

Abstract
While no genome-wide pharmacogenetics study has yet been published, the field of pharmacogenetics is moving towards exploratory, large-scale analyses of the interaction between genetic variation and drug treatment. The Drug Metabolizing Enzymes and Transporters (DMET) platform offers a standardized set of 1936 variants in 225 genes related to drug absorption, distribution, metabolism and elimination that is useful to scan the genome for previously unknown associations between variation in absorption, distribution, metabolism and elimination genes and pharmacokinetic and pharmacodynamic outcomes of drug treatment. The purpose of this review is to put the DMET platform into context within the current study designs that have been used in pharmacogenetics, and to explore the role that DMET has played – and will play – in future pharmacogenetics studies.
Keywords: DMET, genome-wide association study, pharmacogenetics, pharmacokinetics, toxicity
Pharmacogenetics & personalized medicine
Personalized medicine is the use of both a patient’s genotypic and phenotypic data to choose a treatment or therapy that will best help the patient by maximizing benefit and minimizing harm [1]. Thus, personalized medicine holds the promise of increasing the efficacious properties of pharmacotherapy, while minimizing adverse events [2]. Two general methods have historically been employed to determine which drug therapy is appropriate. The first is based on trial and error, where different first-line drugs are given until the most effective treatment is found. The second method employs standard treatments based on the diagnosis, so that patients with the same disease receive much the same treatment [1]. Personalized therapy can guide therapeutic decisions in order to select the appropriate therapy at the appropriate dose in order to mitigate the problems brought by a one-size-fits-all approach.
In addition to identifying the optimal therapy, personalized approaches to medicine may also be employed to determine the optimal dose of a drug. Traditionally, individualized dosing in medical oncology has been based on body surface area, although this method does not account for interindividual differences in pharmacokinetics (PK) and pharmacodynamics (PD) [3]. Phenotypic tests, such as the uracil breath test [4] and the erythromycin breath test [5], that measure an individual’s metabolism via key enzymatic pathways, have also been useful in optimizing dose before administration of certain drugs, while therapeutic dose monitoring can further determine the safest and most effective dose for an individual [1]. Incorporation of genetic information to inform clinical decisions is a rather new practice, but it may add to the aforementioned strategies in order to provide the clinician further information for both the choice of therapy and the dosage at which therapy should begin. It is hoped that such strategies will lead to the quicker identification of safer, more effective therapies and improve existing treatments [1].
Many approaches have been devised to determine associations between genetic variation and interindividual differences in drug pharmacokinetics and pharmacodynamics. These include both candidate gene approaches and the analysis of genes that paraticipate in drug-related pathways. However, exploratory approaches that scan large portions of the genome to determine the most strongly associated variants have been rare in pharmacogenetics [6–9]. The Affymetrix Drug Metabolizing Enzymes and Transporters (DMET) genotyping platform, which offers the ability to scan 1936 variants in 225 genes related to drug metabolism and disposition (see Box 1), has recently been introduced in experimental medicine. The purpose of this review is to put the DMET platform into context within the current study designs that have been used in pharmacogenetics and to explore the role that DMET has played – and will play – in future pharmacogenetics studies.
Box 1. Genes covered by the DMET platform.
Phase I enzymes
CYP1A1, CYP1A2, CYP1B1, CYP2A6, CYP2A7, CYP2A13, CYP2B6, CYP2B7, CYP2B7P1, CYP2C8, CYP2C9, CYP2C18, CYP2C19, CYP2D6, CYP2E1, CYP2F1, CYP2J2, CYP2S1, CYP3A4, CYP3A5, CYP3A7, CYP3A43, CYP4A11, CYP4B1, CYP4F2, CYP4F3, CYP4F8, CYP4F11, CYP4F12, CYP4Z1, CYP7A1, CYP7B1, CYP8B1, CYP11A1, CYP11B1, CYP11B2, CYP17A1, CYP19A1, CYP20A1, CYP21A2, CYP24A1, CYP26A1, CYP27A1, CYP27B1, CYP39A1, CYP46A1, CYP51A1
Phase II enzymes
ADH1A, ADH1B, ADH1C, ADH4, ADH5, ADH6, ADH7, ALDH1A1, ALDH2, ALDH3A1, ALDH3A2, CHST1, CHST2, CHST3, CHST4, CHST5, CHST6, CHST7, CHST8, CHST9, CHST10, CHST11, CHST13, COMT, DPYD, FMO1, FMO2, FMO3, FMO4, FMO5, FMO6, GSTA1, GSTA2, GSTA3, GSTA4, GSTA5, GSTM1, GSTM2, GSTM3, GSTM4, GSTM5, GSTO1, GSTP1, GSTT1, GSTT2, GSTZ1, MAOA, MAOB, NAT1, NAT2, NNMT, NQO1, SULT1A1, SULT1A2, SULT1A3, SULT1B1, SULT1C1, SULT1C2, SULT1E1, SULT2A1, SULT2B1, SULT4A1, TPMT, UGT1A1, UGT1A3, UGT1A4, UGT1A5, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, UGT2A1, UGT2B4, UGT2B7, UGT2B11, UGT2B15, UGT2B17, UGT2B28, UGT8
Transporters
ABCB1, ABCB4, ABCB7, ABCB11, ABCC1, ABCC2, ABCC3, ABCC4, ABCC5, ABCC6, ABCC8, ABCC9, ABCG1, ABCG2, ATP7A, ATP7B, SLCA13, SLC10A1, SLC10A2, SLC13A1, SLC15A1, SLC15A2, SLC16A1, SLC19A1, SLC22A1, SLC22A11, SLC22A12, SLC22A14, SLC22A2, SLC22A3, SLC22A4, SLC22A5, SLC22A6, SLC22A7, SLC22A8, SLC28A1, SLC28A2, SLC28A3, SLC29A1, SLC29A2, SLC5A6, SLC6A6, SLC7A5, SLC7A7, SLC7A8, SLCO1A2, SLCO1B1, SLCO1B3, SLCO2B1, SLCO3A1, SLCO4A1, SLCO5A1
Other
ABP1, AHR, AKAP9, ALB, AOX1, ARNT, ARSA, CBR1, CBR3, CDA, CES2, CROT, DCK, EPHX1, EPHX2, FAAH, G6PD, HMGCR, HNMT, MAT1A, METTL1, NR1I2, NR1I3, NR3C1, ORM1, ORM2, PNMT, PON1, PON2, PON3, POR, PPARD, PPARG, PTGIS, RALBP1, RPL13, RXRA, SEC15L1, SERPINA7, SETD4, SPG7, TBXAS1, TPSG1, TYMS, VKORC1, XDH
DMET: Drug Metabolizing Enzymes and Transporters. Modified from [11].
Absorption, distribution, metabolism & excretion
So far many of the most relevant allelic variants in drug treatment have been in the genes encoding enzymes and transporters involved in drug absorption, distribution, metabolism and excretion (ADME). Enzymes involved in the biotransformation of substrates are classified as phase I or phase II. Phase I enzymes catalyze hydrolysis, reduction and oxidation reactions, and phase II enzymes catalyze conjugation reactions such as sulfation, acetylation and glucuronidation [10]. The majority of phase I reactions are catalyzed by the cytochrome P450 (CYP) enzymes [11]. There are 18 families of CYPs, that are further divided into 44 subfamilies consisting of 57 total genes. However, only three of those families, CYP1, CYP2 and CYP3, catalyze most phase I reactions of drugs [2]; over 75% of prescribed drugs are metabolized at least in part by three subfamilies, CYP3A, CYP2D6 and CYP2C [12]. Unlike most phase I reactions, phase II reactions typically significantly enable the excretion of drugs by considerably increasing the hydrophilicity of the substrate or deactivate highly reactive species [10]. Key phase II enzymes include N-acetyltransferases 1 and 2 (NAT1 and NAT2), thiopurine S-methyltransferase (TPMT), and the uridine disphosphate glucoronosyltransferase (UGT) family; polymorphisms in these genes have been shown to have clinical implications for a variety of conditions [1].
Transporters play a critical role in ADME and, as such, must be considered in combination with metabolic enzymes. Although some drugs may passively diffuse across membranes, many drugs are effluxed and/or influxed via active transport or facilitated diffusion; thus, transporters affect drug uptake, bioavailability, targeting, efficacy, toxicity and clearance [13]. Two types of transport superfamilies, ATP-binding cassette (ABC) proteins and solute-linked carrier (SLC) proteins, are responsible for the majority of drug transport, although it is important to note that these are also involved in the transport of many endogenous substrates [1]. ABC transporters are generally efflux pumps [13], while SLC proteins are typically influx transporters that mediate facilitated diffusion of their substrates [14]. Many transporters have a broad range of substrates; ABCB1, also known as P-glycoprotein and MRD1, transports several classes of drugs, including anticancer agents, antibiotics, immunosuppressants and statins [13].
While understanding genetic variation in ADME genes is essential in personalizing therapy, consideration of these genes alone is not sufficient. Polymorphisms in genes not directly responsible for drug metabolism or transport, such as drug targets and nuclear receptors, may also affect a patient’s response to treatment. For example, there is some evidence that an EGFR SNP found in tumor tissue may lead to better overall survival in patients with colorectal cancer receiving irinotecan and cetuximab, an anti-EGFR antibody [15]. Also, it has been shown that patients with varying numbers of repeats in the promoter region of ALOX5 do not respond as well as wild-type individuals to treatment with an ALOX5 inhibitor as an anti-asthma treatment [16]. The nuclear receptors pregnane X receptor (PXR) and consititutive active/androstane receptor both play critical roles in drug response by binding a wide range of xenobiotics and regulating expression of many ADME genes, including transporters and phase I and phase II enzymes [17]. Genes regulated by PXR include ABCB1, ABCC2, CYP2C8, CYP3A4, UGTs and sulfotransferases (SULTs). Many SNPs in PXR are associated with altered expression levels of both PXR and downstream genes such as CYP3A4 [18].
To truly personalize medicine, genetic data must be evaluated in the context of the individual; gene–gene, gene–drug and gene–environment interactions all influence the course of a disease, including response to treatment. In one study investigating the pharmacogenetics of bronchodilator drug response in different ethnic populations, no single SNP investigated in the study correlated with response. However, when considered together, SNPs in both IL-6 and IL-6R consistently correlated with drug response, suggesting gene–gene interaction [19]. Also, SNPs in genes with regulatory functions may affect downstream genes, such as PXR and CYP3A4 expression levels, as discussed previously. There is a plethora of gene–drug interactions in the literature, many with significant clinical implications. For example, individuals homozygous for the CYP2C9*3 allele are only 10% as efficient as wild-type individuals in warfarin clearance and, because of the drug’s narrow therapeutic index, are four-times more likely to experience a bleeding complication [1]. Also, a form of para-aminobenzoic acid, a compound found in sunscreens, has been shown to irreversibly inactivate NAT1 [20]. In addition to gene–gene and gene–drug interactions, gene–environment interactions affect disease development. Environmental factors can be found from the cellular level, such as epigenetic modifications and different intracellular components in various cell types, to the organismal level, such as diet and behavior. In nonpolarized CD41 TH2 cells, a SNP in the promoter regions of IL-13, a key component in the asthma inflammation cascade, is associated with decreased expression levels; however, the same SNP is associated with increased expression in polarized CD41 TH2 cells [21]. Further, there is evidence that individuals with certain polymorphisms in 5-lipoxygenase (5-LO) are at higher risk for developing atherosclerosis than wild-type individuals if they consume high amounts of arachidonic acid. However, the proatherogenic effect of the polymorphisms diminishes as the amount of arachidonic acid consumed decreases [22]. Thus, an individual’s environment modifies the effects of genetic variations, and gene–gene, gene–drug and gene–environment interactions must be considered in pharmacogenetics studies.
Candidate gene, pathway-based & genome-wide pharmacogenetics approaches
While many methodologies have been employed to characterize the pharmacogenetics of various agents, studies are typically designed in three different ways: candidate gene approaches, pathway-based approaches and genome-wide analyses [23]. Each type of technique is useful in certain contexts, although each is also limited in certain ways. A brief overview is provided below.
Most pharmacogenetics studies have employed the ‘candidate gene approach’ to detect associations between known SNPs and clinical or pharmacological end points. This practice continues to yield promising results. The first such studies utilized candidate variants investigating 6-mercaptopurine toxicity in individuals carrying certain low-activity TPMT variants [24–26]. Other examples include the interaction between irinotecan and UGT1A1 [27–30], thymidylate synthase in 5-fluorouracil treatment [31,32], DNA repair genes in cisplatin treatment [33–38], CYP2D6 variants and tamoxifen efficacy [39], and numerous other studies. Perhaps the most useful aspects of the candidate gene approach are its hypothesis-driven nature and the fact that studies can often be scaled to acquire sufficient statistical power. However, this methodology has been rather inconsistent in validating genetic markers, especially in cases where allelic variants are not highly penetrant, making the results of these studies difficult to interpret [23]. In addition, much focus has been placed on SNPs that are perhaps irrelevant to drug treatment. For example, much research has been conducted to determine if variants in CYP3A4, a liver detoxifying enzyme involved in the metabolism of roughly 50% of marketed drugs [12], are also associated with drug pharmacokinetics and pharmacodynamics [12,40]. These studies have been very inconsistent, and much effort has been expended while few clear-cut clinical applications are apparent. Overall, the candidate gene approach is useful for studying cases where there is a major drug metabolism or target gene that has a polymorphism that significantly changes its function [23].
The genome-wide approach is useful to determine the most significant SNPs associated with a phenotype amongst a high-density set of polymorphisms. However, this approach suffers from being discovery-driven rather than hypothesis-driven, resulting in weak statistical signals and false positives (i.e., Type I error) [23]. Given the number of hypotheses evaluated, such approaches are nearly impossible to scale for sufficient statistical power, and given the sample size, cost and computing power required for these studies, they are impractical in many cases. Nonetheless, testable hypotheses can be formulated to validate and characterize useful candidates identified from genome-wide studies, and validation sets are often used to detect similar signals in two separate cohorts, thus decreasing the impact of Type I error [41]. For this reason, genome-wide studies require a two-stage design where discoveries are made using a high-density SNP array and are then validated using additional patient sets and a more hypothesis-driven approach. The genome-wide approach is most useful to discover hitherto unknown SNP associations where prior knowledge (i.e., mechanism, inheritance pattern, protein interactions and so on) is not available. These approaches have primarily led to the identification of risk variants in certain diseases. Examples of such associations include the 8q24 variant in prostate malignancies and the TERT-CLPTM1L locus in lung cancer [42–44]. No pharmacogenetics study has yet been completed using a true genomewide approach, as fewer patients are available to study. However, opting for a lower resolution genome-wide approach that is appropriate for the sample size of most pharmacogenetics studies may be more useful to identify potentially important gene regions in the genome for fine mapping in future pharmacogenetics studies [23].
The pathway-based approach, which utilizes foreknowledge of both genetic variants, genes and the pathways that these genes participate in, has been particularly useful in identifying and characterizing pharmacogenetics end points given that studies are conducted to test the interaction between genes, rather than assuming that each SNP confers a monogenic trait [23]. However, the incorporation of interaction testing has required the utilization of machine learning techniques, and these techniques can often be complex and require larger sample sizes than candidate gene approaches. Moreover, validation of gene–gene interactions is often difficult because a fundamental understanding of the biology of the interactions is required, but the current knowledge base is often incomplete [45]. Examples of pathway-based approaches include the study of VKORC1 and CYP2C9 polymorphisms in warfarin treatment, polymorphic variation in DNA repair pathways (i.e., ERCC1, XPD, XRCC1) in DNA damaging chemotherapy, and the study of multiple polymorphisms in the docetaxel metabolism and elimination pathway [46].
Available genotyping technologies that are designed to detect associations between polymorphisms and drug ADME or outcome have primarily been pathway-based; that is, they determine the genotype at multiple loci from drug-metabolizing enzymes, transporters, targets and the cellular machinery that regulates expression of these factors. However, most of the available platforms are small-scale, only evaluating genetic variants in a small subset of genes [11]. The Affymetrix DMET platform was designed to capture a larger subset of 1936 SNPs in 225 genes that are known to contribute to drug metabolism, and in this way, the DMET platform can be thought of as a low- to midscale pathway analysis genotyping platform that allows for a more comprehensive exploration of pharmacogenetics associations within a large number of known ADME pathways.
DMET
Selection of genes, genetic markers & mode of operation
The gene selection of DMET was primarily based on a core list of genes identified by major academic, pharmaceutical industry and genomic technology representatives participating in the PharmaADME consortium [101] on the basis that many of these genes may be involved in ADME properties of drugs. The consortium ranked over 9000 SNPs and many complex mutations within these genes (i.e., triallelic markers, small indel mutations, gene conversion and/or whole deletion alleles) according to clinical research utility. Currently, PharmaADME genes represent 95% (45/47) of the phase I enzymes, 93% (74/80) of the phase II enzymes, 98% (51/52) of the transporters, and 52% (24/46) of ‘other genes’ on the DMET array (see Box 1). The DMET panel was modified from this prioritized marker set to include 31 additional genes (i.e., 225 genes total), mostly comprising genes that regulate intracellular processes that facilitate ADME (i.e., scaffolding proteins, nuclear receptors, serum binding proteins and so on). We have included a list of selected genes on the DMET platform and corresponding selected drug pathways to highlight the platform’s wide-ranging pharmacologic significance (see Box 1). The genes presented were selected by their ‘VIP’ status on PharmGKB [102], and the selected drug pathways listed are also those identified by PharmGKB. It should be noted that only selected associated drug pathways are presented here; we did not include endogenous compounds or nontherapeutic xenobiotics.
The DMET platform is designed to capture genetic variants important in the ADME properties of drugs, and since many types of genetic variants may influence ADME, the panel consists of both common and rare variants. This is in contrast to genotyping arrays that select alleles that are useful to define genomic regions where a causative genetic variant may exist. The DMET array interrogates several types of markers, including copy-number variations, insertions/deletions, biallelic and triallelic SNPs. While quality testing of genotyping arrays is usually accompanied by less than 100% representation due to inappropriate SNP detection, or gene homology, the DMET platform is quite comprehensive; often it includes more variants than were previously investigated in other studies. Recently, Affymetrix has added additional content relevant to drug ADME, and a tool to identify haplotypes amongst 779 polymorphisms in a core set of 61 genes identified by the PharmaADME consortium to be of high-importance in drug metabolism. Moreover, the platform was used to identify additional haplotypes that were not observed in populations previously explored by the HapMap project. Further information regarding DMET coverage and haplotype determination is published online [103,104]. Finally, a brief overview of the mode of operation of the platform is provided in Figure 1.
Figure 1. Mode of operation for the DMET array.
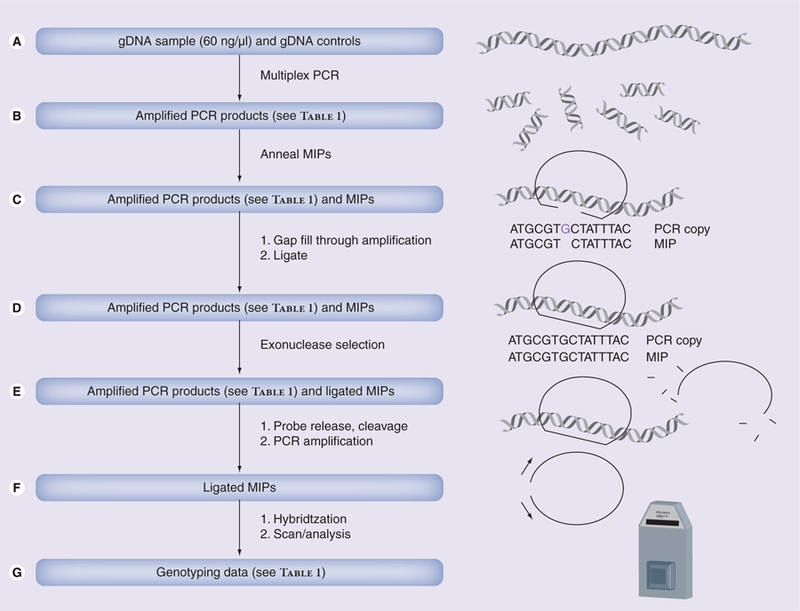
Data is generated by (A) amplifying genomic DNA (60 ng/µl) and controls at regions of interest using multiplex PCR. (B) Heat denaturing a mixture of PCR products and MIPs, and bringing the mixture to annealing temperature such that MIPs anneal at SNP sites. (C) Adding thermostable DNA polymerase, unlabeled dNTPs and ligase to extend the MIP probes and ligate them into circular form. (D) Adding exonuclease to eliminate any unligated MIPs. (E) Releasing the MIP probe from the DNA, cleaving the probes at common cleavage sites, and amplifying the remaining MIPs with labeled dNTPs. (F) Hybridizing the MIPs to the DMET array and reading the signal emitted from the labeled, hybridized MIP probes, followed by (G) interpreting the signal to generate genotyping data.
DMET: Drug Metabolizing Enzymes and Transporters; gDNA: Genomic DNA; MIP: Molecular inversion probe.
Applications of DMET in the literature
The DMET platform has now been used by several investigators to conduct correlative pharmacogenetic research. We used the DMET platform to determine if interindividual genetic variation was associated with the efficacy and toxicity of docetaxel-based therapy alone (n = 14) or in combination with thalidomide (n = 33) in men with advanced prostate cancer [47,48]. This exploratory study determined that ten SNPs in three genes (PPAR-δ, SULT1C2 and CHST3) were related to therapeutic response, while 11 SNPs in seven genes (SPG7, CHST3, CYP2D6, NAT2, ABCC6, ATP7A, CYP4B1 and SLC10A2) were related to toxicity, and some associations were restricted to only patients receiving the combination (i.e., PPAR-δ) [48]. As is the case with many pharmacogenetics studies that apply genotyping data in many genes to a small cohort of patients, the results may contain false positives, are difficult to explain, and are not generalizable to docetaxel-based therapy. Nonetheless, this study identified many novel candidate genes that may be related to docetaxel and/or thalidomide therapy, and studies are underway to validate and characterize these findings.
Caldwell et al. applied the DMET genotyping platform to identify a single polymorphism in CYP4F2 that was very strongly related to warfarin dose requirements in a relatively large cohort of patients being treated at the Marshfield Clinic (WI, USA) (n = 436) [49]. The investigators then validated the initial finding in three sets of individuals treated at the same institution (n = 61), at Washington University in St Louis, MO, USA (n = 269), and the University of Florida, FL, USA (n = 295). Interestingly, CYP4F2 increased the predictive power of clinical variables and CYP2C9 and VKORC1 genotypes that are sometimes used as predictors of warfarin dose [49,50].
Finally, Mega et al. have utilized DMET technology to investigate a cohort of healthy individuals (n = 162) and a cohort of patients being treated with clopidogrel, an antiplatelet agent, for a previous myocardial infarction (n = 1477) [51]. The primary end points included comparison of a specific set of 54 variants in six genes (CYP2C19, CYP2C9, CYP2B6, CYP3A4, CYP3A5 and CYP1A2) versus plasma concentration of the active metabolite of clopidogrel in healthy individuals, and platelet inhibition response and adverse events in patients who had experienced a myocardial infarction. This study found that carriers of reduced-function CYP2C19 alleles had significantly lower levels of the active metabolite, decreased platelet aggregation inhibition and an increased number of adverse events. Mega et al. also determined that those carrying reduced-function CYP2B6 alleles exhibited lower plasma concentrations of the active metabolite and reduced inhibition of platelet aggregation. These data have led the US FDA to review if CYP2C19 genotyping should be conducted prior to starting antiplatelet therapy.
Published genome-wide association studies versus DMET
There are few examples of successful genomewide association studies (GWAS) that have studied clinical pharmacology. While a preponderance of factors have led to this problem, it has historically been due to low sample sizes in drug trials, large degrees of interindividual variability in clinical outcome measures, and the polygenetic basis for variation in PK/PD. As such, successful GWAS in clinical pharmacology have demonstrated some common characteristics where few variants with high penetrance have withstood the strict multiple-testing requirements of such studies, and these variants were associated with well-defined adverse drug reaction (ADR) clinical end points. Here we highlight three studies that have successfully applied a GWAS approach in clinical pharmacology.
The first study evaluated 550,000 polymorphisms in 181 patients receiving warfarin, along with two replication populations of similar patients (n = 374). The study identified previously reported variants in CYP2C19 and VKORC1 in association with warfarin dose requirements, and the authors concluded that only CYP2C9 and VKORC1 variants had effect sizes that were large enough to be discovered by GWAS [6]. Another study applied a 300K marker array to compare 85 individuals with definite or incipient myopathy following therapy with simvastatin and 90 controls from a clinical trial consisting of 12,000 participants. Replication was conducted in a study consisting of 20,000 participants. This study revealed that a single common SNP in SLCO1B1 (~15% allele frequency) was strongly related to the development of myopathy (odds ratio: 17.4 for homozygous variants), and that this SNP was in near complete linkage with another SNP that was previously linked to statin pharmacokinetics [8]. Interestingly, both of the afore-mentioned studies were confirmatory and did not reveal novel SNPs worthy of further exploration. Another study determined that the HLA-B*5701 genotype is involved in drug-induced liver injury (DILI) following flucloxacillin after interrogating 866,399 markers in 51 cases of DILI and 282 sex- and ancestry-matched controls, followed by a second cohort of 23 cases and 64 controls [7]. This study identified a region in chromosome 6 where several associations with DILI were noted, the strongest of which (HLA-B*5701) conferred an 80- to 100-fold increase in DILI risk.
It is perhaps not surprising that most successful genome-wide pharmacogenetics studies have investigated ADRs, since in ADR research it is assumed that genetic predictors of risk have high penetrance [9], and this has been borne out in the above three examples. However, ADR studies are still limited, since some ADR traits are polygenic, have low penetrance or are dependent on rare variants that are very difficult to detect when only a section of the population will experience an ADR. Moreover, this selection begs the question whether or not confounding factors influence the results, given that early clinical trials are seldom randomized.
Thus, while it is exceedingly difficult to hypothetically compare study designs that depend on a multitude of factors, the previously reported literature suggests that DMET has higher power to detect associations, but may also miss clinically meaningful associations. Although some have quantified the power of different GWAS designs via simulation with some interesting results [9], a direct comparison of power is difficult to ascertain given the number of variables that must be taken into account. For example, comparisons of GWAS versus DMET would be difficult to understand without specific parameter values in a study of A outcome(s), for which each of B SNPs, with common allele frequency C, has penetrance D% would have E% power to detect at least F of the SNPs in a population of size G, where each individual SNP is tested at a Type I error level of H. Additionally, DMET samples variants that may alter drug effect, while GWAS arrays are typically designed to capture areas of genetic linkage to identify chromosomal regions; thus, the two technologies evaluate two different types of genetic markers. However, it is clear that since DMET interrogates fewer variants, there is a significant power advantage (α/n = 2.5 × 10−5) over large-scale genotyping arrays (α/n = 10−6 for 50K SNPs, and α/n = 10−7 for 500K SNPs), although power can be optimized in some cases depending on study design [9]. Studies in DMET have revealed clinically meaningful results in fewer patients, and this is most likely due to the power advantage in selecting a pathway of genes involved in ADME to examine clinical pharmacology end points. It is also clear that while several investigators have examined warfarin dose requirements via genome-wide approaches [6], the association with CYP4F2 had been missed until DMET was applied, perhaps because the effect size was smaller than that of VKORC1 and CYP2C9 [49]. Moreover, studies by Mega et al. have demonstrated that the DMET array can be applied to actual pharmacokinetics end points where multiple genes influence interindividual variation [51]. However, DMET would not have identified some of the aforementioned the HLA-B*5701 determined by the GWAS approach.
In summary, it appears that GWAS are particularly useful in cases where there are a large number of patients to evaluate, clinical end points are simple to define without variability, and the genetic factors associated with the end points are highly penetrant. The DMET approach is also useful in the aforementioned scenarios, although it may miss highly significant, causative genetic variants. However, DMET is particularly useful for exploration of clinical pharmacology in smaller patient populations that have more variable end points and polygenetic traits. Ultimately, the individual investigator must weigh the objectives of the study and the samples that are available in order to determine which platform is appropriate on a study-by-study basis.
Strengths of DMET
The DMET platform has therefore already proven useful in investigating alleles with known ADME properties and has been applied to studies that have purely investigated pharmacodynamic end points, therapeutic dose monitoring and pharmacokinetics. While DMET could certainly be used in several types of investigations (i.e., risk studies, detecting differences in genotype distributions amongst racial backgrounds and so on), perhaps the main strength of the technology will be its application in early studies evaluating clinical pharmacology end points in clinical trials of investigational agents [11]. While the field is in its infancy, exploratory pharmacogenetic profiling would improve the success of many early clinical trials, especially in agents with narrow therapeutic indices (i.e., many anticancer agents), in order to understand interindividual differences in drug efficacy, toxicity and pharamacokinetics [1]. Such testing may identify subgroups of nonresponders and toxic responders that should be treated differently than the rest of the population, in addition to identifying appropriate doses based on genetic makeup.
The DMET platform seems to be particularly suited for this application for several reasons. First, the DMET technology allows for a hypothesis-driven pathway-based approach that is also exploratory in nature. In this way, the DMET technology can pare down the number of alleles that are investigated in a single study, thereby reducing Type I error from multiple testing. So far, the field of pharmacogenetics has not produced a definitive study exploring all or most of the SNPs in the human genome versus drug outcome. The primary reason for this is the number of patients required for such a study is very large, requiring collaborations across many institutes, and is also very expensive. The DMET platform is the first of its kind to investigate such a large number of SNPs in clinical pharmacology (albeit some GWAS have investigated significant ADRs), and as can be seen from initial investigations applying the technology, the number of patients required to generate valid candidates is much lower.
Another benefit of the DMET platform is that it can allow a direct comparison of the same set of alleles across multiple cohorts of individuals receiving the same drug or drugs under different circumstances. This has the potential to greatly benefit drug development and clinical trials investigating new agents and new uses for established agents. The candidate gene approach has led to a number of disparate SNPs that have been related to multiple end points in widely varying populations with multiple ethnic backgrounds [1]. This has significantly muddied the waters of the literature, and has made it difficult to determine which SNPs and haplotypes are truly associated with a given phenotype, and to what extent [1]. Moreover, candidate gene and small-scale pathway-based approaches are not conducive to understanding unknown pathways that may influence the PK/PD of investigational agents without significant representation in the literature. The DMET platform can overcome both of the aforementioned issues. Applying the DMET technology in clinical investigations could identify the most penetrant SNPs that are consistently associated with effects across a wide range of drugs that interact with similar pathways and determine if a given SNP is important in studies investigating multiple applications of the same drug. The DMET platform would also potentially identify polymorphisms that explain drug PK/PD in early Phase I clinical trials, and that information could be used to determine the recommended dose for later Phase II and Phase III trials. Thus, it could lead to better trial design and a lower attrition rate for new agents that do not work well in the general population, but significantly benefit a smaller percentage of individuals [11].
Weaknesses of DMET
One major obstacle to utilizing the DMET platform in prospective studies that will employ the DMET technology in order to inform clinical decisions is that the technology is not yet FDA approved, or Clinical Laboratory Improvement Amendments (CLIA) certified (although CLIA certification may be realized within the next year). While this would not hinder using the platform in the context of drug development to find novel genetic markers that are related to drug treatment, and then validate those markers using FDA-approved methods, FDA approval would be required in order to use the DMET platform to conduct large-scale genotyping in known markers to make dose adjustments and other decisions regarding drug treatment. For example, one could use DMET to conduct a prospective study to identify new markers that predict tamoxifen treatment efficacy, but one could not use DMET to make dose adjustments or recommend alternative therapies based on CYP2D6 genotypes where the FDA has recommended genotyping efforts prior to drug administration [11]. Thus, the DMET technology only applies in experimental, but not clinical applications of personalized medicine.
Another weakness of the DMET technology is that it does not include polymorphisms in many drug targets or in genes that are related to environmental exposures that could influence drug metabolism. Targeted therapy is becoming more and more prevalent as newer drugs are being developed, and medicine is moving away from drugs that cause indiscriminant effects in the body [52]. Although the DMET platform is not designed for the purpose of evaluating drug targets (albeit there are some molecular targets on DMET, such as VKORC, that are not apparent from the name of the platform), DMET will not be sufficient to evaluate many alleles that may influence PK/PD, and separate studies will be required. This could be impractical in some cases where variation in a target is more important than the genetic influence on ADME properties. For example, genetic variation in the VEGF pathway may influence the therapeutic outcome of treatment with antiangiogenesis agents (e.g., bevacizumab) [53], and this would not be detected by DMET.
Next, while the DMET technology significantly limits Type I error due to exploration in a large set of polymorphisms, it will still be difficult to translate the results to inform clinical decisions, and preclinical studies and large validation sets are still required to carry the results of DMET from the bench to the bedside. Indeed, all of the studies that have evaluated patients with DMET so far [48,49,51] have many validation steps that are required to translate the results of the DMET study. Early clinical trials are typically very small in size, and the DMET would therefore be impractical in many Phase I studies. This is not a shortcoming of DMET specifically, but is a shortcoming of scaling up genotyping studies in early clinical trials. Perhaps opting for smaller genotyping platforms may be more desirable in many cases, and the trade-off between more SNPs for better coverage versus less SNPs for more translatable results needs to be clarified by experts in the fields of genomics applications in clinical pharmacology and pharmacogenomics. Moreover, the DMET technology is not customizable, and technologies such as Illumina (CA, USA) can accomplish the same end as DMET with variants selected by the investigator for the purposes of a particular study.
Conclusion
The DMET platform represents a significant step forward in scaling up pathway-based pharmacogenetics studies, although it is still a relatively new addition to the armamentarium of personalized medicine. At this point, the platorm has been utilized to its full extent in two studies [11,49], and has been used to focus on several specific genes in another study [51]. In each type of study design, it has yielded several previously unknown associations between polymorphisms and therapy with widely used drugs (i.e., docetaxel, warfarin and clopidogrel, respectively) that were then validated in separate cohorts. Thus, the DMET platform represents an exploratory, pathway-based platform that scans the genome for SNPs and haplotypes in ADME genes that may correlate with interindividual variation in drug treatment. Like the genomewide approach, it offers a more comprehensive analysis of the genome, but lessens the possibility of Type I error associated with GWAS. It is also similar to the pathway-based approach in that it can be used to test certain hypotheses, but is less likely to overlook an important variant. To our knowledge, the DMET platform is the only such technology that crosses over both study designs, with the exception of Illumina technology that allows for moderate-throughput genotyping in a similar number of customizable SNPs [11]. However, the DMET technology has the potential to standardize pharmacogenetics studies, and the SNP set is curated such that the major SNPs in biologically relevant ADME genes are covered. The DMET platform also has an allele translation tool that is useful to determine which haplotypes are most important in drug treatment [103]. While future studies will determine the ultimate utility of the DMET platform, it is hoped that the technology will improve clinical therapy by unlocking many of the genetic-based mysteries of pharmacokinetic and pharmacodynamic variation in both investigational and well-used pharmacological agents alike.
Table 1.
Selected genes covered by the DMET platform and selected associated drug pathways.
| Gene | Selected associated drug pathways |
|---|---|
| Phase I enzymes | |
| CYP2A6 | Coumarin |
| Sm-12502 | |
| Tegafur | |
| CYP2B6 | 17-α-ethynylestradiol |
| Artemisinin | |
| Bupropion | |
| Clopidogrel | |
| Cyclophosphamide | |
| Diazepam | |
| Efavirenz | |
| Ifosfamide | |
| Ketamine | |
| Methadone | |
| Meperidine | |
| Mephenytoin | |
| Midazolam | |
| Nevirapine | |
| Propofol | |
| Selegiline | |
| Tamoxifen | |
| Thiotepa | |
| Ticlopidine | |
| CYP2C9 | Celecoxib |
| Cyclophosphamide | |
| Flurbiprofen | |
| Fluvastatin | |
| Glipizide | |
| Ibuprofen | |
| Ifosfamide | |
| Indomethacin | |
| Lornoxicam | |
| Phenytoin | |
| Raldecoxib | |
| Tolbutamide | |
| Warfarin | |
| CYP2C19 | Amitriptyline |
| Carbamazepine | |
| Carisoprodol | |
| Chloramphenicol | |
| Cimetidine | |
| Citalopram | |
| Clomipramine | |
| Clopidogrel | |
| Cyclophosphamide | |
| Felbamate | |
| Fluoxetine | |
| Fluvoxamine | |
| Hexobarbital | |
| Imipramine | |
| Indomethacin | |
| Ketoconazole | |
| Lansoprazole | |
| Lansoprazole | |
| Mephenytoin | |
| Mephobarbital | |
| Moclobemide | |
| Modafinil | |
| Nelfinavir | |
| Nilutamide | |
| Omeprazole | |
| Oxcarbazepine | |
| Pantoprazole | |
| Phenobarbitone | |
| Phenytoin | |
| Primidone | |
| Probenecid | |
| Progesterone | |
| Proguanil | |
| Propranolol | |
| Rabeprazole | |
| Rifampin | |
| Teniposide | |
| Ticlopidine | |
| Topiramate | |
| Warfarin | |
| CYP2D6 | Amitriptyline |
| Atomoxetine | |
| Carvedilol | |
| Chlorpheniramine | |
| Chlorpromazine | |
| Citalopram | |
| Clomipramine | |
| Clozapine | |
| Codeine | |
| Debrisoquine | |
| Desipramine | |
| Dextromethorphan | |
| Dihydrocodeine | |
| Doxepin | |
| Flecainide | |
| Fluoxetine | |
| Fluvoxamine | |
| Gefitinib | |
| Haloperidol | |
| Imipramine | |
| Maprotiline | |
| Metoprolol | |
| Mexiletine | |
| Mianserin | |
| Morphine | |
| Nortriptyline | |
| Paroxetine | |
| Perhexiline | |
| Perphenazine | |
| Propafenone | |
| Risperidone | |
| Sparteine | |
| Tamoxifen | |
| Thioridazine | |
| Timolol | |
| Tolterodine | |
| Tramadol | |
| Yohimbine | |
| Zuclopenthixol | |
| CYP3A4/5 | Alfentanil |
| Alprazolam | |
| Amlodipine | |
| Aripiprazole | |
| Astemizole | |
| Atorvastatin | |
| Buspirone | |
| Cafergot | |
| Cerivastatin | |
| Chlorpheniramine | |
| Cilostazol | |
| Cisapride | |
| Clarithromycin | |
| Codeine | |
| Ciclosporin | |
| Dapsone | |
| Dextromethorphan | |
| Diazepam | |
| Diltiazem | |
| Docetaxel | |
| Domperidone | |
| Eplerenone | |
| Erythromycin | |
| Estradiol | |
| Felodipine | |
| Fentanyl | |
| Finasteride | |
| Gleevec | |
| Haloperidol | |
| Hydrocortisone | |
| Indinavir | |
| Irinotecan | |
| Laam | |
| Lercanidipine | |
| Lidocaine | |
| Lovastatin | |
| Methadone | |
| Midazolam | |
| Nateglinide | |
| Nelfinavir | |
| Nifedipine | |
| Nisoldipine | |
| Nitrendipine | |
| Ondansetron | |
| Pimozide | |
| Progesterone | |
| Quinine | |
| Ritonavir | |
| Salmeterol | |
| Saquinavir | |
| Sildenafil | |
| Simvastatin | |
| Sirolimus | |
| Tacrolimus (FK506) | |
| Tamoxifen | |
| Taxol | |
| Telithromycin | |
| Terfenadine | |
| Terfenidine | |
| Testosterone | |
| Trazodone | |
| Triazolam | |
| Verapamil | |
| Vincristine | |
| Zaleplon | |
| Zolpidem | |
| Phase II enzymes | |
| ALDH1A1 | Cyclophosphamide |
| Ifosfamide | |
| Retinaldehyde | |
| COMT | Levodopa |
| DPYD | 5-fluorouracil |
| GSTP1 | Cisplatin |
| Cyclophosphamide | |
| Doxorubicin | |
| Etoposide | |
| Oxaliplatin | |
| Pyrimethamine | |
| NQO1 | Cisplatin |
| Dicumarol | |
| Doxorubicin | |
| TPMT | 6-mercaptopurine |
| 6-thioguanine | |
| UGT1A1 | Atazanavir |
| Etoposide | |
| Irinotecan | |
| Tamoxifen | |
| Transporters | |
| ABCB1 | Amitriptyline |
| Atorvastatin | |
| Clopidogrel | |
| Ciclosporin | |
| Daunorubicin | |
| Digoxin | |
| Doxorubicin | |
| Etoposide | |
| Exofenadine | |
| Indinavir | |
| Irinotecan | |
| Loperamide | |
| Paclitaxel | |
| Rhodamine 123 | |
| Ritonavir | |
| Saquinavir | |
| Tacrolimus | |
| Talinolol | |
| Topotecan | |
| Verapamil | |
| Vinblastine | |
| SLC19A1 | 10-edam |
| 5,10-dideazatetrahydrofolate | |
| Gw1843u89 | |
| Leucovorin | |
| Methotrexate | |
| Pemetrexed | |
| Raltrexed | |
| SLCO1B1 | Atorvastatin |
| Atrasentan | |
| Benzylpenicillin | |
| Bosentan | |
| Caspofungin | |
| Cerivastatin | |
| Clotrimazole | |
| Ciclosporin A | |
| Enalaprilat | |
| Irinotecan | |
| Lovastatin | |
| Methotrexate | |
| Mifepristone | |
| Olmesartan | |
| Paclitaxel | |
| Pioglitazone | |
| Pitavastatin | |
| Pravastatin | |
| Repaglinide | |
| Repaglinide | |
| Rifampicin | |
| Rifamycin SV | |
| Rosiglitazone | |
| Rosuvastatin | |
| Temocapril | |
| Troglitazone | |
| Troglitazone | |
| Valsartan | |
| Rifampicin | |
| Rifamycin SV | |
| Rosiglitazone | |
| Rosuvastatin | |
| Temocapril | |
| Troglitazone | |
| Troglitazone | |
| Valsartan | |
| Other | |
| HMGCR | Pravastatin |
| Atorvastatin | |
| Fluvastatin | |
| Lovastatin | |
| Simvastatin | |
| PTGIS | Celecoxib |
| TYMS | 5-fluoruracil |
| Gemcitabine | |
| Methotrexate | |
| Tomudex | |
| VKORC1 | Acenocoumarol |
| Coumarin | |
| Warfarin | |
Genes were selected if they were identified as VIP genes, and were related to the ADME properties, or pathways of therapeutics by PharmGKB [102].
ADME: Absorption, distribution, metabolism and excretion; DMET: Drug Metabolizing Enzymes and Transporters.
Future perspective.
The field of pharmacogenomics began by exploring candidate variants in relation to drug outcome, and has since moved towards larger, primarily pathway-based studies. No whole-genome approach has yet been published. The DMET platform has recently been applied to larger-scale pathway-based analyses than have been previously published, and has already elucidated some novel findings in clopidogrel, warfarin, docetaxel and thalidomide. While future studies will ultimately clarify the utility of this technology, the DMET platform is poised to determine the most important genetic variants in future clinical trials. Once these variants have been identified, preclinical investigations into the molecular basis behind the findings on DMET and prospective clinical validation cohorts will be required in order to translate the results for FDA approval and ultimately for clinical use. It is hoped that discovery-based applications of DMET will identify variants that can be genotyped a priori in order to improve therapy with current widely prescribed agents, and improve drug development in investigational agents by identifying nonresponders and toxic responders leading to a more personalized approach to therapy.
Executive summary.
Pharmacogenetics is useful in personalizing the practice of medicine, such that it can be used to guide therapeutic decisions of dose and choice of therapy. Pharmacogenetics research is expanding to involve more discovery-based approaches, and the Drug Metabolizing Enzymes and Transporters (DMET) platform is poised to become an important research tool.
The DMET platform interrogates genetic variation in genes involved in the absorption, distribution, metabolism and elimination (ADME) of therapeutics, in addition to a number of genes involved in ADME through indirect relationships.
As compared with the current strategies for genetic association studies, the DMET array can be thought of as a pathway-based approach, and the DMET array could be applied to a very wide range of therapeutics.
Despite its recent introduction, the DMET platform has already been applied in clinical pharmacology with promising results. When these results are compared with published genome-wide association studies, it is evident that while the DMET platform may miss certain associations outside of the ADME pathway, DMET is more applicable to the nature of the samples and end points evaluated in clinical pharmacology.
Strengths include increased power, reduction in Type I error, standardization of exploratory pharmacogenetics and better clinical trial design.
Progress will depend on improving the coverage of DMET as targeted therapeutics emerge, validating the results from DMET exploration (both prospectively and retrospectively), understanding the molecular basis behind the findings and translating the results from the DMET platform into US FDA-approved, clinically applicable strategies for treatment.
Acknowledgments
Financial
Tristan Sissung, William Figg and John Deeken served as consultants for Affymetrix and reviewed the accuracy and relevance of genes and genetic variants included on the DMET Plus panel prior to its public release in 2008. Tristan Sissung and William Figg complied with NIH policy regarding compensation to federal employees. No further ongoing financial relationships exist between the authors and Affymetrix. This study was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Bethesda, MD, USA. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organization imply endorsement by the US Government.
No writing assistance was utilized in the production of this manuscript.
Footnotes
Competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Bibliography
Papers of special note have been highlighted as:
▪ of interest
▪▪ of considerable interest
- 1.Zhou SF, Di YM, Chan E et al. : Clinical pharmacogenetics and potential application in personalized medicine. Curr. Drug Metab 9, 738–784 (2008).▪▪ Thorough review of the current status of personalized medicine.
- 2.Zanger U, Turpeinen M, Klein K, Schwab M: Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Anal. Bioanal. Chem 392, 1093–1108 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Walko C, McLeod H: Pharmacogenomic progress in individualized dosing of key drugs for cancer patients. Nat. Clin. Pract. Oncol 6, 153–162 (2009). [DOI] [PubMed] [Google Scholar]
- 4.Mattison L, Fourie J, Hirao Y et al. : The uracil breath test in the assessment of dihyrdropyrimidine dehydrogenase activity: pharmacokinetic relationship between expired 13CO2 and plasma [2–13C]dihydrouracil. Clin. Cancer Res 12, 549–555 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Kurnik D, Wood A, Wilkinson G: The erythromycin breath test reflects P-glycoprotein function independently of cytochrome P450 3A activity. Clin. Pharmacol. Ther 80, 238–234 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Cooper GM, Johnson JA, Langaee TY et al. : A genome-wide scan for common genetic variants with a large influence on warfarin maintenance dose. Blood 112, 1022–1027 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Daly AK, Donaldson PT, Bhatnagar P et al. : HLA-B*5701 genotype is a major determinant of drug-induced liver injury due to flucloxacillin. Nat. Genet 41, 816–819 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Link E, Parish S, Armitage J et al. : SLCO1B1 variants and statin-induced myopathy –a genomewide study. N. Engl. J. Med 359, 789–799 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Nelson MR, Bacanu SA, Mosteller M et al. : Genome-wide approaches to identify pharmacogenetic contributions to adverse drug reactions. Pharmacogenomics J 9, 23–33 (2009). [DOI] [PubMed] [Google Scholar]
- 10.Parkinson A: Biotransformation of xenobiotics. In: Casarett and Doull’s Toxicology: The Basic Science of Poisons Klaassen CD (Ed.). McGraw-Hill, NY, USA: 133–224 (2001). [Google Scholar]
- 11.Deeken J: The Affymetrix DMET platform and pharmacogenetics in drug development. Curr. Opin. Mol. Ther 11, 260–268 (2009). [PubMed] [Google Scholar]
- 12.van Schaik RH: CYP450 pharmacogenetics for personalizing cancer therapy. Drug resistance update: reviews and commentaries in antimicrobial and anticancer chemotherapy. Drug Resist. Updat 11, 77–98 (2008).▪▪ Comprehensive review of CYP450s in pharmacogenetics.
- 13.Sissung TM, Gardner ER, Gao R, Figg WD: Pharmacogenetics of membrane transporters: a review of current approaches. Methods Mol. Biol 448, 41–62 (2008). [DOI] [PubMed] [Google Scholar]
- 14.Kindla J, Fromm MF, Konig J: In vitro evidence for the role of OATP and OCT uptake transporters in drug–drug interactions. Expert Opin. Drug Metab. Toxicol 5, 489–500 (2009). [DOI] [PubMed] [Google Scholar]
- 15.Heist RS, Christiani D: EGFR-targeted therapies in lung cancer: predictors of response and toxicity. Pharmacogenomics 10, 59–68 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Drazen JM, Yandava CN, Dube L et al. : Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat. Genet 22, 168–170 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Timsit YE, Negishi M: CAR and PXR: the xenobiotic-sensing receptors. Steroids 72, 231–246 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang B, Xie W, Krasowski MD: PXR: a xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenomics 9, 1695–1709 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Corvol H, De Giacomo A, Eng C et al. : Genetic ancestry modifies pharmacogenetic gene–gene interaction for asthma. Pharmacogenet. Genomics 19, 489–496 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rodrigues-Lima F, Dairou J, Dupret JM: Effect of environmental substances on the activity of arylamine N-acetyltransferases. Curr. Drug Metab 9, 505–509 (2008). [DOI] [PubMed] [Google Scholar]
- 21.von Mutius E: Gene–environment interactions in asthma. J. Allergy Clin. Immunol 123, 3–11; quiz 2–3 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Lusis AJ, Fogelman AM, Fonarow GC: Genetic basis of atherosclerosis: part II: clinical implications. Circulation 110, 2066–2071 (2004). [DOI] [PubMed] [Google Scholar]
- 23.Wu X, Gu J, Spitz MR: Strategies to Identify Pharmacogenomic Biomarkers: Candidate Gene, Pathway-Based, and Genome-Wide Approaches 1st Edition Humana Press, NJ, USA: (2008).▪▪ Comprehensive book chapter comparing candidate gene, pathway based and genome-wide association study approaches in genetic association studies.
- 24.Evans WE, Horner M, Chu YQ, Kalwinsky D, Roberts WM: Altered mercaptopurine metabolism, toxic effects, and dosage requirement in a thiopurine methyltransferase-deficient child with acute lymphocytic leukemia. J. Pediatr 119, 985–989 (1991). [DOI] [PubMed] [Google Scholar]
- 25.Lennard L, Lewis IJ, Michelagnoli M, Lilleyman JS: Thiopurine methyltransferase deficiency in childhood lymphoblastic leukaemia: 6-mercaptopurine dosage strategies. Med. Pediatr. Oncol 29, 252–255 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Relling MV, Hancock ML, Boyett JM, Pui CH, Evans WE: Prognostic importance of 6-mercaptopurine dose intensity in acute lymphoblastic leukemia. Blood 93, 2817–2823 (1999). [PubMed] [Google Scholar]
- 27.Ando Y, Saka H, Ando M et al. : Polymorphisms of UDP-glucuronosyltransferase gene and irinotecan toxicity: a pharmacogenetic analysis. Cancer Res 60, 6921–6926 (2000). [PubMed] [Google Scholar]
- 28.Innocenti F, Undevia SD, Iyer L et al. : Genetic variants in the UDP-glucuronosyltransferase 1A1 gene predict the risk of severe neutropenia of irinotecan. J. Clin. Oncol 22, 1382–1388 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Marcuello E, Altes A, Menoyo A, Del Rio E, Gomez-Pardo M, Baiget M: UGT1A1 gene variations and irinotecan treatment in patients with metastatic colorectal cancer. Br. J. Cancer 91, 678–682 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rouits E, Boisdron-Celle M, Dumont A, Guerin O, Morel A, Gamelin E: Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity: a molecular and clinical study of 75 patients. Clin. Cancer Res 10, 5151–5159 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Pullarkat ST, Stoehlmacher J, Ghaderi V et al. : Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenomics J 1, 65–70 (2001). [DOI] [PubMed] [Google Scholar]
- 32.Villafranca E, Okruzhnov Y, Dominguez MA et al. : Polymorphisms of the repeated sequences in the enhancer region of the thymidylate synthase gene promoter may predict downstaging after preoperative chemoradiation in rectal cancer. J. Clin. Oncol 19, 1779–1786 (2001). [DOI] [PubMed] [Google Scholar]
- 33.Goekkurt E, Hoehn S, Wolschke C et al. : Polymorphisms of glutathione S-transferases (GST) and thymidylate synthase (TS) – novel predictors for response and survival in gastric cancer patients. Br. J. Cancer 94, 281–286 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gurubhagavatula S, Liu G, Park S et al. : XPD and XRCC1 genetic polymorphisms are prognostic factors in advanced non-small-cell lung cancer patients treated with platinum chemotherapy. J. Clin. Oncol 22, 2594–2601 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Park DJ, Stoehlmacher J, Zhang W, Tsao-Wei DD, Groshen S, Lenz HJ: A Xeroderma pigmentosum group D gene polymorphism predicts clinical outcome to platinum-based chemotherapy in patients with advanced colorectal cancer. Cancer Res 61, 8654–8658 (2001). [PubMed] [Google Scholar]
- 36.Ruzzo A, Graziano F, Kawakami K et al. : Pharmacogenetic profiling and clinical outcome of patients with advanced gastric cancer treated with palliative chemotherapy. J. Clin. Oncol 24, 1883–1891 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Stoehlmacher J, Ghaderi V, Iobal S et al. : A polymorphism of the XRCC1 gene predicts for response to platinum based treatment in advanced colorectal cancer. Anticancer Res 21, 3075–3079 (2001). [PubMed] [Google Scholar]
- 38.Zhou W, Gurubhagavatula S, Liu G et al. : Excision repair cross-complementation group 1 polymorphism predicts overall survival in advanced non-small cell lung cancer patients treated with platinum-based chemotherapy. Clin. Cancer Res 10, 4939–4943 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Borges S, Desta Z, Li L et al. : Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin. Pharmacol. Ther 80, 61–74 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Anglicheau D, Legendre C, Beaune P, Thervet E: Cytochrome p450 3A polymorphisms and immunosuppressive drugs: an update. Pharmacogenomics 8, 835–849 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Ryan SG: Regression to the truth: replication of association in pharmacogenetic studies. Pharmacogenomics 4, 201–207 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Eeles RA, Kote-Jarai Z, Giles GG et al. : Multiple newly identified loci associated with prostate cancer susceptibility. Nat. Genet 40, 316–321 (2008). [DOI] [PubMed] [Google Scholar]
- 43.Rafnar T, Sulem P, Stacey SN et al. : Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat. Genet 41, 221–227 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thomas G, Jacobs KB, Yeager M et al. : Multiple loci identified in a genome-wide association study of prostate cancer. Nat. Genet 40, 310–315 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Cordell HJ: Epistasis: what it means, what it doesn’t mean, and statistical methods to detect it in humans. Hum. Mol. Genet 11, 2463–2468 (2002). [DOI] [PubMed] [Google Scholar]
- 46.Baker SD, Verweij J, Cusatis GA et al. : Pharmacogenetic pathway analysis of docetaxel elimination. Clin. Pharmacol. Ther 85, 155–163 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dahut WL, Gulley JL, Arlen PM et al. : Randomized phase II trial of docetaxel plus thalidomide in androgen-independent prostate cancer. J. Clin. Oncol 22, 2532–2539 (2004). [DOI] [PubMed] [Google Scholar]
- 48.Deeken JF, Cormier T, Price DK et al. : A pharmacogenetic study of docetaxel and thalidomide in patients with castration-resistant prostate cancer using the DMET genotyping platform. Pharmacogenomics J (In press). [DOI] [PMC free article] [PubMed]
- 49.Caldwell MD, Awad T, Johnson JA et al. : CYP4F2 genetic variant alters required warfarin dose. Blood 111, 4106–4112 (2008).▪▪ First discovery of the role of variants in CYP4F2 in association with warfarin dosing.
- 50.Borgiani P, Ciccacci C, Forte V et al. : CYP4F2 genetic variant (rs2108622) significantly contributes to warfarin dosing variability in the Italian population. Pharmacogenomics 10, 261–266 (2009). [DOI] [PubMed] [Google Scholar]
- 51.Mega JL, Close SL, Wiviott SD et al. : Cytochrome p-450 polymorphisms and response to clopidogrel. N. Engl. J. Med 360, 354–362 (2009).▪▪ First discovery of the role of variants in CYP2C19 and CYP2B6 in clopidogrel therapy.
- 52.Drews J: Drug discovery: a historical perspective. Science 287, 1960–1964 (2000). [DOI] [PubMed] [Google Scholar]
- 53.Schneider BP, Wang M, Radovich M et al. : Association of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 genetic polymorphisms with outcome in a trial of paclitaxel compared with paclitaxel plus bevacizumab in advanced breast cancer: ECOG 2100. J. Clin. Oncol 26, 4672–4678 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
Websites
- 101.Website that defined the initial SNP selection of DMET www.pharmaadme.org/joomla/ (Accessed 28 October, 2009)
- 102.A comprehensive source that curates pharmacogenetics relationships www.pharmgkb.org (Accessed 28 October, 2009)
- 103.DMET™ Plus Premier Pack allele translation reports: summary of comprehensive drug disposition genotyping into commonly recognized allele names www.affymetrix.com/support/technical/whitepapers/dmet_plus_translation.pdf (Accessed 28 October, 2009)
- 104.Single-sample analysis methodology for the DMET™ Plus Product www.affymetrix.com/support/technical/whitepapers/dmet_plus_algorithm_whitepaperv1.pdf (Accessed 28 October, 2009)