

Introduction:
Cystic fibrosis (CF) is the most common autosomal recessive genetic disorder in Caucasians2, 3 and is also one of the most lethal.4 It is caused by a mutation in the gene coding for the CF transmembrane conductance regulator (CFTR) protein on chromosome 7.3 With advances in medical care, the life expectancy of patients with CF has increased from 16 years in 1970 to 47.7 years in 2016.5 Given this improved longevity, the extrapulmonary manifestations related to CF are identified more often. While the leading cause of death in CF patients continues to be from complications of pulmonary disease, followed by complications of lung transplantation, liver disease has been identified as the third most common and the most important non-pulmonary cause of death in these patients.5 In the current era of CF management, the prevalence of cystic fibrosis liver disease (CFLD) has been described to approach 40% in patients with CF6, 7 and accounts for 2–5% of overall CF mortality.8–10
CFLD is well described in pediatric patients and studies have demonstrated that most patients present with evidence of CFLD before puberty.7, 11 A large retrospective study by Boelle et al evaluating 3,328 CF patients born after 1985 demonstrated that the incidence of CFLD increased by approximately 1% each year after the age of 5 and reached 10% by the age of 30.12 Risk factors identified for CFLD in this study included male sex, CFTR F508del homozygosity, and history of meconium ileus.12 With improved life expectancy, a larger proportion of patients with CF now consist of adults over the age of 18 (52% in 2016 compared to just 29% in 1986).5 A recent study involving a longitudinal cohort of adult patients followed over a median of 24.5 years at the National Institutes of Health (NIH) Clinical Center in the United States (US) demonstrated that adult onset CFLD occurred at a median age of 37 in patients who did not have evidence of CFLD during childhood.1
Pathophysiology:
The CFTR gene encodes for a protein that is found on the apical surface of cholangiocytes and gallbladder epithelia (Figure 1).2, 13 This CFTR protein is responsible for regulating the fluid and electrolyte content of bile by increasing apical biliary chloride secretion to create a transmembrane gradient of Cl- which can then be used to increase bile acid independent bile flow via the Cl-/HCO3- exchanger along with passive movement of water.2, 14 This leads to increased fluidity of bile as well as alkalinisation of the bile. Thus, mutations in the CFTR protein can lead to impaired secretion of Cl- and thus lead to the development of viscous bile with reduced flow and alkalinity.14, 15 While the mechanism of the development of cirrhosis in CF is still unclear, it is felt that these changes can lead to stagnation of the bile which leads to accumulation of toxic bile acids and increased infections. As such, liver biopsies early in pediatric patients have demonstrated mucus-plugging in cholangiocytes.16 These changes can lead to periductal inflammation, damage to cholangiocytes, bile duct proliferation, and periportal fibrosis (Figure 2a).14 For this reason, CFLD typically presents as a cholestatic liver disease with the typical, well described hepatic lesion of focal biliary cirrhosis, particularly in the pediatric population and in patients with more severe mutations (Figure 2b).2, 17 In addition to these changes, a recent study demonstrated that CFTR regulates toll-like receptor 4 (TLR-4)-dependent inflammatory responses by inhibiting Rous sarcoma oncogene cellular homologue (Src) activity, and mutations in CFTR lead to self-activation of Src leading to increased inflammatory cytokines and disruption of the epithelial barrier.9, 18 This in turns can lead to translocation of bacteria into the portal circulation, hepatic inflammation, and fibrosis.19 In addition to focal biliary cirrhosis, many patients with CF can present with hepatic steatosis (Figure 2c). Traditionally, hepatic steatosis in the CF population has been associated with nutritional deficiencies, particularly essential fatty acids.20 More recently it has been described that steatosis in patients with CF is multifactorial and includes etiologies similar to those in the general population such as non-alcoholic fatty liver disease (NAFLD) and alcoholic liver disease. A questionnaire-based study performed in the United Kingdom found that 83% of patients with CF drink alcohol, with 13% falling in the excessive or at-risk category, though this was less than the general population which was found to be 23%.21 Also, patients with CF are not immune to the obesity epidemic. With improvements in nutritional support, according to the 2016 CF foundation annual report, the median BMI for adult patients with CF has increased by 3 points over the past 20 years. Of adult patients, 18.3% are obese with body mass index greater than 30, with the majority having CF mutations other than F508del.5 In addition, a study characterizing adult patients with CF demonstrated that these patients also develop other metabolic risk factors typically associated with NAFLD, including diabetes mellitus or impaired glucose tolerance and hypertriglyceridemia, particularly with increasing age.22 It remains unclear whether or not these metabolic risk factors also predispose CF patients to the development of non-alcoholic steatohepatitis (NASH) similar to the general population. While hepatic steatosis and focal biliary cirrhosis and are more common findings in pediatric patients with CF, focal biliary cirrhosis is not believed to be as common in adult patients with CF. Several studies have recently demonstrated that the liver disease and portal hypertension that is associated with CFLD is not entirely due to the development of fibrosis or cirrhosis, but that non-cirrhotic portal hypertension (NCPH) also plays a role. Studies have shown that only 20–27% of patients with CF presenting with signs of portal hypertension actually have underlying cirrhosis.9, 23 Portal hypertension in these patients is felt to be due to NCPH and explants have demonstrated evidence of presinusoidal-type portal hypertension due to obliterative venopathy with fibrosis within portal vein branches that is consistent with this diagnosis.24 Biopsies from patients with CFLD have also shown evidence of nodular regenerative hyperplasia (NRH) which is a type of NCPH and may be related to recurrent vascular and infectious complications and possibly drug-induced liver injury (Figure 2d).1
Figure 1:

Schematic demonstrating the proposed pathogenesis of liver disease related to cystic fibrosis. Defective functioning of cystic fibrosis transmembrane conductance regulators (CFTR) expressed in the intra- and extra-hepatic cholangiocytes results in impaired Cl− secretion across the apical membrane, leading to decreased bile flow and increased bile precipitation. This results in ductular obstruction with subsequent periportal inflammation and fibrosis and focal biliary cirrhosis. Progressive disease may manifest as multifocal biliary cirrhosis and portal hypertension.
Figure 2:
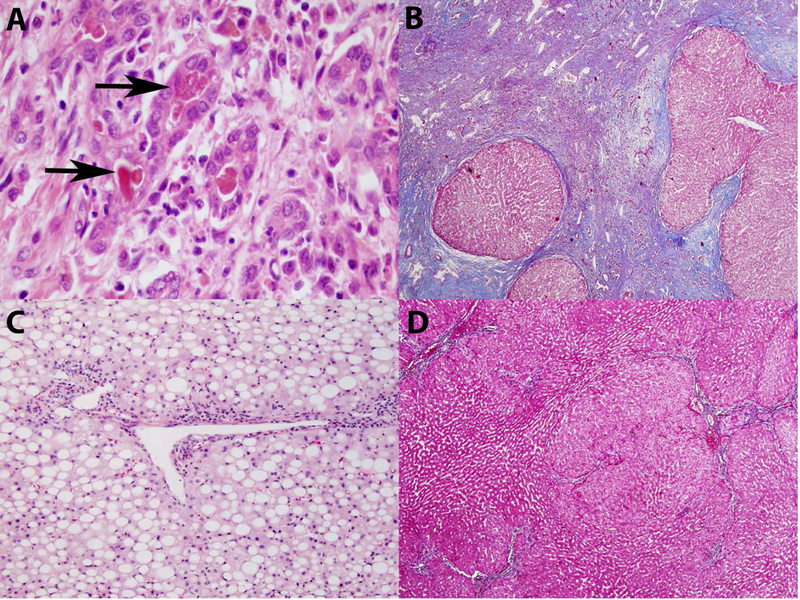
Liver histology demonstrating: a) ductular reaction with bile stasis, neutrophilic infiltration, and granular debris (arrows). b) focal cirrhosis changes. c) marked, pan-acinar steatosis. d) nodular regenerative hyperplasia
Finally, another notable condition that leads to chronic liver injury in patients with CF is the development of hepatolithiasis due to changes in the composition of and stagnation of bile. This can lead to stricturing biliary diseases and sclerosing cholangitis which can itself lead to a secondary biliary cirrhosis. A prospective study evaluating 27 patients with CF via MRCP demonstrated that of 9 patients who met clinical, biochemical, or ultrasonographic criteria of liver disease, all had abnormal MRCP results, with features resembling primary sclerosing cholangitis (PSC) in 5 and simple biliary lesions in the remaining 4, and 9 patients who didn’t meet criteria for liver disease also had abnormal findings (5 PSC-like and 4 simple biliary lesions).25 In addition, a recent study demonstrated that 19% of patients with PSC carry mutations in CFTR and that 50% had CFTR polymorphisms.26
Clinical Presentation and Diagnosis:
Patients with CFLD may have a wide array of presentations, from asymptomatic and mild elevations in hepatic and biliary enzymes, to steatosis and hepatosplenomegaly, to findings of decompensated cirrhosis with portal hypertension, ascites, and variceal bleeding. While hepatomegaly and splenomegaly are common in patients with CFLD, traditional physical examinations are typically inaccurate in identifying subtle changes in organomegaly. Also, although elevated hepatic enzymes are common in patients with CF, these alone have been not shown to be accurate in diagnosing CFLD. Studies have demonstrated that 53–93% of patients with CF have at least one abnormal value of aspartate aminotransferase (AST) or alanine aminotransferase (ALT), while over one-third have abnormal levels of gamma glutamyl-transferase (GGT) by the age of 21.9, 20, 27 These frequently abnormal liver tests may be due to the multiple infections and medications that patients with CF experience. Additionally, while not specifically studied in CF patients, a large study evaluating the sensitivity and specificity of ALT thresholds currently used by children’s hospitals in detecting NAFLD, HBV, and HCV, demonstrated sensitivity of only 32–48% and specificity of 92% and 96% in boys and girls respectively.28 However, Woodruff et al demonstrated that AST > 1.5 X ULN or GGT >1.5X ULN were strong predictors of underlying liver disease in patients with CF27 and Bodewes et al demonstrated a strong correlation between persistently elevated GGT and the development of cirrhosis within 2 years.29 Therefore, patients presenting with significant or persistently elevated liver biochemistries warrant further investigation for evidence of CFLD. A proposed algorithm for the evaluation of CFLD patients with abnormal liver tests is presented in Figure 3. With the evolution and advancements in hepatic imaging, subtle changes that occur early in the course of CFLD have become more easily identifiable. A recent study proposing a classification system based on a short unenhanced MRI protocol demonstrated a sensitivity of 94.1% and specificity of 84.6% when discriminating CFLD from controls, and that disease severity significantly corresponded to these findings.30 This study also demonstrated that the albumin-bilirubin score (ALBI) was better than Child-Pugh score in assessing the severity of liver disease in these patients. Interestingly, this study did not find any significant difference in hepatic fat fraction in CF patients compared to controls, suggesting that these findings were more related to other patient characteristics such as history of diabetes or medication exposure. Statistically significant differences between CF patients and controls were noted in amount of periportal fat deposition, periportal tracking on diffusion weighted images, bile duct abnormalities, heterogeneous liver parenchyma, gallbladder alterations, periportal fibrosis, and widening of fissures and hilum.30
Figure 3:

Algorithmic evaluation for cystic fibrosis liver disease in patients with cystic fibrosis and abnormal liver tests.
In addition to simple laboratory values and imaging, other non-invasive measures of liver disease have been utilized in staging and diagnosing CFLD. Two lab-based markers, AST-to-platelet ratio index (APRI) and the Fibrosis-4 score (FIB-4) have been described to correlate with the presence of CFLD as diagnosed by dual-pass liver biopsies, with AUROC of 0.75 for APRI and 0.60 for FIB-4.31 Another non-invasive measure of liver disease that has been studied in CFLD includes vibration controlled transient elastography (VCTE). VCTE, particularly Fibroscan® (Echosens, Paris), has been approved by the FDA for use in staging of other liver diseases such as hepatitis C, and as such it is becoming increasingly available. While official cut-offs have yet to be established in CFLD, a study by Kitson et al demonstrated that liver stiffness measurements (LSM) by Fibroscan® greater than 6.8 kPa had a 76% sensitivity and 92% specificity for detecting CFLD.32 In addition, liver stiffness has been shown to directly correlate with the severity of CFLD and was felt to be better than APRI with AUROC of 0.91 vs 0.78 respectively.33 A recent study by Gominon et al demonstrated that while the median LSM progressed by 0.23 kPa/year in patients with CF, it progressed at a much faster rate (0.94 kPa/year, p=0.02) in those who develop CFLD.34 The authors of this study concluded that in addition to the baseline LSM, the rate of change in liver stiffness can be used to predict the development of CFLD. With the expanding body of literature exploring VCTE in CF patients, there may be clinical utility in performing annual LSM as a screening tool in CF patients. Due to the challenges in diagnosing CFLD, which can lead to delayed medical management, patients with CF should be screened for signs of liver disease with yearly physical examinations, liver enzyme testing including GGT, monitoring for declining platelets counts, and abdominal imaging. If available, non-invasive measurements of fibrosis including VCTE and APRI should be performed in at risk patients.
Diagnostic Criteria
Given the wide spectrum of diseases within CFLD, there is currently no clear method for diagnosing CFLD. While liver biopsy has been described to be the gold standard of diagnosing underlying CFLD, it’s use is limited by the inherited risks of performing an invasive liver biopsy, as well as the patchy nature of liver involvement in CFLD which may be underrepresented or missed by a liver biopsy sample. Because of this, several diagnostic criteria have been proposed for the diagnosis of CFLD. The most commonly used is one that was proposed by Debray et al in 2011 which involved at least 2 of following after ruling out other causes of liver disease: hepatomegaly or splenomegaly, abnormal LFTs above the upper limits of normal at least 3 consecutive determinations over 12 months, or ultrasound evidence of liver involvement, with liver biopsies being performed if there was any doubt.17 Another commonly used definition was one that was proposed by the CF Foundation after a meeting was convened in 2007, which proposed classifying CFLD as either 1) CF related liver disease with clinical exam, imaging, histologic, or laparoscopic findings of cirrhosis or portal hypertension, or 2) liver involvement without cirrhosis consisting of at least one of: persistent or intermittent elevations of AST, ALT, GGT at least 2 X ULN, steatosis or fibrosis on histology, cholangiopathy based on imaging, or US findings other than those consistent with cirrhosis, or 3) pre-clinical with no exam, radiologic, or biochemical evidence of liver disease.35 More recently, a new diagnostic criteria was proposed based on the findings of a longitudinal study at the NIH which incorporated the use of non-invasive fibrosis markers of liver disease such as transient elastography, APRI, and FIB-4 with previously proposed criteria.1 This latter criteria was shown to be able to detect more cases of CFLD compared to previously proposed criteria by Debray et al (47% vs 22% respectively), suggesting that prior studies may be underestimating the prevalence of actual disease. A recent longitudinal study by Alexopoulou et al (in press) evaluating a cohort of 62 patients with CF compared the old criteria (Debray) and new criteria (NIH) and found similar findings, with 25.8% and 43.5% of patients meeting criteria for CFLD respectively. However, the clinical significance of this higher rate of diagnosis still needs to be determined. Table 1 describes the features of each of these diagnostic criteria.
Table 1:
Features of each of these diagnostic criteria
| Debray Criteria | NIH Criteria | CF Foundation Classification | |
|---|---|---|---|
| Any of: | Radiologic evidence of diffuse liver disease, cirrhosis, or portal hypertension |
Evidence of cirrhosis or portal HTN based on any of: Clinical exam Imaging Histology Laparoscopy |
|
| Liver biopsy consistent with CFLD (performed if any doubt) | Liver biopsy consistent with CFLD (performed for any reason) | ||
| At least 2 of: | Hepatomegaly or splenomegaly on exam and confirmed by ultrasound | Hepatomegaly or splenomegaly on imaging |
Liver involvement without cirrhosis with at least 1 of: Persistent/Intermittent elevations of AST, ALT, GGT >2x ULN Steatosis/fibrosis on histology Cholangiopathy on imaging US findings other than cirrhotic features |
| Abnormal ALT, AST, or GGT above the ULN at least 3 consecutive determinations over 12 months | At least 2 persistently abnormal ALT, AST, GGT, or ALP over 2 years | ||
| US evidence of liver involvement or portal hypertension | |||
| Persistently abnormal APRI, FIB-4, or AAR | |||
| Abnormal Fibroscan® at any time | |||
| Pre-clinical with no exam, radiologic, or biochemical evidence of liver disease | |||
Differential Diagnosis:
Aside from the more common presentations of CFLD, patients with CF are at risk for other causes of chronic liver injury. Therefore, patients with CF presenting with abnormal liver enzymes or abnormal hepatic imaging should be screened for other causes of chronic liver disease. As CFLD is primarily a cholestatic liver disease, diseases such as PSC and primary biliary cholangitis need to be ruled out. Also, patients need to be screened for biliary diseases such as choledocholithiasis and disease related to an obstructive process; MRCP/MRI may be indicated in these patients. Drug-induced liver injury (DILI) is another important etiology of liver disease in these patients and often leads to intermittent or persistently elevated liver function tests. This includes antibiotics that are well established as causes of DILI, as well as newly developed medications that are used to directly modulate the CFTR protein such as ivacaftor and lumacaftor. These newer medications have demonstrated an ability to cause elevations in serum aminotransferases up to 3 times ULN in up to 25% of patients.36 Finally, all patients need to be screened for concomitant alcohol abuse (screening questionnaires such as CAGE), viral hepatitis such as hepatitis A, B and C (serum serologies and/or PCR), autoimmune hepatitis (ANA, ASMA, anti-LKM, and total immunoglobulins), Wilson disease (ceruloplasmin), celiac disease (anti-transglutaminase antibody and/or duodenal biopsies), alpha-1-antitrypsin deficiency (A1AT level and phenotype), as well as metabolic risk factors for NAFLD (diabetes or glucose intolerance, central adiposity, hypertension, hypertriglyceridemia, low HDL).
Management:
Once CFLD has been diagnosed, the goals of treatment consist of managing the sequelae associated with each specific disease, particularly those associated with portal hypertension, and treatment to delay the progression of disease. All patients with CFLD should have monitoring of liver enzymes and markers associated with hepatic synthetic function including coagulation (PT/INR) and albumin every 3–6 months.
Portal Hypertension
The diagnosis and management of portal hypertension is crucial given the potential severe consequences associated with esophageal or gastric variceal bleeding. However, there are no clear guidelines as to when and how to initiate screening or how often to perform this. A retrospective study evaluating episodes of GI bleeding at a tertiary referral center in London, England which is also a referral center for cystic fibrosis patients described 18 patients with CFLD who presented with gastrointestinal bleeding, with the median age of first bleed at 20 years (range 9.7 to 30.9).37 This wide age range suggests that screening for portal hypertension should occur once the diagnosis of CFLD has been made, and that screening should occur annually.17
With respect to how the screening should be performed, while transjugular measurements of hepatic portal pressures is the current gold standard in detecting and quantifying portal hypertension, this diagnostic modality is invasive and not uniformly available. Additionally, hepatic venous pressure gradient measurements are poor at detecting pre-sinusoidal portal hypertension, which is often a component of different diseases within the NCPH spectrum. While additional studies are still required for validation, several non-invasive measures of portal hypertension have been shown to predict the presence of portal hypertension and esophageal varices in patients with CFLD. For example, progressively lower levels of platelets on longitudinal follow-up have previously been shown to correlate with presence and severity of liver disease and may possibly be associated with increased risks of mortality.1 A Cochrane review evaluating the presence of esophageal varices in patients with liver diseases and portal hypertension from diseases other than CFLD demonstrated that the platelet count-to-spleen length ratio is a more accurate predictor than either alone in adult patients, while other studies have suggested that platelet counts alone may be better in pediatric populations.38, 39 As such, the British Society of Pediatric Gastroenterology, Hepatology and Nutrition (BSPGHAN) recommends screening endoscopies in patients with platelet counts less than 120*109/L.38 As these recommendations were based on studies involving liver diseases other than CFLD, studies are necessary to validate these findings in CFLD. Finally, CT, MRI, and US findings suggestive of portal hypertension, such as recanalization of the umbilical vein and visualization of varices are also very helpful in determining which patients have portal hypertension.
Once the diagnosis of portal hypertension has been made, upper endoscopic evaluation is the method of choice for determining the presence of esophageal and gastric varices, and screening allows for detection of varices prior to bleeding. In patients with high risk varices (ie: grade 2 or 3), band-ligation is preferred to non-selective beta-blockers given the increased risk of pulmonary complications in these patients. Patients with esophageal varices can then be placed in a banding protocol that consists of endoscopies at regular intervals until the varices are successfully eradicated.9, 38 However, given their concomitant underlying pulmonary disease, patients with CF are at increased risk for endoscopic procedures and anesthesia, and the risks of general anesthesia should be weighed against the risks of bleeding. Indeed, one survey demonstrated that pediatric patients with CF are typically screened at lower rates than those with other forms of liver disease.40 Unfortunately there is currently no non-invasive measure that has been validated to help predict who would benefit from endoscopic procedures in patients with CFLD, as is the case of the Baveno VI recommendations in cirrhotic patients of other etiologies.41 However, the non-invasive measures that are used in other causes of portal hypertension may still be helpful in determining which patients would benefit from endoscopic screening.
Patient who fail medical management of portal hypertension and have variceal bleeding or other complications such as ascites, hypersplenism and severe thrombocytopenia may benefit from transhepatic porto-systemic shunts (TIPS), creation of surgical shunts, or splenectomy or partial splenic embolization.42, 43 There is no clear consensus of which modality is best and this typically depends on the patient’s symptoms, underlying anatomy, center experience and transplant candidacy. In the study by Gooding et al, of the 38 episodes of variceal bleeding in patients with CFLD, 30 were controlled endoscopically (band ligation and/or sclerotherapy, with 1 patient requiring glue injection for gastric varices), while 2 patients required TIPS after failed endoscopic treatment and 1 patient requiring surgical splenorenal shunt after failed endoscopic treatment.37 It should be noted that this study was performed in 2005 and a large proportion of patients underwent sclerotherapy rather than band ligation, which is currently the preferred treatment option for esophageal varices. While decompressive shunts are not recommended as primary prophylaxis, they are typically the best option for long-term prevention of recurrent esophageal and gastric variceal rebleeding.42
Medical Management of CFLD
Unfortunately, medical treatments to delay progression or improve CFLD are still lacking. Traditionally, once diagnosed with CFLD, patients have been started on ursodeoxycholic acid (UDCA). While its exact mechanism of action is controversial, UDCA is a hydrophilic secondary bile acid which is felt to increase the fluidity and change the hydrophobicity of bile while also providing cytoprotective and anti-apoptotic effects.44, 45 Most of the data for the use of UDCA comes from its use in other cholestatic liver diseases such as PSC and PBC, and while it’s utility in PBC is well established, its benefit in PSC is more debatable. Similarly, a recent Cochrane review performed on CFLD patients found that the data to support the use of UDCA is limited with mostly low-quality studies and an absence of data regarding long-term outcomes such as death or need for liver transplantation.46 Furthermore, in the study by Boelle et al, UDCA did not prevent the development of severe CFLD.12 On the other hand, a study by van der Feen et al demonstrated a decrease in liver stiffness as measured by transient elastography by 0.70 kPa/year in those with mild liver disease but not in those with CF related cirrhosis.47 Whether or not UDCA is beneficial in CFLD has yet to be proven, though at this time given the lack of alternative therapies and overall good tolerance, particularly at low and medium doses less than 20mg/kg/day, most patients who are diagnosed with CFLD are still started on this medication.7
Recently, new therapies have been approved for patients with CF which directly target the CFTR protein and show promising results in pulmonary function.48 These include Ivacaftor, a CFTR modulator that improves chloride transport through CFTR channels which has been available since 2012, and Lumacaftor, which is used in conjunction with Ivacaftor and increases membrane expression of the CFTR protein. While these medications show promise in other gastrointestinal complications associated with CF, their effects on the development and treatment of CFLD have not been directly evaluated in any trials.49 Unfortunately, specific therapies for the treatment of CFLD are still lacking and further research is certainly needed in this area. Currently, there is a single ongoing phase II clinical trial evaluating different formulations of ursodiol in patients with CFLD (NCT00004315). Additionally, in-vitro studies looking at inhibiting Src using a kinase inhibitor have shown that when used in combination with Ivacaftor and Lumacaftor, cholangiocytes fluid secretion is restored to normal.50 However, human studies have not yet been performed.
Nutrition
Nutrition is a key part of the management of patients with CF and is associated with better lung function and survival in children with CF.43 Malnutrition is multifactorial, including pancreatic insufficiency as well as CFLD which can lead to malabsorption of fats and fat-soluble vitamins. Severe malnutrition itself can also lead to hepatic steatosis, which is in the spectrum of CFLD. Screening for and repleting fat-soluble vitamins is thus important in these patients. Patients also often require higher calorie intakes (110–200% of the general population) along with high fat diets in order to achieve their nutritional goals.43 Patients with pancreatic insufficiency will benefit from pancreatic enzyme replacement therapy.43 Unfortunately, medical treatment of CFLD has not yet been shown to improve nutritional outcomes in patients with CFLD. However, liver transplantation in patients with CFLD has been shown to preserve nutritional status with one study showing mild improvement in median BMI 5 years post-transplantation compared to pre-transplantation (19.6 kg/m2 vs 18.0 kg/m2), though the difference was not statistically significant.51
Cancer Screening
Finally, while there is no clear data on the risk of development of hepatocellular carcinoma (HCC) in patients with CFLD, once patients are found to have advanced fibrosis or cirrhosis, they should also be placed on a screening protocol for HCC with ultrasounds every 6 months based on guidelines for cirrhosis of other etiologies.52
Transplantation:
According to the Cystic Fibrosis Patient Registry, 1,642 patients with CF were reported to have had a solid organ transplant by 2016.5 Lung transplantation is the most common organ transplanted in these patients. However, patients with planned lung transplantations that demonstrate severe portal hypertension from CFLD have been described to benefit from simultaneous liver and lung transplantations.53 Unfortunately, most recent studies evaluating combined liver and lung transplantations within the past decade only consist of single-center experiences. Alternatively, liver transplantation alone has been performed in CF patients with hepatic decompensation and manifestations of portal hypertension such as variceal bleeding who have preserved lung function.53, 54 A study evaluating the effects of liver transplantation on lung function demonstrated that liver transplantation was neither beneficial nor detrimental to pulmonary function, with no difference in the rate of FEV-1 decline after transplantation.55 However, a different study performed in the United Kingdom demonstrated that the rate of decline in FEV1 was lower (−0.74%, p=0.04) compared to a predicted 3% annual decline in patients with CF up to 4 years after transplantation.51 Finally, a single-center retrospective analysis evaluating the long-term outcomes of 9 patients with CF who underwent liver transplantation demonstrated that three required subsequent lung transplantation and one required combined liver and lung transplant within 10 years of the initial liver transplant procedure. In addition, four other patients were undergoing evaluation for lung transplantation.56 Taken together, while liver transplantation may slow decline in pulmonary function, it appears not to halt the disease process and patients may ultimately require lung transplantation as well.
There are currently no clear guidelines to aid in determining which CFLD patients should be evaluated for liver transplantation. In general, patients with CFLD are considered for liver transplant if there is evidence of hepatic decompensation, such as coagulopathy (INR >1.5), ascites, jaundice, or extensive variceal bleeding that cannot be controlled by a portosystemic shunt.8 In addition, there remains controversy regarding the timing of transplantation. The majority of liver transplants performed for CFLD are in children, with 79% (182 out of 230) of liver transplants in CF patients being performed in children between 1987 and 2008.35, 54 Contraindications to isolated liver transplant include active pulmonary infections or poor pulmonary function as expressed by FEV-1 less than 50%, extensive pulmonary fibrosis on imaging, or pulmonary hypertension (>35mmHg).35 In these patients, combined liver and lung transplantations should be considered. However, the rates of combined liver and lung transplantation remains low, consisting of only 6% of transplants between 1987 and 2008 and only 50 cases recorded in the Scientific Registry of Transplant Recipients (SRTR), for all causes, between 2005 to 2015.54, 57 CF patients requiring combined liver-lung transplants must show signs of decreased pulmonary function with FEV-1 below 40% and their diagnosis must have been confirmed by genetic analysis. Exception points can be requested for these patients. Patients who are over the age of 18 are assigned a score that is 3 points below the median allocation MELD at transplant for liver recipients in the same donation service area (DSA). Patients under the age of 12 who meet the requirements for standardized PELD scores, as well as patients between the ages of 12 to 17 are assigned a score equal to the median MELD at transplant for all liver recipients in the same DSA.57
A retrospective study reviewing the United Network for Organ Sharing Data (UNOS) database evaluated long-term outcomes for patients with CF undergoing liver transplantation. While somewhat lower than that of other etiologies of liver disease, the 5-year survival rate of CFLD related transplants was described to be respectable at 85.5% in children and 72.7% in adults. More importantly, this was significantly better than the 5-year survival rates in patients who remained on the transplant list, with hazard ratios of 0.33 and 0.25 in pediatric and adult patients respectively.58 In another study, evaluating isolated liver transplantation as well as combined liver-lung transplantation, 1 and 5-year survivals were not significant different between the two groups (80% and 80% vs 83.9% and 75.7% respectively).54 The major causes of death in these patients is pulmonary disease (22.7%) and hemorrhage (18.2%).54 Thus, in patients with decompensated cirrhosis or uncontrollable bleeding due to portal hypertension that is not amenable to surgical shunts or TIPS, liver transplantation is a viable option, either alone or in combination with lung transplantation.
Conclusion:
Significant advancement in medical management has resulted in dramatically improved life expectancies in patients with CF compared to even just 10 years ago. While prognosis has historically been intertwined with declining pulmonary status in CF patients, CFLD is now the third leading cause of death, the most common non-pulmonary cause of CF related deaths, and accounts for up to 5% of deaths in patients with CF. Up until recently, the majority of patients with CFLD presented in childhood, however, recent evidence seems to suggest a possible second wave of liver disease that becomes evident in adulthood.1 It remains unclear if this liver disease is a result of underlying CF, or if it is the result of secondary complications such as chronic infections, therapeutics, or a yet to be understood process. There remains no consensus methodology for the diagnosis of CFLD, particularly given its patchy nature and variable presentations. However, different diagnostic algorithms have been described, some of which include newer imaging techniques and non-invasive measures of liver disease. Further exploration in this area is needed given the importance of early diagnosis and intervention. Currently, medical treatment for CFLD remains limited, and is a burgeoning area for exploration. For patients with progressive disease, liver transplantation either alone, or in combination with lung transplantation appears to be a feasible alternative and with improved outcomes and prolonged survival.
KEY POINTS.
Cystic fibrosis liver disease is the third leading cause of death in patients with cystic fibrosis.
There are no clear guidelines for diagnosing cystic fibrosis liver disease and several diagnostic criteria have been proposed in the past.
The majority of patients with cystic fibrosis liver disease are diagnosed before puberty, though as life expectancies improve, adult-onset cystic fibrosis liver disease is becoming increasingly recognized.
There are no proven therapies for cystic fibrosis liver disease, though ursodeoxycholic acid is commonly used.
Liver transplant is a viable option, either alone or in combination with lung transplantation for patients with advanced liver disease from cystic fibrosis.
SYNOPSIS.
Cystic fibrosis liver disease (CFLD) remains the third leading cause of death in patients with cystic fibrosis. Although a majority of patients with CFLD present in childhood, recent studies suggest a second wave of liver disease in adulthood.1 There are no clear guidelines for diagnosing CFLD. Treatment options for CFLD remain limited, and while UDCA is widely utilized, its long-term benefit is unclear. Those who develop hepatic decompensation or uncontrolled variceal bleeding may benefit from liver transplant, either alone, or in combination with lung transplant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE STATEMENT
The authors have nothing to disclose.
References:
- 1.Koh C, Sakiani S, Surana P, et al. Adult-onset cystic fibrosis liver disease: Diagnosis and characterization of an underappreciated entity. Hepatology 2017;66:591–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colombo C Liver disease in cystic fibrosis. Curr Opin Pulm Med 2007;13:529–36. [DOI] [PubMed] [Google Scholar]
- 3.Rowe SM, Miller S, Sorscher EJ. Mechanisms of disease: Cystic fibrosis. New England Journal of Medicine 2005;352:1992–2001. [DOI] [PubMed] [Google Scholar]
- 4.Strausbaugh SD, Davis PB. Cystic fibrosis: a review of epidemiology and pathobiology. Clin Chest Med 2007;28:279–88. [DOI] [PubMed] [Google Scholar]
- 5.Cystic Fibrosis Foundation: Patient Registry 2016 Annual Data Report 2016.
- 6.Bhardwaj S, Canlas K, Kahi C, et al. Hepatobiliary Abnormalities and Disease in Cystic Fibrosis Epidemiology and Outcomes Through Adulthood. Journal of Clinical Gastroenterology 2009;43:858–864. [DOI] [PubMed] [Google Scholar]
- 7.Colombo C, Alicandro G. Liver disease in cystic fibrosis: illuminating the black box. Hepatology 2018. [DOI] [PubMed]
- 8.Leung DH, Narkewicz MR. Cystic Fibrosis-related cirrhosis. J Cyst Fibros 2017;16 Suppl 2:S50–S61. [DOI] [PubMed] [Google Scholar]
- 9.Kamal N, Surana P, Koh C. Liver disease in patients with cystic fibrosis. Curr Opin Gastroenterol 2018;34:146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatterjee K, Goyal A, Shah N, et al. Contemporary national trends of cystic fibrosis hospitalizations and co-morbidities in the United States. Adv Respir Med 2016;84:316–323. [DOI] [PubMed] [Google Scholar]
- 11.Lamireau T, Monnereau S, Martin S, et al. Epidemiology of liver disease in cystic fibrosis: a longitudinal study. J Hepatol 2004;41:920–5. [DOI] [PubMed] [Google Scholar]
- 12.Boelle PY, Debray D, Guillot L, et al. Cystic Fibrosis Liver Disease: Outcomes and Risk Factors in a Large Cohort of French Patients. Hepatology 2018. [DOI] [PMC free article] [PubMed]
- 13.Cohn JA, Strong TV, Picciotto MR, et al. Localization of the cystic fibrosis transmembrane conductance regulator in human bile duct epithelial cells. Gastroenterology 1993;105:1857–64. [DOI] [PubMed] [Google Scholar]
- 14.Kobelska-Dubiel N, Klincewicz B, Cichy W. Liver disease in cystic fibrosis. Prz Gastroenterol 2014;9:136–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Spirli C, Granato A, Zsembery K, et al. Functional polarity of Na+/H+ and Cl-/HCO3-exchangers in a rat cholangiocyte cell line. Am J Physiol 1998;275:G1236–45. [DOI] [PubMed] [Google Scholar]
- 16.Colombo C, Battezzati PM, Crosignani A, et al. Liver disease in cystic fibrosis: A prospective study on incidence, risk factors, and outcome. Hepatology 2002;36:1374–82. [DOI] [PubMed] [Google Scholar]
- 17.Debray D, Kelly D, Houwen R, et al. Best practice guidance for the diagnosis and management of cystic fibrosis-associated liver disease. Journal of Cystic Fibrosis 2011;10:S29–S36. [DOI] [PubMed] [Google Scholar]
- 18.Fiorotto R, Villani A, Kourtidis A, et al. The cystic fibrosis transmembrane conductance regulator controls biliary epithelial inflammation and permeability by regulating Src tyrosine kinase activity. Hepatology 2016;64:2118–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flass T, Tong S, Frank DN, et al. Intestinal lesions are associated with altered intestinal microbiome and are more frequent in children and young adults with cystic fibrosis and cirrhosis. PLoS One 2015;10:e0116967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lindblad A, Glaumann H, Strandvik B. Natural history of liver disease in cystic fibrosis. Hepatology 1999;30:1151–8. [DOI] [PubMed] [Google Scholar]
- 21.Mc Ewan FA, Hodson ME, Simmonds NJ. The prevalence of “risky behaviour” in adults with cystic fibrosis. J Cyst Fibros 2012;11:56–8. [DOI] [PubMed] [Google Scholar]
- 22.Georgiopoulou VV, Denker A, Bishop KL, et al. Metabolic abnormalities in adults with cystic fibrosis. Respirology 2010;15:823–9. [DOI] [PubMed] [Google Scholar]
- 23.Hillaire S, Cazals-Hatem D, Bruno O, et al. Liver transplantation in adult cystic fibrosis: Clinical, imaging, and pathological evidence of obliterative portal venopathy. Liver Transpl 2017;23:1342–1347. [DOI] [PubMed] [Google Scholar]
- 24.Witters P, Libbrecht L, Roskams T, et al. Liver disease in cystic fibrosis presents as non-cirrhotic portal hypertension. J Cyst Fibros 2017;16:e11–e13. [DOI] [PubMed] [Google Scholar]
- 25.Durieu I, Pellet O, Simonot L, et al. Sclerosing cholangitis in adults with cystic fibrosis: a magnetic resonance cholangiographic prospective study. J Hepatol 1999;30:1052–6. [DOI] [PubMed] [Google Scholar]
- 26.Werlin S, Scotet V, Uguen K, et al. Primary sclerosing cholangitis is associated with abnormalities in CFTR. J Cyst Fibros 2018;17:666–671. [DOI] [PubMed] [Google Scholar]
- 27.Woodruff SA, Sontag MK, Accurso FJ, et al. Prevalence of elevated liver enzymes in children with cystic fibrosis diagnosed by newborn screen. J Cyst Fibros 2017;16:139–145. [DOI] [PubMed] [Google Scholar]
- 28.Schwimmer JB, Dunn W, Norman GJ, et al. SAFETY study: alanine aminotransferase cutoff values are set too high for reliable detection of pediatric chronic liver disease. Gastroenterology 2010;138:1357–64, 1364 e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bodewes FA, van der Doef HP, Houwen RH, et al. Increase of Serum gamma-Glutamyltransferase Associated With Development of Cirrhotic Cystic Fibrosis Liver Disease. J Pediatr Gastroenterol Nutr 2015;61:113–8. [DOI] [PubMed] [Google Scholar]
- 30.Poetter-Lang S, Staufer K, Baltzer P, et al. The Efficacy of MRI in the diagnostic workup of cystic fibrosis-associated liver disease: A clinical observational cohort study. Eur Radiol 2018. [DOI] [PMC free article] [PubMed]
- 31.Leung DH, Khan M, Minard CG, et al. Aspartate aminotransferase to platelet ratio and fibrosis-4 as biomarkers in biopsy-validated pediatric cystic fibrosis liver disease. Hepatology 2015;62:1576–83. [DOI] [PubMed] [Google Scholar]
- 32.Kitson MT, Kemp WW, Iser DM, et al. Utility of transient elastography in the non-invasive evaluation of cystic fibrosis liver disease. Liver Int 2013;33:698–705. [DOI] [PubMed] [Google Scholar]
- 33.Aqul A, Jonas MM, Harney S, et al. Correlation of Transient Elastography With Severity of Cystic Fibrosis-related Liver Disease. J Pediatr Gastroenterol Nutr 2017;64:505–511. [DOI] [PubMed] [Google Scholar]
- 34.Gominon AL, Frison E, Hiriart JB, et al. Assessment of Liver Disease Progression in Cystic Fibrosis Using Transient Elastography. J Pediatr Gastroenterol Nutr 2018;66:455–460. [DOI] [PubMed] [Google Scholar]
- 35.Flass T, Narkewicz MR. Cirrhosis and other liver disease in cystic fibrosis. J Cyst Fibros 2013;12:116–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.LiverTox: CYSTIC FIBROSIS AGENTS - IVACAFTOR AND LUMACAFTOR
- 37.Gooding I, Dondos V, Gyi KM, et al. Variceal hemorrhage and cystic fibrosis: outcomes and implications for liver transplantation. Liver Transpl 2005;11:1522–6. [DOI] [PubMed] [Google Scholar]
- 38.Davison S Assessment of liver disease in cystic fibrosis. Paediatr Respir Rev 2018;27:24–27. [DOI] [PubMed] [Google Scholar]
- 39.Colli A, Gana JC, Yap J, et al. Platelet count, spleen length, and platelet count-to-spleen length ratio for the diagnosis of oesophageal varices in people with chronic liver disease or portal vein thrombosis. Cochrane Database Syst Rev 2017;4:CD008759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jeanniard-Malet O, Duche M, Fabre A. Survey on Clinical Practice of Primary Prophylaxis in Portal Hypertension in Children. J Pediatr Gastroenterol Nutr 2017;64:524–527. [DOI] [PubMed] [Google Scholar]
- 41.Maurice JB, Brodkin E, Arnold F, et al. Validation of the Baveno VI criteria to identify low risk cirrhotic patients not requiring endoscopic surveillance for varices. J Hepatol 2016;65:899–905. [DOI] [PubMed] [Google Scholar]
- 42.Marti J, Gunasekaran G, Iyer K, et al. Surgical management of noncirrhotic portal hypertension. Clinical Liver Disease 2015;5:112–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bolia R, Ooi CY, Lewindon P, et al. Practical approach to the gastrointestinal manifestations of cystic fibrosis. J Paediatr Child Health 2018;54:609–619. [DOI] [PubMed] [Google Scholar]
- 44.Paumgartner G, Beuers U. Ursodeoxycholic acid in cholestatic liver disease: mechanisms of action and therapeutic use revisited. Hepatology 2002;36:525–31. [DOI] [PubMed] [Google Scholar]
- 45.Kappler M, Espach C, Schweiger-Kabesch A, et al. Ursodeoxycholic acid therapy in cystic fibrosis liver disease--a retrospective long-term follow-up case-control study. Aliment Pharmacol Ther 2012;36:266–73. [DOI] [PubMed] [Google Scholar]
- 46.Cheng K, Ashby D, Smyth RL. Ursodeoxycholic acid for cystic fibrosis-related liver disease. Cochrane Database Syst Rev 2017;9:CD000222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van der Feen C, van der Doef HP, van der Ent CK, et al. Ursodeoxycholic acid treatment is associated with improvement of liver stiffness in cystic fibrosis patients. J Cyst Fibros 2016;15:834–838. [DOI] [PubMed] [Google Scholar]
- 48.Ramsey BW, Davies J, McElvaney NG, et al. A CFTR potentiator in patients with cystic fibrosis and the G551D mutation. N Engl J Med 2011;365:1663–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Houwen RHJ, van der Woerd WL, Slae M, et al. Effects of new and emerging therapies on gastrointestinal outcomes in cystic fibrosis. Curr Opin Pulm Med 2017;23:551–555. [DOI] [PubMed] [Google Scholar]
- 50.Fiorotto R, Amenduni M, Mariotti V, et al. Src kinase inhibition reduces inflammatory and cytoskeletal changes in DeltaF508 human cholangiocytes and improves cystic fibrosis transmembrane conductance regulator correctors efficacy. Hepatology 2018;67:972–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dowman JK, Watson D, Loganathan S, et al. Long-term impact of liver transplantation on respiratory function and nutritional status in children and adults with cystic fibrosis. Am J Transplant 2012;12:954–64. [DOI] [PubMed] [Google Scholar]
- 52.Marrero JA, Kulik LM, Sirlin CB, et al. Diagnosis, Staging, and Management of Hepatocellular Carcinoma: 2018 Practice Guidance by the American Association for the Study of Liver Diseases. Hepatology 2018;68:723–750. [DOI] [PubMed] [Google Scholar]
- 53.Halldorson J, AlQahtani K. Outcomes of Combined Liver/Lung Transplantation for Cystic Fibrosis Using SRTR Analysis. Am J Transplant 2017;17. [Google Scholar]
- 54.Arnon R, Annunziato RA, Miloh T, et al. Liver and combined lung and liver transplantation for cystic fibrosis: analysis of the UNOS database. Pediatr Transplant 2011;15:254–64. [DOI] [PubMed] [Google Scholar]
- 55.Miller MR, Sokol RJ, Narkewicz MR, et al. Pulmonary function in individuals who underwent liver transplantation: from the US cystic fibrosis foundation registry. Liver Transpl 2012;18:585–93. [DOI] [PubMed] [Google Scholar]
- 56.Sivam S, Al-Hindawi Y, Di Michiel J, et al. Liver and lung transplantation in cystic fibrosis: an adult cystic fibrosis centre’s experience. Intern Med J 2016;46:852–4. [DOI] [PubMed] [Google Scholar]
- 57.OPTN/UNOS Liver Review Board Policy 2017.
- 58.Mendizabal M, Reddy KR, Cassuto J, et al. Liver transplantation in patients with cystic fibrosis: analysis of United Network for Organ Sharing data. Liver Transpl 2011;17:243–50. [DOI] [PubMed] [Google Scholar]