


Abstract
Proper repair of oxidatively damaged DNA bases is essential to maintain genome stability. 8-Oxoguanine (7,8-dihydro-8-oxoguanine, 8-oxoG) is a dangerous DNA lesion because it can mispair with adenine (A) during replication resulting in guanine to thymine transversion mutations. MUTYH DNA glycosylase is responsible for recognizing and removing the adenine from 8-oxoG:adenine (8-oxoG:A) sites. Biallelic mutations in the MUTYH gene predispose individuals to MUTYH-associated polyposis (MAP), and the most commonly observed mutation in some MAP populations is Y165C. Tyr165 is a ‘wedge’ residue that intercalates into the DNA duplex in the lesion bound state. Here, we utilize single molecule fluorescence microscopy to visualize the real-time search behavior of Escherichia coli and Mus musculus MUTYH WT and wedge variant orthologs on DNA tightropes that contain 8-oxoG:A, 8-oxoG:cytosine, or apurinic product analog sites. We observe that MUTYH WT is able to efficiently find 8-oxoG:A damage and form highly stable bound complexes. In contrast, MUTYH Y150C shows decreased binding lifetimes on undamaged DNA and fails to form a stable lesion recognition complex at damage sites. These findings suggest that MUTYH does not rely upon the wedge residue for damage site recognition, but this residue stabilizes the lesion recognition complex.
INTRODUCTION
Base excision repair (BER), a pathway conserved from bacteria to humans, is implicated in cancer initiation and tumor progression ((1–7), and for reviews see (8,9)). The BER pathway is responsible for the removal of the majority of endogenous DNA lesions (for reviews see (10–13)), including oxidized guanine. Guanine has the lowest redox potential of the four DNA bases and it is readily oxidized to 7,8-dihydro-8-oxoguanine (8-oxoG). When paired with cytosine (C), this mutated base is directly recognized and removed by hOGG1 DNA glycosylase (for a review see (14)) before it is repaired by downstream enzymes in the short patch repair pathway. However, if 8-oxoG is present during replication, replicative polymerases will frequently insert adenine opposite the damaged residue (for a review see (15)). If the adenine strand then undergoes a further replication event, a G to T transversion mutation will occur. When 8-oxo-dGTP is present in the nucleotide pool, polymerases can also insert 8-oxoG opposite adenine (A) or C and contribute to downstream mutation events (16–19).
The mutational consequences of 8-oxoG:A mismatch are so severe that organisms have a glycosylase, MUTYH, that is highly specific for removal of adenine paired with 8-oxoG (for reviews see (20,21)). MUTYH is a monofunctional glycosylase and does not nick the DNA backbone. The resulting apurinic (AP) site opposite 8-oxoG is a high risk for further degradation and DNA strand breaks, and it has been proposed that MUTYH remains tightly bound to this product until coordinated handoff to AP endonuclease (APE) in the next step of the BER pathway (22–24). After cleavage of the DNA backbone by APE, downstream BER polymerase beta inserts cytosine opposite 8-oxoG, and ligase seals the backbone. This temporary conversion to 8-oxoG:C gives hOGG1 another chance to remove 8-oxoG and prevent a G to T transversion mutation (20,24). MUTYH is highly conserved amongst living organisms; the Escherichia coli MutY protein is 41% similar to human (25,26) while the Mus musculus protein MUTYH is 86% similar to human (25).
A critical first step in BER is the glycosylase search for and recognition of damage sites (27–30), a feat that glycosylases accomplish using only thermal energy. This search is complicated by the fact that oxidatively-damaged DNA bases often differ only slightly from their normal counterparts and are present in a vast excess of undamaged bases. Glycosylase search plays a significant role in overall enzyme activity but is difficult to observe in traditional bulk solution assays that rely on short oligonucleotide (oligo) substrates. It has recently been shown through single molecule fluorescence microscopy (SMFM) studies with long DNA tightrope substrates that glycosylases scan rotationally along the DNA backbone for damages until recognition or dissociation occurs (31,32). SMFM studies have also shown that two structurally distinct glycosylase families, the Nth (endonuclease III) or helix-hairpin-helix superfamily, to which the MutY homologs belong, as well as the Fpg/Nei (formamidopyrimidine DNA glycosylase/endonuclease VIII) family, both share similar diffusive behavior while scanning DNA (33,34). The scanning is random, bidirectional, and highly redundant. Furthermore, all of the E. coli glycosylases studied use a ‘wedge’ amino acid residue to periodically insert into the DNA and interrogate for damage (33,34). Previous work was limited to E. coli glycosylases that are believed to play a ‘housekeeping’ role in damage removal and are likely to have a large search burden. However, the role of the wedge residue in mammalian glycosylases such as MUTYH, which may be cell cycle regulated (16) and therefore have different search requirements and search mechanisms, has not been investigated using SMFM.
In humans, biallelic mutations in the gene encoding MUTYH glycosylase are responsible for a subset of classic familial adenomatous polyposis called MUTYH-associated polyposis (MAP) (for reviews see (20,35,36)). One of the most common mutations observed in Caucasian MAP patients is Y165C (37–40), the wedge residue of MUTYH. A crystal structure of the bacterial ortholog MutY shows the corresponding wedge tyrosine residue inserted into the DNA helix directly 5′ to 8-oxoG (41). This insertion helps to stabilize adenine extrusion from the duplex into the enzyme active site (Figure 1A), a common feature observed in DNA glycosylases. Bulk ensemble studies indicate that MUTYH wedge variant enzymes have reduced catalytic activity (23,42–49) and decreased binding affinity for DNA damage sites (23,42–44,47), but bulk assays are unable to resolve whether these deficiencies may be due to inadequate DNA scanning, inefficient lesion recognition, or fast release of the lesion. The structural similarities in the position of the MutY wedge residue as compared to E. coli Fpg, Nei, and Nth raise the question of whether the MutY/MUTYH wedge residue plays a similar role in base interrogation during damage search (33,34). To date all SMFM studies of glycosylases on damaged substrates relied upon randomly distributed damage within lambda (λ) DNA and were unable to pinpoint spatially where the damage resided in the tightrope. The studies herein utilize a newly developed DNA tightrope substrate that contains damage sites at known locations to allow for direct observation of MUTYH glycosylase interactions with damage.
Figure 1.

MutY structure, single-molecule assay, and model for MUTYH diffusion. A) Crystal structure of B. stearothermophilus MutY (green) (PDB 1RRQ) crosslinked to an 8-oxoG:A site showing the enzyme encircling the DNA (blue). The wedge residue (green) intercalates 5′ to the damage (red), and the adenine (orange) is everted from the base stack into the active pocket. B) DNA molecules (blue) extended by hydrodynamic flow and non-specifically attached between immobile 3 μm polylysine-coated silica beads in a flow cell to resemble tightropes for observation of glycosylase (green) scanning. Qdot-glycosylase complex constructed by targeting the hexahistidine tag on the C-terminus of the glycosylase with a biotinylated antihistidine IgG linked to the streptavidin-coated Qdot. (C) DNA tightropes consist of concatemerized plasmids containing regularly spaced Cy5 fiducial markers 2727 base pairs apart. (D) Tightrope substrates for single molecule experiments consist of a combination of Cy5 marked plasmids and plasmids that contain a single damage site (8-oxoG:A or 8-oxoG:C). The substrates concatemerize with randomly distributed Cy5 markers, and the position of the damage site can be determined using the plasmid repeat distance (905 nm). (E) Example kymograph of MUTYH scanning along a concatemer tightrope that contains 8-oxoG damage sites. (F) Model for MUTYH scanning along tightrope DNA.
We use SMFM on two types of DNA tightrope substrates to investigate the role of the tyrosine wedge residue in the search and damage site recognition behavior of both wild-type (WT) and wedge variant (i.e. E. coli (Y82C) and M. musculus (Y150C)) orthologs of human MUTYH. We found that on undamaged DNA, the MutY/MUTYH wedge residue does not play a role in a slow search mode that was observed for other glycosylases. On λ DNA tightrope substrates that contain randomly distributed AP product analog sites, WT enzymes demonstrate long-lived pauses during diffusional search, while wedge variants do not. On plasmid concatemer DNA tightropes that contain specific 8-oxoG:A sites in precise marked locations, we directly observe extremely efficient recognition and binding of 8-oxoG:A by MUTYH WT. We also observe that MUTYH WT recognizes and pauses at 8-oxoG:C sites. In contrast, the MUTYH Y150C wedge variant has decreased binding affinity to both undamaged and damage-containing DNA, and once bound to the damage-containing DNA is inefficient at both detecting damage and remaining bound to it. These MUTYH Y150C DNA scanning characteristics may explain the reported inefficient damage site repair (23). These studies represent the first direct, real time observation of a glycosylase as it interacts with a specific damage substrate at a known location and provide new insights into specific search deficiencies of a glycosylase cancer variant.
MATERIAL AND METHODS
Glycosylase preparation and activity
MutY and MUTYH were cloned with a single C-terminal hexahistidine tag by insertion of the glycosylase cDNA into pET30a. The form of the murine enzyme used is N-terminally truncated and starts at Ser29 as reported in (22). The bacterial Y82C and murine Y150C wedge substitutions were generated using site directed mutagenesis (Agilent Technologies). All mutations were confirmed through sequencing by the UVM Advanced Genome Technology Core. The active fractions of MutY and MUTYH WT and variant enzymes were determined using burst phase analysis in the presence of excess concentrations of substrate as described (50). Briefly, 20 nM of a 35-base pair oligo containing A opposite 8-oxoG and 5′ end labeled with (γ-32P) on the adenine-containing strand was incubated with MutY or MUTYH enzyme. These measurements were taken in the presence of three different concentrations of total enzyme (not exceeding 4 nM active enzyme) to allow for fitting of the burst phase for active fraction (50). MutY assays were carried out at 37°C in 50 mM Tris, pH 8.0, 150 mM potassium glutamate, 1 mM DTT, and 1 mg/mL BSA. MUTYH assays were carried out at 37°C in 50 mM Tris, pH 8.0, 30 mM potassium glutamate, 1 mM DTT, and 1 mg/mL BSA. 10 μL portions of the reaction were quenched into 2 μL 2N NaOH at 0.25, 0.5, 1, 2, 5, 15, 30, and 60 min and heated to 95°C for 5 min to allow for backbone cleavage of excised strand. 12 μL of formamide loading dye was added to each sample, and the cleaved strands were resolved from the non-repaired strands on a 12% (w/v) polyacrylamide sequencing gel. The gel was dried on Whatman 3M paper, exposed to a phosphor image screen, and imaged on a BioRad Pharos FX Plus phosphoimager and analyzed using Quantity One Software (Bio-Rad). Burst phase analysis was also used to ensure that conjugation to Qdot did not affect activity of MutY WT and MUTYH WT enzymes (Supplementary Figure S11).
Single molecule concatemer substrates
To insert 8-oxoG damage sites opposite an A residue, a concatemerized plasmid system was developed (51–53). First, excision of a single strand region was necessary to allow for insertion of an oligo opposite a specific base in the plasmid. Three Nt.BspQI (NEB) nicking sites were cloned into a 39 basepair region of pUC19 (NEB) plasmid (Supplementary Figure S4). Once nicked, two short single stranded oligos are melted away and replaced by 10-fold molar excess over plasmid of a 39 bp oligo (Tm ∼ 73.5°C) containing the damage (Supplementary Figure S5A). To minimize production of nonspecific damage, annealing was accomplished by heating the plasmid to 80°C for one minute and cooling to 4°C over 10 minutes. Following insertion of the 39 bp oligo, the nicked sites are sealed using T4 DNA ligase (NEB). To remove randomly occurring AP sites, the ligated plasmid was treated with a 5-fold molar excess of APE for 1 hour at 37°C. Nicked plasmids (either from APE treatment or failed ligation) were degraded using Exonuclease V (NEB) followed by removal of the exonuclease using a Monarch PCR cleanup kit (NEB). Plasmids were digested with BsaI (NEB), and the enzyme was removed using a Monarch PCR cleanup kit (NEB). Linear plasmids were stored at -20°C before concatemerization at a plasmid molar concentration of 50 nM for 30 minutes at 16°C using T4 DNA ligase (NEB). When linearized plasmid containing 8-oxoG:A is concatemerized using T4 DNA Ligase, the resulting tightrope contains single damage sites separated by exactly 2726 bp of intervening undamaged DNA (Supplementary Figures S5 and S6). Within a tightrope, all nicking sites, and accordingly all damages, exist on one strand of the DNA. To visualize the position of the damage sites in the pNIKCAT concatemer tightropes during single-molecule experiments, a mixture of 20% Cy5 pNIKCAT was combined with 80% undamaged pNIKCAT or 8-oxoG pNIKCAT to create tightropes that contain small percentage of a randomly occurring fiducial dye marker (Supplementary Figures S5A and S5B). Importantly, preparations of 8-oxoG and Cy5 pNIKCAT plasmids showed 90% of incorporation of 8-oxoG- or Cy5-containing oligos (Supplementary Figures S5C and S6A). When combined in an 80:20 ratio (8-oxoG:Cy5), the resulting concatemer tightropes consist of 72% 8-oxoG pNIKCAT, 18% Cy5 pNIKCAT, and 10% undamaged pNIKCAT. The damage sites lie within an integer multiple of the plasmid length from these fiducial markers (2727 bp) (Supplementary Figure S6).
Single molecule sample preparation
Our standard buffers used for SMFM experiments are SMFM elongation buffer (50 mM Tris, pH 8.0, 50–150 mM potassium glutamate, and 2 mM DTT) and SMFM glycosylase buffer (50 mM Tris, pH 8.0, 50–150 mM potassium glutamate, 1 mg/mL BSA and 2 mM DTT). The potassium glutamate concentrations during imaging differ for MutY (150 mM) and MUTYH (50 mM) because MUTYH Y150C shows little glycosylase activity above 50 mM NaCl. For λ DNA substrates, 200 μL of 1.3 μg/mL λ was introduced into the chamber and elongated by 100 μL cycles of infusion and withdrawal at a flow rate of 500 μL/min over 5 minutes to allow for non-specific adhesion of the DNA to polylysine coated silica beads. The chamber was then rinsed with 200 μL single-molecule elongation buffer and equilibrated using three additions of 200 μL single molecule glycosylase buffer. When imaging with λ, the glycosylase was conjugated under conditions that were shown in previous experiments to give a single antibody conjugated to each Qdot, and thus only a single glycosylase per Qdot. Briefly, 5 μL of Penta-his biotinylated antibody (Qiagen) was combined with 1 μL streptavidin coated Qdot 655 for 10 minutes on ice. Equal volumes of this mixture were combined with 200 nM active MutY or MUTYH and allowed to bind for 10 minutes on ice to give a molar ratio of 1 active glycosylase: 5 antibodies: 1 Qdot. This solution was then diluted to a final concentration of 30 pM glycosylase: 30 pM Qdot in single-molecule glycosylase buffer for introduction into the chamber and imaging. For conditions where no catalytic reaction was possible (undamaged λ, λ with rAP sites), the chamber was periodically rinsed with 50 mM Tris, pH 8.0, 800 mM potassium glutamate buffer to remove glycosylase before rinsing with three 200 μL volumes of single-molecule glycosylase buffer and reintroduction with freshly diluted glycosylase solution. These salt rinses did not affect the DNA-strings or the scanning behavior, and were even used as a pretreatment for the concatemer substrates as described below. We found that YOYO-1 was disruptive to MutY scanning along undamaged λ DNA, and all λ substrates were stained with 5 nM YOYO-1 at the end of an experiment.
For plasmid concatemer substrates, after attachment to the pump the chamber was blocked against residual small plasmid monomer fragments using 200 μL single-molecule glycosylase buffer. The chamber was rinsed with 200 μL single-molecule elongation buffer before 200 uL of ∼3 μg/mL plasmid concatemer was stretched as described above for λ. After the infusion/withdrawal cycles, the chamber was rinsed with 200 μL single-molecule elongation buffer, followed by a rinse with 200 μL 50 mM Tris, pH 8.0, 800 mM potassium glutamate. The chamber was then equilibrated using three additions of 200 μL single molecule glycosylase buffer. The labeling was slightly different for plasmid concatemer substrates. To prevent any unobserved enzyme-DNA binding events, the conjugation was held to a strict 1 total enzyme: 1 antibody: 1 Qdot ratio. Under this scheme, it was necessary to first incubate the enzyme with the antibody for 10 minutes on ice before adding the Qdot. The glycosylase was then introduced into the chamber at a final concentration of 30–50 pM in single-molecule glycosylase buffer for imaging. To stabilize the Cy5 dyes, imaging of plasmid concatemers took place in the presence of oxygen scavengers at a final concentration of 45 μg/mL catalase, 70 μg/mL glucose oxidase, and 5.83 mg/mL glucose. Plasmid concatemer substrates were not rinsed with high salt to remove glycosylases, and YOYO-1 was not used on plasmid concatemers.
Image acquisition and analysis
Movies of λ DNA substrates were recorded on a microscope system previously described (34). Movies of plasmid concatemer substrates were recorded on a customized Nikon TE2000U microscope, equipped with through-the-objective (Nikon PlanApo 100 ×, 1.49 N.A.) excitation light from a 639nm 1000mW laser (Coherent). Excitation light was adjusted to a subcritical, near-TIRF angle. Images were recorded using an intensified CCD camera (XR Turbo G, running Piper Control, version 2.7.00.08, software; Standard Photonics, Palo Alto CA) where typically 5,000 images (99 nm per pixel) were captured with 66ms integration time (15 frames per second).
All glycosylases that bound to a DNA tightrope and could be observed for greater than 2 frames (133 ms) were catalogued and included in further analysis. Qdot-labeled glycosylases interacting with DNA tightropes for greater than 24 frames were tracked using ImageJ (National Institutes of Health, Bethesda, MD) with the SpotTracker 2D plug-in (54). Cy5 fiducial dyes were localized by analyzing a single image, formed by integrating the initial 500 frames of a recording, and then analyzed using the ImageJ plugin ThunderSTORM (55). The position of damages relative to these dye fiducials is determined by the plasmid repeat unit (Figure 1, Supplementary Figures S5 and S6). Stage drift that occurred between localization of Cy5 fiducial dyes and subsequent recording of Qdot-labeled glycosylase motion was determined by maximizing the cross-correlation of 40 × 40 pixel windows centered on the 3-micron beads which support the DNA tightropes, cropped from recordings of Cy5 fiducial dyes and Qdot-labeled glycosylases, respectively. Measured drift occurring between these recordings were then subtracted from glycosylase trajectory, with these drift values typically less than 2 pixels (200nm). Using this method, the combined localization error for damage or Cy5 dye position was 21.7 nm.
Data analysis
We used two different approaches to characterize diffusion constants, and each method required a minimum observation time. Time-weighted (sliding window) diffusion constants for any trajectory that was greater than 60 frames in duration were calculated by determining an MSD within a 60-frame-wide sliding window. Diffusion constants (D) were calculated by characterizing the displacement vs. time trajectories using MSD analysis (33,34). MSDs were defined as follows:
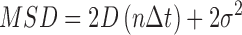 |
For 1<n<N/4, where
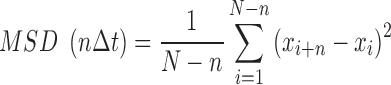 |
where Δt is the time interval between frames (66 ms), xi is the position of the glycosylase at time i, and σ is the tracking error, which was found to be 34.6 nm for the experimental tracking data. A diffusion constant for each complete trajectory (Dtraj) with a duration greater than 24 frames was calculated from the slope of the MSD of the first 25% of the MSD vs. nΔt plot, where N is the total number of frames in the trajectory. Information about number of tightropes included, molecules observed, and total number of data frames analyzed is provided in Supplementary Table S1.
Damage site encounter lifetimes were counted as the number of consecutive frames that the enzyme remained within 100 nm of a position-mapped lesion (red points in Figure 3A and B). This wide margin was chosen to be approximately 3 times the standard deviation of the tracking error so as to avoid inadvertently sub-dividing individual damage encounter events due to single-frame spurious spot localizations. If the enzyme departs the site (black points in Figure 3A and B), and then re-visits the same location, they are considered separate events. The distribution of encounter event lifetimes was fit with a double exponential distribution:
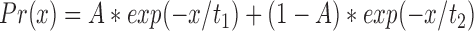 |
Figure 3.

Representative displacement trajectories showing MUTYH Y150C interactions with 8-oxoG:A damage. Segments of three individual scanning events are shown in (A). Red portions of the trace represent positions within +/- 100 nm of the damage site. (B) When portions of the trajectories from panel (A) are displayed with an expanded x-axis, MUTYH Y150C shows complex interactions with damage sites, including transient pauses, bypass events, and a combination of bind/release damage site revisitation behaviors. Bypass is indicated where interaction with the damage site is shorter than the 1.4 s mean transit lifetime for fast diffusion in the absence of damage. 8-oxoG:A damage sites are marked as black lines. Dye site is marked as a green line. (C) The survival fraction of MUTYH Y150C encounter event lifetimes (red circles) within ±100 nm of a known damage site. The population was fit with a double exponential to give lifetimes of 1.6 ± 0.3s (89%) and 10 ± 5s (11%), (120 damage encounter events, 5 trajectories). The survival fraction of MUTYH Y150C (green diamonds) and MUTYH WT (blue squares) encounters with random undamaged sites show single populations with lifetimes of 1.1 ± 0.1s (131 encounters, 5 trajectories) and 1.4 ± 0.2s (70 encounters, 3 trajectories), respectively. Lifetime values represent mean and standard deviation. See Supplementary Table S1 for sample statistics.
Where A is the amplitude of the first (fast) phase, and t1 and t2 are time constants for the fast and slow populations, respectively. This distribution was fit using maximum likelihood fitting of univariate distributions, as implemented in the ‘fitdistr’ function in the MASS package for the statistical programming language R (Figure 3C) (56). For controls at undamaged locations, we also collected encounter lifetimes of MUTYH Y150C and MUTYH WT at undamaged locations on the tightrope (as determined relative to fiducial markers), which were analyzed by the same methodologies. These control data sets were also fit with the same double exponential model, but in both cases, fitted results indicated the presence of only a single population (A∼1.0) (Figure 3C, Supplementary Figure S12).
Overall enzyme binding lifetimes (Figure 4E–G) were calculated for all molecules according to the survival estimator methodologies described by Kaplan and Meier (57), as implemented in the ‘survival’ package of the statistical programming language ‘R’ (http://www.R-project.org/) (58). Statistical significance was established using a t test on log-transformed data for diffusion constants, and the log-rank test for binding lifetime data.
Figure 4.
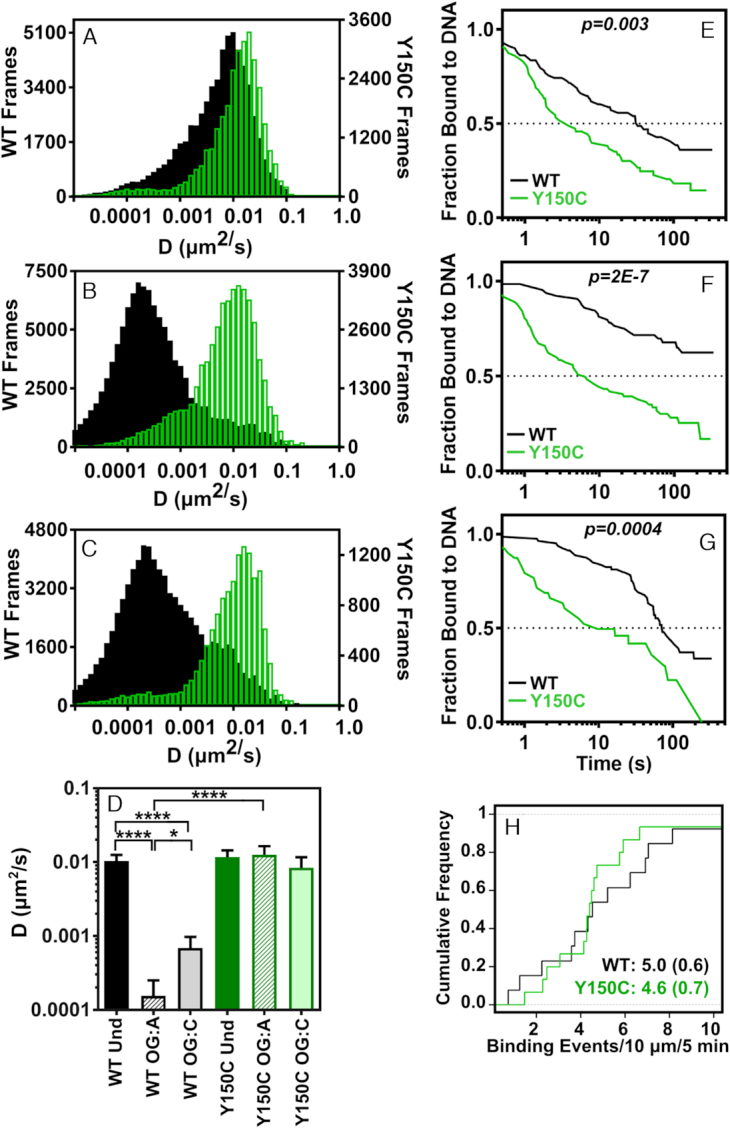
Diffusion and binding behavior of MUTY WT and Y150C on DNA concatemer tightropes. Compiled time-weighted distributions of diffusion constants from sliding window (60 frames per window) analysis of displacement trajectories for diffusion of MUTYH WT (black bars) and MUTYH Y150C (green bars) on concatemer tightropes containing (A) undamaged, (B) 8-oxoG:A, (C) 8-oxoG:C sites. (D) Mean value of diffusion constants for trajectories (Dtraj), determined by fitting the first 25% of the MSD plot for each trajectory (error bars represent SEM), (**** p < 0.0001, two tailed). Binding lifetime survival curves are shown for MUTYH WT (black traces) and MUTYH Y150C (green traces) on (E) undamaged, (F) 8-oxoG:A, and (G) 8-oxoG:C sites. p-values describing significance between the traces were determined using a log-rank (Mantel-Haenszel) test. Number of molecules for each condition corresponds to the information given in Supplementary Table S1. (H) Binding frequency of MUTYH WT (black trace, n = 26) and MUTYH Y150C (green trace, n = 30) to undamaged concatemer DNA under equivalent enzyme and DNA tightrope concentrations with mean and SD in parentheses. See Supplementary Table S1 for sample statistics.
To determine the fraction of enzymes that bypass a lesion, we measured the total end-to-end distance scanned (i.e. total scanning range) for each trajectory as a function of total observation time (Figure 5). First, trajectories that persist for <10s were omitted from this analysis, as they did not persist long enough to have traveled at least 450 nm (the farthest possible distance from the nearest damage site), which was calculated using Mean First Passage time:
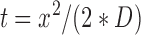 |
where t is time, x is the distance traveled and D is the diffusion constant for diffusion along DNA. The proportion of encounter events that exceed 1000 nm (the length of one plasmid plus tracking error) in total scanning range (implying the molecule bypassed a lesion) is reported in Figure 5F and Supplementary Figure S9. Chi square analysis was used to determine if the difference in bypass frequency was significant as indicated by p-values less than 0.05 (Figure 5F).
Figure 5.

Total scanning distance range of glycosylases on concatemer substrates as a function of observation time. The total distance of DNA scanned is plotted as a function of how long the glycosylase was observed (A) MUTYH WT on undamaged, (B) MUTYH WT on 8-oxoG:A, (C) MUTYH WT on 8-oxoG:C, (D) MUTYH Y150C on undamaged, (E) MUTYH Y150C on 8-oxoG:A. In plots A-E, n represents the total number of tracked glycosylase trajectories. The dotted line is plotted at t = 10s, and gray points are molecules that were bound for less than 10s. A histogram of the time observed is shown above plots A-E, and a histogram of total scanning range for molecules observed for >10s is to the right side. The shaded green region indicates the length of one plasmid concatemer. For molecules that were bound for longer than 10s, the fraction of molecules in each condition that scanned more than 1000 nM is shown in (F). Significance was determined using chi square analysis (*p < 0.05, ****p < 0.0001).
RESULTS
MutY and MUTYH recognize apurinic sites in λ DNA tightropes
E. coli MutY and murine MUTYH show some significant differences in rates of catalysis, binding affinities, and substrate recognition that may indicate differences in interactions with undamaged DNA (25). To compare MutY and MUTYH search behavior on undamaged DNA to that observed for E. coli Fpg, Nei, and Nth glycosylase WT and wedge variants in previous studies, we utilized our SMFM assay with λ DNA tightropes to characterize MutY and MUTYH diffusion (Figure 1B, Supplementary Figure S1) (33,34). Briefly, the position of the Qdot-labeled glycosylase on the tightrope was observed over time, and the resulting displacement trajectory acquired through the SpotTracker plug-in for ImageJ (54). Trajectories were analyzed using mean squared displacement analysis to fit for an apparent one-dimensional diffusion constant across the entire trajectory (Dtraj) (Supplementary Figures S1E and S1F, Supplementary Table S1). As was observed in previous studies of glycosylase diffusion, MutY and MUTYH show complex diffusion behavior on these tightrope substrates with periods of diffusion that range across three orders of magnitude. Therefore, diffusion constants were fitted using a 60-frame sliding window across a trajectory (Supplementary Figure S2). The diffusion constants from all trajectories were then compiled into frequency distributions that describe overall diffusive behavior for each variant under each condition (Supplementary Figures S1A–S1D, Figures 2 and 4A–C).
Figure 2.

Representative sample trajectories and diffusive behavior of individual MUTYH WT and Y150C enzymes on concatemer tightrope substrates. Displacement trajectories are shown in the left column for (A) MUTYH WT on undamaged concatemer, (B) MUTYH WT on 8-oxoG:A containing concatemer, (C), MUTYH Y150C on undamaged concatemer, and (D) MUTYH Y150C on 8-oxoG:A containing concatemer. Dotted lines represent the location of 8-oxoG:A damage sites. The center column shows the distribution of diffusion constants determined using a 60-frame sliding window analysis of the corresponding trajectory in the left column. Kymographs of the corresponding trajectory are shown to the right. The first portion of each kymograph shows the position of the dye markers and damage sites, which are indicated with black or red labels. The green arrow in the kymograph of panel (B) indicates the location of the plotted trajectory. See Supplementary Table S1 for sample statistics.
As has been observed for E. coli Fpg, Nei and Nth, MutY WT and MUTYH WT have a fast mode of diffusive-like motion along undamaged λ DNA with a diffusion constant of ∼0.01 μm2s−1 (Supplementary Figure S1). Our SMFM assay does not have the resolution to directly distinguish rotation from linear scanning along the DNA axis, but by modeling apparent one-dimensional diffusion and correcting for Stokes drag from the Qdot, it was shown that diffusion constants of 0.01–0.05 μm2s−1 are consistent with rotational diffusion along the DNA backbone (33). We find that the profile of the diffusion constant distribution of MutY Y82C and MUTYH Y150C differ only slightly from their WT counterparts on undamaged λ DNA substrates (Supplementary Figure S1A).
Since MutY and MUTYH both show extremely high binding affinity for their AP site product, a chemically inert reduced AP (rAP) site analog was introduced into λ DNA to observe how the enzymes interact with product sites (59,60). These substrates contain randomly distributed rAP damage sites with a mean separation distance of 1600 base pairs (bp) (Supplementary Figure S3). Diffusion constant distributions from time weighted diffusion analysis show that the WT enzymes spend the majority of the time paused with an apparent diffusion constant less than 0.001 μm2s−1 (Supplementary Figures S1C and S1D, Supplementary Table S1). In comparison to the WT enzymes, MutY Y82C and MUTYH Y150C wedge variants spend significantly less time paused in the presence of rAP sites (Supplementary Figures S1C and S1D). However, the wedge variants do show decreased diffusion constants on rAP λ DNA sites versus diffusion along undamaged DNA (Supplementary Figures S1E and S1F).
Single glycosylase diffusion trajectories show pauses in the presence of 8-oxoG damage sites
Since murine MUTYH behaved so similarly to E. coli MutY in our assays and given the much greater structural similarity of the murine MUTYH to the human enzyme, we focused on the behavior of the murine MUTYH on specific 8-oxoG:A damage. To visualize MUTYH behavior at precisely located 8-oxoG:A sites, which cannot be generated using chemical or radiative methods, a plasmid concatemer substrate was constructed (51–53) (Figure 1C and D). These tightropes consist of repeating units of a pUC19 derivative (2727 bp) that each contain a single 8-oxoG:A damage mismatch site. Within a tightrope, all damages exist on one strand of the DNA. During the SMFM experiment, the tightropes used are comprised of a mixture of 72% damage-containing (or undamaged control) plasmids and 18% plasmids that contain a fluorescent Cy5 dye at the same location, with the balance (10%) containing undamaged DNA at this location (see Materials and Methods). Inclusion of fluorescently labeled plasmids allows for determination of the precise location of damage sites using the periodic plasmid repeat of 2727 bp or 905 nm (Figure 1C and Supplementary Figure S6). This approach allowed for the identification of glycosylase diffusive behavior that occurs in regions of the tightrope corresponding to damage (Figure 2).
As was observed on undamaged λ DNA tightropes, individual MUTYH WT and MUTYH Y150C molecules show primarily fast diffusive-like motion along undamaged concatemer substrates (Figure 2A and C, Supplementary Figures S7 and S8). In stark contrast, when MUTYH WT is observed on an 8-oxoG:A concatemer substrate, trajectories show very short periods of fast diffusion followed by long pause events (Figure 2B, Supplementary Figures S7 and S8), the majority of which persist beyond the duration of the recordings (300 s). Spatial alignment of these pause events relative to the location of known dye molecules shows that many of these pauses occur at positions that are indicated as damage sites. Trajectories of MUTYH Y150C on 8-oxoG:A concatemer substrates show few long duration (>10 s) pause events (Figures 2D and 3). However, some of these trajectories do show repeated brief pauses at damage site locations, suggesting the variant can transiently recognize the 8-oxoG:A lesion (Figure 3A and B). To characterize the timescales of variant damage interactions, we compiled 120 damage encounters of MUTYH Y150C on 8-oxoG:A sites and fit the distribution of residence lifetimes at these locations with a double exponential model (Figure 3C). The results of this fit give a short-lived dwell time population with a mean lifetime of 1.6 ± 0.3s (89%) and a longer-lived population with mean lifetime of 10 ± 5s (11%). As controls (see Materials and Methods), we compared these values to residence lifetimes for MUTYH Y150C at specific undamaged locations 300 nm away from the mapped damage site locations (1.1 ± 0.1s, n = 131), and MUTYH WT at specific locations on undamaged concatemers (1.4 ± 0.2s, n = 70) (Supplementary Figure S12). As the short-lived population is common to all these experimental conditions, regardless of the presence or absence of damage, we attribute these short dwell times (1.1–1.6s) to the diffusive behavior of the enzyme on DNA. The long-lived events (∼10s) of MUTYH Y150C at 8-oxoG:A-containing locations we attribute to transient damage recognition by the variant enzyme.
To characterize the behavior of all observed molecules under a given condition, diffusion constant distributions were compiled for molecules that bound to DNA for longer than 60 frames. These distributions show that overall, MUTYH WT and Y150C diffuse rapidly on undamaged DNA indicated by a single population with a diffusion constant centered at 0.01 um2s−1 (Figure 4A). However, MUTYH WT spends the majority of time stopped or slowly diffusing in the presence of 8-oxoG:A sites indicated by a single population of diffusion constants centered around 0.0001 um2s−1 (Figure 4B). Although transient pauses are observed for MUTYH Y150C in the presence of 8-oxoG:A sites (Figure 3B), these pauses represent a minor contribution to the overall diffusive behavior, and the variant shows primarily a fast diffusion similar to that observed on undamaged DNA (Figure 4B). The diffusive behavior represented in these distributions is consistent with the whole trajectory analysis (Figure 4D, Supplementary Table S1).
We also investigated the diffusive behavior of MUTYH WT and Y150C on 8-oxoG:C sites (Figure 4C). In ensemble experiments, all orthologs of MUTYH are catalytically inactive for cleavage of a cytosine (C) opposite 8-oxoG (61,62). However, ensemble experiments showed that murine MUTYH has significant binding affinity for 8-oxoG:C sites with KD values comparable to those on non-cleavable adenine sites (25). In the SMFM displacement trajectories of MUTYH WT on 8-oxoG:C, pauses of significant duration are observed (Supplementary Figures S7 and S8), and some trajectories show sequential pauses at positions separated by one plasmid length. The diffusion constant distribution shows that MUTYH WT remains primarily paused in the presence of 8-oxoG:C sites (Figure 4C), and Dtraj falls between the values measured for undamaged and 8-oxoG:A sites (Figure 4D). Thus, MUTYH WT shows significant recognition of 8-oxoG:C.
MUTYH Y150C releases more quickly from undamaged tightrope DNA than MUTYH WT
Our single molecule studies also helped characterize DNA binding and release behavior for MUTYH enzymes. There has been some question about overall enzymatic stability of MUTYH Y150C (23). To investigate whether the variant enzyme is able to find and bind undamaged DNA as effectively as the WT enzyme, we did a side by side comparison of MUTYH WT and Y150C binding frequency per base of DNA at identical enzyme and undamaged substrate concentrations in a SMFM chamber. Under these conditions, we observe that MUTYH WT binds to DNA at a rate of 0.50 (±0.06) events per μm of DNA over 5 minutes and Y150C enzymes bind to DNA at a rate of 0.46 (±0.07) events per μm over 5 minutes (Figure 4H). These values indicate that the DNA binding efficacy for the MUTYH Y150C variant is not compromised under the SMFM conditions.
Binding affinity of the enzyme for a substrate is dependent on the ratio of the ‘on’ binding rates and ‘off’ release rates. Kinetic assays in bulk solution show increased rates of substrate release by MutY Y82C, but product release kinetics for MUTYH Y150C were difficult to characterize due to enzyme instability over long periods at 37°C (23). With our SMFM experiments carried out over much shorter time periods and at room temperature, and we sought to observe altered lesion binding lifetimes for MUTYH Y150C on the damage site tightrope substrates as compared to MUTYH WT. In the SMFM assay MutY and MUTYH have much longer binding lifetimes than have been observed for E. coli Fpg, Nth, and Nei (31,33,34), and under some conditions MUTYH molecules remain bound to a damaged substrate for longer than the 300 s duration of recording. Therefore we estimated MUTYH binding lifetimes using the Kaplan-Meier approach (57). These survival curves show significantly decreased median binding lifetime for MUTYH Y150C when compared with MUTYH WT on each substrate (Figure 4E and F). On undamaged DNA, MUTYH WT shows a median binding lifetime of 30.9 s, which is 10-fold greater than that of the variant (3.3 s) (Figure 4E). In the presence of 8-oxoG:A, the difference is even greater, with Y150C showing a median binding lifetime of 5.1 s, while WT has a median binding lifetime greater than 300 s (Figure 4F). Overall, we conclude that MUTYH Y150C is fully capable of binding to undamaged DNA as effectively as WT, but the variant has an enhanced detachment rate on all substrates.
MUTYH WT does not bypass 8-oxoG:A sites
MUTYH is selective for removal of adenine from mispaired substrates (25,50,61–66). Efficient adenine base removal is predicated on efficient damage site recognition, which requires not only fast DNA search rates but also rapid and effective binding to the lesion. To determine whether MUTYH commits to long-lived binding when a lesion is first encountered, or if MUTYH is able to bypass lesions, we examined the total scanning range (see Materials and Methods) as a function of time observed on undamaged, 8-oxoG:A, or 8-oxoG:C containing concatemers. According to a simple model of one-dimensional (1D) diffusion, total scanning range should increase with the square root of observation time (67), which was observed for both MUTYH WT and Y150C on undamaged DNA (Figure 5A and C) where 89% and 96% of molecules bound for >10s scan more than one plasmid, respectively. However, clear evidence for significantly more restricted diffusion is seen when MUTYH WT is presented with 8-oxoG:A or 8-oxoG:C containing concatemers. In the presence of 8-oxoG:A, only 29% of MTUYH WT molecules (Figure 5B) scan more than one plasmid of DNA, consistent with the proportion of constituent plasmids that do not contain damage sites (28%, see Materials and Methods), indicating that the WT enzyme does not bypass 8-oxoG:A sites. In the presence of 8-oxoG:C sites, 44% of MUTYH WT molecules scan more than one plasmid, intermediate between the 8-oxoG:A and undamaged conditions (Figure 5F) and indicating that lesion bypass must be possible for 8-oxoG:C substrates. We note that some MUTYH WT trajectories on 8-oxoG:C containing concatemers show prolonged pauses, followed by diffusion and subsequent pausing at other locations one plasmid length away (∼900 nm, Supplementary Figure S8). This behavior was not observed on 8-oxoG:A containing concatemers. These results indicate that WT enzymes bypass or release 8-oxoG:C sites at a greater rate that 8-oxoG:A sites. In contrast, 85% of MUTYH Y150C molecules diffuse farther than one plasmid length in the presence of 8-oxoG:A sites, which does not differ from the undamaged condition and demonstrates minimal lesion recognition and binding (Figure 5F).
Our previous studies of the role of the wedge residue in glycosylase scanning examined diffusive behavior of alanine substituted wedge variants. To investigate whether the cysteine wedge residue that is present in the variant enzymes contributes any residual damage site recognition or binding behavior, we also utilized our SMFM assay to investigate interaction of MUTYH Y150A with concatemer tightropes that contain 8-oxoG:A and 8-oxoG:C sites (Supplementary Figure S10). Overall, the diffusion constants, diffusive behavior, and binding lifetimes for MUTYH Y150A were consistent with those of MUTYH Y150C. Thus, these data suggest that cysteine provides no search or lesion stabilization functionality to the MUTYH Y150C variant beyond what would be contributed by alanine.
DISCUSSION
MUTYH WT and Y150C interactions with undamaged DNA
The first step in the glycosylase search for damage sites is a non-specific binding and sliding interaction with DNA (Figure 1F). This fast sliding facilitates diffusion to distant regions in the DNA to help increase lesion recognition efficiency. Although this step is essential for the initiation of proper repair of damage sites, bulk gel assays are often unable to measure binding constants for glycosylases on undamaged substrates due to the dynamic nature of enzyme-DNA interactions. SMFM studies represent one of the first methods to allow for characterization of how glycosylases interact with undamaged DNA. We found that MutY and MUTYH WT enzymes primarily exhibit a fast diffusion mode along undamaged DNA (Figure 4A, Supplementary Figures S1A and S1B), which differs from the combination of fast and slow scanning modes observed for E. coli Fpg, Nei, and Nth WT enzymes on undamaged λ DNA (34). The lack of a slow diffusion mode is also reflected in significantly higher overall diffusion constants (Dtraj) for MutY and MUTYH as compared to the previous measurements (Supplementary Table S1) (34). This fast mode of diffusion with diffusion constants of approximately 0.01–0.05 μm2s−1 appears to be a common feature of glycosylase interactions with DNA and has been attributed to fast sliding along the DNA backbone (33). More recent studies have suggested that single molecule methods lack the time resolution necessary to capture microscopic bind and release events or ‘hops’ which may be present in apparent one-dimensional diffusion (68), and the studies herein do not rule out the presence of short hopping events. The fact that the murine MUTYH moves rapidly along undamaged DNA in a way similar to E. coli MutY is interesting based on the finding that MUTYH may associate with PCNA and the replication fork (16,69–72). Our results would suggest that MUTYH is capable of engaging in a fully mobile search for damage sites when other binding partners are absent.
In the SMFM assay, MUTYH WT enzymes have a significantly longer binding lifetime to undamaged DNA as compared to E. coli Fpg, Nei, and Nth (33,34). Although the catalytic domain of MutY and MUTYH are in the helix-hairpin-helix superfamily of glycosylases that includes Nth, MutY contains an extra C-terminal region that primarily interacts with the 8-oxoG strand (41,73–76). This C-terminal domain is even longer in mammalian MUTYH enzymes. Crystal structures show that together, the N- and C-terminal domains of bacterial MutY almost fully encircle the DNA strand (41). In contrast, a crystal structure of a Schiff base complex between E. coli Nth and DNA shows that Nth has a smaller contact circumference around the DNA (77). It is possible that the difference in binding affinity between MUTYH and Nth derives from an increased MUTYH contact footprint on the DNA strand, which may result in stabilization of the non-specific MUTYH-DNA interrogation complex. A longer binding lifetime for MUTYH would help to ensure a more thorough search for 8-oxoG:A, which would be important because MUTYH lacks a backup repair system to help with lesion recognition and removal.
Our SMFM studies show that MUTYH Y150C has an impaired binding affinity for undamaged DNA as compared to MUTYH WT (Figure 4). This result differs from previous SMFM studies that showed Fpg, Nei, and Nth wedge variant enzymes did not have decreased binding lifetimes on undamaged DNA (34). In our studies, the MUTYH Y150C variant binds to DNA effectively but has an increased release rate from undamaged DNA (Figure 4E–G). This observation suggests that the tyrosine somehow stabilizes enzyme interactions with tightrope DNA or overall structural stability of the enzyme. The decreased binding lifetime of MUTYH Y150C must necessarily have negative consequences for the efficiency of the variant's search for damage site by impeding both the variant's ability to find the site and the ability to properly handoff to downstream BER enzymes. The variant enzyme may not remain bound to the DNA for long enough to find the damage, which would force the enzyme to switch from a facilitated diffusion search method to one closer to random three-dimensional search (28,78,79).
MUTYH WT lesion recognition and binding
As a glycosylase encounters a lesion, the enzyme must transition from a non-specific interaction with DNA to a lesion recognition complex (Figure 1F). This process is dynamic, which makes it difficult to study using heterogeneous bulk assays and static crystallographic snapshots. Our real-time observation of a glycosylase on a substrate with specific damage sites at known positions along the DNA also gives new insight into how the enzyme interacts with these sites in the context of a large excess of undamaged DNA competitor. MUTYH does not bypass 8-oxoG:A damage sites (Figure 5B), and the high efficiency of MUTYH damage site recognition indicates the enzyme has directional and configurational flexibility in orientation of the enzyme with respect to the DNA strand during the damage search. Various structural studies of MUTYH bound to a lesion site show that the C-terminal domain of the enzyme is largely responsible for contacting the damage-containing DNA strand, while the N-terminal catalytic domain makes contacts with the adenine strand (11,75,76,80). Although our tightropes assemble with no directional preference relative to bead attachment points, our concatemers are designed such that all 8-oxoG sites are contained within one single strand of the tightrope. If we hypothesize that the enzyme randomly loads onto the DNA and moves like a locomotive during the scan with the N-terminal domain of MUTYH remaining in contact with only one strand while the C-terminal domain tracks solely along the other strand, only half of the enzymes would be capable of recognizing 8-oxoG:A sites. As is evident in Figure 4B, the majority of enzymes are able to detect the damage site within 2727 basepairs of scanning distance. Therefore, our data suggest the enzyme searches along DNA with low affinity contacts that allow for frequent reorientation to allow for detection and binding to 8-oxoG:A sites. Our findings are consistent with a recent crystal structure of the N-terminal domain of B. stearothermophilus MutY crosslinked to undamaged DNA which showed fewer interactions between the MutY N-terminal amino acids and the DNA backbone, suggesting a relatively ‘loose’ binding interaction as compared to the complex of MutY on a lesion site (75).
It has been suggested that interactions with PCNA might direct MUTYH to orient toward the newly replicated daughter strand to aid in efficient repair of the post replicative adenine mismatch (16,47). Our studies show that MUTYH has the ability of to find any 8-oxoG:A lesion at a first pass and does not require interactions with PCNA or other proteins in order to find the damage mismatch site. Accordingly, any strand specific activity of MUTYH would necessarily be dictated by interactions with other proteins and not due to an intrinsic strand specificity of the enzyme. Thus, in the case where 8 oxo-dGTP has been inserted by polymerases into the parental strand, MUTYH would require interactions with PCNA or other replication factors to orient toward the daughter strand and prevent repair that could result in an A to C transversion (16–19).
MUTYH is a special glycosylase in that base excision of an undamaged adenine base is accompanied by recognition of an 8-oxoG damaged base, posing a unique challenge for damage site search and recognition. Previous SMFM studies of Fpg, Nei, and Nth enzymes revealed that these glycosylases rely upon the wedge for a slow diffusive search mode along undamaged DNA (33,34) and specific damage recognition and binding (34,81–83). Contrary to these earlier studies, we do not observe a slow mode of diffusion for the MutY and MUTYH wedge variants along undamaged λ DNA or undamaged concatemer DNA (Figure 3, Supplementary Figure S1). This result begs the question of how MUTYH searches for the 8-oxoG:A damage site? When paired with adenine, 8-oxoG exposes a hydrogen bonding network, including the 2-amino group, to the major groove side of the duplex. Structural studies show that B. stearothermophilus MutY makes contact with this face of the damaged base through a histidine residue that lies on a short C-terminal loop (41), and it has been suggested that this loop might be in proximity of the major groove in an undamaged DNA substrate (75). A study of E. coli MutY function on various substrate analogs found that the 2-amino group of 8-oxoG is critical for early recognition of an 8-oxoG:A damage site (84). Therefore, it is likely that MUTYH rapidly slides along DNA until the C-terminal domain encounters the 2-amino group of 8-oxoG, whereupon the enzyme stops, repositions, and further interrogates the damage site using the wedge residue. According to this model, MUTYH wedge insertion primarily facilitates proper adenine eversion into the enzymatic pocket, which serves as the second step in lesion discrimination to prevent accidental cleavage of cytosine. MUTYH is also capable of removing adenine from G:A pairs at a much slower rate than that observed for 8-oxoG:A pairs (25). It is possible and we cannot rule out that MUTYH may also detect adenine mismatches using other mechanisms.
MUTYH WT binding to 8-oxoG:C damage
Displacement trajectories of MUTYH WT in the presence of 8-oxoG:C reveal pausing (Supplementary Figures S7 and S8) and decreased diffusion constants indicative of lesion binding (Figure 4D, Supplementary Table S1). However, these WT pause events are shorter-lived than those in the presence of 8-oxoG:A sites. MUTYH Y150C does not pause at all on 8-oxoG:C sites. These SMFM results are consistent with electrophoretic mobility shift assays (EMSA) assays showing some interaction between mammalian MUTYH and 8-oxoG:C (24,25). The crystal structure of B. stearothermophilus (Bs) MutY crosslinked to 8-oxoG:C showed the enzyme is in a ‘nearly identical’ conformation to that observed for Bs MutY bound to an 8-oxoG paired with a non-cleavable adenine analog (76). Taken together the SMFM, EMSA, and structural studies of MUTYH on 8-oxoG:C lesions reinforce the suggestion that initial 8-oxoG:A damage site recognition primarily relies upon the 8-oxoG residue. The extent to which MUTYH WT recognizes and binds 8-oxoG:C lesions suggests that the enzyme will have significant interactions with any 8-oxoG lesion site and may have an additional role in repair of 8-oxoG damage. MUTYH does not interfere with the activity of hOGG1 on 8-oxoG:C sites, indicating that MUTYH binding to 8-oxoG:C does not prevent repair (24). However, if MUTYH interacts with proteins at the replication fork, perhaps a transient complex between MUTYH and 8-oxoG:C is enough to prevent replication at the site before hOGG1 arrives to excise the damaged base.
MUTYH Y150C search and binding deficiencies
Characterizing interaction deficiencies between glycosylase variants and damage sites using traditional gel based assays can be complicated by decreased catalytic efficiency or enzyme stability. The SMFM studies herein reveal that MUTYH Y150C retains some ability to find 8-oxoG:A sites, but is much less efficient at lesion binding than the WT enzyme (Figure 3). Although MUTYH Y150C appears to frequently bypass lesion sites without an observable pause, the high redundancy of search would predict that the enzyme would have multiple opportunities to find the lesion. Accordingly, the trajectories show the variant ‘revisiting’ damage sites with repeated pause events (Figure 3). However, in the context of the high efficiency of lesion detection by MUTYH WT and the possibility that MUTYH engages in replication coupled repair, lesion bypass would be more detrimental for MUTYH than other housekeeping glycosylases like hOGG1 or hNTH. Even when the lesion is discovered by MUTYH Y150C, the short residence time on damage sites suggests that the variant would not have time to excise adenine or handoff to APE. Using previously determined catalytic rates, we can estimate whether MUTYH Y150C could undergo catalysis on our tightrope substrates. Gel-based studies show that MUTYH Y150C removes adenine from an oligonucleotide substrate with an excision rate of 0.26 min−1 (23). This catalytic rate is relatively slow compared with the short-lived pauses we observe (10 ± 5s, Figure 3C), and therefore we conclude that the majority of the transient pauses in MUTYH Y150C displacement trajectories do not proceed to the catalytic step. By investigating the search behavior of MUTYH Y150C in the context of large amounts of competing undamaged DNA, our studies indicate that proper 8-oxoG:A lesion binding and repair by the variant would be greatly decreased in a genomic context, leading to increased genome instability.
General model for MUTYH search
These results suggest that MUTYH WT diffuses rapidly along the DNA backbone with a relatively loose interaction to allow for repositioning at the first encounter with a damage site (Figure 1F). MUTYH is not relying upon the tyrosine wedge residue for interrogation of DNA for damage, but likely utilizes the additional mammalian C-terminal region to detect the 8-oxoG damaged base regardless of whether it is paired with adenine or cytosine. Once an 8-oxoG site is encountered, intercalation of the wedge residue is critical for forming a stable complex between MUTYH and the damage site. If MUTYH WT interacts with an 8-oxoG:A site, the adenine is able to enter the enzymatic pocket to undergo cleavage, at which time the enzyme forms a high affinity product-bound state. Unlike the WT enzyme, MUTYH Y150C has difficulty remaining bound to undamaged DNA during the damage search. Once a damage site is encountered, the variant inefficiently recognizes the site and likely releases the site before catalysis can occur.
Conclusions
Our SMFM studies demonstrate that individual MUTYH WT enzymes are capable of a highly efficient bidirectional search for 8-oxoG:A sites, while the variant has compromised binding affinity for undamaged DNA that must negatively impact damage site search. Although MUTYH Y150C retains some ability to recognize the 8-oxoG:A site, the variant is unable to form a stable complex with the lesion site. Therefore, the MUTYH Y150C wedge variant has a combination of perturbed interactions with DNA that lead to a persistence of damage mismatch sites and accumulation of mutation. As more genetic information about germline and somatic mutations becomes available, elucidating the molecular basis for MUTYH functional impairment will help in deciphering the physiological consequences of these mutations. One important factor in BER is the transient non-covalent interactions between glycosylases, DNA, and other repair and regulatory factors, all of which can be investigated using single molecule methods similar to the ones reported here.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Guy Kennedy from the Instrumentation and Model Facility at UVM for his microscopy expertise, and the members of Prof. Sheila David's laboratory at UC-Davis for helpful discussions. We also thank Dr. Andrew Dunn for helpful discussions.
Supplementary DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health [CA P01098893 to A.J.L., S.S.W.]; UVM Summer Research Internship [to T.S.H.]; UVM Department of Microbiology and Molecular Genetics Nicole J. Ferland Summer Research Award [to T.S.H.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Donigan K.A., Hile S.E., Eckert K.A., Sweasy J.B.. The human gastric cancer-associated DNA polymerase beta variant D160N is a mutator that induces cellular transformation. DNA Repair (Amst.). 2012; 11:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nemec A.A., Donigan K.A., Murphy D.L., Jaeger J., Sweasy J.B.. Colon cancer-associated DNA polymerase beta variant induces genomic instability and cellular transformation. J. Biol. Chem. 2012; 287:23840–23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Eckenroth B.E., Towle-Weicksel J.B., Sweasy J.B., Doublie S.. The E295K cancer variant of human polymerase beta favors the mismatch conformational pathway during nucleotide selection. J. Biol. Chem. 2013; 288:34850–34860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Galick H.A., Kathe S., Liu M., Robey-Bond S., Kidane D., Wallace S.S., Sweasy J.B.. Germ-line variant of human NTH1 DNA glycosylase induces genomic instability and cellular transformation. Proc. Natl. Acad. Sci. U.S.A. 2013; 110:14314–14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Donigan K.A., Sun K.W., Nemec A.A., Murphy D.L., Cong X., Northrup V., Zelterman D., Sweasy J.B.. Human POLB gene is mutated in high percentage of colorectal tumors. J. Biol. Chem. 2012; 287:23830–23839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Prakash A., Carroll B.L., Sweasy J.B., Wallace S.S., Doublie S.. Genome and cancer single nucleotide polymorphisms of the human NEIL1 DNA glycosylase: activity, structure, and the effect of editing. DNA Repair (Amst.). 2014; 14:17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamtich J., Nemec A.A., Keh A., Sweasy J.B.. A germline polymorphism of DNA polymerase beta induces genomic instability and cellular transformation. PLoS Genet. 2012; 8:e1003052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nemec A.A., Wallace S.S., Sweasy J.B.. Variant base excision repair proteins: contributors to genomic instability. Semin. Cancer Biol. 2010; 20:320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wallace S.S., Murphy D.L., Sweasy J.B.. Base excision repair and cancer. Cancer Lett. 2012; 327:73–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Duclos S., Doublié S., Wallace S.S.. Greim H, Albertini RJ. The Cellular Response to the Genotoxic Insult: The Question of Threshold for Genotoxic Carcinogens. 2012; Cambridge: The Royal Society of Chemistry; 115–159. [Google Scholar]
- 11. Fromme J.C., Verdine G.L.. Base excision repair. Adv. Prot. Chem. 2004; 69:1–41. [DOI] [PubMed] [Google Scholar]
- 12. Wallace S.S. Biological consequences of free radical-damaged DNA bases. Free Radical Biol. Med. 2002; 33:1–14. [DOI] [PubMed] [Google Scholar]
- 13. Mitra S., Boldogh I., Izumi T., Hazra T.K.. Complexities of the DNA base excision repair pathway for repair of oxidative DNA damage. Environ. Mol. Mutag. 2001; 38:180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hazra T.K., Hill J.W., Izumi T., Mitra S.. Multiple DNA glycosylases for repair of 8-oxoguanine and their potential in vivo functions. Prog. Nucleic Acid Res. Mol. Biol. 2001; 68:193–205. [DOI] [PubMed] [Google Scholar]
- 15. Amoroso A., Crespan E., Wimmer U., Hubscher U., Maga G.. DNA polymerases and oxidative damage: friends or foes. Curr. Mol. Pharmacol. 2008; 1:162–170. [DOI] [PubMed] [Google Scholar]
- 16. Boldogh I., Milligan D., Lee M.S., Bassett H., Lloyd R.S., McCullough A.K.. hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res. 2001; 29:2802–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Colussi C., Parlanti E., Degan P., Aquilina G., Barnes D., Macpherson P., Karran P., Crescenzi M., Dogliotti E., Bignami M.. The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Curr. Biol. 2002; 12:912–918. [DOI] [PubMed] [Google Scholar]
- 18. Larson E.D., Iams K., Drummond J.T.. Strand-specific processing of 8-oxoguanine by the human mismatch repair pathway: inefficient removal of 8-oxoguanine paired with adenine or cytosine. DNA Repair (Amst.). 2003; 2:1199–1210. [DOI] [PubMed] [Google Scholar]
- 19. Minnick D.T., Pavlov Y.I., Kunkel T.A.. The fidelity of the human leading and lagging strand DNA replication apparatus with 8-oxodeoxyguanosine triphosphate. Nucleic Acids Res. 1994; 22:5658–5664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. David S.S., O'Shea V.L., Kundu S.. Base-excision repair of oxidative DNA damage. Nature. 2007; 447:941–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Markkanen E., Dorn J., Hubscher U.. MUTYH DNA glycosylase: the rationale for removing undamaged bases from the DNA. Front. Genet. 2013; 4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang H., Clendenin W.M., Wong D., Demple B., Slupska M.M., Chiang J.H., Miller J.H.. Enhanced activity of adenine-DNA glycosylase (Myh) by apurinic/apyrimidinic endonuclease (Ape1) in mammalian base excision repair of an A/GO mismatch. Nucleic Acids Res. 2001; 29:743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pope M.A., Chmiel N.H., David S.S.. Insight into the functional consequences of hMYH variants associated with colorectal cancer: distinct differences in the adenine glycosylase activity and the response to AP endonucleases of Y150C and G365D murine MYH. DNA Repair (Amst.). 2005; 4:315–325. [DOI] [PubMed] [Google Scholar]
- 24. Tominaga Y., Ushijima Y., Tsuchimoto D., Mishima M., Shirakawa M., Hirano S., Sakumi K., Nakabeppu Y.. MUTYH prevents OGG1 or APEX1 from inappropriately processing its substrate or reaction product with its C-terminal domain. Nucleic Acids Res. 2004; 32:3198–3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pope M.A., David S.S.. DNA damage recognition and repair by the murine MutY homologue. DNA Repair (Amst.). 2005; 4:91–102. [DOI] [PubMed] [Google Scholar]
- 26. Slupska M.M., Baikalov C., Luther W.M., Chiang J.H., Wei Y.F., Miller J.H.. Cloning and sequencing a human homolog (hMYH) of the Escherichia coli mutY gene whose function is required for the repair of oxidative DNA damage. J. Bacteriol. 1996; 178:3885–3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zharkov D.O., Grollman A.P.. The DNA trackwalkers: principles of lesion search and recognition by DNA glycosylases. Mutat. Res. 2005; 577:24–54. [DOI] [PubMed] [Google Scholar]
- 28. Lee A.J., Warshaw D.M., Wallace S.S.. Insights into the glycosylase search for damage from single-molecule fluorescence microscopy. DNA Repair (Amst.). 2014; 20:23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Verdine G.L., Bruner S.D.. How do DNA repair proteins locate damaged bases in the genome. Chem. Biol. 1997; 4:329–334. [DOI] [PubMed] [Google Scholar]
- 30. Friedman J.I., Stivers J.T.. Detection of damaged DNA bases by DNA glycosylase enzymes. Biochemistry. 2010; 49:4957–4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Blainey P.C., van Oijen A.M., Banerjee A., Verdine G.L., Xie X.S.. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:5752–5757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Blainey P.C., Luo G., Kou S.C., Mangel W.F., Verdine G.L., Bagchi B., Xie X.S.. Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 2009; 16:1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dunn A.R., Kad N.M., Nelson S.R., Warshaw D.M., Wallace S.S.. Single Qdot-labeled glycosylase molecules use a wedge amino acid to probe for lesions while scanning along DNA. Nucleic Acids Res. 2011; 39:7487–7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nelson S.R., Dunn A.R., Kathe S.D., Warshaw D.M., Wallace S.S.. Two glycosylase families diffusively scan DNA using a wedge residue to probe for and identify oxidatively damaged bases. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:E2091–E2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cheadle J.P., Sampson J.R.. MUTYH-associated polyposis–from defect in base excision repair to clinical genetic testing. DNA Repair (Amst.). 2007; 6:274–279. [DOI] [PubMed] [Google Scholar]
- 36. Jasperson K.W., Tuohy T.M., Neklason D.W., Burt R.W.. Hereditary and familial colon cancer. Gastroenterology. 2010; 138:2044–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Al-Tassan N., Chmiel N.H., Maynard J., Fleming N., Livingston A.L., Williams G.T., Hodges A.K., Davies D.R., David S.S., Sampson J.R. et al.. Inherited variants of MYH associated with somatic G: C → T: A mutations in colorectal tumors. Nat. Genet. 2002; 30:227–232. [DOI] [PubMed] [Google Scholar]
- 38. Sieber O.M., Lipton L., Crabtree M., Heinimann K., Fidalgo P., Phillips R.K., Bisgaard M.L., Orntoft T.F., Aaltonen L.A., Hodgson S.V. et al.. Multiple colorectal adenomas, classic adenomatous polyposis, and germ-line mutations in MYH. New Engl. J. Med. 2003; 348:791–799. [DOI] [PubMed] [Google Scholar]
- 39. Sampson J.R., Jones S., Dolwani S., Cheadle J.P.. MutYH (MYH) and colorectal cancer. Biochem. Soc. Trans. 2005; 33:679–683. [DOI] [PubMed] [Google Scholar]
- 40. Poulsen M.L., Bisgaard M.L.. MUTYH Associated Polyposis (MAP). Curr. Genomics. 2008; 9:420–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fromme J.C., Banerjee A., Huang S.J., Verdine G.L.. Structural basis for removal of adenine mispaired with 8-oxoguanine by MutY adenine DNA glycosylase. Nature. 2004; 427:652–656. [DOI] [PubMed] [Google Scholar]
- 42. Ali M., Kim H., Cleary S., Cupples C., Gallinger S., Bristow R.. Characterization of mutant MUTYH proteins associated with familial colorectal cancer. Gastroenterology. 2008; 135:499–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Chmiel N.H., Livingston A.L., David S.S.. Insight into the functional consequences of inherited variants of the hMYH adenine glycosylase associated with colorectal cancer: Complementation assays with hMYH variants, and pre-steady-state kinetics of the corresponding mutated E-coli enzymes. J. Mol. Biol. 2003; 327:431–443. [DOI] [PubMed] [Google Scholar]
- 44. D’Agostino V.G., Minoprio A., Torreri P., Marinoni I., Bossa C., Petrucci T.C., Albertini A.M., Ranzani G.N., Bignami M., Mazzei F.. Functional analysis of MUTYH mutated proteins associated with familial adenomatous polyposis. DNA Repair (Amst.). 2010; 9:700–707. [DOI] [PubMed] [Google Scholar]
- 45. Kundu S., Brinkmeyer M.K., Livingston A.L., David S.S.. Adenine removal activity and bacterial complementation with the human MutY homologue (MUTYH) and Y165C, G382D, P391L and Q324R variants associated with colorectal cancer. DNA Repair (Amst.). 2009; 8:1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Livingston A.L., Kundu S., Pozzi M.H., Anderson D.W., David S.S.. Insight into the roles of tyrosine 82 and glycine 253 in the Escherichia coli adenine glycosylase MutY. Biochemistry. 2005; 44:14179–14190. [DOI] [PubMed] [Google Scholar]
- 47. Parker A.R., Sieber O.M., Shi C., Hua L., Takao M., Tomlinson I.P., Eshleman J.R.. Cells with pathogenic biallelic mutations in the human MUTYH gene are defective in DNA damage binding and repair. Carcinogenesis. 2005; 26:2010–2018. [DOI] [PubMed] [Google Scholar]
- 48. Molatore S., Russo M.T., D’Agostino V.G., Barone F., Matsumoto Y., Albertini A.M., Minoprio A., Degan P., Mazzei F., Bignami M. et al.. MUTYH mutations associated with familial adenomatous polyposis: functional characterization by a mammalian cell-based assay. Hum. Mutat. 2010; 31:159–166. [DOI] [PubMed] [Google Scholar]
- 49. Wooden S.H., Bassett H.M., Wood T.G., McCullough A.K.. Identification of critical residues required for the mutation avoidance function of human MutY (hMYH) and implications in colorectal cancer. Cancer Lett. 2004; 205:89–95. [DOI] [PubMed] [Google Scholar]
- 50. Porello S.L., Leyes A.E., David S.S.. Single-turnover and pre-steady-state kinetics of the reaction of the adenine glycosylase MutY with mismatch-containing DNA substrates. Biochemistry. 1998; 37:14756–14764. [DOI] [PubMed] [Google Scholar]
- 51. Ghodke H., Wang H., Hsieh C.L., Woldemeskel S., Watkins S.C., Rapic-Otrin V., Van Houten B.. Single-molecule analysis reveals human UV-damaged DNA-binding protein (UV-DDB) dimerizes on DNA via multiple kinetic intermediates. Proc. Natl. Acad. Sci. USA. 2014; 111:E1862–E1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Luzzietti N., Brutzer H., Klaue D., Schwarz F.W., Staroske W., Clausing S., Seidel R.. Efficient preparation of internally modified single-molecule constructs using nicking enzymes. Nucleic Acids Res. 2011; 39:e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Luhnsdorf B., Kitsera N., Warken D., Lingg T., Epe B., Khobta A.. Generation of reporter plasmids containing defined base modifications in the DNA strand of choice. Anal. Biochem. 2012; 425:47–53. [DOI] [PubMed] [Google Scholar]
- 54. Sage D., Neumann F.R., Hediger F., Gasser S.M., Unser M.. Automatic tracking of individual fluorescence particles: Application to the study of chromosome dynamics. IEEE Trans. Image Process. 2005; 14:1372–1383. [DOI] [PubMed] [Google Scholar]
- 55. Ovesny M., Krizek P., Borkovec J., Svindrych Z.K., Hagen G.M.. ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics. 2014; 30:2389–2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Venables W.N., Ripley B.D.. Modern Applied Statistics with S. 2002; NY: Springer. [Google Scholar]
- 57. Kaplan E.L., Meier P.. Nonparametric estimation from incomplete observations. J. Am. Stat. Assoc. 1958; 53:457–481. [Google Scholar]
- 58. Team R.C. R: A language and environment for statistical computing. 2013; Vienna: R Foundation for Satistical Computing. [Google Scholar]
- 59. Jorgensen T.J., Kow Y.W., Wallace S.S., Henner W.D.. Mechanism of action of Micrococcus-Luteus Gamma-Endonuclease. Biochemistry. 1987; 26:6436–6443. [DOI] [PubMed] [Google Scholar]
- 60. Kow Y.W. Mechanism of action of Escherichia coli exonuclease III. Biochemistry. 1989; 28:3280–3287. [DOI] [PubMed] [Google Scholar]
- 61. Parker A., Gu Y., Lu A.L.. Purification and characterization of a mammalian homolog of Escherichia coli MutY mismatch repair protein from calf liver mitochondria. Nucleic Acids Res. 2000; 28:3206–3215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lu A.L., Fawcett W.P.. Characterization of the recombinant MutY homolog, an adenine DNA glycosylase, from yeast Schizosaccharomyces pombe. J. Biol. Chem. 1998; 273:25098–25105. [DOI] [PubMed] [Google Scholar]
- 63. Au K.G., Clark S., Miller J.H., Modrich P.. Escherichia-Coli muty gene encodes an adenine glycosylase active on G-a mispairs. Proc. Natl. Acad. Sci. U.S.A. 1989; 86:8877–8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Tsaiwu J.J., Liu H.F., Lu A.L.. Escherichia-Coli muty protein has both N-Glycosylase and apurinic apyrimidinic endonuclease activities on a-Circle-C and a-Circle-G mispairs. Proc. Natl. Acad. Sci. U.S.A. 1992; 89:8779–8783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lu A.L., Tsaiwu J.J., Cillo J.. DNA Determinants and substrate specificities of Escherichia-Coli muty. J. Biol. Chem. 1995; 270:23582–23588. [DOI] [PubMed] [Google Scholar]
- 66. Porello S.L., Williams S.D., Kuhn H., Michaels M.L., David S.S.. Specific recognition of substrate analogs by the DNA mismatch repair enzyme MutY. J. Am. Chem. Soc. 1996; 118:10684–10692. [Google Scholar]
- 67. Berg H.C. Random Walks in Biology. 1983; Princeton: Princeton University Press. [Google Scholar]
- 68. Rowland M.M., Schonhoft J.D., McKibbin P.L., David S.S., Stivers J.T.. Microscopic mechanism of DNA damage searching by hOGG1. Nucleic Acids Res. 2014; 42:9295–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Parker A., Gu Y., Mahoney W., Lee S.H., Singh K.K., Lu A.L.. Human homolog of the MutY repair protein (hMYH) physically interacts with proteins involved in long patch DNA base excision repair. J. Biol. Chem. 2001; 276:5547–5555. [DOI] [PubMed] [Google Scholar]
- 70. Matsumoto Y. Molecular mechanism of PCNA-dependent base excision repair. Prog. Nucleic Acid Res. Mol. Biol. 2001; 68:129–138. [DOI] [PubMed] [Google Scholar]
- 71. Hayashi H., Tominaga Y., Hirano S., McKenna A.E., Nakabeppu Y., Matsumoto Y.. Replication-associated repair of adenine:8-oxoguanine mispairs by MYH. Curr. Biol. 2002; 12:335–339. [DOI] [PubMed] [Google Scholar]
- 72. Brinkmeyer M.K., David S.S.. Distinct functional consequences of MUTYH variants associated with colorectal cancer: Damaged DNA affinity, glycosylase activity and interaction with PCNA and Hus1. DNA Repair (Amst.). 2015; 34:39–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Gogos A., Cillo J., Clarke N.D., Lu A.L.. Specific recognition of A/G and A/7,8-dihydro-8-oxoguanine (8-oxoG) mismatches by Escherichia coli MutY: removal of the C-terminal domain preferentially affects A/8-oxoG recognition. Biochemistry. 1996; 35:16665–16671. [DOI] [PubMed] [Google Scholar]
- 74. Chmiel N.H., Golinelli M.P., Francis A.W., David S.S.. Efficient recognition of substrates and substrate analogs by the adenine glycosylase MutY requires the C-terminal domain. Nucleic Acids Res. 2001; 29:553–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wang L., Chakravarthy S., Verdine G.L.. Structural basis for the Lesion-scanning mechanism of the MutY DNA glycosylase. J. Biol. Chem. 2017; 292:5007–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Wang L., Lee S.J., Verdine G.L.. Structural basis for avoidance of promutagenic DNA repair by MutY adenine DNA glycosylase. J. Biol. Chem. 2015; 290:17096–17105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fromme J.C., Verdine G.L.. Structure of a trapped endonuclease III-DNA covalent intermediate. EMBO J. 2003; 22:3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Halford S.E., Marko J.F.. How do site-specific DNA-binding proteins find their targets. Nucleic Acids Res. 2004; 32:3040–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Berg O.G., Winter R.B., von Hippel P.H.. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry. 1981; 20:6929–6948. [DOI] [PubMed] [Google Scholar]
- 80. Lee S., Verdine G.L.. Atomic substitution reveals the structural basis for substrate adenine recognition and removal by adenine DNA glycosylase. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:18497–18502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Banerjee A., Santos W.L., Verdine G.L.. Structure of a DNA glycosylase searching for lesions. Science. 2006; 311:1153–1157. [DOI] [PubMed] [Google Scholar]
- 82. Qi Y., Nam K., Spong M.C., Banerjee A., Sung R.J., Zhang M., Karplus M., Verdine G.L.. Strandwise translocation of a DNA glycosylase on undamaged DNA. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:1086–1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Koval V.V., Kuznetsov N.A., Ishchenko A.A., Saparbaev M.K., Fedorova O.S.. Real-time studies of conformational dynamics of the repair enzyme E. coli formamidopyrimidine-DNA glycosylase and its DNA complexes during catalytic cycle. Mutat. Res. 2010; 685:3–10. [DOI] [PubMed] [Google Scholar]
- 84. Manlove A.H., McKibbin P.L., Doyle E.L., Majumdar C., Hamm M.L., David S.S.. Structure-Activity relationships reveal key features of 8-Oxoguanine: A mismatch detection by the MutY glycosylase. ACS Chem. Biol. 2017; 12:2335–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.