

Short abstract
Previously we reported that a group of inflammatory mediators significantly enhanced resurgent currents in dorsal root ganglion neurons. To understand the underlying intracellular signaling mechanism, we investigated the effects of inhibition of extracellular signal-regulated kinases and protein kinase C on the enhancing effects of inflammatory mediators on resurgent currents in rat dorsal root ganglion neurons. We found that the extracellular signal-regulated kinases inhibitor U0126 completely prevented the enhancing effects of the inflammatory mediators on both Tetrodotoxin-sensitive and Tetrodotoxin-resistant resurgent currents in both small and medium dorsal root ganglion neurons. U0126 substantially reduced repetitive firing in small dorsal root ganglion neurons exposed to inflammatory mediators, consistent with prevention of resurgent current amplitude increases. The protein kinase C inhibitor Bisindolylmaleimide I also showed attenuating effects on resurgent currents, although to a lesser extent compared to extracellular signal-regulated kinases inhibition. These results indicate a critical role of extracellular signal-regulated kinases signaling in modulating resurgent currents and membrane excitability in dorsal root ganglion neurons treated with inflammatory mediators. It is also suggested that targeting extracellular signal-regulated kinases-resurgent currents might be a useful strategy to reduce inflammatory pain.
Keywords: Resurgent currents, inflammatory mediators, dorsal root ganglion, extracellular signal-regulated kinases, protein kinase C, Tetrodotoxin-sensitive, Tetrodotoxin-resistant
Background
Resurgent sodium currents are activated during the repolarization phase of action potentials where classic sodium channels become inactivated.1,2 Therefore, resurgent currents can provide a depolarizing drive for the generation of subsequent action potentials and can contribute to the repetitive firing of neurons. For example, co-expression of Nav1.6 and sodium channel β4 subunits, which increases Nav1.6-mediated resurgent currents, caused spontaneous firing and increased multiple evoked firing in dorsal root ganglion (DRG) neurons compared to expression of Nav1.6 only.3 An Nav1.7 sodium channel mutant associated with paroxysmal extreme pain disorder (PEPD) enhanced resurgent currents in cultured DRG neurons and increased the frequency of action potentials in simulated DRG neurons.4 On the other hand, reduced resurgent currents caused by knocking down sodium channel β4 subunits decreased membrane excitability of cerebellar neurons.5 and of DRG neurons.6 Therefore, modulation of resurgent currents can be a useful approach to modulate neuronal excitability.
Since its first description, resurgent currents have been found in a variety of neuronal tissues in central and peripheral nervous system.7 The Tetrodotoxin–sensitive (TTX-S) resurgent current has been reported in a variety of neuronal tissues including cerebellum,8 perirhinal cortex,9,10 hippocampal area,11 and DRG.12 In addition to this TTX-S resurgent current, we recently found that a Tetrodotoxin-resistant (TTX-R) resurgent current expressed in DRG neurons.13 The TTX-R resurgent currents are slower than TTX-S resurgent currents, are mediated by Nav1.8, and contribute to membrane excitability of DRG neurons in the presence of TTX. We also found that both TTX-S and TTX-R resurgent currents were enhanced by an inflammatory soup of classic inflammatory mediators.13 Inflammatory mediators released during tissue injury can often increase membrane excitability of DRG neurons and cause peripheral sensitization14,15; therefore, it is likely that both TTX-S and TTX-R resurgent currents can contribute to enhanced membrane excitability and increased pain sensitivity induced by inflammatory mediators under inflammatory pain conditions.
It is well known that multiple intracellular signal pathways are activated by inflammatory mediators.16,17 Extracellular signal-regulated kinases (ERK) and protein kinase C (PKC) are two important protein kinases activated by inflammatory mediators.18,19 In the current study, we examined the role of ERK and PKC activation in the enhancing effects of inflammatory mediators on TTX-S and TTX-R resurgent currents in small and medium DRG neurons. We found that the ERK inhibitor U0126 completely prevented the enhancing effects of inflammatory mediators on both TTX-S and TTX-R resurgent currents in small and medium DRG neurons. In addition, to a lesser extent, we found PKC inhibitor Bisindolylmaleimide I (BIM I) also partially prevented the enhancement in resurgent currents caused by the same inflammatory mediators.
Materials and methods
Cell culture
DRG neurons were dissociated from adult rats and were cultured as previously described.13,20 Animal procedures were approved by the Indiana University School of Medicine Institutional Animal Care and Use Committee. Briefly, adult male Sprague Dawley rats weighing 120 to 180 g were rendered unconscious by exposure to CO2 and decapitated. Lumbar DRG (L1-L6) were collected and then incubated in Dulbecco’s Modified Eagle Medium (DMEM) containing collagenase (1 mg/ml) and protease (1 mg/ml) for 40 min at 37°C. The ganglia were then triturated with fire-polished Pasteur pipets in DMEM supplemented with 10% fetal bovine serum (FBS). Dissociated cells were seeded on glass coverslips coated with poly-D-lysine and laminin for 10 min before DMEM/FBS supplemented with 30 ng/ml nerve growth factor was introduced. Cell cultures were maintained in regular 95% air and 5% CO2 in an incubator at 37°C.
Electrophysiology
DRG neurons were recorded after culturing for 16 to 28 h as previously described.13 Briefly, small (<30 µm) and medium (30–45 µm) diameter DRG neurons were chosen for whole-cell patch clamping at room temperature of about 22°C. The extracellular solution for voltage-clamp recordings of sodium currents consisted of: 130 mM NaCl, 30 mM TEA chloride, 1 mM MgCl2, 3 mM KCl, 1 mM CaCl2, 0.05 mM CdCl2, 10 mM HEPES, and 10 mM D-glucose, pH 7.3. DRG neurons were recorded with fire-polished, wax-coated glass patch pipettes (0.6–1.0 MΩ) fabricated from 1.7 mm capillary glass using a Sutter P-97 puller (Novato). The pipette solution for voltage-clamp recordings contained 140 mM CsF, 10 mM NaCl, 1.1 mM EGTA, and 10 mM HEPES, pH 7.3. The junction potential, fast capacitance, slow capacitance, 80% to 90% of series resistance were compensated. Electrical signals were amplified, digitized, stored using a HEKA EPC-10 amplifier, Pulse software (version 8.80; HEKA Elektronik), and a Window-based Pentium IV computer.
For voltage-clamp recordings, DRG neurons were held at −100 mV. Membrane currents were sampled at 20 kHz, filtered online at 5 kHz, and further filtered at 1 kHz digitally in Pulsefit. Leak currents were canceled by a P/5 subtraction protocol. Sodium currents were recorded 10 to 15 min after formation of whole-cell to ensure equilibration between pipette solution and cytoplasmic milieu and to allow adequate rundown of Nav1.9-mediated persistent TTX-R sodium currents. Resurgent currents were recorded using a two voltage step protocol. A 20 ms, +30 mV voltage pulse was first used to inactivation transient sodium currents. Then, a series of repolarization pulses were used for activation of resurgent currents. The repolarization pulses were 400 ms long and decreased from +15 to −70 mV in 5 mV decrements.
Resurgent currents were recorded from small (<30 µm) and medium (35–45 µm) DRG neurons using standard extracellular solution without addition of TTX. The peak resurgent currents were also normalized to peak transient current amplitude, recorded using a standard steady-state inactivation protocol, to calculate the ratio value for resurgent currents. This measure of the ratio resurgent current value allows the size of the resurgent current relative to the peak transient current to be readily compared between treatment groups. For the medium neurons that expressed only fast resurgent currents, single component steady-state inactivation curves were obtained indicating those neurons were only expressing TTX-S fast sodium currents. For the medium neuron that only expressed TTX-R resurgent currents, as well as small DRG neurons, two component steady-state inactivation curves were recorded suggesting that those neurons expressed both TTX-S fast, and TTX-R slow sodium currents. In those neurons, a prepulse inactivation was used to separate the TTX-R and TTX-S current components and to calculate the peak transient TTX-S currents.21
Perforated whole-cell patch clamp was used to conduct current-clamp recording of membrane excitability of DRG neurons. Amphotericin B (240 µg/mL) was included in the pipette solution for membrane perforation. The extracellular solution for current-clamp recordings of sodium currents consisted of 140 mM NaCl, 3 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 10 mM HEPES, pH 7.3. DRG neurons were recorded from using fire-polished, Borosilicate glass patch pipettes (4–6 MΩ). The pipette solution contained 30 mM KCl, 110 mM potassium gluconate, 0.5 mM EGTA, 5 mM HEPES, and 3 mM Mg-ATP, pH 7.3.
Inhibitors of ERK and PKC and control chemicals (U0126, U0124, BIM I, dimethyl sulfoxide (DMSO)) were pre-treated in culture media for 15 min and were continuously kept in the recording chamber. An inflammatory soup including a group of inflammatory mediators (1 µM bradykinin, 10 µM 5-HT, 10 µM histamine, 10 µM PGE2, and 5 µM ATP),13 phorbol myristate acetate (PMA), and 4α-PMA were pretreated for 5 min in the recording chamber before the operation of patch clamping starts. The total time of each coverslip of cells stayed in the recording chamber was less than 90 min.
Results
In this study, we tested the effects of ERK and PKC inhibitors on the enhancing effects of the inflammatory soup on TTX-S and TTX-R resurgent currents. In the small neurons (Figure 1(a)), fast resurgent currents (TTX-S) were not observed and only slow resurgent currents (TTX-R) were recorded in these cells. For the medium neurons, about half expressed only fast resurgent currents (Figure 1(b)). Among the other half, >80% only displayed slow resurgent currents (Figure 1(c)), while <20% expressed a combination of fast and slow resurgent currents (Figure 1(d)). The medium neurons that expressed a combination of fast and slow resurgent currents were excluded from further quantitative analysis in this study due to their small number and the difficulty accurately measuring the fast and slow resurgent current components separately in these cells.
Figure 1.
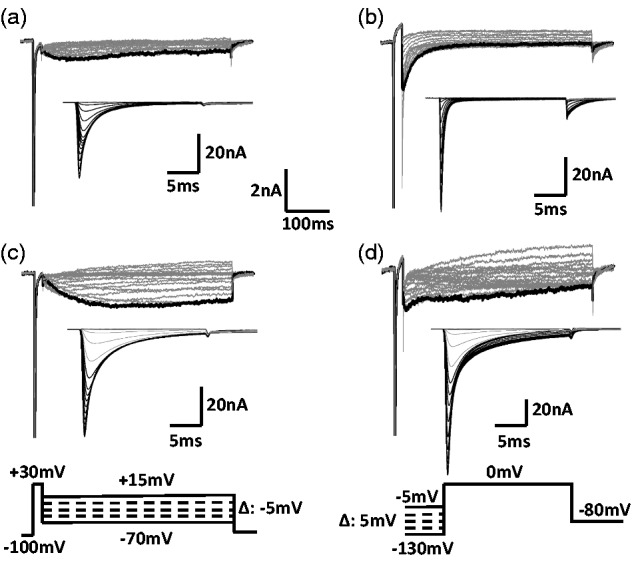
Expression pattern of resurgent currents in DRG neurons. Resurgent and regular sodium current were recorded from small (diameter: 20 to 30 µm) and medium (diameter: 35 to 45 µm) DRG neurons dissociated from adult rats. In all the small neurons, both fast (TTX-S) and slow (TTX-R) regular sodium currents were expressed (a, inset). However, only a slow (TTX-R) resurgent current was recorded from small DRG neurons. In the medium DRG neurons that expressed only fast (TTX-S) regular sodium currents ((b), inset), a fast (TTX-S) resurgent current was recorded (b). In the medium DRG neurons that expressed both fast (TTX-S) and slow (TTX-R) regular sodium currents ((c) and (d) insets), two types of expression pattern of resurgent currents were recorded. In the majority (>80%) of such neurons, a single slow (TTX-R) resurgent current was recorded (c). In other cells (<20%), both a fast (TTX-S) and a slow (TTX-R) components of resurgent currents were recorded (d). The voltage protocol for recording resurgent currents (Bottom, left) includes a series of repolarization voltage steps (+15 to −70 mV decreased by 5-mV steps, 450 ms) from a depolarization step (−100 mV to +30 mV, 20 ms). The regular sodium currents were recorded using a standard steady-state inactivation protocol that depolarized neurons to 0 mV after 500 ms pre-holding at −130 to −5 mV with an increment of 5 mV (Bottom, right). Scales for the regular sodium currents: horizontal scale, 5 ms; vertical scale, 20 nA.
TTX-S resurgent currents in medium DRG neurons
We tested the effects of the ERK inhibitor U0126 on the TTX-S resurgent currents in medium diameter DRG neurons that showed a single fast component in their steady-state inactivation curve which indicated expression of TTX-S sodium channels only (Figure 1(b)). The TTX-S resurgent currents are mostly mediated by Nav1.6, the dominant sodium channel isoform in the medium-to-large DRG neurons.12 To compare the resurgent currents, both ratio resurgent currents (ratio of resurgent currents normalized to peak transient currents) and resurgent current density (resurgent currents normalized to cell capacitance) are shown in Figure 2 and Table 1, respectively. In the presence of U0124 (pretreatment in culture wells for 15 min and maintained in the recording chamber), the inactive control for U0126, the inflammatory soup significantly increased TTX-S, ratio resurgent currents in medium diameter DRG neurons (Figure 2(a), (b), and (e)). This increase was completely prevented by U0126 (Figure 2(c) to (e)). This complete preventative effect suggests that ERK activation during inflammatory soup application is necessary for inflammatory mediators to increase TTX-S resurgent currents in medium diameter DRG neurons. A similar preventative effect of U0126 on the density of TTX-S resurgent currents is shown in Table 1. Moreover, U0126 significantly inhibited baseline density of TTX-S resurgent currents (Table 1). Inflammatory mediators did not produce significant differences in the half activation voltage, half inactivation voltage, and current density of the peak transient TTX-S sodium current under either U0124 or U0126 conditions (Table 1).
Figure 2.

Effects of ERK inhibition on TTX-S resurgent currents in DRG neurons. Representative TTX-S, fast resurgent currents were recorded from medium DRG neurons ((a) to (d)). The resurgent currents were elicited by a series of repolarization voltage steps (+15 to −70 mV decreased by 5-mV steps, 450 ms) from a depolarization step (−100mV to +30 mV, 20 ms). ERK inhibitor U0126 (10 µM) and its inactive control U0124 (10 µM) were pretreated for 15 min in culture medium and were maintained in the recording chamber. (a and b) In the presence of U0124, the TTX-S resurgent currents were enhanced by a group of inflammatory mediators (IMs) including 1 µM bradykinin, 10 µM 5-HT, 10 µM histamine, 10 µM PGE2, and 5 µM ATP. (c and d) U0126 completely prevented the enhancing effects of inflammatory mediators on the TTX-S resurgent currents. Note that U0126 also significantly reduced TTX-S resurgent currents compared to U0124. (e) Statistical data of ratio resurgent currents were compared between groups with or without inflammatory mediators. Ratio resurgent currents were defined as peak resurgent currents normalized to peak TTX-S transient currents (maximal steady-state inactivation currents) in the same cells (the same below). The data were presented as mean ± standard error of the mean. Student’s t-test was used to compare the difference, *P < 0.05 (vs. U0124).
Table 1.
Effects of ERK inhibition on TTX-S resurgent and transient sodium currents in medium neurons.
| U0124 | U0216 | IM+U0124 | IM+U0126 | ||
|---|---|---|---|---|---|
| INaRn (N) | (pA/pF) | 39.6±3.018 (3) | 21.8±2.0#15 (3) | 48.6±3.6*17 (3) | 26.9±3.913 (2) |
| V0.5_mn (N) | (mV) | −38.6±2.218 (3) | −33.2±2.015 (3) | −39.9±1.517 (3) | −37.0±1.913 (2) |
| V0.5_hn (N) | (mV) | −58.7±1.018 (3) | −51.4±3.2#15 (3) | −59.3±1.617 (3) | −59.5±1.813 (2) |
| INaTn (N) | (nA/pF) | 1.13±0.0918 (3) | 0.95±0.0715 (3) | 1.18±0.1217 (3) | 0.95±0.0813 (2) |
Data are expressed as mean ± SEM. Significant level was set at P < 0.05. A level of P < 0.1 is regarded as a high probability event and might indicate a trend change. INaR: resurgent current; n (N): cell number (culture number); V0.5_m: half activation voltage; V0.5_h: half inactivation voltage; INaT: transient current; ANOVA: analysis of variance.
*P < 0.1; Student’s t-test, IM+U0124 versus U0124.
#P < 0.05; post hoc Tukey’s test, one-way ANOVA.
We then tested the effects of PKC inhibitor BIM I on the TTX-S resurgent currents in medium diameter DRG neurons. In the control DMSO condition, the inflammatory soup significantly increased TTX-S resurgent currents in medium diameter DRG neurons (Figure 3(a), (b), and (e), Table 2). This increase seemed to be partially prevented by pretreatment with 1 µM BIM I (Figure 3(c) to (e), Table 2). The partial prevention of this enhancement of TTX-S resurgent currents suggests that PKC activation during inflammatory soup application is involved in the modulation of inflammatory mediators on TTX-S resurgent currents in medium diameter DRG neurons. Inflammatory mediators negatively shifted the half activation voltage of TTX-S currents in the presence of DMSO, the negative control for BIM I. BIM I prevented this shift in the half activation voltage induced by inflammatory mediators (Table 2). On the other hand, inflammatory mediators did not produce significant differences in the half inactivation voltage, or in the current density of the peak transient TTX-S sodium current under either DMSO or BIM I conditions (Table 2).
Figure 3.

Effects of PKC inhibition on TTX-S resurgent currents in DRG neurons. Representative TTX-S, fast resurgent currents were recorded from medium DRG neurons (a–d). The resurgent currents were elicited by the same voltage protocol as described in Figure 1. PKC inhibitor BIM I (1 µM) and its control DMSO (1:1000) were pretreated for 15 min in culture medium and were maintained in the recording chamber. (a and b) In DMSO control condition, the TTX-S resurgent currents were enhanced by inflammatory mediators (IMs). (c and d) BIM I seemed to partially prevent the enhancing effects of inflammatory mediators on the TTX-S resurgent currents. (e) The data were presented as mean ± standard error of the mean. Student’s t-test was used to compare the difference, *P < 0.05 (vs. DMSO). DMSO: dimethyl sulfoxide; BIM I: bisindolylmaleimide I.
Table 2.
Effects of PKC inhibition on TTX-S resurgent and transient sodium currents in medium neurons.
| DMSO | BIM I | IM | IM+BIM I | ||
|---|---|---|---|---|---|
| INaRn (N) | (pA/pF) | 40.3±2.711 (4) | 45.1±6.810 (3) | 55.8±5.5**19 (4) | 52.1±4.29 (4) |
| V0.5_mn (N) | (mV) | −37.4±2.511 (4) | −40.2±2.410 (3) | −44.7±1.9**19 (4) | −38.9±2.99 (4) |
| V0.5_hn (N) | (mV) | −58.9±0.611 (4) | −58.4±1.310 (3) | −58.6±0.816 (4) | −59.9±1.58 (4) |
| INaTn (N) | (nA/pF) | 1.44±0.0811 (4) | 1.37±0.1310 (3) | 1.30±0.0819 (4) | 1.48±0.139 (4) |
INaR: resurgent current; n (N): cell number (culture number); V0.5_m: half activation voltage; V0.5_h: half inactivation voltage; INaT: transient current; DMSO: dimethyl sulfoxide; BIM I: bisindolylmaleimide I. Data are expressed as mean ± SEM.
**P < 0.05; Student’s t-test, IM versus DMSO.
TTX-R resurgent currents in medium DRG neurons
The effects of the ERK inhibitor U0126 on the TTX-R resurgent currents were tested in medium diameter DRG neurons that showed fast and slow components in their steady-state inactivation curve which indicated expression of both TTX-S and TTX-R sodium channels (Figure 1(c)). The TTX-R resurgent currents are mediated by Nav1.8, the dominant TTX-R sodium channel isoform in the small-to-medium diameter DRG neurons.13 In the presence of U0124, the inflammatory soup significantly increased TTX-R resurgent currents in medium diameter DRG neurons (Figure 4(a), (b), and (e), Table 3). This increase was completed blocked by U0126 (Figure 4(c) to (e), Table 3). This complete prevention suggests that ERK activation during inflammatory soup application is necessary for inflammatory mediators to increase TTX-R resurgent currents in medium DRG neurons. Inflammatory mediators did not produce significant differences in the half activation voltage, half inactivation voltage (both TTX-S and TTX-R components), or the current density of the peak transient sodium currents (both TTX-S and TTX-R components) under either U0124 or U0126 conditions (Table 3).
Figure 4.

Effects of ERK inhibition on TTX-R resurgent currents in medium DRG neurons. Representative TTX-R, slow resurgent currents were recorded from medium DRG neurons (a–d). The resurgent currents were elicited by the same voltage protocol as described in Figure 1. ERK inhibitor U0126 (10 µM) and its inactive control U0124 (10 µM) were pretreated for 15 min in culture medium and were maintained in the recording chamber. (a and b) In the presence of U0124, the TTX-R resurgent currents were enhanced by inflammatory mediators (IMs). (c and d) U0126 completely prevented the enhancing effects of inflammatory mediators on the TTX-R resurgent currents. Note that U0126 also reduced TTX-R resurgent currents compared to U0124 insignificantly (P < 0.1). (e) The data were presented as mean ± standard error of the mean. Student’s t-test was used to compare the difference, *P < 0.05 (vs. U0124).
Table 3.
Effects of ERK inhibition on TTX-R resurgent and transient sodium currents in medium neurons.
| U0124 | U0216 | IM+U0124 | IM+U0126 | ||
|---|---|---|---|---|---|
| INaRn (N)n_s+f | (pA/pF) | 13.7±2.014 (3)1 | 16.4±6.613 (3)2 | 19.7±2.3*13 (3)2 | 11.3±2.89 (2)3 |
| V0.5_mn (N) | (mV) | −27.4±2.314 (3) | −24.1±3.713 (3) | −29.6±3.812 (3) | −31.5±2.88 (2) |
| V0.5_h_TTXSn (N) | (mV) | −70.6±1.914 (3) | −64.1±1.612 (3) | −67.0±2.611 (3) | −65.4±2.48 (2) |
| V0.5_h_TTXRn (N) | (mV) | −35.1±2.513 (3) | −35.3±1.211 (3) | −29.7±1.810 (3) | −37.8±2.27 (2) |
| INaT_TTXSn (N) | (nA/pF) | 0.45±0.0614 (3) | 0.44±0.0713 (3) | 0.53±0.0913 (3) | 0.45±0.089 (2) |
| INaT_TTXRn (N) | (nA/pF) | 0.55±0.0614 (3) | 0.62±0.1013 (3) | 0.97±0.1513 (3) | 0.55±0.129 (2) |
Data are expressed as mean ± SEM. Significant level was set at P < 0.05. A level of P < 0.1 is regarded as a high probability event and might indicate a trend change. INaR: resurgent current; n (N): cell number (culture number); n_s + f: number of medium neurons expressing both slow and fast resurgent currents; V0.5_m: half activation voltage; V0.5_h_TTXS: half inactivation voltage of the TTX-S currents; V0.5_h_TTXR: half inactivation voltage of the TTX-R currents; INaT_TTXS: TTX-S transient current; INaT_TTXR: TTX-R transient current.
*P < 0.1; Student’s t-test, IM+U0124 versus U0124.
We next tested the effects of PKC inhibitor BIM I on the TTX-R resurgent currents in medium diameter DRG neurons. In the presence of DMSO, the inflammatory soup increased TTX-R resurgent currents in medium diameter DRG neurons (Figure 5(a), (b), and (e), Table 4). This increase seemed to be partially prevented by BIM I (Figure 5(c) to (e), Table 4). The partial prevention of the enhancing effects suggests PKC activation during inflammatory soup application is involved in the modulation of inflammatory mediators on TTX-R resurgent currents in medium diameter DRG neurons. Inflammatory mediators did not produce significant differences in the half activation voltage, half inactivation voltage (both TTX-S and TTX-R components), or the current density of the peak transient sodium current (both TTX-S and TTX-R components) under either DMSO or BIM I conditions (Table 4) in medium diameter DRG neurons that express both TTX-S and TTX-R sodium currents.
Figure 5.

Effects of PKC inhibition on TTX-R resurgent currents in medium DRG neurons. Representative TTX-R, slow resurgent currents were recorded from medium DRG neurons (a–d). The resurgent currents were elicited by the same voltage protocol as described in Figure 1. PKC inhibitor BIM I (1 µM) and control DMSO (1:1000) were pretreated for 15 min in culture medium and were maintained in the recording chamber. (a and b) In DMSO control condition, the TTX-R resurgent currents were enhanced by inflammatory mediators (IMs). (c and d) BIM I seemed to partially prevent the enhancing effects of inflammatory mediators on the TTX-R resurgent currents. (e) The data were presented as mean ± standard error of the mean. Student’s t-test was used to compare the difference, *P < 0.05 (vs. DMSO). DMSO: dimethyl sulfoxide; BIM I: bisindolylmaleimide I.
Table 4.
Effects of PKC inhibition on TTX-R resurgent and transient sodium currents in medium neurons.
| DMSO | BIM I | IM | IM+BIM I | ||
|---|---|---|---|---|---|
| INaRn (N)n_s+f | (pA/pF) | 10.2±3.411 (4)2 | 12.7±5.07 (2)1 | 24.4±6.3*13 (4)1 | 18.3±5.89 (4)1 |
| V0.5_mn (N) | (mV) | −44.5±3.69 (4) | −41.2±4.27 (2) | −46.0±5.711 (4) | −45.5±3.49 (4) |
| V0.5_h_TTXSn (N) | (mV) | −60.0±0.78 (3) | −62.6±2.27 (2) | −62.3±1.08 (3) | 64.8±2.19 (3) |
| V0.5_h_TTXRn (N) | (mV) | −31.3±1.97 (3) | −30.0±1.87 (2) | −32.3±0.87 (3) | −31.8±0.99 (3) |
| INaT_TTXSn (N) | (nA/pF) | 0.61±0.0711 (4) | 0.56±0.117 (2) | 0.81±0.1513 (2) | 0.56±0.109 (2) |
| INaT_TTXRn (N) | (nA/pF) | 0.75±0.1011 (4) | 0.78±0.207 (2) | 0.75±0.2013 (4) | 0.65±0.099 (4) |
Data are expressed as mean ± SEM. INaR: resurgent current; n (N): cell number (culture number); n_s + f: number of medium neurons expressing both slow and fast resurgent currents; V0.5_m: half activation voltage; V0.5_h_TTXS: half inactivation voltage of the TTX-S currents; V0.5_h_TTXR: half inactivation voltage of the TTX-R currents; INaT_TTXS: TTX-S transient current; INaT_TTXR: TTX-R transient current; DMSO: dimethyl sulfoxide; BIM I: bisindolylmaleimide I.
*P < 0.1; Student’s t-test, IM versus DMSO.
TTX-R resurgent currents in small DRG neurons
The effects of the ERK inhibitor U0126 on the TTX-R resurgent currents were also tested in small diameter DRG neurons, all of which expressed fast and slow components in their steady-state inactivation curves but only expressed TTX-R resurgent currents (Figure 1(a)). TTX-S resurgent currents were not observed in small diameter DRG neurons in the present study. In the presence of U0124, the inflammatory soup significantly increased TTX-R resurgent currents in small diameter DRG neurons (Figure 6(a), (b), and (e), Table 5). This increase was completely blocked by U0126 (Figure 6(c) to (e), Table 5). The complete prevention of the enhancing effects suggests ERK activation during inflammatory soup application is necessary for inflammatory mediators to increase TTX-R resurgent currents in small diameter DRG neurons. Inflammatory mediators significantly shifted the half activation voltage negatively in the presence of U0124 (Table 5). U0126 prevented the effect of inflammatory mediators on the half activation voltage (Table 5). On the other hand, inflammatory mediators did not produce significant differences in the half inactivation voltage (both TTX-S and TTX-R components), or the current density of the peak transient sodium currents (both TTX-S and TTX-R components) under either U0124 or U0126 conditions (Table 5).
Figure 6.

Effects of ERK inhibition on TTX-R resurgent currents in small DRG neurons. Representative TTX-R, slow resurgent currents were recorded from small DRG neurons (a–d). The resurgent currents were elicited by the same voltage protocol as described in Figure 1. DRG neurons-exhibited slow resurgent currents (TTX-R) were chosen to study. ERK inhibitor U0126 (10 µM) and its inactive control U0124 (10 µM) were pretreated for 15 min in culture medium and were maintained in the recording chamber. (a and b) In the presence of U0124, the TTX-R resurgent currents were enhanced by inflammatory mediators (IMs). (c and d) U0126 completely prevented the enhancing effects of inflammatory mediators on the TTX-R resurgent currents. (e) The data were presented as mean ± standard error of the mean. Student’s t-test was used to compare the difference, *P < 0.05 (vs. U0124).
Table 5.
Effects of ERK inhibition on TTX-R resurgent and transient sodium currents in small neurons.
| U0124 | U0216 | IM+U0124 | IM+U0126 | ||
|---|---|---|---|---|---|
| INaRn (N) | (pA/pF) | 15.2±2.810 (3) | 9.3±2.210 (3) | 30.1±6.9**7 (2) | 20.0±5.99 (2) |
| V0.5_mn (N) | (mV) | −22.3±2.710 (3) | −21.5±2.910 (3) | −30.6±3.3*7 (2) | −24.1±3.29 (2) |
| V0.5_h_TTXSn (N) | (mV) | −70.6±2.410 (3) | −72.1±1.68 (3) | −67.5±1.07 (2) | −68.7±1.09 (2) |
| V0.5_h_TTXRn (N) | (mV) | −34.3±1.910 (3) | −37.9±2.210 (3) | −37.7±1.17 (2) | −40.6±2.69 (2) |
| INaT_TTXSn (N) | (nA/pF) | 0.86±0.1610 (3) | 0.51±0.1110 (3) | 1.24±0.217 (2) | 1.13±0.289 (2) |
| INaT_TTXRn (N) | (nA/pF) | 1.27±0.2310 (3) | 1.21±0.1710 (3) | 1.47±0.247 (2) | 1.25±0.269 (2) |
Data are expressed as mean ± SEM. INaR: resurgent current; n (N): cell number (culture number); V0.5_m: half activation voltage; V0.5_h_TTXS: half inactivation voltage of the TTX-S currents; V0.5_h_TTXR: half inactivation voltage of the TTX-R currents; INaT_TTXS: TTX-S transient current; INaT_TTXR: TTX-R transient current.
*P < 0.1; Student’s t-test, IM+U0124 versus U0124.
**P < 0.05; Student’s t-test, IM+U0124 versus U0124.
We next tested the effects of PKC inhibitor BIM I on the TTX-R resurgent currents in small diameter DRG neurons. In the presence of DMSO, the inflammatory soup significantly increased TTX-R resurgent currents in small diameter DRG neurons (Figure 7(a), (b), and (e), Table 6). This increase was completely prevented by BIM I (Figure 7(c) to (e), Table 6). The complete prevention of the enhancing effects suggests PKC activation during inflammatory soup application is necessary in the modulation of inflammatory mediators on TTX-R resurgent currents in small diameter DRG neurons. Inflammatory mediators did not produce significant differences in the half activation voltage, half inactivation voltage (both TTX-S and TTX-R components), or the current density of the peak transient sodium current (both TTX-S and TTX-R components) under either DMSO or BIM I conditions (Table 6). However, the lack of statistical difference in peak transient, TTX-S currents between U0126 and inflammatory mediators (IM)+U0126 groups might be due to a relatively small sample size compared to the variability of current amplitude among cells.
Figure 7.

Effects of PKC inhibition on TTX-R resurgent currents in small DRG neurons. Representative TTX-R, slow resurgent currents were recorded from small DRG neurons (a–d). The resurgent currents were elicited by the same voltage protocol as described in Figure 1. PKC inhibitor BIM I (1 µM) and control DMSO (1:1000) were pretreated for 15 min in culture medium and were maintained in the recording chamber. (a and b) In DMSO control condition, the TTX-R resurgent currents were enhanced by inflammatory mediators (IMs). (c and d) BIM I completely prevented the enhancing effects of inflammatory mediators on the TTX-R resurgent currents. (e) The data were presented as mean ± standard error of the mean. Student’s t-test was used to compare the difference, **P < 0.01 (vs. DMSO). DMSO: dimethyl sulfoxide; BIM I: bisindolylmaleimide I.
Table 6.
Effects of PKC inhibition on TTX-R resurgent and transient sodium currents in small neurons.
| DMSO | BIM I | IM | IM+BIM I | ||
|---|---|---|---|---|---|
| INaRn (N) | (pA/pF) | 32.8±8.812 (3) | 28.4±9.58 (3) | 64.7±11.9**8 (2) | 28.8±8.910 (2) |
| V0.5_mn (N) | (mV) | −31.9±4.010 (3) | −39.0±3.57 (3) | −37.1±4.88 (2) | 35.6±2.29 (2) |
| V0.5_h_TTXSn (N) | (mV) | −72.4±1.98 (3) | −72.6±1.27 (3) | −72.1±1.28 (2) | 73.8±1.29 (2) |
| V0.5_h_TTXRn (N) | (mV) | −35.0±1.410 (3) | −34.3±0.97 (3) | −35.8±1.18 (2) | 35.9±0.99 (2) |
| INaT_TTXSn (N) | (nA/pF) | 1.55±0.2312 (3) | 1.09±0.238 (3) | 1.67±0.258 (2) | 1.30±0.2610 (2) |
| INaT_TTXRn (N) | (nA/pF) | 1.22±0.1612 (3) | 1.01±0.128 (3) | 1.34±0.228 (2) | 1.05±0.1910 (2) |
Data are expressed as mean±SEM. INaR: resurgent current; n (N): cell number (culture number); V0.5_m: half activation voltage; V0.5_h_TTXS: half inactivation voltage of the TTX-S currents; V0.5_h_TTXR: half inactivation voltage of the TTX-R currents; INaT_TTXS: TTX-S transient current; INaT_TTXR: TTX-R transient current; DMSO: dimethyl sulfoxide; BIM I: bisindolylmaleimide I.
**P < 0.05; Student’s t-test, IM versus DMSO.
Repetitive action potential firing in small DRG neurons
The effects of the ERK inhibitor U0126 on repetitive action potential firing was tested in small diameter DRG neuron in the presence of the inflammatory mediator soup. Compared to U0124, U0126 significantly reduced the number of action potentials triggered by a suprathreshold current injection (3× current threshold) (Figure 8(a) to (c)). However, U0126 did not change the resting membrane potentials (Figure 8(d)) or rheobases (Figure 8(e)) significantly.
Figure 8.

Effects of ERK inhibition on repetitive firing of small DRG neurons in the presence of inflammatory mediators. Small DRG neurons were recorded under whole-cell current clamp conditions. A three-time rheobase current was injected and the representative responses from neurons in the presence of U0124 and U0126 were shown (a and b). U0126 significantly reduced the number of action potentials (APs) (a–c) without changing rest membrane potential (RMP) (d) or rheobase (e), significantly. The data were presented as mean ± standard error of the mean. The data were from 20 neurons and 5 cell cultures for each group. Student’s t-test was used to compare the difference, *P < 0.05.
Effects of PKC activation on resurgent currents in DRG neurons
Our data using the PKC inhibitor BIM I indicate that PKC activation is involved in the enhancing effects of inflammatory mediators on resurgent currents in DRG neurons. To directly test if PKC activation alone can modulate resurgent currents, we tested the effects of the PKC activator (PMA) and compared PMA to its inactive control 4α-PMA. As shown in Tables 7 to 9, PMA did not change density of TTX-S (medium neurons) or TTX-R (both medium and small neurons) resurgent currents significantly. However, PMA significantly inhibited peak transient sodium currents in medium DRG neurons that express TTX-S currents only (Table 7). On the other hand, PMA did not significantly change peak transient currents (both TTX-S and TTX-R) in either medium or small DRG neurons that express both TTX-S and TTX-R currents (Tables 8 and 9). In addition, PMA did not significantly change the half activation voltage or half inactivation voltage for both TTX-S and TTX-R currents in the three groups of DRG neurons tested (Tables 7 to 9).
Table 7.
Effects of PMA on TTX-S resurgent and transient sodium currents in medium neurons.
| 4α-PMA | PMA | ||
|---|---|---|---|
| INaRn (N) | (pA/pF) | 39.7±4.69 (3) | 43.5±4.112 (3) |
| V0.5_mn (N) | (mV) | −35.8±3.210 (3) | −41.7±1.812 (3) |
| V0.5_hn (N) | (mV) | −50.4±2.811 (3) | −55.7±2.811 (3) |
| INaTn (N) | (nA/pF) | 1.61±0.1211 (3) | 1.11±0.10*12 (3) |
Data are expressed as mean ± SEM. INaR: resurgent current; n (N): cell number (culture number); V0.5_m: half activation voltage; V0.5_h: half inactivation voltage; INaT: transient current; PMA: phorbol myristate acetate.
*P < 0.01 (PMA vs. 4α-PMA); Student’s t-test.
Table 9.
Effects of PMA on TTX-R resurgent and transient sodium currents in small neurons.
| 4α-PMA | PMA | ||
|---|---|---|---|
| INaRn (N) | (pA/pF) | 15.7±3.712 (3) | 14.8±1.811 (3) |
| V0.5_mn (N) | (mV) | −22.9±0.812 (3) | −23.1±0.612 (3) |
| V0.5_h_TTXSn (N) | (mV) | −64.9±0.812 (3) | −64.8±0.712 (3) |
| V0.5_h_TTXRn (N) | (mV) | −33.7±0.512 (3) | −37.3±1.512 (3) |
| INaT_TTXSn (N) | (nA/pF) | 1.06±0.1412 (3) | 1.42±0.2212 (3) |
| INaT_TTXRn (N) | (nA/pF) | 0.87±0.1212 (3) | 0.59±0.1312 (3) |
Data are expressed as mean±SEM. INaR: resurgent current; n (N): cell number (culture number); V0.5_m: half activation voltage; V0.5_h_TTXS: half inactivation voltage of the TTX-S currents; V0.5_h_TTXR: half inactivation voltage of the TTX-R currents; INaT_TTXS: TTX-S transient current; INaT_TTXR: TTX-R transient current; PMA: phorbol myristate acetate.
Table 8.
Effects of PMA on TTX-R resurgent and transient sodium currents in medium neurons.
| 4α-PMA | PMA | ||
|---|---|---|---|
| INaRn (N)n_s+f | (pA/pF) | 21.6±3.110 (4)1 | 22.2±3.16 (3)1 |
| V0.5_mn (N) | (mV) | −42.6±6.69 (3) | −37.8±4.67 (3) |
| V0.5_h_TTXSn (N) | (mV) | −55.0±3.312 (4) | −61.7±3.08 (3) |
| V0.5_h_TTXRn (N) | (mV) | −28.5±2.212 (4) | −30.2±1.88 (3) |
| INaT_TTXSn (N) | (nA/pF) | 1.58±0.2112 (4) | 1.07±0.198 (3) |
| INaT_TTXRn (N) | (nA/pF) | 1.20±0.1112 (4) | 0.92±0.198 (3) |
Data are expressed as mean ± SEM. INaR: resurgent current; n (N): cell number (culture number); n_s + f: number of medium neurons expressing both slow and fast resurgent currents; V0.5_m: half activation voltage; V0.5_h_TTXS: half inactivation voltage of the TTX-S currents; V0.5_h_TTXR: half inactivation voltage of the TTX-R currents; INaT_TTXS: TTX-S transient current; INaT_TTXR: TTX-R transient current; PMA: phorbol myristate acetate.
Discussion
Our results show that ERK inhibition reduces baseline TTX-S resurgent currents in medium diameter DRG neurons and completely prevented the enhancing effects of inflammatory mediators on TTX-S resurgent currents in medium diameter DRG neurons, on TTX-R resurgent currents in medium diameter DRG neurons, and on TTX-R resurgent currents in small diameter DRG neurons. Those results suggest that ERK activation mediates the enhancing effects of inflammatory mediators on resurgent currents in DRG neurons. On the other hand, PKC inhibition only partially prevented the enhancing effects of inflammatory mediators on TTX-S and TTX-R resurgent currents in medium diameter DRG neurons. However, PKC inhibition completely prevented the enhancing effects of inflammatory mediators on TTX-R resurgent currents in small diameter DRG neurons. These results suggest that while PKC activation is also likely involved in the enhancing effects of inflammatory mediators on resurgent currents in DRG neurons, PKC activation plays a greater role in small diameter neurons than in medium diameter neurons. It is hypothesized that ERK activation leading to enhanced resurgent currents plays an important role in inflammatory and other chronic pain conditions. Assuming this hypothesis is correct in some chronic pain conditions, targeting ERK enhancement of resurgent currents could be a useful approach to treat chronic pain.
Possible mechanisms of ERK and PKC modulating resurgent currents
Our current studies did not suggest whether the effects of ERK and PKC inhibition were via a direct effect of protein kinases phosphorylating sodium channels or if they were mediated by intermediate molecules in DRG neurons. However, direct modulation of DRG sodium channels by ERK and PKC has been reported. Stamboulian et al. reported a direct phosphorylation and modulation of Nav1.7 by pERK1/2.22 They showed that pERK1 phosphorylated specific residues within intracellular loop 1 (L1) of Nav1.7, that inhibition of pERK1/2 caused a depolarizing shift of activation and fast inactivation of Nav1.7, and that mutation of these phosphoacceptor sites abrogated the effects of pERK1/2 on this channel. The same group of researchers also showed that activated p38 mitogen-activated protein kinase directly phosphorylated Nav1.6 and Nav1.8 sodium channels and regulated their current densities.23,24 Although no one has demonstrated a direct phosphorylation of ERK on Nav1.6 and Nav1.8, which is most relevant in this study, such a possibility can be suggested based on the results observed in the present research.
A direct phosphorylation by PKC of sodium channels has been well shown.25,26 We reported recently that the PKC activator PMA can phosphorylate a conserved PKC phosphorylation site of Nav1.7 in HEK293 cells, increasing Nav1.7-mediated resurgent currents without affecting peak transient current density.27 However, in this study in DRG neurons, we found that PMA did not change resurgent currents significantly in medium or small neurons. On the other hand, PMA significantly decreased peak transient TTX-S current in medium neurons that express only TTX-S currents. Those results suggest that different PKC isoforms might be expressed in HEK293 and DRG neurons, and activation of different PKC isoforms might have distinct effects on sodium currents. Basal PKC and activated PKC by PMA or inflammatory mediators used in this study might be different in DRG neurons than in HEK293 cells. It is also important to note that while PEPD mutations in Nav1.7 can enhance resurgent currents, the predominant isoform underlying the TTX-S resurgent current in medium diameter DRG neurons is likely to be dependent on Nav1.6, and Nav1.6, Nav1.7, and Nav1.8 channels are likely to be differentially regulated by PKC. Nevertheless, the effects of PKC inhibition in this study could be caused by a direct effect on sodium channels. On the other hand, an indirect effect of PKC on sodium channels cannot be excluded, especially as a possibility of PKC activation causing ERK activation can be envisioned. For instance, our studies found that both PKC and ERK inhibitors completely prevented inflammatory mediator–induced increase in resurgent currents in small DRG neurons. This can be explained by that PKC activity is necessary for ERK activation and subsequent increase of resurgent currents induced by inflammatory mediators in the small DRG neurons. On the other hand, it is also possible that a double activation of PKC and ERK is required to increase resurgent currents in the small DRG neurons.
An inflammatory soup consisting of five inflammatory mediators was used in this study in order to mimic a collective effect of inflammatory mediators. However, the complex components of the inflammatory soup complicate the identification of an explanation for the precise mechanism on ERK and PKC action, especially for the identification of a bridge between inflammatory mediators and PKC and ERK action. For example, many receptors have been reported to link inflammatory mediators and PKC/ERK activation in DRG neurons. EP1-PKC and EP4-ERK pathways contributed to the PGE2-induced TRPV1 externalization.28 In Sham-operated mice, bradykinin-induced nociceptive responses were associated with B2-ERK pathway in unmyelinated DRG neurons.29 In contrast, these behaviors were associated with B1-ERK pathway in myelinated DRG neurons in nerve-injured animals. In addition, 5-HT2-PKC and H1R-PKC pathways contributed to the enhanced TRPV1 function induced by serotonin and histamine, respectively.30–32 Therefore, future studies using single inflammatory mediators will be needed to determine the contribution of specific pathways to the enhancing effects of inflammatory mediators on resurgent currents in DRG neurons.
ERK inhibition, sodium channel gating, and membrane excitability of DRG neurons
A previous study has found that ERK inhibition caused (1) a depolarizing shift of activation and inactivation of Nav1.7 expressed in HEK293 cells and (2) membrane hyperpolarization and decreased firing frequency of action potentials in DRG neurons.22 In the present study, we found that ERK inhibition caused (1) a depolarizing shift of inactivation of TTX-S currents in medium DRG neurons expressing TTX-S currents only and (2) decreased firing frequency of action potentials in DRG neurons in the presence of a group of inflammatory mediators. The partial similar effects of ERK inhibition between the previous and the current studies suggest that ERK inhibition reduces membrane excitability through modulating sodium channels in DRG neurons. However, the differences in cell types or subtypes (HEK 293 vs. DRG neurons; different subpopulation of DRG neurons), microenvironment (in the presence or absence of inflammatory mediators), sodium channel isoform expression, and/or recording methods of patch clamping (conventional vs. perforated) likely contribute to the differences in effects of ERK inhibition observed between the previous and the current study. The results from this study suggest that the inhibitory effects of ERK inhibition on membrane excitability of DRG neurons in the presence of inflammatory mediators are mediated by the inhibitory effects of ERK inhibition on resurgent currents.
ERK activation, resurgent currents, and inflammatory pain
Our previous and current studies demonstrated that inflammatory mediators enhance TTX-S and TTX-R resurgent currents in small-to-medium diameter DRG neurons.13 The enhancing effects of inflammatory mediators on DRG resurgent currents are dependent on ERK activation. It has been well documented that ERK activation, including ERK activation at the DRG level, plays an important role in development of inflammatory and neuropathic pain.18,19 In fact, our recent study found that TTX-S resurgent currents in medium diameter DRG neurons were up-regulated in a low back inflammatory pain model: local injection of zymosan to DRG (LID).6 Blockade of this increase in TTX-S resurgent currents by in vivo knockdown of the β4 subunit in DRG completely reversed the LID-induced mechanical hyperalgesia and mechanical allodynia.6 It would be interesting for future studies to test whether the LID-induced increase in pain behavior is caused by ERK-dependent increase in DRG resurgent currents. It would be also interesting to test this hypothesis in other inflammatory pain models or other chronic pain models that have a significant inflammatory component.
Acknowledgment
The authors thank Mr. Wentong Zhang for his contribution to cell culture.
Author contributions
TRC, ESN, JLK, JSM, KLK, and ZYT conceived the study; ZYT designed the experiments; BW and ZYT performed the experiments; BW and ZYT analyzed the data; and TRC, ESN, JLK, JSM, KlK, and ZYT wrote the manuscript.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: KLK and JSM are employees of Eli Lilly and Co. JLK and ESN were employees of Eli Lilly at the time the research was performed. The remaining authors declare no competing financial interests.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: ZYT was supported by Indiana Spinal Cord and Brain Injury Research Fund Grants (2015 and 2017) and by a Showalter Trustee Fund Research Grant (068650-00002B). TRC was supported by a Lilly Research Award and National Institutes of Health grants (NS053422 and NS055860).
References
- 1.Theile JW, Cummins TR. Recent developments regarding voltage-gated sodium channel blockers for the treatment of inherited and acquired neuropathic pain syndromes. Front Pharmacol 2011; 2: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lewis AH, Raman IM. Resurgent current of voltage-gated Na+ channels. J Physiol 2014; 592: 4825–4838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barbosa C, Tan ZY, Wang R, Xie W, Strong JA, Patel RR, Vasko MR, Zhang JM, Cummins TR. Navbeta4 regulates fast resurgent sodium currents and excitability in sensory neurons. Mol Pain. 2015; 11: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jarecki BW, Piekarz AD, Jackson JO, 2nd, Cummins TR. Human voltage-gated sodium channel mutations that cause inherited neuronal and muscle channelopathies increase resurgent sodium currents. J Clin Invest 2010; 120: 369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bant JS, Raman IM. Control of transient, resurgent, and persistent current by open-channel block by Na channel beta4 in cultured cerebellar granule neurons. Proc Natl Acad Sci U S A 2010; 107: 12357–12362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie W, Tan ZY, Barbosa C, Strong JA, Cummins TR, Zhang JM. Upregulation of the sodium channel NaVbeta4 subunit and its contributions to mechanical hypersensitivity and neuronal hyperexcitability in a rat model of radicular pain induced by local dorsal root ganglion inflammation. Pain 2016; 157: 879–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci 1997; 17: 4517–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Afshari FS, Ptak K, Khaliq ZM, Grieco TM, Slater NT, McCrimmon DR, Raman IM. Resurgent Na currents in four classes of neurons of the cerebellum. J Neurophysiol 2004; 92: 2831–2843. [DOI] [PubMed] [Google Scholar]
- 9.Hargus NJ, Nigam A, Bertram EH, 3rd, Patel MK. Evidence for a role of Nav1.6 in facilitating increases in neuronal hyperexcitability during epileptogenesis. J Neurophysiol 2013; 110: 1144–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nigro MJ, Quattrocolo G, Magistretti J. Distinct developmental patterns in the expression of transient, persistent, and resurgent Na+ currents in entorhinal cortex layer-II neurons. Brain Res 2012; 1463: 30–41. [DOI] [PubMed] [Google Scholar]
- 11.Castelli L, Nigro MJ, Magistretti J. Analysis of resurgent sodium-current expression in rat parahippocampal cortices and hippocampal formation. Brain Res 2007; 1163: 44–55. [DOI] [PubMed] [Google Scholar]
- 12.Cummins TR, Dib-Hajj SD, Herzog RI, Waxman SG. Nav1.6 channels generate resurgent sodium currents in spinal sensory neurons. FEBS Lett 2005; 579: 2166–2170. [DOI] [PubMed] [Google Scholar]
- 13.Tan ZY, Piekarz AD, Priest BT, Knopp KL, Krajewski JL, McDermott JS, Nisenbaum ES, Cummins TR. Tetrodotoxin-resistant sodium channels in sensory neurons generate slow resurgent currents that are enhanced by inflammatory mediators. J Neurosci 2014; 34: 7190–7197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richardson JD, Vasko MR. Cellular mechanisms of neurogenic inflammation. J Pharmacol Exp Ther 2002; 302: 839–845. [DOI] [PubMed] [Google Scholar]
- 15.Song XJ, Zhang JM, Hu SJ, LaMotte RH. Somata of nerve-injured sensory neurons exhibit enhanced responses to inflammatory mediators. Pain 2003; 104: 701–709. [DOI] [PubMed] [Google Scholar]
- 16.Petho G, Reeh PW. Sensory and signaling mechanisms of bradykinin, eicosanoids, platelet-activating factor, and nitric oxide in peripheral nociceptors. Physiol Rev 2012; 92: 1699–1775. [DOI] [PubMed] [Google Scholar]
- 17.Cheng JK, Ji RR. Intracellular signaling in primary sensory neurons and persistent pain. Neurochem Res 2008; 33: 1970–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cruz CD, Cruz F. The ERK 1 and 2 pathway in the nervous system: from basic aspects to possible clinical applications in pain and visceral dysfunction. Curr Neuropharmacol 2007; 5: 244–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Obata K, Noguchi K. MAPK activation in nociceptive neurons and pain hypersensitivity. Life Sci 2004; 74: 2643–2653. [DOI] [PubMed] [Google Scholar]
- 20.Cummins TR, Black JA, Dib-Hajj SD, Waxman SG. Glial-derived neurotrophic factor upregulates expression of functional SNS and NaN sodium channels and their currents in axotomized dorsal root ganglion neurons. J Neurosci 2000; 20: 8754–8761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cummins TR, Waxman SG. Downregulation of tetrodotoxin-resistant sodium currents and upregulation of a rapidly repriming tetrodotoxin-sensitive sodium current in small spinal sensory neurons after nerve injury. J Neurosci 1997; 17: 3503–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stamboulian S, Choi JS, Ahn HS, Chang YW, Tyrrell L, Black JA, Waxman SG, Dib-Hajj SD. ERK1/2 mitogen-activated protein kinase phosphorylates sodium channel Na(v)1.7 and alters its gating properties. J Neurosci 2010; 30: 1637–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hudmon A, Choi JS, Tyrrell L, Black JA, Rush AM, Waxman SG, Dib-Hajj SD. Phosphorylation of sodium channel Na(v)1.8 by p38 mitogen-activated protein kinase increases current density in dorsal root ganglion neurons. J Neurosci 2008; 28: 3190–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wittmack EK, Rush AM, Hudmon A, Waxman SG, Dib-Hajj SD. Voltage-gated sodium channel Nav1.6 is modulated by p38 mitogen-activated protein kinase. J Neurosci 2005; 25: 6621–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Catterall WA. Voltage-gated sodium channels at 60: structure, function and pathophysiology. J Physiol 2012; 590: 2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dib-Hajj SD, Cummins TR, Black JA, Waxman SG. Sodium channels in normal and pathological pain. Annu Rev Neurosci 2010; 33: 325–347. [DOI] [PubMed] [Google Scholar]
- 27.Tan ZY, Priest BT, Krajewski JL, Knopp KL, Nisenbaum ES, Cummins TR. Protein kinase C enhances human sodium channel hNav1.7 resurgent currents via a serine residue in the domain III-IV linker. FEBS Lett 2014; 588: 3964–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma W, St-Jacques B, Rudakou U, Kim YN. Stimulating TRPV1 externalization and synthesis in dorsal root ganglion neurons contributes to PGE2 potentiation of TRPV1 activity and nociceptor sensitization. Eur J Pain 2017; 21: 575–593. [DOI] [PubMed] [Google Scholar]
- 29.Rashid MH, Inoue M, Matsumoto M, Ueda H. Switching of bradykinin-mediated nociception following partial sciatic nerve injury in mice. J Pharmacol Exp Ther 2003; 308: 1158–1164. [DOI] [PubMed] [Google Scholar]
- 30.Kajihara Y, Murakami M, Imagawa T, Otsuguro K, Ito S, Ohta T. Histamine potentiates acid-induced responses mediating transient receptor potential V1 in mouse primary sensory neurons. Neurosci 2010; 166: 292–304. DOI: 10.1016/j.neuroscience.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 31.Ohta T, Ikemi Y, Murakami M, Imagawa T, Otsuguro K, Ito S. Potentiation of transient receptor potential V1 functions by the activation of metabotropic 5-HT receptors in rat primary sensory neurons. J Physiol 2006; 576: 809–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sugiuar T, Bielefeldt K, Gebhart GF. TRPV1 function in mouse colon sensory neurons is enhanced by metabotropic 5-hydroxytryptamine receptor activation. J Neurosci 2004; 24: 9521–9530. [DOI] [PMC free article] [PubMed] [Google Scholar]