

Autophagy regulates social behaviors by affecting the ability of brain cells to receive inhibitory signaling.
Abstract
Dysfunctional mTOR signaling is associated with the pathogenesis of neurodevelopmental and neuropsychiatric disorders. However, it is unclear what molecular mechanisms and pathogenic mediators are involved and whether mTOR-regulated autophagy continues to be crucial beyond neurodevelopment. Here, we selectively deleted Atg7 in forebrain GABAergic interneurons in adolescent mice and unexpectedly found that these mice showed a set of behavioral deficits similar to Atg7 deletion in forebrain excitatory neurons. By unbiased quantitative proteomic analysis, we identified γ-aminobutyric acid receptor–associated protein-like 2 (GABARAPL2) to differentially form high–molecular weight species in autophagy-deficient brains. Further functional analyses revealed a novel pathogenic mechanism involving the p62-dependent sequestration of GABARAP family proteins, leading to the reduction of surface GABAA receptor levels. Our work demonstrates a novel physiological role for autophagy in regulating GABA signaling beyond postnatal neurodevelopment, providing a potential mechanism for the reduced inhibitory inputs observed in neurodevelopmental and neuropsychiatric disorders with mTOR hyperactivation.
INTRODUCTION
Recent genetic studies indicate the involvement of dysregulated mechanistic target of rapamycin (mTOR) signaling, a central regulator of various cellular processes including protein translation, actin dynamics, and autophagy, in neurodevelopmental [e.g., autism spectrum disorder (ASD) and intellectual disability (ID)] and neuropsychiatric [e.g., schizophrenia (SZ) and bipolar disorder (BD)] disorders (1). For instance, mutations in several genes such as TSC1, TSC2, NF1, and PTEN result in hyperactivated mTOR and are associated with multiple dimensions of psychiatric symptoms including social deficits. Many behavioral deficits observed in these models can be alleviated with mTOR inhibitors such as rapamycin, thus highlighting the predominant contribution of mTOR dysregulation to the abnormalities. Most studies to date have focused on delineating the consequences of hyperactivated mTOR and its influence on protein synthesis and synaptic plasticity (2, 3). Yet, how the dysregulation of other downstream signaling pathways like autophagy may contribute to the manifestation of behavioral abnormalities in neurodevelopmental and neuropsychiatric disorders remains poorly understood (4). Moreover, the mediators and molecular mechanisms therein remain unclear.
Deficits in γ-aminobutyric acid (GABA) signaling have also been closely implicated in the pathogenesis of neurodevelopmental and neuropsychiatric disorders. For example, Prader-Willi syndrome, Angelman syndrome, and cases of SZ are associated with 15q11-q13 copy number variations, a chromosomal region that encodes for multiple subunits of the GABAA receptor including GABRB3, GABRA5, and GABRG3 (5). Postmortem studies using brain samples from ASD (6) and SZ (7) patients have revealed alterations in glutamic acid decarboxylase (GAD65 and GAD67) expression, further implicating defective GABAergic signaling in pathogenesis. GABA neurotransmission was also highlighted in BD pathology, as polymorphisms in multiple GABAA receptor subunits have been identified in BD patients (8). Furthermore, deletion of a number of genes associated with neurodevelopmental and neuropsychiatric disorders in mice has revealed specific deficits in the development of GABAergic interneurons or GABA neurotransmission (9, 10). Collectively, there is overwhelming evidence that links GABA signaling deficits to neurodevelopmental and neuropsychiatric disorders.
Despite the wealth of experimental evidence highlighting the critical roles of both mTOR and GABAergic signaling in the pathogenesis of neurodevelopmental and neuropsychiatric disorders, we know little about whether they may converge and modulate neuronal functions beyond postnatal neurodevelopment. In this study, we specifically impaired autophagic function in forebrain GABAergic interneurons of adolescent mice to evaluate the impact of this major downstream pathway regulated by mTOR and revealed a common pathogenic mechanism by which autophagy deficiency affects GABA signaling in both GABAergic inhibitory neurons and excitatory neurons.
RESULTS
Overlapping behavioral deficits displayed by mice with autophagy deficiency in forebrain GABAergic interneurons or excitatory neurons
To evaluate the contribution of disrupted mTOR-autophagic signaling in forebrain GABAergic interneurons to the manifestation of behavioral abnormalities associated with neurodevelopmental or neuropsychiatric disorders, we generated and examined the conditional deletion of Atg7 by Dlx5-creERT2. Alterations in mTOR signaling were previously shown to affect neurogenesis and migration of forebrain GABAergic interneurons (11), and autophagy was proposed recently to disrupt synaptic pruning (4, 12). Thus, Atg7 deletion was initiated in adolescent mice to exclude those effects to specifically examine the consequence of autophagy deficiency in mature GABAergic interneurons. We first confirmed the reduction of Atg7 expression and observed the accumulation and aggregate formation of p62, an adaptor protein responsible for recognizing and loading of cargo substrates into autophagosomes, as a classical marker of autophagy suppression/disruption (13, 14) in all major classes of forebrain GABAergic interneurons without detectable neurodegeneration at 6- to 8-month post-Cre recombinase induction, when all experiments were performed (Fig. 1, A and B, and fig. S1, A to D). We then performed a battery of behavioral tests (see Methods for details) on Dlx5-creERT2 Atg7 conditional knockout (cKO) and control (Dlx5-control) mice and examined whether their phenotypes in each test are similar to or different from those in CaMKII-cre Atg7 cKO/control (CaMKII-control) mice. We found that both Atg7 cKO mice shared a set of behavioral abnormalities including reduced social interaction, as examined by resident-intruder (Fig. 1, C and D) and three-chamber social interaction tests (Fig. 1, E and F), disturbed nesting behavior (Fig. 1, G and H), and increased anxiety as demonstrated by elevated plus maze (Fig. 1, I and J). In contrast, no deficits in motor function, exploratory activity, or stress vulnerability were observed for both Atg7 cKO mice by open-field activity test and tail suspension test (fig. S1, E to H).
Fig. 1. Autophagy deficiency in forebrain GABAergic inhibitory or excitatory neurons resulted in an overlapping set of ASD-like behavioral deficits.
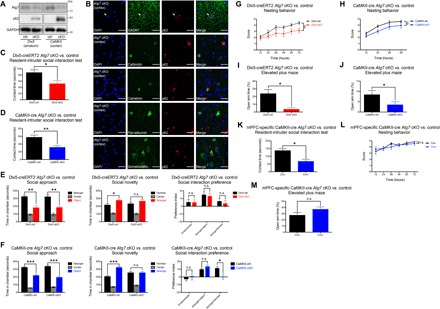
(A) Atg7 conditional deletion by Dlx5-creERT2 and CaMKII-cre led to increased p62 levels in affected brain regions. (B) p62+ aggregate formation was observed in all major subclasses of cortical GABAergic interneurons of Dlx5-creERT2 Atg7 cKO mice. (C and D) Resident-intruder social interaction test showed deficits in both Dlx5-creERT2 and CaMKII-cre Atg7 cKO mice. n = 10 (Dlx5-control), n = 8 (Dlx5-cKO), n = 16 (CaMKII-control), n = 16 (CaMKII-cKO). (E and F) Three-chamber social interaction test revealed a difference in Dlx5-creERT2 and CaMKII-cre Atg7 cKO mice for social novelty (middle) but not social approach (left). A social interaction preference index was also calculated, which showed a statistical significant difference for social novelty across genotypes for CaMKII-cre but not Dlx5-creERT2 Atg7 cKO animals (right). n = 12 (Dlx5-control), n = 10 (Dlx5-cKO), n = 16 (CaMKII-control), n = 16 (CaMKII-cKO). (G and H) Deficits in nesting behavior were observed for both Dlx5-creERT2 and CaMKII-cre Atg7 cKO mice. n = 12 (Dlx5-control), n = 10 (Dlx5-cKO), n = 14 (CaMKII-control), n = 10 (CaMKII-cKO). (I and J) Elevated plus maze showed that both Dlx5-creERT2 and CaMKII-cre Atg7 cKO mice have increased anxiety. n = 5 (Dlx5-control), n = 4 (Dlx5-cKO), n = 14 (CaMKII-control), n = 16 (CaMKII-cKO). (K) Localized Atg7 deletion in mPFC excitatory neurons in 12-week-old Atg7flox/flox mice led to deficits in social behavior as measured by the resident-intruder social interaction test. n = 7 animals per genotype. (L and M) mPFC-specific autophagy deficiency in excitatory neurons did not affect nesting or anxiety-related behaviors. n = 7 animals per genotype. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, unpaired two-tailed Student’s t test; §P < 0.05, between genotypes, two-way repeated-measures analysis of variance (ANOVA). n.s., not statistically significant.
Recent studies suggested that behavioral abnormalities in mice with autophagy deficiency in forebrain excitatory neurons or microglia were caused by synaptic pruning deficits during postnatal neurodevelopment; however, the molecular mechanism was not fully examined (4, 12). As we have observed similar behavioral deficits in Dlx5-cre ERT2 Atg7 cKO mice, although Atg7 deletion was initiated beyond the peak of synaptic pruning in adolescent mice, we asked whether this finding was specific to GABAergic interneurons or whether conditional Atg7 deletion in forebrain excitatory neurons at a similar stage may also disrupt social behaviors. To test this hypothesis, we performed adeno-associated virus (AAV) injections to locally delete Atg7 in excitatory neurons of the medial prefrontal cortex (mPFC) of adolescent Atg7flox/flox mice (12 weeks old) (fig. S1, I and J), a brain region previously shown to be critical for social interactions (15, 16). We observed that mice with autophagy deficiency, specifically in mPFC excitatory neurons, similarly exhibited social interaction deficits compared to controls (Fig. 1K), while other behavioral abnormalities were not observed (Fig. 1, L and M, and fig. S1, K and L). Thus, autophagy in forebrain excitatory neurons continues to play an important role in regulating social behavior beyond postnatal development. Furthermore, despite deleting Atg7 from distinct neuronal populations, the similar set of behavioral deficits observed in both Atg7 cKO mice suggested that a common mechanism may be involved.
Aggregation and mislocalization of GABARAPs to p62+ aggregates in autophagy-deficient neurons of Dlx5-creERT2 and CaMKII-cre Atg7 cKO mice
We reasoned that because autophagy deficiency in neurons accumulates p62+ and ubiquitin+ aggregates (17), the soluble pool of affected proteins could be substantially reduced by enhanced protein aggregation and, consequently, be compromised in their functions. To identify proteins that were differentially aggregated into high–molecular weight species (>250 kDa) in Atg7 cKO brains, we performed an unbiased quantitative proteomic screen by combining semidenaturing detergent agarose gel electrophoresis (SDD-AGE) (18) with spike-in stable isotope labeling of amino acids in mammals (SILAM) (Fig. 2A) (19). To maximize the sample size and homogeneity for reliable, quantitative mass spectrometric (MS) analysis, we carried out this experiment using cortical brain homogenates from CaMKII-cre Atg7 cKO and control mice (6 months old). We specifically searched for proteins similar to p62, which were absent in the high–molecular weight fraction from control brains but had increased amounts in Atg7 cKO brains (fig. S1B), because their functions may be most severely affected by autophagy deficiency. We identified 15 proteins that fit our criteria (SILAM score > 1.5 in both of two independent experiments) (Fig. 2, B and C, and fig. S2A). When we extended this experiment with Atg7 cKO and control brains from other ages (3 and 12 months old), only three proteins including p62 consistently met the selection criteria (Fig. 2D and fig. S2B). Among them, we were interested in GABA receptor–associated protein-like 2 (GABARAPL2), which is involved in autophagy as an Atg8-like protein (20) and whose paralog GABARAP was suggested to mediate trafficking of GABAA receptors (21). However, the neuronal functions of GABARAP family proteins remain poorly understood, although they are all highly expressed in the brain (22). All three family members showed increased protein levels in Atg7 cKO brains (Fig. 2E), in contrast to other Atg8-like proteins like LC3 that do not substantially accumulate under autophagy-deficient conditions (17). We next examined by native polyacrylamide gel electrophoresis (PAGE) whether the identification of GABARAPL2 in our MS-based screen was due to the increased amounts of GABARAPL2 in Atg7 cKO brains or was actually caused by a change from its native conformation to high–molecular weight species. We found that, in addition to accumulating in Atg7 cKO brains, the migration of GABARAPL2 and GABARAP under native conditions appears to have shifted to a high–molecular weight region (Fig. 2F), indicating potential changes in self-assembly or interactions with other proteins such as formation of homo-/hetero-oligomeric protein complexes. To gain further insights, we examined the subcellular localization of GABARAPL2 and its paralogs, GABARAPL1 and GABARAP, by immunofluorescence in Dlx5-creERT2, full, and mPFC-specific CaMKII-cre Atg7 cKO mice. We found that all three proteins were sequestered into the p62+ aggregates formed following Atg7 deletion (Fig. 2, G to I, and fig. S2, C to J). The mislocalization of GABARAPs to p62+ aggregates and their coaggregation, in addition to the observed shift from monomeric species to high–molecular weight complexes, suggest that the functions of GABARAPs could be compromised as a result of autophagy deficiency.
Fig. 2. Mislocalization and altered conformational status of GABARAPs in Atg7 cKO brains.
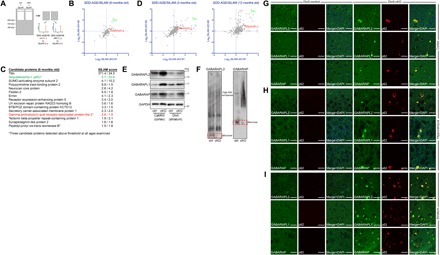
(A) Schematic diagram of SDD-AGE/SILAM experiment. (B) Abundance of proteins identified in high molecular weight fraction (>250 kDa) only in CaMKII-cre Atg7 cKO but not control brains by SDD-AGE/SILAM experiments (6-month-old mice). (C) Candidate proteins meeting the selection criteria are listed with SILAM scores representing relative abundance to SILAM-labeled sample. Selection criteria are depicted schematically in fig. S2B. Candidate proteins identified by these criteria at all three ages examined are specifically marked by asterisks. (D) GABARAPL2 and p62 were consistently identified by SDD-AGE/SILAM in CaMKII-cre Atg7 cKO brain homogenates at 3 months (left) and 12 months (right) of age. (E) p62-like accumulation of GABARAPs was observed in Dlx5-creERT2 and CaMKII-cre Atg7 cKO mice. (F) Native PAGE analysis revealed shifts of GABARAPL2 and GABARAP into high–molecular weight (MW) complexes in the brains of CaMKII-cre Atg7 cKO mice (cortex). (G to I) GABARAPs were observed to mislocalize to p62+ aggregates in affected neurons of Dlx5-creERT2 Atg7 cKO mice. Scale bars, 20 μm (G to I).
Because GABARAP was originally identified as a GABAA receptor–interacting protein (23) and suggested to mediate the trafficking of GABAA receptors (21), we examined whether surface expression of GABAA receptors may be altered in Atg7 cKO mice. To this end, we performed in situ surface receptor biotinylation on striatal and hippocampal tissue dissected from Dlx5-creERT2 and CaMKII-cre Atg7 cKO and control mice, respectively, to maximize sample homogeneity, as those tissues are predominantly populated by Cre recombinase–expressing Dlx5+ or CaMKIIα+ neurons. While no significant differences were observed for surface fractions of both α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-d-aspartate (NMDA) receptors, a specific reduction of surface GABAA receptors was detected in both tissues lacking Atg7 (Fig. 3A and fig. S3A). Next, to investigate whether the reduced surface GABAA receptor levels would impair electrophysiological functions, we performed recordings of both miniature inhibitory and excitatory postsynaptic currents (mIPSC and mEPSC, respectively) from CA1 pyramidal cells (Atg7 intact) of Dlx5-creERT2 Atg7 cKO and control mice. We detected a significant increase in mIPSC frequency without any changes in amplitude (Fig. 3B), suggesting that autophagy deficiency in GABAergic interneurons enhanced GABA release onto their target cells. In contrast, mEPSC recordings showed no differences (fig. S3B), indicating that there were no non–cell-autonomous effects on excitatory neurons by Atg7 deletion in GABAergic interneurons. Together, these results revealed that GABAergic interneurons in Dlx5-creERT2 Atg7 cKO mice are potentially hyperactive, consistent with the lower surface expression of GABAA receptors and thus reduced inhibitory input. From these experiments, we revealed that GABARAPs were functionally compromised because of the formation of high–molecular weight species in autophagy-deficient neurons, which led to reduced trafficking of GABAA receptors in affected cells and altered GABAergic signaling in the brain.
Fig. 3. Autophagy deficiency led to a reduction of surface GABAA receptors and increased GABA release by Atg7 cKO GABAergic interneurons.
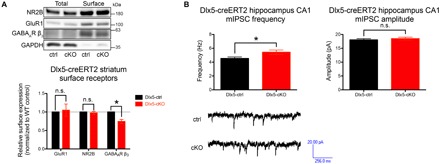
(A) Significant reductions in surface expression of GABAA receptors (GABAAR) were observed in striata of Dlx5-creERT2 Atg7 cKO mice (see fig. S3A). n = 4 animals per genotype. (B) mIPSCs recorded from CA1 pyramidal cells in Dlx5-creERT2 Atg7 cKO hippocampi showed a significant increase in frequency (left) but no differences in amplitude (right). Representative traces are shown at the bottom. n = 35 cells recorded from five different animals. Data are presented as means ± SEM. * P < 0.05, paired (A) or unpaired (B) two-tailed Student’s t test.
Reduced GABARAPL2 function contributes to reduced GABAA receptor trafficking in autophagy-deficient cortical GABAergic interneurons
To examine the molecular mechanism underlying this observation in detail, we examined autophagy deficiency in vitro using cultured cortical GABAergic interneurons derived from the embryonic medial ganglionic eminence (MGE). Similar to autophagy-deficient brain tissue, we observed the accumulation of GABARAPs and p62 as a result of Atg7 deletion (Fig. 4A). Consistent with results from in situ surface receptor biotinylation, we found a specific reduction of surface GABAA receptors in Atg7 cKO GABAergic interneurons without alterations in the total protein level, while AMPA and NMDA receptors remained unchanged in both total and surface fractions (Fig. 4B and fig. S4A). We further confirmed the reduced surface expression of GABAA receptors by nonpermeable immunofluorescence staining. Using an antibody specific to the extracellular domain of β2/3 subunit, we observed a reduction of surface-exposed GABAA receptors in autophagy-deficient GABAergic interneurons (Fig. 4C and fig. S4B). To directly assess whether the reduction of surface GABAA receptors in Atg7 cKO neurons affected their activity, we recorded spontaneous action potentials from Atg7 cKO and control neurons. Consistent with hyperactivity of GABAergic interneurons in the hippocampi of Dlx5-creERT2 Atg7 cKO mice (Fig. 3B), we observed a significant increase in the frequency of spontaneous action potentials (Fig. 4D, top, and fig. S4C) in cultured Atg7 cKO neurons without any changes in resting membrane potential (Fig. 4D, bottom).
Fig. 4. GABAA receptor trafficking was disrupted by alterations of GABARAPL2 due to autophagy deficiency.
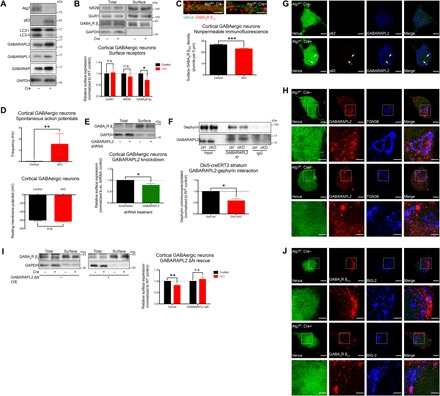
(A) Atg7 deletion in cultured cortical GABAergic interneurons (DIV15) derived from the MGEs of Atg7flox/flox embryos followed by infection with lentivirus expressing Venus reporter with or without Cre recombinase. Disrupted LC3 processing and accumulations of p62 and GABARAPs were observed as a result of autophagy deficiency induced by Atg7 deletion. (B) A significant reduction of surface GABAA receptors was observed in Atg7 cKO cultured cortical GABAergic interneurons, whereas NMDA and AMPA receptors were unchanged in both total and surface fractions. n = 4 independent experiments using distinct preparations of primary cultured neurons. (C) Immunofluorescence under nonpermeable conditions showed a significant reduction of surface GABAA receptors in Atg7 cKO cultured cortical GABAergic interneurons. Venus expression (green fluorescence) was used as an indicator of infected neurons. n = 30 cells analyzed per genotype. (D) A significant increase in the frequency of spontaneous action potentials fired by Atg7 cKO cultured cortical GABAergic interneurons was observed compared to controls (top). n > 25 cells per genotype; data shown for n = 10 and 9 cells for control and Atg7 cKO, respectively, from three distinct preparations of primary cultured neurons, which fired spontaneous action potentials. Resting membrane potential was not altered by Atg7 deletion in cultured cortical GABAergic interneurons (bottom). n > 30 cells per genotype from three distinct preparations of primary cultured neurons. (E) Gabarapl2 knockdown in WT cultured cortical GABAergic interneurons resulted in a significant reduction of surface GABAA receptors. n = 3 independent experiments using distinct preparations of primary cultured neurons. (F) GABARAPL2-gephyrin interaction was significantly reduced in the brains of Dlx5-creERT2 (striatum) Atg7 cKO mice (see fig. S4D). Quantifications shown represent the amount of gephyrin coimmunoprecipitated normalized by total gephyrin and amount of GABARAPL2 immunoprecipitated from individual samples. n = 3 independent experiments using tissue homogenates from distinct animals. (G) GABARAPL2 was mislocalized to p62+ aggregates formed in Atg7 cKO cultured cortical GABAergic interneurons in a similar manner as in affected neurons of Atg7 cKO brains. (H) GABARAPL2 was observed by immunofluorescence to localize to the TGN marked by TGN38 in controls, but this localization was reduced in Atg7 cKO cultured cortical GABAergic interneurons as the protein became mislocalized into foci. Venus signal was reduced in merge images to allow better visualization of signals from other channels. (I) GABARAPL2 ΔN overexpression (O/E) reversed the reduction of surface GABAA receptors observed in Atg7 cKO cultured cortical GABAergic interneurons. n = 5 independent experiments using distinct preparations of primary cultured neurons. (J) Intracellular accumulations of GABAA receptors were observed in BIG-2+ structures of Atg7 cKO cultured cortical GABAergic interneurons but not in controls. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, paired (B, E, F, and I) or unpaired (C) two-tailed Student’s t test or Kolmogorov-Smirnov test (D). Scale bars, 2 μm (C), 5 μm (G), 5 μm (H and J), and 2 μm (insets).
While GABARAP was reported to mediate GABAA receptor trafficking (21), whether GABARAPL2 and GABARAPL1 also have this function has not been addressed. In particular, GABARAPL2 was previously reported to show no interaction with GABAA receptors or gephyrin, a scaffolding protein involved in clustering and stabilization of GABAA receptors (24). We first examined whether GABARAPL2 may also be involved with GABAA receptor trafficking by short hairpin RNA (shRNA)–mediated knockdown. As shown in Fig. 4E, Gabarapl2 knockdown in wild-type (WT) GABAergic interneurons (fig. S4D) resulted in a significant reduction of surface GABAA receptors, indicating that it is also involved in GABAA receptor trafficking. Since GABARAPs, as Atg8 homologs, have the ability to bind to p62 (25), we hypothesized that the accumulated p62 in Atg7 cKO neurons may outcompete their normal physiological interactors, such as gephyrin, and contribute to the reduction in surface GABAA receptors. Because we found that GABARAPL2 may also be involved in GABAA receptor trafficking, we next performed immunoprecipitation experiments to examine the GABARAPL2-gephyrin interaction. We found that GABARAPL2 does interact with gephyrin in the brain but, more importantly, observed a significant reduction in the relative amount of coimmunoprecipitated gephyrin from Atg7 cKO brains (Fig. 4F and fig. S4E). Similar to Atg7 cKO brains, we observed by immunofluorescence in Atg7 cKO neurons that GABARAPL2 is mislocalized to p62+ aggregates (Fig. 4G and fig. S4F), with reduced localization to the trans-Golgi network (TGN, as marked by TGN38) compared to control neurons (Fig. 4H). Together, these data revealed that GABARAPL2 is involved in GABAA receptor trafficking, and this function was disrupted in autophagy-deficient neurons as it had mislocalized to p62+ aggregates, away from the TGN.
Because GABARAPL2 appears to have a physiological role in mediating GABAA receptor trafficking and this function may be compromised in autophagy-deficient neurons, we attempted to replenish the functional protein pool in Atg7 cKO neurons via GABARAPL2 overexpression and reverse the reduction of surface GABAA receptor expression. Because ectopically expressed GABARAPL2 was likely to interact with and be sequestered by p62+ aggregates in a manner similar to the endogenous protein, we instead used an N-terminal truncation mutant of GABARAPL2 lacking its first eight amino acids (GABARAPL2 ΔN), corresponding to a region in LC3 critical for interactions with p62 (fig. S4G) (25). After confirmation that the N-terminal deletion similarly disrupts the interaction between GABARAPL2 and p62 (fig. S4H), we attempted to rescue the reduced surface expression of GABAA receptors in Atg7 cKO neurons by overexpression of GABARAPL2 ΔN (fig. S4I). While Atg7 cKO neurons with Venus overexpression in parallel showed a significant reduction in GABAA receptors, GABARAPL2 ΔN overexpression resulted in no significant difference between Atg7 cKO and control neurons (Fig. 4I). Together, these results demonstrated that the loss of GABARAPL2 functions was causal for the reduced GABAA receptor trafficking in Atg7 cKO neurons.
Conversely, a number of other proteins including BIG-2 (ARFGEF2) and NSF are also known to be involved in GABAA receptor trafficking. We next examined whether autophagy deficiency led to the mistrafficking of GABAA receptors by specifically affecting GABARAPs or via a more general effect involving other proteins in the process as well. By both immunofluorescence and immunoblotting experiments, we observed that BIG-2 and NSF did not mislocalize to p62+ aggregates (fig. S5, A and B) or accumulate in Atg7 cKO neurons or brain homogenates like p62 and GABARAPs (fig. S5, C and D). When we examined the localization of GABAA receptors with BIG-2 (primarily in the TGN of both Atg7 cKO and control neurons), we observed large clusters of GABAA receptors colocalized with BIG-2+ structures in Atg7 cKO neurons not present in controls (Fig. 4J), consistent with our hypothesis that GABAA receptors may accumulate at the TGN when functions of GABARAPs are disrupted.
The findings described above suggest that the reduced surface expression of GABAA receptors observed in autophagy-deficient neurons is primarily due to a partial loss of function of GABARAPs as a result of formation of high–molecular weight species and/or sequestration by p62+ aggregates. We investigated whether endosomal/lysosomal trafficking of GABAA receptors may have been altered due to the disruption of autophagic signaling to examine its potential contributions to the reduction of surface GABAA receptors. Immunofluorescence analyses of the subcellular localization of GABAA receptors in various endosomal (Rab5+, Rab11+, or Rab7+ endosomes) and lysosomal (LAMP-1+) compartments in Atg7 cKO and control neurons suggested that only a minor population (10 to 15%) of GABAA receptors reside in these compartments and that this localization was not altered by autophagy deficiency (fig. S6, A to D). Thus, the reduction of surface GABAA receptors in Atg7 cKO neurons was not associated with any changes in their endocytic flux.
Excessive p62 accumulation in autophagy-deficient and mTOR-hyperactivated neurons results in reduced GABAA receptor surface expression due to mislocalized GABARAPs
We next asked whether the observed disruption of GABAA receptor trafficking is specific to autophagy-deficient conditions or is due to a general increase in p62 levels. First, by immunofluorescence, we found that p62 overexpression in WT neurons led to the formation of p62+ aggregates that also sequestered GABARAPs (Fig. 5A and fig. S7, A to C). Second, we observed a significant reduction in surface GABAA receptors in WT neurons with p62 overexpression (Fig. 5B). Third, if the reduced surface expression of GABAA receptors was caused by sequestration of GABARAPs by p62, a reduction of p62 in Atg7 cKO neurons would be expected to restore such deficits. To test this hypothesis, we performed surface receptor biotinylation experiments on Atg7 cKO and control neurons with or without Sqstm1 (p62) knockdown. Consistent with our prediction, Sqstm1 knockdown in Atg7 cKO neurons reversed the reduction of surface GABAA receptors to control levels (Fig. 5C). Together, this series of experiments suggests that the pathologic accumulation of p62 in Atg7 cKO neurons sequesters GABARAPs and disrupts the normal functions of GABARAPs, resulting in a reduction of surface GABAA receptor levels.
Fig. 5. Surface GABAA receptor expression depended on p62 levels.
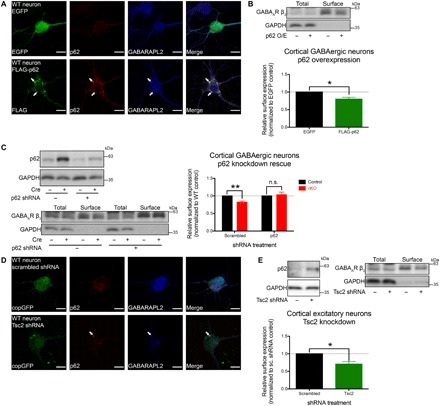
(A) p62 overexpression in WT cultured cortical GABAergic interneurons resulted in aggregates that recruited GABARAPL2 (see fig. S7, B and C). EGFP, enhanced green fluorescent protein. (B) p62 overexpression in WT cultured cortical GABAergic interneurons led to a significant reduction of surface GABAA receptors. n = 3 independent experiments using distinct preparations of primary cultured neurons. (C) Sqstm1 knockdown in Atg7 cKO cultured cortical GABAergic interneurons rescued the reduction in surface GABAA receptors. n = 5 independent experiments using distinct preparations of primary cultured neurons (D) mTOR hyperactivation via Tsc2 knockdown in WT cultured cortical excitatory neurons resulted in p62+ aggregates that sequestered GABARAPL2 (see fig. S7, E and F). (E) Tsc2 knockdown resulted in a significant reduction of surface GABAA receptors in WT cultured cortical excitatory neurons. n = 4 independent experiments using distinct preparations of primary cultured neurons. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, paired two-tailed Student’s t test. Scale bars, 15 μm (A and D).
While we have demonstrated that pathologic p62 accumulation in autophagy-deficient neurons affects GABA signaling via a disruption of GABARAP functions, we questioned whether this pathogenic mechanism may have even broader implications. It was previously reported that Tsc1 heterozygous excitatory neurons are hyperexcitable in a cell-autonomous manner due to a specific reduction in inhibitory input (26). As the Tsc1/2 complex is known to negatively regulate mTOR via the guanosine triphosphate (GTP)–binding protein Rheb and, in turn, promotes autophagic signaling, we asked whether the reported reduction of inhibitory input due to the derepression of mTOR activity (26) may have resulted from the mechanism described here. To address this question, we tested whether mTOR hyperactivation in cortical excitatory neurons due to a disruption of Tsc1/2 signaling may also lead to a reduction of surface GABAA receptor expression. To achieve mTOR hyperactivation, we used an shRNA-mediated knockdown approach against the mTOR negative regulator Tsc2 in WT cultured cortical excitatory neurons (fig. S7D). Consistent with previous reports (27), mTOR hyperactivation due to Tsc2 knockdown resulted in autophagy suppression, as indicated by increased p62 levels (fig. S7D). We revealed that similar to p62+ aggregates formed in Atg7 cKO neurons and WT neurons overexpressing p62, GABARAPs were recruited to the p62+ aggregates formed in WT neurons with derepressed mTOR activity (Fig. 5D and fig. S7, E and F). Surface receptor biotinylation experiments revealed that Tsc2 knockdown also resulted in a significant reduction of GABAA receptors in WT neurons (Fig. 5E). Together, our results suggest a general mechanism that autophagy suppression due to mTOR hyperactivation, similar to autophagy deficiency in the case of Atg7 deletion, disrupts the trafficking of GABAA receptors via the sequestration of GABARAPs by p62+ aggregates.
Increased aggregation of GABARAPs in postmortem ASD brain samples
Given that both Dlx5-creERT2 and CaMKII-cre Atg7 cKO mice exhibited social interaction deficits, we sought to determine whether GABARAPs are affected in ASD patients. To this end, we examined the expression and solubility of GABARAP, GABARAPL1, and GABARAPL2 in frozen BA40 brain samples from age- and gender-matched ASD patients and controls. Significant increases of all three proteins in the detergent-insoluble (0.2% sarkosyl) fraction were observed in ASD patients compared to controls (Fig. 6, A and B) without any significant differences in total tissue homogenates (Fig. 6, A and C). Furthermore, we observed a trend for increased insoluble p62 and a significant reduction in TSC2 expression in ASD patients (Fig. 6, D and E). TSC2 expression was negatively correlated with the insoluble amounts of not only p62 but also GABARAP family proteins (Fig. 6, F to I). These results are consistent with previous reports that autophagy may be suppressed in at least a subpopulation of ASD patients (4, 28), and that the function of GABARAPs in mediating GABAA receptor trafficking could also be compromised.
Fig. 6. Increased aggregation of GABARAPs in human ASD brain samples.
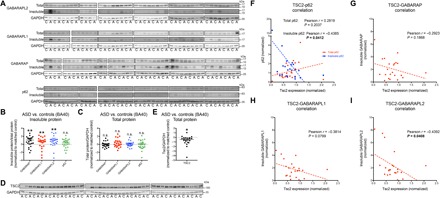
(A to C) No significant difference was observed in total expression of GABARAPs between ASD and matched (age, sex, and ethnicity) control brains (BA40); however, significant increases in the detergent-insoluble fraction were observed for all three proteins. Insoluble p62 also showed a trend for increase in ASD brain samples. n = 22 pairs of ASD and matched controls. (D and E) A significant reduction in TSC2 expression was detected in ASD brain samples. n = 22 pairs of ASD and matched controls. (F to I) Negative correlations were observed between insoluble GABARAPs, insoluble p62, and TSC2 expression. No correlation with age was observed. Data are presented as means ± SEM. *P < 0.05, **P < 0.01, paired two-tailed Student’s t test.
DISCUSSION
Previous studies have shown ~20 to 35% reductions in surface GABAA receptor expression in several disease-relevant brain regions in ASD patients (29), and these reductions have been demonstrated to cause significant deficits in inhibitory synaptic strength (30). However, the underlying molecular mechanisms therein have remained unclear. In this study, we have uncovered a previously unknown pathogenic role of excessive p62 accumulation in autophagy-deficient neurons, which, via the sequestration and mislocalization of GABARAPs, disrupts the trafficking of GABAA receptors to the cell surface. We have also demonstrated a novel physiological role for GABARAPL2 in GABAA receptor trafficking, which was previously reported to show no interaction with GABAA receptor γ2 subunit or gephyrin by yeast two-hybrid assay (24). However, using brain homogenates from Atg7 cKO and control mice, we demonstrated not only that GABARAPL2 interacted with gephyrin but also that the interaction was reduced in Atg7 cKO brains. Furthermore, the role of GABARAPL2 in GABAA receptor trafficking was confirmed by knockdown experiments in WT neurons. Thus, despite some sequence differences between the GABARAPs, all three proteins are likely to be involved in trafficking of GABAA receptors, which may explain why Gabarap deletion alone did not significantly affect surface GABAA receptor expression (31).
The present study indicates that the disruption of GABAA receptor trafficking due to the sequestration of GABARAPs may represent a general pathogenic mechanism that contributes to various neurodevelopmental and neuropsychiatric disorders associated with dysregulated mTOR. This convergence between GABA and mTOR signaling might offer a mechanistic explanation for long-standing unresolved observations that increased epileptic activity in human patients and mutant mice with mTOR hyperactivation can be reversed by pharmacologic mTOR inhibition. As we have demonstrated here, derepression of mTOR signaling due to reduced Tsc2 leads to a reduction of surface GABAA receptors expressed on the affected neurons, which may be the underlying mechanism for the reduced inhibitory inputs into Tsc1 mutant neurons described previously (26).
Consistent with a reduction of surface GABAA receptors in autophagy-deficient neurons, we observed the affected GABAergic interneurons to be hyperactive and increased GABA release onto their postsynaptic target cells as a result of disinhibition from reduced inhibitory inputs. Conversely, the same p62/GABARAP-dependent pathogenic mechanism affecting GABAA receptor trafficking was observed when Atg7 was deleted in excitatory neurons by CaMKII-cre and would consequently result in hyperactivity of those cells in a similar fashion, in line with previous observations that CaMKII-cre Atg7 cKO mice are more prone to develop spontaneous epileptic seizures (28). Our present findings, together with previous studies, suggest that there is an overall disruption in the excitatory-inhibitory (E-I) balance of autophagy-deficient neurons. Several recent studies have shown that disturbances in E-I balance are causal to social interaction deficits associated with various neurodevelopmental and neuropsychiatric disorders (15, 16). Thus, we surmise that the altered E-I balance, albeit in opposite directions by affecting either forebrain GABAergic interneurons or excitatory neurons, may have altered gamma oscillations or other types of network-level neurotransmission and caused the observed defects in social behaviors common to both Atg7 cKO mice we have examined in this study (32).
Conversely, we note that social behavioral deficits appear to be more severe for CaMKII-cre Atg7 cKO mice compared to Dlx5-cre Atg7 cKO animals, as demonstrated by the social interaction preference index in the three-chamber social interaction test. This differential effect on social approach and novelty may be due to a difference in the relative proportion of affected cells, as only about half of all cortical GABAergic interneurons were observed to form p62+ aggregates in Dlx5-creERT2 Atg7 cKO mice, while nearly all forebrain excitatory neurons affected by full or mPFC-specific CaMKII-cre Atg7 deletion contained p62+ aggregates (4, 17, 28). The difference is likely caused by a reduced recombination efficiency associated with age, as reported previously for Dlx5-creERT2 mice (33). In addition, we believe that other factors may have also contributed to this behavioral difference. First, overlapping but distinct regions of the forebrain were affected between Dlx5-creERT2 (cortex, hippocampus, and striatum) and CaMKII-cre (cortex and hippocampus). Second, the complex effect of deleting Atg7 from distinct subclasses of GABAergic interneurons [some of which function to suppress the activity of other inhibitory neurons, thereby producing an overall disinhibition effect (34–36)] may have resulted in distinct outcomes at the network and behavioral level. Thus, while we have uncovered that autophagy is required in forebrain GABAergic interneurons for normal regulation of social behaviors, future studies using subclass-specific Cre mouse lines will provide additional insights into their distinct contributions to the behavioral phenotypes reported in our current study on Dlx5-creERT2 Atg7 cKO mice.
The novel pathogenic mechanism we described here is distinct from that proposed previously concerning defective synaptic pruning in the developing brain as the primary cause of behavioral abnormalities due to autophagy deficiency (4, 12). While our model does not preclude that both mechanisms can operate independently to contribute to the observed behavioral deficits, our results suggest that synaptic pruning deficits may play a secondary role in the observed pathology. Although Atg7 deletion by Dlx5-creERT2 and mPFC-specific CaMKII-cre was initiated from adolescence, when most of the synaptic pruning had already occurred, both mice unexpectedly showed social deficits. Thus, our findings highlight the possibility that behavioral deficits arising from autophagy deficiency may not necessarily be related to specific defects during neurodevelopment per se, but rather a disruption in neurotransmission regardless of age. Local administration of the GABA antagonist bicuculline methiodide to WT animals after development to reduce GABA neurotransmission was previously shown to impair sociability (37). Together, our findings demonstrate that a disruption of GABA neurotransmission independent of defects during neurodevelopment may result in behavioral abnormalities including social deficits. While mutations contributing to neurodevelopmental and neuropsychiatric disorders in human patients may cause distinct neuronal deficits during and following neurodevelopment, their effects on mature neurons are rarely examined. It is important to note that several studies using mouse models of neurodevelopmental disorders have demonstrated that reexpression of the deleted genes after postnatal development can partially reverse neuronal and behavioral deficits (38, 39). These findings suggest not only that neurodevelopmental defects can be reversed but also that the impairment of neuronal functions and behaviors in adolescence and adulthood is contributed by the loss of functional protein of interest in mature neurons.
In addition to the p62-dependent pathogenic mechanism described here, our experiments using human brain samples indicated that insoluble GABARAPs levels are increased in at least a subpopulation of ASD patients. In line with this observation, several genetic studies have previously identified deletions or microdeletions affecting ATG7 (40) or GABARAP (41) in patients with ASD or ID, potentially implicating disrupted autophagy and altered GABARAPs as pathogenic factors. Furthermore, gene expression analyses using postmortem human brain samples of schizophrenics suggest possible deficits in autophagic signaling (42). Although it remains unclear whether GABARAPs are directly involved, it was recently demonstrated that the trafficking of GABAA receptor subunits is altered in schizophrenic patients (43). Thus, our study offers a potential molecular mechanism for these previous reports using human brain samples. A recent study on Ulk2 (44), an autophagic regulator previously shown to be partially duplicated in a single SZ patient, suggested an effect on GABA signaling and behavior in Ulk2 heterozygous mice, although the deficits were attributed to an ambiguous mechanism specific to excitatory neurons in the PFC. We have demonstrated that the p62-dependent sequestration of GABARAPs affects both GABAergic interneurons and excitatory neurons in a similar manner to produce common behavioral abnormalities. Furthermore, our findings using Tsc2 knockdown neurons provide broader implications for neurodevelopmental and neuropsychiatric disorders associated with mTOR dysregulation. Thus, this study provokes further studies on postmortem human brain samples for mTOR-related disorders to determine whether insoluble GABARAPs and disrupted autophagy may represent a more general defect affecting GABA neurotransmission and contribute to the manifestation of psychiatric symptoms.
METHODS
Animal care
All mice used were maintained on a C57BL/6J genetic background. Conditional deletion in forebrain GABAergic inhibitory neurons was achieved by genetic cross between Atg7flox/flox mice (45) and Dlx5-CreERT2 mice (33). Conditional Atg7 deletion in forebrain excitatory neurons by CaMKII-cre (transgenic) was as previously described by Nilsson and colleagues (17). Genotypes of all animals were confirmed using polymerase chain reaction (PCR) from genomic tail DNA samples, as described previously (17, 33). For Dlx5-creERT2 animals, Atg7flox/+, Dlx5creERT2/+ and Atg7flox/flox, Dlx5creERT2/+ were considered as Dlx5-control and cKO, respectively, to ensure equivalent gene dosages of Dlx5 in both genotypes (targeted mutation), whereas for CaMKII-cre animals, Atg7flox/flox, Cre− and Atg7flox/flox, Cre+ were considered as CaMKII-control and cKO, respectively. All experiments described were performed at 6 to 8 months after tamoxifen administration (Dlx5-creERT2) or 6 to 8 months of age (CaMKII-cre).
For CreERT2 induction, tamoxifen (2 μg per day) was administered by intraperitoneal injection during 8 and 10 weeks of age (four consecutive days during each week). Tamoxifen was first dissolved in ethanol (40 mg/ml) and subsequently in two volumes of canola oil. Ethanol was then removed in a centrifuge evaporator, resulting in a final tamoxifen concentration of 20 mg/ml. Dissolved tamoxifen was stored at −30°C until use and warmed to 37°C before injection.
Breeding of Dlx5-creERT2 animals was conducted using Atg7flox/+, Dlx5creERT2/+ males and Atg7flox/flox, Dlx5+/+ females or Atg7flox/flox, Dlx5+/+ males and Atg7flox/+, Dlx5creERT2/+ females. Neither parents were treated with tamoxifen. Breeding of CaMKII-cre animals was conducted using Atg7flox/flox, Cre+ males and Atg7flox/flox, Cre− females to avoid the possibility of abnormal rearing behaviors.
Preparation of primary neuronal cultures
Primary cortical GABAergic interneuron and excitatory neuron cultures were prepared from embryonic day 15 (E15) embryos (vaginal plug = E0) according to standard protocols. Briefly, Atg7flox/flox or WT embryos were dissected from pregnant dam, with cortex and MGE subdissected and pooled in ice-cold Hanks’ balanced salt solution (HBSS). Dissected tissues were washed three times with ice-cold HBSS, followed by trypsinization for 15 min at 37°C. Trypsinization was terminated by the addition of equal volume of 10% fetal bovine serum (FBS)–Dulbecco’s modified Eagle’s medium (DMEM), and dissected tissues were triturated gently. Cell suspension was filtered through a 40-μm cell strainer to remove undigested tissues and plated onto polyethylenimine-coated cell culture plates or glass coverslips. Primary neuronal cultures were maintained in MACS Neuro Medium (130-093-570, Miltenyi Biotec Inc.) supplemented with MACS NeuroBrew-21 (130-093-566, Miltenyi Biotec Inc.), 0.5 mM GlutaMAX (35050061, Thermo Fisher Scientific Inc.), and 1% penicillin/streptomycin mix (09367-34, Nacalai Tesque Inc.) in a 37°C humidified incubator. All experiments were performed after 15 days in vitro (DIV15), with lentiviral infection performed on DIV0 for Atg7 deletion by Cre recombinase expression or GABARAPL2 ΔN overexpression, DIV5 for p62 overexpression or Tsc2 knockdown, and DIV10 for GABARAPL2 knockdown. For biochemical experiments, cells were plated on coated six-well plates at a density of 2.0 × 106 cells per well and infected with 20 to 25 μl of lentivirus per well. For immunofluorescence and electrophysiology experiments, cells were plated on coated glass coverslips at a density of 4.0 × 104 cells per well, maintained in 24-well plates, and infected with 1 μl of lentivirus per well.
Preparation of lentivirus
Lentivirus was produced by transfection of CSII-CMV-MCS-IRES2-Venus, CSII-CMV-Cre-HA-IRES2-Venus, CSII-CMV-FLAG-GABARAPL2 ΔN-IRES2-mRFP, CSII-CMV-FLAG-GABARAPL2 ΔN-IRES2-Cre-HA, CSII-CMV-FLAG-p62 plasmids, or pSIH1-copGFP/mRFP plasmids with shRNA sequences (table S1), with pCAG-HIVgp and pCAG-VSV-G plasmids into Lenti-X 293 T cells grown on 10-cm dishes and maintained in 8 ml of DMEM supplemented with 10% FBS and 1% penicillin/streptomycin mix at 37°C. Cultured medium was collected 2 days after transfection and filtered through a 0.45-μm syringe filter, and then lentivirus was precipitated using Lenti-X Concentrator (631232, Clontech Laboratories Inc.) according to the manufacturer’s instructions. Precipitated lentivirus was resuspended in 250 μl of neuronal culture medium and kept at 4°C protected from light until use.
Preparation of AAV
AAV was produced by transfection of pAAV2-CaMKII promoter-MCS-IRES2-Venus or pAAV2-CaMKII promoter-CreHA-IRES2-Venus plasmids into AAV-293 cells with pAdΔF6 and pAAV2/1 plasmids. AAVs were purified by a discontinuous iodixanol gradient, as described previously (46). AAV titer was determined by quantitative PCR. Stereotaxic injections were bilaterally placed at 2.43 mm anterior to the bregma, 0.3 mm lateral to the sagittal suture, and 1.2 mm below the skull surface in 12-week-old CaMKII-cre Atg7flox/flox mice. One microliter of lentivirus (5.0 × 1012 genome copy/ml) was injected at a rate of 0.1 μl/min by a microinjector (Narishige).
Immunoblotting
Mouse brain regions of interest (i.e., striatum, cortex, and hippocampus) were homogenized in homogenization buffer containing 10 mM tris (pH 7.5), 500 mM NaCl, 10% sucrose, 1 mM EDTA, and a protease inhibitor mix (03969-34, Nacalai Tesque Inc.), followed by brief sonication. Tissue homogenates were centrifuged at 1500g for 5 min at 4°C to remove debris, with protein concentration measured using bicinchoninic acid (BCA) reagent (23227, Thermo Fisher Scientific Inc.) using bovine serum albumin (BSA) as standard. Ten micrograms of total proteins per lane was separated by SDS-PAGE in precast 5 to 20% gradient acrylamide gels (2331730, Atto Corp.) and transferred to polyvinylidene difluoride (PVDF) membrane by semidry electrophoretic transfer. PVDF membranes were blocked using 1:1 mix of tris-buffered saline with 0.05% Tween 20 (TBS-T) and Blocking One (03953-95, Nacalai Tesque Inc.) and then incubated with primary antibodies diluted using TBS-T/Blocking One overnight with gentle agitation at 4°C overnight. PVDF membranes were washed three times with TBS-T and incubated with secondary antibodies using TBS-T/Blocking One for 2 hours with gentle agitation at 4°C. After incubation with secondary antibodies, PVDF membranes were washed three times with TBS-T. Antibodies were detected using luminol-based chemiluminescence assay kits [Chemi-Lumi One L (07880) or Chemi-Lumi One Ultra (11644), Nacalai Tesque Inc.] and ImageQuant LAS 3000 Mini (GE Healthcare). No manipulations other than global contrast and brightness adjustments were performed on gel images in Adobe Photoshop. Primary antibodies used in this study are listed in table S2.
Immunoprecipitation
Animals were anesthetized using phenobarbital and perfused transcardially with phosphate-buffered saline (PBS). Brains were removed, with cortex, hippocampus, and striatum subdissected and then flash-frozen using liquid nitrogen until experiment. Brain samples were homogenized as described above, and protein concentration was measured by BCA assay. Tissue homogenate (500 μg of total protein) was mixed in immunoprecipitation (IP) buffer containing 50 mM tris (pH 7.4), 150 mM NaCl, 1 mM MgCl2, 1 mM EDTA, 1% NP-40, 0.25% SDS, and a protease inhibitor mix with either protein-specific antibody or whole immunoglobulin G (IgG) (both 1 μg) prebound to protein G magnetic beads (2 hours) at 4°C on a rotator for 2 hours. Protein G magnetic beads were then isolated by magnet and washed three times with IP buffer, followed by elution in SDS sample buffer, and boiled at 100°C for 10 min. Samples were analyzed by immunoblotting, as described above.
Native PAGE analysis
Tissue homogenates were prepared as described above, and protein concentration was measured by BCA assay. Ten micrograms of total protein was mixed with sample buffer and a final concentration of 1% sarkosyl or digitonin for analysis of GABARAPL2 and GABARAP, respectively. Samples were separated on NativePAGE 3-12% Bis-Tris gels and processed in the same manner as SDS-PAGE.
Sedimentation assay using postmortem human brain samples
Frozen postmortem human brain samples (BA40) were obtained via the National Institutes of Health (NIH) NeuroBioBank and stored at −80°C until use. Patient information provided by the NIH NeuroBioBank is listed in table S3. Frozen tissue samples were dissected to obtain approximately 50 mg of brain tissue, and tissue homogenates were generated as above for mouse brain tissues. Sarkosyl was added to each sample (500 μg of total proteins in 200 μl of total volume) for a 0.2% final concentration. The diluted protein samples were then incubated at 4°C for 30 min before ultracentrifugation at 100,000g for 1 hour at 4°C. Following ultracentrifugation, the supernatants were removed with the pellets resuspended and sonicated briefly in radioimmunoprecipitation assay (RIPA) buffer [50 mM tris (pH 7.4), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and a protease inhibitor mix]. Proteins from total tissue homogenates and detergent-insoluble pellets were separated by SDS-PAGE as described above. All paired samples (paired by NIH NeuroBioBank for age, sex, and ethnicity) were loaded side by side for comparison in the order listed in table S3.
Immunofluorescence
Animals were anesthetized using phenobarbital and perfused transcardially with PBS. Brains were removed and immersion-fixed by 4% paraformaldehyde (PFA) overnight without subdissection. PFA immersion-fixed brain tissues were processed for either cryostat or paraffin sectioning. For cryostat sectioning, brain tissues were cryoprotected in PBS containing 30% sucrose overnight at 4°C and frozen in optimal cutting temperature (OCT) compound (4583, Sakura Finetek Japan Co. Ltd.). Frozen sections were cut at a thickness of 10 μm at −22°C and stored at −30°C until use after drying or as 30-μm floating sections transferred to PBS. Paraffin-embedded brain tissues were cut at a thickness of 7 μm and stored at room temperature until use. For some antibodies, antigen retrieval (0.1% Triton X-100 in 10 mM citric acetate, pH 6.0, at 121°C for 3 min) before blocking was required for optimal staining. Tissue sections were blocked with blocking solution (5% goat serum and 0.3% Tween 20 in PBS) for 30 min at 4°C and incubated with primary antibodies diluted in blocking solution overnight at 4°C. Following overnight incubation, tissue sections were washed three times with blocking solution followed by incubation with secondary antibodies diluted in blocking solution for 2 hours at 4°C. Tissue sections were washed three times with PBS and mounted using VECTASHIELD mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (H-1200, Vector Laboratories) or ProLong Diamond Antifade Mountant (P36965, Thermo Fisher Scientific Inc.) for observation by confocal microscopy (Nikon C2 si and Leica TCS SP8). Base images were captured and exported as TIFF files, and figures were created using Adobe Photoshop. Deconvolution was performed for some images using Huygens Professional (Scientific Volume Imaging). No manipulations other than global contrast and brightness adjustments were performed on the images obtained.
Nonpermeable immunofluorescence
For nonpermeable immunofluorescence experiments, procedures were similar to regular immunofluorescence as above, with the exception that both blocking and antibody incubations were performed in PBS with 5% goat serum only. Ten individual neurons, each from three independent preparations of cortical GABAergic interneurons, were used to derive the data presented. Between two and five distinct 10-μm segments from each neuron were randomly chosen and quantified for the number of nonpermeable immunofluorescence puncta, as detected by an antibody recognizing the extracellular domain of GABAA receptor β//2/3 subunit.
Surface receptor biotinylation
Primary neuronal cultures were washed twice with ice-cold PBS containing 0.5 mM MgCl2 and 1 mM CaCl2 (PBS/Mg2+/Ca2+). Surface proteins were biotinylated using EZ-Link sulfo-NHS-SS-biotin (0.25 mg/ml) (21331, Thermo Fisher Scientific Inc.) for 20 min at 4°C with gentle agitation. Neurons were washed three times with ice-cold PBS/Mg2+/Ca2+ supplemented with 50 mM glycine and 0.5% BSA and twice with PBS/Mg2+/Ca2+ before lysis by the addition of RIPA buffer. Biotinylated proteins were enriched using streptavidin-conjugated magnetic beads (LSKMAGT10, EMD Millipore Co.) by mixing 50 μg of total proteins with 25 μl of magnetic beads. Streptavidin magnetic beads were washed three times using RIPA buffer after a 2-hour incubation period at 4°C on a rotator, and biotinylated proteins were eluted by heating the sample in SDS sample buffer for 10 min at 100°C. The eluted proteins were separated by SDS-PAGE and analyzed by immunoblotting, along with total lysate between treatment groups for comparison. In situ tissue surface protein biotinylation was performed using the same protocol except that freshly dissected tissue was biotinylated with sulfo-NHS-SS-biotin (1.0 mg/ml) for 45 min. The levels of surface biotinylated receptors were normalized by the levels of the receptor of interest and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in total lysate or homogenate and then compared across genotypes or treatment groups.
Semidenaturing detergent agarose gel electrophoresis and spike-in stable isotope labeling by amino acids in mammals
Animals were anesthetized at indicated ages using phenobarbital and perfused transcardially with PBS. Brains were removed, with cortex and hippocampus subdissected and flash-frozen using liquid nitrogen. Cortex and hippocampus were combined and homogenized in homogenization buffer, followed by brief sonication. Tissue homogenate was centrifuged at 1500g for 5 min to remove cell debris, and protein concentration was measured with BCA reagent using BSA as standard. Experimental tissue homogenate (1000 μg) was mixed with 500 μg of brain homogenate from mice fed with feed containing l-lysine (13C6) (MT-LYSC6-MB-PK, Cambridge Isotope Laboratories Inc.) for spike-in SILAM analysis. SDD-AGE sample buffer was added to the mixed sample and incubated at room temperature for 10 min before loading onto a 1.6% low melting point agarose gel (01146, Nacalai Tesque Inc.) containing 1% SDS in tris-acetate-EDTA (TAE) buffer. After electrophoresis in TAE buffer containing 0.1% SDS, the gel piece above 250 kDa was excised, melted, and digested by thermostable β-agarase (31107121, Nippon Gene Co.). Proteins in this “high–molecular weight” fraction were prepared for enzymatic digestion by using the filter-aided sample preparation, as described previously (47). The protein sample was digested overnight at 37°C using Lys-C, with digested peptides eluted from the filter by centrifugation. For MS, 2 μg of peptides was analyzed by liquid chromatography–electrospray ionization–MS/MS (LC-ESI-MS/MS).
Mass spectrometry
Protein analysis by LC-MS/MS
The enzymatically digested protein fragments were applied to a liquid chromatograph (EASY-nLC 1000; Thermo Fisher Scientific Inc.) coupled to a Q Exactive hybrid quadrupole-orbitrap mass spectrometer (Thermo Fisher Scientific Inc.) with a nanospray ion source in positive mode. The peptides derived from protein fragments were separated on a nano–high-performance liquid chromatography capillary column C18 (0.075 mm inside diameter × 150 mm length, 3 mm particle size, Nikkyo Technos Co. Ltd.). The mobile phase “A” was water with 0.1% formic acid, and the mobile phase “B” was acetonitrile with 0.1% formic acid. Two different slopes were used for a 60-min gradient at a flow rate of 300 ml/min: 5 to 35% B in 48 min, and then 35 to 65% B in 12 min. The Q Exactive MS was operated in top 10 data-dependent scan mode. The parameters of Q Exactive were as follows: spray voltage, 2.3 kV; capillary temperature, 275°C; mass range, 350 to 1800 m/z (mass/charge ratio); normalized collision energy, 28%. Raw data were acquired with Xcalibur software.
Protein identification
The MS and MS/MS data were searched against the Swiss-Prot database using Proteome Discoverer (Thermo Fisher Scientific Inc.) with the MASCOT search engine software (Matrix Science Inc.). The search parameters were as follows: enzyme, Lys-C protease static modifications, carbamidomethyl (Cys); dynamic modifications, oxidation (Met); precursor mass tolerance, ± 6 ppm (parts per million); fragment mass tolerance, ± 20 mDa; maximum missed cleavages, 1. The proteins were considered identified when their false discovery rates were less than 5%.
Behavioral tests
All behavioral tests were performed using male mice during the light cycle. For each genotype, at least two independent cohorts of mice were tested. All experiments and data analyses were performed in a blinded manner.
Open-field activity test
The experimental animal was placed in the center of an open-field apparatus (50 × 50 × 40 cm3) illuminated by a light-emitting diode and allowed to move freely for 15 min. Parameters including distance traveled, time spent in the central area of the open field (30% of total area), and rearing time were measured every minute. Data were recorded and analyzed by using TimeOFCR4 (O’Hara & Co. Ltd.).
Resident-intruder social interaction test
The experimental animal was acclimated to its cage before the experiment. A distinct 6-week-old WT male DBA/2CrSlc (Japan SLC Inc.) was introduced to the experimental animal for a 10- or 5-min period for Dlx5-creERT2 and CaMKII-cre mice, respectively. Interaction between the animals was recorded using a digital video camera, with total contact time and number of contacts initiated by the experimental animal quantified.
Three-chamber social interaction test
The animal was first acclimated to the test apparatus made of clear fiberglass and consisted of three chambers with small entrances in between them for 10 min with objects in the opposing chambers. Following acclimatization, an unfamiliar mouse (stranger) and an object were introduced into opposing chambers, with the experimental mouse allowed to explore for an additional 10 min to evaluate the social approach. Social novelty was examined by replacing the object with a second unfamiliar mouse into the opposite chamber, with the experimental mouse allowed to explore for 10 min. The first stranger mouse was considered to be the familiar mouse in the last stage of the test. Time spent in each chamber was quantified and presented. Distinct 6-week-old WT male C57BL/6JJmsSlc mice (Japan SLC Inc.) were used as unfamiliar mice in this experiment. Time spent in the left and right chambers were compared for each genotype separately to examine social approach (object versus mouse) and social novelty (familiar versus stranger mouse), as detailed previously by Silverman and colleagues (48). The social interaction preference index was also calculated to compare across genotypes. This was defined as a percentage of the time in a selected chamber (e.g., chamber with mouse) relative to the total time spent in both chambers (e.g., sum of time in chamber with mouse and time in chamber with object). Therefore, the percentage of time in the selected chamber will be 50 if there is no preference.
Elevated plus maze test
The animal was placed in the center of a plus-shaped maze with two opposing ends lined by clear fiberglass (closed arms) and the two other ends without any barriers (open arms). The animal was allowed to move freely on the elevated maze for a period of 5 min. Parameters such as total distance traveled, distance traveled and time spent in open and closed arms, and time in center were measured. Data were recorded and analyzed by TimeEP2 (O’Hara & Co. Ltd.).
Tail suspension test
The experimental animal was suspended by holding its tail onto a force measurement apparatus for a 10-min period. The duration of immobility was recorded every minute. Data were recorded and analyzed by using TimeFZ2 (O’Hara & Co. Ltd.).
Nesting behavior test
Nesting behavior was examined and quantified according to the scoring system, as described previously (49). Briefly, mice were placed into individual cages with corncob bedding and a nestlet before the dark phase. Photos of the cages were taken every 12 hours for 2 or 3 days, and a score at each time point was assigned according to the rating scale.
Electrophysiology
Electrophysiology experiments were performed as described previously (50), with details provided below.
Slice preparation
Mice were deeply anaesthetized with halothane (2-bromo-2-chloro-1,1,1-trifluoroethane, Takeda Pharmaceutical Co. Ltd.) or isoflurane [(2RS)-2-chloro-2-(difluoromethoxy)-1,1,1-trifluoroethane, Pfizer Inc.] and sacrificed by decapitation. The whole brain was quickly removed into an ice-cold cutting solution comprising 200 mM sucrose, 4 mM KCl, 1 mM NaH2PO4, 0.2 mM CaCl2, 10 mM MgCl2, 26.2 mM NaHCO3, 11 mM d-(+)-glucose, 0.1 mM l(+)-ascorbic acid, and 0.5 mM sodium pyruvate, saturated with 95% O2 and 5% CO2. Each brain hemisphere glued on a piece of agar block (4% in saline) was sliced along the longer axis of hippocampus at a thickness of 400 μm, using a microslicer (LinearSlicer Pro7, Dosaka EM Co. Ltd.). Hippocampi were dissected out of whole-brain slices and recovered in a submerged holding chamber filled with a physiological medium [artificial cerebrospinal fluid (aCSF)] comprising 119 mM NaCl, 2.5 mM KCl, 1 mM NaH2PO4, 2.5 mM CaCl2, 1.3 mM MgCl2, 26.2 mM NaHCO3, 11 mM d-(+)-glucose, 0.1 mM l(+)-ascorbic acid, and 0.5 mM sodium pyruvate, continuously bubbled with 95% O2 and 5% CO2. The slices, which had been equilibrated for more than 2 hours, were transferred to an immersion-type recording chamber and superfused with well-bubbled aCSF at a rate of 2.5 ml/min. All physiological experiments were carried out at 25°C.
Patch clamp (acute hippocampal slices)
Whole-cell voltage clamp configuration was obtained by a blind access to CA1 pyramidal cell layer with a patch pipette (4 to 7 megohms, Harvard Apparatus). The composition of the pipette solution for EPSCs recording was the following: 122.5 mM Cs gluconate, 17.5 mM CsCl, 10 mM Hepes, 0.2 mM EGTA, 8 mM NaCl, 2 mM Mg-ATP, and 0.3 mM Na3-GTP (pH 7.3; 290 to 300 mOsm). For IPSC recording, Cs gluconate was partially replaced with CsCl as follows: 40 mM Cs gluconate, 100 mM CsCl, 10 mM Hepes, 0.2 mM EGTA, 8 mM NaCl, 2 mM Mg-ATP, and 0.3 mM Na3-GTP (pH 7.3; 290 to 300 mOsm). AMPA receptor– and NMDA receptor–mediated currents (AMPAR- and NMDAR-EPSCs) were recorded at −70 and +40 mV, respectively, in the presence of picrotoxin (100 μM) and CGP55845 (1 μM). NMDAR-EPSCs were recorded after pharmacologic isolation with 2,3-dihydroxy-6-nitro-7-sulfamoyl-benzo[f]quinoxaline (NBQX) (20 μM). Schaffer collateral fibers were surgically cut and stimulated at 0.1 Hz. The amplitude of the EPSC was analyzed for the comparison between genotypes. IPSCs mediated by GABAA receptors were recorded under blockade of NBQX (10 μM) and D-AP5 (25 μM). mEPSC and mIPSC were recorded at −70 mV in the presence of tetrodotoxin (TTX; 0.5 μM). Amplitude and frequency for miniature currents were analyzed using Mini Analysis Program version 6 (Synaptosoft Inc.) for the comparison between genotypes. Series resistances were less than 30 megohms, and there was no significant difference in series resistance between experimental groups. The monitored synaptic responses were filtered at 1 kHz using an Axon 200B or 700B amplifier and digitized at 10 kHz using Clampex software of pClamp 9 or 10 suite (Molecular Devices LLC). Membrane potentials in whole-cell patch clamp were represented without being compensated for liquid junction potential.
Patch clamp (primary cultured cortical GABAergic neurons)
Whole-cell patch-clamp recordings were carried out using DIV14 to 21 primary cortical GABAergic interneuron cultures placed on the stage of a Zeiss Axio Examiner upright microscope using Axopatch 200B and Multiclamp amplifiers (Axon Instruments). The recording chamber was continuously perfused with an extracellular solution containing 130 mM NaCl, 2.5 mM KCl, 2.2 mM CaCl2, 1.5 mM MgCl2, 10 mM d-glucose, and 10 mM Hepes (pH 7.35; osmolarity adjusted to 290 mOsm). The micropipettes were made from borosilicate glass capillaries, with a resistance in the range of 3 to 5 megohms. The intracellular solution contained 100 mM k-gluconate, 17 mM KCl, 5 mM NaCl, 5 mM MgCl2, 10 mM Hepes, 0.5 mM EGTA, 4 mM ATPK2, and 0.5 mM GTP-Na (pH 7.3; osmolarity adjusted to 280 mOsm). All recordings were performed at room temperature (21° to 24°C). Data were recorded at 10 kHz and analyzed offline. All analysis was performed in Clampfit and Excel.
Drug
Drugs were purchased from the following sources: picrotoxin (28004-71, Nacalai Tesque Inc.; sc-202765, Santa Cruz Biotechnology Inc.), TTX (32775-51, Nacalai Tesque Inc.; 1078, Tocris Bioscience), CGP55845 (1248, Tocris Bioscience), D-AP5 (0106, Tocris Bioscience), and NBQX (0373, Tocris Bioscience).
Statistical analyses
The statistical significance of the data was examined by Student’s t test or Kolmogorov-Smirnov test for comparisons between two groups, or by one-way analysis of variance (ANOVA) with Bonferroni’s multiple comparison test for analyses of three or more groups using Microsoft Excel 16 or GraphPad Prism 6 unless otherwise indicated. Data are presented as means ± SEM. Results were considered statistically significant if P < 0.05.
Ethical considerations
All experimental procedures were in accordance with guidelines for animal care and human tissue sample use of RIKEN Center for Brain Science.
Supplementary Material
Acknowledgments
We thank members of the Laboratory for Protein Conformation Diseases, S. Itohara, and C. Yokoyama for discussion, as well as H. Fujimura and N. Takahashi for technical assistance. We thank M. Komatsu for providing the p62 expression plasmid. We are grateful to the Support Unit for Bio-Material Analysis in RIKEN CBS’s Research Resources Division for technical help with MS sample analysis, DNA sequencing, and FANTOM clone distribution. Postmortem human brain samples used in this study were obtained via the NIH NeuroBioBank, specifically the University of Maryland Brain and Tissue Bank, which provided the samples. Funding: This work was supported by Grant-in-Aid for Scientific Research on Innovative Areas from Japan Society for the Promotion of Science (26116044 to M.T. and JP18dm0107083 to T.Y.), RIKEN Pioneering Projects, Cellular Evolution (M.T.), the RIKEN Cellular Evolution project (M.T.), the Strategic Research Program for Brain Sciences from the Japan Agency for Medical Research and Development (JP18H05435 to T.Y.), RIKEN CBS/BSI internal funds (Y.G. and M.T.), the Postdoctoral Fellowship Program for Foreign Researchers from Japan Society for the Promotion of Science (23-01512 to K.K.H.), and the RIKEN Foreign Postdoctoral Researcher program (K.K.H.). Author contributions: K.K.H. and M.T. conceived experiments, wrote the manuscript, and secured funding. K.K.H., N.T., A.W., T.E.C., H.M., and Y.N.-M. performed the experiments. P.N. and T.C.S. provided Atg7 floxed mice, expertise, and feedback. R.E., Y.G., and T.Y. provided expertise and feedback. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaau8237/DC1
Fig. S1. Atg7 deletion in forebrain GABAergic neurons by Dlx5-creERT2.
Fig. S2. GABARAPs mislocalized to p62+ aggregates in affected neurons of Dlx5-creERT2 and CaMKII-cre Atg7 cKO mice.
Fig. S3. Autophagy deficiency led to a reduction of surface GABAA receptors but no change in excitatory signaling.
Fig. S4. Reduction of GABAA receptors in cortical GABAergic interneurons due to compromised functions of GABARAPL2 by autophagy deficiency.
Fig. S5. Levels and localizations of BIG-2 and NSF were not affected by autophagy deficiency.
Fig. S6. GABAA receptors are not substantially localized to endosomal and lysosomal compartments in cortical GABAergic interneurons and are not altered by autophagy deficiency.
Fig. S7. Reduced surface GABAA receptor expression by manipulation of p62 levels.
Table S1. shRNA sequences used in this study.
Table S2. Antibodies used in this study.
Table S3. Patient information for frozen postmortem human brain samples.
REFERENCE AND NOTES
- 1.Costa-Mattioli M., Monteggia L. M., mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat. Neurosci. 16, 1537–1543 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Gkogkas C. G., Khoutorsky A., Ran I., Rampakakis E., Nevarko T., Weatherill D. B., Vasuta C., Yee S., Truitt M., Dallaire P., Major F., Lasko P., Ruggero D., Nader K., Lacaille J.-C., Sonenberg N., Autism-related deficits via dysregulated eIF4E-dependent translational control. Nature 493, 371–377 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Santini E., Huynh T. N., MacAskill A., Carter A. G., Pierre P., Ruggero D., Kaphzan H., Klann E., Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493, 411–415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tang G., Gudsnuk K., Kuo S.-H., Cotrina M. L., Rosoklija G., Sosunov A., Sonders M. S., Kanter E., Castagna C., Yamamoto A., Yue Z., Arancio O., Peterson B. S., Champagne F., Dwork A. J., Goldman J., Sulzer D., Loss of mTOR-dependent macroautophagy causes autistic-like synaptic pruning deficits. Neuron 83, 1131–1143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coghlan S., Horder J., Inkster B., Mendez M. A., Murphy D. G., Nutt D. J., GABA system dysfunction in autism and related disorders: From synapse to symptoms. Neurosci. Biobehav. Rev. 36, 2044–2055 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fatemi S. H., Halt A. R., Stary J. M., Kanodia R., Schulz S. C., Realmuto G. R., Glutamic acid decarboxylase 65 and 67 kDa proteins are reduced in autistic parietal and cerebellar cortices. Biol. Psychiatry 52, 805–810 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Curley A. A., Arion D., Volk D. W., Asafu-Adjei J. K., Sampson A. R., Fish K. N., Lewis D. A., Cortical deficits of glutamic acid decarboxylase 67 expression in schizophrenia: Clinical, protein, and cell type-specific features. Am. J. Psychiatry 168, 921–929 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.The Wellcome Trust Case Control Consortium , Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Han S., Tai C., Jones C. J., Scheuer T., Catterall W. A., Enhancement of inhibitory neurotransmission by GABAA receptors having α2,3-subunits ameliorates behavioral deficits in a mouse model of autism. Neuron 81, 1282–1289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fazzari P., Paternain A. V., Valiente M., Pla R., Luján R., Lloyd K., Lerma J., Marín O., Rico B., Control of cortical GABA circuitry development by Nrg1 and ErbB4 signalling. Nature 464, 1376–1380 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Fu C., Cawthon B., Clinkscales W., Bruce A., Winzenburger P., Ess K. C., GABAergic interneuron development and function is modulated by the Tsc1 gene. Cereb. Cortex 22, 2111–2119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim H.-J., Cho M.-H., Shim W. H., Kim J. K., Jeon E.-Y., Kim D.-H., Yoon S.-Y., Deficient autophagy in microglia impairs synaptic pruning and causes social behavioral defects. Mol. Psychiatry 22, 1576–1584 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Komatsu M., Waguri S., Koike M., Sou Y.-s., Ueno T., Hara T., Mizushima N., Iwata J.-i., Ezaki J., Murata S., Hamazaki J., Nishito Y., Iemura S.-i., Natsume T., Yanagawa T., Uwayama J., Warabi E., Yoshida H., Ishii T., Kobayashi A., Yamamoto M., Yue Z., Uchiyama Y., Kominami E., Tanaka K., Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Yoshii S. R., Kuma A., Akashi T., Hara T., Yamamoto A., Kurikawa Y., Itakura E., Tsukamoto S., Shitara H., Eishi Y., Mizushima N., Systemic analysis of Atg5-null mice rescued from neonatal lethality by transgenic ATG5 expression in neurons. Dev. Cell 39, 116–130 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Wang F., Zhu J., Zhu H., Zhang Q., Lin Z., Hu H., Bidirectional control of social hierarchy by synaptic efficacy in medial prefrontal cortex. Science 334, 693–697 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Yizhar O., Fenno L. E., Prigge M., Schneider F., Davidson T. J., O’Shea D. J., Sohal V. S., Goshen I., Finkelstein J., Paz J. T., Stehfest K., Fudim R., Ramakrishnan C., Huguenard J. R., Hegemann P., Deisseroth K., Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature 477, 171–178 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nilsson P., Loganathan K., Sekiguchi M., Matsuba Y., Hui K., Tsubuki S., Tanaka M., Iwata N., Saito T., Saido T. C., Aβ secretion and plaque formation depend on autophagy. Cell Rep. 5, 61–69 (2013). [DOI] [PubMed] [Google Scholar]
- 18.Kryndushkin D. S., Alexandrov I. M., Ter-Avanesyan M. D., Kushnirov V. V., Yeast [PSI+] prion aggregates are formed by small Sup35 polymers fragmented by Hsp104. J. Biol. Chem. 278, 49636–49643 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Rayavarapu S., Coley W., Cakir E., Jahnke V., Takeda S., Aoki Y., Grodish-Dressman H., Jaiswal J. K., Hoffman E. P., Brown K. J., Hathout Y., Nagaraju K., Identification of disease specific pathways using in vivo SILAC proteomics in dystrophin deficient mdx mouse. Mol. Cell. Proteomics 12, 1061–1073 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weidberg H., Shvets E., Shpilka T., Shimron F., Shinder V., Elazar Z., LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 29, 1792–1802 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leil T. A., Chen Z.-W., Chang C.-S. S., Olsen R. W., GABAA receptor-associated protein traffics GABAA receptors to the plasma membrane in neurons. J. Neurosci. 24, 11429–11438 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lein E. S., Hawrylycz M. J., Ao N., Ayres M., Bensinger A., Bernard A., Boe A. F., Boguski M. S., Brockway K. S., Byrnes E. J., Chen L., Chen L., Chen T.-M., Chi Chin M., Chong J., Crook B. E., Czaplinska A., Dang C. N., Datta S., Dee N. R., Desaki A. L., Desta T., Diep E., Dolbeare T. A., Donelan M. J., Dong H.-W., Dougherty J. G., Duncan B. J., Ebbert A. J., Eichele G., Estin L. K., Faber C., Facer B. A., Fields R., Fischer S. R., Fliss T. P., Frensley C., Gates S. N., Glattfelder K. J., Halverson K. R., Hart M. R., Hohmann J. G., Howell M. P., Jeung D. P., Johnson R. A., Karr P. T., Kawal R., Kidney J. M., Knapik R. H., Kuan C. L., Lake J. H., Laramee A. R., Larsen K. D., Lau C., Lemon T. A., Liang A. J., Liu Y., Luong L. T., Michaels J., Morgan J. J., Morgan R. J., Mortrud M. T., Mosqueda N. F., Ng L. L., Ng R., Orta G. J., Overly C. C., Pak T. H., Parry S. E., Pathak S. D., Pearson O. C., Puchalski R. B., Riley Z. L., Rockett H. R., Rowland S. A., Royall J. J., Ruiz M. J., Sarno N. R., Schaffnit K., Shapovalova N. V., Sivisay T., Slaughterbeck C. R., Smith S. C., Smith K. A., Smith B. I., Sodt A. J., Stewart N. N., Stumpf K.-R., Sunkin S. M., Sutram M., Tam A., Teemer C. D., Thaller C., Thompson C. L., Varnam L. R., Visel A., Whitlock R. M., Wohnoutka P. E., Wolkey C. K., Wong V. Y., Wood M., Yaylaoglu M. B., Young R. C., Youngstrom B. L., Yuan X. F., Zhang B., Zwingman T. A., Jones A. R., Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 (2007). [DOI] [PubMed] [Google Scholar]
- 23.Wang H., Bedford F. K., Brandon N. J., Moss S. J., Olsen R. W., GABAA-receptor-associated protein links GABAA receptors and the cytoskeleton. Nature 397, 69–72 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Kneussel M., Haverkamp S., Fuhrmann J. C., Wang H., Wässle H., Olsen R. W., Betz H., The γ-aminobutyric acid type A receptor (GABAAR)-associated protein GABARAP interacts with gephyrin but is not involved in receptor anchoring at the synapse. Proc. Natl. Acad. Sci. U.S.A. 97, 8594–8599 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shvets E., Fass E., Scherz-Shouval R., Elazar Z., The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J. Cell Sci. 121, 2685–2695 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Bateup H. S., Johnson C. A., Denefrio C. L., Saulnier J. L., Kornacker K., Sabatini B. L., Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 78, 510–522 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Di Nardo A., Wertz M. H., Kwiatkowski E., Tsai P. T., Leech J. D., Greene-Colozzi E., Goto J., Dilsiz P., Talos D. M., Clish C. B., Kwiatkowski D. J., Sahin M., Neuronal Tsc1/2 complex controls autophagy through AMPK-dependent regulation of ULK1. Hum. Mol. Genet. 23, 3865–3874 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McMahon J., Huang X., Yang J., Komatsu M., Yue Z., Qian J., Zhu X., Huang Y., Impaired autophagy in neurons after disinhibition of mammalian target of rapamycin and its contribution to epileptogenesis. J. Neurosci. 32, 15704–15714 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oblak A. L., Gibbs T. T., Blatt G. J., Reduced GABAA receptors and benzodiazepine binding sites in the posterior cingulate cortex and fusiform gyrus in autism. Brain Res. 1380, 218–228 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ge Y., Kang Y., Cassidy R. M., Moon K.-M., Lewis R., Wong R. O. L., Foster L. J., Craig A. M., Clptm1 limits forward trafficking of GABAA receptors to scale inhibitory synaptic strength. Neuron 97, 596–610.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Sullivan G. A., Kneussel M., Elazar Z., Betz H., GABARAP is not essential for GABAA receptor targeting to the synapse. Eur. J. Neurosci. 22, 2644–2648 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Cao W., Lin S., Xia Q.-q., Du Y.-l., Yang Q., Zhang M.-y., Lu Y.-q., Xu J., Duan S.-m., Xia J., Feng G., Xu J., Luo J.-h., Gamma oscillation dysfunction in mPFC leads to social deficits in neuroligin 3 R451C knockin mice. Neuron 97, 1253–1260.e7 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Taniguchi H., He M., Wu P., Kim S., Paik R., Sugino K., Kvitsani D., Fu Y., Lu J., Lin Y., Miyoshi G., Shima Y., Fishell G., Nelson S. B., Huang Z. J., A resource of Cre driver lines for genetic targeting of GABAergic neurons in cerebral cortex. Neuron 71, 995–1013 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang W., Zhang L., Liang B., Schroeder D., Zhang Z.-w., Cox G. A., Li Y., Lin D.-T., Hyperactive somatostatin interneurons contribute to excitotoxicity in neurodegenerative disorders. Nat. Neurosci. 19, 557–559 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu H., Jeong H.-Y., Tremblay R., Rudy B., Neocortical somatostatin-expressing GABAergic interneurons disinhibit the thalamorecipient layer 4. Neuron 77, 155–167 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pfeffer C. K., Xue M., He M., Huang Z. J., Scanziani M., Inhibition of inhibition in visual cortex: The logic of connections between molecularly distinct interneurons. Nat. Neurosci. 16, 1068–1076 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paine T. A., Swedlow N., Swetschinski L., Decreasing GABA function within the medial prefrontal cortex or basolateral amygdala decreases sociability. Behav. Brain Res. 317, 542–552 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mei Y., Monteiro P., Zhou Y., Kim J.-A., Gao X., Fu Z., Feng G., Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530, 481–484 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sztainberg Y., Chen H.-m., Swann J. W., Hao S., Tang B., Wu Z., Tang J., Wan Y.-W., Liu Z., Rigo F., Zoghbi H. Y., Reversal of phenotypes in MECP2 duplication mice using genetic rescue or antisense oligonucleotides. Nature 528, 123–126 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poultney C. S., Goldberg A. P., Drapeau E., Kou Y., Harony-Nicolas H., Kajiwara Y., De Rubeis S., Durand S., Stevens C., Rehnström K., Palotie A., Daly M. J., Ma’ayan A., Fromer M., Buxbaum J. D., Identification of small exonic CNV from whole-exome sequence data and application to autism spectrum disorder. Am. J. Hum. Genet. 93, 607–619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carvalho C. M. B., Vasanth S., Shinawi M., Russell C., Ramocki M. B., Brown C. W., Graakjaer J., Skytte A.-B., Vianna-Morgante A. M., Krepischi A. C. V., Patel G. S., Immken L., Aleck K., Lim C., Cheung S. W., Rosenberg C., Katsanis N., Lupski J. R., Dosage changes of a segment at 17p13.1 lead to intellectual disability and microcephaly as a result of complex genetic interaction of multiple genes. Am. J. Hum. Genet. 95, 565–578 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Barnes M. R., Huxley-Jones J., Maycox P. R., Lennon M., Thornber A., Kelly F., Bates S., Taylor A., Reid J., Jones N., Schroeder J., Scorer C. A., Davies C., Hagan J. J., Kew J. N. C., Angelinetta C., Akbar T., Hirsch S., Mortimer A. M., Barnes T. R. E., de Belleroche J., Transcription and pathway analysis of the superior temporal cortex and anterior prefrontal cortex in schizophrenia. J. Neurosci. Res. 89, 1218–1227 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Mueller T. M., Remedies C. E., Haroutunian V., Meador-Woodruff J. H., Abnormal subcellular localization of GABAA receptor subunits in schizophrenia brain. Transl. Psychiatry 5, e612 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sumitomo A., Yukitake H., Hirai K., Horike K., Ueta K., Chung Y., Warabi E., Yanagawa T., Kitaoka S., Furuyashiki T., Narumiya S., Hirano T., Niwa M., Sibille E., Hikida T., Sakurai T., Ishizuka K., Sawa A., Tomoda T., Ulk2 controls cortical excitatory–inhibitory balance via autophagic regulation of p62 and GABAA receptor trafficking in pyramidal neurons. Hum. Mol. Genet. 27, 3165–3176 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Komatsu M., Waguri S., Chiba T., Murata S., Iwata J.-i., Tanida I., Ueno T., Koike M., Uchiyama Y., Kominami E., Tanaka K., Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 (2006). [DOI] [PubMed] [Google Scholar]
- 46.Zolotukhin S., Byrne B. J., Mason E., Zolotukhin I., Potter M., Chesnut K., Summerford C., Samulski R. J., Muzyczka N., Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 6, 973–985 (1999). [DOI] [PubMed] [Google Scholar]
- 47.Wiśniewski J. R., Zougman A., Nagaraj N., Mann M., Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Silverman J. L., Yang M., Lord C., Crawley J. N., Behavioural phenotyping assays for mouse models of autism. Nat. Rev. Neurosci. 11, 490–502 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deacon R. M. J., Assessing nest building in mice. Nat. Protoc. 1, 1117–1119 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Matsukawa H., Akiyoshi-Nishimura S., Zhang Q., Luján R., Yamaguchi K., Goto H., Yaguchi K., Hashikawa T., Sano C., Shigemoto R., Nakashiba T., Itohara S., Netrin-G/NGL complexes encode functional synaptic diversification. J. Neurosci. 34, 15779–15792 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/4/eaau8237/DC1
Fig. S1. Atg7 deletion in forebrain GABAergic neurons by Dlx5-creERT2.
Fig. S2. GABARAPs mislocalized to p62+ aggregates in affected neurons of Dlx5-creERT2 and CaMKII-cre Atg7 cKO mice.
Fig. S3. Autophagy deficiency led to a reduction of surface GABAA receptors but no change in excitatory signaling.
Fig. S4. Reduction of GABAA receptors in cortical GABAergic interneurons due to compromised functions of GABARAPL2 by autophagy deficiency.
Fig. S5. Levels and localizations of BIG-2 and NSF were not affected by autophagy deficiency.
Fig. S6. GABAA receptors are not substantially localized to endosomal and lysosomal compartments in cortical GABAergic interneurons and are not altered by autophagy deficiency.
Fig. S7. Reduced surface GABAA receptor expression by manipulation of p62 levels.
Table S1. shRNA sequences used in this study.
Table S2. Antibodies used in this study.
Table S3. Patient information for frozen postmortem human brain samples.