

Abstract
Erythropoietin is a glycoproteic hormone that regulates hematopoiesis by acting on its specific receptor (EpoR). The expression of EpoR in the central nervous system (CNS) suggests a role for this hormone in the brain. Recently, we developed a new Epo variant without hematopoietic activity called EpoL, which showed marked neuroprotective effects against oxidative stress in brain ischemia related models. In this study, we have evaluated the neuroprotective effects of EpoL against oxidative stress induced by chronic treatment with Aβ. Our results show that EpoL was neuroprotective against Aβ-induced toxicity by a mechanism that implicates EpoR, reduction in reactive oxygen species, and reduction in astrogliosis. Furthermore, EpoL treatment improved calcium handling and SV2 levels. Interestingly, the neuroprotective effect of EpoL against oxidative stress induced by chronic Aβ treatment was achieved at a concentration 10 times lower than that of Epo. In conclusion, EpoL, a new variant of Epo without hematopoietic activity, is of potential interest for the treatment of diseases related to oxidative stress in the CNS such as Alzheimer disease.
Keywords: Erythropoietin, Glycosylation, Amyloid beta, Oxidative stress, Neuroprotection, Alzheimer disease
Graphical abstract
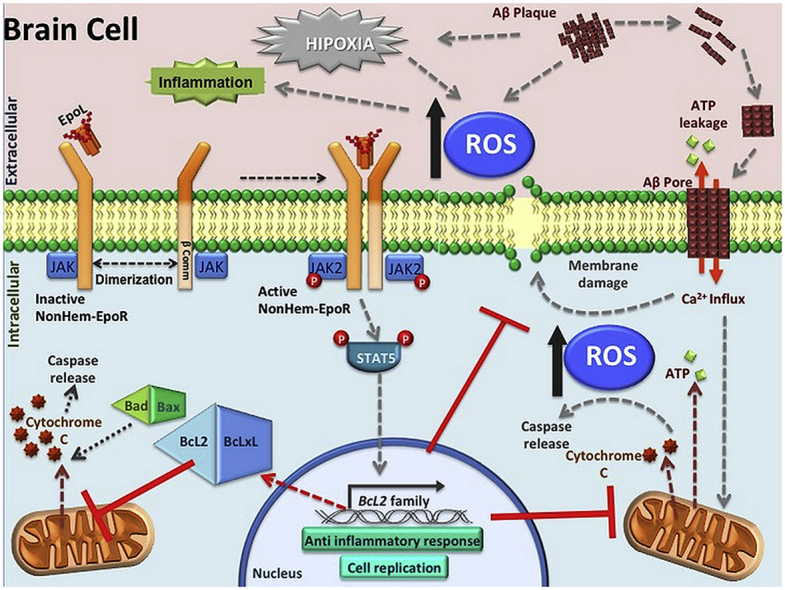
1. Introduction
Erythropoietin (Epo) is a glycoprotein hormone belonging to the superfamily of type I cytokines and is mainly responsible for the proliferation, differentiation and maturation of erythroid cells, in both embryonic and adult stages [1]. This hormone is a glycoprotein characterized by having a glycosylation pattern conformed by 4 glycosylated chains, 3 of them being N-glycosylations and one an O-glycosylation [2], responsible for 40% of its molecular weight. Glycosylation distribution gives a great heterogeneity to the mature protein, while the absence of glycosylation reduces the stability of intermediate species and causes changes in the binding kinetics of the erythropoietin receptor (EpoR) [3]. Additionally, the oligosaccharide chains attached to Epo increase its molecular size and thereby prevent glomerular filtration, while the presence of terminal neuraminic acids prevent the hormone uptake by hepatic transporters, increasing their plasma half-life [1,[4], [5], [6]]. Epo effects produced by Epo/EpoR interaction activates signaling cascades that act on the control of apoptosis.
The expression of EpoR has also been observed in non-erythroid tissues explaining the pleiotropic effects described for Epo. This receptor is expressed in the central nervous system (CNS) as a heterodimer, which suggests a specific role of Epo in this tissue [[6], [7], [8]]. The involvement of EpoR is critical for neuroprotection in vitro. For example, in Alzheimer's Disease (AD), the accumulation of beta amyloid peptide (Aβ1-42) is directly related to cellular apoptosis and DNA fragmentation [9]. Those processes have been stopped with Epo pre-treatment for 1 h (h) [10]. These results correlate with an increase in cognitive function, as evaluated by the Morris water maze test [11]. Additionally, Epo treatment has been associated with stimulation of neuronal proliferation in the dentate gyrus of the hippocampus in an AD animal model [12,13]. These data correlate with results that show an increase in EpoR expression in brain lysates from Alzheimer's patients as compared to brain lysates of healthy patients [14].
The Aβ peptide in AD is responsible for the loss of ionic homeostasis due to its ability to perforate the cell membrane forming a pore that allows the massive entrance of cations, especially calcium, inducing apoptosis and an imbalance of intracellular ATP [15,16]. There are several molecules that prevent the activation of the apoptotic pathway in cellular models pretreated with amyloid beta peptide, including erythropoietin. Activation of EpoR initiates Bcl2 gene transcription, which blocks apoptosis and prevents cell death [[17], [18], [19]]. Consequently, Epo-induced neuroprotection mediated by EpoR activation causes an inhibition of apoptotic proteins and blocks the activation of caspases in AD models [11,20,21]. The blockade in caspase activity is directly related with other pathways activated by EpoR, e.g. the activation of Bcl-xL inducing a decrease in reactive oxygen species (ROS) [17,22,23]. The relationship between these two effects, and the activation of EpoR has been demonstrated through the silencing of EpoR in astroglia using small interfering RNA (siRNA) techniques [24].
Current evidence suggests that Epo treatments can reduce the damage induced by different stresses such as inflammatory or ischemic brain processes, or neurodegenerative diseases [[24], [25], [26], [27]]. Therefore, the development of new Epo isoforms lacking hematopoietic effects could be considered as new potential targets which exert neuroprotection without the risk of vascular damage. In previous work, we used a new variant of Epo without hematopoietic activity (EpoL), but with neuroprotective effects on stress mediated by ROS [17]. Here, we evaluate if EpoL can provide similar neuroprotective effects against oxidative stress mediated by the Aβ peptide as compared to the hematopoietic Epo variant as control.
2. Materials and methods
Animal care and protocols were in accordance with the National Institutes of Health recommendations and approved by the Ethics Committee at the University of Concepcion and Autonomous University of Madrid.
2.1. Epo expression and purification
Recombinant human erythropoietin (Epo) was obtained from the genetically transformed Chinese ovarian hamster cell line (CHO; ATCC CCL-61, USA). The supernatant culture was centrifuged at 390 g (10 min) and purified by Blue-Sepharose and a chelating affinity chromatography column. Epo samples were quantified by a commercial human Epo ELISA kit (R&D, USA) and stored at −80 °C.
2.2. Goat mammary gland adenoviral mediated Epo expression
Three nulliparous Saanen goats (Capra hircus) of around 1.8 years old were used during the first month of natural lactation. The expression of recombinant human erythropoietin was carried out by the recombinant adenoviral infusion method described elsewhere [35]. Milk collection started 48 h after adenoviral vector inoculation and lasted for 12 days.
2.3. EpoL purification from milk
The recombinant human EpoL was purified from skimmed goat milk corresponding to the milk from days 2–6 in the mammary glands infused with the AdhEpo vector, as was previously described [35]. Milk was diluted in 100 mM EDTA containing 150 mM NaCl, at pH 4.0 and stirred at 4 °C. The insoluble fraction was removed by centrifugation and the supernatant was precipitated with 30% ammonium sulfate for 1 h at 4 °C. The supernatant was dialyzed overnight at 4 °C using 20 mM Tris–HCl, 1% Tween 20 at pH 7.4 and loaded into a Blue-Sepharose and chelating affinity chromatography column. EpoL was quantified by a commercial human Epo ELISA kit (R&D, USA) and stored at −80 °C.
2.4. Cell culture assays
2.4.1. PC-12 cell line
(ATCC CRL-1721, USA): Cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (Hyclone, USA) with 5% fetal bovine serum, 5% horse serum, 100 U/ml penicillin, 100 g/ml streptomycin, and 2 mM l-glutamine and were incubated under standard conditions (37 °C, 5% CO2).
2.4.2. Primary culture of hippocampal neurons
Hippocampal neurons were obtained from 18-day-old Sprague–Dawley rat embryos. The primary hippocampal cultures were maintained in a neuronal feeding medium consisting of 90% minimal essential medium (HyClone, USA), 5% heat-inactivated horse serum (HyClone, USA), 5% fetal bovine serum (Gibco, USA), and a mixture of nutrient supplements. Cultures at 14–15 days in vitro (DIV) were treated with Epo or EpoL at neuroprotective concentrations in co-incubation with Aβ oligomers at 37 °C and 5% CO2.
2.4.3. Epo pre-treatment assay
PC-12 cells at 85% confluence were pretreated with Epo or EpoL for 1 h and then stressed with Aβ40 peptide oligomers during 24 h using the same medium with Epo or EpoL. Subsequently, the percentage of live cells was quantified.
2.5. Soluble oligomers of Aβ
The human Aβ1–40 was dissolved in Dimethyl sulfoxide (DMSO, Sigma- Aldrich) at a concentration of 80 μM and stored at −20 °C. The soluble oligomer solution was freshly prepared from a stock external solution and aggregated under standard conditions of 200 rpm at 37 °C for 2 h [28]. The final concentrations obtained for oligomers were 0.5 μM, 1 μM, and 5 μM. The oligomer species were previously described and confirmed [29,30]. Cells were treated for 24 h alone or co-incubated with Epo or EpoL extracts at different concentrations. The Aβ25-35 peptide was obtained from Sigma (Madrid, Spain), dissolved in phosphate buffer solution (PBS) and incubated with organotypic hippocampus cultures at different concentrations, according to the protocol previously described [31].
2.5.1. Cell viability assays
PC-12 cell cultures were seeded at a density of 90,000 cells/well and used 24 h after plating. After exposing the cells to each experimental condition, they were incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (1 mg/ml) for 30 min, and, thereafter, precipitated MTT was dissolved using isopropanol cooled for 15 min. The tetrazolium ring of MTT can be cleaved by active dehydrogenases in order to produce a precipitated formazan compound. Absorbance was measured in a multiplate reader (NovoStar, LabTech BMG, Germany) at two wavelengths: 560 nm and 620 nm, and the difference was quantified using NovoStar Software for the different experimental conditions.
2.5.2. Quantitative real time PCR
Total RNA of PC-12 cells treated was purified using TRIZOL (Sigma, USA), and the reaction was performed with the commercial kit KAPA SYBR FAST qPCR (KapaBiosystems, USA) and the equipment for Stratagene MX3000P (ThermoFisher, USA) real-time PCR. The qPCR was performed using RNA as a template, and the primers were ordered from Integrated DNA Technologies (Coralville, USA): BcL-2 (Forward: GATGACTGAGTA CCTGAACCG, Reverse: CAGAGACAGCCAGGAGAAATC) and β-actin (Forward: CACTTTCTACAATGAGCTGCG, Reverse: CTGGATGGCTACGTACATGG). The comparative threshold cycle values were normalized for the β-actin reference gene and the results were expressed as CT relative quantification by the 2-ΔΔCT method.
2.6. Organotypic hippocampal cultures
Organotypic hippocampal cultures were obtained from brains of 8–10 day old Sprague Dawley rats. Hippocampal slices (300 μm thick) were prepared and separated in ice-cold Hank's balanced salt solution (HBSS) composed of: glucose 15 mM, CaCl2 1.3 mM, KCl 5.36 mM, NaCl 137.93 mM, KH2PO4 0.44 mM, Na2HPO4 0.34 mM, MgCl2 0.49 mM, MgSO4 0.44 mM, NaHCO3 4.1 mM, HEPES 25 mM, 100 U/ml of penicillin, and 0.100 mg/ml of gentamicin. Four slices were placed on Millicell 0.4 μm culture inserts (Millipore, Spain) within each well of a six well culture plate with culture media. The culture media was composed of 50% minimal essential medium (MEM), 25% Hank's balanced salt solution, and 25% heat-inactivated horse serum (Life Technologies, Spain). After 4 days in culture, the slices were treated with β-amiloyd25-35 (Sigma-Aldrich, Spain) for 4 days, with or without Epo or EpoL at previously determined neuroprotective concentrations (Castillo et al., 2018). Cultures were maintained at 37 °C and 5% CO2, and the medium was changed twice a week.
2.6.1. Cell death measurement of organotypic cultures
At the end of each experiment, organotypic cultures were loaded with 1 μg/ml propidium iodide (PI) and Hoechst 33342 (Hoechst) for 30 min at 37 °C and 5% CO2. PI and Hoechst fluorescence from the cornu Ammonis 1 (CA1) region was measured using a Fluorescence inverted NIKON eclipse T2000-U microscope. Wavelengths of excitation and emission for PI and Hoechst were 530 or 350, and 580 or 460 nm, respectively. Fluorescence analysis was performed using Image J version 5.0 software. The amount of cell death was determined by the ratio between PI signal and Hoechst fluorescence. Data were normalized using basal values as 100%.
2.6.2. ROS measurement in organotypic cultures
The fluorescence probe CM-H2DCFDA (2′, 7′-dichlorodihydrofluorescein diacetate) was used to determine ROS. After the treatment, cultures were loaded with 5 μM CM-H2DCFDA and 1 μg/ml of Hoechst for 30 min at 37 °C and 5% CO2. CM-H2DCFDA crosses the cell membrane and is hydrolyzed by intracellular esterase to the non-fluorescent form, dichlorodihydrofluorescein. This reacts with ROS to form dichlorofluorescein, a green fluorescent dye. Fluorescence was measured on an inverted fluorescence microscope (inverted NIKON eclipse T2000-U microscope). Fluorescence analysis was performed using Image J version 5.0 software. ROS increase production in the CA1 region was determined by the ratio between DCFDA and Hoechst fluorescence. Data were normalized using basal values as 100%.
2.6.3. Immunofluorescence of hippocampus organotypic cultures
After treatments, hippocampal cultures were detached from the insert culture to evaluate differences in the expression of microglia (using ionized calcium binding adaptor molecule-1 protein, Iba-1, as a marker) and astrocytes (using glial fibrillary acidic protein, GFAP, as a marker). Slices were fixed using paraformaldehyde (4%) for 1 h and the sections were washed three times for 5 min with PBS. The slices were permeabilized using Triton (0.1%) solution for 10 min and blocked with normal goat serum and BSA for 1 h. The cultures were incubated with anti-Iba-1 (Wako Chemicals, Rafer S.L) or with an anti-GFAP (Millipore, Spain) antibody over-night at a dilution of 1:500. After washing with PBS, sections were incubated with anti-rabbit IgG or anti-mouse IgG secondary antibodies, respectively, for 2 h and 30 min at a dilution of 1:800. Then, slices were washed for three times for 5 min with PBS, applying in the second wash 1 μg/ml Hoechst 33342. The montage was made with Dako mounting medium (Agilent, USA). Images were taken on a confocal microscope (TCS SPE; Leica, Wetzlar, Germany) and analyzed using the software Image J and plugin Neuron, version 5.0.
2.6.4. Calcium transients
Calcium (Ca+2) transients were measured in cells loaded with Fluo4-AM (Invitrogen), a non-fluorescent acetoxymethyl ester calcium indicator. Hippocampal neurons were cultured on coverslips coated with poly-l-lysine (Sigma-Aldrich) and incubated with Fluo4-AM (5 μM) for 30 min at 37 °C. Thereafter, the neurons were washed twice and mounted on a perfusion chamber on a TE2000 microscope (Nikon). Fluo4-AM was excited at 480 nm wavelength, and the emission was recorded at 535 nm. Changes in cytosolic Ca+2 were registered with an EM CCD camera (iXon ANDOR) and a Lambda 10B (Sutter Instruments) interface. Regions of interest were simultaneously selected on neuronal somata containing Fluo-4 fluorescence in an optical field having usually more than 10 cells. The Ca+2 spikes were obtained as previously determined [32]; briefly, the spikes were analyzed individually from each neuron with an Imaging Workbench 6.0 software (Indec System), and the frequency for each experimental condition was obtained from the number of Ca+2 increases in the cell body during the recording time (200 s). The external solution used to incubate the dye and measure the frequency of Ca2+ oscillations contained (in mM): 150 NaCl, 5.4 KCl, 2.0 CaCl2, 1.0 MgCl2, 10 glucose, and 10 HEPES (pH 7.4, 330 mOsmol).
2.6.5. Statistical analysis
Data in the graphics is expressed as mean ± SEM. Statistical analyses of the results were performed using one-way ANOVA with Tukey test or Kruskal-Wallis and Dunns test, as appropriate. Statistical significance was considered as: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001 versus stressed cells (Aβ treatments); +p < 0.05, ++p < 0.01 and +++p < 0.001, ++++p < 0.0001 versus control group; #p < 0.05, ##p < 0.001, ###p < 0.001, ####p < 0.0001 versus treated cells (+Epo, +EpoL or + EpoL + Anti-EpoR).
3. Results
3.1. EpoL has neuroprotective effect against stress induced by chronic treatment with Aβ40
The neuroprotective effect was evaluated by the MTT assay in PC 12 cells co-incubated with soluble oligomers of Aβ40 (1 μM) for 24 h and increasing concentrations of EpoL or Epo (10; 50; 100 ng/mL). Aβ alone reduced cellular viability to 59.4% (±2.57). Co-incubations with EpoL or Epo achieved minimum neuroprotective effects at 10 and 100 ng/mL, respectively; whereas the maximum effect of EpoL was at 50 ng/mL with a 76.08% (±6.72) of cellular viability, and the maximum effect for Epo was at 100 ng/mL with a 66.27% (±6.12) of neuroprotection (Fig. 1A). These concentrations were selected to evaluate the involvement of the EpoR in neuroprotective effects. We then co-incubated neuronal cultures with an antibody (Ab) to block the interaction between hormone variants and the receptor (Fig. 1B). The neuroprotective effect of Epo and EpoL against Aβ was dependent on the interaction with the EpoR. Furthermore, we evaluated the anti-apoptotic pathways activated by Epo and EpoL co-incubation using qRT-PCR. The results demonstrated that neurons treated with Aβ in the presence of Epo or EpoL showed a significant increase in the expression of the antiapoptotic gene BcL-2, as measured by qPCR (Fig. 1C).
Fig. 1.

EpoL induced a protective effect against stress induced by chronic Aβ treatment. (A) Viability assay in PC12 cells treated with oligomers of Aβ40 (1uM) for 24 h and Epo or EpoL at different concentrations. The percentage of viability response was evaluated by MTT. (B) Viability assay using a specific antibody to block interaction between EpoR and EpoL or EpoL. (C) Evaluation of the relative expression of the Bcl-2 gene using β-actin as a housekeeping gene after 24 h of treatment with Aβ and co-treatment with Epo or EpoL, at the neuroprotective concentrations observed. Values are mean ± SEM, n = 3 using one-way ANOVA and Dunns test. *:p < 0.05; **:p < 0.01 versus PC12 cells treated with Ab; ++++:p < 0.0001 versus control cells.
3.2. EpoL co-incubation prevents the decrease in spontaneous intracellular calcium oscillations induced by Aβ40 treatment
To evaluate the effect of hormone variants on spontaneous calcium handling, primary hippocampal cultures were co-incubated with Epo or EpoL at neuroprotective concentrations previously determined, and Aβ oligomers for 24 h. The cells were then loaded with Fluo 4AM dye (Fig. 2A). Quantification of oscillation frequency, expressed as percentage of control (Fig. 2B), showed that Aβ treatment caused a significant decrease in this parameter to 33.96% (±4.45), whereas in cells co-incubated with Aβ and EpoL, the oscillation frequency percentage was restored to control levels (115% ± 5.9). Co-incubation with Epo did not show total recovery of the response (72.42% ± 1.2) as observed for EpoL (representative lines are shown in Fig. 2C).
Fig. 2.

EpoL treatment restored calcium homeostasis in neuronal cells exposed to Aβ. (A) Hippocampal neurons were treated for 24 h with oligomers of Aβ40 and co-incubated with Epo or EpoL at neuroprotective concentrations, as represented in the scheme on the top part of figure. (B) Quantification of percentage frequency of calcium transients using Fluo4AM loaded cells treated for 24 h with the different treatments. (C) Representative images of calcium transients over a 200 s period in cells exposed to the different indicated treatments. Values are mean ± SEM, n = 3 using one-way ANOVA and Dunns test. ***:p < 0.001; ****:p < 0.0001 versus neuronal cells treated with Aβ; ++++:p < 0.0001 versus control cells; ###:p < 0.001; ####:p < 0.0001 versus EpoL or Epo treatment.
Moreover, when cells were co-incubated with anti-EpoR plus EpoL and Aβ in the way that is described in Fig. 3A, the recovery of the intracellular calcium response previously described was lost. The quantification of the frequency calcium transients using anti-EpoR was 46.5% ± 14.08, similar to that observed with Aβ treatments (33.96% ± 4.45) (Fig. 3B), suggesting a relevant role for EpoR on EpoL effects. Representative time course of one neuron for each condition are shown in panel C of Fig. 3.
Fig. 3.

The intracellular calcium modulating effects of EpoL in Aβ treated neurons is EpoR dependent. (A) Hippocampal neurons were treated for 24 h with oligomers of Aβ40 and co-incubated with EpoL and a specific antibody at neuroprotective concentrations, as represented in the schematic. (B) Quantification of percentage frequency of calcium transients in Fluo4AM loaded cells treated for 24 h with Aβ or Aβ+EpoL or Aβ+ EpoL + Aβ, respectively. (C) Representative images of calcium transients during 200 s, under the indicated treatments. Values are mean ± SEM, n = 3 using one-way ANOVA and Dunns test. ****:p < 0.0001 versus neuronal culture treated with Aβ; ++++:p < 0.0001 versus control cells; ####:p < 0.0001 versus EpoL or Epo treatment.
3.3. EpoL treatment preserves key elements of synaptic architecture against Aβ40 synaptotoxicity
It has been widely observed that neurotoxicity is one of the most common signals for synaptic failure, and these events are associated to changes in protein levels of the synaptic machinery. For example, changes in levels of presynaptic proteins such as synaptic vessel 2 proteins (SV2) affect neurotransmitter release and could be correlated with the decrease in spontaneous calcium transients which reflects a post-synaptic response that could be a result of the toxicity induced by Aβ40 [33]. To test this hypothesis, we used an immunocytochemistry approach to measure the immunoreactivity of SV2 proteins (Fig. 4, left panel, green color). A decrease in SV2 levels were observed when the neurons were treated with Aβ (1 μM, 24h), and these results could help to explain part of the changes in Ca2+ frequency, as previously described [34]. Therefore, we decided to evaluate the effect of EpoL co-incubation on SV2 levels using immunofluorescence to determine the variations of this protein in neurons. Quantification of SV2 on Map-2 reactive cells (Fig. 4, left panel, red color) was performed. We observed a decrease in SV2 to 68.04% (±5.34) in Aβ treated neurons with respect to control conditions. When neurons were co-incubated with EpoL and Aβ, SV2 levels were similar to control values (99.83% ± 7.76), suggesting a protective effect (Fig. 4, right panel).
Fig. 4.

Neuroprotective effect of EpoL on synaptic markers induced by chronic treatments of Aβ. (A) Confocal images of neuronal cultures treated for 24 h with oligomers of Aβ40 and co-incubated with EpoL at neuroprotective concentrations to analyze Map2 and SV2. (B) The number of SV2 punctas observed in the first 20 μm of each neuron after 24 h of co-incubation is represented. Values are mean ± SEM, n = 3 using one-way ANOVA and Dunns test. ***:p < 0.001 versus neuronal culture treated with Aβ; +++:p < 0.001 versus control cells.
3.4. EpoL prevents ROS increase induced by Aβ25-35 chronic treatment in hippocampal organotypic cultures
Organotypic hippocampal cultures, obtained as previously described, were exposed for 4 days to Aβ25-35 (0.5 μM) and showed an increase in local ROS as measured by changes in the fluorescence of DFCDA, about 148 ± 4.1% in the CA1 region, with respect to control conditions (Fig. 5A, second frame). Using the same experimental approach, co-incubation with neuroprotective concentrations of Epo or EpoL (100 ng/mL) demonstrated a significant reduction in ROS induced by Aβ; while Epo elicited values near control conditions (114 ± 11.5%), EpoL maintained ROS production about 78 ± 4.14%, suggesting more efficient antioxidant properties. (Fig. 5 B). This antioxidant protection could represent a key event to avoid cell death in the hippocampus and induce a delay in neuronal network degeneration in relevant areas, such as CA1.
Fig. 5.

EpoL treatment decreased oxidative stress in hippocampal organotypic cultures. (A) Epifluorescence images of hippocampal organotypic cultures treated for 4 days with Aβ25-35 (0.5 μM) and co-incubated with EpoL or Epo at neuroprotective concentrations. (B) Images showing the fluorescence intensity of DCF-DA/Hoechst signal in CA1 region. Values are mean ± SEM, n = 5 using one-way ANOVA and Tukey test. ****:p < 0.0001 versus neuronal culture treated with Aβ; ++++:p < 0.001 versus control cells; ###:p < 0.001 versus Epo treatment.
3.5. EpoL prevents cell death induced by Aβ25-35 chronic treatment in hippocampal organotypic cultures
To corroborate the previous observation related with the antioxidant properties of EpoL, we analyzed cell death in hippocampal organotypic cultures treated for 4 days with Aβ25-35 (0.5 μM) oligomers using PI dye as a cell death indicator (see materials and methods). Using the same experimental conditions for Fig. 5, we observed that the treatment with Aβ alone induced an increment in PI intensity about 176 ± 17.4%, representing close to 80% more death than control conditions (Fig. 6A). On the other hand, co-incubation of the slices with EpoL reduced PI fluorescence to levels similar to control cultures (98 ± 14.1%). Surprisingly, in this model, co-incubation with Epo did not provide protection (191.9 ± 20.2%), showing very similar values to the Aβ condition and higher than control (Fig. 6B).
Fig. 6.

Neuroprotective effect of EpoL in hippocampal organotypic cultures against stress induced by chronic treatments of Aβ. (A) Epifluorescence images of hippocampal organotypic cultures treated for 4 days with Aβ25-35 (0.5 μM) and co-incubated with EpoL or Epo at neuroprotective concentrations. (B) Representative fluorescence images of PI/Hoechst signal of the CA1 region. Values are mean ± SEM, n = 5 using one-way ANOVA and Tukey test. ****:p < 0.0001 versus neuronal culture treated with Aβ; ++++:p < 0.001 versus control cells; ###:p < 0.001 versus Epo treatment.
3.6. The neuroprotective effect of EpoL in organotypic cultures is related to EpoL-EpoR interaction
To corroborate that these important effects observed in organotypic tissues were related with the interaction of EpoL with the receptor (EpoR), as was demonstrated in cell cultures, the same experimental approaches used in Fig. 5, Fig. 6 were replicated, using a specific anti-EpoR antibody (Ab) with the aim of blocking the interaction between EpoL and EpoR. As represented in Fig. 7, co-incubation with the anti EpoR antibody prevented the neuroprotective effects of EpoL in terms of its capacity to reduce ROS production (Fig. 7A), avoid neuronal death, and neuroprotection (Fig. 7B), showing no statistical differences with the Aβ condition. These data suggest that the EpoR could be an important part of the neuroprotective mechanism of EpoL.
Fig. 7.

The neuroprotective effect of EpoL in hippocampal organotypic cultures against Aβ stress depended on EpoR interaction: (A) Quantification of fluorescence signal intensity of DCF-DA/Hoechst on the CA1 region of hippocampus obtained by fluorescence images of hippocampi organotypic cultures treated for 4 days with Aβ25-35 (0.5 μM) and co-incubated with EpoL or EpoL + Anti-EpoR (Ab). (B) Quantification of PI/Hoechst signal of the CA1 region, as the same conditions used in A. Values are mean ± SEM, n = 7 using One-way ANOVA and Tukey test. ***:p < 0.001, “ns” versus Aβ; ###:p < 0.001 versus ctrl.
3.7. EpoL prevents glial activation induced by Aβ treatment in hippocampal organotypic cultures
It has been demonstrated in the CNS that an important factor to induce ROS is glial activation. Hence, we decided to examine the changes in astrogliosis levels to correlate with our previous observation regarding the capacity of EpoL to reduce ROS in cells treated with Aβ25-35. To evaluate this, we analyzed the morphological changes associated to astrogliosis induced by chronic treatment with Aβ25-35 in hippocampal organotypic cultures [31]. Immunofluorescence was assessed to determine variations on glial fibrillary acidic protein (GFAP) intensity signal according to the protocol shown in Fig. 8A. Immunofluorescence quantification of tissues treated with Aβ showed an increment of 60.2 ± 3% with respect to control conditions. However, co-incubation with EpoL and Aβ demonstrated a reduction to 27.7 ± 2%. A similar effect was found for Epo that showed a 26.5 ± 2% increase with respect to control (Fig. 8 B). These values were obtained from the analysis of the images shown in Fig. 8C, and correlate with results found in tissues under different experimental conditions shown in Fig. 8D. Taken together, these data suggest that the Anti-ROS effects of EpoL could be associated to the reduction in glial activation, and also to the intrinsic effects on neurons associated to one survival pathway through EpoR activation.
Fig. 8.

EpoL and Epo prevented astrogliosis induced by chronic treatment with Aβ. (A) Immunofluorescence confocal images of organotypic hippocampal cultures treated for 4 days with Aβ25-35 (0.5 μM) and co-incubated with EpoL or Epo at a neuroprotective concentration to analyze GFAP intensity signal. (B) Scheme of protocol used. (C) Mean intensity of GFPA fluorescence in the CA1 observed under each experimental condition using ImageJ software. Values are mean ± SEM, n = 3 using one-way ANOVA and Dunns test. ***:p < 0.001 versus culture treated with Aβ25-35; ++++:p < 0.001 versus control cells.
4. Discussion
The method used to produce erythropoietin in this study resulted in a new variant of Epo with a different glycosylation pattern characterized by neutral, bi-antennary and a-sialylated structures, termed EpoL. These characteristics demonstrated a marked reduction in hematopoietic activity as previously described [17,35]. In spite of this property, EpoL interacted with EpoR in the same way as Epo and activated the JAK/STAT pathway leading to overexpression of Bcl-2 family genes, similarly to Epo [17,24]. In our previous work, we demonstrated an improved effect of EpoL to protect CNS cells treated with oxidative agents in in vitro and ex vivo models, showing that EpoL was more potent than Epo to provide neuroprotection [17]. In this study, by using different chronic Aβ models, we have been able to show a similar neuroprotective profile of EpoL when compared to Epo. It is notable to point out that the concentrations required to achieve a similar neuroprotective pattern for EpoL was 10 times lower than that of Epo. Furthermore, the protective actions of EpoL were mediated by the receptor as demonstrated by the use of the anti-EpoR antibody. This result, together with our previous work performed in oxidative stress brain ischemia-models [17], further supports the notion that the neuroprotection actions of EpoL are receptor mediated.
Considering that EpoL and NonHem-EpoR interaction (expressed in CNS) is similar to the Epo/EpoR interaction, we decided to evaluate the effects of EpoL and EpoR activation on presynaptic (SV2 levels) and postsynaptic (Ca2+ levels) parameters as indicators of neuronal network functionality in primary hippocampal neurons [33,36]. We and others have established that cell death is recorded as a sustained calcium increase [37]. While calcium transients are an indicator of synaptic activity, as reflected in the activity induced through neurotransmitter release, Ca+2 entry into the post-synaptic component through ligand-activated and voltage dependent channels involves its access [33,38,39]. Therefore, calcium transients are correlated tightly with the health of the presynaptic component and together represents indicators of synaptic communication and network activity. It has been described that SV2 levels are altered in in vitro AD models [15], which represents a good indicator for presynaptic dysfunction, while a diminished frequency in calcium transients induced by chronic Aβ treatment has been correlated with neurotoxicity and neuronal death [15,28]. Nevertheless, Epo or EpoL co-incubations prevented this alteration in Ca+2 handling. In addition, EpoL showed a neuroprotective effect represented by preservation of normal intracellular Ca2+ oscillations, and its oscillation frequency was similar to control conditions. Interestingly, higher concentrations of Epo, compared to EpoL, were required to maintain an oscillation frequency similar to control, suggesting a major efficiency of EpoL to activate the EpoR. Because the anti-EpoR antibody abolished the effects of EpoL, we believe that the EpoR is relevant for the neuroprotective effects of EpoL (Fig. 3). Furthermore, this effect could be associated to the neuroprotective mechanism of EpoL, since it would reduce calcium overloading due to chronic exposure to Aβ.
The decreased frequency in calcium transients could be correlated to a diminished SV2 signal in neuronal cells treated with Aβ oligomers [30,40]. These effects were probably related to variations in intracellular ionic homeostasis and release of neurotransmitters. We observed neuroprotective effects of EpoL on SV2 levels in neurons co-treated with Aβ (Fig. 4). These results suggest another common neuroprotective mechanism between Epo and EpoL, according to the demonstrated effect of Epo on neurotoxicity induced by glutamate release [8,41]. Correlated with this evidence, the hippocampal organotypic cultures treated with Aβ showed decreased oxidative stress and cellular death when EpoL was co-incubated (Fig. 5, Fig. 6, respectively). Furthermore, EpoL prevented the increase in basal ROS induced in this ex vivo model because incubation with the non-hematopoietic variant significantly decreased the DCF-DA signal, with respect to control slices without EpoL treatment. Moreover, cell death was decreased with EpoL co-incubation, as measured by PI fluorescence; and this protective effect could be related to the induction of the antiapoptotic protein Bcl-2 (Fig. 1). As previously described with neuroprotective compounds that decrease ROS in organotypic cultures, Epo and EpoL could reduce oxidative stress by the activation of antioxidant pathways such as Nrf2 [22,31,42,43].
On the other hand, the amino acid composition of erythropoietin is the same as EpoL; it has a hydrophobic core which may alter the interaction of Aβ oligomers with the cellular membrane blocking the pore formation process, similar to other neuroprotective agents reported in in vitro treatments [44].
Finally, another indicator of oxidative stress and inflammatory process related to chronic Aβ exposure is astrogliosis, which is characterized by an atypical increase in the number and morphology of astrocytes due to nearby neuronal death [45]. Using the same previous experimental conditions, we evaluated different parameters of glial activation as a sign of chronic inflammatory processes related to neurodegenerative diseases and other diseases where there exists an increased oxidative stress, such as cerebrovascular infarct [44,[46], [47], [48]]. Here, we analyzed astrogliosis signs through the measurement of GFAP, which is altered with Aβ treatments. We found that EpoL was able to reduce astrogliosis, similar to our previous results using Epo, in agreement with Nrf2 and antiapoptotic pathways induced by neuroprotector agents (Fig. 8) [[49], [50], [51]].
5. Conclusions
In conclusion, the EpoL variant induced several beneficial effects in the CNS such as reducing redox imbalance, neuronal cell death, and diminished other toxicity markers like chronic inflammation and synaptic dysfunction. All these effects were mediated by the interaction with the EpoR and subsequent activation. Taken together, the post-transductional modifications of the EpoL variant promoted a specific interaction with the neuronal EpoR receptor, and represents a relevant finding that could lead to the development of a new pharmacological strategy associated to the first biological drugs designed to modulate neuronal EpoR in the fight against the deleterious effects of AD on the neuronal network. Therefore, our finding can help open a new chemical and biological space to develop anti-AD Drugs.
Acknowledgments
Grant Innova-Corfo “13IDL2-18688”; Conicyt Grant “Beca Doctorado Nacional no. 21130386”; Fondecyt Grant “1161078”.
Contributor Information
J.R. Toledo, Email: jotoledo@udec.cl.
J. Fuentealba, Email: jorgefuentealba@udec.cl.
References
- 1.Bunn H.F. Erythropoietin. Cold Spring Harb. Perspect. Med. 2013;3:a011619. doi: 10.1101/cshperspect.a011619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sinclair A.M. Erythropoiesis stimulating agents: approaches to modulate activity. Biol. Targets & Ther. 2013;7:161–174. doi: 10.2147/BTT.S45971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang J., Tian F., Cai Y., Qian X., Costello C.E., Ying W. Site-specific qualitative and quantitative analysis of the N- and O-glycoforms in recombinant human erythropoietin. Anal. Bioanal. Chem. 2014;406:6265–6274. doi: 10.1007/s00216-014-8037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jelkmann W. The enigma of the metabolic fate of circulating erythropoietin (Epo) in view of the pharmacokinetics of the recombinant drugs rhEpo and NESP. Eur. J. Haematol. 2002;69:265–274. doi: 10.1034/j.1600-0609.2002.02813.x. [DOI] [PubMed] [Google Scholar]
- 5.Jelkmann W. 'O', Erythropoietin carbamoylation versus carbamylation. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transp. Assoc. - Eur. Renal Assoc. 2008;23:3033. doi: 10.1093/ndt/gfn342. author reply 3033-4. [DOI] [PubMed] [Google Scholar]
- 6.Jelkmann W., Bohlius J., Hallek M., Sytkowski A.J. The erythropoietin receptor in normal and cancer tissues. Crit. Rev. Oncol.-Hematol. 2008;67:39–61. doi: 10.1016/j.critrevonc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Brines M., Cerami A. Emerging biological roles for erythropoietin in the nervous system. Nat. Rev. Neurosci. 2005;6:484–494. doi: 10.1038/nrn1687. [DOI] [PubMed] [Google Scholar]
- 8.Yu D., Fan Y., Sun X., Yao L., Chai W. Effects of erythropoietin preconditioning on rat cerebral ischemia-reperfusion injury and the GLT-1/GLAST pathway. Exp. Therapeut. Med. 2016;11:513–518. doi: 10.3892/etm.2015.2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumar A., Singh A., Ekavali A review on Alzheimer's disease pathophysiology and its management: an update. Pharmacol. Rep. PR. 2015;67:195–203. doi: 10.1016/j.pharep.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 10.Chong Z.Z., Li F., Maiese K. Erythropoietin requires NF-kappaB and its nuclear translocation to prevent early and late apoptotic neuronal injury during beta-amyloid toxicity. Curr. Neurovascular Res. 2005;2:387–399. doi: 10.2174/156720205774962683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Esmaeili Tazangi P., Moosavi S.M., Shabani M., Haghani M. Erythropoietin improves synaptic plasticity and memory deficits by decrease of the neurotransmitter release probability in the rat model of Alzheimer's disease. Pharmacol. Biochem. Behav. 2015;130:15–21. doi: 10.1016/j.pbb.2014.12.011. [DOI] [PubMed] [Google Scholar]
- 12.Arabpoor Z., Hamidi G., Rashidi B., Shabrang M., Alaei H., Sharifi M.R., Salami M., Dolatabadi H.R., Reisi P. Erythropoietin improves neuronal proliferation in dentate gyrus of hippocampal formation in an animal model of Alzheimer's disease. Adv. Biomed. Res. 2012;1:50. doi: 10.4103/2277-9175.100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Armand-Ugon M., Aso E., Moreno J., Riera-Codina M., Sanchez A., Vegas E., Ferrer I. Memory improvement in the AbetaPP/PS1 mouse model of familial Alzheimer's disease induced by carbamylated-erythropoietin is accompanied by modulation of synaptic genes. J. Alzheimer's Dis. JAD. 2015;45:407–421. doi: 10.3233/JAD-150002. [DOI] [PubMed] [Google Scholar]
- 14.Assaraf M.I., Diaz Z., Liberman A., Miller W.H., Jr., Arvanitakis Z., Li Y., Bennett D.A., Schipper H.M. Brain erythropoietin receptor expression in Alzheimer disease and mild cognitive impairment. J. Neuropathol. Exp. Neurol. 2007;66:389–398. doi: 10.1097/nen.0b013e3180517b28. [DOI] [PubMed] [Google Scholar]
- 15.Sepulveda F.J., Fierro H., Fernandez E., Castillo C., Peoples R.W., Opazo C., Aguayo L.G. Nature of the neurotoxic membrane actions of amyloid-beta on hippocampal neurons in Alzheimer's disease. Neurobiol. Aging. 2014;35:472–481. doi: 10.1016/j.neurobiolaging.2013.08.035. [DOI] [PubMed] [Google Scholar]
- 16.Saez-Orellana F., Godoy P.A., Bastidas C.Y., Silva-Grecchi T., Guzman L., Aguayo L.G., Fuentealba J. ATP leakage induces P2XR activation and contributes to acute synaptic excitotoxicity induced by soluble oligomers of beta-amyloid peptide in hippocampal neurons. Neuropharmacology. 2016;100:116–123. doi: 10.1016/j.neuropharm.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 17.Castillo C., Zaror S., Gonzalez M., Hidalgo A., Burgos C.F., Cabezas O.I., Hugues F., Jimenez S.P., Gonzalez-Horta E., Gonzalez-Chavarria I., Gavilan J., Montesino R., Sanchez O., Lopez M.G., Fuentealba J., Toledo J.R. Neuroprotective effect of a new variant of Epo nonhematopoietic against oxidative stress. Redox Biol. 2018;14:285–294. doi: 10.1016/j.redox.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen J., Wu Y., Xu J.Y., Zhang J., Sinclair S.H., Yanoff M., Xu G., Li W., Xu G.T. ERK- and Akt-dependent neuroprotection by erythropoietin (EPO) against glyoxal-AGEs via modulation of Bcl-xL, Bax, and BAD. Investig. Ophthalmol. Vis. Sci. 2010;51:35–46. doi: 10.1167/iovs.09-3544. [DOI] [PubMed] [Google Scholar]
- 19.Sargin D., Friedrichs H., El-Kordi A., Ehrenreich H. Erythropoietin as neuroprotective and neuroregenerative treatment strategy: comprehensive overview of 12 years of preclinical and clinical research. Best practice & research. Clinical Anaesthesiol. 2010;24:573–594. doi: 10.1016/j.bpa.2010.10.005. [DOI] [PubMed] [Google Scholar]
- 20.Ehrenreich H., Aust C., Krampe H., Jahn H., Jacob S., Herrmann M., Siren A.L. Erythropoietin: novel approaches to neuroprotection in human brain disease. Metab. Brain Dis. 2004;19:195–206. doi: 10.1023/b:mebr.0000043969.96895.3c. [DOI] [PubMed] [Google Scholar]
- 21.Ehrenreich H., Degner D., Meller J., Brines M., Behe M., Hasselblatt M., Woldt H., Falkai P., Knerlich F., Jacob S., von Ahsen N., Maier W., Bruck W., Ruther E., Cerami A., Becker W., Siren A.L. Erythropoietin: a candidate compound for neuroprotection in schizophrenia. Mol. Psychiatr. 2004;9:42–54. doi: 10.1038/sj.mp.4001442. [DOI] [PubMed] [Google Scholar]
- 22.Buendia I., Gomez-Rangel V., Gonzalez-Lafuente L., Parada E., Leon R., Gameiro I., Michalska P., Laudon M., Egea J., Lopez M.G. Neuroprotective mechanism of the novel melatonin derivative Neu-P11 in brain ischemia related models. Neuropharmacology. 2015;99:187–195. doi: 10.1016/j.neuropharm.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 23.Chen J., Yang Z., Zhang X. Carbamylated erythropoietin: a prospective drug candidate for neuroprotection. Biochem. Insights. 2015;8:25–29. doi: 10.4137/BCI.S30753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma R., Hu J., Huang C., Wang M., Xiang J., Li G. JAK2/STAT5/Bcl-xL signalling is essential for erythropoietin-mediated protection against apoptosis induced in PC12 cells by the amyloid beta-peptide Abeta25-35. Br. J. Pharmacol. 2014;171:3234–3245. doi: 10.1111/bph.12672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lourhmati A., Buniatian G.H., Paul C., Verleysdonk S., Buecheler R., Buadze M., Proksch B., Schwab M., Gleiter C.H., Danielyan L. Age-dependent astroglial vulnerability to hypoxia and glutamate: the role for erythropoietin. PLoS One. 2013;8 doi: 10.1371/journal.pone.0077182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Millet A., Bouzat P., Trouve-Buisson T., Batandier C., Pernet-Gallay K., Gaide-Chevronnay L., Barbier E.L., Debillon T., Fontaine E., Payen J.F. Erythropoietin and its derivates modulate mitochondrial dysfunction after diffuse traumatic brain injury. J. Neurotrauma. 2016;33:1625–1633. doi: 10.1089/neu.2015.4160. [DOI] [PubMed] [Google Scholar]
- 27.Rodriguez Cruz Y., Strehaiano M., Rodriguez Obaya T., Garcia Rodriguez J.C., Maurice T. An intranasal formulation of erythropoietin (Neuro-EPO) prevents memory deficits and amyloid toxicity in the APPSwe transgenic mouse model of Alzheimer's disease. J. Alzheimer's Dis. JAD. 2017;55:231–248. doi: 10.3233/JAD-160500. [DOI] [PubMed] [Google Scholar]
- 28.Sepulveda F.J., Parodi J., Peoples R.W., Opazo C., Aguayo L.G. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS One. 2010;5 doi: 10.1371/journal.pone.0011820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuentealba J., Dibarrart A.J., Fuentes-Fuentes M.C., Saez-Orellana F., Quinones K., Guzman L., Perez C., Becerra J., Aguayo L.G. Synaptic failure and adenosine triphosphate imbalance induced by amyloid-beta aggregates are prevented by blueberry-enriched polyphenols extract. J. Neurosci. Res. 2011;89:1499–1508. doi: 10.1002/jnr.22679. [DOI] [PubMed] [Google Scholar]
- 30.Parodi J., Sepulveda F.J., Roa J., Opazo C., Inestrosa N.C., Aguayo L.G. Beta-amyloid causes depletion of synaptic vesicles leading to neurotransmission failure. J. Biol. Chem. 2010;285:2506–2514. doi: 10.1074/jbc.M109.030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buendia I., Parada E., Navarro E., Leon R., Negredo P., Egea J., Lopez M.G. Subthreshold concentrations of melatonin and galantamine improves pathological AD-hallmarks in hippocampal organotypic cultures. Mol. Neurobiol. 2016;53:3338–3348. doi: 10.1007/s12035-015-9272-5. [DOI] [PubMed] [Google Scholar]
- 32.Fuentealba J., Dibarrart A., Saez-Orellana F., Fuentes-Fuentes M.C., Oyanedel C.N., Guzman J., Perez C., Becerra J., Aguayo L.G. Synaptic silencing and plasma membrane dyshomeostasis induced by amyloid-beta peptide are prevented by Aristotelia chilensis enriched extract. J. Alzheimer's Dis. JAD. 2012;31:879–889. doi: 10.3233/JAD-2012-120229. [DOI] [PubMed] [Google Scholar]
- 33.Andoh T., Echigo N., Kamiya Y., Hayashi M., Kudoh I., Goto T. Effects of erythropoietin on intracellular calcium concentration of rat primary cortical neurons. Brain Res. 2011;1387:8–18. doi: 10.1016/j.brainres.2011.02.077. [DOI] [PubMed] [Google Scholar]
- 34.Aly K.A., Beebe E.T., Chan C.H., Goren M.A., Sepulveda C., Makino S., Fox B.G., Forest K.T. Cell-free production of integral membrane aspartic acid proteases reveals zinc-dependent methyltransferase activity of the Pseudomonas aeruginosa prepilin peptidase PilD. MicrobiologyOpen. 2013;2:94–104. doi: 10.1002/mbo3.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toledo J.R., Sanchez O., Segui R.M., Garcia G., Montanez M., Zamora P.A., Rodriguez M.P., Cremata J.A. High expression level of recombinant human erythropoietin in the milk of non-transgenic goats. J. Biotechnol. 2006;123:225–235. doi: 10.1016/j.jbiotec.2005.10.019. [DOI] [PubMed] [Google Scholar]
- 36.Assandri R., Egger M., Gassmann M., Niggli E., Bauer C., Forster I., Gorlach A. Erythropoietin modulates intracellular calcium in a human neuroblastoma cell line. J. Physiol. 1999;516(Pt 2):343–352. doi: 10.1111/j.1469-7793.1999.0343v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fuchsberger T., Martinez-Bellver S., Giraldo E., Teruel-Marti V., Lloret A., Vina J. Abeta induces excitotoxicity mediated by APC/C-Cdh1 depletion that can Be prevented by glutaminase inhibition promoting neuronal survival. Sci. Rep. 2016;6:31158. doi: 10.1038/srep31158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Simms B.A., Zamponi G.W. Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron. 2014;82:24–45. doi: 10.1016/j.neuron.2014.03.016. [DOI] [PubMed] [Google Scholar]
- 39.Turner R.W., Zamponi G.W. T-type channels buddy up. Pflueg. Arch. Eur. J. Physiol. 2014;466:661–675. doi: 10.1007/s00424-013-1434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramirez A.E., Pacheco C.R., Aguayo L.G., Opazo C.M. Rapamycin protects against Abeta-induced synaptotoxicity by increasing presynaptic activity in hippocampal neurons. Biochim. Biophys. Acta. 2014;1842:1495–1501. doi: 10.1016/j.bbadis.2014.04.019. [DOI] [PubMed] [Google Scholar]
- 41.Gu L., Xu H., Wang F., Xu G., Sinha D., Wang J., Xu J.Y., Tian H., Gao F., Li W., Lu L., Zhang J., Xu G.T. Erythropoietin exerts a neuroprotective function against glutamate neurotoxicity in experimental diabetic retina. Investig. Ophthalmol. Vis. Sci. 2014;55:8208–8222. doi: 10.1167/iovs.14-14435. [DOI] [PubMed] [Google Scholar]
- 42.Calabrese V., Cornelius C., Dinkova-Kostova A.T., Calabrese E.J., Mattson M.P. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxidants Redox Signal. 2010;13:1763–1811. doi: 10.1089/ars.2009.3074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buendia I., Egea J., Parada E., Navarro E., Leon R., Rodriguez-Franco M.I., Lopez M.G. The melatonin-N,N-dibenzyl(N-methyl)amine hybrid ITH91/IQM157 affords neuroprotection in an in vitro Alzheimer's model via hemo-oxygenase-1 induction. ACS Chem. Neurosci. 2015;6:288–296. doi: 10.1021/cn5002073. [DOI] [PubMed] [Google Scholar]
- 44.Peters C., Fernandez-Perez E.J., Burgos C.F., Espinoza M.P., Castillo C., Urrutia J.C., Streltsov V.A., Opazo C., Aguayo L.G. Inhibition of amyloid beta-induced synaptotoxicity by a pentapeptide derived from the glycine zipper region of the neurotoxic peptide. Neurobiol. Aging. 2013;34:2805–2814. doi: 10.1016/j.neurobiolaging.2013.06.001. [DOI] [PubMed] [Google Scholar]
- 45.Magaki S.D., Williams C.K., Vinters H.V. Glial function (and dysfunction) in the normal & ischemic brain. Neuropharmacology. 2018 May 15;134(Pt B):218–225. doi: 10.1016/j.neuropharm.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Iwai M., Stetler R.A., Xing J., Hu X., Gao Y., Zhang W., Chen J., Cao G. Enhanced oligodendrogenesis and recovery of neurological function by erythropoietin after neonatal hypoxic/ischemic brain injury. Stroke. 2010;41:1032–1037. doi: 10.1161/STROKEAHA.109.570325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zurita M.P., Munoz G., Sepulveda F.J., Gomez P., Castillo C., Burgos C.F., Fuentealba J., Opazo C., Aguayo L.G. Ibuprofen inhibits the synaptic failure induced by the amyloid-beta peptide in hippocampal neurons. J. Alzheimer's Dis. JAD. 2013;35:463–473. doi: 10.3233/JAD-122314. [DOI] [PubMed] [Google Scholar]
- 48.Pasqualetti G., Brooks D.J., Edison P. The role of neuroinflammation in dementias. Curr. Neurol. Neurosci. Rep. 2015;15:17. doi: 10.1007/s11910-015-0531-7. [DOI] [PubMed] [Google Scholar]
- 49.Egea J., Buendia I., Parada E., Navarro E., Rada P., Cuadrado A., Lopez M.G., Garcia A.G., Leon R. Melatonin-sulforaphane hybrid ITH12674 induces neuroprotection in oxidative stress conditions by a 'drug-prodrug' mechanism of action. Br. J. Pharmacol. 2015;172:1807–1821. doi: 10.1111/bph.13025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nekoui A., Blaise G. Erythropoietin and nonhematopoietic effects. Am. J. Med. Sci. 2017;353:76–81. doi: 10.1016/j.amjms.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 51.Wang R., Wu X., Liang J., Qi Z., Liu X., Min L., Ji X., Luo Y., Zhao H. Intra-artery infusion of recombinant human erythropoietin reduces blood-brain barrier disruption in rats following cerebral ischemia and reperfusion. Int. J. Neurosci. 2015;125:693–702. doi: 10.3109/00207454.2014.966354. [DOI] [PubMed] [Google Scholar]