

Iron cycling, including reduction of Fe(III) and oxidation of Fe(II), involves the formation, transformation, and dissolution of minerals and dissolved iron-organic matter compounds. It has been shown previously that Fe can be cycled so rapidly that no measurable changes in Fe(II) and Fe(III) concentrations occur, leading to a so-called cryptic cycle. Cryptic Fe cycles have been shown to be driven either abiotically by a combination of photochemical reduction of Fe(III)-OM complexes and reoxidation of Fe(II) by O2, or microbially by a combination of Fe(III)-reducing and Fe(II)-oxidizing bacteria. Our study demonstrates a new type of light-driven cryptic Fe cycle that is relevant for the photic zone of aquatic habitats involving abiotic photochemical reduction of Fe(III)-OM complexes and microbial phototrophic Fe(II) oxidation. This new type of cryptic Fe cycle has important implications for biogeochemical cycling of iron, carbon, nutrients, and heavy metals and can also influence the composition and activity of microbial communities.
KEYWORDS: geomicrobiology, iron cycling, iron oxidation
ABSTRACT
Fe-organic matter (Fe-OM) complexes are abundant in the environment and, due to their mobility, reactivity, and bioavailability, play a significant role in the biogeochemical Fe cycle. In photic zones of aquatic environments, Fe-OM complexes can potentially be reduced and oxidized, and thus cycled, by light-dependent processes, including abiotic photoreduction of Fe(III)-OM complexes and microbial oxidation of Fe(II)-OM complexes, by anoxygenic phototrophic bacteria. This could lead to a cryptic iron cycle in which continuous oxidation and rereduction of Fe could result in a low and steady-state Fe(II) concentration despite rapid Fe turnover. However, the coupling of these processes has never been demonstrated experimentally. In this study, we grew a model anoxygenic phototrophic Fe(II) oxidizer, Rhodobacter ferrooxidans SW2, with either citrate, Fe(II)-citrate, or Fe(III)-citrate. We found that strain SW2 was capable of reoxidizing Fe(II)-citrate produced by photochemical reduction of Fe(III)-citrate, which kept the dissolved Fe(II)-citrate concentration at low (<10 μM) and stable concentrations, with a concomitant increase in cell numbers. Cell suspension incubations with strain SW2 showed that it can also oxidize Fe(II)-EDTA, Fe(II)-humic acid, and Fe(II)-fulvic acid complexes. This work demonstrates the potential for active cryptic Fe cycling in the photic zone of anoxic aquatic environments, despite low measurable Fe(II) concentrations which are controlled by the rate of microbial Fe(II) oxidation and the identity of the Fe-OM complexes.
IMPORTANCE Iron cycling, including reduction of Fe(III) and oxidation of Fe(II), involves the formation, transformation, and dissolution of minerals and dissolved iron-organic matter compounds. It has been shown previously that Fe can be cycled so rapidly that no measurable changes in Fe(II) and Fe(III) concentrations occur, leading to a so-called cryptic cycle. Cryptic Fe cycles have been shown to be driven either abiotically by a combination of photochemical reduction of Fe(III)-OM complexes and reoxidation of Fe(II) by O2, or microbially by a combination of Fe(III)-reducing and Fe(II)-oxidizing bacteria. Our study demonstrates a new type of light-driven cryptic Fe cycle that is relevant for the photic zone of aquatic habitats involving abiotic photochemical reduction of Fe(III)-OM complexes and microbial phototrophic Fe(II) oxidation. This new type of cryptic Fe cycle has important implications for biogeochemical cycling of iron, carbon, nutrients, and heavy metals and can also influence the composition and activity of microbial communities.
INTRODUCTION
The biogeochemical cycling of elements plays an important role in shaping the modern environment and has contributed to the evolution of the Earth system throughout its history (1, 2). However, many important microbial element cycles are said to be “cryptic” due to the fact that rapid production and consumption rates of the relevant compounds maintain extremely low and stable concentrations despite rapid turnover (3). In the last decade, several studies have identified cryptic cycles in different environments, illustrating that cryptic elemental cycles are widely distributed and can influence, or even drive, the cycling of other elements (4–8). So far, the majority of studies on cryptic elemental cycles have focused on sulfur and carbon (7). Cryptic cycling of Fe has only been observed in a few aqueous environments and can involve combinations of abiotic or biotic reactions. For example, it was suggested that abiotic cryptic Fe cycling in the oxic surface ocean was driven by the simultaneous photochemical reduction of Fe(III)-OM complexes, followed by the abiotic reoxidation of Fe(II) by oxygen/reactive oxygen species (9–13).
Fe-OM complexes, including both Fe(II)-OM and Fe(III)-OM complexes, are widespread in the environment (10, 14–19). For example, almost all of the dissolved Fe (>99%) in seawater was shown to be associated with organics, according to modeling and speciation analysis (11, 20, 21). Additionally, organic matter and the formation of OM-metal complexes were shown to determine the speciation, concentration, and distribution of Fe in soils and seawater (10, 22–25). In photic zones, Fe(III)-OM complexes can be photochemically reduced, forming Fe(II), a process which was suggested to be one of the reasons for the higher-than-expected abundance of Fe(II) in many oxic natural aquatic environments (20, 26, 27). Depending on the conditions, e.g., the wavelength of the light and type of organic ligands (28), photochemical Fe(III) reduction can happen either directly or indirectly (20). In the direct photochemical Fe(III) reduction, an electron is directly transferred from the photoexcited organic ligand to Fe(III) (29), whereas in the indirect photochemical Fe(III) reduction, Fe(III) is reduced by reactive radical species that are produced photochemically by ligands, e.g., the superoxide radical (O2−) (30). Independent of whether photochemical Fe(III) reduction occurs directly or indirectly, both processes are accompanied by the transformation or loss of functional groups of the organic matter (20). Therefore, photochemical Fe(III) reduction was also suggested to play an important role in the degradation of organic matter (11, 31–33).
Fe can also be oxidized or reduced by bacteria, but biotically catalyzed cryptic Fe cycling is much less well characterized. In photic environments, phototrophic Fe(II)-oxidizing bacteria can anaerobically oxidize Fe(II) and use the electrons for carbon fixation. Together with Fe(III)-reducing bacteria, which reduce Fe(III) using organic or inorganic compounds as electron donors, phototrophic Fe(II)-oxidizing bacteria were suggested to be one of the key players in cryptic Fe cycling in a few redox-stratified lakes by reoxidation of the Fe(II) produced from microbial Fe(III) reduction (4, 34). However, it is currently unknown whether cryptic Fe cycling could also occur from the combined action of phototrophic Fe(II)-oxidizing bacteria and photochemical reduction of Fe(III)-OM complexes.
In this study, we therefore used a model anoxygenic phototrophic Fe(II) oxidizer (Rhodobacter ferrooxidans SW2) to determine whether these bacteria can oxidize different Fe(II)-OM complexes and whether they can reoxidize the Fe(II) species produced from abiotic photochemical reduction of Fe(III)-OM, thus closing a cryptic iron cycle under photic conditions.
RESULTS
Oxidation of Fe(II)-OM complexes by Rhodobacter ferrooxidans strain SW2.
In order to better understand the role of microorganisms in the cryptic Fe cycle and to evaluate whether cryptic light-dependent Fe cycling could occur with different Fe(II)-OM complexes, we performed cell suspension experiments with the phototrophic Fe(II) oxidizer R. ferrooxidans strain SW2. We incubated 1.5 × 108 cells/ml of strain SW2 with either Fe(II)-citrate, Fe(II)-EDTA, Fe(II)-PPHA [Fe(II)-Pahokee peat humic acid], or Fe(II)-SRFA [Fe(II)-Suwannee River fulvic acid] complexes. This nongrowth medium contains only those Fe(II)-OM complexes plus 1 mM NaHCO3, 20 mM piperazine-N,N′-bis(2-ethanesulfonic acid) (PIPES), and NaCl, i.e., no other salts, nutrients, or vitamins. This guarantees that cells are actively metabolizing but no cell growth is possible, and that therefore, changing cell numbers does not influence the quantification of metabolic rates.
In these cell suspension experiments, we observed that all Fe(II)-OM complexes tested were oxidized by strain SW2, while the Fe(II) oxidation rates varied for the different Fe(II)-OM complexes (Fig. 1). The Fe(II)-EDTA complex showed the highest oxidation rates (ca. 48 μM/h, or 32.1 fM/h per cell) followed by Fe(II)-citrate (ca. 31 μM/h, or 20.7 fM/h per cell), Fe(II)-SRFA (ca. 28 μM/h, or 18.6 fM/h per cell), and Fe(II)-PPHA (ca. 14 μM/h, or 9.6 fM/h per cell). With the exception of Fe(II)-PPHA, the Fe(II) oxidation rates for all Fe(II)-OM complexes were higher than the rate for non-OM-bound free Fe(II) (ca.24 μM/h, or 15.8 fM/h per cell). Increased light intensities also increased the oxidation rates of Fe(II)-PPHA (Fig. 4).
FIG 1.
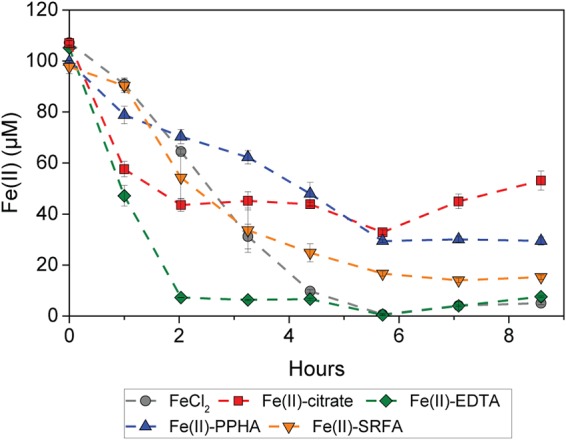
Oxidation of Fe(II) by R. ferrooxidans SW2. Fe(II) was present in the form of either non-OM-bound free Fe(II), Fe(II)-citrate, or Fe(II)-EDTA, Fe(II)-PPHA, or Fe(II)-SRFA complexes (yellow triangle). The results are reported as an average, and error bars indicate standard errors calculated from two independent parallel experiments. There was no Fe(II) oxidation in abiotic controls (Fig. S6).
FIG 4.
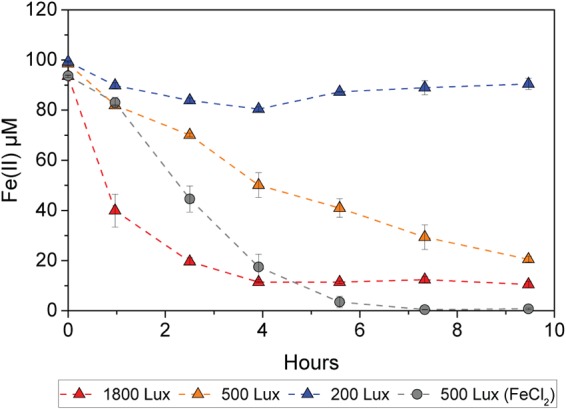
Oxidation of Fe(II)-PPHA complexes by R. ferrooxidans SW2 at different light intensities of ca. 1,800 lx, 500 lx, and 200 lx. For comparison, at 500 lx, duplicate treatments were amended with FeCl2 without PPHA. Error bars indicate standard errors calculated from two independent parallel experiments.
Not only the rates but also the extent of Fe(II) oxidation varied in experiments with these different Fe(II)-OM complexes (Fig. 1). Although initially, the oxidation of Fe(II)-citrate was faster than that of the non-OM-bound free Fe(II), the decrease in Fe(II) concentration virtually halted after 2 h, with a remaining 40% of Fe(II) after more than 8 h of incubation. In addition to Fe(II)-citrate, Fe(II)-PPHA and Fe(II)-SRFA showed a lower extent of Fe(II) oxidation than did the non-OM-bound free Fe(II). About 30% and 15% of Fe(II) still remained in the treatments with Fe(II)-PPHA and Fe(II)-SRFA, respectively. Only Fe(II)-EDTA showed a high extent of Fe(II) oxidation as the non-OM-bound Fe(II) [with <5% remaining Fe(II)].
Abiotic photoreduction of Fe(III)-OM complexes.
Photochemical Fe(II) reduction represents the second half of the cryptic photic Fe cycle [i.e., Fe(II) formation]. To quantify photochemical Fe(III) reduction in the presence of different OM, we incubated non-OM-bound Fe(III), Fe(III)-citrate, Fe(III)-EDTA, Fe(III)-PPHA, and Fe(III)-SRFA under the same conditions present in the cell suspension experiment (pH 7, light intensity of 550 lx) but in the absence of microbes. We found that the rates and extents of photochemical Fe(III) reduction by OM vary for the four different types of OM (Fig. 2). Fe(III)-citrate had the highest photochemical Fe(III) reduction rates among all the Fe(III)-OM complexes. About 20% of the Fe(III) (ca. 20 μM) was reduced to Fe(II) within 8 h of incubation (ca. 2.5 μM/h), and Fe(III) photoreduction continued until all the Fe(III) was reduced after 2 days. Fe(III)-EDTA had the second highest Fe(III) reduction rates, with about 5.6% and 30% of the Fe(III) being reduced after 8 h and 2 days, respectively (ca. 0.7 μM/h). From Fe(III)-PPHA and Fe(III)-SRFA complexes, ca. 12 and 14 μM Fe(II) [12 and 14% of the initial Fe(III)] was produced within 2 days (ca. 0.25 and 0.3 μM/h), respectively. This was less Fe(II) than that formed from Fe(III)-citrate and Fe(III)-EDTA but still more than in the OM-free controls that showed only 7 μM free Fe(II) within 2 days (0.15 μM/h).
FIG 2.
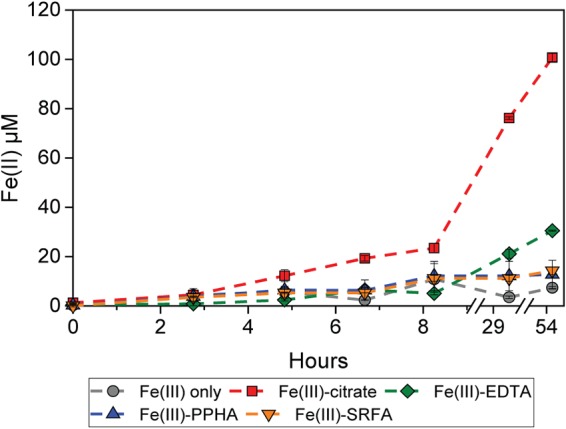
Abiotic photochemical reduction of Fe(III). Fe(III) was photochemically reduced to Fe(II) in the absence of organic chelators or in the presence of citrate, EDTA, Fe(II)-PPHA complexes, or Fe(II)-SRFA complexes. Error bars indicate standard errors calculated from two independent parallel experiments.
Cryptic cycling of Fe-citrate complexes by Rhodobacter ferrooxidans strain SW2.
In order to determine if Fe(II) and Fe(III) can be recycled by microbial phototrophic oxidation of Fe(II)-OM complexes and abiotic photoreduction of Fe(III)-OM complexes, we incubated the phototrophic Fe(II)-oxidizing bacterium R. ferrooxidans SW2 with either Fe(II)-citrate, Fe(III)-citrate, or citrate only and determined the concentrations of Fe(II) and cell numbers over time. We chose the Fe citrate system because Fe(II)-citrate showed one of the two highest microbial Fe(II) oxidation rates (Fig. 1), Fe(III)-citrate showed the highest photochemical reduction rate (Fig. 2), and citrate is an important environmental ligand which is known to be produced and released in many biotic systems, particularly by bacteria (35).
The presence of bacteria had a significant impact on the Fe(II) concentration over time in the treatments with Fe(III)-citrate (Fig. 3). We found that in the absence of phototrophic Fe(II)-oxidizing bacteria, i.e., in the abiotic controls, almost all of the Fe(III) present as an Fe(III)-OM complex was photochemically reduced to Fe(II) within 3 days of incubation. After these 3 days, the concentration of Fe(II) remained constant at ca. 1.8 mM (Fig. 3A). In contrast, when Fe(II)-oxidizing bacteria were present initially, there was no significant accumulation of Fe(II) during the incubation, and Fe(II) stayed constant at a very low concentration (10 to 30 μM). When strain SW2 was incubated with Fe(II)-citrate, Fe(II) was oxidized to Fe(III) within 3 days of incubation, and then Fe(II) remained constant at a low-micromolar concentration, similar to the treatments with Fe(III)-citrate and strain SW2.
FIG 3.

Fe(II) concentrations and cell abundances in R. ferrooxidans SW2 growth experiments (20°C, light incubation). (A) Concentration of Fe(II) over time in the presence of either Fe(III)-citrate or Fe(II)-citrate. The gray symbols represent abiotic controls with Fe(III)-citrate without R. ferrooxidans SW2. (B) Cell abundance of R. ferrooxidans SW2 in the growth experiment with Fe(III)-citrate, Fe(II)-citrate, and only citrate without Fe(II). The data are shown as averages of duplicate flow cytometry measurements. Error bars indicate standard errors calculated from two independent parallel experiments. There was no Fe(II) oxidation in abiotic controls with Fe(II)-citrate (Fig. S6).
Although the steady-state concentration of Fe(II) was low, we observed significant growth of strain SW2 cells independent of whether the growth medium was initially amended with Fe(III)-citrate or with Fe(II)-citrate (Fig. 3B). In both cases, the cell abundances increased 24- and 23-fold within 14 days of incubation and reached their maximum numbers of 6 × 108 and 9 × 108 cells/ml in the treatments with Fe(III)-citrate and Fe(II)-citrate, respectively. In addition to the slightly different maximum cell numbers, the growth rates and the length of the lag phases were also different depending on whether Fe(II)-citrate or Fe(III)-citrate was provided. Growth of strain SW2 in the treatment with Fe(III)-citrate started much earlier (within 3 days of incubation) than in the treatments with Fe(II)-citrate, where an obvious increase in cell numbers was observed only after 3 days (Fig. 3B). However, despite the longer lag phase of cell growth in the treatment with Fe(II)-citrate, during the exponential-phase strain SW2 grew significantly faster than in the treatment with Fe(III)-citrate. The doubling time of strain SW2 in the treatment with Fe(II)-citrate was about 2.9 days in the exponential phase, which is more than 2-fold faster than the doubling time of SW2 in the treatment with Fe(III)-citrate (ca. 6.5 days).
In the absence of Fe(II) or Fe(III) (citrate only), strain SW2 did not show any growth, demonstrating that growth in the Fe(II)-/Fe(III)-citrate-amended treatments was due to enzymatic Fe(II) oxidation and not based on utilization of the citrate (Fig. 3B).
DISCUSSION
Cryptic Fe cycling composed of photochemical reduction of Fe(III)-OM complexes and phototrophic Fe(II)-oxidizing bacteria.
Redox cycling of Fe plays an important role for many other biogeochemical cycles and can strongly influence the fate of pollutants and nutrients (2, 36). However, Fe cycling may be masked by balanced oxidation/reduction rates, which lead to low and stable iron concentrations which can be difficult to measure. Two types of cryptic Fe cycles have been identified before. First, a purely abiotic Fe cycle consisting of photochemical reduction of Fe(III)-OM and chemical Fe(II) oxidation by O2 has been proposed (11). Second, a biotic cryptic Fe cycle combining Fe(III)-reducing and Fe(II)-oxidizing bacteria has been observed in a stratified lake (4). Based on the results of the present study, we propose a new type of cryptic Fe cycle where Fe is cycled in photic zones by photochemical reduction of Fe(III)-OM complexes and microbial phototrophic Fe(II) oxidation (Fig. 4). The potential for this cryptic Fe cycle is demonstrated by the fact that multiple Fe(III)-OM complexes can be photochemically reduced to Fe(II) (Fig. 2), and the corresponding Fe(II)-OM complexes can be oxidized by phototrophic Fe(II)-oxidizing bacteria (Fig. 1). In our experiments, the Fe(II)-/Fe(III)-citrate system showed the highest Fe turnover rates, in which the phototrophic Fe(II) oxidizer R. ferrooxidans strain SW2 showed significant cell growth, while the steady-state concentration of Fe(II) remained stable in the low-micromolar range (Fig. 3). Additionally, we were also able to confirm growth by cryptic Fe cycling when both Fe(II)-citrate and Fe(III)-citrate complexes were initially present together in the medium (see Fig. S1 in the supplemental material).
Consequences of photochemical reduction of Fe(III)-OM.
Several abiotic and biotic reactions are involved in the photic cryptic Fe cycle with OM and bacteria. First of all, abiotic photochemical Fe(III) reduction drives the reductive side of the cryptic Fe cycle. In our study, different Fe(III)-OM complexes showed different rates of photochemical Fe(III) reduction (Fig. 2), supporting previous findings that different Fe(III)-OM complexes have different half-life times in light (13, 37, 38). These different photochemical reduction rates for different Fe(III)-OM complexes could be due to several reasons, such as the type and abundance of different functional groups, as well as the structure of the Fe-OM complexes (20). For example, the abundance of highly reactive alpha-hydroxyl carboxylate groups (28, 39) present in citrate could explain why Fe(III)-citrate had higher photochemical reduction rates than those of the other Fe(III)-OM complexes in our experiments.
Regardless of how fast the reduction of different Fe(III)-OM was, Fe(III) reduction reactions are accompanied with the transformation or loss of functional groups of the organic matter (20). For example, during the reduction of Fe(III)-citrate to Fe(II), citrate is oxidized, forming acetone and carbon dioxide via several intermediates, e.g., α-ketoglutarate (40–42). The general reaction could be represented as
where h is Planck's constant and ν is the light frequency.
These organic compounds formed as the products of OM photolysis could also serve as a carbon and electron source for bacteria, as was previously demonstrated in Rhodobacter capsulatus SB1003, a bacterium which could not oxidize dissolved non-OM-complexed Fe(II) (43) but could grow on Fe(III)-citrate utilizing the products of Fe(III)-citrate photochemical reactions (44). However, control experiments showed that R. ferrooxidans SW2 did not grow on 1 mM acetone (Fig. S2), the product of photochemical citrate degradation. Therefore, it is not possible that the cells grow by oxidizing only the OM without microbial oxidation of Fe(II). If the microbes did not reoxidize Fe(II) to Fe(III), there would be Fe(II) accumulation as shown in our abiotic controls with Fe(III)-citrate without cells (Fig. 3). This clearly showed that the Fe(II) is microbially reoxidized, thus closing the cryptic iron cycle. Overall, those organic compounds formed by photochemical Fe(III) reduction may also have stimulated the growth of some other heterotrophic bacteria in the environment, but it did not prevent the phototrophic Fe(II) oxidation microbes from oxidizing Fe(II) (Fig. 3A).
Microbial oxidation of Fe(II) in the presence of OM.
So far, it was unknown whether phototrophic Fe(II)-oxidizing bacteria, e.g., those that can oxidize and grow on non-OM-complexed Fe(II), are also capable of oxidizing Fe(II)-OM complexes. The results of this study (Fig. 1 and 3) demonstrated that Fe(II)-OM complexes can be oxidized by the phototrophic Fe(II)-oxidizing bacterium R. ferrooxidans strain SW2. This is interesting because strain SW2 was proposed to oxidize Fe(II) intracellularly with a putative iron oxidase FoxE embedded in the periplasm of the cell (43, 45). The observed oxidation of large Fe(II)-OM complexes, such as the Fe(II)-PPHA and Fe(II)-SRFA complexes, which were determined to be in the colloidal size range (46), suggests that strain SW2 may also be able to oxidize Fe(II) extracellularly, similar to many other bacteria with enzymatic Fe(II) oxidation pathways (47), since the large Fe(II)-PPHA and Fe(II)-SRFA complexes may not be easily transported into the periplasm.
The slightly slower oxidation of Fe(II)-PPHA is probably due to the dark color of the Fe(II)-PPHA complex, which absorbs light, thus lowering the light intensity that is available for the phototrophic microorganisms (Fig. 4). The different rates of Fe(II) oxidation for the different Fe(II) complexes [compared to free Fe(II)] may also have several other explanations. For example, the reactivity of Fe(II) and Fe(II)-OM complexes with the Fe(II)-oxidizing proteins may be different (48), or complexation of Fe(II) by OM may be beneficial for other microbial processes involved in Fe(II) oxidation as well. A possible benefit of the presence of organic ligands for the bacteria could be that complexation of the product of Fe(II) oxidation, i.e., the Fe(III), by the OM keeps the Fe(III) in solution and thus may negate the need to protect the cell from Fe(III) mineral encrustation. The cells thus may not need to synthesize organic polymer fibers which keep the Fe(III) minerals away from the membrane (49), as the Fe(III)-OM complexes are water soluble.
In addition to the rates, also the extents of microbial Fe(II) oxidation were different for the different Fe(II)-OM complexes (Fig. 1). This can have several reasons, since many parameters change when Fe(II) is complexed by different types of OM and at different concentrations of Fe(II)-OM and Fe(III)-OM complexes. These parameters include potential differences in Fe speciation and/or in the structure and size of the Fe(II)-OM complexes (46), varied redox potentials, different availability of Fe(II)- and Fe(III)-binding functional groups of the humic and fulvic acids (50), and different Fe(III)-OM photoreduction rates (26). All these differences are expected to influence not only the rates but also the extent of microbial Fe(II) oxidation, as also shown in an experiment with different concentrations of Fe(II)-OM complexes (Fig. S3).
Environmental/geochemical implications.
It is likely that the photic cryptic Fe cycle we describe here also exists and is widespread in many natural habitats. First, microbial Fe(II) oxidation is widespread, and phototrophic Fe(II)-oxidizing bacteria were commonly found in many environments, including lakes, soils, and freshwater and marine sediments (36, 51–54). Second, Fe-OM complexes are abundant in the environment, and many biologically produced Fe-chelating siderophores contain alpha-hydroxy carboxylate groups which can reduce Fe(III) to Fe(II) in the light (11, 28, 55–58). While in our batch growth experiment, Fe cycling is limited by the amount of initially present citrate [that allows photoreduction and thus, the regeneration of Fe(II) as the substrate for the Fe(II)-oxidizing microorganisms], in the environment, the organic ligands (e.g., citrate) are constantly produced and therefore would allow continuous Fe cycling (59, 60). Future studies determining the concentration of organic ligands needed for maintaining photochemical Fe(III) reduction are required to evaluate the relevance of cryptic cycling of Fe(III)-OM in different environments.
In the environment, cryptic cycling of Fe could also greatly influence other biogeochemical cycles (61), such as cycling of Mn species and As species which cooccur with Fe in many anoxic environments (62, 63). The cryptic cycling of Fe may also influence the transport of toxic metals in the form of Fe-OM-metal complexes, as the Fe(III)-/Fe(II)-OM complexes may have different binding capacities and binding mechanisms to heavy metals (64, 65). The reoxidation of Fe(II)-OM by phototrophic Fe(II)-oxidizing bacteria could not only increase the extent of Fe(III) photochemical reduction but also result in a larger extent of photolysis of OM and generation of labile OM for the growth of heterotrophic bacteria (Fig. 5). This could potentially influence microbial community composition as a whole, since the cryptic cycling of Fe could not only support the growth and carbon fixation of Fe(II)-oxidizing bacteria in habitats with low Fe(II) concentration but also continuously promote the release of labile carbon for a heterotrophic bacterial community. Overall, the results of this study suggest that, despite low steady-state concentrations of Fe, Fe cycling in photic zones of aqueous and terrestrial habitats could be more prominent and have a larger influence on carbon cycling than expected based on the low Fe concentrations alone.
FIG 5.
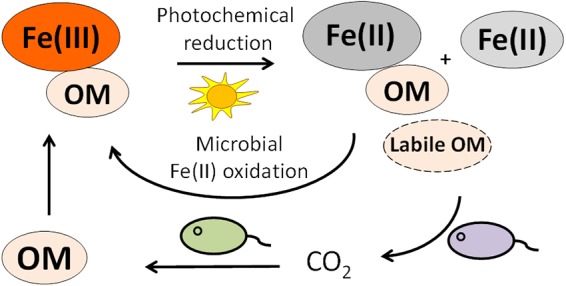
Proposed mechanism of cryptic Fe cycling by R. ferrooxidans SW2. Fe(III) is photochemically reduced to Fe(II) in the presence of OM-producing Fe(II)-OM complexes and labile OM. The Fe(II) produced, including free Fe(II) and Fe(II)-OM complexes, can be reoxidized to Fe(III) by microbial phototrophic Fe(II) oxidation. The electrons from microbial Fe(II) oxidation are used to fix CO2, therefore potentially further increasing the content of OM and thus stimulating further photochemical Fe(III) reduction. During Fe(III) photochemical reduction of Fe(III)-OM complexes, part of the photolyzed OM may become labile and can serve as a source of carbon and energy for the growth of bacteria.
MATERIALS AND METHODS
Bacterial strain and precultivation.
The phototrophic Fe(II)-oxidizing bacterium R. ferrooxidans SW2 was isolated from a freshwater pond (52) and routinely cultivated in the author’s lab with 10 mM FeCl2 in a basal medium which was prepared anoxically, as described previously (66). In order to remove remaining Fe(III) minerals from the inoculum prior to the experiment, strain SW2 was transferred 3 to 4 times in the basal medium with H2:CO2 (80:20) in the headspace as an electron donor instead of Fe(II).
Medium and chemicals.
Different media were made for different experiments. In total, three different types of medium were prepared. First, for the nongrowth cell suspension experiment, an anoxic PIPES-buffered medium (pH 7) with Fe(II)-OM complexes was prepared following a slightly modified method published previously (67). To guarantee complexation of most of the Fe(II) present by the different sources of OM tested, the final concentrations of Fe(II), citrate, EDTA, PPHA, and SRFA were 0.1 mM, 0.2 mM, 0.12 mM, 0.2 mg/ml, and 0.2 mg/ml, respectively. Speciation calculation using an Fe(II)-OM geochemical model (68) showed that more than 99% of the Fe(II) was present as Fe(II)-OM complexes with these chosen concentrations. Medium was dispensed into 15-ml Hungate tubes with a final volume of 5 ml and amended with 1 mM NaHCO3 and 0.5% CO2–99.5% N2 in the headspace. Second, for photochemical Fe(III) reduction experiments, 0.1 mM FeCl3, instead of FeCl2, was added to the anoxic PIPES medium with the same concentrations of OM, NaHCO3, and CO2. Third, for the growth experiment, 25 ml anoxic basal medium (66) was dispensed into 50-ml serum bottles and was amended with 4 mM HOC(COONa)(CH2COONa)2·2H2O (sodium citrate), either without Fe or with 2 mM FeCl2 or FeCl3.
Experimental design.
To evaluate the possibility of cryptic Fe cycling with Fe-OM complexes, we first performed independent SW2 cell suspension experiments with different Fe(II)-OM complexes and photochemical Fe(III)-OM reduction experiments. For the cell suspension experiment, the bacteria were first cultured to the late-exponential phase. Cells were harvested by centrifugation (7,000 × g, 20 min, 25°C), washed twice, and resuspended in 20 mM PIPES buffer containing 20 mM NaCl. An aliquot of the cell suspension was added to the nongrowth medium in Hungate tubes containing different Fe(II)-OM complexes, including Fe(II)-citrate, Fe(II)-EDTA, Fe(II)-PPHA, and Fe(II)-SRFA. The final cell number was ca. 1.5 × 108 cells/ml. The Hungate tubes with the cell suspension were placed horizontally under a 40-W incandescent light bulb and incubated at 20°C and a light intensity of ca. 550 lx. Abiotic photochemical Fe(III) reduction experiments were carried out in the same way as the cell suspension experiments but without cells and with Fe(II) replaced by Fe(III) (added as FeCl3). Additionally, to evaluate the effect of the dark color of humic acids on Fe(II) oxidation (humic acids may absorb part of the light and decrease light intensity for the phototrophic bacteria; Fig. S4), we performed a cell suspension experiment to determine the oxidation of Fe(II)-PPHA complexes by R. ferrooxidans SW2 at different light intensities of ca. 1,800 lx, 500 lx, and 200 lx (Fig. S5).
To determine if the anoxygenic phototrophic strain SW2 can grow by cryptic Fe-OM cycling, we performed a growth experiment. Strain SW2 was inoculated in the anoxic basal medium containing either citrate only, citrate with Fe(II), or citrate with Fe(III), using an initial cell number of ca. 3 × 107 cells/ml. The initial concentrations of Fe were about 2 mM for Fe(II) and Fe(III), and the initial concentration of citrate was 4 mM. According to the stoichiometry of Fe(III) reduction to Fe(II) by citrate (40, 41), 1 mol citrate can reduce 2 mol Fe(III), meaning that Fe(III) can be cycled a maximum of 4 times in our experiment with initial concentrations of 2 mM Fe and 4 mM citrate. This also implies that after each Fe cycle, the remaining citrate could still form a complex with Fe(III). The cells in the serum bottles were incubated at 25°C with a light intensity of ca. 1,000 lx generated by a 40-W light bulb.
Sample analysis.
Samples were taken in an anoxic glove box (100% N2, UNIlab Plus; MBraun, Germany) every 3 to 4 days for the growth experiment or every 1 to 2 h for cell suspension and abiotic photochemical Fe(III) reduction experiments. Fe(II) concentrations were determined anoxically using the ferrozine assay (69) modified as in Peng et al. (67). The quantification of Fe(II) in samples without PPHA and SRFA was performed using 1 M anoxic HCl and anoxic ferrozine solution (0.1% [wt/vol]) dissolved in ammonium acetate (C2H7NO2, 50% [wt/vol]). Samples with PPHA and SRFA were first diluted with anoxic Milli-Q H2O and immediately mixed with ferrozine solutions without 1 M HCl to avoid potential redox reactions of Fe with PPHA and SRFA during acidification. All ferrozine measurements were conducted in triplicate and the results reported as an average. Maximum rates of Fe(II) oxidation for the individual experiments were calculated from the slopes of the linear fits of Fe(II) concentrations at the steepest part of the experiments; at least three data points were used in the calculation of the Fe(II) oxidation rates. Cell numbers were quantified using an Attune NxT flow cytometer (Thermo Fisher Scientific) equipped with a blue laser beam as an excitation source (488 nm). Prior to flow cytometry, an aliquot of the cells was stained using BacLight green stain (Thermo Fisher Scientific). Cells were distinguished from debris by their properties in the side-scatter and fluorescence parameters. All measurements were conducted in duplicate and the results reported as an average.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the German Research Foundation (DFG) grant KA 1736/36-1.
We thank Ellen Röhm and Lars Grimm for assistance in the laboratory.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.02826-18.
REFERENCES
- 1.Druschel GK, Kappler A. 2015. Geomicrobiology and microbial geochemistry. Elements 11:389–394. doi: 10.2113/gselements.11.6.389. [DOI] [Google Scholar]
- 2.Borch T, Kretzschmar R, Kappler A, Cappellen PV, Ginder-Vogel M, Voegelin A, Campbell K. 2010. Biogeochemical redox processes and their impact on contaminant dynamics. Environ Sci Technol 44:15–23. doi: 10.1021/es9026248. [DOI] [PubMed] [Google Scholar]
- 3.Kappler A, Bryce C. 2017. Cryptic biogeochemical cycles: unravelling hidden redox reactions. Environ Microbiol 19:842–846. doi: 10.1111/1462-2920.13687. [DOI] [PubMed] [Google Scholar]
- 4.Berg JS, Michellod D, Pjevac P, Martinez-Perez C, Buckner CRT, Hach PF, Schubert CJ, Milucka J, Kuypers MMM. 2016. Intensive cryptic microbial iron cycling in the low iron water column of the meromictic Lake Cadagno. Environ Microbiol 18:5288–5302. doi: 10.1111/1462-2920.13587. [DOI] [PubMed] [Google Scholar]
- 5.Canfield DE, Stewart FJ, Thamdrup B, De Brabandere L, Dalsgaard T, Delong EF, Revsbech NP, Ulloa O. 2010. A cryptic sulfur cycle in oxygen-minimum–zone waters off the Chilean coast. Science 330:1375–1378. doi: 10.1126/science.1196889. [DOI] [PubMed] [Google Scholar]
- 6.Durham BP, Sharma S, Luo H, Smith CB, Amin SA, Bender SJ, Dearth SP, Van Mooy BAS, Campagna SR, Kujawinski EB, Armbrust EV, Moran MA. 2015. Cryptic carbon and sulfur cycling between surface ocean plankton. Proc Natl Acad Sci U S A 112:453–457. doi: 10.1073/pnas.1413137112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hansel CM, Ferdelman TG, Tebo BM. 2015. Cryptic cross-linkages among biogeochemical cycles: novel insights from reactive intermediates. Elements 11:409–414. doi: 10.2113/gselements.11.6.409. [DOI] [Google Scholar]
- 8.Holmkvist L, Ferdelman TG, Jørgensen BB. 2011. A cryptic sulfur cycle driven by iron in the methane zone of marine sediment (Aarhus Bay, Denmark). Geochim Cosmochim Acta 75:3581–3599. doi: 10.1016/j.gca.2011.03.033. [DOI] [Google Scholar]
- 9.Emmenegger L, Schonenberger R, Sigg L, Sulzberger B. 2001. Light-induced redox cycling of iron in circumneutral lakes. Limnol Oceanogr 46:49–61. doi: 10.4319/lo.2001.46.1.0049. [DOI] [Google Scholar]
- 10.Boyd PW, Ellwood MJ. 2010. The biogeochemical cycle of iron in the ocean. Nat Geosci 3:675–682. doi: 10.1038/ngeo964. [DOI] [Google Scholar]
- 11.Barbeau K, Rue EL, Bruland KW, Butler A. 2001. Photochemical cycling of iron in the surface ocean mediated by microbial iron(III)-binding ligands. Nature 413:409. doi: 10.1038/35096545. [DOI] [PubMed] [Google Scholar]
- 12.Sunda W, Huntsman S. 2003. Effect of pH, light, and temperature on Fe-EDTA chelation and Fe hydrolysis in seawater. Mar Chem 84:35–47. doi: 10.1016/S0304-4203(03)00101-4. [DOI] [Google Scholar]
- 13.Voelker BM, Morel FMM, Sulzberger B. 1997. Iron redox cycling in surface waters: effects of humic substances and light. Environ Sci Technol 31:1004–1011. doi: 10.1021/es9604018. [DOI] [Google Scholar]
- 14.Bhattacharyya A, Schmidt MP, Stavitski E, Martínez CE. 2018. Iron speciation in peats: chemical and spectroscopic evidence for the co-occurrence of ferric and ferrous iron in organic complexes and mineral precipitates. Org Geochem 115:124–137. doi: 10.1016/j.orggeochem.2017.10.012. [DOI] [Google Scholar]
- 15.Hopwood MJ, Statham PJ, Skrabal SA, Willey JD. 2015. Dissolved iron(II) ligands in river and estuarine water. Mar Chem 173:173–182. doi: 10.1016/j.marchem.2014.11.004. [DOI] [Google Scholar]
- 16.Luther GW, Shellenbarger PA, Brendel PJ. 1996. Dissolved organic Fe(III) and Fe(II) complexes in salt marsh porewaters. Geochim Cosmochim Acta 60:951–960. doi: 10.1016/0016-7037(95)00444-0. [DOI] [Google Scholar]
- 17.Neubauer E, Köhler SJ, von der Kammer F, Laudon H, Hofmann T. 2013. Effect of pH and stream order on iron and arsenic speciation in boreal catchments. Environ Sci Technol 47:7120–7128. doi: 10.1021/es401193j. [DOI] [PubMed] [Google Scholar]
- 18.Sundman A, Karlsson T, Laudon H, Persson P. 2014. XAS study of iron speciation in soils and waters from a boreal catchment. Chem Geol 364:93–102. doi: 10.1016/j.chemgeo.2013.11.023. [DOI] [Google Scholar]
- 19.Yu C, Virtasalo JJ, Karlsson T, Peltola P, Österholm P, Burton ED, Arppe L, Hogmalm JK, Ojala AEK, Åström ME. 2015. Iron behavior in a northern estuary: large pools of non-sulfidized Fe(II) associated with organic matter. Chem Geol 413:73–85. doi: 10.1016/j.chemgeo.2015.08.013. [DOI] [Google Scholar]
- 20.Barbeau K. 2006. Photochemistry of organic iron(III) complexing ligands in oceanic systems. Photochem Photobiol 82:1505–1516. doi: 10.1562/2006-06-16-IR-935. [DOI] [PubMed] [Google Scholar]
- 21.Rue EL, Bruland KW. 1995. Complexation of iron(III) by natural organic ligands in the Central North Pacific as determined by a new competitive ligand equilibration/adsorptive cathodic stripping voltammetric method. Mar Chem 50:117–138. doi: 10.1016/0304-4203(95)00031-L. [DOI] [Google Scholar]
- 22.Liu X, Millero FJ. 2002. The solubility of iron in seawater. Mar Chem 77:43–54. doi: 10.1016/S0304-4203(01)00074-3. [DOI] [Google Scholar]
- 23.Kuma K, Nishioka J, Matsunaga K. 1996. Controls on iron(III) hydroxide solubility in seawater: the influence of pH and natural organic chelators. Limnol Oceanogr 41:396–407. doi: 10.4319/lo.1996.41.3.0396. [DOI] [Google Scholar]
- 24.Kraemer SM. 2004. Iron oxide dissolution and solubility in the presence of siderophores. Aquat Sci 66:3–18. doi: 10.1007/s00027-003-0690-5. [DOI] [Google Scholar]
- 25.Kraemer SM, Butler A, Borer P, Cervini-Silva J. 2005. Siderophores and the dissolution of iron-bearing minerals in marine systems. Rev Mineral Geochem 59:53–84. doi: 10.2138/rmg.2005.59.4. [DOI] [Google Scholar]
- 26.Kuma K, Nakabayashi S, Suzuki Y, Kudo I, Matsunaga K. 1992. Photo-reduction of Fe(III) by dissolved organic substances and existence of Fe(II) in seawater during spring blooms. Mar Chem 37:15–27. doi: 10.1016/0304-4203(92)90054-E. [DOI] [Google Scholar]
- 27.Borer PM, Sulzberger B, Reichard P, Kraemer SM. 2005. Effect of siderophores on the light-induced dissolution of colloidal iron(III) (hydr)oxides. Mar Chem 93:179–193. doi: 10.1016/j.marchem.2004.08.006. [DOI] [Google Scholar]
- 28.Barbeau K, Rue EL, Trick CG, Bruland KW, Butler A. 2003. Photochemical reactivity of siderophores produced by marine heterotrophic bacteria and cyanobacteria based on characteristic Fe(III) binding groups. Limnol Oceanogr 48:1069–1078. doi: 10.4319/lo.2003.48.3.1069. [DOI] [Google Scholar]
- 29.Lever ABP. 1974. Charge transfer spectra of transition metal complexes. J Chem Educ 51:612. doi: 10.1021/ed051p612. [DOI] [Google Scholar]
- 30.Voelker BM, Sedlak DL. 1995. Iron reduction by photoproduced superoxide in seawater. Mar Chem 50:93–102. doi: 10.1016/0304-4203(95)00029-Q. [DOI] [Google Scholar]
- 31.Fisher JM, Reese JG, Pellechia PJ, Moeller PL, Ferry JL. 2006. Role of Fe(III), phosphate, dissolved organic matter, and nitrate during the photodegradation of domoic acid in the marine environment. Environ Sci Technol 40:2200–2205. doi: 10.1021/es051443b. [DOI] [PubMed] [Google Scholar]
- 32.Lockhart HB, Blakeley RV. 1975. Aerobic photodegradation of iron (III)-(ethylenedinitrilo) tetraacetate (ferric EDTA). Implications for natural waters. Environ Sci Technol 9:1035–1038. doi: 10.1021/es60110a009. [DOI] [PubMed] [Google Scholar]
- 33.Martin JD, Ito Y, Homann VV, Haygood MG, Butler A. 2006. Structure and membrane affinity of new amphiphilic siderophores produced by Ochrobactrum sp. SP18. J Biol Inorg Chem 11:633–641. doi: 10.1007/s00775-006-0112-y. [DOI] [PubMed] [Google Scholar]
- 34.Llirós M, García–Armisen T, Darchambeau F, Morana C, Triadó–Margarit X, Inceoğlu Ö, Borrego CM, Bouillon S, Servais P, Borges AV, Descy JP, Canfield DE, Crowe SA. 2015. Pelagic photoferrotrophy and iron cycling in a modern ferruginous basin. Sci Rep 5:13803. doi: 10.1038/srep13803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sandy M, Butler A. 2009. Microbial iron acquisition: marine and terrestrial siderophores. Chem Rev 109:4580–4595. doi: 10.1021/cr9002787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Melton ED, Swanner ED, Behrens S, Schmidt C, Kappler A. 2014. The interplay of microbially mediated and abiotic reactions in the biogeochemical Fe cycle. Nat Rev Microbiol 12:797–808. doi: 10.1038/nrmicro3347. [DOI] [PubMed] [Google Scholar]
- 37.Faust BC, Zepp RG. 1993. Photochemistry of aqueous iron(III)-polycarboxylate complexes: roles in the chemistry of atmospheric and surface waters. Environ Sci Technol 27:2517–2522. doi: 10.1021/es00048a032. [DOI] [Google Scholar]
- 38.Svenson A, Kaj L, Björndal H. 1989. Aqueous photolysis of the iron (III) complexes of NTA, EDTA and DTPA. Chemosphere 18:1805–1808. doi: 10.1016/0045-6535(89)90464-5. [DOI] [Google Scholar]
- 39.Miller CJ, Rose AL, Waite TD. 2013. Hydroxyl radical production by H2O2-mediated oxidation of Fe(II) complexed by Suwannee River fulvic acid under circumneutral freshwater conditions. Environ Sci Technol 47:829–835. doi: 10.1021/es303876h. [DOI] [PubMed] [Google Scholar]
- 40.Buchanan DNE. 1970. Mössbauer spectroscopy of radiolytic and photolytic effects on ferric citrate. J Inorg Nucl Chem 32:3531–3533. doi: 10.1016/0022-1902(70)80161-0. [DOI] [Google Scholar]
- 41.Frahn J. 1958. The photochemical decomposition of the citrate-ferric iron complex: a study of the reaction products by paper ionoporesis. Aust J Chem 11:399–405. doi: 10.1071/CH9580399. [DOI] [Google Scholar]
- 42.Abrahamson HB, Rezvani AB, Brushmiller JG. 1994. Photochemical and spectroscopic studies of complexes, of iron(III) with citric acid and other carboxylic acids. Inorganica Chim Acta 226:117–127. doi: 10.1016/0020-1693(94)04077-X. [DOI] [Google Scholar]
- 43.Croal LR, Jiao Y, Newman DK. 2007. The fox operon from Rhodobacter strain SW2 promotes phototrophic Fe(II) oxidation in Rhodobacter capsulatus SB1003. J Bacteriol 189:1774–1782. doi: 10.1128/JB.01395-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Caiazza NC, Lies DP, Newman DK. 2007. Phototrophic Fe(II) oxidation promotes organic carbon acquisition by Rhodobacter capsulatus SB1003. Appl Environ Microbiol 73:6150–6158. doi: 10.1128/AEM.02830-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pereira L, Saraiva IH, Oliveira ASF, Soares CM, Louro RO, Frazão C. 2017. Molecular structure of FoxE, the putative iron oxidase of Rhodobacter ferrooxidans SW2. Biochim Biophys Acta Bioenerg 1858:847–853. doi: 10.1016/j.bbabio.2017.07.002. [DOI] [PubMed] [Google Scholar]
- 46.Liao P, Li W, Jiang Y, Wu J, Yuan S, Fortner JD, Giammar DE. 2017. Formation, aggregation, and deposition dynamics of NOM-iron colloids at anoxic-oxic interfaces. Environ Sci Technol 51:12235–12245. doi: 10.1021/acs.est.7b02356. [DOI] [PubMed] [Google Scholar]
- 47.Shi L, Dong H, Reguera G, Beyenal H, Lu A, Liu J, Yu H-Q, Fredrickson JK. 2016. Extracellular electron transfer mechanisms between microorganisms and minerals. Nat Rev Microbiol 14:651. doi: 10.1038/nrmicro.2016.93. [DOI] [PubMed] [Google Scholar]
- 48.Liu J, Wang Z, Belchik SM, Edwards MJ, Liu C, Kennedy DW, Merkley ED, Lipton MS, Butt JN, Richardson DJ, Zachara JM, Fredrickson JK, Rosso KM, Shi L. 2012. Identification and characterization of MtoA: a decaheme c-type cytochrome of the neutrophilic Fe(II)-oxidizing bacterium Sideroxydans lithotrophicus ES-1. Front Microbiol 3:37. doi: 10.3389/fmicb.2012.00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miot J, Benzerara K, Obst M, Kappler A, Hegler F, Schädler S, Bouchez C, Guyot F, Morin G. 2009. Extracellular iron biomineralization by photoautotrophic iron-oxidizing bacteria. Appl Environ Microbiol 75:5586–5591. doi: 10.1128/AEM.00490-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tipping E, Lofts S, Sonke JE. 2011. Humic ion-binding model VII: a revised parameterisation of cation-binding by humic substances. Environ Chem 8:225–235. doi: 10.1071/EN11016. [DOI] [Google Scholar]
- 51.Bryce C, Blackwell N, Schmidt C, Otte J, Huang Y-M, Sara K, Tomaszewski E, Schad M, Warter V, Peng C, Byrne J, Kappler A. 2018. Microbial anaerobic Fe(II) oxidation—ecology, mechanisms and environmental implications. Environ Microbiol 20:3462–3483. doi: 10.1111/1462-2920.14328. [DOI] [PubMed] [Google Scholar]
- 52.Ehrenreich A, Widdel F. 1994. Anaerobic oxidation of ferrous iron by purple bacteria, a new type of phototrophic metabolism. Appl Environ Microbiol 60:4517–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Laufer K, Nordhoff M, Røy H, Schmidt C, Behrens S, Jørgensen BB, Kappler A. 2016. Coexistence of microaerophilic, nitrate-reducing, and phototrophic Fe(II) oxidizers and Fe(III) reducers in coastal marine sediment. Appl Environ Microbiol 82:1433–1447. doi: 10.1128/AEM.03527-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Otte JM, Harter J, Laufer K, Blackwell N, Straub D, Kappler A, Kleindienst S. 2018. The distribution of active iron-cycling bacteria in marine and freshwater sediments is decoupled from geochemical gradients. Environ Microbiol 20:2483–2499. doi: 10.1111/1462-2920.14260. [DOI] [PubMed] [Google Scholar]
- 55.Haygood MG, Holt PD, Butler A. 1993. Aerobactin production by a planktonic marine Vibrio sp. Limnol Oceanogr 38:1091–1097. doi: 10.4319/lo.1993.38.5.1091. [DOI] [Google Scholar]
- 56.Reid RT, Live DH, Faulkner DJ, Butler A. 1993. A siderophore from a marine bacterium with an exceptional ferric ion affinity constant. Nature 366:455. doi: 10.1038/366455a0. [DOI] [PubMed] [Google Scholar]
- 57.Martinez JS, Zhang GP, Holt PD, Jung H-T, Carrano CJ, Haygood MG, Butler A. 2000. Self-assembling amphiphilic siderophores from marine bacteria. Science 287:1245–1247. doi: 10.1126/science.287.5456.1245. [DOI] [PubMed] [Google Scholar]
- 58.Barbeau K, Zhang G, Live DH, Butler A. 2002. Petrobactin, a photoreactive siderophore produced by the oil-degrading marine bacterium marinobacter hydrocarbonoclasticus. J Am Chem Soc 124:378–379. doi: 10.1021/ja0119088. [DOI] [PubMed] [Google Scholar]
- 59.Lehmann J, Kleber M. 2015. The contentious nature of soil organic matter. Nature 528:60. doi: 10.1038/nature16069. [DOI] [PubMed] [Google Scholar]
- 60.Mimmo T, Del Buono D, Terzano R, Tomasi N, Vigani G, Crecchio C, Pinton R, Zocchi G, Cesco S. 2014. Rhizospheric organic compounds in the soil-microorganism-plant system: their role in iron availability. Eur J Soil Sci 65:629–642. doi: 10.1111/ejss.12158. [DOI] [Google Scholar]
- 61.Raiswell R, Canfield DE. 2012. The iron biogeochemical cycle past and present. Geochem Perspect 1:1–220. doi: 10.7185/geochempersp.1.1. [DOI] [Google Scholar]
- 62.Thamdrup B. 2000. Bacterial manganese and iron reduction in aquatic sediments, p 41–84. In Schink B. (ed), Advances in microbial ecology, vol 16 Springer, Boston, MA. [Google Scholar]
- 63.Zhu Y-G, Xue X-M, Kappler A, Rosen BP, Meharg AA. 2017. Linking genes to microbial biogeochemical cycling: lessons from arsenic. Environ Sci Technol 51:7326–7339. doi: 10.1021/acs.est.7b00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Catrouillet C, Davranche M, Dia A, Bouhnik-Le Coz M, Demangeat E, Gruau G. 2016. Does As (III) interact with Fe (II), Fe (III) and organic matter through ternary complexes? J Colloid Interface Sci 470:153–161. doi: 10.1016/j.jcis.2016.02.047. [DOI] [PubMed] [Google Scholar]
- 65.Sharma P, Rolle M, Kocar B, Fendorf S, Kappler A. 2011. Influence of natural organic matter on As transport and retention. Environ Sci Technol 45:546–553. doi: 10.1021/es1026008. [DOI] [PubMed] [Google Scholar]
- 66.Hegler F, Posth NR, Jiang J, Kappler A. 2008. Physiology of phototrophic iron(II)-oxidizing bacteria: implications for modern and ancient environments. FEMS Microbiol Ecol 66:250–260. doi: 10.1111/j.1574-6941.2008.00592.x. [DOI] [PubMed] [Google Scholar]
- 67.Peng C, Sundman A, Bryce C, Catrouillet C, Borch T, Kappler A. 2018. Oxidation of Fe(II)–organic matter complexes in the presence of the mixotrophic nitrate-reducing Fe(II)-oxidizing bacterium Acidovorax sp. BoFeN1. Environ Sci Technol 52:5753–5763. doi: 10.1021/acs.est.8b00953. [DOI] [PubMed] [Google Scholar]
- 68.Catrouillet C, Davranche M, Dia A, Bouhnik-Le Coz M, Marsac R, Pourret O, Gruau G. 2014. Geochemical modeling of Fe(II) binding to humic and fulvic acids. Chem Geol 372:109–118. doi: 10.1016/j.chemgeo.2014.02.019. [DOI] [Google Scholar]
- 69.Stookey LL. 1970. Ferrozine–a new spectrophotometric reagent for iron. Anal Chem 42:779–781. doi: 10.1021/ac60289a016. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.