

Abstract
Background
Opioid drugs, including fentanyl, are commonly used to treat neuropathic pain, and are considered effective by some professionals. Most reviews have examined all opioids together. This review sought evidence specifically for fentanyl, at any dose, and by any route of administration. Other opioids are considered in separate reviews.
Objectives
To assess the analgesic efficacy of fentanyl for chronic neuropathic pain in adults, and the adverse events associated with its use in clinical trials.
Search methods
We searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE, and Embase from inception to June 2016, together with the reference lists of retrieved articles, and two online study registries.
Selection criteria
We included randomised, double‐blind studies of two weeks' duration or longer, comparing fentanyl (in any dose, administered by any route, and in any formulation) with placebo or another active treatment in chronic neuropathic pain.
Data collection and analysis
Two review authors independently searched for studies, extracted efficacy and adverse event data, and examined issues of study quality and potential bias. We did not carry out any pooled analyses. We assessed the quality of the evidence using GRADE.
Main results
Only one study met our inclusion criteria. Participants were men and women (mean age 67 years), with postherpetic neuralgia, complex regional pain syndrome, or chronic postoperative pain. They were experiencing inadequate relief from non‐opioid analgesics, and had not previously taken opioids for their neuropathic pain. The study used an enriched enrolment randomised withdrawal design. It was adequately blinded, but we judged it at unclear risk of bias for other criteria.
Transdermal fentanyl (one‐day fentanyl patch) was titrated over 10 to 29 days to establish the maximum tolerated and effective dose (12.5 to 50 µg/h). Participants who achieved a prespecified good level of pain relief with a stable dose of fentanyl, without excessive use of rescue medication or intolerable adverse events ('responders'), were randomised to continue with fentanyl or switch to placebo for 12 weeks, under double‐blind conditions. Our prespecified primary outcomes were not appropriate for this study design, but the measures reported do give an indication of the efficacy of fentanyl in this condition.
In the titration phase, 1 in 3 participants withdrew because of adverse events or inadequate pain relief, and almost 90% experienced adverse events. Of 258 participants who underwent open‐label titration, 163 were 'responders' and entered the randomised withdrawal phase. The number of participants completing the study (and therefore continuing on treatment) without an increase of pain by more than 15/100 was 47/84 (56%) with fentanyl and 28/79 (35%) with placebo. Because only 63% responded sufficiently to enter the randomised withdrawal phase, this implies that only a maximum of 35% of participants entering the study would have had useful pain relief and tolerability with transdermal fentanyl, compared with 22% with placebo. Almost 60% of participants taking fentanyl were 'satisfied' and 'very satisfied' with their treatment at the end of the study, compared with about 40% with placebo. This outcome approximates to our primary outcome of moderate benefit using the Patient Global Impression of Change scale, but the group was enriched for responders and the method of analysis was not clear. The most common adverse events were constipation, nausea, somnolence, and dizziness.
There was no information about other types of neuropathic pain, other routes of administration, or comparisons with other treatments.
We downgraded the quality of the evidence to very low because there was only one study, with few participants and events, and there was no information about how data from people who withdrew were analysed.
Authors' conclusions
There is insufficient evidence to support or refute the suggestion that fentanyl works in any neuropathic pain condition.
Plain language summary
Fentanyl for neuropathic pain in adults
Bottom line
There is no good evidence to support or refute the suggestion that fentanyl works in any neuropathic pain condition.
Background
Neuropathic pain is pain coming from a damaged nervous system. It is different from pain messages that are carried along healthy nerves from damaged tissue (e.g. from a fall or cut, or an arthritic knee). Neuropathic pain is often treated by different medicines (drugs) from those used for pain from damaged tissue, which we often think of as painkillers. There are different types of neuropathic pain, with different causes. Some medicines that are used to treat depression or epilepsy can be very effective in some people with neuropathic pain. Sometimes opioid painkillers are used to treat neuropathic pain.
Opioid painkillers are drugs such as morphine. Morphine is derived from plants, but many opioids are also made by chemical synthesis rather than being extracted from plants. Fentanyl is one of these synthetic opioids. It is available in numerous countries for use as a painkiller and, when used to treat chronic pain, is usually given through an adhesive patch, so it is taken into the body through the skin.
Study characteristics
In January 2016 we searched for clinical trials where fentanyl was used to treat neuropathic pain in adults. We found one small study that did this and met our requirements for the review. The study had a complicated design. Study participants first received fentanyl (as one‐day skin patches) for one month. Those who responded to therapy (achieved a predetermined level of pain relief) were then randomly allocated to continue receiving fentanyl or placebo for 12 weeks. The participants had one of three different types of neuropathic pain and had not taken opioids before. There were only 163 people in the 12‐week comparison with placebo.
Key results
The study found that more people taking fentanyl had pain relief than those taking placebo. About 1 in 7 participants stopped taking fentanyl because of side effects, and 1 in 5 did not get a good level of pain relief in the first part of the study. Almost half of those who continued into the second part of the study also stopped. The most common side effects were constipation, nausea (feeling sick), somnolence (feeling sleepy), and dizziness. These are typical side effects with opioids such as fentanyl. There was so little information from this single study that we concluded there was no convincing evidence to support or reject a meaningful benefit for fentanyl over placebo.
Quality of the evidence
We rated the quality of the evidence as very low because there was only one study, with few participants and events, and an unusual design. Very‐low‐quality evidence means that we are very uncertain about the results.
Summary of findings
for the main comparison.
| Transdermal fentanyl compared with placebo for neuropathic pain | ||||||
|
Patient or population: Adults with chronic neuropathic pain Settings: Community Intervention: Fentanyl 1‐day adhesive patch 12.5 to 50 µg/h Comparison: Placebo patch | ||||||
| Outcomes | Probable outcome with intervention | Probable outcome with comparator | RR (95% CI) | No of studies, participants | Quality of the evidence (GRADE) | Comments |
| Moderate benefit: at least 30% reduction in pain, or PGIC much or very much improved |
Pain treatment: satisfied, very satisfied 49/84 |
Pain treatment: satisfied, very satisfied 32/79 |
Not calculated | 1 study, 163 participants, 81 events | Very low | Downgraded three times: single study, few participants and events, approximates to prespecified outcome, imputation for withdrawals not specified, unusual mix of pain conditions |
| Substantial benefit: at least 50% reduction in pain, or PGIC much improved |
No data | No data | ‐ | ‐ | ‐ | ‐ |
| Lack of efficacy withdrawal in randomised double‐blind phase | 19/84 | 39/79 | Not calculated | 1 study, 163 participants, 81 events | Very low | Downgraded three times: single study, few participants and events |
| Adverse event withdrawal in randomised double‐blind phase | 14/84 | 4/79 | Not calculated | 1 study, 163 participants, 81 events | Very low | Downgraded three times: single study, few participants and events |
| Serious adverse events | 13/258 during titration phase 8/84 in randomised double‐blind phase |
4/79 in randomised double‐blind phase | Not calculated | Titration: 1 study, 258 participants, 13 events Randomised double‐blind phase: 1 study, 163 participants, 12 events |
Very low | Downgraded three times: single study, few participants and events |
| Deaths | None | None | ‐ | 1 study, 258 participants, 0 events | Very low | Downgraded three times: estimated incidence not more frequent than 1 in 861 |
| CI: Confidence interval; PGIC: Patient Global Impression of Change; RR: Risk Ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Eypasch E, Lefering R, Kum CK, Troidl H. Probability of adverse events that have not yet occurred: a statistical reminder. BMJ 1995;311(7005):619‐20.
Background
This review is based on a template for reviews of drugs used to relieve neuropathic pain. The aim is for all reviews to use the same methods, based on new criteria for what constitutes reliable evidence in chronic pain (Moore 2010a; Appendix 1).
Description of the condition
The 2011 International Association for the Study of Pain definition of neuropathic pain is "pain caused by a lesion or disease of the somatosensory system" (Jensen 2011), and is based on a definition agreed at an earlier consensus meeting (Treede 2008). Neuropathic pain may be caused by nerve damage, but is often followed by changes in the central nervous system (Moisset 2007). The origin of neuropathic pain is complex (Baron 2010; Baron 2012; Tracey 2011; von Hehn 2012), and neuropathic pain features can be found in people with joint pain (Soni 2013).
Many people with neuropathic pain conditions are significantly disabled with moderate or severe pain for many years. Chronic pain conditions comprised 5 of the 11 top‐ranking conditions for years lived with disability in 2010 (Vos 2012), and are responsible for considerable loss of quality of life and employment, and increased healthcare costs (Moore 2014a).
Neuropathic pain is usually divided according to the cause of nerve injury. There may be many causes, but some common causes of neuropathic pain include diabetes (painful diabetic neuropathy (PDN)), shingles (postherpetic neuralgia (PHN)), amputation (stump and phantom limb pain), neuropathic pain after surgery or trauma, stroke or spinal cord injury, trigeminal neuralgia, and HIV infection. Sometimes the cause is unknown.
In systematic reviews, the overall prevalence of neuropathic pain in the general population is reported to be between 7% and 10% (van Hecke 2014), and about 7% in a systematic review of studies published since 2000 (Moore 2014a). In individual countries, prevalence rates have been reported as 3.3% in Austria (Gustorff 2008), 6.9% in France (Bouhassira 2008), and up to 8% in the UK (Torrance 2006). Some forms of neuropathic pain, such as PDN and postsurgical chronic pain (which is often neuropathic in origin), are increasing in prevelance (Hall 2008).
Estimates of incidence vary between individual studies for particular origins of neuropathic pain, often because of small numbers of cases. In primary care in the UK, between 2002 and 2005, the incidences (per 100,000 person‐years' observation) were 28 (95% confidence interval (CI) 27 to 30) for PHN, 27 (26 to 29) for trigeminal neuralgia, 0.8 (0.6 to 1.1) for phantom limb pain, and 21 (20 to 22) for PDN (Hall 2008). Others have estimated an incidence of 4 in 100,000 per year (Katusic 1991; Rappaport 1994) for trigeminal neuralgia, and of 12.6 per 100,000 person‐years for trigeminal neuralgia and 3.9 per 100,000 person‐years for PHN in a study of facial pain in the Netherlands (Koopman 2009). One systematic review of chronic pain demonstrated that some neuropathic pain conditions, such as PDN, can be more common than other neuropathic pain conditions, with prevalence rates up to 400 per 100,000 person‐years (McQuay 2007).
Neuropathic pain is difficult to treat effectively, with only a minority of people experiencing a clinically relevant benefit from any one intervention. A multidisciplinary approach is now advocated, combining pharmacological interventions with physical or cognitive (or both) interventions. Conventional analgesics such as paracetamol and nonsteroidal anti‐inflammatory drugs are not thought to be effective, but are frequently used (Di Franco 2010; Vo 2009). Some people may derive some benefit from a topical lidocaine patch or low‐concentration topical capsaicin, although evidence about benefits is uncertain (Derry 2012; Derry 2014). High‐concentration topical capsaicin may benefit some people with PHN (Derry 2013).
Treatment is often by so‐called 'unconventional analgesics', such as antidepressants (duloxetine and amitriptyline; Lunn 2014; Moore 2012a; Sultan 2008), or antiepileptics (gabapentin or pregabalin; Moore 2009; Moore 2014b; Wiffen 2013). The proportion of people who achieve worthwhile pain relief (typically at least 50% pain intensity reduction; Moore 2013a) is small, generally only 10% to 25% more than with placebo, with numbers needed to treat for an additional beneficial outcome (NNT) usually between 4 and 10 (Kalso 2013; Moore 2013b). Neuropathic pain is not particularly different from other chronic pain conditions in that only a small proportion of trial participants have a good response to treatment (Moore 2013b).
One overview of treatment guidelines pointed out some general similarities between recommendations, but guidelines are not always consistent with one another (O'Connor 2009). The current National Institute for Health and Care Excellence (NICE) guidance suggests offering a choice of amitriptyline, duloxetine, gabapentin, or pregabalin as initial treatment for neuropathic pain (with the exception of trigeminal neuralgia), with switching if the first, second, or third drugs tried are not effective or not tolerated (NICE 2013).
Description of the intervention
Fentanyl was first synthesised in the 1950s and was found to be significantly more potent than commonly used opioids, such as morphine. It was initially used for intravenous anaesthesia and analgesia in the 1960s and became a mainstay of intraoperative and perioperative analgesia and both conscious and deep sedation in the in‐hospital setting. Peak analgesic effects of intravenous fentanyl last for 30 to 60 minutes, but onset of analgesia is rapid. Fentanyl is approximately 80 to 100 times more potent than morphine, is highly lipophilic, and binds strongly to plasma proteins (Trescot 2008). Fentanyl is associated with possible hypoxaemia (low oxygen levels in the blood) after surgery (McQuay 1979).
Fentanyl undergoes extensive metabolism in the liver, and is subject to first‐pass metabolism in the liver and possibly small intestine, though hepatic extraction from blood may be more complicated (Bullingham 1984). Various formulations of fentanyl have been studied, including a rapid transmucosal formulation for buccal absorption or intranasal sprays for acute breakthrough pain, and transdermal formulations for chronic pain (Lötsch 2013; Nelson 2009). The transdermal formulation has a lag time of 6 to 12 hours to onset of action after application, and typically reaches steady state in three to six days. When a patch is removed, a subcutaneous reservoir remains, and drug clearance may take up to 24 hours.
Fentanyl patches are available as generic formulations, and brand names include Fentalis®, Matrifen®, Mezolar®, Osmanil®, Tilofyl®, Victanyl®, Durogesic®, and DTrans®. They are available as 12, 25, 50, 75, and 100 μg/h transdermal patches. The 25, 50, 75, and 100 μg/h patches were first licensed in 1994, and a 12 μg patch followed in 2005. Transdermal fentanyl provides 'rate controlled' drug delivery over 72 hours, although shorter and longer delivery periods are under development. A 24‐hour patch has been licensed for some pain conditions in Japan. Surface area exposed, skin permeability, and local blood flow determine absorption (Heiskanen 2009). Absorption was impaired in 10 cachectic (weak and underweight) compared with 10 normal weight cancer patients, (Heiskanen 2009). The 'reservoir patch' is being phased out and replaced with a matrix design, as it was likely to leak if damaged or cut. Trials have demonstrated no difference in pain intensity reduction or overall adverse effects between the reservoir and matrix patches. Satisfaction was improved, and wearability, adhesion, and comfort were improved with the matrix patches (Cachia 2011).
Fentanyl patches have been suggested to have some benefits over more traditional opioids such as oral morphine. There is a favourable safety profile in people with renal insufficiency, as this does not affect fentanyl elimination, while renal insufficiency or failure cause a build‐up of active metabolites of opioids such as morphine. Fentanyl is also considered to cause less constipation than morphine. However, fentanyl patches are not generally recommended in clinical practice for people who are opioid naïve. Moreover, exposure to heat through fever, sunbathing, hot showers or baths, and warm weather can cause more fentanyl to be released into the skin and cause serious, or even fatal, adverse events.
How the intervention might work
Opioids such as fentanyl bind to specific opioid receptors in the nervous system and other tissues; there are three principal classes of receptors (mu, kappa, and delta) though others have been suggested, and subtypes of receptors are considered to exist. Binding of opioid agonists such as fentanyl to receptors brings about complex cellular changes, the outcomes of which include decreased perception of pain, decreased reaction to pain, and increased pain tolerance. Opioids from plant sources have been used for thousands of years to treat pain.
Why it is important to do this review
One UK survey found that weak and strong opioids were used frequently for treating neuropathic pain (Hall 2013). Fentanyl patches can be useful in people who cannot tolerate oral opioids. Titrating the dose of fentanyl patches can be difficult and it is probably better to convert from a dose of morphine or other oral opioid that is effective but not tolerated. Since the early 2000s, a marked increase in prescribing of opioids for non‐cancer pain in general, despite a relatively modest evidence base, has in some countries led to widespread diversion with consequent abuse, misuse, and mortality (Franklin 2014; Weisberg 2014; Zin 2014). There were 1.2 million prescriptions for fentanyl in primary care in England in 2014 at a cost of almost GBP57 million (PCA 2015) and the amount of fentanyl prescribed has been rising substantially (Zin 2014), although not all this prescribing will be for neuropathic pain.
Concurrently, suspicion has arisen that opioid‐induced hyperalgesia, together with tolerance to the analgesic effects of opioids, may in reality result in a lesser degree of benefit for opioids in neuropathic pain than previously assumed.
The standards used to assess evidence in chronic pain trials have evolved substantially in recent years, with particular attention being paid to trial duration, withdrawals, and statistical imputation following withdrawal, all of which can substantially alter estimates of efficacy. The most important change is the move from using mean pain scores, or mean change in pain scores, to the number of people who have a large decrease in pain (by at least 50%) and who continue in treatment, ideally in trials of 8 to 12 weeks' duration or longer. A pain intensity reduction of 50% or more correlates with improvements in comorbid symptoms, function, and quality of life. These standards are set out in the Cochrane Pain, Palliative and Supportive Care Group (PaPaS) Author and Referee Guidance for pain studies (PaPaS 2012).
This Cochrane review assessed evidence using methods that make both statistical and clinical sense, and used developing criteria for what constitutes reliable evidence in chronic pain (Moore 2010a). To be included, trials had to meet a minimum of reporting quality (blinding, randomisation), validity (duration, dose and timing, diagnosis, outcomes, etc.), and size (ideally at least 500 participants in a comparison in which the NNT is 4 or above; Moore 1998). This approach sets high standards for the demonstration of efficacy and marks a departure from how reviews were conducted previously.
Taking this newer, more rigorous approach is particularly important for opioids in chronic non‐cancer pain. Opioids in clinical trials in non‐cancer pain are associated with very high withdrawal rates of up to 60% over about 12 weeks (Moore 2010b). Many withdrawals occur within the first few weeks, when participants experience pain relief but cannot tolerate the drug. The common practice of using the last observed results carried forward to the end of the trial many weeks later (last observation carried forward (LOCF)) can, therefore, produce results based largely on people who are no longer in the trial, and who in the real world could not achieve pain relief because they could not take the tablets. The newer standards, outlined in Appendix 1, would not allow this and can produce very different results. For example, one large analysis of pooled data from trials in osteoarthritis and chronic low back pain conducted over about 12 weeks judged oxycodone effective, but an analysis of the same data using the new clinically meaningful standards showed it to be significantly worse than placebo (Lange 2010).
One previous Cochrane review demonstrated the limitations of our knowledge about opioids in neuropathic pain except in short duration studies of 24 hours or less (McNicol 2013). These limitations were confirmed by reviews specific to buprenorphine and oxycodone (Gaskell 2014; Wiffen 2015). A review specific to fentanyl is timely.
Objectives
To assess the analgesic efficacy of fentanyl for chronic neuropathic pain in adults, and the adverse events associated with its use in clinical trials.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials with double‐blind assessment of participant outcomes following two weeks or more of treatment, although the emphasis of the review was on studies with a duration of eight weeks or longer. We required full journal publication, with the exception of online clinical trial results summaries of otherwise unpublished clinical trials and abstracts with sufficient data for analysis. We did not include short abstracts (usually meeting reports). We excluded studies that were non‐randomised, studies of experimental pain, case reports, and clinical observations.
Types of participants
We included studies involving adults aged 18 years and above with one or more chronic neuropathic pain condition including (but not limited to):
cancer‐related neuropathy;
central neuropathic pain;
complex regional pain syndrome (CRPS) Type II;
human immunodeficiency virus (HIV) neuropathy;
painful diabetic neuropathy (PDN);
phantom limb pain;
postherpetic neuralgia (PHN);
postoperative or traumatic neuropathic pain;
spinal cord injury;
trigeminal neuralgia.
Where studies included participants with more than one type of neuropathic pain, we planned to analyse results according to the primary condition.
Types of interventions
Fentanyl at any dose, by any route, administered for the relief of neuropathic pain and compared with placebo or any active comparator.
Types of outcome measures
We anticipated that studies would use a variety of outcome measures, with most studies using standard subjective scales (numerical rating scale (NRS) or visual analogue scale (VAS)) for pain intensity (where higher numbers indicate more pain) or pain relief (where higher numbers indicate more relief), or both. We were particularly interested in Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) definitions for moderate and substantial benefit in chronic pain studies (Dworkin 2008). These are defined as:
at least 30% pain relief over baseline (moderate benefit);
at least 50% pain relief over baseline (substantial benefit);
much or very much improved on Patient Global Impression of Change scale (PGIC; moderate benefit); and
very much improved on PGIC (substantial benefit).
These outcomes are different from those used in most earlier reviews, concentrating as they do on dichotomous outcomes where pain responses do not follow a normal (Gaussian) distribution. People with chronic pain desire high levels of pain relief, ideally more than 50% pain intensity reduction, and ideally having no worse than mild pain (Moore 2013a; O'Brien 2010).
We have included a 'Summary of findings' table as set out in the PaPaS author guide (PaPaS 2012), to include outcomes of at least 30% and at least 50% pain intensity reduction, much or very much improvement on PGIC, withdrawals due to adverse events, serious adverse events, and death. We used the GRADE approach to assess the quality of evidence related to each of the key outcomes listed below (Chapter 12, Higgins 2011), as appropriate.
Primary outcomes
Participant‐reported pain relief of 30% or greater
Participant‐reported pain relief of 50% or greater
PGIC much or very much improved
PGIC very much improved
Secondary outcomes
Any pain‐related outcome indicating some improvement
Withdrawals due to lack of efficacy, adverse events, and for any cause
Participants experiencing any adverse event
Participants experiencing any serious adverse event. Serious adverse events typically include any untoward medical occurrence or effect that at any dose results in death, is life‐threatening, requires hospitalisation or prolongation of existing hospitalisation, results in persistent or significant disability or incapacity, is a congenital anomaly or birth defect, is an 'important medical event' that may jeopardise the person, or may require an intervention to prevent one of the above characteristics or consequences
Specific adverse events, particularly somnolence and dizziness
Search methods for identification of studies
Electronic searches
We searched the following databases, without language restrictions.
Cochrane Central Register of Controlled Trials (CENTRAL, via the Cochrane Register of Studies Online database (CRSO)) to 14 June 2016.
MEDLINE (via Ovid) from 1946 to 14 June 2016.
Embase (via Ovid) from 1974 to 14 June 2016.
The search strategies for CENTRAL, MEDLINE, and Embase are listed in Appendix 2, Appendix 3, and Appendix 4, respectively.
Searching other resources
We reviewed the bibliographies of relevant studies and review articles, and searched two clinical trial registries, (ClinicalTrials.gov (ClinicalTrials.gov) and the World Health Organization International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/)), to identify additional published or unpublished data. We contacted Janssen‐Cilag Ltd who were able to clarify the publication status of the included study shortly before full publication. We did not contact investigators or other study sponsors.
Data collection and analysis
We planned to perform separate analyses according to particular neuropathic pain conditions, combining different neuropathic pain conditions in analyses for exploratory purposes only.
Selection of studies
We determined eligibility by reading the abstract of each study identified by the search. We eliminated studies that clearly did not satisfy the inclusion criteria, and obtained full copies of the remaining studies. Two review authors made the decisions, reading these studies independently and reaching agreement by discussion. We did not anonymise the studies in any way before assessment. We have included a PRISMA flow chart (Figure 1).
1.
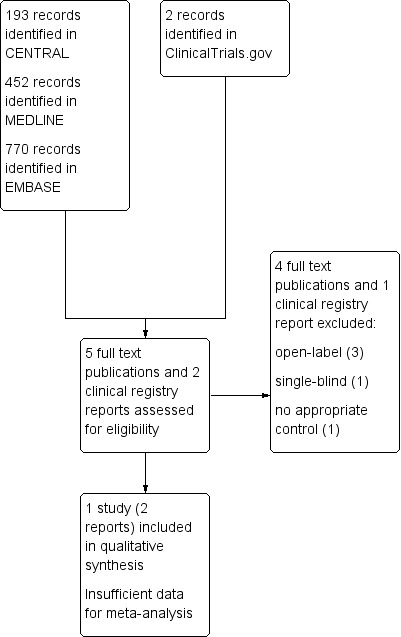
Study flow diagram.
Data extraction and management
Two review authors extracted data independently using a standard form and checked for agreement before entry into Review Manager 5 (RevMan 2014) or any other analysis tool. We included information about the pain condition and number of participants treated, drug and dosing regimen, control intervention, study design, study duration and follow up, analgesic outcome measures and results, withdrawals, and adverse events (participants experiencing any adverse event or serious adverse event).
Assessment of risk of bias in included studies
We used the Oxford Quality Score as the basis for inclusion (Jadad 1996), limiting inclusion to studies that were randomised and double‐blind as a minimum.
Two review authors independently assessed the risk of bias for each study, using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Chapter 8, Higgins 2011), and adapted from those used by the Cochrane Pregnancy and Childbirth Group, with any disagreements resolved by discussion. We assessed the following for each study.
Random sequence generation (checking for possible selection bias). We assessed the method used to generate the allocation sequence as: low risk of bias (any truly random process, random number table, computer random number generator); unclear risk of bias (when the method used to generate the sequence is not clearly stated). We excluded studies at a high risk of bias that used a non‐random process (odd or even date of birth, hospital or clinic record number).
Allocation concealment (checking for possible selection bias). The method used to conceal allocation to interventions prior to assignment determines whether intervention allocation could have been foreseen in advance of, or during, recruitment, or changed after assignment. We assessed the methods as: low risk of bias (telephone or central randomisation, consecutively numbered, sealed, opaque envelopes); unclear risk of bias (when method was not clearly stated). We excluded studies that did not conceal allocation and were, therefore, at a high risk of bias (open list).
Blinding of outcome assessment (checking for possible detection bias). We planned to assess the methods used to blind study participants and outcome assessors from knowledge of which intervention a participant received. We assessed the methods as: low risk of bias (study stated that it was blinded and described the method used to achieve blinding, identical tablets, matched in appearance and smell); unclear risk of bias (study stated that it was blinded but did not provide an adequate description of how it was achieved). We excluded studies at a high risk of bias that were not double‐blind.
Incomplete outcome data (checking for possible attrition bias due to the amount, nature, and handling of incomplete outcome data). We assessed the methods used to deal with incomplete data as: low risk of bias (fewer than 10% of participants did not complete the study, or used 'baseline observation carried forward' analysis (BOCF), or both); unclear risk of bias (used LOCF analysis); or high risk of bias (used 'completer' analysis).
Size of study (checking for possible biases confounded by small size). We assessed studies as being at low risk of bias (200 participants or more per treatment arm); unclear risk of bias (50 to 199 participants per treatment arm); or high risk of bias (fewer than 50 participants per treatment arm).
Measures of treatment effect
We planned to calculate NNTs as the reciprocal of the absolute risk reduction (McQuay 1998). For unwanted effects, the NNT becomes the number needed to treat for an additional harmful outcome (NNH) and is calculated in the same manner. We planned to use dichotomous data to calculate risk ratios (RRs) with 95% confidence intervals (CIs) using a fixed‐effect model unless we found significant statistical heterogeneity (see Assessment of heterogeneity). We planned not to use continuous data in analyses, and intended to extract and use continuous data, which probably reflect efficacy and utility poorly, only if useful for illustrative purposes.
Unit of analysis issues
We accepted randomisation to the individual participant only. We planned to split the control treatment arm between active treatment arms in a single study if the active treatment arms were not combined for analysis.
Dealing with missing data
We planned to use intention‐to‐treat (ITT) analysis where the ITT population consisted of participants who were randomised, took at least one dose of the assigned study medication, and provided at least one post‐baseline assessment. We would assign zero improvement to missing participants wherever possible.
Assessment of heterogeneity
We planned to deal with clinical heterogeneity by combining studies that examined similar conditions, and to assess statistical heterogeneity visually (L'Abbé 1987) and with the use of the I2 statistic. When the I2 value was greater than 50%, we would consider possible reasons for this.
Assessment of reporting biases
The aim of this review was to use dichotomous outcomes of known utility and of value to patients with pain (Hoffman 2010; Moore 2010c; Moore 2010d; Moore 2010e; Moore 2013a). The review would not depend on what the authors of the original studies chose to report or not, though clearly difficulties would arise in studies that did not report any dichotomous results.
We planned to assess publication bias using a method designed to detect the amount of unpublished data with a null effect required to make any result clinically irrelevant (usually taken to mean an NNT of 10 or higher; Moore 2008).
Data synthesis
We planned to use a fixed‐effect model for meta‐analysis. We would have used a random‐effects model for meta‐analysis if there was significant clinical heterogeneity and it was considered appropriate to combine studies. We planned to analyse data for each painful condition separately.
Quality of the evidence
We used the GRADE system to assess the quality of the evidence related to the key outcomes listed in Types of outcome measures, as appropriate (Appendix 5; Chapter 12.2, Higgins 2011). Two review authors independently rated the quality of evidence for each outcome.
We paid particular attention to:
inconsistency, where point estimates vary widely across studies, or CIs of studies show minimal or no overlap (Guyatt 2011);
potential for publication bias, based on the amount of unpublished data required to make the result clinically irrelevant (Moore 2008).
In addition, there may be circumstances where the overall rating for a particular outcome needs to be adjusted as recommended by GRADE guidelines (Guyatt 2013a). For example, if there are so few data that the results are highly susceptible to the random play of chance, or if a study used LOCF imputation in circumstances where there are substantial differences in adverse event withdrawals, one would have no confidence in the result and would need to downgrade the quality of the evidence by three levels to very low quality. In circumstances where no data were reported for an outcome, we would report the level of evidence as very low quality (Guyatt 2013b).
'Summary of findings' table
We have included a 'Summary of findings' table to present the main findings in a transparent and simple tabular format. In particular, we have included key information concerning the quality of evidence, the magnitude of effect of the interventions examined, and the sum of available data on the outcomes of 'moderate' and 'substantial' benefit, withdrawal due to lack of efficacy, withdrawal due to adverse events, serious adverse events, and death.
Subgroup analysis and investigation of heterogeneity
We planned all analyses to be according to individual neuropathic pain conditions, because placebo response rates for the same outcome can vary between conditions, as can the drug‐specific effects (Moore 2009).
We did not plan subgroup analyses since our experience of previous reviews indicates that there would be too few data for any meaningful subgroup analysis (Gaskell 2014; McNicol 2013).
Sensitivity analysis
We planned no sensitivity analysis because the evidence base was known to be too small to allow reliable analysis. We planned to examine details of dose‐escalation schedules in the unlikely situation that this could provide some basis for a sensitivity analysis, but this was not possible.
Results
Description of studies
Results of the search
Searches identified 193 potentially relevant records in CENTRAL, 452 in MEDLINE, and 770 in Embase, and two in clinical trial registries. After deduplication and reading the titles and abstracts we obtained and read the full texts of five published records and two clinical trial registry reports. We excluded five studies, and included one (two reports) (Figure 1).
Included studies
We included one study, identified as a registry report (NCT01008553) and in a published pooled analysis (Arai 2015). This study used an enriched enrolment randomised withdrawal (EERW) design in which 258 participants underwent open‐label titration over 10 to 29 days with fentanyl one‐day patches to determine the maximum tolerated dose. It was not clear whether participants stopped or continued with previously inadequate medication.
Those who had pain intensity below 45/100 in the last three days of this open‐label titration, an improvement from pre‐treatment of at least 15/100, achieved a stable dosage of fentanyl, and required fewer than two doses per day of rescue medication, were classified as 'responders' (163 participants; 63% of those entering the open titration period) and were randomised to double‐blind treatment with either the same dose of fentanyl or placebo for 12 weeks. Of 84 receiving fentanyl, 47 completed the 12 weeks, compared with 28/79 receiving placebo (Arai 2015).
Fentanyl was administered as a one‐day patch, and all participants started with fentanyl 12.5 µg/h, increasing to a maximum of 50 µg/h over 10 to 29 days. Morphine hydrochloride was available as rescue medication (5 mg per fentanyl 12.5 µg/h).
Participants were opioid‐naïve and had pain that was not adequately controlled with non‐opioid analgesics. They were adults (mean age 67 years) with PHN (51%), CRPS (type not specified, 20%), or chronic postoperative pain (for a duration of 12 weeks or more, but no further details given; 29%). There were approximately equal numbers of men and women. Mean baseline pain intensity was 74/100 before treatment and 40/100 at the end of titration. For those entering the double‐blind period, baseline pain was 30/100 in the fentanyl group and 28/100 in the placebo group.
Excluded studies
We excluded five studies. Three were open‐label studies, two of which were in mixed pain conditions, one was a single‐blind study (ongoing), and one compared fentanyl with fentanyl plus gabapentin (no appropriate control). See the Characteristics of excluded studies table.
Risk of bias in included studies
See Figure 2 for a summary of our assessment of the risk of bias in the included study. Studies with EERW designs are likely to have additional sources of bias, or may require somewhat different assessments (Moore 2015), but we have not included those here.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
The study did not report the methods used to generate the randomisation sequence or to conceal allocation (unclear risk of bias).
Blinding
Blinding was achieved by using a placebo patch that was indistinguishable from the fentanyl patch. There was a period of down‐titration for the placebo group, which would help to maintain the blinding (low risk of bias).
Incomplete outcome data
Study did not mention how missing data were handled. The study's primary outcome (median time to withdrawal) appears robust, but other outcomes are unclear.
Other potential sources of bias
We judged the study to be at unclear risk of bias due to its size (84 and 79 participants in treatment arms for the randomised phase).
Effects of interventions
See: Table 1
Since we identified only one study for inclusion, we were unable to carry out any analyses.
Efficacy
Details of efficacy outcomes are provided in Appendix 6.
Participants with at least 30% or at least 50% pain relief
These outcomes were not reported.
PGIC much or very much improved
The study reported the participants' assessment of 'treatment satisfaction for pain' on a five‐point scale (very dissatisfied, dissatisfied, neither satisfied or dissatisfied, satisfied, very satisfied) after the double‐blind phase. We judged that the categories of 'satisfied' and 'very satisfied' approximate to our PGIC outcome of moderate benefit. In the fentanyl group, 49/84 (58%) participants were either satisfied or very satisfied, and corresponding data in the placebo group were 32/79 (41%). It is not clear how data from participants who withdrew due to adverse events were analysed for this outcome, but some form of imputation must have been used for participants who withdrew as the numbers are greater than the number who completed the study.
Other pain‐related outcomes
The primary outcome chosen in the study was median time to withdrawal from the double‐blind phase, where withdrawal occurred when there was worsening of pain of 15/100 (mean increase over three consecutive days) from entry into this phase, use of three or more doses of rescue medication per day for five or more days, the participant requested it because of a lack of efficacy, or the participant requested an increase in study drug dosage. The median time to withdrawal for the placebo group was 45 days, but could not be estimated for the fentanyl group because fewer than half the participants withdrew over the 12 weeks; the assumption must be that the median time to withdrawal was greater than 42 days.
The study did not report the number of participants who remained 'responders' at the end of the 12‐week double‐blind treatment period as a treatment outcome. However, Arai 2015 reports the number of participants completing the study (and therefore continuing on treatment) without an increase in pain of more than 15/100. For fentanyl, this was 47/84 (56%) and for placebo it was 28/79 (35%). The implication, then, is that because only 63% responded sufficiently to enter the randomised withdrawal phase, only a maximum of 35% of participants entering the study would have useful pain relief and tolerability with transdermal fentanyl, compared with 22% with placebo.
The group mean change (increase) in pain intensity from randomisation to the end of the double‐blind phase (mean of last three days) was 0.5/100 (from 30.1 to 29.6) in the fentanyl group, and 9.6/100 (from 27.5 to 37.1) in the placebo group. This mean change is unlikely to be of clinical significance, but probably masks larger changes in some individuals. It is not clear how data from participants who withdrew during treatment were analysed for this outcome.
We downgraded the quality of the evidence for efficacy to very low because there was only one study, with a small number of participants, low numbers of events, and there was no information about how participants who withdrew from the study were analysed (see Table 1).
Withdrawals
Details of withdrawals are provided in Appendix 7.
Withdrawals due to lack of efficacy
Following the titration period, 50/258 participants were classified as non‐responders to fentanyl. During the double‐blind period, one participant taking fentanyl withdrew because of perceived lack of efficacy, and a further 18/84 were withdrawn because they experienced a greater than 15/100 increase in pain intensity. With placebo, 4/79 withdrew because of perceived lack of efficacy, and a further 35/79 were withdrawn because they experienced a greater than 15/100 increase in pain intensity.
Withdrawals due to adverse events
During titration 39/258 participants withdrew due to adverse events. During the double‐blind period 14/84 participants taking fentanyl and 4/79 taking placebo withdrew due to adverse events.
Other withdrawals
During titration 12/258 participants withdrew because of physician (2) or participant (6) decision, participant was determined to increase dosage (3), and participant was considered not appropriate for the study (1). During the double‐blind period 4/84 participants taking fentanyl withdrew due to physician (1) or participant (2) decision, and an inability to perform required tests (1), and 8/79 taking placebo withdrew due to physician (2) or participant (2) decision, and excessive use of rescue medication (4).
We downgraded the quality of the evidence for withdrawals to very low because there was only one study, with a small number of participants per treatment arm in the randomised withdrawal phase, and low numbers of events.
Adverse events
Details of adverse events are provided in Appendix 7.
Any adverse event
During the titration phase, 231/258 (90%) participants experienced at least one adverse event. During the double‐blind phase, 72/84 (86%) and 56/79 (71%) participants experienced at least one adverse event with fentanyl and placebo, respectively. Generally, participants experienced fewer adverse events during the double‐blind period.
Most adverse events were of mild or moderate intensity.
Serious adverse events
During the titration phase, 13/258 participants experienced a serious adverse event. During the double‐blind phase, 8/84 and 4/79 participants experienced a serious adverse event with fentanyl and placebo, respectively. No serious adverse event occurred in more than one participant, and no deaths were reported.
Specific adverse events
Constipation (124/258), nausea (103/258), somnolence (118/258), and dizziness (52/258) were the most common adverse events reported during the titration period. As would be expected, the rates of these events were lower in the double‐blind period. Among the 84 participants taking fentanyl in the double‐blind phase, 12 reported constipation, 12 nausea, 12 somnolence, and 6 dizziness, and among 79 taking placebo, 10 reported constipation, 10 nausea, 5 somnolence, and 3 dizziness.
A small number of participants experienced application‐site reactions during titration: pruritus (15/258), erythema (8/258), dermatitis (4/258), rash (3/258). Few participants in either group reported these events during the double‐blind period.
We downgraded the quality of the evidence for adverse events to very low because there was only one study, with a small number of participants per treatment arm in the randomised withdrawal phase, and low numbers of events.
Discussion
Summary of main results
We found only one study to include in this review. The study assessed the efficacy of fentanyl, using the transdermal route of administration, for the treatment of neuropathic pain in opioid‐naïve participants; 258 participants entered the dose‐titration period, and 163 'responders' entered the double‐blind phase. The results indicated a reduction in pain intensity with open‐label fentanyl that was better maintained with fentanyl than placebo in the randomised, double‐blind withdrawal period. However, during titration, 1 in 3 (101/258) participants withdrew overall, mainly due to intolerable adverse events (1 in 7; 39/258) or not achieving a sufficiently good level of pain relief to proceed to the double‐blind phase (1 in 5; 50/258). Of those who did enter the double‐blind phase, only about 1 in 2 (37/84) in the fentanyl group and 2 in 3 (51/79) in the placebo group maintained good pain relief and were able to tolerate adverse events over 12 weeks.
Taking the pool of participants originally recruited, therefore, after 12 weeks treatment a maximum of about 3 in 10 would have maintained low pain and continued with the treatment, compared with 2 in 10 with placebo.
Most (90%) participants experienced adverse events during the titration phase, and the majority continued to do so in the double‐blind phase, although most participants experienced fewer events. Most adverse events were of mild or moderate intensity, and the most common were constipation, nausea, somnolence, and dizziness, which are typically associated with opioids.
Overall completeness and applicability of evidence
The amount of evidence we have is small, from a single study. Although participants had three different types of neuropathic pain, the study was underpowered to demonstrate a differential response. Participants were opioid‐naïve at screening, so high numbers of adverse events and withdrawals are to be expected; lower levels might be seen in an unselected or opioid‐tolerant population. Fentanyl patches are generally not recommended for opioid‐naïve patients.
About half of participants had PHN, 20% had CRPS, and 29% had chronic postoperative pain at enrolment. The study did not specify CRPS type II as an inclusion criterion, and provided no further details about the nature of the surgery leading to postoperative pain. We have no information about the efficacy of fentanyl in other types of neuropathic pain, such as diabetic neuropathy, or about routes of administration other than transdermal, or comparisons with other active treatments. The particular formulation used in this study is not commonly used; in most countries a 72‐hour (three‐day) patch is available.
As best we know, there is insufficient evidence to support or refute the use of fentanyl for treating neuropathic pain. This is despite the fact that a UK survey found that weak and strong opioids were used frequently for treating neuropathic pain, either alone or in combination with other drugs (Hall 2013). The lack of evidence for long‐term benefit with fentanyl reflects similar findings for oxycodone, buprenorphine, and other opioids (Gaskell 2014; McNicol 2013; Wiffen 2015). This lack of evidence of efficacy combined with substantial evidence of harm has led to calls for referral to a pain management specialist (ideally with expertise in opioid use) if daily dosing exceeds 80 to 100 mg morphine equivalents, particularly if pain and function are not substantially improved (Franklin 2014).
The number of participants in the single included study (258 screened, 163 in the randomised double‐blind phase) contrasts with 1096 who participated in randomised open studies and 1393 in observational studies of various designs (Appendix 8). Despite there being 2489 participants in these studies, with chronic pain of mostly mixed origins, no useful conclusions can be drawn because of problems in design, in outcomes, and in comparators used. Although some people had useful pain relief with fentanyl, there is no additional evidence that the proportion would be any higher than with no treatment.
One study did provide a useful insight into the relative efficacy of transdermal fentanyl and oral pregabalin in neuropathic cancer pain (Raptis 2014). The study had a randomised but open parallel design and was conducted over four weeks with initial titration for both drugs; initial pain relief was high, about 7/10 on a numerical rating scale. Of 60 participants given transdermal fentanyl, 22 (37%) had pain intensity reduction of 30% or more. Of 60 participants given oral pregabalin, 44 (73%) had that degree of pain reduction. All‐cause withdrawals were 10/60 for fentanyl and 3/60 for pregabalin. Pregabalin has a well‐established evidence base in neuropathic pain (Moore 2009), and the evidence from this open, but otherwise well‐conducted study, indicates fentanyl to be much less effective.
Quality of the evidence
The methods used in the included study are fundamentally sound, but the study is substantially underpowered, particularly for the randomised, double‐blind, withdrawal phase, and does not specify the imputation method(s) used for withdrawals (Moore 2015). It does not report the most useful outcome from the double‐blind phase, the number of participants who maintained therapeutic efficacy and were able to continue taking the medication (with tolerable adverse events), although we were able to estimate this.
These factors downgrade the evidence for all outcomes to very low quality, which means that further research is very likely to have an important impact on our confidence in our understanding of the effect.
Potential biases in the review process
We know of no potential biases in the review process. It is unlikely that there is a large body of unpublished evidence showing a large effect from fentanyl in neuropathic pain.
Agreements and disagreements with other studies or reviews
This review agrees with previous reviews and Cochrane reviews that there appears to be no body of good clinical studies assessing the efficacy of fentanyl, at any dose or in formulation, for neuropathic pain (McNicol 2013). The one study in this review was published after McNicol 2013. A recent review of all pharmacotherapy for neuropathic pain in adults did not mention fentanyl (Finnerup 2015).
Authors' conclusions
Implications for practice.
For people with neuropathic pain
There is insufficient evidence to support or refute the suggestion that fentanyl has any efficacy in any neuropathic pain condition.
For clinicians
There is insufficient evidence to support or refute the suggestion that fentanyl has any efficacy in any neuropathic pain condition.
For policy makers
There is insufficient evidence to support or refute the suggestion that fentanyl has any efficacy in any neuropathic pain condition. In the absence of any supporting evidence, it should probably not be recommended, except at the discretion of a pain specialist with particular expertise in opioid use.
For funders
There is insufficient evidence to support or refute the suggestion that fentanyl has any efficacy in any neuropathic pain condition. In the absence of any supporting evidence, it should probably not be recommended, except at the discretion of a pain specialist with particular expertise in opioid use.
Implications for research.
Large, robust, randomised trials with patient‐centred outcomes would be required to produce evidence to support or refute the efficacy of fentanyl in neuropathic pain. The necessary design of such trials is well established, but, for opioids in neuropathic pain, the main outcomes should be those of at least a 30% and at least a 50% reduction in pain intensity over baseline at the end of a trial of 12 weeks' duration in participants continuing on treatment. Withdrawal for any reason should be regarded as treatment failure, and LOCF analysis should not be used. The reason for this is that, in chronic pain, opioids frequently produce withdrawal rates of 50% or more, meaning that LOCF analysis can overstate treatment efficacy to a large extent. BOCF should be used in preference to LOCF as it provides a more relevant estimate of efficacy for the real world.
What's new
| Date | Event | Description |
|---|---|---|
| 29 May 2019 | Amended | Contact details updated. |
| 11 October 2017 | Review declared as stable | See Published notes. |
Notes
No new studies likely to change the conclusions are expected. Therefore, this review has now been stabilised following discussion with the authors and editors. If appropriate, we will update the review if new evidence likely to change the conclusions is published, or if standards change substantially which necessitate major revisions.
Acknowledgements
Institutional support was provided by the Oxford Pain Relief Trust.
The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Pain, Palliative and Supportive Care Review Group (PaPaS). Disclaimer: the views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the NIHR, National Health Service (NHS), or the Department of Health.
The protocol was based on a template developed in collaboration with the Cochrane Neuromuscular Diseases and Musculoskeletal Review Groups. The editorial process was managed by PaPaS.
Appendices
Appendix 1. Methodological considerations for chronic pain
There have been several changes in how the efficacy of conventional and unconventional treatments is assessed in chronic painful conditions. The outcomes are now better defined, particularly with new criteria for what constitutes moderate or substantial benefit (Dworkin 2008); older trials may report only participants with 'any improvement'. Newer trials tend to be larger, avoiding problems from the random play of chance. Newer trials also tend to be of longer duration, up to 12 weeks, and longer trials provide a more rigorous and valid assessment of efficacy in chronic conditions. New standards have evolved for assessing efficacy in neuropathic pain, and we are now applying stricter criteria for the inclusion of trials and assessment of outcomes, and are more aware of problems that may affect our overall assessment. To summarise some of the recent insights that must be considered in this new review.
Pain results tend to have a U‐shaped distribution rather than a bell‐shaped distribution. This is true in acute pain (Moore 2011a; Moore 2011b), back pain (Moore 2010d), and arthritis (Moore 2010e), as well as in fibromyalgia (Straube 2010); in all cases average results usually describe the experience of almost no‐one in the trial. Data expressed as averages are potentially misleading, unless they can be proven to be suitable.
As a consequence, we have to depend on dichotomous results (the individual either has or does not have the outcome) usually from pain changes or patient global assessments. The Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials (IMMPACT) group has helped with their definitions of minimal, moderate, and substantial improvement (Dworkin 2008). In arthritis, trials of less than 12 weeks' duration, and especially those shorter than eight weeks, overestimate the effect of treatment (Moore 2010d); the effect is particularly strong for less‐effective analgesics, and this may also be relevant in neuropathic‐type pain.
The proportion of people with at least moderate benefit can be small, even with an effective medicine, falling from 60% with an effective medicine in arthritis to 30% in fibromyalgia (Moore 2009; Moore 2010d; Moore 2010e; Moore 2013b; Moore 2014c; Straube 2008; Sultan 2008). One Cochrane review of pregabalin in neuropathic pain and fibromyalgia demonstrated different response rates for different types of chronic pain (higher in diabetic neuropathy and postherpetic neuralgia and lower in central pain and fibromyalgia) (Moore 2009). This indicates that different neuropathic pain conditions should be treated separately from one another, and that pooling should not be done unless there are good reasons for doing so.
Individual patient analyses indicate that people who get good pain relief (moderate or better) have major benefits in many other outcomes, affecting quality of life in a significant way (Moore 2010c; Moore 2014a).
Imputation methods such as last observation carried forward (LOCF), used when participants withdraw from clinical trials, can overstate drug efficacy, especially when adverse event withdrawals with drug are greater than those with placebo (Moore 2012b).
Appendix 2. Search strategy for CENTRAL via CRSO
MESH DESCRIPTOR Neuralgia EXPLODE ALL TREES (610)
MESH DESCRIPTOR Peripheral Nervous System Diseases EXPLODE ALL TREES (2590)
MESH DESCRIPTOR Somatosensory Disorders EXPLODE ALL TREES (710)
((pain* or discomfort*) adj10 (central or complex or nerv* or neuralg* or neuropath*)):TI,AB,KY (3306)
((neur* or nerv*) adj6 (compress* or damag*)):TI,AB,KY (635)
1 OR 2 OR 3 OR 4 OR 5 (6357)
MESH DESCRIPTOR Fentanyl EXPLODE ALL TREES (3897)
(fentanyl or fentanil* or Abstral or Actiq or DTrans or Durogesic or Fentalis or Matrifen or Mezolar or Osmanil or Sublimaze or Tilofyl or Victanyl):TI,AB,KY (9907)
6 AND 9 (193)
Appendix 3. Search strategy for MEDLINE via Ovid
exp NEURALGIA/ (15118)
exp PERIPHERAL NERVOUS SYSTEM DISEASES/ (124585)
exp SOMATOSENSORY DISORDERS/ (17961)
((pain* or discomfort*) adj10 (central or complex or nerv* or neuralg* or neuropath*)).mp. (43123)
((neur* or nerv*) adj6 (compress* or damag*)).mp. (52215)
1 or 2 or 3 or 4 or 5 (200106)
Fentanyl/ (11986)
(fentanyl or fentanil* or Abstral or Actiq or DTrans or Durogesic or Fentalis or Matrifen or Mezolar or Osmanil or Sublimaze or Tilofyl or Victanyl).mp. (17707)
7 or 8 (17707)
randomized controlled trial.pt. (417624)
randomized.ab. (309508)
placebo.ab. (159698)
drug therapy.fs. (1862631)
randomly.ab. (219030)
trial.ab. (322047)
groups.ab. (1378466)
10 or 11 or 12 or 13 or 14 or 15 or 16 (3483323)
6 and 9 and 17 (452)
Appendix 4. Search strategy for Embase via Ovid
exp NEURALGIA/ (81076)
exp PERIPHERAL NERVOUS SYSTEM DISEASES/ (55241)
exp SOMATOSENSORY DISORDERS/ (72226)
((pain* or discomfort*) adj10 (central or complex or nerv* or neuralg* or neuropath*)).mp. (80613)
((neur* or nerv*) adj6 (compress* or damag*)).mp. (73746)
1 or 2 or 3 or 4 or 5 (294021)
Fentanyl/ (50046)
(fentanyl or fentanil* or Abstral or Actiq or DTrans or Durogesic or Fentalis or Matrifen or Mezolar or Osmanil or Sublimaze or Tilofyl or Victanyl).mp. (54103)
7 or 8 (54103)
random*.ti,ab. (1042778)
factorial*.ti,ab. (26694)
(crossover* or cross over* or cross‐over*).ti,ab. (79506)
placebo*.ti,ab. (230284)
(doubl* adj blind*).ti,ab. (163796)
assign*.ti,ab. (276944)
allocat*.ti,ab. (99797)
RANDOMIZED CONTROLLED TRIAL.sh. (390913)
DOUBLE‐BLIND PROCEDURE.sh. (127382)
CROSSOVER PROCEDURE.sh. (45369)
10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 (1482600)
6 and 9 and 20 (770)
Appendix 5. GRADE: criteria for assigning grade of evidence
The GRADE system uses the following criteria for assigning a quality level to a body of evidence (Chapter 12, Higgins 2011).
High: randomised trials; or double‐upgraded observational studies.
Moderate: downgraded randomised trials; or upgraded observational studies.
Low: double‐downgraded randomised trials; or observational studies.
Very low: triple‐downgraded randomised trials; or downgraded observational studies; or case series/case reports.
Factors that may decrease the quality level of a body of evidence are:
limitations in the design and implementation of available studies suggesting high likelihood of bias;
indirectness of evidence (indirect population, intervention, control, outcomes);
unexplained heterogeneity or inconsistency of results (including problems with subgroup analyses);
imprecision of results (wide confidence intervals);
high probability of publication bias.
Factors that may increase the quality level of a body of evidence are:
large magnitude of effect;
all plausible confounding would reduce a demonstrated effect or suggest a spurious effect when results show no effect;
dose‐response gradient.
Appendix 6. Summary of outcomes: efficacy
| Study | Treatment | Pain outcome | Other efficacy outcome |
| Arai 2015 |
Titration: Fentanyl 1‐day patch, 12.5 to 50 µg/h Randomised withdrawal: (1) Fentanyl patch (titrated dose) (2) Placebo (downtitration) |
Titration: 163/258 completed Mean PI reduced from 73.5 (SD 12.8) to 39.5 (20.0) (mean of last 3 days) (scale 0 to 100) Randomised withdrawal: Mean change in PI from randomisation to last 3 days of double‐blind period (scale 0 to 100) (1) 0.5 (30.1, SD 11.5 to 29.6, SD 21.7) (2) 9.6 (27.5, SD 11.2 to 37.1, SD 20.2) PGIC (satisfied, very satisfied with pain treatment; 5‐point scale) (1) 49/84 (2) 32/79 |
Randomised withdrawal: Median time to withdrawal due to insufficient analgesic efficacy (1) not estimated (2) 45 days Use of rescue medication (mean doses/day) (1) 0.4 (SD 0.68) (2) 0.7 (SD 0.86) Measures of health disability using SF‐36 did not change substantially during treatment |
| PGIC: Patient Global Impression of Change; PI: pain intensity; SD: standard deviation; SF‐36: Short‐Form 36‐Item Health Survey | |||
Appendix 7. Summary of outcomes: adverse events and withdrawals
| Study | Treatment | Adverse events | Serious AEs | Withdrawals |
| Arai 2015 |
Titration: Fentanyl 1‐day patch, 12.5 to 50 µg/h Randomised withdrawal: (1) Fentanyl patch (titrated dose) (2) Placebo (down titration) |
Titration:
Any AE 231/258 (most mild or moderate) Most common were: constipation (124/258), nausea (103/258), somnolence (118/258), and dizziness (52/258) Application‐site reactions during titration: pruritus (15/258), erythema (8/258), dermatitis (4/258), rash (3/258) Randomised withdrawal: (1) 72/84 (2) 56/79 Most mild or moderate Fentanyl: 12 reported constipation, 12 nausea, 12 somnolence, and 6 dizziness Placebo: 10 reported constipation, 10 nausea, 5 somnolence, and 3 dizziness Few participants in either group reported application site reactions during the double‐blind period |
No deaths reported Titration: 13/258 Randomised withdrawal: (1) 8/84 (2) 4/79 |
Titration: AE: 39/258 Participant decision: 6/258 Physician decision: 2/258 Participant determined to increase dose: 3/258 Participant not appropriate for study: 1/258 Non‐responders: 50/258 Randomised withdrawal: Fentanyl: 37/84 AE: 14/84 LoE: 1/84 > 15/100 increase in PI: 18/84 Physician or participant decision: 3/84 Other: 1/84 Placebo: 51/79 AE: 4/79 LoE: 4/79 > 15/100 increase in PI: 35/79 Physician or participant decision: 4/79 Other: 4/79 |
| AE: adverse event; LoE: lack of efficacy; PI: pain intensity | ||||
Appendix 8. Details of studies of fentanyl not included in this review
| Study | Participants | Design and duration | Number | Intervention | Efficacy data | Adverse event data |
| Randomised open studies | ||||||
| Allan 2001 | CNCP requiring strong opioids Moderate control with oral opioids | Randomised, open, cross‐over 2 x 4 weeks | N = 256 | TD fentanyl compared with CR morphine | 212/251 assessed for preference: fentanyl 65% morphine 28% no preference 7% Mean pain score lower with fentanyl (58/100 vs 63/100) Better satisfaction with fentanyl |
More withdrawals (38 vs 22) and AE withdrawals (27 vs 10) with fentanyl More nausea, less constipation with fentanyl |
| Allan 2005 | CLBP naïve to strong opioid | Randomised, open, parallel 13 months | N = 680 | TD fentanyl compared with CR morphine | Similar pain relief, less constipation with fentanyl | Similar numbers completed (48% vs 53%) AE withdrawals: 125 fentanyl, 104 morphine LoE withdrawals: 18 fentanyl, 15 morphine |
| Canneti 2013 | Peripheral neuropathic pain with advanced AIDS | Randomised, open, parallel 12 months | N = 40 | TD fentanyl compared with TD buprenorphine | Mean PI reduced significantly in both groups Buprenorpine slightly better | Both well tolerated |
| Raptis 2014 | Neuropathic cancer pain | Randomised, open, parallel 4 weeks | N = 120 | TD fentanyl compared with pregabalin Increasing doses over 4 weeks | ≥ 30% PI reduction: pregabalin 73%, fentanyl 37% % mean change from baseline: pregabalin 46%, fentanyl 22% Participant satisfaction more frequent with pregabalin | AEs more frequent with fentanyl (34 vs 16) Most common: nausea, somnolence, dizziness with fentanyl, nausea, somnolence with pregabalin AE withdrawals: fentanyl 10/60, pregabalin 3/60 |
| Observational studies | ||||||
| Agarwal 2007 | Peripheral neuropathic pain, CRPS type I, postamputation pain PI ≥ 3/10 | Open, cohort 16 weeks | N = 53 51 entered titration 44 entered maintenance 40 completed | TD fentanyl (3‐day) 6‐week titration 25‐150 μg/h 8‐week maintenance Stable, non‐opioid medicines continued, opioid wash out | > 30% pain reduction: 30/53 > 50% pain reduction: 21/53 Mean reduction in PI (0 to 10): 2.94 ± 0.27 % pain relief: 34 ± 14% (48% in completers) Increase in average daytime activity: 37% | Drowsiness (47%) Nausea/vomiting (28%) Constipation (9%) Skin reactions (9%) Withdrawals due to AEs |
| Dellemijn 1998 | Neuropathic pain ‐ mixed | Open‐label extension 24 months | N = 48 | TD fentanyl ‐ titrated over 12 weeks to max 100 μg/h or max tolerated
Then tapered by 25 μg/h weekly, and substituted with morphine SR 60 mg/d, tapering over 10 days 2 weeks with no opioid, then offered 2‐year extension |
12‐week dose escalation period: 16 withdrawals due to LoE, AEs 17/30 who completed chose not to continue with extension (AEs not justified by benefit (13), pain relief persisted (4)) 3 discontinued in first year, 1 in second year Sedation, nausea, constipation most frequent AEs | |
| Franco 2002 | CNCP | Open‐label, observational 6 months | N = 236 (120 neuropathic pain) | TD fentanyl | 34% participants had PI < 3/10 after 6 months. Greatest reduction in PI in first 3 months | Somnolence, vomiting, dizziness most common AEs |
| Milligan 2001 | CNCP moderate or severe | Open‐label, observational 12 months | N = 532 *103 participated in Allan 2001 | TD fentanyl | 67% had ≥ moderate pain control on treatment Global satisfaction 42% | 231 withdrew: 130 due to AEs, 39 LoE (most in first few months) Nausea, constipation, somnolence most common |
| Mitra 2011 | Persistent pain ‐ mixed | Open‐label, observational 12 months | N = 46 | TD fentanyl vs TD buprenorphine | Equivalent for efficacy Tolerance after 6 months | More AE withdrawals with fentanyl early on, buprenorphine later |
| Mystakidou 2003 | CNCP | Open‐label, observational Long‐term ‐ up to 4 years for a few participants |
N = 529 | TD fentanyl | Improvements in pain and QoL Median duration of effective pain management 10 months No difference between NP and nociceptive | 55 withdrew, all but one within 4 weeks. 24 AE withdrawals, 24 LoE withdrawals Constipation, nausea, sleepiness most common |
| Park 2011 | CNCP (most NP), requiring opioids PI moderate or severe | Open‐label, observational | N = 65 41 evaluated | TD fentanyl titrated 12 weeks | Mean PI decreased by 62% (6.7/10 to 2.6/10) | 24 withdrawals: 1 LoE, 18 AE Nausea, dizziness, drowsiness, constipation, vomiting most common |
| AE: adverse event; CLBP: chronic low back pain; CNCP: chronic non‐cancer pain; CR: controlled release; CRPS: complex regional pain syndrome; LoE: lack of efficacy; N: number of participants in study; NP: neuropathic pain; PI: pain intensity; QoL: quality of life; SR: sustained release TD: transdermal | ||||||
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Arai 2015.
| Methods | Study N02. Multicentre, EERW, open‐label titration phase (10 to 29 days), and parallel group, double‐blind withdrawal phase (12 weeks) | |
| Participants | PHN, CRPS, postoperative pain syndrome > 12 weeks, mean PI > 50/100, opioid‐naïve, inadequate PR with non‐opioid analgesics Excluded: pain from other causes, asthma, bradyarrythmia, severe respiratory function disorders, hepatic dysfunction, renal impairment, hypersensitivity to fentanyl or other opioids N = 258 (titration phase) M 134, F 124 Mean age 67 years (SD 14) Mean baseline PI 74/100 (SD 13) N = 163 (double‐blind phase) M 83, F 80 Mean age 67 years (SD 14) Mean baseline PI 29/100 (SD 11) |
|
| Interventions | Titration phase: fentanyl 1‐day adhesive patch 12.5 µg/h for minimum 2 days. Increased by 12.5 µg/h based on VAS PI and use of rescue medication to maximum 50 µg/h, over 10 to 29 days Double‐blind phase: Fentanyl 1‐day adhesive patch 12.5 to 50 µg/h, n = 84 Placebo patch, n = 79 Patches applied to chest abdomen, upper arm or thigh, replaced every day for 12 weeks Rescue medication: morphine hydrochloride (5 mg per fentanyl 12.5 µg/h) |
|
| Outcomes | Participants responding during titration period Median time to withdrawal due to loss of analgesic efficacy Median change in PI from randomisation to last 3 days of double‐blind period (VAS) Satisfaction scores |
|
| Notes | Oxford Quality Score: R1, DB2, W1 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of sequence generation not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | "Placebo patch indistinguishable from one‐day adhesive transdermal patch containing fentanyl" |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | "Placebo patch indistinguishable from one‐day adhesive transdermal patch containing fentanyl" |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Analyses used full analysis set, but no mention of imputation methods. Study's primary outcome seems robust, but other outcomes unclear |
| Size | Unclear risk | 50 to 199 participants per treatment arm |
CRPS: complex regional pain syndrome; DB: double‐blind; EERW: enriched enrolment randomised withdrawal; F: female; M: male; N: number of participants in study; n: number of participants in treatment arm; PHN: postherpetic neuralgia; PI: pain intensity; PR: pain relief; R: randomised; SD: standard deviation; VAS: visual analogue scale; W: withdrawals.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Canneti 2013 | Open‐label study |
| Ding 2014 | Compares fentanyl with fentanyl + gabapentin |
| Kalso 2007 | Open‐label, mixed conditions, secondary analysis of Allan 2005 |
| NCT01127100 | Single‐blind (outcomes assessor), ongoing study |
| Park 2010 | Open‐label cohort, mixed conditions |
Differences between protocol and review
In the protocol the inclusion criteria included both CRPS types I and II as a diagnosis of neuropathic pain. We have now removed CRPS type I because it is no longer considered to be neuropathic pain. We identified no studies in CRPS type I.
Contributions of authors
SD and RAM wrote the protocol.
SD and PC searched for and selected studies for inclusion and carried out data extraction.
All review authors were involved in the analysis and in writing the full review.
Sources of support
Internal sources
-
Oxford Pain Relief Trust, UK.
General institutional support
External sources
-
The National Institute for Health Research (NIHR), UK.
NIHR Cochrane Programme Grant: 13/89/29 ‐ Addressing the unmet need of chronic pain: providing the evidence for treatments of pain
Declarations of interest
SD: none known.
CS: none known. She is a specialist pain physician and manages patients with chronic pain.
PC received support from Boston Scientific (2014) for travel and accommodation at a scientific meeting; Boston Scientific does not market drugs. PC is a specialist pain physician and manages patients with chronic pain.
PW: none known.
RK has consulted for Grunenthal Ltd (2014‐15) and MundiPharma Research (2015), and received lecture fees from Grunenthal Ltd (2013‐14) and Pfizer Ltd (2013‐14). He is an Associate Professor in Clinical Pharmacy Practice and Advanced Pharmacy Practitioner.
DA has received lecture fees from Grünenthal (2013, 2014, 2015) and Pfizer (2013, 2016). He is a specialist pain physician and manages patients with chronic pain.
RAM has received grant support from RB relating to individual patient‐level analyses of trial data on ibuprofen in acute pain and the effects of food on drug absorption of analgesics (2013), and from Grünenthal relating to individual patient‐level analyses of trial data regarding tapentadol in osteoarthritis and back pain (2015). He has received honoraria for attending boards with Menarini concerning methods of analgesic trial design (2014), with Novartis about the design of network meta‐analyses (2014), and RB on understanding the pharmacokinetics of drug uptake (2015).
Stable (no update expected for reasons given in 'What's new')
References
References to studies included in this review
Arai 2015 {published data only}
- Arai T, Kashimoto Y, Ukyo Y, Tominaga Y, Imanaka K. Two placebo‐controlled, randomized withdrawal studies to evaluate the fentanyl 1 day patch in opioid‐naïve patients with chronic pain. Current Medical Research and Opinion 2015;31(12):2207‐18. [DOI: 10.1185/03007995.2015.1092127; JNS020‐JPN‐N02] [DOI] [PubMed] [Google Scholar]
- Janssen Pharmaceutical KK (Sponsors). A confirmatory study of fentanyl in participants with post‐herpetic neuralgia, complex regional pain syndrome or postoperative pain syndrome. clinicaltrials.gov/ct2/show/record/NCT01008553 (first received 5 November 2009). [CTG: NCT01008553]
References to studies excluded from this review
Canneti 2013 {published data only}
- Canneti A, Luzi M, Marco P, Cannata F, Pasqualitto F, Spinoglio A, et al. Safety and efficacy of transdermal buprenorphine and transdermal fentanyl in the treatment of neuropathic pain in AIDS patients. Minerva Anestesiologica 2013;79(8):871‐83. [PUBMED: 23558760] [PubMed] [Google Scholar]
Ding 2014 {published data only}
- Ding Y, Yao P, Lan P, Ma J, Wang Z, Hong T. Assessment of transdermal fentanyl combined with gabapentin for malignant neuropathic pain treatment. [Chinese]. Chinese Journal of Clinical Oncology 2014;41:1307‐11. [EMBASE ID: 2014953970] [Google Scholar]
Kalso 2007 {published data only}
- Kalso E, Simpson K, Slappendel R, Dejonckheere J, Richarz U. Predicting long‐term response to strong opioids in patients with low back pain: findings from a randomized, controlled trial of transdermal fentanyl and morphine. BMC Medicine 2007;5:39. [DOI: 10.1186/1741-7015-5-39] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT01127100 {published data only}
- NCT01127100. Gabapentin versus transdermal fentanyl matrix for chronic neuropathic pain. clinicaltrials.gov/ct2/show/NCT01127100 (first received 19 May 2010). [CTG: NCT01127100]
Park 2010 {published data only}
- Park JH, Kim JH, Yun SC, Roh SW, Rhim SC, Kim CJ, et al. Evaluation of efficacy and safety of fentanyl transdermal patch (Durogesic D‐TRANS) in chronic pain. Acta Neurochirurgica (Wien) 2011;153(1):181‐90. [DOI: 10.1007/s00701-010-0785-4] [DOI] [PubMed] [Google Scholar]
Additional references
Agarwal 2007
- Agarwal S, Polydefkis M, Block B, Haythornthwaite J, Raja SN. Transdermal fentanyl reduces pain and improves functional activity in neuropathic pain states. Pain Medicine 2007;8(7):554‐62. [DOI: 10.1111/j.1526-4637.2006.00246.x] [DOI] [PubMed] [Google Scholar]
Allan 2001
- Allan L, Hays H, Jensen NH, Waroux BL, Bolt M, Donald R, et al. Randomised crossover trial of transdermal fentanyl and sustained release oral morphine for treating chronic non‐cancer pain. BMJ 2001;322(7295):1154‐8. [DOI: 10.1136/bmj.322.7295.1154] [DOI] [PMC free article] [PubMed] [Google Scholar]
Allan 2005
- Allan L, Richarz U, Simpson K, Slappendel R. Transdermal fentanyl versus sustained release oral morphine in strong‐opioid naïve patients with chronic low back pain. Spine (Phila Pa 1976) 2005;30(22):2484‐90. [PUBMED: 16284584] [DOI] [PubMed] [Google Scholar]
Baron 2010
- Baron R, Binder A, Wasner G. Neuropathic pain: diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurology 2010;9(8):807‐19. [DOI: 10.1016/S1474-4422(10)70143-5] [DOI] [PubMed] [Google Scholar]
Baron 2012
- Baron R, Wasner G, Binder A. Chronic pain: genes, plasticity, and phenotypes. Lancet Neurology 2012;11(1):19‐21. [DOI: 10.1016/S1474-4422(11)70281-2] [DOI] [PubMed] [Google Scholar]
Bouhassira 2008
- Bouhassira D, Lantéri‐Minet M, Attal N, Laurent B, Touboul C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain 2008;136(3):380‐7. [DOI: 10.1016/j.pain.2007.08.013] [DOI] [PubMed] [Google Scholar]
Bullingham 1984
- Bullingham RE, Moore RA, Symonds HW, Allen MC, Baldwin D, McQuay HJ. A novel form of dependency of hepatic extraction ratio of opioids in vivo upon the portal vein concentration of drug: comparison of morphine, diamorphine, fentanyl, methadone and buprenorphine in the chronically cannulated cow. Life Sciences 1984;34(21):2047‐56. [DOI] [PubMed] [Google Scholar]
Cachia 2011
- Cachia E, Ahmedzai SH. Transdermal opioids for cancer pain. Current Opinion in Supportive and Palliative Care 2011;5(1):15‐9. [DOI: 10.1097/SPC.0b013e3283437a39] [DOI] [PubMed] [Google Scholar]
Dellemijn 1998
- Dellemijn PL, Duijn H, Vanneste JA. Prolonged treatment with transdermal fentanyl in neuropathic pain. Journal of Pain and Symptom Management 1998;16(4):220‐9. [DOI: 10.1016/S0885-3924(98)00070-0] [DOI] [PubMed] [Google Scholar]
Derry 2012
- Derry S, Moore RA. Topical capsaicin (low concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD010111] [DOI] [PMC free article] [PubMed] [Google Scholar]
Derry 2013
- Derry S, Sven‐Rice A, Cole P, Tan T, Moore RA. Topical capsaicin (high concentration) for chronic neuropathic pain in adults. Cochrane Database of Systematic Reviews 2013, Issue 2. [DOI: 10.1002/14651858.CD007393.pub3] [DOI] [PubMed] [Google Scholar]
Derry 2014
- Derry S, Wiffen PJ, Moore RA, Quinlan J. Topical lidocaine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2014, Issue 7. [DOI: 10.1002/14651858.CD010958.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Di Franco 2010
- Franco M, Iannuccelli C, Atzeni F, Cazzola M, Salaffi F, Valesini G, et al. Pharmacological treatment of fibromyalgia. Clinical and Experimental Rheumatology 2010;28(6 Suppl 63):S110‐6. [PubMed] [Google Scholar]
Dworkin 2008
- Dworkin RH, Turk DC, Wyrwich KW, Beaton D, Cleeland CS, Farrar JT, et al. Interpreting the clinical importance of treatment outcomes in chronic pain clinical trials: IMMPACT recommendations. Journal of Pain 2008;9(2):105‐21. [DOI: 10.1016/j.jpain.2007.09.005] [DOI] [PubMed] [Google Scholar]
Finnerup 2015
- Finnerup NB, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin RH, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta‐analysis. Lancet Neurology 2015;14(2):162‐73. [DOI: 10.1016/S1474-4422(14)70251-0] [DOI] [PMC free article] [PubMed] [Google Scholar]
Franco 2002
- Franco ML, Jesus Berro M, Calvo I, Pezonaga L, Sulleiro R, Seoane A, et al. Usefulness of transdermal fentanyl in the management of nonmalignant chronic pain: A prospective, observational, multicenter study. Pain Clinic 2002;14(2):99‐112. [Google Scholar]
Franklin 2014
- Franklin GM, American Academy of Neurology. Opioids for chronic noncancer pain: a position paper of the American Academy of Neurology. Neurology 2014;83(14):1277‐84. [DOI: 10.1212/WNL.0000000000000839] [DOI] [PubMed] [Google Scholar]
Gaskell 2014
- Gaskell H, Moore RA, Derry, S, Stannard C. Oxycodone for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2014, Issue 6. [DOI: 10.1002/14651858.CD010692.pub2] [DOI] [PubMed] [Google Scholar]
Gustorff 2008
- Gustorff B, Dorner T, Likar R, Grisold W, Lawrence K, Schwarz F, et al. Prevalence of self‐reported neuropathic pain and impact on quality of life: a prospective representative survey. Acta Anaesthesiologica Scandinavica 2008;52(1):132‐6. [DOI: 10.1111/j.1399-6576.2007.01486.x] [DOI] [PubMed] [Google Scholar]
Guyatt 2011
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 7. Rating the quality of evidence – inconsistency. Journal of Clinical Epidemiology 2011;64(12):1294‐302. [DOI: 10.1016/j.jclinepi.2011.03.017] [DOI] [PubMed] [Google Scholar]
Guyatt 2013a
- Guyatt G, Oxman AD, Sultan S, Brozek J, Glasziou P, Alonso‐Coello P, et al. GRADE guidelines: 11. Making an overall rating of confidence in effect estimates for a single outcome and for all outcomes. Journal of Clinical Epidemiology 2013;66(2):151‐7. [DOI: 10.1016/j.jclinepi.2012.01.006] [DOI] [PubMed] [Google Scholar]
Guyatt 2013b
- Guyatt GH, Oxman AD, Santesso N, Helfand M, Vist G, Kunz R, et al. GRADE guidelines: 12. Preparing summary of findings tables‐binary outcomes. Journal of Clinical Epidemiology 2013;66(2):158‐72. [DOI: 10.1016/j.jclinepi.2012.01.012] [DOI] [PubMed] [Google Scholar]
Hall 2008
- Hall GC, Carroll D, McQuay HJ. Primary care incidence and treatment of four neuropathic pain conditions: a descriptive study, 2002‐2005. BMC Family Practice 2008;9:26. [DOI: 10.1186/1471-2296-9-26] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hall 2013
- Hall GC, Morant SV, Carroll D, Gabriel ZL, McQuay HJ. An observational descriptive study of the epidemiology and treatment of neuropathic pain in a UK general population. BMC Family Practice 2013;14:28. [DOI: 10.1186/1471-2296-14-28] [DOI] [PMC free article] [PubMed] [Google Scholar]
Heiskanen 2009
- Heiskanen T, Mätzke S, Haakana S, Gergov M, Vuori E, Kalso E. Transdermal fentanyl in cachectic cancer patients. Pain 2009;144(1‐2):218‐22. [DOI: 10.1016/j.pain.2009.04.012] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hoffman 2010
- Hoffman DL, Sadosky A, Dukes EM, Alvir J. How do changes in pain severity levels correspond to changes in health status and function in patients with painful diabetic peripheral neuropathy?. Pain 2010;149(2):194‐201. [DOI: 10.1016/j.pain.2009.09.017] [DOI] [PubMed] [Google Scholar]
Jadad 1996
- Jadad AR, Moore RA, Carroll D, Jenkinson C, Reynolds DJ, Gavaghan DJ, et al. Assessing the quality of reports of randomized clinical trials: is blinding necessary?. Controlled Clinical Trials 1996;17(1):1‐12. [DOI: 10.1016/0197-2456(95)00134-4] [DOI] [PubMed] [Google Scholar]
Jensen 2011
Kalso 2013
- Kalso E, Aldington DJ, Moore RA. Drugs for neuropathic pain. BMJ 2013;347:f7339. [DOI: 10.1136/bmj.f7339] [DOI] [PubMed] [Google Scholar]
Katusic 1991
- Katusic S, Williams DB, Beard CM, Bergstralh EJ, Kurland LT. Epidemiology and clinical features of idiopathic trigeminal neuralgia and glossopharyngeal neuralgia: similarities and differences, Rochester, Minnesota,1945‐1984. Neuroepidemiology 1991;10:276‐81. [DOI: 10.1159/000110284] [DOI] [PubMed] [Google Scholar]
Koopman 2009
- Koopman JS, Dieleman JP, Huygen FJ, Mos M, Martin CG, Sturkenboom MC. Incidence of facial pain in the general population. Pain 2009;147(1‐3):122‐7. [DOI: 10.1016/j.pain.2009.08.023] [DOI] [PubMed] [Google Scholar]
L'Abbé 1987
- L'Abbé KA, Detsky AS, O'Rourke K. Meta‐analysis in clinical research. Annals of Internal Medicine 1987;107:224‐33. [DOI] [PubMed] [Google Scholar]
Lange 2010
- Lange B, Kuperwasser B, Okamoto A, Steup A, Häufel T, Ashworth J, et al. Efficacy and safety of tapentadol prolonged release for chronic osteoarthritis pain and low back pain. Advances in Therapy 2010;27(6):381‐99. [DOI: 10.1007/s12325-010-0036-3] [DOI] [PubMed] [Google Scholar]
Lunn 2014
- Lunn MP, Hughes RA, Wiffen PJ. Duloxetine for treating painful neuropathy, chronic pain or fibromyalgia. Cochrane Database of Systematic Reviews 2014, Issue 1. [DOI: 10.1002/14651858.CD007115.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lötsch 2013
- Lötsch J, Walter C, Parnham MJ, Oertel BG, Geisslinger G. Pharmacokinetics of non‐intravenous formulations of fentanyl. Clinical Pharmacokinetics 2013;52(1):23‐36. [DOI: 10.1007/s40262-012-0016-7] [DOI] [PubMed] [Google Scholar]
McNicol 2013
- McNicol ED, Midbari A, Eisenberg E. Opioids for neuropathic pain. Cochrane Database of Systematic Reviews 2013, Issue 8. [DOI: 10.1002/14651858.CD006146.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
McQuay 1979
- McQuay HJ, Moore RA, Paterson GM, Adams AP. Plasma fentanyl concentrations and clinical observations during and after operation. British Journal of Anaesthesia 1979;51(6):543‐50. [DOI] [PubMed] [Google Scholar]
McQuay 1998
- McQuay H, Moore R. An Evidence‐Based Resource for Pain Relief. Oxford (UK): Oxford University Press, 1998. [ISBN: 0‐19‐263048‐2] [Google Scholar]
McQuay 2007
- McQuay HJ, Smith LA, Moore RA. Chronic pain. In: Stevens A, Raftery J, Mant J, Simpson S editor(s). Health Care Needs Assessment, 3rd Series. Oxford (UK): Radcliffe Publishing, 2007:519‐99. [ISBN: 978‐1‐84619‐063‐6] [Google Scholar]
Milligan 2001
- Milligan K, Lanteri‐Minet M, Borchert K, Helmers H, Donald R, Kress HG, et al. Evaluation of long‐term efficacy and safety of transdermal fentanyl in the treatment of chronic noncancer pain. Journal of Pain 2001;2(4):197‐204. [DOI: 10.1054/jpai.2001.25352] [DOI] [PubMed] [Google Scholar]
Mitra 2011
- Mitra F, Chowdhury S, Williams G, Shelley M. Comparison of transdermal fentanyl and buprenorphine on clinical outcomes, acceptability and side‐effects on prospective one‐year follow‐up of long‐term persistent pain patients. Australian Pain Society Conference. DArwin, NT Australia: Australian Society of Anaesthetists, 2011; Vol. 39 (4):747‐8.
Moisset 2007
- Moisset X, Bouhassira D. Brain imaging of neuropathic pain. Neuroimaging 2007;37(Suppl 1):S80‐8. [DOI: 10.1016/j.neuroimage.2007.03.054] [DOI] [PubMed] [Google Scholar]
Moore 1998
- Moore RA, Gavaghan D, Tramèr MR, Collins SL, McQuay HJ. Size is everything ‐ large amounts of information are needed to overcome random effects in estimating direction and magnitude of treatment effects. Pain 1998;78(3):209‐16. [DOI: 10.1016/S0304-3959(98)00140-7] [DOI] [PubMed] [Google Scholar]
Moore 2008
- Moore RA, Barden J, Derry S, McQuay HJ. Managing potential publication bias. In: McQuay HJ, Kalso E, Moore RA editor(s). Systematic Reviews in Pain Research: Methodology Refined. Seattle (WA): IASP Press, 2008:15‐24. [ISBN: 978‐0‐931092‐69‐5] [Google Scholar]
Moore 2009
- Moore RA, Straube S, Wiffen PJ, Derry S, McQuay HJ. Pregabalin for acute and chronic pain in adults. Cochrane Database of Systematic Reviews 2009, Issue 3. [DOI: 10.1002/14651858.CD007076.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2010a
- Moore RA, Eccleston C, Derry S, Wiffen P, Bell RF, Straube S, et al. "Evidence" in chronic pain ‐ establishing best practice in the reporting of systematic reviews. Pain 2010;150(3):386‐9. [DOI: 10.1016/j.pain.2010.05.011] [DOI] [PubMed] [Google Scholar]
Moore 2010b
- Moore RA, Straube S, Derry S, McQuay HJ. Chronic low back pain analgesic studies ‐ a methodological minefield. Pain 2010;149(3):431‐4. [DOI: 10.1016/j.pain.2010.02.032] [DOI] [PubMed] [Google Scholar]
Moore 2010c
- Moore RA, Straube S, Paine J, Phillips CJ, Derry S, McQuay HJ. Fibromyalgia: moderate and substantial pain intensity reduction predicts improvement in other outcomes and substantial quality of life gain. Pain 2010;149(2):360‐4. [DOI: 10.1016/j.pain.2010.02.039] [DOI] [PubMed] [Google Scholar]
Moore 2010d
- Moore RA, Smugar SS, Wang H, Peloso PM, Gammaitoni A. Numbers‐needed‐to‐treat analyses ‐ do timing, dropouts, and outcome matter? Pooled analysis of two randomized, placebo‐controlled chronic low back pain trials. Pain 2010;151(3):592‐7. [DOI: 10.1016/j.pain.2010.07.013] [DOI] [PubMed] [Google Scholar]
Moore 2010e
- Moore RA, Moore OA, Derry S, Peloso PM, Gammaitoni AR, Wang H. Responder analysis for pain relief and numbers needed to treat in a meta‐analysis of etoricoxib osteoarthritis trials: bridging a gap between clinical trials and clinical practice. Annals of the Rheumatic Diseases 2010;69(2):374‐9. [DOI: 10.1136/ard.2009.107805] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2011a
- Moore RA, Straube S, Paine J, Derry S, McQuay HJ. Minimum efficacy criteria for comparisons between treatments using individual patient meta‐analysis of acute pain trials: examples of etoricoxib, paracetamol, ibuprofen, and ibuprofen/paracetamol combinations after third molar extraction. Pain 2011;152(5):982‐9. [DOI] [PubMed] [Google Scholar]
Moore 2011b
- Moore RA, Mhuircheartaigh RJ, Derry S, McQuay HJ. Mean analgesic consumption is inappropriate for testing analgesic efficacy in post‐operative pain: analysis and alternative suggestion. European Journal of Anaesthesiology 2011;28(6):427‐32. [DOI: 10.1097/EJA.0b013e328343c569] [DOI] [PubMed] [Google Scholar]
Moore 2012a
- Moore RA, Derry S, Aldington D, Cole P, Wiffen PJ. Amitriptyline for neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2012, Issue 12. [DOI: 10.1002/14651858.CD008242.pub2] [DOI] [PubMed] [Google Scholar]
Moore 2012b
- Moore RA, Straube S, Eccleston C, Derry S, Aldington D, Wiffen P, et al. Estimate at your peril: imputation methods for patient withdrawal can bias efficacy outcomes in chronic pain trials using responder analyses. Pain 2012;153(2):265‐8. [DOI: 10.1016/j.pain.2011.10.004] [DOI] [PubMed] [Google Scholar]
Moore 2013a
- Moore RA, Straube S, Aldington D. Pain measures and cut‐offs ‐ 'no worse than mild pain' as a simple, universal outcome. Anaesthesia 2013;68(4):400‐12. [DOI: 10.1111/anae.12148] [DOI] [PubMed] [Google Scholar]
Moore 2013b
- Moore A, Derry S, Eccleston C, Kalso E. Expect analgesic failure; pursue analgesic success. BMJ 2013;346:f2690. [DOI: 10.1136/bmj.f2690] [DOI] [PubMed] [Google Scholar]
Moore 2014a
- Moore RA, Derry S, Taylor RS, Straube S, Phillips CJ. The costs and consequences of adequately managed chronic non‐cancer pain and chronic neuropathic pain. Pain Practice 2014;14(1):79‐94. [DOI: 10.1111/papr.12050] [DOI] [PubMed] [Google Scholar]
Moore 2014b
- Moore RA, Wiffen PJ, Derry S, McQuay HJ. Gabapentin for chronic neuropathic pain and fibromyalgia in adults. Cochrane Database of Systematic Reviews 2014, Issue 4. [DOI: 10.1002/14651858.CD007938.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2014c
- Moore RA, Cai N, Skljarevski V, Tölle TR. Duloxetine use in chronic painful conditions ‐ individual patient data responder analysis. European Journal of Pain 2014;18(1):67‐75. [DOI: 10.1002/j.1532-2149.2013.00341.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
Moore 2015
- Moore RA, Wiffen PJ, Eccleston C, Derry S, Baron R, Bell RF, et al. Systematic review of enriched enrolment, randomised withdrawal trial designs in chronic pain: a new framework for design and reporting. Pain 2015;156(8):1382‐95. [DOI: 10.1097/j.pain.0000000000000088] [DOI] [PubMed] [Google Scholar]
Mystakidou 2003
- Mystakidou K, Parpa E, Tsilika E, Mavromati A, Smyrniotis V, Georgaki S, et al. Long‐term management of noncancer pain with transdermal therapeutic system‐fentanyl. Journal of Pain 2003;4(6):298‐306. [DOI: 10.1016/S1526-5900(03)00632-1] [DOI] [PubMed] [Google Scholar]
Nelson 2009
- Nelson L, Schwaner R. Transdermal fentanyl: pharmacology and toxicology. Journal of Medical Toxicology 2009;5(4):230‐41. [DOI] [PMC free article] [PubMed] [Google Scholar]
NICE 2013
- National Institute for Health and Care Excellence (NICE). Neuropathic pain ‐ pharmacological management: the pharmacological management of neuropathic pain in adults in non‐specialist settings, 2013. www.nice.org.uk/guidance/cg173 (accessed 6 October 2016).
O'Brien 2010
- O'Brien EM, Staud RM, Hassinger AD, McCulloch RC, Craggs JG, Atchison JW, et al. Patient‐centered perspective on treatment outcomes in chronic pain. Pain Medicine 2010;11(1):6‐15. [DOI: 10.1111/j.1526-4637.2009.00685] [DOI] [PubMed] [Google Scholar]
O'Connor 2009
- O'Connor, AB, Dworkin RH. Treatment of neuropathic pain: an overview of recent guidelines. American Journal of Medicine 2009;122(10 Suppl):S22‐32. [DOI: 10.1016/j.amjmed.2009.04.007] [DOI] [PubMed] [Google Scholar]
PaPaS 2012
- Cochrane Pain, Palliative and Supportive Care Group (PaPaS) author and referee guidance. papas.cochrane.org/papas‐documents (accessed 6 October 2016).
Park 2011
- Park JH, Kim JH, Yun SC, Roh SW, Rhim SC, Kim CJ, et al. Evaluation of efficacy and safety of fentanyl transdermal patch (Durogesic D‐TRANS) in chronic pain. Acta Neurochirurgica (Wien) 2011;153(1):181‐90. [DOI: 10.1007/s00701-010-0785-4] [DOI] [PubMed] [Google Scholar]
PCA 2015
- Prescribing and Medicines Team, Health and Social Care Information Centre. Prescription Cost Analysis, England 2014. content.digital.nhs.uk/catalogue/PUB17274/pres‐cost‐anal‐eng‐2014‐rep.pdf (accessed 6 October 2016):119‐21. [ISBN: 978‐1‐78386‐365‐5]
Rappaport 1994
- Rappaport ZH, Devor M. Trigeminal neuralgia: the role of self‐sustaining discharge in the trigeminal ganglion. Pain 1994;56:127‐38. [DOI] [PubMed] [Google Scholar]
Raptis 2014
- Raptis E, Vadalouca A, Stavropoulou E, Argyra E, Melemeni A, Siafaka I. Pregabalin vs. opioids for the treatment of neuropathic cancer pain: a prospective, head‐to‐head, randomized, open‐label study. Pain Practice 2014;14(1):32‐42. [DOI: 10.1111/papr.12045] [DOI] [PubMed] [Google Scholar]
RevMan 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Soni 2013
- Soni A, Batra R, Gwilym S, Spector T, Hart D, Arden N, et al. Neuropathic features of joint pain: a community‐based study. Arthritis & Rheumatism 2013;65(7):1942‐9. [DOI: 10.1002/art.37962] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2008
- Straube S, Derry S, McQuay HJ, Moore RA. Enriched enrolment: definition and effects of enrichment and dose in trials of pregabalin and gabapentin in neuropathic pain. A systematic review. British Journal of Clinical Pharmacology 2008;66(2):266‐75. [DOI: 10.1111/j.1365-2125.2008.03200] [DOI] [PMC free article] [PubMed] [Google Scholar]
Straube 2010
- Straube S, Derry S, Moore RA, Paine J, McQuay HJ. Pregabalin in fibromyalgia ‐ responder analysis from individual patient data. BMC Musculoskeletal Disorders 2010;11:150. [DOI: 10.1186/1471-2474-11-150] [DOI] [PMC free article] [PubMed] [Google Scholar]
Sultan 2008
- Sultan A, Gaskell H, Derry S, Moore RA. Duloxetine for painful diabetic neuropathy and fibromyalgia pain: systematic review of randomised trials. BMC Neurology 2008;8:29. [DOI: 10.1186/1471-2377-8-9] [DOI] [PMC free article] [PubMed] [Google Scholar]
Torrance 2006
- Torrance N, Smith BH, Bennett MI, Lee AJ. The epidemiology of chronic pain of predominantly neuropathic origin. Results from a general population survey. Journal of Pain 2006;7(4):281‐9. [DOI: 10.1016/j.jpain.2005.11.008] [DOI] [PubMed] [Google Scholar]
Tracey 2011
- Tracey I. Can neuroimaging studies identify pain endophenotypes in humans?. Nature Reviews Neurology 2011;7(3):173‐81. [DOI: 10.1038/nrneurol.2011.4] [DOI] [PubMed] [Google Scholar]
Treede 2008
- Treede RD, Jensen TS, Campbell JN, Cruccu G, Dostrovsky JO, Griffin JW, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology 2008;70(18):1630‐5. [DOI: 10.1212/01.wnl.0000282763.29778.59] [DOI] [PubMed] [Google Scholar]
Trescot 2008
- Trescot AM, Datta S, Lee M, Hansen H. Opioid pharmacology. Pain Physician 2008;11:S133‐53. [PubMed] [Google Scholar]
van Hecke 2014
- Hecke O, Austin SK, Khan RA, Smith BH, Torrance N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 2014;155(4):654‐62. [DOI: 10.1016/j.pain.2013.11.013] [DOI] [PubMed] [Google Scholar]
Vo 2009
- Vo T, Rice AS, Dworkin RH. Non‐steroidal anti‐inflammatory drugs for neuropathic pain: how do we explain continued widespread use?. Pain 2009;143(3):169‐71. [DOI: 10.1016/j.pain.2009.03.013] [DOI] [PubMed] [Google Scholar]
von Hehn 2012
- Hehn CA, Baron R, Woolf CJ. Deconstructing the neuropathic pain phenotype to reveal neural mechanisms. Neuron 2012;73(4):638‐52. [DOI: 10.1016/j.neuron.2012.02.008] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vos 2012
- Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990‐2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012;380(9859):2163‐96. [DOI: 10.1016/S0140-6736(12)61729-2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Weisberg 2014
- Weisberg DF, Becker WC, Fiellin DA, Stannard C. Prescription opioid misuse in the United States and the United Kingdom: cautionary lessons. International Journal on Drug Policy 2014;25(6):1124‐30. [DOI: 10.1016/j.drugpo.2014.07.009] [DOI] [PubMed] [Google Scholar]
Wiffen 2013
- Wiffen PJ, Derry S, Moore RA, Aldington D, Cole P, Rice ASC, et al. Antiepileptic drugs for neuropathic pain and fibromyalgia ‐ an overview of Cochrane reviews. Cochrane Database of Systematic Reviews 2013, Issue 11. [DOI: 10.1002/14651858.CD010567.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiffen 2015
- Wiffen PJ, Derry S, Moore RA, Stannard C, Aldington D, Cole P, et al. Buprenorphine for neuropathic pain in adults. Cochrane Database of Systematic Reviews 2015, Issue 9. [DOI: 10.1002/14651858.CD011603.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Zin 2014
- Zin CS, Chen LC, Knaggs RD. Changes in trends and pattern of strong opioid prescribing in primary care. European Journal of Pain 2014;18(9):1343‐51. [DOI: 10.1002/j.1532-2149.2014.496.x] [DOI] [PMC free article] [PubMed] [Google Scholar]