

Breast cancer angiogenesis and metastasis are major, unresolved clinical challenges. We studied EPAC1, and elucidate tumor secretome-driven metastatic properties in the tumor-endothelium. Using a primary human tumor culture, we show that iMVD is dynamic under drug pressure and may associate with treatment response.
Abstract
Breast cancer is a leading cause of cancer-related mortality in women. Triple-negative breast cancer (TNBC; HER2−, ER−/PR−) is an aggressive subtype prone to drug resistance and metastasis, which is characterized by high intratumor microvascular density (iMVD) resulting from angiogenesis. However, the mechanisms contributing to the aggressive phenotypes of TNBC remain elusive. We recently reported that down-regulation of exchange factor directly activated by cyclic AMP (cAMP), also known as EPAC1, leads to a reduction in metastatic properties including proliferation and cell migration in TNBC cell lines. Here, we report that EPAC1 supports TNBC-induced angiogenesis, tumor cell migration and invasiveness as well as pro-metastatic phenotypes in endothelial cells induced through the tumor secretome. Using an approach that integrates proteomics with bioinformatics and gene ontologies, we elucidate that EPAC1 supports a tumor-secreted network of angiogenic, cell adhesion and cell migratory pathways. Using confocal microscopy, we show that signaling molecules involved in focal adhesion, including Paxillin and MENA, are down-regulated in the absence of EPAC1, and electric cell substrate impedance sensing technique confirmed a role for EPAC1 on TNBC-induced endothelial cell permeability. Finally, to provide a translational bridge, we studied iMVD and therapy response using a primary human tumor explant assay, CANscriptTM, which suggests a link between therapy-modulated neovascularization and drug sensitivity. These data provide mechanistic insight into the role of EPAC1 in regulating the tumor microenvironment, iMVD and cancer cell-induced angiogenesis, a dynamic mechanism under drug pressure that may associate to treatment failure.
Introduction
Breast cancer is the most common cancer in women, behind skin cancer, and remains the second leading cause of cancer-related mortality (1). This aggressive tumor type is classified into four broad subtypes: estrogen receptor positive (ER+), progesterone receptor positive (PR+), HER2 positive (HER2+) and triple-negative breast cancer (TNBC) characterized by an absence of ER, PR and HER2 expression. Indeed, TNBC has one of the highest metastatic potentials of all the subtypes, which is tightly associated to the high degree of vascularity feeding the tumor, known as intratumor microvascular density (iMVD) (2). In the absence of abundant cell surface receptors for targeted therapy, key intracellular molecular players that drive TNBC are being sought as druggable targets.
In an effort to study the drivers of TNBC aggressiveness, emerging evidence points to a role for cyclic AMP (cAMP), a key secondary messenger, in breast cancer progression (3). cAMP is involved in several signaling pathways, including those responsible for the regulation of cell proliferation, differentiation and migration (4,5). cAMP can act through two signaling axes: the PKA-dependent and the PKA-independent (6). Indeed, the effect of PKA-independent pathway modulation through exchange factor directly activated by cAMP (EPAC1) is a well-established mode of regulating the networks (7). EPAC1 is a guanine nucleotide exchange factor, which is involved in activity of Ras family proteins, specifically Rap1 and Rap2 (7,8). Furthermore, EPAC1 is implicated in several biological functions such as cell survival, cell junction interactions, secretions, cell–cell adhesion, differentiation and cell migration (7–10). However, the role of EPAC1 in metastasis is yet to be resolved.
As an important driver of metastasis, new blood supply to the tumor via neo-angiogenesis is a fundamental biological process that cancer cells can hijack (10). Cancer cell-secreted factors in the tumor microenvironment can cause induction of new blood vessels and tumor cell dissemination (11–13). Since the spread of tumors at a distant secondary site necessarily requires the initiation of angiogenesis to sustain the tumor mass, anti-angiogenic therapy has been employed to target highly metastatic cancers such as TNBC (14). Despite these advances, little progress has been made to thwart metastasis, pin-point drivers and ‘lynch-pins’ of angiogenesis, and target these pathways to improve survival.
In this study, we interrogated the role of EPAC1 on the angiogenic and metastatic potential of TNBC using an endothelial cell in vitro model. We hypothesized that EPAC1 contributes to several phenotypic alterations in tumor cells including migratory and invasive features, as well as angiogenic potential of the tumor microenvironment and vasculature via the secretome. To test this hypothesis, we used gene knockdown studies and interrogated the effect of tumor cell secretome on iMVD, in vitro. Using gene ontology, we report a cell signaling network between cAMP-modulated EPAC1 and secreted molecular proteins that regulate neo-vascularization, migration, locomotion and blood vessel development. We further demonstrate a potential role for cell adhesion and matrix proteins including Paxillin, MENA and tubulin. With the help of a primary human tumor explant platform, we determined that iMVD is dynamic under drug pressure, and associates to predicted clinical response to therapy. Together, these findings demonstrate that EPAC1 supports the microvascular density of aggressive breast cancers, as well as the angiogenic and metastatic properties within the tumor microenvironment, which may underpin response or resistance to therapy.
Material and methods
Antibodies and reagents
Antibodies purchased from commercial sources were EPAC1 (Cat no. 4155S, CST, 1:1000), Paxillin (Cat no. 612405, BD Biosciences, 1:500), Phalloidin-Actin (Cat. no. 8953, CST, 1:500), MENA (Cat no. NBPI-87914, NOVUS, 1:1000), Tubulin (Cat no. T5168, Sigma, 1:2000), secondary anti-mouse (Cat no. 554002, BD, 1:2000) and rabbit HRP (Cat no. 554021, BD, 1:2000). Cell culture insert (Cat no. MCEP12H48, Millipore), Calcein –AM (Cat no. 564061, BD Biosciences) and Matrigel (Cat no. 356237, BD Biosciences) were purchased for co-culture and angiogenesis studies. HUVEC were purchased from LONZA (C2519A), Lipofectamine 2000 (Cat no.11668027), Pen-Strep (Cat no. 15070063), trypsin (Cat no. 25200056), Antifade reagent with DAPI (Cat no P36935) and CFSE (Cat no. C34570) were purchased from Invitrogen (Thermo). MDA-MB-231 cells were purchased from NCCS, India. L-15 Media (Cat no. Al011A), FBS (RM10681) were acquired from HiMEDIA Laboratories India. 8-O-Me-cAMP (Cat no. 4853, Tocris) and Microchips (8W10E) for vascular permeability analysis were purchased from ibidi. CD34 antibody was obtained from the commercial vendor abcam, clone EP373Y.
Cell culture conditions
MDA-MB-231 cells were cultured in 25 cm2 vented culture flasks in L-15 medium supplemented with 10% fetal bovine serum. Cells were passaged twice a week and media was changed before each experiment. HUVEC cells were grown and cultured as per guided by manufacturers.
In vitro gene transfection
MDA-MB-231 cells were grown in a six-well plate for 24 h before transfection. siEPAC1 and all-star negative control siRNA were used with Lipofectamine-2000 for transfection. Cells were washed thrice with Opti-Mem and incubated with transfection mix for 4 h. After 4 h, media was replaced with fresh complete L-15 media and cells were kept for 48 h without disturbance. Cells were harvested and lysed for the analysis of Protein expression through western blot.
Preparation of conditioned media (CM)
MDA-MB-231 cells were grown in a six-well plate overnight (O/N) and transfected with siEPAC1 and control siRNA as discussed in above section. Conditioned media (CM) was collected and stored immediately at −80°C and used further for other experiments. Conditioned media (4 ml) was concentrated to 200 µl using 3 kDa filter and 1× phosphate buffer saline (PBS) buffer. Firstly, 3 kDa filter was equilibrated with 1× PBS buffer by centrifugation (4000 rpm for 30 min) and then conditioned and unconditioned media were added along with 1× PBS buffer in separate filters in 1:3 ratio (1 ml sample and 3 ml 1× PBS buffer) and centrifuged (4000 rpm for 30 min.). Flow through were discarded and same procedure was repeated three times until all media were passed. Finally, media were concentrated to 200 µl. After concentrating both conditioned and unconditioned media protein amount was quantified by Bradford assay and approximately 200 µg equivalent of protein lysate was used for angiogenesis array experiment.
Western blot analysis
Transfected cells were analyzed for the expression of EPAC1 and other proteins. MDA-MB-231 cells were harvested and lysed with lysis buffer (0.5 % SDS, 50 mM Tris–HCl, 1 mM EDTA). Cell lysate were loaded at 20 μg/well and separated by 10 and 12.5% SDS-PAGE for 3 h at 120 V. Gel was transferred on to the polyvinylidene difluoride (PVDF) membrane for 1.5 h and membrane was blocked with blocking buffer (5% skim milk in PBS with 0.1% Tween-20) for 1 h. Membrane was incubated with primary antibodies (material section) in PBST O/N at 4°C. Next day, membrane was washed with PBST and incubated with secondary antibodies. Membrane was imaged with Alpha Imager system (Protein Simple). Results were analyzed and intensities were calculated using ImageJ software (15).
Immunofluorescence assay
To explore the effect of down regulation of EPAC1 on MDA-MB-231 cells, we performed cellular imaging. Cells were seeded on coverslips and transfected with siEPAC1 as mentioned above. After 48 h, cells were washed with cold 1× PBS and fixed with 4% formaldehyde (Sigma, Cat no 47408) for 10 minutes. 1× PBS wash was given after every step until mounting with DAPI. Cells were permeabilized and blocked with TritonX-100 and 1 % FBS-1× PBS, respectively. Cells were incubated with primary antibodies (material section) for 2 h and respective secondary antibodies (Alexa Fluor 488 cat no. A11029, molecular probes and Alexa Fluor 594 cat no. A11012) for 2 h in dark. Later, coverslips were sealed with vector shield DAPI and imaged with Nikon confocal A1microscope. Green laser (excitation 490 and emission 525) and Red laser (excitation 591 and emission 614) were used to capture the images.
In vitro invasion assay
Invasive phenotype of cancer cells were analyzed with the culture insert (Corning). Culture inserts were coated with Matrigel (50 µg/ml/well) and incubated overnight at 37°C in CO2 incubator. Transfected MDA-MB-231 cells were trypsinized and seeded in a density of 2.5 × 105 cells/ml in 200 µl media on to the insert with incomplete L-15 medium. Lower chamber of insert was filled with L-15 media (10 % FBS). After 24 h, cells were washed with 1× PBS. Fixing and staining was performed with methanol and crystal violet, respectively. Images of the invaded cells were captured by Leica Microscope. A total of three random areas from each well were selected and the experiment was repeated in triplicates.
2D angiogenesis assay
Tumor-induced angiogenesis assay was performed with primary cells (HUVEC). Initially matrigel was thawed overnight at 4°C and coated (100 µl/well) in 96-well plate. After the incubation of plate at 37°C, HUVECs were trypsinized and mixed with CM of transfected MDA-MB-231 cells and cultured for next 8 h. There were four experimental groups for the assay namely (A) HUVEC cells with EBM media, (B) HUVEC cells with growth factor Midkine (positive control), (C) HUVEC cells with siEPAC1 transfected CM from MDA-MB-231 cells and (D) HUVEC cells with CM from scrambled siRNA transfected MDA-MB-231. Images were taken after 8 h incubation. Capillary structure was captured and tube length, nodes were quantified using AngioTool (16).
3D co-culture assay
To investigate the angiogenic tube formation in a tumor microenvironment condition, we performed a 3D co-culture assay. Before starting the assay, we transfected MDA-MB-231 cells with siEPAC1 as mentioned in above sections and loaded with CFSE dye for 30 min in dark. HUVECs and MDA-MB-231 cells were trypsinized and 5000 cells/100 µl was inter mixed with 100 µl matrigel and incubated in dark in a chamber slide. A media change was given carefully after 50 min and cells were kept in incubator for 2 days while media was changed every alternate day. Images were captured by Leica microscope and angiogenic tube sprouting of HUVEC in culture with MDA-MB-231 was quantified manually.
Angiogenesis profile assay assessment
Tissue culture media (4 ml) of transfected MDA-MB-231 cells was collected and concentrated as mentioned above. Concentrated protein was quantified by Bradford assay and pour on to the membranes following manufacturer instructions. Approximately 200 µg equivalent of protein was taken for array experiment from both control and siEPAC1 transfected tissue culture media. Proteome profiling for angiogenic proteins was performed according to manufacturer’s protocol. Results were analyzed using ImageJ software (15). Angiogenic proteome profiling was done two times (biological duplicate).
Electric cell-substrate impedance sensing (ECIS)-based analysis
To analyze the effect of cancer cells on vascular permeability in primary cells, we performed ECIS assay. Electric Cell-substrate Impedance Sensing (Applied Biophysics) is a real-time analysis of cellular activities in culture system. It is an impedance-based system to check the morphological, migratory and vascular leakage properties of cultured cells (17). HUVECs were plated on 8W10E microchips, pre-coated with gelatin. After 24 h, CM from siEPAC1 transfected cells along with control transfected cells were poured in to the wells. HUVECs without any CM were used as control for treated cells. In one well, 8-O-ME cAMP treatment was given for 20 min and after removal of 8-O-ME cAMP, CM of EPAC1 transfected cells was used. Effect on changes in electric impedance of the monolayer was monitored at every 10 s. Endothelial cell permeability in the in vitro mimic condition of cancer metastasis was observed until 48 h and analyzed by ECIS software.
Human explant studies
Anonymous human TNBC tissues were assessed by CANscriptTM using fresh specimen. All samples were taken from patients refractory to taxane-containing regimens and varying stages of disease were obtained from Mitra Biotech collected under IRB approval from HCG Bangalore Institute of Oncology with due consent. Fresh tumor tissues were collected immediately after surgical resection. The tumor samples were transported to the laboratory at 4°C, in appropriate transport buffer within 60 min post-resection, for ex vivo studies and molecular and pathological evaluation. Tissues were cut into thin sections and cultured in tissue culture plates using optimized conditions with matrix proteins that match the tumor indication and grade. Tumors were treated with a taxane (docetaxel) at clinical max concentration (Cmax) for 72 h. DMSO was used as a vehicle control. After treatment, tumor cell viability was measured. Immunohistochemistry (IHC) was performed as described in methods. As a quality control measure, each tumor tissue contains at least 20% tumor content, which is evaluated by clinical pathologists. Predicting response to therapy was performed using a clinically trained algorithm that was described previously (18).
Immunohistochemical analysis
Changes in CD34+ was determined prior to and after drug treatment evaluated by IHC using specific antibodies. Initial antigen retrieval of FFPE sections was done in Antigen Unmasking Solution (Citrate based, Vector Laboratories) by exposure to microwave heating for 30 min. Quenching of endogenous peroxidase was done by 3% H2O2 for 15 min. Protein blocking was carried out at room temperature (RT) for 1 h with 10% goat serum. FFPE sections were incubated with primary antibodies at RT for 1 h. Rabbit monoclonal CD34 antibody (Abcam) was used at a 1:200 dilution for 1 h at RT-all followed by incubation with HRP-conjugated secondary antibody (SignalStain® Boost IHC Detection Reagent; Cell Signaling Technology) for 1 h at RT. Chromogenic development of signal was done using 3,3ʹ-diaminobenzidine (DAB Peroxidase Substrate Kit; Vector Laboratories). Tissues were counterstained with Hematoxylin (Papanicolaous solution 1a). Scoring and calculations were performed by quantifying the number of contiguous CD34+ ‘nodes’ in the vehicle-treated and drug-treated IHC in individual fields of view from each IHC slide and expressed as CD34+/field.
Results
Downregulation of EPAC1 in TNBC cells reduces angiogenesis in HUVEC culture models, in vitro
We first asked whether TNBC-related EPAC1 contributes to angiogenesis. To address this, we employed an in vitro model that harnesses human vascular endothelial cells (HUVEC) grown in culture with media that was pre-conditioned overnight by the TNBC cell line, MDA-MB-231. We began with four experimental groups: (i) HUVEC cells with EBM media, (ii) HUVEC cells with growth factor Midkine (positive control), (iii) HUVEC cells with siEPAC1 transfected CM from MDA-MB-231 cells and (iv) HUVEC cells with CM from scrambled siRNA transfected MDA-MB-231. Using an endothelial in vitro cell culture approach, we quantified the number of sprouted ‘tubes’ and ‘nodes’, which characterize the degree of angiogenesis and therefore neovascular likelihood (19). As expected, Midkine, the positive control, resulted in statistically increased angiogenic properties of HUVEC compared to unconditioned media, as indicated by sprouted tubes and nodes. Indeed, increased tubes and nodes in HUVEC culture was also observed when media from control siRNA transfected MDA-MB-231 cells was introduced to culture suggesting the secretome of TNBC cells induces angiogenic features in endothelial cell culture (Figure 1A and C). Interestingly, we observed an arrest in formation of tubes and nodes in HUVEC cells under the effect of siEPAC1 transfected MDA-MB-231 cells media (Figure 1C, *P < 0.05, N = 3). Taken together, these data indicate that EPAC1 mediates a key component of the signaling axis related to TNBC-induced angiogenesis in endothelial cells. siRNA-mediated downregulation of EPAC1 is shown by western blot (Figure 1B).
Figure 1.

Down regulation of EPAC1 reduces angiogenesis. (A) Representative images from tube formation assay was performed to check the effect of silencing EPAC1 in MDA MB 231 cells on endothelial cells. EPAC1 was silenced by siRNA in MDA-MB-231 and tissue culture media (TCM) was collected and incubated with HUVEC’s. Midkine was used as a positive control, unconditioned media was used to demonstrate vascular growth in the absence of tumor cells. (B) siRNA downregulation of EPAC1 shown by western blot and relative intensity quantitation of EPAC1 western blot shown by bar graph. (C) Tube length and nodes formation quantified and represented. Scale bar indicates 100 µm. Total tube length and number of nodes are calculated using AngioTool, which indicate a significant inhibition in vascular development (*P < 0.05 compared to control, N = 3).
Downregulation of tumor cell EPAC1 reduces invasion and angiogenic sprouting in vitro
Next, we wanted to interrogate the role of EPAC1 as it confers angiogenic properties in a tumor-endothelial cell co-culture experiment. To do this, we employed a 3-D co-culture assay, which was performed with HUVECs in the presence of siEPAC1- and control siRNA-transfected MDA-MB-231 cells that are pre-labeled with a fluorescent dye (e.g. CSFE). Incubating cells together, we determined that fewer endothelial cell sprouting events occurred in HUVEC cells (CSFEneg) when cultured with siEPAC1-transfected MDA-MB-231 cells with respect to control cells (Figure 2A, *P < 0.05, N = 3). To support these findings, we analyzed tumor cell invasion using a trans-well assay (see Methods for details). While our observations were consistent with previous evidence that TNBC are highly invasive, we determined, based on the number of invading MDA-MB-231 through micropores, EPAC1 knockdown led to diminished invasion compared to wild-type (siRNA control) conditions (Figure 2B, *P < 0.05 N = 3). This finding suggests that EPAC1 may play a critical role in both neovascularization (via the secretome) and invasive properties of tumor cells into the vasculature, two features that are key in driving metastasis.
Figure 2.

Down regulation of EPAC1 reduces Invasion and sprouting in a 3D co-culture model. (A) Representative image showing the sprouting assay, which was tested in co-culture of HUVEC and MDA-MB-231 in the presence or absence of EPAC1 (siRNA) (*P < 0.05). MDA-MB-231 cells were stained by CFSE dye to differentiate between cell lines (green signal). Right panel shows quantification (*P < 0.05, N = 3). (B) Representative images from transwell migration assay show siRNA transfected cells were analyzed for their invasive properties by Transwell migration assay. Right panel shows number of invading cells determined by visual quantification (*P < 0.05, N = 3).
Loss of EPAC1 leads to dysregulation of pro- and anti-angiogenic proteins in the tumor secretome
Based on the evidence above, we suspected extracellular signals derived from tumor cells mediate the angiogenic behavior we observed in HUVEC. Therefore, we interrogated the secretome of tumor cells treated with siEPAC1 or a scrambled control. We employed a proteome array to profile angiogenic proteins from CM of siEPAC1 and control siControl MDA-MB-231 cells. As shown in the waterfall and volcano plots, we identified numerous statistically significantly downregulated proteins in the TNBC secretome after silencing EPAC1 including fibroblast growth factor family members (FGF-4 and FGF-a) and transforming growth factor (TGF) family member TGFb and complement protein, amphiregulin (Figure 3A–C). Furthermore, we identified a small number of proteins were increased (most notably SERPINE1 and Thrombospondin-1) (Figure 3B and C). To understand the relationship between the proteins dysregulated in the siEPAC1 TNBC secretome, we performed gene ontology (GO) analysis. Based on a statistically significant false discovery rate (FDR < 0.001), we determined that downregulation of EPAC1 leads to an increase in proteins that are involved in negative regulation of adhesion, blood vessel morphogenesis and angiogenesis (Figure 3C). Proteins that positively regulate cell migration and locomotion were found to be significantly downregulated after silencing EPAC1 (Figure 3D, *P < 0.05).
Figure 3.

Angiogenic proteome profiling of tissue culture media (TCM). (A) Representative image of proteome array. (B) Waterfall plot shows Log2 fold change in the optical density of protein spots as quantified using ImageJ software (siEPAC1 vs. scrambled control). (C) Volcano plot shows the proteins that are statistically significantly upregulated by a log2 fold change of 0.1 or higher (blue bars). Red bar indicates the cut-off for statistical significance, P < 0.05. (D) Gene ontology networks were constructed using the highest (upper panel) and lowest (lower panel) differentially expressed proteins between EPAC1 and control determined using the STRING consortium open source software, false discovery rate was set P < 0.001. Proteome array was performed in duplicate and biological replicate on two independent occasions (N = 4).
EPAC1 downregulation affects cellular adhesion, blood vessel development and intracellular network scaffolding properties
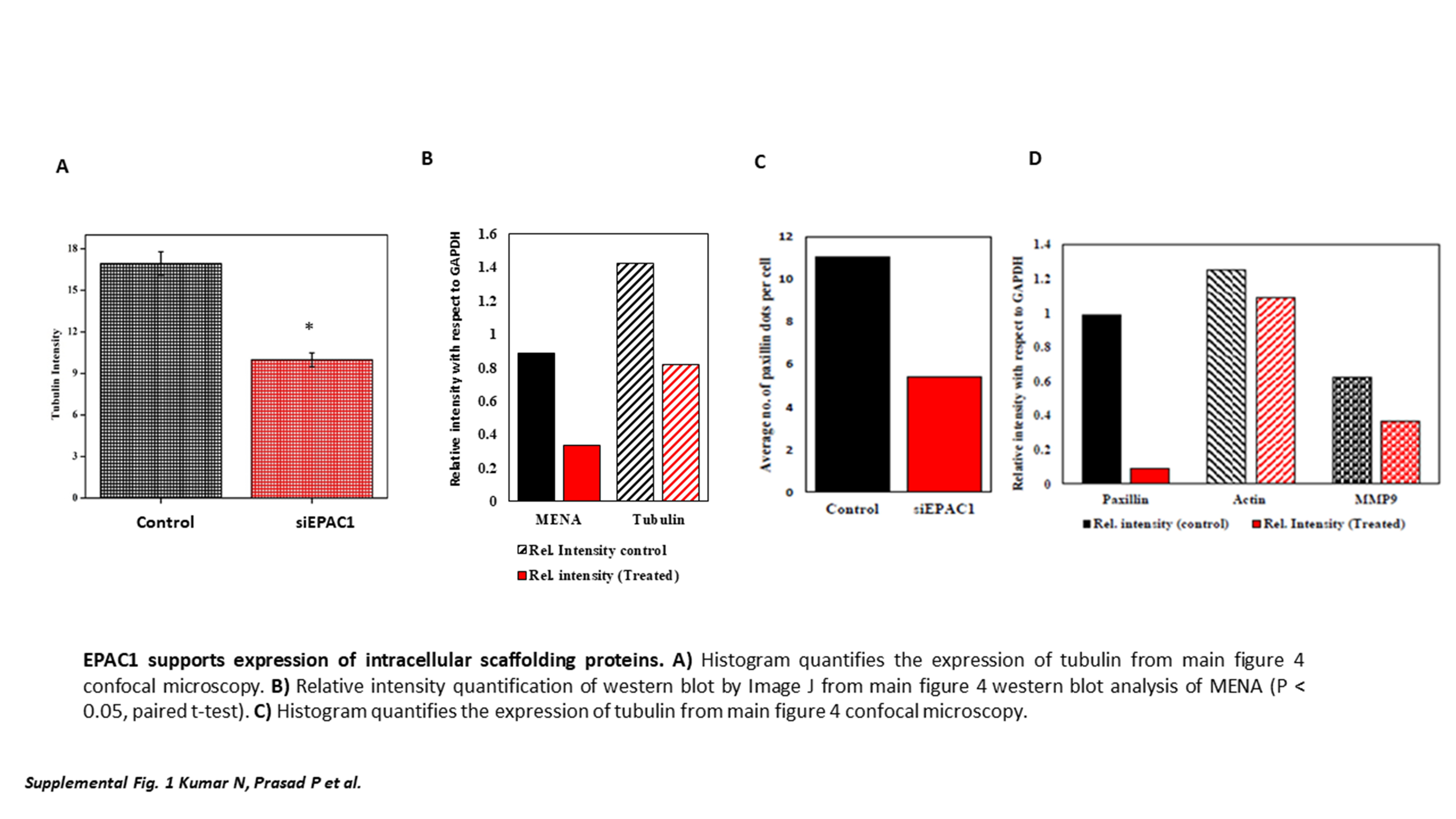
Next, we sought to interrogate proteins that are associated with the intracellular scaffold network, which may drive cell migration and locomotion. Using confocal microscopy, we profiled focal adhesion proteins including MENA and Paxillin, which are associated with malignant migratory features (20,21). MENA protein spots were found to be localized at the periphery in siRNA control cells, which are diminished when EPAC1 is downregulated (Figure 4A). This evidence was confirmed by western blot analysis (Figure 4B). Confocal analysis also indicated the dysregulation of the cytoskeleton marker tubulin. ImageJ analysis of tubulin was done to confirm the difference in control and siEPAC1 treated cells (*P < 0.05, N > 3 replicates from multiple fields) (Figure 4A; Supplementary Figure 1A, available at Carcinogenesis Online), which was confirmed by western blot (Figure 4B; Supplementary Figure 1B, available at Carcinogenesis Online). Furthermore, we identified a diminished intensity and lower incidence of Paxillin observed by confocal imaging (Figure 4C; Supplementary Figure 1C, available at Carcinogenesis Online), which were also confirmed by western blot analysis (Figure 4D; Supplementary Figure 1D, available at Carcinogenesis Online).
Figure 4.

Down regulation of EPAC1 modulates intracellular scaffolding support. (A) Representative confocal image depicts expression and localization of tubulin, MENA in siRNA-treated cells. (a) Overlay of Tubulin and MENA (control); enlarged inset image (1) Immunostained by MENA; (2) Immunostained by Tubulin (3) Merged MENA and Tubulin. (b) Overlay of Tubulin and MENA (siEPAC1); Enlarged inset image (1) Immunostained by MENA; (2) Immunostained by Tubulin (3) Merged image of MENA and Tubulin. (B) Western blot analysis was performed on siEPAC1 and siControl TNBC cells to profile expression of MENA and tubulin. (C) Representative immunofluorescence of actin and paxillin: (a) overlay of paxillin and actin (Control); Enlarged inset (1) immunostained by actin; (2) immunostained by paxillin; (3) Merged image of actin and paxillin. (b) Overlay of actin and paxillin (siEPAC1); Enlarged inset (1) Immunostained by paxillin; (2) Immunostained by actin; (3) Merged image of paxillin and actin. (D) Western blot analysis was performed on siEPAC1 and siControl TNBC cells to profile expression of MMP9, actin and paxillin.
Role of EPAC1 and the cytoskeleton in HUVEC in vascular permeability
In order to study dynamic changes in cell motility and locomotion, we interrogated the permeability of endothelial cells, which infers changes to the intracellular cytoskeleton and can be performed using ECIS (17). We plated endothelial cells (HUVEC) on an ECIS microchip following standard protocol to interrogate metastasis (22). The cells were allowed to spread and form junctions and cell–cell contact on the chip. Conditioned media of control siRNA transfected MDA-MB-231 cells changed the junctional properties and increased the vascular permeability of the endothelial cell monolayer (Figure 5, magenta trace). We activated signaling by addition of 8-pCPT-2-O-Me-cAMP-AM and found tightening of endothelial cell monolayer which is evidenced by increased normalized resistance (Figure 5, red trace). Interestingly, silencing EPAC1 modulated this increased permeabilization (Figure 5, blue trace).
Figure 5.

Down-regulation of EPAC1 in tumor cells prevents the increase in permeability in primary endothelial cells (ECIS). Representative trace showing HUVECs plated on 8W10E microchips, pre-coated with gelatin. After 24 h conditioned media of EPAC1, siRNA transfected cells along with negatively transfected cells were poured in the wells. HUVEC without any conditioned media were used as control for treated cells. In one well 8-OME cAMP treatment was given for 20 min and after removal of 8-OME cAMP, CM of EPAC1 transfected cells were used. Effect on changes in electric impedance of the monolayer was taken at every sec. Cell permeability analysis was observed until 48 h.
Ex vivo tumor culture associates microvascular density with response to therapy
To this point, we have developed comprehensive evidence to support EPAC1 as a putative ‘lynch-pin’ for microvascular density, invasion and angiogenesis, which contribute to metastatic features of the tumor-endothelium. To provide a clinical translation of iMVD in the context of breast cancer treatment, we first studied the role of microvascular density in primary human tumor tissue. Recent evidence indicates that iMVD may be prognostic following treatment with conventional anticancer agents, including cytotoxic chemotherapy (23). Therefore, we employed a recently-described human cancer explant assay that closely represents clinical response and resistance to therapy (CANscriptTM) (18). Explants were generated from fresh tumor biopsy isolated from TNBC and hormone receptor positive patients, and cultured ex vivo on matched tumor matrix proteins supplemented with autologous patient plasma. The explants were treated with cytotoxic chemotherapies at clinically reported Cmax doses for 72 h, and evaluated for: (1) predicted response to therapy using a clinically trained algorithm (i.e. M-Score) (18), and (2) CD34+ neovasculature nodes by IHC (24) (Figure 6A and B). Primarily, we confirmed a high incidence of iMVD in TNBC tissue compared to hormone receptor positive samples (Figure 6C and D *P < 0.05). By quantifying incidence of CD34+ expression from IHC, our preliminary evidence suggests that response to therapy may be linked to baseline expression of CD34+ vascular nodes (Figure 6E). Indeed, we determined that a reduction in CD34+ nodes associates to poor response to therapy, which was predicted by the clinically trained algorithm (Figure 6E).
Figure 6.

Ex vivo evidence associates microvascular density and therapy response. A human tumor explant assay was used to study intratumor microvascular density iMVD in the presence or absence of conventional breast cancer therapy. (A) Schematic representing the tumor culture assay. The explant live tissue assay reliably recreates the entire tumor ecosystem including microenvironment components, stroma and immune contexture in which drugs are introduced (1). A multi-dimensional phenotypic interrogation is performed to predict the clinical response to therapy (2). (B) Schematic illustrates the steps taken to study neovascularization in the explant model. Briefly, IHC is performed to study CD34+ microvascular nodes, which are quantified per field in the vehicle control cohort and the drug treated cohort (docetaxel). (C) Table represents patient indications, subtypes and other demographic information. (D) Representative IHC are shown in the left panel, quantitated CD34+ nodes/field are shown in the right panel. N = 4 biological replicates, P < 0.05 by student’s t-test. (E) Predicted responders (M-score ≥26) and predicted non-responders (M-score ≤25) were quantified for CD34+ per field, and represented as pairwise between the vehicle control and drug-treatment. Lower panel compares the change of CD34+/field between responders and non-responders. N = 5 biological replicates.
Discussion
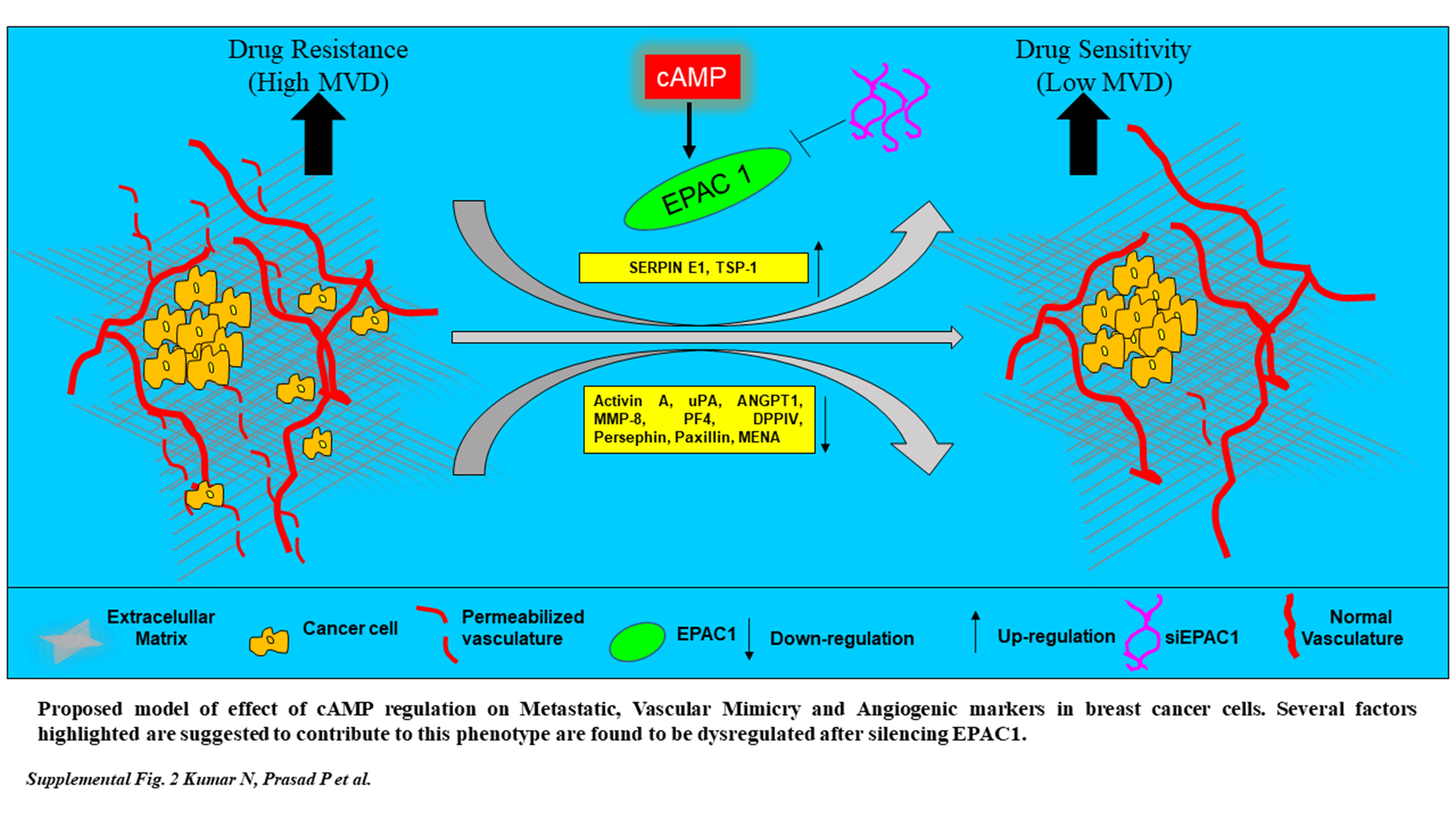
Neovascularization and angiogenesis are key features of metastasis and cancer-related mortality. Our recent evidence that EPAC1 associates to tumor dissemination lead us to study its effect on angiogenic and invasive properties. To do this, we employed molecular techniques that allow us to modulate expression of EPAC1 and interrogate phenotypic effects using in vitro models of TNBC. Using a human breast cancer ex vivo assay, we also provide evidence that dynamic alterations to iMVD and neo-vascularization under drug pressure, determined using CD34+ expression and a quantitative clinical pathology approach, may provide clues to therapy response and failure. Together, these findings provide evidence that link biomarkers of metastasis with iMVD and therapy failure, features that may be supported by a key ‘lynch-pin’, EPAC1 (see proposed schematic, Supplementary Figure 2, available at Carcinogenesis Online).
Vascular mimicry is a process in which cancer cells acquire endothelial cell-like properties to aid metastatic potential (25). Interestingly, serine proteases (i.e. SERPIN family of proteins) are known to program cancer cells for vascular mimicry (26). Indeed, our proteome profiling analysis identified a link between SERPIN (E1) and EPAC1. Although our profile did not provide a statistical link between SERPINE1 overexpression, when we combined that evidence with our identification of thrombospondin 1, we determined there is a statistically significant network of proteins that negatively regulate migration and angiogenesis. Indeed, these features are highly associated with high iMVD and breast cancer metastasis (27). Future examination of SERPINE1 may elucidate a signaling network that underpins aggressive features of angiogenesis, and therefore a potential druggable target.
Interestingly, evidence in other tumor types, such as pancreatic ductal adenocarcinoma, suggests that EPAC1 is involved in migration and invasion (4). In fact, conditioned medium of melanoma cells with high expression of EPAC1 causes increased migration of HUVEC through Fibroblast growth factors (FGF2) (28). Similarly, our study has provided preliminary evidence that FGF may play a role, in-concert with other proteins, to drive EPAC1-mediated angiogenesis and metastatic phenotypes. Indeed, several FGF family members are modulated in EPAC1-diminished MDA-MB-231 cells, which classifies some of the earliest angiogenic-factors described in breast cancer and is also involved in metastasis progression (29). In addition to these growth factors, we noted statistically significantly downregulated proteins in siRNA EPAC1 treated MDA-MB-231 cells including IGFBP3, Activin-A and Amphiregulin, which have been loosely associated with poor prognosis for metastatic cancer (30). We speculate that these growth factors may harmonize to provide a substrate for angiogenic potential, which is dysregulated in TNBC versus luminal breast cancers. Further examination of these associations may be warranted.
Excitingly, our preliminary evidence from ex-vivo culture suggests a link between CD34+ incidence and neovascularization (angiogenesis), which may predispose response to therapy, thus bridging prognosis of aggressive cancers, such as TNBC, with iMVD and metastasis. Indeed, previous studies have shown that iMVD can be reliably measured by quantifying the occurrence of CD34 marker, and that MVD can be used to evaluate the rate of neoangiogenesis in breast cancer (31). Our evidence that CD34 is dynamically altered under drug pressure, and is potentially tied to clinical response, is a key finding that may link the expression of EPAC1. However, more work needs to be done in order to pinpoint this relationship. These findings have now driven us to explore how microvascular density and other precursor biomarkers of angiogenesis may contribute to treatment response or failure. Such an ex vivo assay may help elucidate the role of antiangiogenic drugs, such as bevacizumab, in breast cancer and other tumor indications.
In addition, our study provides a comprehensive look into the mechanisms of EPAC1-related tumor metastasis. Key protein networks that underpin this effect may provide clues and new druggable targets. With the use of CANscript and tumor modeling ex vivo, we are able to study how the entire tumor ecosystem, biomarkers of neovascularization and dynamic changes to the microenvironment contribute to early dissemination of tumor cells and treatment response. Together with angiogenic molecular profiling, we anticipate a concerted effort to target these proteins could overcome the challenges associated with metastatic breast cancer.
Supplementary material
Supplementary material can be found at Carcinogenesis online.
{kind=link}
{kind=link}
Acknowledgments
Seema Sehrawat is the recipient of BioCARe Award from Department of Biotechnology, Ministry of Science and Technology, Govt. of India. Shiv Nadar Foundation is acknowledged for providing the PhD fellowship to Mr. Naveen Kumar, Mr. Peeyush Prasad. Aaron Goldman is supported by the Breast Cancer Alliance Young Investigator Award.
Conflict of Interest: AG is an employee of Mitra Biotech and holds equity. All other authors declare no conflict of interest.
Abbreviations
- cAMP
cyclic AMP
- ECIS
electric cell-substrate impedance sensing
- IHC
immunohistochemistry
- iMVD
intratumor microvascular density
- PBS
phosphate buffer saline
- TNBC
triple-negative breast cancer
References
- 1. Sauer A.G., et al. (2017)Updated review of prevalence of major risk factors and use of screening tests for cancer in the United States. Cancer Epidemiol. Biomarkers Prev., 26, 1192–1208. [DOI] [PubMed] [Google Scholar]
- 2. Hudis C.A., et al. (2011)Triple-negative breast cancer: an unmet medical need. Oncologist, 16 (Suppl. 1), 1–11. [DOI] [PubMed] [Google Scholar]
- 3. Yan K., et al. (2016)The cyclic AMP signaling pathway: exploring targets for successful drug discovery (review). Mol. Med. Rep., 13, 3715–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Almahariq M., et al. (2013)A novel EPAC-specific inhibitor suppresses pancreatic cancer cell migration and invasion. Mol. Pharmacol., 83, 122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grandoch M., et al. (2009)Epac inhibits migration and proliferation of human prostate carcinoma cells. Br. J. Cancer, 101, 2038–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Seino S., et al. (2005)PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol. Rev., 85, 1303–1342. [DOI] [PubMed] [Google Scholar]
- 7. de Rooij J., et al. (1998)Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature, 396, 474–477. [DOI] [PubMed] [Google Scholar]
- 8. Kawasaki H., et al. (1998)A family of cAMP-binding proteins that directly activate Rap1. Science, 282, 2275–2279. [DOI] [PubMed] [Google Scholar]
- 9. Kumar N., et al. (2017)Role of exchange protein directly activated by cAMP (EPAC1) in breast cancer cell migration and apoptosis. Mol. Cell. Biochem., 430, 115–125. [DOI] [PubMed] [Google Scholar]
- 10. Schneider B.P., et al. (2005)Angiogenesis of breast cancer. J. Clin. Oncol., 23, 1782–1790. [DOI] [PubMed] [Google Scholar]
- 11. Longatto Filho A., et al. (2010)Angiogenesis and breast cancer. J. Oncol., 2010, 576384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bielenberg D.R., et al. (2015)The contribution of angiogenesis to the process of metastasis. Cancer J., 21, 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Vasudev N.S., et al. (2014)Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis, 17, 471–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Le Tourneau C., et al. (2007)Sunitinib: a novel tyrosine kinase inhibitor. A brief review of its therapeutic potential in the treatment of renal carcinoma and gastrointestinal stromal tumors (GIST). Ther. Clin. Risk Manag., 3, 341–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schindelin J., et al. (2012)Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zudaire E., et al. (2011)A computational tool for quantitative analysis of vascular networks. PLoS One, 6, e27385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wegener J., et al. (2000)Electric cell-substrate impedance sensing (ECIS) as a noninvasive means to monitor the kinetics of cell spreading to artificial surfaces. Exp. Cell Res., 259, 158–166. [DOI] [PubMed] [Google Scholar]
- 18. Majumder B., et al. (2015)Predicting clinical response to anticancer drugs using an ex vivo platform that captures tumour heterogeneity. Nat. Commun., 6, 6169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. DeCicco-Skinner K.L., et al. (2014)Endothelial cell tube formation assay for the in vitro study of angiogenesis. J. Vis. Exp., 91, e51312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gurzu S., et al. (2013)The possible role of mena protein and its splicing-derived variants in embryogenesis, carcinogenesis, and tumor invasion: a systematic review of the literature. Biomed. Res. Int., 2013, 365192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. López-Colomé A.M., et al. (2017)Paxillin: a crossroad in pathological cell migration. J. Hematol. Oncol., 10, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pouliot N, Pearson HB, Burrows A. (2000–2013) Investigating metastasis using in vitro platforms. In: Madame Curie Bioscience Database [Internet]. Landes Bioscience, Austin, TX. [Google Scholar]
- 23. Weidner N. (2008)Measuring intratumoral microvessel density. Methods Enzymol., 444, 305–323. [DOI] [PubMed] [Google Scholar]
- 24. Sidney L.E., et al. (2014)Concise review: evidence for CD34 as a common marker for diverse progenitors. Stem Cells, 32, 1380–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Angara K., et al. (2017)Vascular mimicry: a novel neovascularization mechanism driving Anti-Angiogenic Therapy (AAT) resistance in glioblastoma. Transl. Oncol., 10, 650–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wagenblast E., et al. (2015)A model of breast cancer heterogeneity reveals vascular mimicry as a driver of metastasis. Nature, 520, 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Guidi A.J., et al. (1994)Microvessel density and distribution in ductal carcinoma in situ of the breast. J. Natl. Cancer Inst., 86, 614–619. [DOI] [PubMed] [Google Scholar]
- 28. Baljinnyam E., et al. (2014)Epac1 increases migration of endothelial cells and melanoma cells via FGF2-mediated paracrine signaling. Pigment Cell Melanoma Res., 27, 611–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Korc M., et al. (2009)The role of fibroblast growth factors in tumor growth. Curr. Cancer Drug Targets, 9, 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Weidner N., et al. (1991)Tumor angiogenesis and metastasis–correlation in invasive breast carcinoma. N. Engl. J. Med., 324, 1–8. [DOI] [PubMed] [Google Scholar]
- 31. Bottini A., et al. (2002)Changes in microvessel density as assessed by CD34 antibodies after primary chemotherapy in human breast cancer. Clin. Cancer Res., 8, 1816–1821. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.