

SUMMARY
Alternative polyadenylation (APA) produces mRNA isoforms with different 3’UTR lengths. Previous studies indicated that 3’ end processing and mRNA export are intertwined in gene regulation. Here, we show that mRNA export factors generally facilitate usage of distal cleavage and polyadenylation sites (PASs), leading to long 3’UTR isoform expression. By focusing on the export receptor NXF1, which exhibits the most potent effect on APA in this study, we reveal several gene features that impact NXF1-dependent APA, including 3’UTR size, gene size and AT content. Surprisingly, NXF1 downregulation results in RNAP II accumulation at the 3’ end of genes, correlating with its role in APA regulation. Moreover, NXF1 cooperates with CFI-68 to facilitate nuclear export of long 3’UTR isoform with UGUA motifs. Together, our work reveals important roles of NXF1 in coordinating transcriptional dynamics, 3’ end processing, and nuclear export of long 3’UTR transcripts, implicating NXF1 as a nexus of gene regulation.
3’ end processing and mRNA export are intertwined in gene regulation. Chen et al., show that the mRNA export receptor NXF1 globally impacts RNAP II dynamics, enhances expression of long 3’UTR isoforms, and facilitates their nuclear export, implicating NXF1 as a nexus for gene expression.
Keywords: Alternative polyadenylation, 3’UTR length, mRNA export, NXF1, transcriptional dynamics
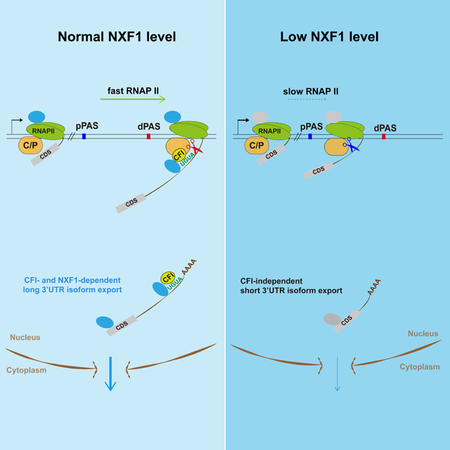
Graphical abstract
INTRODUCTION
Expression of protein-coding genes in eukaryotes involves multiple steps, including transcription, processing of the nascent transcripts, nuclear export, translation, and mRNA decay, which are often coupled to ensure streamlined regulation. Nuclear export of mRNAs has increasingly been appreciated as a key step of gene regulation (Izaurralde, 2002; Muller-McNicoll et al., 2016; Reed and Magni, 2001; Rodriguez-Navarro and Hurt, 2011; Wickramasinghe and Laskey, 2015). It requires the evolutionarily conserved transport receptor NXF1-NXT1 (Bjork and Wieslander, 2014; Walsh et al., 2010), a heterodimer with weak and nonspecific RNA binding activities. A set of adaptors facilitate the interactions between exporting mRNAs and NXF1-NXT1. One of the key NXF1 adaptors is the highly conserved transcription-export (TREX) complex, which contains ALYREF, UAP56 and the THO sub-complex (THOC1/2/3/6/7) (Bjork and Wieslander, 2014; Strasser et al., 2002). In addition, SR proteins have also been implicated as important NXF1 adaptors (Huang et al., 2003; Muller-McNicoll et al., 2016).
Cleavage and polyadenylation (CPA) of pre-mRNAs involves an endonucleolytic cleavage of the nascent RNA at the polyadenylation site (PAS), followed by synthesis of the poly(A) tail (Shi and Manley, 2015). Mammalian CPA complex includes four sub-complexes, namely, the cleavage and polyadenylation specificity factor (CPSF), cleavage stimulation factor (CstF), cleavage factor I (CFI), and cleavage factor II (CFII) (Shi and Manley, 2015). Most eukaryotic genes harbor multiple PASs, leading to alternative polyadenylation (APA) (Tian and Manley, 2017). The majority of APA sites are located in the last exon of genes, resulting in expression of mRNA isoforms with different 3’UTR lengths (Fu et al., 2011; Hoque et al., 2013; Shepard et al., 2011). APA isoforms can have substantial differences in mRNA localization, translation, and decay (Mayr, 2016; Tian and Manley, 2017). Recent studies have found that long 3’UTR isoforms are relatively more abundant in the nucleus as compared to short ones (Djebali et al., 2012; Neve et al., 2016), suggesting a possible 3’UTR size-based nuclear export control.
A growing number of mechanisms have been found to regulate APA (Lackford et al., 2014; Li et al., 2015; Zheng and Tian, 2014). One of the most prominent APA regulators is CFI. Two of its constituent proteins, CFI-25 and CFI-68, have displayed widespread regulation of APA (Li et al., 2015; Martin et al., 2012; Masamha et al., 2014; Zhu et al., 2018), through binding with UGUA motifs near distal PASs and facilitating CPSF recruitment (Zhu et al., 2018). In contrast, proximal PASs are depleted of the UGUA motif, leading to selective enhancement of CPA at distal PAS by CFI (Li et al., 2015). In addition, various aspects of the transcription process influence APA, including promoter activities (Ji et al., 2011; Nagaike et al., 2011) and transcription elongation rate (Liu et al., 2017).
Multiple lines of evidence indicate that CPA and nuclear export are interconnected. For examples, Yra1, the yeast ALYREF counterpart, interacts with the CPA and termination factor Pcf11 and impacts APA. Further, human THO proteins have been shown to influence APA events (Katahira et al., 2013; Tran et al., 2014). In addition, CFI-68 interacts with NXF1 and has been implicated as an NXF1 adaptor (Ruepp et al., 2009). Notably, SR proteins also regulate APA and impact nuclear export of APA isoforms (Muller-McNicoll et al., 2016). However, how mRNA export factors in general impact APA and how nuclear export of APA isoforms are controlled to modulate gene expression remain unknown. Here we report that mRNA export factors generally promote usage of distal PASs, with NXF1 showing the most potent effect. Using chromatin IP (ChIP) of RNAP II and analysis of nascent RNAs, we show that NXF1 significantly impacts RNAP II distribution at the 3’ end of genes, correlating to its roles in promoting distal PAS usage. Further, NXF1 cooperates with CFI-68 to promote long 3’UTR isoform export. Together, our work positions NXF1 at the nexus for gene regulation, coordinating transcription, pre-mRNA processing, and nuclear export.
RESULTS
Knockdown of nuclear export factors generally leads to 3’UTR shortening
To investigate how mRNA export factors affect APA regulation, we carried out siRNA-mediated knockdown (KD) of the mRNA export receptor NXF1 and several components of TREX, including ALYREF, UAP56 and THOC2, in HeLa cells. Western blot analysis showed that protein levels of these factors were knocked down by 90% (Figure 1A). To obtain a global view of APA, we subjected total RNAs from KD cells to 3’READS+, an updated version of the 3’ Region Extraction And Deep Sequencing method for APA analysis (Zheng et al., 2016).
Figure 1. mRNA export factors generally promote expression of long 3’UTR isoforms, See alsoFigures S1, S2, S7 and Table S1.

(A) Western blot analysis of knockdown efficiencies of ALYREF (72 hr KD), UAP56 (40 hr KD), THOC2 (72 hr KD), NXF1 (40 hr KD).
(B) Schematic of analysis of 3’UTR APA.
(C) Degrees of 3’UTR length changes in KD samples as indicated by the median RED values. Error bars are based on random sampling of data for 20 times (see Materials and Methods for detail). Statistically significant level was indicated based on FDR cutoffs (** < 5%, * < 10%).
(D) 3’UTR APA changes in cells treated with siNXF1. Genes with significant 3’UTR lengthening (red) or shortening (blue) are indicated. The numbers of genes and ratio are shown. Significant genes are those with P < 0.05 (Fisher’s exact test) and isoform abundance change > 5%.
(E) Top, schematic illustrating primer sets targeting alternative 3’UTR (aUTR, red) or common region (common, black). Bottom, relative ratio of aUTR to common region. Data are shown as mean ± s.d..
(F) Western blot examining the expression of siRNA-resistant NXF1. * indicates non-specific bands.
(G) Genes were divided into two groups based on 3’UTR length changes. Median values are 0.02 for genes with shortened 3’UTRs and −0.02 for other genes. P = 0.02 (Wilcoxon test). Gene expression was based on RNA-seq reads mapped to the CDSs of genes.
(H) Cntl or NXF1 KD cells were treated with ActD (20μg/ml), followed by 3’ end RT-qPCR at indicated time points.
(I) Analysis of APA in Cntl and VSV M-overexpression cells. APA is represented by the ratio of abundance between aUTR and common region. Data are shown as mean ± s.d..
In both Cntl and KD samples, 60–68% of protein-coding genes displayed APA, with an average of 2.3–2.8 APA isoforms per gene detected in each sample (Table S1). To examine 3’UTR APA, we focused on the top two 3’UTR APA isoforms based on their expression values. They were named proximal PAS (pPAS) and distal PAS (dPAS) isoforms based on their relative positions to the promoter (Figure 1B).
We calculated a relative expression difference (RED) score for each gene, defined as Δlog2 (dPAS expression level/pPAS expression level) between KD and Cntl samples. Therefore, a positive RED score indicates 3’UTR lengthening whereas a negative one 3’UTR shortening. Interestingly, all KDs led to global 3’UTR shortening, with NXF1 and UAP56 KDs being more significant than ALYREF and THOC2 KDs (FDR = 10%, Figure 1C). This trend could also be detected by analyzing average 3’UTR length of a gene weighted over all isoforms (Figure S1A). Note that some genes may have an opposite trend of regulation, such as the 184 genes showing 3’UTR lengthening upon NXF1 KD (Figure 1D). In NXF1 KD cells, genes with shortened 3’UTRs outnumbered those with lengthened 3’UTRs by 5.9-fold (Figure 1D). This result indicates that nuclear export factors have a general role in enhancing long 3’UTR isoform expression, with NXF1 showing the most potent effect.
A large fraction of APA sites are located in introns (Hoque et al., 2013; Tian et al., 2007). We next examined intronic polyadenylation (IPA) events in the KD cells. We grouped all isoforms using IPA sites together and compared their expression with that of isoforms using last exon PASs (Figure S1B). Interestingly, no significant regulation of IPA events could be discerned with these nuclear export factor KDs (FDR = 10%, Figure S1C). Therefore, our data indicate that APA regulation by mRNA export factors is restricted mostly to the last exon. Because NXF1 KD showed the most prominent effect, we focused on NXF1 in the rest of our study.
APA changes observed in NXF1 KD is mainly due to altered PAS choice
Using RT-qPCR and primers targeting alternative 3’UTR (aUTR) sequences and common regions of APA isoforms (illustrated in Figure 1E), we confirmed our 3’READS+ results with a number of genes that displayed significant 3’UTR shortening in NXF1 KD cells (Figures 1E, S1D). Expression of an siRNA-resistant NXF1 in NXF1 KD cells led to reversal of APA changes for all the genes tested (Figure 1E, F), indicating that the changes are not due to siRNA off-target effect.
3’UTR shortening could be due to selective degradation of long isoforms. If true, one would expect overall downregulation of genes with shortened 3’UTRs. However, gene expression analysis using RNA-seq revealed that NXF1 KD in fact led to slight upregulation of genes with shortened 3’UTRs as compared to other genes (median = 0.02 vs. −0.02, p = 0.02, Wilcox test, Figure 1G), refuting the possibility that 3’UTR shortening by NXF1 KD is through long isoform degradation. This conclusion was also validated by detailed half-life analysis of short and long 3’UTR isoforms of two genes (SIAH2 and NFKB1) in Cntl and NXF1 KD cells. NXF1 KD had no effect on the mRNA decay rate of either isoform (Figure 1H).
Because reduced expression of multiple mRNA export factors all caused global APA changes, we next asked whether mRNA export blockage per se might lead to isoform abundance changes by overexpression of the Vesicular Stomatitis Virus (VSV) M protein, which suppresses mRNA export by targeting Nup98 and Rae1 (Faria et al., 2005; von Kobbe et al., 2000) (Figure S2A). However, no apparent effect on relative expression of APA isoforms was detectable with most genes we examined (Figure 1I), even though their transcripts overall were more enriched in the nucleus in VSVM overexpressing cells (Figure S2B, C). Together, these data indicate that NXF1 facilitates the expression of long 3’UTR isoforms not through controlling mRNA stability or nuclear export per se.
Common and distinct APA events regulated by NXF1 and CFI-68
We next sought to explore the mechanism(s) by which NXF1 impacts APA site choice. Because NXF1 physically interacts with CFI-68 (Ruepp et al., 2009; Figure S3), a CPA factor with a prominent role in APA (Li et al., 2015; Martin et al., 2012; Masamha et al., 2014; Zhu et al., 2018), one possible mechanism is through regulation of CFI activity. To examine this, we carried out the same 3’READS+ analysis for siCFI-68 KD cells (Figure 2A). Consistent with previous studies (Li et al., 2015; Martin et al., 2012; Masamha et al., 2014; Zhu et al., 2018), CFI-68 KD led to drastic 3’UTR shortening, with 8.6-fold more genes showing 3’UTR shortening vs. lengthening and a median RED value of −0.97 (Figure 2B, C). Therefore, the extent of 3’UTR shortening caused by CFI-68 KD appeared to exceed that of NXF1 KD (median RED = −0.53), even though we cannot rule out the possibility that differential KD efficiencies may contribute to the difference. Importantly, while a set of APA events were commonly affected by both KDs (Figure 2C), and a modest correlation of 3’UTR APA change profiles was discernable between NXF1 and CFI-68 KD cells (r = 0.46, Pearson Correlation Coefficient, Figure 2C), ~38% of the APA events significantly regulated by NXF1 did not show apparent changes in CFI-68 KD cells (Figure 2C). Using RT-qPCR, we validated APA events commonly or differentially regulated by siNXF1 and siCFI-68 (Figures 2D, S1D).
Figure 2. NXF1 regulates APA largely via a CFI-68-independent mechanism, See alsoFigure S1.

(A) Western blot analysis of knockdown efficiency of CFI-68 (72 hr KD) in HeLa cells.
(B) 3’UTR APA changes in cells treated with siCFI-68. Genes with significant 3’UTR lengthening (red) or 3’UTR shortening (blue) are indicated. The numbers of genes and ratio are shown. Significant genes are those with P < 0.05 (Fisher’s exact test) and isoform abundancechange >5%. Only the two most abundant isoforms for each gene were analyzed.
(C) Correlation of 3’UTR APA regulation between CFI-68 KD and NXF1 KD cells. RED value is used to indicate the extent of APA regulation. A Venn diagram is shown on the right to indicate the numbers of genes in different groups. Sh, genes with shortened 3’UTRs.
(D) Left, genes commonly regulated by NXF1 and CFI-68; Middle, NFAT5 is regulated by NXF1 only; right, TATDN3 and RBM18 are regulated by CFI-68 only. Ratios of RT-qPCR signal of aUTR to that of common region are shown. Data are shown as mean ± s.d..
(E) Additive effects of CFI-68 and NXF1 KDs. Ratios of RT-qPCR signal of aUTR to that of common region are shown. Data are shown as mean ± s.d..
(F) Overexpression of CFI-68 in NXF1 KD cells does not rescue APA events commonly regulated by CFI-68 and NXF1. Data are shown as mean ± s.d..
(G) Western blot examining overexpression of CFI-68.
(H) Overexpression of NXF1 in CFI-68 KD cells does not rescue APA events commonly regulated by CFI-68 and NXF1. Data are shown as mean ± s.d..
(I) Western blot analysis examining overexpression of NXF1.
We next co-knocked down NXF1 and CFI-68. For some genes, co-KD did not shorten 3’UTRs beyond the level of single KD, e.g., RAB10 and RPL22 (Figure 2E), suggesting overlapping functions between NXF1 and CFI-68. However, for most genes we examined, co-KD led to additive effects on 3’UTR shortening, e.g., JMY, MARCH5, NHP2L1, SIAH2, TMCC, and UMTD2 (Figure 2E). Consistently, CFI-68 overexpression in NXF1 KD cells did not reverse the APA changes (Figure 2F, G), and vice versa (Figure 2H, I). Together, these data indicate that NXF1 and CFI-68 employ largely distinct mechanisms to regulate APA.
Genomic features governing APA events regulated by CFI-68 and NXF1
We next investigated features associated with APA events regulated by NXF1 and/or CFI-68 (Figure 3A). Consistent with previous findings (Li et al., 2015; Martin et al., 2012; Zhu et al., 2018), 3’UTR shortening events regulated by CFI-68 only or by both CFI-68 and NXF1 showed significant UGUA-motif enrichment in the upstream region of distal PASs (region a, Figure 3A). By contrast, no such enrichment could be discerned for those regulated by NXF1 KD only (Figure 3A).
Figure 3. Feature analysis of APA events regulated by NXF1 and/or CFI-68.

(A) Frequency of UGUA in each surrounding region of the PAS of a gene set (3’UTR shortening by siCFI-68 only, or by siNXF1 only, or by both) is normalized to that of other APA genes.
(B) Top, schematic showing features. aUTR, alternative UTR; cUTR, common UTR. ***P < 0.001; n.s., P > 0.05. Note that the AT content is based on the last exon (normalized by its length), and UGUA is the frequency of UGUA in the −100 nt to −40 nt region upstream of the distal PAS. Bottom, Pearson correlation coefficient for correlation between a feature and 3’UTR RED values.
(C) Genes were divided into five bins based on aUTR size. Average RED value for each aUTR size bin is shown for siNXF1 (red) or siCFI-68 (blue) sample. Error bars are standard error of mean.
(D) Similar to 3C, except that genes were divided based on gene size (TSS to last PAS). Error bars are standard error of mean.
(E) Genes were divided into three bins based on gene size and AT content separately. Average RED value for each gene bin is shown for siNXF1 (red) or siCFI-68 (blue). Error bars are standard deviation based on two biological replicates. Statistical significance was based on the Kolmogorov-Smirnov test. ***p < 0.001; n.s., not significant.
Using a linear regression model, we systematically identified gene features correlating with APA regulation by NXF1 or CFI-68 (see Methods for detail). The features we used included gene size, 3’UTR size, cUTR (common 3’UTR) size, aUTR size, AT content of the last exon (AT content in this study), and UGUA near distal PAS (Figure 3B). In line with our previous finding (Li et al., 2015), aUTR size was an important feature correlating APA changes in CFI-68 KD cells (Figure 3B). Interestingly, it was also the most prominent feature for APA regulation by NXF1 (Figure 3B). As such, the longer the aUTR size, the greater 3’UTR shortening was in both KD samples (Figure 3C).
Strikingly, we found that AT content and gene size were also significantly correlated with APA regulation by both CFI-68 and NXF1 (Figure 3B, D). Note that these features have never been reported before for APA regulation. Interestingly, AT content played a more apparent role in APA regulation in short genes than in long genes in NXF1 KD cells (Figure 3E). By contrast, its impact on APA was similar for genes of all sizes in CFI-68 KD cells. Thus, these results revealed CFI-related and -nonrelated APA events regulated by NXF1. Also notable is that CFI-68 KD elicited a conspicuous activation of IPA events, in stark contrast to NXF1 KD (Figure S1C), further supporting the notion that these two proteins have distinct mechanisms to regulate APA.
NXF1 regulates RNAP II occupancy on different gene sets
To ask whether NXF1 had a direct role in CPA reaction, we carried out an in vitro cleavage assay using HeLa nuclear extract and a PAS-containing RNA substrate, 3M-SVL. Compared to the extract from Cntl, those from CFI-68 KD showed a decreased cleavage activity (Figure 4A), consistent with its direct role in CPA reaction (Shi et al., 2009). By contrast, siNXF1 did not affect in vitro cleavage to a discernable level (Figure 4A), indicating that NXF1 does not participate in the CPA reaction per se. In line with this notion, the previously purified CPA complex included CFI-68 but not NXF1 (Shi et al., 2009).
Figure 4. NXF1 regulates RNAP II distribution on genes, See alsoFigure S4 and Table S2.

(A) In vitro transcribed 3M-SVL RNA was incubated under cleavage conditions in the absence or presence of nuclear extracts prepared from indicated cells.
(B) Metagene analysis of normalized RNAP II signals (log2 (IP/input)) on genes based on ChIP-seq data. ChIP-seq signal at each position of a gene is divided by the sum of signal of the gene to normalize each gene’s contribution to the plot.
(C) Same as (B), except that genes are divided into short, medium, and long groups based on gene size. Gene size median is indicated for each group. Gene size ranges are the same as those in Figure 3E.
(D) PAS to gene body ratio of RNAP II signals in different gene size groups.
(E) Gene body to TSS ratio of RNAP II signals in different gene size groups.
(F) Difference (KD vs. Cntl) in PAS region to TSS region ratio of RNAP II signals for genes with shortened 3’UTRs and other genes. P-value was based on the Kolmogorov-Smirnov test.
We then reasoned that NXF1 might affect PAS choice through alteration of transcription dynamics, which has recently been shown to regulate APA in Drosophila (Liu et al., 2017). To examine this possibility, we carried out a genome-wide analysis of RNAP II distribution using ChIP-seq. Using an antibody to all forms of RNAP II, we found that overall, RNAP II signals were decreased around the transcription start site (TSS) and increased in gene body (GB) and around the PAS in NXF1 KD cells (Figure 4B), as compared to Cntl cells, suggesting that decreased NXF1 level might slow down transcriptional elongation, leading to enrichment of RNAP II in GB and PAS. However, this profile change could also be explained by greater promoter clearance of RNAP II, leading to more rapid RNAP II transitioning from the promoter into GB (see Discussion).
Considering the significant impact of gene size on NXF1-mediated APA regulation, we next divided genes into three groups based on gene size and examined the impact of NXF1 KD on RNAP II distribution along the gene. Strikingly, large genes displayed much more pronounced differences in RNAP II distribution than short genes (Figure 4C). Based on the RNAP II signal ratio between the PAS region and GB, NXF1 KD caused significant enrichment of RNAP II signals around the PAS (Figure 4D). The impact was more pronounced on large genes than short ones based on p-values (compare 3.4 ×10−11 to 1.1 ×10−10 and 2.5 ×10−4, Figure 4D). Using TSS as reference, RNAP II in NXF1 KD cells was enriched in GB as well (Figure 4E). This trend again was more apparent with large genes (Figure 4E). When genes were grouped by the AT content, RNAP II signals in NXF1 KD cells were more significantly enriched in GB and the PAS region in genes with high AT content (Figure S4, A–C). Importantly, genes with shortened 3’UTRs in NXF1 KD cells showed significantly higher RNAP II signals at the PAS region vs. TSS (P = 1.6 × 10−3, Figure 4F), consistent with the notion that NXF1-regulated APA change is a consequence of altered RNAP II dynamics. Together, our data indicate that NXF1 has a significant role in RNAP II distribution, especially for large genes and in high AT content regions.
Nascent RNA analysis corroborates the role of NXF1 in RNAP II distribution
Because ChIP-seq data do not directly indicate transcribing RNAP II, we next metabolically labelled RNAs with 4-thiouridine (4sU) for 5 min, extracted 4sU-labeled RNAs from the chromatin fraction, and subjected them for RNA-seq analysis using a method which resulted in reads biased to the 3’ end of RNA (Figure 5A and see Methods for details). As such, this method could reveal nascent RNA production at the loci of transcribing RNAP II, akin to NET-seq and TT-seq methods (Mayer et al., 2015; Schwalb et al., 2016). Consistent with ChIP-seq data, nascent transcripts were globally increased in the 3’ region upon NXF1 KD, and concomitantly decreased in the 5’ region (Figure 5B, C). These changes in nascent RNAs were more pronounced in large genes (P = 4.5 × 10−15, K-S test, Figure 5D) than in medium-sized genes (P = 7.6 × 10−6) or short genes (P = 0.2). A similar trend was observed by analyzing nascent RNA signals in genes with high, medium, and low AT contents (Figure 5E). Similar to the RNAP II ChIP-seq data, nascent RNA signal changes were more significant in genes with shortened 3’UTRs than other genes in NXF1 KD cells (P = 5.7 × 10−10, Figure 5F). Thus, both ChIP-seq and nascent RNA-seq data indicate that NXF1 is important for regulating RNAP II distribution on genes.
Figure 5. Nascent RNA analysis indicates changes of transcriptional dynamics in NXF1 KD cells, See alsoFigure S4 and Table S2.

(A) Schematic of RNA-seq analysis of nascent RNAs..
(B) Nascent RNA signals along the gene. Gene is divided into 10 fractions based on their gene size (from the TSS to PAS). Each fraction represents 10% of each gene..
(C) Metagene analysis of nascent RNA-seq reads along the gene. The zoom inset shows RNAP II distribution at the PAS region..
(D) Violin plot showing ratio of nascent RNA reads in the 3’ region (last 30% of gene region) to that in the 5’ region (first 30% of gene region) in genes with different sizes. P-value was based on the Kolmogorov-Smirnov test.
(E) Same as (D), except that genes are grouped according to dinucleotide AT content in the last exon..
(F) Difference (KD vs. Cntl) in ratio of nascent RNA reads in 3’ region to 5’ region for genes with shortened 3’UTRs and other genes. P-value was based on the Kolmogorov-Smirnov test..
(G) Co-precipitations of RNAP II and CFI-68 with Flag-NXF1 or Flag-eIF4A3 (Cntl) in the absence or presence of RNase A..
(H) Co-precipitations of Flag-NXF1 by the RNAP II antibody or IgG in the absence or presence of RNase A..
(I) ChIP-PCR to examine Flag-NXF1 binding along RAB10 and TMCC1 genes, as well as an intergenic region 3’ of TMCC1. The schematic on top denotes the locations of primer sets. Data are shown as mean ± s.d..
NXF1 associates with RNAP II through protein-protein interactions
We next asked how NXF1 regulates RNAP II dynamics. Considering the interactions between NXF1 and SR proteins (Huang et al., 2003; Muller-McNicoll et al., 2016), we first examined whether NXF1 regulates RNAP II distribution through splicing regulation. Using the RNA-seq data, we indeed detected overall suppression of skipped exon inclusion in NXF1 KD cells (Figure S4, D–F) (see Discussion). However, RNAP II (PAS/GB) distribution change did not correlate with splicing regulation (Figure S4E), refuting the possibility that splicing alternation leads to RNAP II distribution.
We then asked whether NXF1 associates with RNAP II by carrying out Flag immunoprecipitation from cells stably expressing Flag-NXF1 at the physiological level (Figure S4G). Significantly, Flag-NXF1 associated CFI-68 and RNAP II, and these associations were partially resistant to RNase A treatment (Figure 5G). As a control, the association of CFI-68 with eIF4A3, a component of the exon-junction complex that was not known to interact with CFI-68, was abolished upon RNase A treatment (Figure 5G). Conversely, the RNAP II antibody, but not the control IgG, co-precipitated Flag-NXF1 even in the presence of RNase A (Figure 5H). These data indicate that NXF1 associates with RNAP II through protein-protein interactions. In further support of this, ChIP-PCR data showed that Flag-NXF1 could be readily detected in the GB of two genes that showed 3’UTR shortening upon NXF1 KD, RAB10 and TMCC1 (Figure 5I). Together, these data indicate that NXF1 might regulate transcriptional elongation by interacting with transcribing RNAP II.
NXF1 and CFI-68 both promote cytoplasmic accumulation of long 3’UTR isoforms
We next wanted to examine whether NXF1 plays a role in differentiating nuclear export of long and short 3’UTR isoforms. To this end, we carried out 3’READS+ analysis of nuclear and cytoplasmic RNAs from Cntl and NXF1 KD cells (Figure 6A). Western blot analysis confirmed the purity of nuclear and cytoplasmic fractions (Figure 6B). We next calculated the ratio of RNA abundance in the cytoplasm vs. nucleus, named the C/N index, for each transcript with a defined PAS. As reported previously (Neve et al., 2016), long 3’UTR isoforms generally had a lower C/N than short ones in both cells (Figure 6C). Importantly, the C/N difference between two APA isoforms (ΔC/N) correlated with the aUTR size (Figure 6D), indicating that 3’UTR size in general negatively impacts mRNA export. Interestingly, the ΔC/N values were more conspicuous in NXF1 KD cells (Figure 6E), indicating that nuclear export of long 3’UTR isoforms requires NXF1 to a greater extent than short isoforms.
Figure 6. Differential impacts of NXF1 on nuclear export of different transcripts, See alsoFigure S5.

(A) Schematic of experimental design.
(B) Western blot analysis to examine purity of nuclear and cytoplasmic fractions. MTR4 and Tubulin served as nuclear and cytoplasmic markers, respectively.
(C) 3’UTR APA isoform abundance difference between cytoplasm and nucleus (C/N, log2) in siCntl, siNXF1, and siCFI-68 cells.
(D) Genes were divided into five bins based on aUTR size. Median Δlog2 (C/N) between dPASand pPAS isoforms in each gene bin is shown for siNXF1 (red), siCFI-68 (blue) or siCntl (gray) cells. Data are shown as mean ± s.d..
(E) Cumulative distribution of C/N difference between 3’UTR isoforms in siNXF1 (red line), siCFI-68 (blue line) or siCntl (black line) cells.
(F) Correlation between gene features and Δlog2 (C/N) of transcripts in siNXF1 or siCFI-68 cells.
Because NXF1 physically interacts with CFI-68 (Ruepp et al., 2009; Figure S3), which preferentially binds the UGUA motif in long 3’UTR isoforms (Li et al., 2015; Zhu et al., 2018), we reasoned that CFI-68 and NXF1 might cooperate to facilitate long 3’UTR isoform export. 3’READS+ analysis of the nuclear and cytoplasmic RNAs from CFI-68 KD cells indeed showed that the ΔC/N values were more prominent in CFI-68 KD cells than Cntl cells (Figure 6, C-E). Note that CFI-68 KD did not affect the nuclear localization of NXF1, and vice versus (Figure S5A, B).
Using a linear regression model to analyze transcript features associated with decreased C/N ratios in KD cells, we indeed found 3’UTR size as one of the top features related to decreased nuclear export (Figure 6F). Interestingly, all top features identified with APA regulation by CFI-68 (Figure 3B) were also found associated with decreased nuclear export (Figure 6F), indicating CFI-68 impacts PAS usage and mRNA export in a concerted fashion. Several other features were associated with increased C/N after NXF1 KD, including EJC density, number of coding exons, transcript length, and CDS size (Figure 6F), suggesting a connection between NXF1-mediated nuclear export and splicing (see Discussion). Using a NXF1 iCLIP data set previously generated in mouse cells (Muller-McNicoll et al., 2016), we found that NXF1 binding to transcripts is greater in aUTRs than cUTRs (Figure S5C), and NXF1 binding is enriched near the PAS, especially the dPAS (Figure S5D). Together, these data indicate that both NXF1 and CFI-68 play a role in nuclear export of long 3’UTR transcripts.
NXF1 is especially required for nuclear export of long 3’UTR isoforms regulated by CFI-68
To corroborate our global findings, we constructed a set of Smad reporter plasmids containing the same CDS with variable 3’UTR lengths (Figure 7A). When transfected into HeLa cells, cytoplasmic abundance of a transcript progressively decreased and its nuclear abundance concomitantly increased as 3’UTR size increased (Figure 7A), indicating that long 3’UTRs indeed have a negative role in mRNA export. This result was also confirmed with a similar set of β-globin reporters (Figure S6A). Importantly, the same sequences inserted into the 5’ region of the reporter did not affect mRNA export (Figure S6B), indicating that the export alteration was not caused by sequence per se. We next wanted to examine whether nuclear export of long 3’UTR isoforms are more sensitive to NXF1 expression levels. Because a full depletion of NXF1 would block all mRNAs from export, we partially knocked down NXF1 (less than 30% KD, Figure 7B, C). While nuclear export of wS mRNA decreased by 2.82-fold after NXF1 KD, the effect was much stronger with the wS-3UTR mRNA (8.74-fold, Figure 7B), indicating that long 3’UTR transcripts are more reliant on NXF1 for nuclear export than short 3’UTR transcripts.
Figure 7. NXF1 and CFI-68 cooperatively facilitate nuclear export of mRNAs with long 3’UTRs, See alsoFigure S6.

(A) Top, schematic of Smad constructs. Bottom, FISH to detect the N/C distribution of Smad reporter mRNAs at 8 hr after transfection. The graph shows N/C ratios of different reporters. Data are represented as mean ± s.d..
(B) DNA constructs were microinjected into the nuclei of Cntl or NXF1 KD (24 hr) cells, followed by FISH and DAPI staining 2 hr post-injection. The graph shows N/C ratios of mRNAs in different cells…
(C) Western blot examining the protein level of NXF1 in cells transfected with siCntl or siNXF1 for 24 hr.
(D) Top, schematic of Smad constructs. DNA constructs were transfected into HeLa cells, followed by FISH and DAPI staining 8 hr post-transfection. The graph shows N/C ratios of mRNAs in different cells..
(E) The wS-3UTR-UGUA construct was microinjected into the nuclei of Cntl or CFI-25 and CFI-68 double KD cells, followed by FISH and DAPI staining 2 hr post-injection. The graph shows N/C ratios of the mRNA in different cells..
(F) Western blot examining the knockdown efficiency of CFI factors. (G) DNA constructs were microinjected into the nuclei of Cntl or NXF1 KD (24 hr), followed by FISH and DAPI staining 2 hr post-injection. The graph shows N/C ratios of mRNAs in different cells..
(H) A model summarizing the roles of NXF1 in coordinating transcriptional dynamics, APA and mRNA export.
To confirm the role of CFI in long 3’UTR isoform export, we inserted five UGUA tandem repeats into the wS-3UTR construct (Figure 7D). Consistent with our global result, this insertion significantly enhanced nuclear export of the wS-3UTR mRNA (Figure 7D). Importantly, this enhancement was inhibited when CFI was knocked down (Figure 7E, F), indicating that UGUA facilitates mRNA export via CFI. Further, long 3’UTR transcripts with UGUA motifs depended on NXF1 for nuclear export to a greater extent, because a partial KD of NXF1 blocked nuclear export of the wS-3UTR-UGUA mRNA, but not the wS mRNA (Figure 7G). Together, these results depict a mechanism by which NXF1 promotes nuclear export of long 3’UTR isoforms, especially those with UGUA motifs that are recognized by CFI-68.
DISCUSSION
In this study, we report that mRNA export receptor NXF1 enhances expression of long 3’UTR isoforms and facilitates their nuclear export. We show NXF1-mediated APA regulation is attributable to its impact on RNAP II dynamics, likely through direct interactions between NXF1 and RNAP II. In addition, the enhancement of nuclear export of long 3’UTRs by NXF1 is mediated by its interaction with CFI-68, which preferentially binds to UGUA motifs enriched in long 3’UTR isoforms. Therefore, our work implicates NXF1 as a nexus for gene expression, coordinating transcriptional dynamics, pre-mRNA processing, and nuclear export.
NXF1-mediated APA regulation
Accumulating evidence indicates that mRNA export is intertwined with 3’ end processing (Johnson et al., 2009; Johnson et al., 2011; Katahira et al., 2013; Tran et al., 2014). Here we show a general role of mRNA export factors in promoting distal PAS usage. Differential KD efficiencies notwithstanding, the mRNA export receptor NXF1 had the most significant effect on APA among all the export factors analyzed. Whether other mRNA export factors regulate APA through NXF1 requires further investigation.
We show that NXF1 and CFI-68 use different mechanisms to regulate APA. Notably, SR proteins, another group of NXF1 adaptors, have been found to regulate APA (Muller-McNicoll et al., 2016). Using RNA-seq data previously generated from SRSF3 KD cells, which showed general 3’UTR shortening (Muller-McNicoll et al., 2016), we found that SRSF3-regulated APA events only marginally overlapped with those regulated by NXF1 or CFI-68 (Figure S7), indicating that SR proteins may use another distinct mechanism to regulate APA.
Both RNAP II ChIP-seq and nascent RNA-seq data indicate enhanced enrichment of RNAP II at the 3’ end of genes in NXF1 KD cells. While we cannot rule out the possibility of impaired RNAP II recruitment at the promoter, a more plausible explanation is slowdown of RNAP II elongation along the gene. A slower RNAP II would make proximal PAS more likely to be used, leading to 3’UTR shortening. This effect would make long genes more susceptible because of their greater dependence on elongation to complete the transcriptional cycle. Similarly, transcription of genes in high AT content regions, which have a high elongation rate at the baseline level (Veloso et al., 2014), may also be more sensitive to elongation rate alternation in NXF1 KD cells. Therefore, the effect of NXF1 on RNAP II dynamics could also explain features associated with APA regulation by NXF1.
It is to be noted that while ChIP-seq and nascent RNA-seq provide similar conclusions, they interrogate distinct aspects of RNAP II. Whereas ChIP-seq identifies RNAP II positions along the genome, it does not immediately reveal whether the RNAP II is engaged in transcription. In contrast, nascent RNA-seq identifies transcriptionally engaged RNAP II. We note that metagene profiles based on ChIP-seq are slightly different than those from nascent RNA-seq, especially at the TSS region. This may be due to the fact that RNAP II population enriched at the TSS is largely in a paused state or with a slow elongation rate, making the corresponding nascent RNAs refractory to the short time 4sU labelling used in our protocol.
We show that NXF1 interacts with RNAP II. However, how this interaction translates into regulation of RNAP II elongation remains unknown. NXF1 is localized at the nuclear pore and associates with multiple nuclear pore complex (NPC) factors (Bachi et al., 2000; Forler et al., 2004; Katahira et al., 1999; Segref et al., 1997) (Table S2), which have been implicated in transcription regulation (Casolari et al., 2004; Pascual-Garcia et al., 2017; Tan-Wong et al., 2009; Texari et al., 2013; D’Angelo, 2018; Rohner et al., 2013; Sood and Brickner, 2014). It is thus possible that NXF1 regulates RNAP II elongation through NPC factors. In line with this, Mex67, the yeast NXF1 counterpart, targets actively transcribed genes to nuclear pores (Dieppois et al., 2006). Alternatively, NXF1 may regulate RNAP II elongation through other factors. In this vein, it is worth mentioning that NXF1 has been found to interact with the transcription factor IRF5 (Fu et al., 2017). More future studies are needed to further unravel the mechanism(s) by which NXF1 alters RNAP II dynamics.
NXF1-mediated nuclear export control of long 3’UTR isoforms
Here, we show that long 3’UTRs generally inhibit mRNA export. While the underlying mechanism remains unclear, we found that NXF1 plays an important role in long 3’UTR isoform export. NXF1 interacts with adaptors that mediate its recruitment to mRNAs. One of such adaptors is CFI-68, which specifically binds the UGUA motif (Ruepp et al., 2009), typically enriched for distal PASs (Wang et al., 2018), and promotes long 3’UTR isoform export. We note that other RBPs may have similar functions. For example, both SR proteins and ALYREF have been reported to bind to 3’UTRs of mRNAs (Muller-McNicoll et al., 2016; Shi et al., 2017), which may also facilitate long 3’UTR mRNA export.
Consistent with the functional coupling between mRNA export and splicing, several splicing-related features were found to be associated with NXF1-mediated nuclear export (Figure 6F). Intriguingly, transcripts with more exons appear to have a higher C/N ratio in NXF1 KD cells. One possibility is that NXF1 KD leads to greater intron retention which results in increased nuclear degradation of mRNAs, and transcripts with more introns have a greater chance of having retained introns.
In summary, we propose a model in which NXF1 coordinates transcriptional elongation, 3’ end processing, and nuclear export (Figure 7H): in normal cells, NXF1 promotes RNAP II elongation, leading to preferential usage of the distal PASs. The resultant long 3’UTR isoforms are exported with the help of NXF1 and CFI-68 owing to the UGUA motif. When the NXF1 level is low, RNAP II elongation rate is decreased, leading to preferential usage of proximal PASs and production of short 3’UTR isoforms. Short 3’UTR isoforms are exported into cytoplasm with less dependence on NXF1 or CFI. The coordinated regulation of transcription, RNA processing and export appears to be a common theme in yeast (Minocha et al., 2018). It would be interesting in the future to examine how this mechanism is executed in cells under different conditions, such as cell differentiation and stress, when cells display global 3’UTR length changes (Tian and Manley, 2017), and in cells in which NXF1 is inhibited.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to the lead contact Hong Cheng (hcheng@sibcb.ac.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
HeLa and HEK293 cells were cultured in DMEM supplemented with 10% FBS (Biochrom). For establishment of Flag-NXF1 and Flag-Cntl (Flag-eIF4A3) stable expression cell lines, HAGE-Flag-NXF1 or HAGE-Flag-eIF4A3 plasmid together with psPAX2 and pMD2.G plasmids were co-transfected into HEK293 cells. The media containing viruses were harvested after 48 hr and added to HeLa cells, and then single green fluorescence cell was sorted by Flow Cytometer (AriaII, BD).
METHOD DETAILS
Plasmids
The 3M-SVL plasmid was a kind gift of Yongsheng Shi (Shi et al., 2009). The VSVM-GFP plasmid was kindly provided by Elisa Izaurralde (von Kobbe et al., 2000). To make the Flag-NXF1 and Flag-CFI-68 constructs, the coding regions of NXF1 and CFI-68 were separately cloned into p3xFlag-CMV10 (Sigma-Aldrich). To make the Flag-NXF1 siRNA resistant construct, the siRNA target sequence GCGCCATTCGCGAACGATTTT was replaced with AGTGCAATACGAGAGCGTTTC by mutagenesis. To make GST-NXF1 constructs, the coding region of NXF1 was cloned into pGEX-4T1 (Pharmacia). For HA-CFI-68 construct, the coding region of CFI-68 was cloned into HA-pcDNA3.0 (Invitrogen). For HAGE-Flag-NXF1 and HAGE-Flag-eIF4A3 constructs, the coding regions of NXF1 and eIF4A3 were separately cloned into pHAGE-fEF1a-IRES-ZsGreen. For wG and wS constructs with different 3’UTR lengths, different copies of the β-globin cDNA sequence were cloned into the 3’UTR of β-globin and Smad constructs, with 1 UTR, 2 UTR and 3 UTR indicating 1, 2, or 3 copies of β-globin cDNAs. Plasmids encoding GST-eIF4A3, β-globin WT, Smad WT were previously described (Chi et al., 2013).
RNAi and transfection
siRNA and DNA transfections were performed with RNAi MAX (Invitrogen) and Lipofectamine 2000 (Invitrogen), respectively, according to the manufacturer’s protocols. siRNA targeting sequences are shown in Table S3. Note that for UAP56 KD, cells were treated with both UAP56 and URH49 siRNAs. For rescue experiments, siRNAs were transfected into HeLa cells and expression plasmids were transfected 6 hr post transfection of siRNAs.
Reverse transcription and PCRs
For reverse transcription, total RNA was treated with RNase-free RQ DNase I (Promega) for 2 hr at 37 °C, and cDNA was synthesized from 1μg of RNA with an oligo dT primer using M-MLV reverse transcriptase (Promega). For 3’ end RT-qPCR, cDNA was synthesized using a P7-t25-vn oligo-dT primer, and PCR was performed using P7 and gene-specific primers listed in Table S3. Quantitative PCR was carried out using the GoTaq qPCR Master Mix (Promega) according to manufacturer’s protocol. Primer sequences are listed in Table S3.
DNA microinjections, FISH and immunofluorescence
Microinjections were carried out as previously described (Shi et al., 2017). HeLa cells were plated on fibronectin-coated 20-mm coverslips at the bottom of 35-mm dishes. Plasmid DNA (50 ng/μl) was co-injected with FITC-conjugated 70-kDa dextran. For each experiment, 80–100 cells were microinjected. Fifteen min post-injection, transcription was terminated with α-amanitin (1 μg/ml; Sigma-Aldrich), and incubation continued for the desired period of time before fixation. To detect poly(A)+ RNAs, HeLa cells were fixed with 1% formaldehyde/Acetic Acid in phosphate buffered saline (PBS). Cells were washed with PBS three times and permeabilized with 0.5% Triton in PBS for 5 min, followed by washes with 2 × saline-sodium citrate buffer (SSC) twice and incubation at 42 °C with a high performance liquid chromatography-purified Alexa 548-conjugated oligo dT(70) probe for 16 hr. Cells were then washed with 0.5 × SSC twice and 2 × SSC twice, followed by DAPI staining. Images were captured with a DP72-CCD camera (Olympus) on an inverted microscope using the DP-BSW software (Olympus).
To analyze the distribution of β-globin and Smad reporter mRNAs, an HPLC-purified Alexa 548-conjugated 70 nt probe (Vector probe) that hybridizes to the pcDNA3 vector sequence was used. HeLa cells transfected with β-globin and Smad constructs were fixed with 4% PFA in 1× PBS for 15 min, followed by washes with PBS three times and permeabilization with PBS/0.1% Triton for 15 min. Cells were then washed with 1× PBS three times and 1× SSC/50% formamide twice, and were incubated with the vector probe at 37 °C for 12–16 hr. The cells were then washed with 1× SSC/50% formamide four times, followed by DAPI staining. Images were captured on an inverted confocal microscope (Olympus). Alexa 548 conjugated vector probe was used to detect the distribution of β-globin or Smad mRNA. Images were captured with a DP72-CCD camera (Olympus). FISH quantitation was carried out using the ImageJ software (National Institutes of Health). N/C ratios were calculated as described (Valencia et al., 2008).
For immunofluorescence, cells were rinsed with 1 × PBS and fixed with 4% paraformaldehyde for 15 min at room temperature, followed by permeabilization with 0.1% Triton X-100. The cells were subjected to immunofluorescence staining with CFI-68 (1:200) or Flag (1:1000) antibody at room temperature for 1 hr. After three washes with 1 × PBS, the cells were incubated with Alexa 546-labeled anti-rabbit or anti-mouse secondary antibody (1:1000) in blocking buffer (1× PBS, 0.1% Triton X-100, and 2 mg/ml BSA) at room temperature for 1 hr. Then cells were washed with 1 × PBS three times for 15 min each, followed by DAPI staining. Images were captured with a DP72-CCD camera (Olympus) on an inverted microscope using the DP-BSW software (Olympus).
Nuclear and cytoplasmic RNA preparation
For nuclear and cytoplasmic RNA preparation, 1 × 107 HeLa cells were washed with ice-cold PBS and harvested on ice. Cells were suspended in hypotonic buffer (10mM HEPES, pH 7.9/1.5mM MgCl2/10mM KCl/0.2mM PMSF/0.5mM DTT) and incubated for 10 min on ice. The swollen cells were dounce homogenized, followed by centrifugation. The supernatant and pellet were cytoplasmic and nuclear extracts, respectively. RNAs from nuclear or cytoplasmic fractions were extracted using TRIzol (Invitrogen).
ChIP
HeLa and Flag-NXF1 stable expression cells were crosslinked with 1% formaldehyde at room temperature for 10 min. Crosslinking was quenched in 125 mM glycine at room temperature for 5 min, and cells were washed twice with cold PBS and then collected by centrifugation (3000 rpm, 5 min). Cells were re-suspended and sonicated in lysis buffer (50 mM HEPES ph7.6, 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.1% DOC, 1% SDS) for 20 min. Chromatin was centrifuged at 14,000 rpm for 15 min, and supernatants were diluted 10-fold in dilution buffer (50 mM Tris-Cl, 167 mM NaCl, 1% Triton X-100, 0.1% DOC and 1 mM EDTA). Supernatant was pre-cleared in 50μl of protein G Dynabeads, which were pre-incubated in 1mg/ml herring sperm DNA and 1% BSA in 40 °C overnight. Lysates were incubated with 4μg of RNAP II antibody (Santa Cruz), Flag antibody (Sigma-Aldrich) or IgG (Santa Cruz) at 4 °C overnight. 50 μl of beads were added and rotated at room temperature for 3 hr. Beads were washed once in wash buffer (10 mM Tris PH 8.0, 1 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.1% DOC, 0.2% Sarkosyl), once in high salt buffer (10 mM Tris PH 8.0, 1 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.1% DOC, 0.2% Sarkosyl), once in LiCl buffer (10 mM Tris PH8.0, 1 mM EDTA, 250 mM LiCl, 1% NP40, 1% DOC), and twice in TE buffer (10 mM Tris, 1 mM EDTA, pH 8.0). Input and beads were re-suspended in PK buffer (100 mM Tris pH 8.0, 12.5 mM EDTA pH 8.0, 200 mM NaCl, 1% SDS) with 1μl RNase A (10 mg/ml). After incubation at 37 °C for 1 hr, 10 μl PK (10 mg/ml) was added. Crosslinking was reversed in 200 mM NaCl at 60 °C for 7 hr. DNA was phenol-chloroform-extracted and ethanol-precipitated. For sequencing, 5 ng of DNA fragments were generated using Next Ultra DNA Library Prep Kit for Illumina (NEB) according to the manufacturer’s instructions. Briefly, DNA fragments were repaired by End Prep Enzyme Mix and were ligated to adaptors by Ligase Master Mix. Libraries with approximately 500-bp were purified by AMPure XP Beads, followed by PCR amplification and gel purification. The barcoded DNA libraries were quantified with Qubit and Agilent Bioanalyzer 2100, and sequenced on an Illumina Hiseq X ten using a double-read protocol of 300 cycles at Geneseeq Technology, Nanjing, China. For qPCRs, 0.3 ng of DNA and the GoTaq qPCR Master Mix (Promega) were used according to manufacturer’s protocol. Primer sequences are listed in Table S3.
GST pull downs
35S-labeled CFI-68 was produced using the TNT T7 quick-coupled transcription/translation kit (Promega). 10 μl of in vitro translation mixtures was rotated at 4 °C overnight with 8μg of purified GST fusion proteins and 20μl of Glutathione Sepharose 4 Fast Flow beads in pull-down buffer (1× PBS/0.1% Triton/0.2 mM PMSF/protease inhibitor). The beads were then washed with pull-down buffer five times. Proteins were eluted with SDS loading buffer, separated by SDS-PAGE, and visualized by autoradiography.
Immunoprecipitations
About 4 × 106 of cells were harvested and re-suspended in 1 ml of 1 × TBS (Tris-buffered saline)/0.5% Triton X-100. After sonication and centrifugation, lysates were incubated at 30 °C for 20 min in the presence or absence of 50 ng/μl of RNase A. After centrifugation, lysates were incubated with 5μg of RNAP II antibody (BioLegend), Flag antibody (Sigma-Aldrich) or IgG (Santa Cruz) at 4 °C overnight, followed by rotation with nProteinA Sepharose beads (GE) at 4 °C for an additional 2 hr. The samples were washed four times and eluted with SDS loading buffer for Western blot analysis.
3’READS+
3’READS+ was carried out as previously described (Zheng et al., 2016). Briefly, RNA samples (total or nuclear or cytoplasmic RNA) were prepared from HeLa cells with or without siRNA treatment. Poly(A)+ RNA in 10 μg of total RNA was captured using oligo (dT)25 magnetic beads (NEB) in the binding buffer (10 mM Tris-Cl, pH 7.5, 150 mM NaCl, 1 mM EDTA, and 0.05% TWEEN 20) and fragmented on the beads using RNase III (NEB) at 37 °C for 15 min. After washing with the binding buffer, poly(A)+ fragments were eluted from the beads with TE buffer (10 mM Tris-Cl, 1 mM EDTA, pH 7.5) and precipitated with ethanol, followed by ligation to heat-denatured 5’adaptor (5’-CCUUGGCACCCGAGAAUUCCANNNN, Sigma-Aldrich). The ligation products were captured by biotin-T15-(+TT)5 oligo attached to Dynabeads MyOne Streptavidin C1 (Life Technologies). After washing with washing buffer (10 mM Tris-Cl, pH 7.5, 1 mM NaCl, 1 mM EDTA, and 0.05% TWEEN 20), RNA fragments on the beads were incubated with RNase H (Epicentre) at 37 °C for 30 min. Following washing with RNase H buffer, RNA fragments were eluted from the beads in elution buffer (1 mM NaCl, 1 mM EDTA, and 0.05% TWEEN 20) at 50 °C, precipitated with ethanol, and then ligated to heat-denatured 5’ adenylated 3’ adapter (5’-rApp/NNNGATCGTCGGACTGTAGAACTCTGAAC/3ddC, where N is a random nucleotide). The ligation products were then precipitated and reverse transcribed using M-MLV reverse transcriptase (Promega), followed by PCR amplification using the Phusion high-fidelity DNA polymerase (NEB) and bar-coded PCR primers for 12–18 cycles. PCR products were size-selected twice with AMPure XP beads (Beckman Coulter). The size and quantity of the libraries eluted from the AMPure beads were examined using a high sensitivity DNA kit on an Agilent Bioanalyzer (Agilent). Libraries were sequenced on an Illumina HiSeq 2000 or NextSeq machine.
RNA-seq of total RNA
For RNA sequencing, 5 ug of total nuclear or cytoplasmic RNA was used for poly(A)+ RNA selection. Stranded cDNA libraries were generated with TruSeq Stranded Total RNA Sample Prep Kit (Illumina) according to the manufacturer’s instructions. The cDNA libraries were sequenced on an Illumina Hiseq X ten using a double-read protocol with 300 cycles at Geneseeq Technology, Nanjing, China.
RNA-seq of nascent RNAs
Nascent RNA sequencing was largely adapted from the NET-seq (Mayer et al., 2015) and TT-seq methods (Schwalb et al., 2016), with some modifications. Briefly, cells at 75~90% confluency on a 15-cm dish were treated with 500 μM 4-thiouridine (4sU) for 5 min before harvest. Cells were washed with ice-cold PBS buffer, and scraped off from the dish in 10 ml of ice-cold PBS while on ice. After centrifugation at 420 × g at 4 °C for 2 min, cells were lysed by incubation with 4 ml of ice-cold HLB+N buffer (10 mM Tris-HCl (pH 7.5), 10 mM NaCl, 2.5 mM MgCl2 and 0.5% (vol/vol) NP-40) on ice for 5 min. One ml of ice-cold HLB+NS buffer (10 mM Tris-HCl (pH 7.5), 10 mM NaCl, 2.5 mM MgCl2, 0.5% (vol/vol) NP-40 and 10% (wt/vol) sucrose) was underlaid to the cell lysate, and the lysate was centrifuged at 420× g at 4 °C for 5 min. After removal of the supernatant, the pelleted nuclei were re-suspended in 125 l of ice-cold NUN1 buffer (20 mM Tris-HCl (pH 7.9), 75 mM NaCl, 0.5 mM EDTA and 50% (vol/vol) glycerol) by pipetting up and down, mixed with 1.2 ml of ice-cold NUN2 buffer (20 mM HEPES-KOH (pH 7.6), 300 mM NaCl, 0.2 mM EDTA, 7.5 mM MgCl2, 1% (vol/vol) NP-40 and 1 M urea) by pulse vortexing, and incubated on ice for 5 min. The mixture was centrifuged at 14,000× g at 4 °C for 2 min to pellet chromatin. After removal of the supernatant completely, RNA was extracted from the chromatin pellet using 1 ml TRIzol.
Extracted chromatin RNA from each sample (4 μg) was fragmented at 94°C for 1 min using NEB Next Magnesium RNA Fragmentation Module (NEB). The reaction was then diluted to 100 μl with nuclease-free H2O, mixed with 5 μg of glycogen, 10 μl of 3M Sodium Acetate (pH 5.5), and 300 μl of ethanol. After precipitation at −80 °C for 45 min, the RNA was pelleted by centrifugation at 14,000 × g at 4 °C for 20 min. The pellet was washed with cold 75% ethanol, air-dried, and dissolved in 5 μl of 2 μM of 5’ adenylated 3’ blocked 3’ adapter (5’-Aden/NNNNNNGATCGTCGGACTGTAGAACTCTGAAC/3ddC, where N is a random nucleotide). After incubation at 70 °C for 2 min, the sample was immediately chilled on ice, and incubated with ligation mi× at 25 °C for 2 hr. The ligation mix contained 2 μl of nuclease-free H2O, 6 μl of 50% PEG 8000, 1.5 μl of 10× T4 RNA ligase buffer, 0.25 μl of SuperaseIn (20 U/μl), and 0.25 μl of truncated RNA ligase 2 (KQ, 200 U/μl). To biotinylate 4sU-labeled RNA fragments, the ligation reaction was mixed with 125 μl of nuclease-free H2O, 20 μl of 10 × biotinylation buffer (100 mM Tris pH 7.4, 10 mM EDTA), and 40 μl of biotin-HPDP (1 μg/μl in DMF). The mixture was incubated at room temperature on a rotator for 1.5 hr. The reaction was extracted with 200 μl of chloroform three times and the RNA in the aqueous phase was precipitated.
Biotinylated RNA pellet was dissolved in 50 μl of nuclease-free H2O and denatured by incubation at 70 °C for 2 min. After chilling on ice, the RNA was mixed with 50 μl of Streptavidin C1 Dynabeads in 2 × B&W buffer (10 mM Tris-HCl (pH 7.5), 1 mM EDTA, and 2 M NaCl) supplemented with 0.5 μl of SuperaseIn (20 U/μl). The mixture was incubated on a rotator at room temperature for 15 min. After removal of the supernatant on a magnetic stand, the beads were washed three times with 0.5 ml of 1 × B&W buffer (5 mM Tris-HCl (pH 7.5), 0.5 mM EDTA, 1 M NaCl) at 65 °C, followed by three washes with 1 × B&W buffer at room temperature. After complete removal of the washing buffer, the beads were re-suspended with 30 μl of the PNK reaction mix, which contained 3 μl of 10 × PNK buffer, 22 μl of H2O, 3 μl of 10 mM ATP, 1.5 μl of PNK (10U/μl), and 0.5 μl of SuperaseIn (20 U/μl). The mixture was rotated in an oven at 37 °C for 30 min and then mixed with 170 μl of 1 × B&W buffer containing 100 mM DTT and incubated at room temperature for 3 min to elute 4sU-labled RNA. Isolated 4sU-labeled RNA was then subject to 5’ adapter ligation, reverse transcription and cDNA library construction, similar to the 3’READS+ method.
FISH quantitation
FISH quantitation was carried out using ImageJ software (National Institutes of Health). N/C ratios were calculated as described (Valencia et al., 2008). Measurements were obtained for background fluorescence (Sb), fluorescence in the nucleus (Sn), total fluorescence of the cell (St), area of the nucleus (An), and area of the cell (At). The cytoplasmic (C) signal of mRNA was calculated as: C = At (St-Sb) − An (Sn-Sb), N/C ratios were calculated as N/C = An (Sn-Sb)/C. For each sample, at least 30 cells were analyzed for statistical test.
Analysis of 3’READS+ data
The sequence corresponding to the 5’ adapter was first removed from raw 3’READS+ reads using Cutadapt (Martin, 2011) (Table S4). Reads with short inserts (<23 nt) were discarded. The retained reads were then mapped to the human genome (hg19) using bowtie2 (local mode)(Langmead and Salzberg, 2012). The six random nucleotides at the 5’ end (derived from the 3’ adapter) were removed before mapping using the setting “−5 6” in bowtie2. Reads with a mapping quality score (MAPQ) ≥10 were kept for further analysis. Reads with ≥2 non-genomic 5’Ts after alignment were called PAS reads. PASs within 24 nt from each other were clustered as previously described (Hoque et al., 2013). PASs with less than 5 PAS reads across all the samples were not used. The retained PASs were then annotated with the RefSeq database. The PAS read counts mapped to genes were normalized by the median ratio method in DESeq (Anders and Huber, 2010). Only APA isoforms with read count greater than 5 in at least one pair of comparing samples were used. The two most abundant APA isoforms (based on PAS reads) with PASs in the 3’UTR of the last exon were selected for 3’UTR APA analysis. Significant APA events were those with relative abundance change > 5% and p-value < 0.05 (Fisher’s exact test) in at least one replicate and with consistent regulation across all replicates. The Kolmogorov–Smirnov (K-S) test was used to compare data distribution between samples. The aUTR size was the distance between proximal and distal PASs in the 3’UTR. Relative Expression Difference (RED) was calculated as the differencein log2 (ratio) of abundances of two PAS isoforms (dPAS vs. pPAS) between two samples. The weighted mean of 3’UTR size for each gene was based on 3’UTR sizes of all 3’UTR APA isoforms, weighted by the expression level of each isoform based on the number of PAS reads. To statistically assess the significance of overall APA differences between samples with different sequencing depths, we used the Global Analysis of Alternative Polyadenylation (GAAP) methodwe previously developed (Li et al., 2015). Briefly, one million PAS reads from each sample were randomly sampled through bootstrapping, and a median RED (mRED) was calculated each time using sampled reads. Sampling was carried out 20 times and the mean mRED and standard deviation were calculated. False discovery rate (FDR) was based on how many times the mRED was below or above zero, depending on the mean.
Analysis of ChIP-seq data
ChIP-seq reads were mapped to the hg19 genome assembly using BWA-MEM (0.7.15) (Li and Durbin, 2009) (Table S5). The read count of unique reads mapped to each position was normorlized to one million (UPM). The genomic position corresponding to the 5’ end of each read was identified by a Python script using the HTSeq software package (Anders et al., 2015). For metagene analysis, normalized reads density in genomic bins of each gene was first calculated as UPM divided by the genomic size of the bin, and the sum of the density of all bins in each gene was normalized to 1. The read density of IP samples was normalized to corresponding density of input samples. The transcription start site (TSS) position of each gene was obtained from RefSeq. The last PAS position was obtained from both 3’READS data and RefSeq.
Analysis of cytoplasm/nucleus fractionation data
Cytoplasm vs. nucleus distribution for each PAS isoform was calculated by log2 (C/N), where C and N were the expression values in cytoplasmic and nuclear fractions, respectively.
Analysis of nascent RNA-seq data
Nascant RNA reads were first trimmed by Cutadapt (Martin, 2011) for nucleotides derived from 5’ and 3’ adapters (Table S6). Trimmed raw reads were mapped to the hg19 genome assembly using STAR (v2.5.2) (Dobin et al., 2013). Only mapped reads without any soft clipping at the 5’ end of read were used for further analysis. The genomic position corresponding to the 5’ end of each read was identified by a Python script using the HTSeq software package (Anders et al., 2015). The 5’ position of a read was used to reflect RNAP II position, and reads mapped to the same genomic position were combined. Unique read count for each position was normorlized to 1 million (UPM) per sample. For metagene analysis, normalized read density in genomic bins of each gene was first calculated as UPM divided by the genomic length of each bin, and the sum of all bins was normalized to 1.
Linear regression analysis using gene features
A linear regression model based on the python scikit-learn library (Pedregosa et al., 2011) was used to examine correlation between gene features and 3’UTR REDs or log2 (C/N) of transcripts. Gene features were extracted from the hg19 RefSeq annotations and 3’READS data including size features (gene size, CDS size, intron size, 5’UTR size, 3’UTR size, aUTR size, cUTR size, transcript length), the dinucleotide AT content features (in gene body, TSS and last 3’ exon), number of coding exons, EJC density (number of exon-exon junctions per length unit), number of UGUA in aUTR (for RED correlation) or in 3’UTR (for log2(C/N) correlation). The significance of each feature was assessed by its individual R2, cumulative R2, and p-value of the model. Features with top individual R2 and p-value are selected and shown.
Gene expression, alternative splicing and 3’UTR APA analysis of RNA-seq
Raw reads from RNA-seq were first mapped to the hg19 genome assembly using STAR (v2.5.2) (Dobin et al., 2013) with default parameters (Table S7). The read count of the coding sequence (CDS) of each gene was summed and then normalized by the median ratio method in DESeq (Dobin et al., 2013). Only genes with more than five reads in all samples were used for further gene expression analysis. Alternative splicing (AS) was analyzed using rMATS (v4.0.2)(Shen et al., 2014) with default parameters. Significant AS events were those with a delta PSI > 5% and p-value < 0.05. Only AS events with at least five reads mapped on splice junctions were used for further analysis.
For 3’UTR APA analysis, the first (proximal) and last (distal) mammal-conserved PASs in the 3’-most exon were extracted from PolyA_DB version 3.2 (Wang et al., 2018). Only genes with more than five reads in both aUTR and cUTR regions were used for further APA analysis. Ratio of read density in cUTR (CDS end to proximal PAS) vs. aUTR (proximal to distal) of each gene was calculated and is named relative expression difference (RED). Difference in RED between knockdown and control samples was used to represent APA changes. Significant APA events were those with a relative abundance change > 5% and p-value < 0.05 (Fisher’s exact test).
Analysis of NXF1 iCLIP data
Genomic positions and corresponding read counts of NXF1 iCLIP sites were obtained from GSE69689 (Müller-McNicoll et al., 2016). Assignment of NXF1 binding sites to cUTRs and aUTRs was based on mammal-conserved 3’UTR PASs in PolyA_DB version 3.2 (Wang et al., 2018). For metagene analysis of iCLIP reads around the first and last PASs, iCLIP reads were mapped to the ±500 bp region of each PAS. The sum of reads was then normalized across genes.
QUANTIFICATION AND STATISTICAL ANALYSIS
FISH quantitation was carried out using ImageJ software (National Institutes of Health). For all RT-qPCR, ChIP-qPCR, and FISH analyzes, the experiments were biologically repeated for three times. All grouped data are presented as mean ± s.d.. Student’s t test was used to determine statistical significance between groups. Plots and indicated statistical analysis were based on Prism software (GraphPad Prism 6.0).
Fisher’s exact test was used to determine the significant level of APA changes. FDR based on data randomization was used to evaluate the significance level of global 3’UTR APA and intronic APA in KD samples. K-S test was used to compare the RED distributions, ChIP-seq signals, nascent RNA levels in different gene sets. The Mann-Whitney-Wilcoxon test was used to compare expression levels of genes with different APA changes. If exact p values are not shown or indicated in legends, they are represented in all figures as follows: *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., p > 0.05, except for Figure 1C and S1C.
DATA AND SOFTWARE AVAILABILITY
All scripts used for data processing and statistical analyses were written in Python, Perl, or R, and are available upon request. Sequencing datasets generated in this study, including those by 3’READS, RNA-seq, ChIP-seq and nascent RNA-seq, have been deposited into the GEO database under the accession number GSE117701.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal Anti-UAP56 | (Chi et al., 2013) | N/A |
| Rabbit polyclonal Anti-ALYREF | (Chi et al., 2013) | N/A |
| Rabbit polyclonal Anti-THOC2 | (Chi et al., 2013) | N/A |
| Rabbit polyclonal Anti-MTR4 | (Fan et al., 2017) | N/A |
| Rabbit polyclonal Anti-CFI-25 | Proteintech | Cat#10322-l-AP |
| Rabbit polyclonal Anti-CFI-68 | Abcam | Cat#ab99347; RRID: AB_10675900 |
| Rabbit polyclonal Anti-NXFl | Proteintech | Cat#10328-l-AP |
| Rabbit polyclonal Anti-RNAP II | SANTA CRUZ | Cat#sc-899; RRID: AB_632359 |
| Mouse monoclonal Anti-RNAP II | BioLegend | Cat#664912 |
| Rabbit IgG | SANTA CRUZ | Cat#sc-2027; RRID: AB_737197 |
| Mouse monoclonal Anti-Tubulin | Sigma-Aldrich | Cat#T5168; RRID: AB_477579 |
| Mouse monoclonal Anti-Flag | Sigma-Aldrich | Cat#F3165; RRID: AB_259529 |
| Mouse monoclonal Anti-GAPDH | Abcam | Cat#ab8245 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Uridine 5’-Triphosphate, [α−32P] | PerkinElmer | Cat#BLU007X250UC |
| Methionine, L-[35S] | PerkinElmer | Cat#NEG709A500UC |
| Actinomycin D | Sigma-Aldrich | Cat#A4262 |
| 4sU | Sigma-Aldrich | Cat#T4509 |
| α-amanitin | Sigma-Aldrich | Cat#A2263 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| Lipofectamine® RNAiMAX | Invitrogen | Cat# 13778150 |
| protein G Dynabeads | Invitrogen | Cat#10004D |
| nProteinA Sepharose 4 Fast Flow beads | GE Healthcare | Cat# 17–5280–02 |
| Glutathione Sepharose 4 Fast Flow beads | GE Healthcare | Cat# 17–5132–02 |
| Critical Commercial Assays | ||
| TNT T7 quick-coupled transcription/translation kit | Promega | Cat#L1170 |
| Deposited Data | ||
| 3’READS+data | This paper | GSE117701 |
| ChIP-seq data | This paper | GSE117701 |
| Nascent RNA-seq data | This paper | GSE117701 |
| RNA-seq of total RNA data | This paper | GSE117701 |
| Experimental Models: Cell Lines | ||
| HeLa | (Chi et al., 2013) | N/A |
| HEK293 | (Chi et al., 2013) | N/A |
| Flag-NXFl stable expression cells | This paper | N/A |
| Oligonucleotides | ||
| siRNAs used in this study see Table S3 | This paper | N/A |
| Cloning primers see Table S3 | This paper | N/A |
| RT-qPCR primers see Table S3 | This paper | N/A |
| 3’ end RT-qPCR primers see Table S3 | This paper | N/A |
| ChIP qPCR primers see Table S3 | This paper | N/A |
| 5’ adaptor for making sequencing libraries: 5 ‘-CCUUGGCACCCGAGAAUUCCANNNN | Sigma-Aldrich | N/A |
| 5’ adenylated 3’ blocked 3’ adapter for making sequencing libraries: 5 ‘-Aden/NNNNNNGATCGTCGGACTGTAGAA CTCTGAAC/3ddC, | Bioo Scientific | N/A |
| Reverse PCR primer for making sequencing: 5’-AATGATACGGCGACCACCGAGATCTAC ACGTTCAGAGTTCTACAGTCCGA | Sigma-Aldrich | N/A |
| Indexed forward PCR primers (index region in bracket): 5’-CAAGCAGAAGA CGGCATACGAGAT[NNNNNN]GTGACTG GAGTT CCTTGGCACCCGAGAATTCCA | Sigma-Aldrich | N/A |
| Vector probe: Alexa 548-conjugated 70 nt probe 5 ‘-AAGGCACGGGGGAGGGGCAAACAACAG ATGGCTGGCAACTAGAAGGCACAGTCGAG GCTGATC AGCGGGT-3’ | This paper | N/A |
| Poly(A)+ RNAs probe: Alexa 548-conjugated oligo dT(70) probe | (Chi et al., 2013) | N/A |
| Recombinant DNA | ||
| Plasmid: Flag-NXFl | This paper | N/A |
| Plasmid: Flag-NXFlR | This paper | N/A |
| Plasmid: Flag-CFI-68 | This paper | N/A |
| Plasmid: HA-CF-68 | This paper | N/A |
| Plasmid: HAGE-Flag-NXFl | This paper | N/A |
| Plasmid: HAGE-Flag-eIF4A3 | This paper | N/A |
| Plasmid: psPAX2 | (Tiscornia et al., 2006) | Addgene #12260 |
| Plasmid: pMD2.G | (Tiscornia et al., 2006) | Addgene #12259 |
| Plasmid: GFP | Clontech | Cat#6080-l |
| Plasmid: VSVM-GFP | (von Kobbe et al., 2000) | N/A |
| Plasmid: GST-eIF4A3 | (Chi et al., 2013) | N/A |
| Plasmid: GST-NXF1 | This paper | N/A |
| Plasmid: 3M-SVL | (Shi et al., 2009) | N/A |
| Plasmid: wS | (Chi et al., 2013) | N/A |
| Plasmid: wS-lUTR | This paper | N/A |
| Plasmid: wS-2UTR | This paper | N/A |
| Plasmid: wS-WT 3UTR | This paper | N/A |
| Plasmid: wS-3UTR-UGUA | This paper | N/A |
| Plasmid: wG | (Chi et al., 2013) | N/A |
| Plasmid: wG-1 UTR | This paper | N/A |
| Plasmid: wG −2 UTR | This paper | N/A |
| Plasmid: wG −3 UTR | This paper | N/A |
| Plasmid: 3 UTR-wG | This paper | N/A |
| Software and Algorithms | ||
| HTSeq | (Anders et al., 2015) | https://htseq.readthedocs.io/ |
| STAR(v2.5.2) | (Dobin et al., 2013) | https://eithub.com/alexdobin/STAR |
| DESeq & DEXSeq | (Anders and Huber, 2010) | https://bioconductor.org/packages/devel/bioc/htm1/DESeq.html |
| PolyA_DB version 3.2 | http://exon.nims.rutgers.edu/polya_db/v3/ | |
| rMATS (v4.0.2) | (Shen et al., 2014) | http://rnaseq-mats.sourceforge.net/ |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtiebio.sourceforge.net/bowtie2/index.shtml |
| BWA-MEM (0.7.15) | (Li and Durbin, 2009) | http://bio-bwa.sourceforge.net/ |
| Cutadapt | (Martin, 2011) | http://cutadapt.readthedocs.io/en/stable/guide.html |
| Scikit-learn | (Pedregosa et al., 2011) | http://scikitlearn.ors/stable/index.html |
| ImageJ | NIH | N/A |
| Other datasets | ||
| RNA-seq data of siSRSF3 | (Muller-McNicoll et al.,2016) | GSE69733 |
| iCLIPdataofNXFl | (Muller-McNicoll etal.,2016) | GSE69689 |
Highlights.
mRNA export factors generally promote expression of long 3’UTR isoforms.
Gene size and AT content impact alternative polyadenylation of 3’UTRs.
NXF1 regulates RNA polymerase II distribution along the gene.
NXF1 together with CFI-68 facilitates nuclear export of long 3’UTR transcripts.
ACKNOWLEDGMENTS
We thank Yongsheng Shi and Elisa Izaurralde for the 3M-SVL and VSVM-GFP plasmids, respectively, Feilong Meng for technical assistance in isolating chromatin RNAs, and members of BT and HC labs for helpful discussions. This work was funded by the National Key R&D Program of China [2017YFA0504400] and National Natural Science Foundation of China [31570822, 91540104 and 31770880] to HC and NIH (GM084089) to BT.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachi A, Braun IC, Rodrigues JP, Pante N, Ribbeck K, von Kobbe C, Kutay U, Wilm M, Gorlich D, Carmo-Fonseca M, et al. (2000). The C-terminal domain of TAP interacts with the nuclear pore complex and promotes export of specific CTE-bearing RNA substrates. RNA 6, 136–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork P, and Wieslander L (2014). Mechanisms of mRNA export. Semin Cell Dev Biol 32, 47–54. [DOI] [PubMed] [Google Scholar]
- Casolari JM, Brown CR, Komili S, West J, Hieronymus H, and Silver PA (2004). Genome-wide localization of the nuclear transport machinery couples transcriptional status and nuclear organization. Cell 117, 427–439. [DOI] [PubMed] [Google Scholar]
- Chi B, Wang Q, Wu G, Tan M, Wang L, Shi M, Chang X, and Cheng H (2013). Aly and THO are required for assembly of the human TREX complex and association of TREX components with the spliced mRNA. Nucleic Acids Res 41, 1294–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo MA (2018). Nuclear pore complexes as hubs for gene regulation. Nucleus 9, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dieppois G, Iglesias N, and Stutz F (2006). Cotranscriptional recruitment to the mRNA export receptor Mex67p contributes to nuclear pore anchoring of activated genes. Mol Cell Biol 26, 7858–7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Kuai B, Wu G, Wu X, Chi B, Wang L, Wang K, Shi Z, Zhang H, Chen S, et al. (2017). Exosome cofactor hMTR4 competes with export adaptor ALYREF to ensure balanced nuclear RNA pools for degradation and export. EMBO J 36, 2870–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria PA, Chakraborty P, Levay A, Barber GN, Ezelle HJ, Enninga J, Arana C, van Deursen J, and Fontoura BM (2005). VSV disrupts the Rae1/mrnp41 mRNA nuclear export pathway. Mol Cell 17, 93–102. [DOI] [PubMed] [Google Scholar]
- Forler D, Rabut G, Ciccarelli FD, Herold A, Kocher T, Niggeweg R, Bork P, Ellenberg J, and Izaurralde E (2004). RanBP2/Nup358 provides a major binding site for NXF1-p15 dimers at the nuclear pore complex and functions in nuclear mRNA export. Mol Cell Biol 24, 1155–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu B, Zhao M, Wang L, Patil G, Smith JA, Juncadella IJ, Zuvela-Jelaska L, Dorf ME, and Li S (2017). RNAi Screen and Proteomics Reveal NXF1 as a Novel Regulator of IRF5 Signaling. Sci Rep 7, 2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sun Y, Li Y, Li J, Rao X, Chen C, and Xu A (2011). Differential genome-wide profiling of tandem 3’ UTRs among human breast cancer and normal cells by high-throughput sequencing. Genome Res 21, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, Park JY, Yehia G, and Tian B (2013). Analysis of alternative cleavage and polyadenylation by 3’ region extraction and deep sequencing. Nat Methods 10, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Gattoni R, Stevenin J, and Steitz JA (2003). SR splicing factors serve as adapter proteins for TAP-dependent mRNA export. Mol Cell 11, 837–843. [DOI] [PubMed] [Google Scholar]
- Izaurralde E (2002). Nuclear export of messenger RNA. Results Probl Cell Differ 35, 133–150. [DOI] [PubMed] [Google Scholar]
- Ji Z, Luo W, Li W, Hoque M, Pan Z, Zhao Y, and Tian B (2011). Transcriptional activity regulates alternative cleavage and polyadenylation. Mol Syst Biol 7, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Cubberley G, and Bentley DL (2009). Cotranscriptional recruitment of the mRNA export factor Yra1 by direct interaction with the 3’ end processing factor Pcf11. Mol Cell 33, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SA, Kim H, Erickson B, and Bentley DL (2011). The export factor Yra1 modulates mRNA 3’ end processing. Nat Struct Mol Biol 18, 1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katahira J, Okuzaki D, Inoue H, Yoneda Y, Maehara K, and Ohkawa Y (2013). Human TREX component Thoc5 affects alternative polyadenylation site choice by recruiting mammalian cleavage factor I. Nucleic Acids Res 41, 7060–7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katahira J, Strasser K, Podtelejnikov A, Mann M, Jung JU, and Hurt E (1999). The Mex67p-mediated nuclear mRNA export pathway is conserved from yeast to human. EMBO J 18, 2593–2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackford B, Yao C, Charles GM, Weng L, Zheng X, Choi EA, Xie X, Wan J, Xing Y, Freudenberg JM, et al. (2014). Fip1 regulates mRNA alternative polyadenylation to promote stem cell self-renewal. EMBO J 33, 878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You B, Hoque M, Zheng D, Luo W, Ji Z, Park JY, Gunderson SI, Kalsotra A, Manley JL, et al. (2015). Systematic profiling of poly(A)+ transcripts modulated by core 3’ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet 11, e1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Freitas J, Zheng D, Oliveira MS, Hoque M, Martins T, Henriques T, Tian B, and Moreira A (2017). Transcription elongation rate has a tissue-specific impact on alternative cleavage and polyadenylation in Drosophila melanogaster. RNA 23, 1807–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Gruber AR, Keller W, and Zavolan M (2012). Genome-wide analysis of pre-mRNA 3’ end processing reveals a decisive role of human cleavage factor I in the regulation of 3’ UTR length. Cell Rep 1, 753–763. [DOI] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. 2011 17. [Google Scholar]
- Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, and Wagner EJ (2014). CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510, 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer A, di Iulio J, Maleri S, Eser U, Vierstra J, Reynolds A, Sandstrom R, Stamatoyannopoulos JA, and Churchman LS (2015). Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution. Cell 161, 541–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C (2016). Evolution and Biological Roles of Alternative 3’UTRs. Trends Cell Biol 26, 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minocha R, Popova V, Kopytova D, Misiak D, Huttelmaier S, Georgieva S, and Strasser K (2018). Mud2 functions in transcription by recruiting the Prp19 and TREX complexes to transcribed genes. Nucleic Acids Res 46, 9890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-McNicoll M, Botti V, de Jesus Domingues AM, Brandl H, Schwich OD, Steiner MC, Curk T, Poser I, Zarnack K, and Neugebauer KM (2016). SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev 30, 553–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaike T, Logan C, Hotta I, Rozenblatt-Rosen O, Meyerson M, and Manley JL (2011). Transcriptional activators enhance polyadenylation of mRNA precursors. Mol Cell 41, 409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve J, Burger K, Li W, Hoque M, Patel R, Tian B, Gullerova M, and Furger A (2016). Subcellular RNA profiling links splicing and nuclear DICER1 to alternative cleavage and polyadenylation. Genome Res 26, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Garcia P, Debo B, Aleman JR, Talamas JA, Lan Y, Nguyen NH, Won KJ, and Capelson M (2017). Metazoan Nuclear Pores Provide a Scaffold for Poised Genes and Mediate Induced Enhancer-Promoter Contacts. Mol Cell 66, 63–76 e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, and Dubourg V (2011). Scikit-learn: Machine learning in Python. Journal of machine learning research 12, 2825–2830. [Google Scholar]
- Reed R, and Magni K (2001). A new view of mRNA export: Separating the wheat from the chaff. Nat Cell Biol 3, E201–E204. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Navarro S, and Hurt E (2011). Linking gene regulation to mRNA production and export. Curr Opin Cell Biol 23, 302–309. [DOI] [PubMed] [Google Scholar]
- Rohner S, Kalck V, Wang X, Ikegami K, Lieb JD, Gasser SM, and Meister P (2013). Promoter- and RNA polymerase II-dependent hsp-16 gene association with nuclear pores in Caenorhabditis elegans. J Cell Biol 200, 589–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp MD, Aringhieri C, Vivarelli S, Cardinale S, Paro S, Schumperli D, and Barabino SM (2009). Mammalian pre-mRNA 3’ end processing factor CF I m 68 functions in mRNA export. Mol Biol Cell 20, 5211–5223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalb B, Michel M, Zacher B, Fruhauf K, Demel C, Tresch A, Gagneur J, and Cramer P (2016). TT-seq maps the human transient transcriptome. Science 352, 1225–1228. [DOI] [PubMed] [Google Scholar]
- Segref A, Sharma K, Doye V, Hellwig A, Huber J, Luhrmann R, and Hurt E (1997). Mex67p, a novel factor for nuclear mRNA export, binds to both poly(A)+ RNA and nuclear pores. EMBO J 16, 3256–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, and Xing Y (2014). rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci U S A 111, E5593–5601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard PJ, Choi EA, Lu J, Flanagan LA, Hertel KJ, and Shi Y (2011). Complex and dynamic landscape of RNA polyadenylation revealed by PAS-Seq. RNA 17, 761–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi M, Zhang H, Wu X, He Z, Wang L, Yin S, Tian B, Li G, and Cheng H (2017). ALYREF mainly binds to the 5’ and the 3’ regions of the mRNA in vivo. Nucleic Acids Res 45, 9640–9653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR 3rd, Frank J, and Manley JL (2009). Molecular architecture of the human pre-mRNA 3’ processing complex. Mol Cell 33, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, and Manley JL (2015). The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes Dev. 29, 889–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood V, and Brickner JH (2014). Nuclear pore interactions with the genome. Curr Opin Genet Dev 25, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strasser K, Masuda S, Mason P, Pfannstiel J, Oppizzi M, Rodriguez-Navarro S, Rondon AG, Aguilera A, Struhl K, Reed R, et al. (2002). TREX is a conserved complex coupling transcription with messenger RNA export. Nature 417, 304–308. [DOI] [PubMed] [Google Scholar]
- Tan-Wong SM, Wijayatilake HD, and Proudfoot NJ (2009). Gene loops function to maintain transcriptional memory through interaction with the nuclear pore complex. Genes Dev 23, 2610–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Texari L, Dieppois G, Vinciguerra P, Contreras MP, Groner A, Letourneau A, and Stutz F (2013). The nuclear pore regulates GAL1 gene transcription by controlling the localization of the SUMO protease Ulp1. Mol Cell 51, 807–818. [DOI] [PubMed] [Google Scholar]
- Tian B, and Manley JL (2017). Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol 18, 18–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, Pan Z, and Lee JY (2007). Widespread mRNA polyadenylation events in introns indicate dynamic interplay between polyadenylation and splicing. Genome Res. 17, 156–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiscornia G, Singer O, and Verma IM (2006). Production and purification of lentiviral vectors. Nat Protoc 1, 241–245. [DOI] [PubMed] [Google Scholar]
- Tran DD, Saran S, Williamson AJ, Pierce A, Dittrich-Breiholz O, Wiehlmann L, Koch A, Whetton AD, and Tamura T (2014). THOC5 controls 3’end-processing of immediate early genes via interaction with polyadenylation specific factor 100 (CPSF100). Nucleic Acids Res 42, 12249–12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia P, Dias AP, and Reed R (2008). Splicing promotes rapid and efficient mRNA export in mammalian cells. Proc Natl Acad Sci U S A 105, 3386–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veloso A, Kirkconnell KS, Magnuson B, Biewen B, Paulsen MT, Wilson TE, and Ljungman M (2014). Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res 24, 896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Kobbe C, van Deursen JM, Rodrigues JP, Sitterlin D, Bachi A, Wu X, Wilm M, Carmo-Fonseca M, and Izaurralde E (2000). Vesicular stomatitis virus matrix protein inhibits host cell gene expression by targeting the nucleoporin Nup98. Mol Cell 6, 1243–1252. [DOI] [PubMed] [Google Scholar]
- Walsh MJ, Hautbergue GM, and Wilson SA (2010). Structure and function of mRNA export adaptors. Biochem Soc Trans 38, 232–236. [DOI] [PubMed] [Google Scholar]
- Wang R, Zheng D, Yehia G, and Tian B (2018). A compendium of conserved cleavage and polyadenylation events in mammalian genes. Genome Res 28, 1427–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickramasinghe VO, and Laskey RA (2015). Control of mammalian gene expression by selective mRNA export. Nat Rev Mol Cell Biol 16, 431–442. [DOI] [PubMed] [Google Scholar]
- Zheng D, Liu X, and Tian B (2016). 3’READS+, a sensitive and accurate method for 3’ end sequencing of polyadenylated RNA. RNA 22, 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng D, and Tian B (2014). RNA-binding proteins in regulation of alternative cleavage and polyadenylation. Adv Exp Med Biol 825, 97–127. [DOI] [PubMed] [Google Scholar]
- Zhu Y, Wang X, Forouzmand E, Jeong J, Qiao F, Sowd GA, Engelman AN, Xie X, Hertel KJ, and Shi Y (2018). Molecular Mechanisms for CFIm-Mediated Regulation of mRNA Alternative Polyadenylation. Mol Cell 69, 62–74 e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All scripts used for data processing and statistical analyses were written in Python, Perl, or R, and are available upon request. Sequencing datasets generated in this study, including those by 3’READS, RNA-seq, ChIP-seq and nascent RNA-seq, have been deposited into the GEO database under the accession number GSE117701.