

Abstract
Background and purpose
The citrus flavanone hesperetin has been proposed for the treatment of several human pathologies, but its cardiovascular actions remain largely unexplored. Here, we evaluated the effect of hesperetin on cardiac electrical and contractile activities, on aortic contraction, on the wild‐type voltage‐gated NaV1.5 channel, and on a channel mutant (R1623Q) associated with lethal ventricular arrhythmias in the long QT syndrome type 3 (LQT3).
Experimental approach
We used cardiac surface ECG and contraction force recordings to evaluate the effects of hesperetin in rat isolated hearts and aortic rings. Whole‐cell patch clamp was used to record NaV1.5 currents (I Na) in rat ventricular cardiomyocytes and in HEK293T cells expressing hNaV1.5 wild‐type or mutant channels.
Key results
Hesperetin increased the QRS interval and heart rate and decreased the corrected QT interval and the cardiac and aortic contraction forces at concentrations equal or higher than 30 μmol·L−1. Hesperetin blocked rat and human NaV1.5 channels with an effective inhibitory concentration of ≈100 μmol·L−1. This inhibition was enhanced at depolarized holding potentials and higher stimulation frequency and was reduced by the disruption of the binding site for local anaesthetics. Hesperetin increased the rate of inactivation and preferentially inhibited I Na during the slow inactivation phase, these effects being more pronounced in the R1623Q mutant.
Conclusions and implications
Hesperetin preferentially inhibits the slow inactivation phase of I Na, more markedly in the mutant R1623Q. Hesperetin could be used as a template to develop drugs against lethal cardiac arrhythmias in LQT3.
Abbreviations
- hNaV1.5
human voltage‐gated sodium 1.5 channel
- INa
sodium current
- INa‐10 ms
sodium current at 10 ms after depolarization
- INa‐500 ms
sodium current at 500 ms after depolarization
- LQT3
long QT syndrome type 3
- TdP
torsades de pointes
What is already known
Lethal arrhythmias of genetic origin remain a major cause of mortality worldwide.
Hesperetin is a natural flavonoid with potential therapeutic value in the prevention of cardiovascular diseases.
What this study adds
Hesperetin blocks the cardiac voltage‐gated Na+ channel NaV1.5, interacting with the local anaesthetic binding site.
Hesperetin increases the inactivation rate of the LQT3 human mutant NaV1.5 channel R1623Q.
What is the clinical significance
Hesperetin could be used as template molecule for developing new drugs against arrhythmias in LQT3.
1. INTRODUCTION
Rapid heart rhythms, such as torsades de pointes (TdP) culminating in ventricular fibrillation, are still some of leading causes of morbidity and mortality including sudden cardiac death (Janse & Rosen, 2006). Despite the progress, treatment of lethal ventricular arrhythmias is still problematic. Current antiarrhythmic drugs have limited effectiveness and may be proarrhythmogenic (Frommeyer & Eckardt, 2016). Lethal arrhythmias of genetic origin have received special attention because they represent a special framework to study the interplay between molecular and clinical events. Of the 15 variants of the inherited long QT syndrome, long QT syndrome type 3 (LQT3) is the third most frequent (Schwartz, Crotti, & Insolia, 2012) and exhibits the highest percentage of lethal events (Splawski et al., 2000).
The LQT3 disorders involve different gain‐of‐function mutations in NaV1.5 channels (Zimmer & Surber, 2008). Mutations, such as ΔKPQ (Fredj, Sampson, Liu, & Kass, 2006), are characterized by a late Na+ current (I Na‐Late). Others, like the R1623Q variant, show a very slow I Na inactivation phenotype with a small I Na‐Late (Kambouris et al., 1998). However, all LQT3 phenotypes show an increased net influx of Na+ that lengthens the action potential duration and could give rise to early afterdepolarizations and triggered activity inducing TdP and ventricular fibrillation in LQT3 (Veerman, Wilde, & Lodder, 2015).
Targeting these mechanistic features in LQT3 mutations could be an effective strategy to reduce the Na+ overload, thus suggesting a novel therapeutic approach to treat LQT3 patients (Remme & Wilde, 2014). However, compounds currently used as antiarrhythmics to treat this syndrome, such as the I Na‐Late blockers ranolazine, mexiletine, and propranolol, also block some other major currents, which may constitute undesirable effects (Antzelevitch et al., 2004; Gao et al., 2013; Miller, Wang, & Zhong, 2014). Thus, there is still a need for new compounds acting on the slowly inactivating or late I Na for the treatment of arrhythmias in LQT3 (Antzelevitch et al., 2014).
Flavonoids constitute a large group of natural compounds with multiple cellular actions having a potential therapeutic value in the prevention and treatment of cardiovascular diseases (Habauzit & Morand, 2012). The citrus flavonoid naringin blocks the fast I Na in rodent cardiomyocytes and also has a strong negative inotropic action (Alvarez‐Collazo, López‐Medina, Rodríguez, & Alvarez, 2014). Its aglycone, naringenin, acts on ionic currents that determine the cardiac action potential (Scholz, Zitron, Katus, & Karle, 2010). Hesperetin, a flavanone closely related to naringenin, is considered as a phytoestrogen of good bioavailability and has anticarcinogenic, antioxidant, and lipid‐lowering properties (Erlund, Meririnne, Alfthan, & Aro, 2001; Kay, Pereira‐Caro, Ludwig, Clifford, & Crozier, 2017; Roohbakhsh, Parhiz, Soltani, Rezaee, & Iranshahi, 2015). Hesperetin blocks the hERG channel, heterologously expressed in Xenopus oocytes, with an effective inhibitory concentration (IC50) ≈270 μmol·L−1 (Scholz et al., 2007). Moreover, hesperetin moderately inhibited NaV1.5 channels expressed in HEK cells and I Na in human atrial cardiomyocytes (Wang et al., 2016). In our opinion, however, a clear lack of control of membrane potential during the flow of large Na+ currents precludes any meaningful analysis of the experiments presented in that study.
Here, we aimed at screening the effects of hesperetin on cardiac and vascular functions and on I Na in single ventricular rat cardiomyocytes. We also aimed at determining the effects of hesperetin on the human cardiac NaV1.5 channel and on its mutant R1623Q, which features a slower I Na inactivation that is associated with a severe LQT3 arrhythmic phenotype (Kambouris et al., 1998). Additionally, we explored whether hesperetin interacts with the local anaesthetic binding site. We found that hesperetin blocked NaV1.5 channels and increased the rate of its inactivation. The latter is more marked in the slow inactivation phase of the R1623Q mutant. These effects suggest that this flavanone may be useful as a template for the development of new compounds that would target the slow inactivation responsible for arrhythmias in this LQT3 mutation.
2. METHODS
2.1. Animals
Animal care and experiments were in accordance to the guidelines and procedures approved by the National Center for Laboratory Animal Reproduction. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny, Browne, Cuthill, Emerson, & Altman, 2010). Experimental models using rats reproduce closely many cardiovascular physiopathological conditions seen in humans (Hearse & Sutherland, 2000). Therefore, experiments were performed using healthy adult male Wistar rats (7–8 weeks, 180–200 g; RRID:RGD_13508588) provided by the National Center for Laboratory Animal Reproduction (Havana, Cuba). All animals were housed (two rats per type III filtered top cage containing shredded aspen shavings as bedding material) in a conventional facility with controlled temperature, humidity, and dark–light cycles and were provided with food and water ad libitum. Animals were killed by pentobarbital anaesthesia (3 mg per 100 g of body weight).
2.2. Recording of electrical and mechanical activities in isolated hearts
Rat hearts were carefully dissected and mounted on a Langendorff column to record the surface ECG and the force of contraction as previously described (Galán, Talavera, Vassort, & Alvarez, 1998). Corrected QT (QTc) was estimated according to Bazett's formula, QTc = QT/√RR interval. The composition of the Tyrode solution was (in mmol·L−1): 140 NaCl, 2.5 KCl, 0.5 MgCl2, 2 CaCl2, 10 tris‐hydroxymethylaminomethane, and 5 glucose (pH 7.4, adjusted with NaOH; gassed with O2; T = 35°C). Hearts were allowed to stabilize for at least 30 min before beginning the recordings.
2.3. Aortic rings
To screen for the vascular actions of hesperetin, we used rings of rat abdominal aorta. To reduce the number of animals, the abdominal aorta was dissected from rats, immediately after the heart was excised. Rings (approximately 2 mm wide) were cut and the endothelium removed by gently rubbing the lumen using a glass rod with a rounded tip. Rings were fixed to a force transducer and placed in an organ bath continuously perfused at a rate of 10 mL·min−1 with the same Tyrode solution used for isolated hearts (gassed with O2; T = 35°C) and stabilized under a load of 0.5 g for 30 min before the beginning of the recordings. Contraction was induced by isosmotically replacing 60 mmol·L−1 NaCl, by KCl in the Tyrode solution, or by applying 10 μmol·L−1 phenylephrine. Only preparations in which ACh (10 μmol·L−1) produced no relaxation of the KCl‐ or phenylephrine‐induced contraction were included in the study.
2.4. Enzymic isolation of ventricular cardiomyocytes
To evaluate whether hesperetin could have an effect on native cells, we used rat ventricular cardiomyocytes, which were isolated as previously described (Alvarez‐Collazo, Díaz‐García, López‐Medina, Vassort, & Alvarez, 2012). Briefly, hearts were quickly removed and dissected in a cold Ca2+‐free Tyrode solution of the following composition (in mmol·L−1): 117 NaCl, 4 KCl, 1.5 KH2PO4, 4.4 NaHCO3, 1.7 MgCl2, 10 HEPES, and 10 glucose, pH 7.4 (adjusted with NaOH). The hearts, mounted on a Langendorff column, were washed for ≈2 min with Tyrode solution containing EGTA (0.23 mmol·L−1) at 35°C and then perfused with Tyrode containing Ca2+ (≈100 μmol·L−1) and collagenase (1 mg·mL−1; CLS2, Worthington, USA), with recirculation of perfusate for 7–10 min. After this period, hearts were cut into small pieces and gently agitated for 2–3 min in fresh enzyme solution but supplemented with Ca2+ (300 μmol·L−1) and BSA (1 mg·mL−1). The solution was then filtered through a nylon mesh (250 μm) and centrifuged at 7× g for 2 min. The resulting pellet was washed with a K+–Tyrode solution without enzymes containing 0.5 mmol·L−1 Ca2+ and BSA. The solution was left for decantation for 5–10 min, and the pellet was again washed with Tyrode solution containing 1 mmol·L−1 Ca2+ and BSA. Myocytes thus obtained were kept at room temperature (21 ± 2°C) and used for experiments for 6 hr.
2.5. Patch‐clamp recordings
For rat ventricular cardiomyocytes, isolated as described above, whole‐cell currents were recorded at room temperature using an RK‐300 patch clamp amplifier (Biologic, France), a Labmaster DMA TL‐1 125 (Mentor, OH, USA), and the ACQUIS1 software (CNRS License, France). Currents were filtered at 3 kHz and digitized at 50‐μs intervals, stored on a computer, and analysed offline with the ACQUIS1 software. To study voltage‐dependent Na+ currents, K+ currents were blocked by substituting all K+ by Cs+ in the extracellular and intracellular solutions and using tetraethylammonium chloride (in the extracellular solution). The L‐type Ca2+ current was blocked with nifedipine (10 μmol·L−1). The extracellular solution contained (in mmol·L−1): 10 NaCl, 107 tetraethylammonium chloride, 20 CsCl, 10 HEPES, 2 CaCl2, 1.8 MgCl2, and 10 glucose, pH 7.4 (adjusted with CsOH). The standard pipette (intracellular) solution contained (in mmol·L−1): 130 CsCl, 0.4 Na2GTP, 5 Na2ATP, 1 Na2–creatine phosphate, 11 EGTA, 4.7 CaCl2, and 10 HEPES, with pH adjusted to 7.2 with CsOH. The low extracellular Na+ concentration was chosen in order to decrease I Na density and avoid, as much as possible, voltage‐clamp errors when huge Na+ currents flow at normal extracellular solutions (Goudet et al., 2001). The pipette resistance was 1.0–1.2 MΩ, and the liquid junction potential was compensated before establishing the gigaseal. No leakage or capacity transient subtraction was used in cardiomyocytes, and only cells with stable gigaseals were selected for patch clamping. Membrane capacitance (C m), membrane time constant (τ m), and series resistance (R s) were determined on voltage‐clamped cardiomyocytes using 10‐ms hyperpolarizing 2‐mV pulses from a holding potential (HP) of −80 mV according to the equations:
where I 0 is the maximum membrane current, I ∞ is the current at the end of the 10‐ms pulse, and V m is the amplitude of the voltage step (2 mV). Average C m and uncompensated R s were 171 ± 12 pF and 3.6 ± 0.4 MΩ respectively (n = 12). The R s could be compensated up to 50% without ringing and was continually monitored during the experiment. Cells in which R s could not be compensated, showed poor seals or showed inadequacies in membrane voltage control, were discarded.
HEK cells (Thermo Fisher Scientific, MA, USA), HEK293T (RRID:CVCL_0063), were seeded on 18‐mm glass coverslips previously coated with poly‐l‐lysine (0.1 mg·L−1). Cell media consisted of a DMEM containing 10% of human serum, 2 U·mL−1 penicillin, 2 mg·mL−1 streptomycin, and 2 mmol·L−1 l‐glutamine. The cells were stored at 37°C in a humidity‐controlled incubator with 10% CO2. The genes encoding human NaV1.5 wild‐type (WT) or the R1623Q mutant as well as the F1760A mutant were cloned into the pCAGGS/IRES‐GFP vector and transiently transfected in HEK293T cells using the TransIT‐293 reagent (Mirus, Madison, MI, USA; MIR 2706). Although auxiliary β‐units could influence function and expression of NaV channel α subunit in heart, their functional roles are quite complex and not completely understood. Their functions are regulated by glycosylation, phosphorylation, and proteolytic processing, but it is uncertain if all these processes still occur (or are the same) in heterologous expression systems (Calhoun & Isom, 2014). For this reason, we only expressed the pore‐forming α subunit of the NaV1.5 channel, which suffices to generate I Na, similar to the native channels (Wang, Nie, George, & Bennett, 1996). Additionally, this approach is commonly reported in the literature and allows us to relate to other results. Transfected cells were identified by the GFP expression during the patch‐clamp experiments, performed 24–48 hr after transfection. Longer culture periods resulted in huge Na+ currents, making difficult to control membrane potential in patch‐clamp experiments. Whole‐cell currents were recorded in only one cell per coverslip. Measurements were performed at room temperature using an EPC7 patch‐clamp amplifier (LIST Electronics, Darmstadt, Germany), a TL‐1 DMA interface (Axon Instruments), and the pClamp software (Version 9.0, Axon Instruments, Foster City, CA, USA; RRID:SCR_011323). Currents were filtered at 3 kHz, digitized at 50‐μs intervals, stored on a computer, and analysed offline with the WinASCD software (KU Leuven License, Belgium). The extracellular solution for HEK293T cells contained (in mmol·L−1): 140 NaCl, 10 HEPES, 2 CaCl2, 1 MgCl2, and 10 glucose, pH 7.4, with NaOH, and the pipette solution contained (in mmol·L−1): 130 CsCl, 5 Na2ATP, 5 Na2–creatine phosphate, 5 EGTA, 1 MgCl2, 1 CaCl2 (free Ca2+, ≈0.4 nmol·L−1), and 10 HEPES, pH adjusted to 7.2 with CsOH. Patch pipettes (1.5–2 MΩ) were pulled from borosilicate capillary tubes. In HEK293T cells, R s (3.3 ± 0.7 MΩ) and C m (23.2 ± 0.8 pF; n = 128) could be determined by the usual routines and the built‐in compensation circuits of the EPC‐7 amplifier. In all recordings, R s was electronically compensated up to 50% without ringing and was continually monitored during the experiment. Liquid junction potential was compensated before establishing the gigaseal, and capacity transients were cancelled using the pClamp P/4 protocol. A more physiological HP of −100 mV was chosen for both cardiomyocytes and HEK293T cells because I Na availability curves were similar at HP of −120 and −100 mV, and no differences in peak I Na densities were found at these HP (Figure S1). I Na was monitored using 50‐ms voltage pulses to −40 mV (cardiomyocytes) or −20 mV (HEK293T cells), applied at 0.25 Hz from the HP of −100 mV.
In cardiomyocytes, peak I Na amplitude was measured as the difference between the peak inward current and the current at the end of the voltage‐clamp pulse. Although Zygmunt, Eddlestone, Thomas, Nesterenko, and Antzelevitch (2001) showed a sustained I Na in dog mid‐myocardial cells 300 ms after the activating pulse, we never found such a current component in our recordings. Instead, we recorded a very small steady outward current that may be related to the Na+–Ca2+ exchanger, which generates an outward current at the test potentials and Na+ and Ca2+ concentrations used in this study (Bers, 2001; Figure S2). In HEK293T cells, peak I Na amplitude was measured as the difference between peak inward current and the zero current level. To take into account the variability in cell membrane area, current amplitudes of each cell were normalized to the C m and expressed as current densities (pA·pF−1). Due to the presence of a residual R s, an adequate characterization of the fast I Na activation is not possible. Thus, we only estimated the time to peak of the current. We also measured the I Na at 10 ms after the depolarizing step (I Na‐10 ms) and the I Na at the end of the 50‐ or 500‐ms voltage‐clamp pulses. For this purpose, in order to improve the signal‐to‐noise ratio, we averaged the last 5 ms of 10 traces in each condition, and the mean current value was taken as I Na‐Late. We calculated the inhibition percentage of hesperetin on I Na to determine the concentration–effect relationships. Using the routines of Origin 9.0 (Microcal OriginLab Corporation, Northampton, MA, USA; RRID:SCR_002815), data were fit by a Hill function of the form:
where [x] is the concentration of the compound used, IC50 is the effective inhibitory concentration, and H is the Hill number. This equation was chosen because it is useful to quantitatively characterize concentration‐dependent effects, but due to its limitations for the interpretation of molecular events, no attempt was made to relate the fitting results to any binding model. The fitting parameters can be compared in Table S1.
Availability curves and current–voltage relationships (I–V) were obtained by clamping the cells at an HP of −100 mV and using a standard double‐pulse voltage‐clamp protocol (frequency, 0.25 Hz). Availability curves were obtained from the normalization of the amplitude of currents recorded at a given test pulse by the maximal current (I Max). These curves were fit by the equation:
where V inac is the voltage for half‐maximal availability and s inac is the slope factor.
I–V relationships were obtained from the amplitude of currents elicited by prepulse potentials and were fit by the equation:
where G Max is the maximal whole‐cell conductance, V Na is the equilibrium potential for Na+, V act is the voltage for half‐maximal activation, and s act is the slope factor. Activation curves were obtained for each cell by dividing the experimental current amplitudes, I(V), by the corresponding values of G Max(V − V Na). Activation curves were fit by the equation:
where V act is the voltage for half‐maximal activation and s act is the slope factor. The fittings were performed using Origin 9.0, and the fitting parameter results can be compared in Tables S2 and S3.
In cardiomyocytes, care was taken to choose only cells in which membrane voltage control was appropriate, and for this reason, n is limited to 5. At the test potential, the time course of the inactivation phase of the I Na traces was fitted by a double exponential using the fitting procedures of ACQUIS1 for cardiomyocytes and Origin 9.0 for HEK293T cells.
This procedure yielded two inactivation time constants, τ fast and τ slow, and the amplitudes of the two components (A fast and A slow), from which we calculated the relative amplitude of the slow component. k is a constant that represents the steady‐state current amplitude. This value was either negligible (<−6 × 10−6 pA·pF−1) in transfected HEK293T cells or slightly positive (<0.05 pA·pF−1) in rat cardiomyocytes due to the possible contribution of the electrogenic Na+–Ca2+ exchanger. It should be noted that in cardiomyocytes, at test potentials of −20 mV or more positive, the time course of the inactivation phase of the I Na traces could be fitted by a single exponential. However, due to the impossibility of obtaining reliable I Na recordings in cardiomyocytes at normal extracellular Na+ solution, we cannot exclude that this is an artefact related to the low extracellular Na+ condition used. The fitting parameter results can be compared in Table S4.
In order to investigate tonic and use‐dependent blocks of I Na by hesperetin, cells were voltage clamped with 50‐ms pulses from −100 to −20 mV at a frequency of 0.25 Hz. Stimulation was stopped, and perfusion with hesperetin was then initiated. After 1 min, stimulation was resumed (in the presence of hesperetin) at frequencies of 0.25, 1, or 5 Hz and then switched to 0.25 Hz. Tonic block was estimated as the difference between the peak control I Na and the I Na at the first pulse after resumption of stimulation during drug superfusion. Use‐dependent block was considered to be the difference between the peak I Na at the first and the 15th pulse during drug exposure. Tonic and use‐dependent blocks are expressed as percentage of total block, which was estimated from the difference between peak I Na currents recorded in control and in the presence of hesperetin.
2.6. Molecular modelling and docking simulations
We used the software Modeller (University of California, USA; Webb & Sali, 2016; RRID:SCR_008395) to generate a model of the structure of human NaV1.5 (hNaV1.5) channel, using its bacterial homologue NavAB (PDB code: 3RVY; Payandeh, Scheuer, Zheng, & Catterall, 2011) as template. This model was further refined, using NAMD (University of Illinois, USA; Phillips et al., 2005; RRID:SCR_014894), by running 10‐ns molecular dynamics simulations to obtain an energetically stable conformation. The final model is composed by the four repeats of the segments of S5, pore forming, and S6 (S5–pore forming–S6), with Repeat I comprising the residues from 231 to 415, Repeat II 820 to 941, Repeat III 1,315 to 1,472, and Repeat IV 1,638 to 1,774. This model was then used as receptor for in silico docking to analyse its interaction with hesperetin using the program AutoDock Vina (University of California, USA; Trott & Olson, 2010; RRID:SCR_011958). The complete pore was used as search space, allowing flexibility of the amino acid side chains pointing inwards as well as for the ligand hesperetin.
2.7. Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al.,2018). Data were analysed using the statistical routines of Origin 9.0 and expressed as means with SEM, from N hearts or aortic rings or n cells. Group data subjected to analysis had N and n values ≥5 independent samples per group, and compared groups had equal size. All data were normally distributed and were confirmed with the Shapiro–Wilk test (95% confidence level). Statistical significance was evaluated with a paired sample t test within groups or two‐sample t test between groups. For differences between multiple groups of data, we used an ANOVA, according to the experimental situation, followed by Tukey's post hoc test. Tests used for statistical comparison between groups (as well as the exact N and n) are indicated either in the text or in the figure legends. Differences were considered statistically significant if P < 0.05. Data recordings were randomized, thus, during experiments, drug concentrations were randomly applied on randomly selected cardiomyocytes or HEK293T cells expressing the WT or mutant channel, but the operators were not blinded to allocation during experiments. Analysis of the data was blinded.
2.8. Materials
Hesperetin ((S)‐2,3‐dihydro‐5,7‐dihydroxy‐2‐(3‐hydroxy‐4‐methoxyphenyl)‐4H‐1‐benzopyran‐4‐one; C16H14O6; PubChem CID: 72281; CAS Number: 520‐33‐2; >98% purity) was purchased from Sigma‐Aldrich (Bornem, Belgium) and was prepared in DMSO as stock solution (100 mmol·L−1) and kept at 4°C. The solvent DMSO had no effect on I Na at the maximal concentration used (0.28 ± 0.13% of inhibition). Dilutions at desired concentrations were freshly prepared from the stock, in the bath or extracellular solutions. All other chemicals were also purchased from Sigma‐Aldrich.
2.9. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18 (Alexander et al., 2017).
3. RESULTS
3.1. Effects of hesperetin on cardiac electrical and mechanical activities
Hesperetin mildly affected the spontaneous electrical activity of rat isolated hearts. The QRS duration was only significantly increased at hesperetin concentrations equal or higher than 30 μmol·L−1 (P < 0.05; Figure 1a,d). Hesperetin only caused significant changes on the QTc and the heart rate (RR interval) at 100 μmol·L−1 (P < 0.05; Figure 1b–d). No spontaneous arrhythmias were observed during hesperetin perfusion even at the highest concentration. Hesperetin exerted significant inhibition of the force of contraction in isolated hearts only at 100 μmol·L−1 (36.7 ± 12%; P < 0.05; Figure 1d). The actions of hesperetin on the ECG and force of contraction were reversible within ≈2 min upon washout with normal Tyrode solution.
Figure 1.
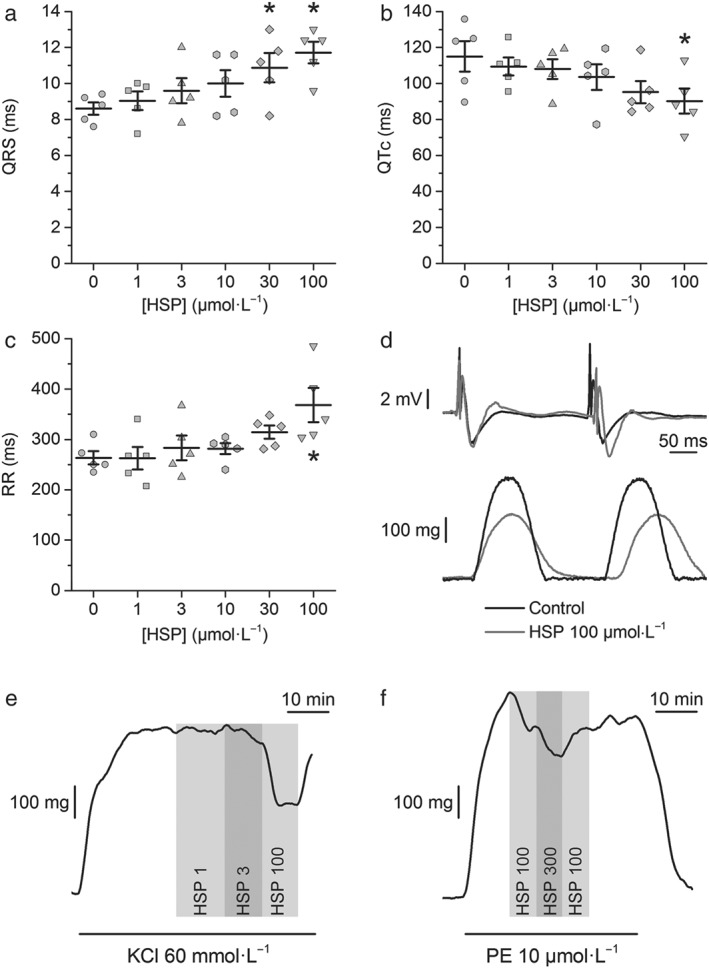
Hesperetin (HSP) barely affects the electrical and contractile activities of isolated hearts and the contraction of aortic rings. Dot and line plots of the effect of hesperetin (HSP) on the (a) QRS duration, (b) corrected QT (QTc), and (c) RR intervals. Hesperetin (100 μmol·L−1) significantly changed the QTc and RR; the QRS was significantly affected by the two higher concentrations of hesperetin studied (N = 5, *P < 0.05, one‐way repeated measures ANOVA with Tukey's post hoc test). (d) Example recordings of ECG (upper panel) and force of contraction (lower panel) in control condition and under the action of hesperetin (100 μmol·L−1). Hesperetin had a significant inhibitory effect on the force of contraction at 100 μmol·L−1 (N = 5, P < 0.05, one‐way repeated measures ANOVA with Tukey's post hoc test). (e,f) Representative recordings of the effects of different concentrations of hesperetin (in μmol·L−1) on aortic ring contraction induced by 60 mmol·L−1 KCl or by 10 μmol·L−1 phenylephrine (PE)
3.2. Vasorelaxing action of hesperetin in rat abdominal aortic rings
Screening experiments showed that hesperetin exerted a modest relaxing action. It decreased the KCl‐induced contraction with an IC50 = 21 ± 11 μmol·L−1 (H = 0.8 ± 0.2; N = 5) and a maximal inhibition of 78 ± 13% (Figure 1e). Hesperetin was less efficacious in decreasing the aortic ring contraction induced by phenylephrine with a maximal inhibition of only 45 ± 9% and an IC50 = 26 ± 16 μmol·L−1 (H = 0.8 ± 0.4; N = 5; Figure 1f).
3.3. Effects of hesperetin on native and recombinant NaV1.5 channels
We first screened for effects of hesperetin on I Na in rat ventricular cardiomyocytes. In control condition, the peak inward I Na recorded at −40 mV was −23 ± 4 pA·pF−1 (n = 10). At this test potential, inactivation time constants τ fast and τ slow were 1.30 ± 0.06 and 9.3 ± 0.3 ms and a relative amplitude of the slow component of 2.6 ± 0.1%. At −20 mV, single inactivation time constant of I Na was 0.86 ± 0.04 ms. hesperetin decreased I Na in a concentration‐dependent manner with an IC50 = 100 ± 14 μmol·L−1 (H = 0.9 ± 0.1; Figure 2). Hesperetin at 100 μmol·L−1, decreased slightly but significantly the slow inactivation time constant of I Na at −40 mV (1.27 ± 0.06 and 8.9 ± 0.3 ms), the relative amplitude of the slow inactivation component (2.0 ± 0.1%; n = 10), and the inactivation time constant of I Na at −20 mV (0.74 ± 0.04 ms; n = 5, P < 0.05, paired t test). Next, we determined the effects of 100 μmol·L−1 hesperetin on the I–V relationship, availability, and activation curves of I Na (Figure 2c,d). V inac was shifted from −71.1 ± 0.2 to −75.1 ± 0.1 mV (P < 0.05). Hesperetin did not change the voltages for half‐maximal activation (V act), the activation curves slope factors, nor the availability curves slope factors (in control condition: V act = −37.1 ± 0.6 mV, s act = 4.4 ± 0.5 mV, and s inac = 7.3 ± 0.2 mV; in the presence of hesperetin: V act = −36.4 ± 0.5 mV, s act = 4.9 ± 0.2 mV, and s inac = 6.9 ± 0.2 mV).
Figure 2.
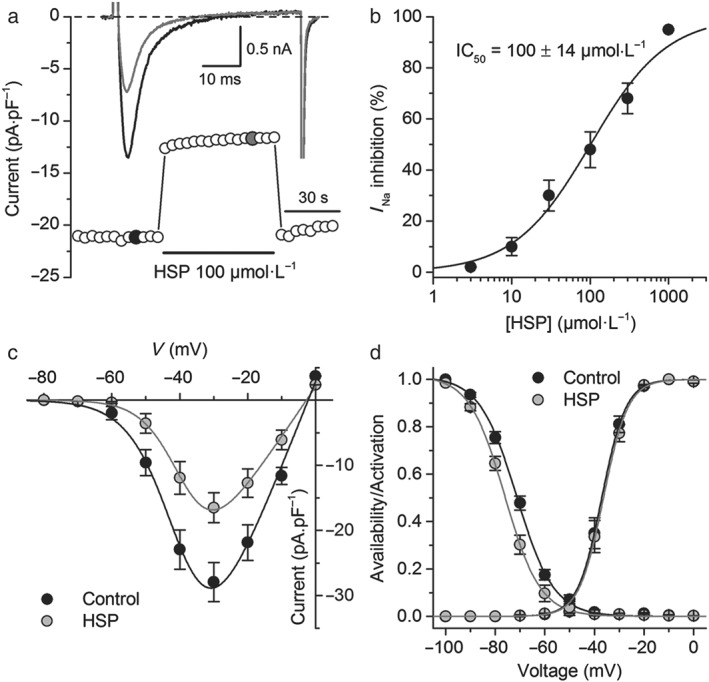
Hesperetin (HSP) inhibits I Na in rat ventricular isolated cardiomyocytes. (a) Time course of the I Na in rat ventricular cardiomyocytes in control condition and in the presence of hesperetin (100 μmol·L−1). Representative current traces in each condition are shown in the inset and correspond to the coloured data points. (b) Concentration–effect relationship for the inhibitory action of hesperetin on I Na. The dots represent the mean ± SEM inhibition percentage during the application of different concentrations of hesperetin (n = 10 cells isolated from six hearts). The line represents the Hill fit of the data, and the inset shows the IC50. (c) Current–voltage relationships obtained in control condition and in the presence of hesperetin (100 μmol·L−1). The dots represent the mean ± SEM (n = 5 cells from five hearts). Hesperetin significantly decreased the current compared with control in the range of voltage from −50 to +10 mV (P < 0.05, paired t test). (d) Availability and activation curves obtained in control condition and in the presence of hesperetin (100 μmol·L−1). The dots represent the mean ± SEM (n = 5 cells from five hearts). Hesperetin significantly shifted the availability curve compared with control (P < 0.05, paired t test)
Although this approach allowed us to determine that hesperetin could inhibit NaV1.5 currents in cardiomyocytes, the results face the constraint that experiments had to be done at low extracellular Na+ concentration. The HEK293T cells overexpressing the hNaV1.5 channel offer the advantages of studying the mechanisms of action of hesperetin on the hNaV1.5 channel at physiological extracellular Na+ concentration. In control condition, the peak inward I Na density (I Na‐Peak) at −20 mV was −84 ± 8 pA·pF−1, and the time to peak was 1.05 ± 0.03 ms (n = 42). Inactivation time constants τ fast and τ slow were 0.46 ± 0.02 and 3.6 ± 0.1 ms (n = 12) with a relative amplitude of the slow component of 3.2 ± 0.1%. Hesperetin reversibly decreased hNaV1.5 currents in HEK293T cells in a concentration‐dependent manner, with an IC50 = 130 ± 10 μmol·L−1 and (H = 0.8 ± 0.1; Figure 3a,b).
Figure 3.
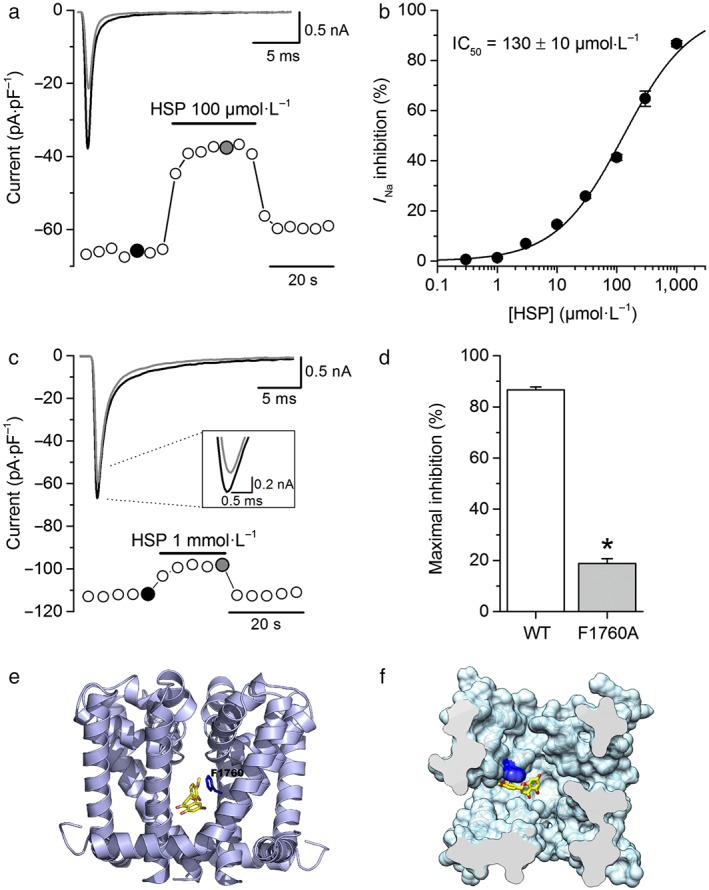
Hesperetin (HSP) inhibits I Na in HEK293T cells expressing the human NaV1.5 channel by interacting with the local anaesthetic binding site. (a) Time course of the I Na from wild‐type (WT) hNaV1.5 expressing HEK293T cells. Hesperetin (100 μmol·L−1) was applied as indicated. Representative current traces in each condition are shown in the inset and correspond with the coloured data points. For the sake of clarity, only the first 25 ms of the current traces is shown. (b) Concentration–effect relationship for the inhibitory action of hesperetin on hNaV1.5 current. The dots represent the mean ± SEM inhibition percentage during the application of different concentrations of hesperetin (n = 12). The line represents the Hill fit of the data, and IC50 value is shown on the inset. (c) Time course of the I Na in a HEK293T cell transiently expressing the hNaV1.5 channel with the mutation F1760A. The inset shows the poor inhibition of peak I Na by hesperetin as well as representative current traces in each condition corresponding with the coloured data points. For the sake of clarity, only the first 25 ms of the current traces is shown. (d) Maximal inhibition of peak I Na by hesperetin (1 mmol·L−1) in WT and F1760A mutated channel. Maximal inhibition of I Na was reduced when the local anaesthetic binding site F1760 is mutated (n = 9, *P < 0.05, two‐sample t test). (e) Full side view and (f) top surface view of a molecular model of the structure of the NaV1.5 channel. Molecular docking simulations ran on this model showed that hesperetin (in yellow) was bound at the NaV1.5 channel binding site for local anaesthetics (residue F1760 in blue)
3.4. Disruption of the local anaesthetic binding site reduces the effect of hesperetin on hNaV1.5 channels
The residue F1760, located in the pore‐forming segment IVS6 of voltage‐gated Na+ channels, is important for the binding of local anaesthetics (Wang, Quan, & Wang, 1998). We determined whether this residue is also relevant for the inhibitory effect of hesperetin on hNaV1.5 channels. Currents recorded at −20 mV in HEK293T cells overexpressing the hNaV1.5 channel F1760A mutant had a maximal peak density of 33 ± 4 pA·pF−1 (n = 9). Hesperetin was largely ineffective on this channel, with a maximal inhibition of less than 20% at 1 mmol·L−1, compared with ≈90% for the WT channel (P < 0.05; Figure 3c,d). Hesperetin (1 mmol·L−1) had no statistically significant effect on the inactivation time constants of the F1760A mutant currents (0.78 ± 0.02 and 7.9 ± 0.1 vs. 0.78 ± 0.01 and 8.2 ± 0.4 ms in control and in hesperetin, respectively; n = 9) nor on the relative amplitude of the slow inactivation component (1.3 ± 0.1% vs. 1.4 ± 0.1% in control and in hesperetin, respectively). Hesperetin had also no significant effect on the V inac of the F1760A mutant channel (−73.2 ± 0.3 and −74.7 ± 0.4 mV in control and in hesperetin, respectively; n = 6). Moreover, nine docking simulations were run on a molecular model of the structure of hNaV1.5 channels. In three of them, hesperetin was bound at the lateral pore fenestrations located at the interface of the domain (D) and segment (S) pairs DIIIS6–DIVS5 and DIVS6–DIS5. In five other simulations, hesperetin was located in a hydrophobic pocket around F1760. The best ranking solution is shown in Figure 3e,f.
3.5. Comparison of the effects of hesperetin on WT hNaV1.5 channels and the LQT3 mutant R1623Q
In order to determine the actions of hesperetin in a more pathologically relevant condition, we studied its effects on the LQT3 mutant channel R1623Q. In control condition, peak R1623Q mutant current density at −20 mV was −90 ± 10 pA·pF−1, and the time to peak was 1.04 ± 0.02 ms (n = 48). These values were not significantly different from those of the WT channel. In contrast, the time constants of inactivation τ fast and τ slow were significantly larger: 2.9 ± 0.1 and 10.2 ± 0.4 ms, respectively, as was the relative amplitude of the slow inactivation component (7.2 ± 0.4%; n = 12; P < 0.05, two‐sample t test). Hesperetin induced a concentration‐dependent decrease of peak R1623Q mutant currents, which was similar to that found for the WT channel (IC50 = 136 ± 17 μmol·L−1 and H = 0.8 ± 0.1; Figure 4a,b).
Figure 4.
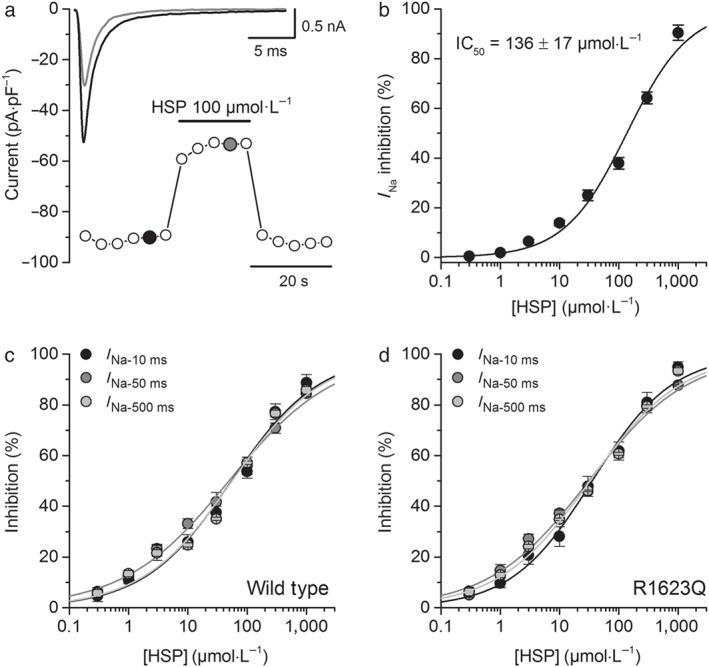
Hesperetin (HSP) inhibits I Na in HEK293T cells expressing the LQT3 R1623Q mutant channels but preferentially inhibits the later phase of the current. (a) Time course of the I Na from R1623Q hNaV1.5 expressing HEK293T cells in control condition and in presence of hesperetin (100 μmol·L−1). Representative current traces in control (black) and upon the application of hesperetin (grey) are shown in the insets and correspond with the coloured data points. For the sake of clarity, only the first 25 ms of the current traces is shown. (b) Concentration–effect curves for the action of hesperetin on R1623Q hNaV1.5 current. The dots represent the mean ± SEM inhibition percentage during the application of different concentrations of hesperetin (n = 12). The line represents the Hill fit of the data, and the calculated IC50 value is shown on the inset. (c,d) Concentration–effect curves for the effect of hesperetin on the current measured 10 ms after the beginning of the voltage‐clamp pulse (I Na‐10 ms) and the I Na measured at the last 5 ms of the 50‐ms (I Na‐50 ms) or 500‐ms (I Na‐500 ms) voltage‐clamp pulse in wild type and R1623Q. I Na‐50 ms IC50 was 49.2 ± 4.7 μmol·L−1 (H = 0.49 ± 0.03) and 31.1 ± 4.0 μmol·L−1 (H = 0.52 ± 0.04) in the WT and the R1623Q respectively. Similarly, IC50 for hesperetin inhibition of the I Na‐500 ms was 55.6 ± 8.9 μmol·L−1 (H = 0.57 ± 0.05) and 32.5 ± 4.3 μmol·L−1 (H = 0.56 ± 0.04) in the WT and the R1623Q respectively
As the main characteristic of the R1623Q mutant is a very slow inactivation phase, we first studied the effects of hesperetin on the amplitude of the inward current at 10 ms after the beginning of the voltage‐clamp depolarization. I Na‐10 ms amplitudes were 3.2 ± 0.2 pA·pF−1 for the WT and 6.3 ± 0.3 pA·pF−1 for the R1623Q mutant. The IC50 for hesperetin‐induced inhibition on I Na‐10 ms in WT and R1623Q (Figure 4c,d) was 54.4 ± 8.6 μmol·L−1 (H = 0.58 ± 0.05) and 35.4 ± 4.2 μmol·L−1 (H = 0.64 ± 0.05). This indicates that hesperetin was significantly more potent on I Na‐10 ms than on peak I Na and significantly more potent in R1623Q channels (P < 0.05). We also measured the I Na at the end of the 50‐ms (I Na‐50 ms) or 500‐ms (I Na‐500 ms) voltage‐clamp pulses. I Na‐50 ms was 1.5 ± 0.1 and 3.4 ± 0.1 pA·pF−1 in the WT and R1623Q channels respectively. I Na‐500 ms was 1.3 ± 0.2 and 3.2 ± 0.3 pA·pF−1 in the WT and R1623Q channels respectively. The results show that hesperetin was significantly more potent on I Na at 50 and 500 ms than on peak I Na and more potent on these currents in R1623Q channels (Figure 4c,d; n = 12, P < 0.05, two‐way ANOVA with Tukey's post hoc test). However, IC50 for I Na‐10 ms, I Na‐50 ms, and I Na‐500 ms was not different.
We next compared the effects of hesperetin on the kinetic properties of the WT and R1623Q mutant currents. For both WT and R1623Q channels, there was no statistically significant effect of 0.3–1,000 μmol·L−1 hesperetin on the time to peak I Na. The inactivation time course of WT currents was little affected by hesperetin, with significantly lower values of τ slow only at concentrations above 100 μmol·L−1 (P < 0.05; Figure 5a,c). Contrastingly, hesperetin markedly reduced both τ fast and τ slow of the R1623Q mutant (P < 0.05; Figures 5b–d and S3). Hesperetin decreased the relative amplitude of the slow inactivation component of WT currents in a concentration‐dependent manner, with a 43.7% decrease at the maximal concentration tested (1,000 μmol·L−1; Figure 5e). This effect was significantly stronger for the R1623Q mutant (n = 12; P < 0.05; two‐way ANOVA with Tukey's post hoc test), with 91.6% decrease in the relative amplitude of the slow inactivation component at 1,000 μmol·L−1 (Figure 5e). In order to highlight the physiological relevance of these findings, we calculated the time integral of the I Na traces to estimate the total charge (Na+) transported during the voltage‐clamp pulse. The results show, first, that in the R1623Q mutant, the amount of Na+ entering the cell was significantly increased by 146 ± 8% with respect to WT (P < 0.05) and, second, that hesperetin was significantly more effective in decreasing the amount of Na+ flowing through R1623Q channels (by 62 ± 3%) than in WT channels (by 50 ± 3%) during a voltage‐clamp pulse (Figure 5f). Taken together, these findings indicate that the hesperetin‐induced increase of I Na inactivation rate in the R1623Q mutant was greater than in the WT channel.
Figure 5.
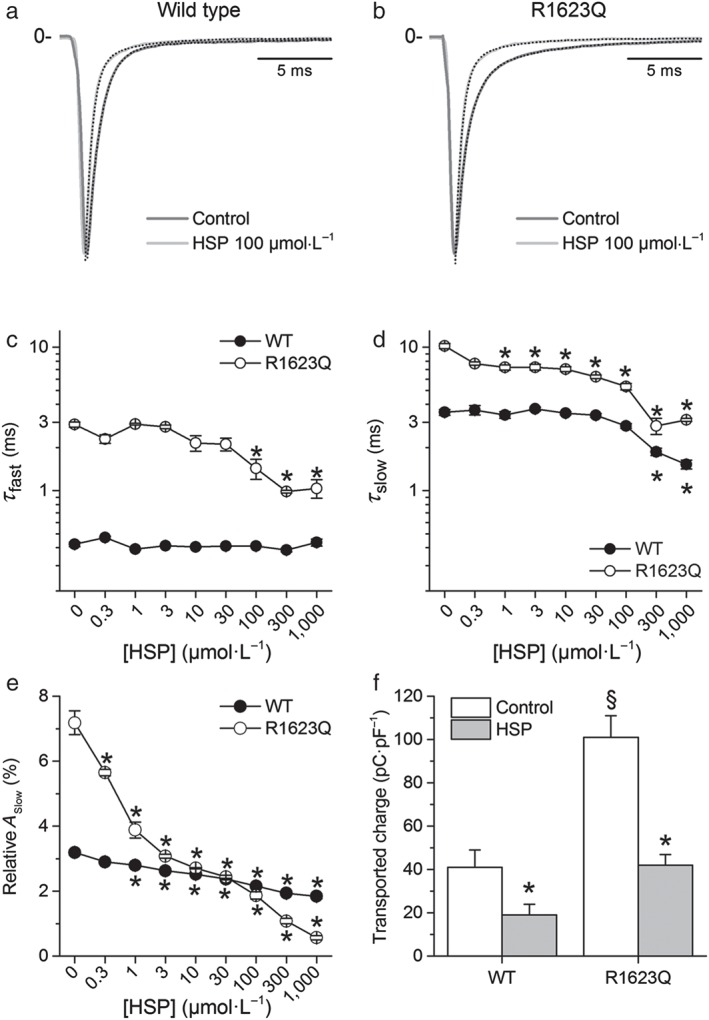
Hesperetin (HSP) decreases cell Na+ load. (a,b) Effects of hesperetin (100 μmol·L−1) on the inactivation time course of I Na in the wild‐type (WT) and R1623Q channels. Current traces were normalized to peak amplitude in order to get a better comparison of the effects on I Na inactivation. For the sake of clarity, only the first 20 ms of the current traces is shown. The dotted lines represent the fit of the inactivation phase of the current with a double‐exponential function with fast and slow components. (c,d) Effects of different concentrations of hesperetin on inactivation time constants of I Na. *P < 0.05 compared with its own control and with WT, n = 12, two‐way ANOVA with Tukey's post hoc test. (e) Concentration‐dependent effects of hesperetin on the relative amplitude of the slow component of I Na in the WT and R1623Q channels. Hesperetin significantly decreased the relative amplitude of the slow inactivation component. *P < 0.05 with respect to its own control, n = 12, one‐way ANOVA with Tukey's post hoc test. (f) hesperetin (100 μmol·L−1) reduced the amount of transported Na+, during the flow of I Na, in R1623Q to the same level as in WT under control condition. Current traces were integrated, and data were expressed as charge normalized to membrane capacitance (pC·pF−1). *P < 0.05 with respect to its own control; § P < 0.05 with respect to WT, n = 32, two‐way ANOVA with Tukey's post hoc test
Using the protocol to examine use‐dependent effects, we found that at stimulation frequencies of 0.25 and 1 Hz, hesperetin blocked both WT and R1623Q channels in a mostly tonic fashion with a small use dependency. However, at a higher stimulation frequency (5 Hz), a use‐dependent block of I Na by hesperetin became more evident in WT or R1623Q expressing cells (Figure S4). We further analysed the action of hesperetin by determining its effects on the I–V, activation, and availability curves (n = 13). In control conditions, there were no major differences in I–V curves (peak inward current at ≈−20 mV) or in availability and activation curves between WT and R1623Q channels (Figures 6a–d and S5). Inhibition of I Na by hesperetin (100 μmol·L−1) was accompanied by a significant (P < 0.05) leftward shift of the availability curves in WT and mutant channels. On the other hand, the activation curves were not affected by hesperetin (100 μmol·L−1 ; Figures 6c–f and S1d).
Figure 6.
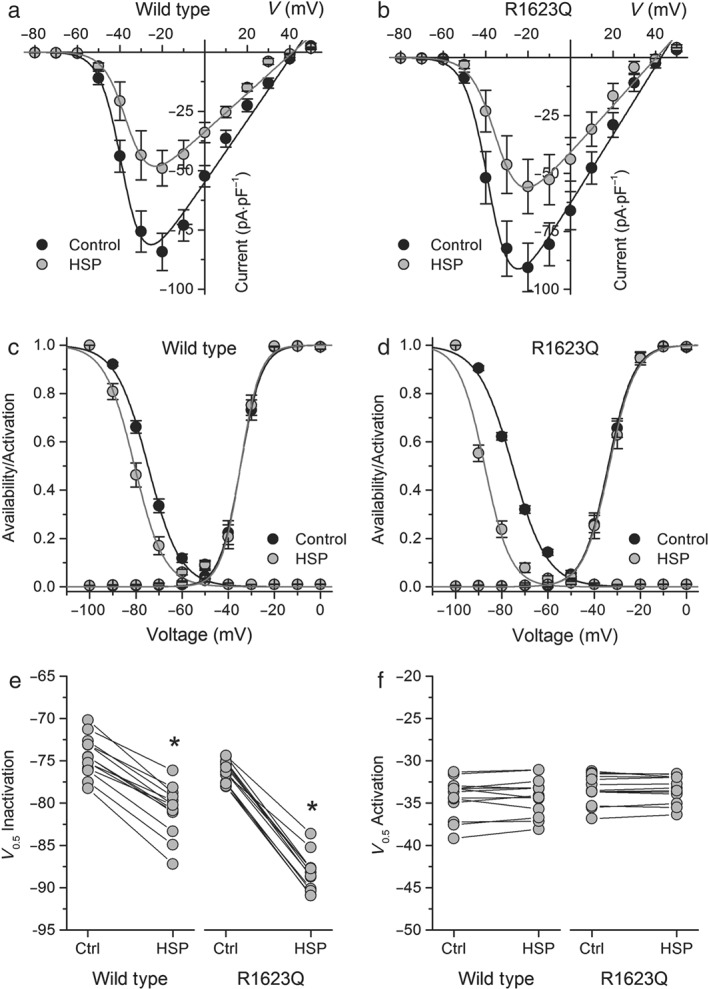
Effects of hesperetin (HSP) on voltage‐dependent kinetics of I Na in the wild‐type (WT) and R1623Q mutants. Current–voltage relationships of I Na in (a) WT and (b) R1623Q under control condition and in the presence of hesperetin (100 μmol·L−1). The dots represent the mean ± SEM (n = 13). Hesperetinsignificantly decreased the current compared with control in the range of voltage from −40 to +20 mV in both the WT and R1623Q channels (P < 0.05, paired t test). Availability and activation curves for (c) WT and (d) R1623Q in control and in the presence of hesperetin (100 μmol·L−1). The dots represent the mean ± SEM (n = 13). Hesperetin significantly shifted the availability curve compared with control in both the WT and R1623Q channels (P < 0.05, paired t test). Dot and line graphs showing the changes in (e) V inac and (f) V act induced by hesperetin (100 μmol·L−1) in both WT and R1623Q mutated channels. Note that hesperetin was more effective in shifting to more hyperpolarized potentials the V inac in the mutant R1623Q channel (n = 13, two‐way ANOVA with Tukey's post hoc test). V inac (and s inac) was −74.8 ± 0.3 mV (6.9 ± 0.3 mV) and −80.6 ± 0.4 mV (6.4 ± 0.4 mV) in WT channel and −75.4 ± 0.5 mV (7.5 ± 0.4 mV) and −87.4 ± 0.8 mV (5.6 ± 0.7 mV) in R1623Q channel in control and hesperetin respectively. V act (and s act) were −34.7 ± 0.4 mV (4.6 ± 0.3 mV) and −34.6 ± 0.4 mV (4.3 ± 0.3 mV) in WT channel and −33.9 ± 0.2 mV (5.5 ± 0.2 mV) and −33.5 ± 0.3 mV (5.6 ± 0.2 mV) in R1623Q channel in control and hesperetin respectively
Finally, we found that the inhibitory effect of hesperetin on peak I Na is strongly dependent on the HP for both WT and R1623Q channels (Figure 7a,b). The voltage for half‐maximal effect V HP and the associated slope factor s HP was similar for peak current amplitudes of WT and R1623Q currents (Figure 7c,d). We also analysed the dependence on the HP of the inhibitory action of hesperetin on I Na‐10 ms. For WT currents, the HP dependency of the hesperetin inhibitory effect on I Na‐10 ms was shifted in ≈6 mV to more negative potentials compared with that of the inhibitory effect on peak currents. Notably, this shift was significantly (P < 0.05) more pronounced (≈12 mV) for the R1623Q currents. Taken together, these results support the notion that hesperetin has a stronger effect on I Na inactivation kinetics than on peak current and that this effect is more evident in the R1623Q currents.
Figure 7.
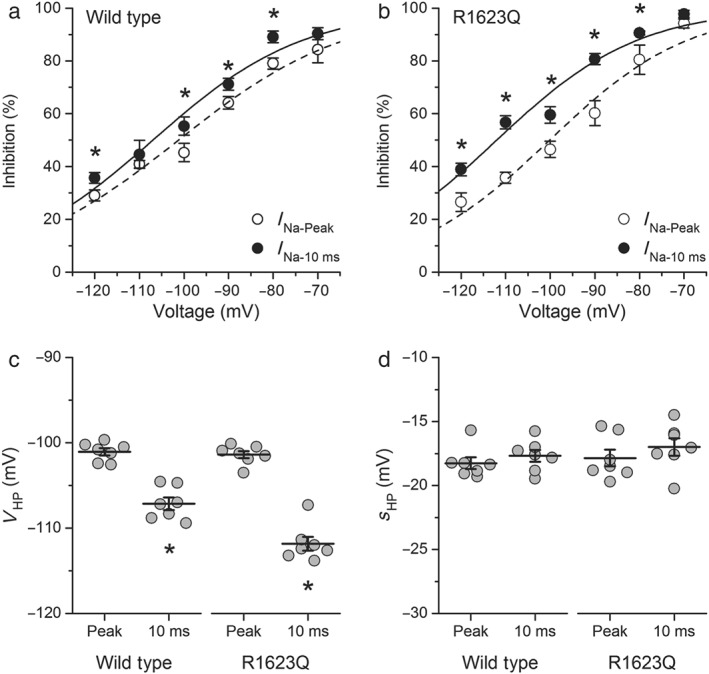
The inhibitory effect of hesperetin is strongly dependent on the holding potential in both the wild‐type and R1623Q channels. Boltzmann functions describing the holding potential dependency of hesperetin (100 μmol·L−1) effect on I Na‐Peak and I Na‐10 ms in (a) wild‐type and (b) R1623Q channels. The dots represent the mean ± SEM of seven cells, *P < 0.05 with respect to I Na‐peak (paired t test). The Boltzmann functions were characterized by (c) a voltage for half‐maximal effect V HP and (d) an associated slope factor s HP. Note that changes in V HP were more marked for I Na‐10 ms (n = 7, *P < 0.05, paired t test)
4. DISCUSSION
The main findings of the present study are that hesperetin blocked the rat and human NaV1.5 channels and that its inhibition was abolished by disruption of the binding site for local anaesthetics. More importantly, hesperetin induced an increase in the rate of inactivation of I Na in the WT, an effect that was more pronounced in the LQT3‐associated R1623Q mutant channel.
Like its congeners quercetin (Wallace, Baczkó, Jones, Fercho, & Light, 2006) and naringin (Alvarez‐Collazo et al., 2014), hesperetin blocked peak Na+ current in rat ventricular cardiomyocytes with an IC50 similar to other known antiarrhythmics such as lidocaine, disopyramide (Barber, Wendt, Starmer, & Grant, 1992), and flecainide (Belardinelli et al., 2013). Our estimated IC50 for hesperetin block of I Na was slightly higher than the one reported by Wang et al. (2016). However, the maximal effect of hesperetin reported in their study was only ≈40% or less. We here report >90% of inhibition, which explains the differences on the estimated IC50. We showed that hesperetin increased the rate of I Na inactivation, and its action was frequency dependent. The effect of hesperetin was voltage dependent, shifting the availability curve to more hyperpolarized potentials. However, to ensure a better voltage control during the flow of I Na, experiments had to be done at low extracellular Na+ concentrations. The heterologous expression system HEK293T offered the advantage of studying the action of hesperetin on human NaV1.5 WT channels at normal extracellular Na+. Hesperetin blocked the WT NaV1.5 channel with an IC50 slightly greater than that for cardiomyocytes, and it was accompanied by a clear decrease of inactivation time constants of I Na and a marked voltage dependency. These differences with respect to cardiomyocytes may be due to the fact that the experiments in HEK293T cells were performed at physiological extracellular Na+ concentration (see Barber et al., 1992).
The aromatic ring in the structure of hesperetin, common in local anaesthetics such as lidocaine, suggested that hesperetin could be interacting with the binding site for local anaesthetics. We studied the F1760A substitution in the human NaV1.5 channel and found that the block of I Na by hesperetin, at the highest concentration used (1 mmol·L−1), was reduced to only ≈20% and lost its voltage dependency. Additionally, hesperetin showed a use‐dependent behaviour on hNaV1.5 channels, which is typical for local anaesthetics such as lidocaine (Wang et al., 1996), thus indicating that hesperetin and lidocaine could share the same binding site within the pore. The notion that hesperetin interacts with the F1760 residue is also supported by the results of our molecular docking simulations. Furthermore, they suggest that hesperetin accesses this binding site via the lateral fenestrations of the hNaV1.5 pore, as previously indicated for local anaesthetics (Carnevale, 2018; O'Reilly et al., 2012; Payandeh et al., 2011). This is the first study suggesting that a flavonoid can bind to the local anaesthetic site (F1760) in the hNaV1.5 channel.
A key finding of the present study is that we also provide direct evidence that hesperetin decreased peak I Na and strongly decreased the amplitude and increased the rate of the slow inactivation phase in the LQT3 mutant R1623Q. The most salient difference of the mutated R1623Q NaV1.5 channels with the WT phenotype, expressed in HEK293T cells, was that the inactivation time constants in R1623Q channels were three to four times larger than in the WT. This slow inactivation is sufficient to ensure the longer action potential duration characteristic of LQT3 and constitute the basis for lethal arrhythmias in this syndrome (Remme, 2013). Hesperetin also blocked the R1623Q channels and its effect was accompanied by a marked decrease of inactivation time constants. However, the effects of hesperetin on the rate of inactivation of the R1623Q mutant were more marked, with significant reductions in τ slow and in the relative amplitude of the slow component already achieved in the low micromolar concentration range. The marked effects of hesperetin on I Na inactivation yielded lower IC50 values for the block of I Na‐10 ms, I Na‐50 ms, and I Na‐500 ms inhibition. When total transported charge (Na+) was estimated, the increase in rate of I Na inactivation by hesperetin, together with the decrease in peak I Na, resulted in a decreased amount of transported Na+ through the cell membrane in cells expressing the WT channel, which was more marked in cells expressing the R1623Q mutant. The marked effect of I Na inactivation in the R1623Q mutant by hesperetin is an important finding because this effect is relevant as an antiarrhythmic mechanism. This effect of hesperetin on I Na inactivation could be also advantageous to decrease the Na+ overload of the cells in LQT3 as a therapeutic strategy for the treatment of this syndrome (Miller et al., 2014).
Hesperetin block of I Na was HP dependent, with significant stronger effects on I Na and I Na‐10 ms at more depolarized HP. This could be physiologically relevant because the resting membrane potential of cardiomyocytes is more depolarized (around −80 mV) than in our experimental conditions. Moreover, we found that block of I Na‐10 ms by hesperetin was more sensitive to the HP compared with that of peak I Na and that this effect was two times more pronounced for the R1623Q currents. A consequence of this voltage‐dependent action is a shift to more negative potentials of the availability curve, an action that could be important in controlling action potential duration in several pathological states but particularly in LQT3 syndromes.
The control of action potential duration as an antiarrhythmic mechanism by blocking late or non‐inactivating Na+ channels has been recognized for a long time (see Coraboeuf, Deroubaix, & Coulombe, 1979). It has been commonly considered that most of the antiarrhythmic action of ‘Class I’ antiarrhythmics (local anaesthetic drugs) is linked to their use‐dependent behaviour. Here, we showed that block of I Na by hesperetin in WT and R1623Q channels was use‐dependent at higher stimulation frequency, which can be favourable for its antiarrhythmic action in LQT3. On the other hand, flavonoid consumption is associated with a decrease in the incidence of cardiovascular diseases due to their antioxidant and lipid‐lowering actions (Habauzit & Morand, 2012). The targeting of the slow inactivating phase of I Na would also contribute to cardioprotection and preventing Na+ overload and could be linked to the beneficial effects of citrus flavanones (e.g., Wallace et al., 2006). In this context, hesperetin exhibits a good bioavailability with maximal plasma concentration ranging from 0.2 to 5.5 μmol·L−1 in a time of 6 hr after ingestion (Erlund et al., 2001; Kay et al., 2017). However, it is rather difficult to translate the pharmacokinetic data obtained in plasma to tissues because hesperetin is a highly lipid soluble compound (logP = 2.6) and could accumulate in tissues at concentrations much higher than those observed in plasma (Takumi, Mukai, Ishiduka, Kometani, & Terao, 2011). It is thus possible that the concentrations used in the present study could be relevant to clinical values and/or to consumption as a human dietary supplement. Hesperetin also shows a better drug profile than other related flavonoids like naringin and naringenin because it shows less negative inotropic action, exerted a mild effect on the cardiac electrical properties in normal hearts, and exhibits a modest vasodilatory action in aortic rings (i.e., at concentrations of 30 μmol·L−1 or higher). The latter effect could be mediated by the inhibitory effect of hesperetin on voltage‐dependent Ca2+ and K+ channels (Liu et al., 2014) as well as TRPM3 (Naylor et al., 2010; Straub et al., 2013).
We must stress that the IC50 of hesperetin for inhibition of peak I Na is in the same order of magnitude as that of other antiarrhythmic compounds such as lidocaine or disopyramide (Barber et al., 1992), flecainide (Belardinelli et al., 2013), and mexiletine (Bankston et al., 2007). However, as for these antiarrhythmics, the IC50 for the decrease of I Na‐10 ms or I Na‐Late by hesperetin was much lower and comparable with those of mexiletine and ranolazine on the I Na‐Late enhanced or not by ATx II (Bankston et al., 2007; Belardinelli et al., 2013). We must also note that hesperetin action was increased at depolarized potentials and was more potent on the mutation R1623Q. More importantly, hesperetin markedly enhanced I Na inactivation, especially in the LQT3 mutant where the effects were statistically significant at concentrations as low as 1 μmol·L−1. The consequence of this action of hesperetin was a significant decrease in the net influx of Na+, an action that could prevent the action potential duration increase. Taking all these into account, hesperetin could be thus an interesting template molecule to develop effective antiarrhythmic drugs to prevent lethal TdP in the LQT3 syndrome.
In conclusion, hesperetin blocked I Na by interacting with the local anaesthetic site. It preferentially increased inactivation rate of NaV1.5 channels and reduced cell Na+ load. The effects on I Na inactivation rate were more marked in the LQT3 mutant R1623Q. These effects not only contributed to a cardioprotective effect of this flavanone but also might be important in the control of action potential duration in the LQT3 syndrome, placing this molecule as a possible template in the design of new drugs for the treatment of this lethal syndrome.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTIONS
J.A.‐C., J.L.A., and K.T. contributed in the conception and design of the study; J.A.‐C., A.L.‐R., L.G., and A.T. in the acquisition of the data; J.A.‐C., L.G., A.T., J.L.A., and K.T. in the analysis and interpretation of the data; J.A.‐C., J.L.A., and K.T. in drafting the manuscript; J.A.‐C., A.L.‐R., A.T., J.L.A., and K.T in the critical revision of the manuscript for important intellectual content; and J.A.‐C., A.L.‐R., L.G., A.T., J.L.A., and K.T. in the final approval of the version to be published.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Design & Analysis, and Animal Experimentation, and as recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1.
Peak INa is saturated at a holding potential of ‐100 mV.
Figure S2. Absence of INa‐Late in rat ventricular cardiomyocytes.
Figure S3. Voltage dependency of inactivation time constants of INa.
Figure S4. Tonic and use‐dependent block of hesperetin on INa in WT and R1623Q mutant hNaV1.5 channels.
Figure S5. Hesperetin does not affect the voltage‐dependence of channel activation.
Table 1: Summary table comparing the inhibition potency of hesperetin on the different experimental models studied.
Table 2: Summary table comparing the values of the parameters obtained from the modified Boltzmann fit of the I‐V relationships on the different experimental models studied.
Table 3: Summary table comparing the effects of hesperetin on the INa activation and availability curves on the different experimental models studied.
Table 4: Summary table comparing the effects of hesperetin on the time course of the inactivation phase of the INa traces evoked at ‐40 mV (cardiomyocytes) or ‐20 mV (hNaV1.5).
ACKNOWLEDGEMENTS
The authors wish to thank Prof. Angelika Lampert (Institute of Physiology, RWTH Aachen University) for graciously providing us the hNaV1.5 clone and Melissa Benoit for excellent assistance and support. Research was supported by grants from the Research Council of the KU Leuven (GOA/14/011) and the Fund for Scientific Research Flanders (FWO: G.0765.13).
Alvarez‐Collazo J, López‐Requena A, Galán L, Talavera A, Alvarez JL, Talavera K. The citrus flavanone hesperetin preferentially inhibits slow‐inactivating currents of a long QT syndrome type 3 syndrome Na+ channel mutation. Br J Pharmacol. 2019;176:1090–1105. 10.1111/bph.14577
REFERENCES
- Alexander, S. P. , Striessnig, J. , Kelly, E. , Marrion, N. V. , Peters, J. A. , Faccenda, E. , … CGTP Collaborators . (2017). The Concise Guide to PHARMACOLOGY 2017/18: Voltage‐gated ion channels. British Journal of Pharmacology, 174, S160–S194. 10.1111/bph.13884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez‐Collazo, J. , Díaz‐García, C. M. , López‐Medina, A. I. , Vassort, G. , & Alvarez, J. L. (2012). Zinc modulation of basal and β‐adrenergically stimulated L‐type Ca2+ current in rat ventricular cardiomyocytes: Consequences in cardiac diseases. Pflugers Pflügers Archiv / European Journal of Physiology, 464, 459–470. 10.1007/s00424-012-1162-3 [DOI] [PubMed] [Google Scholar]
- Alvarez‐Collazo, J. , López‐Medina, A. I. , Rodríguez, A. A. , & Alvarez, J. L. (2014). Mechanism of the negative inotropic effect of naringin in mouse heart. Journal of Pharmacy & Pharmacognosy Research, 2, 148–157. [Google Scholar]
- Antzelevitch, C. , Belardinelli, L. , Zygmunt, A. C. , Burashnikov, A. , Di Diego, J. M. , Fish, J. M. , … Thomas, G. (2004). Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation, 110, 904–910. 10.1161/01.CIR.0000139333.83620.5D [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch, C. , Nesterenko, V. , Shryock, J. C. , Rajamani, S. , Song, Y. , & Belardinelli, L. (2014). The role of late I Na in development of cardiac arrhythmias. Handbook of Experimental Pharmacology, 221, 137–168. 10.1007/978-3-642-41588-3_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankston, J. R. , Yue, M. , Chung, W. , Spyres, M. , Pass, R. H. , Silver, E. , … Kass, R. S. (2007). A novel and lethal de novo LQT‐3 mutation in a newborn with distinct molecular pharmacology and therapeutic response. PLoS One, 2, e1258 10.1371/journal.pone.0001258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber, M. J. , Wendt, D. J. , Starmer, C. F. , & Grant, A. O. (1992). Blockade of cardiac sodium channels. Competition between the permeant ion and antiarrhythmic drugs. The Journal of Clinical Investigation, 90, 368–381. 10.1172/JCI115871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belardinelli, L. , Liu, G. , Smith‐Maxwell, C. , Wang, W.‐Q. , El‐Bizri, N. , Hirakawa, R. , … Shryock, J. C. (2013). A novel, potent, and selective inhibitor of cardiac late sodium current suppresses experimental arrhythmias. The Journal of Pharmacology and Experimental Therapeutics, 344, 23–32. 10.1124/jpet.112.198887 [DOI] [PubMed] [Google Scholar]
- Bers, D. M. (2001). Excitation–contraction coupling and cardiac contractile force (Second ed.). Dordrecht, Springer Netherlands: Kluwer Academic Press. [Google Scholar]
- Calhoun, J.D. , and Isom, L.L. (2014). The role of non‐pore‐forming β subunits in physiology and pathophysiology of voltage‐gated sodium channels. pp 51–89. [DOI] [PubMed]
- Carnevale, V. (2018). Protonation underlies tonic vs. use‐dependent block. Proceedings of the National Academy of Sciences of the United States of America, 115, 3512–3514. 10.1073/pnas.1802178115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coraboeuf, E. , Deroubaix, E. , & Coulombe, A. (1979). Effect of tetrodotoxin on action potentials of the conducting system in the dog heart. The American Journal of Physiology, 236, H561–H567. 10.1152/ajpheart.1979.236.4.H561 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlund, I. , Meririnne, E. , Alfthan, G. , & Aro, A. (2001). Plasma kinetics and urinary excretion of the flavanones naringenin and hesperetin in humans after ingestion of orange juice and grapefruit juice. The Journal of Nutrition, 131, 235–241. 10.1093/jn/131.2.235 [DOI] [PubMed] [Google Scholar]
- Fredj, S. , Sampson, K. J. , Liu, H. , & Kass, R. S. (2006). Molecular basis of ranolazine block of LQT‐3 mutant sodium channels: Evidence for site of action. British Journal of Pharmacology, 148, 16–24. 10.1038/sj.bjp.0706709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommeyer, G. , & Eckardt, L. (2016). Drug‐induced proarrhythmia: Risk factors and electrophysiological mechanisms. Nature Reviews. Cardiology, 13, 36–47. 10.1038/nrcardio.2015.110 [DOI] [PubMed] [Google Scholar]
- Galán, L. , Talavera, K. , Vassort, G. , & Alvarez, J. L. (1998). Characteristics of Ca2+ channel blockade by oxodipine and elgodipine in rat cardiomyocytes. European Journal of Pharmacology, 357, 93–105. 10.1016/S0014-2999(98)00543-3 [DOI] [PubMed] [Google Scholar]
- Gao, Y. , Xue, X. , Hu, D. , Liu, W. , Yuan, Y. , Sun, H. , … Yan, G. X. (2013). Inhibition of late sodium current by mexiletine: A novel pharmotherapeutical approach in timothy syndrome. Circulation. Arrhythmia and Electrophysiology, 6, 614–622. 10.1161/CIRCEP.113.000092 [DOI] [PubMed] [Google Scholar]
- Goudet, C. , Ferrer, T. , Galàn, L. , Artiles, A. , Batista, C. F. V. , Possani, L. D. , … Tytgat, J. (2001). Characterization of two Bunodosoma granulifera toxins active on cardiac sodium channels. British Journal of Pharmacology, 134, 1195–1206. 10.1038/sj.bjp.0704361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habauzit, V. , & Morand, C. (2012). Evidence for a protective effect of polyphenols‐containing foods on cardiovascular health: An update for clinicians. Therapeutic Advances in Chronic Disease, 3, 87–106. 10.1177/2040622311430006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearse, D. J. , & Sutherland, F. J. (2000). Experimental models for the study of cardiovascular function and disease. Pharmacological Research, 41, 597–603. 10.1006/phrs.1999.0651 [DOI] [PubMed] [Google Scholar]
- Janse, M. J. , & Rosen, M. R. (2006). History of arrhythmias. Handbook of Experimental Pharmacology, 1–39. [DOI] [PubMed] [Google Scholar]
- Kambouris, N. G. , Nuss, H. B. , Johns, D. C. , Tomaselli, G. F. , Marban, E. , & Balser, J. R. (1998). Phenotypic characterization of a novel long‐QT syndrome mutation (R1623Q) in the cardiac sodium channel. Circulation, 97, 640–644. 10.1161/01.CIR.97.7.640 [DOI] [PubMed] [Google Scholar]
- Kay, C. D. , Pereira‐Caro, G. , Ludwig, I. A. , Clifford, M. N. , & Crozier, A. (2017). Anthocyanins and flavanones are more bioavailable than previously perceived: A review of recent evidence. Annual Review of Food Science and Technology, 8, 155–180. 10.1146/annurev-food-030216-025636 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. , Emerson, M. , & Altman, D. (2010). Improving bioscience research reporting: The ARRIVE guidelines for reporting animal research. Journal of Pharmacology and Pharmacotherapeutics, 1, 94–99. 10.4103/0976-500X.72351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y. , Niu, L. , Cui, L. , Hou, X. , Li, J. , Zhang, X. , & Zhang, M. (2014). Hesperetin inhibits rat coronary constriction by inhibiting Ca2+ influx and enhancing voltage‐gated K+ channel currents of the myocytes. European Journal of Pharmacology, 735, 193–201. 10.1016/j.ejphar.2014.03.057 [DOI] [PubMed] [Google Scholar]
- Miller, D. , Wang, L. , & Zhong, J. (2014). Sodium channels, cardiac arrhythmia, and therapeutic strategy. Advances in Pharmacology, 70, 367–392. 10.1016/B978-0-12-417197-8.00012-2 [DOI] [PubMed] [Google Scholar]
- Naylor, J. , Li, J. , Milligan, C. J. , Zeng, F. , Sukumar, P. , Hou, B. , … Beech, D. J. (2010). Pregnenolone sulphate‐ and cholesterol‐regulated TRPM3 channels coupled to vascular smooth muscle secretion and contraction. Circulation Research, 106, 1507–1515. 10.1161/CIRCRESAHA.110.219329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly, A. O. , Eberhardt, E. , Weidner, C. , Alzheimer, C. , Wallace, B. a. , & Lampert, A. (2012). Bisphenol A binds to the local anesthetic receptor site to block the human cardiac sodium channel. PLoS One, 7(7), e41667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh, J. , Scheuer, T. , Zheng, N. , & Catterall, W. A. (2011). The crystal structure of a voltage‐gated sodium channel. Nature, 475, 353–358. 10.1038/nature10238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips, J. C. , Braun, R. , Wang, W. , Gumbart, J. , Tajkhorshid, E. , Villa, E. , … Schulten, K. (2005). Scalable molecular dynamics with NAMD. Journal of Computational Chemistry, 26, 1781–1802. 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remme, C. A. (2013). Cardiac sodium channelopathy associated with SCN5A mutations: Electrophysiological, molecular and genetic aspects. The Journal of Physiology, 591, 4099–4116. 10.1113/jphysiol.2013.256461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remme, C. A. , & Wilde, A. A. (2014). Targeting sodium channels in cardiac arrhythmia. Current Opinion in Pharmacology, 15, 53–60. 10.1016/j.coph.2013.11.014 [DOI] [PubMed] [Google Scholar]
- Roohbakhsh, A. , Parhiz, H. , Soltani, F. , Rezaee, R. , & Iranshahi, M. (2015). Molecular mechanisms behind the biological effects of hesperidin and hesperetin for the prevention of cancer and cardiovascular diseases. Life Sciences, 124, 64–74. 10.1016/j.lfs.2014.12.030 [DOI] [PubMed] [Google Scholar]
- Scholz, E. P. , Zitron, E. , Katus, H. A. , & Karle, C. A. (2010). Cardiovascular ion channels as a molecular target of flavonoids. Cardiovascular Therapeutics, 28, 46–52. [DOI] [PubMed] [Google Scholar]
- Scholz, E. P. , Zitron, E. , Kiesecker, C. , Thomas, D. , Kathöfer, S. , Kreuzer, J. , … Greten, J. (2007). Orange flavonoid hesperetin modulates cardiac hERG potassium channel via binding to amino acid F656. Nutrition, Metabolism, and Cardiovascular Diseases, 17, 666–675. 10.1016/j.numecd.2006.06.002 [DOI] [PubMed] [Google Scholar]
- Schwartz, P. J. , Crotti, L. , & Insolia, R. (2012). Long‐QT syndrome: From genetics to management. Circulation. Arrhythmia and Electrophysiology, 5, 868–877. 10.1161/CIRCEP.111.962019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski, I. , Shen, J. , Timothy, K. W. , Lehmann, M. H. , Priori, S. , Robinson, J. L. , … Keating, M. T. (2000). Spectrum of mutations in long‐QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation, 102, 1178–1185. 10.1161/01.CIR.102.10.1178 [DOI] [PubMed] [Google Scholar]
- Straub, I. , Mohr, F. , Stab, J. , Konrad, M. , Philipp, S. , Oberwinkler, J. , & Schaefer, M. (2013). Citrus fruit and fabacea secondary metabolites potently and selectively block TRPM3. British Journal of Pharmacology, 168, 1835–1850. 10.1111/bph.12076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumi, H. , Mukai, R. , Ishiduka, S. , Kometani, T. , & Terao, J. (2011). Tissue distribution of hesperetin in rats after a dietary intake. Bioscience, Biotechnology, and Biochemistry, 75, 1608–1610. 10.1271/bbb.110157 [DOI] [PubMed] [Google Scholar]
- Trott, O. , & Olson, A. J. (2010). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31, 455–461. 10.1002/jcc.21334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veerman, C. C. , Wilde, A. A. M. , & Lodder, E. M. (2015). The cardiac sodium channel gene SCN5A and its gene product NaV1.5: Role in physiology and pathophysiology. Gene, 573, 177–187. 10.1016/j.gene.2015.08.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, C. H. R. , Baczkó, I. , Jones, L. , Fercho, M. , & Light, P. E. (2006). Inhibition of cardiac voltage‐gated sodium channels by grape polyphenols. British Journal of Pharmacology, 149, 657–665. 10.1038/sj.bjp.0706897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. W. , Nie, L. , George, A. L. , & Bennett, P. B. (1996). Distinct local anesthetic affinities in Na+ channel subtypes. Biophysical Journal, 70, 1700–1708. 10.1016/S0006-3495(96)79732-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, G. K. , Quan, C. , & Wang, S. (1998). A common local anesthetic receptor for benzocaine and etidocaine in voltage‐gated μ1 Na+ channels. Pflügers Archiv, 435, 293–302. [DOI] [PubMed] [Google Scholar]
- Wang, H. , Wang, H.‐F. , Zhang, H. , Wang, C. , Chen, Y.‐F. , Ma, R. , … Tang, Q. (2016). Inhibitory effects of hesperetin on NaV1.5 channels stably expressed in HEK 293 cells and on the voltage‐gated cardiac sodium current in human atrial myocytes. Acta Pharmacologica Sinica, 37, 1563–1573. 10.1038/aps.2016.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb, B. , & Sali, A. (2016). Comparative protein structure modeling using MODELLER In Current protocols in bioinformatics (pp. 5.6.1–5.6.37). Hoboken, NJ, USA: John Wiley & Sons, Inc. 10.1002/cpbi.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer, T. , & Surber, R. (2008). SCN5A channelopathies—An update on mutations and mechanisms. Progress in Biophysics and Molecular Biology, 98, 120–136. 10.1016/j.pbiomolbio.2008.10.005 [DOI] [PubMed] [Google Scholar]
- Zygmunt, A. C. , Eddlestone, G. T. , Thomas, G. P. , Nesterenko, V. V. , & Antzelevitch, C. (2001). Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. American Journal of Physiology. Heart and Circulatory Physiology, 281, H689–H697. 10.1152/ajpheart.2001.281.2.H689 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1.
Peak INa is saturated at a holding potential of ‐100 mV.
Figure S2. Absence of INa‐Late in rat ventricular cardiomyocytes.
Figure S3. Voltage dependency of inactivation time constants of INa.
Figure S4. Tonic and use‐dependent block of hesperetin on INa in WT and R1623Q mutant hNaV1.5 channels.
Figure S5. Hesperetin does not affect the voltage‐dependence of channel activation.
Table 1: Summary table comparing the inhibition potency of hesperetin on the different experimental models studied.
Table 2: Summary table comparing the values of the parameters obtained from the modified Boltzmann fit of the I‐V relationships on the different experimental models studied.
Table 3: Summary table comparing the effects of hesperetin on the INa activation and availability curves on the different experimental models studied.
Table 4: Summary table comparing the effects of hesperetin on the time course of the inactivation phase of the INa traces evoked at ‐40 mV (cardiomyocytes) or ‐20 mV (hNaV1.5).