

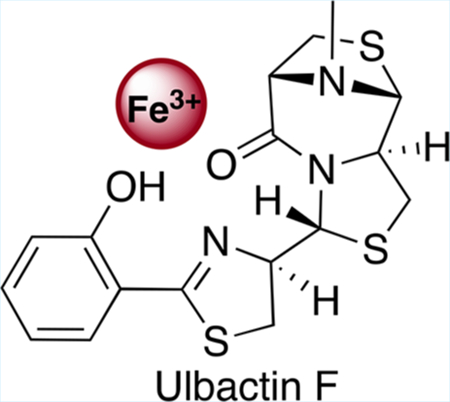
Abstract
Natural products from environmental microbiomes provide exquisite templates for elucidating biological activity in the search for new drugs. A recently discovered marine Brevibacillus sp. metabolite, ulbactin F, was found to inhibit tumor cell migration and invasion at IC50 < 3 μM. Herein, we disclose the first total synthesis of ulbactin F and epi-ulbactin F, which was modeled after the biosynthetic pathway. The scaffold bears structural similarity to siderophores of human pathogens but contains a novel tricyclic ring system derived from cysteine. We have found that ulbactin F forms low-affinity metal complexes, with a preference for Fe3+ and Cu2+, which may hint both at its environmental role and its antimetastatic mechanism of action.
Graphical Abstract
The chemical diversity of marine microbiomes has long been enticing for natural product chemists in the discovery of new scaffolds with novel biological activity.1,2 The fluctional chemical composition, irregular nutrient availability, and complex symbiotic networks of organisms create high selective pressure for bacteria to evolve biosynthetic pathways toward unique metabolites that allow them to establish an ecological niche. Since the 2004 introduction of the first FDA approved marine natural product (Ziconotide),3 efforts to mine the ocean to drive drug discovery have seen renewed interest. As advanced culturing techniques allow the sequencing and fermentation of more microbes, new opportunities arise to use structurally interesting molecules to gain insight into innovative biochemical space.
In 2016, two new natural products fermented from a sponge-derived Brevibacillus sp. were discovered and characterized.4 The compounds were found to contain a phenolate-thiazoline-thiazolidine core scaffold, shared by marine natural products ulbactins A–E,5 and were thus dubbed ulbactins F (l) and G (2) (Figure 1). This core ulbactin structure is also shared by the Pseudomonas aeruginosa siderophore pyochelin (3), a virulence factor relevant to human infection with pFe = 16.0.6 However, ulbactins F-3″S-trans and G-3″R-cis also contain a unique 6,9-imino-1H,3H,5H-thiazolo[4,3-c][1,4]Thiazepin-5-one. This tricycle has only been reported once before in ulbactin D,7 but lacked stereochemical assignment and the bridge N-methyl group. While various ulbactins A—E are reported as UV-absorbers and antimicrobials, the isolation paper found no antimicrobial activity but rather that ulbactins F and G inhibit the migration of epidermoid carcinoma A431 cells at noncytotoxic concentrations. The primary metabolite, ulbactin F, was more rigorously tested and found to inhibit the migration of esophageal squamous carcinoma EC109 cells at IC50 = 2.1 μM and the invasion by murine colon carcinoma 26- L5 cells at IC50 = 1.7 μM. Based on similarity to pyochelin, we hypothesized that these compounds may selectively chelate iron and that this would not only play a role in their antimetastatic properties but also give them previously undisclosed microbial activity in iron-deplete conditions. To test this hypothesis and lay the groundwork for future analog development, we set forth on a total synthesis of this unique scaffold.
Figure 1.
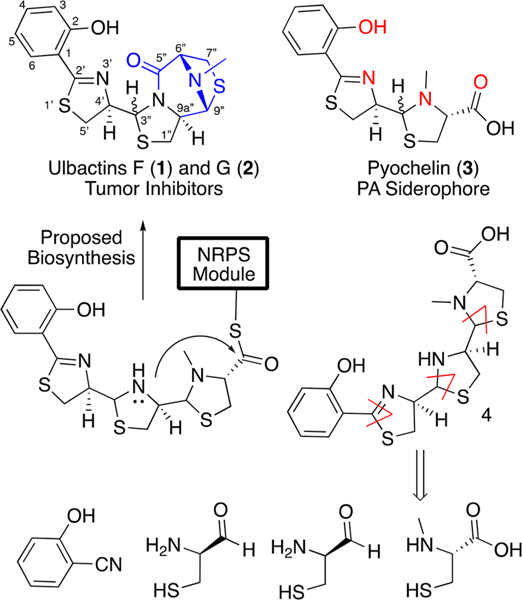
(Top) Structures of ulbactins F-3″S-trans (1), ulbactin G-3″R-cis (2), and pyochelin (3). Structurally novel fused heterocycle in blue. Iron chelating atoms of pyochelin in red. (Bottom) Proposed biosynthesis from putative gene annotation and retrosynthetic analysis.
As ulbactin F is the major metabolite reported (17:1 ulbactin F/G), we targeted our synthetic route toward this diastereomer. The route was designed to be concise and convergent, running three synthetic pathways in parallel and merging them into the final compound. This permitted the usage of simple molecules for the majority of the synthesis culminating with the incorporation of the structural complexity in the final step. Toward this end, we looked to the biosynthesis for inspiration (Figure 1). Ulbactin F is constructed by a nonribosomal peptide synthetase, which operates in an assembly line-like fashion by covalently loading monomers onto enzyme modules and catalytically linking them together into elongated scaffolds (Scheme 1). Salicylate and three units of cysteine (which are modified by cyclization, reduction, and methylation) are assembled into a linear enzyme-bound precursor, which is presumably cleaved from the assembly line by an intramolecular cyclization of the thiazolidine onto the terminal thioester to release the final compound.8 We drew on this chemical logic, targeting a linear ulbactin precursor as the key intermediate. We hypothesized that ulbactin F was preferred over G due to thermodynamic stability of the tricycle, which could be leveraged to selectively construct the natural product.
Scheme 1.
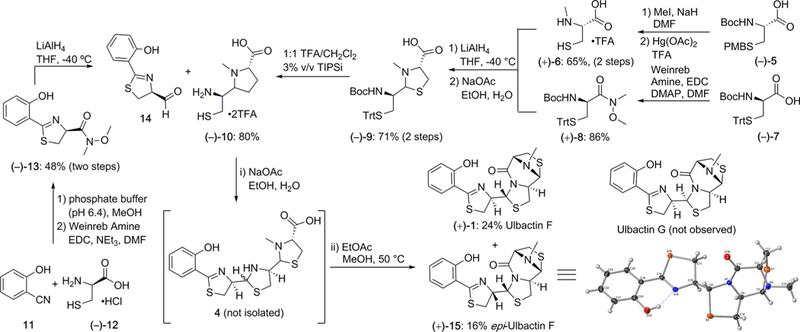
Total Synthesis of Ulbactin F (1) and epi-Ulbactin F (12)
The forward synthesis commenced through the implementation of known chemistry from Mislin and co-workers in the synthesis of pyochelin analogs.9 Salicylnitrile was converted to aldehyde 14 in three steps through Weinreb amide (–)-13. Aldehyde 14, which is prone to epimerization, was taken on crude as precedented in the literature. In parallel, commercially available (–)-5 was selectively N-methylated with excess NaH and methyl iodide.10 Global deprotection occurred using neat TFA with mercury acetate to give N-methyl-L-cysteine salt (+)-6.11 Concurrently, commercially available (–)-7 was converted to the Weinreb amide (+)-8 and was then reduced to the corresponding aldehyde with LiAlH4.9 The aldehyde, which was taken on crude to avoid degradation during purification, was reacted with (+)-6 in a 3-to-1 mixture of ethanol/water with sodium acetate buffer (pH = 6.5) to give protected thiazolidine (–)-9.9 This was then treated with 1:1 TFA/CH2Cl2 with 3% triisopropylsilane as a cation scavenger to give the double-deprotected thiazolidine salt (–)-10.12 Thiazolidine (–)-10 was then mixed with freshly prepared aldehyde 14 in 3-to-1 ethanol/water with sodium acetate buffer (pH = 7.5) in an attempt to synthesize the linear ulbactin precursor 4.
At this stage, we expected to isolate the linear precursor, but to our surprise, small amounts of ulbactin F ([M + H] = 394) were observed by MS analysis in addition to the linear acid ([M + H] = 412). Inspired by this discovery, we sought to optimize the late-stage one-step coupling/cyclization cascade. Initial attempts to thermodynamically drive the reaction to completion resulted in the formation of a number of undesired side products. Ultimately, we found that after workup we could gently heat the crude material in a mixture of ethyl acetate and methanol and saw full conversion to the final ulbactin mass, which was then purified by preparative HPLC. The mild conditions needed to affect the transformation suggest a low energy barrier for the cyclization of the linear acid. Although the cascade reaction proceeds in a modest overall yield, it results in the formation of three new bonds and two heterocycles thereby greatly expediting the total synthesis. Ulbactin F was synthesized in 12% overall yield in a longest linear sequence of six steps.
While we expected thermodynamics to favor ulbactin F (1), somewhat surprisingly we observed no ulbactin G in the final reaction mixture. Instead, an unknown diastereomer with [M + H] = 394 and very similar 1H NMR signals to ulbactin F was isolated as the minor product with a ratio of 3:2. Ulbactins F and G are distinguished largely by the 1H NMR splitting and chemical shifts of H4′ and H3″ (on the adjacent thiazoline and thiazolidine rings, respectively), whereas the unknown diastereomer showed a chemical shift of H3″ very similar to that of ulbactin F. We hypothesized that the stereochemistry at C9a″ and C6″ would be unchanged from starting materials. Additionally, while C9″ is formed racemically to make (–)-10, the final amide coupling requires cis stereochemistry with the carbonyl, and thus, we hypothesized that this sets the overall 3D conformation of the tricycle. With this unchanged, we theorized that the difference was in the stereochemistry at the thiazoline (H4′). This hypothesis was confirmed by X-ray crystallography, which showed the same connectivity as ulbactin F but was epimeric at H4′ and was thus referred to as epi-ulbactin F (15). We deduced this epimerization occurred during the reduction from thiazoline Weinreb amide to aldehyde 14, as 10% epimerization was observed in the synthesis of des-methyl pyochelin by Mislin.9 Our attempts to modify this step with alternative reducing reagents,13 reducing to the primary alcohol and oxidizing to the aldehyde,14 or forming the bisulfite adduct for purification,13 were unsuccessful. When compared with the crystal structure of ulbactin G from the isolation paper,4 epi-ulbactin F shows interesting similarities. The stereochemistry between ulbactin G and epi-ulbactin F are cis at H4′ and H3″, whereas ulbactin F is trans, and while the crystal structure of ulbactin F is folded placing the phenol and the tricycle in close proximity, both ulbactin G and epi-ulbactin F are flat. This presumably indicates that the stereochemistry plays a role in the intramolecular hydrogen bonding network of the molecule and potentially the final cyclization.
We were initially surprised that the tricycle could form without traditional amide coupling reagents. Mechanistically, we propose that this can occur through simple nucleophilic attack of the secondary amine into the carbonyl. It is also possible that the phenol can intramolecularly esterify the terminal carboxylate to form a macrocycle, which then situates the amine in position for nucleophilic attack on the carbonyl, displacing the phenol and forming the final tricycle (Scheme 2). This would explain the selectivity for ulbactin F (and epi-F) over G, as the chairlike transition state strongly favors (R)- stereochemistry at H3″ over (S). Energy minimization of diastereomeric macrocycle structures shows a shorter distance from secondary amine to carbonyl to form ulbactin F (3.9 Å) and epi-ulbactin F (3.7 Å) than to form ulbactin G (4.3 Å) or epi-ulbactin G (5.0 Å) (Supplementary Figure 1). The proposed macrocyclic intermediate opens a low energy pathway to the product, and forming the polycyclic final structure provides a strong thermodynamic driving force toward the final scaffold.
Scheme 2. Proposed Mechanism for Stereoselectivity in Ulbactin Formationa.
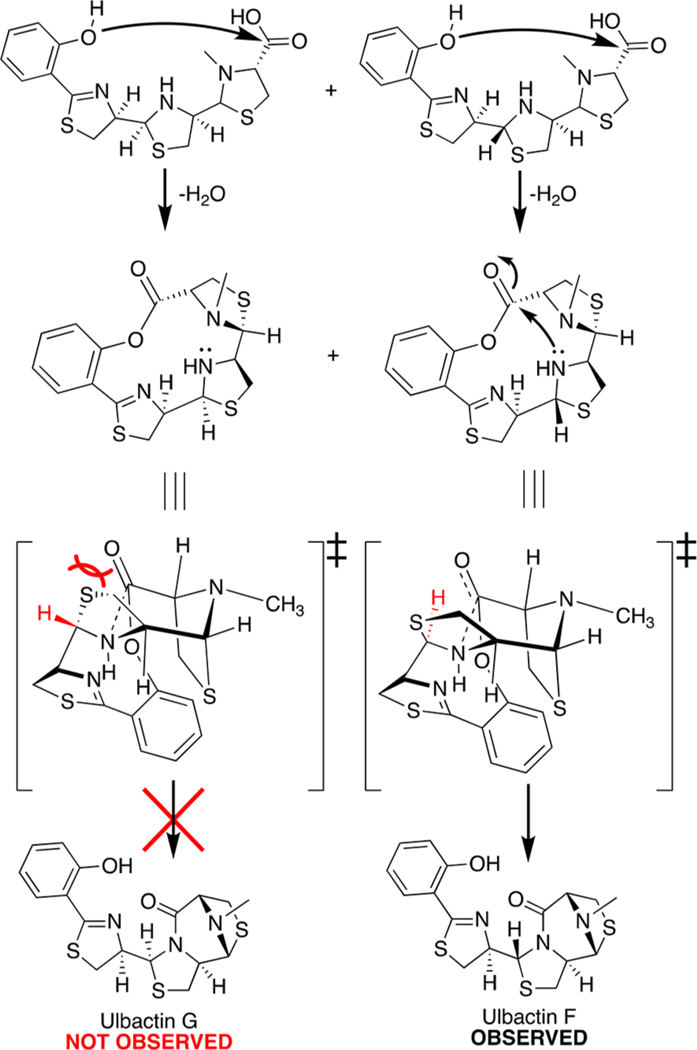
aThe reaction is selective for ulbactin F over G due to a favored chairlike transition state. A similar model applies to epi-ulbactin F over epi-ulbactin G.
With ulbactin F in hand, we proceeded to test its physicochemical properties. Due to its structural similarity to pyochelin, we hypothesized that it could function as an iron-binding molecule. Initial experiments to form an iron complex using iron(III) acetylacetonate resulted in no spectrophoto-metric change (Supplementary Figure 2), indicating no iron chelation. Assays in liquid and solid CAS media supported this initial result (Supplementary Figure 3). However, upon treatment of the natural product with iron(III) trichloride a color change was observed (Supplementary Figure 4).
Despite the lack of a colorimetric indication of iron chelation, titration of both ulbactin F and epi-ulbactin F with iron(lIl) acetylacetonate showed fluorescence quenching, indicating a 1:1 ligand/metal ratio, which was further confirmed by titration of ulbactin F with iron(III) trichloride (Figure 2A). This was somewhat surprising, given the precedent that 1-to-1 chelators have high binding affinity.15 However, we hypothesize that, while ulbactin F has many atoms with the ability to donate electron density to a metal center, many of these ligands are fairly weak donors, and that steric bulk allows the ligand to surround the metal preventing multiligand chelation. There is precedence in the literature for strong iron chelators showing a linear decay in fluorescence toward their final stoichiometric ligand–metal ratio;16 however, ulbactin F shows an exponential decay, indicating that the ligand passes through multiple binding modes as the ratio between total iron(III) and ligand increases. The phenolate thiazoline (high affinity bidentate chelator common in siderophores15) may initially chelate iron in a multiligand fashion, but as iron concentration increases, it becomes more energetically favorable for a single ulbactin F molecule to displace other ligands to form a 1-to-1 complex that excludes other chelators by the steric bulk of the tricycle.
Figure 2.
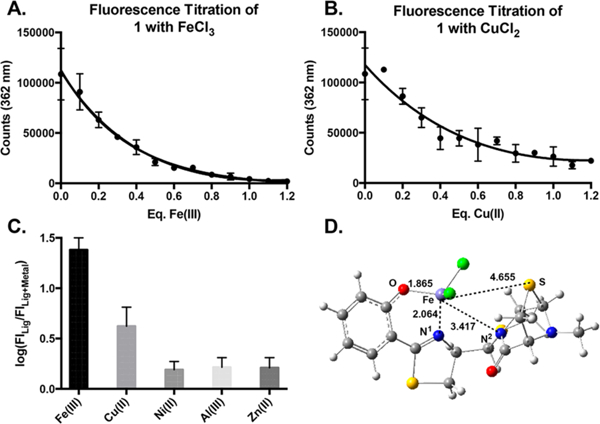
Fluorescence titration of ulbactin F (1) with (A) iron chloride and (B) copper chloride. (C) Metal selectivity of 1, measured on a log scale as the ratio of fluorescence of free ligand over fluorescence of ligand in the presence of one molar equivalent of metal. (D) Free energy minimization of proposed 1-Fe3+ structure.
In proposing an iron-chelation mode, we considered two major possibilities; in-tact ulbactin F coordinating the metal or ring reopening and binding by the linear precursor. In samples treated with excess iron and iron + citrate (competitive chelator), HRMS revealed ulbactin F ([M + H] = 394) as the major ion in addition to the ulbactin F iron complex ([M + H] = 448) (Supplementary Figure 5). The linear precursor was not detected in apo- ([M + H] = 412) or iron-bound ([M + H] = 465) form. These results suggest that the tricycle persists during chelation.
To test the chelation selectivity of the natural product, fluorescence titrations were conducted with a panel of metals (Figure 2,Supplementary Figure 6). Ulbactin F was found to have a distinct preference for Fe3+ and Cu2+ over other metals like Ni2+, Zn2+, and Al3+ (16.7 and 2.8 times higher, respectively, as measured by ratio of fluorescence before and after addition of one molar equivalent of metal). This indicates that charge and size of the metal are not the only factors in determining metal selectivity. Using the stoichiometry determined from fluorescence titrations, HRMS data, information gleaned from metal selectivity, and the crystal structure from the isolation paper as a framework, an iron(III) ulbactin F complex was proposed (utilizing the phenolatethiazoline, amide nitrogen N4″, and S8″), the geometric structure of which was optimized by using density functional theory (here, we used B3LYP functional with double-ζ basis sets augmented with polarization functions: see Supporting Information for more details and Figure 2D). The computed structure of iron(III) ulbactin F complex shows short bond distances from Fe3+ to the phenol and thiazoline, but longer distances to amide nitrogen N4″ and S″, supporting our hypothesis of weak Fe3+ affinity of ulbactin F. This hypothesis is also supported by our finding that the formation of the complex from free ligand and iron trichloride is only slightly thermodynamically favorable (). By contrast, we calculated the reaction of ulbactin F with iron(III) acetylacetonate to be slightly uphill in energy (). These computations validate our observation of a spontaneous color change upon treatment with iron(III) chloride but not with iron(III) acetylacetonate.
Further physicochemical characterization was hindered by very poor aqueous solubility. Whereas pyochelin is reported to be freely soluble to 10% methanol/water,17 ulbactin F was observed to precipitate at up to 50% methanol or DMSO in water. This can be rationalized by the differences in scaffold and polar moieties (hydrogen bonding capabilities) between pyochelin and ulbactin F. Insolubility of the complex prevented traditional methods of KFe determination (spectrophotometric pH titration, EDTA competition assays, etc.), but we were able to show that free catechol can abstract iron(III) from the complex in methanol by UV–vis spectroscopy (SupplementaryFigure 7). Unfortunately, the overlap in absorption wavelength of Fe(cat)3 and Fe(ulbactin) prevented quantification.
Because of the structural similarity to the known siderophore pyochelin and our newly discovered iron-affinity, we attempted to investigate microbial activity of the isolated Fe3+-ulbactin F complex in P. aeruginosa. However, the complex was equally insoluble and failed to promote the growth under iron deficient conditions compared to an FeCl3 control (Supplementary Figure 8). The lack of microbial biological activity may be explained by poor solubility preventing the molecule from ever reaching a molecular target. However, while preliminary testing was conducted against P. aeruginosa due to structural similarity to pyochelin, it may be better suited for different microbes; certain Vibrio species also produce thiazoline siderophores and may be more susceptible to ulbactin. Future analog design will incorporate polar groups to increase aqueous solubility and potentially increase microbial and cancer activity.
Herein, we have disclosed the first total synthesis and metal-binding properties of a marine natural product with potent antimetastatic activity, ulbactin F, as well as its diastereomer epi-ulbactin F. The synthesis is concise, convergent, and builds most of the structural complexity in the final step with a powerful thermodynamic driving force to form the novel fused tricyclic ring system. Furthermore, we show that ulbactin F preferentially forms a colored complex with iron(III) in similar fashion to pyochelin6 and conducted computational analysis to substantiate these findings. The ability of marine bacteria to produce unique structural scaffolds with novel bioactivity is a thriving area of chemical research but remains relatively underexplored, and our methods will form the basis for further derivatization to explore the role of metal chelation in the activity of these structurally complex and biologically active marine natural products.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully acknowledge funding from the National Institute of General Medical Sciences (GM119426 for W.M.W.) and the National Science Foundation (CHE1755698 for W.M.W., CHE-0958205 for D.G.M.), the use of the resources of the Cherry Emerson Center for Scientific Computation, and CHE1531620 for the NMR instrumentation used in this work. We also thank Dr. John Bacsa, Emory X-ray Crystallography Facility, and Thomas C. Pickel for the X-ray structural analysis, and the use of the Rigaku SYNERGY diffractometer, supported by NSF grant CHE1626172. We also acknowledge Nathaniel S. Greenwood (Temple University) for synthetic contributions.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACSPublicationswebsite at DOI: 10.1021/acs.or-glett.8b02599.
Experimental details, spectroscopic data of new compounds, and additional data/figures (PDF)
Accession Codes
CCDC 1861468 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Molinski TF; Dalisay DS; Lievens SL; Saludes JP Nat. Rev. Drug Discovery 2009, 8 (1), 69–85. [DOI] [PubMed] [Google Scholar]
- (2).Rossiter SE; Fletcher MH; Wuest WM Chem. Rev. 2017, 117 (19), 12415–12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).McGivern JG Neuropsychiatr. Dis. Treat. 2007, 3 (1), 69–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Igarashi Y; Asano D; Sawamura M; In Y; Ishida T; Imoto M Org. Lett. 2016, 18 (7), 1658–1661. [DOI] [PubMed] [Google Scholar]
- (5).Eleftherianos I; Dowling A; Wilkinson P; Parkhill J; et al. Proc. Natl. Acad. Sci. U. S. A. 2009, 106 (6), 6–12.19118201 [Google Scholar]
- (6).Brandel J; Humbert N; Elhabiri M; Schalk IJ; Mislin GLA; Albrecht-Gary, A. M. Dalt. Trans. 2012, 41 (9), 2820–2834. [DOI] [PubMed] [Google Scholar]
- (7).Kikuchi K; Chen C; Adachi K; Nishijima M; Nishida F; Takadera T; Sano H Japan Patent No. 200902146486659064, 1997. [Google Scholar]
- (8).Komaki H; Hosoyama A; Ichikawa N; Igarashi Y Gene Reports 2016, 5, 140–143. [Google Scholar]
- (9).Noël S; Guillon L; Schalk IJ; Mislin GL A. Org. Lett. 2011, 13 (5), 844–847. [DOI] [PubMed] [Google Scholar]
- (10).Malkov AV; Vranková K; Černý M; Kočovský PJ Org. Chem. 2009, 74 (21), 8425–8427. [DOI] [PubMed] [Google Scholar]
- (11).Macmillan D; Anderson DW Org. Lett. 2004, 6 (25), 4659–4662. [DOI] [PubMed] [Google Scholar]
- (12).Giustiniano M; Tortorella P; Agamennone M; Di Pizio A; et al. J. Amino Acids 2013, 2013, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Bailey CL; Clary JW; Tansakul C; Klabunde L; Anderson CL; Joh AY; Lill AT; Peer N; Braslau R; Singaram B Tetrahedron Lett. 2015, 56 (5), 706–709. [Google Scholar]
- (14).De Luca L; Giacomelli G; Porcheddu AJ Org. Chem. 2001, 66 (23), 7907–7909. [DOI] [PubMed] [Google Scholar]
- (15).Hider RC; Kong X Nat. Prod. Rep. 2010, 27 (5), 637–657. [DOI] [PubMed] [Google Scholar]
- (16).Bohac TJ; Shapiro JA; Wencewicz TA ACS Infect. Dis. 2017, 3 (11), 802–806. [DOI] [PubMed] [Google Scholar]
- (17).Cox CD; Graham RJ Bacteriol. 1979, 137 (1), 357–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.