

Abstract
Pigmentation is often used to understand how natural selection affects genetic variation in wild populations since it can have a simple genetic basis, and can affect a variety of fitness-related traits (e.g., camouflage, thermoregulation, and sexual display). In gray wolves, the K locus, a β-defensin gene, causes black coat color via a dominantly inherited KB allele. The allele is derived from dog-wolf hybridization and is at high frequency in North American wolf populations. We designed a DNA capture array to probe the geographic origin, age, and number of introgression events of the KB allele in a panel of 331 wolves and 20 dogs. We found low diversity in KB, but not ancestral ky, wolf haplotypes consistent with a selective sweep of the black haplotype across North America. Further, North American wolf KB haplotypes are monophyletic, suggesting that a single adaptive introgression from dogs to wolves most likely occurred in the Northwest Territories or Yukon. We use a new analytical approach to date the origin of the KB allele in Yukon wolves to between 1,598 and 7,248 years ago, suggesting that introgression with early Native American dogs was the source. Using population genetic simulations, we show that the K locus is undergoing natural selection in four wolf populations. We find evidence for balancing selection, specifically in Yellowstone wolves, which could be a result of selection for enhanced immunity in response to distemper. With these data, we demonstrate how the spread of an adaptive variant may have occurred across a species’ geographic range.
Keywords: CBD103, gray wolf, allele age, adaptive introgression, melanism, sequence capture
Introduction
Understanding how polymorphisms are generated and maintained is a key goal of evolutionary biology. One of the most distinct polymorphic traits in nature is coloration (Wright 1917), which is generally determined by the spatial distribution of pigmented hairs, feathers, or scales across the body and by pigment type (Wright 1917; Protas and Patel 2008; Manceau et al. 2010). In mammals, two main types of pigments control coloration, eumelanin (black/brown) and pheomelanin (red/yellow) (Protas and Patel 2008). Switching between these two pigment types is often controlled by the Agouti (ligand)–Mc1r (receptor) pathway (Protas and Patel 2008). Coloration can serve several functions, including camouflage, intraspecific and interspecific signaling, and thermoregulation (Wright 1917; Little 1958; Protas and Patel 2008), and can be subject to positive selection (e.g., Hoekstra and Nachman 2003). Additionally, melanin-based coloration genes are associated with physiological and behavioral traits through pleiotropic effects (Ducrest et al. 2008). Although the evolution of color polymorphism is inherently interesting for the varied functions mentioned above, it is also key to understanding evolutionary processes in general (Svensson 2017). If the causative alleles for phenotypic variation are known, the adaptive function of such alleles could potentially be determined in a natural evolutionary context (e.g., Barrett and Hoekstra 2011).
Melanism in gray wolves (Canis lupus) represents a novel system for understanding how demography and selection can change the patterns of adaptive variation across the entire geographic range of a species. Although many melanistic phenotypes are caused by mutations in Agouti or Mc1r, in the gray wolf melanism is manifested by a novel mutation in the β-defensin CBD103 gene (also referred to as the K locus). This gene encodes an alternative ligand for Mc1r that outcompetes a functioning Agouti ligand if present (Candille et al. 2007; Kerns et al. 2007; Anderson et al. 2009). The KB allele contains a 3 bp deletion that confers a dominantly inherited black (melanistic) coat color phenotype, whereas the wild-type ky allele confers a gray (agouti) coat color in homozygotes (Anderson et al. 2009). A previous study of 47 Arctic and Yellowstone wolves from forest and tundra/taiga habitats by Anderson et al. (2009) used a data set of 52 SNPs to show that the K locus experienced a strong selective sweep in black wolves after an initial introgression event from dogs >500 years ago (Leonard et al. 2002; Anderson et al. 2009). The evidence for a selective sweep, detailed in Anderson et al. (2009), consisted of a near absence of polymorphism within 60 kb of the KB allele in wolves, yet the wild-type ky allele was highly polymorphic. Additionally, the authors compared coalescent times, using a molecular clock approach, as a function of increasing distance from the K locus in dogs and wolves, and showed that the KB allele was introduced into North American wolves from dogs (Anderson et al. 2009). Research over the last several years has clarified the genetic basis and potential fitness advantage of melanism in wolves (e.g., Anderson et al. 2009; Coulson et al. 2011; Stahler et al. 2013; Cassidy et al. 2017), yet details of the evolutionary history, such as the number, timing, and geographic origin of introgression events, remain unknown.
In wolves, the target of selection for black coat color seems unrelated to camouflage, intraspecific signaling, or thermoregulation (e.g., Anderson et al. 2009; Coulson et al. 2011). Specifically, wolf coat color varies from black to white, with tawny variations, and with higher frequencies of lighter phenotypes in the high Arctic where the background vegetation is paler (Musiani et al. 2007). However, gray and black individuals often exist within the same populations and at similar frequencies over time, suggesting that mechanisms such as fitness tradeoffs maintain the polymorphism (Anderson et al. 2009; Coulson et al. 2011; Hedrick et al. 2016). In Yellowstone National Park, where wolf coat color has been intensively studied, the estimated frequency of black wolves in the population remained constant over a 19-year period (1996–2014) at 0.45 (Hedrick et al. 2016). The maintenance of these frequencies has been explained by a strong heterozygous advantage for the KB allele (Coulson et al. 2011; Hedrick et al. 2014), and recent evidence suggests strong disassortative mating for coat color (Hedrick et al. 2016). Demonstrated fitness differences among color genotypes in the Yellowstone population includes highest annual survival, lifespan, and lifetime reproductive success for black heterozygotes and lowest survival, reproductive success, and frequency for black homozygotes (Coulson et al. 2011). In contrast, gray homozygous females show higher annual reproductive success (Stahler et al. 2013). Such fitness variation among all three genotypes suggests pleiotropic effects of variants at the coat color locus (e.g., Ducrest et al. 2008) and that coat color may not be the proximate object of selection given the difference in fitness between the heterozygous and homozygous black coat color genotypes. In this regard, the K locus is a member of the β-defensin family of antimicrobial peptides (Pazgier et al. 2006) and may be involved in adaptive immune response (Yang et al. 1999). K locus mRNA is expressed in canine skin and respiratory tract (Candille et al. 2007; Erles and Brownlie 2010), and mRNA levels are correlated with increased antimicrobial activity against Bordetella bronchiseptica, a respiratory pathogen (Erles and Brownlie 2010). Thus, fitness trade-offs involving the costs and benefits of immunity under varying environmental conditions (Coulson et al. 2011; Stahler et al. 2013) and the fitness of specific mating pairs (Hedrick et al. 2016) may determine the frequency of each genotype. Additionally, the frequency of a SNP in linkage disequilibrium (LD) with the 3 bp deletion is significantly and positively associated with maximum temperature of warmest month across North American wolf ecotypes (Schweizer, vonHoldt, et al. 2016), and temperature and pathogen prevalence are positively related in a multitude of species (Allen et al. 2002; Guernier et al. 2004; Dionne et al. 2007), further supporting the notion that selection within the K locus likely involves immunity.
Initial studies of the K locus have raised further questions concerning the geographic origin, number, and age of introgression events that could not be addressed previously due to limited genomic and geographic sampling. Consequently, we designed a custom hybridization capture array to perform extensive resequencing of a five megabase (Mb) region surrounding the K locus, as well as neutral and control genic regions to assess genomic changes due to demography alone. We sequenced these regions in a large geographic sample of North American wolves and 20 domestic dog breeds. We assessed patterns of nucleotide and haplotype diversity, population-specific decay in linkage disequilibrium (LD), and hierarchical patterns of genetic divergence among populations, and used a novel method to determine when the KB allele likely originated in each of our sampled populations. With these data, we reconstruct the evolutionary history of the black genotype and find a single origin, followed by a rapid spread across North America. We hypothesize that this spread may have been accelerated by the presence of dogs and mesocarnivores as reservoirs for pathogens to which the black locus provided enhanced immunity. Lastly, our population genetic simulations demonstrate that coat color is currently undergoing natural selection in four different wolf populations.
Results
Targeted Capture Sequencing of Dogs and Wolves
We designed a custom hybridization capture array to assay sequence variation surrounding the K locus, and in neutral, genic, telomeric, and nontelomeric regions (fig. 1, see Methods and Supplementary Material online). These presumed neutral, telomeric and nontelomeric regions provide an empirical background for summary statistics, since the K locus is located close to the telomere on chromosome 16 and may show a positional bias in genetic diversity. After target enrichment, sequencing, and alignment to the CanFam3.1 dog reference, all individuals sequenced (n = 382) retained approximately ≥60% of the target regions with at least 25× coverage (mean ± 1SD: 150× ± 61×) (supplementary table S1 and fig. S1, Supplementary Material online). After genotype filtering and quality control, 10,922,248 positions with 162,815 variants and 12,774 insertions/deletions were retained. Based on data from 109 individuals previously genotyped on the Affymetrix Dog SNP array, we calculated 99.7% genotype concordance for heterozygotes (supplementary table S2, Supplementary Material online). After additional quality control filters (see Supplementary Material online), 353 samples were used for further analysis (table 1 and supplementary fig. S2, Supplementary Material online). Pedigree data from 203 Yellowstone wolves was used to aid phasing of haplotypes among all 353 samples using SHAPEIT software (supplementary fig. S3 and table S3, Supplementary Material online).
Fig. 1.

Resequencing strategy and heterozygosity values within sequenced regions. (A) Capture of the K locus indel mutation (triangle above chromosome) and surrounding region with parallel telomeric and nontelomeric regions. (i) A 200 kb core region (shaded section of chromosome below KB indel) and 1 kb fragments spaced every 10 kb to capture variation up to 5 Mb surrounding the K locus. Regions sequenced by Anderson et al. (2009) are represented by rectangles below 200 kb core region, (ii) 1 kb segments sampling a similar core as in (i) and located in 20 telomeric and nontelomeric regions. (B) Genome-wide heterozygosity values for filtered sites measured over six types of genomic region, with standard deviation bars shown. All populations are gray wolves unless otherwise noted.
Table 1.
Sampling Information for Wolves and Dogs Sequenced on the Capture Array.
| # Sequenced | # Used for Analyses | KB Haplotypes | ky Haplotypes | Black | Gray | White | Other | Unknown | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Gray wolf | 358 | 331 | 117 | 544 | 101 | 138 | 25 | 0 | 67 | |
| United States | 246 | 234 | 103 | 365 | 94 | 129 | 0 | 0 | 11 | |
| Alaska | 34 | 33 | 8 | 58 | 6 | 16 | 0 | 0 | 11 | |
| Yellowstone | 212 | 201 | 95 | 307 | 88 | 113 | 0 | 0 | 0 | |
| Canada | 101 | 89 | 12 | 165 | 5 | 7 | 25 | 0 | 52 | |
| Alberta | 8 | 8 | 1 | 15 | 1 | 3 | 3 | 0 | 1 | |
| British Columbia | 2 | 2 | 0 | 4 | 0 | 0 | 0 | 0 | 2 | |
| Manitoba | 4 | 0 | 0 | 0 | ||||||
| Newfoundland | 4 | 2 | 0 | 4 | 0 | 0 | 0 | 0 | 2 | |
| Northwest Territories | 32 | 32 | 9 | 55 | 4 | 4 | 13 | 0 | 11 | |
| Nunavut | 29 | 28 | 0 | 56 | 0 | 0 | 9 | 0 | 19 | |
| Ontario | 2 | 0 | 0 | 0 | ||||||
| Quebec | 8 | 7 | 0 | 14 | 0 | 0 | 0 | 0 | 7 | |
| Saskawatchen | 5 | 4 | 0 | 8 | 0 | 0 | 0 | 0 | 4 | |
| Yukon | 7 | 6 | 2 | 9 | 0 | 0 | 0 | 0 | 6 | |
| Old World | ||||||||||
| Italy | 11 | 8 | 2 | 14 | 2 | 2 | 0 | 0 | 4 | |
| Domestic dog | 21 | 20 | 16 | 24 | 5 | 0 | 1 | 14 | 0 | |
| Breeds fixed for KB | 8 | 8 | 16 | 0 | 5 | 0 | 0 | 3 | 0 | |
| Breeds fixed for ky | 6 | 6 | 0 | 12 | 0 | 0 | 0 | 6 | 0 | |
| Breeds variable for ky and kbr | 4 | 3 | 0 | 6 | 0 | 0 | 0 | 3 | 0 | |
| Breeds with previously unknown K alleles | 3 | 3 | 0 | 6 | 0 | 0 | 1 | 2 | 0 | |
| Admixed | Italy | 3 | 2 | 0 | 4 | 0 | 0 | 0 | 0 | 2 |
| Total | 382 | 353 | 133 | 572 | 106 | 138 | 26 | 14 | 69 |
Note.—Breeds fixed for KB: Dalmation, German Shorthaired Pointer, Irish Water Spaniel, Labrador Retriever, Newfoundland, Standard Poodle. Breeds fixed for ky: Basset Hound, Collie, Doberman Pinscher, Rottweiler, Siberian Husky, Yorkshire Terrier. Breeds with unknown K alleles: American Eskimo Dog, Ibizan Hound, Pharaoh Hound. Breeds variable for ky and Kbr: Boxer, Mastiff, Whippet.
To further assess sequencing quality and to characterize genetic patterns, we partitioned our sequence data into six genomic categories: 1) neutral; 2) the K locus core (200 kb region); 3) genic (all exons and introns); 4) exonic; 5) telomeric; and 6) nontelomeric regions (supplementary table S3, Supplementary Material online). Heterozygosity values measured across all six partitions (fig. 1B andsupplementary table S4, Supplementary Material online) were similar to those based on SNP array data and expectations based on population histories (Gray et al. 2009; vonHoldt, Pollinger, et al. 2010; vonHoldt et al. 2011). Dog heterozygosity values were lower than those in many wolf populations (fig. 1B) which is consistent with a bottleneck in the history of dog domestication (e.g., vonHoldt, Pollinger, et al. 2010; Freedman et al. 2014). Moreover, among wolves and dogs, heterozygosity was higher in telomeric regions than in nontelomeric regions (Ho, telomeric: 0.176%±0.031%; Ho, nontelomeric: 0.121%±0.03%; P value <1e-15), which is consistent with telomeric regions in dogs and humans having higher recombination rates and higher diversity (Myers et al. 2005; Auton et al. 2013). Compared with other genic regions, the 200 kb K locus in wolves and in dogs showed high heterozygosity (Ho: 0.229% ± 0.103%; P value <1e-15), which is unexpected for a selective sweep scenario at the K locus. However, the 200 kb region includes several genic and nongenic regions, which may elevate heterozygosity relative to purely genic regions.
Haplotype Diversity of KB versus ky in Dogs and Wolves
KB haplotypes appeared less variable than ky in all wolf populations, and variability was diminished in KB wolves relative to KB dogs (supplementary fig. S5, Supplementary Material online). Yellowstone wolf haplotypes appeared to be largely composed of a single KB haplotype, with very little haplotype diversity, and Italian wolf KB haplotypes appear more similar to the dogs than to other wolf haplotypes. To further dissect regional genetic variation, we calculated three measurements of diversity (π, θw, and D) in nonoverlapping windows along each of the K locus core, neutral, nontelomeric, and telomeric regions (fig. 2 and supplementary fig. S6, Supplementary Material online; see Methods and Supplementary Material online). We predicted that statistics calculated using the neutral, telomeric, and nontelomeric regions might reflect aspects of population history and genome-wide patterns of variation, whereas statistics summarizing the K locus region might demonstrate evidence of selection and population origin.
Fig. 2.

Nucleotide diversity and Tajima’s D calculated for dogs and North American wolves. Across 5 Mb surrounding the K locus core, nucleotide diversity (A) and Tajima’s D (B) were calculated in 10 kb windows with 1 kb step size for KB and ky haplotypes in wolves and dogs (see keys). Red horizontal lines in (A) and (B) indicate windows containing the 3 bp deletion. (C) Density distributions of nucleotide diversity calculated from nonoverlapping 1 kb windows for KB and ky haplotypes, telomeric regions, and nontelomeric regions in wolves and dogs (see keys). (D) Same as (C) but for Tajima’s D. For each region, the phased sites were concatenated into a single sequence, with telomeric regions ordered from smallest chromosome to largest chromosome.
For the K locus core region, measurements of diversity were consistently lower in wolves than in dogs (e.g. πwolf,KB: 0.00035 ± 0.00045; πdog,KB: 0.00117 ± 0.00105; P value < 1e-15, Wilcoxon signed rank test; fig. 2A and table 2; supplementary table S5, Supplementary Material online), and lower in KB-containing haplotypes than in ky-containing haplotypes (e.g., πwolf,KB: 0.00035 ± 0.00045; πwolf,ky: 0.00216 ± 0.00121; P value <1e-15; table 2; supplementary table S5, Supplementary Material online). Similarly, Tajima’s D within wolf KB haplotypes was negative (mean D: −1.305; fig. 2B), which indicates an excess of rare alleles at low frequency, and is consistent with a selective sweep. In contrast, diversity patterns across the telomeric, nontelomeric, and neutral regions in wolves were similar or higher than diversity in dogs (fig. 2C and D and table 2;supplementary fig. S6, Supplementary Material online), consistent with a domestication bottleneck in the latter. Telomeric regions had higher values of π and θw, perhaps because recombination rate increases towards telomeres and as a result diversity in those regions is higher (Auton et al. 2013; table 2). In dog and wolf ky haplotypes, diversity was significantly higher than that of the other telomeric regions (e. g., πdog,ky: 0.002 ± 0.0013; πdog, Tel: 0.00139 ± 0.0009; P value: 0.00059; table 2 and supplementary table S5, Supplementary Material online), whereas diversity in the KB haplotypes was consistently lower than the other telomeric regions, although this pattern was more pronounced in wolves than in dogs (table 2 and supplementary table S5, Supplementary Material online). These results suggest that the patterns of diversity measured at the K locus, especially with regards to the KB haplotype, are unusual, even in comparison to similarly positioned and sized regions in other chromosomes. In aggregate, these results suggest that the low levels of diversity found within KB haplotypes, especially in wolves, are not characteristic of telomeric regions in general, and could not have been predicted solely from the genomic location of the K locus.
Table 2.
Haplotype Diversity within Dogs and North American Wolves, for Each of Four Types of Region within the Genome.a
| Region Type | Group | π | θw | Tajima's D |
|---|---|---|---|---|
| K locus Core | Dog KB | 0.00117 (0.00105) | 0.00115 (0.00096) | 0.02901 (0.91231) |
| Dog ky | 0.002 (0.0013) | 0.00174 (0.00095) | 0.51471 (1.32856) | |
| Wolf KB | 0.00035 (0.00045) | 0.00093 (0.00068) | −1.30547 (0.59812) | |
| Alaska | 0.00035 (0.00085) | 0.00039 (0.0009) | −0.50833 (1.22007) | |
| NWT | 0.00094 (0.00142) | 0.00066 (0.00097) | 1.64671 (0.82527) | |
| YNP | 0.00011 (0.0003) | 0.0003 (0.00047) | −1.13885 (0.86024) | |
| Yukon | 0.00119 (0.0015) | 0.00119 (0.0015) | NA | |
| Wolf ky | 0.00216 (0.00121) | 0.0017 (0.00076) | 0.60353 (1.00967) | |
| Alaska | 0.00235 (0.00135) | 0.00188 (0.0009) | 0.62744 (0.99614) | |
| NWT | 0.0022 (0.0012) | 0.00197 (0.00098) | 0.32539 (0.82504) | |
| YNP | 0.00187 (0.00121) | 0.00132 (0.00066) | 0.85937 (1.1648) | |
| Yukon | 0.00166 (0.00128) | 0.0016 (0.00112) | 0.06557 (1.0714) | |
| Nontelomeric | Dogs | 0.0009 (0.00085) | 0.00082 (0.00064) | 0.15874 (1.11635) |
| Wolves | 0.00131 (0.00093) | 0.00108 (0.00053) | 0.33245 (0.99481) | |
| Telomeric | Dogs | 0.00139 (0.00099) | 0.00132 (0.00078) | 0.1472 (0.97505) |
| Wolves | 0.00166 (0.00096) | 0.00144 (0.00065) | 0.30451 (0.92183) | |
| Alaska | 0.0016 (0.00104) | 0.00139 (0.00075) | 0.35304 (0.98058) | |
| NWT | 0.00182 (0.00112) | 0.0016 (0.00082) | 0.29791 (0.85435) | |
| YNP | 0.0015 (0.00091) | 0.00095 (0.00051) | 1.06465 (1.05498) | |
| Yukon | 0.00166 (0.0011) | 0.00174 (0.00104) | −0.22621 (0.73964) | |
| Neutral | Dogs | 0.00111 (0.00039) | 0.00101 (0.00031) | 0.30352 (0.62597) |
| Wolves | 0.00146 (0.00041) | 0.00117 (0.00027) | 0.70033 (0.57855) |
Note.—Italics text indicates values for the K locus region that are significantly different from Telomeric regions (P < 0.05 with Wilcoxon rank sum test). See supplementary table S5, Supplementary Material online for P values. Θw, Watterson's theta; π, nucleotide diversity.
Summary statistics were calculated from 1 kb nonoverlapping windows (K locus Core: 103, Nontelomeric: 96, Telomeric: 75) or 10 kb nonoverlapping windows (Neutral: 491), and the mean and standard deviation are provided.
Extended Haplotype Homozygosity of the K Locus
Using the extended haplotype homozygosity (EHH) score, which ranges from 0 (no homozygosity) to 1 (complete homozygosity) (Sabeti et al. 2002), we found that the haplotype containing the derived KB allele had more extensive homozygosity in wolves than in dogs (blue lines in fig. 3A and B;supplementary fig. S7A, Supplementary Material online). These data suggest a recent and dramatic selective sweep for the KB allele in wolves. These patterns are not driven by uneven sampling of the KB haplotype among wolf populations or by relatedness among individuals, as the patterns remain when the Yellowstone wolf population (comprising over half our samples) is subsampled to only 35 unrelated individuals (supplementary fig. S8, Supplementary Material online), and when all wolves are subsampled to be unrelated (supplementary fig. S8, Supplementary Material online).
Fig. 3.

Extended haplotype homozygosity (EHH) decay for the 200 kb core region in dogs and North American wolves. EHH scores along the 200 kb K locus region show the decay of EHH with increasing distance from the core allele (vertical dashed line), for both ancestral ky (red, dashed line) and derived KB (blue, solid line) haplotypes in (A) dogs, (B) all North American wolves, (C) Alaska wolves, (D) Yellowstone wolves, (E) Northwest Territories wolves, and (F) Yukon wolves. Positions on Chromosome 16 are provided.
Comparison of Genetic Variation among Wolf Populations Containing the KB Allele
Using population-wide summary statistics, we inferred the geographic origin of the KB allele. The region of origin should have the highest nucleotide and haplotype diversity, and the highest proportion of ancestral haplotypes (e.g., Tishkoff et al. 2001; Gray et al. 2010) because more time has elapsed for new mutations and recombination events to occur (e.g., Shannon et al. 2015). Additionally, populations moving outside of the origin may be expected to carry a subset of the diversity of the ancestral population (Ramachandran et al. 2005). Given that Alberta and Newfoundland each only had a single KB haplotype, we focused on relative diversity in Alaska, the Northwest Territories, Yellowstone, and Yukon 200 kb K locus regions. For each of the four populations, the diversity of the KB haplotype was significantly lower than the diversity of the telomeric regions (fig. 4 and table 2). We found that KB haplotypes from Yukon had the greatest diversity, followed by Northwest Territories (fig. 4). Yellowstone wolves showed the lowest KB haplotype diversity, despite including the largest number of sampled KB haplotypes (table 1). Importantly, the Yellowstone population is otherwise genetically diverse (fig. 1B), having been derived from three distinct populations from Montana, Alberta, and British Columbia. Values of Tajima’s D for KB haplotypes in Yellowstone wolves were negative (−1.13885 ± 0.86024; table 2), and Alaska KB haplotypes also had a slightly negative Tajima’s D (−0.50833 ± 1.22007; table 2). Although the sample size of Yukon wolves was too low to calculate D, the values of π and θw indicate the highest KB diversity was found in Northwest Territories or Yukon suggesting that region as an origin, with lower diversity in Alaska and Yellowstone.
Fig. 4.
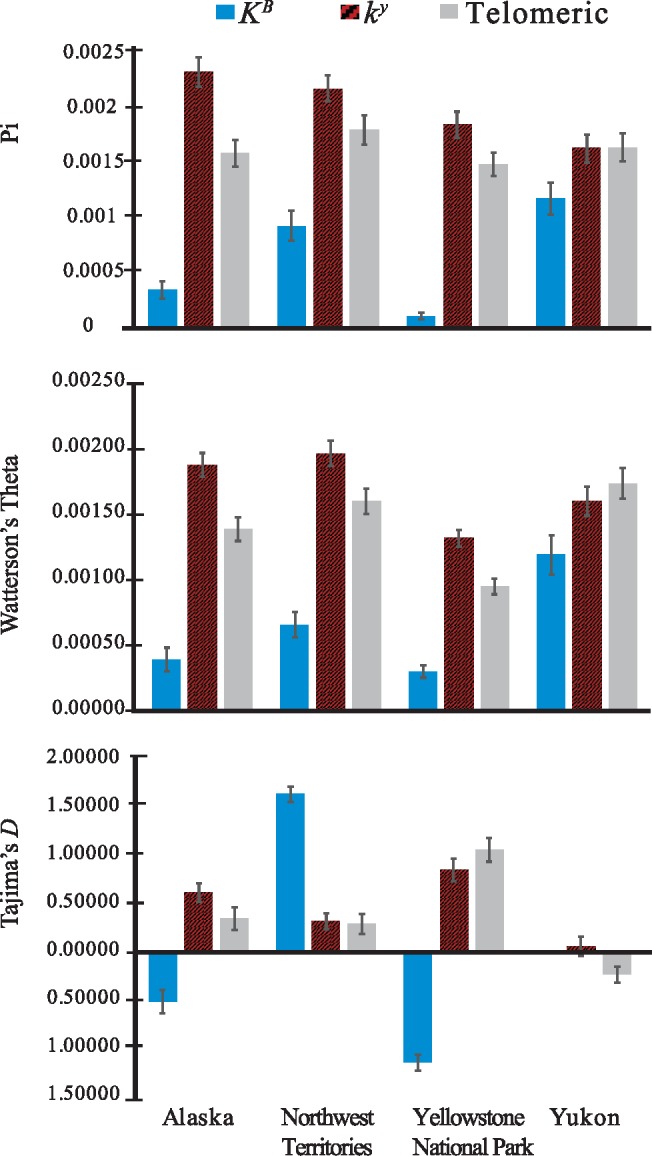
Nucleotide diversity (pi), Watterson’s Theta, and Tajima’s D for North American wolves containing KB and ky haplotypes, separated by geographic location. Error bars show the standard error of the mean for 103 1-kb (K locus core) or 75 1-kb (Telomeric) nonoverlapping windows from phased haplotypes.
Levels of LD were much higher for KB haplotypes than ky haplotypes for the K locus region (fig. 5 and supplementary fig. S9, Supplementary Material online), which had overall higher levels of LD than those of the 10 parallel telomeric regions (supplementary fig. S10, Supplementary Material online). For the ky haplotypes (fig. 5A), most populations had LD below r2 = 0.2 for the 5 Mb region at a distance extending from 20 kb to 1 Mb from the 3 bp deletion (supplementary table S6, Supplementary Material online). Patterns of LD in the Northwest Territories suggest that it was one of the most diverse populations that also contained KB haplotypes. For KB haplotypes (fig. 5B), only dogs, and wolves from the Northwest Territories and Alaska, ever reached r2 = 0.2 within the sampled interval (supplementary table S6, Supplementary Material online). Similar patterns are observed in heatmap triangle plots of pairwise LD (supplementary fig. S9, Supplementary Material online). In contrast, LD decayed rapidly over the ∼200 kb reference telomeric regions, with LD being only slightly higher in dogs than in wolves (supplementary fig. S10, Supplementary Material online).
Fig. 5.

Decay of linkage disequilibrium (measured by r2) for the K locus core plus surrounding 5 Mb region, in (A) ky-containing haplotypes and (B) KB-containing haplotypes. Each population has been downsampled to eight haplotypes other than Yukon KB (n = 3) and Newfoundland ky (n = 4), and variants were filtered for a minor allele frequency >0.05. Note the log scale and different y-axis scales. NWT: Northwest Territories.
Extended Haplotype Homozygosity of the K Locus among Wolf Populations
For each wolf population that contained individuals with KB haplotypes, we calculated the EHH statistic to assess the decay of homozygosity (fig. 3C–F andsupplementary table S7, Supplementary Material online), and found that Alaska and Yellowstone wolves had the most extensive EHH in KB haplotypes (blue lines in fig. 3C and D). In contrast, Northwest Territories (fig. 3E) and Yukon (fig. 3F) populations had EHH scores that declined more rapidly (fig. 3 and supplementary table S7, Supplementary Material online). The nonsymmetrical decay of EHH on either side of the deletion, especially in the Northwest Territories population, may reflect fewer recombination events downstream of the K locus. These uneven patterns upstream and downstream of the K locus may be a result of selection at neighboring loci, although sequence data did not show any fixed, functional differences between KB or ky haplotypes in dogs or wolves other than the 3 bp deletion (Supplementary Material online).
To explore whether patterns of EHH within the reintroduced Yellowstone population were a result of limited diversity within the founders or a subsequent sweep, we measured EHH solely within 12 founders, of which five were KB/ky and seven were ky/ky. Decay of EHH surrounding the core mutation occurred more rapidly in Yellowstone founders (supplementary fig. S11, Supplementary Material online) than in the Yellowstone population (fig. 3D andsupplementary table S7, Supplementary Material online). Over the 5 Mb region, EHH decayed to zero in KB haplotypes ∼1 Mb upstream (supplementary fig. S11 and supplementary table S7, Supplementary Material online), whereas in the current Yellowstone population the EHH score remained above zero even ∼4 Mb upstream (supplementary fig. S7 and supplementary table S7, Supplementary Material online). Given that the Yellowstone population is not inbred (vonHoldt et al. 2008; vonHoldt, Stahler, et al. 2010), these results suggest that an advantageous haplotype rose in frequency subsequent to the foundation of the population. This pattern, in association with results from neutral regions, suggests that selection may be especially strong at the K locus in Yellowstone wolves relative to other wolf populations.
Simulations of Allele Frequency Change in Yellowstone Wolves
We sought to determine whether the present-day allele frequency of the KB allele (22%) in Yellowstone wolves could be explained simply by genetic drift and negative selection or whether other nonneutral processes would have to be considered. We chose to focus our simulations on the Yellowstone population, since its demographic history is known and can be incorporated into models to test for selection. We performed forward simulations based on the initial allele frequency (20%) and demography of the wolves, adjusting Ne for nonrandom mating (see Materials and Methods). Our null model included a lethal recessive condition such that KB homozygotes would not survive. We found that the final allele frequency of 22% was unlikely to be seen under this null model (P value = 0.018; fig. 6A). This result suggests that a nonneutral process (e.g., positive selection, balancing selection) has altered the frequency of the KB allele since the founding of the Yellowstone population in 1995.
Fig. 6.

(A) Null distribution of allele frequencies based on the Yellowstone National Park wolf reintroduction demographic history. The dashed red line denotes the observed value, which corresponds to a P value of 0.018. (B) Prior and posterior distributions of the dominance (h) coefficient for the KB allele. The mean posterior value was −0.179 (95% HPD: −0.483, −0.0040).
To better understand the magnitude and nature of selection, we used approximate Bayesian computation (ABC) to infer the dominance coefficient (h) of the KB allele. We used a uniform prior over (−2, 2) and rejection sampling, retaining simulations where the final allele frequency in each simulation replicate was within 1e-3 of the final observed allele frequency. We found that the mean of the posterior distribution was −0.179 (95% HPD: −0.483, −0.0040; fig. 6B), suggesting that balancing selection maintains variation at this locus. Importantly, the posterior distribution excludes 0, further arguing against the KB allele being a simple recessive lethal.
Hierarchical Patterns of Divergence among Populations
Neighbor joining (NJ) trees generated using pairwise nucleotide divergence values from K locus haplotypes were compared with those based on the neutral data, since the former should provide insight into the specific history at the selected K locus, and the latter should reflect population history (vonHoldt, Pollinger, et al. 2010; vonHoldt et al. 2011; Pilot et al. 2014; Schweizer, vonHoldt, et al. 2016). NJ trees of the neutral regions showed groupings that were concordant with major geographic or taxonomic differences (supplementary fig. S12, Supplementary Material online). For instance, dogs formed a single grouping near Italian wolves, which is concordant with likely European or Asian origins of domestication (Savolainen et al. 2002; Thalmann et al. 2013; Freedman et al. 2014; Fan et al. 2016). Next, we generated NJ trees using data from a 26 kb region surrounding the K locus where there was no evidence of recombination (fig. 7A andsupplementary fig. S13, Supplementary Material online; see Methods). North American KB haplotypes form a single cluster, suggesting a single introgression event (fig. 7A). This cluster also contains a North American dog breed, the Newfoundland, as well as Labrador Retrievers, German Shorthaired Pointer and Portuguese Water Dog, and a Siberian Husky ky haplotype was sister to this group (fig. 7A). A separate cluster of KB haplotypes from Italian wolves are related to European dog breeds, suggesting a separate introgression event. NJ trees generated using pairwise nucleotide divergence between larger regions were not informative due to recombination events further upstream and downstream of the K locus, most notably observed in Northwest Territory wolves (results not shown).
Fig. 7.

Neighbor joining tree and haplotype network of the K locus suggest a single introgression event. (A) Partial neighbor-joining tree of 26 kb core nonrecombining region, based on pairwise nucleotide distances between haplotypes from 172 unrelated individuals. Haplotypes are labeled as belonging to a dog breed (“D”) or wolf population (“W”) and colored according to KB (blue) or ky (red). All nodes had 100% support from 5,000 bootstrap replicates. (B) Haplotype network of all KB haplotypes sampled. Each haplotype (n = 134) consists of a 11,754 bp phased haplotype representing sites that passed filters within the 26 kb nonrecombining region around the K locus deletion. Seven unique KB haplotypes are represented by roman numerals, with the number of haplotypes within each type indicated. Nodes are colored and shaped according to geographic origin and species (see key). Haplotype I includes wolves from Alaska (n = 8), Alberta (n = 1), Northwest Territories (n = 9), Yellowstone (n = 90), and Yukon (n = 2), and dogs (n = 1 for each of German short-haired pointer, labrador retriever, and Portuguese water dog). Haplotype IV includes Dalmatians (n = 2), German Shorthaired Pointers (n = 1), Irish Water Spaniels (n = 2), Portuguese Water Dogs (n = 1), Standard Poodles (n = 2), and Italian wolves (n = 2).
We built haplotype networks using the same nonrecombining 26 kb region surrounding the K locus (see Methods; fig. 7B and supplementary fig. S14, Supplementary Material online). Among ky haplotypes, most sampled dog breeds lie on a single branch of the network whereas North American and Italian wolf haplotypes are represented on other branches of the network (supplementary fig. S14, Supplementary Material online), which is consistent with the evolutionary history of these populations. Among the KB haplotype network, North American wolves mostly form a single cluster, with a few dog haplotypes nested within the cluster (supplementary fig. S14, Supplementary Material online). Italian wolf KB haplotypes group most closely to dog haplotypes rather than wolf haplotypes, which again suggests a different introgression event than that which occurred in North American wolves (supplementary fig. S14, Supplementary Material online). A closer examination of the wolf populations and dog breeds represented by the KB haplotypes (fig. 7B) shows that the Newfoundland dog haplotype (haplotype VI) is close to the most common haplotype (haplotype I) in North American wolf populations.
Age of Introgression in North American Wolves
To understand the timing and spread of the dog KB allele into wolf populations in North America, we estimated time to the common ancestor (TMRCA) for the deletion in each of the four populations containing more than one KB haplotype (Alaska, Northwest Territories, Yellowstone, and Yukon). We used a new method (Smith et al. 2018) which leverages both the decay in LD between the selected allele and nearby sites, as well as the accumulation of new mutations on the selected allele's ancestral haplotype. We find that the oldest TMRCA estimates are found in the Yukon with posterior means ranging from 1598 to 7248 ya, depending on the mutation rate used (fig. 8 and table 3). The youngest TMRCA values are consistently found in Yellowstone, ranging from 202 to 1942 ya. Estimates for Alaska, Northwest Territories, and two Yellowstone posteriors have significant overlap and all fall roughly between 694 and 2135 ya.
Fig. 8.

TMRCA estimates of the KB allele in the four North American populations using four different mutation rates assuming a generation time of 3 years. The violin plots are samples from the posterior distribution of TMRCAs drawn from a Markov chain Monte Carlo run for 50,000 iterations with a standard deviation of 10 for the proposal distribution. The locus includes 3 Mbp of flanking sequence around the selected site.
Table 3.
Mean Posteriors and Credible Intervals for the KB Allele Age Estimates Shown in Figure 8.
| Population | Mutation Rate (/bp/gen.) | TMRCA (years) | 95% Credible Interval |
|---|---|---|---|
| Yellowstone National Park | 0.3 × 10−8 | 202 | 133–285 |
| 0.4 × 10−8 | 250 | 162–352 | |
| 0.45 × 10−8 | 1,513 | 982–2,030 | |
| 1 × 10−8 | 1,942 | 1,346–2,503 | |
| Northwest Territories | 0.3 × 10−8 | 1,301 | 822–1,931 |
| 0.4 × 10−8 | 1,635 | 932–2,333 | |
| 0.45 × 10−8 | 1,704 | 1,137–2,234 | |
| 1 × 10−8 | 2,155 | 1,105–3,250 | |
| Alaska | 0.3 × 10−8 | 694 | 376–1,100 |
| 0.4 × 10−8 | 1,477 | 831–2,329 | |
| 0.45 × 10−8 | 1,801 | 826–3,263 | |
| 1 × 10−8 | 2,135 | 1,602–2,951 | |
| Yukon | 0.3 × 10−8 | 1,598 | 393–3,389 |
| 0.4 × 10−8 | 3,500 | 2,338–4,398 | |
| 0.45 × 10−8 | 4,378 | 2,979–6,169 | |
| 1 × 10−8 | 7,248 | 6,219–8,963 |
In some cases, the choice of different mutation rates did not affect the variability of time estimates (table 3). Among the Alaska and Northwest Territories samples, mutation rates from 0.4 × 10−8 to 0.45 × 10−8 impose similar time estimates. The same is true for the Yellowstone samples at mutation rates of 0.3 × 10−8 and 0.4 × 10−8. However, all estimates using a mutation rate of 0.3 × 10−8 did result in younger TMRCA values.
Demographic Inference and Simulations of Selection in Alaska, Northwest Territories, and Yukon Wolves
To gain insight into the processes that maintain variation at the K locus, we performed demographic inferences from the neutral genetic variation data in the Alaska, Northwest Territories, and Yukon wolf populations. We used a diffusion approximation based method (Gutenkunst et al 2009) and inferred a single population size change demographic model for each population. After incorporating ancestral sequence misidentification into the model, we fit the site frequency spectra across all frequency bins (supplementary fig. S15, Supplementary Material online). We infer that all three populations underwent a contraction at some point in the past, with the Yukon population undergoing the most severe contraction (supplementary table S8, Supplementary Material online).
Armed with this information, we sought to understand the patterns of selection at the K locus more closely. Specifically, we tested whether a null model with complex demography and a recessive lethal mutation introduced through recent admixture could be compatible with the data. In contrast to the Yellowstone population, we do not know the timing of introgression nor the frequency of introgression. Therefore, we used ABC to infer the unknown parameters (time of introgression and introgression frequency). We then asked whether these inferred values are sensible given the other known information. We found that in order to match the final allele frequency data, introgression must have occurred very recently and at nearly 100% frequency in every population (fig. 9). This timing is not supported by our TMRCA estimates (fig. 8), which indicates the allele is much older than a few generations ago. Further, the unreasonably large introgression frequency suggests that the null scenario of complex demography combined with introgression and purifying selection on a recessive lethal allele is unlikely. Instead, our results strongly favor some type of balancing selection maintaining the frequency of the KB allele in these populations.
Fig. 9.

Posterior densities for introgression time (in generations) and introgression frequency in null simulations of (A) Alaska, (B) Northwest Territories, and (C) Yukon wolf populations. Red indicates higher probability, whereas blue indicates lower probability (see key for each plot). Our null model includes negative selection and a demography estimated from the data. Under this null model, there must be very recent introgression at a very high frequency, providing support for an alternate model with positive selection.
Discussion
Melanism in gray wolves is caused by a dominantly inherited 3 bp deletion at the K locus, and the region surrounding the mutation shows signatures of strong selection within KB haplotypes (Anderson et al. 2009). Using a resequencing approach (fig. 1A), we demonstrate that haplotypes containing the KB allele showed signals of a selective sweep in North American wolves more so than dogs (figs. 1B, 2, and 3). We also find support for a single introgression event, likely in Yukon or Northwest Territories. These results are corroborated by the finding that the KB allele age is oldest in the Yukon wolf population (fig. 8). Italian wolves that contain the KB mutation represent a separate introgression with European dog breeds that we speculate may be more recent based on patterns of genetic diversity and linkage disequilibrium.
Selection on the KB Allele in Wolves
Using multiple diversity statistics, LD decay, and EHH, we find compelling evidence of a partial selective sweep of the KB haplotypes and diversity differences between wolves and dogs point to a recent selective sweep within wolves (figs. 2 and 3; supplementary table S5, Supplementary Material online). Given slightly lower diversity in the dog KB haplotype than the dog ky haplotype, it is possible that the dog KB allele underwent an older selective event, perhaps due to selective breeding by humans or natural selection. The target of natural selection on the KB allele may not be melanistic coat color, but rather immunological differences between alleles, due to the previously identified role of β-defensin genes in innate and adaptive immunity (Yang et al. 1999). In dogs, the K locus demonstrates antimicrobial activity against respiratory pathogens (Erles and Brownlie 2010), and there is ample evidence for differential selection in Yellowstone wolves (Coulson et al. 2011; Hedrick 2015). Black heterozygote wolves have a significantly higher overall survival rate than black homozygote wolves (which are rare in the population), implying that the selective effects of a KB allele may have to do with disease rather than pigmentation, since both genotypes have the same coat color (Anderson et al. 2009; Coulson et al. 2011). In fact, black heterozygote wolves of Yellowstone National Park showed higher annual survivorship following the three documented distemper outbreaks than gray homozygous wolves (Yellowstone National Park Wolf Project, unpublished data). Given that multispecies carnivore populations are a reservoir for canine distemper (Almberg et al. 2010), there is a possibility that dogs provide the resistance allele to distemper common in the dog population and in other carnivore species.
With the exception of the Yellowstone wolves, the demographic history of the populations we sampled is uncertain, but population specific demographic events may have reduced haplotype variation that could be confused with selective sweeps. Consequently, we sampled ∼3.74 Mb genic sequence, ∼86.9 kb sequence in similar telomeric positions as the K locus, and ∼5.03 Mb neutral sequence (supplementary table S3, Supplementary Material online), to determine baseline genome wide and genic patterns of variation. Our results show that the K locus core has dramatically reduced variation relative to other genomic regions suggesting this pattern is due to selection, as selection is expected to be locus specific whereas demographic effects should be manifest genome wide. In fact, our specific demographic modeling of the Yellowstone population indicates that selection can explain even changes in haplotype frequency of <10% from an initial founding value (fig. 6). Additionally, population genetic modeling of the three populations with complex demography shows that patterns of diversity cannot be solely explained by inferred demography and purifying selection alone, suggesting balancing selection is required to explain observed diversity (fig. 9).
A Single Origin of Black Coat Color in North American Wolves
Wolves in North America are unique for having a high frequency of black coat color, approaching 50% of individuals in some populations except those in the high Arctic (Anderson et al. 2009). In the Old World, black wolves are rare and generally restricted to regions where dogs and wolves frequently hybridize (but see Galaverni et al. 2017). The high abundance of black wolves throughout North America suggests that a selective sweep has occurred. Our extensive geographic sampling of wolves from 10 states and provinces within North America implies that the admixture between dogs and wolves first occurred in Northern Canada, and most likely in the Yukon Territory. The ancestral population with regards to KB introgression is predicted to have had the most time for recombination to shorten haplotype blocks and therefore generate greater sequence and haplotype diversity. Wolves within the Northwest Territories demonstrated some of the highest levels of KB haplotype diversity (fig. 4), the lowest LD (fig. 5), and extent of haplotype homozygosity (fig. 3). Wolves from Yukon demonstrate similar patterns of diversity, and so are also candidates for KB origin in wolves, especially given estimated KB allele ages (see below). Additional sampling of wolf populations and dog breeds, especially of Arctic breeds and village dogs, may further elucidate the geographic origin and timing of introgression. Samples from Alaska and Yellowstone lack genetic diversity at the KB haplotype, have high levels of LD within KB haplotypes, and have long tracts of extended homozygosity, which suggest the presence of KB allele is more recent in these populations. The diversity of KB haplotypes and their grouping in phylogenetic trees and network is consistent with a single, ancient origin from Native (pre-Columbian) dogs.
Higher levels of haplotype variation and lower levels of LD suggest that the KB allele was first transferred to wolves in Yukon or Northwest Territories. However, this result is not confirmed by phylogenetic analysis which may be underpowered given the very recent transfer of the KB allele to wolves which we estimate occurred between 1,598 and 7,248 years ago (fig. 8 and table 3). Essentially, few if any phylogenetically informative mutations have appeared subsequent to this event that might provide a well-resolved phylogeny. Therefore, we follow previous analyses which have used patterns of LD and haplotype variation to infer origin of introgression events (e.g. Tishkoff et al. 2001; Gray et al. 2010). Conceivably, demographic events might have occurred in some wolf populations which lowered KB diversity and obscured an origin of introgression there. However, genic and genome-wide patterns of variation outside the K locus do not show lower variation in other populations outside Yukon and Northwest Territories. For example, Yellowstone wolves have high genome-wide diversity but the lowest KB diversity (fig. 1B), suggesting the patterns of variation at the K locus are not due to demography.
Several studies exploring geographic origins of Native American dog breeds have suggested that Arctic dog breeds, such as the Inuit sled dog, the Canadian Eskimo dog and the Greenland dog, have archaic mitochondrial DNA haplotypes and show evidence of ancient admixture with wolves (Brown et al. 2013; van Asch et al. 2013). Furthermore, dogs within these populations represent the only living dog breeds with mitochondrial haplotypes that are unique to New World wolves, and black coat color in these breeds are determined by mutations at the K locus. Based on these findings we hypothesize that the original introgression event likely occurred in the High Arctic regions of Northern America, where dogs and native people first coexisted with large wolf populations that shared similar prey (e.g., caribou). Future efforts focused on increasing the sampling of dog breeds may yield insight into the specific dog breed or group of breeds involved in the introgression event. Within our data set, it is notable that the dog breed with the closest KB haplotype, the Labrador Retriever, was established in the high latitudes of North America, and may share some ancestry with native North American dogs that were related to the first black dogs which successfully crossed with gray wolves.
The most ancient KB allele occurs in Yukon wolves, regardless of the mutation rate used (fig. 8). Using the most current estimates of dog mutation rates (0.3–0.4 × 10−8; Skoglund et al. 2015; Frantz et al. 2016) would place the KB origin in Yukon population at around 1,598–3,500 years ago. This date is much more recent than the arrival of the first dogs 8,500–12,000 years ago (Leonard et al. 2002), and on the more recent end of previously estimated range of 500 – 14,000 years ago (Anderson et al. 2009). Contact from Europeans was limited before the early 19th century, so it is likely that Yukon wolves may have interacted exclusively with dogs of First Nations people. Humans, and likely their dogs, were at relatively low population density, so that encounter rates between dogs and wolves may have been infrequent such that only a single introgression event was successful.
Limited Genetic Diversity of the K Locus in Yellowstone Wolves
We found genetic evidence for balancing selection in Yellowstone wolves based on multiple summary statistics and simulations. The Yellowstone population was originally founded in 1995 with 31 wolves from two discrete populations in Alberta and British Columbia, and 10 additional wolves from Montana added the following year (Bangs and Fritts 1996). Given this history, we would expect an increase in heterozygosity and diversity of KB haplotypes relative to a single wild population from southwestern Canada. However, the Yellowstone wolf KB haplotypes demonstrate very low levels of diversity (supplementary fig. S5, Supplementary Material online and fig. 4), high LD (fig. 5B), and an almost completely monomorphic core region (fig. 3D). This pattern might reflect lower diversity in the founding stock, but we found that other genomic regions of the Yellowstone population had average levels of heterozygosity (fig. 1B). We also show that the few Yellowstone founders that we sequenced had substantially higher diversity than the current population (supplementary fig. S7, Supplementary Material online), and consequently, diversity within the K locus region has been lost relative to other genomic regions. Results from our simulations show that the K locus is under balancing selection in the Yellowstone wolf population. Yellowstone may be unique among North American wolf populations in that it surrounded by a high density of carnivore groups, such as mustelids, canids, ursids, felids, and raccoons, that are hosts for disease (Almberg et al. 2010). These other carnivore species are potentially a reservoir for canine disease such as distemper which causes substantial mortality in the Yellowstone wolf population (Stahler et al. 2013) and could be the cause underlying current balancing selection in Yellowstone wolves. Elsewhere, as represented by our wolf populations, the density of host species is lower and the encounter probabilities less.
Conclusions
Using population genetic simulations, we demonstrate evidence of selection at the K locus in North American wolf populations, and confidently rule out genomic location or sampling effects as explanation of previous results. We find that there was likely a single successful introgression event in the Canadian Arctic, that thereafter spread rapidly across the entire continent leaving other wolf populations with lower diversity at the KB haplotype. We apply a new approach using both recombination and mutation rate to estimate the time of introgression, and find that the event is very recent, suggesting one of the most rapid spread of an adaptive variant known in vertebrates. This spread was likely facilitated by the great dispersal abilities of gray wolves, which commonly disperse over 50 km before establishing new territories (Mech and Boitani 2003). Further, although the specific focus of selection is uncertain, the canonical function of the K locus is in immunity, and hence the presence of dogs and other carnivores as reservoirs of disease may have augmented the selective coefficient at this locus. Conceivably, the KB allele may have first evolved in dogs and persisted because of enhanced immunity in dense dog populations where disease thrives, and then was preadapted for selection in adjoining wolf populations that may have also been infected by diseases from a carnivore reservoir. This hypothesis requires conformation with functional tests, such as with a CRISPR modified cell line (Shalem et al. 2014). Finally, consistent with this hypothesis, we detect a signature of balancing selection since the founding of the Yellowstone National Park population, which has been known to suffer from recurrent zoonotic disease. These zoonotics include introduced disease such as mange, and also distemper, a disease common in sympatric dog and wild carnivore populations with which wolves may interact in open range country surrounding the park.
Materials and Methods
Sample Selection and Array Design
We selected wolf samples to maximize the following parameters: 1) the geographic distribution of samples across regions and ecotypes (Schweizer, Robinson, et al. 2016; Schweizer, vonHoldt, et al. 2016); 2) the number of samples with known coat color phenotype and life history data; and 3) the quality and quantity of DNA. A total of 382 samples were sequenced, and, after genotype and sample quality filtering, 353 samples were retained for analysis (table 1).
The capture array was designed to reconstruct the origin and spread of the KB allele. We extracted putatively “neutral” regions from the dog reference genome (CanFam3.1; fig. 1A) for background estimates of individual relatedness and population demography. Details of the design of these regions have been described elsewhere (Freedman et al. 2014; Schweizer, Robinson, et al. 2016) and follow guidelines set by previous studies in humans (Wall et al. 2008). To characterize genetic variation around the K locus, we designed an extensive resequencing approach for almost 5 Mb surrounding the 3 bp causative deletion on chromosome 16. This design included the 200 kb “core” region partially sequenced previously (Anderson et al. 2009), plus 1 kb segments spaced every 10 kb, extending to the end of the chromosome ∼560 kb downstream of the mutation (the K locus is near the telomeres) and 4.2 Mb upstream of the mutation. To test how the proximity of the K locus to the end of the chromosome impacts decay of LD and diversity statistics, we sequenced ten 1 kb fragments spaced on a 200 kb segment in nontelomeric regions for five larger and five smaller chromosomes, plus chromosome 16. Given that dog and wolf chromosomes are acrocentric, we chose the midpoint for each nontelomeric region. Finally, as an empirical background for selection at the K locus, we assessed decay of LD in other telomeric regions by similarly designing ten 1 kb fragments spaced on a 200 kb segment in telomeric regions for five larger and five smaller chromosomes. Each telomeric region began at the same relative distance from the end of the chromosome as the K locus mutation on chromosome 16. In total, we captured approximately 10 Mb of sequence from each individual. To do this, 105,000 120 bp RNA baits were developed by MYcroarray (Ann Arbor, Michigan) so as to maximize specificity of bait hybridization and unique mapping within the genome. These baits covered approximately 91% of the regions we aimed to capture.
Library Prep, Target Enrichment, and Sequencing
Samples were prepared as previously described (Schweizer, Robinson, et al. 2016). Briefly, we extracted genomic DNA from blood or tissue, and sheared DNA of high quality and quantity using a Biorupter NGS Sonication System (Diagenode). DNA samples were sheared to approximately 300–450 bp, and before sequencing were randomized with respect to extraction, library prep and enrichment dates to prevent batch effects. Preparation of sequencing libraries followed the with-bead library preparation protocol of Faircloth (2015), and each sample was barcoded with a unique 6 bp index sequence during adapter ligation so as to enable pooling of 24–25 individuals per lane (Faircloth and Glenn 2012). Subsequent to library preparation, samples were target enriched following the manufacturer’s protocol. To check that libraries were enriched for target regions, we performed enrichment qPCR on all samples prior to sequencing (see Supplementary Material online). Enriched libraries were quantified, pooled in equimolar amounts, and sequenced on a HiSeq 2000 with 100 bp paired-end reads by the QB3 Vincent J. Coates Genomics Sequencing Laboratory (Berkeley, California, USA).
Sequence Alignment, Processing, and Genotype Filtration
Sequence alignment and processing followed the Broad Genome Analysis ToolKit “Best Practices” pipeline (https://www.broadinstitute.org/gatk/guide/best-practices), with the specifics of our processing protocol published elsewhere (Schweizer, Robinson, et al. 2016). In short, demultiplexed fastq reads passing the Illumina filter were trimmed for remaining adapter sequences, then forward and reverse reads were aligned and mapped to the reference boxer genome (CanFam3.1) using bwa aln and bwa sampe (Li 2014). Alignment of wolf sequences to CanFam3.1 produces high quality genotype calls and minimal reference bias due to the very short sequence divergence of wolves and dogs (∼0.1%; [Freedman et al. 2014]). A recently published de novo wolf genome might provide a unique future reference for our analysis (Gopalakrishnan et al. 2017). However, the development of this genome is at early stage relative to the dog genome that we used, which was based on Sanger sequencing, is in its third release, and provides chromosome level information and annotation. After duplicate removal with samtools rmdup (Li et al. 2009), local realignment with GATK, and fixing mate information with picard (http://broadinstitute.github.io/picard/), we ran GATK Base Quality Score Recalibration using a previously generated set of “known” variant sites (Schweizer, Robinson, et al. 2016). The GATK UnifiedGenotyper algorithm was used to call SNPs and indels (insertions and deletions) over the capture array intervals with a padding of 1,000 bp. Variant positions identified by the Unified Genotyper were filtered with GATK VariantFiltration using ten annotation values, as recommended by the Broad Best Practices pipeline. We also removed variant positions with genotype quality (GQ) < 30 and all positions with depth of coverage (DP) < 10.
Data Quality Control
Genotype concordance was assessed for 109 individuals and 204 sites that overlapped between the Affymetrix dog SNP array v2 and the capture array target intervals (Schweizer, vonHoldt, et al. 2016). Using the vcftools package (Danecek et al. 2011), we calculated the sequence-wide heterozygosity, transition/transversion ratio, site missingness and individual missingness for each set of regions. Significant differences in heterozygosity among region types were tested in R using a Mann–Whitney test. The K locus indel genotype had previously been determined for 235 individuals using either Sanger sequence (Anderson et al. 2009) or high resolution melt curve analysis (Coulson et al. 2011; Schweizer & B.M. vonHoldt, unpublished data). We used these data for quality control in our sequence samples.
We performed principal components analysis (PCA) within the smartpca package of eigenstrat (Price et al. 2006). First, using neutral sequence data, we verified that samples grouped according to that expected based on previous studies (vonHoldt et al. 2011; Pilot et al. 2014; Schweizer, vonHoldt, et al. 2016). Samples that did not group according to their expected population or species were dropped from further analysis, as they likely represent bad sequencing runs or poor quality samples. For this analysis, we generated a set of LD-pruned SNPs using the “–indep-pairwise 50 5 0.5” option in PLINK (Purcell et al. 2007). We also generated a subset of unrelated individuals using PRIMUS (Staples et al. 2013) and a maximum identity-by-descent of 0.1, as in (Schweizer, Robinson, et al. 2016).
Summary Statistics on Phased Haplotypes
To phase haplotypes, we used SHAPEITv2 since it can handle varying levels of relatedness among individuals (O'Connell et al. 2014). We filtered sites for a 100% call rate in all individuals, a DP ≥10 and variants with GQ ≥ 30. The remaining 7,761,114 sites were phased by individual chromosome with the following parameters: –burn 10 –prune 10 –main 20 –states 200 –window 0.1 –rho 0.001 –effective-size 20000 –duohmm. Pedigree information from Yellowstone wolves was used by SHAPEIT to improve the accuracy of phasing (O'Connell et al. 2014). We also phased genome data from a Kenyan golden jackal (Canis aureus) for use as the ancestral state (Koepfli et al. 2015).
Using the phased haplotype data, we calculated summary statistics to investigate patterns of polymorphism and divergence within the K locus core and surrounding regions, neutral regions, genic regions, parallel telomeric, and nontelomeric regions. These statistics included the following: π (the average pairwise differences), Watterson’s theta (θw, the average number of segregating sites [Watterson 1975]), and Tajima’s D (a measure of the skew in the site frequency spectrum, which is an indicator of both selection and population history [Tajima 1989]). These statistics were calculated in nonoverlapping windows of 1 kb (for telomeric, nontelomeric, and K locus) or 10 kb (for neutral), in combinations of each sequence type, population, and KB versus ky haplotype. All calculations were performed within the Python EggLib package (De Mita and Siol 2012), and significance of differences in means was tested using a Wilcox signed rank test or a Wilcoxon rank sum test, with continuity correction, in R.
Comparison of Selection Signals in KB versus ky
We used two methods to test for signals of selection in dogs or wolves, and whether the partial sweep occurred on the KB or ky containing haplotypes. First, we visualized large-scale patterns of variation in haplotypes by generating haplotype structure plots in R 3.1.3 (http://www.R-project.org) with the rehh package (Gautier and Vitalis 2012) that showed the ancestral and derived state of each polymorphic position. These plots were also useful for visualizing regions where recombination events may have occurred. Next, we implemented the extended haplotype homozygosity (EHH) test, which measures the relationship between the frequency of an allele of interest and the amount of LD surrounding it (Sabeti et al. 2002). Once a core mutation is identified (here, the KB mutation), increasingly distant SNPs are used to measure the decay of LD from the core haplotype. This measure of decay is called the EHH score and provides the probability that two randomly chosen chromosomes out of a population are identical between the core haplotype and the increasingly distant SNP. The phased Kenyan golden jackal sample was used to infer the ancestral state for each variant, and EHH tests were implemented with the rehh package (Gautier and Vitalis 2012).
Comparison of Diversity among Populations Containing the KB Allele
Using EggLib, we calculated LD decay by measuring the square Pearson’s correlation coefficient (r2) between each polymorphic SNP, after filtering for a minimum allele frequency of 0.05 in each population. Genomic distances were binned by 30 kb for telomeric regions, and by exponentially increasing distances for the surrounding 5 Mb region, and the decay of r2 was plotted in R. Given that sample size can affect LD decay, we performed calculations using a random subset of at most eight haplotypes. For a few populations with small sample sizes, there were fewer than eight haplotypes (see Results). LD in the K locus core and parallel telomeric regions was calculated separately.
Forward Single-Locus Simulations for Yellowstone Wolves
We used historical census data to obtain a demographic model from which to simulate. We modeled alpha-mating as a reduction in Ne, using values suggested in (vonHoldt et al. 2008). We assumed that KB homozygous individuals did not survive and used a custom Wright–Fisher forward simulator, sampling the allele frequency from a binomial distribution every generation. Specifically, we simulated eight generations from the founding till the present day. We matched the initial allele frequency and sampled the frequency each generation, retaining the allele frequency from the final generation. We ran 10,000 simulations and used a one-sided test to determine the probability of observing an allele frequency as high or higher than the observation from the data. We used approximate Bayesian computation to infer the dominance coefficient at this locus. We set s = −1 and drew h from a uniform distribution over (−2, 2) to include over and underdominance. We used a rejection sampling approach and kept 500 simulations where the final allele frequency was within 1e-3 of the observed value.
Patterns of Genetic Divergence among Populations
To identify a region showing no signal of past recombination events, we calculated the minimum number of recombinations using the four gamete test of Hudson and Kaplan (1985), implemented in EggLib, in sliding windows of 1,000 bp with 10 bp step size. We visually confirmed that there were no recombinant haplotypes within this region. We used neighbor-joining trees to infer the hierarchy between haplotypes from different geographic localities and the subsequent implications for introgression history. We used EggLib to calculate the pairwise number of differences between each haplotype for the nonrecombining 26 kb K locus haplotype and neutral data sets. We constructed neighbor joining trees with 5,000 bootstraps using the package ape 3.1-2 in R (Paradis et al. 2004). A haplotype network of the nonrecombining region was constructed within the pegas package of R (Paradis 2010) separately for KB and ky containing haplotypes.
Estimating Age of the KB Allele among North American Wolves
To describe the relative timing of the KB allele's spatial spread and/or timing of selection among wolf populations in North America, we estimated time to the common ancestor (TMRCA) for the deletion in each of the four samples. We used a recent method (Smith et al. 2018) which leverages both the decay in LD between the selected allele and nearby sites, as well as the accumulation of new mutations on the selected allele's ancestral haplotype. If assumptions of the model are reasonably approximated by the data, this approach leads to dramatic gains in accuracy relative to more commonly used Approximate Bayesian Computation methods. Specifically, the method assumes a “star-shaped” genealogy among sites linked to the selected allele's ancestral haplotype. This is a reasonable assumption in cases where the focal allele is subject to strong positive selection; which for our case is appropriate given the results of reduced polymorphism and Tajima's D, and elevated EHH scores observed at the selected locus.
A second assumption of this method involves the specification of an appropriate reference panel of haplotypes that do not have the selected allele. This reference panel should approximate the background haplotypes with which the selected haplotype recombined during its increase in frequency. Simulation results show that a reference panel which is too similar to the selected haplotype can lead to underestimates of the true TMRCA while a misspecified reference panel that is too diverged from the true reference panel will overestimate the TMRCA. For this reason, we excluded the sample of dogs for which a suitable reference panel was not available. We used the local reference panel of haplotypes without the selected allele in each of the four samples from natural populations of North American wolves (see Results).
TMRCA estimates can vary depending on the mutation and recombination rates used. To account for locus specific variation in recombination rates across the K locus region, we used a recombination map inferred for dogs based on patterns of LD (Auton et al. 2013). This recombination map, however, does not include the entire sequenced region downstream of the selected site. We predicted the unobserved recombination rates at these sites using the adjacent 4 Mb of observed recombination rates using the smooth spline function in R with the smoothing parameter set to 0.95.
Several estimates of the per basepair per generation mutation rate have been inferred using different approaches. Skoglund et al. (2015) use ancient DNA from a 35,000 year old wolf and to infer a rate of 0.4 × 10−8. Frantz et al. (2016) calibrate a molecular clock using radiocarbon dating on an ancient dog to infer a similar mutation rate between 0.3 × 10−8and 0.45 × 10−8.We report TMRCA estimates using these values in addition to one higher rate of 1 × 10−8 (as used in Freedman et al. 2014).
Demographic Inference and Simulations of Selection in Alaska, Northwest Territories, and Yukon Wolves
To further test whether patterns of genetic diversity at the K locus are unusual, we inferred the demographic history of the three remaining wolf populations where black wolves are found in our sampling (i.e., Alaska, Northwest Territories, and Yukon). We used ANGSD v 0.920 (Korneliussen et al. 2014) to generate site frequency spectra for each population from 5,055,985 neutral sites, using the Kenyan golden jackal as the ancestral reference sequence (Koepfli et al. 2015). We used (Gutenkunst et al. 2009) to set up a model where a population undergoes a size change T generations ago to size N. We included a parameter for ancestral state misidentification and ran the inference seven different times to ensure we are not optimizing to local minima. We converted time to generations using a mutation rate 0.4 × 10−8 (Skoglund et al. 2015) and used 5,055,985 sites as the sequence length.
Next, we simulated data under these demographies using forward simulations as described above for the Yellowstone population. Since we do not know the initial introgression frequency (i.e., the starting frequency of the KB allele) nor the timing of the introgression, we estimated these parameters using an ABC rejection sampling approach. We drew these parameters from uniform prior distributions and kept simulations where the error was under 0.1, defined as
where is the observed present day allele frequency and is the simulated final allele frequency. We then simulated data using a forward simulation algorithm that samples the allele frequency for each generation using binomial sampling. We apply selection every generation, assuming a recessive lethal condition for the derived allele. We retained 100 pairs of posterior parameter estimates for each population and plotted their joint distribution using a two-dimensional kernel density estimate (Venables and Ripley 2002).
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We are grateful to Diana Dreger, Romolo Caniglia, and Marco Musiani for sample contributions. We thank Jaqueline Robinson and Rachael Treadwell for sample processing, and Victoria Sork, Eleazar Eskin, and two anonymous reviewers for their comments on earlier versions of this manuscript. Pauline Charruau, Bridgett vonHoldt, and Erin Stahler assisted with pedigree reconstruction. This work was supported by the National Science Foundation (DEB-1021397, OPP-0733033 to J.N. and R.K.W; DEB-1245373 to D.R.S. and D.W.S.); National Science Foundation Graduate Research Fellowship (DGE-1144087, DGE-0707424 to R.M.S.); the National Park Service (to D.R.S.and D.W.S.); Yellowstone Forever (to D.R.S. and D.W.S.); the Tapeats Fund (to D.R.S. and D.W.S.); the Perkin-Prothro Foundation (to D.R.S. and D.W.S.); an anonymous donor (to D.R.S. and D.W.S.); the National Human Genome Research Institute (to E.A.O.); the National Science Center, Poland (2015/19/P/NZ7/03971 to O.T.); EU’s Horizon 2020 program under the Marie Skłodowska-Curie grant (665778 to O.T.); NIH/NIGMS (R35GM119856 to K.E.L.); and scholarship support from the University of California, Los Angeles (R.M.S.). This work used the Vincent J. Coates Genomics Sequencing Laboratory at UC Berkeley, supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303.
Author Contributions
Conceived and designed the experiments: R.M.S., M.G., O.T., R.E.G., J.N., R.K.W.; Performed sequence capture and processed data: R.M.S.; Analyzed the data: R.M.S., A.D., J.S., S.H.V., K.L., R.E.G., K.E.L., J.N., R.K.W.; Contributed reagents/materials/analysis tools: R.M.S., A.D., J.S., S.H.V., D.R.S., M.G., D.W.S., E.R., E.A.O., K.E.L., R.E.G., J.N., R.K.W.; Wrote the paper: R.M.S., A.D., J.S., D.R.S., R.K.W., with input from all authors.
References
- Allen AP, Brown JH, Gillooly JF.. 2002. Global biodiversity, biochemical kinetics, and the energetic-equivalence rule. Science 2975586: 1545–1548. [DOI] [PubMed] [Google Scholar]
- Almberg ES, Cross PC, Smith DW.. 2010. Persistence of canine distemper virus in the Greater Yellowstone Ecosystem's carnivore community. Ecol Appl. 207: 2058.. [DOI] [PubMed] [Google Scholar]
- Anderson TM, vonHoldt BM, Candille SI, Musiani M, Greco C, Stahler DR, Smith DW, Padhukasahasram B, Randi E, Leonard JA et al. , 2009. Molecular and evolutionary history of melanism in North American gray wolves. Science 3235919: 1339–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auton A, Rui Li Y, Kidd J, Oliveira K, Nadel J, Holloway JK, Hayward JJ, Cohen PE, Greally JM, Wang J.. 2013. Genetic recombination is targeted towards gene promoter regions in dogs. PLoS Genet. 9:e1003984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangs EE, Fritts SH.. 1996. Reintroducing the gray wolf to central Idaho and Yellowstone National Park. Wildl Soc Bull. 24:402–413. [Google Scholar]
- Barrett RDH, Hoekstra HE.. 2011. Molecular spandrels: tests of adaptation at the genetic level. Nat Rev Genet. 1211: 767–780. [DOI] [PubMed] [Google Scholar]
- Brown SK, Darwent CM, Sacks BN.. 2013. Journal of Archaeological Science. J. Archaeol. Sci. 402: 1279–1288. [Google Scholar]
- Cassidy KA, Mech LD, MacNulty DR, Stahler DR, Smith DW.. 2017. Sexually dimorphic aggression indicates male gray wolves specialize in pack defense against conspecific groups. Behav Process. 136:64–72. [DOI] [PubMed] [Google Scholar]
- Candille SI, Kaelin CB, Cattanach BM, Yu B, Thompson DA, Nix MA, Kerns JA, Schmutz SM, Millhauser GL, Barsh GS.. 2007. A β-defensin mutation causes black coat color in domestic dogs. Science 3185855: 1418–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulson T, Macnulty DR, Stahler DR, vonHoldt BM, Wayne RK, Smith DW.. 2011. Modeling effects of environmental change on wolf population dynamics, trait evolution, and life history. Science 3346060: 1275–1278. [DOI] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST et al. , 2011. The variant call format and VCFtools. Bioinformatics 2715: 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Mita S, Siol M.. 2012. EggLib: processing, analysis and simulation tools for population genetics and genomics. BMC Genet. 13:27.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionne M, Miller KM, Dodson JJ, Caron F, Bernatchez L.. 2007. Clinal variation in MHC diversity with temperature: evidence for the role of host-pathogen interaction on local adaptation in Atlantic salmon. Evolution 619: 2154–2164. [DOI] [PubMed] [Google Scholar]
- Ducrest A-L, Keller L, Roulin A.. 2008. Pleiotropy in the melanocortin system, coloration and behavioural syndromes. Trends Ecol Evol. 23:502–510. [DOI] [PubMed] [Google Scholar]
- Erles K, Brownlie J.. 2010. Expression of Î2-defensins in the canine respiratory tract and antimicrobial activity against Bordetella bronchiseptica. Vet Immunol Immunopathol. 135(1-2): 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faircloth BC. 2015. Illumina TruSeq library prep for target enrich- ment. Available from http://ultraconserved.org; last accessed 15 March 15, 2018. [Google Scholar]
- Faircloth BC, Glenn TC.. 2012. Not all sequence tags are created equal: designing and validating sequence identification tags robust to indels. PLoS One 7:e42543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z, Silva P, Gronau I, Wang S, Armero AS, Schweizer RM, Ramirez O, pollinger J, Galaverni M, Ortega-Del Vecchyo D et al. , 2016. Worldwide patterns of genomic variation and admixture in gray wolves. Genome Res. 262: 163–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frantz LAF, Mullin VE, Pionnier-Capitan M, Lebrasseur O, Ollivier M, Perri A et al. , 2016. Genomic and archaeological evidence suggest a dual origin of domestic dogs. Science 3526290: 1228–1231. [DOI] [PubMed] [Google Scholar]
- Freedman AH, Gronau I, Schweizer RM, Ortega-Del Vecchyo D, Han E, Silva PM, Galaverni M, Fan Z, Marx P, Lorente-Galdos B et al. , 2014. Genome sequencing highlights the dynamic early history of dogs. PLoS Genet. 10:e1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galaverni M, Caniglia R, Pagani L, Fabbri E, Boattini A, Randi E.. 2017. Disentangling timing of admixture, patterns of introgression, and phenotypic indicators in a hybridizing wolf population. Mol Biol Evol. 34:2324–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier M, Vitalis R.. 2012. rehh: an R package to detect footprints of selection in genome-wide SNP data from haplotype structure. Bioinformatics 288: 1176–1177. [DOI] [PubMed] [Google Scholar]
- Gopalakrishnan S, Castruita JAS, Sinding M-HS, Kuderna LFK, Räikkönen J, Petersen B et al. , 2017. The wolf reference genome sequence (Canis lupus lupus) and its implications for Canis spp. population genomics. BMC Genomics 18:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MM, Granka JM, Bustamante CD, Sutter NB, Boyko AR, Zhu L, Ostrander EA, Wayne RK.. 2009. Linkage disequilibrium and demographic history of wild and domestic canids. Genetics 1814: 1493–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MM, Sutter NB, Ostrander EA, Wayne RK.. 2010. The IGF1 small dog haplotype is derived from Middle Eastern grey wolves. BMC Biol. 8:16.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guernier V, Hochberg ME, Guégan J-F.. 2004. Ecology drives the worldwide distribution of human diseases. PLoS Biol. 26: e141.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst R, Hernandez R, Williamson S, Bustamante C.. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5:e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedrick PW. 2015. Heterozygote Advantage: The Effect of Artificial Selection in Livestock and Pets. J Hered. 106:141–154. [DOI] [PubMed] [Google Scholar]
- Hedrick PW, Smith DW, Stahler DR.. 2016. Negative-assortative mating for color in wolves. Evolution 704: 757–766. [DOI] [PubMed] [Google Scholar]
- Hedrick PW, Stahler DR, Dekker D.. 2014. Heterozygote Advantage in a Finite Population: black Color in Wolves. J Hered. 105:457–465. [DOI] [PubMed] [Google Scholar]
- Hoekstra HE, Nachman M.. 2003. Different genes underlie adaptive melanism in different populations of rock pocket mice. Mol Ecol. 125: 1185–1194. [DOI] [PubMed] [Google Scholar]
- Hudson RR, Kaplan NL.. 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111:147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerns JA, Cargill EJ, Clark LA, Candille SI, Berryere TG, Olivier M, Lust G, Todhunter RJ, Schmutz SM, Murphy KE et al. , 2007. Linkage and segregation analysis of black and brindle coat color in domestic dogs. Genetics 1763: 1679–1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koepfli K-P, pollinger J, Godinho R, Robinson J, Lea A, Hendricks S et al. , 2015. Genome-wide evidence reveals that african and eurasian golden jackals are distinct species. Curr Biol. 2516:2158–2165. [DOI] [PubMed] [Google Scholar]
- Korneliussen TS, Albrechtsen A, Nielsen R.. 2014. ANGSD: analysis of next generation sequencing data. BMC Bioinformatics 15:356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard JA, Wayne RK, Wheeler J, Valadez R, Guillén S, Vilà C.. 2002. Ancient DNA evidence for Old World origin of New World dogs. Science 2985598: 1613–1616. [DOI] [PubMed] [Google Scholar]
- Li H. 2014. Towards better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 3020: 2843–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R.. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2516: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little C. 1958. Coat color genes in rodents and carnivores. Q Rev Biol. 332: 103–137. [DOI] [PubMed] [Google Scholar]
- Manceau M, Domingues VS, Linnen CR, Rosenblum EB, Hoekstra HE.. 2010. Convergence in pigmentation at multiple levels: mutations, genes and function. Philos Trans R Soc Lond B Biol Sci. 3651552: 2439–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mech D, Boitani L.. 2003. Wolves: behavior, ecology, and conservation. Chicago (IL: ): University of Chicago Press. [Google Scholar]
- Musiani M, Leonard JA, Cluff HD, Gates CC, Mariani S, Paquet PC, Vilà C, Wayne RK.. 2007. Differentiation of tundra/taiga and boreal coniferous forest wolves: genetics, coat colour and association with migratory caribou. Mol Ecol. 1619: 4149–4170. [DOI] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P.. 2005. A fine-scale map of recombination rates and hotspots across the human genome. Science 3105746: 321–324. [DOI] [PubMed] [Google Scholar]
- O'Connell J, Gurdasani D, Delaneau O, Pirastu N, Ulivi S, Cocca M, Traglia M, Huang J, Huffman JE, Rudan I.. 2014. A general approach for haplotype phasing across the full spectrum of relatedness. PLoS Genet. 10:e1004234–e1004234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis E, Claude J, Strimmer K.. 2004. APE: analyses of phylogenetics and evolution in R language. Bioinformatics 202: 289–290. [DOI] [PubMed] [Google Scholar]
- Paradis E. 2010. pegas: an R package for population genetics with an integrated-modular approach. Bioinformatics 263: 419–420. [DOI] [PubMed] [Google Scholar]
- Pazgier M, Hoover DM, Yang D, Lu W, Lubkowski J.. 2006. Human beta-defensins. Cell Mol Life Sci. 6311: 1294–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilot M, Greco C, vonHoldt BM, Jędrzejewska B, Randi E, Jędrzejewski W, Sidorovich VE, Ostrander EA, Wayne RK.. 2014. Genome-wide signatures of population bottlenecks and diversifying selection in European wolves. Heredity 1124: 428–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price A, Patterson N, Plenge R, Weinblatt M, Shadick N, Reich D.. 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 388: 904–909. [DOI] [PubMed] [Google Scholar]
- Protas ME, Patel NH.. 2008. Evolution of coloration patterns. Annu Rev Cell Dev Biol. 24:425–446. [DOI] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D, Maller J, Sklar P, de Bakker PIW, Daly MJ, Sham PC.. 2007. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 813: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran S, Deshpande O, Roseman CC, Rosenberg NA, Feldman MW, Cavalli-Sforza LL.. 2005. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci U S A. 10244: 15942–15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabeti PC, Reich DE, Higgins JM, Levine HZP, Richter DJ, Schaffner SF, Gabriel SB, Platko JV, Patterson NJ, McDonald GJ et al. , 2002. Detecting recent positive selection in the human genome from haplotype structure. Nature 4196909: 832–837. [DOI] [PubMed] [Google Scholar]
- Savolainen P, Zhang Y-P, Luo J, Lundeberg J, Leitner T.. 2002. Genetic evidence for an East Asian origin of domestic dogs. Science 2985598: 1610–1613. [DOI] [PubMed] [Google Scholar]
- Schweizer RM, Robinson J, Harrigan RJ, Silva P, Galverni M, Musiani M, Green RE, Novembre J, Wayne RK.. 2016. Targeted capture and resequencing of 1040 genes reveal environmentally driven functional variation in grey wolves. Mol Ecol. 251: 357–379. [DOI] [PubMed] [Google Scholar]
- Schweizer RM, vonHoldt BM, Harrigan RJ, Knowles JC, Musiani M, Coltman D, Novembre J, Wayne RK.. 2016. Genetic subdivision and candidate genes under selection in North American grey wolves. Mol Ecol. 251: 380–402. [DOI] [PubMed] [Google Scholar]
- Shannon LM, Boyko RH, Castelhano M, Corey E, Hayward JJ, McLean C, White ME, Abi Said M, Anita BA, Bondjengo NI et al. , 2015. Genetic structure in village dogs reveals a Central Asian domestication origin. Proc Natl Acad Sci U S A. 11244: 13639–13644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG et al. , 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 3436166: 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoglund P, Ersmark E, Palkopoulou E, Dalén L.. 2015. Ancient wolf genome reveals an early divergence of domestic dog ancestors and admixture into high- latitude breeds. Curr Biol. 25(11):1515–1519. [DOI] [PubMed] [Google Scholar]
- Smith J, Coop G, Stephens M, Novembre J.. 2018. Estimating time to the common ancestor for a beneficial allele. Mol Biol Evol. 355:1003–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahler DR, MacNulty DR, Wayne RK, vonHoldt B, Smith DW.. 2013. The adaptive value of morphological, behavioural and life-history traits in reproductive female wolves. J Anim Ecol. 821: 222–234. [DOI] [PubMed] [Google Scholar]
- Staples J, Nickerson DA, Below JE.. 2013. Utilizing graph theory to select the largest set of unrelated individuals for genetic analysis. Genet Epidemiol. 372: 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson EI. 2017. Back to basics: using colour polymorphisms to study evolutionary processes. Mol Ecol. 268: 2204–2211. [DOI] [PubMed] [Google Scholar]
- Tajima F. 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thalmann O, Shapiro B, Cui P, Schuenemann VJ, Sawyer SK, Greenfield DL, Germonpre MB, Sablin MV, Lopez-Giraldez F, Domingo-Roura X et al. , 2013. Complete mitochondrial genomes of ancient canids suggest a European origin of domestic dogs. Science 3426160: 871–874. [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Varkonyi R, Cahinhinan N, Abbes S, Argyropoulos G, Destro-Bisol G, Drousiotou A, Dangerfield B, Lefranc G, Loiselet J et al. , 2001. Haplotype diversity and linkage disequilibrium at human G6PD: recent origin of alleles that confer malarial resistance. Science 2935529: 455–462. [DOI] [PubMed] [Google Scholar]
- van Asch B, Zhang A-B, Oskarsson MCR, Klütsch CFC, Amorim A, Savolainen P.. 2013. Pre-Columbian origins of Native American dog breeds, with only limited replacement by European dogs, confirmed by mtDNA analysis. Proc Biol Sci. 280:20131142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venables WN, Ripley BD. (2002) Modern applied statistics with S, 4th edn New York: Springer. ISBN 0-387-95457-0. [Google Scholar]
- vonHoldt BM, Pollinger JP, Earl DA, Knowles JC, Boyko AR, Parker H, Geffen E, Pilot M, Jedrzejewski W, Jedrzejewska B et al. , 2011. A genome-wide perspective on the evolutionary history of enigmatic wolf-like canids. Genome Res. 218: 1294–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vonHoldt BM, Pollinger JP, Lohmueller KE, Han E, Parker HG, Quignon P, Degenhardt JD, Boyko AR, Earl DA, Auton A et al. , 2010. Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 4647290: 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- vonHoldt BM, Stahler DR, Bangs EE, Smith DW, Jimenez MD, Mack CM, Niemeyer CC, Pollinger JP, Wayne RK.. 2010. A novel assessment of population structure and gene flow in grey wolf populations of the Northern Rocky Mountains of the United States. Mol Ecol. 1920: 4412–4427. [DOI] [PubMed] [Google Scholar]
- vonHoldt BM, Stahler DR, Smith DW, Earl DA, Pollinger JP, Wayne RK.. 2008. The genealogy and genetic viability of reintroduced Yellowstone grey wolves. Mol Ecol. 171: 252–274. [DOI] [PubMed] [Google Scholar]
- Wall JD, Cox MP, Mendez FL, Woerner A, Severson T, Hammer MF.. 2008. A novel DNA sequence database for analyzing human demographic history. Genome Res. 188: 1354–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watterson G. 1975. On the number of segregating sites in genetical models without recombination. Theor Popul Biol. 72: 256–276. [DOI] [PubMed] [Google Scholar]
- Wright S. 1917. Color inheritance in mammals: results of experimental breeding can be linked up with chemical researches on pigments—coat colors of all mammals classified as due to variations in action of two enzymes. J Hered. 8:224–235. [Google Scholar]
- Yang D, Chertov O, Bykovskaia S, Chen Q, Buffo M, Shogan J, Anderson M, Schröder J, Wang J, Howard O.. 1999. β-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science 2865439: 525–528. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.