

The crystal structure of phosphoribulokinase from the cyanobacterium Synechococcus sp. PCC 6301 is presented at 2.4 Å resolution. A disulfide bond is found between Cys40 and the P-loop residue Cys18, revealing the structural basis for the redox inactivation of the enzyme. A second disulfide bond appears to rigidify the dimer interface.
Keywords: phosphoribulokinase, Calvin–Benson–Bassham cycle, photosynthesis, dark reaction, redox regulation, transferases
Abstract
Phosphoribulokinase (PRK) catalyses the ATP-dependent phosphorylation of ribulose 5-phosphate to give ribulose 1,5-bisphosphate. Regulation of this reaction in response to light controls carbon fixation during photosynthesis. Here, the crystal structure of PRK from the cyanobacterium Synechococcus sp. strain PCC 6301 is presented. The enzyme is dimeric and has an α/β-fold with an 18-stranded β-sheet at its core. Interestingly, a disulfide bond is found between Cys40 and the P-loop residue Cys18, revealing the structural basis for the redox inactivation of PRK activity. A second disulfide bond appears to rigidify the dimer interface and may thereby contribute to regulation by the adaptor protein CP12 and glyceraldehyde-3-phosphate dehydrogenase.
1. Introduction
Photosynthetic CO2 fixation by the enzyme ribulose-1,5-bisphosphate carboxylase/oxygenase (Rubisco) in the Calvin–Benson–Bassham (CBB) cycle requires ribulose 1,5-bisphosphate (RuBP), which is generated by phosphoribulokinase (PRK; EC 2.7.1.19) from ribulose 5-phosphate (Ru5P) and ATP. The CBB cycle is fuelled by ATP and NADPH produced by the light reactions of photosynthesis and is rapidly shut down in the absence of light irradiance. The enzyme activity of PRK controls the flux through the CBB cycle.
PRK belongs to the nucleoside/nucleotide kinase (NK) superfamily of proteins, which catalyse phosphate-group transfer reactions to nucleoside, nucleotide or sugar substrates. PRK enzymes from plants, green and red algae, cyanobacteria, proteobacteria and archaea have been studied and classified into three phylogenetic groups (Kobayashi et al., 2003 ▸; Kono et al., 2017 ▸; Miziorko, 2000 ▸). Group I enzymes are found in plants, green algae, red algae and cyanobacteria. Group II PRK enzymes exist in bacteria, with the PRK from the proteobacterium Rhodobacter sphaeroides (RsPRK) as a structural and mechanistic paradigm (Harrison et al., 1998 ▸). The archaeal PRK orthologues form the third group (Kono et al., 2017 ▸). Crystal structures have been solved for the latter two groups, revealing Rossmann-like α/β/α sandwich folds with a Walker A motif (P-loop) serving as an ATP-binding site (Harrison et al., 1998 ▸; Kono et al., 2017 ▸). The enzymatic mechanism by which PRK catalyses the phosphorylation of Ru5P to RuBP has been studied mainly using RsPRK: a catalytic residue in the enzyme deprotonates the O1 hydroxyl oxygen of Ru5P and activates it for nucleophilic attack on the γ-phosphoryl group of ATP (Harrison et al., 1998 ▸). As the γ-phosphoryl group is transferred from ATP to Ru5P, its stereochemistry inverts (Miziorko & Eckstein, 1984 ▸). Thus, the catalytic mechanism of PRK does not involve a phosphoryl-enzyme intermediate (Miziorko & Eckstein, 1984 ▸). However, the exact residues directly involved in the catalytic mechanism remain uncertain owing to a lack of PRK–substrate co-crystal structures. Apart from the predicted catalytic module, the crystal structures of RsPRK (PDB entry 1a7j; Harrison et al., 1998 ▸) and PRK from the archaeon Methanospirillum hungatei (MhPRK; PDB entry 5b3f; Kono et al., 2017 ▸) are highly dissimilar. While RsPRK forms homooctamers and, like other prokaryotic PRKs, is regulated by allostery (Miziorko, 2000 ▸), MhPRK is a functional homodimer (Kono et al., 2017 ▸).
The activity of β-cyanobacterial and plant PRK enzymes is regulated through the reversible oxidation/reduction of cysteine sulfhydryl groups (Hurwitz et al., 1956 ▸; Kobayashi et al., 2003 ▸; Porter et al., 1988 ▸). Catalytic inactivation occurs by disulfide-bond formation induced by the oxidizing cellular or chloroplastic conditions associated with the absence of light (Knuesting & Scheibe, 2018 ▸), and is mediated by thioredoxin f (Brandes et al., 1996 ▸). The structural basis for this reversible inactivation is unknown. Sequences from β-cyanobacteria, green algae and plants contain two pairs of conserved cysteine residues: one close to the presumed ATP-binding pocket and one close to the C-terminus (Thieulin-Pardo et al., 2015 ▸). Whereas thiol modification of Cys16 (but not the C16S mutation) of the N-terminal pair in PRK from green algae and plants strongly impairs the enzyme activity, modification or mutation of the C-terminal pair has no effect on enzyme activity in vitro (Milanez et al., 1991 ▸; Thieulin-Pardo et al., 2015 ▸). Oxidized PRK from these organisms has been reported to become inactive (Porter et al., 1990 ▸) and to subsequently bind another CBB cycle enzyme, glyceraldehyde-3-phosphate dehydrogenase (GAPDH), in a process which is mediated by the small intrinsically disordered protein CP12 in a redox-dependent manner (Clasper et al., 1991 ▸; Howard et al., 2008 ▸; Thieulin-Pardo et al., 2015 ▸; Avilan et al., 1997 ▸; Lebreton et al., 1997 ▸).
Here, we revisit the phylogenetic relations of PRK homologs and find a complex ancestry for the eukaryotic PRK sequences, which group together with the β-cyanobacterial sequences. We report the crystal structure of PRK from the β-cyanobacterium Synechococcus sp. strain PCC 6301 and show that it is a dimer in solution. The crystal structure reveals a Rossmann-like α/β/α sandwich fold, in common with the known PRK structures, and a novel structural organization for the C-terminal third of the protein, which defines the interface in the PRK dimer. Furthermore, we identify disulfide bonds across the predicted active-site pockets and close to the protein–protein interface in the dimer, providing evidence of how the formation of the former disulfide bond blocks the enzyme. Sequence conservation indicates that our structure provides a valid model for PRK enzymes from red algae, green algae and higher plants in addition to β-cyanobacteria.
2. Materials and methods
2.1. Multiple sequence alignment and phylogenetic analysis
Eukaryotic PRK genes were trimmed of chloroplast transit peptide sequences by manual editing guided by the ChloroP software (Emanuelsson et al., 1999 ▸). A selection of 37 PRK and uridine kinase sequences were analysed using the PhyloBot web portal (Hanson-Smith & Johnson, 2016 ▸; Supplementary Fig. S1). The sequences were aligned using the MSAprobs algorithm and a maximum-likelihood phylogenetic tree was generated using the PROCATWAG model with the uridine kinase sequences as an outgroup to root the tree. Tree visualization was performed using AQUAPONY (http://www.atgc-montpellier.fr/aquapony/; Cazaux et al., 2017 ▸).
2.2. Protein production
Synechococcus sp. strain PCC 6301 phosphoribulokinase (Syn6301-PRK) encoded by the vector pHue-prkA (Table 1 ▸) was expressed in Escherichia coli BL21(DE3) cells and purified as described previously (Wilson et al., 2018 ▸), with the exception that the cells were grown in rich 2YT medium to prevent cell death from the production of toxic ribulose 1,5-bisphosphate (RuBP; Wilson & Whitney, 2017 ▸). Purified PRK was further purified and buffer exchanged on a Superdex 200 (GE Healthcare) column equilibrated with 10 mM Tris pH 8.0, 50 mM NaCl. Syn6301-PRK was concentrated to 23 mg ml−1 using a 10 kDa molecular-weight cutoff Vivaspin 20 centrifugal concentrator (GE Healthcare) and snap-frozen in liquid nitrogen.
Table 1. Protein-production information.
| Source organism | Synechococcus sp. strain PCC 6301 |
| Expression vector | pHUE-prkA |
| Expression host | E. coli BL21 |
| Complete amino-acid sequence of the construct produced | MSKPDRVVLIGVAGDSGCGKSTFLNRLADLFGTELMTVICLDDYHSLDRKGRKEAGVTALDPRANNFDLMYEQVKALKNGETIMKPIYNHETGLIDPPEKIEPNRIIVIEGLHPLYDERVRELLDFSVYLDIDDEVKIAWKIQRDMAERGHSYEDVLASIEARRPDFKAYIEPQRGHADIVIRVMPTQLIPNDTERKVLRVQLIQREGRDGFEPAYLFDEGSTIQWTPCGRKLTCSYPGIRLAYGPDTYYGHEVSVLEVDGQFENLEEMIYVEGHLSKTDTQYYGELTHLLLQHKDYPGSNNGTGLFQVLTGLKMRAAYERLTSQAAPVAASV |
2.3. Size-exclusion chromatography coupled to multi-angle static light scattering (SEC-MALS)
Purified Syn6301-PRK (∼70 µg) was analysed using static and dynamic light scattering by auto-injection of the sample onto an SEC column (5 µm, 4.6 × 300 mm column; Wyatt Technology product No. WTC-030N5) at a flow rate of 0.35 ml min−1 in buffer (20 mM MOPS–KOH pH 7.5, 50 mM KCl, 10 mM MgCl2) at 25°C. The column is in line with the following detectors: a variable UV-absorbance detector set at 280 nm (Agilent 1100 series), a DAWN EOS MALS detector (Wyatt Technology, 690 nm laser) and an Optilab rEX refractive-index detector (Wyatt Technology, 690 nm laser; Wyatt, 1993 ▸). Molecular masses were calculated using the ASTRA software (Wyatt Technology) with the dn/dc value set to 0.185 ml g−1. Bovine serum albumin (Thermo) was used as the calibration standard.
2.4. Native mass spectrometry
Purified Syn6301-PRK (20 µM) was incubated in buffer (40 mM Tris–HCl pH 7.0, 50 mM NaCl) with or without 20 mM dithiothreitol (DTT) at room temperature for 1 h. Syn6301-PRK was then buffer-exchanged into 100 mM ammonium acetate pH 7.0 (Fluka) and 0.5 mM β-mercaptoethanol using Micro Bio-Spin 6 size-exclusion columns (Bio-Rad). The PRK concentration was measured from the sample absorption at 280 nm using a NanoDrop 1000 (ThermoFisher) and a molar extinction coefficient of 36 580 M −1 cm−1, and was followed by dilution to 10 µM. Sample analyses were performed immediately after buffer exchange using a quadrupole IM time-of-flight hybrid mass spectrometer with a Z-spray nano-ESI source (Synapt G2-Si, Waters, USA) in positive-ion mode. Gold-plated 10 µm Nano-ESI-Pipettes 8 (Mascom, Bremen) were used as capillaries. The optimized capillary and sample cone voltages were 1.95 kV and 130–150 V, respectively. Spectra were acquired and processed using MassLynx 4.1 (Waters, USA). Spectra were calibrated with 30 mg ml−1 caesium iodide dissolved in 1:1 acetonitrile:water.
2.5. Determination of the PRK cysteine state
Purified Syn6301-PRK (50 µM) was incubated in buffer (40 mM Tris–HCl pH 7.0, 50 mM NaCl) with or without 20 mM dithiothreitol (DTT) at room temperature for 1 h. Syn6301-PRK was then transferred into 100 mM ammonium acetate pH 7.0. The concentration of Syn6301-PRK was adjusted to 45 µM and the free thiol concentration was determined using Ellman’s reagent (Thermo Scientific) according to the manufacturer’s instructions.
2.6. Crystallization
Syn6301-PRK was crystallized by the sitting-drop vapour-diffusion method using the robotics setup at the Crystallization Facility at the Max Planck Institute of Biochemistry, Martinsried, Germany (Table 2 ▸). For vitrification, the crystals were sequentially incubated in reservoir solution additionally containing 7.5 and 15% glycerol for 15 min each and were then rapidly cooled in liquid nitrogen.
Table 2. Crystallization.
| Native | Osmate derivative | |
|---|---|---|
| Method | Sitting-drop vapour diffusion | Sitting-drop vapour diffusion |
| Plate type | 96-well 2-drop MRC Crystallization Plates (Swissci) UVB | 96-well 2-drop MRC Crystallization Plates (Swissci) UVB |
| Temperature (K) | 277 | 277 |
| Protein concentration (mg ml−1) | 23 | 23 |
| Buffer composition of protein solution | 10 mM Tris pH 8.0, 50 mM NaCl | 10 mM Tris pH 8.0, 50 mM NaCl |
| Composition of reservoir solution | 20% PEG 3350, 200 mM ammonium fluoride | 20% PEG 3350, 200 mM ammonium formate |
| Volume and ratio of drop | 200 nl (1:1) | 200 nl (1:1) |
| Volume of reservoir (µl) | 150 | 150 |
2.7. Data collection and processing
X-ray diffraction data were collected at 100 K using the oscillation method on beamline ID29 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France (Table 3 ▸). Three 360° data sets for the osmate derivative were collected at the Os L III edge using κ angles of 0, 20 and 40°. These data sets were merged to maximize the anomalous signal. All data were integrated and scaled with XDS (Kabsch, 2010 ▸). POINTLESS (Evans, 2006 ▸), AIMLESS (Evans & Murshudov, 2013 ▸) and CTRUNCATE (French & Wilson, 1978 ▸), as implemented in the CCP4i graphical user interface (Potterton et al., 2003 ▸), were used for data reduction.
Table 3. Data collection and processing.
Values in parentheses are for the outer shell.
| Native | Osmate derivative | |
|---|---|---|
| Diffraction source | Beamline ID29, ESRF | Beamline ID29, ESRF |
| Wavelength (Å) | 1.03320 | 1.13937 |
| Temperature (K) | 100 | 100 |
| Detector | PILATUS 6M, Dectris | PILATUS 6M, Dectris |
| Crystal-to-detector distance (mm) | 454.54 | 484.71 |
| Rotation range per image (°) | 0.05 | 0.15 |
| Total rotation range (°) | 100 | 3 × 360† |
| Exposure time per image (s) | 0.04 | 0.04 |
| Space group | H32 | H32 |
| a, b, c (Å) | 140.82, 140.82, 205.71 | 141.26, 141.26, 206.22 |
| α, β, γ (°) | 90, 90, 120 | 90, 90, 120 |
| Mosaicity (°) | 0.111 | 0.166 |
| Resolution range (Å) | 49.12–2.40 (2.49–2.40) | 49.33–2.77 (2.92–2.77) |
| Total No. of reflections | 171405 (18652) | 1062487 (68326) |
| No. of unique reflections | 30873 (3223) | 20468 (2938) |
| Completeness (%) | 99.6 (100) | 100 (100) |
| Multiplicity | 5.6 (5.8) | 51.9 (23.3) |
| 〈I/σ(I)〉 | 12.0 (1.3)‡ | 17.4 (1.7)§ |
| R r.i.m. ¶ | 0.081 (1.561) | 0.228 (2.517) |
| R p.i.m. †† | 0.034 (0.646) | 0.030 (0.502) |
| Overall B factor from Wilson plot (Å2) | 62.7 | 75.0 |
Recorded at κ = 0, 20 and 40°.
The crystal exhibited anisotropic X-ray diffraction, with resolutions of 2.71 and 2.4 Å in the hk plane and along the l axis, respectively (according to the CC1/2 > 0.5 criterion of AIMLESS). The mean I/σ(I) in the outer shell falls below 2.0 at 2.57 Å resolution.
The crystal exhibited anisotropic X-ray diffraction, with resolutions of 2.85 and 2.77 Å in the hk plane and along the l axis, respectively (according to the CC1/2 > 0.5 criterion of AIMLESS). The mean I/σ(I) in the outer shell falls below 2.0 at 2.87 Å resolution.
R
r.i.m. = 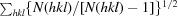
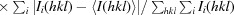 , where 〈I(hkl)〉 is the mean of the observations Ii(hkl) of reflection hkl.
, where 〈I(hkl)〉 is the mean of the observations Ii(hkl) of reflection hkl.
R
p.i.m. = 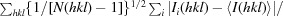
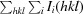 .
.
2.8. Structure solution and refinement
Initially, we attempted to solve the structure by molecular replacement using the coordinates of PRK from M. hungatei (MhPRK; PDB entry 5b3f; Kono et al., 2017 ▸) with MOLREP (Vagin & Isupov, 2001 ▸), but without success. Using a model containing only the homologous residues 1–216, 244–248 and 259–263 of MhPRK, one copy could be located in the asymmetric unit. Additional density in the 2F o − F c map was of too poor quality for model extension. The structure was solved at 3.8 Å resolution using osmium single-wavelength anomalous diffraction (SAD) data from an osmate (K2OsO4) derivative with SHELXC/D/E (Sheldrick, 2010 ▸) as implemented in the HKL2MAP graphical user interface (Pape & Schneider, 2004 ▸). Two osmium binding sites were identified. The map calculated after density modification assuming 75% solvent content (the actual solvent content was 53%) revealed clear features of secondary-structure elements; maps at higher solvent content were noisier. Compared with the molecular-replacement solution, additional β-strands from a second copy of PRK projecting away from the β-sheet of the first PRK molecule were visible. Manual model building was performed with Coot (Emsley & Cowtan, 2004 ▸). The resulting partial model was used to solve the native structure by molecular replacement at 2.4 Å resolution. The phases derived from the solution were used as input for automated model building with ARP/wARP (Perrakis et al., 1999 ▸). The resulting partial model was completed by manual model building in Coot. The model was refined with REFMAC5 using local noncrystallographic symmetry (NCS) restraints and translation–libration–screw (TLS) parametrization of B factors (Murshudov et al., 2011 ▸). The final models contained two copies of PRK per asymmetric unit. The chain identities were consistent between the two models. Residues 1–3 and 333 were disordered in both copies. In chain B, residues 15–20 and 133–160 were also missing. Residues facing solvent channels with disordered side chains were truncated after Cβ. Refinement statistics are summarized in Table 4 ▸. Structure factors and model coordinates have been deposited in the wwPDB as entries 6hzk and 6hzl.
Table 4. Structure solution and refinement.
Values in parentheses are for the outer shell.
| Native | Osmate derivative | |
|---|---|---|
| PDB code | 6hzk | 6hzl |
| Resolution range (Å) | 30.0–2.40 (2.46–2.40) | 30.0–2.77 (2.84–2.77) |
| Completeness (%) | 99.8 | 99.9 |
| σ Cutoff | F > 0.000σ(F) | F > 0.000σ(F) |
| No. of reflections, working set | 27844 (1395) | 18412 (1343) |
| No. of reflections, test set | 1503 (108) | 1017 (66) |
| Final R work † | 0.222 (0.337) | 0.235 (0.338) |
| Final R free † | 0.257 (0.382) | 0.259 (0.414) |
| Cruickshank DPI | 0.3942 | 0.3879 |
| No. of non-H atoms | ||
| Protein | 4954 | 4954 |
| Ligand | 0 | 2 |
| Solvent | 21 | 1 |
| Total | 4975 | 4957 |
| R.m.s. deviations | ||
| Bonds (Å) | 0.008 | 0.004 |
| Angles (°) | 1.218 | 0.897 |
| Average B factors (Å2) | ||
| Protein | 101‡ | 116§ |
| Ligand | — | 169 |
| Solvent | 64 | 58 |
| Ramachandran plot | ||
| Most favoured (%) | 97.7 | 98.5 |
| Allowed (%) | 1.8 | 1.5 |
| Outliers (%) | 0.5 | 0 |
R
work = 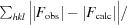
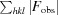 ; R
free is the R factor for a pre-selected subset (5%) of reflections that were not included in refinement.
; R
free is the R factor for a pre-selected subset (5%) of reflections that were not included in refinement.
The protein chains have very different average B factors: 69.4 Å for chain A and 136 Å for chain B.
The protein chains have very different average B factors: 79.7 Å for chain A and 156 Å for chain B.
3. Results and discussion
3.1. Phylogenetic analysis of PRK sequences
During our initial assessment of PRK sequences, we made the surprising discovery that the sequences from cyanobacteria did not present themselves as a homogeneous group. We therefore performed a new phylogenetic analysis using exemplary PRK sequences from all branches of photosynthetic organisms, phototrophic and nonphotosynthetic proteobacteria, and archaea, with several uridine kinase sequences as an outgroup (Supplementary Fig. S1). The analysis identified three classes of PRK sequences: eukaryotic PRK sequences belonging to class I, archaeal sequences belonging to class II and proteobacterial sequences belonging to class III (Fig. 1 ▸). Interestingly, the archaeal PRK sequences (class II) are more closely related to those of eukaryotic PRKs (class I) than to those of PRKs from proteobacteria (class III). Surprisingly, the α-cyanobacterial sequences were grouped together with those of PRKs from proteobacteria (class III), while the β-cyanobacterial sequences are more closely related to those of PRKs from green and red algae and plants (class IA). Eukaryotic PRK sequences from unicellular chromista, dinoflagellates and haptophytes form a distinct clade (class IB). PRK sequences of class IA show the conservation of four cysteine residues: two at the active site (with the exception of Cyanidioschyzon merolae) and two close to the C-terminus (Supplementary Fig. S1). In class IB sequences the active-site cysteine residues are conserved, but the cysteines close to the C-terminus are missing in most branches.
Figure 1.

Maximum-likelihood phylogenetic tree of PRK sequences. PRKs can be classified into three clear major groupings. Class I covers most photosynthetic organisms and can be further subdivided into two groups. Class IA contains vascular land plants, β-cyanobacteria and red algae, which have conserved cysteine pairs both in the active site and close to the C-terminus. Genomes with a class IA PRK also contain the CP12 adaptor protein. Class IB includes diatoms, dinoflagellates, haptophytes and other members of a vaguely defined ‘chromista’ clade. In contrast to class IA, most class IB PRKs lack the conserved cysteine residues close to the C-terminus and most of the source-organism genomes lack CP12. Class II enzymes are found in archaea and are also dimeric (Kono et al., 2017 ▸). Class III PRKs are distal to the other classes, do not feature redox regulation and have an octameric rather than a dimeric holoenzyme state. Uridine kinases, which belong to the same structural superfamily as PRK, form an outgroup in the phylogenetic tree.
Organisms harbouring class IA PRKs, including red algae, also contain the adaptor protein CP12. In contrast, members of class IB appear to lack CP12, and CP12 is also not present in class II and III organisms. CP12 is predicted to be an intrinsically disordered protein containing conserved cysteine residues that, under oxidizing conditions, form an intramolecular disulfide bridge and other secondary structures which enable CP12 to interact with GAPDH and PRK to form an inactive complex (Gontero & Maberly, 2012 ▸; Gardebien et al., 2006 ▸; Graciet et al., 2003 ▸). Disulfide disruption of the C-terminal Cys243–Cys249 pair (Chlamydomonas reinhardtii numbering) in PRK, via mutation of either cysteine residue to serine, prevents the formation of this complex (Thieulin-Pardo et al., 2015 ▸).
In summary, phylogenetic analysis suggests that in class I organisms the redox regulation of the active site of PRK evolved first and an additional, more multifarious form of regulation via complex formation with CP12 and GAPDH evolved later in class IA PRKs and co-evolved with the C-terminal disulfide bridge.
3.2. PRK from Synechococcus sp. strain PCC 6301 is a dimeric enzyme
We cloned the class IA PRK from Synechococcus sp. strain PCC 6301 (Syn6301-PRK), recombinantly expressed it in E. coli and performed structural analysis of the purified protein. Analysis of Syn6301-PRK using Ellman’s assay (Riddles et al., 1983 ▸) showed no free thiol groups, indicating that the cysteine residues became thiol-oxidized during the purification procedure. Size-exclusion chromatography coupled to multi-angle light scattering (SEC-MALS) indicated that thiol-oxidized Syn6301-PRK forms dimers with a hydrodynamic radius of ∼3.8 nm in solution (Fig. 2 ▸ a). Analysis by native mass spectrometry (native MS) confirmed that thiol-oxidized Syn6301-PRK forms a stable dimer (75 640 ± 28 Da; theoretical mass 75 420 Da; Fig. 2 ▸ b). To determine whether reduction of the disulfide bonds, particularly the disulfide bond at the C-terminal dimer interface, would lead to dissociation into monomers, we incubated PRK in 20 mM dithiothreitol for 1 h, which resulted in the reduction of ∼75% of the disulfide bonds, as judged by Ellman’s assay. The reduced Syn6301-PRK remained dimeric, as revealed by native MS (76 098 ± 28 Da; theoretical mass 75 420 Da; Fig. 2 ▸ c). It is therefore likely that Syn6301-PRK remains a dimer in the active state, similar to eukaryotic PRKs, suggesting that all class IA PRK enzymes appear as functional dimers.
Figure 2.

Oligomeric state of Syn6301-PRK. (a) SEC-MALS analysis of thiol-oxidized Syn6301-PRK. The chromatographic absorbance trace at 280 nm wavelength is shown. For the protein peak, the molecular mass determined by static light scattering is indicated. (b) Native MS spectrum of thiol-oxidized Syn6301-PRK. The charge states of the peaks are indicated. (c) Native MS spectrum of Syn6301-PRK after the reduction of disulfide bonds by treatment with 20 mM DTT for 1 h.
3.3. Crystal structure of PRK from Synechococcus sp. strain PCC 6301
The crystal structure of thiol-oxidized Syn6301-PRK was solved by osmium SAD at 3.8 Å resolution. The experimental electron-density map revealed features of secondary-structure elements (Fig. 3 ▸ a). A partial model built according to this map was used to solve the native structure of Syn6301-PRK at 2.4 Å resolution. The crystal structure contained noncrystallographic dimers of PRK, which had approximate twofold rotational symmetry (Fig. 3 ▸ b). However, most crystal contacts are established by one subunit (chain A; shown on the left in Fig. 3 ▸ b); the other subunit (chain B) is mainly fixed via the dimer interface and consequently exhibited increasing disorder when moving away from this interface. This was reflected by decreasing map quality and dramatically increasing B factors in chain B of the model (Fig. 3 ▸ c). This explains why chain B could not be located unambiguously by molecular replacement using the archaeal MhPRK structure (PDB entry 5b3f; Kono et al., 2017 ▸), despite considerable sequence homology (42% identity in the conserved residue range 1–186) between the two enzymes. Because of the disorder in chain B, the structural features of the Syn6301-PRK protomer are discussed based on the structure of chain A in the following sections.
Figure 3.

Crystal structure of Syn6301-PRK. (a) Stereo representation of a portion of the experimental electron-density map calculated with osmium SAD-derived phases after density modification with SHELXE (Sheldrick, 2010 ▸). Electron density is depicted as a blue mesh at 1.5σ. The protein backbone is shown as a Cα trace. All structural figures were drawn with PyMOL (v.2.0; Schrödinger). (b) Perpendicular views of the asymmetric unit in the Syn6301-PRK crystal structure. PRK chains A and B are shown as green and yellow ribbons, respectively. Disulfide bonds are represented as red sticks. Disordered regions are indicated by dotted lines. The N- and C-termini are indicated. (c) B-factor distribution in the asymmetric unit. Increasing B factors are represented as a rainbow colour gradient from blue to red.
3.4. Crystal structure of the Syn6301-PRK protomer
At the core of the Syn6301-PRK protomer is a nine-stranded, mixed β-sheet (Fig. 4 ▸). The five N-terminal strands are arranged in parallel (topology βB–βE–βA–βF–βG) and form the core of an α/β-fold (strands βC and βD form a β-hairpin insertion). α-Helices α1, α3 and α4 run along the parallel β-strands; α1 is preceded by a Walker A motif (P-loop). α-Helices α5 and α6 form a helical hairpin structure arranged almost perpendicular to the β-sheet. Together, these elements probably form the catalytic module of PRK, comprising the binding sites for ATP and Ru5P. The four C-terminal strands form a β-sheet extension with an antiparallel architecture (topology βH–βK–βJ–βI). At the dimer interface, β-strands βI from opposing protomers pair in an antiparallel fashion, creating an 18-stranded, continuous β-sheet in the complex (Fig. 3 ▸ b). Three successive α-helices form the C-terminal segment of the protomer; the final helix α9 aligns with β-strands βF and βG, and thus structurally belongs to the catalytic module. An overview of the secondary-structure topology of Syn6301-PRK is shown in Fig. 4 ▸(c).
Figure 4.

Structure of the Syn6301-PRK protomer. (a, b) Perpendicular views of the Syn6301-PRK protomer. Syn6301-PRK chain A is shown in ribbon representation; the disulfide bonds are shown as sticks. Secondary-structure elements and chain termini are indicated. (c) Secondary-structure topology of Syn6301-PRK. α-Helices and β-strands are represented as red cylinders and green arrows, respectively.
A striking feature of our Syn6301-PRK structure is the presence of two disulfide bonds (red sticks in Figs. 4 ▸ a and 4 ▸ b). The first connects Cys18 in the Walker A motif to Cys40 in β-strand βB (Figs. 4 ▸ a and 4 ▸ b, left). Note that the Walker A motif is disordered in chain B. The second disulfide bond is located in the βI–βJ loop, connecting Cys229 and Cys235, close to the dimer interface. The disulfide bonds Cys18–Cys40 and Cys229–Cys235 are located in positions which should directly affect the catalytic activity and the rigidity of the PRK dimer, respectively.
3.5. Comparison of the Syn6301-PRK protomer with structural homologs
The closest sequence homolog to Syn6301-PRK in the PDB is the archaeal MhPRK (PDB entry 5b3f; Kono et al., 2017 ▸; Fig. 5 ▸). Their catalytic modules (residues 1–186 in Syn6301-PRK) share 44% sequence identity. Consequently, the root-mean-square deviation (r.m.s.d.) is 1.51 Å for 202 matching Cα atoms determined using a distance cutoff of 3.5 Å (these also include residues from βH, βK and α9). The remaining structure strongly diverges: the structure of MhPRK lacks β-strand βH in the main β-sheet and the connection of βK to α9 is remodelled compared with our structure. Consequently, the dimer interfaces differ strongly apart from the fact that a continuous β-sheet connects the subunits in the dimer of both enzymes. Structural homology to the prokaryotic RsPRK (PDB entry 1a7j; Harrison et al., 1998 ▸) is limited to the core of the catalytic module (r.m.s.d. of 1.68 Å for only 138 matching Cα atoms). A more distant homolog to Syn6301-PRK is uridine kinase. The structure of uridine kinase from Thermus thermophilus in complex with AMP-PCP and cytidine (PDB entry 3w8r; Tomoike et al., 2013 ▸) exhibits an r.m.s.d. of 1.54 Å for 119 matching Cα atoms, which is essentially the catalytic module only.
Figure 5.


Comparison of Syn6301-PRK and MhPRK protomers. (a) Stereo representation of superposition of the protomers of Syn6301-PRK and MhPRK. Two perpendicular views are shown. The Cα traces of Syn6301-PRK and MhPRK are shown in green and purple, respectively. The same orientations as in Fig. 4 ▸ are shown. (b) Structure-based sequence alignment of Syn6301-PRK and MhPRK. Secondary-structure elements of the Syn6301-PRK and MhPRK structures are indicated above and below the sequences, respectively. Similar residues are shown in red and identical residues are shown in white on a red background. Green numbers below the sequence indicate the disulfide bonds in redox-blocked Syn6301-PRK. The Walker A motif is indicated in cyan. This figure was prepared using ESPript (Gouet et al., 1999 ▸).
3.6. The active site of Syn6301-PRK
A closer inspection of the predicted active-site pocket revealed that it is remodelled in Syn6301-PRK compared with MhPRK (Figs. 6 ▸ a and 6 ▸ b). In MhPRK, residues 21 and 22 of the Walker A motif located at the N-terminal end of helix α2 (α1 in Syn6301-PRK) form a so-called basket for the triphosphate moiety of ATP (red arrow in Fig. 6 ▸ b). The P-loop orientation found in MhPRK is similar to that in substrate-bound uridine kinase, in which only the triphosphate mimic of AMP-PCP is ordered (purple sticks in Fig. 6 ▸ c). In contrast to MhPRK and uridine kinase, the first turn of helix α1 in Syn6301-PRK is unwound to enable disulfide-bond formation between Cys18 and Cys40 (red sticks in Fig. 6 ▸ a). This rearrangement in thiol-oxidized Syn6301-PRK would prevent ATP binding and thus obliterate the enzyme activity. The importance of the P-loop conformation for enzyme activity is consistent with the sensitivity of eukaryotic PRKs towards the covalent modification of Cys16 (Cys18 in Syn6301-PRK; Milanez et al., 1991 ▸; Thieulin-Pardo et al., 2015 ▸). Conversely, complex formation with ATP would preclude disulfide-bond formation between Cys18 and Cys40, which is consistent with previous in vitro analysis of PRK from Synechococcus sp. strain PCC 7942 (Kobayashi et al., 2003 ▸). Similarly, ATP binding prevented the modification of Cys16 by glutathione in eukaryotic PRKs (Marri et al., 2014 ▸). Moreover, the α5–α6 helical hairpin is reoriented in Syn6301-PRK compared with the other structures, presumably as a consequence of a tight contact in the crystal structure. In molecule B most of the α5–α6 helical hairpin is disordered; the same conformation as observed in molecule A would provoke a clash with a symmetry mate in the crystal structure. The α5–α6 helical hairpin therefore appears to be flexibly linked to the bulk of PRK. In uridine kinase these helices provide contacts to the triphosphate and the ribose moiety of cytidine (cyan sticks in Fig. 6 ▸ c). We predict that Ru5P binds in PRK at a location equivalent to that of the ribose moiety of cytidine in uridine kinase, placing its 1-hydroxyl group for nucleophilic attack on the γ-phosphate of ATP. Comparison of all structures suggests that the α5–α6 helical hairpin might act as a mobile lid that locks the bound substrates during catalysis. In MhPRK these helices were found in a similar location to that in uridine kinase; the loop at the tip was disordered in MhPRK. From the structural comparison, we expect that Lys141 and Arg144 in helix α5 of Syn6301-PRK would assist in polarization of the ATP γ-phosphate. The binding site for Ru5P is presumably formed by residues at the tip of the βC–βD hairpin (Tyr88, His90 and Gly93) and in helix α2 (Arg49 and Arg52), and by His45 and Leu112 at the C-terminal ends of βB and βE, respectively. This assignment would be consistent with the H45N and R49Q mutations in RsPRK (Arg49 in RsPRK corresponds to Arg52 in Syn6301-PRK), which strongly increase the K m for Ru5P (Sandbaken et al., 1992 ▸). The 1-hydroxyl group in Ru5P, which corresponds to the 5′-hydroxyl group of cytidine in the uridine kinase complex structure, is presumably activated by Asp42 in the predicted Walker B motif, which is consistent with the mutation of Asp42 in RsPRK leading to a severe reduction in the catalytic rate (Charlier et al., 1994 ▸). All of these residues are highly conserved in class IA PRK sequences (Supplementary Fig. S2, Fig. 7 ▸ a).
Figure 6.

Comparison of the active sites in Syn6301-PRK, MhPRK and uridine kinase. (a) Ribbon representation of Syn6301-PRK in the redox-blocked state. The Cys18–Cys40 disulfide bond is represented by red sticks. Secondary-structure elements are indicated. The red arrow indicates the Walker A motif (P-loop). (b) Ribbon representation of MhPRK (PDB entry 5b3f; Kono et al., 2017 ▸) in the apo state. The same view as in (a) is shown. The red arrow indicates the Walker A motif (P-loop). (c) Ribbon representation of uridine kinase from T. thermophilus (PDB entry 3w8r; Tomoike et al., 2013 ▸) in the substrate-bound state. The substrates cytidine (Ctn) and AMP-PCP (ATP) are presented as cyan and purple sticks, respectively. The helical hairpin acting as a lid above the substrate-binding sites is indicated.
Figure 7.

Surface conservation and the dimer interface in Syn6301-PRK. (a) Surface conservation in the Syn6301-PRK protomer. Three perpendicular views are shown. Chain A is shown in surface representation, with the residue-similarity score from an alignment of class IA PRK sequences depicted as a colour gradient from cyan to magenta, indicating increasing sequence conservation. The active-site cleft is indicated. Chain B is shown in ribbon representation; disulfide bonds are shown as red sticks. (b) Detailed view of the dimer interface in Syn6301-PRK. The region that is boxed in (a) is magnified. The left panel shows the surface conservation in chain A and key contact residues in chain B highlighted in stick representation. The right panel shows the hydrogen-bond network at the dimer interface. Key contact residues are indicated and shown in stick representation. Putative hydrogen bonds are represented as dotted lines. The Cys229–Cys235 disulfide bond is depicted as red sticks. Secondary-structure elements and loops involved in the dimer interaction are indicated. The position of the local twofold symmetry axis is indicated.
3.7. The dimer interface in Syn6301-PRK
The second site of continuous surface conservation in PRK sequences from class IA organisms is the interface with the second protomer (Supplementary Fig. S2, Fig. 7 ▸). Probably all class IA PRK enzymes form dimers, as observed in Syn6301-PRK and as previously suggested for the β-cyanobacterial PRK from Anabaena cylindrica (Serra et al., 1989 ▸). The PRK dimer interface is surprisingly small, with a buried surface area of only 1785 Å2. Besides the antiparallel β-sheet contacts in the core, interdigitation of the side chains in the β-sheets is found (Fig. 7 ▸ b). The phenyl group of Phe218 points into a hydrophobic pocket formed by the side chains of Leu217, Phe218, Ile224, Trp226 and His275 (Fig. 7 ▸ b, left). The carboxyl group of Asp219 forms hydrogen bonds to Tyr271 and His275 in helix α7; Ser222 forms hydrogen bonds to the indole amide of Trp226 (Fig. 7 ▸ b, right). Important additional contacts are formed by the guanidine moiety of Arg231, which reaches into a negatively charged cavity on the opposing subunit formed by Glu207, Glu220, Glu253 and Ser255 (Fig. 7 ▸ b, right). Furthermore, a contact of Arg231 with the backbone at residue 254 is found. Arg231 belongs the to βI–βJ loop, the conformation of which is stabilized by the disulfide bond between Cys229 and Cys235 (red sticks in Fig. 7 ▸ b). In the thiol state, the loop is expected to become more mobile and the protomer association is likely to be more flexible. The increased mobility of the adjacent helix α7 could allow crosstalk with the active site via helix α9, which might be needed for optimal PRK enzyme activity.
4. Conclusion
Our crystal structure of β-cyanobacterial PRK in the thiol-oxidized state revealed the structural basis for the inactivation of the class IA PRK enzymes under low light conditions. The formation of the Cys18–Cys40 disulfide bond at the low cellular concentrations of reducing agents and ATP in the absence of light remodels the active site and blocks substrate binding. The sequences of PRKs from green algae and plants contain a conserved 17-residue insertion between helix α1 and β-sheet βB (Supplementary Fig. S1), which might add an additional layer of regulation of PRK activity in eukaryotes. In archaeal PRKs (class II), direct active-site remodelling by reversible thiol/disulfide exchange is not possible owing to the absence of such cysteine residues (Fig. 1 ▸). The second disulfide bond, Cys229–Cys235, close to the dimer interface probably rigidifies the interactions in the class IA dimer. This might have direct consequences for catalysis via allosteric signalling to the active site via helices α7 and α9. In addition, oxidation of Cys229 and Cys235 appears to favour complex formation with CP12 and GAPDH, thus shutting down PRK and GAPDH simultaneously (Thieulin-Pardo et al., 2015 ▸). In the complex, other structural components of the catalytic pocket of PRK, such as the mobile helices α5 and α6, might become dislocated, similar to the packing restraints exerted by the crystal structure. Resolving this question will require structure solution of the complete regulatory complex consisting of PRK, CP12 and GAPDH.
Supplementary Material
PDB reference: redox-inhibited phosphoribulokinase from Synechococcus sp. strain PCC 6301, 6hzk
PDB reference: osmate derivative, 6hzl
Supplementary Figures.. DOI: 10.1107/S2053230X19002693/no5156sup1.pdf
Acknowledgments
We thank F. Ulrich Hartl and Gabriel Thieulin-Pardo for critical reading of the manuscript, and the JSBG group at the ESRF in Grenoble, France and the staff at the MPIB Crystallization Facility for their excellent support.
References
- Avilan, L., Gontero, B., Lebreton, S. & Ricard, J. (1997). Eur. J. Biochem. 250, 296–302. [DOI] [PubMed]
- Brandes, H. K., Larimer, F. W. & Hartman, F. C. (1996). J. Biol. Chem. 271, 3333–3335. [DOI] [PubMed]
- Cazaux, B., Castel, G. & Rivals, E. (2017). AQUAPONY: Visualization and Interpretation of Phylogeographic Information on Phylogenetic Trees. https://hal-lirmm.ccsd.cnrs.fr/lirmm-01702654v2. [DOI] [PubMed]
- Charlier, H. A. Jr, Runquist, J. A. & Miziorko, H. M. (1994). Biochemistry, 33, 9343–9350. [DOI] [PubMed]
- Clasper, S., Easterby, J. S. & Powls, R. (1991). Eur. J. Biochem. 202, 1239–1246. [DOI] [PubMed]
- Emanuelsson, O., Nielsen, H. & von Heijne, G. (1999). Protein Sci. 8, 978–984. [DOI] [PMC free article] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- French, S. & Wilson, K. (1978). Acta Cryst. A34, 517–525.
- Gardebien, F., Thangudu, R. R., Gontero, B. & Offmann, B. (2006). J. Mol. Graph. Model. 25, 186–195. [DOI] [PubMed]
- Gontero, B. & Maberly, S. C. (2012). Biochem. Soc. Trans. 40, 995–999. [DOI] [PubMed]
- Gouet, P., Courcelle, E., Stuart, D. I. & Métoz, F. (1999). Bioinformatics, 15, 305–308. [DOI] [PubMed]
- Graciet, E., Lebreton, S., Camadro, J. M. & Gontero, B. (2003). Eur. J. Biochem. 270, 129–136. [DOI] [PubMed]
- Hanson-Smith, V. & Johnson, A. (2016). PLoS Comput. Biol. 12, e1004976. [DOI] [PMC free article] [PubMed]
- Harrison, D. H., Runquist, J. A., Holub, A. & Miziorko, H. M. (1998). Biochemistry, 37, 5074–5085. [DOI] [PubMed]
- Howard, T. P., Metodiev, M., Lloyd, J. C. & Raines, C. A. (2008). Proc. Natl Acad. Sci. USA, 105, 4056–4061. [DOI] [PMC free article] [PubMed]
- Hurwitz, J., Weissbach, A., Horecker, B. L. & Smyrniotis, P. Z. (1956). J. Biol. Chem. 218, 769–783. [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Knuesting, J. & Scheibe, R. (2018). Trends Plant Sci. 23, 769–782. [DOI] [PubMed]
- Kobayashi, D., Tamoi, M., Iwaki, T., Shigeoka, S. & Wadano, A. (2003). Plant Cell Physiol. 44, 269–276. [DOI] [PubMed]
- Kono, T., Mehrotra, S., Endo, C., Kizu, N., Matusda, M., Kimura, H., Mizohata, E., Inoue, T., Hasunuma, T., Yokota, A., Matsumura, H. & Ashida, H. (2017). Nature Commun. 8, 14007. [DOI] [PMC free article] [PubMed]
- Lebreton, S., Gontero, B., Avilan, L. & Ricard, J. (1997). Eur. J. Biochem. 250, 286–295. [DOI] [PubMed]
- Marri, L., Thieulin-Pardo, G., Lebrun, R., Puppo, R., Zaffagnini, M., Trost, P., Gontero, B. & Sparla, F. (2014). Biochimie, 97, 228–237. [DOI] [PubMed]
- Milanez, S., Mural, R. J. & Hartman, F. C. (1991). J. Biol. Chem. 266, 10694–10699. [PubMed]
- Miziorko, H. M. (2000). Adv. Enzymol. Relat. Areas Mol. Biol. 74, 95–127. [DOI] [PubMed]
- Miziorko, H. M. & Eckstein, F. (1984). J. Biol. Chem. 259, 13037–13040. [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Pape, T. & Schneider, T. R. (2004). J. Appl. Cryst. 37, 843–844.
- Perrakis, A., Morris, R. & Lamzin, V. S. (1999). Nature Struct. Biol. 6, 458–463. [DOI] [PubMed]
- Porter, M. A., Potter, M. D. & Hartman, F. C. (1990). J. Protein Chem. 9, 445–451. [DOI] [PubMed]
- Porter, M. A., Stringer, C. D. & Hartman, F. C. (1988). J. Biol. Chem. 263, 123–129. [PubMed]
- Potterton, E., Briggs, P., Turkenburg, M. & Dodson, E. (2003). Acta Cryst. D59, 1131–1137. [DOI] [PubMed]
- Riddles, P. W., Blakeley, R. L. & Zerner, B. (1983). Methods Enzymol. 91, 49–60. [DOI] [PubMed]
- Sandbaken, M. G., Runquist, J. A., Barbieri, J. T. & Miziorko, H. M. (1992). Biochemistry, 31, 3715–3719. [DOI] [PubMed]
- Serra, J. L., Llama, M. J., Rowell, P. & Stewart, W. D. P. (1989). Plant Sci. 59, 1–9.
- Sheldrick, G. M. (2010). Acta Cryst. D66, 479–485. [DOI] [PMC free article] [PubMed]
- Thieulin-Pardo, G., Remy, T., Lignon, S., Lebrun, R. & Gontero, B. (2015). Mol. Biosyst. 11, 1134–1145. [DOI] [PubMed]
- Tomoike, F., Kuramitsu, S. & Masui, R. (2013). Extremophiles, 17, 505–514. [DOI] [PubMed]
- Vagin, A. A. & Isupov, M. N. (2001). Acta Cryst. D57, 1451–1456. [DOI] [PubMed]
- Wilson, R. H., Martin-Avila, E., Conlan, C. & Whitney, S. M. (2018). J. Biol. Chem. 293, 18–27. [DOI] [PMC free article] [PubMed]
- Wilson, R. H. & Whitney, S. M. (2017). Directed Enzyme Evolution: Advances and Applications, edited by M. Alcalde, pp. 101–126. Cham: Springer International.
- Wyatt, P. J. (1993). Anal. Chim. Acta, 272, 1–40.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: redox-inhibited phosphoribulokinase from Synechococcus sp. strain PCC 6301, 6hzk
PDB reference: osmate derivative, 6hzl
Supplementary Figures.. DOI: 10.1107/S2053230X19002693/no5156sup1.pdf