

Abstract
Pigmentary glaucoma (PG) is a common glaucoma subtype that results from release of pigment from the iris, called pigment dispersion syndrome (PDS), and its deposition throughout the anterior chamber of the eye. Although PG has a substantial heritable component, no causative genes have yet been identified. We used whole exome sequencing of two independent pedigrees to identify two premelanosome protein (PMEL) variants associated with heritable PDS/PG. PMEL encodes a key component of the melanosome, the organelle essential for melanin synthesis, storage and transport. Targeted screening of PMEL in three independent cohorts (n = 394) identified seven additional PDS/PG-associated non-synonymous variants. Five of the nine variants exhibited defective processing of the PMEL protein. In addition, analysis of PDS/PG-associated PMEL variants expressed in HeLa cells revealed structural changes to pseudomelanosomes indicating altered amyloid fibril formation in five of the nine variants. Introduction of 11-base pair deletions to the homologous pmela in zebrafish by the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 method caused profound pigmentation defects and enlarged anterior segments in the eye, further supporting PMEL’s role in ocular pigmentation and function. Taken together, these data support a model in which missense PMEL variants represent dominant negative mutations that impair the ability of PMEL to form functional amyloid fibrils. While PMEL mutations have previously been shown to cause pigmentation and ocular defects in animals, this research is the first report of mutations in PMEL causing human disease.
Introduction
Glaucoma is a leading cause of blindness worldwide with an estimated 65 million people affected (1). Pigmentary glaucoma (PG) is a secondary glaucoma subtype estimated to account for 1–1.5% of all glaucoma cases and represents a major cause of blindness in young adults (2). Patients may initially present with pigment dispersion syndrome (PDS) in which pigmented material shed from the posterior surface of the iris is deposited into the anterior chamber. Patients with PDS exhibit corneal endothelial pigment deposition (Krukenberg spindle), radial mid-peripheral iris transillumination defects and dense trabecular meshwork (TM) pigmentation (Fig. 1A), this deposited pigment does not directly obscure vision. Gaps in the iris pigment epithelium (IPE) lacking melanocytes (specialized pigmented cells) or containing dying melanocytes correlate with depigmented zones of the iris (3–7). Normally, aqueous humor produced by the ciliary epithelium flows into the anterior chamber and exits through the TM via the conventional aqueous outflow pathway. Pigment deposition in the TM is associated with impaired aqueous outflow through physical resistance to fluid flow, TM cell death or other still-to-be-identified mechanisms (8,9). In a proportion of PDS patients, compromised aqueous humor outflow elevates the intraocular pressure (IOP), a major risk factor for glaucoma. A subset of these cases progress to PG with characteristic retinal ganglion cell death (glaucomatous optic neuropathy) (10).
Figure 1.
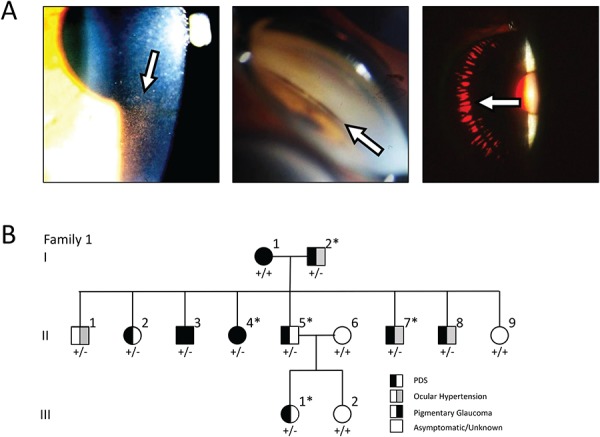
Clinical features of PDS/PG and pedigree of affected Family 1. (A) Clinical features of PDS and PG. Pigment deposition onto the inner surface of the cornea causes the appearance of Krukenberg spindle (left panel) and pigment also accumulates in the TM (center panel). Radial iris transillumination defects also occur (right panel). (B) Pedigree of Family 1 segregating PDS/PG (filled symbols) as analyzed in this study. The individuals marked with an asterisk (*) were analyzed by whole exome sequencing. Genotyping of the PMEL variant p.Gly175Ser is indicated below each pedigree symbol (+, WT; −, mutant).
The available data indicate the inheritance of PDS/PG is likely to be genetically heterogeneous and could also be influenced by diverse environmental factors. Deciphering the genetic basis of PDS/PG has been complicated by several factors: first, only an estimated 15–20% of PDS cases progress to PG over a 15-year period, (11–13) and second, the phenotypic features of PDS are transitory and may only be observed during the third and fourth decades of life (10). Together, the late age-of-onset, the transient intermediate phenotype of PDS and the low conversion rate to PG have made assessment of PG inheritance in pedigrees challenging. Risk factors for conversion also include vigorous exercise and ethnic background with observed conversion rates varying by as much as 32% over 15 years between populations (11,13–17). Twenty-six to forty-eight percent of affected individuals report a positive family history for glaucoma supporting a genetic basis for PG (11). As with other forms of glaucoma, rare variants with large effect sizes may provide significant insight into the genetic etiology and pathophysiology of PDS/PG. Known glaucoma-causing genes such as MYOC, FOXC1 and PITX2 have shown no causative association with PDS/PG but potentially act as modifiers (18–24). In this study, we used whole exome sequencing (WES) of families affected by PDS/PG to identify PMEL as a PDS/PG candidate gene and show that PMEL variants lead to protein dysfunction.
Results
Detection of PMEL variants in PDS/PG index cases and families
Family 1 (US cohort) exhibits inheritance of PDS as an autosomal dominant trait with or without PG. To identify the causative gene variant in this family, WES was completed for five affected members (Fig. 1B). Variants were filtered to retain those with presumed functional effects (nonsense, missense or splice site mutations) that were common to all five patients. Further filtering to select variants with minor allele frequencies <1% in the Exome Aggregate Consortium database (ExAC) and predicted pathogenicity identified the novel missense allele p.G175S in the gene PMEL. Using Sanger sequencing, we evaluated PMEL in the rest of Family 1. The p.G175S heterozygous variant segregated with all affected individuals in generations 2 and 3. In generation 1, individual I:1 was diagnosed with PDS + PG but does not have a mutation in PMEL, which suggests that more than one disease risk variant may be segregating throughout the family, potentially modifying the risk and/or severity of the disease. In that light, individual II:1, who does possess the p.G175S variant in PMEL, did not exhibit pigment dispersion and glaucoma. However, it is also possible that he represents a case where PG had ‘burned out’ before examination (25).
Affected first cousins from Family 2, a Mennonite kindred from western Canada had a diagnosis of PG and exhibited classical features of PDS. Additional members of this kindred were unavailable for study, but had no reported occurrence of PDS or PG, nor were there any known concerns of consanguinity. Therefore, we approached the genetic analysis with the hypothesis that the cousins were affected with an autosomal dominant form of the disease, with reduced penetrance. Genomic DNA from these cousins was sent for WES and their exomes were compared against an unaffected ethnically matched control individual. PDS/PG-associated exonic variants were filtered as above and yielded a set of 11 candidate genes that included PMEL (Supplementary Material, Table S1). None of the candidate genes are located in the reported PDS linkage regions, (26,27) and only PMEL had a reported gene ontology association with pigmentation. PG in Family 2 is thus associated with the rare non-synonymous PMEL variant p.A340V in a heterozygous state, although we cannot rule out the possibility of the disease being modified by heterozygous or homozygous mutations in other genes.
To identify additional PDS/PG-associated PMEL variants, we expanded our genetic study to screen three independent cohorts of PDS/PG patients. In a US cohort of 146 total PDS/PG cases, three rare non-synonymous PMEL variants, p.G325V, p.L389P and p.S641_S642del, were identified in three separate cases (Table 1). In a Canada (CA)/United Kingdom (UK) cohort of 113 additional cases of PDS/PG, one additional variant, p.E370D and one rare variant, p.N111S, were detected in two separate cases (Table 1). Additionally, the rare variant, p.L389P was detected again in two cases. In an Australian cohort of 135 PDS/PG cases, the rare variants p.V332I and p.S371T were found in two separate cases. The p.E370D variant was observed again in two additional cases in this cohort. Overall, nine different non-synonymous PDS/PG-associated PMEL variants were identified in a total of 13 patients, with six located in the essential structural domain called ‘RPT’ (Fig. 2A), and three located in other functional domains. Notably, p.N111S is situated at a confirmed N-linked glycosylation site (28). All variants were found in a heterozygous state in the individual patients supporting a dominant mode of inheritance. Allele frequencies of all PDS/PG-associated PMEL variants are shown in Table 2.
Table 1.
Clinical features of affected individuals
| Subject | Mutation | Age Dx | K-spindle | Angle pigm. | IOP > 21 | VF defects | Refraction | Surgical tx and other conditions | |
|---|---|---|---|---|---|---|---|---|---|
| Family 1 | |||||||||
| I-1 | – | 51 | Y | Y | Y | ||||
| I-2 | p.G175S | 60 | Y | Y | NA | ||||
| II-1 | p.G175S | 45 | NA | NA | Y | NA | |||
| II-2 | p.G175S | 41 | Y | Y | −6.75 OU | Laser tx | |||
| II-3 | p.G175S | 40 | Y | Y | Y | Y | −7.00 OU | Multiple surgeries | |
| II-4 | p.G175S | 45 | NA | NA | NA | NA | Y | ||
| II-5 | p.G175S | 37 | Y | Y | Y | ||||
| II-7 | p.G175S | 37 | Y | Y | Y | −2.00 OU | |||
| II-8 | p.G175S | 35 | Y | Y | Y | −7.00/−7.25 | Laser tx | ||
| III-1 | p.G175S | 19 | Y | NA | |||||
| Family 2 | |||||||||
| II-1 | p.A340V | 40 | Y | Y | Y | Y | NA | Laser tx | |
| II-2 | p.A340V | 47 | Y | Y | Y | Y | NA | Laser tx | |
| Singleton patients from Panels | |||||||||
| CA/UK | p.N111S | NA | Y | Y | NA | ||||
| US | p.G325V | 40 | Y | Y | Y | Y | −7.00/−7.00 | Laser tx OU | |
| AU | p.V332I | NA | Y | Y | NA | NA | NA | ||
| CA/UK | p.E370D | NA | Y | Y | Y | NA | |||
| AU | p.E370D | NA | Y | Y | NA | NA | NA | ||
| AU | p.E370D | NA | Y | Y | NA | NA | NA | ||
| AU | p.S371T | NA | Y | Y | NA | NA | NA | ||
| CA/UK | p.L389P | NA | Y | Y | NA | ||||
| CA/UK | p.L389P | NA | Y | Y | NA | ||||
| US | p.L389P | 18 | Y | Y | −1.00/−2.00 | Laser tx OS | |||
| US | p.Ser641_S642del | 42 | Y | Y | Y | Y | −4.75/−4.25 | ||
Abbreviations: Age Dx, age at diagnosis; K-spindle, presence of Krukenberg spindle, yes (Y) or no (blank); Angle Pigm., pigment visible in the TM located in the ocular angle, yes (Y) or no (blank); IOP >21; IOP greater than 21 mmHg, yes (Y) or no (blank); VF defects, Visual field defects, yes (Y) or no (blank); refraction, spherical equivalent refractive error, right eye/left eye, + values indicate hyperopia, − values indicated myopia; not available (NA); surgical tx and other conditions, surgical procedures (if any) and other ocular or systemic conditions.
Figure 2.
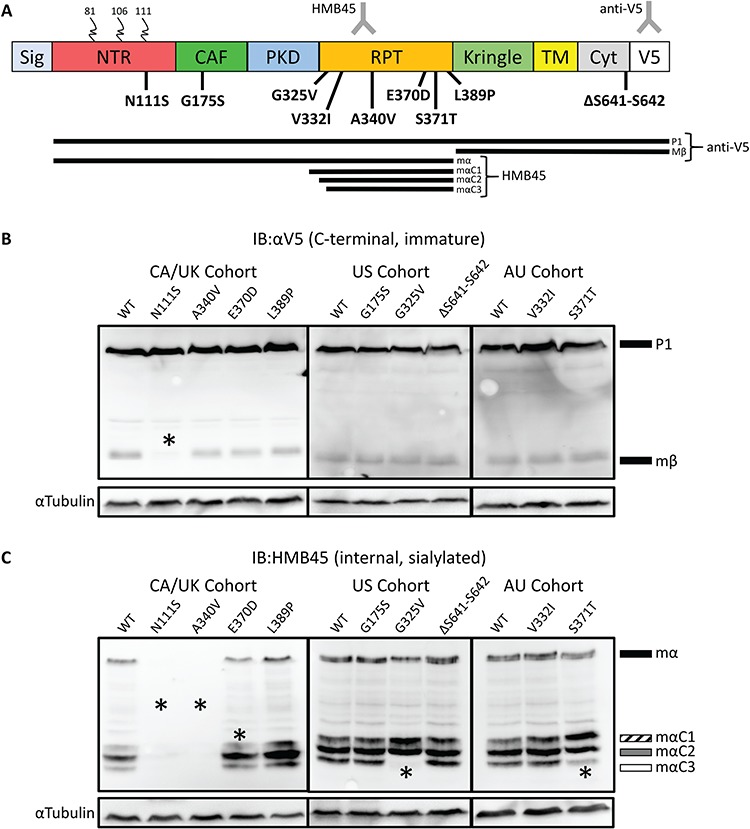
Processing defects apparent in PMEL patient variants. (A) Schematic representation of PMEL protein, the patient variants reported here, and PMEL processing intermediates. Colored blocks indicate protein domains. Curled lines indicate N-linked glycosylation. Sig, Signal domain; NTR, CAF, PKD, RPT, Kringle, Kringle-like Domain; TM, transmembrane domain; Cyt, cytoplasmic domain; V5, V5 tag. The bottom of panel A presents processing intermediates detected by IB, including the C-terminal anti-V5 and internal sialylation specific HMB45 antibodies. The lines represent intermediate processing forms detected by the different antibodies. The HMB45 antibody binds in the RPT domain and is specific to mature sialylated PMEL while anti-V5 binds to the C-terminal tag. Nine missense mutations in PMEL were discovered in individuals affected with PDS: one in the NTR, one in the CAF, six in the RPT and one in the Cyt. (B) Probing transfected HeLa cell lysates with anti-V5 shows that the Mβ band for the p.N111S sample has significantly lower relative intensity compared to WT (P < 0.05). (C) Probing transfected HeLa cell lysates with HMB45 did not detect p.N111S or p.A340V. MαC3 was not detected in p.G325V and had low intensity in p.S371T samples. A significantly lower amount of MαC1 was observed for p.E370D. Asterisks denote statistically significant changes, as quantified in Supplementary Material, Fig. S1B.
Table 2.
Non-synonymous PMEL variants in PDS/PG cases
| Allele frequency | ||||||
|---|---|---|---|---|---|---|
| Protein NP_001186983.1 |
Mutation | cDNA NM_001200054.1 |
dbSNP ID | gnomAD MAF§ | Current study (# alleles / 792) |
PMEL domain* |
| p.Asn111Ser | N111S | c.332A > G | rs773241850 | 0.00000879 | 0.0013 (1) | NTR |
| p.Gly175Ser | G175S | c.523G > A | n/a | n/a | 0.0013 (1) | CAF |
| p.Gly325Val | G325V | c.974G > T | rs148258956 | 0.00003191 | 0.0013 (1) | RPT |
| p.Val332Ile | V332I | c.994G > A | rs748713829 | 0.00007064 | 0.0013 (1) | RPT |
| p.Ala340Val | A340V | c.1019C > T | rs756974126 | 0.00007002 | 0.0013 (1) | RPT |
| p.Glu370Asp | E370D | c.1110G > C | rs17118154 | 0.00247690 | 0.0038 (3) | RPT |
| p.Ser371Thr | S371T | c.1112G > C | rs770516374 | 0.00000879 | 0.0013 (1) | RPT |
| p.Leu389Pro | L389P | c.1166T > C | rs142410496 | 0.00075229 | 0.0038 (3) | RPT |
| p.Ser641_Ser642del | ΔS641-S642 | c.1926T > C | rs765828110 | 0.00005281 | 0.0013 (1) | Cyt |
Targeted resequencing of PMEL confirmed the p.G175S and p.A340V variants found in Families 1 and 2 and identified seven other variants in 11 isolated PDS/PG cases. The cDNA and protein notations shown conform to the Human Genome Variation Society recommendations. gnomAD, Genome Aggregation Database (v2.1); MAF, minor allele frequency; PDS, PG. §European (Non-Finnish) MAF. *See Fig. 2 for PMEL protein domain structure and abbreviations.
To determine if these variants were specifically associated with PDS/PG, we evaluated the prevalence of the identified variants in a control population. The 1799 exomes from an ophthalmologically examined population not known to be affected by PDS/PG were screened for the PDS/PG-associated variants observed in the pedigrees and cohorts (Supplementary Material, Table S5). Of the nine variants we found in patients, only p.E370D and p.L389P (the most common ones) were observed in the control population, matching trends observed on larger population scales in gnomAD (Table 2).
PMEL variants cause processing defects
To investigate whether the PMEL variants identified above alter protein processing, PMEL-expressing plasmids (with a C-terminal V5 epitope tag) were transiently transfected into HeLa cells. Despite not being melanocytic in origin, HeLa cells have been widely used to study the post-translational processing of recombinant PMEL protein as its extensive modifications and essential intracellular trafficking steps are largely conserved in this cell line (29–32). PMEL is extensively modified both proteolytically and by glycosylation (N- and O-linked) and as such immunoblots (IB) with certain antibodies show multiple bands representing different processing intermediates. Immunoblot analysis of cell lysates with the anti-V5 antibody directed against the C-terminal tag revealed the two expected immature PMEL peptide intermediates P1 and Mβ in wild-type (WT) (Fig. 2A). P1, an intact form of PMEL modified with initial N-linked glycosylation was observed for WT PMEL and all variants (Fig. 2B). P1 is proteolytically processed by proprotein convertase in the Golgi to generate two products, Mα (N-terminal) and Mβ (C-terminal) (33). The mature Mβ form was observed for WT PMEL and all variants, however, only weakly for p.N111S. We quantified the signal intensity of the Mβ band expressed as a percentage of total PMEL intensity in each lane to express the efficiency of processing. The p.N111S variant was the only significantly different variant (P < 0.0001, n = 3 biological replicates, Supplementary Material, Fig. S1A) with Mβ accounting for only ~3% of total intensity (compared to 28% for WT PMEL) (Fig. 2B, Supplementary Material, Fig. S1A). No accumulation of the short-lived protein intermediate P2 was observed suggesting that this is not due to inefficient proprotein convertase cleavage but instead that p.N111S prevents efficient endoplasmic reticulum (ER) protein folding and reduces export from the ER.
To further characterize PMEL protein processing, cell lysates were also probed with the HMB45 antibody that is specific to mature sialylated PMEL processing intermediate Mα and three smaller proteolytically processed forms, MαC1, MαC2 and MαC3, containing slight variations of the repeat domain (RPT) (Fig. 2C). Three of the nine variants (p.L389P, p.G175S and p.V332I) were detected and processed normally. However, HMB45-reactive bands for p.N111S and p.A340V were not observed, suggesting these variants impair glycosyl group maturation or render the RPT-containing peptides unable to be detected by standard immunoblotting (Fig. 2C). An inclusion body solubilisation buffer was used to address whether these variants resulted in differences in protein solubility, but it did not change the results (data not shown) suggesting that the biochemical defects are in post-translational modification, protein stability or that HMB45 reactive fragments of these variants form non-fibrillar aggregates insensitive to highly denaturing conditions. In a subset of variants, altered ratios of the MαC1, MαC2 and MαC3 subtypes were observed (Fig. 2C). Significantly, less MαC1 relative to Mα was calculated for p.E370D (Supplementary Material, Fig. S1B), indicating that the variant impairs the production of this product. For p.G325V and p.S371T, significantly more MαC1 and a concomitant decrease in MαC3 was observed suggesting these variants prevent or reduce the production of MαC3 and lead to an accumulation of MαC1. These data are consistent with PDS/PG-associated PMEL variants affecting protein processing.
PDS/PG-associated PMEL variants do not cause trafficking defects
The immunoblotting results revealed that five of the nine tested variants (p.N111S, p.G325V, p.A340V, p.E370D and p.S371T) result in PMEL processing defects. In animal models, several pathological PMEL mutations (34,35) lead to altered subcellular localization. To determine if PDS/PG-associated PMEL variants may cause similar effects, we examined protein trafficking using immunofluorescence microscopy of transiently transfected HeLa cells expressing PDS/PG-associated PMEL variants. Previous research has shown that while antibodies directed against the C-terminus of PMEL (V5 epitope in this study) mainly detect signals in the ER and Golgi, probing with HMB45 detects puncta, which primarily colocalize with endosomes that contain mature PMEL, marking the final step in trafficking to the endosome (33,36,37). No differences between WT PMEL and PDS/PG-associated variants were observed when comparing the appearance of the anti-V5 and HMB45 immunostaining (Supplementary Material, Fig. S2). Extensive subcellular colocalization experiments with a subset of variants comparing the localization of PMEL to several important subcellular compartments (ER, Golgi and endosomes) confirmed normal trafficking (Supplementary Material, Fig. S3). Together these data suggest that all variants traffic normally and thus the defects in protein processing for p.N111S, p.G325V, p.A340V, p.E370D and p.S371T variants cannot be explained by trafficking defects.
Ultrastructural analysis reveals PDS/PG-associated PMEL variants cause fibrillogenesis defects
The essential function of PMEL is dependent on its ability to form amyloid fibrils in the melanosome. Ultrastructural analysis of transfected HeLa cells through transmission electron microscopy (TEM) has been extensively used to assess PMEL fibrillization potential (31). Exogenous expression of PMEL induces ectopic pseudomelanosomes in endosomes containing processed fibrillar PMEL. PMEL expression constructs containing WT or PDS/PG-associated PMEL variants were transfected into HeLa cells to examine their effects on the ultrastructure of the induced pseudomelanosomes (Fig. 3). An artificial deletion variant lacking the RPT domain, previously shown to lack fibril-forming capacity in Hela cells, was used as a comparator (38). Differences in fibril formation, fibril organization and organelle shape were qualitatively assessed and scored for all observed pseudomelanosomes in WT or variant PMEL expressing cells (Supplementary Material, Fig. S4). Generally, although a range of structures were observed, WT pseudomelanosomes tended to have one set of straight evenly spaced fibrils in ellipsoid organelles, as expected. A significant fraction of pseudomelanosomes with abnormal fibril structure (jagged and densely packed) and/or multiple sets of fibrils were observed in cells expressing p.N111S, p.G325V, p.V332I, p.E370D and p.L389P suggesting that melanosomes in cells expressing these variants are different from cells expressing WT PMEL (Fig. 3). Although p.N111S does not change the primary sequence of the RPT domain, the previous observation that p.N111S is inefficiently processed may have downstream consequences causing disordered fibrils. Intriguingly, these data may indicate that some PDS/PG-associated PMEL variants are gain-of-function mutations.
Figure 3.
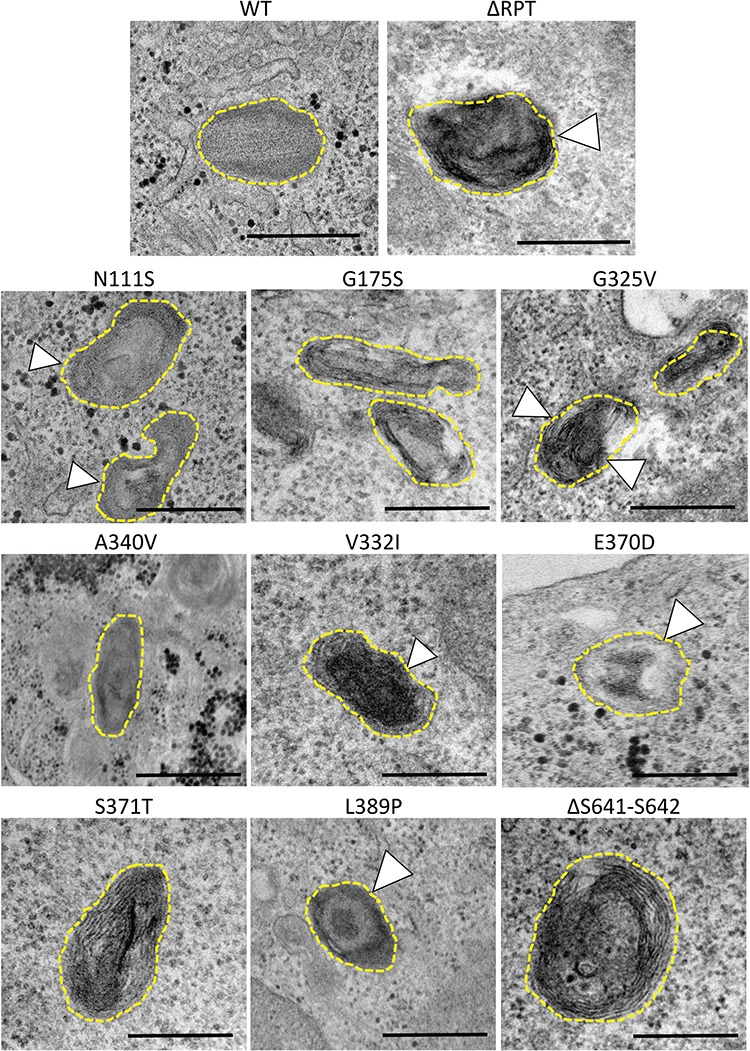
PMEL patient variants exhibit ultrastructural defects in fibril formation and organization. PDS/PG-associated PMEL variants were transfected into HeLa cells where PMEL expression induces the formation of ectopic pseudomelanosomes in endosomes. These pseudomelanosomes were identified based on appearance and scored for several features including fibril appearance, organization and overall organelle shape. Variants were compared to both WT PMEL and p.ΔRPT, a synthetic deletion lacking the RPT that has previously been shown to have significant fibrillogenesis defects. Representative images for all PMEL variants are shown. WT pseudomelanosomes mostly appeared to have straight fibrils with regular spacing and an overall ellipsoid shape. Abnormal fibrils or abnormal fibril organization were observed for five PDS/PG-associated variants (white arrowheads): p.N111S, p.V332I, p.E370D, p.L389P and p.G325 V. Overall fibrillogenesis defects were observed for five of the nine variants analyzed. Quantification of these defects is reported in Supplementary Material, Figure S4. Scale bar, 500 nm.
Disruption of the PMEL homologue in zebrafish causes pigmentation and ocular structure defects
The data above support a causal link between PMEL mutations and PG at the level of biochemistry and cell biology. To further test that PMEL disruption causes PG-like phenotypes in an in vivo system, the homologue of PMEL was disrupted in the zebrafish animal model. Mutations were engineered in zebrafish using clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 and guide RNA targeted to the C-terminal region of the zebrafish homologue pmela (i.e. in the cytoplasmic domain, Fig. 4A). A resulting mutant allele, designated pmelaua5022, was isolated and found to possess an 11-base pair (bp) deletion in this target region (Fig. 4A). This mutation results in a frameshift and is thereby predicted to eliminate 17 amino acids from the C-terminus of the zebrafish Pmela protein. This C-terminal region of PMEL is conserved across vertebrates (e.g. in the 11 most C-terminal residues, 7 are perfectly conserved between zebrafish and human PMEL; these are all lost in the predicted mutant protein). In addition to these predicted disruptions of Pmela protein structure/function, a dramatic reduction in pmela gene product was apparent—the pmela transcript was 20-fold less abundant in the mutant larvae compared to WT (Fig. 4H), presumably reduced via nonsense-mediated decay. Zebrafish larvae that were homozygous for this pmelaua5022 mutation had reduced global pigmentation (Fig. 4B–F), following recessive Mendelian inheritance. This reduced pigmentation was similar to observations when larval zebrafish were injected with a translation-blocking morpholino targeting pmela (Supplementary Material, Fig. S5), as had been reported previously (53), and similar to other mutant alleles of pmela (54). Intriguingly, by 8 days of development the pmelaua5022 homozygous mutants also presented with enlarged anterior segments (Fig. 4C and G), microphthalmia (Supplementary Material, Fig. S6B and C) and eyes that are more spherical in shape (Supplementary Material, Fig. S6D) implicating high IOP. These ocular phenotypes were not as severe at a younger age (Supplementary Material, Fig. S6E).
Figure 4.
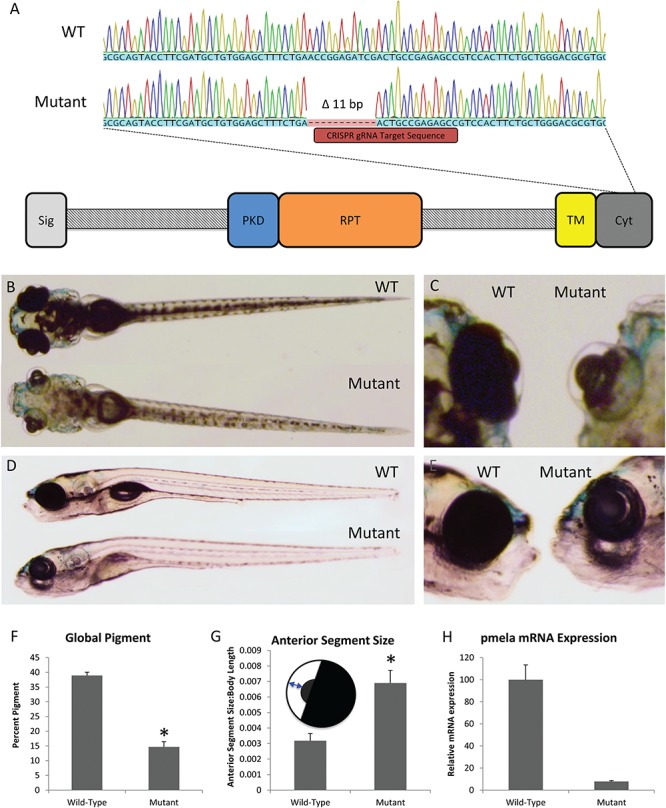
Disrupting the homologue of PMEL in zebrafish demonstrates its requirement for normal pigmentation and ocular health. (A) CRISPR-generated 11-bp deletion in the zebrafish homologue of PMEL and a schematic of where the deletion can be found in the Pmela protein. (B–E) Representative dorsal and lateral images of 8 dpf sibling WT and pmela mutant zebrafish larvae. (F) Zebrafish bearing pmela mutation has reduced pigmentation at 8 dpf. (G) Ocular deformities were apparent in pmela mutants by 8 dpf, including being more spherical. Also, anterior segment size was greater in mutants compared to WT sibling zebrafish larvae. Inset schematic sketches how the anterior segment size was measured (plotted relative to body length). *P < 0.005 using one-tailed t-tests; WT, n = 14; mutant, n = 3. (H) pmela transcripts were substantially less abundant in pmela mutants compared to WT siblings at 3 dpf, as measured by RT-qPCR (plotting three biological replicates consisting of pools of larvae, which were each assessed in three technical replicates). Results plotted are means ± standard error. See also Supplementary Material, Figs S5 and S6.
Discussion
This report establishes variation in PMEL as a cause of PDS/PG based on several mutually supporting lines of evidence. First, WES of two families affected by PDS/PG and having pedigrees consistent with dominant modes of inheritance led to identification of heterozygous non-synonymous variants in PMEL. Second, expanding this genetic study to include three large independent cohorts of PDS/PG patients identified seven different additional non-synonymous PMEL variants associated with this disease. Third, only the most common variants were observed in a large (n = 1799) ophthalmologically examined control population supporting the association of the rare variants with PDS/PG in our affected pedigrees and cohorts. Fourth, seven of the nine PDS/PG-associated PMEL variants caused significant biochemical defects in protein processing, fibril formation or both. And fifth, CRISPR-Cas9 gene editing in zebrafish confirmed that pmela plays a critical role in pigmentation, ocular structure and function. Thus, taken together these data strongly support PMEL genetic variation being a cause of PDS/PG in humans.
Interestingly, mutation of glycoprotein non-metastatic melanoma B (Gpnmb), a close homologue of PMEL with conservation of many functional domains (39), induces iris pigment dispersion in the DBA/2J murine model of glaucoma (40). DBA/2J mice are homozygous for mutations in both Gpnmb (R150X) and tyrosinase-related protein 1 (b allele). In mice, >10 genes involved in melanosome biogenesis, melanin synthesis or melanosome transfer have been implicated in iris pigment dispersion and iris atrophy (40–44). However, association between TYRP1 and GPNMB variants and PDS/PG has not been observed in humans (40). Consistent with our data, PMEL variants have been shown to be responsible for impaired pigmentation and developmental ocular defects in animals. Silver dapple horses present with multiple congenital ocular anomalies (MCOAs) (45), blue merle dogs exhibit dapple pigmentation and MCOAs (46), silver mice have misshapen melanosomes of the retinal pigment epithelium (RPE) and uveal melanocytes (47) and the fading vision zebrafish displays reduced pigmentation in the body and RPE accompanied by disrupted photoreceptor morphology and visual deficits as measured by optokinetic response (48). Melanin synthesis is a tightly regulated process that can generate cytotoxic melanin synthesis intermediates (49), and melanosomal dysfunction could lead to melanocyte death in the IPE.
Indeed, when an 11-bp deletion was introduced to the cytoplasmic domain of a zebrafish homologue of PMEL, pigmentation defects were seen in both the body and the RPE in homozygous mutants. When messenger RNA (mRNA) levels of pmela were analyzed, the abundance of transcripts was dramatically reduced, implying that the mutation has changed the sequence to a degree where the process of nonsense-mediated decay occurs and the protein product would be mostly absent. Upon closer inspection at 8 days post-fertilization (dpf), well beyond the time morpholinos injected into embryos would have been turned over, the larval zebrafish also developed ocular structure defects not apparent in 3 and 6 dpf morphants or homozygous mutant larvae. The sequence of these events is important in determining whether the loss of pigment due to mutations in pmela could cause the ocular defects in the zebrafish and therefore if mutations in PMEL could cause PDS/PG in humans. Intriguingly, the eyes of pmela mutant zebrafish had enlarged anterior segments and were more spherical compared to WT siblings; this is consistent with pmela disruption producing increased IOP and thus reminiscent of a key risk factor in glaucoma.
We hypothesized that the observed variants in PMEL may cause defects in PMEL protein processing, trafficking or function ultimately resulting in defective melanocytes. PMEL is initially synthesized as an integral membrane protein in the ER but is extensively post-translationally modified. During trafficking to endosomes/stage 1 melanosomes PMEL is proteolytically processed and modified with both N- and O-linked glycosylation (Fig. 2A) (29,50,51). Within these organelles, mature PMEL fragments oligomerize to form intraluminal fibers that give melanosomes their distinct ellipsoidal morphology (36,52,53). These fibers uniquely represent an example of a functional amyloid in physiological conditions (31,54–57). The following three PMEL domains (Fig. 2A) comprise these fibrils: the core amyloid fragment (CAF), the polycystic kidney disease domain (PKD) and the RPT so named because it consists of 13 imperfect 10-amino acid repeats (30,31,55). PMEL fibers function as a scaffold for melanin synthesis with reaction intermediates adsorbing onto the fibers and greatly increasing the efficiency of synthesis (56,58,59). As such, PMEL protects melanocytes from harmful melanin synthesis intermediates that generate reactive oxygen species (ROS) when released into the cytosol (29,35,40,47).
Of the nine different PMEL variants detected in patients here, one is in the CAF domain and six are in the RPT domain, which are both integrated into the mature heteropolymeric melanosome fibrils (Table 1, Fig. 2A). One other variant, p.N111S, is predicted to abolish an N-linked glycosylation site in the N-terminal region (NTR) (UniProt). The NTR domain coordinates processing, trafficking and assembly of PMEL fibrils despite not participating in the final structure (37,54). Expression of the p.N111S construct resulted in a reduction of proteolytic products (Fig. 2) but it is not clear whether the loss of glycosylation or the presence of the serine residue is responsible. We used electron microscopy to determine that five of the PMEL variants studied here produce disordered fibrils in pseudomelanosomes or affect fibril organization in transfected HeLa cells, confirming that organellar structure is sensitive to missense mutations in critical residues of this gene. We hypothesize that iris melanocyte health or longevity is affected by melanosome function and that pigment integrity in the lifespan of a human eye is thus susceptible to PMEL variation. We expected to observe relatively subtle defects in fibrillogenesis because PDS/PG is not associated with non-ocular pigmentary defects. The functional impairments described in this study are consistent with the hypothesis that the observed heterozygous PDS/PG-associated PMEL variants are gain-of-function mutations that cause PDS/PG with dominant inheritance, albeit with incomplete penetrance over the timescales observed. While our zebrafish model clearly shows that loss of PMEL impacts ocular function, and that some disease causing variants may be loss-of-function, our data also supports a hypothesis that aberrant protein fibrils or aggregates are also an important mechanism of melanocyte damage.
The principal defect in PDS/PG is thought to be detachment of pigmented cells from the posterior iris with deposition of pigmented material in the TM leading to increased IOP and ultimately glaucoma (10). An initial mechanical explanation proposed that posterior iris bowing created inappropriate contact with the lens zonule, leading to abrasion and liberation of pigment from the IPE. Although irido-zonular contact was indeed demonstrated in a subset of PDS patients (4,5), it remained puzzling why this would occur in a disorder characterized by increased irido-lenticular separation (myopia), yet this is never observed in either hyperopia or microphthalmia. Since surgical intervention to relieve posterior iris bowing does not prevent progression to glaucoma, (60,61) other mechanisms are likely to contribute. Our data are consistent with the hypothesis that the primary cause of pigment release is melanocyte cell death and consequent detachment in accordance with previous observations in mice (62). Given that human ocular pigment deposition is commonly juvenile onset and progresses gradually over decades, we hypothesize that dysfunctional melanosomes lead to the accumulation of ROS and/or cytotoxic melanin synthesis intermediates in iris melanocytes. Iris melanocytes represent a unique population that undergo continuous melanosome biogenesis but do not transfer those melanosomes to keratinocytes as in the hair or skin. This may make them particularly susceptible to mutations that affect melanosome function, as mildly damaging effects can accumulate over time. Accordingly, the relatively more pigmented melanocytes of the posterior IPE are predicted to be the first iris melanocytes to show signs of damage. Other melanocytes in the stroma and anterior IPE would also be affected but over longer timescales proportional to the degree of pigmentation. Although the structural role of PMEL in melanosomes has been well established, it has previously been suggested that the primary function of PMEL is to sequester these ROS and cytotoxic melanin synthesis intermediates to protect the cell (63). Since the factors regulating pigmentation and ROS response are diverse [several of which have been previously implicated in glaucoma (64–66)], this process could be influenced by many genetic and environmental factors explaining the observed genetic heterogeneity for PDS/PG. As a corollary to this model, we anticipate that genetic variants that dramatically affect these processes could lead to simpler PDS/PG inheritance modes due to a larger pathological contribution, whereas combinations of genetic variants with subtle effects could underlie complex inheritance modes. The PMEL variants characterized here cause defects in processing and/or fibrillogenesis that likely impair PMEL’s ability to protect melanocytes from ROS generated during melanin synthesis and storage. Simultaneously, PMEL variants that cause a transition from functional to pathogenic amyloid may also cause cell death through an alternative pathway due to the formation of protein aggregates. The most likely mechanism by which PMEL variants contribute to PDS/PG is melanocyte dysfunction causing an increase in pigment-containing cellular debris in the aqueous humor. Phagocytosis of this material by TM cells causes the accumulation of pigmented material in the TM, which is known to cause TM cell detachment and/or death (67–71). The resultant loss of aqueous humor homeostasis is then associated with increased IOP-mediated retinal ganglion cell death and high-pressure glaucoma. Considering the important role of melanosomes in protecting the RPE from ROS generated during photoreceptor outer segment turnover, it is intriguing to consider whether PMEL variants also simultaneously alter RPE function thereby contributing to PDS/PG retinal phenotypes as has been suggested previously (72,73). Further study is expected to elucidate how these variants contribute to the pathology of PDS/PG and impact melanocyte health.
Materials and Methods
Subjects and clinical evaluation
All study subjects were Caucasian with European ancestry and provided written informed consent. All probands and isolated cases underwent a complete ocular examination. The diagnosis of PDS was based on the presence of characteristic iris transillumination defects detected by slit lamp examination. Other features of PDS including corneal endothelial cell pigment (Krukenberg spindle), zonular pigment deposition and excess pigmentation of the TM on gonioscopy were noted but were not required for case definition. As PDS is a predisposing condition for glaucoma (PG), clinical features defining glaucoma were also recorded including elevated IOP above 21 mmHg and presence of visual field defects characteristic of glaucoma-related optic neuropathy. Clinical information for some family members was obtained from clinical notes. Other clinical features, including refractive error, were noted if available. The research was approved by the following separate Institutional Health Research Ethics Boards: Flinders Medical Centre, Massachusetts Eye and Ear Infirmary and the University of Alberta.
Family 1 and the US cohort (28 additional multiplex families and 118 isolated cases) were comprised of Caucasian PDS/PG patients with European ancestry and were recruited from the Massachusetts Eye and Ear Infirmary, the New York Eye and Ear Infirmary of Mount Sinai and Bascom Palmer Eye Institute.
Two Canadian Mennonite patients, first cousins (Family 2), were assessed on multiple occasions at the Royal Alexandra Hospital Eye Clinic (Edmonton, AB), with comprehensive ophthalmological assessments that included automated perimetry (visual field testing) and disc imaging (optical coherence tomography and Heidelberg retina tomograph (HRT)) (Table 1). Both patients fulfilled the diagnostic criteria for PDS and PG. The second cohort consisted of PDS/PG patients recruited in CA and the UK (three additional families and 110 isolated cases). The third cohort (Australian) consisted of 135 PDS/PG cases recruited from the Australian and New Zealand Registry of Advanced Glaucoma.
Whole exome sequencing
WES was performed on selected members of Family 1 with an Agilent SureSelect V5 + UTR (74.4 Mb target sequence) library preparation kit and the HiSeq2000 sequencer (Illumina). Reads were aligned to the human reference sequence (hg19) with Burrows–Wheeler Aligner v0.6.2-r126, (74) and SAMtools v0.1.18 or r982:295 (75) was used to remove potential duplicates and to make initial single nucleotide polymorphism (SNP) and indel calls that were refined using a custom program (76). Resulting variant calls were annotated using a custom human bp codon resource (76,77). Overall, between 19 200–22 254 exonic sequence alterations were identified in each family member. The average sequencing depth was 110× and 99% of the targeted regions were covered with a minimum read depth of 40×.
Genomic DNA from Family 2 was submitted to the Beijing Genome Institute for WES with the Ion Proton AmpliSeq Exome RDY kit, followed by standard bioinformatics analysis to provide lists of annotated variants. Between 20 573–20 652 exonic variants were detected in each individual (BGI Americas). Ninety-two percent of targeted exons were sequenced with an average of 203× coverage, with 80% of bases at a quality score of Q20 or higher.
WES variants and PMEL screening of the US and Australian cohort was completed using Sanger sequencing as previously described (78).
Targeted high-throughput sequencing
To rapidly and economically genotype the CA/UK cohort a high-throughput targeted next generation sequencing approach was developed. Polymerase chain reaction (PCR) primers were designed using Primer3 (79) and SNPCheck3 to amplify all coding exons of PMEL, avoiding or minimizing the number of polymorphic base-pairing sites (Supplementary Material, Table S2). Primary PCR products were amplified in 20 μl reactions using 0.2 μm of each forward and reverse primer, 1× FailSafe Premix J (Epicentre Biotechnologies), 40 ng genomic DNA and 1 U Taq polymerase (New England Biolabs), using a standard touchdown cycling protocol as follows: denaturation at 95°C for 3 min followed by five cycles of 95°C for 30 s, 64–56°C for 30 s (2°C decrease per cycle), 68°C for 30 s and then 24–33 cycles (optimized per primer pair to produce all bands of similar intensity) of 95°C for 30 s, 54°C for 30 s and 68°C for 30 s, with a final extension at 68°C for 5 min. After confirmation of successful amplification using agarose gels, PCR products were pooled for each individual, purified enzymatically (ExoSAP-IT, Affymetrix USB) and subjected to secondary ‘barcoding’ PCR with unique pairs of Nextera XT v2 index primers (Illumina) as follows: denaturation at 95°C for 3 min followed by eight cycles of 95°C for 30 s, 55°C for 30 s, 68°C for 30 s, with a final extension at 68°C for 5 min. Samples were then bead purified (AMPure XP, Agencourt), with confirmation on a QIAxcel (Qiagen). Samples were then pooled and the library was quantified with a Qubit 2.0 fluorometer (ThermoFisher Scientific) and sized with a Bioanalyzer 2100 (Agilent Technologies) before being analyzed on a MiSeq desktop sequencer (Illumina) at The Applied Genomic Core at the University of Alberta. The 360 Mb of sequence was generated with 90% of the 1.4 million reads (2 × 250 bp) at a Phred quality score of >Q30. The FASTQ files were aligned to the human genome (GRCh38) with the Burrows–Wheeler Aligner v0.6.1-r104 (74) and indexed with SAMtools v1.3.1. (75). Variants were called with VarScan v2.4.2 (80) using a minimum variant allele frequency threshold of 0.35 and annotated with Ensembl’s Variant Effect Predictor (81). The identified variants were confirmed by conventional Sanger sequencing.
Cloning
A complementary DNA (cDNA) encoding WT PMEL17-i, the most abundant protein isoform (NP_008859.1) (38) was purchased from the DNASU Plasmid Repository and subcloned into pGEM-T plasmid (Promega) with an in-frame C-terminal V5 epitope (GKPIPNPLLGLDST). Site-directed mutagenesis was performed (QuikChange Lightning Site-Directed Mutagenesis Kit, Agilent Technologies) to generate plasmids for each patient variant. All plasmid inserts were then subcloned into the pCI mammalian expression vector (Promega) and the full insert sequences were confirmed.
Cell culture
HeLa cells were grown in high-glucose Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and 1× antibiotic-antimycotic solution (ThermoFisher Scientific). Cells were transfected with the plasmid constructs using Lipofectamine 2000 (ThermoFisher Scientific), per the manufacturer’s protocol.
Immunoblotting
HeLa cell lysates were harvested 48 h post-transfection using lysis buffer [0.1% sodium dodecyl sulfate (SDS), 0.5% sodium deoxycholate, 1% IGEPAL CA-630, in phosphate buffered saline] for analysis by denaturing SDS-polyacrylamide gel electrophoresis (PAGE). Insoluble fractions were suspended using inclusion body solubilisation buffer (8 M Urea, 100 mm β-ME, 100 mm Tris/HCl). PMEL proteins were detected by immunoblotting using anti-V5 antibody (Sigma-Aldrich) or HMB45 (Novus Biologicals) and visualized by chemiluminescence (SuperSignal West Femto Maximum Sensitivity Substrate, ThermoFisher Scientific).
Immunofluorescence and colocalization analysis
HeLa cells, grown and transfected on glass coverslips, were fixed with 2% paraformaldehyde 48 h post-transfection, then incubated with the indicated cell localization markers for 1 h: ER (anti-Calreticulin, a generous gift from Dr. Marek Michalak), Golgi (anti-GM130, product #AF8199, Novus Biologicals) and endosomes (anti-LAMP1, product #GR314073, Abcam). Fluorescent secondary antibodies (Jackson ImmunoResearch Laboratories, Inc.) were incubated at 1:5000 dilution for 1 h. Nuclei were visualized by staining with 4′, 6-diamidine-2-phenylindole (DAPI) dye (10 μg/ml). Slides were imaged using a confocal microscope [Olympus IX-81 microscope, Yokagawa CSU 10 spinning disk confocal, Lumen Dynamics X-Cite 120, Hamamatsu EMCCD (C9100–13), acquisition using Perkin Elmer’s Volocity].
Transmission electron microscopy
Ultra-thin sections with a thickness of 60 nm were generated using a Leica UC7 ultramicrotome (Leica Microsystems, Inc.) and contrasted with 2% uranyl acetate and Reinold’s lead citrate. Sections were imaged using a Hitachi H-7650 transmission electron microscope (Hitachi-High Technologies) at 60 kV and a 16-mega pixel TEM camera (XR111, Advanced Microscopy Techniques). Images were qualitatively assessed by scoring each observed pseudomelanosome for the following three morphological features using WT PMEL and p.RPTdel as known normal or aberrant examples, respectively: 1) fibril appearance, where WT fibrils appear straight and uniform, 2) fibril organization, where WT fibrils have regular spacing and only one cluster of fibrils per pseudomelanosomes and 3) organelle shape, where WT organelles appear ellipsoid. If pseudomelanosome features were ambiguous, they were assumed to be normal. A Student’s t-test (two-tailed, P < 0.05) was used to compare PDS/PG-associated PMEL variants to WT using the percent of pseudomelanosomes observed to be abnormal for those features.
Zebrafish CRISPR and reverse transcription quantitative PCR
All zebrafish husbandry and experimentation were completed under Protocol #AUP00000077 approved by the University of Alberta Animal Care and Use Committee: Biosciences under the auspices of the Canadian Council on Animal Care. Zebrafish (WT AB strains) were maintained at 28.5°C in standard conditions (82,83).
Antisense morpholino oligonucleotides (MOs) purchased from Gene Tools, LLC were delivered to embryos using procedures described previously (83). The MOs used in the experiments are shown in Supplementary Material, Table S3. Quantification and statistical analysis of pigmentation was performed by binarization of images, as described previously (84).
To produce guide RNA, the CRISPR binding sequence for the cytoplasmic domain of pmela (Supplementary Material, Table S3) was designed in Geneious 9.1.8. An SP6 promoter was added to this sequence and then annealed to the constant oligomer (Supplementary Material, Table S4) (85). The oligonucleotides were ordered from Integrated DNA Technologies, Inc. The resulting oligonucleotide was then transcribed into guide RNA with T4 DNA polymerase (New England Biolabs), purified (QIAquick PCR Purification Kit, Qiagen) and further transcribed and purified using the mMESSAGE mMACHINE SP6 transcription kit (ThermoFisher Scientific). Zebrafish embryos at the one-cell stage were injected with 1 nL of a cocktail containing 1 μl of guide RNA (at >2000 ng/μl) mixed with 2 μl of Cas9 protein stock (New England Biolabs), 0.5 μl Cas9 buffer (New England Biolabs) and 1.5 μl 1.5 M KCl. After cutting of the target genomic region was confirmed in injected embryos, other injected embryos were raised and their progeny were assessed for mutations surrounding the target region using primers listed in Supplementary Material, Table S4.
The morphology of zebrafish was assessed and documented with a Leica MZ16F stereo-dissection microscope with a mounted 12.8 megapixel digital camera (DP72, Olympus). Quantification and statistical analysis of pigmentation was performed by binarization of images, as described previously (84). Quantification of morphology utilized ImageJ (Wayne Rasband, National Institutes of Health).
Reverse transcription quantitative PCR (RT-qPCR) on zebrafish larvae followed the MIQE guidelines (86) for primer validation and RNA quality control. RNA was extracted from 3 dpf WT and mutant embryos with an RNeasy Minikit (Qiagen) and DNase I (Qiagen) and assessed for high quality on an RNA 6000 NanoChip and 2100 Bioanalyzer (Agilent Technologies). The RNA was then synthesized into cDNA with qScript Supermix (Quantabio). The SYBR green system was used for qPCR on a 7500 Real-Time PCR system (Applied Biosystems) using primers (Supplementary Material, Table S4) validated to produce a single clean peak and maintain a linear amplification over a broad dilution range. qPCR was performed in three technical replicates on each biological replicate. Transcript abundance was reported as relative to β-actin levels (87).
Supplementary Material
Acknowledgements
Thanks to Dr Marek Michalak for his generous gift of the αCalreticulin antibody; to Dr A. Phil Oel and Michele G. Duval for their advice with zebrafish experiments; and to Dr Lance Doucette, Allison Lewis, Fahed Elian, and Elizabeth Yan for their comments on the manuscript. We also thank the technical cores at the University of Alberta for their assistance with sequencing (The Applied Genomics Core) and microscopy (Cell Imaging Center).
Conflict of Interest statement. None declared.
Web resources
ExAC – http://exac.broadinstitute.org
Primer3 – http://bioinfo.ut.ee/primer3
SNPcheck – https://secure.ngrl.org.uk/SNPCheck/snpcheck.htm
Variant Effect Predictor – https://uswest.ensembl.org/info/docs/tools/vep/index.html
gnomAD – http://gnomad.broadinstitute.org
Funding
This work was supported by Alberta Vision Network; the Glaucoma Research Society of Canada; Maternal and Child Health Student Support; and the Canadian Institutes of Health Research. Collection and characterization of the US cohort was supported by the BrightFocus Foundation; the March of Dimes Foundation; and the National Institutes of Health/National Eye Institute [P30 EY014104].
References
- 1. Mantravadi A.V. and Vadhar N. (2015) Glaucoma. Prim. Care, 42, 437–449. [DOI] [PubMed] [Google Scholar]
- 2. Yang J.W., Sakiyalak D. and Krupin T. (2001) Pigmentary glaucoma. J. Glaucoma, 10, S30–S32. [DOI] [PubMed] [Google Scholar]
- 3. Scheie H.G. and Fleischhauer H.W. (1958) Idiopathic atrophy of the epithelial layers of the iris and ciliary body; a clinical study. Trans. Am. Ophthalmol. Soc., 55, 369–388. [PMC free article] [PubMed] [Google Scholar]
- 4. Potash S.D., Tello C., Liebmann J. and Ritch R. (1994) Ultrasound biomicroscopy in pigment dispersion syndrome. Ophthalmology, 101, 332–339. [DOI] [PubMed] [Google Scholar]
- 5. Campbell D.G. (1979) Pigmentary dispersion and glaucoma. Arch. Ophthalmol., 97, 1667. [DOI] [PubMed] [Google Scholar]
- 6. Kupfer C., Kuwabara T. and Kaiser-Kupfer M. (1975) The histopathology of pigmentary dispersion syndrome with glaucoma. Am. J. Ophthalmol., 80, 857–862. [DOI] [PubMed] [Google Scholar]
- 7. Kampik A., Green W.R., Quigley H.A. and Pierce L.H. (1981) Scanning and transmission electron microscopic studies of two cases of pigment dispersion syndrome. Am. J. Ophthalmol., 91, 573–587. [DOI] [PubMed] [Google Scholar]
- 8. Shimizu T., Hara K. and Futa R. (1981) Fine structure of trabecular meshwork and iris in pigmentary glaucoma. Albrecht Von Graefes Arch. Klin. Exp. Ophthalmol., 215, 171–180. [DOI] [PubMed] [Google Scholar]
- 9. Alvarado J.A. and Murphy C.G. (1992) Outflow obstruction in pigmentary and primary open angle glaucoma. Arch. Ophthalmol., 110, 1769. [DOI] [PubMed] [Google Scholar]
- 10. Ritch R. (1996) A unification hypothesis of pigment dispersion syndrome. Trans. Am. Ophthalmol. Soc., 94, 381–405. [PMC free article] [PubMed] [Google Scholar]
- 11. Siddiqui Y., Ten Hulzen R.D., Cameron J.D., Hodge D.O. and Johnson D.H. (2003) What is the risk of developing pigmentary glaucoma from pigment dispersion syndrome? Am. J. Ophthalmol., 135, 794–799. [DOI] [PubMed] [Google Scholar]
- 12. Okafor K., Vinod K. and Gedde S.J. (2017) Update on pigment dispersion syndrome and pigmentary glaucoma. Curr. Opin. Ophthalmol., 28, 154–160. [DOI] [PubMed] [Google Scholar]
- 13. Gomez Goyeneche H.F., Hernandez-Mendieta D.P., Rodriguez D.A., Sepulveda A.I. and Toledo J.D. (2015) Pigment dispersion syndrome progression to pigmentary glaucoma in a Latin American population. J. Curr. Glaucoma Pract., 9, 69–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Migliazzo C.V., Shaffer R.N., Nykin R. and Magee S. (1986) Long-term analysis of pigmentary dispersion syndrome and pigmentary glaucoma. Ophthalmology, 93, 1528–1536. [DOI] [PubMed] [Google Scholar]
- 15. Richter C.U., Richardson T.M. and Grant W.M. (1986) Pigmentary dispersion syndrome and pigmentary glaucoma. Arch. Ophthalmol., 104, 211. [DOI] [PubMed] [Google Scholar]
- 16. Farrar S.M., Shields M.B., Miller K.N. and Stoup C.M. (1989) Risk factors for the development and severity of glaucoma in the pigment dispersion syndrome. Am. J. Ophthalmol., 108, 223–229. [DOI] [PubMed] [Google Scholar]
- 17. Shah I.A., Shah S.A., Nagdev P.R., Abbasi S.A., Abbasi N.A. and Katpar S.A.. Determination of association of pigmentary glaucoma with pigment dispersion syndrome. J. Ayub Med. Coll. Abbottabad, 29, 412–414. [PubMed] [Google Scholar]
- 18. Vincent A.L., Billingsley G., Buys Y., Levin A.V., Priston M., Trope G., Williams-Lyn D. and Héon E. (2002) Digenic inheritance of early-onset glaucoma: CYP1B1, a potential modifier gene. Am. J. Hum. Genet., 70, 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Faucher M., Anctil J.-L., Rodrigue M.-A., Duchesne A., Bergeron D., Blondeau P., Côté G., Dubois S., Bergeron J., Arseneault R. et al. (2002) Founder TIGR/myocilin mutations for glaucoma in the Québec population. Hum. Mol. Genet., 11, 2077–2090. [DOI] [PubMed] [Google Scholar]
- 20. Alward W.L.M., Kwon Y.H., Khanna C.L., Johnson A.T., Hayreh S.S., Zimmerman M.B., Narkiewicz J., Andorf J.L., Moore P.A., Fingert J.H. et al. (2002) Variations in the myocilin gene in patients with open-angle glaucoma. Arch. Ophthalmol., 120, 1189–1197. [DOI] [PubMed] [Google Scholar]
- 21. Wolf C., Gramer E., Müller-Myhsok B., Pasutto F., Gramer G., Wissinger J.B. and Weisschuh N. (2010) Lysyl oxidase-like 1 gene polymorphisms in German patients with normal tension glaucoma, pigmentary glaucoma and exfoliation glaucoma. J. Glaucoma, 19, 136–141. [DOI] [PubMed] [Google Scholar]
- 22. Rao K.N., Ritch R., Dorairaj S.K., Kaur I., Liebmann J.M., Thomas R. and Chakrabarti S. (2008) Exfoliation syndrome and exfoliation glaucoma-associated LOXL1 variations are not involved in pigment dispersion syndrome and pigmentary glaucoma. Mol. Vis., 14, 1254–1262. [PMC free article] [PubMed] [Google Scholar]
- 23. Giardina E., Oddone F., Lepre T., Centofanti M., Peconi C., Tanga L., Quaranta L., Frezzotti P., Novelli G. and Manni G. (2014) Common sequence variants in the LOXL1 gene in pigment dispersion syndrome and pigmentary glaucoma. BMC Ophthalmol., 14, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gould D.B., Smith R.S. and John S.W.M. (2004) Anterior segment development relevant to glaucoma. Int. J. Dev. Biol., 48, 1015–1029. [DOI] [PubMed] [Google Scholar]
- 25. Niyadurupola N. and Broadway D.C. (2008) Pigment dispersion syndrome and pigmentary glaucoma—a major review. Clin. Exp. Ophthalmol., 36, 868–882. [DOI] [PubMed] [Google Scholar]
- 26. Andersen J.S., Pralea A.M., DelBono E.A., Haines J.L., Gorin M.B., Schuman J.S., Mattox C.G. and Wiggs J.L. (1997) A gene responsible for the pigment dispersion syndrome maps to chromosome 7q35-q36. Arch. Ophthalmol., 115, 384–388. [DOI] [PubMed] [Google Scholar]
- 27. Wagner S.H., Delbono E., Greenfield D.S., Parrish R.K., Haines J.L. and Wiggs J.L. (2005) A second locus for pigment dispersion syndrome maps to chromosome 18q21. Invest. Ophthalmol. Vis. Sci., 46, 29. [Google Scholar]
- 28. Yasumoto K., Watabe H., Valencia J.C., Kushimoto T., Kobayashi T., Appella E. and Hearing V.J. (2004) Epitope mapping of the melanosomal matrix protein gp100 (PMEL17): rapid processing in the endoplasmic reticulum and glycosylation in the early Golgi compartment. J. Biol. Chem., 279, 28330–28338. [DOI] [PubMed] [Google Scholar]
- 29. Rochin L., Hurbain I., Serneels L., Fort C., Watt B., Leblanc P., Marks M.S., De Strooper B., Raposo G. and Niel G. (2013) BACE2 processes PMEL to form the melanosome amyloid matrix in pigment cells. Proc. Natl. Acad. Sci. U. S. A., 110, 10658–10663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Valencia J.C., Rouzaud F., Julien S., Chen K.G., Passeron T., Yamaguchi Y., Abu-Asab M., Tsokos M., Costin G.E., Yamaguchi H. et al. (2007) Sialylated core 1 O-glycans influence the sorting of Pmel17/gp100 and determine its capacity to form fibrils. J. Biol. Chem., 282, 11266–11280. [DOI] [PubMed] [Google Scholar]
- 31. Hoashi T., Muller J., Vieira W.D., Rouzaud F., Kikuchi K., Tamaki K. and Hearing V.J. (2006) The repeat domain of the melanosomal matrix protein PMEL17/GP100 is required for the formation of organellar fibers. J. Biol. Chem., 281, 21198–21208. [DOI] [PubMed] [Google Scholar]
- 32. Theos A.C., Truschel S.T., Tenza D., Hurbain I., Harper D.C., Berson J.F., Thomas P.C., Raposo G. and Marks M.S. (2007) A novel pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell, 10, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Leonhardt R.M., Vigneron N., Rahner C. and Cresswell P. (2011) Proprotein convertases process Pmel17 during secretion. J. Biol. Chem., 286, 9321–9337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Watt B., Tenza D., Lemmon M.A., Kerje S., Raposo G., Andersson L. and Marks M.S. (2011) Mutations in or near the transmembrane domain alter PMEL amyloid formation from functional to pathogenic. PLoS Genet., 7, e1002286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Theos A.C., Berson J.F., Theos S.C., Herman K.E., Harper D.C., Tenza D., Sviderskaya E.V., Lamoreaux M.L., Bennett D.C., Raposo G. et al. (2006) Dual loss of ER export and endocytic signals with altered melanosome morphology in the silver mutation of Pmel17. Mol. Biol. Cell, 17, 3598–3612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harper D.C., Theos A.C., Herman K.E., Tenza D., Raposo G. and Marks M.S. (2008) Premelanosome amyloid-like fibrils are composed of only Golgi-processed forms of Pmel17 that have been proteolytically processed in endosomes. J. Biol. Chem., 283, 2307–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Leonhardt R.M., Vigneron N., Hee J.S., Graham M. and Cresswell P. (2013) Critical residues in the PMEL/Pmel17 N-terminus direct the hierarchical assembly of melanosomal fibrils. Mol. Biol. Cell, 24, 964–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hoashi T., Tamaki K. and Hearing V.J. (2010) The secreted form of a melanocyte membrane-bound glycoprotein (Pmel17/gp100) is released by ectodomain shedding. FASEB J., 24, 916–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Theos A.C., Watt B., Harper D.C., Janczura K.J., Theos S.C., Herman K.E. and Marks M.S. (2013) The PKD domain distinguishes the trafficking and amyloidogenic properties of the pigment cell protein PMEL and its homologue GPNMB. Pigment Cell Melanoma Res., 26, 470–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Anderson M.G., Smith R.S., Hawes N.L., Zabaleta A., Chang B., Wiggs J.L. and John S.W.M. (2002) Mutations in genes encoding melanosomal proteins cause pigmentary glaucoma in DBA/2J mice. Nat. Genet., 30, 81–85. [DOI] [PubMed] [Google Scholar]
- 41. Anderson M.G., Hawes N.L., Trantow C.M., Chang B. and John S.W.M. (2008) Iris phenotypes and pigment dispersion caused by genes influencing pigmentation. Pigment Cell Melanoma Res., 21, 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Trantow C.M., Mao M., Petersen G.E., Alward E.M., Alward W.L.M., Fingert J.H. and Anderson M.G. (2009) Lyst mutation in mice recapitulates iris defects of human exfoliation syndrome. Invest. Ophthalmol. Vis. Sci., 50, 1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nair K.S., Cosma M., Raghupathy N., Sellarole M.A., Tolman N.G., Vries W., Smith R.S. and John S.W.M. (2016) YBR/EiJ mice: a new model of glaucoma caused by genes on chromosomes 4 and 17. Dis. Model. Mech., 9, 863–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swaminathan S., Lu H., Williams R.W., Lu L. and Jablonski M.M. (2013) Genetic modulation of the iris transillumination defect: a systems genetics analysis using the expanded family of BXD glaucoma strains. Pigment Cell Melanoma Res., 26, 487–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Andersson L.S., Wilbe M., Viluma A., Cothran G., Ekesten B., Ewart S. and Lindgren G. (2013) Equine multiple congenital ocular anomalies and silver coat colour result from the pleiotropic effects of mutant PMEL. PLoS One, 8, 4–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clark L.A., Wahl J.M., Rees C.A. and Murphy K.E. (2006) Retrotransposon insertion in SILV is responsible for merle patterning of the domestic dog. Proc. Natl. Acad. Sci. U. S. A., 103, 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hellström A.R., Watt B., Fard S.S., Tenza D., Mannström P., Narfström K., Ekesten B., Ito S., Wakamatsu K., Larsson J. et al. (2011) Inactivation of Pmel alters melanosome shape but has only a subtle effect on visible pigmentation. PLoS Genet., 7, e1002285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Schonthaler H.B., Lampert J.M., Lintig J., Schwarz H., Geisler R. and Neuhauss S.C.F. (2005) A mutation in the silver gene leads to defects in melanosome biogenesis and alterations in the visual system in the zebrafish mutant fading vision. Dev. Biol., 284, 421–436. [DOI] [PubMed] [Google Scholar]
- 49. Urabe K., Aroca P., Tsukamoto K., Mascagna D., Palumbo A., Prota G. and Hearing V.J. (1994) The inherent cytotoxicity of melanin precursors: a revision. Biochim. Biophys. Acta, 1221, 272–278. [DOI] [PubMed] [Google Scholar]
- 50. Maresh G.A., Wang W.C., Beam K.S., Malacko A.R., Hellstrom I., Hellstrom K.E. and Marquardt H. (1994) Differential processing and secretion of the melanoma-associated Me20 antigen. Arch. Biochem. Biophys., 311, 95–102. [DOI] [PubMed] [Google Scholar]
- 51. Berson J.F., Theos A.C., Harper D.C., Tenza D., Raposo G. and Marks M.S. (2003) Proprotein convertase cleavage liberates a fibrillogenic fragment of a resident glycoprotein to initiate melanosome biogenesis. J. Cell Biol., 161, 521–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhou B.K., Kobayashi T., Donatien P.D., Bennett D.C., Hearing V.J. and Orlow S.J. (1994) Identification of a melanosomal matrix protein encoded by the murine si (silver) locus using ‘organelle scanning’. Proc. Natl. Acad. Sci. U. S. A., 91, 7076–7080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hurbain I., Geerts W.J.C., Boudier T., Marco S., Verkleij A.J., Marks M.S. and Raposo G. (2008) Electron tomography of early melanosomes: implications for melanogenesis and the generation of fibrillar amyloid sheets. Proc. Natl. Acad. Sci. U. S. A., 105, 19726–19731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Watt B., Niel G., Fowler D.M., Hurbain I., Luk K.C., Stayrook S.E., Lemmon M.A., Raposo G., Shorter J., Kelly J.W. et al. (2009) N-terminal domains elicit formation of functional Pmel17 amyloid fibrils. J. Biol. Chem., 284, 35543–35555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. McGlinchey R.P., Shewmaker F., Hu K.N., McPhie P., Tycko R. and Wickner R.B. (2011) Repeat domains of melanosome matrix protein Pmel17 orthologs form amyloid fibrils at the acidic melanosomal pH. J. Biol. Chem., 286, 8385–8393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fowler D.M., Koulov A.V., Alory-Jost C., Marks M.S., Balch W.E. and Kelly J.W. (2005) Functional amyloid formation within mammalian tissue. PLoS Biol., 4, e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Allison W., DuVal M., Nguyen-Phuoc K. and Leighton P. (2017) Reduced abundance and subverted functions of proteins in prion-like diseases: gained functions fascinate but lost functions affect aetiology. Int. J. Mol. Sci., 18, 2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lee Z.H., Hou L., Moellmann G., Kuklinska E., Antol K., Fraser M., Halaban R. and Kwon B.S. (1996) Characterization and subcellular localization of human Pmel 17/silver, a 110-kDa (pre)melanosomal membrane protein associated with 5,6,-dihydroxyindole-2-carboxylic acid (DHICA) converting activity. J. Invest. Dermatol., 106, 605–610. [DOI] [PubMed] [Google Scholar]
- 59. Chakraborty A.K., Platt J.T., Kim K.K., Kwon B.S., Bennett D.C. and Pawelek J.M. (1996) Polymerization of 5,6-dihydroxyindole-2-carboxylic acid to melanin by the pmel 17/silver locus protein. Eur. J. Biochem., 236, 180–188. [DOI] [PubMed] [Google Scholar]
- 60. Michelessi M. and Lindsley K. (2016) Peripheral iridotomy for pigmentary glaucoma. Cochrane Database Syst. Rev., 2016, CD005655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Costa V.P., Gandham S., Spaeth G.L., Moster M.R., Katz L.J., Wilson R.P. and SMITH M. (1994) The effect of Nd-YAG laser iridotomy on pigmentary glaucoma patients: a prospective study. Invest. Ophthalmol. Vis. Sci., 35, 1852. [Google Scholar]
- 62. Brunberg E., Andersson L., Cothran G., Sandberg K., Mikko S. and Lindgren G. (2006) A missense mutation in PMEL17 is associated with the silver coat color in the horse. BMC Genet., 7, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Simon J.D., Peles D., Wakamatsu K. and Ito S. (2009) Current challenges in understanding melanogenesis: bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res., 22, 563–579. [DOI] [PubMed] [Google Scholar]
- 64. Izzotti A., Bagnis A. and Saccà S.C. (2006) The role of oxidative stress in glaucoma. Mutat. Res., 612, 105–114. [DOI] [PubMed] [Google Scholar]
- 65. Saccà S.C. and Izzotti A. (2008) Oxidative stress and glaucoma: injury in the anterior segment of the eye. Prog. Brain Res., 173, 385–407. doi: 10.5772/54653 [DOI] [PubMed] [Google Scholar]
- 66. Ito Y.A. and Walter M.A. (2013) Genetics and environmental stress factor contributions to anterior segment malformations and glaucoma. Glaucoma—Basic and Clinical Aspects. IntechOpen Limited, London, UK. [Google Scholar]
- 67. Porter K.M., Epstein D.L., Liton P.B., Godeau G. and Pellat B. (2012) Up-regulated expression of extracellular matrix remodeling genes in phagocytically challenged trabecular meshwork cells. PLoS One, 7, e34792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Sherwood M.E. and Richardson T.M. (1988) Phagocytosis by trabecular meshwork cells: sequence of events in cats and monkeys. Exp. Eye Res., 46, 881–895. [DOI] [PubMed] [Google Scholar]
- 69. Buller C., Johnson D.H. and Tschumper R.C. (1990) Human trabecular meshwork phagocytosis. Observations in an organ culture system. Invest. Ophthalmol. Vis. Sci., 31, 2156–2163. [PubMed] [Google Scholar]
- 70. Johnson D.H., Richardson T.M. and Epstein D.L. (1989) Trabecular meshwork recovery after phagocytic challenge. Curr. Eye Res., 8, 1121–1130. [DOI] [PubMed] [Google Scholar]
- 71. Shirato S., Murphy C.G., Bloom E., Franse-Carman L., Maglio M.T., Polansky J.R. and Alvarado J.A. (1989) Kinetics of phagocytosis in trabecular meshwork cells. Flow cytometry and morphometry. Invest. Ophthalmol. Vis. Sci., 30, 2499–2511. [PubMed] [Google Scholar]
- 72. Weseley P., Liebmann J., Walsh J.B. and Ritch R. (1992) Lattice degeneration of the retina and the pigment dispersion syndrome. Am. J. Ophthalmol., 114, 539–543. [DOI] [PubMed] [Google Scholar]
- 73. Greenstein V.C., Seiple W., Liebmann J. and Ritch R. (2001) Retinal pigment epithelial dysfunction in patients with pigment dispersion syndrome. Arch. Ophthalmol., 119, 1291. [DOI] [PubMed] [Google Scholar]
- 74. Li H. and Durbin R. (2009) Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. and 1000 Genome Project Data Processing Subgroup (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Falk M.J., Zhang Q., Nakamaru-Ogiso E., Kannabiran C., Fonseca-Kelly Z., Chakarova C., Audo I., Mackay D.S., Zeitz C., Borman A.D. et al. (2012) NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet., 44, 1040–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Consugar M.B., Navarro-Gomez D., Place E.M., Bujakowska K.M., Sousa M.E., Fonseca-Kelly Z.D., Taub D.G., Janessian M., Wang D.Y., Au E.D. et al. (2015) Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet. Med., 17, 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gaier E.D., Boudreault K., Nakata I., Janessian M., Skidd P., DelBono E., Allen K.F., Pasquale L.R., Place E., Cestari D.M. et al. (2017) Diagnostic genetic testing for patients with bilateral optic neuropathy and comparison of clinical features according to OPA1 mutation status. Mol. Vis., 23, 548–560. [PMC free article] [PubMed] [Google Scholar]
- 79. Untergasser A., Cutcutache I., Koressaar T., Ye J., Faircloth B.C., Remm M. and Rozen S.G. (2012) Primer3—new capabilities and interfaces. Nucleic Acids Res., 40, e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L. and Wilson R.K. (2012) VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res., 22, 568–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P. and Cunningham F. (2016) The Ensembl Variant Effect Predictor. Genome Biol., 17, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Acharya M., Huang L., Fleisch V.C., Allison W.T. and Walter M.A. (2011) A complex regulatory network of transcription factors critical for ocular development and disease. Hum. Mol. Genet., 20, 1610–1624. [DOI] [PubMed] [Google Scholar]
- 83. Kaiser D.M., Acharya M., Leighton P.L.A., Wang H., Daude N., Wohlgemuth S., Shi B. and Allison W.T. (2012) Amyloid beta precursor protein and prion protein have a conserved interaction affecting cell adhesion and CNS development. PLoS One, 7, e51305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Hagerman G.F., Noel N.C.L., Cao S.Y., DuVal M.G., Oel A.P. and Allison W.T. (2016) Rapid recovery of visual function associated with blue cone ablation in zebrafish. PLoS One, 11, e0166932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gagnon J.A., Valen E., Thyme S.B., Huang P., Ahkmetova L., Pauli A., Montague T.G., Zimmerman S., Richter C. and Schier A.F. (2014) Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One, 9, e98186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Bustin S.A., Benes V., Garson J.A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M.W., Shipley G.L. et al. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem., 55, 611–622. [DOI] [PubMed] [Google Scholar]
- 87. Leighton P.L.A., Kanyo R., Neil G.J., Pollock N.M. and Allison W.T. (2018) Prion gene paralogs are dispensable for early zebrafish development and have nonadditive roles in seizure susceptibility. J. Biol. Chem., 293, 12576–12592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.