

Abstract
The privileged uptake of nucleosides into cells has generated interest in the development of nucleoside-analog libraries for mining new inhibitors. Of particular interest are applications in the discovery of substrate mimetic inhibitors for the growing number of identified glycan-processing enzymes in bacterial pathogens. However, the high polarity and the need for appropriate protecting group strategies for nucleosides challenges the development of synthetic approaches. Here we report an accessible, user-friendly synthesis that branches from a common solid phase-immobilized uridinyl-amine intermediate, which can be used as a starting point for diversity-oriented synthesis. We demonstrate the generation of five series of uridinyl nucleoside analogs for investigating inhibitor structure-activity relationships. This library was screened for inhibition of representative enzymes from three functional families including a phosphoglycosyl transferase, a UDP-amino sugar acetyl-transferase, and a glycosyltransferase. These candidates were taken from the Gram-negative bacteria Campylobacter concisus and Campylobacter jejuni, and the Gram-positive bacterium Clostridium difficile, respectively. Inhibition studies show that specific compound series preferentially inhibit selected enzymes, with IC50 values ranging from 35 ± 7 μM to 174 ± 21 μM. Insights from the screen provide a strong foundation for further structural elaboration, to improve potency, which will be enabled by the same synthetic strategy. The solid-phase strategy was also used to synthesize pseudouridine analogues of lead compounds. Finally, the compounds were found to be non-toxic to mammalian cells, further supporting the opportunities for future development.
Graphical Abstract
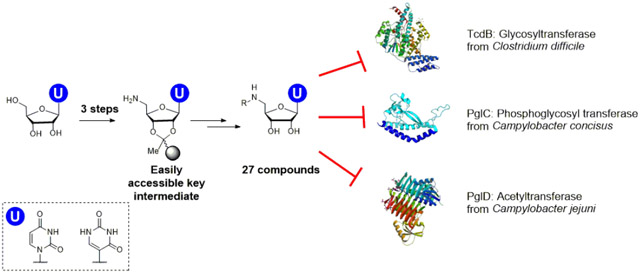
Nucleoside analogs, including both complex natural products and simpler synthetic derivatives, represent an important class of compounds with significant potential as selective tools for biological research and as therapeutic agents. Synthetic nucleosides, such as those that cause premature termination of DNA and RNA polymerases, represent a mainstay of current antiviral and antitumor therapy regimes.1-3 Nucleoside natural products show equally fascinating biological activities, exhibiting, for example, antibacterial activity by inhibition of peptidoglycan and O-antigen biosynthesis.4 A significant impediment however, to exploiting these useful bioactivities is the complexity of the natural product structures, which makes chemical manipulation to reduce toxicity and improve selectivity and pharmacokinetic properties, a major challenge. Nevertheless, nucleosides show advantageous properties for intracellular delivery.5 For example, it is known that nucleoside analogs used in the clinic are absorbed across the intestinal epithelium by nucleoside transporters,6 and many uridine derivatives, such as tunicamycin and mureidomycin, are known to be taken up into bacterial and mammalian cells.7, 8 Additionally, nucleoside derivatives often show advantageous binding of the constituent nucleobases to target proteins.9 As a result, nucleosides represent important targets, and their synthetic analogues are recurrent in the structures of antiviral, antibiotic, and antifungal agents based on competition with the binding of uridine-based substrates.10, 11
As understanding of the biological significance of bacterial glycan-modifying and processing enzymes in virulence and pathogenesis grows,12, 13 so does the need for versatile methodologies for the synthesis of small molecule inhibitors targeted at these processes. However, although the potential of selective inhibitors of these enzymes both as tools and therapeutics is clear, very few compounds have advanced beyond research applications. In many cases, inhibitor discovery for glycan-modifying and processing enzymes is hampered by a lack of structural information about the target, low target specificity, class promiscuity, and poor physicochemical properties for biological applications. A noteworthy exception is the glycosyl transferase (GT) inhibitor N-butyl deoxynojirimycin (Zavesca), which is important for the treatment of Type I Gaucher’s disease.14 However, in many cases, although many glycan-targeted natural products show excellent bioactivity, their clinical application is limited. For example, the potent bacterial GT inhibitor moenomycin shows very slow development of antibiotic resistance in in vitro experiments, but its pharmacokinetic properties limit development, and it has been primarily used in animal feed stocks.15
Many bacterial GTs, phosphoglycosyl transferases (PGTs), and sugar-modifying enzymes utilize uridine diphosphate (UDP)-activated sugar substrates, therefore there is considerable interest in UDP-sugar analogs as substrate mimetics.16 A review by Gloster and Vocadlo examined classes of carbohydrate-like and medicinal chemistry-like inhibitors for glycosyl hydrolases and transferases, delineating the advantages and disadvantages of various approaches.17 Polar molecules tend to have reduced nonspecific protein binding, and the carbohydrate-like inhibitors can display excellent potency against glycan-processing enzymes, albeit often accompanied by class promiscuity with off-target effects that can be challenging to overcome. Additionally, high aqueous solubility may limit permeability through cellular membranes. Alternatively, inhibitors derived from library-screening approaches do not depend on the same protein interactions exploited by native substrates and are less likely to display class promiscuity. However, compounds from such screens are often highly lipophilic and poorly soluble, which limits use in cells and in vivo and furthermore, show a higher likelihood of general target promiscuity.18 As an alternative approach, inhibitor discovery efforts have focused on analogs of donor or acceptor substrates of these enzymes, wherein the negatively-charged phosphate functionality is replaced with an isostere. Additional review articles have compared substrate-like and nonsubstrate-like inhibitors in the context of GTs.19, 20
The screening of modified uridine-based compounds in a variety of organisms and pathways has been the subject of several reports. Approaches developed through solution-phase21-27 and solid-phase28-30 chemistry, have resulted in multiple lead compounds. In particular, solid-phase approaches have ranged from small, focused libraries for specific enzymes29 to large libraries prepared using microscale approaches that were not subject to inhibition analysis.28, 30
Here we present development of a versatile solid-phase synthesis (SPS) platform that can be customized to target nucleoside analog inhibitors of GTs, PGTs and other sugar-modifying enzymes, which builds on advantageous aspects of previous approaches. The SPS methodology accommodates phosphate mimetics, is free from multiple purification steps and protecting group manipulations and is readily adapted to provide compounds for analysis with diverse enzymes that act on UDP-sugars. The SPS approach proceeds through three to five reactions from a common building block. Following assembly, compounds are cleaved from the solid support and purified by reverse phase HPLC (RP-HPLC), yielding desired products in good overall yields (4-25%). Compounds are then assayed in the context of selected glycan-processing enzymes for inhibitory activity, yielding multiple potential leads.
RESULTS AND DISCUSSION
Description of enzyme targets and SPS strategy design.
We present the SPS of five series of uridine analogs and examine activity with three types of bacterial glycan biosynthesis enzymes of interest to the community. GTs such as TcdA and TcdB from C. difficile play crucial roles in toxin release.31 PGTs such as PglC from C. jejuni are essential for bacterial N-linked protein glycosylation,32 which is important for adhesion to and invasion into human intestinal epithelial cells.33 Finally, UDP-aminosugar acetyltransferases such as PglD from the C. jejuni pathway are also important for virulence.34 PglD has recently been the subject of small molecule, non-nucleoside, inhibitor development efforts.35
Each of the series of uridine analogs was designed to investigate multiple structure-activity relationships (SAR) (Figure 1). These include: 1) Variation of the distance between the uridine and an aryl group that was designed to occupy the sugar-binding site of the UDP-sugar; 2) functionality, provided by amino acid side chains, that include metal ion-binding or metal ion-displacing groups; 3) a bifunctional squaramide moiety, which represents a well-studied diphosphate mimetic; 4) variation of the aryl (sugar-binding site occupant) group including size, electron density and hydrogenbonding properties, which is introduced using copper-catalyzed azide-alkyne cycloaddition (CuAAC). By exploiting several points of diversification, the goal is to access different sets of inhibitors, which are active in distinct glycan-modifying enzymes. If the different sets of molecules are effective inhibitors of specific enzyme families, this would expand the utility and customizability of the SPS method towards given targets. If however, the same set of compounds are found to be effective towards different enzymes, such ‘frequent hitters’ would be avoided, as this promiscuity would suggest non-specific activity, and would not be valuable as leads for optimization.36
Figure 1.

Library of uridine analogues showing the scaffolds of series 1-5 compounds and details of the variable R groups (upper right).
The SPS methodology was also applied to the synthesis of compounds containing pseudouridine, thus expanding the repertoire and potential scope of the approach. As the most abundant modification in RNA,37 there is considerable interest in the properties of this nucleoside in therapeutics, and recently pseudouridimycin was found to inhibit bacterial RNA polymerase in otherwise drug-resistant pathogens.2 Since its discovery in the 1950s, pseudouridinyl analogs have been relatively understudied, due to cost and poor synthetic accessibility.
Solid-phase synthesis of uridine analog library.
The synthesis is based on the approach developed by Epple et al.30 The optimized route features resin immobilization via the ribose 2’ and 3’ hydroxyl groups as a stable aliphatic acetal and an efficient, resin-based route from the 5’-hydroxyl intermediate to the 5’-amino-uridine, 10. In general, uracil protecting groups were avoided, to simplify the overall schemes; thus the initial steps were optimized to minimize side reactions at the N-3 position. This immobilized intermediate serves as the common starting point for diversification and has been prepared on a 3.0 mmol scale, which provides sufficient material for the preparation of ten nucleoside analogs that were aliquoted into 10 mM stock solutions sufficient for hundreds of assays. The purity of each resin-bound intermediate was assessed by LC-MS analysis. To prepare 10, the keto ester 7 was synthesized by O-alkylation of 4-(4-hydroxyphenyl)-2-butanone 6 with 5-bromovaleric acid methyl ester (Scheme 1). The ketone was activated with trimethylorthoformate (TMOF) and p-toluenesulfonic acid (pTsOH), which underwent condensation with the ribose diol to yield the acetal 8. Ester hydrolysis provided the uridine-carboxylate 9. The carboxylate was immobilized through amide bond formation with H-Ala-Wang polystyrene resin using 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU) and N,N-diisopropylethylamine (DIPEA). The 5’-OH of the uridine was then converted to the corresponding 5’-NH2, by means of a Mitsunobu reaction with 3,4,5,6-tetrachlorophthalimide (TCP), followed by treatment with ethylenediamine (EDA) in dimethylformamide (DMF) to afford the on-resin 5’-NH2 uridine 10 (see Scheme 1).
Scheme 1.
Synthesis of common intermediate 10 from 4-(4-hydroxyphenyl)-2-butanonea

aReagents and conditions: (a) 5-bromovaleric acid methyl ester, K2CO3, 50 °C, DMF, 96%; (b) i: trimethylorthoformate, p-TsOH, MeOH; ii: Uridine, 2M HCl in dioxane, DMF; (c) LiOH, MeOH/H2O, 97%, 2 steps; (d) H-Ala-Wang resin, HBTU, DIPEA, DMF; (e) DIAD, PPh3, tetrachlorophthalimide, DMF/THF; (f) EDA, DMF.
Starting from 10, nucleoside analogs 1-5 were assembled in either three or five on-resin manipulations (Scheme 2). Series 1 and 2 (compounds 1a-e and 2a-e) were synthesized by standard amide coupling of 10 with either α-azido-Lys(Boc)-OH or 2-azidoacetic acid, then CuAAC using the tris-(3-hydroxylpropyltriazolyl)methylamine (THPTA) ligand and commercially-available terminal alkynes. Finally, global deprotection with 95% TFA/2.5% triiso-propylsilane (TIS)/2.5% H2O afforded the two series with the shortest linker lengths. Series 3 and 4 (compounds 3a-e and 4a-e) were synthesized by amide coupling of 10 to N-α-Fmoc-protected amino acids (here Fmoc-Lys(Boc)-OH or Fmoc-Glu(OtBu)-OH), Fmoc deprotection with 20% piperidine in DMF, followed by amide coupling with 2-azidoacetic acid, then CuAAC with diverse terminal alkynes, and finally deprotection from the resin. The squaramide series 5 (compounds 5a-c and 5f) was generated via two sequential nucleophilic substitutions on diethyl squarate, first with the 5’-amino-uridine 10, followed by addition of a benzyl or homobenzylic primary amine. Resin deprotection was carried out as with Series 1-4.
Scheme 2.
Synthesis of analogs 1-5 from common intermediate 10

aReagents and conditions: (a) Fmoc-Lys(Boc)-OH or Fmoc-Glu(OtBu)-OH, HBTU, DIPEA, DMF; (b) 20% piperidine in DMF; (c) 2-Azido-acetic acid or α-azido-Lys(Boc)-OH, HBTU, DIPEA, DMF; (d) THPTA, CuSO4, sodium ascorbate, alkyne-R; (e) 95/2.5/2.5 TFA:TIS:H2O; (f) diethyl squarate, Et3N, DMF; (g) R-NH2, Et3N, CH2Cl2.
All crude products were then precipitated from the cleavage cocktail with cold ether, resuspended in 20% acetonitrile in water, and purified by RP-HPLC to yield the final products 1-5. Overall compound yields were 4-25% based on the initial loading of carboxylate 9 on the resin.
Enzyme inhibition by uridine analog library.
Purified compounds were assayed for inhibition activity against the three target enzymes using convenient assays in a multiwell plate format. The assays included the UDP-Glo GT assay (Promega), which detects production of UDP, the UMP/CMP-Glo PGT assay (Promega)38 which detects UMP release, and the amino-sugar acetyltransferase assay based on Ellman’s reagent (DTNB), which is applied to monitor the release of coenzyme A (CoASH) upon acyl transfer.39
Inhibition of PglC Measured by Luminescence Using the UMP-Glo Assay.
Bacterial PGTs catalyze transfer of a phosphosugar from a UDP-sugar to undecaprenol phosphate (Und-P) with the release of UMP.40 In C. jejuni, the monotopic PGT PglC catalyzes the first membrane-committed step in N-linked glycosylation, and involves transfer of di-N-acetylbacillosamine (diNAcBac) phosphate from UDP-diNAcBac to Und-P (Figure S1). Recently, the structure of a PglC homolog from C. concisus, which shares 72% sequence homology with the C. jejuni enzyme has been reported.41 Therefore, in the interests of later applying structure-driven nucleoside analog inhibitor optimization, the C. concisus enzyme was pursued in these inhibition studies. The UMP-Glo assay is based on a previously-reported procedure,23 and begins with pre-incubation of PglC in the reaction buffer with Und-P and 100 μM inhibitor for 10 minutes at room temperature (RT). Upon addition of UDP-diNAcBac, the reaction was allowed to proceed for 15 min before quenching with the detection reagent and was transferred into a 96-well plate and luminescence data was collected. Assay results were plotted as the percentage of remaining activity compared to the positive control (no inhibitor).
We note that the Promega Glo assays detect free UMP or UDP generated during the course of the PGT or GT reactions. However, due to the structural similarity between synthesized inhibitors and the UDP and UMP-detecting enzymes used in the Glo assays, we also controlled for off-target inhibition of the Glo reagent enzymes. Glo assay control experiments were first conducted in the presence and absence of inhibitors at 2 μM UMP (for PglC) or 1 μM UDP (for TcdB). These concentrations of UMP and UDP represent the amount of nucleotide released in a typical assay.38 Using this information the percent of background inhibition was used to adjust the luminescence readout.
Figure 2 shows the background-corrected inhibition of PglC in the presence of the 24 inhibitors. A clear grouping from the glutamic acid Series 4 (4a-e) show marked inhibition of PglC relative to the other compound series. In particular, compounds 4b and 4e (solid bars), are the most active. Further evaluation of inhibition over a range of concentrations showed IC50 values of 72 ± 7 μM for compound 4b and 116 ± 18 μM for compound 4e (Figure S2). Additionally, Lineweaver-Burk analysis supported competitive binding of 4b (Figure S3). This observation is consistent with previous work which showed that negatively-charged metal-coordinating groups provide greater inhibitory activity. 23 This informs us that further improvement of potency might be achieved through optimization of the metal-binding moiety. Along with the recently obtained crystal structure of PglC,41 a more refined SAR around the sugar mimetic should also allow for the synthesis of more potent inhibitors, readily obtained from the SPS strategy. Finally, this information, could potentially provide access to small molecules that disrupt protein glycosylation in related pathogens.
Figure 2.
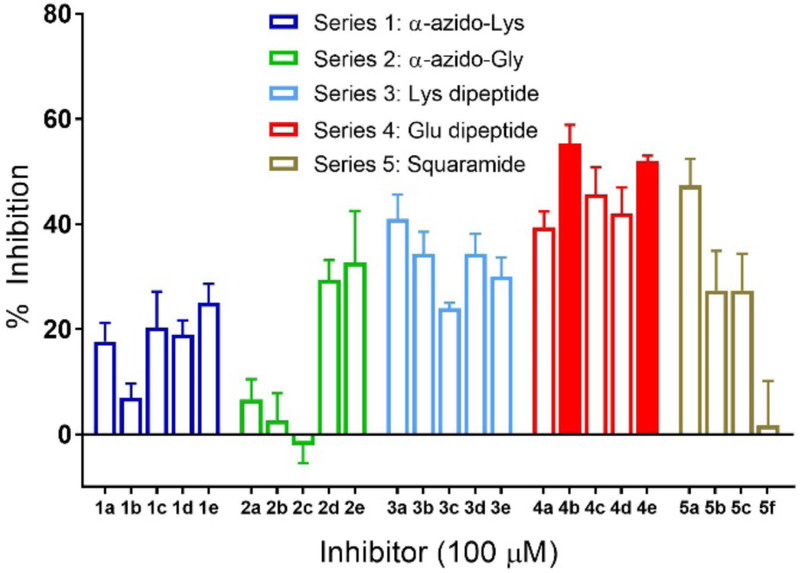
PglC activity was measured by UMP-Glo monitoring luminescence after 10 min pre-incubation with inhibitor and a 15-min reaction with UDP-diNAcBac, in reference to control with no inhibitor. Solid bars indicate compounds with the highest inhibitory activity. Error bars indicate mean ± SD; n=3.
Inhibition of PglD Measured Using the Ellman’s Reagent Assay.
PglD from C. jejuni is a prokaryote-specific UDP-amino sugar acetyltransferase (Figure S1).39 For inhibition studies, PglD was pre-incubated in the reaction buffer with 100 μM inhibitor for 20 minutes at RT, followed by addition of UDP-4-amino-NAcBac, acetyl-CoA, and DTNB. Initial rates were determined by monitoring absorbance at 412 nm in the 6-minute linear portion of the reaction curve, and compared against a positive control without inhibitor. Figure 3 shows the activity of PglD in the presence of the 24 inhibitors. In this case, compound 2b emerged as the best lead. Assays in the presence of a range of concentrations of 2b afforded an IC50 of 35 ± 7 pM (Figure S2). However, Lineweaver-Burk analysis in this case suggests that 2b is a non-competitive inhibitor of PglD (Figure S4, Table S1). Co-crystallization of 2b with PglD will be valuable to understand the binding mode, which can then be used to refine the design of structurally-targeted inhibitors. The ability of compound 2b to disrupt enzymatic activity provides multiple points of variation for the next generation of inhibitors, which would readily accessible through the SPS method. Combined with the recent development of potent nM inhibitors of the acetyl-CoA binding pocket,35 a new class of inhibitors bearing a nucleoside, which may exhibit privileged entry into target bacterial cells, may advance in vivo inhibition studies with C. jejuni.
Figure 3.

PglD activity measured by CoASH release using DTNB. Percent inhibition was determined after 20 min pre-incubation with inhibitor and a 6-min reaction with UDP-4-amino sugar, in reference to control with no inhibitor. Solid bars indicate compounds with the highest inhibitory activity. Error bars indicate mean ± SD; n=3.
Inhibition of TcdB-GTD Measured by Luminescence Using the UDP-Glo Assay.
The primary virulence factors of C. difficile are Toxins A and B (TcdA and TcdB) proteins, which belong to the family of multidomain clostridial glucosylating toxins that inactivate small GTPases, which are key players in eukaryotic signaling.31 Inactivation is the direct consequence of threonine glycosylation in RhoA-C, RhoG, Rac1, and Cdc42 proteins by the N-terminal glucosyltransferase domain (GTD) of the toxins.31 In the inhibition studies we used the catalytically-active TcdB-GTD domain with Cdc42-His6 as the glycosyl acceptor and followed previously-reported assay conditions.42 TcdB-GTD was pre-incubated with Cdc42-His6 and 100 μM inhibitor for 10 minutes at RT. Upon addition of UDP-Glc, the reaction was allowed to proceed for 5 min before quenching with detection reagents and was transferred into a 96-well plate and luminescence data collected. Assay results were plotted as the percentage of remaining activity compared to the positive control (no inhibitor).
Figure 4 shows the percent activity of each GT reaction in the presence of the 24 inhibitors. In this case, two of the squaric acid derivatives (5c and 5f) from series 5 showed the most significant inhibition. Additionally, a single member of series 2 (2e), was also active. The lowest IC50 value was 174 ± 21 μM for squaric compound 5c (Figure S2), for which a Lineweaver-Burk plot indicated competitive binding (Figure S5). Analysis of the crystal structure of TcdB-GTD shows a very deep and structured binding site with little space for extra substitution near the 5’-substituent of the uridine, but with more open space near the glucose binding site of the UDP-glucose substrate.43 From this we can reason that extended structures, as in series 3 and 4 and analogs with longer amino acid side chains, as in series 1, 3, and 4, might not compete efficiently with the native substrate. Conversely, analogs with unsubstituted amino acids (series 2 and 5) featured a few compounds with inhibitory potential, and would be a focus of further syntheses. TcdB-GTD contains the GT-A specific DXD motif, where two catalytically-essential aspartates bind Mn2+, which then binds to the diphosphate of UDP-Glc.44 An inhibitor scaffold which binds divalent cations would then be highly desirable, making squaramide compounds promising, and informing on future directions. This approach opens the possibility of developing competitive inhibitors of the glycosyltransferase domain in C. difficile toxins, to complement other efforts that have focused on noncompetitive inhibitors.42
Figure 4.
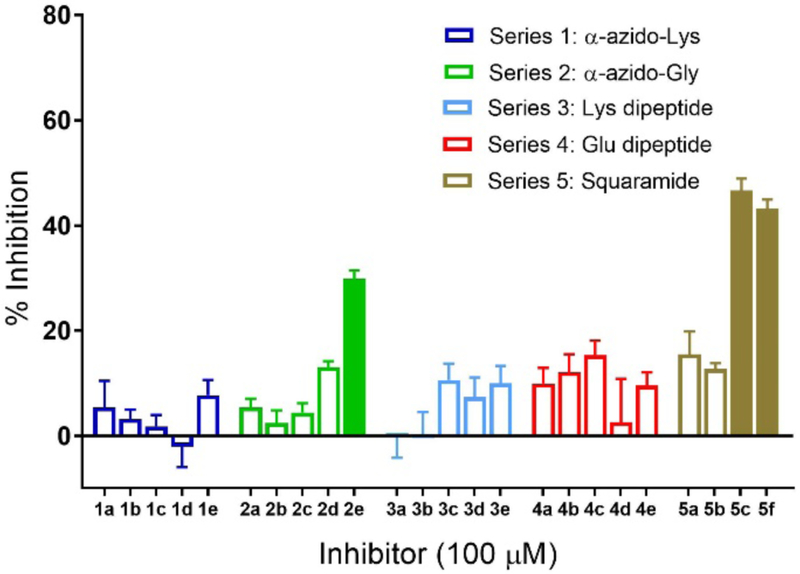
TcdB-GTD activity was measured by UDP-Glo monitoring luminescence. Substrate conversion was measured after 10 min pre-incubation with inhibitor followed by a 5-min reaction time with UDP-Glc. Conversions are normalized to control reaction without inhibitor. Solid bars indicate compounds with the highest inhibitory activity. Error bars indicate mean ± SD; n=3.
Synthesis of Pseudouridine-containing Inhibitor Compounds.
Following screening of the 24-membered nucleoside analog library at a fixed concentration, three of the promising leads were resynthesized as the corresponding pseudouridinyl variants (Scheme S1). The SPS and purification followed the established protocols for the uridine analogs (Scheme S1). Overall, the three compounds, PU2e, PU4b and PU4e, showed similar inhibitory activity relative to the corresponding uridine analogs (Figure 5). This indicates that the uracil binding sites of these enzymes may allow for this modification without a detrimental impact on binding, perhaps by binding to the face of the uracil that shares a similar hydrogen-bond “acceptor-donor-acceptor” pattern. This finding gives way to the opportunity to pursue pseudouridinyl variants of any potent hits identified in the future, which could provide a means of reducing off-target effects in inhibitor development for cell-based studies.
Figure 5.

A. Structures of pseudouridinyl inhibitors. B. Comparison of uridinyl vs pseudouridinyl inhibitors for three selected analogs referenced to control with no inhibitor. Error bars indicate mean ± SD; n=3.
Mammalian Cell Viability Measured by ATP-Glo Assay in Presence of Inhibitors.
The cytotoxicity of the presented nucleoside analogs was measured in a mammalian cell model. As membrane permeation of nucleoside analogs can occur via active transport, this is a major advantage in bacterial target accessibility, but may also be a concern as inhibitors could affect functionally-related glycan-modifying enzymes containing structurally similar active sites.16 One such example is the potent antibiotic tunicamycin which competitively inhibits WecA, but has not been used as an antibiotic in humans because of cytotoxicity in mammalian cells, due to off-target inhibition of the PGT at the inception of the dolichol pathway.40, 45 Nucleoside analogs can also act as antimetabolites that chain terminate DNA and RNA polymerization particularly with 3’-OH substitution with H, N3, F, or alkylation.1 However, the latter is unlikely to be a concern as all of the analogs are modified at ribose C-5’ site and therefore cannot be activated as a polymerase building block.
To probe for general toxicity, we examined the nucleoside analogs through the use of a well-established cell line, IMR-90 human lung fibroblasts. This cell line was chosen because it is a non-carcinoma line that grows well in culture, is often used for proof-of-principle experiments, and is a robust model with a clear cell rounding index for future toxicity-related studies.42, 46 Cell toxicity was measured using Cell Titer Glo (Promega), which is a luciferin-based assay that reports on the number of viable cells based on quantification of ATP, thus indicating the number of metabolically active cells. For the assay a 96-well plate with 20,000 cells per well was incubated overnight to allow adherence, followed by incubation with 50 μM inhibitor in complete media for 24 or 48 hours. Control wells contained either media, untreated cells, cells treated with 0.5% DMSO (inhibitor vehicle), or cells treated with 1 μg mL−1 doxorubicin (a known DNA-damage agent). The Cell Titer Glo reagent was added to induce lysis, and ATP measured by luminescence. Data was plotted as percentage of cell death compared to cells treated with vehicle (Figure S6). No significant cell death was observed after 48 h with the nucleoside analog library, indicating a lack of cytotoxicity in this cell model. In contrast, 50% of cells treated with doxorubicin were dead at 24 h, with near complete cell death at 48 h. Cells treated with 10% DMSO showed complete rounding and detachment from wells after 24 h. Cells treated with 0.5% DMSO were basically unaffected and used to normalize the data from cells treated with nucleoside analogs. Different sets of plates were used at the 24 h and 48 h time points to maintain sterility, with minimal plate variation observed in reference to controls.
Conclusions.
In summary, we have established a practical and versatile SPS method for the facile generation of uridine-based nucleoside analogs. The synthetic route will be highly accessible to a broad range of laboratories, owing to the limited number of simple steps, minimal purification of intermediates, and lack of protecting groups manipulations. The SPS approach has also been shown to be compatible with the synthesis of pseudouridine analogues, which have recently gained interest among researchers, due to promising effects against drug-resistant pathogens. The representative library of 27 uridine or pseudouridine-based compounds were assayed in plate-based formats against three different glycan-modifying enzymes: the phosphoglycosyl transferase PglC from C. concisus, the UDP-4-amino-sugar acetyltransferase PglD from C. jejuni, and the glycosyltransferase domain of TcdB from C. difficile. These three enzymes were chosen based on available structural information, to help facilitate future efforts to optimize lead compounds via a structure-driven approach.
Individual compounds exhibited increased inhibitory activity for each enzyme; the more effective compounds for TcdB did not contain amino acid side chains, whereas glutamic acid-containing constructs inhibited PglC to a greater degree, and one 3-fluoro compound showed good inhibition of PglD, with an IC50 of 35 ± 7 μM. The identification of distinct leads in each enzyme class tested is a good sign towards the minimization of class promiscuity that can plague many enzyme-targeted approaches. Each enzyme is also highly relevant to the pathogenicity of the organisms. Additional support towards minimized off-target effects was given by the lack of compound cytotoxicity in human lung fibroblasts upon 48h exposure.
These studies now provide the springboard for further adaptation of specific analogs, and can guide understanding of structural features of glycan-processing enzymes. Together, the ease of access to a broad class of customizable nucleoside analogs, along with the identification of multiple leads for further inhibitor development, provides a platform for small molecule inhibitor discovery for sugar-nucleotide modifying enzymes that have not been commonly pursued.
METHODS
UMP-Glo Assay.
PglC assays were performed using the Promega UMP-Glo assays, which detects UMP. Due to the structural similarity between the synthesized inhibitors and UMP, we also controlled for off-target inhibition of the Glo reagent enzymes by the nucleoside analogues and subtracted it from the total inhibition. The quenching solution was prepared as described by Promega. Assays contained 20 μM Und-P, 10 % DMSO, 0.1 % Triton X-100, 50 mM HEPES pH 7.5, 100 mM NaCl, 5 mM MgCl2, 20 μM UDP-diNAcBac39 and 0.2 nM PglC in a final volume of 20 uL. Inhibitors were added from a 2 mM stock in DMSO, at a final concentration of 100 μM. PglC was pre-incubated in the reaction mixture lacking UDP-diNAcBac for 10 minutes at RT. The reaction rate was predetermined to be linear over 20 minutes at the given concentrations. Upon addition of UDP-diNAcBac, the reaction was allowed to proceed for 15 min before addition of quenching solution. The reaction mixture was transferred to a 96-well plate (white, nonbinding surface, Corning). The plate was shaken at low speed for 16 min, incubated for 44 min at RT and luminescence was read using a Synergy H1 hybrid plate reader (Biotek). Data was plotted using GraphPad Prism, as percentage remaining activity compared to the positive control (no inhibitor).
Ellman’s Reagent Assay.
PglD assays were performed by monitoring the production of CoASH over the course of the reaction using DTNB. Absorbance was measured in continuous mode at 412 nm. Using a 96-well clear bottom plate, the inhibitors were added from a DMSO stock solution at a final concentration of 100 μM, followed by PglD in 50 mM HEPES, pH 7.4, 1 mM MgCl3, 0.05 mg mL−1 BSA, 0.001% Triton X-100. Inhibitors and enzyme were preincubated 20 min at RT, followed by addition of substrate and Ellman’s reagent cocktail to final concentrations of 6 nM PglD, 274 μM UDP-4-amino-NAcBac,47 295 μM AcCoA, 2 mM DTNB, and 3% DMSO in 100 μL volume. Initial rates were measured in the linear portion of the reaction curve over a 6-min time period at RT, and absorbance was read at 412 nm on the plate reader.
UDP-Glo Assay.
TcdB assays were performed using the Promega UDP-Glo assay, which detects UDP generated over the course of the reaction. Due to the structural similarity between the inhibitors and UDP, we also corrected for off-target inhibition of the Glo reagent enzymes by the nucleoside analogues. The quenching solution was prepared as described by Promega. Assays contained 30 μM Cdc42, 5% DMSO, 0.1 % Triton X-100, 50 mM HEPES pH 7.5, 100 mM KCl, 4 mM MgCl2, 1 mM MnCl2, 20 μM UDP-Glc, and 0.5 nM TcdB-GTD in a final volume of 20 μL. Inhibitors were added from a 2 mM stock in DMSO, at a final concentration of 100 μM. TcdB-GTD was pre-incubated in the reaction mixture lacking UDP-Glc for 10 minutes at RT. The reaction rate was pre-determined to be linear over 15 minutes at the given concentrations. Upon addition of UDP-Glc, the reaction was allowed to proceed for 5 min before addition of quenching solution. The reaction mixture was transferred to a 96-well plate (white, nonbinding surface, Corning). The plate was shaken at low speed for 30 s, and incubated for 1 h at RT, and luminescence was read on the plate reader. Data were plotted as in the UMP-Glo assay.
Expression and Purification of Enzymes.
PglC from C. concisus was expressed and purified as described previously.38 PglD from C. jejuni was expressed and purified following the reported procedure.35 For cloning, expression, and purification of TcdB-GTD from Clostridium difficile, see Supporting Information.
Mammalian Cell Viability.
Mammalian Cell Viability. IMR-90 human lung fibroblasts were obtained from the American Tissue Culture Collection (ATCC) and maintained in Eagle’s Modified Essential Medium (VWR) supplemented with 10% (v/v) fetal bovine serum (FBS) and Pen-Strep (P/S), in a humidified incubator with 5% CO2 and 37 °C. Proliferating cells were sub-cultured at a ratio of 1:3 when confluent, and plated in 96-well TC-treated white clear-bottom plates with lids at 20,000 cells per well, as determined by hemocytomer counting. Overnight incubation with 200 μL complete media allowed adherence of cells to the plate, which were fully confluent by microscope inspection. Growth media was removed from cells by aspiration, followed by the addition of 50 μM nucleoside analogs 1-5 in growth media with 0.5% DMSO. Additional wells instead received either 1.7 μM doxorubicin hydrochloride, 10% DMSO, or 0.5% DMSO in complete media to serve as controls for cell toxicity. Plates were then incubated for 24 h and 48 h time points. Media was then removed by aspiration, 100 μL complete media was added to each well, and 100 μL Cell Titer Glo reagent (Promega) was added to induce lysis. One row of media without cells was used as a background measurement.
Supplementary Material
ACKNOWLEDGMENT
Financial support from the NIH (R01-GM097241 to B.I.) and the MIT UROP program (to G.M.) is gratefully acknowledged. We also acknowledge the MIT Chemistry Department DCIF for NMR and HR-MS analyses.
Footnotes
Supporting Information
Experimental procedures and methods, compound characterization data, and supporting figures (PDF).
Notes
The authors declare no competing financial interests.
REFERENCES
- 1.Dasari A, Choi JS, and Berdis AJ (2016) Chapter 7 - Chemotherapeutic intervention by inhibiting DNA polymerases A2 - Kelley, Mark R, In DNA Repair in Cancer Therapy (Second Edition) (Fishel ML, Ed.), pp 179–224, Academic Press, Boston. [Google Scholar]
- 2.Maffioli SI, Zhang Y, Degen D, Carzaniga T, Del Gatto G, Serina S, Monciardini P, Mazzetti C, Guglierame P, Candiani G, Chiriac AI, Facchetti G, Kaltofen P, Sahl HG, Deho G, Donadio S, and Ebright RH (2017) Antibacterial Nucleoside-Analog Inhibitor of Bacterial RNA Polymerase, Cell 169, 1240–1248.e1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shelton J, Lu X, Hollenbaugh JA, Cho JH, Amblard F, and Schinazi RF (2016) Metabolism, biochemical actions, and chemical synthesis of anticancer nucleosides, nucleotides, and base analogs, Chem. Rev. 116, 14379–14455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ichikawa S, and Matsuda A (2007) Nucleoside natural products and related analogs with potential therapeutic properties as antibacterial and antiviral agents, Exp. Opin. Ther. Pat. 17, 487–498. [Google Scholar]
- 5.Mackey JR, Baldwin SA, Young JD, and Cass CE (1998) Nucleoside transport and its significance for anticancer drug resistance, Drug Resist. Updat. 1, 310–324. [DOI] [PubMed] [Google Scholar]
- 6.Ward JL, and Tse CM (1999) Nucleoside transport in human colonic epithelial cell lines: evidence for two Na+-independent transport systems in T84 and Caco-2 cells1, Biochim. Biophys. Acta - Biomembranes 1419, 15–22. [DOI] [PubMed] [Google Scholar]
- 7.Reiling JH, Clish CB, Carette JE, Varadarajan M, Brummelkamp TR, and Sabatini DM (2011) A haploid genetic screen identifies the major facilitator domain containing 2A (MFSD2A) transporter as a key mediator in the response to tunicamycin, Proc. Natl. Acad. Sci. USA 108, 11756–11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inukai M, Isono F, and Takatsuki A (1993) Selective inhibition of the bacterial translocase reaction in peptidoglycan synthesis by mureidomycins, Antimicrob. Agents Chemother. 37, 980–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jones S, Daley DTA, Luscombe NM, Berman HM, and Thornton JM (2001) Protein–RNA interactions: A structural analysis, Nucleic Acid. Res. 29, 943–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barb AW, Leavy TM, Robins LI, Guan Z, Six DA, Zhou P, Bertozzi CR, and Raetz CRH (2009) Uridine-based inhibitors as new leads for antibiotics targeting Escherichia coli LpxC, Biochemistry 48, 3068–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hartmann HR, and Matter A (1982) Antiproliferative action of a novel fluorinated uridine analog, 5'-deoxy-5-fluorouridine, measured in vitro and in vivo on four different murine tumor lines, Cancer Res. 42, 2412–2415. [PubMed] [Google Scholar]
- 12.Szymanski CM, and Wren BW (2005) Protein glycosylation in bacterial mucosal pathogens, Nat. Rev. Microbiol. 3, 225–237. [DOI] [PubMed] [Google Scholar]
- 13.Messner P, Schaffer C, and Kosma P (2013) Chapter 6 - Bacterial cell-envelope glycoconjugates, In Advances in Carbohydrate Chemistry and Biochemistry (Horton D, Ed.), pp 209–272, Academic Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Platt FM, Neises GR, Dwek RA, and Butters TD (1994) N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis, J. Biol. Chem. 269, 8362–8365. [PubMed] [Google Scholar]
- 15.Zuegg J, Muldoon C, Adamson G, McKeveney D, Le Thanh G, Premraj R, Becker B, Cheng M, Elliott AG, Huang JX, Butler MS, Bajaj M, Seifert J, Singh L, Galley NF, Roper DI, Lloyd AJ, Dowson CG, Cheng TJ, Cheng WC, Demon D, Meyer E, Meutermans W, and Cooper MA (2015) Carbohydrate scaffolds as glycosyltransferase inhibitors with in vivo antibacterial activity, Nat. Comm. 6, 7719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Videira PA, Marcelo F, and Grewal RK (2018) Glycosyltransferase inhibitors: a promising strategy to pave a path from laboratory to therapy, In Carbohydrate Chemistry, pp 135–158, The Royal Society of Chemistry. [Google Scholar]
- 17.Gloster TM, and Vocadlo DJ (2012) Developing inhibitors of glycan processing enzymes as tools for enabling glycobiology, Nat. Chem. Biol. 8, 683. [DOI] [PubMed] [Google Scholar]
- 18.Bruns RF, and Watson IA (2012) Rules for identifying potentially reactive or promiscuous compounds, J. Med. Chem. 55, 9763–9772. [DOI] [PubMed] [Google Scholar]
- 19.Tedaldi L, and Wagner GK (2014) Beyond substrate analogues: new inhibitor chemotypes for glycosyltransferases, MedChemComm 5, 1106–1125. [Google Scholar]
- 20.Wang S, and Vidal S (2013) Recent design of glycosyltransferase inhibitors, In Carbohydrate Chemistry, pp 78–101, The Royal Society of Chemistry. [Google Scholar]
- 21.Moukha-chafiq O, and Reynolds RC (2014) Parallel solution-phase synthesis and general biological activity of a uridine antibiotic analog library, ACS Comb. Sci. 16, 232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Winans KA, and Bertozzi CR (2002) An inhibitor of the human UDP-GlcNAc 4-epimerase identified from a uridine-based Library: A strategy to inhibit O-linked glycosylation, Chem. Biol. 9, 113–129. [DOI] [PubMed] [Google Scholar]
- 23.Walvoort MTC, Lukose V, and Imperiali B (2016) A modular approach to phosphoglycosyltransferase inhibitors inspired by nucleoside antibiotics, Chem. Eur. J. 22, 3856–3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trunkfield AE, Gurcha SS, Besra GS, and Bugg TDH (2010) Inhibition of Escherichia coli glycosyltransferase MurG and Mycobacterium tuberculosis Gal transferase by uridine-linked transition state mimics, Bioorg. Med. Chem. 18, 2651–2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang R, Steensma DH, Takaoka Y, Yun JW, Kajimoto T, and Wong CH (1997) A search for pyrophosphate mimics for the development of substrates and inhibitors of glycosyltransferases, Bioorg. Med. Chem. 5, 661–672. [DOI] [PubMed] [Google Scholar]
- 26.Wang S, Cuesta-Seijo Jose A, Lafont D, Palcic Monica M, and Vidal S (2013) Design of Glycosyltransferase Inhibitors: Pyridine as a Pyrophosphate Surrogate, Chem. Eur. J. 19, 15346–15357. [DOI] [PubMed] [Google Scholar]
- 27.Vembaiyan K, Pearcey JA, Bhasin M, Lowary TL, and Zou W (2011) Synthesis of sugar–amino acid–nucleosides as potential glycosyltransferase inhibitors, Bioorg. Med. Chem. 19, 58–66. [DOI] [PubMed] [Google Scholar]
- 28.Sun D, Jones V, Carson EI, Lee RE, Scherman MS, McNeil MR, and Lee RE (2007) Solid-phase synthesis and biological evaluation of a uridinyl branched peptide urea library, Bioorg. Med. Chem. Lett. 17, 6899–6904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bozzoli A, Kazmierski W, Kennedy G, Pasquarello A, and Pecunioso A (2000) A solid-phase approach to analogues of the antibiotic mureidomycin, Bioorg. Med. Chem. Lett. 10, 2759–2763. [DOI] [PubMed] [Google Scholar]
- 30.Epple R, Kudirka R, and Greenberg WA (2003) Solid-phase synthesis of nucleoside analogues, J. Comb. Chem. 5, 292–310. [DOI] [PubMed] [Google Scholar]
- 31.Voth DE, and Ballard JD (2005) Clostridium difficile Toxins: Mechanism of Action and Role in Disease, Clin. Microbiol. Rev. 18, 247–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Glover KJ, Weerapana E, Chen MM, and Imperiali B (2006) Direct biochemical evidence for the utilization of UDP-bacillosamine by PglC, an essential glycosyl-1-phosphate transferase in the Campylobacter jejuni N-linked glycosylation pathway, Biochemistry 45, 5343–5350. [DOI] [PubMed] [Google Scholar]
- 33.Szymanski CM, Burr DH, and Guerry P (2002) Campylobacter protein glycosylation affects host cell interactions, Infect. Immun. 70, 2242–2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kelly J, Jarrell H, Millar L, Tessier L, Fiori LM, Lau PC, Allan B, and Szymanski CM (2006) Biosynthesis of the N-linked glycan in Campylobacter jejuni and addition onto protein through block transfer, J. Bacteriol. 188, 2427–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Schutter JW, Morrison JP, Morrison MJ, Ciulli A, and Imperiali B (2017) Targeting bacillosamine biosynthesis in bacterial pathogens: Development of inhibitors to a bacterial amino-sugar acetyltransferase from Campylobacter jejuni, J. Med. Chem. 60, 2099–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rishton GM (2003) Nonleadlikeness and leadlikeness in biochemical screening, Drug Disc. Today 8, 86–96. [DOI] [PubMed] [Google Scholar]
- 37.Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, Helm M, Bujnicki JM, and Grosjean H (2013) MODOMICS: a database of RNA modification pathways--2013 update, Nucleic Acid. Res. 41, D262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das D, Walvoort MTC, Lukose V, and Imperiali B (2016) A rapid and efficient luminescence-based method for assaying phosphoglycosyltransferase enzymes, Sci. Rep. 6, 33412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olivier NB, Chen MM, Behr JR, and Imperiali B (2006) In vitro biosynthesis of UDP-N,N'-diacetylbacillosamine by enzymes of the Campylobacter jejuni general protein glycosylation system, Biochemistry 45, 13659–13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lukose V, Walvoort MTC, and Imperiali B (2017) Bacterial phosphoglycosyl transferases: initiators of glycan biosynthesis at the membrane interface, Glycobiology 27, 820–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ray LC, Das D, Entova S, Lukose V, Lynch AJ, Imperiali B, and Allen KN (2018) Membrane association of monotopic phosphoglycosyl transferase underpins function, Nat. Chem. Biol. 14, 538–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tam J, Beilhartz GL, Auger A, Gupta P, Therien AG, and Melnyk RA (2015) Small molecule inhibitors of Clostridium difficile toxin B-induced cellular damage, Chem. Biol. 22, 175–185. [DOI] [PubMed] [Google Scholar]
- 43.Reinert DJ, Jank T, Aktories K, and Schulz GE (2005) Structural Basis for the Function of Clostridium difficile Toxin B, J. Mol. Biol. 351, 973–981. [DOI] [PubMed] [Google Scholar]
- 44.Alvin JW, and Lacy DB (2017) Clostridium difficile toxin glucosyltransferase domains in complex with a non-hydrolyzable UDP-glucose analogue, J. Struct. Biol. 198, 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Duksin D, and Mahoney WC (1982) Relationship of the structure and biological activity of the natural homologues of tunicamycin, J. Biol. Chem. 257, 3105–3109. [PubMed] [Google Scholar]
- 46.Rudolph-Owen LA, Salvato K, Cheng H, Wu J, Towle MJ, and Littlefield BA (2004) A 96-well plate cell-based assay to quantify undesired cytotoxic effects against quiescent non-dividing cells, Cancer Research 64, 264. [Google Scholar]
- 47.Olivier NB, Chen MM, Behr JR, and Imperiali B (2006) In vitro biosynthesis of UDP-N,N’-diacetylbacillosamine by enzymes of the Campylobacter jejuni general protein glycosylation system, Biochemistry 45, 13659–13669. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.