

Abstract
Objective
To update the congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency clinical practice guideline published by the Endocrine Society in 2010.
Conclusions
The writing committee presents updated best practice guidelines for the clinical management of congenital adrenal hyperplasia based on published evidence and expert opinion with added considerations for patient safety, quality of life, cost, and utilization.
Update of best practice guidelines for the clinical management of congenital adrenal hyperplasia with added considerations for patient safety, quality of life, cost, and utilization.
List of Recommendations
Newborn screening
Cost-effectiveness
1.1 We recommend that all newborn screening programs incorporate screening for congenital adrenal hyperplasia due to 21-hydroxylase deficiency. (1|⊕⊕⊕○)
1.2 We recommend that first-tier screens use 17-hydroxyprogesterone assays standardized to a common technology with norms stratified by gestational age. (1|⊕⊕⊕○)
Technical remark: Clinicians should be aware that immunoassays are still in use and remain a source of false-positive results. Specificity may be improved with organic extraction to remove cross-reacting substances.
1.3 We recommend that screening laboratories employ a second-tier screen by liquid chromatography–tandem mass spectrometry in preference to all other methods (e.g., genotyping) to improve the positive predictive value of congenital adrenal hyperplasia screening. (1|⊕⊕○○)
Technical remark: Laboratories utilizing liquid chromatography–tandem mass spectrometry should participate in an appropriate quality assurance program. Additionally, clinicians should realize that immunoassays lead to more false-positive results. Thus, if laboratory resources do not include liquid chromatography–tandem mass spectrometry, a cosyntropin stimulation test should be performed to confirm diagnosis prior to initiation of corticosteroid treatment.
Prenatal treatment of congenital adrenal hyperplasia
2.1 We advise that clinicians continue to regard prenatal therapy as experimental. Thus, we do not recommend specific treatment protocols. (Ungraded Good Practice Statement)
2.2 In pregnant women at risk for carrying a fetus affected with congenital adrenal hyperplasia and who are considering prenatal treatment we recommend obtaining prenatal therapy only through protocols approved by Institutional Review Boards at centers capable of collecting outcomes from a sufficiently large number of patients, so that risks and benefits can be defined more precisely. (1|⊕⊕⊕○)
2.3 We advise that research protocols for prenatal therapy include genetic screening for Y-chromosomal DNA in maternal blood to exclude male fetuses from potential treatment groups. (Ungraded Good Practice Statement)
Diagnosis of congenital adrenal hyperplasia
3.1 In infants with positive newborn screens for congenital adrenal hyperplasia we recommend referral to pediatric endocrinologists (if regionally available) and evaluation by cosyntropin stimulation testing as needed. (1|⊕⊕⊕○)
3.2 In symptomatic individuals past infancy, we recommend screening with an early-morning (before 8 am) baseline serum 17-hydroxyprogesterone measurement by liquid chromatography–tandem mass spectrometry. (1|⊕⊕⊕○)
3.3 In individuals with borderline 17-hydroxyprogesterone levels, we recommend obtaining a complete adrenocortical profile after a cosyntropin stimulation test to differentiate 21-hydroxylase deficiency from other enzyme defects. (1|⊕⊕⊕○)
3.4 In individuals with congenital adrenal hyperplasia, we suggest genotyping only when results of the adrenocortical profile after a cosyntropin stimulation test are equivocal, or cosyntropin stimulation cannot be accurately performed (i.e., patient receiving glucocorticoid), or for purposes of genetic counseling. (2|⊕⊕⊕○)
Technical remark: Genotyping at least one parent aids in the interpretation of genetic test results because of the complexity of the CYP21A2 locus.
Treatment of classic congenital adrenal hyperplasia
4.1 In growing individuals with classic congenital adrenal hyperplasia, we recommend maintenance therapy with hydrocortisone. (1|⊕⊕⊕○)
4.2 In growing individuals with congenital adrenal hyperplasia, we recommend against the use of oral hydrocortisone suspension and against the chronic use of long-acting potent glucocorticoids. (1|⊕⊕⊕○)
4.3 In the newborn and in early infancy, we recommend using fludrocortisone and sodium chloride supplements to the treatment regimen. (1|⊕⊕⊕○)
4.4 In adults with classic congenital adrenal hyperplasia, we recommend using daily hydrocortisone and/or long-acting glucocorticoids plus mineralocorticoids, as clinically indicated. (1|⊕⊕⊕○)
4.5 In all individuals with classic congenital adrenal hyperplasia, we recommend monitoring for signs of glucocorticoid excess, as well as for signs of inadequate androgen normalization, to optimize the adrenal steroid treatment profile. (1|⊕⊕⊕○)
4.6 In all individuals with classic congenital adrenal hyperplasia, we recommend monitoring for signs of mineralocorticoid deficiency or excess. (1|⊕⊕⊕○)
Stress dosing
4.7 In all patients with congenital adrenal hyperplasia who require glucocorticoid treatment, for situations such as febrile illness (>38.5°C), gastroenteritis with dehydration, major surgery accompanied by general anesthesia, and major trauma we recommend increasing the glucocorticoid dosage. (1|⊕⊕⊕○)
4.8 In patients with congenital adrenal hyperplasia under everyday mental and emotional stress and minor illness and/or before routine physical exercise we recommend against the use of increased glucocorticoid doses. (1|⊕⊕○○)
4.9 In patients with congenital adrenal hyperplasia who require treatment, we recommend always wearing or carrying medical identification indicating that they have adrenal insufficiency. (1|⊕⊕⊕○)
4.10 In patients with congenital adrenal hyperplasia, we recommend educating patients and their guardians and close contacts on adrenal crisis prevention and increasing the dose of glucocorticoid (but not mineralocorticoid) during intercurrent illness. (1|⊕⊕⊕○)
4.11 We recommend equipping every patient with congenital adrenal hyperplasia with a glucocorticoid injection kit for emergency use and providing education on parenteral self-administration (young adult and older) or lay administration (parent or guardian) of emergency glucocorticoids. (1|⊕⊕⊕○)
Monitoring therapy
4.12 In patients ≤18 months with congenital adrenal hyperplasia, we recommend close monitoring in the first 3 months of life and every 3 months thereafter. After 18 months, we recommend evaluation every 4 months. (1|⊕⊕○○)
4.13 In pediatric patients with congenital adrenal hyperplasia, we recommend conducting regular assessments of growth velocity, weight, blood pressure, as well as physical examinations in addition to obtaining biochemical measurements to assess the adequacy of glucocorticoid and mineralocorticoid. (1|⊕⊕○○)
4.14 In pediatric patients with congenital adrenal hyperplasia under the age of 2 years, we advise annual bone age assessment until near-adult height is attained. (Ungraded Good Practice Statement)
4.15 In adults with congenital adrenal hyperplasia, we recommend annual physical examinations, which include assessments of blood pressure, body mass index, and Cushingoid features in addition to obtaining biochemical measurements to assess the adequacy of glucocorticoid and mineralocorticoid replacement. (1|⊕⊕○○)
4.16 In adults with congenital adrenal hyperplasia, we recommend monitoring treatment through consistently timed hormone measurements relative to medication schedule and time of day. (1|⊕⊕○○)
4.17 In adults with congenital adrenal hyperplasia, we recommend that clinicians do not completely suppress endogenous adrenal steroid secretion to prevent adverse effects of over treatment. (1|⊕⊕⊕○)
Treatment of nonclassic congenital adrenal hyperplasia
5.1 In children and adolescents with inappropriately early onset and rapid progression of pubarche or bone age and in adolescent patients with overt virilization we suggest glucocorticoid treatment of nonclassic congenital adrenal hyperplasia. (2|⊕⊕○○)
Technical remark: Risks and benefits of glucocorticoid therapy should be considered and discussed with the patient’s family.
5.2 In asymptomatic nonpregnant individuals with nonclassic congenital adrenal hyperplasia we recommend against glucocorticoid treatment. (1|⊕⊕⊕○)
5.3 In previously treated patients with nonclassic congenital adrenal hyperplasia we suggest giving the option of discontinuing therapy when adult height is attained or other symptoms resolve. (2|⊕⊕⊕○)
5.4 In adult women with nonclassic congenital adrenal hyperplasia who also have patient-important hyperandrogenism or infertility we suggest glucocorticoid treatment. (2|⊕⊕○○)
5.5 In most adult males with nonclassic congenital adrenal hyperplasia, we suggest that clinicians generally not prescribe daily glucocorticoid therapy. (2|⊕○○○)
Technical remark: Exceptions include infertility, testicular adrenal rest tumors or adrenal tumors, and phenotypes that are intermediate between classic and nonclassic phenotypes.
5.6 In patients with nonclassic congenital adrenal hyperplasia, we suggest hydrocortisone stress dosing for major surgery, trauma, or childbirth only if a patient has a suboptimal (<14 to 18 μg/dL, <400 to 500 nmol/L) cortisol response to cosyntropin or iatrogenic adrenal suppression. (2|⊕○○○)
Technical remark: A range is given for cortisol cut points due to greater specificity of newer cortisol assays (see below).
Long-term management of patients with congenital adrenal hyperplasia
Transition to adult care
6.1 In adolescent patients with congenital adrenal hyperplasia, we suggest that the transition to adult care begins several years prior to dismissal from pediatric endocrinology. (2|⊕○○○)
Technical remark: We advise the use of joint clinics comprised of pediatric, reproductive, and adult endocrinologists and urologist during this transition.
6.2 In adolescent females with congenital adrenal hyperplasia, we suggest a gynecological history and examination to ensure functional female anatomy without vaginal stenosis or abnormalities in menstruation. (2|⊕⊕○○)
Genetic counseling
6.3 In children with congenital adrenal hyperplasia, adolescents transitioning to adult care, adults with nonclassic congenital adrenal hyperplasia upon diagnosis, and partners of patients with congenital adrenal hyperplasia who are planning a pregnancy, we recommend that medical professionals familiar with congenital adrenal hyperplasia provide genetic counseling. (1|⊕⊕○○)
Fertility counseling
6.4 In individuals with congenital adrenal hyperplasia and impaired fertility we suggest referral to a reproductive endocrinologist and/or fertility specialist. (2|⊕⊕○○)
Management of congenital adrenal hyperplasia and nonclassic congenital adrenal hyperplasia during pregnancy
6.5 In women with nonclassic congenital adrenal hyperplasia who are infertile or have a history of prior miscarriage, we recommend treatment with a glucocorticoid that does not traverse the placenta. (1|⊕⊕○○)
6.6 In women with congenital adrenal hyperplasia who are pregnant, we advise management by an endocrinologist familiar with congenital adrenal hyperplasia. (Ungraded Good Practice Statement)
6.7 In women with congenital adrenal hyperplasia who become pregnant we recommend continued prepregnancy doses of hydrocortisone/prednisolone and fludrocortisone therapy, with dosage adjustments if symptoms and signs of glucocorticoid insufficiency occur. (1|⊕⊕○○)
Technical remark: Clinicians should evaluate the need for an increase in glucocorticoid during the second or third trimester and administer stress doses of glucocorticoids during labor and delivery.
6.8 In women with congenital adrenal hyperplasia who are pregnant, or trying to become pregnant, we recommend against using glucocorticoids that traverse the placenta, such as dexamethasone. (1|⊕⊕○○)
6.9 In women with congenital adrenal hyperplasia who are pregnant, we advise that the birthing plan includes an obstetric specialist. (Ungraded Good Practice Statement)
Surveillance for long-term complications of congenital adrenal hyperplasia and its treatment
6.10 For patients with congenital adrenal hyperplasia, we suggest introducing counseling regarding healthy lifestyle choices at an early age to maintain body mass index within the normal range to avoid metabolic syndrome and related sequelae. (2|⊕○○○)
6.11 In adult patients with congenital adrenal hyperplasia, we suggest screening of bone mineral density in anyone subjected to a prolonged period of higher-than-average glucocorticoid dosing, or who has suffered a nontraumatic fracture. (2|⊕○○○)
6.12 In adults with classic congenital adrenal hyperplasia, we recommend against routine adrenal imaging. (1|⊕○○○)
Technical remark: Reserve adrenal imaging for individuals with classic congenital adrenal hyperplasia who have clinical evidence of an adrenal mass, poor disease control, a lapse in treatment of several years, or lack of response to intensified therapy.
6.13 In males with classic congenital adrenal hyperplasia, we recommend periodic testicular ultrasound to assess for the development of testicular adrenal rest tumors. (1|⊕⊕○○)
6.14 In patients with congenital adrenal hyperplasia, we recommend against routine evaluation for cardiac and metabolic disease beyond that recommended for the general population. (1|⊕⊕○○)
Technical remark: Clinicians should use their own judgment for the above procedures.
Restoring functional anatomy by surgery in individuals with congenital adrenal hyperplasia
7.1 In all pediatric patients with congenital adrenal hyperplasia, particularly minimally virilized girls, we advise that parents be informed about surgical options, including delaying surgery and/or observation until the child is older. (Ungraded Good Practice Statement)
Technical remark: Surgeries should be performed only in centers with experienced pediatric surgeons/urologists, pediatric endocrinologists, pediatric anesthesiologists, behavioral/mental health professionals, and social work services. Extensive discussions regarding risks and benefits, shared decision-making, review of potential complications, and fully informed consent need to occur prior to surgery. The option to forgo surgery should be considered.
7.2 In severely virilized females, we advise discussion about early surgery to repair the urogenital sinus. (Ungraded Good Practice Statement)
7.3 In the treatment of minors with congenital adrenal hyperplasia, we advise that all surgical decisions remain the prerogative of families (i.e., parents and assent from older children) in joint decision-making with experienced surgical consultants. (Ungraded Good Practice Statement)
7.4 In female patients with congenital adrenal hyperplasia for whom surgery is chosen, we suggest vaginoplasty using urogenital mobilization and, when chosen, neurovascular-sparing clitoroplasty for severe clitoromegaly. (2|⊕○○○)
Experimental therapies and future directions
General considerations and unmet clinical needs
8.1 In patients with congenital adrenal hyperplasia, we advise against using experimental treatment approaches outside of formally approved clinical trials. (Ungraded Good Practice Statement)
Adrenalectomy
8.2 In patients with congenital adrenal hyperplasia, we suggest that bilateral adrenalectomy not be performed. (2|⊕○○○)
Mental health
9.1 For individuals with congenital adrenal hyperplasia and their parents, we recommend behavioral/mental health consultation and evaluation to address any concerns related to congenital adrenal hyperplasia. (1|⊕⊕○○)
Technical remark: Clinicians should be aware that individuals with congenital adrenal hyperplasia may be at risk for developing mental health problems and should have a low threshold for referral to psychological or psychiatric treatment. Mental health practitioners should have specialized expertise in assessing and managing congenital adrenal hyperplasia–related psychosocial problems.
Introduction
Summary of changes in 2018 congenital adrenal hyperplasia guidelines
Since the publication of the 2010 Endocrine Society clinical practice guideline for congenital adrenal hyperplasia [CAH (1)], there have been several changes. Neonatal diagnosis methods have been refined to use gestational age in addition to birth weight for cut-point interpretation or to employ liquid chromatography–tandem mass spectrometry (LC-MS/MS) as a secondary screening test. The standard for confirming a diagnosis of CAH continues to be serum 17-hydroxyprogesterone (17OHP) measurements, most often with cosyntropin stimulation. The advent of commercially available serum 21-deoxycortisol measurements may simplify identification of CAH carriers. The use of this analyte, or of steroid profiling to monitor treatment, has yet to be tested.
New human and animal data convey further concerns regarding prenatal dexamethasone (Dex) treatment. No international registry has yet been established for long-term outcomes of individuals treated prenatally with Dex. Although noninvasive prenatal diagnosis of fetal sex is now commonly performed, CAH genotype has been reported only in a proof-of-concept study and is not routinely available. This guideline now includes more detailed protocols for adults, especially pregnant women. We suggest more moderate use of stress dosing during minor illness or minor surgery in patients with CAH.
Over time, the approach to genital reconstructive surgery has changed, incorporating more shared decision-making among parents, patients, surgeons, endocrinologists, mental health providers, and support groups. A systematic review and meta-analysis of published literature on surgery for females with CAH through early 2017 could not identify enough scientifically rigorous studies delineating a favorable benefit-to-risk ratio for either early or late elective genital reconstructive surgery for females with CAH. We maintain that CAH should not be equated with other, rarer 46,XX or XY disorders of sex development (DSD) in formulating treatment guidelines and policies. Our goals have been consistently directed at preserving functional anatomy and fertility.
In another new meta-analysis, investigators found no direct well-controlled evidence of cardiovascular or metabolic morbidity and mortality associated with CAH. Thus, we recommend that individuals with CAH should be monitored according to conventional guidelines for monitoring CAH-unaffected children, adolescents, and adults. Retaining patients with CAH after “graduation” from pediatric care is an important goal, and we have stressed the need for improved mental health monitoring. Finally, in this guideline, we discuss potential new therapies and future ways to improve quality of life (QOL) for individuals with CAH.
Definition, pathophysiology, and morbidities of CAH
CAH is a group of autosomal recessive disorders characterized by impaired cortisol synthesis. Based on neonatal screening and national case registries, the worldwide incidence in most studies ranges from ∼1:14,000 to 1:18,000 births, but the condition is more prevalent in small, genetically isolated groups with a smaller gene pool, particularly in remote geographic regions [e.g., Alaskan Yupiks, among others; Table 1 (2–23)]. CAH is caused in ∼95% of cases by mutations in CYP21A2, the gene encoding adrenal steroid 21-hydroxylase (P450c21) (24, 25). This enzyme converts 17OHP to 11-deoxycortisol and progesterone to deoxycorticosterone, with these products being precursors for cortisol and aldosterone. The blockage of cortisol synthesis leads to corticotropin stimulation of the adrenal cortex, with accumulation of cortisol precursors that are diverted to sex hormone biosynthesis (Fig. 1). A cardinal feature of classic or severe virilizing CAH in newborn females is abnormal development of the external genitalia with variable extent of virilization. Evaluation for CAH needs to be considered for infants found to have bilateral nonpalpable gonads. In 75% of cases with severe enzyme deficiency, inadequate aldosterone production causes salt wasting, failure to thrive, and potentially fatal hypovolemia and shock. Distinctions between various CAH phenotypes are detailed in White and Speiser (27). Newborn screening, now universal in the United States (28) and in many other developed countries (19), can mitigate these complications. Missed diagnosis of salt-losing CAH is associated with increased risk for early neonatal morbidity and mortality. If simple virilizing CAH is not recognized and treated, both girls and boys may undergo rapid postnatal growth and virilization.
Table 1.
Comparative Incidence of Classic CAH in Different Populations
| Country | Complete National Data? | Sample Size | 1/Incidence | PPV % (Term Infants or Overall) | Reference |
|---|---|---|---|---|---|
| Argentina (Buenos Aires) | No | 80,436 | 8937 | 50 | (2) |
| Australia (Western Australia)a | No | 550,153 | 14,869 | N/A | (3) |
| Australia (New South Wales) | No | 185,854 | 15,488 | 1.8 | (4) |
| Australiaa | Yes | 18,034 | N/A | (4) | |
| Brazil | No | 748,350 | 14,967 | (5) | |
| Brazil (state of Goias) | No | 82,603 | 10,325 | 28.6 | (6) |
| Brazil (state of Minas Gerais) | No | 159,415 | 19,927 | 2.1 | (7) |
| Brazil (state of Rio Grande do Sul) | No | 108,409 | 13,551 | 1.6 | (8) |
| China | No | 30,000 | 6084 | (9) | |
| Croatia | Yes | 532,942 | 14,403 | (10) | |
| Cuba | Yes | 621,303 | 15,931 | 0.3 | (11) |
| Czech Republic | Yes | 545,026 | 11,848 | 1.6 | (12) |
| France | Yes | 6,012,798 | 15,699 | 2.3 | (13) |
| Germany (Bavaria) | No | 1,420,102 | 12,457 | 5 | (14) |
| India | No | 55,627 | 6334 | (15) | |
| Japan (Sapporo) | No | 498,147 | 20,756 | 8 | (16) |
| Japan (Tokyo) | No | 2,105,108 | 21,264 | 25.8 | (17) |
| New Zealand | Yes | 1,175,988 | 26,727 | (18) | |
| Sweden | Yes | 2,737,932 | 14,260 | 25.1 | (19) |
| United Kingdoma | Yes | 18,248 | N/A | (20) | |
| United Arab Emirates | Yes | 750,365 | 9030 | (21) | |
| Uruguay | Yes | 190,053 | 15,800 | (22) |
Data are from newborn screening except those designated as coming from national case registries. Data are from studies published in 2008 and later. Earlier studies are summarized by van der Kamp and Wit 2004 (23) and Gidlof et al. 2014 (19).
Abbreviations: N/A, not available; PPV, positive predictive value (for newborn screening; see section 1).
Data are from national case registries.
Figure 1.

(a) Normal fetal adrenal steroidogenesis. Because the fetal adrenal has low levels of 3β-hydroxysteroid dehydrogenase, most steroidogenesis is directed toward dehydroepiandrosterone (DHEA) and thence to DHEA sulfate, but small amounts of steroids enter the pathways toward aldosterone and cortisol. The adrenal 21-hydroxylase P450c21 is essential in both pathways. The adrenal can synthesize small amounts of testosterone via 17βHSD5 (AKR1C3). Included to the lower right is the 11-oxyandrogen pathway, in which androstenedione is converted in the adrenal to 11β-hydroxyandrostenedione (11OHA4) and then in the adrenal and/or peripheral tissues to 11-ketoandrostenedione and ultimately 11-ketotestosterone (11KT). The production of 11OHA4 and 11KT is an important pathway in postnatal life and may also occur in the fetal adrenal. (b) In the absence of the 21-hydroxylase activity of P450c21, three pathways lead to androgens. First, the pathway from cholesterol to DHEA remains intact. Although much DHEA is inactivated to DHEA sulfate, the increased production of DHEA will lead to some DHEA being converted to testosterone and dihydrotestosterone (DHT). Second, although minimal amounts of 17OHP are converted to androstenedione in the normal adrenal, the massive amounts of 17OHP produced in CAH permit some 17OHP to be converted to androstenedione and then to testosterone. Third, the alternative pathway depends on the 5α and 3α reduction of 17OHP to 17OH-allopregnanolone. This steroid is readily converted to androstanediol, which can then be oxidized to DHT by an oxidative 3α-hydroxysteroid dehydrogenase (3αHSD) enzyme. The role of the alternative pathway in CAH is evidenced by increased levels of metabolites of its unique steroidal intermediates in the urine of infants, children, and adults with CAH (26).
In addition to the “classic salt-wasting” and “simple virilizing” forms of CAH diagnosed in infancy, there is also a mild or “nonclassic” form, which features variable degrees of postnatal androgen excess but is sometimes asymptomatic (29). The mild subclinical impairment of cortisol synthesis in nonclassic CAH (NCCAH) generally does not lead to Addisonian crises. Based on haplotype association studies, nonclassic forms of CAH were estimated to have a prevalence of 1:500 to 1:1000 in the general white population but up to 1:50 to 1:100 among populations with high rates of consanguineous marriages (30). More recent CYP21A2 genotype analysis indicates that NCCAH has an overall frequency of ∼1:200 (95% confidence level, 1:100 to 1:280) in the US population (31).
Disease severity correlates with CYP21A2 allelic variation. Genotyping individuals with CAH is fraught with error due to the complexity of gene duplications, deletions, and rearrangements within chromosome 6p21.3 (32). Almost 300 CYP21A2 mutations are known (33), but large deletions and a splicing mutation (intron 2, IVS-13 A/C→G, −13 nucleotides from the splice acceptor site) that ablate enzyme activity comprise ∼50% of classic CAH alleles (34–36). Approximately 5% to 10% of patients with salt-wasting CAH have the hypermobility form of Ehlers-Danlos syndrome due to haploinsufficiency of tenascin-X, encoded by the TNXB gene, which overlaps CYP21A2 (37).
A nonconservative amino substitution in exon 4 (p.Ile172Asn) that preserves ∼1% to 2% of enzyme function is associated with simple virilizing classic CAH (38). A point mutation in exon 7 (p.Val281Leu) that preserves 20% to 50% of enzyme function (38) accounts for most NCCAH alleles (31, 39, 40). Because many compound heterozygous patients carry more than one mutation on either or both CYP21A2 alleles, there is a wide spectrum of phenotypes (35).
Commissioned Systematic Review
The writing committee commissioned two systematic reviews: one concerning the cardiac and metabolic morbidities associated with CAH (41), and the second to determine whether and when clinicians should perform genital surgery (42).
The first review (41) summarized 20 observational studies and demonstrated small but significant increases in systolic and diastolic blood pressure, insulin resistance, and carotid intima thickness in individuals with CAH compared with non-CAH controls. The quality of evidence (i.e., certainty in these estimates) was low due to the observational nature of the evidence, risk of bias, and heterogeneity. Furthermore, population-based studies found higher prevalence of hypertension, hyperlipidemia, and type 2 diabetes in adults with CAH than in non-CAH controls.
The second review (42) summarized 29 observational studies evaluating patients who had undergone surgery at a mean age of 3 years. The studies evaluated various surgical techniques and reported good overall patient and surgeon satisfaction with cosmetic and functional outcomes. The review also provided estimates of surgical complication rates and sexual function. Such evidence was also of low quality and carried a high risk of bias.
1. Newborn Screening
Cost-effectiveness
1.1 We recommend that all newborn screening programs incorporate screening for CAH due to 21-hydroxylase deficiency (21OHD). (1|⊕⊕⊕○)
Evidence
Early recognition and treatment of CAH can prevent serious morbidity and mortality. Currently, all 50 states in the United States, 35 other countries, and portions of 17 additional countries screen for CAH. According to screening results, the incidence of classic CAH in most populations is ∼1:14,000 to 1:18,000. Table 1 summarizes data from 2008 onward; data collected from 1997 to 2007 are similar (23, 43, 44).
Screening markedly reduces the time to diagnosis of infants with CAH (45–48), consequently reducing morbidity and mortality. A retrospective analysis of neonatal dried blood samples that were not screened for CAH from cases of sudden infant death in the Czech Republic and Austria identified three genotypically confirmed cases of classic CAH among 242 samples screened (49). In contrast, a large population-based study in the Manchester area of the United Kingdom found no CAH cases among 1198 dried blood samples from infants who had died between 5 days and 6 months of age (50). Males with salt-wasting CAH are more likely than females to suffer from delayed or incorrect diagnosis, as there is no genital ambiguity. Thus, a relative paucity of salt-wasting males in a patient population may be taken as indirect evidence of unreported deaths from salt-wasting crises. In fact, females outnumber males in some (10, 51–53), although not all (50, 54), retrospective studies in which CAH is clinically diagnosed, and this preponderance of females vanishes when newborn screening is introduced (54). Some researchers have reported a death rate of ∼10% among infants with salt-wasting CAH in the absence of screening (55), but recent estimates from advanced economies are lower, at 0% to 4% (56).
In regard to morbidity, infants diagnosed through screening have less severe hyponatremia (48) and tend to have shorter hospitalizations (46, 48, 50, 57) than infants diagnosed later. Learning disabilities may occur in patients who have had salt-wasting crises (58). Although salt-wasting males would seem to derive the greatest benefit from screening programs, the delay before correct sex assignment of severely virilized females is also reduced (43, 48). Moreover, males with simple virilizing disease may otherwise not be diagnosed until rapid growth and accelerated skeletal maturation are observed in later childhood, leading to compromised adult stature.
Several recent reviews have attempted cost-benefit analysis of newborn screening for CAH. Such estimates generally assume that the only adverse outcome of late diagnosis of CAH is death. It is conventionally assumed that screening for a particular disease is cost-effective at <$50,000 per life-year per quality-adjusted life year (59). Recent estimates have ranged widely from $20,000 (59) to $250,000 to $300,000 (60) per quality-adjusted life year.
Initial screening methodology
1.2 We recommend that first-tier screens use 17OHP assays standardized to a common technology with norms stratified by gestational age. (1|⊕⊕⊕○)
Technical remarks: Clinicians should be aware that immunoassays are still in use and remain a source of false-positive results. Specificity may be improved with organic extraction to remove cross-reacting substances.
Evidence
First-tier screens for CAH employ immunoassays to measure 17OHP in dried blood spots on the same filter paper (“Guthrie”) cards used for other newborn screening tests (46, 59, 61). The automated time-resolved dissociation-enhanced lanthanide fluoroimmunoassay (62) has almost completely supplanted the original radioimmunoassay (63) and other types of assays.
Several technical factors limit the accuracy of these tests. First, 17OHP levels are normally high at birth and decrease rapidly during the first few postnatal days in healthy infants. In contrast, 17OHP levels increase with time in infants affected with CAH. Thus, diagnostic accuracy is poor in the first 2 days unless robust mechanisms exist to obtain follow-up samples. Second, female infants have lower mean 17OHP levels than do males, slightly reducing the sensitivity of newborn screening in some reports (64). This reduced sensitivity is generally not a major problem because almost all females with salt-wasting CAH are virilized, and thus are brought to prompt medical attention. Third, premature, sick, or stressed infants have higher levels of 17OHP than do term infants, generating many false positives. For example, in 26 years of operation of the Swedish screening program, the positive predictive value was 25% for full-term infants but only 1.4% for preterm infants, and the predictive value correlated very strongly with gestational age (19). Finally, immunoassays may lack specificity. There are no universally accepted standards for stratifying infants, but most laboratories use a series of birth weight–adjusted cut-offs (65–67).
Screening a second sample several days later also improves both sensitivity and positive predictive value (47, 61, 68). A recent study suggested that preterm infants should have additional samples rescreened at 2 and 4 weeks of age, which is practical in hospitalized patients (67). Similarly, Brazilian investigators used 99.8 percentile 17OHP values, adjusted for birth weight, to achieve 5.6% and 14.1% positive predictive value at two sampling time points (48 to <72 hours and ≥72 hours, respectively) (69). Moreover, a comparison of one-screen vs two-screen state programs found a higher incidence of CAH when a second screen was employed (1:17,500 vs 1:9500) (70).
Stratifying subjects by actual gestational age rather than birth weight might also improve the specificity of newborn screening, as 17OHP levels are much better correlated with the former variable than with the latter (71). In the Netherlands, adopting gestational age criteria improved the positive predictive value of screening from 4.5% to 16% (57).
With regard to assays, elevated levels of adrenal steroids are not due solely to cross-reaction in immunoassays. Steroid profiles in preterm infants suggest a functional deficiency of several adrenal steroidogenic enzymes with a nadir at 29 weeks gestation (72). However, immunoassays are still in use but may be a source of false-positive results due to cross-reactivity with other steroids, for example, 17-OH-pregnenolone sulfate (73). Immunoassay specificity may be improved with organic extraction to remove cross-reacting substances, such as steroid sulfates.
The dissociation-enhanced lanthanide fluoroimmunoassay was reformulated in late 2009 to reduce its sensitivity to cross-reacting compounds in premature infants (74). This change improved the positive predictive value from 0.4% to 3.7% for the first screen alone (61).
Finally, antenatal corticosteroids may reduce 17OHP levels, potentially increasing the likelihood of false-negative screens. Studies have reported inconsistent effects of antenatal corticosteroid administration in practice (75, 76). As previously noted, testing of later samples would minimize this problem.
Second-tier screening
1.3 We recommend that screening laboratories employ a second-tier screen by LC-MS/MS in preference to all other methods (e.g., genotyping) to improve the positive predictive value of CAH screening. (1|⊕⊕○○)
Technical remark: Laboratories utilizing LC-MS/MS should participate in an appropriate quality assurance program. Additionally, clinicians should realize that immunoassays lead to more false-positive results. Thus, if laboratory resources do not include LC-MS/MS, a cosyntropin stimulation test should be performed to confirm diagnosis prior to initiation of corticosteroid treatment.
Evidence
Decreasing cut-off levels may increase sensitivity, but at a cost of a decreasing positive predictive value. In the United States, the cut-off levels for 17OHP are typically set low enough that clinicians report ∼1% of all tests as positive, with the aim of identifying all children with salt-wasting disease and almost all simple virilizing disease. Because CAH is a rare disease, the positive predictive value is very low, although the specificity and sensitivity are very high (77). In contrast, cut-off values that still identified all infants with salt-wasting CAH but only ∼80% of cases of simple virilizing CAH yielded a positive predictive value of 25% in term infants (19). We could avoid much of the expense and parental anxiety of following up positive newborn screening tests with a specific and sensitive second-level screen. Both biochemical and molecular genetic approaches can be used.
Biochemical second screens.
Limitations of immunoassays for 17OHP include true elevation of levels in premature infants or those who are sick or stressed, and lack of antibody specificity. Organic solvent extraction to increase immunoassay specificity is currently mandated as a second screen in several US states.
However, direct biochemical analysis of steroids using LC-MS/MS is more effective than immunoassays in addressing these issues (78, 79). The run times for individual samples in such assays are 6 to 12 minutes, impractical for screening large numbers of samples, but suitable for the smaller numbers subjected to a second-tier screen using the original dried blood samples (78, 80). It is noteworthy that ∼40% of samples that are positive in first-tier screens with immunoassays actually have normal 17OHP levels as measured by LC-MS/MS, consistent with suboptimal antibody specificity. Measuring steroid ratios may further improve the screening specificity of LC-MS/MS. One approach has examined a ratio defined as the sum of 17OHP and androstenedione levels divided by the cortisol level (81). This strategy was used in actual practice as the second-tier screen in Minnesota for 3 years (204,000 births) and improved the positive predictive value of the CAH screen (82), but subsequent reports from the same center suggested that this approach was inferior to testing a second sample by routine immunoassay (67, 68). However, others have reported markedly superior results with LC-MS/MS (83, 84). The consistency of results might be improved by mandating participation in national proficiency testing programs (85).
Measuring additional analytes or ratios of analytes can also improve screening outcomes. For instance, 21-deoxycortisol (produced by 11β-hydroxylation of 17OHP) is normally not secreted in large amounts (even in preterm infants), and thus elevated levels are highly specific for 21OHD. A modified LC-MS/MS protocol used a ratio defined as the sum of 17OHP and 21-deoxycortisol levels divided by the cortisol level, and this parameter correctly identified all affected children with no false positives, for a positive predictive value of 100% (80). The ratio of the urinary metabolites pregnanetriolone and 6α-hydroxytetrahydrocortisone, measured by gas chromatography and tandem mass spectrometry, also gave excellent specificity, even in preterm infants (86).
Molecular genetic second screens.
CYP21A2 mutations can be detected in DNA extracted from the same dried blood spots used for hormonal screening. Detection methods include dot-blotting protocols (87), ligation detection assays (88, 89), real-time quantitative PCR (90, 91), full sequencing (92), and minisequencing (93). Because >90% of mutant alleles carry one or more of a discrete number of mutations, if no mutations are detected, one can assume that the individual is unaffected. If at least one mutation is detected, the patient requires additional evaluation. The carrier rate for classic CAH in the general population is ∼2%; if this rate were not increased among infants with a positive first screen (90), and 1% of all first screens were positive, then 0.02% (1/5000) of all infants would have a positive second screen by this strategy. Because the actual frequency of CAH is ∼0.006% (∼1/16,000), the positive predictive value of this approach should be ∼30%. There are two reasons why we cannot use the analysis of a single sample to actually diagnose CAH. First, a heterozygous carrier of a known mutation for classic 21OHD could have an undetected novel mutation in the other allele. Second, many CAH alleles carry more than one deleterious mutation, making it impossible to set phase (i.e., to determine whether two mutations are on different alleles or the same allele) without genotyping at least one parent.
Several studies on the genotyping of samples from screening programs have suggested that this approach is a potentially useful adjunct to hormonal measurements (6, 87, 88, 90, 92, 94), but to the best of our knowledge there has been no large-scale study of its efficacy as a second-tier screen in actual use.
Genotyping remains more costly and time-consuming than LC-MS/MS on a per-sample basis. Although the equipment for LC-MS/MS is expensive, many screening programs already have it available for other tests.
Balance of benefits and harms
The writing committee placed a higher value on the benefits of complete ascertainment of infants affected with classic CAH and minimizing the consequences of neonatal salt-wasting crises than on the additional expense of following up false-positive screens.
2. Prenatal Treatment of CAH
2.1 We advise that clinicians continue to regard prenatal therapy as experimental. Thus, we do not recommend specific treatment protocols. (Ungraded Good Practice Statement)
2.2 In pregnant women at risk for carrying a fetus affected with CAH and who are considering prenatal treatment, we recommend obtaining prenatal therapy only through protocols approved by Institutional Review Boards at centers capable of collecting outcomes from a sufficiently large number of patients, so that risks and benefits can be defined more precisely. (1|⊕⊕⊕○)
2.3 We advise that research protocols for prenatal therapy include genetic screening for Y-chromosomal DNA in maternal blood to exclude male fetuses from potential treatment groups. (Ungraded Good Practice Statement)
Evidence
The Endocrine Society’s 2010 CAH guidelines summarized the physiology of prenatal treatment of CAH and the results of studies published through the end of 2009 (1). Prenatal treatment with Dex aims to reduce female genital virilization and its associated risk of social stigma (95), the need for reconstructive surgery, and the emotional distress associated with the birth of a child with atypical sexual development. Prenatal treatment is inappropriate for male fetuses, as this form of CAH does not disrupt the development of male genitalia. Prenatal treatment does not change the need for lifelong hormonal replacement therapy, the need for careful medical monitoring, or the risk of life-threatening salt-losing crises if therapy is interrupted. Some researchers have suggested that prenatal Dex may reduce potential androgenization of the fetal female brain, but such effects are difficult to measure and have not been studied systematically. The following paragraphs describe relevant considerations regarding prenatal Dex treatment.
Prenatal treatment has been suggested for women who have previously delivered a child with CAH and are pregnant again via the same partner. The fetus will have a 1:4 chance of having CAH and a 1:2 chance of being female; thus, there is a 1:8 chance that the fetus will be female and have CAH. Because the period during which fetal genitalia may become virilized begins ∼6 weeks after conception, treatment must be started by 6 to 7 weeks. Because genetic diagnosis by chorionic villous biopsy cannot be performed until 10 to 12 weeks, all pregnancies at risk for CAH would need to be treated, although the treatment is directed at only one in eight fetuses.
Specialized laboratories can perform sex determination from fetal Y-chromosome DNA in maternal blood with 99% accuracy (96), which could improve the probability of treating an affected female fetus from 1:8 to 1:4. In a study of prenatal treatment in 258 fetuses at risk for CAH from 2002 to 2011, testing for Y-chromosome DNA prevented treatment with Dex in only 68% of male fetuses, although the percentage rose during the course of the study (97). We suggest that prenatal sex determination be incorporated into all prenatal treatment research protocols; however, prenatal sex determination is illegal in some countries to prevent female feticide (98). We think that prenatal diagnosis and treatment research should not be performed in such countries. By using blood from both parents and an affected proband and applying massively parallel DNA sequencing coupled with extensive analysis of single nucleotide polymorphisms near the CYP21A2 gene, it was possible to determine the CYP21A2 genotype in 14 of 14 at-risk fetuses within 3 weeks of obtaining fetal DNA in maternal blood samples (99). This approach currently requires expensive equipment and very skillful personnel that are found only in advanced research centers, but the approach holds promise for the future.
In contrast to cortisol and prednisolone, Dex is not inactivated by placental 11βHSD2 and readily reaches the fetus. Therefore, virtually all reports of prenatal treatment use Dex, typically at doses of 20 μg/kg prepregnancy maternal body weight, to a maximum of 1.5 mg/d. In normal, untreated pregnancies, fetal cortisol levels are low in very early gestation and rise during weeks 8 to 12, while the external genitalia are differentiating (100); fetal cortisol is only ∼10% of maternal levels during midgestation (101) and then increases during the third trimester. If Dex freely traverses the placenta, a dose of 20 μg/kg maternal body weight could achieve effective glucocorticoid (GC) levels that exceed typical midgestation fetal levels by ∼60-fold (102). No studies have systematically tested reducing the dose in late gestation.
Efficacy.
Available evidence regarding fetal outcomes and maternal sequelae of prenatal Dex treatment is of low or very low quality due to methodological limitations and small sample sizes. A systematic review and meta-analysis of reports of prenatal treatment published through August 2009 found only four studies that included a control group and provided sufficient data to analyze (103). Among 325 pregnancies treated with Dex, affected female fetuses had a weighted mean difference of −2.33 (95% CI, −3.38 to −1.27) on the Prader scale. Data concerning stillbirths, spontaneous abortions, fetal malformations, and neuropsychological outcomes were sparse, and long-term follow-up data were not reported. Aside from individual case reports, only two series of prenatal treatment of CAH have appeared since August 2009. An update of an ongoing practice in New York reported 63 treated female fetuses with classic CAH, of whom 15 had normal female genitalia, 26 had mild (Prader stages 1 to 2) virilization, and 17 had severe (Prader stages 3 to 5) virilization (mean score, 1.7) (104). In a 10-year French study, among 112 treated female fetuses, 14 had 21-hydroxylase–deficient CAH and three had 11-hydroxylase–deficient CAH; of these 17 girls, 12 had normal female genitalia at birth and 3 had moderate virilization, whereas 2 who were treated late were severely virilized (97). Thus, prenatal Dex is effective in reducing genital virilization of affected female fetuses. Poor results are typically ascribed to delayed treatment or noncompliance (105).
Maternal safety.
Some studies have reported increased pregnancy-associated weight gain, striae, edema, gastric distress, and mood swings but minimal hypertension and gestational diabetes (94, 103). Some women reported Cushingoid features and increased appetite, and many women indicated that they would decline prenatal treatment of a subsequent pregnancy. Thus, prenatal treatment is associated with modest but manageable maternal complications that do not appear to pose a major risk to the mother.
Fetal safety.
The US Food and Drug Administration classifies Dex as a category C drug whose safety in pregnancy is not established: according to the US Food and Drug Administration, “Animal reproduction studies have shown an adverse effect on the fetus and there are no adequate and well-controlled studies in humans, but potential benefits may warrant use of the drug in pregnant women despite potential risks” (106). The Endocrine Society’s 2010 CAH guideline reviewed earlier studies concerning the safety of prenatal Dex and other transplacental GCs (1); only the most important are reiterated here. Recent studies address four areas of concern: potential teratogenicity, birth weight, brain/behavior, and potential long-term effects.
Teratogenicity.
Consistent with animal data that Dex can cause cleft palate, the National Birth Defects Prevention Study reviewed 1769 infants with cleft lip with/without cleft palate born to women who received GCs during the first trimester, finding statistically increased risks of orofacial clefts compared with 4143 controls (107). A recent case report cited the first known instance of an orofacial cleft in a girl affected with CAH treated prenatally with Dex (108). Acute encephalopathy was reported in two infants who had received prenatal Dex, but it is not clear whether this condition was related to prenatal steroid exposure (109). Teratogenic effects of Dex observed in animal models include renal dysgenesis, reduced pancreatic β cell numbers, impaired glucose tolerance, and increased systolic and diastolic blood pressure, all discussed previously (1). Evidence continues to accumulate implicating Dex in numerous developmental defects: exposing fetal rats to Dex altered hepatic programming and increased lipid accumulation (110). Impaired thyroid development with reduced numbers of follicular cells and C cells was observed in another study (111). Incubation of 8- to 11-week-postfertilization human fetal ovaries with Dex doses corresponding to prenatal CAH treatment reduced germ cell density by increasing apoptosis in oogonia (112). Only ∼800 fetuses receiving Dex in the first trimester have been reported to date, and potential teratogenicity was not evaluated in all fetuses.
Birth weight and sequelae.
Multiple doses of antenatal betamethasone can improve pulmonary outcome in preterm infants but are associated with decreased newborn weight, length, and head circumference (113–116). Similarly, newborns prenatally treated with Dex for potential CAH have nominally normal birth weights but nevertheless weigh ∼400 g less than controls (117). Reduced birth weight increases adult risk for chronic disorders, including hypertension, type 2 diabetes, and cardiovascular disease (118); one study associated fetal malnutrition with exposure to GCs (119). Young adults exposed to antenatal GCs have increased aortic stiffness (120). Human placental chorionic plate arteries abundantly express GC receptors; chronic GC exposure in vitro alters vasoreactivity, increasing vascular resistance and potentially contributing to hypertension (121). These observations concerning the developmental origins of adult disease have raised concerns about potential prenatal “programming” by fetal exposure to Dex (119, 122–124).
Brain and behavior.
Adverse effects of GCs on brain development have been reported in human and animal studies (125–127). In rodents, Dex inhibits hippocampal neuronal maturation in vitro (128) and in vivo (129–131), and Dex limits proliferation of neural progenitor cells in cultured embryonic mouse neurospheres (132).
A small, well-designed Swedish study found no differences in intelligence, learning, or long-term memory between children who were prenatally exposed to Dex and those who were not, but the former group had reduced verbal working memory, reduced self-perception of scholastic competence, and increased self-rated social anxiety (133); in contrast, their parents described them as being more sociable than controls (134). Prenatally treated boys had reduced masculine and increased neutral behavior, suggesting unexpected effects on gender-role behavior (135). A follow-up study by the Swedish group found that the negative effects of Dex were sex-specific. Unaffected Dex-treated girls scored lower than did control girls on the Wechsler Intelligence Test for Children III and in visual–spatial working memory. In contrast, boys showed no cognitive impairment (136). The basis for a putative sex-specific effect of Dex is unknown.
Systematic review and meta-analysis of several studies did not detect significant differences in behavior or temperament depending on prenatal Dex exposure (103, 137, 138). Another study did not find effects on working memory in short-term exposed unaffected children or short-term exposed boys with CAH, but girls with CAH treated throughout pregnancy had slower mental processing than did controls by several assessments (139). A very small study from Poland reported improved neurocognitive function among girls with CAH who had received prenatal Dex, although the unaffected, Dex-treated girls had reduced visual perception and visual memory (140). Differences among studies may reflect inadequate sample size, inappropriate controls, or the effects of postnatal infant–mother bonding, which can partially ameliorate the effects of fetal exposure to GCs (141). Although data are inconclusive, any adverse effects of Dex on brain development would be unacceptable.
The long-term effects of fetal GC exposure have been studied in infants whose mothers received Dex or betamethasone to promote fetal lung maturation (142). In this setting, prenatal GC exposure alters the hypothalamic–pituitary–adrenal axis, augmenting the cortisol response to stress (143) with adverse mental health sequelae in childhood and adolescence (144). Long-term effects of postnatal GC on the human brain include decreased memory and hippocampal volume (145), decreased cerebellar cortical volume (146), diminished cognitive function (147–149), increased psychopathology, and reduced QOL (148, 150)
Two confounding factors should be considered in future studies of side effects. First, individuals with CAH who were not prenatally exposed to Dex may have reduced working memory or short-term memory (137, 138). Second, women with CAH had reduced test scores for working memory, processing speed, digit span, and matrix reasoning compared with controls (151). Brain MRI showed effects on the white matter, hippocampus, thalamus, cerebellum and brainstem; magnetic resonance spectroscopy also showed reduced choline content in the temporal lobe. Patients treated with higher GC doses had greater abnormalities (151).
Potential long-term effects.
Long-term effects of fetal exposure to GCs are well described (116). A retrospective epidemiological study found that antenatal Dex used to induce late-gestation pulmonary maturation was an independent risk factor for development of asthma at 3 to 6 years (152). Among 24 prematurely born children who received prenatal Dex for pulmonary maturation, the incidence of asthma and allergic disorders was higher at ages 2 to 5 than among 16 matched controls (153). Studies in rats receiving Dex during gestation showed decreased GC responsiveness and receptor expression (154), as well as suppression of innate cytokines with induction of adaptive cytokines (155).
Individuals who had received antenatal betamethasone 30 years earlier had increased insulin resistance, and 7% had elevated basal cortisol (156). Antenatal synthetic GCs alter fetal rodent DNA methylation, permanently affecting the expression of genes involved in carbohydrate homeostasis and the programming of the hypothalamic–pituitary–adrenal axis (157). Brief maternal exposure to Dex reduced adrenal expression of steroidogenic enzymes during adulthood in mice (158). Altered DNA methylation apparently underlies the long-term effects of both GCs and maternal stress on the fetus (129–131). Effects on subsequent generations may reflect effects on precursor germ cells in the developing gonad (112). Whether and to what extent the alterations observed in the rodent model of prenatal Dex exposure occur in humans cannot be readily determined.
Balance of benefits and harms
Antenatal treatment with CAH remains controversial and poses unresolved ethical questions. Consequently, the 2010 Endocrine Society practice guidelines recommended that “prenatal therapy continue to be regarded as experimental” (1). Since then, the group studying prenatal treatment in Sweden has discontinued this treatment because of “possible adverse side effects” (159). The German Society of Pediatric Endocrinology and Diabetology in conjunction with five other German medical societies concluded that “Prenatal CAH therapy is still an experimental therapy” (160). A “Clinical Opinion” in the American Journal of Obstetrics and Gynecology concluded that the “risks outweigh the benefits” (102). Risk-benefit analysis must consider the need to treat multiple unaffected fetuses, however briefly, without direct benefit to treat one affected female; accumulating data suggesting potential long-term risks from fetal Dex therapy render this approach problematic. Therefore, in validating earlier expert opinion, this writing committee placed a higher value on preventing unnecessary prenatal exposure of the fetus and mother to Dex and avoiding potential harms associated with this exposure than on minimizing the emotional toll of atypical external genital development on parents and patients. Preimplantation genetic diagnosis and other evolving assisted reproductive technologies are additional options (161, 162) but carry their own risk and ethical controversies (163), but this is beyond the context of this guideline.
3. Diagnosis of CAH
3.1 In infants with positive newborn screens for CAH we recommend referral to pediatric endocrinologists (if regionally available) and evaluation by cosyntropin stimulation testing as needed. (1|⊕⊕⊕○)
3.2 In symptomatic individuals past infancy, we recommend screening with an early morning (before 8 am) baseline serum 17OHP measurement by LC-MS/MS. (1|⊕⊕⊕○)
3.3 In individuals with borderline 17OHP levels, we recommend obtaining a complete adrenocortical profile (defined below) after a cosyntropin stimulation test to differentiate 21OHD from other enzyme defects. (1|⊕⊕⊕○)
3.4 In individuals with CAH, we suggest genotyping only when results of the adrenocortical profile after a cosyntropin stimulation test are equivocal, or cosyntropin stimulation cannot be accurately performed (i.e., patient receiving GC), or for purposes of genetic counseling. (2|⊕⊕⊕○)
Technical remark: Genotyping at least one parent aids in the interpretation of genetic test results because of the complexity of the CYP21A2 locus.
Evidence
In neonates with a positive screen, the decision of whether to inform only the infant’s primary physician or a pediatric endocrinologist depends on the availability of subspecialists (47). Usually, the primary care provider follows up moderately elevated 17OHP with a repeat filter paper specimen and evaluates higher values with serum electrolytes and 17OHP levels. If these measurements are abnormal, the clinician refers the infant to a pediatric endocrinologist.
Second-tier screening with LC-MS/MS can efficiently measure a panel of steroids and permit diagnosis of other forms of CAH, as has been shown for 11β-hydroxylase deficiency (164, 165). If basal serum or filter paper results are not fully informative, it is necessary to evaluate the patient with a cosyntropin stimulation test (166). Extant norms are for tests employing a pharmacological dose of 0.25 mg given intravenously (in infants with very low birth weight, the dose may be reduced to 0.125 mg) of cosyntropin (ACTH [1–24]), which maximally stimulates the adrenal cortex. This diagnostic test is distinguished from the low-dose ACTH stimulation test used to evaluate the integrity of the hypothalamic–pituitary–adrenal axis (167). Samples should be obtained at baseline and 60 minutes after administering cosyntropin. At minimum, cortisol and 17OHP should be measured, but 17OHP may be elevated in the presence of other enzymatic defects, particularly 11β-hydroxylase deficiency and, more rarely, 3β-hydroxysteroid dehydrogenase deficiency or P450 oxidoreductase deficiency. To fully differentiate the various enzymatic defects potentially causing CAH, clinicians should ideally send samples to an endocrine reference laboratory for measurement of 17OHP, cortisol, 11-deoxycorticosterone, 11-deoxycortisol, 17-OH-pregnenolone, dehydroepiandrosterone, and androstenedione by LC-MS/MS. If blood volume or venous access are at issue in small infants, a sample can be collected only at 60 minutes following intravenous or intramuscular cosyntropin administration. Product ratios are particularly useful in distinguishing enzymatic defects (164, 165). As an alternative to blood sampling, urine samples can be analyzed at a few special centers using gas chromatography–mass spectrometry or LC-MS/MS; this approach provides a similarly accurate biochemical diagnosis (168).
Cosyntropin stimulation tests should be deferred until after the first 24 to 48 hours of life. There is a high incidence of both false-positive and false-negative results when clinicians obtain samples immediately after birth. Other steroids whose levels are usually elevated in 21OHD include 21-deoxycortisol, androstenedione, and testosterone.
In symptomatic individuals past infancy, LC-MS/MS on serum samples obtained prior to 8 am should be used to screen for CAH. In menstruating females, we recommend sampling in the early follicular phase. Figure 2 contains a sample diagnostic strategy (169–172). Cosyntropin stimulation is needed for patients with indeterminate baseline 17OHP levels. For patients with nondiagnostic stimulated 17OHP values, particularly those receiving GC therapy, genotyping (171, 176, 177) may confirm the diagnosis.
Figure 2.

Diagnosis of 21OHD. Reference standards for hormonal diagnosis were derived from Refs. (170, 171, 173, 174). These diagnostic thresholds appear similar for LC-MS/MS assays from limited data (175). Note that randomly measured 17OHP levels can be normal in NCCAH; hence, 17OHP levels should be screened in the early morning (before 8 am). For menstruating females, steroid measurements should be obtained in the follicular phase and may differ depending on the assay employed. Individuals with classic CAH, including both salt-wasting and simple virilizing forms of 21OHD, typically have unstimulated 17OHP values of several thousand. Note that it is sometimes difficult to distinguish clinically between non–salt-wasting classic and nonclassic forms of CAH.
Hormonal phenotypes correlate quite well with CYP21A2 genotypes; however, genotyping cannot detect salt wasting. For example, genotyping may reveal the IVS2 the salt-wasting and simple virilizing forms (35, 178, 179). Heterozygotes have slightly elevated 17OHP after ACTH stimulation, but there is overlap with unaffected subjects (173). Other analytes have been used as markers of heterozygosity (180, 181), but genotyping is a superior method of heterozygote detection. Heterozygotes do not require medical treatment but should have genetic counseling (see section 6.3).
4. Treatment of Classic CAH
4.1 In growing individuals with classic CAH, we recommend maintenance therapy with hydrocortisone (HC). (1|⊕⊕⊕○)
4.2 In growing individuals with CAH, we recommend against the use of oral HC suspension and against the chronic use of long-acting potent GCs. (1|⊕⊕⊕○)
4.3 In the newborn and in early infancy, we recommend using fludrocortisone and sodium chloride supplements to the treatment regimen. (1|⊕⊕⊕○)
4.4 In adults with classic CAH, we recommend using daily HC and/or long-acting GCs plus mineralocorticoids (MCs), as clinically indicated. (1|⊕⊕⊕○)
4.5 In all individuals with classic CAH, we recommend monitoring for signs of GC excess, as well as for signs of inadequate androgen normalization, to optimize the adrenal steroid treatment profile. (1|⊕⊕⊕○)
4.6 In all individuals with classic CAH, we recommend monitoring for signs of MC deficiency or excess. (1|⊕⊕⊕○)
Evidence
Proper treatment with GCs prevents adrenal crisis and virilization, allowing nearly normal growth and development during childhood. Management of classic CAH is a difficult balance between hyperandrogenism and hypercortisolism. For infant patients, clinicians may exceed the recommended GC doses to reduce markedly elevated adrenal hormone levels, but it is important to rapidly reduce the dose when target levels are achieved. Frequent reassessment is needed. Attempts to completely normalize 17OHP levels typically result in overtreatment with features of Cushing syndrome. Infants with classic 21OHD require GCs in addition to MC treatment and supplemental sodium chloride. The requirement for sodium in normally growing infants is ∼1 mmol/kg per day, the amount provided by human milk. However, in patients with salt-wasting CAH, this amount is insufficient, and sodium chloride supplements are recommended (182). Ideally, a standardized salt solution prepared by a pharmacy or standard sodium chloride tablets should be used for salt supplementation. Sodium chloride supplementation may not be needed if high-dose fludrocortisone is used (183); however, it is particularly important to monitor blood pressure in infants who require treatment with high doses of MC, owing to the variable capacity of the immature renal tubules to reabsorb sodium. Clinicians should reassess MC and sodium doses periodically based on blood pressure, serum sodium, potassium, and plasma renin activity (PRA).
Although the defect in aldosterone biosynthesis is clinically apparent only in the salt-wasting form, subclinical aldosterone deficiency is present in all forms of 21OHD (184, 185) and is best evaluated by the aldosterone-to-PRA ratio (184). Consequently, all individuals with classic CAH benefit from fludrocortisone therapy and adequate dietary sodium beginning in infancy. Maintaining sodium balance is essential for euvolemia and for reducing vasopressin and ACTH, allowing reduced GC doses and thus leading to better growth (186).
During childhood, the preferred GC is HC because its short half-life minimizes the adverse side effects typical of longer-acting, more potent GCs, especially growth suppression (Table 2) (187). In one trial, the estimated growth-suppressive effect of prednisolone was ∼15-fold more potent than that of HC (188); Dex may be up to 70- to 80-fold more potent than HC (189). Although free-alcohol HC suspensions achieve cortisol levels comparable to those achieved by HC tablets (190), HC cypionate oral suspensions were inadequate to control CAH in children (191) due to uneven distribution in liquid form. Good control can be achieved by orally administering crushed, weighed HC tablets mixed with a small volume of liquid, if needed, immediately before administration. Compounding pharmacies should be chosen for reliability in preparing very small doses or special drug formulations, as there have been reports of variable dose accuracy in compounded preparations (192–194).
Table 2.
Maintenance Therapy in Growing Patients with CAH
| Drugs | Total Daily Dose | Daily Distribution |
|---|---|---|
| GCs: HC tablets | 10–15 mg/m2⋅d | 3 times⋅d |
| MCs: fludrocortisone tablets | 0.05–0.2 mg/d | 1–2 times/d⋅d |
| Sodium chloride supplements | 1–2 g/d (17–34 mEq/d) in infancy | Divided into several feedings |
These doses and schedules are meant as examples and should not be construed as a restrictive menu of choices for the individual patient.
Insufficient data exist to recommend fractional distribution of doses throughout the day or empiric dosing in the very early morning hours (195). When doses exceed 20 mg/m2 per day in infants or 15 to 17 mg/m2 per day in adolescents, there is a decrease in height standard deviation score (SDS), leading to a decreased adult height SDS (196–199).
Table 2 provides suggested dosing guidelines. Differences in HC absorption and half-life occur, which may influence HC dosing requirements (200). Although prednisolone and Dex treatments are effective in suppressing adrenal androgens in children with CAH, these more potent drugs are more likely than HC to impede statural growth and cannot be routinely recommended. During puberty, even if replacement therapy and compliance are adequate, control may be suboptimal because of increased cortisol clearance (201). The adult height of patients with CAH correlates negatively with the dose of GC administered in early puberty; patients treated with <20 mg of HC/m2 per day at the start of puberty are significantly taller than those given higher HC doses (187). Therefore, as with younger patients, it is important during puberty to treat with the lowest effective dose to achieve treatment goals.
At or near completion of growth, long-acting GCs may be administered (Table 3), although HC remains the preferred treatment. There are no randomized controlled studies featuring long-term follow-up of adults receiving different modes of treatment of classic CAH, and practice varies (202, 203).
Table 3.
Maintenance Therapy Suggested in Fully Grown Patients
| Type of Long-Acting Corticosteroid | Suggested Dose (mg/d) | Daily Doses |
|---|---|---|
| HC | 15–25 | 2–3 |
| Prednisone | 5–7.5 | 2 |
| Prednisolonea | 4–6 | 2 |
| Methylprednisolone | 4–6 | 2 |
| Dexa | 0.25–0.5 | 1 |
| Fludrocortisone | 0.05–0.2 | 1–2 |
Suspension or elixir may permit improved dose titration for these drugs.
The optimal dose of fludrocortisone substitution in adults (as in infants and children) has not been critically studied. The need for MCs decreases with age, as serum aldosterone is high and renal MC receptor mRNA is low at birth (204), and the salt content in most diets is high. Most nonhypertensive adults with classic CAH benefit from continued fludrocortisone treatment. The requirement for MC replacement should be reassessed during the transition from pediatric to adult care.
Control of hyperandrogenic symptoms in young women may require treatment in addition to GC and MC, such as androgen-receptor antagonists. Oral contraceptives containing drospirenone effectively reduce both adrenal and ovarian androgen synthesis, although not affecting cortisol (205), blood pressure, PRA, or serum potassium levels (206). Oral contraceptives, however, cannot replace GC and MC treatment in classic CAH, although some symptomatic women with NCCAH may prefer such treatment. Spironolactone is relatively contraindicated as an androgen antagonist in salt-wasting CAH, as it is also an MC antagonist and can cause volume depletion. Treatment of hirsutism is beyond the scope of this guideline and has been discussed separately in another Endocrine Society guideline (207).
Balance of benefits and harms
The proposed GC choice places higher value on reducing the negative effects on growing children than on convenience.
Stress dosing
4.7 In all patients with CAH who require GC treatment, for situations such as febrile illness (>38.5°C), gastroenteritis with dehydration, major surgery accompanied by general anesthesia, and major trauma we recommend increasing the GC dosage. (1|⊕⊕⊕○)
4.8 In patients with CAH under everyday mental and emotional stress and minor illness and/or before routine physical exercise we recommend against the use of increased GC doses. (1|⊕⊕○○)
4.9 In patients with CAH who require treatment we recommend always wearing or carrying medical identification indicating that they have adrenal insufficiency. (1|⊕⊕⊕○)
4.10 In patients with CAH, we recommend educating patients and their guardians and close contacts on adrenal crisis prevention and increasing the dose of GC (but not MC) during intercurrent illness. (1|⊕⊕⊕○)
4.11 We recommend equipping every patient with CAH with a GC injection kit for emergency use and providing education on parenteral self-administration (young adult and older) or lay administration (parent or guardian) of emergency GCs. (1|⊕⊕⊕○)
Evidence
Patients with severe forms of 21OHD are unable to produce sufficient cortisol in response to stress, such as febrile illness, gastroenteritis with dehydration, surgery, or trauma, and therefore require increased doses of GC during such episodes (Table 4). In contrast to maintenance treatment given three times daily, we suggest that stress dosing be given every 6 hours (208). In studies of adrenally intact children undergoing anesthesia and minor surgery, serum cortisol does not exceed 10 μg/dL (276 nmol/L) (209). Therefore, the need for stress dosing for minor procedures should be assessed on an individualized basis.
Table 4.
Suggested Stress Doses of GC for Adrenal Crisis
| Patients’ Age | Initial Parenteral HC Dose |
|---|---|
| Infants and preschool children | 25 mg |
| School-age children | 50 mg |
| Adults | 100 mg |
Successive IV HC may be administered as one-quarter of the initial parenteral HC dose (above) given every 6 h.
In a questionnaire-based study of 122 adults with classic CAH, the most common precipitating causes of adrenal crisis were respiratory and gastrointestinal infections (210). In a population-based prospective study of 102 Bavarian children with classic CAH, 27.5% experienced an adrenal crisis or hypoglycemia, mostly during the first 4 years of life, primarily in those with the salt-wasting form of CAH (211). A link to an instructional video for emergency intramuscular injection of HC is provided in the Appendix.
When stress doses of HC are administered, MCs are not needed because HC can activate MC receptors (212). Patients should resume maintenance HC doses when stable and avoid fasting during acute illnesses. Owing to the risk of hypoglycemia and electrolyte imbalance, parents should be instructed to give oral glucose and electrolyte supplementation to young children. Inability to tolerate oral fluids or medication warrants immediate medical attention and parental administration of GCs and isotonic fluids to prevent adrenal crisis. Parenteral GCs are not always carried by emergency service personnel; we recommend that patients be supplied with vials of injectable HC and be taught to administer the drug intramuscularly. Routine exercise and psychological stress (e.g., anxiety and academic examinations) do not require increased GC dosing (213). There is no evidence supporting the use of additional GCs for prolonged extended exercise training.
Adults with classic CAH should continue to carry medical alert identification and injectable HCs for emergencies, as 20% of adrenal crises in patients with CAH occur during adulthood, most commonly during gastrointestinal illnesses (210). A register of 588 individuals with CAH showed a hazard ratio for death of 2.3 (CI, 1.2 to 4.3), equating to a 6.5-year mean reduction in life expectancy (214) attributed to adrenal crises. Separate detailed guidelines are available in a previous Endocrine Society guideline on primary adrenal insufficiency (215).
Monitoring therapy
4.12 In patients ≤18 months with CAH, we recommend close monitoring in the first 3 months of life and every 3 months thereafter. After 18 months, we recommend evaluation every 4 months. (1|⊕⊕○○)
4.13 In pediatric patients with CAH, we recommend conducting regular assessments of growth velocity, weight, blood pressure, as well as physical examinations in addition to obtaining biochemical measurements to assess the adequacy of GC and MC. (1|⊕⊕○○)
4.14 In pediatric patients with CAH under the age of 2 years, we advise annual bone age assessment until near-adult height is attained. (Ungraded Good Practice Statement)
4.15 In adults with CAH, we recommend annual physical examinations, which include assessments of blood pressure, body mass index (BMI), and Cushingoid features in addition to obtaining biochemical measurements to assess the adequacy of GC and MC replacement. (1|⊕⊕○○)
4.16 In adults with CAH, we recommend monitoring treatment through consistently timed hormone measurements relative to medication schedule and time of day. (1|⊕⊕○○)
4.17 In adults with CAH, we recommend that clinicians do not completely suppress endogenous adrenal steroid secretion to prevent adverse effects of over treatment. (1|⊕⊕⊕○)
Evidence
Adjusting medications for CAH is difficult. The challenge in infancy is to find the appropriate fludrocortisone dose without causing hypertension, as MC sensitivity naturally increases in the first year of life. In a prospective study of 33 individuals with classic CAH diagnosed by newborn screening, more than half experienced hypertension in the first 18 months of life (216). In a population-based study of children (n = 716; age range, 3 to 18 years), the dose of fludrocortisone was associated with blood pressure, and children with regularly measured PRA had lower blood pressure than did those without PRA documentation (217).
Pertinent features of the medical history include the age of pubic hair onset, unexpected phallic or body growth, development of adult apocrine odor, and symptoms of salt craving or adrenal crisis. The examination should identify potential accelerated height velocity, signs of virilization, and advanced bone maturation that occur after protracted undertreatment. In contrast, reduced height velocity, accelerated weight gain, and high blood pressure occur after protracted overtreatment. Laboratory data should indicate the need for dose adjustment before changes in growth, bone age, and physical features occur. Bone age is a lagging indicator of past inadequate adrenal suppression and should therefore be used judiciously. Bone age x-rays are not helpful before the age of 2 years; excessive radiation exposure should be avoided. If bone age advances to a pubertal level at an inappropriately early age, testing for secondary central/GnRH-dependent precocious puberty is warranted.
Serum 17OHP and androstenedione are traditional indicators of the adequacy of GC treatment in CAH. More recently, it has been found that metabolites such as 21-deoxycortisol and 11-oxysteroids may provide more direct evidence of adrenal androgen precursor production in CAH (218, 219). Steroids can be measured in blood, saliva (220), urine (86), or dried filter-paper blood samples (221, 222). LC-MS/MS is the gold standard for blood and saliva measurement, whereas gas chromatography–mass spectrometry is the recommended method for urinary measurements of hormones. Circadian rhythm and the timing of GC intake influence steroid measurements (223). Thus, monitoring treatment by consistently timed hormone measurements is recommended. Complete suppression of serum 17OHP level is not a treatment goal but instead indicates overtreatment. Androstenedione levels should be referenced to age- and sex-specific norms. ACTH measurements are not useful in patients with CAH. Acceptably treated patients with CAH generally have upper normal to mildly elevated 17OHP and androstenedione levels when measured in a consistent manner. Clinicians should adjust doses in the context of the overall clinical picture and not solely based on a single laboratory assessment. We do not provide specific target levels for adrenal steroid measurement, because laboratory reference ranges vary, sample timing varies, and one must consider the whole clinical picture.
The prevalence of testicular adrenal rest tumors (TARTs) is variable, increasing after 10 years of age (203, 224). Screening by testicular ultrasound assessments should begin in adolescence. There are no data to suggest how often this should be done, but expert opinion would suggest about every 1 to 2 years in asymptomatic males, or more often in symptomatic patients. Optimization of GC treatment can shrink early stage TARTs and prevent progressive enlargement, resulting in infertility (see also the section on fertility).
The principles of monitoring GC treatment in adult patients with CAH differ somewhat from those employed in children, with attention shifting to reproductive function and chronic complications rather than skeletal maturation. There are neither established optimal biomarkers nor target values, and clinicians should adjust GC doses primarily using clinical indicators. For women, androstenedione and testosterone are good parameters of disease control (202), but additional tests should be considered in the context of menstrual irregularity and signs of androgen excess. For women who experience a delay in conception, GC treatment should aim to achieve a follicular-phase progesterone level <0.6 ng/mL (2 nmol/L), a much tighter control than for women not attempting to conceive, often requiring a bedtime dose of prednisolone (225). The dose of fludrocortisone and/or salt supplementation should be titrated to standing blood pressure, PRA appropriate for age, and serum potassium measurements. For men, suppressed gonadotropins are a reliable sign of infertility, and elevated FSH indicates sustained testicular damage in men with TARTs (226). Men with large TARTs may also have low morning testosterone, indicating poor Leydig cell function (227). The ratio of androstenedione to testosterone is <0.5 in eugonadal men; values >2 indicate poor CAH control with a significant fraction of testosterone of adrenal origin (228). The presence of TARTs does not correlate strictly with the degree of control (229). Table 5 illustrates the use of various analytes in the management of adults with classic CAH. The issue of TARTs is discussed further below (see section 6 on long-term management).
Table 5.
Utility of Various Analytes for Monitoring CAH Treatment
| Patients | Analyte | Physiology | Goals and Comments |
|---|---|---|---|
| All ages | Plasma renin | Volume status | Low to normal unless hypertensive |
| Potassium | MC replacement | Goal is normal | |
| Sodium | GC and MC replacement | Goal is normal | |
| Testosterone | Total androgens | Goal is at or near normal | |
| Androstenedione | Mostly adrenal origin | Goal is at or near normal | |
| Sex hormone–binding globulin | Testosterone-binding protein | For calculation of free and bioavailable testosterone | |
| 17OHP | Variable | Normal values indicate overtreatment | |
| Men | Testosterone | Adrenal or gonadal origin | Interpret abnormal values in context of gonadotropins and androstenedione levels |
| Gonadotropins | Gonadal axis status | Low indicates poor control | |
| Androstenedione | Mainly adrenal | Goal is <0.5× testosterone | |
| Semen analysis | Fertility | Goal is normal | |
| Women | Follicular-phase progesterone | Mainly adrenal origin when elevated | Goal is <0.6 ng/mL (<2 nmol/L) for women trying to conceive |
5. Treatment of NCCAH
5.1 In children and adolescents with inappropriately early onset and rapid progression of pubarche or bone age and in adolescent patients with overt virilization we suggest GC treatment of NCCAH. (2|⊕⊕○○)
Technical remark: Risks and benefits of GC therapy should be considered and discussed with the patient’s family.
5.2 In asymptomatic nonpregnant individuals with NCCAH we recommend against GC treatment. (1|⊕⊕⊕○)
5.3 In previously treated patients with NCCAH we suggest giving the option of discontinuing therapy when adult height is attained or other symptoms resolve. (2|⊕⊕⊕○)
5.4 In adult women with NCCAH who also have patient-important hyperandrogenism or infertility we suggest GC treatment. (2|⊕⊕○○)
5.5 In most adult males with NCCAH, we suggest that clinicians generally not prescribe daily GC therapy. (2|⊕○○○)
Technical remark: Exceptions include infertility, TARTs, or adrenal tumors and phenotypes that are intermediate between classic and nonclassic phenotypes.
5.6 In patients with NCCAH, we suggest HC stress dosing for major surgery, trauma, or childbirth only if a patient has a suboptimal (<14 to 18 μg/dL, <400 to 500 nmol/L) cortisol response to cosyntropin or iatrogenic adrenal suppression. (2|⊕○○○)
Technical remark: A range is given for cortisol cut points due to greater specificity of newer cortisol assays (see below).
Evidence
Expert opinion suggests that individuals with asymptomatic NCCAH do not warrant therapy (230, 231). The writing committee suggests that children should be treated for inappropriately early onset of body hair and odor only when bone maturation is sufficiently accelerated to adversely affect future height. In the presence of premature pubarche without advanced bone age, clinicians can withhold treatment with careful monitoring. In adolescents with irregular menses and acne, symptoms are usually reversed within 3 months of GC treatment, whereas hirsutism remission is more difficult with GC monotherapy. As in other androgenic disorders, an oral contraceptive with or without antiandrogens is likely the best approach for treating hirsutism in women with NCCAH (171, 207, 232, 233). For patients treated in childhood or adolescence, it may be reasonable to consider tapering and discontinuing GC treatment once near-adult height has been reached.
If a woman affected with NCCAH becomes pregnant in the absence of GC treatment, she need not be treated during pregnancy. Two retrospective studies of pregnancies among women with NCCAH found that the majority of pregnancies occurred prior to the mother’s diagnosis of NCCAH (234, 235). GC treatment was given to induce fertility in 23% (234) and 42% (235) of cases. Both studies reported elevated miscarriage rates of ∼25% in those not receiving GC and 6% in those exposed to GC. A third report found no difference in miscarriage rate between GC-treated and untreated women, but the former group had a shorter time to conception (236). Thus, women with subfertility may benefit from GC treatment to conceive and maintain pregnancy.
Available data indicate that TARTs in men with NCCAH are extremely rare (237); consequently, prophylactic GCs do not seem to be warranted in these men. There is no evidence of clinically significant cortisol deficiency or adrenal crisis in NCCAH, and we do not suggest substitution therapy in previously untreated individuals with NCCAH during severe stress unless they have demonstrated a subnormal cortisol response during diagnostic cosyntropin stimulation. Some individuals with NCCAH (60% in one small study) showed an inadequate response to cosyntropin stimulation, but none had frank episodes of adrenal insufficiency (171, 238, 239). A cortisol cut-off range is given as 14 to 18 μg/dL in part due to CBG variability, but also because newer assays with greater specificity run lower (240).
Individuals with the P30L/null genotypes and some de novo mutations comprise a problematic population, as their biochemical and clinical phenotypes straddle the classic/nonclassic boundary. Some of these patients benefit from chronic low-dose HC therapy.
6. Long-Term Management of Patients With CAH
Transition to adult care
6.1 In adolescent patients with CAH, we suggest that the transition to adult care begins several years prior to dismissal from pediatric endocrinology. (2|⊕○○○)
Technical remark: We advise the use of joint clinics comprised of pediatric, reproductive, and adult endocrinologists and urologist during this transition.
6.2 In adolescent females with CAH, we suggest a gynecological history and examination to ensure functional female anatomy without vaginal stenosis or abnormalities in menstruation. (2|⊕⊕○○)
Evidence
Several reviews, but no controlled studies, describe how to transfer patients with CAH from pediatric to adult care. Our suggestions are based on clinical experience (241–244). Adult women with CAH often remember childhood visits to their physician as highly intrusive. Therefore, after follow-up from the initial surgery, clinicians should avoid gynecological examinations unless and until the patient experiences delayed or painful menses, planned sexual activity, or pregnancy.
Adolescent girls with virilizing CAH should be referred to a gynecologist and/or a pediatric surgeon/urologist for a genitourinary examination with sedation or anesthesia when appropriate. The patient and, if appropriate, her family, should discuss whether surgery should be considered. At the appropriate time, the medical/surgical team, ideally including a reproductive endocrinologist, should discuss issues of sexual activity, contraception, and fertility. Obstetricians should be aware that despite an apparent normal pregnancy rate of ∼90%, women with classic CAH have low fecundity (0.25 live births per woman vs 1.8 in the general population) (225). In NCCAH, 72% of pregnancies result in live births (236).
A gradual transition of adolescents to adult care would ideally allow the patient’s relationship with the adult physician to be consolidated before the patient terminates his or her relationship with the pediatric endocrinologist, typically after age 18. At this juncture, patients should be reminded of the importance of continued GC treatment. Poor medical adherence among adults with CAH contributes to depression and increased mortality (245). A baseline bone mineral density (BMD) measurement and, in males, a testicular ultrasound should be considered. Young men should be advised regarding the risk of noncancerous testicular masses (see section 6.5).
Genetic counseling
6.3 In children with CAH, adolescents transitioning to adult care, adults with NCCAH upon diagnosis, and partners of patients with CAH who are planning a pregnancy, we recommend that medical professionals familiar with CAH provide genetic counseling. (1|⊕⊕○○)
Evidence
CAH genotype and phenotype correlate well; siblings with CAH generally, but not always, have similar symptoms and degrees of female virilization. For this autosomal recessive disorder, there is a 25% probability that each subsequent sibling of the index case will have CAH and a 50% probability that each will be an asymptomatic carrier. Based on a classic CAH incidence of 1:10,000 to 1:20,000 (23, 43, 44, 52), 1:50 to 1:71 people in the general population are heterozygotes. Using a median value of 1:60 (∼2%), a patient with classic CAH would have a 1:120 probability of having a child with classic CAH. For NCCAH, almost 70% of diagnosed patients are compound heterozygotes, carrying one allele that causes classic CAH and one that causes NCCAH (171, 246). The milder mutation determines the phenotype, meaning that the patient has NCCAH, but the patient’s offspring have a 50% chance of inheriting the classic CAH allele. Theoretically, and without genotyping, a NCCAH parent has an ∼1:250 risk of having a child with classic CAH [(0.7 × 0.5) × (0.02 × 0.5) = 0.4%]. However, in two retrospective analyses of children born to women with NCCAH, the risk was higher, at 1.5% to 2.5% (234, 235). Similar risks were found in a mixed group of men with CAH and NCCAH (247). To refine the risk, CYP21A2 genotyping is recommended prior to pregnancy planning.
Fertility counseling
6.4 In individuals with CAH and impaired fertility we suggest referral to a reproductive endocrinologist and/or fertility specialist. (2|⊕⊕○○)
Evidence
Fertility in males with CAH is often impaired (226, 237, 248–250). Common factors contributing to male infertility include the presence of TARTs, suppression of gonadotropins, and testicular failure. In one study, males with CAH born after the introduction of neonatal screening had normal fertility (247).
The prevalence of TARTs in boys aged 2 to 18 years who have classic CAH varies from 21% to 28% (227, 251); there have been few studies describing TARTs in males with NCCAH. The prevalence of TARTs increases with age but is highly variable in men with classic CAH. These tumors often regress with intensification of GC therapy if detected early (see section 6.13) (252). The presence of TARTs is a predictor of infertility (226, 248, 253). The prevalence of these tumors varies between 0% and 94%, depending on the study population (254, 255). TARTs are usually small and bilateral, not palpable, but easily detectable by ultrasound (227, 251). TARTs have no malignant features but can lead to obstructive azoospermia and infertility (248). When tumors are unresponsive to intensified GC therapy, testicular sperm extraction can be performed (256). Suppression of gonadotropin secretion by high levels of adrenal androgens also impairs fertility; this is evident when the androstenedione-to-testosterone ratio is >2. Sperm banking may be an option to maintain fertility. One study reported that fewer men with CAH had stable heterosexual relationships than did age-matched controls (255), whereas more recent data showed no such differences in relationships but a reduction in sexual activity among men with CAH (247).
Several studies have shown that, for a variety of reasons, only a minority of women with classic CAH try to conceive (225, 257). Those who wish to conceive can achieve nearly normal pregnancy rates with adequate suppression of follicular-phase progesterone (<0.6 ng/mL = 2 nmol/L, Table 5) (225) by optimizing GC and MC treatments. Factors beyond CAH, such as tubular obstruction and endometriosis, can contribute to infertility and must be excluded. Ovarian adrenal rest tumors are relatively rarely detected compared with TARTs (258). Fertility in the context of NCCAH is discussed in section 5. Ovulation induction and in vitro fertilization, along with other assisted reproductive technologies, may be considered for women in whom these measures prove insufficient.
Management of CAH and NCCAH during pregnancy
6.5 In women with NCCAH who are infertile or have history of prior miscarriage, we recommend treatment with a GC that does not traverse the placenta. (1|⊕⊕○○)
6.6 In women with CAH who are pregnant, we advise management by an endocrinologist familiar with CAH. (Ungraded Good Practice Statement)
6.7 In women with CAH who become pregnant we recommend continued prepregnancy doses of HC/prednisolone and fludrocortisone therapy, with dosage adjustments if symptoms and signs of GC insufficiency occur. (1|⊕⊕○○)
Technical remark: Clinicians should evaluate the need for an increase in GC during the second or third trimester and administer stress doses of GCs during labor and delivery.
6.8 In women with CAH who are pregnant, or trying to become pregnant, we recommend against using GCs that traverse the placenta, such as Dex. (1|⊕⊕○○)
6.9 In women with CAH who are pregnant, we advise that the birthing plan includes an obstetric specialist. (Ungraded Good Practice Statement)
Evidence
Androgen and cortisol levels increase gradually during pregnancy due to increases in sex hormone–binding globulin and corticosteroid-binding globulin. Placental aromatization typically protects the fetus from the potential virilizing effects of maternal androgens (259). Maternal 17OHP is elevated in normal pregnancy and hence cannot be used to monitor GC treatment. High progesterone levels during pregnancy may compete for the MC receptor, theoretically necessitating increased fludrocortisone doses, but this possibility has not been studied. Clinicians should not use Dex, or other steroids that are not inactivated by placental 11β-HSD2, to treat pregnant women affected by CAH. There are no data and no widely accepted recommendations for managing GC doses in pregnancy. Nonspecific symptoms of adrenal insufficiency, including postural hypotension and fatigue, may develop in pregnancy but are not unique to women with classic CAH. GC and/or fludrocortisone doses may be increased if such signs and symptoms occur. In such cases, a GC dosage increase of 20% to 40% from the 24th week onward is often beneficial (215). During labor and delivery, stress doses of GCs should be administered, but there are no controlled studies regarding optimal dosing. Women with CAH may be at increased risk for gestational diabetes (257, 260). Thus, clinicians should monitor glucose tolerance as clinical judgment indicates throughout pregnancy. Treatment should be individualized for pregnant patients with CAH. Caesarean section is the most common method of delivery due to the high prevalence of previous vaginal surgery and cephalopelvic disproportion, although vaginal delivery has been reported in 16% to 42% of women, nearly all of whom had a non–salt-wasting phenotype (225, 257). It is difficult to draw definitive conclusions about the need for GC therapy in women with NCCAH based on limited data (234–236); however, treatment may benefit infertile women with NCCAH or those with a history of miscarriage. Similar principles of pregnancy management apply to women with NCCAH requiring GC treatment during pregnancy.
Surveillance for long-term complications of CAH and its treatment
6.10 For patients with CAH, we suggest introducing counseling regarding healthy lifestyle choices at an early age to maintain BMI within the normal range to avoid metabolic syndrome and related sequelae. (2|⊕○○○)
6.11 In adult patients with CAH, we suggest screening of BMD in anyone subjected to a prolonged period of higher-than-average GC dosing, or who has suffered a nontraumatic fracture. (2|⊕○○○)
6.12 In adults with classic CAH, we recommend against routine adrenal imaging. (1|⊕○○○)
Technical remark: Reserve adrenal imaging for individuals with classic CAH who have clinical evidence of an adrenal mass, poor disease control, a lapse in treatment of several years, or lack of response to intensified therapy.
6.13 In males with classic CAH, we recommend periodic testicular ultrasound to assess for the development of TARTs (see section 6.4). (1|⊕⊕○○)
6.14 In patients with CAH, we recommend against routine evaluation for cardiac and metabolic disease in patients with CAH beyond that recommended for the general population. (1|⊕⊕○○)
Technical remark: Clinicians should use their own judgment for the above procedures.
Evidence
Children and adolescents on standard GC therapy for CAH have no evidence of decreased BMD when assessed by dual-energy x-ray absorptiometry normalized for height, irrespective of duration of treatment, type of GC used, and 17OHP or androgen levels (261–263). The standard of care for good bone health includes age-appropriate vitamin D and calcium intake along with weight-bearing exercise.
In contrast, a retrospective study of 62 adult women with CAH reported that chronic exposure to pharmacological GC doses may lead to bone loss accompanied by an increased incidence of fractures relative to healthy controls (264). Two studies of large cohorts of adults with CAH reported a high prevalence of osteopenia (BMD T-scores, −1.0 to −2.5 SD) and a modestly increased prevalence of osteoporosis (202, 203). No increase in fracture incidence has been observed (265). The occurrence and severity of bone loss did not correlate with CAH genotype or phenotype but appeared to be related to GC exposure. Chakhtoura et al. (266) showed a negative correlation between the cumulative lifetime GC dose and BMD. These data underscore the need to avoid excessive GC exposure.
Adrenal masses affect 1% to 4% of normal men and women (267), and their prevalence increases with age (268). One study using CT imaging in adults with CAH reported a high prevalence of benign adrenal masses, especially among those on inadequate GC therapy (269). Adrenal carcinomas are rarely found in persons with CAH (270), and only a single pediatric case has been reported (271). Massive adrenal myelolipomas have developed in several adults with CAH, requiring surgical removal for mass effects (272). Insufficient data exist to recommend routine screening for adrenal masses.
Children with CAH have a higher BMI than do controls due to increased fat mass (273). Approximately half of pediatric patients are overweight, and 16% to 25% are obese (273–275). The commissioned systematic review included a meta-analysis of 14 observational studies, ranging widely in age (14 months to 63 years, ∼70% <18 years old). The 437 patients with CAH in those 14 studies had mildly increased systolic and diastolic blood pressures (respective mean differences, +4.4 and +2.4 mm Hg), homeostatic model assessment of insulin resistance (+0.5), and carotid intima thickness (+0.08 mm) compared with controls without CAH (41). No statistically significant difference was noted in fasting blood glucose, insulin level, glucose or insulin level 2 hours after a glucose load, or serum lipids. Data on cardiac events were sparse, and most of the literature focused on surrogate outcomes. The commissioned systematic review also summarized evidence from other observational studies that presented data not amenable to meta-analysis, including cohorts from Sweden, the United Kingdom, Germany, and Brazil. These studies suggested that individuals with CAH may have higher frequency of hypertension, hyperlipidemia, atrial fibrillation, venous thromboembolism, obesity, and diabetes. A moderate to high risk of bias was noted among the studies included in the systematic review (41). In view of increased body fat and the potential for cardiac and metabolic consequences, we suggest beginning lifestyle counseling early to counteract these trends.
Women with CAH are often overweight (202, 203, 260), but patients with CAH >30 years of age had similar fat mass to age-matched controls. Few had hypertension, cardiovascular disease, or diabetes. The most significant metabolic abnormality was a 20% prevalence of gestational diabetes, somewhat higher than the prevalence for the general population, which is estimated at 7% to 10% but ranges from 1% to 25% (276).
7. Restoring Functional Anatomy by Surgery in Individuals With CAH
7.1 In all pediatric patients with CAH, particularly minimally virilized girls, we advise that parents be informed about surgical options, including delaying surgery and/or observation until the child is older (Ungraded Good Practice Statement).
Technical remark: Surgeries should be performed only in centers with experienced pediatric surgeons/urologists, pediatric endocrinologists, pediatric anesthesiologists, behavioral/mental health professionals, and social work services. Extensive discussions regarding risks and benefits, shared decision-making, review of potential complications, and fully informed consent need to occur prior to surgery. The option to forgo surgery should be considered.
7.2 In severely virilized females (single urogenital opening, Fig. 3), we advise discussion about early surgery to repair the urogenital sinus (Fig. 4). (Ungraded Good Practice Statement)
7.3 In the treatment of minors with CAH, we advise that all surgical decisions remain the prerogative of families (i.e., parents and assent from older children) in joint decision-making with experienced surgical consultants. (Ungraded Good Practice Statement)
Figure 3.

Lower urogenital anatomy of mild and severe CAH. (a and b) The lower urogenital anatomy of mild and severe CAH is shown. Note the low confluence in (a), where the vagina and urethra meet close to the skin, in contrast to (b), where the confluence of the vagina and urethra is close to the bladder neck. [Illustration ENDOCRINE SOCIETY]
Figure 4.

Partial urogenital mobilization with separation of the urethra and vagina. (a and b) Schematic of partial urogenital sinus mobilization where normal female anatomy is restored. Note the separation in (a) of the vagina and urethra with preparation of the excess common urogenital sinus to form the anterior vaginal wall by anastomosis to the normal anterior vaginal wall (b) and preparation of the posterior perineal skin flap (a) to form the posterior vaginal wall (b). [Illustration ENDOCRINE SOCIETY]
Evidence
There are no randomized controlled studies of either the best age or the best methods for restoring functional female anatomy with a separate urethra and vaginal opening in individuals with CAH. Published literature to date has relied on evaluations in late adolescence or adulthood, often 20 years or more after the initial surgery. During that time, methods for separating the urogenital sinus, bringing the vaginal opening to the perineum, introitoplasty, and treating clitoromegaly have evolved.
Based on present outcome data, we suggest that, for patients with a low urogenital confluence of the vagina and urethra (Fig. 3), experienced surgeons perform complete surgical repair at an early age (separating the urogenital sinus, bringing the vaginal opening to the perineum, introitoplasty, and, if chosen, clitoroplasty) (277–281). For individuals with a high confluence (Fig. 3), the timing is less certain, although retrospective studies report that early surgery has good long-term success in respect to sexual function compared with normal controls (277–281). The unproven surgical advantage of delayed reconstruction is that the risk of vaginal stenosis and the need for subsequent vaginal dilation may be diminished. If clinicians are considering treating infants with severe virilization for clitoromegaly, the advantages of early complete reconstruction is the ability to use the excess common urogenital sinus tissue to reconstruct the anterior vaginal wall (282). One should avoid premenarchal vaginal dilation for stenosis.
In the female patient with CAH with severe virilization in whom no surgery has been performed, there is an ongoing need for observation in regard to possible urinary tract infections and obstruction of menstrual flow at puberty, because the vagina opens into the common urogenital sinus.
In the rare patients with 46,XX CAH and complete virilization (Prader 5/normally formed “penis” with the urethral meatus within the glans) controversy exists with respect to the optimal gender assignment (see section 9 on mental health). Surgical feminization with a female gender identity is especially challenging secondary to extreme virilization. Future fertility is possible as a female. If the same patient is raised as male from infancy or early childhood, surgery to remove the uterus and ovaries prior to puberty, or drugs to suppress pubertal hormones, may be required to avoid gender-incongruent body development. The pros and cons of female vs male gender assignment and the fertility implications must be openly and completely discussed. Lifelong medical treatment with GCs and MCs would still be required, and supplemental testosterone may be required to support male secondary sexual characteristics. Height outcomes for male patients would be short compared with mid-parent height.
Complications following urogenital reconstruction in individuals with CAH may include vaginal stenosis, labial or introital scarring, loss of sexual function, urethra–vaginal fistulae, and urinary incontinence (283). One retrospective study documented a relative decrease in clitoral sensitivity after clitoral surgery, yet other studies have not documented any impairment in sexual function compared with that of age-matched healthy controls (280, 284–286). An enlarged clitoris observed in the newborn may decrease in prominence over time with standard medical treatment. No data exist regarding long-term outcomes for individuals who have not had surgery to separate the urogenital sinus and bring the vagina to the perineum, or those who have not undergone clitoral reduction. Surveys of surgical practice in the United States (287, 288) and internationally (279) also report a preference for early surgery for CAH. Clinicians should share with parents all available information on the timing, risks, benefits, and complications of surgery and counsel them that deferring or forgoing surgery is an option. It is also important to discuss, both with the parents at the time of diagnosis and with the patients as they mature, what is known about the long-term prognosis for sexual and reproductive function. There is no objective evidence at this time as to whether early, late, or no surgery best preserves overall QOL or sexual function.
Recent cohort studies have shown no change in neurocognitive outcomes in children undergoing a single use of anesthesia under the age of 36 months (289). However, animal evidence and retrospective human studies have raised concerns that prolonged or repeated general anesthesia may impair brain development in early life, specifically impairing long-term language abilities and cognition (290).
Balance of benefits and harms
Presumed values in seeking early surgery for virilized females with CAH are restoring female anatomy, preventing urinary tract infection and hydrometrocolpos, reducing parental anxiety regarding the child’s congenital anomaly, avoiding stigmatization of a girl with masculinized genitals, and avoiding the psychological trauma of genital surgery during childhood and adolescence (291, 292). The presumed benefit of late surgery is patient autonomy regarding surgery with a better understanding of the individual’s own preferences (a shared decision-making process as opposed to parental choices) regarding gender identity, risks, benefits, alternatives, and complications.
Genital reconstructive surgery requires the level of surgical experience and endocrine, anesthesiologic, nursing, and psychosocial support that is only found at centers that perform this procedure regularly. Surgical expertise has been defined in one group’s opinion as having performed at least 10 genitoplasties in the preceding 8 years (293).
7.4 In female patients with CAH for whom surgery is chosen, we suggest vaginoplasty using urogenital mobilization and, when chosen, neurovascular-sparing clitoroplasty for severe clitoromegaly. (2|⊕○○○)
Evidence
Total urogenital mobilization (294) signaled a significant advance in the surgical management of CAH. This technique has evolved into the present technique of partial urogenital mobilization, where, instead of a 360° dissection, surgery is avoided superior to the urethra under the pubic bone, a nerve-rich zone that contains the sphincteric musculature necessary for urinary continence (279, 282, 295, 296). Urinary incontinence and vaginal stenosis requiring dilation or reoperation remain as postoperative concerns (297–300). Long-term follow-up studies are now confirming that urinary incontinence is rare, but that a minority of patients will require additional vagina surgery after puberty (277, 282, 301–304). Nerve-sparing clitoroplasty (305) is an optional procedure and should be explained as such.
Balance of benefits and harms
The writing committee shares the stated preference of most patients and clinicians and places a high value on the outcomes of early complete repair performed by surgeons experienced with urogenital mobilization, on the reduced need for complex secondary procedures in adolescence or adulthood, and on maintaining normal perineal and clitoral sensation. Additionally, expert opinion states that, for patients with 46,XX CAH living as females, potential fertility should be preserved to the extent possible. In those living as males, potential options of preservation of ovarian tissues should be discussed with parents (and patients when practical) prior to ovarectomy.
8. Experimental Therapies and Future Directions
General considerations and unmet clinical needs
8.1 In patients with CAH we advise against using experimental treatment approaches outside of formally approved clinical trials. (Ungraded Good Practice Statement)
Evidence
As reviewed in the sections above, despite life-saving GC and fludrocortisone acetate therapies, many children and adults with CAH commonly experience adverse outcomes (202, 203). Therefore, further study of alternative treatment approaches should consider growth, metabolic, reproductive, and neuropsychiatric endpoints.
Improved GC delivery methods
We advocate the development of new treatment approaches that minimize the daily GC dose with the goals of achieving physiological cortisol replacement and preventing excessive androgen secretion. Normal adrenocortical secretion has a circadian rhythm (306, 307). Programmed infusion of HC delivered in a circadian fashion to poorly controlled patients with CAH resulted in nearly normal ACTH and 17OHP (308, 309). In a phase 2 study, eight adults with classic CAH and multiple comorbidities experienced significant reduction in adrenal androgens and significant improvement in QOL parameters and fatigue following 6 months of subcutaneous HC infusion aimed at mimicking physiologic cortisol secretion (310). Although conceptually appealing, parenteral HC hemisuccinate preparations are not designed or approved for subcutaneous administration; infusion site reactions occur, and pump management is complex. Nevertheless, this approach may have value for motivated patients, particularly those who demonstrate rapid HC metabolism.
Once-daily modified-release oral HC in adults with classic CAH afforded serum cortisol concentrations typical of a classic diurnal rhythm; however, serum 17OHP and androstenedione rose to higher levels late in the day than with conventional thrice-daily HC administration (311). A newer version of this product (multiparticulate capsules) was studied in a phase 2 open-label trial of 16 adults with classic CAH (312). Compared with various forms of conventional therapy prior to entry, this approach yielded decreased 17OHP and androstenedione values throughout the day, despite a reduced HC dose equivalent (28 ± 11.8 vs 25.9 ± 7.1 mg/d). A phase 3, parallel-arm, randomized, open-label study is in progress to determine whether this approach improves short-term clinical outcomes (NCT 02716818).
Currently, the lowest dose HC tablet is a 5-mg tablet that is excessive for infants and young children. Availability of pediatric dose formulations would eliminate concerns about improper compounding of HC from tablets (192–194). Another clinical trial examined utility of very low–dose HC granules for treatment of infants with CAH (313).
Androgen/estrogen antagonists and synthesis inhibitors
An alternative approach to optimizing GC exposure is to combine a roughly physiological replacement dose of GC with a second therapy that directly inhibits androgen and estrogen production and/or action, and we think that such approaches deserve further study. The first example of this approach was a four-drug regimen that combined the androgen antagonist flutamide and the aromatase inhibitor testolactone with a reduced dosage of HC (8 mg/m2 per day) and fludrocortisone. Compared with conventional treatment with HC and fludrocortisone, this regimen decreased growth rate, weight velocity, and bone maturation in a crossover study of 12 children (314). In a 2-year randomized parallel study of 28 children, patients receiving the experimental four-drug regimen had normal growth and bone maturation, despite elevated adrenal steroids (315).
All pathways to androgens and estrogens require the enzyme 17-hydroxylase/17,20-lyase (P450c17, CYP17A1) (Fig. 1). Abiraterone acetate is an orally active prodrug of abiraterone, a potent P450c17 inhibitor (316) indicated for treatment of castration-resistant prostate cancer (317, 318). A phase 1, open-label, multiple-dose study of abiraterone acetate enrolled six adult women with classic CAH and high serum androstenedione concentrations (>345 ng/dL or >12 nmol/L) (319). At 250 mg/d, abiraterone acetate normalized the predose androstenedione on day 7 in all participants. Because abiraterone acetate also inhibits gonadal steroid production, this study was limited to adult women taking oral contraceptives. Consequently, abiraterone acetate use in CAH may be limited to prepubertal children and to adults taking gonadal replacement. A phase 1/2 trial of abiraterone acetate in prepubertal children with CAH is in progress (NCT 02574910). Implicit in these new treatment approaches is the goal of normalizing growth and development in children with CAH by reducing GC exposure.
Growth hormone and growth-promoting drugs
A systematic review and meta-analysis of adult height in individuals with classic CAH diagnosed before the age of 5 years was prepared in conjunction with the previous version of these guidelines (186). Of 1016 published reports, only 35 met the eligibility criteria for inclusion in the analysis. All were observational studies with methodological limitations and very low–quality evidence. Again, most patients were diagnosed before the era of newborn screening, fewer than half reported a mean age of diagnosis under 1 year, and most did not give details of GC doses. The pooled data indicated a corrected adult height SDS of −1.05. Subgroup analysis revealed that the addition of MC treatment was associated with increased height outcome.
Individuals with NCCAH can also have compromised adult height, but the height deficit is less severe than with classic CAH. However, there is limited evidence that initiation of GC treatment before puberty may improve adult height (320, 321). A 1- to 2-year nonrandomized study of children with CAH showed improved growth rate and height z score for bone age for growth hormone used alone (n = 12) or in combination with GnRH agonist (n = 8; P < 0.0001) (322). Fourteen patients treated with growth hormone and GnRH plus conventional therapy for ∼4 years had improved adult height (+1.1 SDS) (323) compared with historical controls with CAH treated with conventional therapy alone (SDS of −0.4 vs −1.4, P = 0.01). GnRH analog treatment increases adult height in children with CAH who develop central precocious puberty (324). No randomized study has investigated the effect of a GnRH agonist alone or aromatase inhibitors on adult height in children with CAH and normally timed puberty.
In summary, individuals with CAH can achieve normal adult height through judicious use of standard GC and MC therapies, and height-enhancing drugs are to be considered only for individuals whose heights are, or are expected to be, significantly shorter than those of peers, defined as a height of at least −2.25 SDS. We advocate further prospective, randomized, and carefully controlled studies to determine whether the use of growth-promoting drugs increases adult height in individuals with CAH.
Other medical strategies
An alternative strategy to decrease the adrenal androgen excess is to reduce ACTH production. A single-blind, placebo-controlled, fixed-sequence, single-dose trial of eight women with classic CAH explored the addition of a selective corticotropin-releasing hormone receptor type 1 antagonist, NBI-77860, to conventional therapy (325). The study drug reduced the mean morning increase in ACTH by >40% and that of 17OHP by up to 27% with variable reductions of androstenedione and testosterone.
Another study administered mitotane, a drug that has an adrenolytic effect (used for adrenocortical cancer and Cushing syndrome), to a man with classic CAH and TARTs who was infertile for 2 years (326). Adrenal-derived androgen precursors declined, and TARTs regressed, despite a rise in ACTH. Ultimately, sperm count increased, and paternity was achieved. Mitotane cannot be recommended as routine treatment outside of approved research studies due to its significant toxicities, potential teratogenicity (pregnancy category D), and profound induction of CYP3A4, which markedly increases GC metabolism. A phase 1 trial of ATR-101 (NCT 02804178), which shares some mechanisms with mitotane (327–329), has been completed in adults with classic CAH but is not yet published.
Adrenalectomy
8.2 In patients with CAH we suggest that bilateral adrenaletomy not be performed. (2|⊕○○○)
Evidence
Bilateral adrenalectomy reduces the risk of virilization in females and allows for decreased GC doses. Objections to adrenalectomy are based on surgical risk, possible increased risk of adrenal crisis due to loss of residual adrenal function, and possible loss of hormones that may have beneficial effects.
Among 18 individuals with CAH who underwent bilateral adrenalectomy, 5 patients had one or more adrenal crises, and 2 of the younger patients experienced severe hypoglycemia with illness during ∼5 years of follow-up (330). All patients reported subjective benefits after surgery, including weight loss, a reduced need for frequent monitoring, and reduced signs and symptoms of androgen excess. Eight patients (44%) had elevated steroid precursors postoperatively while on a reduced HC dose, presumably from adrenal rests, which required increased HC doses. However, GC doses were lower after adrenalectomy than before.
Five adult female patients with salt-wasting CAH underwent bilateral adrenalectomy with a mean follow-up time of 4.2 years (331). Two patients underwent adrenalectomy for infertility and became pregnant within 2 years. Three patients underwent adrenalectomy for unsuppressible hyperandrogenism and worsening obesity. All three patients lost weight; however, they all also experienced pigmentation and adrenal crises during follow-up. Adrenalectomy may not totally remove hyperandrogenemia owing to the potential development of adrenal rest tumors in the testes (330), ovaries (332), or retroperitoneum (333). For these reasons, the initial enthusiasm from short-term success has been tempered by long-term complications. Owing to the high risk for significant morbidity and mortality after operation, individuals with a prior history of medical nonadherence are poor candidates for elective adrenalectomy.
Balance of benefits and harms
In recommending further research on experimental therapies in adults, the goal is to improve QOL by maintaining a near-physiological hormone balance. For children, the writing committee placed high value on reducing the impact of GC excess on growth, BMI, and cardiometabolic complications.
Investigation into epinephrine deficiency
We advocate for additional research concerning epinephrine deficiency in the stress response. Individuals with classic CAH have adrenomedullary insufficiency because GCs play essential roles in the development and regulation of the adrenal medulla (334). Combined cortisol and epinephrine deficiency results in glucose, insulin, and leptin dysregulation, shown during short-term high-intensity exercise (335, 336) and long-term moderate-intensity exercise (337). The clinical implications of epinephrine deficiency are not fully known, but it likely contributes to the risk for hypoglycemia during febrile illnesses, especially in young children (211, 338). Epinephrine replacement or supplementation has not been studied.
Preclinical research
Gene therapy temporarily restored adrenal steroidogenesis in 21-hydroxylase–deficient mice (339). The ability to correct the genetic mutations causing CAH by applying gene therapy to an individual’s own adrenal stem cells would theoretically cure CAH and avoid the need for adrenal replacement therapy. Cell-based therapies and gene-editing technology may present novel options for disease remediation or cure in the future (340, 341).
9. Mental Health
9.1 For individuals with CAH and their parents, we recommend behavioral/mental health consultation and evaluation to address any concerns related to CAH. (1|⊕⊕○○)
Technical remark: Clinicians should be aware that individuals with CAH may be at risk for developing mental health problems and should have a low threshold for referral to psychological or psychiatric treatment. Additionally, mental health practitioners should have specialized expertise in assessing and managing CAH-related psychosocial problems.
Evidence
Classic CAH, with the associated risks of potentially fatal electrolyte crises and the effects of hyperandrogenization on the body, brain, and gender-related behavior, may generate anxiety and present challenges to parents and affected individuals (342). In 46,XX newborns with marked genital masculinization, gender assignment is initially in doubt, and parents experience shock. Severely virilized newborns may inadvertently be assigned as males, especially where tradition strongly favors males (343, 344). Once such assignment has been made, it may be difficult to reverse (345). In view of the documentation of good adjustment of male-raised patients with 46,XX CAH with highly masculinized genitalia and the potential risks of feminizing surgery for cosmesis and sexual functioning (see section 7), some experts advise considering deliberate male rearing of newborns with 46,XX CAH with highly masculinized genitalia (346), despite the implied loss of fertility and necessity of lifelong androgen treatment. This argument is supported by the masculinizing effects of prenatal androgen excess in female-raised children with 46,XX CAH on diverse domains of gender-role behaviors (347–351), which may lead to gender questioning and variable transgender identification (352). Nevertheless, most female-raised adolescents and adults with 46,XX CAH end up with a female core gender identity and social role. Of 250 individuals with 46,XX CAH raised female, only 5.2% had serious gender-identity problems (353).
Case reports, but not systematic studies, have documented other psychosocial consequences of atypical genital development (354). These consequences include awareness of the incongruence between the patient’s genital appearance and assigned gender; conflicted gender typing by family members; increased curiosity about the patient’s genitals and increased stigmatization by others; and impaired genital self-image, which may contribute to an overall impaired bodily self-image associated with short stature, increased weight, and hirsutism. Such experiences may result in social withdrawal, especially from situations involving nudity (team sports or medical examinations), and avoidance of romantic interactions and sexual involvement. To prevent adverse psychosocial consequences, clinical management recommendations have typically included corrective genital surgery in early infancy (feminizing or masculinizing, depending on the gender assignment of the child, as discussed in section 7). In view of the potential complications and mixed cosmetic and functional outcomes of such surgery (see section 7), several intersex activist groups, ethicists, and service providers have severely criticized all such surgeries (355–357) or advocated for postponing them until the patient can give informed consent (358–361). Some advocates have called for a moratorium on such surgeries until better empirical evidence of the risks and benefits is available (362). The critics also point out that no controlled observational studies are available to document whether genital surgery prevents the adverse psychosocial consequences of genital ambiguity.
However, surveys of women with CAH showed that most respondents favored genital surgery before adolescence (281, 284, 363–365), as did parents of girls with CAH (281, 285, 366). Moreover, even if the patient has reached the age of consent, obtaining truly informed consent appears unrealistic if the patient is sexually inexperienced. Unfortunately, we lack systematic comparative studies of early vs late (i.e., after attainment of age of consent) genital surgery, or electing to have no surgery at all, in regard to outcomes such as stigma, sexual functioning, and QOL, and we do not yet know whether improvements in surgical techniques in the last decade will yield improvements in outcomes. Parents, therefore, are likely to hear conflicting recommendations (367). In contemplating the pros and cons of early genital surgery, one must also consider that no studies have been conducted to demonstrate that potential adverse psychosocial consequences of gender-incongruent genital appearance can be ameliorated by psychological counseling or psychotherapy. Physicians should inform families of all these concerns and allow them to reach a reasoned decision with input from various sources, including patient and family support groups.
Findings on general QOL in patients with CAH compared with controls vary widely: from better (255, 368) to comparable (369–372) to impaired (202, 284, 373–375). Impaired QOL is more common in adults than in children and is, in some respects, associated with more severe forms of CAH (284, 369, 371) and with increased adiposity, insulin resistance, and use of prednisolone or Dex (376). Inconsistencies in findings are due to a variety of factors such as variations in sample composition, in hormone and surgical treatment regimens, and in assessment tools. DSD-specific tools for the assessment of QOL are yet to be developed. Specific findings on mental health and psychiatric disorders in clinic samples of individuals with CAH are similarly mixed and suffer from comparable methodological problems (342). Epidemiological national registry studies in Sweden have shown that females and males with CAH had an elevated risk of receiving any psychiatric diagnosis [OR, 1.5 (1.1 to 2.2)]. Girls and women with CAH had an increased risk of reaction to extreme stress and adjustment disorders [OR 2.1, (1.3 to 3.6)] and of alcohol abuse [OR, 2.8 (1.7 to 4.7)] compared with those without the disorder, with the highest risk among those with the most severe genotype. For boys and men, there were increased rates of reported suicides and suicide attempts [OR, 2.3 (1.1 to 5.0)] and alcohol abuse [OR, 1.9 (1.0 to 3.5)] (377, 378).
Existing clinical guidelines (379–384) recommend interdisciplinary teams that include mental health professionals with expertise in managing psychosocial problems specific to DSD. Tasks may include (1) parent/family medical education, parent/family counseling regarding psychosocial prognosis, and managing parents’ distress; (2) assisting in gender assignment at birth in cases of marked genital virilization; (3) discussing the pros and cons of gender-confirming (not medically necessitated) genital surgery in infancy and early childhood; and (4) counseling regarding potential gender reassignment of patients with 46,XX CAH after infancy (353). Note that physician-recommended reassignment of inadvertently male-assigned 46,XX patients to female during infancy does not require a psychological gender evaluation, as children’s use of gender labels starts at ∼17 to 24 months, and consistent self-labeling by gender is not attained until ∼3 years of age (385, 386).
Additional DSD-specific items for counseling patients and families include preparation for genital surgery; concerns about inappropriate curiosity or frank stigmatization by other family, peers, lovers, or even medical staff (387) in reaction to gender-atypical somatic features; gender-atypical behavior and related problems with social fit; bisexual and homosexual attractions, which are somewhat increased in women with 46,XX CAH but still limited to a minority (388, 389); sexual functioning; and the impact of the CAH condition and its treatment on QOL. Ideally, mental health staff with expertise in DSD should manage such problems with help from clinical guidelines (342, 379–383, 389–394), educational websites, and long-distance consultations with specialists via the Internet or phone.
Balance of benefits and harms
Because CAH implies multiple emotional stressors and coping challenges for the patients and their families with variable consequences for mental health and QOL, we think that mental health support is a valuable complement to endocrinological and surgical management.
10. Objectives for Future Research
Newborn screening
Determine whether analytes other than 17OHP, either singly or in combination with other biochemical or genetic tests, may improve the sensitivity and specificity of newborn screening programs.
Prenatal treatment
Establish national and international registries of prenatally treated newborns.
Conduct long-term follow-up studies of prenatally treated newborns and control groups through reproductive age.
Diagnosis of CAH
Determine the utility of novel adrenal steroid panels to diagnose CAH.
Treatment of CAH
Determine optimal treatment regimens through prospective trials.
Define the utility of new steroid biomarkers, for example, 21-deoxycortisol, 11-ketotestosterone, and pregnenolone sulfate, in monitoring therapy.
Better delineate situations that require “stress dosing” and the minimal effective GC doses to manage these events.
Determine how GC requirements change throughout pregnancy and delivery.
NCCAH
Rigorously delineate the criteria for diagnosing, treating, and monitoring NCCAH.
Demonstrate the risks and benefits of GC therapy for improving pregnancy outcomes in NCCAH.
Long-term management
Determine the optimal modes of transition of care from pediatric to medical and reproductive endocrinologists.
Conduct long-term studies to assess the risks of cardiovascular disease, tumor formation, infertility, and other comorbidities in adults with CAH.
Develop and implement telemedicine procedures for proper endocrine and psychiatric care of patients and families living in remote areas.
Characterize the long-term implications of genetic findings (e.g., CAH–tenascin-X contiguous deletion) via genetic studies in association with clinical phenotyping.
Surgery
Conduct long-term follow-up studies to assess the outcomes of various surgical approaches compared with delayed surgery or no surgery.
In contrast to other congenital genitourinary abnormalities such as bladder exstrophy, prune belly syndrome, and posterior urethral valves, the incidence of urogenital sinus anomalies associated with CAH has not decreased. Thus, there is a continuing need to derive evidence-based guidelines for surgical treatment of CAH, including ideal timing of surgery, surgical technique, risk of incontinence, risk of additional surgery (such as repair of vaginal stenosis at puberty), risk of loss of sexual function, and extent of clitoral surgery.
Experimental therapies
Develop new treatment approaches that minimize GC exposure.
Further define the clinical implications of epinephrine deficiency.
Mental health
Develop and validate additional tools for evaluating QOL in patients (and their families) to facilitate improved assessment of current and future therapies.
Methodology
Participants
The Writing Committee consisted of 10 content experts representing the following specialties: endocrinology, pediatric urology, and psychology. Two of the committee members brought an international perspective to this guideline topic. The Writing Committee also included a clinical practice guideline methodologist who led the team of comparative effectiveness researchers that conducted the systematic reviews and meta-analyses. The methodologist also supervised application of the GRADE methodological framework for each recommendation, including quality of evidence assessments and strength of recommendation designations.
Guideline development process
The Endocrine Society’s guideline development process combines elements of the GRADE framework (395) with, when relevant, an approach thought to be appropriate for rare endocrine diseases where scientific evidence is limited or nonexistent. The Society applies the steps in the GRADE framework to research questions for which there is an ample body of knowledge of low quality or higher (see Table 6). In these situations, GRADE provides the methodological and statistical rigor that results in robust recommendations that are classified using quality of evidence and strength of recommendation as described in by Guyatt et al. (396) and also represented graphically in Table 6.
Table 6.
GRADE Classification of Guideline Recommendations
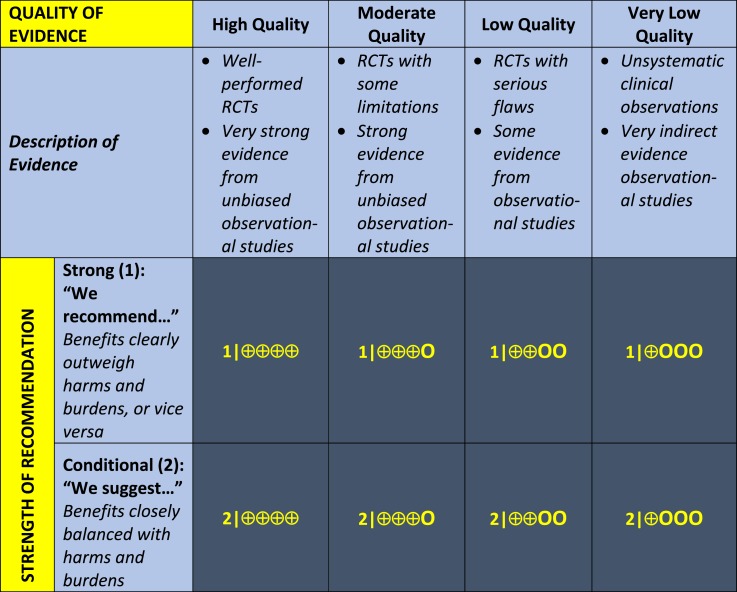
|
Where evidence is extremely limited and/or not systematically analyzed, we provide recommendations based on an expert review of the limited data. This process is less systematic than the GRADE methodological framework; however, these recommendations are also clearly classified using the GRADE classification system.
Some of the Society’s clinical practice guidelines also include ungraded good practice statements (397). This unclassified clinical guidance can include expert opinion statements on good practice, references to recommendations made in other guidelines, and observations on preventive care and shared decision-making.
Guideline recommendations include the relevant population, intervention, comparator, and outcome. When further clarification on implementation is needed, we include technical remarks. These provide supplemental information such as timing, setting, dosing regimens, and necessary expertise. All recommendations are followed by a synopsis of the evidence that underpins it. Authors may also include short statements on patients’ values and preferences, the balance of benefits and harms, and minority opinions, where relevant.
Internal and external review
Approximately 18 months into the development process, Endocrine Society clinical practice guidelines undergo a Comment Review Period (CRP) of 1 month when there is an opportunity for internal and external stakeholders to review the guideline draft and provide comments. These stakeholders include Endocrine Society members, the Society’s Clinical Guidelines Subcommittee (CGS), representatives of any cosponsoring organizations, a representative of Council, and an Expert Reviewer. Following revisions to the guideline manuscript in response to CRP comments, it is returned to CGS, the Council Reviewer, and the Expert Reviewer for a second review and ballot. Finally, the guideline manuscript is subject to JCEM Publisher’s Review prior to publication. This review is undertaken by an individual with expertise in the topic, without relevant conflicts of interest (COIs), and external to the guideline writing committee, CGS, and Council.
COIs
The Endocrine Society’s COI rules for the development of clinical practice guidelines are as follows:
To be considered for membership of a Writing Committee, nominees are required to disclose all relationships with industry for the 12-month period prior to guideline Writing Committee initiation. This is consistent with the reporting time frame for the National Institutes of Health and the Food and Drug Administration.
Potential conflicts of interest that should be declared include all relationships with commercial, noncommercial, institutional, and patient/public organizations that are (or may be) pertinent to the scope of the guideline.
The chair of the Clinical Guidelines Subcommittee reviews all disclosed relationships and determines whether they are relevant to the topic of the guideline and present a potentially relevant conflict of interest.
The chair of the Clinical Guidelines Subcommittee selects Writing Committee Chairs and Co-Chairs, based on COI information, the individuals’ clinical expertise, and other skills. The Endocrine Society Council reviews and endorses the nominees or makes appropriate changes. The three Chairs then select and appoint Writing Committee members.
The Chair and Co-Chair of the Writing Committee must be free of any COI or other biases that could undermine the integrity or credibility of the work.
At least half (≥50%) of the Writing Committee members must be free of relevant COIs.
Following initiation of the Committee, members are asked to disclose relationships with industry at every in-person meeting and on most conference calls.
Writing Committee members with relevant COIs are required to declare the situation and recuse themselves from any relevant discussions, votes, and from drafting recommendations.
All Writing Committee members must refrain from adding new relevant industry relationships throughout the guideline development process.
If a member is aware of another person who might have a conflict and has not declared it for some reason, they are obliged to bring this to the Writing Committee Chair’s attention.
Staff, Writing Committee Chairs, and members must be alert for situations that might present a potential or perceived conflict of interest.
Acknowledgments
Financial Support: This guideline was supported by the Endocrine Society. No other entity provided financial support.
Disclosure Summary: See Appendix B.
Disclaimer: The Endocrine Society’s clinical practice guidelines are developed to be of assistance to endocrinologists by providing guidance and recommendations for particular areas of practice. The guidelines should not be considered inclusive of all proper approaches or methods, or exclusive of others. The guidelines cannot guarantee any specific outcome, nor do they establish a standard of care. The guidelines are not intended to dictate the treatment of a particular patient. Treatment decisions must be made based on the independent judgement of healthcare providers and each patient’s individual circumstances.
The Endocrine Society makes no warranty, express or implied, regarding the guidelines and specifically excludes any warranties of merchantability and fitness for a particular use or purpose. The Society shall not be liable for direct, indirect, special, incidental, or consequential damages related to the use of the information contained herein.
Glossary
Abbreviations:
- 17OHP
17-hydroxyprogesterone
- 21OHD
21-hydroxylase deficiency
- BMI
body mass index
- BMD
bone mineral density
- CAH
congenital adrenal hyperplasia (both classic and nonclassic)
- Dex
dexamethasone
- DSD
disorders of sex development
- GC
glucocorticoid
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- MC
mineralocorticoid
- NCCAH
nonclassic congenital adrenal hyperplasia
- PRA
plasma renin activity
- QOL
quality of life
- SDS
SD score
- TART
testicular adrenal rest tumor
Appendix A
Resources for newborn screening:
Resources for clinical trials on CAH:
Resources for patients and families:
Video demonstration of emergency HC intramuscular injection:
Appendix B.
Appendix B.
Conflict of Interest of CAH Guideline Writing Committee
| Writing Committee Member | Employment | Uncompensated Memberships | Uncompensated Leadership | Personal Financial | Organizational Financial | Spousal/ Family Info. |
|---|---|---|---|---|---|---|
| Phyllis W. Speiser (Chair) | Chief, Division of Pediatrics, and Professor of Pediatric Endocrinology, Cohen Children’s Medical Center of New York, Northwell Health | • American Association of Clinical Endocrinology • Pediatric Endocrine Society | None declared | • Gerson Lehman Group, consultant • Hood Law Firm, medical expert witness | None declared | None declared |
| Wiebke Arlt | Chair of Medicine at the College of Medical and Dental Sciences of the University of Birmingham (UK) and Centre for Endocrinology, Diabetes, and Metabolism | • Society for Endocrinology UK, Chair of Clinical Committee, Member of Council and Nomination Committee • European Network for the Study of Adrenal Tumors, Member of Steering Committee and Adrenocortical Carcinoma Working Group Committee | None declared | • Bayer Health Care Advisory Board • Janssen, Advisory Board • Diurnal Ltd, consultancy and site investigator • Patents on the use of steroid metabolomics for the diagnosis, differential diagnosis, monitoring, and prognostic prediction in steroid-related disorders | None declared | None declared |
| Richard J. Auchus | Professor of Internal Medicine and Pharmacology, University of Michigan | None declared | • American Association of Clinical Endocrinologists, Adrenal Scientific Committee Member and President, Michigan Chapter • Endocrinology Associate Editor • CARES Foundation, Medical Advisory Board | • Novartis Pharmaceuticals, contracted research support and consultant • Strongbridge Biopharma, contracted research support and consultant • Neurocrine Biosciences, contracted research support and consultant • Millendo Therapeutics, contracted research support and consultant • Quest Diagnostics, consultant • Corcept Therapeutics, consultant • Janssen Pharmaceuticals, consultant • Spruce Biosciences, contracted research support and consultant • Diurnal Ltd, consultant • Adrenas Therapeutics, consultant • Selenity Therapeutics, consultant • United States Anti-Doping Agency, consultant | None declared | None declared |
| Laurence Baskin | Professor of Urology and Pediatrics, | None declared | None declared | None declared | None declared | None declared |
| University of California San Francisco | ||||||
| Gerard Conway | University College London Hospitals, London, UK | None declared | None declared | None declared | None declared | None declared |
| Deborah P. Merkea | Senior Investigator, Chief Pediatric Service, | None declared | None declared | Up-to-Date, author | • Diurnal Ltd, Principal Investigator | None declared |
| National Institutes of Health Clinical Center | • Millendo Therapeutics, Principal Investigator | |||||
| Heino Meyer- Bahlberg | Research Scientist, New York State Psychiatric Institute, and Professor of Clinical Psychology (in Psychiatry), Vagelos College of Physicians and Surgeons of Columbia University | • Pediatric Endocrine Society (and 12 other professional nonendocrine societies) • Scientific Advisory Board of dsd-LIFE (research project in six European countries on somatic DSD funded by the European Union) • Consultant, Workgroup on Gender Dysphoria of the American Psychiatric Association • Council on Research and Quality Care member, panel for the 10-year update of the 2005 International Consensus Conference on Intersex Management • Member, Standards of Care Revision (SOC-8) Committee of the World Professional Association for Transgender Health (chapter lead for the intersex-care chapter) • Distinguished Visiting Professor, School of Medicine, Tehran University of Medical Sciences | None declared | None declared | None declared | None declared |
| Walter L. Miller | Retired; Distinguished Professor Emeritus, University of California San Francisco; and Emeritus Chief of Endocrinology, University of California San Francisco Children’s Hospitals | • Pediatric Endocrine Society • Japanese Society for Pediatric Endocrinology (Honorary) • European Society for Pediatric Endocrinology • The CARES Foundation • American Association for the Advancement of Science • American Society for Biochemistry and Molecular Biology | • Chair, History Committee, Pediatric Endocrine Society • Medical Advisory Board, CARES Foundation • Associate Editor for Hormone Research in Paediatrics | • Spruce Biosciences, consultant • Adrenas Therapeutics, consultant • Share of royalties on patents held by the University of California concerning bovine GH | None declared | None declared |
| M. Hassan Murad | Professor of Medicine and | None declared | None declared | None declared | None declared | None declared |
| Director of Evidence-Based Practice Center | ||||||
| Mayo Clinic | ||||||
| Sharon E. Oberfield | Professor of Pediactrics; Director, Division of Pediatric Endocrinology, Diabetes, and Metabolism, | Member of Board of Directors of Androgen Excess and Polycystic Ovary Syndrome Society | • Associate Editor for Hormone Research in Paediatrics | None declared | National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases-B study section | None declared |
| Columbia University Medical Center | ||||||
| Perrin White | Professor of Pediatrics, University of Texas Southwestern Medical Center | None declared | None declared | None declared | • National Institutes of Health, grantee | None declared |
| • Janssen Pharmaceutical, receive study drug gratis |
This work was supported by the Intramural Research Program of the National Institutes of Health.
Footnotes
Cosponsoring Associations: CARES Foundation, European Society of Endocrinology, European Society for Paediatric Endocrinology, Societies for Pediatric Urology, and Pediatric Endocrine Society.
References
- 1. Speiser PW, Azziz R, Baskin LS, Ghizzoni L, Hensle TW, Merke DP, Meyer-Bahlburg HFL, Miller WL, Montori VM, Oberfield SE, Ritzen M, White PC. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2010;95(9):4133–4160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gruñieiro-Papendieck L, Chiesa A, Mendez V, Prieto L. Neonatal screening for congenital adrenal hyperplasia: experience and results in Argentina. J Pediatr Endocrinol Metab. 2008;21(1):73–78. [DOI] [PubMed] [Google Scholar]
- 3. Shetty VB, Bower C, Jones TW, Lewis BD, Davis EA. Ethnic and gender differences in rates of congenital adrenal hyperplasia in Western Australia over a 21 year period. J Paediatr Child Health. 2012;48(11):1029–1032. [DOI] [PubMed] [Google Scholar]
- 4. Gleeson HK, Wiley V, Wilcken B, Elliott E, Cowell C, Thonsett M, Byrne G, Ambler G. Two-year pilot study of newborn screening for congenital adrenal hyperplasia in New South Wales compared with nationwide case surveillance in Australia. J Paediatr Child Health. 2008;44(10):554–559. [DOI] [PubMed] [Google Scholar]
- 5. Nascimento ML, Cristiano AN, Campos T, Ohira M, Cechinel E, Simoni G, Lee J, Linhares RM, Silva PC. Ten-year evaluation of a neonatal screening program for congenital adrenal hyperplasia. Arq Bras Endocrinol Metabol. 2014;58(7):765–771. [DOI] [PubMed] [Google Scholar]
- 6. Silveira EL, Elnecave RH, dos Santos EP, Moura V, Pinto EM, van der Linden Nader I, Mendonca BB, Bachega TA. Molecular analysis of CYP21A2 can optimize the follow-up of positive results in newborn screening for congenital adrenal hyperplasia. Clin Genet. 2009;76(6):503–510. [DOI] [PubMed] [Google Scholar]
- 7. Pezzuti IL, Barra CB, Mantovani RM, Januário JN, Silva IN. A three-year follow-up of congenital adrenal hyperplasia newborn screening. J Pediatr (Rio J). 2014;90(3):300–307. [DOI] [PubMed] [Google Scholar]
- 8. Kopacek C, de Castro SM, Prado MJ, da Silva CM, Beltrão LA, Spritzer PM. Neonatal screening for congenital adrenal hyperplasia in Southern Brazil: a population based study with 108,409 infants. BMC Pediatr. 2017;17(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhong K, Wang W, He F, Wang Z. The status of neonatal screening in China, 2013. J Med Screen. 2016;23(2):59–61. [DOI] [PubMed] [Google Scholar]
- 10. Dumic K, Krnic N, Skrabic V, Stipancic G, Cvijovic K, Kusec V, Stingl K. Classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency in Croatia between 1995 and 2006. Horm Res. 2009;72(5):310–314. [DOI] [PubMed] [Google Scholar]
- 11. González EC, Carvajal F, Frómeta A, Arteaga AL, Castells EM, Espinosa T, Coto R, Pérez PL, Tejeda Y, Del Río L, Segura MT, Almenares P, Robaina R, Fernández JL. Newborn screening for congenital adrenal hyperplasia in Cuba: six years of experience. Clin Chim Acta. 2013;421:73–78. [DOI] [PubMed] [Google Scholar]
- 12. Votava F, Novotna D, Kracmar P, Vinohradska H, Stahlova-Hrabincova E, Vrzalova Z, Neumann D, Malikova J, Lebl J, Matern D. Lessons learned from 5 years of newborn screening for congenital adrenal hyperplasia in the Czech Republic: 17-hydroxyprogesterone, genotypes, and screening performance. Eur J Pediatr. 2012;171(6):935–940. [DOI] [PubMed] [Google Scholar]
- 13. Coulm B, Coste J, Tardy V, Ecosse E, Roussey M, Morel Y, Carel JC; DHCSF Study Group . Efficiency of neonatal screening for congenital adrenal hyperplasia due to 21-hydroxylase deficiency in children born in mainland France between 1996 and 2003. Arch Pediatr Adolesc Med. 2012;166(2):113–120. [DOI] [PubMed] [Google Scholar]
- 14. Odenwald B, Dörr HG, Bonfig W, Schmidt H, Fingerhut R, Wildner M, Nennstiel-Ratzel U. Classic congenital adrenal hyperplasia due to 21-hydroxylase-deficiency: 13 years of neonatal screening and follow-up in Bavaria. Klin Padiatr. 2015;227(5):278–283. [DOI] [PubMed] [Google Scholar]
- 15. Kaur G, Thakur K, Kataria S, Singh TR, Chavan BS, Kaur G, Atwal R. Current and future perspective of newborn screening: an Indian scenario. J Pediatr Endocrinol Metab. 2016;29(1):5–13. [DOI] [PubMed] [Google Scholar]
- 16. Morikawa S, Nakamura A, Fujikura K, Fukushi M, Hotsubo T, Miyata J, Ishizu K, Tajima T. Results from 28 years of newborn screening for congenital adrenal hyperplasia in Sapporo. Clin Pediatr Endocrinol. 2014;23(2):35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsuji A, Konishi K, Hasegawa S, Anazawa A, Onishi T, Ono M, Morio T, Kitagawa T, Kashimada K. Newborn screening for congenital adrenal hyperplasia in Tokyo, Japan from 1989 to 2013: a retrospective population-based study. BMC Pediatr. 2015;15(1):209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Heather NL, Seneviratne SN, Webster D, Derraik JG, Jefferies C, Carll J, Jiang Y, Cutfield WS, Hofman PL. Newborn screening for congenital adrenal hyperplasia in New Zealand, 1994–2013. J Clin Endocrinol Metab. 2015;100(3):1002–1008. [DOI] [PubMed] [Google Scholar]
- 19. Gidlöf S, Wedell A, Guthenberg C, von Döbeln U, Nordenström A. Nationwide neonatal screening for congenital adrenal hyperplasia in Sweden: a 26-year longitudinal prospective population-based study. JAMA Pediatr. 2014;168(6):567–574. [DOI] [PubMed] [Google Scholar]
- 20. Khalid JM, Oerton JM, Dezateux C, Hindmarsh PC, Kelnar CJ, Knowles RL. Incidence and clinical features of congenital adrenal hyperplasia in Great Britain. Arch Dis Child. 2012;97(2):101–106. [DOI] [PubMed] [Google Scholar]
- 21. Al Hosani H, Salah M, Osman HM, Farag HM, El-Assiouty L, Saade D, Hertecant J. Expanding the comprehensive national neonatal screening programme in the United Arab Emirates from 1995 to 2011. East Mediterr Health J. 2014;20(1):17–23. [PubMed] [Google Scholar]
- 22. Larrandaburu M, Matte U, Noble A, Olivera Z, Sanseverino MT, Nacul L, Schuler-Faccini L. Ethics, genetics and public policies in Uruguay: newborn and infant screening as a paradigm. J Community Genet. 2015;6(3):241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van der Kamp HJ, Wit JM. Neonatal screening for congenital adrenal hyperplasia. Eur J Endocrinol. 2004;151(Suppl 3):U71–U75. [DOI] [PubMed] [Google Scholar]
- 24. White PC, New MI, Dupont B. HLA-linked congenital adrenal hyperplasia results from a defective gene encoding a cytochrome P-450 specific for steroid 21-hydroxylation. Proc Natl Acad Sci USA. 1984;81(23):7505–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krone N, Dhir V, Ivison HE, Arlt W. Congenital adrenal hyperplasia and P450 oxidoreductase deficiency. Clin Endocrinol (Oxf). 2007;66(2):162–172. [DOI] [PubMed] [Google Scholar]
- 26. Kamrath C, Hochberg Z, Hartmann MF, Remer T, Wudy SA. Increased activation of the alternative “backdoor” pathway in patients with 21-hydroxylase deficiency: evidence from urinary steroid hormone analysis. J Clin Endocrinol Metab. 2012;97(3):E367–E375. [DOI] [PubMed] [Google Scholar]
- 27. White PC, Speiser PW. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr Rev. 2000;21(3):245–291. [DOI] [PubMed] [Google Scholar]
- 28.National Newborn Screening and Global Resource Center. Newborn Screening Reports and Publications. Incidence reports: 2006. Available at: genes-r-us.uthscsa.edu/newborn_reports. Accessed 28 August 2017.
- 29. Kohn B, Levine LS, Pollack MS, Pang S, Lorenzen F, Levy D, Lerner AJ, Rondanini GF, Dupont B, New MI. Late-onset steroid 21-hydroxylase deficiency: a variant of classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1982;55(5):817–827. [DOI] [PubMed] [Google Scholar]
- 30. Speiser PW, Dupont BO, Rubinstein P, Piazza A, Kastelan A, New MI. High frequency of nonclassical steroid 21-hydroxylase deficiency. Obstet Gynecol Surv. 1986;41(4):244–245. [PMC free article] [PubMed] [Google Scholar]
- 31. Hannah-Shmouni F, Morissette R, Sinaii N, Elman M, Prezant TR, Chen W, Pulver A, Merke DP. Revisiting the prevalence of nonclassic congenital adrenal hyperplasia in US Ashkenazi Jews and Caucasians. Genet Med. 2017;19(11):1276–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang Z, Mendoza AR, Welch TR, Zipf WB, Yu CY. Modular variations of the human major histocompatibility complex class III genes for serine/threonine kinase RP, complement component C4, steroid 21-hydroxylase CYP21, and tenascin TNX (the RCCX module). A mechanism for gene deletions and disease associations. J Biol Chem. 1999;274(17):12147–12156. [DOI] [PubMed] [Google Scholar]
- 33. The Human Gene Mutation Database. Available at: www.hgmd.cf.ac.uk/ac/index.php. Accessed July 27, 2018.
- 34. Krone N, Braun A, Roscher AA, Knorr D, Schwarz HP. Predicting phenotype in steroid 21-hydroxylase deficiency? Comprehensive genotyping in 155 unrelated, well defined patients from southern Germany. J Clin Endocrinol Metab. 2000;85(3):1059–1065. [DOI] [PubMed] [Google Scholar]
- 35. Speiser PW, Dupont J, Zhu D, Serrat J, Buegeleisen M, Tusie-Luna MT, Lesser M, New MI, White PC. Disease expression and molecular genotype in congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Invest. 1992;90(2):584–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Simonetti L, Bruque CD, Fernández CS, Benavides-Mori B, Delea M, Kolomenski JE, Espeche LD, Buzzalino ND, Nadra AD, Dain L. CYP21A2 mutation update: Comprehensive analysis of databases and published genetic variants. Hum Mutat. 2018;39(1):5–22. [DOI] [PubMed] [Google Scholar]
- 37. Miller WL, Merke DP. Tenascin-X, congenital adrenal hyperplasia and the CAH-X syndrome. Horm Res Paediatr. 2018;89(5):352–361. [DOI] [PMC free article] [PubMed]
- 38. Tusie-Luna MT, Traktman P, White PC. Determination of functional effects of mutations in the steroid 21-hydroxylase gene (CYP21) using recombinant vaccinia virus. J Biol Chem. 1990;265(34):20916–20922. [PubMed] [Google Scholar]
- 39. Blanché H, Vexiau P, Clauin S, Le Gall I, Fiet J, Mornet E, Dausset J, Bellanné-Chantelot C. Exhaustive screening of the 21-hydroxylase gene in a population of hyperandrogenic women. Hum Genet. 1997;101(1):56–60. [DOI] [PubMed] [Google Scholar]
- 40. Deneux C, Tardy V, Dib A, Mornet E, Billaud L, Charron D, Morel Y, Kuttenn F. Phenotype-genotype correlation in 56 women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2001;86(1):207–213. [DOI] [PubMed] [Google Scholar]
- 41.Tamhane SU, Rodriguez-Gutierrez R, Iqbal AM, Prokop L, Bancos I, Speiser PW, Murad MH. Cardiovascular and metabolic outcomes in congenital adrenal hyperplasia: a systematic review and meta-analysis. J Clin Endocrinol Metab 2018;103(11):4097–4103. [DOI] [PubMed]
- 42.Almasri J, Zaiem F, Rodriguez-Gutierrez R, Tamhane SU, Iqbal AM, Prokop LJ, Speiser PW, Baskin LS, Bancos I, Murad MH. Genital reconstructive surgery in females with congenital adrenal hyperplasia: a systematic review and meta-analysis. J Clin Endocrinol Metab 2018;103(11):4089–4096. [DOI] [PubMed]
- 43. Pang S, Shook MK. Current status of neonatal screening for congenital adrenal hyperplasia. Curr Opin Pediatr. 1997;9(4):419–423. [DOI] [PubMed] [Google Scholar]
- 44. Therrell BL. Newborn screening for congenital adrenal hyperplasia. Endocrinol Metab Clin North Am. 2001;30(1):15–30. [DOI] [PubMed] [Google Scholar]
- 45. Balsamo A, Cacciari E, Piazzi S, Cassio A, Bozza D, Pirazzoli P, Zappulla F. Congenital adrenal hyperplasia: neonatal mass screening compared with clinical diagnosis only in the Emilia-Romagna region of Italy, 1980–1995. Pediatrics. 1996;98(3 Pt 1):362–367. [PubMed] [Google Scholar]
- 46. Brosnan PG, Brosnan CA, Kemp SF, Domek DB, Jelley DH, Blackett PR, Riley WJ. Effect of newborn screening for congenital adrenal hyperplasia. Arch Pediatr Adolesc Med. 1999;153(12):1272–1278. [DOI] [PubMed] [Google Scholar]
- 47. Therrell BL Jr, Berenbaum SA, Manter-Kapanke V, Simmank J, Korman K, Prentice L, Gonzalez J, Gunn S. Results of screening 1.9 million Texas newborns for 21-hydroxylase-deficient congenital adrenal hyperplasia. Pediatrics. 1998;101(4 Pt 1):583–590. [DOI] [PubMed] [Google Scholar]
- 48. Thilén A, Nordenström A, Hagenfeldt L, von Döbeln U, Guthenberg C, Larsson A. Benefits of neonatal screening for congenital adrenal hyperplasia (21-hydroxylase deficiency) in Sweden. Pediatrics. 1998;101(4):E11. [DOI] [PubMed] [Google Scholar]
- 49. Strnadová KA, Votava F, Lebl J, Mühl A, Item C, Bodamer OA, Torresani T, Bouška I, Waldhauser F, Sperl W. Prevalence of congenital adrenal hyperplasia among sudden infant death in the Czech Republic and Austria. Eur J Pediatr. 2007;166(1):1–4. [DOI] [PubMed] [Google Scholar]
- 50. Hird BE, Tetlow L, Tobi S, Patel L, Clayton PE. No evidence of an increase in early infant mortality from congenital adrenal hyperplasia in the absence of screening. Arch Dis Child. 2014;99(2):158–164. [DOI] [PubMed] [Google Scholar]
- 51. Lebovitz RM, Pauli RM, Laxova R. Delayed diagnosis in congenital adrenal hyperplasia. Need for newborn screening. Am J Dis Child. 1984;138(6):571–573. [DOI] [PubMed] [Google Scholar]
- 52. Nordenström A, Ahmed S, Jones J, Coleman M, Price DA, Clayton PE, Hall CM. Female preponderance in congenital adrenal hyperplasia due to CYP21 deficiency in England: implications for neonatal screening. Horm Res. 2005;63(1):22–28. [DOI] [PubMed] [Google Scholar]
- 53. Thompson R, Seargeant L, Winter JS. Screening for congenital adrenal hyperplasia: distribution of 17α-hydroxyprogesterone concentrations in neonatal blood spot specimens. J Pediatr. 1989;114(3):400–404. [DOI] [PubMed] [Google Scholar]
- 54. Gidlöf S, Falhammar H, Thilén A, von Döbeln U, Ritzén M, Wedell A, Nordenström A. One hundred years of congenital adrenal hyperplasia in Sweden: a retrospective, population-based cohort study. Lancet Diabetes Endocrinol. 2013;1(1):35–42. [DOI] [PubMed] [Google Scholar]
- 55. Watson MS, Mann MY, Lloyd-Puryear MA, Rinaldo P, Howell RR, eds. Newborn screening: toward a uniform screening panel and system [Main report]. Genet Med. 2006;8(Suppl 1):12S–252S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Grosse SD, Van Vliet G. How many deaths can be prevented by newborn screening for congenital adrenal hyperplasia? Horm Res. 2007;67(6):284–291. [DOI] [PubMed] [Google Scholar]
- 57. Van der Kamp HJ, Noordam K, Elvers B, Van Baarle M, Otten BJ, Verkerk PH. Newborn screening for congenital adrenal hyperplasia in the Netherlands. Pediatrics. 2001;108(6):1320–1324. [DOI] [PubMed] [Google Scholar]
- 58. Nass R, Baker S. Learning disabilities in children with congenital adrenal hyperplasia. J Child Neurol. 1991;6(4):306–312. [DOI] [PubMed] [Google Scholar]
- 59. Carroll AE, Downs SM. Comprehensive cost-utility analysis of newborn screening strategies. Pediatrics. 2006;117(5 Pt 2):S287–S295. [DOI] [PubMed] [Google Scholar]
- 60. Yoo BK, Grosse SD. The cost effectiveness of screening newborns for congenital adrenal hyperplasia. Public Health Genomics. 2009;12(2):67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chan CL, McFann K, Taylor L, Wright D, Zeitler PS, Barker JM. Congenital adrenal hyperplasia and the second newborn screen. J Pediatr. 2013;163(1):109–113.e1. [DOI] [PubMed] [Google Scholar]
- 62. Gonzalez RR, Mäentausta O, Solyom J, Vihko R. Direct solid-phase time-resolved fluoroimmunoassay of 17α-hydroxyprogesterone in serum and dried blood spots on filter paper. Clin Chem. 1990;36(9):1667–1672. [PubMed] [Google Scholar]
- 63. Pang S, Hotchkiss J, Drash AL, Levine LS, New MI. Microfilter paper method for 17α-hydroxyprogesterone radioimmunoassay: its application for rapid screening for congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1977;45(5):1003–1008. [DOI] [PubMed] [Google Scholar]
- 64. Varness TS, Allen DB, Hoffman GL. Newborn screening for congenital adrenal hyperplasia has reduced sensitivity in girls. J Pediatr. 2005;147(4):493–498. [DOI] [PubMed] [Google Scholar]
- 65. Allen DB, Hoffman GL, Fitzpatrick P, Laessig R, Maby S, Slyper A. Improved precision of newborn screening for congenital adrenal hyperplasia using weight-adjusted criteria for 17-hydroxyprogesterone levels. J Pediatr. 1997;130(1):128–133. [DOI] [PubMed] [Google Scholar]
- 66. Olgemöller B, Roscher AA, Liebl B, Fingerhut R. Screening for congenital adrenal hyperplasia: adjustment of 17-hydroxyprogesterone cut-off values to both age and birth weight markedly improves the predictive value. J Clin Endocrinol Metab. 2003;88(12):5790–5794. [DOI] [PubMed] [Google Scholar]
- 67. Sarafoglou K, Gaviglio A, Hietala A, Frogner G, Banks K, McCann M, Thomas W. Comparison of newborn screening protocols for congenital adrenal hyperplasia in preterm infants. J Pediatr. 2014;164(5):1136–1140. [DOI] [PubMed] [Google Scholar]
- 68. Sarafoglou K, Banks K, Gaviglio A, Hietala A, McCann M, Thomas W. Comparison of one-tier and two-tier newborn screening metrics for congenital adrenal hyperplasia. Pediatrics. 2012;130(5):e1261–e1268. [DOI] [PubMed] [Google Scholar]
- 69. Hayashi GY, Carvalho DF, de Miranda MC, Faure C, Vallejos C, Brito VN, Rodrigues AS, Madureira G, Mendonca BB, Bachega TA. Neonatal 17-hydroxyprogesterone levels adjusted according to age at sample collection and birthweight improve the efficacy of congenital adrenal hyperplasia newborn screening. Clin Endocrinol (Oxf). 2017;86(4):480–487. [DOI] [PubMed] [Google Scholar]
- 70. Held PK, Shapira SK, Hinton CF, Jones E, Hannon WH, Ojodu J. Congenital adrenal hyperplasia cases identified by newborn screening in one- and two-screen states. Mol Genet Metab. 2015;116(3):133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. van der Kamp HJ, Oudshoorn CGM, Elvers BH, van Baarle M, Otten BJ, Wit JM, Verkerk PH. Cutoff levels of 17-α-hydroxyprogesterone in neonatal screening for congenital adrenal hyperplasia should be based on gestational age rather than on birth weight. J Clin Endocrinol Metab. 2005;90(7):3904–3907. [DOI] [PubMed] [Google Scholar]
- 72. Nomura S. Immature adrenal steroidogenesis in preterm infants. Early Hum Dev. 1997;49(3):225–233. [DOI] [PubMed] [Google Scholar]
- 73. Wong T, Shackleton CHL, Covey TR, Ellis G. Identification of the steroids in neonatal plasma that interfere with 17 α-hydroxyprogesterone radioimmunoassays. Clin Chem. 1992;38(9):1830–1837. [PubMed] [Google Scholar]
- 74. al Saedi S, Dean H, Dent W, Cronin C. Reference ranges for serum cortisol and 17-hydroxyprogesterone levels in preterm infants. J Pediatr. 1995;126(6):985–987. [DOI] [PubMed] [Google Scholar]
- 75. Gatelais F, Berthelot J, Beringue F, Descamps P, Bonneau D, Limal J-M, Coutant R. Effect of single and multiple courses of prenatal corticosteroids on 17-hydroxyprogesterone levels: implication for neonatal screening of congenital adrenal hyperplasia. Pediatr Res. 2004;56(5):701–705. [DOI] [PubMed] [Google Scholar]
- 76. King JL, Naber JM, Hopkin RJ, Repaske DR, Bailey L, Leslie ND. Antenatal corticosteroids and newborn screening for congenital adrenal hyperplasia. Arch Pediatr Adolesc Med. 2001;155(9):1038–1042. [DOI] [PubMed] [Google Scholar]
- 77. White PC. Neonatal screening for congenital adrenal hyperplasia. Nat Rev Endocrinol. 2009;5(9):490–498. [DOI] [PubMed] [Google Scholar]
- 78. Lacey JM, Minutti CZ, Magera MJ, Tauscher AL, Casetta B, McCann M, Lymp J, Hahn SH, Rinaldo P, Matern D. Improved specificity of newborn screening for congenital adrenal hyperplasia by second-tier steroid profiling using tandem mass spectrometry. Clin Chem. 2004;50(3):621–625. [DOI] [PubMed] [Google Scholar]
- 79. Rauh M, Gröschl M, Rascher W, Dörr HG. Automated, fast and sensitive quantification of 17α-hydroxy-progesterone, androstenedione and testosterone by tandem mass spectrometry with on-line extraction. Steroids. 2006;71(6):450–458. [DOI] [PubMed] [Google Scholar]
- 80. Janzen N, Peter M, Sander S, Steuerwald U, Terhardt M, Holtkamp U, Sander J. Newborn screening for congenital adrenal hyperplasia: additional steroid profile using liquid chromatography-tandem mass spectrometry. J Clin Endocrinol Metab. 2007;92(7):2581–2589. [DOI] [PubMed] [Google Scholar]
- 81. Minutti CZ, Lacey JM, Magera MJ, Hahn SH, McCann M, Schulze A, Cheillan D, Dorche C, Chace DH, Lymp JF, Zimmerman D, Rinaldo P, Matern D. Steroid profiling by tandem mass spectrometry improves the positive predictive value of newborn screening for congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2004;89(8):3687–3693. [DOI] [PubMed] [Google Scholar]
- 82. Matern D, Tortorelli S, Oglesbee D, Gavrilov D, Rinaldo P. Reduction of the false-positive rate in newborn screening by implementation of MS/MS-based second-tier tests: the Mayo Clinic experience (2004–2007). J Inherit Metab Dis. 2007;30(4):585–592. [DOI] [PubMed] [Google Scholar]
- 83. Schwarz E, Liu A, Randall H, Haslip C, Keune F, Murray M, Longo N, Pasquali M. Use of steroid profiling by UPLC-MS/MS as a second tier test in newborn screening for congenital adrenal hyperplasia: the Utah experience. Pediatr Res. 2009;66(2):230–235. [DOI] [PubMed] [Google Scholar]
- 84. Seo JY, Park H-D, Kim JW, Oh HJ, Yang JS, Chang YS, Park WS, Lee S-Y. Steroid profiling for congenital adrenal hyperplasia by tandem mass spectrometry as a second-tier test reduces follow-up burdens in a tertiary care hospital: a retrospective and prospective evaluation. J Perinat Med. 2014;42(1):121–127. [DOI] [PubMed] [Google Scholar]
- 85. De Jesús VR, Simms DA, Schiffer J, Kennedy M, Mei JV, Hannon WH. Pilot proficiency testing study for second tier congenital adrenal hyperplasia newborn screening. Clin Chim Acta. 2010;411(21–22):1684–1687. [DOI] [PubMed] [Google Scholar]
- 86. Kamrath C, Hartmann MF, Boettcher C, Zimmer K-P, Wudy SA. Diagnosis of 21-hydroxylase deficiency by urinary metabolite ratios using gas chromatography–mass spectrometry analysis: reference values for neonates and infants. J Steroid Biochem Mol Biol. 2016;156:10–16. [DOI] [PubMed] [Google Scholar]
- 87. Yang YP, Corley N, Garcia-Heras J. Reverse dot-blot hybridization as an improved tool for the molecular diagnosis of point mutations in congenital adrenal hyperplasia caused by 21-hydroxylase deficiency. Mol Diagn. 2001;6(3):193–199. [DOI] [PubMed] [Google Scholar]
- 88. Fitness J, Dixit N, Webster D, Torresani T, Pergolizzi R, Speiser PW, Day DJ. Genotyping of CYP21, linked chromosome 6p markers, and a sex-specific gene in neonatal screening for congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1999;84(3):960–966. [DOI] [PubMed] [Google Scholar]
- 89. Sørensen KM, Andersen PS, Larsen LA, Schwartz M, Schouten JP, Nygren AO. Multiplex ligation-dependent probe amplification technique for copy number analysis on small amounts of DNA material. Anal Chem. 2008;80(23):9363–9368. [DOI] [PubMed] [Google Scholar]
- 90. Kösel S, Burggraf S, Fingerhut R, Dörr HG, Roscher AA, Olgemöller B. Rapid second-tier molecular genetic analysis for congenital adrenal hyperplasia attributable to steroid 21-hydroxylase deficiency. Clin Chem. 2005;51(2):298–304. [DOI] [PubMed] [Google Scholar]
- 91. Olney RC, Mougey EB, Wang J, Shulman DI, Sylvester JE. Using real-time, quantitative PCR for rapid genotyping of the steroid 21-hydroxylase gene in a north Florida population. J Clin Endocrinol Metab. 2002;87(2):735–741. [DOI] [PubMed] [Google Scholar]
- 92. Nordenström A, Thilén A, Hagenfeldt L, Larsson A, Wedell A. Genotyping is a valuable diagnostic complement to neonatal screening for congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1999;84(5):1505–1509. [DOI] [PubMed] [Google Scholar]
- 93. Riepe FG, Krone N, Viemann M, Partsch C-J, Sippell WG. Management of congenital adrenal hyperplasia: results of the ESPE questionnaire. Horm Res. 2002;58(4):196–205. [DOI] [PubMed] [Google Scholar]
- 94. Németh S, Riedl S, Kriegshäuser G, Baumgartner-Parzer S, Concolino P, Neocleous V, Phylactou LA, Borucka-Mankiewicz M, Onay H, Tukun A, Oberkanins C. Reverse-hybridization assay for rapid detection of common CYP21A2 mutations in dried blood spots from newborns with elevated 17-OH progesterone. Clin Chim Acta. 2012;414:211–214. [DOI] [PubMed] [Google Scholar]
- 95. Meyer-Bahlburg HFL, Reyes-Portillo JA, Khuri J, Ehrhardt AA, New MI. Syndrome-related stigma in the general social environment as reported by women with classical congenital adrenal hyperplasia. Arch Sex Behav. 2017;46(2):341–351. [DOI] [PubMed] [Google Scholar]
- 96. Hill M, Finning K, Martin P, Hogg J, Meaney C, Norbury G, Daniels G, Chitty LS. Non-invasive prenatal determination of fetal sex: translating research into clinical practice. Clin Genet. 2011;80(1):68–75. [DOI] [PubMed] [Google Scholar]
- 97. Tardy-Guidollet V, Menassa R, Costa J-M, David M, Bouvattier-Morel C, Baumann C, Houang M, Lorenzini F, Philip N, Odent S, Guichet A, Morel Y. New management strategy of pregnancies at risk of congenital adrenal hyperplasia using fetal sex determination in maternal serum: French cohort of 258 cases (2002–2011). J Clin Endocrinol Metab. 2014;99(4):1180–1188. [DOI] [PubMed] [Google Scholar]
- 98. Eunice M, Ammini AC. Prenatal treatment of mothers with fetuses at risk for congenital adrenal hyperplasia: how relevant is it to Indian context? Indian J Endocrinol Metab. 2013;17(3):373–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. New MI, Tong YK, Yuen T, Jiang P, Pina C, Chan KC, Khattab A, Liao GJ, Yau M, Kim SM, Chiu RW, Sun L, Zaidi M, Lo YM. Noninvasive prenatal diagnosis of congenital adrenal hyperplasia using cell-free fetal DNA in maternal plasma. J Clin Endocrinol Metab. 2014;99(6):E1022–E1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Goto M, Piper Hanley K, Marcos J, Wood PJ, Wright S, Postle AD, Cameron IT, Mason JI, Wilson DI, Hanley NA. In humans, early cortisol biosynthesis provides a mechanism to safeguard female sexual development. J Clin Invest. 2006;116(4):953–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Kari MA, Raivio KO, Stenman UH, Voutilainen R. Serum cortisol, dehydroepiandrosterone sulfate, and steroid-binding globulins in preterm neonates: effect of gestational age and dexamethasone therapy. Pediatr Res. 1996;40(2):319–324. [DOI] [PubMed] [Google Scholar]
- 102. Miller WL, Witchel SF. Prenatal treatment of congenital adrenal hyperplasia: risks outweigh benefits. Am J Obstet Gynecol. 2013;208(5):354–359. [DOI] [PubMed] [Google Scholar]
- 103. Mercè Fernández-Balsells M, Muthusamy K, Smushkin G, Lampropulos JF, Elamin MB, Abu Elnour NO, Elamin KB, Agrwal N, Gallegos-Orozco JF, Lane MA, Erwin PJ, Montori VM, Murad MH. Prenatal dexamethasone use for the prevention of virilization in pregnancies at risk for classical congenital adrenal hyperplasia because of 21-hydroxylase (CYP21A2) deficiency: a systematic review and meta-analyses. Clin Endocrinol (Oxf). 2010;73(4):436–444. [DOI] [PubMed] [Google Scholar]
- 104. New MI, Abraham M, Yuen T, Lekarev O. An update on prenatal diagnosis and treatment of congenital adrenal hyperplasia. Semin Reprod Med. 2012;30(5):396–399. [DOI] [PubMed] [Google Scholar]
- 105. Gorduza D, Tardy-Guidollet V, Robert E, Gay C-L, Chatelain P, David M, Bretones P, Lienhardt-Roussie A, Brac de la Perriere A, Morel Y, Mouriquand P. Late prenatal dexamethasone and phenotype variations in 46,XX CAH: concerns about current protocols and benefits for surgical procedures. J Pediatr Urol. 2014;10(5):941–947. [DOI] [PubMed] [Google Scholar]
- 106.Food and Drug Administration, Health and Human Services. Content and format of labeling for human prescription drug and biological products; requirements for pregnancy and lactation labeling. Fed Regist 2014;79(233):72063–72103. [PubMed]
- 107. Carmichael SL, Shaw GM, Ma C, Werler MM, Rasmussen SA, Lammer EJ; National Birth Defects Prevention Study. Maternal corticosteroid use and orofacial clefts. Am J Obstet Gynecol. 2007;197:585(6).e1–7. [DOI] [PubMed] [Google Scholar]
- 108. Rijk Y, van Alfen-van der Velden J, Claahsen-van der Grinten HL. Prenatal Treatment with dexamethasone in suspected congenital adrenal hyperplasia and orofacial cleft: a case report and review of the literature. Pediatr Endocrinol Rev. 2017;15(1):21–25. [DOI] [PubMed] [Google Scholar]
- 109. Grunt S, Steinlin M, Weisstanner C, Schöning M, Mullis PE, Flück CE. Acute encephalopathy with unilateral cortical-subcortical lesions in two unrelated kindreds treated with glucocorticoids prenatally for congenital adrenal hyperplasia due to 21-hydroxylase deficiency: established facts and novel insight. Horm Res Paediatr. 2013;80(1):57–63. [DOI] [PubMed] [Google Scholar]
- 110. Drake AJ, Raubenheimer PJ, Kerrigan D, McInnes KJ, Seckl JR, Walker BR. Prenatal dexamethasone programs expression of genes in liver and adipose tissue and increased hepatic lipid accumulation but not obesity on a high-fat diet. Endocrinology. 2010;151(4):1581–1587. [DOI] [PubMed] [Google Scholar]
- 111. Manojlović-Stojanoski MN, Filipović BR, Nestorović NM, Šošić-Jurjević BT, Ristić NM, Trifunović SL, Milošević VL. Morpho-functional characteristics of rat fetal thyroid gland are affected by prenatal dexamethasone exposure. Steroids. 2014;84:22–29. [DOI] [PubMed] [Google Scholar]
- 112. Poulain M, Frydman N, Duquenne C, N’Tumba-Byn T, Benachi A, Habert R, Rouiller-Fabre V, Livera G. Dexamethasone induces germ cell apoptosis in the human fetal ovary. J Clin Endocrinol Metab. 2012;97(10):E1890–E1897. [DOI] [PubMed] [Google Scholar]
- 113. Wapner RJ, Sorokin Y, Mele L, Johnson F, Dudley DJ, Spong CY, Peaceman AM, Leveno KJ, Malone F, Caritis SN, Mercer B, Harper M, Rouse DJ, Thorp JM, Ramin S, Carpenter MW, Gabbe SG; National Institute of Child Health and Human Development Maternal-Fetal Medicine Units Network . Long-term outcomes after repeat doses of antenatal corticosteroids. N Engl J Med. 2007;357(12):1190–1198. [DOI] [PubMed] [Google Scholar]
- 114. Murphy KE, Hannah ME, Willan AR, Hewson SA, Ohlsson A, Kelly EN, Matthews SG, Saigal S, Asztalos E, Ross S, Delisle M-F, Amankwah K, Guselle P, Gafni A, Lee SK, Armson BA; MACS Collaborative Group . Multiple courses of antenatal corticosteroids for preterm birth (MACS): a randomised controlled trial. Lancet. 2008;372(9656):2143–2151. [DOI] [PubMed] [Google Scholar]
- 115. Davis EP, Waffarn F, Uy C, Hobel CJ, Glynn LM, Sandman CA. Effect of prenatal glucocorticoid treatment on size at birth among infants born at term gestation. J Perinatol. 2009;29(11):731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Braun T, Challis JR, Newnham JP, Sloboda DM. Early-life glucocorticoid exposure: the hypothalamic-pituitary-adrenal axis, placental function, and long-term disease risk. Endocr Rev. 2013;34(6):885–916. [DOI] [PubMed] [Google Scholar]
- 117. New MI, Carlson A, Obeid J, Marshall I, Cabrera MS, Goseco A, Lin-Su K, Putnam AS, Wei JQ, Wilson RC. Prenatal diagnosis for congenital adrenal hyperplasia in 532 pregnancies. J Clin Endocrinol Metab. 2001;86(12):5651–5657. [DOI] [PubMed] [Google Scholar]
- 118. Barker DJP. In utero programming of chronic disease. Clin Sci (Lond). 1998;95(2):115–128. [PubMed] [Google Scholar]
- 119. Harris A, Seckl J. Glucocorticoids, prenatal stress and the programming of disease. Horm Behav. 2011;59(3):279–289. [DOI] [PubMed] [Google Scholar]
- 120. Kelly BA, Lewandowski AJ, Worton SA, Davis EF, Lazdam M, Francis J, Neubauer S, Lucas A, Singhal A, Leeson P. Antenatal glucocorticoid exposure and long-term alterations in aortic function and glucose metabolism. Pediatrics. 2012;129(5):e1282–e1290. [DOI] [PubMed] [Google Scholar]
- 121. Nugent JL, Wareing M, Palin V, Sibley CP, Baker PN, Ray DW, Farrow SN, Jones RL. Chronic glucocorticoid exposure potentiates placental chorionic plate artery constriction: implications for aberrant fetoplacental vascular resistance in fetal growth restriction. Endocrinology. 2013;154(2):876–887. [DOI] [PubMed] [Google Scholar]
- 122. Seckl JR, Miller WL. How safe is long-term prenatal glucocorticoid treatment? JAMA. 1997;277(13):1077–1079. [PubMed] [Google Scholar]
- 123. Moisiadis VG, Matthews SG. Glucocorticoids and fetal programming part 1: outcomes. Nat Rev Endocrinol. 2014;10(7):391–402. [DOI] [PubMed] [Google Scholar]
- 124. Seckl JR. Prenatal glucocorticoids and long-term programming. Eur J Endocrinol. 2004;151(Suppl 3):U49–U62. [DOI] [PubMed] [Google Scholar]
- 125. Damsted SK, Born AP, Paulson OB, Uldall P. Exogenous glucocorticoids and adverse cerebral effects in children. Eur J Paediatr Neurol. 2011;15(6):465–477. [DOI] [PubMed] [Google Scholar]
- 126. Andela CD, van Haalen FM, Ragnarsson O, Papakokkinou E, Johannsson G, Santos A, Webb SM, Biermasz NR, van der Wee NJ, Pereira AM. mechanisms in endocrinology: Cushing’s syndrome causes irreversible effects on the human brain: a systematic review of structural and functional magnetic resonance imaging studies. Eur J Endocrinol. 2015;173(1):R1–R14. [DOI] [PubMed] [Google Scholar]
- 127. Peffer ME, Zhang JY, Umfrey L, Rudine AC, Monaghan AP, DeFranco DB. Minireview: the impact of antenatal therapeutic synthetic glucocorticoids on the developing fetal brain. Mol Endocrinol. 2015;29(5):658–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Heberden C, Meffray E, Goustard-Langelier B, Maximin E, Lavialle M. Dexamethasone inhibits the maturation of newly formed neurons and glia supplemented with polyunsaturated fatty acids. J Steroid Biochem Mol Biol. 2013;138:395–402. [DOI] [PubMed] [Google Scholar]
- 129. Crudo A, Petropoulos S, Suderman M, Moisiadis VG, Kostaki A, Hallett M, Szyf M, Matthews SG. Effects of antenatal synthetic glucocorticoid on glucocorticoid receptor binding, DNA methylation, and genome-wide mRNA levels in the fetal male hippocampus. Endocrinology. 2013;154(11):4170–4181. [DOI] [PubMed] [Google Scholar]
- 130. Crudo A, Suderman M, Moisiadis VG, Petropoulos S, Kostaki A, Hallett M, Szyf M, Matthews SG. Glucocorticoid programming of the fetal male hippocampal epigenome. Endocrinology. 2013;154(3):1168–1180. [DOI] [PubMed] [Google Scholar]
- 131. Crudo A, Petropoulos S, Moisiadis VG, Iqbal M, Kostaki A, Machnes Z, Szyf M, Matthews SG. Prenatal synthetic glucocorticoid treatment changes DNA methylation states in male organ systems: multigenerational effects. Endocrinology. 2012;153(7):3269–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Samarasinghe RA, Di Maio R, Volonte D, Galbiati F, Lewis M, Romero G, DeFranco DB. Nongenomic glucocorticoid receptor action regulates gap junction intercellular communication and neural progenitor cell proliferation. Proc Natl Acad Sci USA. 2011;108(40):16657–16662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Hirvikoski T, Nordenström A, Lindholm T, Lindblad F, Ritzén EM, Wedell A, Lajic S. Cognitive functions in children at risk for congenital adrenal hyperplasia treated prenatally with dexamethasone. J Clin Endocrinol Metab. 2007;92(2):542–548. [DOI] [PubMed] [Google Scholar]
- 134. Hirvikoski T, Nordenström A, Lindholm T, Lindblad F, Ritzén EM, Lajic S. Long-term follow-up of prenatally treated children at risk for congenital adrenal hyperplasia: does dexamethasone cause behavioural problems? Eur J Endocrinol. 2008;159(3):309–316. [DOI] [PubMed] [Google Scholar]
- 135. Hirvikoski T, Lindholm T, Lajic S, Nordenström A. Gender role behaviour in prenatally dexamethasone-treated children at risk for congenital adrenal hyperplasia—a pilot study. Acta Paediatr. 2011;100(9):e112–e119. [DOI] [PubMed] [Google Scholar]
- 136. Wallensteen L, Zimmermann M, Thomsen Sandberg M, Gezelius A, Nordenström A, Hirvikoski T, Lajic S. Sex-dimorphic effects of prenatal treatment with dexamethasone. J Clin Endocrinol Metab. 2016;101(10):3838–3846. [DOI] [PubMed] [Google Scholar]
- 137. Browne WV, Hindmarsh PC, Pasterski V, Hughes IA, Acerini CL, Spencer D, Neufeld S, Hines M. Working memory performance is reduced in children with congenital adrenal hyperplasia. Horm Behav. 2015;67:83–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Collaer ML, Hindmarsh PC, Pasterski V, Fane BA, Hines M. Reduced short term memory in congenital adrenal hyperplasia (CAH) and its relationship to spatial and quantitative performance. Psychoneuroendocrinology. 2016;64:164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Meyer-Bahlburg HFL, Dolezal C, Haggerty R, Silverman M, New MI. Cognitive outcome of offspring from dexamethasone-treated pregnancies at risk for congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Eur J Endocrinol. 2012;167(1):103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Maryniak A, Ginalska-Malinowska M, Bielawska A, Ondruch A. Cognitive and social function in girls with congenital adrenal hyperplasia—influence of prenatally administered dexamethasone. Child Neuropsychol. 2014;20(1):60–70. [DOI] [PubMed] [Google Scholar]
- 141. Bergman K, Sarkar P, Glover V, O’Connor TG. Maternal prenatal cortisol and infant cognitive development: moderation by infant–mother attachment. Biol Psychiatry. 2010;67(11):1026–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Reynolds RM, Seckl JR. Antenatal glucocorticoid treatment: are we doing harm to term babies? J Clin Endocrinol Metab. 2012;97(10):3457–3459. [DOI] [PubMed] [Google Scholar]
- 143. Alexander N, Rosenlöcher F, Stalder T, Linke J, Distler W, Morgner J, Kirschbaum C. Impact of antenatal synthetic glucocorticoid exposure on endocrine stress reactivity in term-born children. J Clin Endocrinol Metab. 2012;97(10):3538–3544. [DOI] [PubMed] [Google Scholar]
- 144. Khalife N, Glover V, Taanila A, Ebeling H, Järvelin MR, Rodriguez A. Prenatal glucocorticoid treatment and later mental health in children and adolescents. PLoS One. 2013;8(11):e81394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Resmini E, Santos A, Gómez-Anson B, Vives Y, Pires P, Crespo I, Portella MJ, de Juan-Delago M, Barahona MJ, Webb SM. Verbal and visual memory performance and hippocampal volumes, measured by 3-Tesla magnetic resonance imaging, in patients with Cushing’s syndrome. J Clin Endocrinol Metab. 2012;97(2):663–671. [DOI] [PubMed] [Google Scholar]
- 146. Santos A, Resmini E, Crespo I, Pires P, Vives-Gilabert Y, Granell E, Valassi E, Gómez-Anson B, Martínez-Momblán MA, Mataró M, Webb SM. Small cerebellar cortex volume in patients with active Cushing’s syndrome. Eur J Endocrinol. 2014;171(4):461–469. [DOI] [PubMed] [Google Scholar]
- 147. Ragnarsson O, Berglund P, Eder DN, Johannsson G. Long-term cognitive impairments and attentional deficits in patients with Cushing’s disease and cortisol-producing adrenal adenoma in remission. J Clin Endocrinol Metab. 2012;97(9):E1640–E1648. [DOI] [PubMed] [Google Scholar]
- 148. Wagenmakers MAEM, Netea-Maier RT, Prins JB, Dekkers T, den Heijer M, Hermus ARMM. Impaired quality of life in patients in long-term remission of Cushing’s syndrome of both adrenal and pituitary origin: a remaining effect of long-standing hypercortisolism? Eur J Endocrinol. 2012;167(5):687–695. [DOI] [PubMed] [Google Scholar]
- 149. Tiemensma J, Kokshoorn NE, Biermasz NR, Keijser BJ, Wassenaar MJ, Middelkoop HAM, Pereira AM, Romijn JA. Subtle cognitive impairments in patients with long-term cure of Cushing’s disease. J Clin Endocrinol Metab. 2010;95(6):2699–2714. [DOI] [PubMed] [Google Scholar]
- 150. Tiemensma J, Biermasz NR, Middelkoop HAM, van der Mast RC, Romijn JA, Pereira AM. Increased prevalence of psychopathology and maladaptive personality traits after long-term cure of Cushing’s disease. J Clin Endocrinol Metab. 2010;95(10):E129–E141. [DOI] [PubMed] [Google Scholar]
- 151. Webb EA, Elliott L, Carlin D, Wilson M, Hall K, Netherton J, Reed J, Barrett TG, Salwani V, Clayden JD, Arlt W, Krone N, Peet AC, Wood AG. Quantitative brain MRI in congenital adrenal hyperplasia: in vivo assessment of the cognitive and structural impact of steroid hormones. J Clin Endocrinol Metab. 2018;103(4):1330–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Pole JD, Mustard CA, To T, Beyene J, Allen AC. Antenatal steroid therapy for fetal lung maturation: is there an association with childhood asthma? J Asthma. 2009;46(1):47–52. [DOI] [PubMed] [Google Scholar]
- 153. Tseng WN, Chen CC, Yu HR, Huang LT, Kuo HC. Antenatal dexamethasone exposure in preterm infants is associated with allergic diseases and the mental development index in children. Int J Environ Res Public Health. 2016;13(12):E1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Sun Y, Wan X, Ouyang J, Xie R, Wang X, Chen P. Prenatal dexamethasone exposure increases the susceptibility to autoimmunity in offspring rats by epigenetic programing of glucocorticoid receptor. BioMed Res Int. 2016;2016:9409452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Chou MY, Huang LT, Tain YL, Kuo HC, Tiao MM, Sheen JM, Chen CC, Hung PL, Hsieh KS, Yu HR. Age-dependent effects of prenatal dexamethasone exposure on immune responses in male rats. Tohoku J Exp Med. 2017;241(3):225–237. [DOI] [PubMed] [Google Scholar]
- 156. Dalziel SR, Walker NK, Parag V, Mantell C, Rea HH, Rodgers A, Harding JE. Cardiovascular risk factors after antenatal exposure to betamethasone: 30-year follow-up of a randomised controlled trial. Lancet. 2005;365(9474):1856–1862. [DOI] [PubMed] [Google Scholar]
- 157. Iqbal M, Moisiadis VG, Kostaki A, Matthews SG. Transgenerational effects of prenatal synthetic glucocorticoids on hypothalamic-pituitary-adrenal function. Endocrinology. 2012;153(7):3295–3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Quinn TA, Ratnayake U, Castillo-Melendez M, Moritz KM, Dickinson H, Walker DW. Adrenal steroidogenesis following prenatal dexamethasone exposure in the spiny mouse. J Endocrinol. 2014;221(2):347–362. [DOI] [PubMed] [Google Scholar]
- 159. Hirvikoski T, Nordenström A, Wedell A, Ritzén M, Lajic S. Prenatal dexamethasone treatment of children at risk for congenital adrenal hyperplasia: the Swedish experience and standpoint. J Clin Endocrinol Metab. 2012;97(6):1881–1883. [DOI] [PubMed] [Google Scholar]
- 160. Dörr HG, Binder G, Reisch N, Gembruch U, Oppelt PG, Wieacker P, Kratzsch J. Experts’ opinion on the prenatal therapy of congenital adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency—guideline of DGKED in cooperation with DGGG (S1-level, AWMF registry no. 174/013, July 2015). Geburtshilfe Frauenheilkd. 2015;75(12):1232–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Sparrow R. Gender eugenics? The ethics of PGD for intersex conditions. Am J Bioeth. 2013;13(10):29–38. [DOI] [PubMed] [Google Scholar]
- 162. Simpson JL, Rechitsky S. Preimplantation diagnosis and other modern methods for prenatal diagnosis. J Steroid Biochem Mol Biol. 2017;165(Pt A):124–130. [DOI] [PubMed] [Google Scholar]
- 163. Soto-Lafontaine M, Dondorp W, Provoost V, de Wert G. Dealing with treatment and transfer requests: how PGD-professionals discuss ethical challenges arising in everyday practice. Med Health Care Philos. 2018;21(3):375–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Ray JA, Kushnir MM, Yost RA, Rockwood AL, Meikle AW. Performance enhancement in the measurement of 5 endogenous steroids by LC-MS/MS combined with differential ion mobility spectrometry. Clin Chim Acta. 2015;438:330–336. [DOI] [PubMed] [Google Scholar]
- 165. Janzen N, Riepe FG, Peter M, Sander S, Steuerwald U, Korsch E, Krull F, Müller HL, Heger S, Brack C, Sander J. Neonatal screening: identification of children with 11β-hydroxylase deficiency by second-tier testing. Horm Res Paediatr. 2012;77(3):195–199. [DOI] [PubMed] [Google Scholar]
- 166. New MI, Lorenzen F, Lerner AJ, Kohn B, Oberfield SE, Pollack MS, Dupont B, Stoner E, Levy DJ, Pang S, Levine LS. Genotyping steroid 21-hydroxylase deficiency: hormonal reference data. J Clin Endocrinol Metab. 1983;57(2):320–326. [DOI] [PubMed] [Google Scholar]
- 167. Abdu TA, Elhadd TA, Neary R, Clayton RN. Comparison of the low dose short synacthen test (1 μg), the conventional dose short synacthen test (250 μg), and the insulin tolerance test for assessment of the hypothalamo-pituitary-adrenal axis in patients with pituitary disease. J Clin Endocrinol Metab. 1999;84(3):838–843. [DOI] [PubMed] [Google Scholar]
- 168. Caulfield MP, Lynn T, Gottschalk ME, Jones KL, Taylor NF, Malunowicz EM, Shackleton CHL, Reitz RE, Fisher DA. The diagnosis of congenital adrenal hyperplasia in the newborn by gas chromatography/mass spectrometry analysis of random urine specimens. J Clin Endocrinol Metab. 2002;87(8):3682–3690. [DOI] [PubMed] [Google Scholar]
- 169. Azziz R, Hincapie LA, Knochenhauer ES, Dewailly D, Fox L, Boots LR. Screening for 21-hydroxylase-deficient nonclassic adrenal hyperplasia among hyperandrogenic women: a prospective study. Fertil Steril. 1999;72(5):915–925. [DOI] [PubMed] [Google Scholar]
- 170. Armengaud J-B, Charkaluk M-L, Trivin C, Tardy V, Bréart G, Brauner R, Chalumeau M. Precocious pubarche: distinguishing late-onset congenital adrenal hyperplasia from premature adrenarche. J Clin Endocrinol Metab. 2009;94(8):2835–2840. [DOI] [PubMed] [Google Scholar]
- 171. Bidet M, Bellanné-Chantelot C, Galand-Portier M-B, Tardy V, Billaud L, Laborde K, Coussieu C, Morel Y, Vaury C, Golmard J-L, Claustre A, Mornet E, Chakhtoura Z, Mowszowicz I, Bachelot A, Touraine P, Kuttenn F. Clinical and molecular characterization of a cohort of 161 unrelated women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency and 330 family members. J Clin Endocrinol Metab. 2009;94(5):1570–1578. [DOI] [PubMed] [Google Scholar]
- 172. Török D, Halász Z, Garami M, Homoki J, Fekete G, Sólyom J. Limited value of serum steroid measurements in identification of mild form of 21-hydroxylase deficiency. Exp Clin Endocrinol Diabetes. 2003;111(1):27–32. [DOI] [PubMed] [Google Scholar]
- 173. Speiser PW, White PC. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency. Clin Endocrinol (Oxf). 1998;49(4):411–417. [DOI] [PubMed] [Google Scholar]
- 174. Wedell A, Thilén A, Ritzén EM, Stengler B, Luthman H. Mutational spectrum of the steroid 21-hydroxylase gene in Sweden: implications for genetic diagnosis and association with disease manifestation. J Clin Endocrinol Metab. 1994;78(5):1145–1152. [DOI] [PubMed] [Google Scholar]
- 175. Wilson RC, Mercado AB, Cheng KC, New MI. Steroid 21-hydroxylase deficiency: genotype may not predict phenotype. J Clin Endocrinol Metab. 1995;80(8):2322–2329. [DOI] [PubMed] [Google Scholar]
- 176. Bachega TASS, Billerbeck AEC, Marcondes JAM, Madureira G, Arnhold IJP, Mendonca BB. Influence of different genotypes on 17-hydroxyprogesterone levels in patients with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Clin Endocrinol (Oxf). 2000;52(5):601–607. [DOI] [PubMed] [Google Scholar]
- 177. Costa-Barbosa FA, Carvalho VM, Nakamura OH, Bachega TASS, Vieira JGH, Kater CE. Zona fasciculata 21-hydroxysteroids and precursor-to-product ratios in 21-hydroxylase deficiency: further characterization of classic and non-classic patients and heterozygote carriers. J Endocrinol Invest. 2011;34(8):587–592. [DOI] [PubMed] [Google Scholar]
- 178. Balsamo A, Cacciari E, Baldazzi L, Tartaglia L, Cassio A, Mantovani V, Piazzi S, Cicognani A, Pirazzoli P, Mainetti B, Zappulla F. CYP21 analysis and phenotype/genotype relationship in the screened population of the Italian Emilia–Romagna region. Clin Endocrinol (Oxf). 2000;53(1):117–125. [DOI] [PubMed] [Google Scholar]
- 179. Wedell A, Ritzén EM, Haglund-Stengler B, Luthman H. Steroid 21-hydroxylase deficiency: three additional mutated alleles and establishment of phenotype-genotype relationships of common mutations. Proc Natl Acad Sci USA. 1992;89(15):7232–7236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Fiet J, Gueux B, Gourmelen M, Kuttenn F, Vexiau P, Couillin P, Pham-Huu-Trung M-T, Villette J-M, Raux-Demay MC, Galons H, Julien R. Comparison of basal and adrenocorticotropin-stimulated plasma 21-deoxycortisol and 17-hydroxyprogesterone values as biological markers of late-onset adrenal hyperplasia. J Clin Endocrinol Metab. 1988;66(4):659–667. [DOI] [PubMed] [Google Scholar]
- 181. Peter M, Sippell WG, Lorenzen F, Willig RP, Westphal E, Grosse-Wilde H. Improved test to identify heterozygotes for congenital adrenal hyperplasia without index case examination. Lancet. 1990;335(8701):1296–1299. [DOI] [PubMed] [Google Scholar]
- 182. Mullis PE, Hindmarsh PC, Brook CGD. Sodium chloride supplement at diagnosis and during infancy in children with salt-losing 21-hydroxylase deficiency. Eur J Pediatr. 1990;150(1):22–25. [DOI] [PubMed] [Google Scholar]
- 183. Bonfig W, Roehl F, Riedl S, Bramswig J, Richter-Unruh A, Fricke-Otto S, Hubner A, Bettendorf M, Schonau E, Dorr H, Holl RW, Mohnike K. Sodium chloride supplementation is not routinely performed in the majority of German and Austrian infants with classic salt-wasting congenital adrenal hyperplasia and has no effect on linear growth and hydrocortisone or fludrocortisone dose. Horm Res Paediatr. 2018;89(1):7–12. [DOI] [PubMed] [Google Scholar]
- 184. Nimkarn S, Lin-Su K, Berglind N, Wilson RC, New MI. Aldosterone-to-renin ratio as a marker for disease severity in 21-hydroxylase deficiency congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2007;92(1):137–142. [DOI] [PubMed] [Google Scholar]
- 185. Frisch H, Battelino T, Schober E, Baumgartner-Parzer S, Nowotny P, Vierhapper H. Salt wasting in simple virilizing congenital adrenal hyperplasia. J Pediatr Endocrinol Metab. 2001;14(9):1649–1655. [DOI] [PubMed] [Google Scholar]
- 186. Muthusamy K, Elamin MB, Smushkin G, Murad MH, Lampropulos JF, Elamin KB, Abu Elnour NO, Gallegos-Orozco JF, Fatourechi MM, Agrwal N, Lane MA, Albuquerque FN, Erwin PJ, Montori VM. Clinical review: adult height in patients with congenital adrenal hyperplasia: a systematic review and metaanalysis. J Clin Endocrinol Metab. 2010;95(9):4161–4172. [DOI] [PubMed] [Google Scholar]
- 187. Bonfig W, Bechtold S, Schmidt H, Knorr D, Schwarz HP. Reduced final height outcome in congenital adrenal hyperplasia under prednisone treatment: deceleration of growth velocity during puberty. J Clin Endocrinol Metab. 2007;92(5):1635–1639. [DOI] [PubMed] [Google Scholar]
- 188. Punthakee Z, Legault L, Polychronakos C. Prednisolone in the treatment of adrenal insufficiency: a re-evaluation of relative potency. J Pediatr. 2003;143(3):402–405. [DOI] [PubMed] [Google Scholar]
- 189. Rivkees SA, Crawford JD. Dexamethasone treatment of virilizing congenital adrenal hyperplasia: the ability to achieve normal growth. Pediatrics. 2000;106(4):767–773. [DOI] [PubMed] [Google Scholar]
- 190. Sarafoglou K, Gonzalez-Bolanos MT, Zimmerman CL, Boonstra T, Yaw Addo O, Brundage R. Comparison of cortisol exposures and pharmacodynamic adrenal steroid responses to hydrocortisone suspension vs. commercial tablets. J Clin Pharmacol. 2015;55(4):452–457. [DOI] [PubMed] [Google Scholar]
- 191. Merke DP, Cho D, Calis KA, Keil MF, Chrousos GP. Hydrocortisone suspension and hydrocortisone tablets are not bioequivalent in the treatment of children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001;86(1):441–445. [DOI] [PubMed] [Google Scholar]
- 192. Neumann U, Burau D, Spielmann S, Whitaker MJ, Ross RJ, Kloft C, Blankenstein O. Quality of compounded hydrocortisone capsules used in the treatment of children. Eur J Endocrinol. 2017;177(2):239–242. [DOI] [PubMed] [Google Scholar]
- 193. Gudeman J, Jozwiakowski M, Chollet J, Randell M. Potential risks of pharmacy compounding. Drugs R D. 2013;13(1):1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Barillas JE, Eichner D, Van Wagoner R, Speiser PW. Iatrogenic Cushing syndrome in a child with congenital adrenal hyperplasia: erroneous compounding of hydrocortisone. J Clin Endocrinol Metab. 2018;103(1):7–11. [DOI] [PubMed] [Google Scholar]
- 195. German A, Suraiya S, Tenenbaum-Rakover Y, Koren I, Pillar G, Hochberg Z. Control of childhood congenital adrenal hyperplasia and sleep activity and quality with morning or evening glucocorticoid therapy. J Clin Endocrinol Metab. 2008;93(12):4707–4710. [DOI] [PubMed] [Google Scholar]
- 196. Balsamo A, Cicognani A, Baldazzi L, Barbaro M, Baronio F, Gennari M, Bal M, Cassio A, Kontaxaki K, Cacciari E. CYP21 genotype, adult height, and pubertal development in 55 patients treated for 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2003;88(12):5680–5688. [DOI] [PubMed] [Google Scholar]
- 197. Bonfig W, Pozza SB, Schmidt H, Pagel P, Knorr D, Schwarz HP. Hydrocortisone dosing during puberty in patients with classical congenital adrenal hyperplasia: an evidence-based recommendation. J Clin Endocrinol Metab. 2009;94(10):3882–3888. [DOI] [PubMed] [Google Scholar]
- 198. Grigorescu-Sido A, Bettendorf M, Schulze E, Duncea I, Heinrich U. Growth analysis in patients with 21-hydroxylase deficiency influence of glucocorticoid dosage, age at diagnosis, phenotype and genotype on growth and height outcome. Horm Res. 2003;60(2):84–90. [DOI] [PubMed] [Google Scholar]
- 199. Van der Kamp HJ, Otten BJ, Buitenweg N, De Muinck Keizer-Schrama SMPF, Oostdijk W, Jansen M, Delemarre-de Waal HA, Vulsma T, Wit JM. Longitudinal analysis of growth and puberty in 21-hydroxylase deficiency patients. Arch Dis Child. 2002;87(2):139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Hindmarsh PC, Charmandari E. Variation in absorption and half-life of hydrocortisone influence plasma cortisol concentrations. Clin Endocrinol (Oxf). 2015;82(4):557–561. [DOI] [PubMed] [Google Scholar]
- 201. Charmandari E, Hindmarsh PC, Johnston A, Brook CG. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: alterations in cortisol pharmacokinetics at puberty. J Clin Endocrinol Metab. 2001;86(6):2701–2708. [DOI] [PubMed] [Google Scholar]
- 202. Arlt W, Willis DS, Wild SH, Krone N, Doherty EJ, Hahner S, Han TS, Carroll PV, Conway GS, Rees DA, Stimson RH, Walker BR, Connell JM, Ross RJ; United Kingdom Congenital Adrenal Hyperplasia Adult Study Executive (CaHASE) . Health status of adults with congenital adrenal hyperplasia: a cohort study of 203 patients. J Clin Endocrinol Metab. 2010;95(11):5110–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Finkielstain GP, Kim MS, Sinaii N, Nishitani M, Van Ryzin C, Hill SC, Reynolds JC, Hanna RM, Merke DP. Clinical characteristics of a cohort of 244 patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2012;97(12):4429–4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Martinerie L, Viengchareun S, Delezoide AL, Jaubert F, Sinico M, Prevot S, Boileau P, Meduri G, Lombès M. Low renal mineralocorticoid receptor expression at birth contributes to partial aldosterone resistance in neonates. Endocrinology. 2009;150(9):4414–4424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. De Leo V, Morgante G, Piomboni P, Musacchio MC, Petraglia F, Cianci A. Evaluation of effects of an oral contraceptive containing ethinylestradiol combined with drospirenone on adrenal steroidogenesis in hyperandrogenic women with polycystic ovary syndrome. Fertil Steril. 2007;88(1):113–117. [DOI] [PubMed] [Google Scholar]
- 206. de Nadai MN, Nobre F, Ferriani RA, Vieira CS. Effects of two contraceptives containing drospirenone on blood pressure in normotensive women: a randomized-controlled trial. Blood Press Monit. 2015;20(6):310–315. [DOI] [PubMed] [Google Scholar]
- 207. Martin KA, Anderson RR, Chang RJ, Ehrmann DA, Lobo RA, Murad MH, Pugeat MM, Rosenfield RL. Evaluation and treatment of hirsutism in premenopausal women: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2018;103(4):1233–1257. [DOI] [PubMed] [Google Scholar]
- 208. El-Maouche D, Hargreaves CJ, Sinaii N, Mallappa A, Veeraraghavan P, Merke DP. Longitudinal assessment of illnesses, stress dosing and illness sequelae in patients with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2018;103(6):2336–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Taylor LK, Auchus RJ, Baskin LS, Miller WL. Cortisol response to operative stress with anesthesia in healthy children. J Clin Endocrinol Metab. 2013;98(9):3687–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Reisch N, Willige M, Kohn D, Schwarz HP, Allolio B, Reincke M, Quinkler M, Hahner S, Beuschlein F. Frequency and causes of adrenal crises over lifetime in patients with 21-hydroxylase deficiency. Eur J Endocrinol. 2012;167(1):35–42. [DOI] [PubMed] [Google Scholar]
- 211. Odenwald B, Nennstiel-Ratzel U, Dörr HG, Schmidt H, Wildner M, Bonfig W. Children with classic congenital adrenal hyperplasia experience salt loss and hypoglycemia: evaluation of adrenal crises during the first 6 years of life. Eur J Endocrinol. 2016;174(2):177–186. [DOI] [PubMed] [Google Scholar]
- 212. Arriza JL, Weinberger C, Cerelli G, Glaser TM, Handelin BL, Housman DE, Evans RM. Cloning of human mineralocorticoid receptor complementary DNA: structural and functional kinship with the glucocorticoid receptor. Science. 1987;237(4812):268–275. [DOI] [PubMed] [Google Scholar]
- 213. Weise M, Drinkard B, Mehlinger SL, Holzer SM, Eisenhofer G, Charmandari E, Chrousos GP, Merke DP. Stress dose of hydrocortisone is not beneficial in patients with classic congenital adrenal hyperplasia undergoing short-term, high-intensity exercise. J Clin Endocrinol Metab. 2004;89(8):3679–3684. [DOI] [PubMed] [Google Scholar]
- 214. Falhammar H, Frisén L, Norrby C, Hirschberg AL, Almqvist C, Nordenskjöld A, Nordenström A. Increased mortality in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2014;99(12):E2715–E2721. [DOI] [PubMed] [Google Scholar]
- 215. Bornstein SR, Allolio B, Arlt W, Barthel A, Don-Wauchope A, Hammer GD, Husebye ES, Merke DP, Murad MH, Stratakis CA, Torpy DJ. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016;101(2):364–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Bonfig W, Schwarz HP. Blood pressure, fludrocortisone dose and plasma renin activity in children with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency followed from birth to 4 years of age. Clin Endocrinol (Oxf). 2014;81(6):871–875. [DOI] [PubMed] [Google Scholar]
- 217. Bonfig W, Roehl FW, Riedl S, Dörr HG, Bettendorf M, Brämswig J, Schönau E, Riepe F, Hauffa B, Holl RW, Mohnike K, Group ACS; AQUAPE CAH Study Group . Blood pressure in a large cohort of children and adolescents with classic adrenal hyperplasia (CAH) due to 21-hydroxylase deficiency. Am J Hypertens. 2016;29(2):266–272. [DOI] [PubMed] [Google Scholar]
- 218. Turcu AF, Nanba AT, Chomic R, Upadhyay SK, Giordano TJ, Shields JJ, Merke DP, Rainey WE, Auchus RJ. Adrenal-derived 11-oxygenated 19-carbon steroids are the dominant androgens in classic 21-hydroxylase deficiency. Eur J Endocrinol. 2016;174(5):601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Turcu AF, Mallappa A, Elman MS, Avila NA, Marko J, Rao H, Tsodikov A, Auchus RJ, Merke DP. 11-Oxygenated androgens are biomarkers of adrenal volume and testicular adrenal rest tumors in 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2017;102(8):2701–2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. de Groot MJ, Pijnenburg-Kleizen KJ, Thomas CM, Sweep FC, Stikkelbroeck NM, Otten BJ, Claahsen-van der Grinten HL. Salivary morning androstenedione and 17α-OH progesterone levels in childhood and puberty in patients with classic congenital adrenal hyperplasia. Clin Chem Lab Med. 2015;53(3):461–468. [DOI] [PubMed] [Google Scholar]
- 221. Bode HH, Rivkees SA, Cowley DM, Pardy K, Johnson S. Home monitoring of 17 hydroxyprogesterone levels in congenital adrenal hyperplasia with filter paper blood samples. J Pediatr. 1999;134(2):185–189. [DOI] [PubMed] [Google Scholar]
- 222. Wieacker I, Peter M, Borucki K, Empting S, Roehl FW, Mohnike K. Therapy monitoring in congenital adrenal hyperplasia by dried blood samples. J Pediatr Endocrinol Metab. 2015;28(7–8):867–871. [DOI] [PubMed] [Google Scholar]
- 223. Debono M, Mallappa A, Gounden V, Nella AA, Harrison RF, Crutchfield CA, Backlund PS, Soldin SJ, Ross RJ, Merke DP. Hormonal circadian rhythms in patients with congenital adrenal hyperplasia: identifying optimal monitoring times and novel disease biomarkers. Eur J Endocrinol. 2015;173(6):727–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Claahsen-van der Grinten HL, Dehzad F, Kamphuis-van Ulzen K, de Korte CL. Increased prevalence of testicular adrenal rest tumours during adolescence in congenital adrenal hyperplasia. Horm Res Paediatr. 2014;82(4):238–244. [DOI] [PubMed] [Google Scholar]
- 225. Casteràs A, De Silva P, Rumsby G, Conway GS. Reassessing fecundity in women with classical congenital adrenal hyperplasia (CAH): normal pregnancy rate but reduced fertility rate. Clin Endocrinol (Oxf). 2009;70(6):833–837. [DOI] [PubMed] [Google Scholar]
- 226. King TF, Lee MC, Williamson EE, Conway GS. Experience in optimizing fertility outcomes in men with congenital adrenal hyperplasia due to 21 hydroxylase deficiency. Clin Endocrinol (Oxf). 2016;84(6):830–836. [DOI] [PubMed] [Google Scholar]
- 227. Claahsen-van der Grinten HL, Otten BJ, Takahashi S, Meuleman EJ, Hulsbergen-van de Kaa C, Sweep FC, Hermus ARMM. Testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia: evaluation of pituitary-gonadal function before and after successful testis-sparing surgery in eight patients. J Clin Endocrinol Metab. 2007;92(2):612–615. [DOI] [PubMed] [Google Scholar]
- 228. Auchus RJ, Arlt W. Approach to the patient: the adult with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2013;98(7):2645–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Reisch N, Rottenkolber M, Greifenstein A, Krone N, Schmidt H, Reincke M, Schwarz HP, Beuschlein F. Testicular adrenal rest tumors develop independently of long-term disease control: a longitudinal analysis of 50 adult men with congenital adrenal hyperplasia due to classic 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2013;98(11):E1820–E1826. [DOI] [PubMed] [Google Scholar]
- 230. Falhammar H, Nordenström A. Nonclassic congenital adrenal hyperplasia due to 21-hydroxylase deficiency: clinical presentation, diagnosis, treatment, and outcome. Endocrine. 2015;50(1):32–50. [DOI] [PubMed] [Google Scholar]
- 231. Trapp CM, Oberfield SE. Recommendations for treatment of nonclassic congenital adrenal hyperplasia (NCCAH): an update. Steroids. 2012;77(4):342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Spritzer P, Billaud L, Thalabard J-C, Birman P, Mowszowicz I, Raux-Demay M-C, Clair F, Kuttenn F, Mauvais-Jarvis P. Cyproterone acetate versus hydrocortisone treatment in late-onset adrenal hyperplasia. J Clin Endocrinol Metab. 1990;70(3):642–646. [DOI] [PubMed] [Google Scholar]
- 233. Matthews D, Cheetham T. What is the best approach to the teenage patient presenting with nonclassical congenital adrenal hyperplasia: should we always treat with glucocorticoids? Clin Endocrinol (Oxf). 2013;78(3):338–341. [DOI] [PubMed] [Google Scholar]
- 234. Moran C, Azziz R, Weintrob N, Witchel SF, Rohmer V, Dewailly D, Marcondes JAM, Pugeat M, Speiser PW, Pignatelli D, Mendonca BB, Bachega TAS, Escobar-Morreale HF, Carmina E, Fruzzetti F, Kelestimur F. Reproductive outcome of women with 21-hydroxylase-deficient nonclassic adrenal hyperplasia. J Clin Endocrinol Metab. 2006;91(9):3451–3456. [DOI] [PubMed] [Google Scholar]
- 235. Bidet M, Bellanné-Chantelot C, Galand-Portier M-B, Golmard J-L, Tardy V, Morel Y, Clauin S, Coussieu C, Boudou P, Mowzowicz I, Bachelot A, Touraine P, Kuttenn F. Fertility in women with nonclassical congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2010;95(3):1182–1190. [DOI] [PubMed] [Google Scholar]
- 236. Eyal O, Ayalon-Dangur I, Segev-Becker A, Schachter-Davidov A, Israel S, Weintrob N. Pregnancy in women with nonclassic congenital adrenal hyperplasia: time to conceive and outcome. Clin Endocrinol (Oxf). 2017;87(5):552–556. [DOI] [PubMed] [Google Scholar]
- 237. Falhammar H, Nyström HF, Ekström U, Granberg S, Wedell A, Thorén M. Fertility, sexuality and testicular adrenal rest tumors in adult males with congenital adrenal hyperplasia. Eur J Endocrinol. 2012;166(3):441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Nandagopal R, Sinaii N, Avila NA, Van Ryzin C, Chen W, Finkielstain GP, Mehta SP, McDonnell NB, Merke DP. Phenotypic profiling of parents with cryptic nonclassic congenital adrenal hyperplasia: findings in 145 unrelated families. Eur J Endocrinol. 2011;164(6):977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239. Stoupa A, González-Briceño L, Pinto G, Samara-Boustani D, Thalassinos C, Flechtner I, Beltrand J, Bidet M, Simon A, Piketty M, Laborde K, Morel Y, Bellanné-Chantelot C, Touraine P, Polak M. Inadequate cortisol response to the tetracosactide (Synacthen®) test in non-classic congenital adrenal hyperplasia: an exception to the rule? Horm Res Paediatr. 2015;83(4):262–267. [DOI] [PubMed] [Google Scholar]
- 240. Raverot V, Richet C, Morel Y, Raverot G, Borson-Chazot F. Establishment of revised diagnostic cut-offs for adrenal laboratory investigation using the new Roche Diagnostics Elecsys® cortisol II assay. Ann Endocrinol (Paris). 2016;77(5):620–622. [DOI] [PubMed] [Google Scholar]
- 241. Conway GS. Congenital adrenal hyperplasia: adolescence and transition. Horm Res. 2007;68(Suppl 5):155–157. [DOI] [PubMed] [Google Scholar]
- 242. Hughes IA. Congenital adrenal hyperplasia: transitional care. Growth Horm IGF Res. 2004;14(Suppl A):S60–S66. [DOI] [PubMed] [Google Scholar]
- 243. Kruse B, Riepe FG, Krone N, Bosinski HA, Kloehn S, Partsch CJ, Sippell WG, Mönig H. Congenital adrenal hyperplasia—how to improve the transition from adolescence to adult life. Exp Clin Endocrinol Diabetes. 2004;112(7):343–355. [DOI] [PubMed] [Google Scholar]
- 244. Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med. 2003;349(8):776–788. [DOI] [PubMed] [Google Scholar]
- 245. Jenkins-Jones S, Parviainen L, Porter J, Withe M, Whitaker MJ, Holden SE, Morgan CL, Currie CJ, Ross RJM. Poor compliance and increased mortality, depression and healthcare costs in patients with congenital adrenal hyperplasia. Eur J Endocrinol. 2018;178(4):309–320. [DOI] [PubMed] [Google Scholar]
- 246. Finkielstain GP, Chen W, Mehta SP, Fujimura FK, Hanna RM, VanRyzin C, McDonnell NB, Merke DP. Comprehensive genetic analysis of 182 unrelated families with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2011;96(1):E161–E172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Falhammar H, Frisén L, Norrby C, Almqvist C, Hirschberg AL, Nordenskjöld A, Nordenström A. Reduced frequency of biological and increased frequency of adopted children in males with 21-hydroxylase deficiency: a Swedish population-based national cohort study. J Clin Endocrinol Metab. 2017;102(11):4191–4199. [DOI] [PubMed] [Google Scholar]
- 248. Reisch N, Flade L, Scherr M, Rottenkolber M, Pedrosa Gil F, Bidlingmaier M, Wolff H, Schwarz HP, Quinkler M, Beuschlein F, Reincke M. High prevalence of reduced fecundity in men with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2009;94(5):1665–1670. [DOI] [PubMed] [Google Scholar]
- 249. Jääskeläinen J, Kiekara O, Hippeläinen M, Voutilainen R. Pituitary gonadal axis and child rate in males with classical 21-hydroxylase deficiency. J Endocrinol Invest. 2000;23(1):23–27. [DOI] [PubMed] [Google Scholar]
- 250. Stikkelbroeck NMML, Otten BJ, Pasic A, Jager GJ, Sweep CG, Noordam K, Hermus ARMM. High prevalence of testicular adrenal rest tumors, impaired spermatogenesis, and Leydig cell failure in adolescent and adult males with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001;86(12):5721–5728. [DOI] [PubMed] [Google Scholar]
- 251. Martinez-Aguayo A, Rocha A, Rojas N, García C, Parra R, Lagos M, Valdivia L, Poggi H, Cattani A; Chilean Collaborative Testicular Adrenal Rest Tumor Study Group . Testicular adrenal rest tumors and Leydig and Sertoli cell function in boys with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2007;92(12):4583–4589. [DOI] [PubMed] [Google Scholar]
- 252. Stikkelbroeck NMML, Hermus ARMM, Suliman HM, Jager GJ, Otten BJ. Asymptomatic testicular adrenal rest tumours in adolescent and adult males with congenital adrenal hyperplasia: basal and follow-up investigation after 2.6 years. J Pediatr Endocrinol Metab. 2004;17(4):645–653. [DOI] [PubMed] [Google Scholar]
- 253. Bouvattier C, Esterle L, Renoult-Pierre P, de la Perrière AB, Illouz F, Kerlan V, Pascal-Vigneron V, Drui D, Christin-Maitre S, Galland F, Brue T, Reznik Y, Schillo F, Pinsard D, Piguel X, Chabrier G, Decoudier B, Emy P, Tauveron I, Raffin-Sanson ML, Bertherat J, Kuhn JM, Caron P, Cartigny M, Chabre O, Dewailly D, Morel Y, Touraine P, Tardy-Guidollet V, Young J. Clinical outcome, hormonal status, gonadotrope axis, and testicular function in 219 adult men born with classic 21-hydroxylase deficiency. A French national survey. J Clin Endocrinol Metab. 2015;100(6):2303–2313. [DOI] [PubMed] [Google Scholar]
- 254. Cabrera MS, Vogiatzi MG, New MI. Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001;86(7):3070–3078. [DOI] [PubMed] [Google Scholar]
- 255. Jääskeläinen, Voutilainen R. Long-term outcome of classical 21-hydroxylase deficiency: diagnosis, complications and quality of life. Acta Paediatr. 2000;89(2):183–187. [DOI] [PubMed] [Google Scholar]
- 256. Kavoussi PK, Summers-Colquitt RB, Odenwald KC, Kressin M, Kavoussi KM, Pool TB, Kavoussi SK. Sperm retrieval and concomitant tumor resection in azoospermic men with congenital adrenal hyperplasia and bilateral testicular adrenal rest tumors: a case report. J Assist Reprod Genet. 2016;33(4):545–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Hagenfeldt K, Janson PO, Holmdahl G, Falhammar H, Filipsson H, Frisén L, Thorén M, Nordenskjöld A. Fertility and pregnancy outcome in women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Hum Reprod. 2008;23(7):1607–1613. [DOI] [PubMed] [Google Scholar]
- 258. Chen HD, Huang LE, Zhong ZH, Su Z, Jiang H, Zeng J, Liu JC. Ovarian adrenal rest tumors undetected by imaging studies and identified at surgery in three females with congenital adrenal hyperplasia unresponsive to increased hormone therapy dosage. Endocr Pathol. 2017;28(2):146–151. [DOI] [PubMed] [Google Scholar]
- 259. Lo JC, Schwitzgebel VM, Tyrrell JB, Fitzgerald PA, Kaplan SL, Conte FA, Grumbach MM. Normal female infants born of mothers with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1999;84(3):930–936. [DOI] [PubMed] [Google Scholar]
- 260. Falhammar H, Filipsson H, Holmdahl G, Janson PO, Nordenskjöld A, Hagenfeldt K, Thorén M. Metabolic profile and body composition in adult women with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92(1):110–116. [DOI] [PubMed] [Google Scholar]
- 261. Girgis R, Winter JS. The effects of glucocorticoid replacement therapy on growth, bone mineral density, and bone turnover markers in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1997;82(12):3926–3929. [DOI] [PubMed] [Google Scholar]
- 262. Gussinyé M, Carrascosa A, Potau N, Enrubia M, Vicens-Calvet E, Ibáñez L, Yeste D. Bone mineral density in prepubertal and in adolescent and young adult patients with the salt-wasting form of congenital adrenal hyperplasia. Pediatrics. 1997;100(4):671–674. [DOI] [PubMed] [Google Scholar]
- 263. Mora S, Saggion F, Russo G, Weber G, Bellini A, Prinster C, Chiumello G. Bone density in young patients with congenital adrenal hyperplasia. Bone. 1996;18(4):337–340. [DOI] [PubMed] [Google Scholar]
- 264. Falhammar H, Filipsson H, Holmdahl G, Janson P-O, Nordenskjöld A, Hagenfeldt K, Thorén M. Fractures and bone mineral density in adult women with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2007;92(12):4643–4649. [DOI] [PubMed] [Google Scholar]
- 265. Ceccato F, Barbot M, Albiger N, Zilio M, De Toni P, Luisetto G, Zaninotto M, Greggio NA, Boscaro M, Scaroni C, Camozzi V. Long-term glucocorticoid effect on bone mineral density in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Eur J Endocrinol. 2016;175(2):101–106. [DOI] [PubMed] [Google Scholar]
- 266. Chakhtoura Z, Bachelot A, Samara-Boustani D, Ruiz JC, Donadille B, Dulon J, Christin-Maître S, Bouvattier C, Raux-Demay MC, Bouchard P, Carel JC, Leger J, Kuttenn F, Polak M, Touraine P; Centre des Maladies Endocriniennes Rares de la Croissance and Association Surrénales . Impact of total cumulative glucocorticoid dose on bone mineral density in patients with 21-hydroxylase deficiency. Eur J Endocrinol. 2008;158(6):879–887. [DOI] [PubMed] [Google Scholar]
- 267. Barzon L, Sonino N, Fallo F, Palu G, Boscaro M. Prevalence and natural history of adrenal incidentalomas. Eur J Endocrinol. 2003;149(4):273–285. [DOI] [PubMed] [Google Scholar]
- 268. Kloos RT, Gross MD, Francis IR, Korobkin M, Shapiro B. Incidentally discovered adrenal masses. Endocr Rev. 1995;16(4):460–484. [DOI] [PubMed] [Google Scholar]
- 269. Jaresch S, Kornely E, Kley HK, Schlaghecke R. Adrenal incidentaloma and patients with homozygous or heterozygous congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1992;74(3):685–689. [DOI] [PubMed] [Google Scholar]
- 270. Barzon L, Maffei P, Sonino N, Pilon C, Baldazzi L, Balsamo A, Del Maschio O, Masi G, Trevisan M, Pacenti M, Fallo F. The role of 21-hydroxylase in the pathogenesis of adrenal masses: review of the literature and focus on our own experience. J Endocrinol Invest. 2007;30(7):615–623. [DOI] [PubMed] [Google Scholar]
- 271. Varan A, Unal S, Ruacan S, Vidinlisan S. Adrenocortical carcinoma associated with adrenogenital syndrome in a child. Med Pediatr Oncol. 2000;35(1):88–90. [DOI] [PubMed] [Google Scholar]
- 272. Nermoen I, Rørvik J, Holmedal SH, Hykkerud DL, Fougner KJ, Svartberg J, Husebye ES, Løvås K. High frequency of adrenal myelolipomas and testicular adrenal rest tumours in adult Norwegian patients with classical congenital adrenal hyperplasia because of 21-hydroxylase deficiency. Clin Endocrinol (Oxf). 2011;75(6):753–759. [DOI] [PubMed] [Google Scholar]
- 273. Völkl TMK, Simm D, Körner A, Rascher W, Kiess W, Kratzsch J, Dörr HG. Does an altered leptin axis play a role in obesity among children and adolescents with classical congenital adrenal hyperplasia due to 21-hydroxylase deficiency? Eur J Endocrinol. 2009;160(2):239–247. [DOI] [PubMed] [Google Scholar]
- 274. Völkl TM, Simm D, Beier C, Dörr HG. Obesity among children and adolescents with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Pediatrics. 2006;117(1):e98–e105. [DOI] [PubMed] [Google Scholar]
- 275. Cornean RE, Hindmarsh PC, Brook CG. Obesity in 21-hydroxylase deficient patients. Arch Dis Child. 1998;78(3):261–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276. Moyer VA; U.S. Preventive Services Task Force . Screening for gestational diabetes mellitus: U.S. Preventive Services Task Force recommendation statement. Ann Intern Med. 2014;160(6):414–420. [DOI] [PubMed] [Google Scholar]
- 277. Lesma A, Bocciardi A, Corti S, Chiumello G, Rigatti P, Montorsi F. Sexual function in adult life following Passerini-Glazel feminizing genitoplasty in patients with congenital adrenal hyperplasia. J Urol. 2014;191(1):206–211. [DOI] [PubMed] [Google Scholar]
- 278. Houben CH, Tsui SY, Mou JW, Chan KW, Tam YH, Lee KH. Reconstructive surgery for females with congenital adrenal hyperplasia due to 21-hydroxylase deficiency: a review from the Prince of Wales Hospital. Hong Kong Med J. 2014;20(6):481–485. [DOI] [PubMed] [Google Scholar]
- 279. Yankovic F, Cherian A, Steven L, Mathur A, Cuckow P. Current practice in feminizing surgery for congenital adrenal hyperplasia; a specialist survey. J Pediatr Urol. 2013;9(6 Pt B):1103–1107. [DOI] [PubMed] [Google Scholar]
- 280. van der Zwan YG, Janssen EH, Callens N, Wolffenbuttel KP, Cohen-Kettenis PT, van den Berg M, Drop SL, Dessens AB, Beerendonk C; Dutch Study Group on DSD . Severity of virilization is associated with cosmetic appearance and sexual function in women with congenital adrenal hyperplasia: a cross-sectional study. J Sex Med. 2013;10(3):866–875. [DOI] [PubMed] [Google Scholar]
- 281. Binet A, Lardy H, Geslin D, Francois-Fiquet C, Poli-Merol ML. Should we question early feminizing genitoplasty for patients with congenital adrenal hyperplasia and XX karyotype? J Pediatr Surg. 2016;51(3):465–468. [DOI] [PubMed] [Google Scholar]
- 282. Rink RC. Genitoplasty/vaginoplasty. Adv Exp Med Biol. 2011;707:51–54. [DOI] [PubMed] [Google Scholar]
- 283. Crouch NS, Liao LM, Woodhouse CR, Conway GS, Creighton SM. Sexual function and genital sensitivity following feminizing genitoplasty for congenital adrenal hyperplasia. J Urol. 2008;179(2):634–638. [DOI] [PubMed] [Google Scholar]
- 284. Nordenskjöld A, Holmdahl G, Frisén L, Falhammar H, Filipsson H, Thorén M, Janson PO, Hagenfeldt K. Type of mutation and surgical procedure affect long-term quality of life for women with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2008;93(2):380–386. [DOI] [PubMed] [Google Scholar]
- 285. Marei MM, Fares AE, Musa N, Abdelsattar AH, Sharaf A, Hassan MM, Elkotby M, Eltagy G, Hafez M, Elbarbary MM. Timing and outcome concerns regarding feminizing genitoplasty from the perspective of Egyptian families of girls with virilized external genitalia. Horm Res Paediatr. 2016;85(1):49–57. [DOI] [PubMed] [Google Scholar]
- 286. Martínez-Criado Y, Gómez AL, Fernández-Hurtado MA, Barrero R, García-Merino F. [Role of pediatric urologist in the treatment of congenital adrenal hyperplasia: a study of satisfaction and psychosocial aspects]. Cir Pediatr. 2013;26(2):75–80. [PubMed] [Google Scholar]
- 287. Sturm RM, Durbin-Johnson B, Kurzrock EA. Congenital adrenal hyperplasia: current surgical management at academic medical centers in the United States. J Urol. 2015;193(5Suppl):1796–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Roth JD, Casey JT, Whittam BM, Bennett WE Jr, Szymanski KM, Cain MP, Rink RC. Characteristics of female genital restoration surgery for congenital adrenal hyperplasia using a large-scale administrative database. Urology. 2018;115:162–167. [DOI] [PubMed] [Google Scholar]
- 289. Sun LS, Li G, Miller TL, Salorio C, Byrne MW, Bellinger DC, Ing C, Park R, Radcliffe J, Hays SR, DiMaggio CJ, Cooper TJ, Rauh V, Maxwell LG, Youn A, McGowan FX. Association between a single general anesthesia exposure before age 36 months and neurocognitive outcomes in later childhood. JAMA. 2016;315(21):2312–2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Backeljauw B, Holland SK, Altaye M, Loepke AW. Cognition and brain structure following early childhood surgery with anesthesia. Pediatrics. 2015;136(1):e1–e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291. Merke DP, Poppas DP. Management of adolescents with congenital adrenal hyperplasia. Lancet Diabetes Endocrinol. 2013;1(4):341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Szymanski KM, Whittam B, Kaefer M, Frady H, Casey JT, Tran VT, Cain MP, Rink RC. Parental decisional regret and views about optimal timing of female genital restoration surgery in congenital adrenal hyperplasia. J Pediatr Urol. 2018;14(2):156.e1–156.e7. [DOI] [PubMed] [Google Scholar]
- 293. Auchus RJ, Witchel SF, Leight KR, Aisenberg J, Azziz R, Bachega TA, Baker LA, Baratz AB, Baskin LS, Berenbaum SA, Breault DT, Cerame BI, Conway GS, Eugster EA, Fracassa S, Gearhart JP, Geffner ME, Harris KB, Hurwitz RS, Katz AL, Kalro BN, Lee PA, Alger Lin G, Loechner KJ, Marshall I, Merke DP, Migeon CJ, Miller WL, Nenadovich TL, Oberfield SE, Pass KA, Poppas DP, Lloyd-Puryear MA, Quigley CA, Riepe FG, Rink RC, Rivkees SA, Sandberg DE, Schaeffer TL, Schlussel RN, Schneck FX, Seely EW, Snyder D, Speiser PW, Therrell BL, Vanryzin C, Vogiatzi MG, Wajnrajch MP, White PC, Zuckerman AE. Guidelines for the development of comprehensive care centers for congenital adrenal hyperplasia: guidance from the CARES Foundation initiative. Int J Pediatr Endocrinol. 2010;2010(1):275213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Peña A. Total urogenital mobilization–an easier way to repair cloacas. J Pediatr Surg. 1997;32(2):263–267. [DOI] [PubMed] [Google Scholar]
- 295. Kalfa N, Liu B, Cao M, Vilella M, Hsieh M, Baskin LS. 3-Dimensional neuroanatomy of the human fetal pelvis: anatomical support for partial urogenital mobilization in the treatment of urogenital sinus. J Urol. 2008;180(4 Suppl):1709–1714. [DOI] [PubMed] [Google Scholar]
- 296. Poppas DP. Clitoroplasty in congenital adrenal hyperplasia: description of technique. Adv Exp Med Biol. 2011;707:49–50. [DOI] [PubMed] [Google Scholar]
- 297. Peña A. The surgical management of persistent cloaca: results in 54 patients treated with a posterior sagittal approach. J Pediatr Surg. 1989;24(6):590–598. [DOI] [PubMed] [Google Scholar]
- 298. Rink RC, Adams MC. Feminizing genitoplasty: state of the art. World J Urol. 1998;16(3):212–218. [DOI] [PubMed] [Google Scholar]
- 299. Rink RC, Herndon CD, Cain MP, Kaefer M, Dussinger AM, King SJ, Casale AJ. Upper and lower urinary tract outcome after surgical repair of cloacal malformations: a three-decade experience. BJU Int. 2005;96(1):131–134. [DOI] [PubMed] [Google Scholar]
- 300. Rink RC, Metcalfe PD, Cain MP, Meldrum KK, Kaefer MA, Casale AJ. Use of the mobilized sinus with total urogenital mobilization. J Urol. 2006;176(5):2205–2211. [DOI] [PubMed] [Google Scholar]
- 301. Stites J, Bernabe KJ, Galan D, Felsen D, Poppas DP. Urinary continence outcomes following vaginoplasty in patients with congenital adrenal hyperplasia. J Pediatr Urol. 2017;13(1):38.e1–38.e7. [DOI] [PubMed] [Google Scholar]
- 302. Podesta M, Urcullo J. Perineal mobilization of the common urogenital sinus for surgical correction of high urethrovaginal confluence in patients with intersex disorders. J Pediatr Urol. 2008;4(5):352–358. [DOI] [PubMed] [Google Scholar]
- 303. Ludwikowski BM, González R. The surgical correction of urogenital sinus in patients with DSD: 15 years after description of total urogenital mobilization in children. Front Pediatr. 2013;1:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Salle JL, Lorenzo AJ, Jesus LE, Leslie B, AlSaid A, Macedo FN, Jayanthi VR, de Castro R. Surgical treatment of high urogenital sinuses using the anterior sagittal transrectal approach: a useful strategy to optimize exposure and outcomes. J Urol. 2012;187(3):1024–1031. [DOI] [PubMed] [Google Scholar]
- 305. Baskin LS, Erol A, Li YW, Liu WH, Kurzrock E, Cunha GR. Anatomical studies of the human clitoris. J Urol. 1999;162(3 Pt 2):1015–1020. [DOI] [PubMed] [Google Scholar]
- 306. Debono M, Ghobadi C, Rostami-Hodjegan A, Huatan H, Campbell MJ, Newell-Price J, Darzy K, Merke DP, Arlt W, Ross RJ. Modified-release hydrocortisone to provide circadian cortisol profiles. J Clin Endocrinol Metab. 2009;94(5):1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307. Weitzman ED, Fukushima D, Nogeire C, Roffwarg H, Gallagher TF, Hellman L. Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. J Clin Endocrinol Metab. 1971;33(1):14–22. [DOI] [PubMed] [Google Scholar]
- 308. Bryan SM, Honour JW, Hindmarsh PC. Management of altered hydrocortisone pharmacokinetics in a boy with congenital adrenal hyperplasia using a continuous subcutaneous hydrocortisone infusion. J Clin Endocrinol Metab. 2009;94(9):3477–3480. [DOI] [PubMed] [Google Scholar]
- 309. Merza Z, Rostami-Hodjegan A, Memmott A, Doane A, Ibbotson V, Newell-Price J, Tucker GT, Ross RJ. Circadian hydrocortisone infusions in patients with adrenal insufficiency and congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 2006;65(1):45–50. [DOI] [PubMed] [Google Scholar]
- 310. Nella AA, Mallappa A, Perritt AF, Gounden V, Kumar P, Sinaii N, Daley LA, Ling A, Liu CY, Soldin SJ, Merke DP. A phase 2 study of continuous subcutaneous hydrocortisone infusion in adults with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2016;101(12):4690–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311. Verma S, Vanryzin C, Sinaii N, Kim MS, Nieman LK, Ravindran S, Calis KA, Arlt W, Ross RJ, Merke DP. A pharmacokinetic and pharmacodynamic study of delayed- and extended-release hydrocortisone (Chronocort) vs. conventional hydrocortisone (Cortef) in the treatment of congenital adrenal hyperplasia. Clin Endocrinol (Oxf). 2010;72(4):441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Mallappa A, Sinaii N, Kumar P, Whitaker MJ, Daley LA, Digweed D, Eckland DJ, Van Ryzin C, Nieman LK, Arlt W, Ross RJ, Merke DP. A phase 2 study of Chronocort, a modified-release formulation of hydrocortisone, in the treatment of adults with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2015;100(3):1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313. Neumann U, Whitaker MJ, Wiegand S, Krude H, Porter J, Davies M, Digweed D, Voet B, Ross RJ, Blankenstein O. Absorption and tolerability of taste-masked hydrocortisone granules in neonates, infants and children under 6 years of age with adrenal insufficiency. Clin Endocrinol (Oxf). 2018;88(1):21–29. [DOI] [PubMed] [Google Scholar]
- 314. Laue L, Merke DP, Jones JV, Barnes KM, Hill S, Cutler GB Jr. A preliminary study of flutamide, testolactone, and reduced hydrocortisone dose in the treatment of congenital adrenal hyperplasia. J Clin Endocrinol Metab. 1996;81(10):3535–3539. [DOI] [PubMed] [Google Scholar]
- 315. Merke DP, Keil MF, Jones JV, Fields J, Hill S, Cutler GB Jr. Flutamide, testolactone, and reduced hydrocortisone dose maintain normal growth velocity and bone maturation despite elevated androgen levels in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2000;85(3):1114–1120. [DOI] [PubMed] [Google Scholar]
- 316. Garrido M, Peng HM, Yoshimoto FK, Upadhyay SK, Bratoeff E, Auchus RJ. A-ring modified steroidal azoles retaining similar potent and slowly reversible CYP17A1 inhibition as abiraterone. J Steroid Biochem Mol Biol. 2014;143:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317. de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, Chi KN, Jones RJ, Goodman OB Jr, Saad F, Staffurth JN, Mainwaring P, Harland S, Flaig TW, Hutson TE, Cheng T, Patterson H, Hainsworth JD, Ryan CJ, Sternberg CN, Ellard SL, Fléchon A, Saleh M, Scholz M, Efstathiou E, Zivi A, Bianchini D, Loriot Y, Chieffo N, Kheoh T, Haqq CM, Scher HI; COU-AA-301 Investigators . Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364(21):1995–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Ryan CJ, Molina A, Griffin T. Abiraterone in metastatic prostate cancer. N Engl J Med. 2013;368(15):1458–1459. [DOI] [PubMed] [Google Scholar]
- 319. Auchus RJ, Buschur EO, Chang AY, Hammer GD, Ramm C, Madrigal D, Wang G, Gonzalez M, Xu XS, Smit JW, Jiao J, Yu MK. Abiraterone acetate to lower androgens in women with classic 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2014;99(8):2763–2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. New MI, Gertner JM, Speiser PW, Del Balzo P. Growth and final height in classical and nonclassical 21-hydroxylase deficiency. J Endocrinol Invest. 1989;12(8Suppl 3):91–95. [PubMed] [Google Scholar]
- 321. Weintrob N, Dickerman Z, Sprecher E, Galatzer A, Pertzelan A. Non-classical 21-hydroxylase deficiency in infancy and childhood: the effect of time of initiation of therapy on puberty and final height. Eur J Endocrinol. 1997;136(2):188–195. [DOI] [PubMed] [Google Scholar]
- 322. Quintos JBQ, Vogiatzi MG, Harbison MD, New MI. Growth hormone therapy alone or in combination with gonadotropin-releasing hormone analog therapy to improve the height deficit in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001;86(4):1511–1517. [DOI] [PubMed] [Google Scholar]
- 323. Lin-Su K, Vogiatzi MG, Marshall I, Harbison MD, Macapagal MC, Betensky B, Tansil S, New MI. Treatment with growth hormone and luteinizing hormone releasing hormone analog improves final adult height in children with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2005;90(6):3318–3325. [DOI] [PubMed] [Google Scholar]
- 324. Dacou-Voutetakis C, Karidis N. Congenital adrenal hyperplasia complicated by central precocious puberty: treatment with LHRH-agonist analogue. Ann N Y Acad Sci. 1993;687:250–254. [DOI] [PubMed] [Google Scholar]
- 325. Turcu AF, Spencer-Segal JL, Farber RH, Luo R, Grigoriadis DE, Ramm CA, Madrigal D, Muth T, O’Brien CF, Auchus RJ. Single-dose study of a corticotropin-releasing factor receptor-1 antagonist in women with 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2016;101(3):1174–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Bry-Gauillard H, Cartes A, Young J. Mitotane for 21-hydroxylase deficiency in an infertile man. N Engl J Med. 2014;371(21):2042–2044. [DOI] [PubMed] [Google Scholar]
- 327. Cheng Y, Kerppola RE, Kerppola TK. ATR-101 disrupts mitochondrial functions in adrenocortical carcinoma cells and in vivo. Endocr Relat Cancer. 2016;23(4):1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. LaPensee CR, Mann JE, Rainey WE, Crudo V, Hunt SW III, Hammer GD. ATR-101, a selective and potent inhibitor of acyl-CoA acyltransferase 1, induces apoptosis in H295R adrenocortical cells and in the adrenal cortex of dogs. Endocrinology. 2016;157(5):1775–1788. [DOI] [PubMed] [Google Scholar]
- 329. Sbiera S, Leich E, Liebisch G, Sbiera I, Schirbel A, Wiemer L, Matysik S, Eckhardt C, Gardill F, Gehl A, Kendl S, Weigand I, Bala M, Ronchi CL, Deutschbein T, Schmitz G, Rosenwald A, Allolio B, Fassnacht M, Kroiss M. Mitotane inhibits sterol-O-acyl transferase 1 triggering lipid-mediated endoplasmic reticulum stress and apoptosis in adrenocortical carcinoma cells. Endocrinology. 2015;156(11):3895–3908. [DOI] [PubMed] [Google Scholar]
- 330. Van Wyk JJ, Ritzen EM. The role of bilateral adrenalectomy in the treatment of congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2003;88(7):2993–2998. [DOI] [PubMed] [Google Scholar]
- 331. Ogilvie CM, Rumsby G, Kurzawinski T, Conway GS. Outcome of bilateral adrenalectomy in congenital adrenal hyperplasia: one unit’s experience. Eur J Endocrinol. 2006;154(3):405–408. [DOI] [PubMed] [Google Scholar]
- 332. Tiosano D, Vlodavsky E, Filmar S, Weiner Z, Goldsher D, Bar-Shalom R. Ovarian adrenal rest tumor in a congenital adrenal hyperplasia patient with adrenocorticotropin hypersecretion following adrenalectomy. Horm Res Paediatr. 2010;74(3):223–228. [DOI] [PubMed] [Google Scholar]
- 333. Crocker MK, Barak S, Millo CM, Beall SA, Niyyati M, Chang R, Avila NA, Van Ryzin C, Segars J, Quezado M, Merke DP. Use of PET/CT with cosyntropin stimulation to identify and localize adrenal rest tissue following adrenalectomy in a woman with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2012;97(11):E2084–E2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Merke DP, Chrousos GP, Eisenhofer G, Weise M, Keil MF, Rogol AD, Van Wyk JJ, Bornstein SR. Adrenomedullary dysplasia and hypofunction in patients with classic 21-hydroxylase deficiency. N Engl J Med. 2000;343(19):1362–1368. [DOI] [PubMed] [Google Scholar]
- 335. Riepe FG, Krone N, Krüger SN, Sweep FC, Lenders JW, Dötsch J, Mönig H, Sippell WG, Partsch CJ. Absence of exercise-induced leptin suppression associated with insufficient epinephrine reserve in patients with classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Exp Clin Endocrinol Diabetes. 2006;114(3):105–110. [DOI] [PubMed] [Google Scholar]
- 336. Weise M, Mehlinger SL, Drinkard B, Rawson E, Charmandari E, Hiroi M, Eisenhofer G, Yanovski JA, Chrousos GP, Merke DP. Patients with classic congenital adrenal hyperplasia have decreased epinephrine reserve and defective glucose elevation in response to high-intensity exercise. J Clin Endocrinol Metab. 2004;89(2):591–597. [DOI] [PubMed] [Google Scholar]
- 337. Green-Golan L, Yates C, Drinkard B, VanRyzin C, Eisenhofer G, Weise M, Merke DP. Patients with classic congenital adrenal hyperplasia have decreased epinephrine reserve and defective glycemic control during prolonged moderate-intensity exercise. J Clin Endocrinol Metab. 2007;92(8):3019–3024. [DOI] [PubMed] [Google Scholar]
- 338. Kim MS, Ryabets-Lienhard A, Bali B, Lane CJ, Park AH, Hall S, Geffner ME. Decreased adrenomedullary function in infants with classical congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2014;99(8):E1597–E1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Tajima T, Okada T, Ma XM, Ramsey W, Bornstein S, Aguilera G. Restoration of adrenal steroidogenesis by adenovirus-mediated transfer of human cytochrome P450 21-hydroxylase into the adrenal gland of 21-hydroxylase-deficient mice. Gene Ther. 1999;6(11):1898–1903. [DOI] [PubMed] [Google Scholar]
- 340. Ruiz-Babot G, Hadjidemetriou I, King PJ, Guasti L. New directions for the treatment of adrenal insufficiency. Front Endocrinol (Lausanne). 2015;6:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Ruiz-Babot G, Balyura M, Hadjidemetriou I, Ajodha SJ, Taylor DR, Ghataore L, Taylor NF, Schubert U, Ziegler CG, Storr HL, Druce MR, Gevers EF, Drake WM, Srirangalingam U, Conway GS, King PJ, Metherell LA, Bornstein SR, Guasti L. Modeling congenital adrenal hyperplasia and testing interventions for adrenal insufficiency using donor-specific reprogrammed cells. Cell Reports. 2018;22(5):1236–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Meyer-Bahlburg HFL. Psychoendocrinology of congenital adrenal hyperplasia In: New MI, Lekarev O, Parsa A, Yuen TT, O’Malley BW, Hammer GD, eds. Genetic Steroid Disorders. San Diego, CA: Academic Press; 2014:285–300. [Google Scholar]
- 343. Al-Maghribi H. Congenital adrenal hyperplasia: problems with developmental anomalies of the external genitalia and sex assignment. Saudi J Kidney Dis Transpl. 2007;18(3):405–413. [PubMed] [Google Scholar]
- 344. Chowdhury TK, Laila K, Hutson JM, Banu T. Male gender identity in children with 46,XX DSD with congenital adrenal hyperplasia after delayed presentation in mid-childhood. J Pediatr Surg. 2015;50(12):2060–2062. [DOI] [PubMed] [Google Scholar]
- 345. Meyer-Bahlburg HFL. Gender monitoring and gender reassignment of children and adolescents with a somatic disorder of sex development. Child Adolesc Psychiatr Clin N Am. 2011;20(4):639–649. [DOI] [PubMed] [Google Scholar]
- 346. Lee PA, Houk CP, Husmann DA. Should male gender assignment be considered in the markedly virilized patient With 46,XX and congenital adrenal hyperplasia? J Urol. 2010;184(4Suppl):1786–1792. [DOI] [PubMed] [Google Scholar]
- 347. Meyer-Bahlburg HFL, Dolezal C, Baker SW, Ehrhardt AA, New MI. Gender development in women with congenital adrenal hyperplasia as a function of disorder severity. Arch Sex Behav. 2006;35(6):667–684. [DOI] [PubMed] [Google Scholar]
- 348. Hines M, Constantinescu M, Spencer D. Early androgen exposure and human gender development. Biol Sex Differ. 2015;6(1):3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Meyer-Bahlburg HFL, Baratz Dalke K, Berenbaum SA, Cohen-Kettenis PT, Hines M, Schober JM. Gender assignment, reassignment and outcome in disorders of sex development: update of the 2005 consensus conference. Horm Res Paediatr. 2016;85(2):112–118. [DOI] [PubMed] [Google Scholar]
- 350. Hines M, Pasterski V, Spencer D, Neufeld S, Patalay P, Hindmarsh PC, Hughes IA, Acerini CL. Prenatal androgen exposure alters girls’ responses to information indicating gender-appropriate behaviour. Philos Trans R Soc Lond B Biol Sci. 2016;371(1688):20150125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351. Endendijk JJ, Beltz AM, McHale SM, Bryk K, Berenbaum SA. Linking prenatal androgens to gender-related attitudes, identity, and activities: evidence from girls with congenital adrenal hyperplasia. Arch Sex Behav. 2016;45(7):1807–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Pasterski V, Zucker KJ, Hindmarsh PC, Hughes IA, Acerini C, Spencer D, Neufeld S, Hines M. Increased cross-gender identification independent of gender role behavior in girls with congenital adrenal hyperplasia: results from a standardized assessment of 4- to 11-year-old children. Arch Sex Behav. 2015;44(5):1363–1375. [DOI] [PubMed] [Google Scholar]
- 353. Dessens AB, Slijper FM, Drop SL. Gender dysphoria and gender change in chromosomal females with congenital adrenal hyperplasia. Arch Sex Behav. 2005;34(4):389–397. [DOI] [PubMed] [Google Scholar]
- 354. Meyer-Bahlburg HFL. Gender assignment and reassignment in intersexuality: controversies, data, and guidelines for research. Adv Exp Med Biol. 2002;511:199–223. [DOI] [PubMed] [Google Scholar]
- 355. Liao L-M, Wood D, Creighton SM. Parental choice on normalising cosmetic genital surgery. BMJ. 2015;351:h5124. [DOI] [PubMed] [Google Scholar]
- 356. Tamar-Mattis A. Patient advocate responds to DSD surgery debate. J Pediatr Urol. 2014;10(4):788–789. [DOI] [PubMed] [Google Scholar]
- 357.Sytsma SE. Ethics and Intersex. Dordrecht, Netherlands: Springer Netherlands, 2006.
- 358. Feder EK, Dreger A. Still ignoring human rights in intersex care. J Pediatr Urol. 2016;12(6):436–437. [DOI] [PubMed] [Google Scholar]
- 359. Dreger AD. Intersex and human rights: the long view. In: Sytsma SE, ed. Ethics and Intersex. Dordrecht, Netherlands: Springer; 2006:73–86.
- 360. Diamond M, Beh H.G.. The right to be wrong: sex and gender decisions. In: Sytsma SE, ed. Ethics and Intersex. Dordrecht, Netherlands: Springer; 2006:103–114.
- 361. Creighton SM. Adult outcomes of feminizing surgery. In: Sytsma SE, ed. Ethics and Intersex. Dordrecht, Netherlands: Springer; 2006:207–214.
- 362. Diamond M, Sigmundson HK. Management of intersexuality. Guidelines for dealing with persons with ambiguous genitalia. Arch Pediatr Adolesc Med. 1997;151(10):1046–1050. [DOI] [PubMed] [Google Scholar]
- 363. Wisniewski AB, Migeon CJ, Malouf MA, Gearhart JP. Psychosexual outcome in women affected by congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Urol. 2004;171(6 Pt 1):2497–2501. [DOI] [PubMed] [Google Scholar]
- 364. Fagerholm R, Santtila P, Miettinen PJ, Mattila A, Rintala R, Taskinen S. Sexual function and attitudes toward surgery after feminizing genitoplasty. J Urol. 2011;185(5):1900–1904. [DOI] [PubMed] [Google Scholar]
- 365. Zhang H, Pan J, Ji H, Wang Y, Shen W, Liu L, Lu G, Zhou Z. Long-term evaluation of patients undergoing genitoplasty due to disorders of sex development: results from a 14-year follow-up. Sci World J. 2013;2013:298015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366. Dayner JE, Lee PA, Houk CP. Medical treatment of intersex: parental perspectives. J Urol. 2004;172(4 pt 2):1762–1765. [DOI] [PubMed]
- 367. Lundberg T, Lindström A, Roen K, Hegarty P. From knowing nothing to knowing what, how and now: parents’ experiences of caring for their children with congenital adrenal hyperplasia. J Pediatr Psychol. 2017;42(5):520–529. [DOI] [PubMed] [Google Scholar]
- 368. Reisch N, Hahner S, Bleicken B, Flade L, Pedrosa Gil F, Loeffler M, Ventz M, Hinz A, Beuschlein F, Allolio B, Reincke M, Quinkler M. Quality of life is less impaired in adults with congenital adrenal hyperplasia because of 21-hydroxylase deficiency than in patients with primary adrenal insufficiency. Clin Endocrinol (Oxf). 2011;74(2):166–173. [DOI] [PubMed] [Google Scholar]
- 369. Frisén L, Nordenström A, Falhammar H, Filipsson H, Holmdahl G, Janson PO, Thorén M, Hagenfeldt K, Möller A, Nordenskjöld A. Gender role behavior, sexuality, and psychosocial adaptation in women with congenital adrenal hyperplasia due to CYP21A2 deficiency. J Clin Endocrinol Metab. 2009;94(9):3432–3439. [DOI] [PubMed] [Google Scholar]
- 370. Kuhnle U, Bullinger M. Outcome of congenital adrenal hyperplasia. Pediatr Surg Int. 1997;12(7):511–515. [DOI] [PubMed] [Google Scholar]
- 371. Kuhnle U, Bullinger M, Schwarz HP. The quality of life in adult female patients with congenital adrenal hyperplasia: a comprehensive study of the impact of genital malformations and chronic disease on female patients life. Eur J Pediatr. 1995;154(9):708–716. [DOI] [PubMed] [Google Scholar]
- 372. Sanches SA, Wiegers TA, Otten BJ, Claahsen-van der Grinten HL. Physical, social and societal functioning of children with congenital adrenal hyperplasia (CAH) and their parents, in a Dutch population. Int J Pediatr Endocrinol. 2012;2012(1):2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Johannsen TH, Ripa CPL, Mortensen EL, Main KM. Quality of life in 70 women with disorders of sex development. Eur J Endocrinol. 2006;155(6):877–885. [DOI] [PubMed] [Google Scholar]
- 374. Gilban DL, Alves Junior PA, Beserra IC. Health related quality of life of children and adolescents with congenital adrenal hyperplasia in Brazil. Health Qual Life Outcomes. 2014;12(1):107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Strandqvist A, Falhammar H, Lichtenstein P, Hirschberg AL, Wedell A, Norrby C, Nordenskjöld A, Frisén L, Nordenström A. Suboptimal psychosocial outcomes in patients with congenital adrenal hyperplasia: epidemiological studies in a nonbiased national cohort in Sweden. J Clin Endocrinol Metab. 2014;99(4):1425–1432. [DOI] [PubMed] [Google Scholar]
- 376. Han TS, Krone N, Willis DS, Conway GS, Hahner S, Rees DA, Stimson RH, Walker BR, Arlt W, Ross RJ; United Kingdom Congenital adrenal Hyperplasia Adult Study Executive (CaHASE) . Quality of life in adults with congenital adrenal hyperplasia relates to glucocorticoid treatment, adiposity and insulin resistance: United Kingdom Congenital Adrenal Hyperplasia Adult Study Executive (CaHASE). Eur J Endocrinol. 2013;168(6):887–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Engberg H, Butwicka A, Nordenström A, Hirschberg AL, Falhammar H, Lichtenstein P, Nordenskjöld A, Frisén L, Landén M. Congenital adrenal hyperplasia and risk for psychiatric disorders in girls and women born between 1915 and 2010: a total population study. Psychoneuroendocrinology. 2015;60:195–205. [DOI] [PubMed] [Google Scholar]
- 378. Falhammar H, Butwicka A, Landén M, Lichtenstein P, Nordenskjöld A, Nordenström A, Frisén L. Increased psychiatric morbidity in men with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 2014;99(3):E554–E560. [DOI] [PubMed] [Google Scholar]
- 379. Clayton PE, Miller WL, Oberfield SE, Ritzén EM, Sippell WG, Speiser; ESPE/LWPES CAH Working Group . Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrinology and the Lawson Wilkins Pediatric Endocrine Society. Horm Res. 2002;58(4):188–195. [DOI] [PubMed] [Google Scholar]
- 380. Cohen-Kettenis P, Pfafflin S. Transgenderism and Intersexuality in Childhood and Adolescence: Making Choices. Thousand Oaks, CA: Sage Publications; 2003.
- 381. Consortium on the Management of Disorders of Sex Development Handbook for Parents. Available at: www.dsdguidelines.org. Accessed 14 September 2018.
- 382. Consortium on the Management of Disorders of Sex Development Clinical Guidelines for the Management of Disorders of Sexual Differentiation in Childhood. Available at: www.dsdguidelines.org/. Accessed 14 September 2018.
- 383. Hughes IA, Houk C, Ahmed SF, Lee PA; Lawson Wilkins Pediatric Endocrine Society/European Society for Paediatric Endocrinology Consensus Group . Consensus statement on management of intersex disorders. J Pediatr Urol. 2006;2(3):148–162. [DOI] [PubMed] [Google Scholar]
- 384. Brain CE, Creighton SM, Mushtaq I, Carmichael PA, Barnicoat A, Honour JW, Larcher V, Achermann JC. Holistic management of DSD. Best Pract Res Clin Endocrinol Metab. 2010;24(2):335–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385. Martin CL, Ruble DN. Patterns of gender development. Annu Rev Psychol. 2010;61(1):353–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386. Blakemore JE, Berenbaum SA, Liben LS. Gender Development. New York, NY: Psychology Press; /Taylor & Francis Group; 2009. [Google Scholar]
- 387. Meyer-Bahlburg HFL, Khuri J, Reyes-Portillo J, New MI. Stigma in medical settings as reported retrospectively by women with congenital adrenal hyperplasia (CAH) for their childhood and adolescence. J Pediatr Psychol. 2017;42(5):496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388. Meyer-Bahlburg HFL, Dolezal C, Baker SW, New MI. Sexual orientation in women with classical or non-classical congenital adrenal hyperplasia as a function of degree of prenatal androgen excess. Arch Sex Behav. 2008;37(1):85–99. [DOI] [PubMed] [Google Scholar]
- 389. Meyer-Bahlburg HFL. Treatment guidelines for children with disorders of sex development. Neuropsychiatr Enfance Adolesc. 2008;56(6):345–349. [Google Scholar]
- 390. Money J. Sex Errors of the Body and Related Syndromes: A Guide to Counseling Children, Adolescents, and Their Families. 2nd ed. Baltimore, MD: Paul H. Brookes Publishing; 1994. [Google Scholar]
- 391. Hsu C, Rivkees SA. Congenital Adrenal Hyperplasia: A Parents’ Guide. Bloomington, IN: Author House; 2005. [Google Scholar]
- 392. Sandberg DE, Gardner M, Callens N, Mazur T; DSD-TRN Psychosocial Workgroup, the DSD-TRN Advocacy Advisory Network, and Accord Alliance . Interdisciplinary care in disorders/differences of sex development (DSD): the psychosocial component of the DSD–Translational Research Network. Am J Med Genet C Semin Med Genet. 2017;175(2):279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393. Nordenström A, Thyen U. Improving the communication of healthcare professionals with affected children and adolescents. Endocr Dev. 2014;27:113–127. [DOI] [PubMed] [Google Scholar]
- 394. McCauley E. Challenges in educating patients and parents about differences in sex development. Am J Med Genet C Semin Med Genet. 2017;175(2):293–299. [DOI] [PubMed] [Google Scholar]
- 395. Guyatt G, Oxman AD, Akl EA, Kunz R, Vist G, Brozek J, Norris S, Falck-Ytter Y, Glasziou P, DeBeer H, Jaeschke R, Rind D, Meerpohl J, Dahm P, Schünemann HJ. GRADE guidelines: 1. Introduction-GRADE evidence profiles and summary of findings tables. J Clin Epidemiol. 2011;64(4):383–394. [DOI] [PubMed] [Google Scholar]
- 396. Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, Schünemann HJ; GRADE Working Group . GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ. 2008;336(7650):924–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 397. Guyatt GH, Schünemann HJ, Djulbegovic B, Akl EA. Guideline panels should not GRADE good practice statements. J Clin Epidemiol. 2015;68(5):597–600. [DOI] [PubMed] [Google Scholar]