

Abstract
Background
The Southern Ocean is the coldest ocean on Earth but a hot spot of evolution. The bottom-dwelling Eocene ancestor of Antarctic notothenioid fishes survived polar marine glaciation and underwent adaptive radiation, forming >120 species that fill all water column niches today. Genome-wide changes enabling physiological adaptations and the rapid expansion of the Antarctic notothenioids remain poorly understood.
Results
We sequenced and compared 2 notothenioid genomes—the cold-adapted and neutrally buoyant Antarctic toothfish Dissostichus mawsoni and the basal Patagonian robalo Eleginops maclovinus, representing the temperate ancestor. We detected >200 protein gene families that had expanded and thousands of genes that had evolved faster in the toothfish, with diverse cold-relevant functions including stress response, lipid metabolism, protein homeostasis, and freeze resistance. Besides antifreeze glycoprotein, an eggshell protein had functionally diversified to aid in cellular freezing resistance. Genomic and transcriptomic comparisons revealed proliferation of selcys–transfer RNA genes and broad transcriptional upregulation across anti-oxidative selenoproteins, signifying their prominent role in mitigating oxidative stress in the oxygen-rich Southern Ocean. We found expansion of transposable elements, temporally correlated to Antarctic notothenioid diversification. Additionally, the toothfish exhibited remarkable shifts in genetic programs towards enhanced fat cell differentiation and lipid storage, and promotion of chondrogenesis while inhibiting osteogenesis in bone development, collectively contributing to the achievement of neutral buoyancy and pelagicism.
Conclusions
Our study revealed a comprehensive landscape of evolutionary changes essential for Antarctic notothenioid cold adaptation and ecological expansion. The 2 genomes are valuable resources for further exploration of mechanisms underlying the spectacular notothenioid radiation in the coldest marine environment.
Keywords: adaptive radiation, climate change, genome, oxidative stress, Antarctic notothenioids
Introduction
The Southern Ocean (SO) surrounding Antarctica is the coldest body of water on Earth, having been isolated from other world oceans by the Antarctic circumpolar current beginning in the early Oligocene ∼32 million years ago (mya) [1]. The formation of the Antarctic circumpolar current also impeded species dispersal across the Antarctic polar front, and mass extinction of the Antarctic-sequestered fish taxa occurred upon marine glaciation [2]. The rich cosmopolitan fish fauna prior to the isolation of Antarctica is represented today by a single predominant group of related fish species—the Antarctic notothenioids. From a common temperate ancestor, likely a swim bladderless, bottom-dwelling perciform species of the Eocene age [3], the Antarctic notothenioids have evolved to become highly adapted to life in unyielding cold, spectacularly diverse in sizes and morphological innovations, and diversified into all water column habitats, epitomizing an adaptive radiation and a rare marine species flock [4]. Abundant in fish biomass (>90% of random catch) and species (≥128), they are vital in sustaining the contemporary SO food web [2, 5].
What evolutionary processes and mechanisms propelled the Antarctic notothenioid radiation replete with extraordinary trait diversification during its evolutionary history remain fascinating unanswered questions. Two conspicuous trait outcomes—the evolutionary gain of the novel antifreeze glycoprotein (AFGP) gene and function that averted otherwise inescapable death from freezing [6, 7], and exploitation of open niches vacated by extinction of fishes lacking freeze resistance, have been recognized as major contributors to the Antarctic notothenioid radiation [8, 9]. However, little is known of the myriad subtler adaptive changes that must also have evolved in response to challenges from freezing temperatures and the associated high oxygen concentration—the 2 foremost modalities of selection pressure from the SO environments that would pervade all levels of organismal functions, from molecules to cells to system physiology. Another prominent hallmark of notothenioid adaptive radiation is the secondary acquisition of pelagicism in some lineages, enabling their ecological expansion from bottom habitats of their negatively buoyant ancestor to upper water column niches. What evolutionary changes occurred in the cellular and developmental programs that enabled neutral buoyancy and secondary pelagicism are also unknown.
To address these fundamental, system-wide questions about Antarctic notothenioid evolution, whole-genome sequences of multiple and appropriately chosen species from the diverse Antarctic notothenioids are essential. Thus far, whole-genome sequence analysis has been reported for only 1 notothenioid species, the Antarctic rock cod Notothenia coriiceps [10]. A major histocompatibility complex gene locus from Chaenocephalus aceratus was also reported [11]. The N. coriiceps genome provided the key inference that the fast-evolving hemoglobin and mitochondrial proteins are adaptive in increasing efficiency of aerobic cellular respiration in the freezing environment. N. coriiceps is not known to occur in the high-latitude Antarctic coastal waters. Instead, it is widely distributed in the lower latitude waters of the Antarctic Peninsula archipelago and the Scotia Arc islands, reaching localities north of the polar front around sub-Antarctic islands in the Indian Ocean sector [12], a distribution pattern that suggests a considerable degree of thermal plasticity in this species. It is a heavy, bottom fish and one of the hardest boned Antarctic notothenioids [13], reminiscent of the benthic ancestor. To gain insights into evolutionary adaptations in the most cold-adapted and stenothermal Antarctic notothenioids, as well as into the evolutionary changes leading to acquisition of neutral buoyancy that enabled the transition from the ancestral benthic existence to a pelagic life history, a different and more appropriate model Antarctic notothenioid species would be required.
The Antarctic toothfish Dissostichus mawsoni (NCBI:txid6530, Fishbase ID:7039) that grows to giant sizes (2.0 m in length and 140 kg in mass) is an iconic species of the Antarctic notothenioid radiation, with wide distributions in freezing waters of high-latitude Antarctic coasts, as far south as 77.5 S (McMurdo Sound), the southern limit of Antarctic marine life. It thus exemplifies the stenothermal cold-adapted character state. Despite its large size, it is the only notothenioid species that achieved complete neutral buoyancy as adults [14, 15]; thus, this species serves as the best model for examining the evolutionary underpinning of secondary pelagicism in the Antarctic clade. In addition, to discern evolutionary changes from the ancestral temperate state to the derived polar state driven by selection in the cold, oxygen-rich SO environment, a closely related basal non-Antarctic notothenioid comparison species would improve the discriminating power of analyses of genome evolution. The most appropriate species for this purpose is a South American notothenioid, the Patagonian robalo Eleginops maclovinus (NCBI:txid56733, Fishbase ID:466) , which is the sole species in the basal family Eleginopsidae. Also known as the Patagonian blenny, the lineage diverged prior to the isolation of Antarctica, and E. maclovinus is phylogenetically the closest sister species to the modern Antarctic clade [3]. Thus, its genome is the best representative of the temperate character of the most recent common ancestor of the Antarctic notothenioids. We conducted genome sequencing and comparative analyses of these 2 strategically selected species, together with extensive transcriptomic characterizations to profile relevant functional outcomes of the genomic changes. Our results provide several new key insights into evolutionary adaptation and secondary pelagicism of the Antarctic notothenioids in the isolated and extremely cold SO environment.
Materials and Methods
Specimens, sampling, and DNA and RNA isolation
Antarctic toothfish D. mawsoni was collected using vertical setline through drilled hole in sea ice of McMurdo Sound, Antarctica (77 53 S, 166 34.4 E and vicinity), during austral summer field seasons (October through December). Specimens were transported to the aquarium facility in the US National Science Foundation Crary Lab at McMurdo Station and kept in ambient (−1.6°C) flow through seawater tanks, and killed at 2–4 weeks after capture for blood and tissue sampling. The temperate basal notothenioid E. maclovinus was collected by rod and reel in the Patagonian waters of southern Chile during austral winter (June) and transported to the National Science Foundation Research Vessel Laurence Gould at Punta Arenas in a large, aerated Styrofoam cooler of ambient water (∼8°C), where specimens were killed and sampled within a few days prior to southbound transit for winter field season. To obtain tissues from the large-sized D. mawsoni, live specimen was anesthetized with MS222 (tricaine methanesulfonate) inside an ambient seawater–filled floating sheet plastic tubing in the aquarium tank. The anesthetized specimen was then put on a V-shaped trough for dissection. Tissues were quickly removed and cut into small pieces on ice, and immediately immersed and shaken in ≥10 volumes of prechilled (−20°C) 90% ethanol (made with 100% pure ethanol and sterilized MilliQ Type 1 water). The ethanol was replaced with a fresh volume within 10 minutes, and again at 2–3 and 12 hours later. This preservation method serially desiccates the tissue and effectively inactivates tissue nucleases. The tissue samples were kept in a −20°C freezer throughout the serial preservation process and then stored at −20°C until use. To obtain tissues from E. maclovinus, MS222-anesthetized specimen was quickly dissected on ice and preserved at −20°C as described for D. mawsoni. The ethanol-preserved tissues were shipped back to the University of Illinois on dry ice.
RNA for transcriptome sequencing was isolated from −20°C ethanol-preserved tissues using Trizol (Invitrogen, Carlsbad, California, USA)) and quality verified by visualization on gel electrophoresis and an Agilent BioAnalyzer. Collection, handling, and sampling of the Antarctic toothfish and South American E. maclovinus in this study were carried out in compliance with protocol No. 12123 approved by the University of Illinois Institutional Animal Care and Use Committee. Additional juvenile specimens of D. mawsoni were collected by trawl from the waters of the Antarctic Peninsula during the same winter season and sampled on shipboard shortly after capture. The dissected carcasses of E. maclovinus and juvenile D. mawsoni were kept frozen at −80°C, which provided the pelvic bone samples for immunohistochemical detection for expression of candidate genes in bone development. To preserve high molecular weight DNA for genome sequencing, red blood cells of each species were washed with notothenioid saline (0.1 M sodium phosphate buffer, pH 8.0, adjusted to 420 mOsm with NaCl for E. maclovinus, and 540 mOsm for D. mawsoni), and then embedded in 1% melt agarose to provide ∼20 μg DNA per 90-μL block using BioRad plug molds (CHEF Mammalian Genomic DNA Plug Kit No. 1703591, Bio-rad, Hercules, CA, USA). The embedded cells were lysed in situ and the DNA in the agarose blocks was preserved following Amemiya et al (1996) [16]. To recover high molecular weight DNA, the agarose plugs were digested with β-agarase (NEB, Ipswich, MA, USA) followed by phenol extraction and dialysis, and quality verified using pulsed-field electrophoresis.
Sequencing and genome assembly
The sequencing libraries with insert sizes of 170, 250, and 500 base pairs (bp) were prepared for sequencing of the paired-end reads, following a modified version of the manufacturer's protocol (Illumina, San Diego, CA, USA). An integrated protocol from the Mate-Pair Library v2 Sample Preparation Guide (Illumina) and the Paired-End Library Preparation Method Manual (Roche, Branford, Connecticut, USA)) was used to prepare mate-pair libraries with insert sizes of 3, 6, 10, 15, and 20 kb (Additional file 1: Table S1b, S1c). For the transcriptome sequencing, Poly(A)+ messenger RNA (mRNA) was purified using the DynaBeads mRNA Purification kit (Life Technologies, Carlsbad, CA). Paired-end complementary DNA libraries were constructed using the RNA sequencing (RNA-Seq) Next-Generation Sequencing Library Preparation Kit for Whole-Transcriptome Discovery (Gnomegen, San Diego, CA). All of the libraries are sequenced on an Illumina HiSeq 1500 sequencer. The D. mawsoni genome was assembled using SOAPdenovo (SOAPdenovo, RRID:SCR_010752) [17] to build the contigs and SSPACE (SSPACE, RRID:SCR_005056) [18] to scaffold the contigs. The E. maclovinus genome was assembled using Platanus (Platanus, RRID:SCR_015531) to build the contigs, and SSPACE to scaffold the contigs.
Annotation of the genome
We identified repeats, protein-coding genes, and noncoding RNA in the genome assemblies of the 2 species. First, a de novo repeat annotation of D. mawsoni and E. maclovinus genomes was carried out by successively using RepeatModeler (RepeatModeler, RRID:SCR_015027) (version 1.0.8) and RepeatMasker (RepeatMasker, RRID:SCR_012954) (version 4.0.5). De novo repeat libraries of the 2 species were constructed with 2 complementary programs, RECON (RECON, RRID:SCR_006345) [19] and RepeatScout (RepeatScout, RRID:SCR_014653) [20] implemented in the RepeatModeler package. The generated consensus sequences were manually checked by aligning to the Repbase transposable element (TE) library [21] and genes from the NCBI database (nt and nr). The D. mawsoni and E. maclovinus repeat library consisted of 975 and 676 consensus sequences with classification information, respectively, which were used to run RepeatMasker on the assembled scaffolds. Secondly, protein-coding genes were predicted using a combination of homology-based and de novo approaches. GLEAN was used to create a consensus gene set by integrating evidence from each prediction. Then RNA-Seq data were used to rectify gene models. Generated coding genes were aligned to known protein databases, including InterPro [22], Kyoto Encyclopedia of Genes and Genomes (KEGG) [23], and Uniprot [24], and functional assignment was based on that of the best database match. Thirdly, the transfer RNA (tRNA) genes were predicted with tRNAscan-SE (tRNAscan-SE, RRID:SCR_010835) [25]. Aligning the ribosomal rNA (rRNA) template sequences from fishes using BlastN (BlastN, RRID:SCR_001598) with E-value 1e–5 identified the rRNA fragments. The microRNA and small nuclear RNA genes were predicted with INFERNAL (INFERNAL, RRID:SCR_011809) [26] software against the Rfam database (Release 12) [27].
Phylogenetic reconstruction of 10 vertebrate genomes
Protein-coding genes of Atlantic cod (Gadus morhua), tetraodon (Tetraodon nigroviridis), Antarctic notothenioid N. coriiceps, stickleback (Gasterosteus aculeatus), tilapia (Oreochromis niloticus), medaka (Oryzias latipes), zebrafish (Danio rerio), and mouse (Mus musculus) genomes were collected from Ensembl release 84 or NCBI, and D. mawsoni and E. maclovinus genes from this study, were used to build orthologous clusters with OrthoMCL version 2.0.9 (OrthoMCL, RRID:SCR_007839) [28] with default parameters and options. A total of 2,936 one-to-one single-copy genes were identified among the 10 species. Protein-coding sequences of the orthologs were aligned using PRANK (version 140603) [29] under a protein model with default parameters. The coding sequences of the genes were concatenated to a supergene for each species. The supergene sequence data set was subjected to phylogenetic analysis using MrBayes (MrBayes, RRID:SCR_012067) [30], implementing best-fit substitution model (GTR+gamma+I) as determined by Modeltest [31]. The analysis was run 800,000 generations, sampling every 100 generations, with the first 2,000-sample set as burn-in. Branch-specific dN and dS were estimated with codeml of the PAML package [32]. The analyses of changes in gene family size were computed with CAFÉ (CAFE, RRID:SCR_005983) [33].
Gene ontology annotation and identification of positive selection genes
Gene ontology (GO) terms of the D. mawsoni, E. maclovinus, and stickleback orthologs were built with InterProScan (InterProScan, RRID:SCR_005829) [34]. The orthologs of each GO term were concatenated to estimate branch-specific dN and dS using codeml of PAML (PAML, RRID:SCR_014932). A binomial test was used to identify the excess of nonsynonymous changes of GO categories in either D. mawsoni or E. maclovinus lineages referenced to the stickleback. Only the GO terms carrying more than 30 orthologs were put into this calculation. To detect genes evolving under positive selection in D. mawsoni, we used the branch-site model in which likelihood ratio test P values were computed. Fisher exact tests were used to test for overrepresented functional categories among the positive select genes. GO enrichment analyses of the genes under positive selection were performed using a hypergeometric method.
Calling of heterozygous single-nucleotide polymorphisms
All of the paired-end reads were mapped to the assembled scaffolds with the aligner SMALT (SMALT, RRID:SCR_005498) to detect the heterozygous sequence polymorphism in the genomes. The heterozygous single-nucleotide polymorphisms (SNPs) were called with SSAHA_Pileup (version 0.8; [35]). Five thresholds were used to post-filter unreliable SNPs: (i) SSAHA_Pileup SNP score ≥20, (ii) ratio of 2 alleles between 3:17 to 17:3, (iii) the lowest sequencing depth for each allele ≥5, (iv) the minimum distance for adjacent SNPs ≥5 bp, and (v) only 1 polymorphism detected at each SNP position.
Transcriptome analyses
RNA-Seq data derived from liver, gill, stomach, white muscle, red muscle, skin, small intestine, brain, head kidney, caudal kidney, spleen, and ovary were analyzed for variations in gene expression of D. mawsoni and E. maclovinus. RNA-Seq reads were trimmed using Trimmomatic (Trimmomatic, RRID:SCR_011848) (version 0.33) [36] with the parameter set to AVGQUAL at 20, TRAILING at 20, and MINLEN at 50. The cleaned Illumina paired-end reads of each tissue were mapped to the annotated scaffolds of D. mawsoni and E. maclovinus genome using HISAT2 (HISAT2, RRID:SCR_015530) aligner (version 2.0.4) [37]. Cufflinks (Cufflinks, RRID:SCR_014597) (version 2.2.1) [38] normalized gene expressions to the quantified transcription levels (fragments per kilobase million). Differential expressions of the genes were assessed using DEGseq (DEGseq, RRID:SCR_008480) (version 1.28.0) [39] with cutoff at q < 0.001 [40]. GO and KEGG enrichment analyses for the identified differentially expressed genes (DEGs) were performed using cluserProfiler packages [41] with the cutoff at P < 0.05.
Construction of gene collinearity among D. mawsoni, E. maclovinus, and stickleback genomes
The genes of D. mawsoni and E. maclovinus were aligned to the gene model set of stickleback by Blastp (BLASTP, RRID:SCR_001010) with E-value at 1e–20. Two criteria were used to call syntenic gene blocks in the D. mawsoni or E. maclovinus scaffolds: (i) number of the gene on the syntenic block ≥3 and (ii) number of nonsyntenic genes between 2 adjacent syntenic genes ≤10. Each syntenic block was anchored on the stickleback genomes according to the orders of the reference gene.
Western blot analysis of zona pellucida proteins
The zona pellucida proteins (ZPs) were separated on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) at 100V for 90 min in 193 mM glycine and 25 mM Tris (pH 8.8). The resolved proteins were electrophoretically transferred to a nitrocellulose membrane (Millipore) using a Mini-Protean Tetra Cell (BioRad) in a buffer containing 193 mM glycine, 25 mM Tris (pH 8.3), and 20% methanol. The membrane was treated with blocking agent (5% nonfat milk in 1x Tris-buffered saline with Tween 20 [TBST]) for 2 h at room temperature on a shaker. FLAG antibody or β-actin antibody (Hua An Biotechnology Co. Ltd, Hangzhou, China) was added, and the membranes were incubated at room temperature for 1 h. The membrane was then washed with 1x TBST 3 times for 15 min each. The secondary antibody (1:2000 in 1x TBST, Boston Biomedical Inc.) was then added and incubated for 1 h at room temperature. The membrane was washed with 1x TBST twice and 1x TBS once for 15 min each. Color was developed using SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) according to the manufacturer's instructions. Images were acquired using a ChemiDoc MP Imaging System (BioRad).
Assay of Chinese hamster ovary cell survival rate at freezing temperature
DmZPC5 exons were serially deleted using polymerase chain reaction amplification of the DmZPC5 expression vector with primers designed to eliminate desired coding sequences (Fig. 3b). The full-length sequences of 3 DmZPC5 isoforms (DmZPC5-1, DmZPC5-2, and DmZPC5-3) were engineered to contain a FLAG octapeptide and cloned into the expression vector pIRES2-EGFP (Additional file 1: Fig. S5). The 3 constructed vectors and blank control (vector pIRES2-EGFP) were transferred into the Chinese hamster ovary (CHO) cells (American Type Culture Collection CCL-61) obtained from American Type Culture Collection. The CHO cells were cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (Gibco). CHO cells were incubated at 37°C for 2 days and then kept at −2°C for 8 h. The treated CHO cells were collected and washed with Dulbecco's phosphate-buffered saline (PBS) twice. The cells were stained with 10 µg/mL propidium iodide at room temperature for 5 minutes, and numbers of propidium iodide−stained cells (dead cells) were determined by flow cytometry. The survival rate was calculated with the following equation: survival rate = S/(S + D), where S is the number of surviving cells and D is the number the dead cells.
Figure 3:

Evolutionary and functional analyses of the DmZPC5 genes involved in cellular freezing resistance. (A) Duplication of ZPC5gene (DmZPC5) in D. mawsoni. Phylogenetic neighbor-joining tree of ZPC5 genes among D. mawsoni, E. maclovinus, Larimichthys crocea (Lc), T. rubripes (Tr), and O. latipes (Ol). The gene structures are illustrated on the right. The different colored blocks indicate the exons encoding signal peptides (red), zp domains (blue), and the remaining exons (incarnadine). The jagged blocks contain the nonsense mutations in DmZPC5-2a/b and DmZPC5-3 genes that cause premature termination of coding sequences. (B) Western blot analysis of the DmZPC5 isoforms indicated their sizes and temperature-sensitive accumulation. Purified proteins encoded by the 3 DmZPC5 isoforms were detected by an anti-FLAG antibody on the SDS-PAGE gels. All of these 3 DmZPC5 proteins had higher expression levels at 0°C than at 37°C. (C) Assays of cell survival rate under recombinant expression of different DmZPC5 isoforms in CHO cells at a freezing temperature (−2°C for 8 h). The bars represent the mean ± SD (n = 3, biological replicates). The sample pIRES2-EGFP is the expression vector as control. Significant differences in survival rate are indicated by the asterisk (unpaired Student t-test, P < 0.05) and double asterisk (P < 0.01).
Identification of the long interspersed TEs and estimation of their divergence time
The seed sequences of the long interspersed TEs (LINEs) [42] were aligned against the D. mawsoni and E. maclovinus draft genomes, respectively, using BlastN at E-value of 1e-10. According to loci of the alignments, the sequences were extracted from the genomes, which were considered as the candidates of the LINEs. If distance between 2 adjacent candidates was ≤200 bp, these 2 candidates were connected by the sequence between them. All of the candidates and their corresponding seed sequences were mutually aligned by BlastN. Those candidates with >60% identity and >100 bp of alignments to the seed sequences were collected as the LINEs of D. mawsoni and E. maclovinus, respectively.
Alignment of the LINEs was conducted by the multiple sequence aligner ClustalW (ClustalW, RRID:SCR_002909) [43] to cluster any 2 LINEs with highest sequence similarity into a LINE pair in D. mawsoni or E. maclovinus. The evolutionary distance of 2 LINES for each LINE pair was calculated by the Kimura 2-parameter method (EMBOSS distmat, version 6.6.0.0), which reflected the substitution rate per site between the 2 LINEs. According to the calculated synonymous substitution rates for 7,958 D. mawsoni−E. maclovinus orthologous pairs, the mean synonymous substitution rate is ∼0.227. The peak of the substitution rate is at 0.04, which estimated the LINE burst to be ∼6.5 mya when the species divergence time between D. mawsoni and E. maclovinus is ∼37 mya [44].
Tissue fixation and immunohistochemistry
Pieces of pelvic bone (with muscle) and the attached fins (≤20 × 20 × 5 mm) were dissected from frozen specimens of young D. mawsoni and E. maclovinus and immersed in a fixation solution, KINFix, which contains 62.5% (v/v) ethanol, 6.71% (v/v) acetic acid, and 6% (w/v) trehalose [45], for >24 h. Tissues were decalcified in ethylenediaminetetraacetic acid solution (cat. No. 041-22031, WAKO) for ∼2 weeks. Then the specimen was dehydrated in graded ethanol (70%, 80%, 90%, 95%, 1 × 1 h each), 100% ethanol 2 × 1 h at room temperature, xylene for 2 × 1 h, and embedded in low-melting paraffin for 2 × 1 h, and kept overnight at 56°C, then embedded in paraffin. For each tissue, 5-μm thick serial sections were cut with a microtome (RM2245, Leica). Immunostaining was performed using the EnVision detection system (cat. No. K5007, Dako). Slides were deparafinized in xylene and rehydrated in a descending series of ethanol (100%, 95%, 90%, and 70%), and washed in PBS. Endogenous peroxidases were blocked with 3% H2O2 for 10 min, after which the sections were incubated with 5% bovine serum albumin for 35 min. Then, the slides were incubated overnight with the primary connective tissue growth factor (CTGF) antibody (1:400 dilution) (cat. No. ab6992, Abcam) at 4°C. Next, the sections were washed 4 times with PBS for 15 min followed by incubation with a goat anti–rabbit secondary antibody for 35 min at 37°C. After 4 washes with PBS, 3, 3’-diaminobenzidine was added to visualize the immunoreactivity. All slides were counterstained with hematoxylin-eosin. The sections were dehydrated in a mounting series of alcohol (70%, 90%, 100%, and 100%) and in xylene. Finally, slides were mounted using neutral balsam mounting medium, and analyzed under a bright-field microscope (AXIO imager M2, ZEISS).
Results
D. mawsoni and E. maclovinus genome sequencing and assembly
The geographic distributions and sampling locations of D. mawsoni and E. maclovinus are illustrated in Fig. 1a. The genome of 1 D. mawsoni juvenile (12 kg) of undetermined sex and 1 young adult male E. maclovinus (∼100 g) were de novo sequenced using Illumina sequencing platforms. The genomes of both species comprise 24 pairs of chromosomes (2n = 48) [46, 47]. Analyses of 17-mer frequency distribution indicated a genome size of approximately 842 Mb for D. mawsoni and 727 Mb for E. maclovinus (Additional file 1: Fig. S1), consistent with the mean genome sizes of 840 and 780 Mb for the toothfish and robalo, respectively, determined by flow cytometry (Additional file 1: Table S1a). The raw sequence data after cleaning and error correction (Additional file 1: Table S1b, S1c) were assembled using SOAPdenovo [17] for D. mawsoni, and Platanus [48] for E. maclovinus followed by scaffold building with SSPACE [18]. The assembled toothfish genome had a contig N50 length of 23.1 Kb and scaffold N50 length of 2.2 Mb, while those of the robalo were 10.9 Kb and 0.69 Mb, respectively. The assembled toothfish and robalo genomes are approximately 757 and 744 Mb, respectively (Table 1; Additional file 1: Table S2a, S2b), consistent with k-mer and flow cytometry estimates, and achieving >90% and 95% coverage of the genome size based on flow cytometry of the 2 species, respectively. The completeness of both genomes was assessed with benchmarking universal single-copy orthologs (BUSCOs) [49], referencing the lineage data set of actinopterygii_odb9 and orthologs of zebrafish, which reflected the complete BUSCOs at 97.2% for the D. mawsoni genome and 95.0% for the E. maclovinus genome (Additional file 1: Table S2c). The guanine-cytosine content of the D. mawsoni genome is 0.4070, nearly identical to the 0.4066 of E. maclovinus, and both are lower than that of a model fish, the stickleback Gasterosteus aculeatus (Additional file 1: Fig. S2). The accuracy of the genome assembly was assessed by alignment of the scaffolds to publicly available unigenes of D. mawsoni and E. maclovinus, and the coverage of the initial contigs was found to be approximately 98.8% and 99.1%, respectively (Additional file 1: Table S3), suggesting an acceptable quality of the genome assemblies. Alignment of the sequence reads to the assemblies estimated an overall heterozygous rate of approximately 2.58 and 2.40 per Kb for D. mawsoni and E. maclovinus, respectively (Additional file 1: Table S4).
Figure 1:
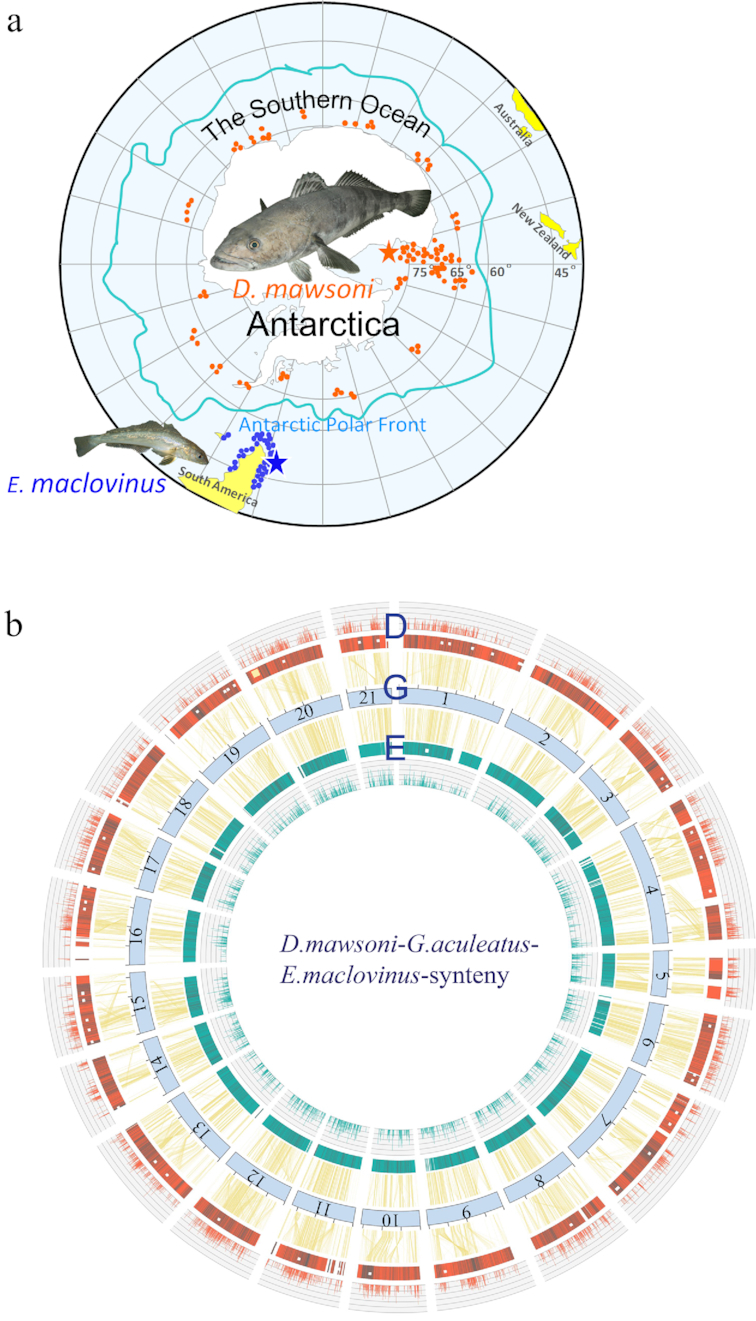
Sequenced species and genome synteny. (A) Sampling location and geographic distribution. The red and blue filled circles indicate the geographic distributions of D. mawsoni and E. maclovinus, respectively (www.fishbase.de version 02/2018 [50] and Hanchet et al. [51]). The red and blue stars show the respective locations where the sequenced individuals were collected. The Antarctic polar front, an approximation of the mean position of the Antarctic circumpolar current, is adopted from Barker and Thomas [52]. The image of D. mawsoni is a courtesy from Elliot DeVries and that of E. maclovinus is from Dirk Schories. (B) Gene collinearity among D. mawsoni, E. maclovinus, and G. aculeatus (stickleback). The scaffolds of D. mawsoni (the circularized red blocks labeled with “D”) and E. maclovinus (the circularized green blocks labeled “E”) are anchored on the 21 stickleback chromosomes (the circularized light blue blocks labeled with “G,” 1–21), according to the gene collinearity (the connecting yellow lines). The black vertical lines within the D. mawsoni and E. maclovinus scaffolds indicate occurrence of LINEs greater than 500 bp in these positions. The sequence length is indicated by the 5-Mb tick marks on the reference stickleback chromosomes. The outermost circle of red vertical lines and the innermost circle of green vertical lines indicate the quantified expression levels (fragments per kilobase million) of the genes located on the corresponding D. mawsoni and E. maclovinus scaffolds, respectively. The expression profiles are derived from the transcriptome data of white muscles (see the transcriptome section). The small white squares and rectangles scattered in the scaffolds show the locations of the selcys-tRNA genes of D. mawsoni and E. maclovinus. The single yellow square shows the location of AFGP genes in the D. mawsoni genome.
Table 1:
Overview of assembly and annotation
| D. mawsoni | E. maclovinus | |
|---|---|---|
| Assembly | ||
| Total length (Mb) | 756.8 | 744.4 |
| Contig N50 length (Kb) | 23.1 | 10.9 |
| Scaffold N50 length (Kb) | 2,216.2 | 694.7 |
| Scaffold N90 length (Kb) | 202.7 | 167.2 |
| Largest scaffold (Mb) | 13.8 | 4.9 |
| Quantity of scaffolds (>N90 length) | 536 | 1,185 |
| Annotation | ||
| Quantity of predicted protein-coding genes | 22,516 | 22,959 |
| Quantity of predicted noncoding RNA genes | 2,434 | 2,185 |
| Content of TEs (%) | 21.38 | 10.02 |
| Heterozygous SNP rate (SNPs per kb) | 2.58 | 2.40 |
Genome annotation and synteny alignment between D. mawsoni and E. maclovinus
A total of 22,516 and 22,959 protein-coding genes were annotated in the D. mawsoni and E. maclovinus genome, respectively, by combining the results from homologous and de novo prediction methods using the gene modeler GLEAN (Additional file 1: Table S5). The protein-coding genes of the toothfish and robalo, along with the sequenced notothenioid N. coriiceps and model species G. aculeatus and zebrafish Danio rerio, were clustered using OrthoMCL [28]. We found 8,825 gene clusters that were common to all 5 species. Genes shared among the notothenioids are similar in quantity, 12,269 between toothfish and robalo and 12,421 between toothfish and N. coriiceps (Additional file 1: Fig. S3). In annotations of conserved noncoding RNA genes, we predicted 1,097 tRNA, 110 rRNA, 422 small nuclear RNA, and 295 microRNA genes in the toothfish genome (Additional file 1: Table S6a), while the robalo genome was annotated to carry 1,037 tRNA, 44 rRNA, 891 small nuclear RNA, and 286 microRNAs (Additional file 1: Table S6b). The much larger number of rRNA copies (2.5-fold) in D. mawsoni than E. maclovinus is consistent with the presence of dual chromosomal loci of recombinant DNA genes detected by in situ fluorescent hybridization in the giant toothfish [46], as opposed to the single recombinant DNA locus in other notothenioids [53]. The 2 species showed largely similar profiles in their microRNAome, with minor differences in the copy number of some individual microRNA (Additional file 1: Table S7). Strikingly, the toothfish genome contains many more selcys-tRNA genes than robalo (84 vs. 1; Additional file 1: Table S8). This extensive duplication of selcys-tRNA genes, accompanied by high expression of selenoproteins in D. mawsoni (detailed in a later section: "Transcriptomic adaptation to the cold environment"), signifies that mitigation of oxidative stress through selenoproteins, many of which are strong antioxidants, is likely an important selection force in the evolution of the Antarctic notothenioid genome in the freezing and oxygen-rich waters.
De novo annotation of repeat sequences revealed >2-fold increase in overall repeat content in D. mawsoni (21.38% of genome) over the basal E. maclovinus (10.02%) (Additional file 1: Table S9). TEs of the toothfish genome, including long terminal repeat retrotransposons, non–long terminal repeat retrotransposons (LINEs and short interspersed TEs [SINEs]), and DNA transposons, was more than twice that of E. maclovinus. Examination of the N. coriiceps genome exhibited similar accumulation of the TEs (23.5%). Among simple sequence repeats, dimer repeats constituted the majority in both genomes, and tetramers and pentamers showed the highest levels of increment in D. mawsoni (Additional file 1: Table S10a and S10b). The doubling of TE content in the D. mawsoni and N. coriiceps genomes suggests higher activity of TEs in the Antarctic species relative to the basal robalo, suggesting a likely contributing factor to the observed trend of increasing genome sizes in more derived Antarctic notothenioid lineages [54].
For global genome alignment, we anchored the D. mawsoni and E. maclovinus scaffolds to the 21 linkage groups of the well-characterized 3-spined stickleback genome according to gene collinearity (Fig. 1b; Additional file 2). Most of the notothenioid scaffolds had extensive collinearity to the corresponding stickleback chromosomes. The 84 duplicated selcys-tRNA genes are widespread throughout the toothfish genome. Inversion and translocation of genome segments have occurred in both D. mawsoni and E. maclovinus relative to stickleback, but the D. mawsoni genome showed more frequent rearrangements. Insertion sites for the expanded TEs of the D. mawsoni genome were random, thereby expanding the lengths of almost all of the linkage groups. Mapping the RNA-Seq transcript data that we obtained from white muscles for the 2 species on their respective synteny showed that the heavier insertion of TEs in the toothfish genome did not appear to adversely affect the expression of the neighboring protein genes because strong gene expression in regions of high TE content was maintained (Fig. 1b).
Burst of LINE expansion in the cold
Involvement of gain (or loss) of mobile element copies in genome size and genome restructuring affecting species differentiation has increasingly gained empirical support [55, 56]. TEs are normally under epigenetic regulation, but waves of TE proliferations could arise from environmental changes that cause physiological stress and disrupt epigenetic control [57]. We therefore examined potential linkage in timing between LINE expansion (2-fold increase in the toothfish vs. robalo) and onset of frigid SO marine conditions. A multiple sequence alignment of several LINEs (LINE/I, LINE/L2, LINE/RTE-BovB, and LINE_Rex-Babar—the most abundant types in the toothfish genome) was made, which clustered 349 D. mawsoni LINE pairs and 213 E. maclovinus LINE pairs, where the 2 LINEs within each pair share the highest sequence similarity, approximating the least divergence time. Calculation of the nucleotide substitution rates for each LINE pair identified a burst of emergence of LINEs in the D. mawsoni genome with the substitution rates centered at 0.04, which corresponded with a divergence time of 6.5 mya (Fig. 2a; see also Materials and Methods) based on the mean substitution rate of the LINEs. A similar trend of LINE expansion was also observed in the N. coriiceps genome. This estimated timing of LINE burst expansion correlated with the radiation of the majority of the modern Antarctic notothenioid clades beginning in the late Miocene when seawater temperatures steadily declined [9]. In contrast, no burst expansion of LINEs was detected in the E. maclovinus genome, supporting an Antarctic/cold-specific LINE burst in the toothfish, and corroborating our prior empirical evidence for an increase in retrotransposition activity of D. mawsoni LINE-1, resulting in more copies in the transfected cells when subjected to stress from nonphysiologically low incubation temperature [42].
Figure 2:

Evolution of the genomes and genes. (A) Timing and frequency of LINE insertion in D. mawsoni, E. maclovinus, and N. coriiceps showing correlation between onset of late Miocene deep cooling and burst LINE insertions in the Antarctic toothfish and bullhead notothen. The black trace indicates global temperature trends during the Oligocene, Miocene, Pliocene (Pli), and Pleistocene (Ple) from 30 to 0 mya, modified from Zachos et al. (2008) [58], Near et al. (2012) [9], and Favre et al. (2015) [59]. The red and blue lines indicate the insertion frequency of LINEs (the percentage of the calculated LINE pairs) in the D. mawsoni and E. maclovinus genomes, respectively, during these periods. (B) Reconstructed phylogeny of 9 teleost fish lineages using 2,936 orthologous genes (mouse serving as outgroup) and the calculated dN/dS ratio for each branch, showing a 2-fold faster evolutionary rate in the Antarctic notothenioids. (C) Comparison of adaptive evolution between D. mawsoni and E. maclovinus genomes. Data points represent the mean dN/dS value of each GO term, each of which consists of ≥30 genes. The red and blue circles show the GO terms with significantly higher dN/dS ratios (P < 0.05, binomial test) in D. mawsoni and E. maclovinus, respectively. The grey circles are those showing no significant difference. GO terms falling on the dashed line of linearity have the same dN/dS ratios in the 2 species. (D) Gene duplication in D. mawsoni. A subset [56] of the 202 gene families detected to contain higher gene copy numbers in the D. mawsoni genome relative to other species are listed on the left, with their respective KEGG pathway listed on the right. The gene copy numbers are measured by color difference. The pathways highlighted in red are especially abundant in D. mawsoni and might be relevant to physiological adaptation of D. mawsoni in the freezing environment. (E) A subset of duplicated gene families in E. maclovinus, showing different KEGG pathways between D. mawsoni and E. maclovinus in terms of gene duplication. The red highlighted pathway (ether lipid metabolism) indicates that a common duplication occurred in the 3 notothenioids.
Accelerated protein evolution in the cold
Ectotherms are vulnerable to disruptive effects on protein structures and reaction rate depression at low temperatures. Antarctic notothenioids in perennially freezing high-latitude waters face the extremes of these effects, as well as an oxidative environment due to high oxygen concentrations resulting from increased gas solubility at low temperatures. We examined evidence for adaptive evolution of proteins in response to these selection pressures. The gene models of D. mawsoni, E. maclovinus, 7 other teleost genomes, and the mouse genome as outgroup were clustered, from which 2,936 one-to-one single-copy orthologs were obtained to reconstruct a phylogenetic tree (Fig. 2b). Stickleback is the closest species to the notothenioid clade as expected, and E. maclovinus is sister to the 2 Antarctic notothenioids, D. mawsoni and N. coriiceps. We found that evolutionary rates of the orthologous genes based on calculated dN/dS values (the ratio of the rate of nonsynonymous substitution to the rate of synonymous substitution) were elevated in the notothenioid lineage compared with the other fish lineages. This faster rate is more pronounced in the 2 Antarctic species, about twice that of the basal E. maclovinus, suggesting intensified selection pressures driving genome evolution in the Antarctic environment. To identify GO categories that were evolving faster in the toothfish or robalo, the dN/dS ratios of 7,584 orthologous genes among the 2 notothenioids and the stickleback were calculated and the mean dN/dS value of the genes associated with each GO term was calculated for each species. These orthologous genes were annotated to 411 GO terms, of which 281 showed significantly higher mean dN/dS ratios in D. mawsoni, while only 19 demonstrated higher mean rates in E. maclovinus (Fig. 2c; Additional file 2). The faster evolving GO processes in D. mawsoni included "gene expression," "protein folding," "tRNA metabolic process," "cell-redox homeostasis," "immune response," "response to stress," "lipid metabolic process," "DNA repair," "vesicle-mediated transport," and others. To assess which D. mawsoni genes experienced positive selection, we tested using the Branch-site model (in PAML) on a reconstructed phylogenetic tree of 6 fish species with the D. mawsoni lineage assigned as the foreground branch (Additional file 1: Fig. S4a). A total of 526 positively selected genes were identified (Additional file 1: Table S11a). The most significantly enriched KEGG pathway (Additional file 1: Table S11b) was “protein digestion and absorption” (P = 5.1e−5) and GO term (Additional file 1: Table S11c) was “protein binding” (P = 6.0e−4), which indicate that maintenance of protein homeostasis played an important role in shaping the D. mawsoni genome. In addition, a complement of genes (sorbs2a, acox1, apoa1a, scp2a, tnni3k, and perilipin-like genes; Additional file 1: Table S11a) involved in the peroxisome proliferator-activated receptor (PPAR) signaling pathway—the key pathway in adipocyte development regulation—were found to be under positive selection, suggesting occurrence of adaptive changes in lipid metabolism in D. mawsoni.
Gene duplication in the freezing environment
Gene duplication plays fundamental roles in the emergence of adaptive features. In the list of predicted protein coding genes from the toothfish and robalo genomes, we identified 202 families that have increased in copy number in D. mawsoni, compared with the other 8 fish genomes (Additional file 3). KEGG enrichment analyses of these expanded gene families yielded enrichment in pathways involved in protein homeostasis and lipid and bone metabolism, such as “protein digestion and absorption,” “regulation of lipolysis in adipocyte,” “fat digestion and absorption,” “ether lipid metabolism,” and “osteoclast differentiation” (Fig. 2d; Additional file 2), suggesting that genomic capacity for these functional pathways had increased in D. mawsoni during evolution in chronic cold conditions. Corroborating this cold-specificity is that the expanded gene families found in the basal E. maclovinus relative to the other fish genomes, including D. mawsoni and N. coriiceps, yielded distinctly different enriched KEGG profiles (Fig. 2e); this analysis indicates that the functional traits gained through gene duplication in E. maclovinus were driven by different selective pressures, consistent with our previous findings [60, 61]. Due to inherent inefficiency in correctly assembling highly similar DNA sequences in the shotgun sequencing strategy, there are likely many more duplicated genes that had eluded detection. For example, many paralogs of ZPs, such as ZPAX1, ZPC1, and ZPC2 in D. mawsoni have been shown, through array-based genome hybridization and quantitative polymerase chain reaction, to be duplicated [60, 62]. Among the set of genes found duplicated in our previous report [42], 23 are identified in this study (Additional file 1: Fig. S4b).
Gene duplication has contributed prominently to the evolutionary gain of freezing avoidance in Antarctic notothenioids. Generally regarded as a key innovation of the Antarctic notothenioid radiation, the AFGP gene evolved from a trypsinogen-like protease ancestor followed by extensive intragenic and whole-gene duplications, generating a large gene family that would provide an abundance of this novel life-saving protein [7]. By referencing the published AFGP haplotypes that were assembled from bacterial artificial chromosome clone sequences of the same individual used in this study (Genbank accessions HQ447059 and HQ447060) [63], we identified and assembled the AFGP loci shotgun reads, recaptured the 2 AFGP haplotypes and integrated them in the draft genome, and localized them to a region syntenic with a scaffold in LG 20 of the stickleback genome (Fig. 1b).
Resistance to freezing extends beyond the AFGPs. We found gene duplication for a protein that a priori would not be expected to function in freeze resistance: ZP protein (or eggshell protein) additionally provided protection against cellular freezing. Products of ZPC5 from D. mawsoni have been shown to enhance freezing resistance of eggs of recipient zebrafish both in vivo and in vitro [62]. In this study, we found 4 copies of ZPC5 in the D. mawsoni genome that encoded 3 different sized ZPC5 proteins in the toothfish ovary—DmZPC5_1, DmZPC5_2a/DmZPC5_2b, and DmZPC_3 in decreasing order of size, corresponding to gradually shortened C-termini from the conserved ZP domain due to nonsense mutations in exons 9 and 10 (Fig. 3a; Additional file 1: Fig. S5a). A single ZPC5 gene in the basal E. maclovinus was found, which corresponded to the full-length DmZPC5_1. We expressed the three DmZPC5 isoforms in CHO cells (Additional file 1: Fig. S5b and 3b) and assayed cell survival rate at freezing temperature (−2°C for 8 h). DmZPC5_3 was the most active isoform in maintaining cell viability while DmZPC5_1 was the least active one (Fig. 3c). Further analyses showed that DmZPC5_3 was more likely retained inside the cell than DmZPC5_2 and DmZPC5_1 (Additional file 1: Fig. S6), and less likely to become polymerized inside the cell compared with DmZPC5_1 (Additional file 1: Fig. S7a), corroborating our previous finding that only unpolymerized ZP proteins are active for the ice-melting promoting activity [62]. We detected DmZPC5 expression across many tissues besides ovary in D. mawsoni (Additional file 1: Fig. S7b), consistent with a distribution expected of a general protective function.
Transcriptomic adaptation to the cold environment
To assess the functional relevance of the detected genomic outcomes to life in freezing conditions, we characterized and compared transcriptomes of 12 tissues including brain, liver, red muscle, white muscle, gill, skin, intestine, stomach, spleen, head kidney, caudal kidney, and ovary between native specimens of D. mawsoni and E. maclovinus. We found that >10,000 genes were differentially expressed in pairwise comparisons between the 2 species, with the toothfish showing substantially more upregulated genes than the robalo. Enrichment testing on KEGG pathways yielded findings that many signaling pathways in the tissues of toothfish were significantly enriched in DEGs, including the transforming growth factor β (TGF-β), AMP-activated protein kinase, and PPAR pathways, known to play essential roles in development, metabolism, and stress responses (Additional file 1: Fig. S8a). GO enrichment analysis of the DEGs demonstrated upregulation of hundreds of GO biological processes, including translation, transferrin transport, cell redox homeostasis, cellular response to unfolded protein, ubiquitin-dependent protein catabolic process, regulation of innate immunity, MAPK cascade, positive regulation of apoptosis pathways, many of the pathways involved in lipid metabolism, and anti–reactive oxygen species pathways represented by “selenium compound metabolic process” and “selenocysteine metabolic process” (Additional file 1: Fig. S8b). These results corroborated the expression profiles in a previous study with the lower depth of sequencing available at that time [60], and additionally revealed further details of transcriptomic cold adaptation from the much deeper sequencing across a comprehensive set of tissue transcriptomes. A striking finding was the greatly increased transcriptional activities across many selenium-containing protein genes in the toothfish tissues compared with robalo (Fig. 4; Additional file 1: Fig. S9). Correspondingly, the genes involved in the translation of selenocysteine-containing proteins were also significantly upregulated in the toothfish (Fig. 4). Selenoproteins have well-known functions in coping with cellular oxidative stress; thus, the great expansion of selcys-tRNA genes (Fig. 1b, Additional file 1: Table S8) and the significantly upregulated expression of many kinds of selenoprotein mRNAs indicate that augmented anti–reactive oxygen species capacity has evolved as an important adaptation to the constantly freezing environment, where saturated levels of O2 and cold-depressed metabolic rates would make oxidative stress a formidable challenge for cellular life. Interestingly, the expression of glutathione peroxidase 4b (gpx4b) (Fig. 4), a selenoprotein uniquely able to reduce lipid hydroperoxides [64, 65], was lower in D. mawsoni, suggesting that alternative lipid metabolic programs may exist in the toothfish. Accordingly, we found that all isoforms of the major players in lipid droplet assembly (PLN2, PLN5, fitm, seipin) important for lipid storage in adipose tissue were upregulated in all toothfish tissues examined relative to E. maclovinus, signifying a shift of lipid distribution towards storage in D. mawsoni, as described in detail below.
Figure 4:

Comparison of gene expression between D. mawsoni and E. maclovinus tissues. The colored symbols represent the genes involved in 3 metabolic processes (listed on the right). The genes with significantly higher expression in D. mawsoni or E. maclovinus are labeled on the corresponding organs.
Altered lipid metabolism in D. mawsoni for neutral buoyancy
In a striking evolutionary departure from the heavy, bottom-water ancestral character (exemplified by E. maclovinus), a handful of Antarctic notothenioids have secondarily acquired neutral or near neutral buoyancy, enabling ecological diversification into and filling of mid-water niches—a distinctive hallmark of the Antarctic notothenioid adaptive radiation. The giant toothfish D. mawsoni, despite growing to massive sizes, being robustly muscled, and lacking a swim bladder, is the only notothenioid that has attained complete neutral buoyancy [14, 15]. Known morphological specializations include extensive lipid (mostly triglycerides) deposits under skin and in the musculature, and a light skeleton of mostly cartilage and little mineralized bone, adaptations that reduce overall density and provide static lift [14]. To understand the genetic basis of the large accumulation of lipids and reduced mineralization in D. mawsoni, we carried out transcriptome comparisons between D. mawsoni and several other notothenioids in which neutral buoyancy is not developed.
We compared gene expression profiles of muscles of D. mawsoni, and of the negatively buoyant Antarctic N. coriiceps and the basal E. maclovinus to elucidate evolutionary differences in the mechanisms of intermuscular lipid deposit. Compared with E. maclovinus, genes involved in triacylglycerol synthesis in the toothfish muscle were overrepresented in DEGs (P < 0.05) and markedly upregulated in transcription, including the key enzymes acylglycerol-3-phosphate O-acyltransferase isoforms and cytidine diphosphate-diacylglycerol synthase (Additional file 1: Fig. S10). An important regulator of this process, lipin1, was downregulated in D. mawsoni. Lipin1 is known to exert dual effects on lipid metabolism—it acts as a phosphatidate phosphatase enzyme to form diacylglycerol required for lipid synthesis but also serves as a transcriptional co-activator to promote fatty acid oxidation [66]. The downregulation of lipin1 was consistent with downregulation of fatty acid oxidation in D. mawsoni because about half of the genes involved in fatty acid oxidation were also downregulated relative to the robalo (Additional file 1: Table S12). At the same time, genes involved in regulation of lipid storage were overrepresented in the DEGs (P < 0.05) and all but 1 gene (MEST) were upregulated in the toothfish (Additional file 1: Table S13). These data strongly suggest a shift of metabolic pathways from lipid breakdown to lipid biosynthesis and lipid storage in D. mawsoni muscle relative to E. maclovinus, favoring deposit of lipids, thus contributing to neutral buoyancy. Compared with N. coriiceps, D. mawsoni muscle showed an overall trend of upregulation of genes involved in glycerolipid biosynthesis and lipid storage, but the differences are not as dramatic as in the D. mawsoni/E. maclovinus comparison. Expression levels of many genes relevant to lipid oxidation were fairly similar between the 2 species, suggesting common downregulation in lipid oxidation in the Antarctic species compared with the temperate E. maclovinus (Additional file 1: Fig. S10 and Tables S12 and S13). Consistent with downregulation of the lipid oxidation in the muscles of the 2 Antarctic fishes were their lower expression levels of lipid oxidation mitigating selenoenzyme gpx4 compared with the robalo. In total, the transcriptome comparisons revealed substantial genetic reprogramming in D. mawsoni muscle that would favor the large lipid deposition in this species.
GO enrichment tests on the muscle DEGs also indicated regulatory change (P = 0.073) in fat cell differentiation between D. mawsoni and E. maclovinus. Adipogenesis in almost all animals is predominantly regulated by PPARγ [67]. Expression of PPARγ in D. mawsoni muscle was upregulated by more than 5.6-fold compared with E. maclovinus (Additional file 1: Table S14). In addition, as many as 16 known pro-adipogenetic factors were upregulated from 1- to 8-fold. Several factors in the TGF-β (TGFB1, smad3), Wnt (Sirt1, sirt2, frizzled-related protein [FRZB]), and Notch (jag1b, Hes1) pathways and Jun dimerization protein 2 (JDP2), reportedly negative regulators of adipogenesis, were also upregulated. When compared with N. coriiceps, about half of the DEGs in the D. mawsoni/E. maclovinus comparison under this GO term were statistically insignificant, but remarkably the comparison yielded significant upregulation of 10 pro-adipogenetic factors in D. mawsoni muscle, including the most important regulator, PPARγ (Fig. 5a), and only 1 negative regulating factor (TGFB1) [68, 69]. These results strongly suggest that regulatory promotion of adipogenesis in D. mawsoni muscle is a key contributing factor to fat deposition and attainment of neutral buoyancy.
Figure 5:
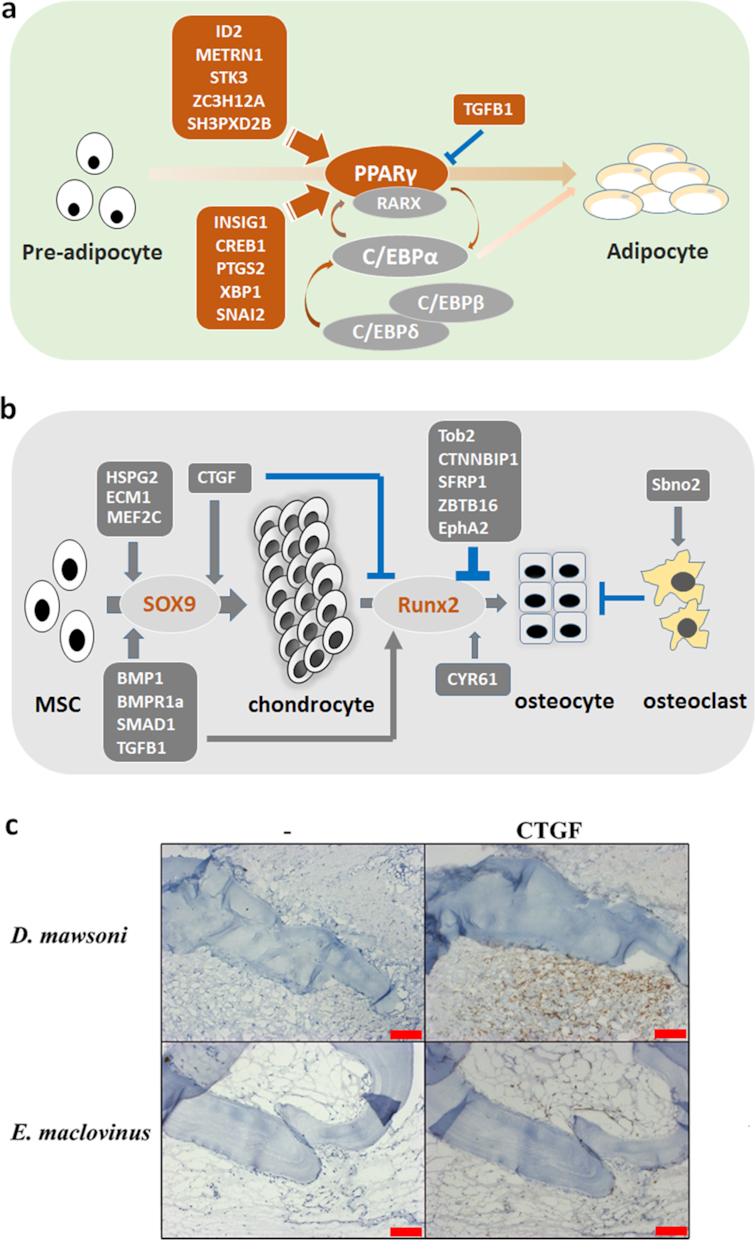
Schematic diagram showing changed regulation of buoyancy-related developmental pathways. (A) Enhanced adipogenetic pathways in D. mawsoni muscle. The genes shadowed in red were upregulated in D. mawsoni while those shadowed in grey were unchanged. (B) Changed osteogenetic regulation in D. mawsoni bone. Genes shadowed in dark grey were upregulated in D. mawsoni while those in light grey were not changed. The arrows (in dark red or dark grey) indicate a positive effect on the process while blocked (in blue) lines indicate inhibitory effect. MSC: mesenchymal stem cell. (C). Immunohistochemical staining to detect the abundance of CTGF in cross sections of pelvic fin of D. mawsoni and E. maclovinus. The left panels of each fish are immunohistochemical staining without the first antibody as negative control. The presence of CTGF is indicated by the brown staining in the tissues shown at right. Scale bar, 50 μm.
Reduction of ossification in D. mawsoni
To reveal the genes involved in the reduced bone ossification in D. mawsoni, we compared the transcriptomes of pelvic girdle bones between D. mawsoni (0% body weight in seawater) and the negatively buoyant Antarctic notothenioids Trematomus bernacchii and Pagothenia borchgrevinki (3.52% and 2.75% of body weight, respectively, in seawater) [14]. A total of 1,733 DEGs showing the same direction of change in the D. mawsoni/P. borchgrevinki and the D. mawsoni/T. bernacchii comparisons were identified and used for further analysis (Additional file 3). We found that 48 genes encoding various ribosomal proteins were significantly reduced in expression in the D. mawsoni bone, suggesting either a lower protein translation activity or fewer metabolically active cells in the pectoral girdle than in the other 2 nototheniids. The DEGs were enriched with hundreds of GO biological processes, including “extracellular matrix,” “ossification,” “response to hypoxia,” “angiogenesis,” and “lipid storage,” indicating that multiple genetic programs were distinctly regulated in the toothfish bone (Additional file 1: Fig. S11). In terms of ossification, it was noteworthy that expressions of the 2 major regulators of vertebrate bone development, sox9 and runx2 [70], were not significantly altered among the 3 notothenioids. However, expression of many genes of the BMP pathways, wnt pathways, and many regulatory factors known to be involved in the process were specifically altered in D. mawsoni, which likely shifted the developmental balance between chondrogenesis and osteogenesis (Fig. 5b; Additional file 1: Table S15). Among these highly upregulated genes (depicted in Fig. 5b), CTGF has been implicated in early events of osteogenic differentiation including proliferation and recruitment of osteoprogenitors; however, when expressed constitutively, CTGF would inhibit both Wnt-3A and BMP-9–induced osteoblast differentiation [71]. HSPG2 (prostaglandin-endoperoxide synthase 2) is required for the chondrogenic and adipogenic differentiation from synovial mesenchymal cells via its regulation of sox9 and PPARγ, but not for osteogenic differentiation via runx2 [72], and ECM1 (extracellular matrix protein 1) interacts with HSPG2 to regulate chondrogenesis [73]. MEF2C, a transcription factor that regulates muscle and cardiovascular development, controls bone development by activating the genetic program for chondrocyte hypertrophy [74]. Some of the upregulated genes are known to inhibit osteoblastogenesis, such as Tob2 [75], CTNNBIP1 [76], secreted frizzled-related protein 1 (SRFP1) [77], and ZBTB16 [78]. Some members of the TGF-β superfamily (BMPR1a, TGF-β1, SMAD1), which were upregulated in D. mawsoni, are known to promote both chondrogenesis and osteoblastogenesis [79]. CYR61 and PTN specifically promote osteoblastogenesis [80, 81], but expression of PTN is drastically reduced, consistent with reduced hard bone formation. A few genes influence ossification via regulating osteoclastogenesis; e.g., Sbno2 promotes osteoclast fusion [82], and activation of the EphA2 signaling on osteoblasts led to bone reabsorption [83]. We found that genes associated with osteoclast differentiation are significantly enriched in the DEGs (P < 0.05) and all were upregulated (Additional file 1: Tables S16 and S17). Overall, the gene expression patterns in the toothfish bone demonstrated a genetic shift to chondrogenesis over osteoblastogenesis in bone development, which would reduce bone density and contribute to achieving neutral buoyancy.
Studies have indicated that the majority of clinical conditions associated with human bone loss are accompanied by increased marrow adiposity, possibly due to shifting of the balance between osteoblast and adipocyte differentiation in bone marrow stromal (skeletal) stem cells [84]. A few signaling pathways such as the TGF-β/BMP pathways and the Wnt pathway (represented by CTNNBIP and SFRP1 in this case) are known to participate in regulation of both bone and adipocyte development in animals. In the toothfish, we found enriched GO terms relevant to regulation of “response to lipid” and “lipid storage” (Additional file 1: Table S12), indicating possible linkage in the regulatory network that orchestrates the loss of ossification and gain of lipids in D. mawsoni bones.
To verify whether the elevated transcription of the regulatory factors indeed resulted in more abundant protein, we selected the factor CTGF for immunohistochemical staining in the bone and surrounding tissues of pelvic fins of D. mawsoni and E. maclovinus because it is the only factor for which an effective monoclonal antibody is currently available. Much stronger signal was detected in the D. mawsoni fin tissue (Fig. 5c), supporting a correlation between protein abundance and mRNA transcription in the case of CTGF. This result further supports the involvement of CTGF in the reduced ossification in D. mawsoni.
Discussion
We sequenced and compared the genomes and transcriptomes of the cold-adapted high-latitude Antarctic toothfish D. mawsoni and the basal temperate relative E. maclovinus representing the ancestral character state to deduce Antarctic-specific evolutionary and adaptive changes supporting physiological activities of notothenioid fishes in freezing and oxygen-rich SO waters, as well as the gain of secondary pelagicism fundamental to Antarctic notothenioid niche expansion and adaptive radiation. The assembled genomes achieved 90% (D. mawsoni) and 95% (E. maclovinus) coverage of the respective genome size estimated by cell flow cytometry, and with greater scaffold N50 than the currently available sole Antarctic notothenioid (N. coriiceps) genome, greatly enhancing comprehensive, genome-wide discovery of evolutionary processes.
We found 2-fold expansion of TEs in the Antarctic toothfish over the temperate robalo E. maclovinus and deduced the timing of a burst of 1 major class of TEs (LINEs) to ∼6.5 mya, temporally correlating with the late Miocene onset of a steady cooling trend of the SO and diversification of the modern Antarctic notothenioid clade, suggesting a role of cold-induced TE expansion in notothenioid speciation. We found that many of the protein coding genes in the toothfish evolved rapidly and experienced positive selection, among which genes relevant to preservation of protein homeostasis were particularly prominent. Multiple gene families have undergone duplication during evolution in the cold, as exemplified by genes that confer resistance to freezing in the cold SO waters: the AFGP family that evolved de novo and confers extracellular freeze avoidance, and duplicated ZP ZPC5 genes that functionally diversified to aid in cellular freezing resistance. Through transcriptome comparisons, we found that functional output of the cellular apparatus for selenoprotein production in the Antarctic toothfish was greatly elevated compared with the basal temperate robalo, suggesting evolutionary mobilization of antioxidant selenoproteins in mitigating intensified oxidative stresses arising from the O2-rich SO environment.
The evolutionary transition from the negatively buoyant ancestral character to complete neutral buoyancy in the Antarctic toothfish entailed remarkable genetic reprograming of fat deposition and bone development. We found upregulation of processes of adipogenesis in skeletal muscle, and triacylglycerol synthesis and fat storage were favored over fatty acid oxidation. In bone development, a regulatory cascade favoring chondrogenesis over osteoblastogenesis was especially evident. The shift in fat synthesis and storage, together with reduction of ossification, is therefore key in evolutionary gain of neutral buoyancy and secondary pelagicism in D. mawsoni, and likely in the handful of other pelagic notothenioids, allowing them to diversify into mid-water niches, a distinctive hallmark of the Antarctic notothenioid adaptive radiation.
The remarkable diversification of Antarctic notothenioids (and several other polar fish lineages) is integral to the conclusion from a recent analysis of latitudinal diversity gradient of marine fishes that high-latitude cold water lineages exhibit exceptionally high rates of speciation compared with tropical lineages, counter to expectation based on latitudinal species richness [85]. Rates of molecular evolution based on phylogenetic tree branch lengths are not found to be slower at high latitudes [85]. We have shown more definitively in this study that in the cold-adapted Antarctic notothenioid fish, evolutionary rates in fact accelerated in thousands of protein-coding genes; extensive cold-specific gene duplication and functional diversification had occurred, such as in the ZP protein gene families; and TE mobility was remarkably elevated, which likely contributed to the observed higher frequency of chromosomal rearrangements. In mammals, ZP3 is known to function in sperm–egg recognition [86] and TE activity is positively related to rate of speciation [87]. How these genomic and functional changes elicited by selective pressures from the cold SO temperatures might have acted as intrinsic factors affecting notothenioid speciation are rich questions for further investigation.
Conclusion
In summary, the results of this study provided robust new insights into genomic and transcriptomic alterations enabling cold adaptation and niche expansion of the predominant and ecologically vital Antarctic fish group in the SO. The genomes also serve as valuable resources for future investigations of genomic and evolutionary changes in the diverse Antarctic notothenioid families driven by paleoclimate changes in the SO, studies that may shed light on questions of why the coldest ocean has been a hot spot of species formation.
Availability of supporting data and materials
All of the Illumina short read sequencing data of this project have been deposited at NCBI under the accession No. BioProject PRJNA401363 (http://www.ncbi.nlm.nih.gov/sra/). The assembled draft genomes and their annotations have been released at the official website of the Shanghai Ocean University (http://202.121.66.128/). The current version of the data set is the first version (v1). Antarctic toothfish genome and transcriptome [88], Patagonian robalo genome and transcriptome [89], and other supporting data are also available via the GigaScience GigaDB repository [90].
Supporting information
URLs: KEGG, http://www.genome.jp/kegg/; KAAS, http://www.genome.jp/tools/kaas/;SSPACE, https://www.baseclear.com/genomics/bioinformatics/basetools/SSPACE; Platanus, http://platanus.bio.titech.ac.jp/; SMALT, http://www.sanger.ac.uk/resources/software/smalt/; SOAPdenovo, http://soap.genomics.org.cn; RepeatModeler, http://www.repeatmasker.org/RepeatModeler.html; RepeatMasker, http://www.repeatmasker.org; Repbase, http://www.girinst.org/repbase/; Timetree, http://www.timetree.org/;Ensembl, ftp://ftp.ensembl.org/pub/; http://evolution.genetics.washington.edu/phylip.html; PAML, http://abacus.gene.ucl.ac.uk/software/paml.html; GLEAN, https://github.com/glean/glean; Interpro, http://www.ebi.ac.uk/interpro/; Infernal, http://eddylab.org/infernal/; Rfam, http://rfam.xfam.org/; OrthoMCL, http://orthomcl.org/orthomcl/; Mrbayes, http://mrbayes.sourceforge.net/; SSAHA_Pileup, ftp://ftp.sanger.ac.uk/pub/zn1/ssaha_pileup/; HISAT2, http://ccb.jhu.edu/software/hisat2/index.shtml.
Additional files
Additional file 1: Figs. S1–S11 and Tables S1–S17.
Additional file 2: supporting data for Fig. 1b, Fig. 2c, Fig. 2d, and Fig. 4.
Additional file 3: list of duplicated protein gene families of D. mawsoni and E. maclovinus.
Additional file 4: list of DEGs between D. mawsoni and 2 negatively buoyant notothenioids.
Abbreviations
AFGP: antifreeze glycoprotein; bp: base pairs; BUSCO: benchmarking universal single-copy orthologs; CHO: Chinese hamster ovary; CTGF: connective tissue growth factor; DEG: differentially expressed genes; ECM1: extracellular matrix protein 1; GO: gene ontology; gpx4: glutathione peroxidase 4; HSPG2: prostaglandin-endoperoxide synthase 2; JDP2: Jun dimerization protein 2; KEGG: Kyoto Encyclopedia of Genes and Genomes; LINE: long interspersed transposable elements; mRNA: messenger RNA; MSC: mesenchymal stem cell; mya: million years ago; NCBI: National Center for Biotechnology Information; PBS: phosphate-buffered saline; PPAR: peroxisome proliferator-activated receptor; RNA-Seq: RNA sequencing; rRNA: ribosomal RNA; SDS-PAGE: sodium dodecyl sulfate polyacrylamide gel electrophoresis; SNP: single-nucleotide polymorphism; SO: Southern Ocean; SRFP1: secreted frizzled-related protein 1; TBST: Tris-buffered saline with Tween 20; TE: transposable element; TGF-β: transforming growth factor β; tRNA: transfer RNA; ZP: zona pellucida protein.
Competing interests
The authors declare they have no competing financial interests.
Funding
The work was supported by grants from the Natural Science Foundation of China (No. 41761134050, 31572611, 31572598) and the Major Science Innovation Grant (2017-01-07-00-10-E00060) from the Shanghai Education Committee, the Key Achievement Supporting Grant from Laboratory for Marine Biology and Biotechnology, Qingdao National Laboratory for Marine Science and Technology to L.C., and the USA National Science Foundation Polar Programs grant ANT1142158 to C.H.C.C.
Author contributions
L.C. and C.H.C.C. conceived and managed the project and its components. C.H.C.C. and M.H. performed fish and tissue collections and sample preparations. K.R.M. and X.Z. contributed to DNA and RNA preparations and genome size determination. Y.L., M.Y., and W.L. performed genome annotation and RNA-Seq data analysis. Y.R. and S.P. conducted de novo genome assembly. K.T.B. confirmed the AFGP loci. Q.X., Y.F., and L.C. designed and performed the biological experiments. Sample preparation and genome sequencing were carried out by S.J., W.Z., and J.W. L.C., Q.X., Y.L., and C.H.C.C. analyzed the data as a whole and wrote the manuscript, and C.H.C.C. and W.W. contributed interpretation of data and edits to the manuscript.
Supplementary Material
10/2/2018 Reviewed
10/3/2018 Reviewed
10/22/2018 Reviewed
ACKNOWLEDGEMENTS
We thank BGI-Shenzhen for their technical support in sequencing of the D. mawsoni genome and Mr. Liping Shu of Wuhan Ice-Harbor Biological Technological Co. Ltd. for his help in the annotation of the E. maclovinus genome.
References
- 1. Livermore R, Nankivell A, Eagles G, et al.. Paleogene opening of Drake Passage. Earth Planet Sci Lett. 2005;236(1–2):459–70. [Google Scholar]
- 2. Eastman JT. The nature of the diversity of Antarctic fishes. Polar Biol. 2005;28(2):93–107. [Google Scholar]
- 3. Near TJ, Dornburg A, Harrington RC, et al.. Identification of the notothenioid sister lineage illuminates the biogeographic history of an Antarctic adaptive radiation. BMC Evol Biol. 2015;15:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eastman JT. Antarctic notothenioid fishes as subjects for research in evolutionary biology. Antarct Sci. 2000;12(3):276–87. [Google Scholar]
- 5. La Mesa M, Eastman JT, Vacchi M. The role of notothenioid fish in the food web of the Ross Sea shelf waters: a review. Polar Biol. 2004;27(6):321–38. [Google Scholar]
- 6. Devries AL, Eastman JT. Lipid sacs as a buoyancy adaptation in an Antarctic fish. Nature. 1978;271(5643):352–3. [Google Scholar]
- 7. Chen LB, DeVries AL, Cheng CHC. Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish. Proc Natl Acad Sci U S A. 1997;94(8):3811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matschiner M, Hanel R, Salzburger W. On the origin and trigger of the notothenioid adaptive radiation. Plos One. 2011;6(4):e18911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Near TJ, Dornburg A, Kuhn KL, et al.. Ancient climate change, antifreeze, and the evolutionary diversification of Antarctic fishes. Proc Natl Acad Sci U S A. 2012;109(9):3434–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shin SC, Ahn DH, Kim SJ, et al.. The genome sequence of the Antarctic bullhead notothen reveals evolutionary adaptations to a cold environment. Genome Biol. 2014;15(9):468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Malmstrom M, Matschiner M, Tørresen OK, et al.. Evolution of the immune system influences speciation rates in teleost fishes. Nat Genet. 2016;48(10):1204–10. [DOI] [PubMed] [Google Scholar]
- 12. Gon O, Heemstra P, . Fishes of the Southern Ocean. Grahamstown, South Africa: J.L.B. Smith Institute of Ichthyology, 1990: 305. [Google Scholar]
- 13. Eastman JT, Witmer LM, Ridgely RC, et al.. Divergence in skeletal mass and bone morphology in Antarctic notothenioid fishes. J Morphol. 2014;275(8):841–61. [DOI] [PubMed] [Google Scholar]
- 14. Eastman JT, Devries AL. Buoyancy adaptations in a swim‐bladderless Antarctic fish. J Morphol. 2010;167(1):91–102. [DOI] [PubMed] [Google Scholar]
- 15. Eastman JT, Devries AL. Buoyancy studies of notothenioid fishes in McMurdo Sound, Antarctica. Copeia. 1982;1982(2):385–93. [Google Scholar]
- 16. Amemiya CT, Ota T, Litman GW. Construction of P1 artificial chromosome (PAC) libraries from lower vertebrates. In: Birren B, Lai E, eds. Nonmammalian Genomic Analysis: A Practical Guide. San Diego: Academic Press; 1996:223–56. [Google Scholar]
- 17. Li RQ, Li YR, Kristiansen K, et al.. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24(5):713–4. [DOI] [PubMed] [Google Scholar]
- 18. Boetzer M, Henkel CV, Jansen HJ, et al.. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27(4):578–9. [DOI] [PubMed] [Google Scholar]
- 19. Bao ZR, Eddy SR. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002;12(8):1269–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Price AL, Jones NC, Pevzner PA. De novo identification of repeat families in large genomes. Bioinformatics. 2005;21:I351–8. [DOI] [PubMed] [Google Scholar]
- 21. Repbase. https://www.girinst.org/repbase/ accessed March, 2018. [Google Scholar]
- 22. Apweiler R, Attwood TK, Bairoch A, et al.. The InterPro database, an integrated documentation resource for protein families, domains and functional sites. Nucleic Acids Res. 2001;29(1):37–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000;28(1):27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bairoch A, Apweiler R, Wu CH, et al.. The universal protein resource (UniProt). Nucleic Acids Res. 2005;33:D154–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25(5):955–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nawrocki EP, Kolbe DL, Eddy SR. Infernal 1.0: inference of RNA alignments. Bioinformatics. 2009;25(10):1335–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nawrocki EP, Burge SW, Bateman A, et al.. Rfam 12.0: updates to the RNA families database. Nucleic Acids Res. 2015;43(D1):D130–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li L, Stoeckert CJ, Roos DS. OrthoMCL: Identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13(9):2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loytynoja A, Goldman N. webPRANK: a phylogeny-aware multiple sequence aligner with interactive alignment browser. BMC Bioinformatics. 2010;11:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huelsenbeck JP, Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17(8):754–5. [DOI] [PubMed] [Google Scholar]
- 31. Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–8. [DOI] [PubMed] [Google Scholar]
- 32. Yang ZH. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 2007;24(8):1586–91. [DOI] [PubMed] [Google Scholar]
- 33. De Bie T, Cristianini N, Demuth JP, et al.. CAFE: a computational tool for the study of gene family evolution. Bioinformatics. 2006;22(10):1269–71. [DOI] [PubMed] [Google Scholar]
- 34. Zdobnov EM, Apweiler R. InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001;17(9):847–8. [DOI] [PubMed] [Google Scholar]
- 35. SSAHA_PILEUP, ftp://ftp.sanger.ac.uk/pub/zn1/ssaha_pileup/, Accessed January, 2018. [Google Scholar]
- 36. Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim D, Landmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–U121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Trapnell C, Roberts A, Goff L, et al.. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7(3):562–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wang LK, Feng ZX, Wang X, et al.. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26(1):136–8. [DOI] [PubMed] [Google Scholar]
- 40. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Series B Stat Methodol. 1995;57(1):289–300. [Google Scholar]
- 41. Yu GC, Wang LG, Han YY, et al.. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16(5):284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen S, Yu M, Chu X, et al.. Cold-induced retrotransposition of fish LINEs. J Genet Genomics. 2017;44(8):385–94. [DOI] [PubMed] [Google Scholar]
- 43. Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22(23):2971–2. [DOI] [PubMed] [Google Scholar]
- 45. Stefanits H, Bienkowski M, Galanski M, et al.. KINFix - a formalin-free non-commercial fixative optimized for histological, immunohistochemical and molecular analyses of neurosurgical tissue specimens. Clin Neuropathol. 2016;35(1):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ghigliotti L, Mazzei F, Ozouf-Costaz C, et al.. The two giant sister species of the Southern Ocean, Dissostichus eleginoides and Dissostichus mawsoni, differ in karyotype and chromosomal pattern of ribosomal RNA genes. Polar Biol. 2007;30(5):625–34. [Google Scholar]
- 47. Mazzei F, Ghigliotti L, Coutanceau J-P, et al.. Chromosomal characteristics of the temperate notothenioid fish Eleginops maclovinus (Cuvier). Polar Biol. 2008;31(5):629–34. [Google Scholar]
- 48. Kajitani R, Toshimoto K, Noguchi H, et al.. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014;24(8):1384–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Simão FA, Waterhouse RM, Ioannidis P, et al.. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31(19):3210–2. [DOI] [PubMed] [Google Scholar]
- 50. Fishbase, http://www.fishbase.org, Accessed March 2018. [Google Scholar]
- 51. Hanchet S, Dunn A, Parker S, et al.. The Antarctic toothfish (Dissostichus mawsoni): biology, ecology, and life history in the Ross Sea region. Hydrobiologia. 2015;761(1):397–414. [Google Scholar]
- 52. Barker PF, Thomas E. Origin, signature and palaeoclimatic influence of the Antarctic Circumpolar Current. Earth Sci Rev. 2004;66(1–2):143–62. [Google Scholar]
- 53. Pisano E, Ozouf-Costaz C. Cytogenetics and evolution in extreme environment: the case of Antarctic fishes. In: Val AL, Kapoor BG, eds. Fish Adaptations. Enfield, NH: Science Publishers Inc; 2003:309–30. [Google Scholar]
- 54. Detrich HW, Stuart A, Schoenborn M, et al.. Genome enablement of the notothenioidei: genome size estimates from 11 species and BAC libraries from 2 representative taxa. J Exp Zool B Mol Dev Evol. 2010;314(5):369–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rebollo R, Horard B, Hubert B, et al.. Jumping genes and epigenetics: towards new species. Gene. 2010;454(1–2):1–7. [DOI] [PubMed] [Google Scholar]
- 56. Auvinet J, Graça P, Belkadi L, et al.. Mobilization of retrotransposons as a cause of chromosomal diversification and rapid speciation: the case for the Antarctic teleost genus Trematomus. BMC Genomics. 2018;19:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chenais B, Caruso A, Hiard S, et al.. The impact of transposable elements on eukaryotic genomes: from genome size increase to genetic adaptation to stressful environments. Gene. 2012;509(1):7–15. [DOI] [PubMed] [Google Scholar]
- 58. Zachos JC, Dickens GR, Zeebe RE. An early Cenozoic perspective on greenhouse warming and carbon-cycle dynamics. Nature. 2008;451(7176):279–83. [DOI] [PubMed] [Google Scholar]
- 59. Favre A, Päckert M, Pauls SU, et al.. The role of the uplift of the Qinghai-Tibetan Plateau for the evolution of Tibetan biotas. Biol Rev Camb Philos Soc. 2015;90(1):236–53. [DOI] [PubMed] [Google Scholar]
- 60. Chen ZZ, Cheng CH, Zhang J, et al.. Transcriptomic and genomic evolution under constant cold in Antarctic notothenioid fish. Proc Natl Acad Sci U S A. 2008;105(35):12944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Xu QH, Cheng CH, Hu P, et al.. Adaptive evolution of hepcidin genes in Antarctic notothenioid fishes. Mol Biol Evol. 2008;25(6):1099–112. [DOI] [PubMed] [Google Scholar]
- 62. Cao LX, Huang Q, Wu Z, et al.. Neofunctionalization of zona pellucida proteins enhances freeze-prevention in the eggs of Antarctic notothenioids. Nat Commun. 2016;7:12987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Nicodemus-Johnson J, Silic S, Ghigliotti L, et al.. Assembly of the antifreeze glycoprotein/trypsinogen-like protease genomic locus in the Antarctic toothfish Dissostichus mawsoni(Norman). Genomics. 2011;98(3):194–201. [DOI] [PubMed] [Google Scholar]
- 64. Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830(5):3289–303. [DOI] [PubMed] [Google Scholar]
- 65. Friedmann Angeli JP, Schneider M, Proneth B, et al.. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chen Y, Rui BB, Tang LY, et al.. Lipin family proteins - key regulators in lipid metabolism. Ann Nutr Metab. 2015;66(1):10–18. [DOI] [PubMed] [Google Scholar]
- 67. Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006;4(4):263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Ignotz RA, Massagué J. Type beta transforming growth factor controls the adipogenic differentiation of 3T3 fibroblasts. Proc Natl Acad Sci U S A. 1985;82(24):8530–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Petruschke T, Röhrig K, Hauner H. Transforming growth factor beta (TGF-beta) inhibits the differentiation of human adipocyte precursor cells in primary culture. Int J Obes Relat Metab Disord. 1994;18(8):532–6. [PubMed] [Google Scholar]
- 70. Long FX, Ornitz DM. Development of the endochondral skeleton. Cold Spring Harb Perspect Biol. 2013;5(1):a008334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Luo Q, Kang Q, Si W, et al.. Connective tissue growth factor (CTGF) is regulated by Wnt and bone morphogenetic proteins signaling in osteoblast differentiation of mesenchymal stem cells. J Biol Chem. 2004;279(53):55958–68. [DOI] [PubMed] [Google Scholar]
- 72. Sadatsuki R, Kaneko H, Kinoshita M, et al.. Perlecan is required for the chondrogenic differentiation of synovial mesenchymal cells through regulation of Sox9 gene expression. J Orthop Res. 2017;35(4):837–46. [DOI] [PubMed] [Google Scholar]
- 73. Mongiat M, Fu J, Oldershaw R, et al.. Perlecan protein core interacts with extracellular matrix protein 1 (ECM1), a glycoprotein involved in bone formation and angiogenesis. J Biol Chem. 2003;278(19):17491–9. [DOI] [PubMed] [Google Scholar]
- 74. Arnold MA, Kim Y, Czubryt MP, et al.. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell. 2007;12(3):377–89. [DOI] [PubMed] [Google Scholar]
- 75. Gamez B, Rodriguez-Carballo E, Bartrons R, et al.. MicroRNA-322 (miR-322) and its target protein Tob2 modulate Osterix (Osx) mRNA stability. J Biol Chem. 2013;288(20):14264–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tago K, Nakamura T, Nishita M, et al.. Inhibition of Wnt signaling by ICAT, a novel beta-catenin-interacting protein. Gene Dev. 2000;14(14):1741–9. [PMC free article] [PubMed] [Google Scholar]
- 77. Yao W, Cheng Z, Shahnazari M, et al.. Overexpression of secreted frizzled-related protein 1 inhibits bone formation and attenuates parathyroid hormone bone anabolic effects. J Bone Miner Res. 2010;25(2):190–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Onizuka S, Iwata T, Park SJ, et al.. ZBTB16 as a downstream target gene of osterix regulates osteoblastogenesis of human multipotent mesenchymal stromal cells. J Cell Biochem. 2016;117(10):2423–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Dexheimer V, Gabler J, Bomans K, et al.. Differential expression of TGF-β superfamily members and role of Smad1/5/9-signalling in chondral versus endochondral chondrocyte differentiation. Sci Rep. 2016;6:36655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Tare RS, Oreffo ROC, Clarke NMP, et al.. Pleiotrophin/osteoblast-stimulating factor 1: dissecting its diverse functions in bone formation. J Bone Miner Res. 2002;17(11):2009–20. [DOI] [PubMed] [Google Scholar]
- 81. Su JL, Chiou J, Tang CH, et al.. CYR61 regulates BMP-2-dependent osteoblast differentiation through the αvβ3 integrin/integrin-linked kinase/ERK pathway. J Biol Chem. 2010;285(41):31325–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Maruyama K, Uematsu S, Kondo T, et al.. Strawberry notch homologue 2 regulates osteoclast fusion by enhancing the expression of DC-STAMP. J Exp Med. 2013;210(10):1947–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Diercke K, Sen S, Kohl A, et al.. Compression-dependent up-regulation of ephrin-A2 in PDL fibroblasts attenuates osteogenesis. J Dent Res. 2011;90(9):1108–15. [DOI] [PubMed] [Google Scholar]
- 84. Abdallah BM, Kassem M. New factors controlling the balance between osteoblastogenesis and adipogenesis. Bone. 2012;50(2):540–5. [DOI] [PubMed] [Google Scholar]
- 85. Rabosky DL, Chang J, Title PO, et al.. An inverse latitudinal gradient in speciation rate for marine fishes. Nature. 2018;559(7714):392–5. [DOI] [PubMed] [Google Scholar]
- 86. Litscher ES, Williams Z, Wassarman PM. Zona pellucida glycoprotein ZP3 and fertilization in mammals. Mol Reprod Dev. 2009;76(10):933–41. [DOI] [PubMed] [Google Scholar]
- 87. Ricci M, Peona V, Guichard E, et al.. Transposable elements activity is positively related to rate of speciation in mammals. J Mol Evol. 2018;86(5):303–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chen L, Lu Y, Li W, et al.. The genome and transcriptome of the Antarctic toothfish Dissostichus mawsoni. GigaScience Database. 2019. 10.5524/102162 [DOI] [Google Scholar]
- 89. Chen L, Lu Y, Li W, et al.. The genome and transcriptome of the Patagonia robalo Eleginops maclovinus. GigaScience Database. 2019. 10.5524/102163 [DOI] [Google Scholar]
- 90. Chen L, Lu Y, Li W, et al.. Supporting data for “Genomic bases for colonizing the freezing Southern Ocean revealed by the genomes of Antarctic toothfish and Patagonia robalo.”. GigaScience Database. 2019. 10.5524/100545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
10/2/2018 Reviewed
10/3/2018 Reviewed
10/22/2018 Reviewed