm1, and Gbp7), and other proteins involved in the anti-viral response (Isg15 and Rsad2) (Figure 2E). These examples identify endothelial processes within (e.g. arterial angiogenesis) or across (e.g. host defense) vessel types. Other signals might reflect cell states specialized for iron handling, calcium signaling, and the stress response (Data S4G).
Functional specializations within endothelial, glial, and other non-neuronal cell classes could be ubiquitous features of the adult mouse brain, or could be enriched in particular brain regions. We compared the relative abundance of cells from each region within fibroblast-like and endothelial subclusters (Figure 2F)(results for other non-neuronal classes in Data S4H). While endothelial subpopulations had similar abundances across regions, fibroblast-like subpopulations exhibited different contributions from cortical and subcortical areas: the cortex and hippocampus contributed disproportionately to the population that more strongly expressed genes with membrane-transport functions (subcluster 6), while collagen-expressing cell populations (subclusters 2 and 3) came largely from the basal ganglia and thalamus (Figure 2F).
A Neuronal Transcriptional Program Related to Axon Function
Cell states involve constellations of co-expressed genes that enact cellular functions. In neurons, the most well-studied state involves the immediate early genes (IEGs), which are transcribed in response to the Ca2+ influx that follows action potentials (Bading, 2013; Hrvatin et al., 2017). IEG expression is largely uniform across neuronal types (Hrvatin et al., 2017) and brain regions and exhibits little background expression, making it straightforward to detect in the Drop-seq data.
Neurons might share other transcriptional dynamics in common. To identify such signatures, we looked for transcriptional patterns that, despite being ascertained in different regions or neuronal classes, involved similar gene combinations. Analysis of pairwise correlations across neuronal IC gene loadings revealed three prominent correlation blocks (Figure 3A). One such block included 15 inter-correlated ICs representing IEG-expression signatures from different regions and neuronal types. A second block arose from diverse thalamic neurons, suggesting a tissue-specific transcriptional program. We focused on the third correlation block, which consisted of ICs from many brain regions and neuronal types.
Figure 3. A prevalent expression program in neurons related to axon structure and presynaptic function.

(A) Hierarchical clustering of pairwise Pearson correlations of gene-loading scores for biological ICs from 45 neuronal subclustering analyses. Right, enlargement of boxed region. Correlation blocks correspond to the immediate early gene (“IEG”) transcriptional state, thalamus-specific ICs (“TH”), or “Neurofilament” ICs, which are contributed from different regions and driven by genes that encode neurofilament subunits and other proteins involved in Ca2+ handling, vesicle exocytosis, and membrane excitability. (B) The Neurofilament transcriptional signal (IC 17) in frontal cortex Sst+/Pvalb+ interneurons (Cluster 2). Left, IC 17 cell-loadings displayed on subcluster tSNE plot. Right, gene-loading plot, with the top 20 genes shown. (C) Color-coded subcluster identities for frontal cortex cluster 2. N=10 subclusters were based on n=9 biological ICs. The graded loading of IC 17 is discretized into subclusters 2–8, 2–7, and 2–9. (D) Single gene expression plots. (E) Comparison of Neurofilament (Syt2, Pvalb, and Nefm) and control gene (Gabra4) single-cell transcript counts across Pvalb+ subclusters from Drop-seq. Transcript means were compared with a one-way Anova. Asterisk, P < 0.05; n.s P > 0.05. Tukey Honest Significance Difference Test. (F-G) Neurofilament gene and control gene in situ transcript count experiments within Pvalb+ frontal cortex cells using smFISH. Left, example single confocal planes. Right, quantification of transcript densities. Pvalb+ cells were split into n=3 groups based on Syt2 levels (low, medium, and high) mimicking subclusters 2–9, 2–7, and 2–8. Differences in transcript densities were statistically tested as in (E). Longer arrows indicate higher Pvalb expression. (F) Experiment 1, Pvalb, Syt2, and Nefm. (G) Experiment 2, Pvalb, Syt2, and Gabra4 (control).
(H) The Neurofilament IC is observed in flash-frozen nuclei from frontal cortex. The Neurofilament (IC 25) cell-loading signal distribution across the Sst+/Pvalb+ interneuron subcluster. Left, cell-loadings displayed on subcluster tSNE plot. Right, gene-loading plots with top 20 genes are shown.
This transcriptional pattern involved a constellation of genes that underlie axonal and pre-synaptic function. We call this signal the “Neurofilament IC,” because three of the most strongly contributing genes encode the neurofilament subunits of the axonal cytoskeleton (Nefl, Nefm, and Nefh) (Yuan et al., 2012). Other co-regulated genes included Syt2, Vamp1, and Cplx1 – which have roles in vesicle exocytosis – and Pvalb and Caln1, which bind presynaptic Ca2+ (Data S5). Genes contributing to this transcriptional pattern appear to maintain axon function and support or tune neurotransmitter release.
Neurofilament ICs were ascertained in all brain regions and appeared to shape gene expression in diverse neuronal populations. The expression of genes with the strongest Neurofilament IC contributions tended to covary both within and across neuronal types. Among interneurons, Neurofilament IC cell loading was most prominent in fast-firing Pvalb+ populations. In frontal cortex interneuron cluster 2, expression levels of the genes strongly contributing to the Neurofilament IC – including Nefh, Kcnc3, Syt2, and Nefm – were continuously distributed and strongly correlated among Pvalb+ interneurons but lower and less correlated among Sst+ interneurons (Figure 3B–D). In the hippocampus, Pvalb± interneurons exhibited high cell loading for the Neurofilament IC, as did the Pvalb± “prototypical” neurons of the GPe (Mallet et al., 2012; Saunders et al., 2016) (Data S5). We ascertained a similar pattern among cortical glutamatergic (Data S5) and subcortical neuromodulatory (Figure 7O) populations.
Figure 7. Eccentric spiny projection neurons represent a third axis of SPN diversity.

(A) tSNE plot of color-coded global clusters (n=15) for striatum dataset. Clusters 10, 11, and 13 are presumed SPNs. (B) Expression plot of pan-SPN marker Ppp1r1b, direct pathway SPN (dSPN) marker Drd1, and indirect pathway SPN (iSPN) marker Adora2a. Ppp1r1b+ cells within Cluster 13 are eccentric SPNs (eSPN). (C) Mean expression comparisons between SPN populations (log-normal scale). Left, cluster 10 vs cluster 11 (iSPN vs dSPNs). Right, cluster 13 vs clusters 10 and 11 (eSPNs vs d/iSPNs). Differentially expressed genes (fold ratio > 2 and P < 10−100 by binomTest (Robinson et al., 2010), STAR Methods) are shown with dark dots and totals listed above. Red arrow indicates selective expression in eSPNs. (D) Expression plot of n=4 genes (Casz1, Otof, Cacng5, and Pcdh8) enriched in eSPNs vs d/iSPNs (red arrow in C). Across all global clusters, genes are highly enriched in cluster 13 (red arrows). (E-F) eSPNs are anatomically dispersed throughout the striatum. (E) Single confocal planes from smFISH experiments validating co-expression of pan-SPN (Ppp1r1b) and highly-selective eSPN markers (Cacng5, Otof, and Casz1) in dorsal striatum. Top, Ppp1r1b, Cacng5, and Otof. Bottom, Ppp1r1b, Cacng5, and Casz1. Arrowhead indicates triple-positive cells. (F) Locations of triple positive Ppp1r1b, Cacng5, and Otof cells on a schematic of coronal striatum. D, dorsal; V, ventral; L, lateral; M, medial. (G) Color-coded subclusters from cluster 13. Subclusters 13–1, 13–2, 13–3, 13–4, and 13–5 correspond to eSPNs (83% of cells, black labels). The identity of other subclusters (17% of cells, gray labels) is described in Figure S7. (H) Expression plot of pan-SPN (Ppp1r1b), pan-eSPN (Otof), dSPN (Drd1), iSPN (Adora2a), subcluster 13–5 (Th, Npffr1) markers. (I-J) Single confocal planes from smFISH experiments validating co-expression of markers in dorsal striatum. Arrowhead indicates triple-positive cells. (I) Co-expression of Otof with Adora2a and Drd1. (J) Co-expression of subcluster 13–5 markers. Triple-positive cells in dorsal striatum are indicated with white arrowheads. Top, Adora2a, Th, Otof. Bottom, Adora2a, Th, Npffr1.
Single-molecule fluorescent in situ hybridization experiments confirmed that, as predicted, expression of Pvalb, Syt2, and Nefm were highly correlated among Pvalb+ neurons in the frontal cortex (Figure 3E–G). Furthermore, Neurofilament IC cell loading was visible among Pvalb+ interneurons following Drop-seq analysis of 28,194 single nuclei isolated from flash-frozen frontal cortex (Figure 3H). These experiments confirm that the Neurofilament signal is present in vivo and not an artifact of cell isolation.
We conclude that diverse neuron types share a coordinated transcriptional program involving genes that facilitate maintenance, elaboration, or subcellular transport to the axon and presynaptic terminal. Neuronal types characterized by extensive axonal arbors, long-distance axonal projections, and/or faster firing rates tended to utilize this transcriptional program more than other neurons. At the same time, the magnitude of expression varied among neurons of the same subtype, suggesting that this transcriptional program contributes to both intra- and inter-type diversity.
Gene-gene Co-expression Relationships Inferred from Hundreds of Cell types and States
Functional imperatives constrain patterns of gene co-expression in unknown ways. To analyze co-expression relationships across diverse brain cell types and states, we utilized gene-expression profiles for the 565 cell populations (averaging 565 cells and 1.9 million UMIs per population); these data are less influenced by biological and statistical noise than single-cell-level data (Figure S3A).
To assess whether gene-gene expression correlations captured known functional relationships, we focused first on subunits of nicotinic acetylcholine receptors (nAChRs, n=16 genes), ligand-gated, pentameric ion channels whose eclectic but well-described subunit combinations are known to vary by region and neuron type (Gotti et al., 2006).
Across cell populations, expression of nAChR genes exhibited two prominent correlation blocks, each containing genes that encode subunits of known heteromeric α/β channels (Zoli et al., 2015)(Figure 4A). For example, expression of Chrna3 and Chrnb4 (known to form functional receptors) was positively correlated across a large range (from 0.01 to 100 transcripts per 100K) (Figure 4B). Expression of other gene pairs encoding heteromeric receptors (Chrna6/Chrnb3 and Chrna4/Chrnb2) was also well correlated, whereas Chrnb1 and Chrnb2 were not, consistent with a lack of β1/β2 channels in brain (Figure 4B). These correlations match prominent subunit combinations associated at a protein level, suggesting nAChR composition is achieved in part by cell-type-specific patterns of RNA expression.
Figure 4. Inferring ion channel gene-gene co-expression relationships across hundreds of brain cell types and states.

(A-B) Nicotinic acetylcholine receptor (nAChR) subunit co-expression correlations across 565 brain cell populations. (A) Hierarchical clustering of pairwise correlations of n=16 nAChR subunit genes (color-coded by family). (B) Scatterplots of subunit expression (log10 scale). (C-E) Correlation structure among voltage-gated (VG) Na and K channels measured from 323 neuronal populations. (C) Hierarchical clustering of pairwise expression correlations. The VGK (n=17) and VGNA (n=1) alpha subunit families are color-coded and labeled. The correlation block containing channels known to control firing rate is shown with an arrow. (D-E) Select pairwise subunit expression correlations. Neuronal populations known to exhibit fast firing rates are shown in red (Figure S3D). Slc6a8 and Hcn2 were frequently correlated with the alpha subunit genes that putatively encode firing rate (Figure S3D). See also Figure S3.
Homomeric nAChR subunits that do not have obligate partners might also lack positive pairwise correlations with other subunit genes. Several α-subunit genes had this property, including those known (Chrna7, Chrna9, and Chrna10) and others not yet known (Chrna1 and Chrna2) to form homomeric channels in brain (Gotti et al., 2006). Gene expression correlations across cell types recapitulated known nAChR receptor subunit combinations (Gotti et al., 2006) more accurately than correlations based on the bulk expression profiles for the nine regions (Figure S3B).
Co-expression relationships for other vital gene families are less understood. Neurons attain type-specific electrophysiological properties by expressing combinations of voltage-gated ion channels (Marder and Goaillard, 2006), but we know little about what combinations of channels enact neurons’ distinct physiological properties (Tripathy et al., 2017). We therefore evaluated expression pattern correlations for 71 voltage-gated potassium (VGK) and sodium (VGNA) channel genes across neuronal populations, focusing on the alpha (pore-forming) subunits. We observed strong correlation blocks involving specific subsets of VGK and VGNA genes (Figure 4C).
To evaluate whether correlation structure relates to electrophysiological properties, we identified neuron populations known to be fast-firing (Figure S3D). One set of co-expressed genes encoded proteins that enable fast and persistent action potentials, including the Kcnc1–3 (Kv3.1), Kcna1 (Kv1.1) and Scn8a (Nav1.6) channels (Chen et al., 2008; Goldberg et al., 2008; Rudy and McBain, 2001) (Figure 4D). Fast-firing cell types expressed high levels of Kcnc2/Kcnc1 and Scn1a/Scn8a, variable levels of Kcnc4, and low levels of Scn2a1, whose expression was inversely correlated with Kcnc1–3 and Scn1a (Figure 4D). These relationships nominate hypotheses about channel contributions to physiological properties: for example, Kcnc4 could be a Kv3 family member that tailors membrane properties orthogonal to firing frequency, and Scn2a1 might undermine fast-firing.
Finally, we asked what other genes were co-expressed with alpha subunits associated with fast-firing (Figure S3E). This collection contained Neurofilament genes (Figure 3), ion channels (e.g. Hcn2), and unexpected genes, such as the transcription factor Foxj1 and the creatine transporter Slc6a8. Indeed, expression of Slc6a8 and Hcn2 were strongly correlated and high in fast-firing cell types (Figure 4E). Our data suggest human neurological symptoms associated with Slc6a8 mutations (van de Kamp et al., 2013) may in part be due to deficits in fast-firing neurons, consistent with cortical GABAergic synapse loss observed in mouse models (Baroncelli et al., 2017). Gene co-expression relationships across large numbers of cell types yield new hypotheses about genes, brain circuitry, and disease.
Cell-type Specialization Between Cortical Poles
The cerebral cortex processes motor, sensory, and associative information and is expanded in primates, especially humans (Buckner and Krienen, 2013). Little is known about what molecular specializations contribute to region-specific cortical function. We first determined how accurately our cortex datasets reflect the true balance of cellular populations in vivo and then identified transcriptional specializations within each non-neuronal cell class and across glutamatergic and GABAergic neurons.
We tested for distortion of cell type abundance by comparing our frontal cortex dataset to tissue (Figure S4D–M and STAR Methods). Neurons were over-represented relative to non-neurons (Drop-seq: 0.76 ± 02 mean±sem; tissue: 0.51 ± 03) and exhibited a twofold greater GABAergic/glutamatergic ratio (ISH: 5.1:1; Drop-seq: 11:1). Both effects could be partially explained by cell-inclusion thresholds, in which small but real transcript libraries had been excluded from downstream analysis (neurons: 5,039±15 mean±sem; non-neurons: 1,696±9; Glu: 5,299±16; GABA: 2,626±21)(Figure S4D–I). Among GABAergic interneurons, Vip+ cells were over-represented and Sst+ and Pvalb+ cells were underrepresented (ISH vs Drop-seq: Vip+, 16% vs 35%; Pvalb+, 31% vs 25%; Sst+, 28% vs 22%), which cannot be explained by higher transcript counts (Pvalb+: 2,996 ± 61; Sst+: 2,758 ± 53; Vip+: 2,236 ± 32), suggesting a preferential depletion (Figure S4K–M). We conclude our data exhibit modest skews in cellular representation driven by transcript abundance and viability.
To identify molecular specializations across cortical regions, we performed semi-supervised ICA on cells from frontal and posterior cortex (grouped together), analyzing each cell class separately. Glutamatergic neurons generated more regionally specialized ICs than GABAergic interneurons and non-neurons (Figure 5A), and exhibited subclusters with skewed regional abundances (Figure 5B–D). Selective markers for these cell subclusters confirmed their asymmetric distributions across the cortical mantle (Figure 5D and Figure S5). Glutamatergic neurons exhibited unusual regional specialization not only in subpopulation representation (subclusters), but in the precise gene expression pattern within each subpopulation: far more genes were differentially expressed when comparing regions within glutamatergic neuron as opposed to interneuron or non-neuronal subpopulations (Figure 5E). All these lines of evidence suggest that regional specializations are driven by glutamatergic neurons, consistent with theory from humans and other primates (Krienen et al., 2016).
Figure 5. Excitatory glutamatergic neurons underlie regional specialization in cortex.

(A) Relative contributions of frontal (FC) vs posterior (PC) cortex cells to biological ICs in six separate cell-class analyses. IC Skew is 1 if only FC cells contribute and 0 if only PC cells contribute; equal contribution is 0.5 (dotted line). (B-D) Subcluster analyses illustrate stronger regionalization for excitatory neurons than other cell classes across cortical regions. (B) Subcluster tSNE plots for six cell classes. Cells are color-coded by region (left) and subcluster (right). Total numbers of subclusters are shown. (C) Representation of FC vs PC cells within subclusters. Dot size denotes fractional representation; asterisks denote significant FC vs PC difference (> 3:1 compositional skew and P < 0.05, Barnard’s test, STAR Methods). (D) Top left, tSNE plot of excitatory neurons color-coded by region. Top right, expression of Sccpdh and Whrn, genes enriched in subclusters disproportionately composed of FC or PC cells, respectively (Figure S5). Bottom, ISH (Allen). High expression, long arrow; Medium expression, short arrow. (E) FC-PC expression differences within cell populations. Barplot shows the number of differentially expressed genes between FC and PC cells within each subcluster (> 2-fold change, P < .05, Bonferroni corrected). See also Figure S4 and Figure S5.
Resolving Neuron Types within the Basal Ganglia
We identified 323 neuronal subclusters across 9 regions (Data S3). Here, we illustrate systematic neuron classification using three disease-relevant nuclei of the basal ganglia (Albin et al., 1989). Neurons of the striatum have been extensively characterized, yet neurons of the globus pallidus externus (GPe) and substantia nigra reticulata (SNr) have received less attention (Figure 1 & Data S1).
To define GPe neuron types, we screened markers of global clusters and subclusters for expression in the GPe (GP/NB dataset). GPe neurons were present in cluster 2 (n=11,103 cells), one of three neuron clusters (Figure 6A). Cluster 1 contained cholinergic neurons (n=437 cells) and cluster 3 contained neurons of the adjacent striatum and basolateral amygdala (n=9,847 cells). Within cluster 2, 8 of 25 subclusters appeared intrinsic to the GPe or adjacent ventral pallidum (VP), of which 4 were GPe-exclusive (Figure 6B–E). The other 17 subclusters mapped to the thalamic reticular nucleus, the substantia innominata, and the lateral olfactory tract (Figure S6A–D).
Figure 6. Transcription-based identification of known and novel neuron type distinctions within the basal ganglia.

(A-E) Globus pallidus externus (GPe). (F-J) Substantia nigra reticulata (SNr). (K-O) Dopaminergic vs acetylcholinergic neuromodulatory neuron populations. (A) tSNE plot of color-coded global clusters (n=11) for GP/NB dataset. Clusters 1, 2, and 3 are neuronal. (B) Subclusters within cluster 2. Black subclusters correspond to those of GP/NB. (C) Subclusters color-coded by candidate anatomical regions, inferred by ISH expression patterns of selective marker genes (Figure S6) and consistent with dissections (Data S1). Ventral pallidum (VP), substantia innominata (SI), striatum (STR), lateral olfactory tract (LOT), rostral entopeduncular nucleus (EP) and the thalamic reticular nucleus (TRN). (D) Dot plot illustrating the expression patterns of neurotransmitter genes, neuron type markers from the literature and novel markers identified here. (E) ISH experiments (Allen) illustrating expression within the GPe and/or VP (sagittal sections). Dotted line approximate boundaries. (F) tSNE plot of color-coded global clusters (n=14) for substantia nigra/VTA. Clusters 1, 3, and 4 are neuronal. (G) Subcluster structure within cluster 3. Black subclusters correspond to those of SNr. (H) Candidate anatomical regions inferred by ISH (Figure S6). Ventral tagmental area (VTA), red nucleus (RN), supramammillary nucleus (SuM), thalamus (TH), and deep mesencephalic nucleus (DpMe). (I) Dot plot as in (D). Genes for neurotransmitters, current SNr markers, and novel markers identified here. (J) ISH experiments (Allen) illustrating expression within the SNr (sagittal sections). (K) Subclusters within Th+/Ddc+ dopaminergic cluster 3 from the SN/VTA dataset. (L-M) Example cluster 3 ICs that encode spatial signals within the SNc/VTA. (L) IC cell loadings displayed on tSNE plot. (K). IC gene-loadings. Top ten genes shown at right.
(L) ISH experiments (sagittal sections) for Lpl (IC 10, top) and Aldh1a1 (IC 12, bottom). IC 10 identifies the dorsal VTA, while IC 12 identifies the ventral VTA and SNc (Allen). (O-P) Minimal heterogeneity identified within Chat+/Slc5a7+ cholinergic cluster 1 from the GP/NB dataset. (O) Plot of IC 4 cell-loadings. Based on IC 4, cells are assigned as subcluster 1–1 or 1–2. (P) IC 4 gene-loading plot. Top ten loading genes suggest a Neurofilament-type signal (Figure 3).
To associate subclusters with putative neuron types, we compared published GPe markers to selective markers we identified (Figure 6D). Only Pvalb and Penk/Foxp2 were selectively expressed in GPe subclusters (Kita, 1994; Voorn et al., 1999), suggesting 2–14 represents fast-spiking “prototypical” neurons, while 2–19 represents the slow-firing “arkypallidal” population (Abdi et al., 2015; Mallet et al., 2012). Interestingly, 2–13 is similar to 2–14, sharing markers like Grem1, but is distinguished by stronger expression of Scn4b, Kcnc3 and other “Neurofilament program” genes (Data S5). The fourth GPe subcluster (2–17) is enriched for Elfn1/Grik3 and has not been characterized. Several subclusters (2–15, 2–21, and 2–18) expressed markers in the VP and bordering GPe (Figure 6D,E), suggesting neuron types shared across regions with different behavioral functions and connectivity (Gittis et al., 2014; Kita, 2007; Smith et al., 2009). Border-spanning subclusters may explain a neuron type that is synaptically incorporated into the GPe, but exhibits VP-like axonal projections (Chen et al., 2015; Saunders et al., 2015).
SNr neurons were identified with a similar procedure (SN/VTA dataset). Of 4 global neuron clusters (Figure 6F), one (cluster 3, n=10,049 cells) contained SNr neurons (Figure 6G). Other clusters contained hippocampal (cluster 1, n=73 cells), thalamic (cluster 2, n=297), and dopaminergic (cluster 4, n=1,841) populations. Within cluster 3, 6 of the 19 subclusters mapped to the SNr (Figure 6H–J and Figure S6E–H). Two subcluster pairs shared selective markers (3–18/3–19: Sema3a, Adarb2; 3–17/3–13: Pax5, Pou6f2), suggesting relatedness, but were distinguished by ion channels (Kcnc3, Kcna1), transmembrane proteins (Tmem132c), and transcription factors (Pou3f1) that could imply a state or subtype distinction (Figure 6I). Of the remaining subclusters, 3–12 expressed Slc17a6±, likely corresponding to the glutamatergic projection to thalamus (Antal et al., 2014). Subcluster 3–2 was Gad2+/Pvalb− and expressed the developmental marker Zfpm2 (Lahti et al., 2016), likely representing a third GABAergic type marked by Sox14 and Cplx3 expression (Figure 6I).
The GPe and SNr abut populations of neuromodulatory neurons that release acetylcholine (ACh) or dopamine (DA), respectively. Molecular specializations related to anatomical location exist for DA neuron subtypes (Lammel et al., 2008; Poulin et al., 2014), yet it is unclear if ACh-releasing neurons exhibit similar heterogeneity and whether this diversity has a spatial component.
Subcluster analyses of the dopaminergic (SN/VTA, cluster 4, 919 cells) and cholinergic (GP/NB, cluster 1, n=218 cells) clusters revealed that dopaminergic neurons were indeed more heterogeneous than the cholinergic neurons (DA: 6 biological ICs, 9 subclusters; ACh: 2 biological ICs, 2 subclusters)(Figure 6A,F). While aspects of DA diversity related to spatial positioning – for example, delineating the dorsal (IC 10) from ventral (IC 12) VTA (Figure 6L–N) - cholinergic neurons exhibited only a Neurofilament-like signal (IC 4), with no spatial component (Figure 6O,P). Thus, DA neurons are more transcriptionally regionalized than cholinergic neurons. Sampling cholinergic neurons from other areas of the basal forebrain (Zaborszky et al., 2013) could reveal additional signals.
Molecular Specializations of Striatal Principal Neurons
Spiny projection neurons (SPNs) represent ~95% of neurons in rodent striatum. Two principal categories distinguish SPN subsets. The first – based on divergent axonal projections and dopamine signaling – assigns SPNs to similarly numerous “direct” (dSPN) and “indirect” (iSPN) pathways (Albin et al., 1989). The second – based on processing limbic versus sensory/motor information – groups SPNs into spatial compartments, the so-called “patch” and “matrix” (Gerfen, 1992; Graybiel and Ragsdale, 1978). Both dSPNs and iSPNs are present in the patch and matrix.
Two large clusters corresponded to dSPNs (Cluster 10, n=30,835 cells) and iSPNs (Cluster 11, n=25,305)(Striatum dataset, Figure 7A,B) and were distinguished by 68 differentially expressed genes, including known and undescribed pathway markers (Figure 7C). To identify cells from patch and matrix, we inspected transcriptional signals from SPN subclustering (dSPN Cluster 10: n=9 biological ICs; iSPN Cluster 11: n=10, Data S6). Each analysis identified a single candidate “patch IC” whose most strongly contributing genes included known patch markers such as Tac1 and Pdyn. Approximately 10% of iSPNs and dSPNs exhibited this patch signal (Figure S7A,B).
To appreciate how the patch/matrix distinction affects pathway specialization, we compared gene loadings across the patch-encoding ICs for dSPNs and iSPNs (Figure S7C). We observed classic (Tac1) and undescribed (Tshz1) pan-patch markers (Figure S7D) as well as genes enriched in either iSPN (Asic4) or dSPN (Necab1) patches (Figure S7E). Our data suggest complex SPN specializations across pathways and compartments: the transcriptional features endowed by patch compartments are not identical for dSPNs and iSPNs, and some of these differences appear to eliminate pathway expression differences found in the matrix dSPNs and iSPNs (Figure S7E).
“Eccentric” SPNs: A novel, third axis of SPN diversity
Surprisingly, about 4% of SPNs (Ppp1r1b+) were observed in a third, smaller cluster that also expressed Adora2a and Drd1 (cluster 13: n=2,744 cells; 4.5% of Ppp1r1b+ neurons; Figure 7A,B). These SPNs differed in expression from dSPNs+iSPNs by 110 genes (more than the 68 that distinguished dSPNs and iSPNs from each other) (Figure 7C), and expressed many genes that had little expression in the rest of the striatum (Figure 7D). Due to their transcriptional divergence from canonical SPNs, we call this population “eccentric” SPNs (eSPNs). eSPNs were intermixed with other SPNs in the striatum with no obvious spatial organization (Figure 7E,F). Our data account for all known striatal interneuron types (3.9% of total neurons)(Tepper and Bolam, 2004), suggesting by exclusion that eSPNs are not interneurons. We conclude that eSPNs are striatal principal neurons.
eSPN subclusters (Figure S7F–H) were divided into two major groups (Figure 7H), separated by a gene set that included markers used to distinguish canonical iSPNs from dSPNs, such as Drd1 and Adora2a (Figure 7I). Expression of markers associated with canonical SPNs suggests eSPNs have been molecularly “camouflaged,” including in studies using mice that have employed Drd1 and Adora2a driven–transgenes to label and manipulate dSPNs or iSPNs (Heiman et al., 2008; Kozorovitskiy et al., 2012; Kravitz et al., 2012). Despite sharing markers, Adora2a+ eSPNs and Drd1+ eSPNs are distinguished from their canonical SPN counterparts by expression levels of many genes (Adora2a+ SPNs: 35 genes; Drd1+ SPNs: 96 genes; Figure S7I). We validated additional eSPN diversity predicted by Drop-seq, including an ultra-rare eSPN Adora2a±/Th±/Npffr1+ subtype (13–5) that accounts for just 0.3% of all SPNs (n=88 cells)(Figure 7J). One clue about the anatomical identity of eSPNs comes from this small Th+ population, as spiny Th+ principal cells with similar spatial arrangement to eSPNs have been observed in striatum and appear to be dynamically regulated by dopamine (Darmopil et al., 2008). Subsets of the eSPN class share markers previously reported to distinguish SPN subtypes (Gökce et al., 2016).
We conclude that 1) eSPNs represent a third axis of SPN diversity, distinct from the dSPN/iSPN and patch/matrix distinctions; 2) eSPNs harbor rare, additional molecular diversity; and 3) by using markers thought to exclusively distinguish iSPNs from dSPNs, functional studies have lumped eSPNs in with canonical SPNs. These results highlight the utility of unbiased, high-throughput single-cell methods for defining neuronal populations.
Discussion
The mammalian brain is a mosaic of spatially intermixed cell classes and types. Since Cajal and Golgi, single-cell analyses of cell morphology, membranes, and synapses have helped illustrate how structure relates to function in neural circuits. High-throughput single-cell-resolution molecular techniques such as Drop-seq allow newly systematic approaches for cataloging the cellular building blocks of the brain.
Here, we analyzed RNA expression in more than 690K individual cells sampled from nine different regions of the adult mouse brain, encompassing all brain cell classes. We developed an ICA-based computational method that we used to disentangle technical effects from endogenous biological signals. We highlighted several ways in which such data identify novel cell types, ascertain cell states, and clarify the molecular basis of regionalization across brain circuits and cell classes.
Transcriptional variation among individual cells has diverse sources. Cluster-groups derived from these and other data should not be reflexively equated with cell “types”. We identified categorically distinct patterns of RNA expression originating from cell types, but also continuously varying patterns that appeared to correspond to spatial locations and cellular states. Our computational approach was critical for recognizing and understanding these diverse effects on RNA expression, all of which can simultaneously affect a cell’s RNA expression profile. This approach allowed us to identify a transcriptional program we believe is enacted to maintain axon and presynaptic function, to different degrees both within and across neuron types. We also resolved signals from striatal SPNs representing differences in pathway (“direct” versus “indirect”), spatial arrangement (“patch” versus “matrix”), and a cryptic molecular SPN distinction (“eccentric” SPNs).
The size and complexity of single-cell datasets can limit their utilization. To enable diverse uses of our atlas, we developed interactive web-based software (DropViz, http://dropviz.org/) that facilitates access and dynamic exploration of the data. We hope that single-cell gene expression profiles, and the patterns present among very many such profiles, can function as a lingua franca for discussing – and functionally dissecting – the cellular diversity of the adult brain.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Requests for further information may be directed to Lead Contact Steven A. McCarroll (mccarroll@genetics.med.harvard.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell suspensions were generated from adult male mice (60–70 days old; C57Blk6/N). Two BAC transgenic mouse lines Ald1l1-EGFP (Jackson Labs, 026033) and Cx3cr1-GFP (Jackson Labs, 005582) used in IHC experiments were also included as single replicates in the Frontal Cortex Drop-seq dataset. Both transgenics were on a C57BL/6N background. To estimate the fraction of cells retained in single-cell suspensions, Nestin-Cre driver mice (Jackson Labs, 003771) were bred lox-stop-lox (lsl) ZsGreen Cre-reporter (Jackson Labs, 007906). Mice were group housed prior to experimentation on a reverse 12-hour light-dark schedule. All experiments were approved by and in accordance with Harvard Medical School IACUC protocol number IS00000055–3.
METHOD DETAILS
Adult mouse brain single-cell suspensions
Cell suspensions were generated by adapting protocols from single-cell patch-clamp recording (Carter and Bean, 2009) and digest times were optimized for each region (Table S1). Mice were anesthetized by isoflurane inhalation and perfused through the heart with ice-cold Sucrose-HEPES “Cutting Buffer” containing (in mM) 110 NaCl, 2.5 KCl, 10 HEPES, 7.5 MgCl2, and 25 glucose, 75 sucrose (~350 mOsm kg−1). The brain was removed and placed in ice-cold Cutting Buffer. Blocking cuts depended on the region of interest and desired slice orientation (Table S1). Blocked brains were then transferred to a slicing chamber containing ice-cold Cutting Buffer. 400 μm thick brain slabs were cut with a Leica VT1000s vibratome. Slabs containing the regions of interest were gently transferred to a dissection dish with ice-cold “Dissociation Buffer” containing (in mM): 82 Na2SO4, 30 K2SO4, 10 HEPES, 10 glucose and 5 MgCl2. Dissociation Buffer avoided activity-induced toxicity by 1) excluding extracellular Ca2+ and 2) utilizing ionic concentrations that maintain voltage-gated Na channels in an inactivated state [Vm = −30.5 mV, estimated by the Goldman-Hodgkin-Katz equation using the following parameters: Inside(mM): K+ = 140, Na+ = 4, Cl− = 24; Outside(mM): K+ = 30, Na+ = 82, Cl− = 5; P: K+ = 1, Na+ = 0.05, Cl− = 0.45; T = 34°C]. Regions of interest were gently dissected under visual guidance of a stereoscope (Leica MZ10). Dissection also served as a wash step between buffers. Chunks of tissue containing the regions of interest were then transferred into 5 ml of “Dissociation + Enzyme Buffer” in a 15 ml falcon tube. “Dissociation + Enzyme Buffer” consists of “Dissociation Buffer” with 3 mg/ml of Protease XXIII (Sigma-Aldrich, P5380) and 10 units/ml of Papain, 0.5 mM L-Cysteine and 0.25 mM EDTA (Worthington, LK003153). Digestion was performed at 34 C using durations that were optimized for each region in a separate set of experiments (Table S1; see below). Tubes containing digested tissue were transferred onto ice and the “Dissociation Buffer + Enzyme” replaced with 10 ml of “Dissociation Buffer + Stop Solution” containing “Dissociation Buffer” and 1 mg/ml Trypsin Inhibitor (Sigma-Aldrich, T6522), 2 mg/ml BSA (Sigma-Aldrich, A2153) and 1 mg/ml Ovomucoid Protease Inhibitor (Worthington, LK003153). Tissue chunks were then carefully titrated with a series of n=4 fire-polished, Pasteur pipets with successively smaller bores. Bubbles were avoided. Falcon tubes containing 10 ml of titurated cells were then centrifuged at 300 g for 10 minutes. The supernatant was removed and discarded, taking care not to disturb the cell pellet. The pellet was then resuspended in 5 ml of “Dissociation Buffer + Stop Solution” and centrifuged again using the same conditions. The supernatant was again removed. The cleaned cell pellet was resuspended in “Dissociation Buffer” containing 0.01% BSA (w/v; “Dissociation Buffer + BSA”). The volume of resuspension depended on the region (Table S1). Suspensions were then passed through a pre-wet 40 μm filter into a new tube on ice. N=2 10 μl samples were then drawn from the tube and mixed 1:1 with 10 μl of 2× dye mix containing Dissociation Buffer and 20 μM EthD-1 (Thermo Fisher Scientific, L-3224), 20 μM Calcein-AM (Thermo Fisher Scientific, L-3224) and 40 μM Hoechst 33342 (Thermo Fisher Scientific, 62249). After 5 minutes incubation, 10 μl from each sample was loaded onto a haemocytometer (Propper, 090001) and imaged using a fluorescent microscope (Zeiss, Axio Observer Z1). For each of the two samples, three random locations were imaged using DIC and three fluorescent channels to capture the dyes. These images were used to calculate cell concentrations and metrics of cell intactness. Drop-seq analysis was performed on 44 cell suspensions, with 3–7 replicates per region.
Characterization of single-cell suspensions
Cell recovery rates from intact tissue
To determine what fraction of total brain cells are retained in a single cell suspension, we compared estimates of total cell number from stereological counts of tissue to cell totals in suspension. For suspension assays, we estimated cell totals from a single 400 μm coronal slab of either Frontal Cortex or dorsal Striatum. Stereological assays were performed on a series of thinner, paraformaldehyde-fixed tissue from the same anterior-posterior location (40 μm; see IHC section for fixation procedure). To get total counts, densities were multiplied by volume estimates calculated using a thickness of 400 μm and surface area calculated from slide scans of the same tissue sections (Olympus VS110). A 10% shrinkage factor due to fixation was taken into account. We aided cell counting by using a double transgenic mouse (male, P59–60) carrying a Nestin-Cre driver (Jackson Labs, 003771) and lox-stop-lox (lsl) ZsGreen Cre-reporter (Jackson Labs, 007906). ZsGreen aids in the identification of single-cells as it is sequestered in the soma and proximal processes of cells. Specifically, confocal stacks were obtained with a 40× objective using the FV1200 (Olympus). ZsGreen+ and DAPI+ cells were randomly sampled within a grid, using bounding box of 5 μm in the Z dimension to avoid over-counting (Cortex, mouse 1: n=22 stacks, n=181 boxes; mouse 2: n=12 stacks, n=108 boxes; Striatum, mouse 1: n=15 stacks, n=1135 boxes; mouse 2: n=16 stacks, n=144 boxes). In both Frontal Cortex and Striatum, ~66% of DAPI+ cells were ZsGreen+ on average. To estimate cell totals in suspensions of acute 400 μm slabs of Frontal Cortex or Striatum were cut from Nestin-Cre;lsl-ZsGreen mice. We controlled the volume of tissue in each tube by processing only one coronal slice / tube at the appropriate anterior-posterior location. We resuspended the pellet in 1 mL of Dissociation Buffer + BSA and then re-measured the volume to account for extra buffer introduced by filter wetting. 20 μl samples were assayed n=3 times on the haemocytometer. Each sample was randomly imaged at n=5 locations. To estimate the concentrations of all cells and ZsGreen+ cells, images containing DIC, DAPI and ZsGreen channel were analyzed using ImageJ. Sample concentrations were converted into an average total number of cells for each experiment using the volume of the suspension.
Cell class, type and subtype acquisition bias
To determine in what ways Drop-seq datasets are or are not representative of brain tissue, we compared cellular representations between tissue and Drop-seq datasets focusing on cell classes, types and subtypes within Frontal Cortex.
To evaluate representations at the level of cell class, we performed two sets of stereological counting experiments using IHC and transgenic mouse lines. In the first experiment, we used the Ald1l1-GFP (Jackson Labs, 026033) line to drive GFP expression in astrocytes and the immuno-labeled Neurons using anti-NeuN (Millipore, MAB377) antibody and Oligodendrocyte/Polydendrocyte using anti-Olig2 (Millipore, AB9610) antibody. We visualized NeuN using a secondary antibody in the red channel (goat-anti mouse 568; Abcam, ab175473) and Olig2 (goat-anti Rabbit 647; Abcam, ab150079) in far-red. GFP was not immune-enhanced. Sections were mounted in media containing DAPI as counterstain (Vector Laboratories, Vectashield). Four sections containing Frontal Cortex were counted from n=1 mouse, using n=34 random fields with n=9 boxes sampled from each. In the second set of experiments, we replaced Ald1l1-GFP with Cx3cr1-GFP mice (Jackson Labs, 005582) to label microglia instead of astrocytes. Three sections were counted from n=2 mice, using n=68 random fields in total with n=9 boxes sampled from each.
To evaluate representations at the level of cell type and subtypes, we used smFISH labeling combined with whole-slide imaging to quantify neuron populations in situ. We sampled the frontal cortex of n=3 p60 male mice by performing smFISH on every 5th cryosection (18 sections per animal) followed by whole-slide imaging on a Zeiss Axioscan z1 at 20× magnification. For quantification, images were down sampled (Zoom factor × 4) and converted to TIFF via the Zeiss CZItoTiffBatchConverter (CZI to Tiff Converter Suite software by Zeiss). Regions of interest that approximated the regions microdissected as inputs for Drop-seq experiments (i.e. frontal cortex) were defined by hand using a custom ROI pipeline in CellProfiler 2.1.1 (Carpenter et al., 2006) and subsequently input into a custom CellProfiler cell-counting pipeline. To evaluate cell type comparisons, we assessed inhibitory/excitatory ratios, using cocktails of smFISH probes to label GABAergic (Gad1/Gad2) and glutamatergic (Slc17a6/Slc17a7/Slc17a8) populations (Advanced Cell Diagnostic Biosystems). We counted a total of 487,003 glutamatergic and GABAergic neurons total from n=3 mice using a custom pipeline in CellProfiler. To evaluate representations at the level of subtypes, we focused on cortical interneurons, using smFISH probes against Pvalb, Sst and Vip. We quantified n=68,734 interneurons, calling cells as single, double and triple-positive population using a second custom CellProfiler pipeline (n= 3 mice).
Nuclei Drop-seq
Frontal cortex sections (400 μm) were flash frozen using liquid nitrogen, minced and resuspended in 4 mL, then passed through a 20 μm then a 5 μm filter before being centrifuged for 10 minutes at 500 g. The pellet was then resuspended in 1 mL of “Dissociating Buffer” (see above). Isolated nuclei were Hoechst stained and counted, then analyzed in standard Drop-seq devices at a concentration of 176 nuclei/μL.
Immunohistochemistry, smFISH and Stereology
smFISH
Mouse brains were extracted, flash frozen in liquid nitrogen, and embedded in OCT (Sakura Tissue-Tek ref 4583). Frozen, 14 μm, coronal sections were cut on a cryostat (Leica CM 1950) and processed for 3-color smFISH according to the ACD RNAScope multiplexed fluorescent protocol for fresh frozen tissue (ACD user manual document numbers 320513 & 320293). Briefly, sections were post-fixed in 4% PFA (Electron Microscopy Sciences) in PBS for 15 minutes, followed by alcohol dehydration. Sections were permeabilized with the proprietary protease cocktail in “pretreat IV” followed by target probe hybridization (Key Resources). For each experiment, ACD 3-plex positive control and 3-plex negative control probes were run alongside target probes to ensure tissue quality and control for background respectively. Probes were visualized with the ACD “Alt-B” color module across all experiments. For puncta counting experiments, stacks were acquired Leica SP8 at 63× magnification (n= 4–10 Z planes, 1 μm steps). In situ expression was quantified by smFISH puncta counting in Pvalb+ cells using maximum projections through confocal stacks acquired in Frontal Cortex. Soma were manually segmented based after Gaussian-filtering Pvalb puncta. The resulting regions of interest (ROIs) were used as masks for analysis of other fluorescent channels. ROIs contained DAPI nuclei and exhibited little overlap. Manual segmentation and automated puncta detection/surface area measurements were implemented in CellProfiler 2.1.1 (Carpenter et al., 2006).
IHC
Mice were deeply anesthetized with isoflurane and transcardially perfused with 4% paraformaldehyde (PFA) in 0.1 M sodium phosphate buffer (1× PBS). Brains were post-fixed for 1–3 days, washed in 1× PBS and sectioned (40 μm) coronally using a Vibratome (Leica). Slices were then immunostained for antibodies described. Slices were incubated in a 1× PBS blocking solution containing 5% normal horse serum and 0.3% Triton X-100 for 1 hour at room temperature. Slices were then incubated overnight at 4°C in the same solution containing primary antibodies at the following concentrations (1:100, NeuN; 1:500, Olig2; see above section “Cell class, type and subtype acquisition bias in Drop-seq datasets” for antibody details). The next morning, sections were washed three times for five minutes in 1× PBS for and then incubated for 1 hour at room temperature in the blocking solution containing donkey anti-goat Alexa 647 or Alexa 568. After drying, slices were mounted on slides (Super Frost) and allowed to dry. ProLong antifade mounting media containing DAPI (Molecular Probes) was applied and slides were coverslipped and sealed.
Drop-seq library preparation and sequencing
Drop-seq libraries were prepared as previously described (Macosko et al., 2015)(Drop-seq protocol v3.1), with full details available online (http://mccarrolllab.com/dropseq/). Deviations from the original protocol are noted. Cell and bead concentrations were matched to two different sets of PDMS devices generating droplet of different volumes such that the lambda loading parameter for cells was 0.08–0.1 and for 0.09–0.13 for beads. Cell suspensions were diluted with using room temperature “Dissociation Buffer + BSA.” For device A (droplet diameter: 125 μm; droplets/μl: 980), cells were loaded at 100 cells/μl (λcell = 100/980=0.1). For device B (droplet diameter: 100 μm; droplets/μl: 2631), cells were loaded at 220 cells/μl (λcell = 220/2631=0.08). Bead concentrations of 125 beads/μl (A) and 250 beads/μl (B) achieved λbeads between or 0.09 (A) and 0.13 (B). Flow rates for cells/beads were 1.8 – 4 mL/hr, adjusted to the highest value possible while ensuring homogenous droplet sizes. Oil flow rates were between 13–15 mL/hr. Cell suspensions were split between two Drop-seq rigs to reduce runtime. Drop-seq encapsulation was performed within 2 hours after suspension preparation. Drop-seq conditions for nuclei paralleled those outlined for cells (λnuclei ~ 0.08). The molecular workflow for Reverse Transcription, cDNA Amplification, Tagmentation and Sequencing follows that of Macosko et al. Note, the number of beads and corresponding STAMPs that were pooled for SMRT cDNA amplification varied between 2–8K beads/reaction.
QUANTIFICATION AND STATISTICAL ANALYSIS
Distinguishing single-cell libraries
To identify Drop-seq beads that hybridized mRNA from intact cells versus mRNAs in solution, we plotted total library size (UMI counts) versus the library fraction corresponding to mitochondrial and ribosomal transcripts. We reasoned that since mitochondrial/ribosomal transcripts are present at high-concentration in the suspension, their abundance could serve as a signature for non-cellular (ie background/ambient) hybridization. To identify beads containing cellular libraries, we plotted the library size versus library fraction attributed to mitochondrial/ribosomal transcripts.
These two-dimensional plots revealed three regions of density:
Large cell libraries with low fractions of mitochondrial and ribosomal transcripts corresponding to cell-bead interaction events
Small cell libraries with high fractions of mitochondrial and ribosomal transcripts corresponding to background-bead interactions
Small libraries with low fractions of mitochondrial and ribosomal transcripts that likely represent libaries that have splintered from a larger, “sibling” library due to mutations incurred in the cell barcode during amplification.
To distinguish these groups, we used an automaton-based approach to distinguish “watershed boundaries” allowing us to define borders for each region of density.
Pre-processing of Drop-seq data
Illumina sequencing data were aligned to a composite genome consisting of GRCm38.81 and transgenes as previously described (Macosko et al., 2015). Full details are available online (http://mccarrolllab.com/wp-content/uploads/2016/03/Drop-seqAlignmentCookbookv1.2Jan2016.pdf). Digital gene expression (DGE) matrices from each sequencing pool were compiled before ICA-based analysis.
ICA based analysis and clustering
Analysis of Drop-seq data was performed using two iterative rounds of independent component analysis (ICA) on each of the nine tissue regions separately (first round, “Global clustering”; second round, “Subclustering”). Function definitions and parameter settings of all operations performed are provided. In the first stage, digital gene expression matrices were column-normalized. Cells with fewer than 400 expressed genes were removed from analysis. To identify a set of highly variable genes, we first calculated the average mean and variance of each gene, and selected genes that were: (1) 0.1 log10 units above the expected variance for a perfectly Poisson-distributed gene of equivalent mean expression; and (2) above a Bonferroni-corrected 99% confidence interval defined by a normal approximation of a Poisson distribution. These selected genes were then centered and scaled across all cells, and ICA was performed with 60 components (except for cerebellum, where only 30 components were used), using the fastICA package in R. Clustering of these components was performed by a very similar process to that of the R package Seurat(Gierahn et al., 2017; Satija et al., 2015): a shared nearest neighbor (SNN) graph was generated, setting the k parameter to 25 from a distance matrix computed in IC space. Next, clustering of this graph was performed with the smart local moving algorithm (SLM)(Waltman and van Eck, 2013), a modularity-based approach to detecting communities, using a resolution setting of 0.01. This produced 11–22 Global clusters across the nine different tissues, partitioning cells into broad “classes.”
To identify finer substructure amongst these classes, classes with more than 200 cells were selected for subclustering. The largest 50% of the cells from each of these clusters was subjected to a variable gene selection, scaling, and independent component analysis. The independent component space is highly dependent on the number of components K that are selected for computation. To automatically nominate a value for K, we took advantage of the fact that the fastICA algorithm begins with a whitening step, in which a singular value decomposition is used to select the top K eigenvectors (i.e. principal components) for maximization of non-Gaussianity(Hyvärinen, 1999). We therefore calculated the number of statistically meaningful principal components using the Jackstraw method (Chung and Storey, 2015), to obtain a suitable value for K. In almost all instances of subclustering, this value was used, with a few exceptions where K was increased slightly. These values of K were then used to compute ICs for each subclustered class.
A total of 1,758 ICs, distributed across 11 cell classes, were then individually manually curated for inclusion in clustering. By studying gene loadings and cell scores, each IC was assigned to one of four “status” categories:
Doublet (n=759), in which top loading genes were identified as markers of another cell class (e.g. microglial genes seen in the analysis of a neuronal cell class);
Outlier (n=99), in which only a small number of cells (less than five) showed high cell scores;
Artifact (n=315), in which either: (a) cell scores showed obvious replicate-specific biases, or (b) the top loading genes largely intersected with those observed to vary strongly with digest time (Figures 3 and S3), or contained many heat-shock proteins;
Biological (n=601), in which the IC signal is believed to be likely biologically meaningful (or cannot be confidently assigned to categories 1, 2, or 3).
To detect and remove cells with high scores on doublet and outlier ICs, we simulated a Gaussian centered at the mode of the IC cell loading distribution, and flagged cells that were situated at the far-right of the distribution. The mode was detected by performing a kernel density estimation of the IC loadings using the density() function in R, and the standard deviation was calculated across all scores for that IC. Doublets and Outliers were identified as cells whose upper-bound p-value was less than 0.01 (FDR-corrected). Only ICs annotated as Biological ICs were included in the generation of the SNN graph for clustering; Technical ICs (Doublet, Outlier, and Artifact) were not included. We note however, additional “technical” influences may exist in Biological ICs. Our goal was to subcluster the data such that, as best as possible, cells with strong cell loading for each Biological IC defined their own particular subcluster. To do this, we clustered the cells across a range of the parameters k (number of nearest neighbors used in SNN generation) and r (resolution parameter in SLM), inspected the resulting clusters for enrichment for specific ICs, and selected a specific pair of parameters k and r that maximized the 1:1 correspondence between IC and subcluster (Figure S2J,K).
Cross-cortex analysis
Drop-seq profiles from six classes—excitatory neuron (n=82,936 cells), inhibitory neuron (n=7,783), oligodendrocyte/polydendrocyte (n=3,505), microglia/macrophage (n=1,027), astrocyte (n=7,782), and fibroblast-like/endothelial (n=3,578) — were individually extracted from the Frontal and Posterior Cortex DGEs, and run through a single round of the ICA analysis pipeline described above. To calculate the regional skew of ICs (Figure 5A), cells from each region were downsampled to equal representation, and each IC that passed curation was manually thresholded (by examining that IC’s cell score distribution) to identify cells with positive IC scores. The skew was the fraction of supra-threshold frontal cortex cells divided by the total number of positive cells (Skew score is 1 if only FC cells contribute and 0 if only PC cells contribute; Equal contribution is 0.5). Biological ICs were used to generate subclusters within each cell class analysis. Subclusters were classified as having FC versus PC skew if they 1) exhibited >3:1 compositional difference between FC and PC and 2) P < 0.05 (Bonferroni-adjusted) using Barnard’s exact test. To quantify the number of differentially expressed genes between frontal and posterior cortical cells within each subcluster, differential expression was performed using the binomTest from the edgeR package (Robinson et al., 2010), using a fold-change threshold of 2, and a Bonferroni-adjusted P-value of 0.05.
Correlation analysis across cell populations
We generated subcluster-level profiles by summing the integer counts of each cell in each subcluster together to form 565 distinct profiles (Figure S7A,B). To compare pairwise correlations across nAChR subunits, all 565 profiles were selected and normalized to 100K UMIs. Pearson correlations were calculated across all genes using linear expression data and grouped via hierarchical clustering using the heatmap.2 function of the gplots R package. Note, Chrna3/Chrnb4/Chrna5 are adjacently located on chromosome 9; Chrna4/Chrnb2 genes are on chromosome 2 and 3, respectively. For voltage gated Na and K alpha subunit analysis, only the 323 profiles corresponding to neurons were used and the gene set was thresholded to include only those genes with > 1 out of 100K transcripts. Scatterplots of profile expression were displayed in log10 space.
Neuron classification in the GP/NB and SN/VTA
In dot plots, dot diameter represents the fraction of cells within a subcluster where a transcript was counted. Colors represent average single-cell scaled expression value (out of 100K UMIs, log10).
Transcriptional diversity of striatal SPNs
To identify differentially expressed genes across SPN populations, we first generated subcluster-level profiles by summing the integer counts of each cell in each SPN subcluster. Differentially expressed genes exhibited >2 fold differences and had P<10−100 based on the binomTest (Robinson et al., 2010) and were visualized in SPN scatterplots with large, dark dots. Non-eSPN subclusters (13–6, 13–7, 13–8, 13–9 and 13–10) were excluded from the differential expression analysis described in Figure S7I.
DATA AND SOFTWARE AVAILABILITY
The accession number for the sequencing data reported in this paper is GEO: GSE116470. Processed sequencing files – including single-cell DGEs for each region and DGEs of cell populations based on subcluster assignments – are also available from the DropViz website (http://dropviz.org/).
ADDITIONAL RESOURCES
Dropviz
To present data for exploration, analysis and sharing, we developed a web-based application called DropViz. The application allows a user to filter cells in the atlas data by brain region, cell class, cell cluster and cell type. The application displays global and cluster-specific t-SNE plots of the filtered cells. Users can also search by gene name and then overlay relative gene expression on t-SNE plots or display a ranked plot of gene expression. The application provides a scatter plot to compare the relative expression between two cell sets, which allows for the identification of differentially expressed genes. The source code is available at https://github.com/broadinstitute/dropviz.
Supplementary Material
Data S1. Region-specific tissue volumes processed as input for Drop-seq. Related to Figure 1.
(A–I) Diagrams of tissue volumes dissected from acute brain slices (400 μm). Areas shaded red indicate approximate tissue volumes used for each Drop-seq dataset. Total Hippocampus (including part of the subiculum) was dissected from sections cut ~15° off horizontal from the nearly the entire ventral dorsal axis. Equivalent tissue is shown on a coronal schematic for clarity. Thalamus datasets included both coronal and sagittal replicates. Schematics were adapted from the Paxinos Mouse Atlas. Brain slicing details are presented in Table S1.
Data S2. ICA-based global clustering is robust. Related to Figure 1.
(A) To determine the global cluster robustness, we compared the global cluster identities of single cells after clustering was performed with all cells or cell subsets (full data, n= 156,167 cells; subsets are in 10% increments from 10 to 90%). Cluster similarity was measured by average Jaccard Distance (color-coded). Only Cluster 11 – corresponding to the Microglia and Macrophage – exhibits mean Jaccard distances <0.85. This is due to Microglia and Macrophage subclusters occasionally being split into distinct Global clusters.
Data S3. Presentation of 323 neuronal subclusters identified across atlas regions. Related to Figure 1.
(A–I) For each atlas region, neuron global clusters and subclusters are presented. Left, color-coded Global cluster tSNE plots. Only neuron clusters are numbered. Total cell numbers are shown for each region. Right, color-coded subcluster assignments identified from independent subclustering analysis of each global neuron cluster. Global clusters with < 200 cells were not subclustered. tSNE plots were only generated for subclusters with > 2 Biological ICs. Total cell numbers used in each subcluster analysis are shown. Candidate anatomical identities and pan-subcluster enriched markers are listed below each neuron subcluster and correspond to the labels used in the Dropviz software.
Data S4. Transcriptional signals in non-neuronal cells from across brain regions. Related to Figure 2.
(A–H) Transcriptional diversity in non-neuronal cell classes throughout the brain. Following the region specific analyses, single-cell profiles assigned to each non-neuronal cell class (astrocytes, microglia & macrophage, oligodendrocytes, polydendrocytes, mural, fibroblast-like and endothelial), were combined, grouped and re-analyzed using our ICA-based system. All ICs curated as Biological during subclustering are presented along with color-coded subcluster assignments and region-enrichment analysis (H).
(A–G) IC cell-loadings displayed on tSNE plots. Gene-loading plots are shown with top ten loading genes. Expression plots are also shown for for the Aldh1a1 gene (Mural) and Tm4sf1, Slc38a5, Mgp and Cp (Endothelial).
(H) Dot plots illustrating region-enrichment across each non-neuronal subcluster. The number of cells contributed by each region were normalized by region totals and the scaled by subcluster totals. The “All” condition illustrates no region enrichment.
Data S5. The Neurofilament signal is prominently but not exclusively associated with fast-spiking Pvalb+ neuron populations. Related to Figure 3.
(A–B) Example neuron subcluster analyses illustrating the distribution of the Neurofilament IC signal and relationship to Pvalb expression.
(A) Left, color-coded subcluster assignments. Middle, cell-loadings for Neurofilament ICs. Right, gene-loadings for each IC, with top ten genes shown at right. tSNE plots are used for display. Hippocampus Cluster 1 contains interneurons. GP/NB Cluster 2 contains neurons of the GPe and basal forebrain. Frontal Cortex Cluster 3 contains neurons of layer 6 and subplate.
(B) Pvalb expression.
Data S6. Transcriptional signals detected in striatal dSPNs (Cluster 10) and iSPNs (Cluster 11). Related to Figure 7.
(A–B) Biological ICs identified during subclustering of Striatal dSPNs (Cluster 10, n=9) and iSPNs (Cluster 11, n=10). These signals include specializations related to cell types, cell states and spatial signals. IC cell-loadings displayed on tSNE plots. Gene-loading plots are shown with top ten loading genes. dSPN IC 15 and iSPN IC 14 represent the patch/matrix distinction and are discussed in Figure S7.
Figure S1. Comprehensive identification of brain cell classes. Related to Figure 1.
(A) Example single-cell suspension generated from frontal cortex that contained little debris and exhibited diverse cellular morphologies. Bottom, enlargements of example cells.
(B) T-distributed stochastic neighbor embedding (tSNE) plot of gene expression relationships amongst the n=113,171 cell hippocampus dataset based on the first round of ICA-based clustering (see Figure 1B). Left, tSNE plot color-coded based on “global” clusters (numbered 1–19). Right, tSNE plot color-coded based on major cell classes of the brain.
(C) Violin plots showing example gene markers that distinguish across and within brain cell classes (log10). Genes are color-coded by cell class. Global clusters are ordered by cell class.
(D) Cartoons of the major cell classes of the brain. Numbers below indicate the corresponding global clusters from hippocampus.
(E) Dot plots displaying the proportional representation of individual cell classes across regions. The major trend suggests that neuronal proportions vary inversely with those of oligodendrocytes, endothelial cells, and mural cells. Fibroblast-like cells comprised a similar proportion of all cells in each region. Polydendrocyte and astrocyte abundance appeared to vary independently of other cell classes, exhibiting enrichment in the GPe as expected based on earlier findings (Cui et al., 2016). Small fractions of choroid plexus and ependymal cells were sampled from ventricle-adjacent regions, while sparse neurogenic populations were observed in regions adjacent to the subventricular and subgranular zones (frontal cortex, striatum, and hippocampus) (Ming and Song, 2011). These trends could be driven both by endogenous regional variation and differences in tissue preparation (Table S1).
(F) Number of subclusters by region.
(G) Transcriptional correlations across atlas subclusters are largely explained by cell class and not region of origin. Hierarchical clustering diagram showing pairwise Pearson correlation scores calculated pairwise between 565 atlas subcluster populations. The analysis was restricted to genes with significantly variable expression (STAR Methods). Color-coded bars at the top of the plot display the ordered region/cell class assignments for the subcluster.
{kind=link}
Figure S2. ICA-based subclustering identifies interpretable transcriptional signals that can be used to remove technical artifacts and create transparent signal-cluster relationships. Related to Figure 1.
(A–B) Comparison of Independent component analysis (ICA) versus principal component analysis (PCA) in encoding cell type/state distinctions from scRNA-seq data. ICs encode transcriptional sources such as cell types with single components; PCs encode the same sources through complex combinations of components. Here we use the ICs/PCs identified from the Frontal Cortex Cplx3+/Synpr+ interneuron subcluster analysis (Cluster 1). Subclusters were first defined by n=9 non-technical ICs (“Biological ICs”; see Figure 1, STAR Methods). On the same dataset, the first n=9 PCs were also identified.
(A) Cell-loadings for IC 10 and PC 9 displayed on an IC-based tSNE plot. Both IC 10 and PC 9 loadings are highest in a group of cells (arrowheads) that we subsequently identify as subcluster 1–7 (Synpr+/Dpy19l1+). High IC 10 loading is almost completely restricted to this group. PC 9, however, exhibits a range of loadings in other parts of the tSNE plot. Thus IC 10 exclusively encodes this particular group of interneurons, whereas PC 9 encodes an abstract signal differentially distributed over different interneuron subsets.
(B) ICs exhibit singular relationships to clusters built from single-cell transcriptomes, whereas PCs are distributed in combinations. IC-based hierarchical clustering of single-cells from subcluster 1–7. Cell-loadings based on n=9 PCs or n=9 ICs are plotted, with high loading in yellow and low loading purple. Results are similar if hierarchical clustering is performed with PCs.
(C) Examples of replicate and doublet “technical” ICs from frontal cortex. Top, example of an independent component (IC 38, cluster 6) representing a replicate-based signal. Left, plot of cell-loading scores (y-axis). On the x-axis, cells are ordered by library size (largest to smallest) within each sequencing pool (color-coded). Sequencing pools from the same mouse are grouped with black and gray bars above. Center, cell-loading scores plotted on the subcluster 6 tSNE (tSNE constructed from all ICs). Higher loadings are shown in darker red. Right, gene-loading plot. The n=10 genes with the highest scores are shown at right. Bottom, example of a component (IC 15, cluster 6) representing a “doublet” signal. Plots and layout are similar to the above. IC 15 loads heavily on a small number of cells across mice and sequencing pools. The top loading genes are markers of the polydendrocyte cell class, suggesting a signal that represents a “doublet” transcriptome consisting of layer 2/3 neurons and polydendrocytes. Inset shows area of the tSNE plot with high IC 15 loading.
(D–G) Experimental identification of transcriptional signals related to preparation of acute brain into single cells and examples of these digest-related “technical” ICs in frontal cortex.
(D) Schematic illustrating “over” and “under” digest experiments. Instead of optimal protease digest times (STAR Methods) for Frontal Cortex (FC) or Hippocampus (HP) (optimal: FC, 2hrs; HP, 1.5), regions were under-digested (1 hr) or over-digested (3 hrs) before Drop-seq library creation.
(E–F) Digest-associated ICs encode a similar transcriptional signal exhibited across regions and cell classes.
(E) Digest-skew plots for subclustering ICs. Under and over-digest Drop-seq libraries were grouped by region co-analyzed using our ICA-based system. For every IC identified through subclustering, cell-loadings scores were grouped by digest condition and compared using a rank-sum test. The resulting P-values are plotted on the y-axis. Each IC is represented by a dot and ordered by P-value within each subcluster analysis (color-coded and labeled by cell class). The size of the dot represents average difference in loading between digest groups. Lower P-values and larger dots are indicative of ICs that encode digest time. Digest-related signals are largely restricted to 1 or 2 ICs with neuron and astrocyte cell classes. A P-value cut-off of –log10 > 20 was used as a threshold to define digest-related ICs (dotted line).
(F) Pairwise mutual information analysis based on n=251 shared genes highlights a similar signal encoded by digest-related ICs. The gene contributions to this signal are characterized in (G) and this signal is also present in our atlas datasets (H).
(G) Example of digest-related ICs from FC co-analysis of under- and over-digested cells. Left, cell loadings color-coded by digest condition. Right, gene-loading plots for the 10 top and bottom loading genes. IC 15 (cluster 2, Syt6+ deep-layer glutamatergic) and IC 68 (cluster 1, Nptxr+ upper-layer glutamatergic) show contributions from similar genes. Genes related to ATP synthesis and Calmodulins (eg., Cox6a1, Ndufa4, Calm2) load on the under-digested cells, while another set of genes including the nuclear-enriched Meg3 and Malat1 load highly onto the over-digested condition.
(H) Digest-related ICs were commonly observed in atlas subclustering analyses, suggesting heterogeneity in cellular transcriptional response to identical preparation conditions. Two examples of digest-related ICs from atlas FC cluster 6 (20 and 23). Left, cell-loading plots and insets highlight the correlation of IC loading to library size. IC 20 tends to load on smallest libraries, while IC 23 loads on the largest. Middle, cell-loadings for IC 20 and 23 demonstrate that digest-related signals contribute to local relationships within the subcluster 6 tSNE plot. Right, gene-loading plots for the 10 top and bottom loading genes indicate a similar signal to that identified in over and under-digested experiments. Digest-related ICs were excluded from downstream atlas subclustering analyses.
(I) Removing technical ICs prevents spurious transcriptional similarities. E xample tSNE cell-loading plot for cluster 6 digest-related IC 23 before and after IC curation. Before curation, cells with high loading of digest-related IC 23 (E, bottom left) are locally grouped. After curation, technical ICs (including IC 23) are removed, creating a different tSNE plot and preventing the digest-related effects from contributing to clustering.
(J–K) Illustration of subclustering strategy based on Biological ICs.
(J) Correspondence between heterogeneous transcriptional signals (Biological ICs) and subclusters identified by modularity clustering (Similar to Figure 1E, but for Frontal Cortex cluster 1). tSNE cell-loading plots for each of the n=11 Biological ICs. Bottom right, the resulting plot in which the n=11 subclusters are identified.
(K) Portfolios of subclustering solutions. We select the solution that most appropriately represents the underlying ICs cell-loading structure (STAR Methods). To generate the portfolio, we varied both the number of neighbors in the underling network graphs (k) as well as the resolution of the (k) of the cluster assignments. Here we present a sample of the portfolio for Frontal Cortex cluster 1 (top, shown in (J)) or cluster 6 (bottom, from Figure 1E). The selected solutions are boxed.
{kind=link}
Figure S3. Identifying ion channel gene-gene co-expression relationships from hundreds of cell populations. Related to Figure 4.
(A) The number of individual single-cell profiles that contribute to each of the 565 transcriptionally-similar cell populations identified in the atlas (median=171 cells, mean=565).
(B) Receiver operating curve analysis of cell populations and synthetic bulk tissue nAChR subunit correlation distributions. Known nAChR subunit interactions (Gotti et al., 2006) were treated as true positives to calculate true positive rate and false positive rate. Red, pairwise nAChR gene correlations calculated from subcluster profiles; green, same correlations computed from “synthetic bulk” tissue, in which each tissue replicate was separately summed (n=42); blue, subcluster profiles in which data are randomly permuted to yield the same mean expression but random values.
(C) Expression levels of the n=17 nAChR subunits across the 565 cell populations. Genes are color-coded by family and labeled according to traditional brain/muscle association.
(D) List of the neuronal populations identified as exhibiting fast action potential firing rates by region.
(E) Genes most frequently correlated with the n=15 genes that make up the correlation block associated with action potential firing rate (Figure 4D). For each gene, the top n=25 most highly correlated genes were identified, producing a total list of 375 genes. The bar plot reflects the gene frequencies, displaying only those genes with three or more counts.
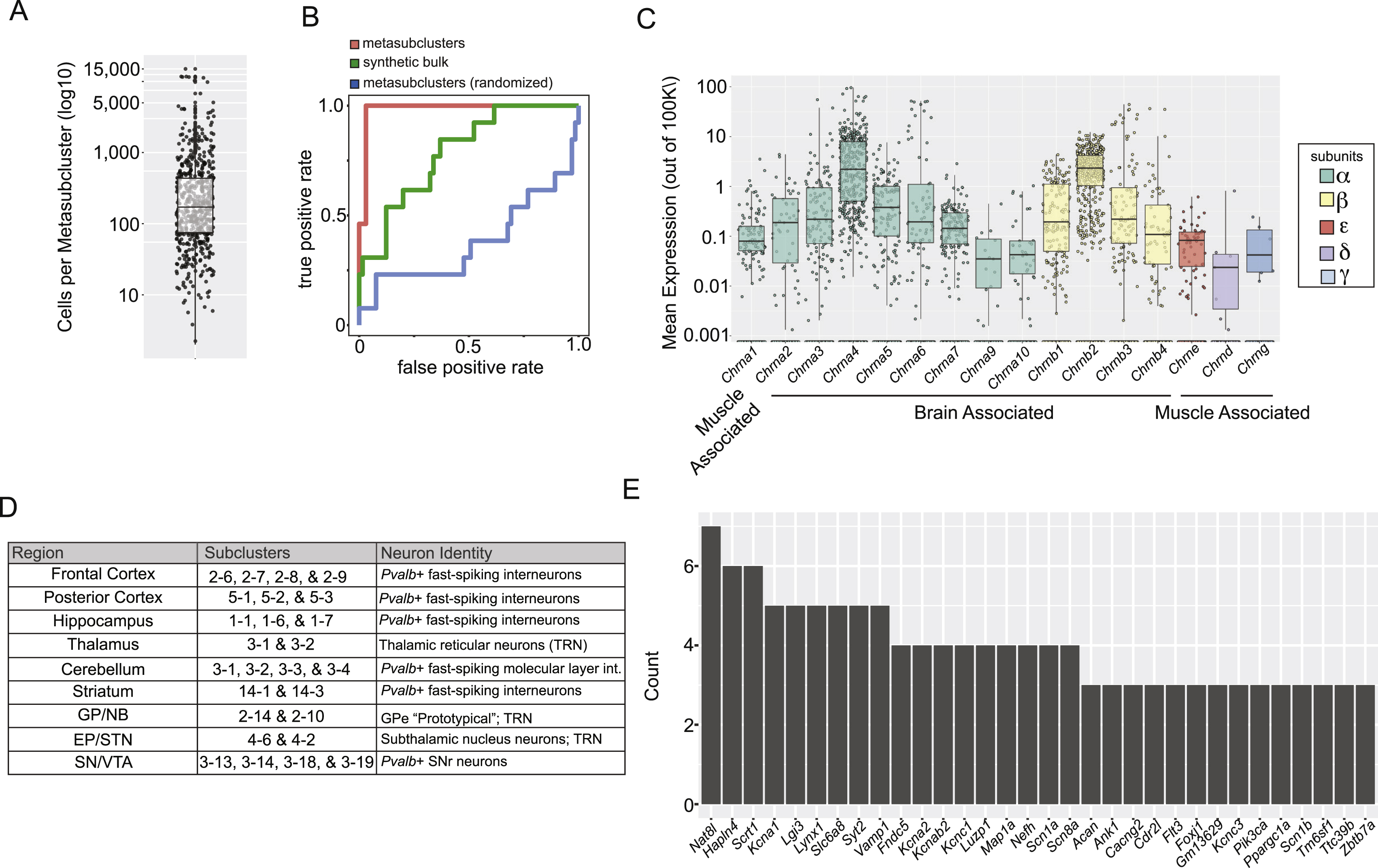{kind=link}
Figure S4. Comparing intact tissue to single-cell suspensions and Drop-seq datasets to determine cell recovery rates and acquisition biases at the level of cell class, cell type and cell subtype. Related to Figure 5.
(A–B) Estimating the fraction of brain cells recovered in single-cell suspensions generated from intact tissue. To aid in the identification of single cells, we used Nestin-Cre;lsl-ZsGreen mice in which the ZsGreen fluorophore is enriched in the soma of heterogenous set of brain cell classes.
(A) ZsGreen reporter fluorescence identifies single cells in suspension (left) and in intact Frontal Cortex (right, sagittal section).
(B) To approximate cell recovery rates from intact tissue, we estimated the total number of ZsGreen+ cells in a fixed volume of intact Frontal Cortex and Striatum using stereological counting and compared those starting totals to the number of cells estimated to be recovered in suspension (STAR Methods)(Tissue, n=2 mice, n=12–22 confocal stacks/mouse, n =108–181 sampling boxes/mouse; Suspension, n=5 mice, n=6 samples/suspension). Boxplot shows estimates per mouse (color-coded).
(C) Recovery fraction (total suspension / total tissue) by region. Error bars indicate standard error of the mean recovery fraction and account for error estimated in the tissue and suspension means.
(D–K) Addressing acquisition bias at the level of cell class, cell type and cell subtype. Biases in acquisition could result from at least two sources: 1) tolerance to dissociation and 2) transcript abundance, as larger libraries are more likely to be distinguished as cells from background (STAR Methods).
(D–F) Cell class acquisition bias. The fraction of neurons, astrocytes, oligodendrocytes/polydendrocytes and microglia were estimated in tissue using stereological counts ascertained from confocal stacks of tissue in which cell classes were labeled with or immunohistochemistry (IHC) or transgenes expressing GFP. Drop-seq estimates were made by calculating the total number of cells assigned to all Global clusters of a given class.
(D) Two sets of experiments (n=3 cell classes/experiment) were used to estimate the relative fraction of neurons, astrocytes, oligodendrocytes/polydendrocytes and microglia in intact tissue. Top, in the first set of experiments, GFP was expressed in astrocytes using the Aldh1l1-GFP line, neurons were stained with NeuN and oligodendrocytes/polydendrocytes were labelled with Olig1 (n=34 imaging fields from n=1 mouse). In the second set of experiments (bottom), the GFP driver line was replaced with Cx3cr1-GFP to target microglia and the IHC labels remained the same (n=36 and 32 imaging fields from n=2 mice).
(E) Box plot comparing cell class fractions per mouse estimated from intact tissue versus Drop-seq.
(F) Transcript abundance distributions differ by cell class. Box plot of single-cell transcript counts (log10) grouped and ordered by cell class. Neurons have larger Drop-seq libraries than non-neurons. Larger libraries could partially account for overrepresentation of neurons.
(G–I) Cell type acquisition bias. The ratio of excitatory (glutamatergic) to inhibitory (GABAergic) neurons was estimated using smFISH and high-throughput imaging. Drop-seq estimates were made by counting the number of cells within glutamatergic or GABAergic subclusters.
(G) Cocktails of ISH probes against genetic markers of glutamatergic (Slc17a6/7/8) and GABAergic (Gad1/2) neurotransmission identify excitatory and inhibitory neuron populations. Automated co-localization analysis performed on slide-scans identified n=487,003 neurons (n=3 mice).
(H) Boxplots of E/I ratios from ratio (10:1 vs 5:1) as measured by smFISH. individual mice estimated from smFISH and Drop-seq. On average, Drop-seq datasets exhibit twice the E/I
(I) Boxplots of single-cell transcript abundance by neuron type (log10). Excitatory neurons have larger transcript abundance distributions than inhibitory interneurons, which could contribute to the increased E/I ratio in Drop-seq datasets.
(K–M) Cell subtype acquisition bias. The ratio of major inhibitory neuron classes was estimated using smFISH and high-throughput imaging. Drop-seq estimates were made by counting the number of cells within subclusters enriched for one or more interneuron markers.
(K) ISH probes against interneuron markers Pvalb, Sst and Vip were used to identify interneuron subpopulations. Automated co-localization analysis performed on slide scans identified n=68,734 interneurons (n=3 mice).
(L) Boxplots of interneuron subtype fractions estimated from smFISH and Drop-seq. Drop-seq datasets are enriched for Vip+ interneurons at the cost of Pvalb+ and Sst+ interneurons.
(M) Boxplots of single-cell transcript abundance by interneuron subtype (log10). Larger libraries in Pvalb+ and Sst+ interneurons cannot explain their relative depletion compared to Vip+ interneurons.
{kind=link}
Figure S5. Cell class based transcriptional comparison of cortical regions. Related to Figure 5.
(A) tSNE plot color-coded by the n=7 excitatory subclusters with greater than 3:1 cell representation skew from FC or PC (and P < 0.05, Barnard’s test, see Figure 5C). The subcluster number is shown, along with a colored dot to represent the skewed region (FC, salmon color; PC, blue).
(B) tSNE based gene plots (top) and ISH stains (bottom, Allen) for markers enriched in each of the skewed clusters. Arrows point to high expression areas. Equivalent data for subclusters 21 and 5 are shown in Figure 5D
{kind=link}
Figure S6. Inferring the anatomical location of subclusters from the GP/NB Cluster 2 and SN/VTA Cluster 3 analysis using ISH patterns of marker genes. Related to Figure 6.
(A) tSNE color-coded by subclusters originating from subcluster analysis of GP/NB Cluster 2, which encodes mostly non-Striatal GABAergic and glutamatergic neurons.
(B) Subclusters color-coded by candidate anatomical regions.
(C) Dot plot illustrating the expression patterns of neurotransmitter marker genes and novel pairs of markers identified across all n=25 subclusters in this analysis. Dot diameter represents the fraction of cells within a subcluster where a transcript was counted. Color scale represents average single-cell scaled expression value (out of 100K UMIs, log10).
(D) ISH experiments (Allen) illustrating the spatial expression patterns of single, selective marker genes for each subcluster. Subclusters are grouped by inferred anatomical location.
(E–H) Same plots and organization as above, but for SN/VTA Cluster 3 that encodes mostly GABAergic and glutamatergic neurons of the SNr and VTA.
{kind=link}
Figure S7. Molecular specializations within spiny projection neurons subtypes. Related to Figure 7.
(A–E) Identification and comparison of patch iSPN and dSPN transcriptomes.
(A) Color-coded subcluster assignments for striatum cluster 10 (dSPNs) and cluster 11 (iSPNs) displayed on tSNE plot.
(B) ICs encoding patch-specific transcriptional signals in dSPNs (IC 15) and iSPNs (IC 20). Left, IC cell-loadings on tSNE plots. Right, gene-loadings with top 10 genes displayed. A full description of biological ICs resulting from SPN subcluster analyses is presented in (Data S6).
(C) Identification of shared and distinct dSPN and iSPN patch-enriched genes. Scatterplot comparing gene loadings between patch ICs for dSPNs (IC 15) and iSPNs (IC 20). Genes with high loading in both ICs are hypothesized to be enriched in both patch iSPNs and dSPNs. This group includes described (Tac1) and previously undescribed (Tshz1) genes enriched in patch dSPNs and iSPNs. Genes that load strongly onto either IC are candidates for patch molecular specialization by SPN pathway. For example, among matrix SPNs, Asic4 was selectively expressed in the dSPNs but not iSPNs; among patch SPNs, however, Asic4 was expressed in both dSPNs and iSPNs. Thus Asic4 helps tailor iSPNs but not dSPNs to the patch habitat.
(D) ISH stains for Tac1 and Tshz1 on coronal sections of striatum (Allen). Arrowheads point to example patches.
(E) Expression tSNE plots for Asic4 and Necab1 illustrating patch enrichment exclusive to iSPNs and dSPNs, respectively.
(F–H) Identifying non-eSPNs in Cluster 13.
(F) Color-coded subcluster identities. Subclusters labels in black correspond to eSPNs; labels in red correspond to non-Striatal neurons
(G) Expression of SPN marker Ppp1r1b is enriched in eSPN subclusters.
(H) Non-eSPN subclusters express unique markers that suggest non-Striatal origins. Left, expression of markers on tSNE plots. Right, ISH stains for each marker gene (Allen). Top, Slc17a7 expression suggests subclusters 13-6, 13-7 and 13-8 originate from adjacent cortex. Middle, Cartpt expression suggests subcluster 13-9 originates from the Bed Nucleus of the Stria Terminalis. Bottom, Nfib expression suggests subcluster 13-10 originates from the Amygdala. Red arrows point to regions of interest.
(I) eSPNs and SPN populations that express the same pathway markers are transcriptionally distinct. Genome-wide gene mean expression comparisons between Drd1+ or Adora2a+ SPN and eSPN subtypes (log normal scale). Left, Drd1+ eSPN subclusters (13-1, 13-2, 13-3 vs Drd1+ SPN Cluster 10. Right, Adora2a+ eSPN subclusters (13-4 & 13-5) vs Adora2a+ Cluster 11. Differentially expressed genes are shown with larger, dark dots (>2 natural log fold difference and P<10−100, binomTest (Robinson et al., 2010) and total number listed above each plot.
{kind=link}
Table S1. Preparation of region-specific single cell suspensions from acute brain tissue. Related to Figure 1.
Sample insights:
Systematic survey of molecular diversity across all cell classes
Neuronal “Neurofilament” program & co-regulation of ion channels
Glutamatergic neurons specialize cortical regions
Neuronal classification within the basal ganglia, including a rare population of striatal SPNs
Highlights.
690K individual cells analyzed from nine regions of adult mouse brain
RNA expression patterns corresponding to cell types, states and locations
Transcriptional programs supporting axonal function and neuronal specializations
Online data resource “DropViz”
Sampling across multiple brain regions identifies hundreds of transcriptionally distinct groups of cells and reveals large-scale features of brain organization and neuronal diversity
Acknowledgments
This work was supported by the Broad Institute’s Stanley Center for Psychiatric Research and by a Helen Hay Whitney Fellowship to A.S. We thank Rahul Satija and Andrew Butler for advice and helpful conversations. M.B. was supported by T32GM007753, T32MH020017, HST Idea^2, The Sackler Scholar Programme in Psychobiology. We thank Dr. Christina Usher for assistance with manuscript preparation and Dr. Gordon Fishell for comments on manuscript drafts.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests
References
- Abbott NJ, Rönnbäck L, and Hansson E (2006). Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7, 41–53. [DOI] [PubMed] [Google Scholar]
- Abdi A, Mallet N, Mohamed FY, Sharott A, Dodson PD, Nakamura KC, Suri S, Avery SV, Larvin JT, Garas FN, et al. (2015). Prototypic and Arkypallidal Neurons in the Dopamine-Intact External Globus Pallidus. J Neurosci 35, 6667–6688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1873.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albin RL, Young AB, and Penney JB (1989). The functional anatomy of basal ganglia disorders. Trends Neurosci 12, 366–375. [DOI] [PubMed] [Google Scholar]
- Andermann ML, and Lowell BB (2017). Toward a Wiring Diagram Understanding of Appetite Control. Neuron 95, 757–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DJ (2012). Optogenetics, sex, and violence in the brain: implications for psychiatry. Bio Psych 71, 1081–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antal M, Beneduce BM, and Regehr WG (2014). The Substantia Nigra Conveys Target-Dependent Excitatory and Inhibitory Outputs from the Basal Ganglia to the Thalamus. J Neurosci 34, 8032–8042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bading H (2013). Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci 14, 593–608. [DOI] [PubMed] [Google Scholar]
- Baroncelli L, Molinaro A, Cacciante F, Alessandrì MG, Napoli D, Putignano E, Tola J, Leuzzi V, Cioni G, and Pizzorusso T (2017). A mouse model for creatine transporter deficiency reveals early onset cognitive impairment and neuropathology associated with brain aging. Hum Mol Genet 25, 4186–4200. [DOI] [PubMed] [Google Scholar]
- Boström K, Zebboudj AF, Yao Y, Lin TS, and Torres A (2004). Matrix GLA Protein Stimulates VEGF Expression through Increased Transforming Growth Factor-β1 Activity in Endothelial Cells. J Biol Chem 279, 52904–52913. [DOI] [PubMed] [Google Scholar]
- Buckner RL, and Krienen FM (2013). The evolution of distributed association networks in the human brain. Trends Cogn Sci 17, 648–665. [DOI] [PubMed] [Google Scholar]
- Campbell JN, Macosko EZ, Fenselau H, Pers TH, Lyubetskaya A, Tenen D, Goldman M, Verstegen AMJ, Resch JM, McCarroll SA, et al. (2017). A molecular census of arcuate hypothalamus and median eminence cell types. Nat Neuro 20, 484–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter AE, Genome TJ, Lamprect M, Clark C, Kang IH, Friman O, Guertin D, Chang JH, Moffat J, Golland P, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Gen Bio 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter BC, and Bean BP (2009). Sodium Entry during Action Potentials of Mammalian Neurons: Incomplete Inactivation and Reduced Metabolic Efficiency in Fast-Spiking Neurons. Neuron 64, 898–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MC, Ferrari L, Sacchet MD, Foland-Ross LC, Qiu M-H, Gotlib IH, Fuller PM, Arrigoni E, and Lu J (2015). Identification of a direct GABAergic pallidocortical pathway in rodents. Eur J Neurosci 41, 748–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yu FH, Sharp EM, Beacham D, Scheuer T, and Catterall WA (2008). Functional properties and differential neuromodulation of Na(v)1.6 channels. Mol Cell Neurosci 38, 607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung NC, and Storey JD (2015). Statistical significance of variables driving systematic variation in high-dimensional data. Bioinformatics 31, 545–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contois LW, Nugent DP, Caron JM, Cretu A, Tweedie E, Akalu A, Liebes L, Friesel R, Rosen C, Vary C, et al. (2012). Insulin-like growth factor binding protein-4 differentially inhibits growth factor-induced angiogenesis. J Biol Chem 287, 1779–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Pitt JE, Pamukcu A, Poulin J-F, Mabrouk OS, Fiske MP, Fan IB, Augustine EC, Young KA, Kennedy RT, et al. (2016). Blunted mGluR Activation Disinhibits Striatopallidal Transmission in Parkinsonian Mice. Cell Reports 17, 2431–2444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmopil S, Muñetón-Gómez VC, de Ceballos ML, Bernson M, and Moratalla R (2008). Tyrosine hydroxylase cells appearing in the mouse striatum after dopamine denervation are likely to be projection neurones regulated by L-DOPA. Eur J Neurosci 27, 580–592. [DOI] [PubMed] [Google Scholar]
- Fu Y, Lai Y, Wang Q, Liu X, He W, Zhang H, Fan C, and Yang G (2013). Overexpression of clusterin promotes angiogenesis via the vascular endothelial growth factor in primary ovarian cancer. Mol Med Rep 7, 1726–1732. [DOI] [PubMed] [Google Scholar]
- Gerfen CR (1992). The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu. Rev. Neurosci 15, 285–320. [DOI] [PubMed] [Google Scholar]
- Gierahn TM, Wadsworth MH, Hughes TK, Bryson BD, Butler A, Satija R, Fortune S, Love JC, and Shalek AK (2017). Seq-Well: portable, low-cost RNA sequencing of single cells at high throughput. Nat Neuro. 14, 395–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gittis AH, Berke JD, Bevan MD, Chan CS, Mallet N, Morrow MM, and Schmidt R (2014). New roles for the external globus pallidus in basal ganglia circuits and behavior. J Neurosci 34, 15178–15183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, and Rudy B (2008). K+ Channels at the Axon Initial Segment Dampen Near-Threshold Excitability of Neocortical Fast-Spiking GABAergic Interneurons. Neuron 58, 387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotti C, Zoli M, and Clementi F (2006). Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends in Pharmacological Sciences 27, 482–491. [DOI] [PubMed] [Google Scholar]
- Gökce Ö, Stanley GM, Treutlein B, Neff NF, Camp JG, Malenka RC, Rothwell PE, Fuccillo MV, Südhof TC, and Quake SR (2016). Cellular Taxonomy of the Mouse Striatum as Revealed by Single-Cell RNA-Seq. Cell Reports 16, 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graybiel AM, and Ragsdale CW (1978). Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc Natl Acad Sci 75, 5723–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guadall A, Orriols M, Rodríguez-Calvo R, Calvayrac O, Crespo J, Aledo R, Martínez-González J, and Rodríguez C (2011). Fibulin-5 Is Up-regulated by Hypoxia in Endothelial Cells through a Hypoxia-inducible Factor-1 (HIF-1α)-dependent Mechanism. J Biol Chem 286, 7093–7103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib N, Li Y, Heidenreich M, Swiech L, Avraham-Davidi I, Trombetta JJ, Hession C, Zhang F, and Regev A (2016). Div-Seq: Single-nucleus RNA-Seq reveals dynamics of rare adult newborn neurons. Science 353, 925–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X, Wang R, Zhou Y, Fei L, Sun H, Lai S, Saadatpour A, Zhou Z, Chen H, Ye F, et al. (2018). Mapping the Mouse Cell Atlas by Microwell-Seq. Cell 172, 1091–1097.e17. [DOI] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, et al. (2008). A Translational Profiling Approach for the Molecular Characterization of CNS Cell Types. Cell 135, 738–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill RA, Tong L, Yuan P, Murikinati S, Gupta S, and Grutzendler J (2015). Regional Blood Flow in the Normal and Ischemic Brain Is Controlled by Arteriolar Smooth Muscle Cell Contractility and Not by Capillary Pericytes. Neuron 87, 95–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochgerner H, Zeisel A, nnerberg PLX, and Linnarsson S (2018). Conserved properties of dentate gyrus neurogenesis across postnatal development revealed by single-cell RNA sequencing. Nat Neuro. 163, 1–15. [DOI] [PubMed] [Google Scholar]
- Hrvatin S, Hochbaum DR, Nagy MA, Cicconet M, Robertson K, Cheadle L, Zilionis R, Ratner A, Borges-Monroy R, Klein AM, et al. (2017). Single-cell analysis of experience-dependent transcriptomic states in the mouse visual cortex. Nat Neuro. 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes S, and Chan-Ling T (2004). Characterization of Smooth Muscle Cell and Pericyte Differentiation in the Rat Retina In Vivo. Invest. Ophthalmol. Vis. Sci 45, 2795–12. [DOI] [PubMed] [Google Scholar]
- Hyvärinen A (1999). Fast and robust fixed-point algorithms for independent component analysis. IEEE Trans. Neural Netw 10, 626–634. [DOI] [PubMed] [Google Scholar]
- Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, Schwartz RS, Keating MT, and Li DY A critical role for elastin signaling in vascular morphogenesis and disease. Development 130, 411–423. [DOI] [PubMed] [Google Scholar]
- Kita H (1994). Parvalbumin-immunopositive neurons in rat globus pallidus: a light and electron microscopic study. Brain Res 657, 31–41. [DOI] [PubMed] [Google Scholar]
- Kita H (2007). Globus pallidus external segment. Prog Brain Res 160, 111–133. [DOI] [PubMed] [Google Scholar]
- Klein AM, Mazutis L, Akartuna I, Tallapragada N, Veres A, Li V, Peshkin L, Weitz DA, and Kirschner MW (2015). Droplet Barcoding for Single-Cell Transcriptomics Applied to Embryonic Stem Cells. Cell 161, 1187–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozorovitskiy Y, Saunders A, Johnson CA, Lowell BB, and Sabatini BL (2012). Recurrent network activity drives striatal synaptogenesis. Nature 485, 646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krabbe S, Gründemann J, and Lüthi A (2017). Amygdala Inhibitory Circuits Regulate Associative Fear Conditioning. Bio Psych 1–10. [DOI] [PubMed] [Google Scholar]
- Kravitz AV, Tye LD, and Kreitzer AC (2012). Distinct roles for direct and indirect pathway striatal neurons in reinforcement. Nat Neuro. 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krienen FM, Yeo BTT, Ge T, Buckner RL, and Sherwood CC (2016). Transcriptional profiles of supragranular-enriched genes associate with corticocortical network architecture in the human brain. Proc Natl Acad Sci 113, E469–E478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahti L, Haugas M, Tikker L, Airavaara M, Voutilainen MH, Anttila J, Kumar S, Inkinen C, Salminen M, and Partanen J (2016). Differentiation and molecular heterogeneity of inhibitory and excitatory neurons associated with midbrain dopaminergic nuclei. Development 143, 516–529. [DOI] [PubMed] [Google Scholar]
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, and Roeper J (2008). Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron 57, 760–773. [DOI] [PubMed] [Google Scholar]
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, et al. (2015). Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell 161, 1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet N, Micklem BR, Henny P, Brown MT, Williams C, Bolam JP, Nakamura KC, and Magill PJ (2012). Dichotomous Organization of the External Globus Pallidus. Neuron 74, 1075–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marder E, and Goaillard J-M (2006). Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci 7, 563–574. [DOI] [PubMed] [Google Scholar]
- Marques S, Zeisel A, Codeluppi S, van Bruggen D, Mendanha Falcão A, Xiao L, Li H, Häring M, Hochgerner H, Romanov RA, et al. (2016). Oligodendrocyte heterogeneity in the mouse juvenile and adult central nervous system. Science 352, 1326–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer C, Jaglin XH, Cobbs LV, Bandler RC, Streicher C, Cepko CL, Hippenmeyer S, and Fishell G (2015). Clonally Related Forebrain Interneurons Disperse Broadly across Both Functional Areas and Structural Boundaries. Neuron 87, 989–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G-L, and Song H (2011). Adult Neurogenesis in the Mammalian Brain: Significant Answers and Significant Questions. Neuron 70, 687–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser EI, Kropff E, and Moser M-B (2008). Place cells, grid cells, and the brain’s spatial representation system. Annu. Rev. Neurosci 31, 69–89. [DOI] [PubMed] [Google Scholar]
- Nehls V, and Drenckhahn D (1991). Heterogeneity of microvascular pericytes for smooth muscle type alpha-actin. J Cell Biol 113, 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Hare JK, Ade KK, Sukharnikova T, Van Hooser SD, Palmeri ML, Yin HH, and Calakos N (2016). Pathway-Specific Striatal Substrates for Habitual Behavior. Neuron 89, 472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulin J-F, Zou J, Drouin-Ouellet J, Kim K-YA, Cicchetti F, and Awatramani RB (2014). Defining midbrain dopaminergic neuron diversity by single-cell gene expression profiling. Cell Reports 9, 930–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg AB, Roco CM, Muscat RA, Kuchina A, Sample P, Yao Z, Gray L, Peeler DJ, Mukherjee S, Chen W, et al. (2018). Single-cell profiling of the developing mouse brain and spinal cord with split-pool barcoding. Science 12, eaam8999–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B, and McBain CJ (2001). Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci 24, 517–526. [DOI] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Huang KW, and Sabatini BL (2016). Globus Pallidus Externus Neurons Expressing parvalbumin Interconnect the Subthalamic Nucleus and Striatal Interneurons. PLoS ONE 11, e0149798–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders A, Oldenburg IA, Berezovskii VK, Johnson CA, Kingery ND, Elliott HL, Xie T, Gerfen CR, and Sabatini BL (2015). A direct GABAergic output from the basal ganglia to frontal cortex. Nature 7, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, Adiconis X, Levin JZ, Nemesh J, Goldman M, et al. (2016). Comprehensive Classification of Retinal Bipolar Neurons by Single-Cell Transcriptomics. Cell 166, 1308–1323.e1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KS, Tindell AJ, Aldridge JW, and Berridge KC (2009). Ventral pallidum roles in reward and motivation. Behav Brain Res 196, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternson SM (2013). Hypothalamic survival circuits: blueprints for purposive behaviors. Neuron 77, 810–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney MD, Ayyadurai S, and Zlokovic BV (2016). Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neuro 19, 771–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, and Regev A (2017). Scaling single-cell genomics from phenomenology to mechanism. Nature 541, 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasic B, Menon V, Nguyen TN, Kim T-K, Jarsky T, Yao Z, Levi B, Gray LT, Sorensen SA, Dolbeare T, et al. (2016). Adult mouse cortical cell taxonomy revealed by single cell transcriptomics. Nat Neuro 19, 335–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper JM, and Bolam JP (2004). Functional diversity and specificity of neostriatal interneurons. Curr Opin Neurobiol 14, 685–692. [DOI] [PubMed] [Google Scholar]
- Tripathy SJ, Toker L, Li B, Crichlow C-L, Tebaykin D, Mancarci BO, and Pavlidis P (2017). Transcriptomic correlates of neuron electrophysiological diversity. PLoS Computational Biology 13, e1005814–e1005828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trost A, Lange S, Schroedl F, Bruckner D, Motloch KA, Bogner B, Kaser-Eichberger A, Strohmaier C, Runge C, Aigner L, et al. (2016). Brain and Retinal Pericytes: Origin, Function and Role. Front Cell Neurosci 10, 3171–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S, Abulhoul L, Grünewald S, Anselm I, Azzouz H, Bratkovic D, de Brouwer A, Hamel B, et al. (2013). Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J Med Genet 50, 463–472. [DOI] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. [DOI] [PubMed] [Google Scholar]
- Voorn P, van de Witte S, Tjon G, and Jonker AJ (1999). Expression of enkephalin in pallido-striatal neurons. Ann N Y Acad Sci 877, 671–675. [DOI] [PubMed] [Google Scholar]
- Wallace ML, Saunders A, Huang KW, Philson AC, Goldman M, Macosko EZ, McCarroll SA, and Sabatini BL (2017). Genetically Distinct Parallel Pathways in the Entopeduncular Nucleus for Limbic and Sensorimotor Output of the Basal Ganglia. Neuron 94, 138–152.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waltman L, and van Eck NJ (2013). A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 86, 75–14. [Google Scholar]
- Wang LP, Li F, Wang D, Xie K, Wang D, Shen X, and Tsien JZ (2011). NMDA receptors in dopaminergic neurons are crucial for habit learning. Neuron 72, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu YE, Pan L, Zuo Y, Li X, and Hong W (2017). Detecting Activated Cell Populations Using Single-Cell RNA-Seq. Neuron 96, 313–328.e317. [DOI] [PubMed] [Google Scholar]
- Yuan A, Rao MV, Veeranna, and Nixon RA (2012). Neurofilaments at a glance. J Cell Sci 125, 3257–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaborszky L, Csordas A, Mosca K, Kim J, Gielow MR, Vadasz C, and Nadasdy Z (2013). Neurons in the Basal Forebrain Project to the Cortex in a Complex Topographic Organization that Reflects Corticocortical Connectivity Patterns: An Experimental Study Based on Retrograde Tracing and 3D Reconstruction. Cerebral Cortex. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel A, Muñoz-Manchado AB, Codeluppi S, Lönnerberg P, La Manno G, Juréus A, Marques S, Munguba H, He L, Betsholtz C, et al. (2015). Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142. [DOI] [PubMed] [Google Scholar]
- Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, et al. (2017). Massively parallel digital transcriptional profiling of single cells. Nature Communications 8, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Pistillo F, and Gotti C (2015). Diversity of native nicotinic receptor subtypes in mammalian brain. Neuropharmacology 96, 302–311. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Region-specific tissue volumes processed as input for Drop-seq. Related to Figure 1.
(A–I) Diagrams of tissue volumes dissected from acute brain slices (400 μm). Areas shaded red indicate approximate tissue volumes used for each Drop-seq dataset. Total Hippocampus (including part of the subiculum) was dissected from sections cut ~15° off horizontal from the nearly the entire ventral dorsal axis. Equivalent tissue is shown on a coronal schematic for clarity. Thalamus datasets included both coronal and sagittal replicates. Schematics were adapted from the Paxinos Mouse Atlas. Brain slicing details are presented in Table S1.
Data S2. ICA-based global clustering is robust. Related to Figure 1.
(A) To determine the global cluster robustness, we compared the global cluster identities of single cells after clustering was performed with all cells or cell subsets (full data, n= 156,167 cells; subsets are in 10% increments from 10 to 90%). Cluster similarity was measured by average Jaccard Distance (color-coded). Only Cluster 11 – corresponding to the Microglia and Macrophage – exhibits mean Jaccard distances <0.85. This is due to Microglia and Macrophage subclusters occasionally being split into distinct Global clusters.
Data S3. Presentation of 323 neuronal subclusters identified across atlas regions. Related to Figure 1.
(A–I) For each atlas region, neuron global clusters and subclusters are presented. Left, color-coded Global cluster tSNE plots. Only neuron clusters are numbered. Total cell numbers are shown for each region. Right, color-coded subcluster assignments identified from independent subclustering analysis of each global neuron cluster. Global clusters with < 200 cells were not subclustered. tSNE plots were only generated for subclusters with > 2 Biological ICs. Total cell numbers used in each subcluster analysis are shown. Candidate anatomical identities and pan-subcluster enriched markers are listed below each neuron subcluster and correspond to the labels used in the Dropviz software.
Data S4. Transcriptional signals in non-neuronal cells from across brain regions. Related to Figure 2.
(A–H) Transcriptional diversity in non-neuronal cell classes throughout the brain. Following the region specific analyses, single-cell profiles assigned to each non-neuronal cell class (astrocytes, microglia & macrophage, oligodendrocytes, polydendrocytes, mural, fibroblast-like and endothelial), were combined, grouped and re-analyzed using our ICA-based system. All ICs curated as Biological during subclustering are presented along with color-coded subcluster assignments and region-enrichment analysis (H).
(A–G) IC cell-loadings displayed on tSNE plots. Gene-loading plots are shown with top ten loading genes. Expression plots are also shown for for the Aldh1a1 gene (Mural) and Tm4sf1, Slc38a5, Mgp and Cp (Endothelial).
(H) Dot plots illustrating region-enrichment across each non-neuronal subcluster. The number of cells contributed by each region were normalized by region totals and the scaled by subcluster totals. The “All” condition illustrates no region enrichment.
Data S5. The Neurofilament signal is prominently but not exclusively associated with fast-spiking Pvalb+ neuron populations. Related to Figure 3.
(A–B) Example neuron subcluster analyses illustrating the distribution of the Neurofilament IC signal and relationship to Pvalb expression.
(A) Left, color-coded subcluster assignments. Middle, cell-loadings for Neurofilament ICs. Right, gene-loadings for each IC, with top ten genes shown at right. tSNE plots are used for display. Hippocampus Cluster 1 contains interneurons. GP/NB Cluster 2 contains neurons of the GPe and basal forebrain. Frontal Cortex Cluster 3 contains neurons of layer 6 and subplate.
(B) Pvalb expression.
Data S6. Transcriptional signals detected in striatal dSPNs (Cluster 10) and iSPNs (Cluster 11). Related to Figure 7.
(A–B) Biological ICs identified during subclustering of Striatal dSPNs (Cluster 10, n=9) and iSPNs (Cluster 11, n=10). These signals include specializations related to cell types, cell states and spatial signals. IC cell-loadings displayed on tSNE plots. Gene-loading plots are shown with top ten loading genes. dSPN IC 15 and iSPN IC 14 represent the patch/matrix distinction and are discussed in Figure S7.
Figure S1. Comprehensive identification of brain cell classes. Related to Figure 1.
(A) Example single-cell suspension generated from frontal cortex that contained little debris and exhibited diverse cellular morphologies. Bottom, enlargements of example cells.
(B) T-distributed stochastic neighbor embedding (tSNE) plot of gene expression relationships amongst the n=113,171 cell hippocampus dataset based on the first round of ICA-based clustering (see Figure 1B). Left, tSNE plot color-coded based on “global” clusters (numbered 1–19). Right, tSNE plot color-coded based on major cell classes of the brain.
(C) Violin plots showing example gene markers that distinguish across and within brain cell classes (log10). Genes are color-coded by cell class. Global clusters are ordered by cell class.
(D) Cartoons of the major cell classes of the brain. Numbers below indicate the corresponding global clusters from hippocampus.
(E) Dot plots displaying the proportional representation of individual cell classes across regions. The major trend suggests that neuronal proportions vary inversely with those of oligodendrocytes, endothelial cells, and mural cells. Fibroblast-like cells comprised a similar proportion of all cells in each region. Polydendrocyte and astrocyte abundance appeared to vary independently of other cell classes, exhibiting enrichment in the GPe as expected based on earlier findings (Cui et al., 2016). Small fractions of choroid plexus and ependymal cells were sampled from ventricle-adjacent regions, while sparse neurogenic populations were observed in regions adjacent to the subventricular and subgranular zones (frontal cortex, striatum, and hippocampus) (Ming and Song, 2011). These trends could be driven both by endogenous regional variation and differences in tissue preparation (Table S1).
(F) Number of subclusters by region.
(G) Transcriptional correlations across atlas subclusters are largely explained by cell class and not region of origin. Hierarchical clustering diagram showing pairwise Pearson correlation scores calculated pairwise between 565 atlas subcluster populations. The analysis was restricted to genes with significantly variable expression (STAR Methods). Color-coded bars at the top of the plot display the ordered region/cell class assignments for the subcluster.
Figure S2. ICA-based subclustering identifies interpretable transcriptional signals that can be used to remove technical artifacts and create transparent signal-cluster relationships. Related to Figure 1.
(A–B) Comparison of Independent component analysis (ICA) versus principal component analysis (PCA) in encoding cell type/state distinctions from scRNA-seq data. ICs encode transcriptional sources such as cell types with single components; PCs encode the same sources through complex combinations of components. Here we use the ICs/PCs identified from the Frontal Cortex Cplx3+/Synpr+ interneuron subcluster analysis (Cluster 1). Subclusters were first defined by n=9 non-technical ICs (“Biological ICs”; see Figure 1, STAR Methods). On the same dataset, the first n=9 PCs were also identified.
(A) Cell-loadings for IC 10 and PC 9 displayed on an IC-based tSNE plot. Both IC 10 and PC 9 loadings are highest in a group of cells (arrowheads) that we subsequently identify as subcluster 1–7 (Synpr+/Dpy19l1+). High IC 10 loading is almost completely restricted to this group. PC 9, however, exhibits a range of loadings in other parts of the tSNE plot. Thus IC 10 exclusively encodes this particular group of interneurons, whereas PC 9 encodes an abstract signal differentially distributed over different interneuron subsets.
(B) ICs exhibit singular relationships to clusters built from single-cell transcriptomes, whereas PCs are distributed in combinations. IC-based hierarchical clustering of single-cells from subcluster 1–7. Cell-loadings based on n=9 PCs or n=9 ICs are plotted, with high loading in yellow and low loading purple. Results are similar if hierarchical clustering is performed with PCs.
(C) Examples of replicate and doublet “technical” ICs from frontal cortex. Top, example of an independent component (IC 38, cluster 6) representing a replicate-based signal. Left, plot of cell-loading scores (y-axis). On the x-axis, cells are ordered by library size (largest to smallest) within each sequencing pool (color-coded). Sequencing pools from the same mouse are grouped with black and gray bars above. Center, cell-loading scores plotted on the subcluster 6 tSNE (tSNE constructed from all ICs). Higher loadings are shown in darker red. Right, gene-loading plot. The n=10 genes with the highest scores are shown at right. Bottom, example of a component (IC 15, cluster 6) representing a “doublet” signal. Plots and layout are similar to the above. IC 15 loads heavily on a small number of cells across mice and sequencing pools. The top loading genes are markers of the polydendrocyte cell class, suggesting a signal that represents a “doublet” transcriptome consisting of layer 2/3 neurons and polydendrocytes. Inset shows area of the tSNE plot with high IC 15 loading.
(D–G) Experimental identification of transcriptional signals related to preparation of acute brain into single cells and examples of these digest-related “technical” ICs in frontal cortex.
(D) Schematic illustrating “over” and “under” digest experiments. Instead of optimal protease digest times (STAR Methods) for Frontal Cortex (FC) or Hippocampus (HP) (optimal: FC, 2hrs; HP, 1.5), regions were under-digested (1 hr) or over-digested (3 hrs) before Drop-seq library creation.
(E–F) Digest-associated ICs encode a similar transcriptional signal exhibited across regions and cell classes.
(E) Digest-skew plots for subclustering ICs. Under and over-digest Drop-seq libraries were grouped by region co-analyzed using our ICA-based system. For every IC identified through subclustering, cell-loadings scores were grouped by digest condition and compared using a rank-sum test. The resulting P-values are plotted on the y-axis. Each IC is represented by a dot and ordered by P-value within each subcluster analysis (color-coded and labeled by cell class). The size of the dot represents average difference in loading between digest groups. Lower P-values and larger dots are indicative of ICs that encode digest time. Digest-related signals are largely restricted to 1 or 2 ICs with neuron and astrocyte cell classes. A P-value cut-off of –log10 > 20 was used as a threshold to define digest-related ICs (dotted line).
(F) Pairwise mutual information analysis based on n=251 shared genes highlights a similar signal encoded by digest-related ICs. The gene contributions to this signal are characterized in (G) and this signal is also present in our atlas datasets (H).
(G) Example of digest-related ICs from FC co-analysis of under- and over-digested cells. Left, cell loadings color-coded by digest condition. Right, gene-loading plots for the 10 top and bottom loading genes. IC 15 (cluster 2, Syt6+ deep-layer glutamatergic) and IC 68 (cluster 1, Nptxr+ upper-layer glutamatergic) show contributions from similar genes. Genes related to ATP synthesis and Calmodulins (eg., Cox6a1, Ndufa4, Calm2) load on the under-digested cells, while another set of genes including the nuclear-enriched Meg3 and Malat1 load highly onto the over-digested condition.
(H) Digest-related ICs were commonly observed in atlas subclustering analyses, suggesting heterogeneity in cellular transcriptional response to identical preparation conditions. Two examples of digest-related ICs from atlas FC cluster 6 (20 and 23). Left, cell-loading plots and insets highlight the correlation of IC loading to library size. IC 20 tends to load on smallest libraries, while IC 23 loads on the largest. Middle, cell-loadings for IC 20 and 23 demonstrate that digest-related signals contribute to local relationships within the subcluster 6 tSNE plot. Right, gene-loading plots for the 10 top and bottom loading genes indicate a similar signal to that identified in over and under-digested experiments. Digest-related ICs were excluded from downstream atlas subclustering analyses.
(I) Removing technical ICs prevents spurious transcriptional similarities. E xample tSNE cell-loading plot for cluster 6 digest-related IC 23 before and after IC curation. Before curation, cells with high loading of digest-related IC 23 (E, bottom left) are locally grouped. After curation, technical ICs (including IC 23) are removed, creating a different tSNE plot and preventing the digest-related effects from contributing to clustering.
(J–K) Illustration of subclustering strategy based on Biological ICs.
(J) Correspondence between heterogeneous transcriptional signals (Biological ICs) and subclusters identified by modularity clustering (Similar to Figure 1E, but for Frontal Cortex cluster 1). tSNE cell-loading plots for each of the n=11 Biological ICs. Bottom right, the resulting plot in which the n=11 subclusters are identified.
(K) Portfolios of subclustering solutions. We select the solution that most appropriately represents the underlying ICs cell-loading structure (STAR Methods). To generate the portfolio, we varied both the number of neighbors in the underling network graphs (k) as well as the resolution of the (k) of the cluster assignments. Here we present a sample of the portfolio for Frontal Cortex cluster 1 (top, shown in (J)) or cluster 6 (bottom, from Figure 1E). The selected solutions are boxed.
Figure S3. Identifying ion channel gene-gene co-expression relationships from hundreds of cell populations. Related to Figure 4.
(A) The number of individual single-cell profiles that contribute to each of the 565 transcriptionally-similar cell populations identified in the atlas (median=171 cells, mean=565).
(B) Receiver operating curve analysis of cell populations and synthetic bulk tissue nAChR subunit correlation distributions. Known nAChR subunit interactions (Gotti et al., 2006) were treated as true positives to calculate true positive rate and false positive rate. Red, pairwise nAChR gene correlations calculated from subcluster profiles; green, same correlations computed from “synthetic bulk” tissue, in which each tissue replicate was separately summed (n=42); blue, subcluster profiles in which data are randomly permuted to yield the same mean expression but random values.
(C) Expression levels of the n=17 nAChR subunits across the 565 cell populations. Genes are color-coded by family and labeled according to traditional brain/muscle association.
(D) List of the neuronal populations identified as exhibiting fast action potential firing rates by region.
(E) Genes most frequently correlated with the n=15 genes that make up the correlation block associated with action potential firing rate (Figure 4D). For each gene, the top n=25 most highly correlated genes were identified, producing a total list of 375 genes. The bar plot reflects the gene frequencies, displaying only those genes with three or more counts.
Figure S4. Comparing intact tissue to single-cell suspensions and Drop-seq datasets to determine cell recovery rates and acquisition biases at the level of cell class, cell type and cell subtype. Related to Figure 5.
(A–B) Estimating the fraction of brain cells recovered in single-cell suspensions generated from intact tissue. To aid in the identification of single cells, we used Nestin-Cre;lsl-ZsGreen mice in which the ZsGreen fluorophore is enriched in the soma of heterogenous set of brain cell classes.
(A) ZsGreen reporter fluorescence identifies single cells in suspension (left) and in intact Frontal Cortex (right, sagittal section).
(B) To approximate cell recovery rates from intact tissue, we estimated the total number of ZsGreen+ cells in a fixed volume of intact Frontal Cortex and Striatum using stereological counting and compared those starting totals to the number of cells estimated to be recovered in suspension (STAR Methods)(Tissue, n=2 mice, n=12–22 confocal stacks/mouse, n =108–181 sampling boxes/mouse; Suspension, n=5 mice, n=6 samples/suspension). Boxplot shows estimates per mouse (color-coded).
(C) Recovery fraction (total suspension / total tissue) by region. Error bars indicate standard error of the mean recovery fraction and account for error estimated in the tissue and suspension means.
(D–K) Addressing acquisition bias at the level of cell class, cell type and cell subtype. Biases in acquisition could result from at least two sources: 1) tolerance to dissociation and 2) transcript abundance, as larger libraries are more likely to be distinguished as cells from background (STAR Methods).
(D–F) Cell class acquisition bias. The fraction of neurons, astrocytes, oligodendrocytes/polydendrocytes and microglia were estimated in tissue using stereological counts ascertained from confocal stacks of tissue in which cell classes were labeled with or immunohistochemistry (IHC) or transgenes expressing GFP. Drop-seq estimates were made by calculating the total number of cells assigned to all Global clusters of a given class.
(D) Two sets of experiments (n=3 cell classes/experiment) were used to estimate the relative fraction of neurons, astrocytes, oligodendrocytes/polydendrocytes and microglia in intact tissue. Top, in the first set of experiments, GFP was expressed in astrocytes using the Aldh1l1-GFP line, neurons were stained with NeuN and oligodendrocytes/polydendrocytes were labelled with Olig1 (n=34 imaging fields from n=1 mouse). In the second set of experiments (bottom), the GFP driver line was replaced with Cx3cr1-GFP to target microglia and the IHC labels remained the same (n=36 and 32 imaging fields from n=2 mice).
(E) Box plot comparing cell class fractions per mouse estimated from intact tissue versus Drop-seq.
(F) Transcript abundance distributions differ by cell class. Box plot of single-cell transcript counts (log10) grouped and ordered by cell class. Neurons have larger Drop-seq libraries than non-neurons. Larger libraries could partially account for overrepresentation of neurons.
(G–I) Cell type acquisition bias. The ratio of excitatory (glutamatergic) to inhibitory (GABAergic) neurons was estimated using smFISH and high-throughput imaging. Drop-seq estimates were made by counting the number of cells within glutamatergic or GABAergic subclusters.
(G) Cocktails of ISH probes against genetic markers of glutamatergic (Slc17a6/7/8) and GABAergic (Gad1/2) neurotransmission identify excitatory and inhibitory neuron populations. Automated co-localization analysis performed on slide-scans identified n=487,003 neurons (n=3 mice).
(H) Boxplots of E/I ratios from ratio (10:1 vs 5:1) as measured by smFISH. individual mice estimated from smFISH and Drop-seq. On average, Drop-seq datasets exhibit twice the E/I
(I) Boxplots of single-cell transcript abundance by neuron type (log10). Excitatory neurons have larger transcript abundance distributions than inhibitory interneurons, which could contribute to the increased E/I ratio in Drop-seq datasets.
(K–M) Cell subtype acquisition bias. The ratio of major inhibitory neuron classes was estimated using smFISH and high-throughput imaging. Drop-seq estimates were made by counting the number of cells within subclusters enriched for one or more interneuron markers.
(K) ISH probes against interneuron markers Pvalb, Sst and Vip were used to identify interneuron subpopulations. Automated co-localization analysis performed on slide scans identified n=68,734 interneurons (n=3 mice).
(L) Boxplots of interneuron subtype fractions estimated from smFISH and Drop-seq. Drop-seq datasets are enriched for Vip+ interneurons at the cost of Pvalb+ and Sst+ interneurons.
(M) Boxplots of single-cell transcript abundance by interneuron subtype (log10). Larger libraries in Pvalb+ and Sst+ interneurons cannot explain their relative depletion compared to Vip+ interneurons.
Figure S5. Cell class based transcriptional comparison of cortical regions. Related to Figure 5.
(A) tSNE plot color-coded by the n=7 excitatory subclusters with greater than 3:1 cell representation skew from FC or PC (and P < 0.05, Barnard’s test, see Figure 5C). The subcluster number is shown, along with a colored dot to represent the skewed region (FC, salmon color; PC, blue).
(B) tSNE based gene plots (top) and ISH stains (bottom, Allen) for markers enriched in each of the skewed clusters. Arrows point to high expression areas. Equivalent data for subclusters 21 and 5 are shown in Figure 5D
Figure S6. Inferring the anatomical location of subclusters from the GP/NB Cluster 2 and SN/VTA Cluster 3 analysis using ISH patterns of marker genes. Related to Figure 6.
(A) tSNE color-coded by subclusters originating from subcluster analysis of GP/NB Cluster 2, which encodes mostly non-Striatal GABAergic and glutamatergic neurons.
(B) Subclusters color-coded by candidate anatomical regions.
(C) Dot plot illustrating the expression patterns of neurotransmitter marker genes and novel pairs of markers identified across all n=25 subclusters in this analysis. Dot diameter represents the fraction of cells within a subcluster where a transcript was counted. Color scale represents average single-cell scaled expression value (out of 100K UMIs, log10).
(D) ISH experiments (Allen) illustrating the spatial expression patterns of single, selective marker genes for each subcluster. Subclusters are grouped by inferred anatomical location.
(E–H) Same plots and organization as above, but for SN/VTA Cluster 3 that encodes mostly GABAergic and glutamatergic neurons of the SNr and VTA.
Figure S7. Molecular specializations within spiny projection neurons subtypes. Related to Figure 7.
(A–E) Identification and comparison of patch iSPN and dSPN transcriptomes.
(A) Color-coded subcluster assignments for striatum cluster 10 (dSPNs) and cluster 11 (iSPNs) displayed on tSNE plot.
(B) ICs encoding patch-specific transcriptional signals in dSPNs (IC 15) and iSPNs (IC 20). Left, IC cell-loadings on tSNE plots. Right, gene-loadings with top 10 genes displayed. A full description of biological ICs resulting from SPN subcluster analyses is presented in (Data S6).
(C) Identification of shared and distinct dSPN and iSPN patch-enriched genes. Scatterplot comparing gene loadings between patch ICs for dSPNs (IC 15) and iSPNs (IC 20). Genes with high loading in both ICs are hypothesized to be enriched in both patch iSPNs and dSPNs. This group includes described (Tac1) and previously undescribed (Tshz1) genes enriched in patch dSPNs and iSPNs. Genes that load strongly onto either IC are candidates for patch molecular specialization by SPN pathway. For example, among matrix SPNs, Asic4 was selectively expressed in the dSPNs but not iSPNs; among patch SPNs, however, Asic4 was expressed in both dSPNs and iSPNs. Thus Asic4 helps tailor iSPNs but not dSPNs to the patch habitat.
(D) ISH stains for Tac1 and Tshz1 on coronal sections of striatum (Allen). Arrowheads point to example patches.
(E) Expression tSNE plots for Asic4 and Necab1 illustrating patch enrichment exclusive to iSPNs and dSPNs, respectively.
(F–H) Identifying non-eSPNs in Cluster 13.
(F) Color-coded subcluster identities. Subclusters labels in black correspond to eSPNs; labels in red correspond to non-Striatal neurons
(G) Expression of SPN marker Ppp1r1b is enriched in eSPN subclusters.
(H) Non-eSPN subclusters express unique markers that suggest non-Striatal origins. Left, expression of markers on tSNE plots. Right, ISH stains for each marker gene (Allen). Top, Slc17a7 expression suggests subclusters 13-6, 13-7 and 13-8 originate from adjacent cortex. Middle, Cartpt expression suggests subcluster 13-9 originates from the Bed Nucleus of the Stria Terminalis. Bottom, Nfib expression suggests subcluster 13-10 originates from the Amygdala. Red arrows point to regions of interest.
(I) eSPNs and SPN populations that express the same pathway markers are transcriptionally distinct. Genome-wide gene mean expression comparisons between Drd1+ or Adora2a+ SPN and eSPN subtypes (log normal scale). Left, Drd1+ eSPN subclusters (13-1, 13-2, 13-3 vs Drd1+ SPN Cluster 10. Right, Adora2a+ eSPN subclusters (13-4 & 13-5) vs Adora2a+ Cluster 11. Differentially expressed genes are shown with larger, dark dots (>2 natural log fold difference and P<10−100, binomTest (Robinson et al., 2010) and total number listed above each plot.
Table S1. Preparation of region-specific single cell suspensions from acute brain tissue. Related to Figure 1.
Data Availability Statement
The accession number for the sequencing data reported in this paper is GEO: GSE116470. Processed sequencing files – including single-cell DGEs for each region and DGEs of cell populations based on subcluster assignments – are also available from the DropViz website (http://dropviz.org/).