

This review discusses the features of TADs across species, and their role in chromosome organization, genome function, and evolution.
Abstract
Understanding the mechanisms that underlie chromosome folding within cell nuclei is essential to determine the relationship between genome structure and function. The recent application of “chromosome conformation capture” techniques has revealed that the genome of many species is organized into domains of preferential internal chromatin interactions called “topologically associating domains” (TADs). This chromosome chromosome folding has emerged as a key feature of higher-order genome organization and function through evolution. Although TADs have now been described in a wide range of organisms, they appear to have specific characteristics in terms of size, structure, and proteins involved in their formation. Here, we depict the main features of these domains across species and discuss the relation between chromatin structure, genome activity, and epigenome, highlighting mechanistic principles of TAD formation. We also consider the potential influence of TADs in genome evolution.
INTRODUCTION
The three-dimensional (3D) folding of the eukaryotic genome in the nucleus is a highly organized process tightly linked to functional DNA-dependent processes, such as DNA replication and transcription. The nuclear positioning of genes within nuclei can correlate with transcription, with active genes being located more often in the nuclear interior compared to repressed or heterochromatic regions, which are found closer to the periphery (1). Chromosomes occupy distinct subnuclear territories (2), with transcriptionally active loci positioned at their surface (3). In the last decade, key features of genome organization have been revealed by chromosome conformation capture methods (4, 5), in particular by their high-throughput version called Hi-C (6), which allows the genome-wide identification of chromatin contacts [for review, see (7)]. Hi-C uncovered general principles of chromosome folding, such as the decay of the frequency of chromosomal contacts following a power law that has a scaling exponent close to −1 in many species (6, 8–12), but genome folding is far from being homogeneous (13). At large scales, chromosomes segregate into regions of preferential long-range interactions that form two mutually excluded types of chromatin, referred to as “A” and “B” compartments (6, 14, 15). A compartments correspond to gene-rich and active chromatin, while B compartments are mostly enriched in repressive chromatin (6, 14). At a scale of tens to hundreds of kilobases, chromosomes fold into domains with preferential intradomain interactions compared to interdomain interactions with the neighboring cis chromatin domains, i.e., spanning domain borders (11, 16–18). These contact domains are now commonly referred to as topologically associating domains (TADs) (Fig. 1) (18). The presence of these domains has been described in many species, indicating that they may represent a conserved feature of genome organization. TADs are architectural chromatin units that define regulatory landscapes, suggesting their fundamental implication in shaping functional chromosomal organization. TAD boundaries correspond to those of replication domains (19), and genes tend to be coregulated during cell differentiation when they are located within the same TAD (18, 20–22). A reporter gene inserted within the genome is subjected to the influence of enhancers over large regulatory domains that correlate strongly with TADs (23), and contacts between enhancers and gene promoters are mainly restricted within TADs (24). This is consistent with TADs representing a functionally privileged scale of chromosome folding (22), and the constraint of functional contacts within TADs appears to be essential to ensure proper gene regulation. Disruption of TAD structures by altering their boundaries can lead to ectopic contacts between cis-regulating elements and gene promoters, and thus gene misexpression, which can contribute to developmental defects and cancer (25–30). Therefore, deciphering the structural and the functional nature of TADs has become crucial to elucidate the rules of higher-order genome organization and regulation, and given their importance in pathology, understanding TADs has acquired medical relevance. However, even if genome folding into self-interacting domains has been a widely adopted strategy in evolution, TADs or contact domains can differ in size, chromatin features, and mechanisms underlying their formation. This suggests that TADs might be subdivided in different subtypes, each of them characterized by specific structural and functional properties. Moreover, the identification of TADs strongly depends on the resolution of Hi-C data and the method of TAD annotation. Increasing sequencing depth and resolution reveals finer patterns of chromatin contacts as well as internal insulation regions (Fig. 1). Thus, the identification of TAD borders has proven to be difficult. Furthermore, to what extent these chromatin domains represent the same layer of genome organization in different species remains unclear. In this review, we will first present the main features of chromosome folding at the submegabase scale, highlighting similarities and differences between TADs observed in various organisms. We will then consider their relationship with physical and functional organization of the chromatin fiber and their potential role in genome evolution.
Fig. 1. Hierarchical folding of the eukaryotic genome.
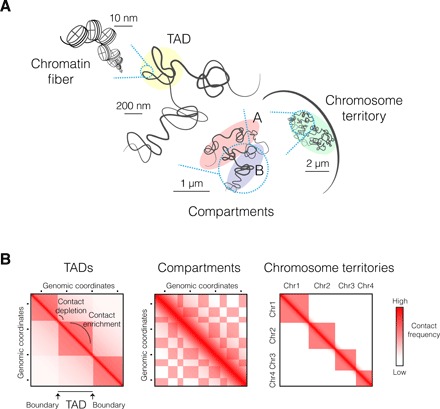
(A) Schematic view of chromosome folding inside the nucleus. The finest layer of chromatin folding is at the DNA-histone association level, forming nucleosomes organized into the ~11-nm chromatin fiber (133). Chromatin is packed at different nucleosome densities depending on gene regulation and folds at the submegabase scale into higher-order domains of preferential internal interactions, referred to as TADs. At the chromosomal scale, chromatin is segregated into active “A” and repressed “B” compartments of interactions, reflecting preferential contacts between chromatin regions of the same epigenetic features. Individual chromosomes occupy their own space within the nucleus, forming chromosome territories. (B) Schematic representation of Hi-C maps at different genomic scales, reflecting the different layers of higher-order chromosome folding. Genomic coordinates are indicated on both axes, and the contact frequency between regions is represented by a color code. At the submegabase scale, TADs appear as squares along the diagonal enriched in interactions, separated by contact depletion zones delimited by TAD boundaries. At the chromosomal scale, chromatin long-range interactions form a characteristic plaid pattern of two mutually excluded A and B compartments. Last, intrachromosomal interactions are overrepresented compared to interchromosomal contacts, consistent with the formation of individual chromosome territories.
TADs ACROSS SPECIES
TADs in mammals
TAD features appear to be strongly conserved in mammals (16, 31). 5C and Hi-C studies first showed the partitioning of the chromosomes into domains of hundreds of kilobases (median size of 880 kb) (16, 18), occupying 91% of the mouse genome. Higher-resolution Hi-C map detected finer domains, also dubbed sub-TADs, with a median size of 185 kb and associated with enrichment of specific chromatin marks (14, 32). A notable feature of most mammalian TAD boundaries is the presence of the CCCTC-binding factor (CTCF) together with the structural maintenance of chromosomes (SMC) cohesin complex (14, 16, 31, 33). These borders are engaged in strong interactions, seen as “corner peaks” on Hi-C maps (Fig. 2), suggesting the formation of loops between CTCF binding sites. Notably, these loop-anchored TADs almost always form between CTCF sites positioned in a convergent orientation, and the removal or change in orientation of a single CTCF site can be sufficient to abolish or shift the position of a TAD boundary (13, 28, 34, 35), demonstrating the crucial role of CTCF in defining mammalian TAD borders. The propensity of CTCF to form homodimers and to bind RNA molecules may be important for this function (36). A linear tracking mechanism, which is referred to as the “loop extrusion model,” has been proposed for the formation of these TADs (13, 37, 38). According to this model, chromatin would be extruded by an engaged cohesin SMC complex until the complex is dissociated or until it encounters two convergent and bound CTCF sites at TAD borders [for review, see (39)]. In line with this, cohesin subunit chromatin immunoprecipitation–sequencing (ChIP-seq) peaks tend to be more interior to the loop relative to CTCF peaks (13, 37). This model has been supported by recent molecular genetic studies. The depletion of CTCF, cohesin, or the cohesin-loading factor Nipbl leads to the disruption of loop domains (40–43). Conversely, the removal of the cohesin release factor Wapl reinforces the strength of the loops at TAD borders (44). The CTCF/cohesin association forming the loop-anchored TADs thus appears to come from an equilibrium between loading and removal of the cohesin complex, with corner peak loops reflecting an increased residence time of the complex at TAD boundaries (45). In agreement, loops disappear when the cohesin complex is not loaded on chromatin or when it does not stop at CTCF borders, while they are stabilized when cohesin stays on chromatin (40–45). Cohesin restoration rapidly reverses these effects, consistent with a model where loop extrusion is a dynamic process (41). Another SMC protein complex, condensin II, together with TFIIIC, has been found enriched at TAD borders (46). Consistent with a potential role of condensin, loop extrusion has been observed in vitro by live imaging, where naked DNA can be extruded (1500 base pairs per second) by this complex (47). This demonstrates the existence of such a mechanism and calls for an analysis of the interplay between condensin and cohesin in TAD formation. However, super-resolution microscopy data suggest that cohesin (and, most likely, CTCF) is required to position TAD boundaries coherently in different cells rather than for TAD formation. Chromatin tracing with sequential fluorescence in situ hybridization (FISH) labeling showed TAD-like structural units in individual cells, in both wild-type and cohesin-depleted conditions, but the position of the boundaries lies at CTCF sites in wild-type cells, whereas it is randomized in cohesin-depleted cells (48). These data suggest that other loop extruding mechanisms might exist in the absence of cohesin or that TADs can form by spontaneous chromatin contact features. In addition to defining boundaries, CTCF is also present at enhancer-promoter pairs within TADs, forming smaller loop domains (14) involving Mediator and cohesin (33), and another protein, YY1, may also contribute to enhancer-promoter interactions, together with cohesin, in a more cell type–specific manner (49). Furthermore, modeling of chromatin fibers suggests that transcription-associated supercoiling could also be involved in the process of loop extrusion (50). Consistently, type II DNA topoisomerase is often found positioned with CTCF and cohesin at domain borders, which may help to solve topological problems (51). If loop-associated domains represent a key feature of TADs in mammals, with approximately 75 to 95% of boundaries being associated with CTCF depending on the cell type (16, 24), then some boundaries are CTCF independent, consistent with approximately 20% of TAD boundaries being resistant to CTCF loss (40). These boundaries are associated with transcription (16, 24) or correspond to a demarcation between active and repressed chromatin regions, i.e., between A and B chromatin type (14, 32). For example, Hi-C profiling of embryonic mouse stem cells and differentiated neuronal progenitor cells revealed the appearance of boundaries at promoters of newly transcribed genes during differentiation in the absence of CTCF binding (24). Mammalian TADs seem therefore not to be always the result of CTCF/cohesin loops and could sometimes rather be defined by chromatin state and transcription (32). However, CRISPR-dCas9–mediated transcriptional activation does not create a new boundary (24). Hi-C analysis in mouse sperm, which is transcriptionally silent, but has bound RNA polymerase II (RNAPII) and active or silent histone modifications, shows similar interaction domains as embryonic stem cells (52). These data suggest that transcription per se is not sufficient and that transcription factors are likely involved in insulating CTCF-independent TAD borders. Last, TADs appear gradually during early mouse embryogenesis (53, 54) and they are still observed after the inhibition of the transcription with α-amanitin (53, 54), whereas blocking of DNA replication with aphidicolin inhibits TAD establishment (54), suggesting a potential role of replication in the primary establishment of TADs.
Fig. 2. Examples of Hi-C profiles from different species.
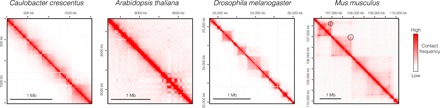
Hi-C maps [visualized with Juicebox (134)] of different species (24, 67, 75, 90, 135) showing more or less pronounced 3D partitioning of the genome. TADs are not obvious in Arabidopsis genome, but boundary-like regions and insulated genome units are discernible. In Drosophila, TADs are well demarcated and correlate well with the epigenetic landscape. A specific feature of mammalian TADs is the presence of “corner peaks,” i.e., peaks of interactions at the edges of TADs (indicated by black circles), revealing the presence of chromatin loops.
TADs in Drosophila
In Drosophila, the presence of TADs has first been identified using Hi-C in whole embryos (11), revealing the presence of discrete interaction domains along chromosomes (Fig. 2). Drosophila TADs appear well correlated with epigenetic states and were classified in four main classes according to their specific chromatin signatures: transcriptionally active TADs, associated with active histone modifications such as trimethylation of histone H3 lysine 4 and 36 (H3K4me3 and H3K36me3) (active TADs); Polycomb-repressed TADs enriched in H3K27me3 and Polycomb group (PcG) proteins (PcG TADs); TADs devoid of known specific marks (null or void TADs); and classical heterochromatin enriched in H3K9me2, HP1, and Su(var)3-9 (heterochromatin TADs) (11). Originally, Hi-Cs in Drosophila revealed approximately 1300 TADs with an average size of nearly 100 kb (11, 17), but recent studies using higher map resolution showed a finer partitioning into >2000 (21, 55) or >4000 TADs (56), where TADs and inter-TAD regions can be subdivided into smaller domains with a median size of few tens of kilobases (56). The calling and the annotation of TADs depend on the computational method and the algorithm used (57), which can explain such variability in the number of identified TADs despite similar Hi-C resolution. Independently on the number of identified TADs, the transcriptionally silent TADs (PcG, null/void, and HP1, i.e., B-type chromatin) occupy the largest portion of the genome and are larger in genomic size than the active ones (11, 17, 21, 56). The large majority of TAD boundaries are present in gene-dense, chromatin-accessible, transcribed regions enriched in active chromatin marks (17, 58–60), most of them occurring at active gene promoters (21). Various insulator proteins have been found enriched at boundaries, including BEAF-32, Chromator, CP190, or M1BP (11, 17, 21, 59, 60), and combination of these proteins such as BEAF-32/Chromator or BEAF-32/CP190 is a good predictor of boundaries (56). Cohesin and condensin II subunits, as well as TFIIIC, were also found enriched at TAD borders (61). However, in contrast to mammals, there is little enrichment of CTCF nor interaction loops at TAD borders. This is a startling observation, because Drosophila CTCF has a conserved Zn finger domain that binds to the same sequence as the mammalian counterpart. The reason why fly CTCF is not a major TAD boundary definition protein and, instead, is rather involved in Hox gene regulation (62) remains to be studied. Despite their enrichment, the role of insulator proteins in Drosophila TAD formation is still unclear, for example, small interfering RNA (siRNA)–mediated depletion of BEAF-32 does not abolish boundaries (21). Whether a total degradation of the protein or whether the depletion of a combination of these factors is required to see clear effects remains to be investigated. Of importance, the description of boundary features largely depends on the calling of the TADs and therefore on the resolution of Hi-C. Using high-resolution Hi-C, it was recently proposed that TAD organization in Drosophila reflects the switch between active and inactive chromatin and that many of the previously identified boundaries actually correspond to small active domains (32, 56). TAD patterning mirrors the transcriptional state, with large inactive regions forming prominent repressed TADs separated by transcribed genes that are often clustered in the genome (32, 60). The size and the degree of transcriptional activity of these active regions correlate with the local strength of compartmentalization, with broader and more active TADs forming the most pronounced A compartment domains (63). To decipher mechanisms driving TAD formation, chromatin interaction profiling has been performed during Drosophila embryogenesis (59, 64). At early stages, before zygotic genome activation (ZGA)—a wave of zygotic transcriptional activation occurring during embryonic development—the genome is mostly unstructured but contains few boundary-like regions enriched in housekeeping genes and associated with RNAPII occupancy. During ZGA, TAD boundaries progressively appear at housekeeping genes concomitantly with de novo recruitment of RNAPII, reaching a plateau after these activation waves. Consistent with the link between transcription and boundaries, α-amanitin or triptolide-induced inhibition of transcription leads to a decrease of TAD insulation, although boundaries do not completely disappear (32, 59), indicating that the reduction of RNAPII and transcription is not sufficient to abolish TAD formation. A role for the Zelda transcription factor was uncovered in establishing insulation at TAD boundaries (59). Zelda may cooperate with other factors, such as BEAF-32 and GAGA factor (GAF), found at TAD borders. Moreover, Zelda at RNAPII-bound sites is also implicated in the formation of active long-range chromatin loops, often spanning multiple TADs. This first wave of active chromatin loops depending on Zelda might correspond to the onset of genome folding in Drosophila. These loops are located close to strong TAD boundaries, a situation reminiscent to CTCF in mammalian nuclei (64). Whether this organization necessitates cohesin or cohesin-like activity remains to be addressed. Later during embryogenesis, TADs and TAD insulation become more and more pronounced, and the formation of chromatin loops in repressive PcG domains, which involves GAF, represents another specific feature of Drosophila TADs (55, 64). However, these loops do not occur between TAD boundaries but are present at the interior of PcG TADs and correspond to contacts between PcG protein binding sites. Given these specificities, these loops do not seem to be a general feature of TAD formation in Drosophila but rather a mechanism involved in PcG gene silencing (64). Of note, no Zelda or GAF homologs have been found in vertebrates, indicating that some of these looping mechanisms maybe peculiar to Drosophila.
TADs in Caenorhabditis elegans
In Caenorhabditis elegans, self-interacting domains of ~1 Mb size are present on the X chromosome but are not a clear feature of autosomal chromosome organization (65). While some boundary-like regions are found in autosomes, they are stronger and more abundant on the X chromosome. The hermaphrodite X chromosome is specifically bound by the dosage compensation complex (DCC), a condensin complex. High-affinity DCC binding sites overlap with X TAD boundaries, and DCC depletion strongly reduces insulation at these boundaries, consistent with a pivotal role of the DCC. Moreover, CRISPR-Cas9–mediated deletion of a binding site of the DCC complex was sufficient to remove its cognate boundary. Intriguingly, these DCC-bound boundaries are also engaged in long-range interactions and it would be important to understand whether these features can be separated or whether they are interdependent.
TADs in plants
Genome compartmentalization into TADs, in the sense of a complete partitioning into adjacent self-interacting domains, was not obvious in the model plant Arabidopsis thaliana (Fig. 2). However, the Arabidopsis genome harbors compacted domains of interactions enriched with repressed chromatin marks such as H3K27me3 or H3K9me2 (9, 66, 67). More than 1000 “boundary-like regions,” defined as starting or ending points of interacting domains, were identified (67). These regions are composed of transcriptionally active and open chromatin that separate inactive genomic regions (32, 67). If TADs are not a clear feature of Arabidopsis genome, then they have been distinctly observed in rice and cotton (68–70), where chromatin at TAD boundaries is highly expressed and enriched in active chromatin marks. Another study described the presence of TAD-like domains in various plant species, including maize, tomato, sorghum, foxtail millet, and rice (71). Similar to Drosophila TADs (11), they can be classified into four chromatin types according to their epigenetic signatures: active domains, repressive domains enriched in DNA methylation, Polycomb domains enriched in H3K27me3, and domains devoid of specific marks. Thus, the link between transcription, epigenetic status, and chromatin topology appears as a main feature of chromosome organization in these species. In plants, no protein with insulator function such as CTCF has been identified. However, DNA GC-rich motifs similar to sequences bound by plant-specific transcription factors belonging to the TCP family have been identified at rice TAD boundaries (69). Studies focusing on the function of these proteins found at boundaries would be required to characterize their potential role in shaping plant TADs.
Self-interacting domains in yeast
Self-interacting domains, called globules, have been identified in Schizosaccharomyces pombe (10). These globules (50 to 100 kb in size) are separated by boundaries enriched in cohesin binding. The partial loss of function of rad21, a cohesin subunit, is associated with a disruption of globules, seen as a loss of insulation at cohesin peaks. The presence of globules and the role of cohesin in their formation are conserved in G1 cells, indicating a different role for cohesin than in sister chromatid cohesion. In Saccharomyces cerevisiae, TAD-like structures were not initially observed using a derivative of the 4C method (72). More recently, a method similar to Hi-C called Micro-C, in which micrococcal nuclease is used instead of restriction enzymes to produce small chromatin fragments to be ligated when close in 3D, allowed the generation of contact maps at single-nucleosome resolution, which revealed the presence of small self-interacting domains (73). These domains generally contain one to five genes and are approximately 5 kb in size. The boundaries between these small domains are enriched for highly expressed gene promoters—although not all promoters form boundaries—transcription-associated marks, the remodeling the structure of chromatin (RSC) adenosine triphosphate (ATP)–dependent chromatin remodeling complex, and the cohesin loading factor ssc2. A recent model proposes that transcription-induced supercoiling, together with the action of topoisomerases at TAD borders, can explain the formation of self-interacting chromatin domains in S. pombe (74).
TAD-like domains in bacteria
Hi-C performed in Caulobacter crescentus revealed the presence of discrete chromosomal interaction domains (CIDs) resembling eukaryote TADs (Fig. 2), ranging from 30 to 420 kb in size (75), with boundaries enriched in highly expressed genes. Inhibition of transcription elongation disrupted CID boundaries, and moving highly expressed genes in a different genomic location led to neo boundaries. This study suggests that regions enriched in plectonemes form CIDs, while boundaries are established by highly expressed genes and the formation of plectoneme-free regions. CIDs of similar genomic sizes have also been identified in Bacillus subtilis (76, 77), and macrodomain-like regions have been reported for the Escherichia coli chromosome (78). Even in Mycoplasma pneumoniae, a model organism with a small genome size and a simplified gene regulatory network, CIDs have been observed. In this case, CIDs range from 15 to 33 kb in size, smaller than those reported for C. crescentus and B. subtilis. Genes within the same domain tend to be coregulated, suggesting that, even in such a small genome, chromosome organization may influence transcriptional regulation (79). A common theme in C. crescentus and M. pneumoniae is that the sharpness of CIDs depends on supercoiling. Moreover, prokaryotic model organisms have provided crucial information on the role of SMC complexes in genome organization. Studies in B. subtilis and C. crescentus revealed that SMC rings are able to encircle DNA and tether chromosome arms, forming processive loops (77, 80, 81) that depend on the adenosine triphosphatase (ATPase) activity of the complex (82). These data thus provide strong evidence that an active loop extrusion mechanism is involved in shaping bacterial chromosome organization. However, SMC-mediated extrusion does not seem to be necessary for CID formation, because SMC depletion in C. crescentus leads to a decrease of inter-arm chromosomal contacts but CID boundaries remain unchanged (75).
General and specific features of TADs
Although TADs or interaction domains emerge as a fundamental component of genome organization, their features are not universally conserved (Fig. 2). Contact domains can be more or less pronounced, and their boundaries can be more or less sharp. In addition, the molecular mechanisms underlying their formation can be diverse, consistent with the existence of different types of TADs. It is therefore unclear whether TADs are the universal unit of higher-order genome organization or whether they emerged repeatedly during evolution as a consequence of the interplay between different molecular engines acting on chromatin. Nevertheless, one notable feature conserved across species is the relationship between gene activity and genome folding. Boundary regions are often found to be highly enriched in active chromatin in Drosophila (11, 17, 21, 58–60), mammals (16, 24), zebrafish (in which TADs are similar to those of mammals) (83), plants (67, 69), S. cerevisiae (73), and Plasmodium falciparum, which also shows domain-like structures of 5 to 10 kb (32, 84). In the bacteria C. crescentus, boundaries are also found at transcribed gene promoters (75). In the fungus Neurospora crassa, the genome is compartmentalized into heterochromatic and euchromatic regions, where gene-rich regions form domains <100 kb in genomic size that are comparable to metazoan TADs or yeast globules and separated by heterochromatic islands enriched in H3K9me3 (8). By comparing different species, including Drosophila, Arabidopsis, N. crassa, C. elegans, and the protozoan P. falciparum, Rowley and colleagues (32) suggested that the transcriptional activity partitions the genome into Hi-C domains, with active genes interacting more frequently with other active genes and forming active domains when they cluster on the genome. Contact domain boundaries would then correspond to the switches between transcribed and inactive genomic regions (32). The distribution and the transcriptional output of transcribed gene clusters along the genome of various species might therefore define the strength of local insulation of their TADs, as recently observed in Drosophila (63). However, transcription per se does not appear to be sufficient to create boundaries (24), and not all transcribed sites make boundaries, indicating that other factors, perhaps DNA binding of transcription factors, insulator/architectural proteins, or a combination of both, are required. Mammalian Hi-C maps display an additional TAD feature, which is the presence of CTCF/cohesin chromatin loops between CTCF convergent sites (14). Enrichment of inverted CTCF sites at TAD boundaries was also observed in zebrafish (83), suggesting that this characteristic is conserved through vertebrate lineage. However, CTCF has not been found in other organisms such as plants, yeast, or C. elegans (85), and consistently, loop-anchored domains are not found in these species. Conversely, other insulator proteins may play a similar role in defining TAD boundaries at transcribed domains in other species, such as BEAF-32 and CP190 in Drosophila (56), or TCP proteins in plants (69). The localization of cohesin depends on transcription in mammals (86), and cohesin-mediated boundaries may form at transcribed sites even in the absence of CTCF-like proteins. For instance, cohesin and its loader Nipped-B are associated with transcriptionally active regions in Drosophila (87, 88). Depletion of cohesin and associated factors in other species than mammals would be interesting to decipher its role in TAD formation, and it would help elucidate whether self-interacting domains emerge from a conserved mechanism regulating DNA-dependent processes during evolution. Alternatively, in some species, TADs could arise from the differential folding of chromatin regions with different epigenomic states and thus rather reflect differential chromatin contacts in regions of different gene expression output.
PHYSICAL NATURE OF TADs
TADs and compartments
The relation between epigenome and genome organization raises the question of how the physical properties of chromatin shape chromosome structure. In Drosophila, active chromatin domains display a weaker contact density in Hi-C (11, 60) or a stronger contact depletion between adjacent active TADs (56) compared to inactive TADs, indicating differential folding. Super-resolution stochastic optical reconstruction microscopy (STORM) revealed that active domains are more decondensed than the inactive ones (89). The classification based on global run-on sequencing (GRO-seq) of the Drosophila genome into active or inactive chromatin states reflects very well the TAD pattern obtained with Hi-C (32). Therefore, the correspondence between interactions obtained with Hi-C and chromatin state (11, 32, 60), together with the different folding of active compared to inactive chromatin, suggested that, in Drosophila, the compartmentalization of the chromosome into TADs may reflect the physical exclusion of active and inactive chromatin. Ulianov and colleagues (60) proposed that Drosophila inactive TADs are condensed chromosomal domains separated by active chromatin regions. Recent super-resolution analysis of chromatin organization accredited this view by showing the partitioning of the chromatin fiber into TAD-based physical domains, where repressed TADs form condensed globular nanocompartments interspersed by more open active regions (90). Similarly, STORM imaging of immunolabeled repressive H3K27me3 or active H3K4me3 marks showed clear separation of these two chromatin types, where active domains were found at the borders of repressed ones (91). This feature of chromosome organization is even observed in endoreplicated Drosophila polytene chromosomes in which TADs correspond to dense bands, while decompacted interbands correspond to inter-TAD regions (60, 92, 93). In Drosophila, chromatin state and genome structure seem therefore tightly linked at both the TAD and compartment levels. These two layers of organization, i.e., TADs enriched in internal interactions and compartments representing long-range interactions between domains of the same epigenetic features, correspond to the folding of genome units that preferentially interact within themselves and with homotypic domains (Fig. 3A) (11, 32, 90). The fact that compartments are not observed in polytene chromosomes presumably reflects the absence of long-range contacts because of the extensive pairing in trans of endoreplicated chromosomes. The correspondence between epigenome and interaction profiles is also observed in plants, where both short- and long-range contacts are correlated with epigenetic profiles (9, 66–68, 71). In species where chromosomal contacts correlate well with the epigenome, the mutual exclusion of different chromatin types may then be sufficient to create a TAD-based pattern for chromosome organization. Gene transcription and delimitation of epigenetic landscapes, for example, mediated by insulator proteins (94), would then provide the framework of genome organization.
Fig. 3. Schematic representation of chromatin folding in Drosophila and mammals.
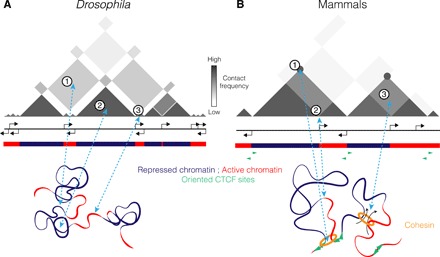
(A) In Drosophila, both TADs and compartments correspond to epigenetic domains that preferentially fold within themselves and in far-cis with homotypic TADs (1). Large repressed chromatin region forms prominent and condensed TADs (2), separated by transcribed genes that can form clusters of small active TAD or inter-TAD–like regions of decondensed chromatin (3). (B) In mammals, the “loop extrusion model” proposed for TAD formation involves a loop extrusion factor, here cohesin, loaded on the chromatin by Nipbl and unloaded by Wapl. Cohesin extrudes chromatin until it dissociates, bumps into another cohesin, or reaches the border of the TAD bound by CTCF proteins in inverted orientation or by other boundary components. These loops are seen as a strong peak of interaction between TAD borders (1). Insulation can also be observed at active transcription start sites (2), and as recently suggested, the loop extrusion process could compete with the local segregation of active and inactive chromatin by mixing them (3) (45).
In mammals, the mutual exclusion of active and inactive chromatin is not sufficient to generate TAD boundaries. These regions are frequently transcribed and often correspond to transitions between different chromatin states (14, 16, 18), but TADs can include multiple chromatin types. Instead, a correspondence between chromatin activity and long-range interactions appears more prominently at the compartment scale (14, 15, 95). The effect of cohesin depletion on Hi-C contact maps as well as on super-resolution imaging maps suggests the presence of two parallel mechanisms of chromatin organization (41, 42, 48). Without cohesin, mammalian TAD-like domains might form spontaneously by chromatin interactions, which probably are preferential for same-type chromatin. However, these interactions rarely form domains with coherent boundaries in every cell. These boundaries are therefore implemented by the action of CTCF/cohesin-mediated loops (48). At the longer range, epigenetic features dominate, with TADs of similar chromatin type interacting preferentially to define chromosome compartments (14, 41, 42). The fact that zygotic maternal chromatin contains TADs and loops, but not compartments, also suggests that TADs and compartments are formed by distinct mechanisms (96). Using polymer simulations, Nuebler and colleagues (45) proposed that chromatin folding in mammals comes from a competition between dynamic loop extrusion and the compartmentalization defined by the epigenetic status, in which the processing of loop extrusion factors counteracts the segregation of compartments (Fig. 3B). In Hi-C data, the preferential interactions at short cis genomic distance of chromatin of the same type would then be blurred by the mechanism of extrusion. Therefore, CTCF/cohesin loops in mammals may correspond to an additional layer of genome organization on top of chromatin compartmentalization defined by its epigenetic state. This view is coherent with the fact that mammalian TADs have been shown to contain subdomains corresponding to active or repressed chromatin (14, 32). This is also consistent with cohesin removal experiments, which abrogate CTCF/cohesin-mediated loops while revealing a finer chromatin compartmentalization that accurately reflects the underlying epigenetic landscape (41, 42). This compartmentalization resembles that of Drosophila Hi-C maps, and this may actually reflect the absence in this species of such a process (Fig. 3).
Concerning the mechanism that segregates active from inactive chromatin, Ulianov and colleagues (60) proposed an attractive “self-assembly” model, whereby the stickiness of nonacetylated (inactive) nucleosomes, as opposed to the absence of bridging ability for acetylated (active) nucleosomes, could explain chromatin partitioning into TADs and TAD boundaries (also called inter-TADs). Moreover, the concentration of repressive histone methylation marks such as H3K9me2/3 and H3K27me3, which can spread over large genomic regions (97), and which serve as a platform to recruit large multimeric complexes, could help in the agglomeration and separation of active and inactive chromatin inside the nucleus. Recently, exclusion and compartmentalization of chromatin domains have been directly investigated on the basis of the physicochemical properties of their components. On the one hand, the classical heterochromatin segregation is driven by phase separation, mediated, at least in part, by HP1a and HP1α multivalent hydrophobic interactions in Drosophila and mammals, respectively (98, 99). On the other hand, active domains may also generate phase-separated compartments. Clusters of enhancers, regulating cooperatively gene expression and defined as super-enhancers, can undergo phase separation by transcriptional coactivators, ensuring local concentration of regulating factors in a segregated 3D environment (100). These observations are in agreement with the visualization of chromatin-associated clusters of RNAPII and Mediator enriched at super-enhancers, behaving as phase-separated condensates (101). Phase separation has also been shown through kinase-mediated hyperphosphorylation of the RNAPII C-terminal repeat domain (CTD) (102). Last, polymer simulations of chromosome folding are consistent with phase-separated A and B compartments in mammals (45). In general, the components involved in phase-separated condensates contain intrinsically disordered protein domains and can exhibit multivalent interactions with each other to create specific environments, in which biochemical reactions and interactions might be highly favored (103, 104). These studies shed new light on how physicochemical properties of chromatin-associated factors can form segregated compartments, and further investigations will be directed at understanding how this can be linked to TAD formation and/or stabilization.
TAD structure and dynamics
It appears clearer and clearer that TADs correspond to a functional subdivision of the genome into regions in which regulatory contacts are spatially confined. The fact that disruptions of TADs lead to de novo enhancer-promoter interactions and gene misexpression emphasizes this crucial role (25–30). This functional property could reflect the formation of physically insulated genomic units or a higher contact probability between gene promoters and cis-regulatory elements confined within a TAD. Hi-C data generally represent averaged interaction profiles coming from millions of cells, making the characterization of the physical nature of TADs difficult. Therefore, whether TADs reflect statistical frequencies of chromatin interactions within cell population or whether they represent genuine physical units in each cell nucleus has been a crucial question recently investigated by numerous studies.
Single-cell Hi-C (scHi-C) has been lately introduced. Although the first study suggested a generally conserved TAD organization at the single-cell level (105), subsequent scHi-C studies reveal substantial heterogeneity in contacts at the TAD scale from cell to cell (96, 106, 107), with domains appearing as mere tendencies that become more visible when averaged over a population of cells. Individual nuclear structures may, however, be difficult to address with scHi-C, given coverage and resolution limitations, and because this technique can identify a maximum of one interaction per genomic fragment at a time without information concerning the relative spatial positioning of each fragment. However, microscopy and polymer modeling are in agreement with scHi-C, suggesting that mammalian TADs can display various conformations, ranging from condensed and globular objects to more stretched configurations (106, 108). This might depend, in part, on the cell-specific transcriptional output, consistent with the finding that different levels of transcriptional activity of Tsix alleles were related to fluctuations in TAD conformations (108). Boundary precision and degree of insulation of TADs can also vary among different cell types (24, 53, 54, 109) or during cell cycle progression (110).
The recent application of super-resolution microscopy, such as STORM or 3D-structured illumination microscopy (3D-SIM), has allowed finer-scale chromatin architecture to be analyzed at the single-cell level, opening the possibility of studying the structural properties of chromosome domains [for review, see (111)]. Using FISH and 3D-SIM, it was shown that, despite heterogeneous folding of individual TADs and diversity in their relative arrangement in 3D space, discrete nanocompartments corresponding to repressed chromatin (repressed TADs) interspersed by decondensed active domains can be observed in individual cells, suggesting that the Drosophila TAD pattern reflects a fairly stable segregation of active and inactive chromatin domains (90). Therefore, a dynamic intra-TAD folding is compatible with a steady separation of autonomous chromosomal units, at least in Drosophila. In mammals, TADs can contain various epigenetic marks and may be more flexible in shape, as suggested by the dynamic binding of CTCF and cohesin at loop anchors (112). However, recent super-resolution microscopy has also revealed the existence of nanosized chromatin domains in mammals (113–115), which correlate with epigenetic features, similarly to Drosophila. In both mammals and fly, H3K27me3 repressed regions form discrete and compacted domains (91, 116), with active chromatin domains located at their periphery (91, 117). Focusing on the imaging of several histone modifications associated with differential epigenetic states in mammals, Xu and colleagues (118) were able to resolve the higher-order chromatin organization into three major structural characteristics, including segregated nanoclusters for lysine acetylation, dispersed nanodomains for active histone methylation, and compact large aggregates for repressive histone methylation. This is consistent with previous observations of large and dense “clutches” of nucleosomes corresponding to heterochromatic regions compared to smaller and less dense RNAPII-associated chromatin (115), and with chromatin decondensation at transcribed sites (119). The combination of super-resolution microscopy and live imaging showed that chromatin nanodomains move coherently and that their structure depends on cohesin and nucleosome-nucleosome interactions (114). These domains have a peak diameter of approximately 160 nm, which was estimated to cover 130 to 200 kb. This estimated genomic size is in good agreement with the nanocompartments (approximately 190 nm for a 200-kb repressed TAD) observed in Drosophila (90), and with that of sub-TADs identified in mammals (median size, 185 kb) (14), but is smaller than in mammalian TADs (average size, 880 kb) (16). However, when STORM super-resolution microscopy was combined with sequential DNA labeling of multi-megabase genomic regions, larger globular nanocompartments of several hundred nanometers, equivalent to full TADs, were observed (48). In the future, it will be important to assess the relation between TADs and sub-TADs in different types of mammalian chromatin to understand whether sub-TADs exist in each cell and to determine their prevalence in each type of chromatin.
Differentiation processes may also represent a source of variability of TAD structures. At the megabase scale, TAD patterns in mammals appear largely conserved in different cell lines and even across species (16, 31), whereas on a submegabase scale, subdomains within a TAD could become merged or disconnected, depending on developmentally regulated events (16, 24, 33, 109, 120). In this case, the dynamics is largely due to the appearance of new regulatory enhancer-promoter contacts involving specific transcription factors, concomitant to gene expression during lineage specification or cell reprogramming (24, 109, 120). It was recently shown that TADs can be variable in different cells (48), but it will be important to study whether the variability depends on specific activities of enhancers and target promoters. Related to this point, recent live-cell imaging methods have started to shed light into the dynamics of functional elements (121–123). The observation of coordinated transcriptional bursts and the fact that enhancer and promoter interactions seem to adopt a “stirring model,” in which the search will be confined and potentiated rather than a conventional stable loop, suggest a dynamic view of enhancer-promoter interactions (122, 123). Moreover, the act of transcription per se might stabilize proximal chromatin conformation, reducing enhancer-promoter distances (121). Last, it has been proposed that transcription-induced supercoiling could participate in the establishment of contact between functional elements within TADs (124). Therefore, TADs may establish a local chromosomal environment in which regulatory signals might act to tune the probability of dynamic interaction among distally located enhancers and promoters.
TADs AND GENOME EVOLUTION
TADs are generally present in metazoans (125) and, despite different mechanisms in TAD formation and the open questions concerning their structure and dynamics, function as regulatory units of the genome, and genes contained within them tend to be coregulated during development (18, 20–22). Furthermore, they define the limits of the chromosomal domains in which gene promoters are contacted by cis-regulatory elements (23, 24). Therefore, TADs appear particularly interesting for the study of genome evolution (125). In particular, they might act as buffering elements, allowing mutations to exert local effects without affecting surrounding extra-TAD loci. As an example of this, CTCF binding sequences were shown to be more prone to changes within TADs than in boundaries, allowing the creation of potential new regulatory contacts within chromosomal domains to emerge in a modular fashion, preventing them from affecting extra-TAD loci (31). Furthermore, TADs appear relatively flexible in size and can tolerate the gain or loss of DNA sequence (126), which can also favor the emergence of novel regulatory effects. The TAD organization could allow the evolution of new cis-regulatory elements by limiting the influence of these regulatory changes to a few genes, namely, those located within the same TAD (125). On the other hand, a subset of TADs is associated with high level of noncoding conservation, which may be important to preserve the expression regulation of key developmental genes. Therefore, these 3D structures may also contribute to the maintenance of selective pressure of internal elements that are necessary for the precise control of specific loci (126).
If TADs are advantageous for genome function, then one might expect their boundaries to be highly conserved. Dixon and colleagues (16) showed that syntenic regions between mouse and human are very similar in chromatin structure and that 75.9% of boundaries in mouse are present in human, while 53.8% of human boundaries are present in mouse. Another study investigated the evolution of chromosomal topology across four mammalian species (mouse, dog, rabbit, and macaque) and again observed the conservation of chromatin structure within syntenic regions (31). Conserved TADs are associated with conserved CTCF binding sites and motif orientation at their borders, while changes in internal domain structures are correlated with changes in binding and orientation of CTCF, indicating a co-evolution of CTCF binding and chromatin structure. In addition, comparison of gibbon and human genomes showed that gibbon breaks of synteny mainly occur at TAD boundaries and that epigenetic landscapes are maintained after rearrangement (127). Consistently, pairs of genes situated within the same TAD in zebrafish are found more often close to each other in other vertebrate genomes than those situated in two neighboring TADs (83). Moreover, the comparison of gene expression data from many mouse and human tissues indicates that genes within TADs have more conserved expression patterns, and disruption of TADs by evolutionary rearrangements is associated with changes in gene expression profiles (128). Therefore, it appears that TADs are maintained as intact modules during evolution, which may help the conservation of functional regulatory landscapes. An example of this conservation can be illustrated with the analysis of the Six homeobox gene cluster in distant species: the echinoderm sea urchin, zebrafish, and mammals. Despite subsequent rounds of whole genomic duplications, this cluster remained organized into two adjacent TADs that have different expression patterns, with borders associated with orientation-inverted CTCF sites (129). In addition to the selective pressure for the maintenance of intact TADs, another force that might contribute to the same result is linked to frequent organization of TAD borders into a locally open chromatin structure, consistent with more frequent DNA double-strand breaks and repair relative to internal TAD sequences, such that TAD boundaries may represent hotspots for genomic rearrangements (127). This feature, together with the fact that the disruption of TAD borders can have detrimental effects by leading to ectopic contacts and gene deregulation, might contribute to the maintenance of TADs during evolution.
On the other side of the coin, changes in TAD architecture could sometimes represent an evolutionary advantage. Some gibbon breaks of synteny did not colocalize with TAD boundaries, indicating that, even if these events are rare, they may play a role in generating new regulatory landscapes (127). Genomic duplications in patient cells can result in the formation of new chromatin domains (neo-TADs). Sometimes, neo-TADs can explain the pathology, but in other cases, they have no phenotype (26). In this last case, the neo-TAD appears well insulated from the rest of the genome, and this may provide a potential window of opportunity for divergent genome evolution. Hox gene regulation represents a remarkable example to illustrate how new regulatory landscape may have arisen from changes in 3D chromatin structures. Important for mouse limb development, the HoxD cluster resides precisely over a TAD boundary flanked by two TADs with distinct regulatory capacities. This specific configuration allows HoxD genes to read regulatory information on both sides, with a switch occurring from the posterior to the anterior genomic regions to ensure proper gene expression pattern (130). The boundary between the two TADs is therefore dynamic during development and corresponds to a transition between active and inactive promoters of the HoxD genes. This bimodal and flexible regulation of HoxD clusters by cis-regulatory elements located in two adjacent TADs is conserved in zebrafish (131), but is absent in the invertebrate Amphioxus, where a unique Hox gene cluster is present within a single TAD (125). The split into two TADs is robust against perturbation, because only a large deletion, including the whole cluster, eventually leads to the fusion of the two TADs (132). The appearance of a new genomic region may therefore have led to novel cis-regulatory inputs in the vertebrate lineage.
CONCLUSIONS AND PERSPECTIVES
The hierarchical folding of chromosomes is a conserved feature of genome 3D organization during evolution. Notably, the segregation of active and repressed chromatin represents a key principle of chromosome organization at multiple scales, from the formation of mutually excluded compartments at the chromosomal scale to the local segregation of submegabase domains, forming TAD-like structures (32). In mammals, the presence of inverted CTCF binding sites is associated with the formation of chromatin loops, acting in addition to the preexisting compartmentalization defined by the chromatin state. This may correspond to an additional layer of organization that is partially overriding homotypic chromatin cis interactions to build large-scale TADs. It will be interesting to study whether this role of CTCF in defining TAD borders might have been specifically gained in the vertebrate lineage or, alternatively, whether it was lost in Drosophila but is still present in other phylogenetic branches characterized by the presence of CTCF proteins. Certainly, it is not a necessary requisite as in other species, such as C. elegans, CTCF is absent, whereas self-interacting domains exist (125). Moreover, other eukaryotes like plants or yeast display TAD-like structures in the absence of CTCF proteins. This indicates that TADs are a more fundamental chromatin architecture that can exist without CTCF in other species, and in these cases, they are more correlated with transcriptional clustering. The distribution and density of transcribed genes and the presence and localization of insulator/architectural proteins may provide a framework to explain the contact patterns observed in these species. Therefore, different mechanisms leading to the compartmentalization of genome into autonomous units could produce similar output, i.e., the definition of regulatory landscapes within chromosomes. The conservation of TAD-like structures during evolution would then be functional, rather than structural.
If our understanding of the 3D genome organization has recently increased drastically, then outstanding questions remain to be addressed. Transcription has been tightly linked to chromosome folding, especially at TAD borders, but neither does its inhibition abolish boundaries (32, 53, 54, 59), nor is its induction sufficient to create insulation (24). Hence, what drives CTCF-independent boundary formation? To what extent do TADs regulate genome activity, as opposed to emerging as a consequence of genome function? Also, if many studies focused on the role of mammalian TADs in transcription through the spatial regulation of contacts between gene promoters and cis-regulatory elements, then it is not clear whether this applies to other organisms. Is the partitioning of genomes into domains generally required to ensure proper gene regulation or are other genome functions the reason to be of TADs, at least in a subset of nonmammalian species? The development of single-cell omics, live imaging, super-resolution microscopy, and modeling of the chromatin fiber, combined with state-of-the-art genome engineering technologies, offers a powerful toolset for addressing these questions in the coming years.
Acknowledgments
Funding: Q.S. was supported by the French Ministry of Higher Education and Research and La Ligue Nationale Contre le Cancer. F.B. was supported by CNRS. Research in the G.C. laboratory was supported by grants from the CNRS, the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme [grant agreements no. 676556 (MuG) and no. 788972 (3DEpi)], the Agence Nationale de la Recherche (ANR-15-CE12-0006 EpiDevoMath), the Fondation pour la Recherche Médicale (DEI20151234396), the INSERM, the French National Cancer Institute (INCa), and the Laboratory of Excellence EpiGenMed. Author contributions: Q.S., F.B., and G.C. wrote the manuscript. Competing interests: The authors declare that they have no competing interest. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the articles cited herein.
REFERENCES AND NOTES
- 1.Bickmore W. A., The spatial organization of the human genome. Annu. Rev. Genomics Hum. Genet. 14, 67–84 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Cremer T., Cremer M., Chromosome territories. Cold Spring Harb. Perspect. Biol. 2, a003889 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shah S., Takei Y., Zhou W., Lubeck E., Yun J., Eng C.-H. L., Koulena N., Cronin C., Karp C., Liaw E. J., Amin M., Cai L., Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell 174, 363–376.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bonev B., Cavalli G., Organization and function of the 3D genome. Nat. Rev. Genet. 17, 661–678 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Dekker J., Rippe K., Dekker M., Kleckner N., Capturing chromosome conformation. Science 295, 1306–1311 (2002). [DOI] [PubMed] [Google Scholar]
- 6.Lieberman-Aiden E., van Berkum N. L., Williams L., Imakaev M., Ragoczy T., Telling A., Amit I., Lajoie B. R., Sabo P. J., Dorschner M. O., Sandstrom R., Bernstein B., Bender M. A., Groudine M., Gnirke A., Stamatoyannopoulos J., Mirny L. A., Lander E. S., Dekker J., Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326, 289–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sati S., Cavalli G., Chromosome conformation capture technologies and their impact in understanding genome function. Chromosoma 126, 33–44 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Galazka J. M., Klocko A. D., Uesaka M., Honda S., Selker E. U., Freitag M., Neurosporachromosomes are organized by blocks of importin alpha-dependent heterochromatin that are largely independent of H3K9me3. Genome Res. 26, 1069–1080 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grob S., Schmid M. W., Grossniklaus U., Hi-C analysis in Arabidopsis identifies the KNOT, a structure with similarities to the flamenco locus of Drosophila. Mol. Cell 55, 678–693 (2014). [DOI] [PubMed] [Google Scholar]
- 10.Mizuguchi T., Fudenberg G., Mehta S., Belton J.-M., Taneja N., Folco H. D., FitzGerald P., Dekker J., Mirny L., Barrowman J., Grewal S. I. S., Cohesin-dependent globules and heterochromatin shape 3D genome architecture in S. pombe. Nature 516, 432–435 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sexton T., Yaffe E., Kenigsberg E., Bantignies F., Leblanc B., Hoichman M., Parrinello H., Tanay A., Cavalli G., Three-dimensional folding and functional organization principles of the Drosophila genome. Cell 148, 458–472 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Zhang Y., McCord R. P., Ho Y.-J., Lajoie B. R., Hildebrand D. G., Simon A. C., Becker M. S., Alt F. W., Dekker J., Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell 148, 908–921 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sanborn A. L., Rao S. S. P., Huang S.-C., Durand N. C., Huntley M. H., Jewett A. I., Bochkov I. D., Chinnappan D., Cutkosky A., Li J., Geeting K. P., Gnirke A., Melnikov A., McKenna D., Stamenova E. K., Lander E. S., Aiden E. L., Chromatin extrusion explains key features of loop and domain formation in wild-type and engineered genomes. Proc. Natl. Acad. Sci. U.S.A. 112, E6456–E6465 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rao S. S. P., Huntley M. H., Durand N. C., Stamenova E. K., Bochkov I. D., Robinson J. T., Sanborn A. L., Machol I., Omer A. D., Lander E. S., Aiden E. L., A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang S., Su J.-H., Beliveau B. J., Bintu B., Moffitt J. R., Wu C.-t., Zhuang X., Spatial organization of chromatin domains and compartments in single chromosomes. Science 353, 598–602 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dixon J. R., Selvaraj S., Yue F., Kim A., Li Y., Shen Y., Hu M., Liu J. S., Ren B., Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485, 376–380 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou C., Li L., Qin Z. S., Corces V. G., Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell 48, 471–484 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nora E. P., Lajoie B. R., Schulz E. G., Giorgetti L., Okamoto I., Servant N., Piolot T., van Berkum N. L., Meisig J., Sedat J., Gribnau J., Barillot E., Blüthgen N., Dekker J., Heard E., Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature 485, 381–385 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pope B. D., Ryba T., Dileep V., Yue F., Wu W., Denas O., Vera D. L., Wang Y., Hansen R. S., Canfield T. K., Thurman R. E., Cheng Y., Gülsoy G., Dennis J. H., Snyder M. P., Stamatoyannopoulos J. A., Taylor J., Hardison R. C., Kahveci T., Ren B., Gilbert D. M., Topologically associating domains are stable units of replication-timing regulation. Nature 515, 402–405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon J. R., Gorkin D. U., Ren B., Chromatin domains: The unit of chromosome organization. Mol. Cell 62, 668–680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ramirez F., Bhardwaj V., Arrigoni L., Lam K. C., Grüning B. A., Villaveces J., Habermann B., Akhtar A., Manke T., High-resolution TADs reveal DNA sequences underlying genome organization in flies. Nat. Commun. 9, 189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhan Y., Mariani L., Barozzi I., Schulz E. G., Blüthgen N., Stadler M., Tiana G., Giorgetti L., Reciprocal insulation analysis of Hi-C data shows that TADs represent a functionally but not structurally privileged scale in the hierarchical folding of chromosomes. Genome Res. 27, 479–490 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Symmons O., Uslu V. V., Tsujimura T., Ruf S., Nassari S., Schwarzer W., Ettwiller L., Spitz F., Functional and topological characteristics of mammalian regulatory domains. Genome Res. 24, 390–400 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonev B., Cohen N. M., Szabo Q., Fritsch L., Papadopoulos G. L., Lubling Y., Xu X., Lv X., Hugnot J.-P., Tanay A., Cavalli G., Multiscale 3D genome rewiring during mouse neural development. Cell 171, 557–572.24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flavahan W. A., Drier Y., Liau B. B., Gillespie S. M., Venteicher A. S., Stemmer-Rachamimov A. O., Suvà M. L., Bernstein B. E., Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529, 110–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franke M., Ibrahim D. M., Andrey G., Schwarzer W., Heinrich V., Schöpflin R., Kraft K., Kempfer R., Jerković I., Chan W. L., Spielmann M., Timmermann B., Wittler L., Kurth I., Cambiaso P., Zuffardi O., Houge G., Lambie L., Brancati F., Pombo A., Vingron M., Spitz F., Mundlos S., Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538, 265–269 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Hnisz D., Weintraub A. S., Day D. S., Valton A.-L., Bak R. O., Li C. H., Goldmann J., Lajoie B. R., Fan Z. P., Sigova A. A., Reddy J., Borges-Rivera D., Lee T. I., Jaenisch R., Porteus M. H., Dekker J., Young R. A., Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 351, 1454–1458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lupiáñez D. G., Kraft K., Heinrich V., Krawitz P., Brancati F., Klopocki E., Horn D., Kayserili H., Opitz J. M., Laxova R., Santos-Simarro F., Gilbert-Dussardier B., Wittler L., Borschiwer M., Haas S. A., Osterwalder M., Franke M., Timmermann B., Mundlos S., Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 161, 1012–1025 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lupiáñez D. G., Spielmann M., Mundlos S., Breaking TADs: How alterations of chromatin domains result in disease. Trends Genet. 32, 225–237 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Weischenfeldt J., Dubash T., Drainas A. P., Mardin B. R., Chen Y., Stütz A. M., Waszak S. M., Bosco G., Halvorsen A. R., Raeder B., Efthymiopoulos T., Erkek S., Siegl C., Brenner H., Brustugun O. T., Dieter S. M., Northcott P. A., Petersen I., Pfister S. M., Schneider M., Solberg S. K., Thunissen E., Weichert W., Zichner T., Thomas R., Peifer M., Helland A., Ball C. R., Jechlinger M., Sotillo R., Glimm H., Korbel J. O., Pan-cancer analysis of somatic copy-number alterations implicates IRS4 and IGF2 in enhancer hijacking. Nat. Genet. 49, 65–74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vietri Rudan M., Barrington C., Henderson S., Ernst C., Odom D. T., Tanay A., Hadjur S., Comparative Hi-C reveals that CTCF underlies evolution of chromosomal domain architecture. Cell Rep. 10, 1297–1309 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rowley M. J., Nichols M. H., Lyu X., Ando-Kuri M., Rivera I. S. M., Hermetz K., Wang P., Ruan Y., Corces V. G., Evolutionarily conserved principles predict 3D chromatin organization. Mol. Cell 67, 837–852.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phillips-Cremins J. E., Sauria M. E. G., Sanyal A., Gerasimova T. I., Lajoie B. R., Bell J. S. K., Ong C.-T., Hookway T. A., Guo C., Sun Y., Bland M. J., Wagstaff W., Dalton S., McDevitt T. C., Sen R., Dekker J., Taylor J., Corces V. G., Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153, 1281–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Wit E., Vos E. S. M., Holwerda S. J. B., Valdes-Quezada C., Verstegen M. J. A. M., Teunissen H., Splinter E., Wijchers P. J., Krijger P. H. L., de Laat W., CTCF binding polarity determines chromatin looping. Mol. Cell 60, 676–684 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Guo Y., Xu Q., Canzio D., Shou J., Li J., Gorkin D. U., Jung I., Wu H., Zhai Y., Tang Y., Lu Y., Wu Y., Jia Z., Li W., Zhang M. Q., Ren B., Krainer A. R., Maniatis T., Wu Q., CRISPR inversion of CTCF sites alters genome topology and enhancer/promoter function. Cell 162, 900–910 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saldaña-Meyer R., González-Buendía E., Guerrero G., Narendra V., Bonasio R., Recillas-Targa F., Reinberg D., CTCF regulates the human p53 gene through direct interaction with its natural antisense transcript, Wrap53. Genes Dev. 28, 723–734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fudenberg G., Imakaev M., Lu C., Goloborodko A., Abdennur N., Mirny L. A., Formation of chromosomal domains by loop extrusion. Cell Rep. 15, 2038–2049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vian L., Pękowska A., Rao S. S. P., Kieffer-Kwon K.-R., Jung S., Baranello L., Huang S.-C., Khattabi L. E., Dose M., Pruett N., Sanborn A. L., Canela A., Maman Y., Oksanen A., Resch W., Li X., Lee B., Kovalchuk A. L., Tang Z., Nelson S., Pierro M. D., Cheng R. R., Machol I., St. Hilaire B. G., Durand N. C., Shamim M. S., Stamenova E. K., Onuchic J. N., Ruan Y., Nussenzweig A., Levens D., Aiden E. L., Casellas R., The energetics and physiological impact of cohesin extrusion. Cell 173, 1165–1178.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fudenberg G., Abdennur N., Imakaev M., Goloborodko A., Mirny L. A., Emerging evidence of chromosome folding by loop extrusion. Cold Spring Harb. Symp. Quant. Biol. 82, 45–55 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nora E. P., Goloborodko A., Valton A.-L., Gibcus J. H., Uebersohn A., Abdennur N., Dekker J., Mirny L. A., Bruneau B. G., Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell 169, 930–944.e22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao S. S. P., Huang S.-C., St. Hilaire B. G., Engreitz J. M., Perez E. M., Kieffer-Kwon K.-R., Sanborn A. L., Johnstone S. E., Bascom G. D., Bochkov I. D., Huang X., Shamim M. S., Shin J., Turner D., Ye Z., Omer A. D., Robinson J. T., Schlick T., Bernstein B. E., Casellas R., Lander E. S., Aiden E. L., Cohesin loss eliminates all loop domains. Cell 171, 305–320.e24 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schwarzer W., Abdennur N., Goloborodko A., Pekowska A., Fudenberg G., Loe-Mie Y., Fonseca N. A., Huber W., Haering C. H., Mirny L., Spitz F., Two independent modes of chromatin organization revealed by cohesin removal. Nature 551, 51–56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wutz G., Várnai C., Nagasaka K., Cisneros D. A., Stocsits R. R., Tang W., Schoenfelder S., Jessberger G., Muhar M., Hossain M. J., Walther N., Koch B., Kueblbeck M., Ellenberg J., Zuber J., Fraser P., Peters J.-M., Topologically associating domains and chromatin loops depend on cohesin and are regulated by CTCF, WAPL, and PDS5 proteins. EMBO J. 36, 3573–3599 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haarhuis J. H. I., van der Weide R. H., Blomen V. A., Yáñez-Cuna J. O., Amendola M., van Ruiten M. S., Krijger P. H. L., Teunissen H., Medema R. H., van Steensel B., Brummelkamp T. R., de Wit E., Rowland B. D., The cohesin release factor WAPL restricts chromatin loop extension. Cell 169, 693–707.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nuebler J., Fudenberg G., Imakaev M., Abdennur N., Mirny L. A., Chromatin organization by an interplay of loop extrusion and compartmental segregation. Proc. Natl. Acad. Sci. U.S.A. 115, E6697–E6706 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yuen K. C., Slaughter B. D., Gerton J. L., Condensin II is anchored by TFIIIC and H3K4me3 in the mammalian genome and supports the expression of active dense gene clusters. Sci. Adv. 3, e1700191 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ganji M., Shaltiel I. A., Bisht S., Kim E., Kalichava A., Haering C. H., Dekker C., Real-time imaging of DNA loop extrusion by condensin. Science 360, 102–105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bintu B., Mateo L. J., Su J.-H., Sinnott-Armstrong N. A., Parker M., Kinrot S., Yamaya K., Boettiger A. N., Zhuang X., Super-resolution chromatin tracing reveals domains and cooperative interactions in single cells. Science 362, eaau1783 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weintraub A. S., Li C. H., Zamudio A. V., Sigova A. A., Hannett N. M., Day D. S., Abraham B. J., Cohen M. A., Nabet B., Buckley D. L., Guo Y. E., Hnisz D., Jaenisch R., Bradner J. E., Gray N. S., Young R. A., YY1 is a structural regulator of enhancer-promoter loops. Cell 171, 1573–1588.e28 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Racko D., Benedetti F., Dorier J., Stasiak A., Transcription-induced supercoiling as the driving force of chromatin loop extrusion during formation of TADs in interphase chromosomes. Nucleic Acids Res. 46, 1648–1660 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Uusküla-Reimand L., Hou H., Samavarchi-Tehrani P., Rudan M. V., Liang M., Medina-Rivera A., Mohammed H., Schmidt D., Schwalie P., Young E. J., Reimand J., Hadjur S., Gingras A.-C., Wilson M. D., Topoisomerase II beta interacts with cohesin and CTCF at topological domain borders. Genome Biol. 17, 182 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jung Y. H., Sauria M. E. G., Lyu X., Cheema M. S., Ausio J., Taylor J., Corces V. G., Chromatin states in mouse sperm correlate with embryonic and adult regulatory landscapes. Cell Rep. 18, 1366–1382 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Du Z., Zheng H., Huang B., Ma R., Wu J., Zhang X., He J., Xiang Y., Wang Q., Li Y., Ma J., Zhang X., Zhang K., Wang Y., Zhang M. Q., Gao J., Dixon J. R., Wang X., Zeng J., Xie W., Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature 547, 232–235 (2017). [DOI] [PubMed] [Google Scholar]
- 54.Ke Y., Xu Y., Chen X., Feng S., Liu Z., Sun Y., Yao X., Li F., Zhu W., Gao L., Chen H., Du Z., Xie W., Xu X., Huang X., 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell 170, 367–381.e20 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Eagen K. P., Aiden E. L., Kornberg R. D., Polycomb-mediated chromatin loops revealed by a subkilobase-resolution chromatin interaction map. Proc. Natl. Acad. Sci. U.S.A. 114, 8764–8769 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Q., Sun Q., Czajkowsky D. M., Shao Z., Sub-kb Hi-C in D. melanogaster reveals conserved characteristics of TADs between insect and mammalian cells. Nat. Commun. 9, 188 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forcato M., Nicoletti C., Pal K., Livi C. M., Ferrari F., Bicciato S., Comparison of computational methods for Hi-C data analysis. Nat. Methods 14, 679–685 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cubeñas-Potts C., Rowley M. J., Lyu X., Li G., Lei E. P., Corces V. G., Different enhancer classes in Drosophila bind distinct architectural proteins and mediate unique chromatin interactions and 3D architecture. Nucleic Acids Res. 45, 1714–1730 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hug C. B., Grimaldi A. G., Kruse K., Vaquerizas J. M., Chromatin architecture emerges during zygotic genome activation independent of transcription. Cell 169, 216–228.e19 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Ulianov S. V., Khrameeva E. E., Gavrilov A. A., Flyamer I. M., Kos P., Mikhaleva E. A., Penin A. A., Logacheva M. D., Imakaev M. V., Chertovich A., Gelfand M. S., Shevelyov Y. Y., Razin S. V., Active chromatin and transcription play a key role in chromosome partitioning into topologically associating domains. Genome Res. 26, 70–84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Van Bortle K., Nichols M. H., Li L., Ong C.-T., Takenaka N., Qin Z. S., Corces V. G., Insulator function and topological domain border strength scale with architectural protein occupancy. Genome Biol. 15, R82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gambetta M. C., Furlong E. E. M., The insulator protein CTCF is required for correct Hox gene expression, but not for embryonic development in Drosophila. Genetics 210, 129–136 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Luzhin A. V., Flyamer I. M., Khrameeva E. E., Ulianov S. V., Razin S. V., Gavrilov A. A., Quantitative differences in TAD border strength underly the TAD hierarchy in Drosophila chromosomes. J. Cell. Biochem. 120, 4494–4503 (2019). [DOI] [PubMed] [Google Scholar]
- 64.Ogiyama Y., Schuettengruber B., Papadopoulos G. L., Chang J.-M., Cavalli G., Polycomb-dependent chromatin looping contributes to gene silencing during Drosophila development. Mol. Cell 71, 73–88.e5 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Crane E., Bian Q., McCord R. P., Lajoie B. R., Wheeler B. S., Ralston E. J., Uzawa S., Dekker J., Meyer B. J., Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 523, 240–244 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Feng S., Cokus S. J., Schubert V., Zhai J., Pellegrini M., Jacobsen S. E., Genome-wide Hi-C analyses in wild-type and mutants reveal high-resolution chromatin interactions in Arabidopsis. Mol. Cell 55, 694–707 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang C., Liu C., Roqueiro D., Grimm D., Schwab R., Becker C., Lanz C., Weigel D., Genome-wide analysis of local chromatin packing in Arabidopsis thaliana. Genome Res. 25, 246–256 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dong Q., Li N., Li X., Yuan Z., Xie D., Wang X., Li J., Yu Y., Wang J., Ding B., Zhang Z., Li C., Bian Y., Zhang A., Wu Y., Liu B., Gong L., Genome-wide Hi-C analysis reveals extensive hierarchical chromatin interactions in rice. Plant J. 94, 1141–1156 (2018). [DOI] [PubMed] [Google Scholar]
- 69.Liu C., Cheng Y.-J., Wang J.-W., Weigel D., Prominent topologically associated domains differentiate global chromatin packing in rice from Arabidopsis. Nat. Plants 3, 742–748 (2017). [DOI] [PubMed] [Google Scholar]
- 70.Wang M., Wang P., Lin M., Ye Z., Li G., Tu L., Shen C., Li J., Yang Q., Zhang X., Evolutionary dynamics of 3D genome architecture following polyploidization in cotton. Nat. Plants 4, 90–97 (2018). [DOI] [PubMed] [Google Scholar]
- 71.Dong P., Tu X., Chu P.-Y., Lü P., Zhu N., Grierson D., Du B., Li P.-h., Zhong S., 3D chromatin architecture of large plant genomes determined by local A/B compartments. Mol. Plant 10, 1497–1509 (2017). [DOI] [PubMed] [Google Scholar]
- 72.Duan Z., Andronescu M., Schutz K., McIlwain S., Kim Y. J., Lee C., Shendure J., Fields S., Blau C. A., Noble W. S., A three-dimensional model of the yeast genome. Nature 465, 363–367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hsieh T.-H. S., Weiner A., Lajoie B., Dekker J., Friedman N., Rando O. J., Mapping nucleosome resolution chromosome folding in yeast by micro-C. Cell 162, 108–119 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Benedetti F., Racko D., Dorier J., Burnier Y., Stasiak A., Transcription-induced supercoiling explains formation of self-interacting chromatin domains in S. pombe. Nucleic Acids Res. 45, 9850–9859 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le T. B. K., Imakaev M. V., Mirny L. A., Laub M. T., High-resolution mapping of the spatial organization of a bacterial chromosome. Science 342, 731–734 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marbouty M., Le Gall A., Cattoni D. I., Cournac A., Koh A., Fiche J.-B., Mozziconacci J., Murray H., Koszul R., Nollmann M., Condensin- and replication-mediated bacterial chromosome folding and origin condensation revealed by Hi-C and super-resolution imaging. Mol. Cell 59, 588–602 (2015). [DOI] [PubMed] [Google Scholar]
- 77.Wang X., le T. B. K., Lajoie B. R., Dekker J., Laub M. T., Rudner D. Z., Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilis. Genes Dev. 29, 1661–1675 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Espeli O., Mercier R., Boccard F., DNA dynamics vary according to macrodomain topography in the E. coli chromosome. Mol. Microbiol. 68, 1418–1427 (2008). [DOI] [PubMed] [Google Scholar]
- 79.Trussart M., Yus E., Martinez S., Baù D., Tahara Y. O., Pengo T., Widjaja M., Kretschmer S., Swoger J., Djordjevic S., Turnbull L., Whitchurch C., Miyata M., Marti-Renom M. A., Lluch-Senar M., Serrano L., Defined chromosome structure in the genome-reduced bacterium Mycoplasma pneumoniae. Nat. Commun. 8, 14665 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Tran N. T., Laub M. T., Le T. B. K., SMC progressively aligns chromosomal arms in Caulobacter crescentus but is antagonized by convergent transcription. Cell Rep. 20, 2057–2071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang X., Brandão H. B., Le T. B. K., Laub M. T., Rudner D. Z., Bacillus subtilis SMC complexes juxtapose chromosome arms as they travel from origin to terminus. Science 355, 524–527 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang X., Hughes A. C., Brandão H. B., Walker B., Lierz C., Cochran J. C., Oakley M. G., Kruse A. C., Rudner D. Z., In vivo evidence for ATPase-dependent DNA translocation by the Bacillus subtilis SMC condensin complex. Mol. Cell 71, 841–847.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kaaij L. J. T., van der Weide R. H., Ketting R. F., de Wit E., Systemic loss and gain of chromatin architecture throughout zebrafish development. Cell Rep. 24, 1–10.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ay F., Bunnik E. M., Varoquaux N., Bol S. M., Prudhomme J., Vert J.-P., Noble W. S., Le Roch K. G., Three-dimensional modeling of the P. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression. Genome Res. 24, 974–988 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ong C.-T., Corces V. G., Insulators as mediators of intra- and inter-chromosomal interactions: A common evolutionary theme. J. Biol. 8, 73 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Busslinger G. A., Stocsits R. R., van der Lelij P., Axelsson E., Tedeschi A., Galjart N., Peters J.-M., Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 544, 503–507 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Misulovin Z., Schwartz Y. B., Li X.-Y., Kahn T. G., Gause M., MacArthur S., Fay J. C., Eisen M. B., Pirrotta V., Biggin M. D., Dorsett D., Association of cohesin and Nipped-B with transcriptionally active regions of the Drosophila melanogaster genome. Chromosoma 117, 89–102 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schaaf C. A., Misulovin Z., Gause M., Koenig A., Gohara D. W., Watson A., Dorsett D., Cohesin and polycomb proteins functionally interact to control transcription at silenced and active genes. PLOS Genet. 9, e1003560 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Boettiger A. N., Bintu B., Moffitt J. R., Wang S., Beliveau B. J., Fudenberg G., Imakaev M., Mirny L. A., Wu C.-t., Zhuang X., Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Szabo Q., Jost D., Chang J.-M., Cattoni D. I., Papadopoulos G. L., Bonev B., Sexton T., Gurgo J., Jacquier C., Nollmann M., Bantignies F., Cavalli G., TADs are 3D structural units of higher-order chromosome organization in Drosophila. Sci. Adv. 4, eaar8082 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cattoni D. I., Cardozo Gizzi A. M., Georgieva M., Di Stefano M., Valeri A., Chamousset D., Houbron C., Déjardin S., Fiche J.-B., González I., Chang J.-M., Sexton T., Marti-Renom M. A., Bantignies F., Cavalli G., Nollmann M., Single-cell absolute contact probability detection reveals chromosomes are organized by multiple low-frequency yet specific interactions. Nat. Commun. 8, 1753 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Eagen K. P., Hartl T. A., Kornberg R. D., Stable chromosome condensation revealed by chromosome conformation capture. Cell 163, 934–946 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Stadler M. R., Haines J. E., Eisen M. B., Convergence of topological domain boundaries, insulators, and polytene interbands revealed by high-resolution mapping of chromatin contacts in the early Drosophila melanogaster embryo. eLife 6, e29550 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Narendra V., Rocha P. P., An D., Raviram R., Skok J. A., Mazzoni E. O., Reinberg D., CTCF establishes discrete functional chromatin domains at the Hox clusters during differentiation. Science 347, 1017–1021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nir G., Farabella I., Estrada C. P., Ebeling C. G., Beliveau B. J., Sasaki H. M., Lee S. D., Nguyen S. C., McCole R. B., Chattoraj S., Erceg J., Abed J. A., Martins N. M. C., Nguyen H. Q., Hannan M. A., Russell S., Durand N. C., Rao S. S. P., Kishi J. Y., Soler-Vila P., Di Pierro M., Onuchic J. N., Callahan S. P., Schreiner J. M., Stuckey J. A., Yin P., Aiden E. L., Marti-Renom M. A., Wu C.-t., Walking along chromosomes with super-resolution imaging, contact maps, and integrative modeling. PLOS Genet. 14, e1007872 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Flyamer I. M., Gassler J., Imakaev M., Brandão H. B., Ulianov S. V., Abdennur N., Razin S. V., Mirny L. A., Tachibana-Konwalski K., Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature 544, 110–114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ho J. W. K., Jung Y. L., Liu T., Alver B. H., Lee S., Ikegami K., Sohn K.-A., Minoda A., Tolstorukov M. Y., Appert A., Parker S. C. J., Gu T., Kundaje A., Riddle N. C., Bishop E., Egelhofer T. A., Hu S. S., Alekseyenko A. A., Rechtsteiner A., Asker D., Belsky J. A., Bowman S. K., Chen Q. B., Chen R. A.-J., Day D. S., Dong Y., Dose A. C., Duan X., Epstein C. B., Ercan S., Feingold E. A., Ferrari F., Garrigues J. M., Gehlenborg N., Good P. J., Haseley P., He D., Herrmann M., Hoffman M. M., Jeffers T. E., Kharchenko P. V., Kolasinska-Zwierz P., Kotwaliwale C. V., Kumar N., Langley S. A., Larschan E. N., Latorre I., Libbrecht M. W., Lin X., Park R., Pazin M. J., Pham H. N., Plachetka A., Qin B., Schwartz Y. B., Shoresh N., Stempor P., Vielle A., Wang C., Whittle C. M., Xue H., Kingston R. E., Kim J. H., Bernstein B. E., Dernburg A. F., Pirrotta V., Kuroda M. I., Noble W. S., Tullius T. D., Kellis M., MacAlpine D. M., Strome S., Elgin S. C. R., Liu X. S., Lieb J. D., Ahringer J., Karpen G. H., Park P. J., Comparative analysis of metazoan chromatin organization. Nature 512, 449–452 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Larson A. G., Elnatan D., Keenen M. M., Trnka M. J., Johnston J. B., Burlingame A. L., Agard D. A., Redding S., Narlikar G. J., Liquid droplet formation by HP1α suggests a role for phase separation in heterochromatin. Nature 547, 236–240 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Strom A. R., Emelyanov A. V., Mir M., Fyodorov D. V., Darzacq X., Karpen G. H., Phase separation drives heterochromatin domain formation. Nature 547, 241–245 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sabari B. R., Dall’Agnese A., Boija A., Klein I. A., Coffey E. L., Shrinivas K., Abraham B. J., Hannett N. M., Zamudio A. V., Manteiga J. C., Li C. H., Guo Y. E., Day D. S., Schuijers J., Vasile E., Malik S., Hnisz D., Lee T. I., Cisse I. I., Roeder R. G., Sharp P. A., Chakraborty A. K., Young R. A., Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cho W.-K., Spille J.-H., Hecht M., Lee C., Li C., Grube V., Cisse I. I., Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science 361, 412–415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lu H., Yu D., Hansen A. S., Ganguly S., Liu R., Heckert A., Darzacq X., Zhou Q., Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature 558, 318–323 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Erdel F., Rippe K., Formation of chromatin subcompartments by phase separation. Biophys. J. 114, 2262–2270 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hnisz D., Shrinivas K., Young R. A., Chakraborty A. K., Sharp P. A., A phase separation model for transcriptional control. Cell 169, 13–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nagano T., Lubling Y., Stevens T. J., Schoenfelder S., Yaffe E., Dean W., Laue E. D., Tanay A., Fraser P., Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature 502, 59–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Stevens T. J., Lando D., Basu S., Atkinson L. P., Cao Y., Lee S. F., Leeb M., Wohlfahrt K. J., Boucher W., O’Shaughnessy-Kirwan A., Cramard J., Faure A. J., Ralser M., Blanco E., Morey L., Sansó M., Palayret M. G. S., Lehner B., Di Croce L., Wutz A., Hendrich B., Klenerman D., Laue E. D., 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature 544, 59–64 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tan L., Xing D., Chang C.-H., Li H., Xie X. S., Three-dimensional genome structures of single diploid human cells. Science 361, 924–928 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Giorgetti L., Galupa R., Nora E. P., Piolot T., Lam F., Dekker J., Tiana G., Heard E., Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcription. Cell 157, 950–963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Stadhouders R., Vidal E., Serra F., di Stefano B., Le Dily F., Quilez J., Gomez A., Collombet S., Berenguer C., Cuartero Y., Hecht J., Filion G. J., Beato M., Marti-Renom M. A., Graf T., Transcription factors orchestrate dynamic interplay between genome topology and gene regulation during cell reprogramming. Nat. Genet. 50, 238–249 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nagano T., Lubling Y., Várnai C., Dudley C., Leung W., Baran Y., Mendelson Cohen N., Wingett S., Fraser P., Tanay A., Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature 547, 61–67 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lakadamyali M., Cosma M. P., Advanced microscopy methods for visualizing chromatin structure. FEBS Lett. 589, 3023–3030 (2015). [DOI] [PubMed] [Google Scholar]
- 112.Hansen A. S., Pustova I., Cattoglio C., Tjian R., Darzacq X., CTCF and cohesin regulate chromatin loop stability with distinct dynamics. eLife 6, e25776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Fang K., Chen X., Li X., Shen Y., Sun J., Czajkowsky D. M., Shao Z., Super-resolution imaging of individual human subchromosomal regions in situ reveals nanoscopic building blocks of higher-order structure. ACS Nano 12, 4909–4918 (2018). [DOI] [PubMed] [Google Scholar]
- 114.Nozaki T., Imai R., Tanbo M., Nagashima R., Tamura S., Tani T., Joti Y., Tomita M., Hibino K., Kanemaki M. T., Wendt K. S., Okada Y., Nagai T., Maeshima K., Dynamic organization of chromatin domains revealed by super-resolution live-cell imaging. Mol. Cell 67, 282–293e7 (2017). [DOI] [PubMed] [Google Scholar]
- 115.Ricci M. A., Manzo C., García-Parajo M. F., Lakadamyali M., Cosma M. P., Chromatin fibers are formed by heterogeneous groups of nucleosomes in vivo. Cell 160, 1145–1158 (2015). [DOI] [PubMed] [Google Scholar]
- 116.Kundu S., Ji F., Sunwoo H., Jain G., Lee J. T., Sadreyev R. I., Dekker J., Kingston R. E., Polycomb repressive complex 1 generates discrete compacted domains that change during differentiation. Mol. Cell 65, 432–446.e5 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Smeets D., Markaki Y., Schmid V. J., Kraus F., Tattermusch A., Cerase A., Sterr M., Fiedler S., Demmerle J., Popken J., Leonhardt H., Brockdorff N., Cremer T., Schermelleh L., Cremer M., Three-dimensional super-resolution microscopy of the inactive X chromosome territory reveals a collapse of its active nuclear compartment harboring distinct Xist RNA foci. Epigenetics Chromatin 7, 8 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Xu J., Ma H., Jin J., Uttam S., Fu R., Huang Y., Liu Y., Super-resolution imaging of higher-order chromatin structures at different epigenomic states in single mammalian cells. Cell Rep. 24, 873–882 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang Y., Maharana S., Wang M. D., Shivashankar G. V., Super-resolution microscopy reveals decondensed chromatin structure at transcription sites. Sci. Rep. 4, 4477 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dixon J. R., Jung I., Selvaraj S., Shen Y., Antosiewicz-Bourget J. E., Lee A. Y., Ye Z., Kim A., Rajagopal N., Xie W., Diao Y., Liang J., Zhao H., Lobanenkov V. V., Ecker J. R., Thomson J. A., Ren B., Chromatin architecture reorganization during stem cell differentiation. Nature 518, 331–336 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen H., Levo M., Barinov L., Fujioka M., Jaynes J. B., Gregor T., Dynamic interplay between enhancer-promoter topology and gene activity. Nat. Genet. 50, 1296–1303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fukaya T., Lim B., Levine M., Enhancer control of transcriptional bursting. Cell 166, 358–368 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gu B., Swigut T., Spencley A., Bauer M. R., Chung M., Meyer T., Wysocka J., Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 359, 1050–1055 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Benedetti F., Dorier J., Stasiak A., Effects of supercoiling on enhancer-promoter contacts. Nucleic Acids Res. 42, 10425–10432 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Acemel R. D., Maeso I., Gómez-Skarmeta J. L., Topologically associated domains: A successful scaffold for the evolution of gene regulation in animals. Wiley Interdiscip. Rev. Dev. Biol. 6, e265 (2017). [DOI] [PubMed] [Google Scholar]
- 126.Harmston N., Ing-Simmons E., Tan G., Perry M., Merkenschlager M., Lenhard B., Topologically associating domains are ancient features that coincide with Metazoan clusters of extreme noncoding conservation. Nat. Commun. 8, 441 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Lazar N. H., Nevonen K. A., O'Connell B., McCann C., O'Neill R. J., Green R. E., Meyer T. J., Okhovat M., Carbone L., Epigenetic maintenance of topological domains in the highly rearranged gibbon genome. Genome Res. 28, 983–997 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Krefting J., Andrade-Navarro M. A., Ibn-Salem J., Evolutionary stability of topologically associating domains is associated with conserved gene regulation. BMC Biol. 16, 87 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gómez-Marín C., Tena J. J., Acemel R. D., López-Mayorga M., Naranjo S., de la Calle-Mustienes E., Maeso I., Beccari L., Aneas I., Vielmas E., Bovolenta P., Nobrega M. A., Carvajal J., Gómez-Skarmeta J. L., Evolutionary comparison reveals that diverging CTCF sites are signatures of ancestral topological associating domains borders. Proc. Natl. Acad. Sci. U.S.A. 112, 7542–7547 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Andrey G., Montavon T., Mascrez B., Gonzalez F., Noordermeer D., Leleu M., Trono D., Spitz F., Duboule D., A switch between topological domains underlies HoxD genes collinearity in mouse limbs. Science 340, 1234167 (2013). [DOI] [PubMed] [Google Scholar]
- 131.Woltering J. M., Noordermeer D., Leleu M., Duboule D., Conservation and divergence of regulatory strategies at Hox loci and the origin of tetrapod digits. PLOS Biol. 12, e1001773 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Rodríguez-Carballo E., Lopez-Delisle L., Zhan Y., Fabre P. J., Beccari L., El-Idrissi I., Huynh T. H. N., Ozadam H., Dekker J., Duboule D., The HoxD cluster is a dynamic and resilient TAD boundary controlling the segregation of antagonistic regulatory landscapes. Genes Dev. 31, 2264–2281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ou H. D., Phan S., Deerinck T. J., Thor A., Ellisman M. H., O’Shea C. C., ChromEMT: Visualizing 3D chromatin structure and compaction in interphase and mitotic cells. Science 357, eaag0025 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Durand N. C., Robinson J. T., Shamim M. S., Machol I., Mesirov J. P., Lander E. S., Aiden E. L., Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Liu C., Wang C., Wang G., Becker C., Zaidem M., Weigel D., Genome-wide analysis of chromatin packing in Arabidopsis thaliana at single-gene resolution. Genome Res. 26, 1057–1068 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]