

Abstract
Background
Female pattern hair loss (FPHL), or androgenic alopecia, is the most common type of hair loss affecting women. It is characterised by progressive shortening of the duration of the growth phase of the hair with successive hair cycles, and progressive follicular miniaturisation with conversion of terminal to vellus hair follicles (terminal hairs are thicker and longer, while vellus hairs are soft, fine, and short). The frontal hair line may or may not be preserved. Hair loss can have a serious psychological impact on women.
Objectives
To determine the efficacy and safety of the available options for the treatment of female pattern hair loss in women.
Search methods
We updated our searches of the following databases to July 2015: the Cochrane Skin Group Specialised Register, CENTRAL in the Cochrane Library (2015, Issue 6), MEDLINE (from 1946), EMBASE (from 1974), PsycINFO (from 1872), AMED (from 1985), LILACS (from 1982), PubMed (from 1947), and Web of Science (from 1945). We also searched five trial registries and checked the reference lists of included and excluded studies.
Selection criteria
We included randomised controlled trials that assessed the efficacy of interventions for FPHL in women.
Data collection and analysis
Two review authors independently assessed trial quality, extracted data and carried out analyses.
Main results
We included 47 trials, with 5290 participants, of which 25 trials were new to this update. Only five trials were at 'low risk of bias', 26 were at 'unclear risk', and 16 were at 'high risk of bias'.
The included trials evaluated a wide range of interventions, and 17 studies evaluated minoxidil. Pooled data from six studies indicated that a greater proportion of participants (157/593) treated with minoxidil (2% and one study with 1%) reported a moderate to marked increase in their hair regrowth when compared with placebo (77/555) (risk ratio (RR) = 1.93, 95% confidence interval (CI) 1.51 to 2.47; moderate quality evidence). These results were confirmed by the investigator‐rated assessments in seven studies with 1181 participants (RR 2.35, 95% CI 1.68 to 3.28; moderate quality evidence). Only one study reported on quality of life (QoL) (260 participants), albeit inadequately (low quality evidence). There was an important increase of 13.18 in total hair count per cm² in the minoxidil group compared to the placebo group (95% CI 10.92 to 15.44; low quality evidence) in eight studies (1242 participants). There were 40/407 adverse events in the twice daily minoxidil 2% group versus 28/320 in the placebo group (RR 1.24, 95% CI 0.82 to 1.87; low quality evidence). There was also no statistically significant difference in adverse events between any of the individual concentrations against placebo.
Four studies (1006 participants) evaluated minoxidil 2% versus 5%. In one study, 25/57 participants in the minoxidil 2% group experienced moderate to greatly increased hair regrowth versus 22/56 in the 5% group (RR 1.12, 95% CI 0.72 to 1.73). In another study, 209 participants experienced no difference based on a visual analogue scale (P = 0.062; low quality evidence). The assessments of the investigators based on three studies (586 participants) were in agreement with these findings (moderate quality evidence). One study assessed QoL (209 participants) and reported limited data (low quality evidence). Four trials (1006 participants) did not show a difference in number of adverse events between the two concentrations (RR 1.02, 95% CI 0.91 to 1.20; low quality evidence). Both concentrations did not show a difference in increase in total hair count at end of study in three trials with 631 participants (mean difference (MD) −2.12, 95% CI −5.47 to 1.23; low quality evidence).
Three studies investigated finasteride 1 mg compared to placebo. In the finasteride group 30/67 participants experienced improvement compared to 33/70 in the placebo group (RR 0.95, 95% CI 0.66 to 1.37; low quality evidence). This was consistent with the investigators' assessments (RR 0.77, 95% CI 0.31 to 1.90; low quality evidence). QoL was not assessed. Only one study addressed adverse events (137 participants) (RR 1.03, 95% CI 0.45 to 2.34; low quality evidence). In two studies (219 participants) there was no clinically meaningful difference in change of hair count, whilst one study (12 participants) favoured finasteride (low quality evidence).
Two studies (141 participants) evaluated low‐level laser comb therapy compared to a sham device. According to the participants, the low‐level laser comb was not more effective than the sham device (RR 1.54, 95% CI 0.96 to 2.49; and RR 1.18, 95% CI 0.74 to 1.89; moderate quality evidence). However, there was a difference in favour of low‐level laser comb for change from baseline in hair count (MD 17.40, 95% CI 9.74 to 25.06; and MD 17.60, 95% CI 11.97 to 23.23; low quality evidence). These studies did not assess QoL and did not report adverse events per treatment arm and only in a generic way (low quality evidence). Low‐level laser therapy against sham comparisons in two separate studies also showed an increase in total hair count but with limited further data.
Single studies addressed the other comparisons and provided limited evidence of either the efficacy or safety of these interventions, or were unlikely to be examined in future trials.
Authors' conclusions
Although there was a predominance of included studies at unclear to high risk of bias, there was evidence to support the efficacy and safety of topical minoxidil in the treatment of FPHL (mainly moderate to low quality evidence). Furthermore, there was no difference in effect between the minoxidil 2% and 5% with the quality of evidence rated moderate to low for most outcomes. Finasteride was no more effective than placebo (low quality evidence). There were inconsistent results in the studies that evaluated laser devices (moderate to low quality evidence), but there was an improvement in total hair count measured from baseline.
Further randomised controlled trials of other widely‐used treatments, such as spironolactone, finasteride (different dosages), dutasteride, cyproterone acetate, and laser‐based therapy are needed.
Keywords: Female, Humans, Alopecia, Alopecia/therapy, Drug Administration Schedule, Finasteride, Finasteride/therapeutic use, Hair, Hair/drug effects, Hair/growth & development, Low‐Level Light Therapy, Minoxidil, Minoxidil/adverse effects, Minoxidil/therapeutic use, Randomized Controlled Trials as Topic
Plain language summary
Treatments for female pattern hair loss
Review question
Which treatments are effective and safe for female pattern hair loss (FPHL)?
Background
The most common type of hair loss in women is FPHL, also known as androgenic alopecia. Unlike men, women do not go bald, but have hair thinning predominantly over the top and front of the head. It can occur at any time, from puberty until later in life. However, it occurs more frequently in postmenopausal women.
The diagnosis is supported by careful history taking (including family history). Other causes should be considered; therefore, a clinical examination and laboratory tests may be necessary. FPHL can have a significant impact on self‐consciousness, and the damage to a woman's self‐confidence can affect her quality of life (QoL), leading to feelings of unattractiveness, shame, discomfort, emotional stress, and low self‐esteem.
Study characteristics
We examined the available evidence up to 7 July 2015. Forty‐seven studies, which included 5290 women, met the inclusion criteria of this Cochrane review. The mean age of participants in the studies varied from 27 to 57 years. We assessed over half of the included studies as at unclear risk of bias, 16 as high risk, and only five studies as low risk of bias. Funding was provided in 26 of the 47 studies, mainly by pharmaceutical companies.
Key results
This Cochrane review found that minoxidil is more effective than placebo. In six studies, the proportion of women that experienced at least moderate hair regrowth was twice as high in the minoxidil group compared to the placebo group. This was confirmed by the investigators assessments in seven studies. In eight studies, there was an important increase in total hair count per cm² in the minoxidil group compared to the placebo group. QoL was only assessed in one study and it was unclear from the data if there was an important improvement. The number of adverse events was similar for both groups. These were mostly mild, consisting of itch, skin irritation, dermatitis, and additional hair growth on areas other than the scalp.
Four studies compared minoxidil (2%) to minoxidil (5%), but none of the studies indicated any benefit of the higher concentration over the lower concentration. The number of adverse events did not differ between the two groups. Minoxidil should not be used in pregnant or lactating women.
Three studies compared finasteride to placebo. Finasteride is only approved in men for treatment of hair loss as well as for enlarged prostate. In one of the three studies the opinion of both the participants and investigators were evaluated but finasteride was shown to be no more effective than placebo. Hair count improved only in the finasteride group in a small study with 12 participants, but not in the other two studies (219 participants). Adverse events were only addressed in one study and these were similar in both groups. The investigators of these studies did not assess QoL.
Laser comb therapy did not appear to be more effective than sham therapy according to the participants in two studies with 141 participants. Nonetheless an important increase in hair growth was reported in both these studies. QoL was not addressed, and adverse events were not reported per intervention group, making these data less usable.
Individual studies investigated most of the other interventions and comparisons, and we could not make any firm conclusions about the efficacy or safety of these other interventions.
Although it is generally acknowledged that renewed hair shedding occurs relatively soon after discontinuation of treatment, none of the included studies reported data on the sustainability of the treatment effect, nor on the possible impact of hair regrowth, reflected by a decrease in time spent by women on hair styling or the use of wigs.
Quality of the evidence
We rated the quality of evidence for most outcomes as moderate or low. The lower quality of evidence was mainly caused by risk of bias in studies (e.g. no blinding) or a small sample size making the results less precise.
Summary of findings
Summary of findings for the main comparison. Minoxidil versus placebo.
| Minoxidil (1%, 2% and 5%) compared to placebo for female pattern hair loss (FPHL) | ||||||
| Patient or population: women with FPHL Intervention: minoxidil (1%, 2% and 5%) Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with minoxidil | |||||
| The proportion of participants with self‐rated clinically significant hair regrowth at the end of the study Assessed with: 3 to 7 point Likert scales Follow‐up: range 24 weeks to 32 weeks | Study population | RR 1.93 (1.51 to 2.47) | 1148 (6 RCTs) | ⊕⊕⊕⊝ moderate1 | 26.5% versus 13.9% experienced moderate to marked hair regrowth | |
| 139 per 1000 | 268 per 1000 (209 to 343) | |||||
| Change in quality of life Assessed with: VAS, 0 = negative, 50 = neutral and 100 = positive Scale from: 0 to 100 | — | — | — | 260 (1 RCT) | ⊕⊕⊝⊝ low2,3 | Standard deviations (SDs) were missing, therefore we could not calculate mean difference (MD). The VAS score was 54.4 for 5% minoxidil, 52.1 for 2% minoxidil, and 46.5 for placebo |
| Adverse events | Study population | RR 1.24 (0.82 to 1.87) | 727 (4 RCTs) | ⊕⊕⊝⊝ low4,5 | These are the data for the minoxidil 2% versus placebo. The RR for minoxidil 1% versus placebo was 1.12 (95% CI 0.61 to 2.06) and for minoxidil 5% versus placebo 2.05 (95% CI 0.96 to 4.37) | |
| 88 per 1000 | 109 per 1000 (72 to 164) | |||||
| Proportion of participants with investigator‐rated clinically significant hair regrowth at the end of the study Assessed with: 4 to 7 point Likert scales Follow‐up: range 24 weeks to 32 weeks | Study population | RR 2.35 (1.68 to 3.28) | 1181 (7 RCTs) | ⊕⊕⊕⊝ moderate6 | 18.4% versus 7.7% had at least moderate hair regrowth | |
| 77 per 1000 | 181 per 1000 (129 to 253) | |||||
| Change from baseline in total hair count Follow‐up: range 24 weeks to 32 weeks | The mean change from baseline in total hair count ranged from −3.25 to 20.4 hairs/cm² | The mean change from baseline in total hair count in the intervention group was 13.18 hairs/cm²higher (10.92 higher to 15.44 higher) | — | 1242 (8 RCTs)7 | ⊕⊕⊝⊝ low8,9 | The impact of excluding Price 1990 from this analysis had a marginal effect on the overall pooled result (RR 12.96, 95% 10.69 to 15.24) |
| Degree of hair shedding from baseline to the end of the study | Study population | Not estimable | 380 (3 RCTs) | ⊕⊝⊝⊝ very low10,11,12 | Pazoki‐Toroudi 2012: MD ‐37.85 hairs, 95% CI −54.22 to −21.48; P < 0.00001) in favour of minoxidil. Number of participants reporting decrease: Whiting 1992: RR 1.34, 95% CI 0.68 to 2.66; Tsuboi 2007: RR 1.13, 95% CI 0.95 to 1.33 | |
| Not pooled | Not pooled | |||||
| Cosmetic appearance of the hair or participant satisfaction Assessed with: VAS with 0 = no benefit, 50 = moderate benefit, and 100 = great benefit Scale from: 0 to 100 | — | — | — | 260 (1 RCT) | ⊕⊕⊝⊝ low2,3 | VAS score was 60.0 (27.6) for 5% minoxidil, 50.5 (35.5) for 2% minoxidil and 41.8 (29.9) for placebo |
| Change in quality (or pattern) of hair regrowth (e.g. thickness) Assessed with: Savin Female Density scale and target area hair width was 0.87 (1.315) mm/cm² | Not estimable | Not estimable | — | 372 (2 RCTs) | ⊕⊕⊝⊝ low2,13 | Investigators in Lucky 2004 reported statistically significant differences in hair density for both minoxidil concentrations compared to placebo. NCT01325350: MD 0.80, 95% CI 0.36 to 1.24; P = 0.004 |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: CI: confidence interval; RR: risk ratio; OR: odds ratio; GRADE: Grading of Recommendations Assessment, Development and Evaluation; FPHL: female pattern hair loss;RCT: randomised controlled trial; MD: mean difference; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1Downgraded 1 level for serious risk of bias as we judged 5/6 studies' key domains of risk of bias, i.e. sequence generation, allocation concealment, and blinding as 'unclear'. Tsuboi 2007 was at 'low risk'. 2Downgraded 1 level for serious imprecision due to small sample size, not meeting optimal information size. 3Downgraded 1 level for serious risk of bias due to high drop‐out rate and per‐protocol analysis. 4Downgraded 1 level for serious risk of bias as all 4 studies were at unclear to high risk of bias. 5Downgraded 1 level for serious imprecision due to low occurrence of events. 6Lucky 2004 had 3 treatment arms. 7Downgraded 1 level for serious risk of bias as we judged 6/7 studies' key domains of risk of bias, i.e. sequence generation, allocation concealment, and blinding as 'unclear'. Tsuboi 2007 was at 'low risk'. 8Downgraded 1 level for serious risk of bias as we judged 7/8 studies' key domains of risk of bias, i.e. sequence generation, allocation concealment, and blinding as 'unclear'. Tsuboi 2007 was at 'low risk'. 9Price 1990 was an outlier, with a small sample size (N = 8). There was possible publication bias, single participant with large treatment effect, and the result may be due to natural sampling variation. 10Downgraded 1 level for serious risk of bias as in 2/3 studies' key domains of risk of bias, i.e. sequence generation, allocation concealment, and blinding were judged 'unclear'. Tsuboi 2007 was at 'low risk'. 11Downgraded 1 level for serious inconsistency as only Pazoki‐Toroudi 2012 showed a difference in favour of minoxidil whilst the other 2 studies did not. 12Downgraded 1 level for serious imprecision due to wide CIs, and the optimal information size is not met. 13Downgraded 1 level for serious risk of bias due to attrition bias in Lucky 2004 and unclear risk of selection, performance, and detection bias in NCT01325350.
Summary of findings 2. Minoxidil 2% versus minoxidil 5%.
| Minoxidil 2% compared to minoxidil 5% for female pattern hair loss (FPHL) | ||||||
| Patient or population: women with FPHL Intervention: minoxidil 2% Comparison: minoxidil 5% | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with minoxidil 5% | Risk with minoxidil 2% | |||||
| Proportion of participants with self‐rated clinically significant hair regrowth Assessed with: 7 point Likert scale in Blume‐Peytavi 2011a, VAS scale 0‐100 in Lucky 2004 Follow‐up: range 24 weeks to 48 weeks | Study population | Not estimable | 322 (2 RCTs) | ⊕⊕⊝⊝ low1,2 | In Blume‐Peytavi 2011a moderate to greatly increased hair regrowth in minoxidil 2% group versus 5% RR 1.12, 95% CI 0.72 to 1.73. VAS in Lucky 2004 62.9 (16.7 standard deviation (SD)) versus 68.1 (17.9 SD), authors' reported P = 0.062 | |
| Not pooled | Not pooled | |||||
| Change in quality of life Assessed with: VAS Scale from: 0 to 100 | — | — | — | 209 (1 RCT) | ⊕⊕⊝⊝ low3,4 | Investigators reported "no statistically significant difference in impact of hair loss on quality of life between the two intervention groups" |
| Adverse events | Study population | RR 1.02 (0.91 to 1.20) | 1006 (4 RCTs) | ⊕⊕⊝⊝ low5,6 | Excluding Lucky 2004 from the analysis, reduced the degree of heterogeneity, with minimal impact on the pooled results | |
| 369 per 1000 | 376 per 1000 (335 to 442) | |||||
| Proportion of participants with investigator‐rated clinically significant hair regrowth Assessed with: 7 point Likert scales in Blume‐Peytavi 2011a and Sheng 2014. VAS scale in Lucky 2004 Follow‐up: range 24 weeks to 52 weeks | Study population | Not estimable | 586 (3 RCTs) | ⊕⊕⊕⊝ moderate7 | These outcomes in the 3 studies were in agreement with the participant assessments that there was no difference between the 2 concentrations of minoxidil | |
| Not pooled | Not pooled | |||||
| Change from baseline to study conclusion in total hair count Follow‐up: range 26 weeks to 52 weeks | The mean change from baseline to study conclusion in total hair count ranged from 23.7 to 31.9 hairs/cm² | The mean change from baseline to study conclusion in total hair count in the intervention group was 2.12 hairs/cm²lower (5.47 lower to 1.23 higher) | — | 631 (3 RCTs) | ⊕⊕⊝⊝ low8,9 | No difference between the 2 concentrations of minoxidil |
| Degree of hair shedding from baseline to the end of the study ‐ not measured | See comment | See comment | Not estimable | (0 studies) | — | This outcome was not assessed in any of the studies |
| Cosmetic appearance of the hair or participant satisfaction | Study population | Not estimable | 322 (2 RCTs) | ⊕⊕⊕⊝ moderate2 | 3/4 in Blume‐Peytavi 2011a were more satisfied after minoxidil 2% versus > 50% on 5%. In Lucky 2004 the scores on the VAS were 50.5 (SD 32.5) in the 2% group versus 60.0 (SD 27.6) in the 5% group | |
| Not pooled | Not pooled | |||||
| Change in quality (or pattern) of hair regrowth (e.g. thickness) | Not estimable | Not estimable | — | 322 (2 RCTs) | ⊕⊕⊕⊝ moderate10 | No differences in both studies between the treatment arms |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: CI: confidence interval; RR: risk ratio; OR: odds ratio; GRADE: Grading of Recommendations Assessment, Development and Evaluation; FPHL: female pattern hair loss;RCT: randomised controlled trial; MD: mean difference; VAS: visual analogue scale | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1Downgraded 1 level for serious risk of imprecision, due to wide CIs in Blume‐Peytavi 2011a. 2Downgraded 1 level for serious risk of bias due to the fact participants were not blinded in Blume‐Peytavi 2011a. 3Downgraded 1 level for serious risk of bias due to high drop‐out rate and per‐protocol analysis. 4Downgraded 1 level for serious imprecision due to small sample size, and not meeting the optimal information size. 5Downgraded 1 level for serious inconsistency, due to Lucky 2004 showing (as only study) a statistically significant difference in favour of minoxidil 2%. 6Downgraded 1 level for serious risk of bias as blinding was unclear in 3/4 studies. 7Downgraded 1 level for serious risk of bias, due to the fact the blinding of the investigators was not assured in Blume‐Peytavi 2011a and Sheng 2014. 8Downgraded 1 level for serious risk of imprecision, due to wide CIs, and the optimal information size is not met. 9Downgraded 1 level for serious risk of bias, due to the fact that outcome assessors in NCT01145625 were not blinded. 10Downgraded 1 level for serious imprecision, due to low sample sizes not meeting optimal information sizes.
Summary of findings 3. Finasteride versus placebo.
| Finasteride compared to placebo for female pattern hair loss (FPHL) | ||||||
| Patient or population: women with FPHL Intervention: finasteride Comparison: placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with finasteride | |||||
| The proportion of participants with self‐rated clinically significant hair regrowth at the end of the study Assessed with: Questionnaire (Barber 1998) Follow‐up: mean 12 months | Study population | RR 0.95 (0.66 to 1.37) | 137 (1 RCT) | ⊕⊕⊝⊝ low1,2 | Finasteride was no more effective than placebo | |
| 471 per 1000 | 448 per 1000 (311 to 646) | |||||
| Change in quality of life ‐ not measured | See comment | See comment | — | (0 studies) | — | This outcome was not assessed in any of the studies |
| Adverse events Follow‐up: mean 12 months | Study population | RR 1.03 (0.45 to 2.34) | 137 (1 RCT) | ⊕⊕⊝⊝ low1,2 | ||
| 786 per 1000 | 809 per 1000 (354 to 1000) | |||||
| Proportion of participants with investigator‐rated clinically significant hair regrowth Assessed with: 7‐point rating scale (‐3 = greatly decreased to +3 = greatly increased) Follow‐up: mean 12 months | Study population | RR 0.77 (0.31 to 1.90) | 137 (1 RCT) | ⊕⊕⊝⊝ low1,2 | Finasteride was no more effective than placebo, this is consistent with the assessments of the participants | |
| 186 per 1000 | 143 per 1000 (58 to 353) | |||||
| Change from baseline to study conclusion in total hair count | Study population | Not estimable | 231 (3 RCTs) | ⊕⊕⊝⊝ low3,4 | In two of the studies (Price 2000 and Whiting 1999) there was no clinically meaningful difference whilst in Keene 2011 there was a difference of around 17 hairs in favour of finasteride | |
| Not pooled | Not pooled | |||||
| Degree of hair shedding from baseline to the end of the study | — | — | 137 (1 RCT) | ⊕⊕⊝⊝ low1,5 | Although the investigators in Price 2000 provided no data, they reported that there was no statistically significant difference in the slowing down of hair loss between the two groups | |
| Cosmetic appearance of the hair or participant satisfaction Follow‐up: mean 12 months | Study population | RR 0.78 (0.40 to 1.53) | 137 (1 RCT) | ⊕⊕⊝⊝ low1,2 | ||
| 229 per 1000 | 178 per 1000 (91 to 350) | |||||
| Change in quality (or pattern) of hair regrowth (e.g. thickness) ‐ not measured | See comment | See comment | Not estimable | (0 studies) | — | This outcome was not assessed in any of the studies |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: CI: confidence interval; RR: risk ratio; OR: odds ratio; GRADE: Grading of Recommendations Assessment, Development and Evaluation; FPHL: female pattern hair loss;RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1Downgraded 1 level for serious risk of bias as we judged key domains of risk of bias, i.e. sequence generation, allocation concealment, and blinding as 'unclear'. 2Downgraded 1 level for serious imprecision due to wide CIs, small sample size, and not meeting the optimal information size. 3Downgraded 1 level for serious risk of bias as in 2 studies key domains of risk of bias, i.e. sequence generation, allocation concealment, and blinding were judged 'unclear'. Furthermore in Whiting 1999 the drop‐out ratio was > 30% and the analysis was per‐protocol. 4Downgraded 1 level for serious inconsistency as in Price 2000 and Whiting 1999 there was no meaningful difference, while Keene 2011 did show a difference of around 19 hairs in favour of finasteride. 5Downgraded 1 level for serious imprecision due to small sample size not meeting the optimal information size.
Summary of findings 4. Low‐level laser comb versus sham device.
| Low‐level laser comb compared to sham device for female pattern hair loss (FPHL) | ||||||
| Patient or population: women with FPHL Intervention: low‐level laser comb Comparison: sham device | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with sham device | Risk with low‐level laser comb | |||||
| Proportion of participants with self‐rated clinically significant hair regrowth Assessed with: 5 point Likert scale. However, minimally improved and improved data are combined by investigators Follow‐up: mean 26 weeks | Study population | Not estimable | 141 (2 RCTs) | ⊕⊕⊕⊝ moderate1,2 | The participants did not consider the low‐level laser comb to be more effective than the sham device (RR 1.54, 95% CI 0.96 to 2.49 for Jimenez 2014a and RR 1.18, 95% CI 0.74 to 1.89 for Jimenez 2014b | |
| Not pooled | Not pooled | |||||
| Change in quality of life ‐ not measured | — | — | — | (0 studies) | — | This outcome was not assessed in any of the studies |
| Adverse events Follow‐up: mean 26 weeks | Study population | Not estimable | 141 (2 RCTs) | ⊕⊕⊝⊝ low3,4 | The investigators reported that there were "laser comb‐related adverse events: dry skin (5.1%), pruritus (2.5%), scalp tenderness (1.3%), irritation (1.3%) & a warm sensation at the site (1.3%)" | |
| Not pooled | Not pooled | |||||
| Participants with investigator‐rated clinically significant hair regrowth ‐ not measured | See comment | See comment | Not estimable | (0 studies) | — | This outcome was not assessed in any of the studies |
| Change from baseline to study conclusion in total hair count Follow‐up: mean 26 weeks | Not estimable | Not estimable | — | 122 (2 RCTs) | ⊕⊕⊝⊝ low2,5 | There were statistically significant differences in favour of low‐level laser comb. MD 17.40 hairs, 95% CI 9.74 to 25.06 (Jimenez 2014a); MD 17.60 hairs, 95% CI 11.97 to 23.23 (Jimenez 2014b) |
| Degree of hair shedding from baseline to the end of the study ‐ not measured | — | — | — | (0 studies) | — | This outcome was not assessed in any of the studies |
| Cosmetic appearance of the hair or participant satisfaction Follow‐up: mean 26 weeks | Study population | Not estimable | 141 (2 RCTs) | ⊕⊕⊕⊝ moderate1,2 | Minimally improved or improved thickness or fullness of the hair: RR 1.46, 95% CI 0.86 to 2.49 for Jimenez 2014a and RR 1.33, 95% CI 0.76 to 2.33 for Jimenez 2014b | |
| Not pooled | Not pooled | |||||
| Change in quality (or pattern) of hair regrowth (e.g. thickness) ‐ not measured | See comment | See comment | Not estimable | (0 studies) | — | This outcome was not assessed in any of the studies |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). Abbreviations: CI: confidence interval; RR: risk ratio; OR: odds ratio; ITT: intention‐to‐treat; GRADE: Grading of Recommendations Assessment, Development and Evaluation; FPHL: female pattern hair loss;RCT: randomised controlled trial; MD: mean difference | ||||||
| GRADE Working Group grades of evidence High quality: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate quality: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low quality: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect. Very low quality: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect. | ||||||
1Not downgraded for risk of bias, due to the fact we recalculated the RR for the intention‐to‐treat (ITT) population, whilst in both studies (Jimenez 2014a; Jimenez 2014b) these were analysed as per‐protocol. 2Downgraded 1 level for serious imprecision, due to wide CIs for both studies, due to small sample size, and not meeting optimal information size. 3Downgraded 1 level for serious risk of bias, due to the fact there was a drop‐out rate of 19.2% in Jimenez 2014a and 9.5% in Jimenez 2014b, and data analysis was per‐protocol. Furthermore, the trials did not provide exact data of adverse events per treatment arm. 4Downgraded 1 level for imprecision, due to small sample size, and not meeting optimal information size. 5Downgraded 1 level for serious risk of bias, due to the fact there was a drop‐out rate of 19.2% in Jimenez 2014a and 9.5% in Jimenez 2014b, and data analysis was per‐protocol.
Background
We have listed unfamiliar terms in the 'Glossary of terms' (Table 5).
1. Glossary of terms.
| Term | Definition |
| Alopecia | Loss of hair from head or body |
| Anagen hair | Active, growing hair |
| Anagen phase | Active growth phase of hair follicles (2 to 7 years) |
| Catagen phase | Involution phase of the hair follicle |
| Ferritin | Iron‐containing proteins that are widely distributed in animals, plants, and micro‐organisms. Their major function is to store iron in a non‐toxic bioavailable form |
| Follicular miniaturisation | The follicles produce hair that is thinner and thinner, until they either stop producing hair or produce hair that is so fine it is barely noticeable |
| Hepatotoxic | Chemical‐driven liver damage |
| Hyperandrogenism | Condition characterised by excessive production/secretion of androgens |
| Hypertrichosis | Excessive (terminal and vellus) hair in non‐androgen dependent body sites; varies in people with different ethnic background without any pathological findings |
| Hirsutism | Excessive hairiness on women in those parts of the body where terminal hair does not normally occur or is minimal ‐ for example, beard or chest hair |
| Ludwig scale | Classification of female pattern hair loss (FPHL) stages I to III (minimal, moderate, intense) (Ludwig 1977) |
| 5‐alpha‐reductase | An enzyme that converts testosterone, the male sex hormone, into the more potent hormone, dihydrotestosterone |
| Sinclair scale | 5‐point scale (1 = normal, 5 = advanced hair loss) used to assess FPHL (Dinh 2007) |
| Telogen hair | Dormant, inactive hair |
| Telogen phase | Resting phase of the hair follicle (3 months) |
| Telogen effluvium | Massive hair loss resulting from the early entry of hairs into the telogen phase |
| Terminal hair | Thicker, longer, and pigmented hair |
| Tincture¹ | An alcoholic extract of a drug derived from a plant |
| Vasodilation¹ | Widening of the blood vessels |
| Vellus hair | Short, fine, light‐coloured, and barely noticeable hair that develops on most of a person's body from childhood |
Abbreviations: FPHL: female pattern hair loss. ¹Definition taken from: Martin 1998
Description of the condition
Definition and clinical features
Female pattern hair loss (FPHL) is an increasingly common clinical problem in women (Bienová 2005; Gan 2005; Hoffmann 2000), with over 21 million affected in the USA alone (Leavitt 2008). However, as the androgen‐dependent (male hormone) nature of the condition has not been clearly established, it has been proposed that in women the commonly‐used term 'androgenic alopecia' (AGA) should be replaced by 'female pattern hair loss' (FPHL) (Olsen 2001; Yip 2011). Both terms can be found in the literature and are often used interchangeably.
Hair growth occurs in cycles of various phases: anagen is the growth phase; catagen is the involuting or regressing phase; and telogen, the resting or quiescent phase (Dinh 2007). FPHL is characterised by the production of shorter and finer hairs due to progressive miniaturisation of hair follicles, so fine vellus hairs are produced instead of thicker terminal hairs (Trüeb 2002). Hair shedding can vary in intensity over time and from individual to individual. The onset of hair loss may precede menarche in young women or occur as late as the sixth decade of life (Olsen 2001; Olsen 2005; Yip 2011). Women who present to their doctor with a reduction in hair density often have thinning and widening of the area of hair loss on the central part of the scalp, which includes a breach of the frontal hairline. This sequence of symptoms is generally described as a 'Christmas tree' pattern (Blume‐Peytavi 2011b; Olsen 2008) (see Figure 1 and Figure 2). The frontal hairline may or may not be preserved; however, as with male pattern hair loss, the degree of loss of hair from the temples does not necessarily correlate with the presence or severity of mid‐frontal scalp hair loss (Sinclair 2005; Yip 2011).
1.
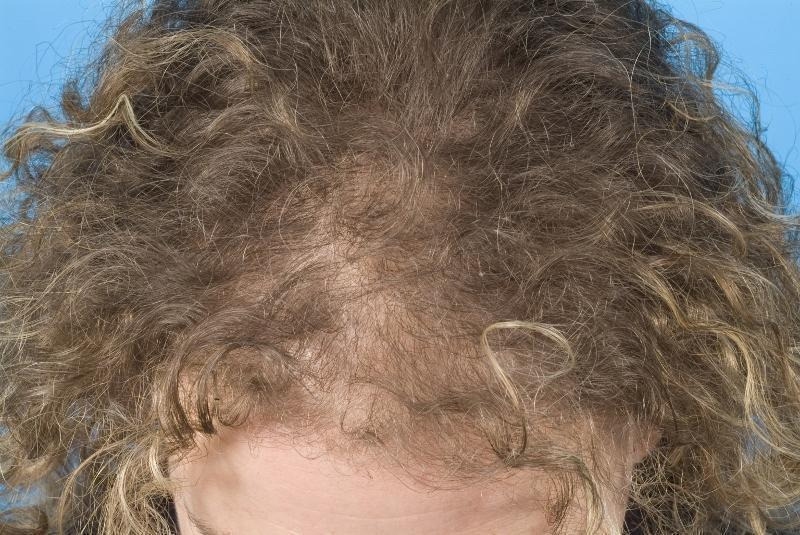
Stage 1 on Ludwig scale (mild female pattern hair loss). Copyright © 2011 Department of Dermatology, Leiden University Medical Centre: reproduced with permission.
2.
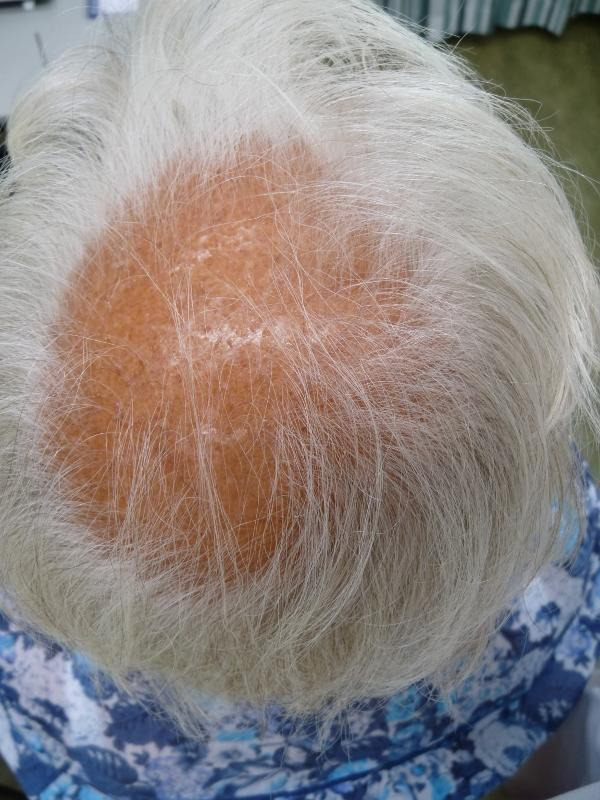
Stage 3 on Ludwig scale (severe female pattern hair loss). Copyright © 2011 Department of Dermatology, Leiden University Medical Centre: reproduced with permission.
The clinical evaluation and definition of the pattern of hair loss in women with FPHL has traditionally relied on the Ludwig (three‐point) classification (Ludwig 1977); however, a five‐point grading scale has been introduced more recently (Dinh 2007; Sinclair 2004). In all three Ludwig stages, there is hair loss in increasing severity on the front and top of the scalp, with relative preservation of the frontal hairline; the back and sides may or may not be involved (Ludwig 1977). In the five‐point mid‐frontal grading scale (visual analogue scale (VAS)), stage one represents the normal female hair pattern; stage two, mild hair loss; and the other stages, more severe hair loss (Gan 2005).
The diagnosis of FPHL in women is supported by a history of gradual thinning of the scalp hair over a period of months to years, which is characterised by a diffuse reduction of hair density over the crown and mid‐frontal scalp region (Atanaskova Mesinkovska 2013; Birch 2002; Dinh 2007; Messenger 2006). In women with FPHL, a family history may not be as clearly defined as in men with AGA (Olsen 2005), and although there is often a positive association between family history and FPHL or AGA, a negative history should not specifically preclude a diagnosis of FPHL (Blume‐Peytavi 2011b). A detailed history, including any family history of FPHL or AGA, and a thorough clinical examination should be undertaken, and this needs to include examination for features of hyperandrogenism (Atanaskova Mesinkovska 2013; Blume‐Peytavi 2011b; Dinh 2007). Clinical evaluation should include examination of the scalp skin, hair density, and facial (including eyebrows and eyelashes) and body hair, as well as signs of acne, hirsutism, or both. If the medical history suggests hyperandrogenism, an examination for cliteromegaly should also be undertaken (Blume‐Peytavi 2011b; Dinh 2007).
Women with menstrual cycle disturbances or those exhibiting marked acne, hirsutism, or both, should be investigated fully (Dinh 2007). The tests include the free androgen index test (FAI), and measurement of the levels of sex hormone‐binding globulin (SHBG) and prolactin (Blume‐Peytavi 2011b). More details on laboratory testing and excluding other causes of FPHL are found in the S1 guideline for diagnostic evaluation in androgenetic alopecia in men, women, and adolescents (Blume‐Peytavi 2011b). Polycystic ovary syndrome is the most common cause of hyperandrogenism, and although virilizing tumours may be implicated, they tend to be rare and characterised by recent onset and rapidly progressing severe hair loss, among other features (Blume‐Peytavi 2011b; Dinh 2007; Sinclair 2011; van Zuuren 2015). However, a lack of clinical evidence of hyperandrogenism does not necessarily rule out the presence of biochemical hyperandrogenism (Dinh 2007).
Loss of hair can also expose the scalp to sun damage and pose an increased risk of skin cancer (Yip 2011).
Other possible causes of hair loss must be considered; thus, the differential diagnosis of FPHL should include telogen effluvium (Sinclair 2005). Chronic telogen effluvium is defined as excessive shedding of hair for at least six months without a noticeable widening of the area of hair loss in the midfrontal scalp region (Dinh 2007). It can occur as a primary idiopathic event; secondary to thyroid disease, systemic lupus erythematosus, or end‐stage renal disease; or it may be due to certain drugs or nutritional deficiencies (Camacho‐Martínez 2009; Dinh 2007; Sinclair 2004). A sudden increase in hair loss is more consistent with a diagnosis of acute telogen effluvium, which may follow childbirth, severe systemic illness, or may be precipitated by certain medications (Dinh 2007). Alopecia areata diffusa is characterised by diffuse, patchy hair shedding in sharply defined areas (Leavitt 2008). It usually affects women over 40 years of age, many of whom are misdiagnosed as having telogen effluvium (Trüeb 2010).
Symptoms
Hair loss can have a significant negative psychological impact on both men and women (Dolte 2000; Hadshiew 2004; Levy 2013; Sinclair 2011). However, because hair has important social and psychological relevance to women, they tend to suffer more than men. A woman's hair is within her control to create her femininity, beauty, and sexuality. It is an "essential part of self‐identity (or 'body‐image')" (Cash 2001). For many people, hair is a "physical attribute that expresses individuality, and it is central to feelings of attractiveness or unattractiveness" (Cash 2001). In women it can be a source of concern in terms of feeling removed from what is considered a 'normal' female appearance (Cash 2001).
Studies have revealed that women with FPHL experience increased levels of self‐consciousness, feelings of unattractiveness, shame, discomfort, and emotional stress; some of which can lead to social withdrawal (Cash 2001; Reid 2011; van der Donk 1991). The Women's Androgenetic Alopecia Quality of Life Questionnaire (WAA‐QOL) is a validated instrument, which has been used to assess the impact of FPHL on quality of life (QoL) in women (Dolte 2000). However, recent research has indicated that the severity of a woman's hair loss is not a reliable predictor of QoL or perception of severity in hair loss (Reid 2011). People may often rate their hair loss more severely than a dermatologist (Biondo 2004); therefore, clinicians should be alert to the possible impact of a woman's perception of hair loss on her QoL.
Epidemiology and causes
Although FPHL is the most common type of hair loss in women, estimates of its true prevalence vary widely (Trüeb 2002). The fact that investigators tend to use different diagnostic criteria (and usually don’t describe them clearly) may contribute to the variation in prevalence figures. However, it is generally recognised that the prevalence of FPHL increases with age (Dinh 2007). The prevalence of FPHL among women aged between 20 to 29 years increases from 12% to approximately 60% for women aged 80 and over (Gan 2005; Yip 2011). It is reported to be lower in Asian women, and although prevalence is considered to be less in African women, very limited data are available to support this contention (Blume‐Peytavi 2011b).
Genetic predisposition as well as hormonal factors are involved in the cause of FPHL (Dinh 2007). Most women with FPHL do not have signs and symptoms of androgen excess, and systemic androgen levels are, in general, normal (Atanaskova Mesinkovska 2013; Blume‐Peytavi 2011b; Olsen 2005; Yip 2011). In these women, the local conversion of testosterone into dihydrotestosterone in the hair follicles is supposed to initiate terminal to vellus transformation (Price 2003).
A complex pattern of inheritance and a number of genes are considered to be associated with FPHL (Ali 2008; Atanaskova Mesinkovska 2013; El‐Samahy 2009; Richeti 2013; Sinclair 2011; Westberg 2001; Yip 2011). A variation of the androgen receptor gene has been identified in postmenopausal women, leading to increased serum levels of androgens (Ali 2008). In premenopausal women, certain variants of the androgen receptor gene and the oestrogen receptor beta gene seem to be involved (Westberg 2001). The role of oestrogens (female sex hormones) are probably of equal importance to that of androgens, but whether oestrogens have a stimulatory or an inhibitory effect is still a matter of debate (Yip 2011).
Low ferritin (iron‐containing proteins) levels have been suggested as possible contributory factors in FPHL (Kantor 2003), although a more recent study did not support this (Olsen 2010).
"The demonstration of thyroid hormone receptor expression in hair follicle cells indicates that thyroid hormone may affect hair growth directly" (Messenger 2000). In view of the "similarity between hair loss in hypothyroidism and FPHL, the implications may extend to other forms of hair loss besides that seen in thyroid deficiency" (Messenger 2000).
Description of the intervention
Current treatment options for women with FPHL are either topical (applied to the scalp) or systemic (taken orally).
Topical: minoxidil, aminexil, oestrogens, or alfatradiol (Atanaskova Mesinkovska 2013; Dinh 2007; Olsen 2005).
Systemic: hormonal contraception, cyproterone acetate, finasteride, spironolactone, and flutamide (Atanaskova Mesinkovska 2013; Bienová 2005; Dinh 2007; Olsen 2005).
Minoxidil is an antihypertensive vasodilator (Atanaskova Mesinkovska 2013). The topical formulation is available in three concentrations (1%, 2%, and 5%), with the 2% concentration applied once daily and the 5% either once or twice daily as the most commonly prescribed treatments (Atanaskova Mesinkovska 2013; Blumeyer 2011; Dinh 2007). Minoxidil as a 1% concentration is less frequently used, and in most countries, the 5% concentration is only registered for the treatment of AGA in men (Rogers 2008). Common side‐effects include scalp irritation and hypertrichosis on the cheeks and forehead (Rogers 2008). Minoxidil is contraindicated in pregnant and lactating women (Rogers 2008).
Aminexil is a derivative of minoxidil, which is available as a shampoo and in vials (Blumeyer 2011), but it has not been approved by either the U.S. Food and Drug Administration (FDA) or the European Medicines Agency (EMA).
Twice daily applications of 1% to 5% tincture of progesterone (a major hormone in the female menstrual cycle) can be used, but not in concentrations greater than 2%, or more than 2 mL per day, as it may cause menstrual irregularities. Topical oestrogens include fulvestrant twice daily or topical estradiol valerate 0.03% once daily (Gassmueller 2008; Georgala 2004). Alfatradiol is a 5‐alpha‐reductase inhibitor, but it is not freely available in many countries (Blume‐Peytavi 2007).
Systemic treatments that focus on antiandrogenic therapy include cyproterone acetate, spironolactone, finasteride, and flutamide (Atanaskova Mesinkovska 2013; Bienová 2005; Blumeyer 2011; Dinh 2007). As all of these treatments carry the risk of malformation in male foetuses, effective contraceptive advice should be provided to women of childbearing age (Blumeyer 2011; Olsen 2005).
Cyproterone acetate treatment is often used as a combination therapy of 2 mg in oral contraceptives plus cyproterone acetate up to 100 mg/day on days five to 15 of the menstrual cycle (Blumeyer 2011; Camacho‐Martínez 2009). Important side‐effects are depression, weight gain, breast tenderness, and loss of libido (Leavitt 2008).
Finasteride can be prescribed in varying doses, between 1 and 5 mg, and is generally well‐tolerated, but some women may experience breast tenderness and increased libido (Dinh 2007). Furthermore, it is not registered by the FDA or EMA for use in women.
Spironolactone (a diuretic, which is also used as an antiandrogen) in a dose of 50 to 200 mg/day, is one of the most frequently prescribed medications for FPHL in the USA (Dinh 2007; Leavitt 2008). Well known side‐effects are electrolyte imbalance, cycle disturbances, fatigue, drowsiness, urticaria, breast tenderness, hypotension, and haematological disturbances (Dinh 2007). Therefore, especially in the first weeks or months, blood pressure and electrolyte screening should be monitored (Dinh 2007).
Flutamide is not a first‐line drug due to its potentially severe hepatotoxic effects, but it has been used as a last‐resort treatment (Yazdabadi 2011).
There has also been a steadily increasing interest in a variety of low‐level laser treatment options over recent years (Jimenez 2014a; Jimenez 2014b; Lanzafame 2014).
Other considered treatments include food and herbal supplements, hair transplantation, and less frequently used medical treatments (e.g. dutasteride, cimetidine, tretinoin, and ketoconazole) (Atanaskova Mesinkovska 2013; Blumeyer 2011; Dinh 2007). Cosmetic aids are other important management options and include hairstyling techniques, hair replacements, camouflage products, and hair accessories (Dinh 2007; Inui 2013).
As soon as treatment is stopped, shedding of hair may resume within weeks Dinh 2007). Women with FPHL need thoughtful evaluation and management as well as reassurance (Dinh 2007; Price 2003), especially when current options for the treatment of this condition do not appear to demonstrate any long‐term or permanent benefits.
How the intervention might work
Strategies to improve scalp hair density include prolongation of anagen duration, reversal of terminal to vellus transformation, or generation of de novo hair induction from the inter‐follicular epidermis (Ellis 2002). Minoxidil has a direct effect on the proliferation and differentiation of follicular keratinocytes (epidermal cells), leading to a prolongation of the anagen phase (Rogers 2008). In essence, it encourages hair to move from the resting stage to the active growth stage (Rogers 2008). Potassium channels found in human hair follicles may play a role in this process, but the exact mechanism of action is still unclear (Shorter 2008). Aminexil, a derivative of minoxidil, has a similar mode of action (Blumeyer 2011).
Cyproterone acetate is a progestin (synthetic hormone) with antiandrogen action. It acts by blocking androgen receptors, which prevents androgens (male hormones) from binding to these receptors and suppresses luteinizing hormone (which in turn reduces testosterone levels) (Dinh 2007; Leavitt 2008). It is often combined with oral contraceptives, especially ethinyl estradiol 35 μg with 2 mg cyproterone acetate (Blumeyer 2011; Dinh 2007; Leavitt 2008). Spironolactone reduces the activity of 5‐alpha‐reductase, inhibits the biosynthesis of androgens, and has a direct antagonistic effect on androgen receptors (Dinh 2007; Leavitt 2008). Finasteride is a selective inhibitor of 5‐alpha‐reductase, which reduces the conversion of testosterone into dihydrotestosterone (DHT) (Bienová 2005; Rogers 2008), thereby lowering serum and scalp levels of DHT, while increasing scalp levels of testosterone. Low‐level laser treatment options may increase anagen hairs by stimulating epidermal stem cells in the hair follicle bulge (Atanaskova Mesinkovska 2013; Avci 2013).
Why it is important to do this review
Although a range of options are available for the treatment of FPHL, it is unclear how effective they are and if any have a long‐term beneficial effect. Many of these interventions may have important and undesirable side‐effects. This Cochrane review is needed to clarify the best approach to treating this condition, to provide reliable decision‐making information to clinicians and people with the condition about the benefits and harms of available treatments, and to be the basis for recommendations for future research.
Objectives
To determine the efficacy and safety of the available options for the treatment of female pattern hair loss in women.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs).
Types of participants
Any woman of any age who had been diagnosed with female pattern hair loss (FPHL) or androgenic alopecia (AGA) by a dermatologist or clinician. We included women with increased circulating androgens, whether due to physiological causes, polycystic ovary syndrome, or any other causes. However, we excluded women with androgen‐producing adrenal or ovary tumours.
Types of interventions
We considered any intervention for FPHL or AGA.
Types of outcome measures
Primary outcomes
The proportion of participants with self‐rated clinically significant hair regrowth at the end of the study.
Change in quality of life (QoL) using any validated and recognised generic or disease‐specific instrument, e.g. the Women's Androgenetic Alopecia Quality of Life Questionnaire (WAA‐QOL) (Dolte 2000).
Adverse effects: safety, tolerability, and any reported adverse events.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth at the end of the study.
Mean change in total hair count from baseline to the end of the study.
Degree of hair shedding from baseline to the end of the study.
Cosmetic appearance of the hair or participant satisfaction.
Change in quality (or pattern) of hair regrowth (e.g. thickness).
We defined 'clinically significant' outcomes as, for example, a single level change on the Sinclair scale (Messenger 2006; Sinclair 2004). We accepted all outcomes measures that used a recognised generic or validated scale (e.g. Ludwig scale, Sinclair scale).
Search methods for identification of studies
We aimed to identify all relevant RCTs regardless of language or publication status (published, unpublished, in press, or in progress).
Electronic searches
Jan Schoones (JS) updated the following searches to 7 July 2015. Prior searches were done by JS and Skin Group's Information Specialist.
The Cochrane Central Register of Controlled Trials (CENTRAL) in the Cochrane Library (2015, Issue 6) using the search strategy in Appendix 1.
MEDLINE via Ovid (from 1946) using the strategy in Appendix 2.
EMBASE via Ovid (from 1974) using the strategy in Appendix 3.
PsycInfo via Ovid (from 1872) using the strategy in Appendix 2.
PubMed (from 1947) using the strategy in Appendix 4.
Web of Science (from 1945) using the strategy in Appendix 5.
The Skin Group's Information Specialist updated the following searches to 15 July 2015.
The Cochrane Skin Group Specialised Register using the following terms: (androgen* AND alopecia) OR (female AND pattern AND hair AND loss) OR (female and baldness) or (female AND pattern AND alopecia).
AMED via Ovid (Allied and Complementary Medicine, from 1985) using the strategy in Appendix 6.
LILACS (Latin American and Caribbean Health Science Information database, from 1982) using the strategy in Appendix 7.
Trial registries
We searched the following trial registries on 24 July 2015 (Esther J van Zuuren (EvZ) and JS) using the search terms: androgenic alopecia, androgenetic alopecia, and FPHL.
The ISRCTN registry (www.controlled‐trials.com).
The US National Institutes of Health Ongoing Trials Register (www.clinicaltrials.gov).
The Australian New Zealand Clinical Trials Registry (www.anzctr.org.au).
The World Health Organization International Clinical Trials Registry platform (www.who.int/trialsearch).
The EU Clinical Trials Register (www.clinicaltrialsregister.eu/).
Searching other resources
Adverse effects
We did not perform a separate search for adverse effects of the target intervention. However, we did examine data on adverse effects from the included studies we identified.
References from published studies
We examined the bibliographies of the included and excluded studies for further references to potentially eligible RCTs.
Correspondence
We contacted the trial investigators and asked them to provide missing data or clarify study details (see Table 6).
2. Contact with investigators.
| Study ID | Response | Additional | Comment |
| Bezzola 2009 | No | No | There were no separate data for women. The primary outcome was diameter of hair, not one of the outcomes for this review. We excluded this study. |
| Blume‐Peytavi 2007 | Yes | Yes | IPD (individual patient data) were unavailable. We included this study. |
| Blume‐Peytavi 2011a | Yes | Yes | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study. We received a response on 16 November 2011: "The allocation concealment was performed using sequentially numbered, sealed, opaque envelopes, and kept by the project manager of the CRC." |
| Bureau 2003 | Yes | No | We could contact one of the investigators, who was unable to provide separate data on women. We included this study. |
| Carmina 2003 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study. |
| DeVillez 1994 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study. |
| Draelos 2005 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study. |
| Farella 1991 | No | No | The Italian Cochrane Centre translated and assessed this study, but it was a controlled clinical trial (CCT), so we excluded it. |
| Fischer 2004 | Yes | Yes | We received information that allowed a change in the assessment for several domains from unclear to low risk of bias. We included this study. |
| Gassmueller 2008 | Yes | Yes | We received information that allowed a change in assessment for several domains from unclear to low risk of bias. We included this study. |
| Georgala 2004 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study. |
| Gehring 2000 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study. |
| Golpour 2013 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. One review author, ZF, translated this study, which only included males, so we excluded this study. |
| Guerrero 2009 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. The data was mixed in terms of gender. We included this study. |
| Hong 2007 | Yes | Yes | The trial conduct was confirmed, i.e. sequence allocation/concealment and blinding. The data was mixed in terms of gender. We included this study. hongck@cau.ac.kr; dermahan@gmail.com |
| Jacobs 1993 | No | No | We were unable to contact the study investigators. We included this study. |
| Jimenez 2014a; Jimenez 2014b | Yes | Yes | The trial conduct was confirmed, i.e. sequence allocation/concealment and blinding. We included this study. dm@hairmax.com; Leonard Stillman. |
| Keene 2011 | Yes | Yes | The trial conduct was confirmed, i.e. sequence allocation/concealment and blinding. We included this study. drkeene@hairrestore.com; andyg@appliedbiology.com |
| Kim 2009 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. skin4u@korea.ac.kr. We included this study. |
| Kim 2013 | Yes | Yes | The trial conduct was confirmed, i.e. sequence allocation, but not for concealment and blinding. We did not receive separate data for women. We included this study. chhuh@snu.ac.kr; seokjong@knu.ac.kr |
| Lanzafame 2014 | Yes | Yes | The trial conduct was confirmed, i.e. sequence allocation, as well as losses to follow‐up. raymond.lanzafame@gmail.com. We included this study. |
| Le Floc'h 2015 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study. npiccardi@rd.loreal.com |
| Li 1996 | Yes | Yes | We excluded this study as it was a CCT. |
| Mazzarella 1997 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. The data was mixed in terms of gender. We included this study. |
| Minozzi 1997 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study. |
| Oura 2008 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study. |
| Pazoki‐Toroudi 2012 | No | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study. hpazooki@farabi.tums.ac.ir |
| Price 1990 | Yes | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We received no response from the investigator. We included this study. |
| Price 2000 | Yes | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We received no response from the investigator. We included this study. |
| Sheng 2014 | Yes | Yes | The trial conduct was confirmed, i.e. sequence allocation/concealment and blinding. We included this study. dxq93216@medmail.com.cn; felix_sheng@medmail.com.cn |
| Shin 2007 | Yes | Yes | The trial conduct was confirmed, i.e. sequence allocation/concealment. We included this study. hceun@snu.ac.kr; reslab@naver.com (Dr Shin Hyoseung); oskwon@snu.ac.kr |
| Sinclair 2002 | Yes | Yes | Information provided to us enabled a change from unclear to high risk of bias. We excluded this study. |
| Thom 2001/Thom 2006 | No | No | The data was mixed in terms of gender. We received no response from the investigator. We included this study. erling.thom@parexel.com; info@pharmamedico.com. |
| Tsuboi 2007 | Yes | Yes | The information we received allowed us to change the assessment for several domains from unclear to low risk of bias. We included this study. |
| Ukşal 1999 | Yes | No | The trial conduct was unconfirmed, i.e. sequence allocation/concealment and blinding. We included this study, but there were missing data. We received no response from the investigator. |
| Vexiau 2002 | Yes | Yes | We received information regarding the hyperandrogenic profile of the women. We included this study. |
| ACTRN12607000027415 | No | No | No further information regarding publication date. yazdaa27@gmail.com; Rod.SINCLAIR@svhm.org.au |
| EUCTR2013‐002740‐85‐ES | No | No | No further information regarding publication date nor if these are the same studies. eduardoanitua@eduardoanitua.com; virginia.cuadrado@bti‐implant.es; alopecia.ccdermatologico@gmail.com. Are EUCTR2013‐002740‐85‐ES and NCT01885676 the same studies, as everything is the same, except contact persons (same inclusion, same number of patients, same treatments, same sponsor) |
| NCT00175617 | No | No | No further information regarding publication date. andreas.finner@vch.ca |
| NCT00197379 | No | No | No further information regarding publication date. itoutai@hama‐med.ac.jp |
| NCT00418249 | No | No | No further information regarding publication date. rlurie@bezeqint.net |
| NCT01145625 | Yes | No | The trial conduct was confirmed, i.e. sequence allocation/concealment. LinkedIn Professor Kendall, and U Doshi and website Johnson & Johnson. Received from both Johnson & Johnson international as NL reply they forwarded it to responsible party, however, no response |
| NCT01189279 | No | No | No further information regarding publication date. clinicaltrials@allergan.com, international and Dutch website |
| NCT01226459 | No | No | No further information regarding publication date. |
| NCT01325350 | No | No | Allergan through website twice, no reply. |
| NCT01655108 | Yes | Yes | The trial conduct was confirmed, i.e. sequence allocation/concealment and blinding and received all possible data of the submitted paper. izelda@unb.br, Barbara Uzel: barbara.uzel@gmail.com |
| NCT01662089 | No | No | No further information regarding publication date. rattapongthuangtong@yahoo.com |
| NCT01686295 | No | No | No further information regarding publication date. candresen@tklresearch.com |
| NCT01885676 | No | No | No further information regarding publication date. eduardoanitua@eduardoanitua.com |
| NCT01900041 | Yes | Yes | The trial conduct was confirmed, i.e. sequence allocation and flow chart. We included this study. Larissa.Cheredeeva@merz.ru |
| NCT01967277 | Yes | Yes | The trial conduct was confirmed, i.e. concealment and blinding (but not sequence generation). We included this study. pschnoor@capillus.com, info@capillus.com |
| NCT02074943 | Yes | No | There was no further information regarding publication date, and the study is not yet finished. Jerry.Shapiro@vch.ca |
Abbreviations: CCT: controlled clinical trial (quasi‐randomised).
Data collection and analysis
We followed the previously published protocol (Cusmanich 2009) for this Cochrane review. This is the first update of the original published Cochrane review (van Zuuren 2012).
Selection of studies
Two review authors (EvZ and Zbys Fedorowicz (ZF)) assessed the titles and abstracts identified from the searches. We only included RCTs that evaluated FPHL in women in this Cochrane review. The two review authors independently assessed each included study to determine whether the predefined selection criteria were met, and they resolved any differences of opinion through discussion within the review team. We have listed the excluded studies and the reasons for their exclusion in the 'Characteristics of excluded studies' section of the review.
Data extraction and management
Two review authors (EvZ and ZF) extracted data using a previously developed data extraction form, and resolved any disagreements on data extraction by consensus. We contacted the trial authors and asked them to provide missing data where possible. Two review authors (EvZ and ZF) checked and entered the data into Review Manager (RevMan) (Review Manager (RevMan) 2014).
Assessment of risk of bias in included studies
Two review authors (EvZ and ZF) independently assessed the risk of bias in the included studies following the domain‐based evaluation described in Chapter 8 of theCochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). They compared the evaluations, and resolved any inconsistencies by discussion.
We rated the following domains separately for each of the included studies as either 'low risk of bias', 'high risk of bias', or 'unclear' if the risk of bias was uncertain or unknown.
Whether the allocation sequence was adequately generated ('sequence generation').
Whether the allocation was adequately concealed ('allocation concealment').
Whether knowledge of the allocated interventions was adequately prevented during the study ('blinding').
Whether incomplete outcome data were adequately addressed.
Whether reports of the study were free of suggestion of selective outcome reporting.
Whether the study was apparently free of other sources of bias that could put it at high risk of bias, e.g. baseline imbalance.
We have reported these assessments in the 'Risk of bias' table for each individual study in the 'Characteristics of included studies' section of the review.
We also categorised and reported the overall risk of bias of each of the included studies according to the following.
Low risk of bias (plausible bias unlikely to seriously alter the results) if all criteria were met.
Unclear risk of bias (plausible bias that raises some doubt about the results) if one or more criteria were assessed as unclear.
High risk of bias (plausible bias that seriously weakens confidence in the results) if one or more criteria were not met.
We reported these assessments in the 'Risk of bias in included studies' section.
Measures of treatment effect
We presented continuous outcomes on the original scale as reported in each individual study. In future updates, if included studies report similar outcomes using different scales, we will standardise these by dividing the estimated coefficient by its standard deviation (SD), to allow us to make comparisons between scales.
We presented dichotomous outcomes data as risk ratios (RR). We reported all outcome data with their associated 95% confidence intervals (CIs) and analysed them in RevMan (Review Manager (RevMan) 2014) using the Mantel‐Haenszel test, unless we stated otherwise.
Unit of analysis issues
Cross‐over studies
We included one cross‐over study (Blume‐Peytavi 2007), but as this study did not report any wash‐out period, we only included data from the first treatment period.
Multi‐armed studies
For continuous outcomes, we included participants from the control arms of within multi‐arm studies approximately equally in the pair‐wise comparisons with the active intervention arms. The mean and SD summary statistics for the placebo participants remained unchanged.
Dealing with missing data
We successfully contacted the investigators of several included trials (see Table 6). We re‐analysed the data according to a treatment by allocation principle, whenever possible, and according to Section 16.2.2 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). If study authors did not report data and had conducted a per‐protocol analysis, we inspected the degree of imbalance in the dropout rate between the trial arms to determine the potential impact of bias. In the absence of a treatment by allocation population, we used an available case population and reported this accordingly.
Assessment of heterogeneity
We assessed clinical heterogeneity by examining the characteristics of the studies, the similarity between the types of participants, and the interventions. Also, we determined the degree of heterogeneity between the studies using the I² statistic. We reported heterogeneity as important if it was at least moderate to substantial by an I² statistic value of greater than 60% (Higgins 2011). If we could explain this by clinical reasoning and could make a coherent argument for combining the studies, we entered these into a meta‐analysis. In cases where we could not adequately explain the heterogeneity, we did not pool the data.
The clinical diversity between the studies included in this review, as well as the limited number of studies that we could combine for each intervention, only allowed us to make assessments of heterogeneity between the studies for two of the comparisons.
Assessment of reporting biases
We performed assessments of reporting bias following the recommendations on testing for funnel plot asymmetry (Egger 1997), as described in Section 10.4.3.1 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011), for primary and secondary outcomes where we performed meta‐analysis (at least three studies needed). We only presented funnel plots where there was some evidence of asymmetry in the plots. We explored the possible sources of asymmetry with an additional sensitivity analysis.
Data synthesis
Two review authors (EvZ and ZF) analysed the data in RevMan (Review Manager (RevMan) 2014) and reported them in accordance with the advice in Chapter 9 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We only performed a meta‐analysis if we were able to identify an adequate number of studies (n ≥ 3) that investigated similar interventions and reported data that exhibited not less than moderate heterogeneity (Treadwell 2006). We used a fixed‐effect model to pool the data into a meta‐analysis, and we fitted a random‐effects model as part of a sensitivity analysis to explore the degree of heterogeneity between studies.
Subgroup analysis and investigation of heterogeneity
We analysed the different concentrations of the interventions as subgroups by comparing the RRs and 95% CI. If we observed differences in effect estimates between the subgroups, we analysed these separately.
In future updates of this Cochrane review and if a sufficient number of studies examining similar comparisons are available, we will consider if any further subgroup analyses are warranted, for example, age groups, pre‐ and postmenopausal, ethnic background, and the presence of hyperandrogenism.
Sensitivity analysis
We performed sensitivity analyses to assess the robustness of the results of this review; thus, we repeated all fixed‐effect meta‐analyses using random‐effects models. We conducted an additional sensitivity analysis, which excluded one study (Price 1990) with suspected reporting bias in comparison one, i.e. minoxidil versus placebo, and a further sensitivity analysis in comparison two i.e. minoxidil (2%) versus minoxidil (5%), which excluded one study, which had twice the number of adverse events in the 5% treatment arm (Lucky 2004).
Results
Description of studies
Results of the search
Our earlier searches retrieved 334 references to studies plus 10 references from other sources. The updated searches provided 563 references to studies in addition to 30 ongoing studies (of which seven appear to be completed and had data available on clinicaltrials.gov and we could include, and 11 were terminated or completed but no data available which are listed under Characteristics of studies awaiting classification) (see Figure 3). The total number of references retrieved for both sets of searches was therefore 937. There were 923 records after removal of duplicates. After examination of the titles and abstracts, we excluded 813 of these references from the review. We obtained full‐text copies of the remaining 110 records for further evaluation. We translated several studies that were not published in the English language — two in Chinese (Li 1996; Sheng 2014), two in Farsi (Enshaieh 2005; Golpour 2013), three in Korean (Hong 2007; Kim 2009; Shin 2007), one in Spanish (Guerrero 2009), one in German (Gehring 2000), and three in Italian (Farella 1991; Minozzi 1997; Policarpi 1993) — prior to assessment for eligibility.
3.
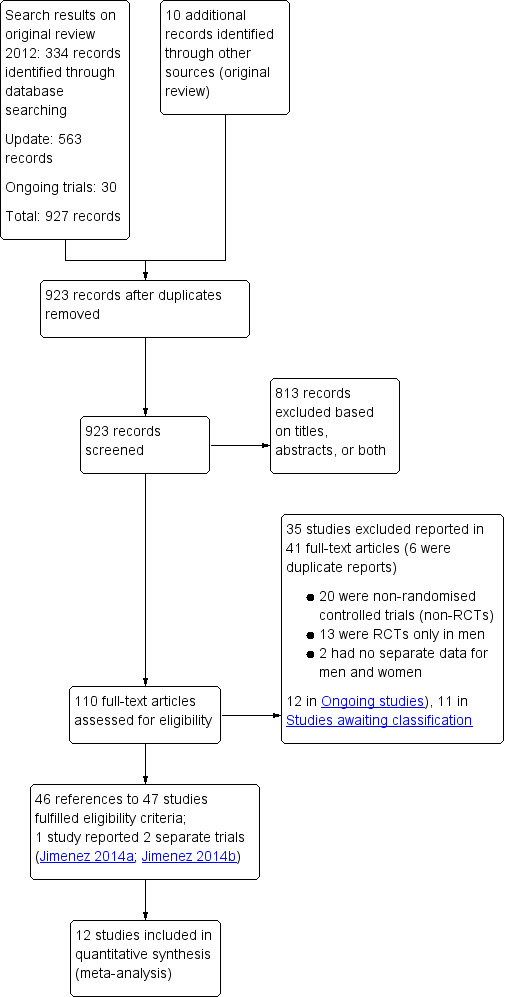
Study flow diagram.
We excluded 35 studies reported in 41 records; six were duplicate reports of studies (see the 'Characteristics of excluded studies' section). We did not include 23 ongoing studies (seven of the 30 ongoing studies were already completed) in the analyses (see the 'Characteristics of ongoing studies' section), but we will include these in future updates of this review when data are available.
Overall, we included 46 references to 47 studies but with two separate trials included in one report (Jimenez 2014a; Jimenez 2014b). For further details, see the 'Study flow diagram' (Figure 3).
Included studies
This update added 25 further studies to the existing 22 studies included in the previous version of this review (van Zuuren 2012). The 47 studies comprised a total of 5290 participants (see the 'Characteristics of included studies' section).
Characteristics of the trial setting and methods
All of the included studies were randomised controlled trials, 29 had a placebo, 14 had an active control treatment arm, and four studies included both a placebo and an active control arm. Eleven studies were conducted prior to the year 2000. The duration of most studies was between six and 12 months, with a mean of 30.4 weeks. Twenty‐two studies were conducted in Europe, 18 in the USA or Canada, two in Central‐ or South America, and 10 in Asia.
Characteristics of the participants
The number of participants included in the individual studies varied widely, from six to 404 women, with a mean of 112 participants. The age of the participants ranged from 18 to 89 years, with most between 18 and 60 years and a mean of 40.5 years. All women had been diagnosed with androgenic alopecia (AGA) or female pattern hair loss (FPHL), and two studies included hyperandrogenic women (Carmina 2003; Vexiau 2002).
Characteristics of the interventions
The included studies evaluated a wide range of interventions: 17 studies assessed minoxidil (Blume‐Peytavi 2007; Blume‐Peytavi 2011a; DeVillez 1994; Jacobs 1993; Lucky 2004; NCT01145625; NCT01226459; NCT01655108; NCT01900041; Olsen 1991; Pazoki‐Toroudi 2012; Price 1990; Rietschel 1987; Sheng 2014; Tsuboi 2007; Vexiau 2002; Whiting 1992), and six studies examined the effects of finasteride (Carmina 2003; Keene 2011; Mazzarella 1997; Price 2000; Ukşal 1999; Whiting 1999). Two studies included cyproterone acetate in one treatment arm (Carmina 2003; Vexiau 2002), and two other studies evaluated flutamide (Carmina 2003; Ukşal 1999). Five studies investigated laser treatment (Jimenez 2014a; Jimenez 2014b; Kim 2013; Lanzafame 2014; NCT01967277). Two studies examined bimatoprost (NCT01189279; NCT01325350).
A total of 17 studies addressed other interventions: alfatradiol (Blume‐Peytavi 2007); essential oil (Bureau 2003); 0.5% octyl nicotinate and 5.0% myristyl nicotinate (Draelos 2005); topical melatonin‐alcohol solution (Fischer 2004); topical fulvestrant solution (Gassmueller 2008); an oral combination product of millet seed extract, L‐cystine, and calcium pantothenate (Gehring 2000); oestrogen ointment (Georgala 2004; Guerrero 2009); cytopurine/pentadecanoic glyceride (Hong 2007); red ginseng powder capsules (Kim 2009); nutritional supplement (Le Floc'h 2015); systemic oestrogens (Minozzi 1997); oral gelatin cystine and lotion based on gelatine cystine and Serenoa repens (Morganti 1998); 0.75% adenosine lotion (Oura 2008); the application of a pulsed electrostatic field (Policarpi 1993); AP‐FHG0604T (plant extracts; Shin 2007) and spironolactone (Ukşal 1999). Several trials compared and evaluated a number of these interventions in the different treatment arms.
Characteristics of the outcome measures
Half of the included studies evaluated 'hair regrowth' as assessed by the participants (Blume‐Peytavi 2011a; Bureau 2003; Carmina 2003; DeVillez 1994; Hong 2007; Jacobs 1993; Jimenez 2014a; Jimenez 2014b; Kim 2013; Le Floc'h 2015; Lucky 2004; Mazzarella 1997; NCT01226459; NCT01325350; NCT01655108; Olsen 1991; Oura 2008; Pazoki‐Toroudi 2012; Policarpi 1993; Price 2000; Shin 2007; Tsuboi 2007; Whiting 1992), which was the primary outcome for this review. However, none of these outcomes were measured or reported according to the definition of 'clinically significant' hair regrowth, which was prespecified for this review (see the 'Types of outcome measures' section).
The outcome measures used to assess hair regrowth consisted of questionnaires that assessed these outcomes using three‐ to seven‐point scales. They included a wide range of scaling items, many of which were inadequately defined, i.e. 'none, mild, moderate improvement' or 'worsened to marked improved', and were not matched across the included studies. Three studies reported that they had applied a "modified version of a validated self‐administered hair growth questionnaire" (Carmina 2003; Oura 2008; Price 2000), which was developed in a previous study (Barber 1998). The investigators provided no details of how and if their 'modified version' was tested prior to its use, and as it was originally designed for the evaluation of interventions for male pattern baldness, its validity as an assessment tool for FPHL is unclear. Two studies utilised a standard 100 mm visual analogue scale (VAS) for participant assessments of hair growth (Lucky 2004; Shin 2007). Benefit from treatment was scored: 0 = "no benefit" to 100 = "great benefit". Lucky 2004 was also the only included study to assess the effects of two of the interventions on quality of life (QoL), a key primary outcome for this review. The study used a six‐item VAS‐based questionnaire for these assessments, but did not indicate if the instrument had been previously tested or validated.
A large proportion of the trials (32) assessed treatment‐associated adverse events either through questionnaires that rated the "tolerability of treatment" (Blume‐Peytavi 2007) or "dermal reactions", such as erythema, scaling, and itching (Gassmueller 2008), or these were reported as incidental events by the participants during the course of the individual study.
Several included studies assessed the secondary outcomes for this review, but in general, the methods of measurement and the timing of the assessments were not uniform across these studies. Over half of the studies included two of the secondary outcomes, i.e. investigator‐rated clinically significant hair regrowth and the change in total hair count from baseline to study conclusion. Eleven studies addressed hair shedding (Carmina 2003; Guerrero 2009; Hong 2007; Kim 2009; Le Floc'h 2015; Mazzarella 1997; Oura 2008; Pazoki‐Toroudi 2012; Tsuboi 2007; Vexiau 2002; Whiting 1992). Twelve studies assessed cosmetic appearance and 'patient' satisfaction (Blume‐Peytavi 2011a; Carmina 2003; Draelos 2005; Kim 2009; Kim 2013; Le Floc'h 2015; Lucky 2004; NCT01655108; Oura 2008; Price 2000; Thom 2001; Thom 2006).
Fourteen studies evaluated one of the secondary outcomes for this review, i.e. quality and pattern of hair regrowth (Blume‐Peytavi 2007; Blume‐Peytavi 2011a; Bureau 2003 ; Carmina 2003; Gassmueller 2008; Jimenez 2014a; Jimenez 2014b; Kim 2009; Kim 2013; Le Floc'h 2015; NCT01325350; NCT01655108; NCT01900041; Oura 2008).
We examined any of the patient‐reported outcomes (PROs) presented in the included studies against the 'checklist for describing and assessing PRO's in clinical trials' (see Table 7), which is provided in Chapter 17.6.a of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011).
3. Checklist for describing and assessing patient‐reported outcomes (PROs) in clinical trials.
| 1. What were PROs measuring? a. What concepts were the PROs used in the study measuring? b. What rationale (if any) for selection of concepts or constructs did the authors provide? c. Were patients involved in the selection of outcomes measured by the PROs? |
| 2. Omissions a. Were there any important aspects of health (e.g. symptoms, function, perceptions) or quality of life (e.g. overall evaluation, satisfaction with life) that were omitted in this study from the perspectives of the patient, clinician, significant others, payers, or other administrators and decision‐makers? |
| 3. If randomised trials and other studies measured PROs, what were the instruments' measurement strategies? a. Did investigators use instruments that yield a single indicator or index number, a profile, or a battery of instruments? b. If investigators measure PROs, did they use specific or generic measures, or both? c. Who exactly completed the instruments? |
| 4. Did the instruments work in the way they were supposed to work ‐ validity? a. Had the instruments used been validated previously (provide reference)? Was evidence of prior validation for use in this population presented? b. Were the instruments re‐validated in this study? |
| 5. Did the instruments work in the way they were supposed to work ‐ ability to measure change? a. Are the PROs able to detect change in patient status, even if those changes are small? |
| 6. Can you make the magnitude of effect (if any) understandable to readers? a. Can you provide an estimate of the difference in patients achieving a threshold of function or improvement, and the associated number needed to treat (NNT)? |
|
Table 17.6.a Patrick D, Guyatt GH, Acquadro C. Chapter 17: Patient‐reported outcomes. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. |
Abbreviations: PRO: patient‐reported outcome.
Excluded studies
We excluded 35 studies, and reported the reasons for their exclusion in the 'Characteristics of excluded studies' tables. We excluded all of these studies only after assessment of the full‐text reports. The most frequent reason for their exclusion was that they were non‐RCTs, or because they included only male participants.
Risk of bias in included studies
We assessed each included study for risk of bias and reported the judgements for the individual domains in the 'Risk of bias' table associated with each study. We have also presented these in the 'Risk of bias' graph in Figure 4 and the 'Risk of bias' summary in Figure 5.
4.
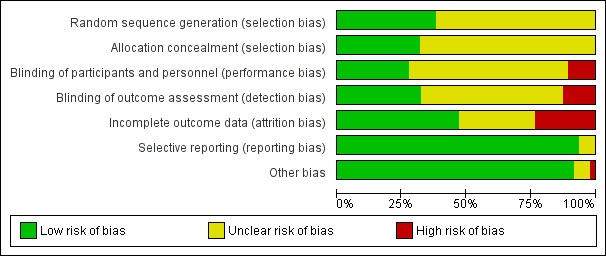
'Risk of bias' graph: review authors' judgements about each 'Risk of bias' item presented as percentages across all included studies.
5.
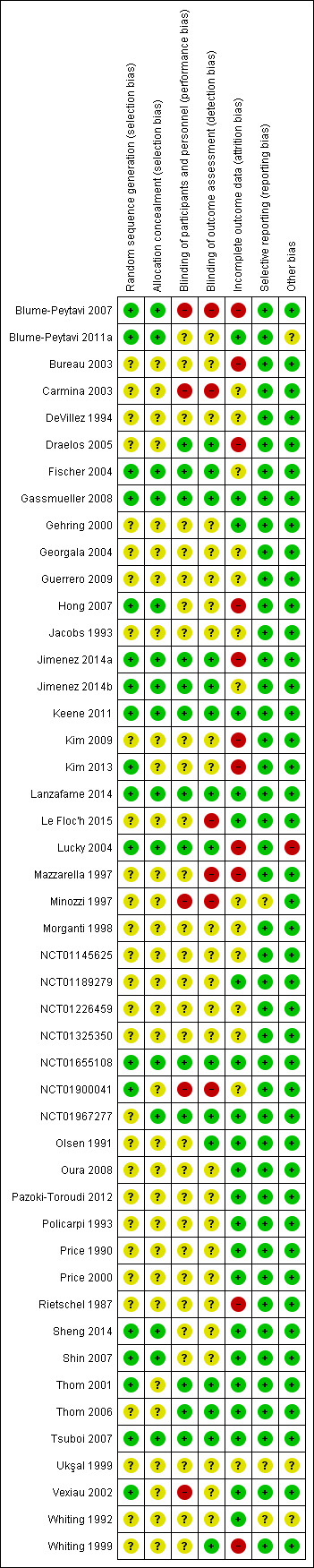
'Risk of bias' summary: review authors' judgements about each 'Risk of bias' item for each included study.
We assessed the overall risk of bias for each included study, and we considered five studies to be at 'low risk of bias' as they met all criteria across all domains in the Cochrane 'Risk of bias' assessment tool (plausible bias unlikely to seriously alter the results) (Gassmueller 2008; Keene 2011; Lanzafame 2014; NCT01655108; Tsuboi 2007). We categorised 16 studies as at high risk of bias (plausible bias that seriously weakens confidence in the results) because one or more domains received a judgement of high risk (Blume‐Peytavi 2007; Bureau 2003; Carmina 2003; Draelos 2005; Hong 2007; Jimenez 2014a; Kim 2009; Kim 2013; Le Floc'h 2015; Lucky 2004; Mazzarella 1997; Minozzi 1997; NCT01900041; Rietschel 1987; Vexiau 2002; Whiting 1999). We rated the remaining 26 studies as at unclear risk of bias (plausible bias that raises some doubt about the result) because we assessed one or more criteria as unclear.
Some of these assessments were, to a certain extent, based on the inadequate reporting of the criteria that are a prerequisite in the evaluation of methodological rigour, in terms of trial design and conduct. Concealment of the allocation sequence and blinding are key domains in the assessment of risk of bias, and a number of included studies provided insufficient detail to enable us to make accurate judgements. Protocol deviation, losses to follow‐up with incomplete data, and subsequent per‐protocol analyses were other important sources of potential bias in a number of the included studies. We were able to amend the judgements for a number of the domains after we successfully contacted several trial investigators. For these and further details, see the 'Risk of bias' tables in the 'Characteristics of included studies' section.
Allocation
The methods used to generate the allocation sequence and how the sequence was concealed, such that participants and investigators enrolling participants could not foresee the upcoming assignment, are the most important and sensitive indicators that bias has been minimised in a clinical trial (Schulz 1995).
Sequence generation
Eighteen studies described in sufficient detail the method used to generate the allocation sequence (Blume‐Peytavi 2007; Blume‐Peytavi 2011a; Fischer 2004;Gassmueller 2008; Hong 2007; Jimenez 2014a; Jimenez 2014b; Keene 2011; Kim 2013; Lanzafame 2014; Lucky 2004; NCT01655108; NCT01900041; Sheng 2014; Shin 2007; Thom 2001; Tsuboi 2007; Vexiau 2002). Therefore, we judged these studies as at low risk of bias for this domain. We considered the remaining 29 studies as at unclear risk of bias.
Allocation concealment
Pharmacy‐controlled, central allocation or sequentially numbered, opaque, sealed envelopes/packets ensured that the intervention allocations could not have been foreseen in advance of, or during, enrolment in 15 studies (Blume‐Peytavi 2007; Blume‐Peytavi 2011a; Fischer 2004; Gassmueller 2008; Hong 2007; Jimenez 2014a; Jimenez 2014b; Keene 2011; Lanzafame 2014; Lucky 2004; NCT01655108; NCT01967277; Sheng 2014; Shin 2007; Tsuboi 2007), which we judged low risk of bias for this domain. The remaining trials did not report the method used to conceal the allocation sequence; thus, they received a judgment of unclear risk of bias for this domain.
Blinding
Thirteen studies described in sufficient detail the measures used to blind study participants and personnel from knowledge of which intervention a participant received (Draelos 2005; Fischer 2004; Gassmueller 2008; Jimenez 2014a; Jimenez 2014b; Keene 2011; Lanzafame 2014; Lucky 2004; NCT01655108; NCT01967277; Thom 2001; Thom 2006; Tsuboi 2007). Blinding was achieved by identical pre‐labelled bottles, tablets, devices, or packages. As five studies were open label (Blume‐Peytavi 2007; Carmina 2003; Minozzi 1997; NCT01900041; Vexiau 2002), the outcome or outcome measurement was likely to be influenced by lack of blinding. In these studies, we judged this domain as at high risk of bias. Inadequate reporting did not permit us to make a clear judgement for this domain in the other 29 studies.
Fifteen studies ensured blinding of outcome assessment (Draelos 2005; Fischer 2004; Gassmueller 2008; Jimenez 2014a; Jimenez 2014b; Keene 2011; Lanzafame 2014; Lucky 2004; NCT01655108; NCT01967277; Olsen 1991; Thom 2001; Thom 2006; Tsuboi 2007; Whiting 1999). In six studies, we judged that there was a high risk of detection bias mainly due to the open‐label design (Blume‐Peytavi 2007; Carmina 2003; Le Floc'h 2015; Mazzarella 1997; Minozzi 1997; NCT01900041), whilst for the remainder (26 studies), we judged this domain as at unclear risk of bias.
Incomplete outcome data
In slightly less than half of the included studies (22), incomplete outcome data appear to have been adequately addressed. The losses to follow‐up were reasonably well‐balanced across intervention groups, with similar reasons for missing data across the groups. However, in 11 studies, the high dropout rate and subsequent per‐protocol analysis of the data resulted in a judgement of high risk of bias for this domain (Blume‐Peytavi 2007; Bureau 2003; Draelos 2005; Hong 2007; Jimenez 2014a; Kim 2009; Kim 2013; Lucky 2004; Mazzarella 1997; Rietschel 1987; Whiting 1999).
The remaining 14 studies reported insufficient information to permit a clear judgment of the risk of bias for this domain (Carmina 2003; DeVillez 1994; Fischer 2004; Georgala 2004; Guerrero 2009; Jacobs 1993; Jimenez 2014b; Minozzi 1997; Morganti 1998; NCT01145625; NCT01226459; NCT01325350; NCT01900041; Ukşal 1999).
Selective reporting
Protocols were available for only 13 included studies (Blume‐Peytavi 2011a; Jimenez 2014a; Jimenez 2014b; Keene 2011; Lanzafame 2014; Le Floc'h 2015; NCT01145625; NCT01189279; NCT01226459; NCT01325350; NCT01655108; NCT01900041; NCT01967277). Based on this information as well as on the details provided in the methods section of the reports, 44 of the 47 studies appear to have reported all prespecified outcomes and we therefore judged them to be free of selective reporting. We considered the remaining three studies to be at unclear risk of bias (Minozzi 1997; Ukşal 1999; Whiting 1992). One of these studies, Ukşal 1999, was reported only as an abstract to conference proceedings, which provided insufficient information to make a clear judgement for this domain. Although Whiting 1992 did not fully report the primary outcomes of participant and investigator assessments of hair regrowth, this did not appear to be intentional; and as the impact of this was unclear, we judged this domain as at unclear risk of bias. The investigators in Minozzi 1997 did not report all of their prespecified outcomes, but it was uncertain to what extent the lack of data for anything other than sex hormone‐binding globulin (SHBG) had any impact on their reported results; therefore, we judged this domain as at unclear risk of bias.
Other potential sources of bias
We judged this domain as at 'low risk of bias' in most of the included studies (43). Although the impact of study sponsorship in Lucky 2004 was unclear, the "protocol‐prohibited concomitant medications" used by a number of participants, mostly in the active intervention group, represented a high risk of bias for this domain in this study. In Blume‐Peytavi 2011a, the baseline imbalance between the intervention groups, i.e. a higher proportion of participants with more extensive hair thinning in the 5% minoxidil group, posed a risk of bias for this domain in this study. In Whiting 1992, the potential impact of the wide range in duration (six months to 25 years) of hair loss at baseline was unclear. In Ukşal 1999, which was reported as an abstract, we had insufficient information to permit a judgement.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4
Fifteen studies provided no usable or retrievable data, and did not contribute further to the results of this review (see Table 8). The main reasons why we could not use data were: studies did not report data separately for men and women, there were very limited data available in abstracts to conference proceedings, or studies did not address any of our outcomes.
4. Included studies with no usable or irretrievable data.
| Study ID | Interventions and comparisons | N | Comments |
| Bureau 2003 | Essential oil solution (E2F7) and electromagnetic pulses versus placebo solution and electromagnetic pulses | 93 | No separate data for men and women Investigators unable to provide these |
| Fischer 2004 | Melatonin–alcohol solution versus alcohol solution | 40 | None of our outcomes were addressed |
| Guerrero 2009 | Minoxidil 2% versus 17α‐estradiol | 40 | No separate data for men and women No response from principal investigator |
| Hong 2007 | Cytopurine, pentadecanoic glyceride, 95% ethanol topical solution versus vehicle solution | 95 | No separate data for men and women |
| Kim 2009 | Korean red ginseng versus placebo | 40 | No separate data for men and women No response from principal investigator |
| Kim 2013 | Low level light therapy versus sham device | 40 | No separate data for men and women No response from principal investigator |
| Mazzarella 1997 | Finasteride 0.005% lotion versus vehicle | 56 | No separate data for men and women No response from principal investigator |
| Minozzi 1997 | Ethinyl estradiol (0.02 mg/day) versus transdermal estradiol (0.05 mg/day) with medroxyprogesterone acetate (MPA) versus ethinyl estradiol (0.02 mg/day) with cyproterone acetate | 63 | Diagnosis of female pattern hair loss (FPHL) was not clearly defined/stated. No response from principal investigator None of our outcomes were assessed |
| Morganti 1998 | Active lotion (gelatine‐cystine and Serenoa repens) versus placebo lotion versus active diet supplement (gelatine‐cystine) versus placebo supplement versus active lotion and active supplement (n = 12) | 60 | No separate data for men and women No response from principal investigator |
| NCT01189279 | Bimatoprost A versus bimatoprost B versus bimatoprost C | 42 | None of our outcomes were addressed, no separate data for men and women. No response from Allergan |
| NCT01226459 | Minoxidil 5% versus vehicle | 404 | Too many inconsistencies regarding numbers that dropped out and number of participants analysed, as well as in calculations made for mean changes from baseline. No response from principal investigators |
| Rietschel 1987 | Minoxidil 2% versus minoxidil 3% versus placebo | 149 | No separate data for men and women No response from principal investigator |
| Thom 2001 | Dietary supplement versus placebo | 60 | No separate data for men and women No response from principal investigator |
| Thom 2006 | Dietary supplement versus placebo | 60 | No separate data for men and women No response from principal investigator |
| Ukşal 1999 | Spironolactone versus flutamide versus finasteride | ? | Poster, limited data, unclear how many participants in each group. No response from principal investigator |
Abbreviations: FPHL: female pattern hair loss, N: Number randomised
We have categorised comparisons as follows.
Treatments with topical minoxidil (comparisons 1 to 8).
Other topical treatments (comparisons 9 to 15).
Oral treatments (comparisons 16 to 18).
Laser‐based treatments and electrostatic field (comparisons 19 to 22).
1. Minoxidil (1%, 2%, and 5%) versus placebo
Nine trials provided data for some outcomes for this comparison (DeVillez 1994; Jacobs 1993; Lucky 2004; NCT01325350; Olsen 1991; Pazoki‐Toroudi 2012; Price 1990; Tsuboi 2007; Whiting 1992). Seven of these trials examined the effects of a 2% concentration of minoxidil, whereas Tsuboi 2007 compared a 1% concentration with placebo, one study included an additional 5% arm (Lucky 2004), and one three‐armed study compared minoxidil 5% with placebo as one of the three comparisons within that study (Pazoki‐Toroudi 2012). See also 'Summary of findings' table 1 (Table 1).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
Six studies (1148 participants) reported participant‐rated clinically significant (moderate to marked) hair regrowth (DeVillez 1994; Jacobs 1993; NCT01325350; Olsen 1991; Pazoki‐Toroudi 2012; Tsuboi 2007). Pooled data, using a fixed‐effect model, from these studies indicated that a greater proportion of participants (157/593) treated with minoxidil reported a statistically significant moderate increase in hair regrowth when compared with placebo (77/555) (risk ratio (RR) 1.93, 95% confidence interval (CI) 1.51 to 2.47; six trials, 1148 participants; P < 0.00001; I² statistic = 24%, Analysis 1.1). We compared the effect size and the precision estimates of 1% minoxidil versus placebo, 2% minoxidil versus placebo, and 5% minoxidil versus placebo. Thus, we conclude that although the difference would appear to favour minoxidil (2%), this was relatively small and provides no evidence of any genuine difference with the 1% concentration, whilst the higher concentration of 5% did not show a statistically significant difference compared to placebo. Although there was little suggestion of heterogeneity, we repeated this analysis using a random‐effects model to assess the extent of the between‐study heterogeneity (see Table 9). One other study, Lucky 2004 (381 participants, data on 260 participants), which also reported data for this outcome, used a VAS rated zero to 100 to measure change in hair growth/scalp coverage, with a higher score indicating more scalp coverage. The VAS score was 58.3 (18.2 standard deviation (SD)) in the placebo group, 62.9 (16.7 SD) in the 2% minoxidil group, and 68.1 (17.9 SD) in the 5% minoxidil group. According to the principal investigators only the self assessments of 5% minoxidil versus placebo reached a statistically significant difference (P < 0.001).
1.1. Analysis.
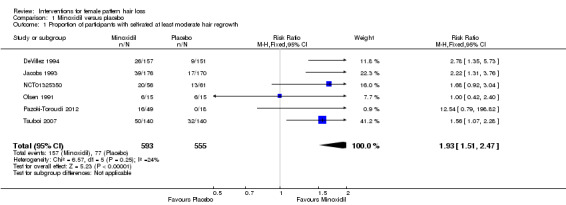
Comparison 1 Minoxidil versus placebo, Outcome 1 Proportion of participants with self‐rated at least moderate hair regrowth.
5. Table of random‐effects sensitivity analyses.
| Analysis | Concentration/subgroups | Risk ratio | 95% confidence interval | P value | I² statistic | Chi² test (P value) |
|
Analysis 1.1 Self‐rated hair regrowth |
Pooled | 1.82 | 1.34 to 2.46 | < 0.00001 | 24% | 0.25 |
|
Analysis 1.2 Adverse events |
Minoxidil (1%) | 1.12 | 0.61 to 2.06 | 0.72 | — | — |
| Minoxidil (2%) | 1.19 | 0.79 to 1.79 | 0.40 | 0% | 0.67 | |
| Minoxidil (5%) | 1.83 | 0.51 to 6.55 | 0.35 | 63% | 0.10 | |
|
Analysis 1.3 Investigator‐rated hair regrowth |
Pooled | 2.17 | 1.53 to 3.06 | < 0.00001 | 2% | 0.41 |
|
Analysis 1.4 Increase in total hair count |
Pooled | 13.24 | 10.80 to 15.65 | < 0.00001 | 9% | 0.36 |
|
Analysis 1.5 Increase in total hair count (sensitivity analysis) |
Pooled | 12.96 | 10.69 to 15.24 | < 0.00001 | 0% | 0.76 |
| Analysis 2.1 | Pooled | 0.96 | 0.70 to 1.30 | 0.77 | 76% | 0.005 |
| Analysis 2.2 | Pooled | 1.12 | 0.93 to 1.35 | 0.22 | 50% | 0.13 |
| Analysis 2.3 | Pooled | −2.12 | −5.47 to 1.23 | 0.22 | 0% | 0.55 |
Change in QoL
Lucky 2004 rated the impact of hair loss on QoL on a VAS (data on 260 participants), and at 48 weeks the mean score in the 2% minoxidil group was 52.1 and 46.5 in the placebo group (study authors reported P = 0.04, Student t‐test), where a score of 50 indicates "neutral" impact, ranging up to 100 as "positive" impact. In the 5% minoxidil group, the mean VAS score was 54.4 (slightly more than no change) compared to 46.5 with placebo (study authors reported P = 0.004, Student t‐test). Although the study investigators reported these scores without SDs and stated them as being statistically significant, the mean differences (MDs) between intervention groups were marginal and can be considered not clinically important. The other included studies did not assess this outcome.
Adverse effects, safety, and tolerability
Comparison of the effect size and precision estimates for minoxidil (1%) versus placebo (RR 1.12, 95% CI 0.61 to 2.06; one trial, 280 participants), minoxidil (2%) versus placebo (RR 1.24, 95% CI 0.82 to 1.87), and minoxidil (5%) versus placebo (RR 2.05, 95% CI 0.96 to 4.37; four trials, 427 participants) reveals that although the difference between the adverse effects favours the minoxidil (1%) concentration, this is small and provides limited evidence of any genuine difference between the three concentrations (see Analysis 1.2). The interaction between the subgroups of dose and effect size did not provide adequate evidence to demonstrate a difference, but this is likely to be due to a lack of power (P value = 0.43).
1.2. Analysis.
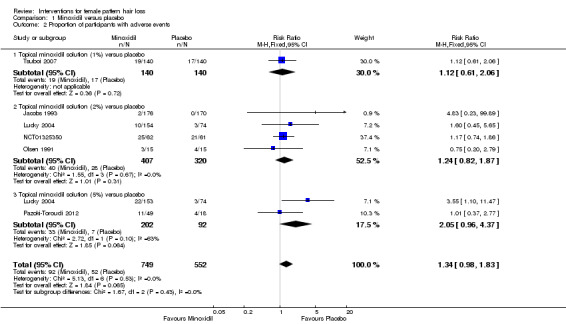
Comparison 1 Minoxidil versus placebo, Outcome 2 Proportion of participants with adverse events.
In most instances, the adverse events reported were mild and consisted of pruritus, skin irritation, and dermatitis. Additional hair growth on areas other than the scalp, e.g. sideburns and forehead, was reported in 71/153 participants in the minoxidil (5%) group compared to 34/154 in the minoxidil (2%) group and 12/74 in the placebo group (Lucky 2004). The data for adverse events were incompletely reported in DeVillez 1994, with the investigators indicating only that "no serious or unexpected medical events were reported during the study", and Whiting 1992 reported that no serious side effects were encountered.
Pooled adverse events data for all of the concentrations of minoxidil versus placebo showed a RR of 1.34 (95% CI 0.98 to 1.83; five trials, 1301 participants; I² statistic = 0%), with no statistically significant difference between groups.
Secondary outcomes
Proportion of participants with investigator‐rated hair regrowth
Seven studies (1181 participants) comparing one or more of these interventions provided data for this outcome (DeVillez 1994; Jacobs 1993; NCT01325350; Olsen 1991; Pazoki‐Toroudi 2012; Tsuboi 2007; Whiting 1992). The investigator‐rated assessments were in agreement with the participant self‐rated assessments, both of which reported and confirmed a statistically significant increase in moderate to marked hair regrowth with minoxidil (112/610) compared to placebo (44/571) (RR 2.35, 95% CI 1.68 to 3.28; seven trials, 1181 participants; P < 0.00001 I² statistic = 2%; Analysis 1.3). A comparison of the effect size and precision of estimates for minoxidil (1%) versus placebo, and minoxidil (2%) versus placebo revealed that there was no appreciable difference between the two concentrations of minoxidil for this outcome. Pazoki‐Toroudi 2012 showed no statistically significant difference between the minoxidil (5%) group and placebo, but the effect estimate was very imprecise with wide CIs, due to a lack of events in the placebo group. One other study, Lucky 2004 (data on 260 participants), used a VAS, rated zero to 100, to evaluate change in hair growth/scalp coverage with a higher score indicating better coverage. The changes were 2.2 (17.9 SD) in the placebo group, 10.3 (17.0 SD) in the minoxidil (2%) group and 11.7 (17.2 SD) in the minoxidil (5%) group.
1.3. Analysis.
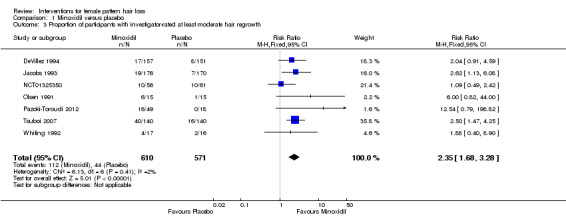
Comparison 1 Minoxidil versus placebo, Outcome 3 Proportion of participants with investigator‐rated at least moderate hair regrowth.
Change from baseline in total hair count
Eight studies including 1242 participants reported data for the mean change in total hair count from baseline (DeVillez 1994; Jacobs 1993; Lucky 2004; NCT01325350; Olsen 1991; Price 1990; Tsuboi 2007; Whiting 1992). The MD across the studies favoured minoxidil and ranged from eight to 42 hairs (total hair count). The pooled data illustrated that the increase in total hair count in the minoxidil group when compared to the placebo group was 13.18 (95% CI 10.92 to 15.44; eight trials, 1242 participants; P < 0.00001; I² statistic = 9%; Analysis 1.4). A comparison of the effect size and precision of the estimates indicated that there was no evidence of any systematic difference between the three concentrations of minoxidil. In addition, we observed funnel plot asymmetry that was attributable to a single study of eight participants (Price 1990) (see Figure 6). It remains unclear whether this asymmetry was the result of publication bias, small‐study effects, or an artefact of natural variability. The impact of excluding this study from Analysis 1.4 had a marginal effect on the overall pooled result (RR 12.96, 95% CI 10.69 to 15.24; seven trials, 1108 participants; P < 0.00001; I² statistic = 0%), which we report as a sensitivity analysis (see Analysis 1.5); however, it did remove any suggestion of heterogeneity. In an attempt to assess the between study heterogeneity, we repeated both of these analyses using a random‐effects model, and presented the results in Table 9. We found little difference between the two sets of analyses.
1.4. Analysis.
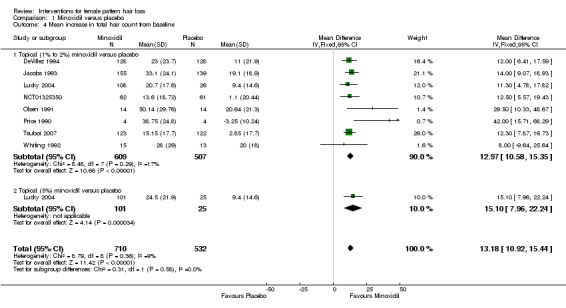
Comparison 1 Minoxidil versus placebo, Outcome 4 Mean increase in total hair count from baseline.
6.
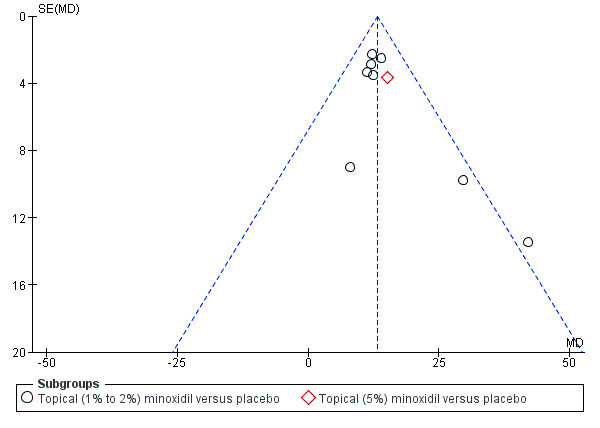
Funnel plot of comparison: 1 Minoxidil versus placebo, outcome: 1.4 Mean increase in total hair count from baseline.
1.5. Analysis.
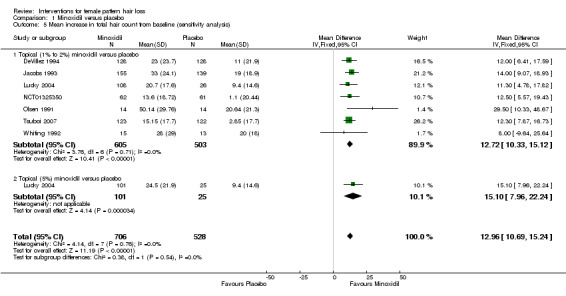
Comparison 1 Minoxidil versus placebo, Outcome 5 Mean increase in total hair count from baseline (sensitivity analysis).
Degree of hair shedding from baseline to the end of the study
Three studies reported data on this outcome (Pazoki‐Toroudi 2012; Tsuboi 2007; Whiting 1992). In Pazoki‐Toroudi 2012 there was a reduction in the number of shed hair of 38.32 (40.98 SD) in the minoxidil (5%) group (43 participants) compared to a reduction of 0.47 (23.50 SD) in the placebo group (18 participants) with a MD of −37.85 (95% CI −54.22 to −21.48; P < 0.00001).
Two thirds (10/17) of the participants in the minoxidil (2%) group reported a decrease in hair shedding compared with less than half (7/16) in placebo group, and that this was more noticeable at the second month of treatment in Whiting 1992 (RR 1.34, 95% CI 0.68 to 2.66; one trial, 33 participants).
However, there was no statistically significant difference in the number of participants in Tsuboi 2007 that reported a decrease in hair loss, and 98/140 participants in the minoxidil (1%) group compared to 87/140 demonstrated a large placebo effect (RR 1.13, 95% CI 0.95 to 1.33, one trial, 180 participants).
Cosmetic appearance of the hair or participant satisfaction
One study measured participants' satisfaction as a 'benefit of treatment' and rated it using a VAS (zero = no benefit, 50 = moderate benefit, and 100 = great benefit) (Lucky 2004). Participant satisfaction in the two minoxidil groups compared to placebo was 18.2 for the minoxidil (5%) group (principal investigators reported P < 0.001, Student t‐test) and 8.7 for the minoxidil (2%) group (P = 0.09, Student t‐test). The score for the minoxidil (5%) group was 60.0 (27.6 SD), for the (2%) group 50.5 (32.5 SD), and 41.8 (29.9 SD) for the placebo group, and was rated using a VAS score (0 = no benefit, 50 = moderate benefit, and 100 = great benefit). The investigators concluded that there was evidence of increased participant satisfaction with the higher, rather than with the lower, concentration. However, there was a 32% loss to follow‐up in this study and the data analysis was per‐protocol.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The mean change in hair density, assessed on the Savin Female Density scale at 48 weeks, was −0.9 in the minoxidil (2%) group compared to −0.4 in the placebo group (investigators reported P = 0.012, Student t‐test), and a lower score represented a more beneficial effect (Lucky 2004). The mean change in hair density in the minoxidil (5%) group was −0.8 (P value = 0.015, Student t‐test) compared to placebo. In NCT01325350 the mean change from baseline in target area hair width was 0.87 (1.315) mm/cm² in the minoxidil (2%) group versus 0.07 (1.183) mm/cm² in the placebo group with a MD of 0.80 (95% CI 0.36 to 1.24; P = 0.0004) which favoured minoxidil (2%).
2. Minoxidil (2%) versus minoxidil (5%)
Four studies compared these interventions (Blume‐Peytavi 2011a; Lucky 2004; NCT01145625; Sheng 2014). One study, Blume‐Peytavi 2011a, used a 2% concentration applied twice daily and the 5% concentration once daily, whereas Lucky 2004 and Sheng 2014 applied both concentrations twice daily. In NCT01145625 both treatment arms received a single dose each day. None of the four studies reported any significant difference in efficacy between either of the two concentrations of minoxidil. See also 'Summary of findings' table 2 (Table 2).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
Two of the studies did not assess this outcome (NCT01145625; Sheng 2014). In Blume‐Peytavi 2011a 44% (25/57) of the participants in the minoxidil (2%) group, as opposed to 39% (22/56) of those in the minoxidil (5%) group, experienced moderate to greatly increased hair regrowth (RR 1.12, 95% CI 0.72 to 1.73; one trial, 113 participants). In Lucky 2004 (209 participants for this comparison) the VAS score showed a change in hair growth/scalp coverage of 62.9 (16.7 SD) in the minoxidil (2%) group, compared to a score of 68.1 (17.9 SD) in the minoxidil (5%) group (investigators reported P = 0.062).
Change in QoL
Only one study evaluated this outcome (Lucky 2004). The trial investigators reported that at week 48 there was no statistically significant difference in impact of hair loss on QoL between the two intervention groups.
Adverse effects, safety, and tolerability
There was no statistically significant difference in the number of adverse events reported in either intervention group in three of the studies (Blume‐Peytavi 2011a; NCT01145625; Sheng 2014). However, in Lucky 2004 the number of participants that reported adverse events appeared to favour the lower concentration. These were reported by 10/154 participants in the minoxidil (2%) twice daily group compared to 22/153 in the minoxidil (5%) twice daily group (RR 0.45, 95% CI 0.22 to 0.92, one trial, 307 participants). Hypertrichosis (more hair growth on areas other than the scalp), dermatitis, and pruritus were also reported more frequently in the minoxidil (5%) group. Pooling of the data indicated a RR of 1.02 (95% CI 0.91 to 1.15; four trials, 1006 participants; I² statistic = 76%; Analysis 2.1). Exclusion of this single study, Lucky 2004, from the analysis slightly altered the pooled results and reduced the degree of heterogeneity (RR 1.10, 95% CI 0.99 to 1.23; three trials, 699 participants; I² statistic = 50%; Analysis 2.2). The twice daily dose of 5% in this study resulted in double the number of adverse events compared to the twice daily dosage of minoxidil (2%). We repeated the analyses using a random‐effects model and found little difference between the two sets of analyses (Table 9).
2.1. Analysis.
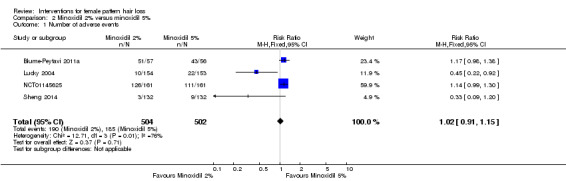
Comparison 2 Minoxidil 2% versus minoxidil 5%, Outcome 1 Number of adverse events.
2.2. Analysis.
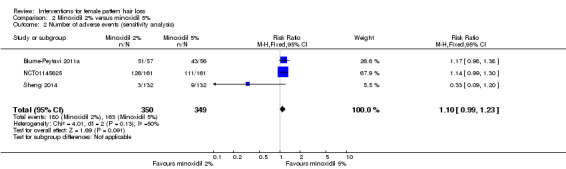
Comparison 2 Minoxidil 2% versus minoxidil 5%, Outcome 2 Number of adverse events (sensitivity analysis).
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
The investigator‐ and participant‐rated assessments were largely in agreement in three of the studies, i.e. there was no evidence of a difference between the two concentrations of minoxidil for stimulating hair growth. In Blume‐Peytavi 2011a the investigator‐rated assessments revealed that 12/57 participants in the minoxidil (2%) group had moderate to greatly increased hair growth compared to 14/56 in the minoxidil (5%) group (RR 0.84, 95% CI 0.43 to 1.66; one trial 113 participants). These results were confirmed by the investigator‐rated VAS scores in Lucky 2004, which showed no statistically significant difference in efficacy between either concentration of minoxidil (principal investigators' reported P = 0.608). In Sheng 2014 27/132 participants in the minoxidil (2%) group had a moderate to marked improvement compared to 34/132 in the minoxidil (5%) group (RR 0.79, 95% CI 0.51 to 1.24; one trial 264 participants). NCT01145625 did not assess this outcome.
Change from baseline to study conclusion in total hair count
The pooled data from three studies (631 participants) (Blume‐Peytavi 2011a; Lucky 2004; NCT01145625) indicated that there was no statistically significant difference in the change in total hair count from baseline to the end of study between the two treatment groups (MD −2.12, 95% CI −5.47 to 1.23; I² statistic = 0%; see Analysis 2.3 and Table 9).
2.3. Analysis.
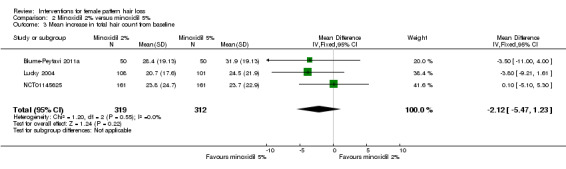
Comparison 2 Minoxidil 2% versus minoxidil 5%, Outcome 3 Mean increase in total hair count from baseline.
Degree of hair shedding from baseline to the end of the study
None of the studies assessed this outcome.
Cosmetic appearance of the hair or participant satisfaction
Almost three quarters of the participants in the minoxidil (2%) group were more satisfied with the appearance of their hair at the end of treatment compared to more than half of those in the minoxidil (5%) group (Blume‐Peytavi 2011a). Assessments of 'benefit of treatment' in Lucky 2004 rated on a VAS scored 50.5 (32.5 SD) in the 2% group versus 60.0 (27.6 SD) in the 5% group (principal investigators' reported P = 0.29, Student t‐test). The two other studies did not assess this outcome (NCT01145625; Sheng 2014).
Change in quality (or pattern) of hair regrowth (e.g. thickness)
There was no statistically significant difference in the non‐vellus cumulative target area hair width (mm/cm² between the minoxidil (2% and 5%) applications in Blume‐Peytavi 2011a, and similarly in Lucky 2004 for hair density, assessed as the mean change from baseline based on the Savin Female Density scale. The other two studies did not assess this outcome (NCT01145625; Sheng 2014).
3. Minoxidil 12.5% + azelaic acid + betamethasone 17‐valerate 0.025% versus placebo
One three‐armed study at unclear risk of bias conducted over a period of 24 weeks, which included 75 participants, evaluated this comparison (Pazoki‐Toroudi 2012).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
In the minoxidil high extra combination (MHEC) group, as named by the principal investigators, 21/57 of participants reported moderate to marked improvement compared to 0/18 in the placebo group (RR 14.09, 95% CI 0.90 to 221.58; one trial, 75 participants).
Change in QoL
This study did not assess this outcome.
Adverse effects, safety, and tolerability
The number of adverse events were 14/57 in the MHEC group and 4/18 in the placebo group (RR 1.11, 95% 0.42 to 2.94; one trial, 75 participants). Adverse events included irritation, hypertrichosis, pruritus, and headache.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
The judgements regarding improvement (moderate and marked) were 100% in agreement with the participant assessments (RR 14.09, 95% CI 0.90 to 221.58; one trial 75 participants).
Change from baseline to study conclusion in total hair count
This study did not assess this outcome.
Degree of hair shedding from baseline to the end of the study
The mean decrease in number of shed hairs was 53.80 (40.94 SD) in the MHEC group versus 0.47 (23.50 SD) in the placebo group with a MD of −53.33 hairs (95% CI −68.66 to −38.00; P < 0.00001) in favour of MHEC.
Cosmetic appearance of the hair or participant satisfaction
This study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
This study did not assess this outcome.
4. Minoxidil 12.5% + azelaic acid + betamethasone 17‐valerate 0.025% versus minoxidil 5%
This is the third comparison, which included 106 participants, that Pazoki‐Toroudi 2012 evaluated.
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
In the MHEC group, 21/57 participants considered themselves to have moderate to marked improvement compared to 16/49 in the minoxidil (5%) group (RR 1.13, 95% CI 0.67 to 1.91; one trial, 106 participants).
Change in QoL
This study did not assess this outcome.
Adverse effects, safety, and tolerability
There were 14 adverse events in 57 participants in the MHEC group and 11/49 in the minoxidil (5%) group (RR 1.09, 95% 0.55 to 2.18; one trial, 106 participants). Adverse events included irritation, hypertrichosis, pruritus, and headache.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
According to the investigators, 21/57 participants in MHEC group versus 16/49 in the minoxidil (5%) group showed a moderate to marked improvement of hair regrowth (RR 1.13, 95% CI 0.67 to 1.91; one trial, 106 participants), which is in concordance with the participants' assessments.
Change from baseline to study conclusion in total hair count
This study did not assess the outcome.
Degree of hair shedding from baseline to the end of the study
The mean decrease in number of shed hairs was 53.80 (40.94 SD) in the MHEC group, which was better than 38.32 (40.98 SD) in the minoxidil (5%) group with a MD of −15.48 hairs (95% CI −31.82 to 0.86).
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The study did not assess this outcome.
5. Minoxidil (2%) versus alfatradiol
One study (103 participants), which we assessed as at high risk of bias, reported limited data for this comparison (Blume‐Peytavi 2007).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
No adverse effects were reported for either intervention in this study (Blume‐Peytavi 2007). "Tolerability of treatment" was participant‐ and investigator‐assessed, and, although inadequately defined, the investigators referred to it in further similar studies as "pruritus and local intolerance". No relevant data were reported at 6 months, and the data at 12 months were incomplete and implausibly analysed (see the 'Risk of bias in included studies' section). Therefore, we have not included these in the meta‐analysis.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change from baseline to study conclusion in total hair count
At 12 months the increase in hair density was 17.20 (SD 32.95) hairs/cm² in the minoxidil group compared to 9.8 (SD 31.79) hairs/cm² in the alfatradiol group with a MD of 7.40 hairs/cm² (95% CI −8.98 to 23.78), which was not statistically significant.
Degree of hair shedding from baseline to the end of the study
The study did not assess this outcome.
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The mean change in cumulative hair thickness from baseline to 12 months was 2.10 (3.10 SD) mm/cm² in the minoxidil group compared to 0.40 (3.31 SD) mm/cm² in the alfatradiol group with a MD of 1.70 mm/cm² (95% CI 0.07 to 3.33, P = 0.04).
6. Intradermal applications (mesotherapy) with minoxidil 0.5% per 2 mL versus intradermal applications (mesotherapy) with saline 0.9%
A single study, NCT01655108, at low risk of bias with 54 participants compared these interventions over 10 sessions and at weekly intervals.
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
In the mesotherapy plus minoxidil group 7/27 participants reported a moderate to sharp increase in hair volume compared to 1/27 in the mesotherapy plus saline group (RR 7.00, 95% CI 0.92 to 53.10; one trial 54 participants).
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
All participants in both groups reported pain, but there were more reports in the mesotherapy plus minoxidil group (authors' reported P < 0.10). The investigators also indicated that there was no difference between the groups regarding the nature of other adverse events such as headache, burning, and itching.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change from baseline to study conclusion in total hair count
The study did not assess this outcome.
Degree of hair shedding from baseline to the end of the study
In the mesotherapy plus minoxidil group 18/27 participants noticed a decrease in the extent of hair loss compared to 8/27 in the mesotherapy plus saline group (RR 2.25, 95% CI 1.19 to 4.27; one trial 54 participants; P = 0.01), which was in favour of mesotherapy plus minoxidil.
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The study did not assess this outcome.
7. Pantovigar one capsule three times a day plus minoxidil 2% twice daily versus minoxidil 2% twice daily
Pantovigar is a natural hair loss supplement. One study, at high risk of bias, evaluated it in combination with topical minoxidil versus minoxidil (2%) monotherapy (NCT01900041). It was conducted over 26 weeks and included 74 participants.
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
In the Pantovigar plus minoxidil group 15 adverse events were reported in 37 participants versus 19 adverse events in 37 participants in the minoxidil "only" group (RR 0.79, 95% CI 0.48 to 1.30; one trial 74 participants).
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change from baseline to study conclusion in total hair count
The study did not assess this outcome.
Degree of hair shedding from baseline to the end of the study
The investigators reported that "changes in percentage telogen rate were comparable between both groups. No statistically significant difference was observed (P = 0.45)."
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The study did not assess this outcome.
8. Minoxidil and oral contraceptive pill (OCP) versus cyproterone acetate and OCP
One study, including 66 participants and assessed as high risk of bias, compared the effects of these interventions (Vexiau 2002).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
Three participants in the minoxidil combined with OCP group reported pruritus, and one reported weight gain. A further participant in the cyproterone acetate group reported weight gain.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change from baseline to study conclusion in total hair count
Minoxidil combined with the OCP was more effective than cyproterone acetate. Mean change from baseline in the combination group was 7.7 hairs (9.3 SD) and in the cyproterone acetate group −0.2 hairs (6.7 SD). The MD in total number of hairs per 0.36 cm² between the minoxidil group was 7.90 (95% CI 3.70 to 12.10).
Degree of hair shedding from baseline to the end of the study
Both groups reported large decreases in self‐assessed mean hair loss; rated on a VAS, these were −28 (24 SD) for the minoxidil group versus −24 (26 SD) mm for the cyproterone acetate group. These found there was no difference in the reduction of hair loss between the 2 treatment groups (MD −4.00, 95% CI −17.52 to 9.52)
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (thickness and density)
The study did not assess this outcome.
9. Estradiol valerate topical ointment (3%) for 12 weeks versus estradiol valerate topical ointment (3%) for 24 weeks versus placebo vehicle only for 24 weeks
A single study (75 participants) at unclear risk of bias provided minimal data for this comparison (Georgala 2004).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
Two of 25 participants in the 12‐week group reported mild pruritus itching on the scalp compared to 4/25 in the 24‐week group and 2/25 in the placebo group. In the 24‐week treatment group, two participants experienced postmenopausal uterine bleeding, which resulted in their withdrawal from the study.
Secondary outcomes
The study did not assess any of our secondary outcomes.
10. Octyl nicotinate (0.5%) and myristyl nicotinate (5%) versus placebo
A single study (60 participants) at unclear risk of bias compared the safety and efficacy of octyl nicotinate (0.5%) and myristyl nicotinate (5%) versus placebo over six months (Draelos 2005).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
Draelos 2005 did not report data separately for adverse events for each intervention, only cumulatively as scalp stinging (9), scalp burning (2), scalp itching (12), scalp redness (4), and eye irritation (7). These occurred in both placebo and active intervention groups, and the study authors concluded that they were related to the volatile vehicle, and not the active constituent.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
At six months, 22/40 participants treated with the combination therapy showed an increase in hair fullness compared to 4/20 of those treated with placebo (RR 2.75, 95% CI 1.10 to 6.90; one trial, 60 participants).
Change from baseline to study conclusion in total hair count
The study did not assess this outcome.
Degree of hair shedding from baseline to the end of the study
The study did not assess this outcome.
Cosmetic appearance of the hair or participant satisfaction
Although no data were reported, the investigators referred to a "positive trend" in the participants' assessments of the appearance of their hair, but indicated that this did not reach significance (investigators' reported P value = 0.05).
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The study did not assess this outcome.
11. Fulvestrant 70 mg/mL versus placebo
A single study (70 participants) at low risk of bias provided limited outcome data for this comparison (Gassmueller 2008), and concluded that fulvestrant was ineffective after 16 weeks in the treatment of FPHL.
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
There was no statistically significant difference in the number of adverse events between the interventions. These were mild, i.e. cold and headache, and similar in the fulvestrant group (10/34) and the placebo group (16/36) (RR 0.66, 95% CI 0.35 to 1.25; one trial, 70 participants). The study reported that both fulvestrant and the vehicle were well‐tolerated.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change from baseline to study conclusion in total hair count
The study did not assess this outcome.
Degree of hair shedding from baseline to the end of the study
The study did not assess this outcome.
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
No statistically significant differences were reported in terms of percentage change from baseline in cumulative hair thickness, nor in hair density. So there was no evidence in favour of fulvestrant over placebo.
12. Adenosine versus placebo
Only one study, which was at unclear risk of bias and included 30 participants, evaluated the effect of this intervention in the treatment of FPHL for a 12‐month duration (Oura 2008).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
The study evaluated self‐assessments with a questionnaire, however the investigators only provided P values. These indicated that for hair growth there was no statistically significant difference between the two treatment groups at month 12 (investigators reported "P value = 0.081, Mann–Whitney U‐test")
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
There were no adverse events in either group.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
The dermatologist‐rated assessments indicated that four out of 15 participants in the adenosine treatment group improved compared to 2/15 in the placebo group (RR 2.00, 95% CI 0.43 to 9.32; one trial, 30 participants), and that these results were reasonably consistent with the investigator‐rated assessments.
Change from baseline to study conclusion in total hair count
The investigators reported that "hair density did not change significantly between the groups at any time point".
Degree of hair shedding from baseline to the end of the study
The investigators reported that at the end of the study there was a statistically significant difference in favour of the adenosine group regarding the prevention of hair loss (authors' reported "P value = 0.036, Mann–Whitney U‐test"). However, they did not provide any data to support this conclusion.
Cosmetic appearance of the hair or participant satisfaction
Although the 'change in appearance' at 12 months appeared to favour adenosine (investigators' reported "P value = 0.048, Mann–Whitney U‐test"), there was no statistically significant difference in satisfaction between the two groups at the end of the study.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The thick hair ratio (number of hairs thicker than 80 μm in diameter/thinner hairs) did not improve in the adenosine group over 12 months, but it did show a decrease in the placebo group (investigators reported "P value = 0.002, Student t‐test") with a difference between the two groups in favour of adenosine (investigators reported "P value = 0.04, Student t‐test").
13. Botanical tincture versus placebo
One study compared a botanical tincture (which contained Thuja occidentalis extract, Swertia extract, pantotenilethylether, 4‐pyrrolidine 2,6‐diaminopyrimidine 1‐oxide, cyanocobalamin, 95% ethanol, saline) to placebo (Shin 2007). Both were applied twice daily for 18 weeks in 33 participants. The limited data that were reported for this study, which we assessed as at unclear risk of bias, indicated a lack of efficacy for this treatment.
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
The improvement score based on a VAS scale, rated from zero to 10, did not show a statistically significant difference between the groups (with a score of 4.4 (2.47 SD) in the botanical tincture group versus 4.5 (2.80 SD) in the placebo group.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
There were no adverse events reported in either group.
Secondary outcomes
Participants with investigator‐rated clinically significant hair regrowth
After 18 weeks the score in the botanical tincture group was 1.0 (0.82 SD) with a score of 1 representing a 0 to 25% improvement. In the placebo group the score was 0.8 (0.80 SD), which indicated no change or worse.
Change from baseline to study conclusion in total hair count
The investigators reported no statistically significant change in hair count compared to baseline for either group.
Degree of hair shedding from baseline to the end of the study
According to the investigators the degree of hair shedding, which was assessed using a VAS, was 5.0 (2.89 SD) in the botanical tincture group compared to 4.4 (3.20 SD) in the placebo group. There was no statistically significant difference between the groups.
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The investigators reported no statistically significant change in hair diameter compared to baseline for either group.
14. Bimatoprost different concentrations versus vehicle
One five‐armed study at unclear risk of bias, which included a total of 306 participants, evaluated the different concentrations of bimatoprost topical applications versus vehicle (NCT01325350). Three arms included bimatoprost formulation A, B, and C without further specification, and the other two arms were minoxidil and vehicle. We have reported the comparisons versus minoxidil under comparison 22, and minoxidil versus vehicle under comparison 1.
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
This was rated on a seven‐point scale of the Subject Self Assessment in Alopecia (SSA) score. Accordingly, 12/61 participants considered they had a moderate to great increase in hair regrowth in bimatoprost formulation A, 14/61 in formulation B, and 14/61 in formulation C versus 13/61 in the vehicle group. None of the comparisons showed a statistically significant difference compared to vehicle or compared to any of the bimatoprost concentration, which suggested that according to participants the treatment is ineffective.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
In the bimatoprost formulation A group 21/61 participants reported adverse events, with five experiencing hypertrichosis; 21/61 in formulation B (one hypertrichosis), 18/61 in formulation C (no hypertrichosis); and 21/61 in the vehicle group (one hypertrichosis). There was no statistically significant difference between any of the treatment arms.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
Based on the Investigator's Global Assessment scale, the investigators judged that 10/61 in the bimatoprost formulation A group had a moderate to greatly increased hair regrowth, 8/61 in formulation B group, 10/61 in formulation C group, and 10/61 in the vehicle group, with no statistically significant difference between any of the groups. These assessments were in agreement with the participants' judgements.
Change from baseline to study conclusion in total hair count
The mean change in hair count was not clinically meaningful in any group. The mean change from baseline in hair count was 0.4 (17.10 SD) in the bimatoprost formulation A group, 3.5 (18.21 SD) in formulation B group, 4.3 (16.82 SD) in the formulation C group, and 1.1 (20.44 SD) in the vehicle group.
Degree of hair shedding from baseline to the end of the study
The study did not assess this outcome.
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The mean change from baseline in target area hair width was 0.13 (1.198 SD) mm/cm² in the bimatoprost formulation A group, 0.19 (1.067 SD) mm/cm² in group B, 0.30 (1.263 SD) mm/cm² in group C, and 0.07 (1.1183 SD) mm/cm² in the vehicle group. No treatment arm showed a statistically significant difference of vehicle or against the other formulations.
15. Bimatoprost different concentrations versus minoxidil
The same five‐armed study as in comparison 21 evaluated the different concentrations of topical applications versus minoxidil 2% (NCT01325350).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
This was rated on a seven‐point scale of the SSA. Accordingly, 12/61 participants considered that they had a moderate to great increase in hair regrowth with the bimatoprost formulation A, 14/61 in formulation B, 14/61 in formulation C, versus 20/61 in the minoxidil group. Although the minoxidil group scored better on the SSA score, there was no statistically significant difference between any of the comparisons.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
Adverse events reported were 21/61 in bimatoprost formulation A, with five experiencing hypertrichosis; 21/61 in formulation B (one hypertrichosis), 18/61 in formulation C (no hypertrichosis); and 25/62 in the minoxidil group (three hypertrichosis). There was no statistically significant difference between any of the treatment arms.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
Based on the Investigator's Global Assessment scale, the investigators judged that 10/61 in the bimatoprost formulation A group had a moderate to greatly increased hair regrowth, 8/61 in formulation B group, 10/61 in formulation C group, and 10/61 in the minoxidil group, with no statistically significant difference between any of the groups. This is in agreement with the participants judgements.
Change from baseline to study conclusion in total hair count
In the bimatoprost formulation A group, the mean change in hair count from baseline was 0.4 (17.10 SD) in the bimatoprost formulation A group, 3.5 (18.21 SD) in formulation B group, 4.3 (16.82 SD) in the formulation C group, and 13.6 (18.72 SD) in the minoxidil group. There was a statistically significant MD for each of the comparisons bimatoprost versus minoxidil in favour of minoxidil. The MD that compared formulation A versus minoxidil was −13.20 (95% CI −19.53, to −6.87; P < 0.0001). For formulation B versus minoxidil the MD was −10.10 (95% CI −16.63 to −3.57; P = 0.002), and for formulation C versus minoxidil the MD was −9.30 (95% CI −15.59 to −3.01; P = 0.004).
Degree of hair shedding from baseline to the end of the study
The study did not assess this outcome.
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The mean change from baseline in target area hair width was 0.13 (1.198 SD) mm/cm² in the bimatoprost formulation A group, 0.19 (1.067 SD) mm/cm² in group B, 0.30 (1.263 SD) mm/cm² in group C, and 0.87 (1.315 SD) mm/cm² in the minoxidil group. There was a statistically significant difference in favour of minoxidil in all comparisons. The MD of bimatoprost formulation A versus minoxidil was −0.74 (95% CI −1.18 to −0.30; P = 0.001), bimatoprost formulation B versus minoxidil MD −0.68 (95% CI −1.10 to −0.26; P = 0.002), and formulation C versus minoxidil MD −0.57 (95% CI −1.01 to −0.13; P = 0.01).
16. Cyproterone versus flutamide versus finasteride
One study that included 36 participants examined these comparisons (Carmina 2003), but we assessed it as at high risk of bias. The investigators reported that flutamide at a dose of 250 mg daily provided a modest improvement in alopecia after one year, whereas cyproterone acetate 50 mg and finasteride 5 mg were not considered effective.
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
Hair regrowth was participant‐assessed with a standardised questionnaire (Barber 1998). Two of 12 participants in the cyproterone group reported improvement in hair growth compared to 3/12 in the flutamide group and 1/12 in the finasteride group.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
No adverse events were reported for these comparisons; however, in the flutamide group, 2/12 participants had a slight increase in liver enzymes, which is considered to be a common side‐effect of this intervention.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change from baseline to study conclusion in total hair count
The study did not assess this outcome.
Degree of hair shedding from baseline to the end of the study
In the flutamide group, 8/12 participants reported an improvement in slowing down of hair loss versus 3/12 in the cyproterone acetate group and 1/12 in the finasteride group.
Cosmetic appearance of the hair or participant satisfaction
Improvement of hair appearance was reported by 3/12 participants in the flutamide group compared to 1/12 in the cyproterone acetate and none in the finasteride group. The flutamide participants were also more satisfied with their therapy (5/12) versus 3/12 in the cyproterone acetate and the finasteride group (1/12).
Change in quality (or pattern) of hair regrowth (e.g. thickness)
Baseline to end of study hair density was investigator‐assessed on a seven‐point scale (−3 = greatly decreased to 3 = greatly increased). In the cyproterone group, this scale was rated after 12 months as 0.5 (0.2 SD) in the flutamide group 0.9 (0.2 SD) and in the finasteride group 0.1 (0.2 SD).
17. Finasteride (1 mg) versus placebo
Three studies examined this comparison (Keene 2011; Price 2000; Whiting 1999). We assessed Keene 2011 as at low risk of bias, Price 2000 at unclear risk of bias, and Whiting 1999 at high risk of bias. See also 'Summary of findings' table 3 (Table 3).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
Participant‐ and investigator‐rated assessments in Price 2000 were largely in agreement that finasteride was no more effective than placebo. In the finasteride group, 30/67 participants considered themselves improved versus 33/70 in the placebo group (RR 0.95, 95% CI 0.66 to 1.37; one trial, 137 participants). Keene 2011 and Whiting 1999 did not assess this outcome.
Change in QoL
None of the studies assessed this outcome.
Adverse effects, safety, and tolerability
Both groups in Price 2000 reported a similar number of adverse events: 53/67 in the finasteride group versus 55/70 in the placebo group (RR 1.03, 95% CI 0.45 to 2.34; one trial, 137 participants). Several of the adverse events reported in this study are common in postmenopausal women and are not necessarily drug‐related. The placebo group reported more adverse events, such as headache and depression. Keene 2011 and Whiting 1999 did not assess this outcome.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
Only one study provided data for this outcome (Price 2000). The investigators reported that 10/67 participants in the finasteride group showed a moderate increase versus 13/70 in the placebo group, which included one participant with a greatly increased change in hair growth (RR 0.77, 95% CI 0.31 to 1.90; one trial, 137 participants). Keene 2011 and Whiting 1999 did not assess this outcome.
Change from baseline to study conclusion in total hair count
At 12 months, both treatment groups in Price 2000 (data on 125 participants for this outcome) demonstrated a similar degree of hair loss by hair count, with a mean decrease from baseline in hair count of 8.7 hairs in the finasteride group versus 6.6 in the placebo group, but no SDs were reported. Individual patient data were provided in Keene 2011 for two different hair counts. The mean increase from baseline in hair count 1 was 17.86 hairs/cm², and 11.86 hairs/cm² in hair count 2 for seven participants on finasteride, and one hair, and ‐4.4 hairs for the two hair counts for the five participants on placebo. In Whiting 1999, there was an increase of 0.2 (0.9 SD) in change from baseline in total hair count of terminal hairs in the finasteride group (44 participants) versus 1.1 (0.9 SD) in the placebo group (50 participants) (MD −0.90, 95% CI −1.2 to −0.54; P < 0.00001), which was in favour of placebo. However, the difference of one hair in a 4 mm punch biopsy is not clinically important.
Degree of hair shedding from baseline to the end of the study
Although the investigators in Price 2000 provided no data, they reported that there was no statistically significant difference in the slowing down of hair loss between the two groups at the end of the study. The other two studies did not assess this outcome.
Cosmetic appearance of the hair or participant satisfaction
Twelve out of 67 participants on finasteride were satisfied with their hair overall compared to 16/70 in the placebo group (RR 0.78, 95% CI 0.40 to 1.53; one trial, 137 participants), which was not a statistically significant difference (Price 2000). The other two studies did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g thickness)
The studies did not assess this outcome
18. Nutritional supplement versus no supplement
One study, which we assessed as at high risk of bias and included 120 participants, investigated the efficacy of a nutritional supplement that contained 460 mg fish oil, 460 mg black currant seed oil, 5 mg vitamin E, 30 mg vitamin C, and 1 mg lycopene versus no supplement during six months (Le Floc'h 2015).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
Only data were reported for the group that received the supplement and 69/80 participants reported a moderate to large increase in hair density, and the investigators reported P < 0.001. These data would suggest a huge beneficial effect for the supplement. However, these numbers are unusable without the data from the comparator group. As we were unable to retrieve these from the investigators, they cannot be further analysed.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
There were no adverse events reported in either group.
Secondary outcomes
Participants with investigator‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change from baseline to study conclusion in total hair count
The study did not assess this outcome.
Degree of hair shedding from baseline to the end of the study
Hair loss according to the self‐assessments of the participants decreased in 71/80 women in the supplement group compared to 27/40 in the group that received no treatment (RR 1.31, 95% CI 1.05 to 1.65; one trial, 120 participants; P = 0.02) in favour of the supplement group.
Cosmetic appearance of the hair or participant satisfaction
Data were only reported for the supplement group, which indicated that 68/80 women were satisfied, but data for the untreated group were missing.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The study assessed hair density on a seven‐point Likert scale, rated from −3 to 3, and showed that 22 women had a moderate increase in hair density and just one patient in the supplement group experienced a "great increase" versus no improvement at all in the 'no‐treatment' group (RR 23.79, 95% CI 1.48 to 381.88; one trial, 120 participants; P = 0.03).
19. Low level laser comb versus sham device
Two studies evaluated laser combs versus sham device therapy for three times a week for 12 minutes for a period of 26 weeks. Jimenez 2014a (78 participants) used a nine‐beam laser comb, and Jimenez 2014b (63 participants) used a dual 12‐beam laser comb. We assessed the studies as at high risk and unclear risk of bias, respectively. Although there appeared to be a statistically significant difference in the increase in hair count, this was not reflected in the participant assessments. See 'Summary of findings' table 4 (Table 4).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
Both studies combined the data that reported minimally improved and improved hair regrowth, and therefore we cannot be certain to what extent participants considered these improvements to be clinically relevant. In Jimenez 2014a 36/53 in the nine‐beam laser comb group reported minimal improvement of their hair loss versus 11/25 in the sham device group (RR 1.54, 95% CI 0.96 to 2.49; one trial, 78 participants). In Jimenez 2014b 26/42 participants in the 12‐beam laser comb group considered their hair loss minimally improved or improved versus 11/21 in the sham device group (RR 1.18, 95% CI 0.74 to 1.89, one trial, 63 participants).
Change in QoL
The studies did not assess this outcome.
Adverse effects, safety, and tolerability
The report in both studies also included two studies in men. The adverse events were not addressed per study, and the investigators reported that there were "no serious adverse events in any study and laser comb‐related adverse events consisted of dry skin (5.1 %), pruritus (2.5 %), scalp tenderness (1.3 %), irritation (1.3 %), and a warm sensation at the site (1.3 %)."
Secondary outcomes
Participants with investigator‐rated clinically significant hair regrowth
The studies did not assess this outcome.
Change from baseline to study conclusion in total hair count
The mean change from baseline in the nine‐beam laser comb group was 20.2 (11.2 SD) hairs/cm² versus 2.8 (16.5 SD) in the sham device group with a MD of 17.40 hairs/cm² (95% CI 9.74, to 25.06; P < 0.00001), which is a statistically significant difference (Jimenez 2014a). The data in Jimenez 2014b were similar with an increase of 20.6 (11.6 SD) hairs/cm² in the 12‐beam laser comb group and 3.0 (9.3 SD) in the sham device group (MD 17.60 hairs/cm², 95% CI 11.97, to 23.23; P < 0.00001).
Degree of hair shedding from baseline to the end of the study
The studies did not assess this outcome.
Cosmetic appearance of the hair or participant satisfaction
At 26 weeks 31/53 participants in the nine‐beam laser comb group felt there was minimally improved or improved thickness or fullness of the hair compared to 10/25 in the sham device group (RR 1.46, 95% CI 0.86 to 2.49; one trial 78 participants) (Jimenez 2014a). In Jimenez 2014b 24/42 in the nine‐beam laser comb group experienced minimal improvement or improvement versus 9/21 in the sham device group (RR 1.33, 95% CI 0.76 to 2.33; one trial, 63 participants).
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The studies did not assess this outcome.
20. Low level laser in bicycle‐helmet like apparatus versus sham helmet
One study at low risk of bias in 47 participants evaluated these interventions for 25 minutes every other day over 16 weeks (Lanzafame 2014). The helmet in the active treatment group contained 21, 5 mW diode lasers (655 ± 5 nm) and 30 LEDS (655 ± 20 nm), and showed that the helmet with the low level laser improved hair count.
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
No adverse events were reported in either intervention arm.
Secondary outcomes
Participants with investigator‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change from baseline to study conclusion in total hair count
The mean change from baseline was 100.3 hairs per 2.85 cm² (53.4 SD) in the laser helmet group versus 23.9 (30.1 SD) in the sham device group with a MD of 76.40 hairs per 2.85 cm² (95% CI 50.91 to 101.89; P < 0.00001), which favours the laser helmet group
Degree of hair shedding from baseline to the end of the study
The study did not assess this outcome.
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The study did not assess this outcome.
21. Handi‐Dome laser versus incandescent red light source
One study in 44 women, which we assessed as at unclear risk of bias, compared these interventions which consisted of a single, 30 minute treatment, every other day over 16 weeks (NCT01967277) .
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
No adverse events were reported in either intervention arm.
Secondary outcomes
Participants with investigator‐rated clinically significant hair regrowth
The study did not assess this outcome.
Change from baseline to study conclusion in total hair count
The Handi‐Dome laser group showed a larger increase in hair count than the red light group with a mean change of 89.9 (63.3 SD) in the Handi‐Dome laser group versus 18.5 (24.4 SD) in the red light group (MD 71.40, 95% CI 41.08 to 101.72; P < 0.00001).
Degree of hair shedding from baseline to the end of the study
The study did not assess this outcome.
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The study did not assess this outcome.
22. Pulsed electrostatic field versus sham
One poorly reported trial, which we assessed as at unclear risk of bias, provided independent patient data (IPD) for the six female participants (Policarpi 1993).
Primary outcomes
Proportion of participants with self‐rated clinically significant hair regrowth
Two out of four participants in the active treatment group showed a "significant improvement" at 36 weeks compared to neither of the two participants in the sham group.
Change in QoL
The study did not assess this outcome.
Adverse effects, safety, and tolerability
The study did not assess this outcome.
Secondary outcomes
Proportion of participants with investigator‐rated clinically significant hair regrowth
The investigator‐rated assessments indicated there was no clinically "significant hair growth" observed in the participants in both treatment arms.
Change from baseline to study conclusion in total hair count
The percentage change in mean hair count from baseline at 36 weeks in the active group was 2.96 in the first participant, and 16.95%, 4.67%, and 3.37% in subsequent participants. In the two participants in the sham group, the percentage change was 1.15% and 1.45%.
Degree of hair shedding from baseline to the end of the study
The study did not assess this outcome.
Cosmetic appearance of the hair or participant satisfaction
The study did not assess this outcome.
Change in quality (or pattern) of hair regrowth (e.g. thickness)
The study did not assess this outcome.
Discussion
Summary of main results
Forty‐seven studies, which examined 5290 participants, met the inclusion criteria of this Cochrane review. Only one included study assessed one of our key patient‐preferred outcomes, 'quality of life' (QoL) (Lucky 2004). Most included studies focused on change in total (non‐vellus) hair count. However, although this may provide a quantifiable, objective, and more readily intelligible outcome, it is considered to be physician‐preferred, rather than an outcome directed towards addressing participants' preferences. Other frequently‐reported outcomes included participant‐assessed improvement of hair regrowth, as well as physician‐assessed improvement, and adverse events. Female pattern hair loss (FPHL) can be distressing, and is known to impact on QoL. Thus, the importance of assessing the efficacy of interventions targeted at improving this key outcome should not be underestimated (Biondo 2010). Pooling of data was only feasible for a limited number of the outcomes reported in the included studies and was confined to those that evaluated the efficacy of minoxidil compared to placebo and minoxidil (2%) versus minoxidil (5%).
Based on the findings of this review, the only interventions that appeared to demonstrate a measure of efficacy were minoxidil (2% and 5% concentrations) with the quality of evidence being mainly moderate to low ('Summary of findings' table 1; 'Summary of findings' table 2). Both concentrations illustrated a good safety profile, but there is wide acknowledgement that doses in excess of 60 mg a day may lead to an increase in the number of adverse effects. Therefore, the application of 1 mL minoxidil 2% (20 mg/mL) twice daily or 1 mL minoxidil 5% (50 mg/mL) once daily should not be exceeded and the threshold is reached quicker with the 5% dosage. After discontinuation of treatment, renewed hair loss is likely to reoccur within three months and all beneficial effects will have disappeared in six months (Olsen 2005; Torres 2015).
Finasteride appeared to have a limited to no effect, but the results were inconsistent across the three studies with the quality of evidence rated low ('Summary of findings' table 3). Finasteride is not approved for use in women and has well known side effects such as libido reduction, breast tenderness, depression, and abnormalities of the external genitalia of a male foetus when used in pregnant women. Low‐level laser therapy showed an increase in total hair count, but this was not supported by both the participant‐assessments of improvement ('Summary of findings' table 4; moderate to low quality evidence).
For further details see the 'Summary of findings' tables (Table 1; Table 2; Table 3; Table 4).
Overall completeness and applicability of evidence
The studies included in this review, which evaluated a range of interventions, did not provide sufficient data to enable fair and reliable comparisons to be made for any one single intervention against another for a specific outcome, with the exception of minoxidil 2% and 5%, which, based on the evidence available, appeared to be safe and effective in the treatment of FPHL.
Most interventions were evaluated in a single study, and none of the studies addressed more than a very limited number of our outcomes, which illustrates gaps in the overall completeness of the evidence. The quality of data reporting and its analysis was variable across the included studies.
Mean change in hair count and adverse events were the most commonly addressed outcomes, followed by participant‐ and physician‐assessments of improvement of hair regrowth. However, there was a lack of consistency in the choice and assessment of other outcomes across the studies. Although it is generally acknowledged that renewed hair shedding occurs relatively soon after discontinuation of treatment, none of the studies reported data on the sustainability of the treatment effect after the end of the study, which is an outcome of some considerable importance to participants. Furthermore, none of the studies, except Lucky 2004, reported the possible impact of hair regrowth reflected by a decrease in time spent by the women on hair styling, or the use of wigs.
We identified several ongoing studies that may eventually help to fill in some of the gaps in the evidence for the efficacy or otherwise of some of the other interventions, e.g. spironolactone, plasma rich in growth factors, and injections of autologous dermal and epidermal cells into the balding scalp (see the 'Characteristics of ongoing studies' section). However, the research institute that was conducting these 10 studies on these cell injections has gone into liquidation and we were unable to access further details.
Quality of the evidence
Limitations in study design and implementation
Although the study design in the included studies appeared to have been at best adequate, our study‐level assessments of the risk of bias for a number of the domains in several of these studies revealed some of the limitations in their implementation, which we have reported in the 'Risk of bias in included studies' section of this review.
There was considerable variation in how well the studies were reported, and in particular the methods trials used to generate the sequence, to conceal the allocation, and the measures taken to blind investigators and participants. These factors, compounded with our unsuccessful attempts to contact many of the investigators for additional information, created difficulties in making accurate assessments of the risk of bias in more than 60% of the included studies.
In many instances, the key outcomes assessed in the included studies provided limited data, much of which we could not pool except for minoxidil 2% and 5%, and, consequently, did not allow any wider assessment or comparison of the effects of the interventions across the studies.
Indirectness of the evidence
The participants in the included studies were, in general, a clinically representative sample matching the inclusion criteria; therefore, we did not have any significant concerns about the appropriateness of participants identified in the review (see the 'Characteristics of included studies' section).
Twenty‐nine of the 47 studies included in this review were placebo‐controlled trials, which may only provide limited evidence on the advantages or disadvantages of new relative to existing interventions. Physicians need to have access to information about the benefits and harms of individual treatments, as well as the comparative efficacy of these interventions, and direct comparison trials are more likely to provide additional evidence that is both relevant and direct.
Patient‐reported outcomes (PROs) are a prerequisite for informing evidence‐based decision‐making, but the importance of PROs — specifically those used in evaluating the impact of interventions on QoL and which are of direct relevance to patients — appears to have been underestimated by the investigators in most included studies. A validated disease‐specific tool for the assessment of QoL in women with FPHL (Dolte 2000) has been available for many years, yet none of the included studies appear to have recognised its value as a reliable instrument that can be used for assessment of this crucially important outcome. The one study that evaluated the impact of the interventions on QoL utilised a simple questionnaire (Lucky 2004), and, as with most of the PROs assessed in the included studies, this did not satisfy some of the more fundamental criteria provided in the 'checklist for describing and assessing PROs in clinical trials' (see Table 7).
Inconsistency of the results
The low number of studies investigating similar interventions, with the exception of minoxidil versus placebo, and minoxidil (2%) versus minoxidil (5%) did not permit pooling of data for most of the comparisons. Therefore, any inferences about the inconsistency of the results could only be drawn from these comparisons. We investigated the heterogeneity between the studies for these comparisons and report these in the Effects of interventions as well as in Sensitivity analysis. sections of this review.
Imprecision of the results
Of the 47 studies included in this review, nine studies provided data for the comparison of minoxidil versus placebo. We downgraded the quality of evidence for serious imprecision for three of the separate outcomes, i.e. QoL, adverse events and degree of hair shedding, due to low sample size or low occurrence of events. In the comparison minoxidil 2% versus minoxidil 5% that covered four studies, we downgraded the quality of evidence for serious imprecision for the proportion of participants with self‐rated clinically significant hair regrowth, QoL, and change from baseline to study conclusion in total hair count for having wide CIs or low sample size. In the comparison finasteride versus placebo and the comparison of low‐level laser comb versus sham device, we downgraded the quality of evidence several times for serious imprecision for not meeting the optimal information size (see the 'Summary of findings' tables; Table 1; Table 2; Table 3; Table 4).
Publication bias
Based on a visual assessment of funnel plots in each case, there was no evidence of asymmetry in Analysis 1.1,Analysis 1.2, and Analysis 1.3. However, in Analysis 1.4, examination of the funnel plot (Figure 6) revealed asymmetry, which was caused by the inclusion of one small study that randomised eight participants and reported extremely positive results favouring minoxidil (Price 1990). After we investigated the individual participant data from the study, it remained unclear if the large treatment effect was the result of publication bias, small‐study effects, or an artefact of natural variability (see Section 10.4.1 of the Cochrane Handbook for Systematic Reviews of Interventions, Higgins 2011). To assess the impact of this study, we performed a sensitivity analysis, which, after exclusion of the study, resulted in little change to the overall treatment effect (see Analysis 1.5).
Potential biases in the review process
We made every attempt to limit bias in the review process by ensuring a comprehensive search for potentially eligible studies. The review authors' independent assessments of eligibility of studies for inclusion in this review and extraction of data minimised the potential for additional bias beyond that detailed in the 'Risk of bias in included studies' tables. The incompleteness of some of the reports and our inability to obtain clarification of certain trial details or to resolve ambiguities in the reports may have contributed to some bias in their assessment, but, where these conditions applied, we explicitly stated this in the review text. The effects of language bias on the identification and selection of studies for inclusion in a systematic review is widely recognised; therefore, we ensured that any studies not in the English language were translated so that we could assess them for eligibility.
Agreements and disagreements with other studies or reviews
We retrieved one systematic review, Hassani 2012, which although published in 2012 only searched up to August 2008, excluded studies with small sample size and studies not in the English language, and did not reflect the extent of our current up‐to‐date searches. The review also lacked methodological rigour i.e. did not report prespecified outcomes, included an incomplete 'Risk of bias' assessment, and lacked a rating of the quality of the evidence.
We also identified several literature reviews (Avci 2013; Birch 2002; Camacho‐Martínez 2009; Dinh 2007; Leavitt 2008; Olsen 2005; Price 2003; Trüeb 2010; Varothai 2014) and three guidelines (Blumeyer 2011; Drake 1996; Lee 2013b) that covered aspects of the diagnosis and management of FPHL in women. Although the reviews were a valuable resource to answer background questions covering the pathogenesis, classification, and epidemiology of the condition, none included a systematic search of the literature, nor a critical appraisal of the studies cited as references in support of the various treatment options described. The two guidelines provided comprehensive clinical recommendations on the effectiveness of a range of interventions for both men and women. However, to ensure that a guideline provides balanced information on the benefits and limitations of the therapeutic interventions being evaluated, its process of development should be transparent, robust, and reproducible; it should also clearly demonstrate that the supporting evidence was systematically reviewed (Nasser 2011).
The earlier of the three guidelines, Drake 1996, which was produced by the American Academy of Dermatology's Guidelines/Outcomes Committee, lacked transparency and reproducibility, in that it did not report on the methodological approach used by its developers but only that the guideline reflected the "best data available at the time the report was prepared". However, the developers wisely cautioned that "the results of future studies may require alteration of its conclusions and recommendations."
The guideline by Lee 2013b targeted Asian participants, both male and female, and provided little to no detail of how the evidence was gathered and summarized, and no indication of how the recommendations made were based on the evidence.
The 'Evidence‐based (S3) guideline for the treatment of androgenetic alopecia in women and in men' (Blumeyer 2011) was commissioned by the European Dermatology Forum to evaluate the "efficacy of the currently available therapeutic options". Although its development relied heavily on a formal consensus process negotiated between members of the guideline group and was therefore deemed reasonably transparent, we are in disagreement over the robustness of the methodological approach used in its development. Lack of clarity in the process, and ultimately its reproducibility, was illustrated by the incomplete reporting of some important steps taken in study assessment, handling of missing trial data, analysis and interpretation of results, and summary of the adverse events.
We recognise an important area of discord with the method of grading of evidence for this guideline, which was based on study design and "summarised in a level of evidence" that combined the study design with a quality measure described by the developers as "mainly consistent results". However, these consistencies or inconsistencies, or indeed how they were defined or assessed in any of the individual studies, were unreported. It remains unclear if these factors were a potential source of bias, because, unlike in our systematic review, no 'Risk of bias' assessments were undertaken and the guideline developers did not report anything. Critically, four of the key studies underpinning the guideline recommendations for minoxidil were graded as "A₂ evidence resulting in an evidence level 1", which was inconsistent with our judgment that they were all categorised as at high risk of bias. A further seven studies were graded as B level evidence ("randomised, clinical studies of lesser quality"), but these quality criteria were also not clearly reported. Also, from the rather limited detail provided by the developers, a number of these assessments were not in agreement with the 'Risk of bias' assessments performed in this Cochrane review (see the 'Assessment of risk of bias in included studies' section).
In making their study level assessments of evidence, the guideline developers also did not appear to have taken into consideration the conceptual differences between methodological quality and reporting quality. Thus, the "level of evidence" in the guidelines was based solely on the methodological quality of the individual trial as reported, with no clear indication if the developers had attempted to contact investigators to clarify missing trial details and data, which would have enabled more robust and exhaustive 'Risk of bias' assessments to be carried out.
We specifically question and are at variance with the guideline developers' decision to summarise studies that combine data from men and women and narratively describe the treatment efficacy directed towards women. More importantly, we draw attention to the data analysis for the efficacy of minoxidil that included only the participants within the active treatment arms, ignorant of any placebo effect, which we have demonstrated can be considerable and is a further point of disagreement with our review. The, possibly unintentional, effect of this (as reported in the guideline) is an implied superiority in efficacy of the minoxidil (2% and 5%) concentrations compared to minoxidil (1%). However, in our systematic review, after accounting for the placebo participants in the analysis, the study that evaluated minoxidil (1%) (Tsuboi 2007) provided results that were the fourth largest treatment effect out of the eight included studies in Analysis 1.4 and, similarly, in Analysis 1.3. Notably, whilst the guideline provided a narrative synthesis of the data, the conclusions in this Cochrane review are inferences derived from a systematic and evidence‐based approach.
In this systematic review, a closer examination of the primary research clearly indicated that the lower concentration of minoxidil was well‐tolerated and without the adverse events associated with the higher concentration (Tsuboi 2007). Benefits and harms are equally important for decision‐making; thus, we noted the rather limited emphasis placed on the discussion of harms in the S3 guideline, in which the adverse events were only reported in a generic narrative as "instruction for use/practicability", lacked a structured analysis, and was in sharp contrast with the more detailed exploration undertaken in this review.
The strength of clinical recommendations in the S3 guideline was based on the level of evidence and a number of other factors, none of which were clearly defined, nor appeared to correspond to the widely‐recognised GRADE (Grading of Recommendations Assessment, Development and Evaluation) approach to developing and presenting recommendations for management of patients (Guyatt 2008). In contrast, we used this method in this review to examine and categorise the quality level of a body of evidence.
Therefore, whilst we concur with the general conclusions reached in both guidelines in terms of direction of treatment effect, we express a level of disagreement with the magnitude, and, more specifically, as reported in the S3 guideline, where it underpins the relevant clinical recommendations for minoxidil.
Authors' conclusions
Implications for practice.
Based on only those studies that are most likely to have provided reliable results (i.e. reproducible, repeatable, and therefore valid), and selecting the most rigorously described and conducted studies, we conclude that there is mainly moderate to low quality evidence to support the efficacy of only one of the interventions for female pattern hair loss (FPHL), notably minoxidil.
Minoxidil (2%) topical solution twice daily appears to be effective and safe, and minoxidil (5%) used once daily may be as effective as minoxidil (2%) used twice daily, which is likely to result in improved adherence. However, the higher concentration of minoxidil (5%) is only registered for therapeutic management of FPHL in a small number of countries worldwide.
Although finasteride continues to be prescribed for treatment of women with FPHL, this therapeutic option does not appear to be supported by current research based on randomised controlled trials. Low‐level laser therapy options are attracting interest but the results so far have been inconsistent.
Clinical decision‐making on the choice of intervention for FPHL should be based on high‐level evidence if it is available, but in the absence of such evidence for any other specific intervention, these decisions should continue to be guided by clinical experience and peoples' individual characteristics and preferences until further evidence for these other interventions becomes available.
In view of the fact that there may be a delay before any treatment effect can be noticed, and as most of the available treatments fail to achieve the desired end result, cosmetic aids and hair transplant surgery need to be included in the decision‐making process. Furthermore, physicians should also try to address the psychosocial impact, coping mechanisms, and QoL issues when treating women with FPHL.
Implications for research.
It is widely perceived that 2% minoxidil is more effective than the 1% concentration, and this is reflected in the fact that the 2% concentration is most frequently registered worldwide for FPHL. However, the results from one study included in this review indicate that 1% minoxidil does not appear any less effective than 2% minoxidil and is also associated with a potentially lower number of adverse events. There was also evidence (mainly moderate to low quality) that 5% minoxidil once daily was as effective as 2% minoxidil twice daily; a factor which may be important in improving adherence in future clinical trials.
There is also an urgent need for high‐quality, well‐designed, and rigorously‐reported studies of other widely‐used treatments, such as spironolactone, finasteride (at different dosages), dutasteride, cyproterone acetate, and laser‐based therapy. Conceivably, some studies listed in the 'Characteristics of ongoing studies' section of this review will be able to provide answers to these remaining questions in the future.
There was wide variability in the conduct and the quality of reporting of many trials. A major area for improvement would be in the standardisation of outcome reporting in any future research. The use of proprietary severity scales and non‐standardised scales significantly hampered our ability to combine study results for a meta‐analysis. Outcomes collected in future trials should be primarily based on a standardised scale of the participant's assessment of the treatment efficacy, and they should also have a greater emphasis on changes in QoL as a result of the interventions. Standardised and uniform scales should be developed and used for physicians' assessments, and these should reliably reflect the proportion of participants with investigator‐rated clinically significant hair regrowth and mean change in total hair count from baseline to the end of the study. Follow‐up studies addressing the sustainability of hair regrowth after discontinuation of treatment should be taken into account as they constitute an important outcome for participants. Another important patient‐reported outcome should be the impact of the hair regrowth reflected by a decrease in the time spent by women on hair styling, including the use of wigs.
Future randomised controlled trials must be well‐designed, well‐conducted, and adequately delivered, with subsequent reporting, including high‐quality descriptions of all aspects of methodology. Rigorous reporting needs to conform to the Consolidated Standards of Reporting Trials (CONSORT) statement, and this will enable appraisal and interpretation of results, and accurate judgements to be made about the risk of bias and the overall quality of the evidence. Although it is uncertain whether reported quality mirrors actual study conduct, it is noteworthy that studies with unclear methodology have been shown to produce biased estimates of treatment effects (Schulz 1995). Adherence to guidelines, such as the CONSORT statement, would help ensure complete reporting.
For further research recommendations based on the EPICOT (evidence, population, intervention, comparison, outcomes, and time) format (Brown 2006), see Table 10.
6. Research recommendations based on a gap in the evidence of the effects of interventions for female pattern hair loss (FPHL).
| Core elements | Issues to consider | Status of research for this review |
| Evidence (E) | What is the current state of the evidence? | This systematic review included 47 RCTs. There is mainly moderate to low quality evidence for the efficacy and safety of topical minoxidil 2% and 5% in the treatment of FPHL. Minoxidil (2%) topical solution twice daily appears to be effective and safe, and minoxidil (5%) used once daily may be as effective as minoxidil (2%) used twice daily, which may result in improved adherence. However, the higher concentration (5%) of minoxidil is only registered for the therapeutic management of FPHL in a small number of countries around the world. |
| Population (P) | Diagnosis, disease stage, comorbidity, risk factors, gender, age, ethnic group, specific inclusion or exclusion criteria, clinical setting | The participants should be aged 18 to 89 years. A distinction between women with and without a hyperandrogenic profile should be made, and between ethnic groups as well as pre‐ and postmenopausal women. Inclusion criteria
Exclusion criteria
|
| Intervention (I) | Type, frequency, dose, duration, prognostic factor | The study duration should be at least 6 months. High‐quality, well‐designed, and rigorously‐reported studies of other widely used treatments, e.g. spironolactone, finasteride (at different dosages), dutasteride, cyproterone acetate, and laser‐based therapy should be included. Information on direct and indirect costs of the interventions should be addressed. |
| Comparison (C) | Type, frequency, dose, duration, prognostic factor | Direct comparison studies of the widely used treatments are warranted. |
| Outcome (O) | Which clinical or patient‐related outcomes will the researcher need to measure, improve, influence, or accomplish? Which methods of measurement should be used? |
Participant's assessment of the treatment efficacy and changes in quality of life using standardised questionnaires, e.g. the Women's Androgenetic Alopecia Quality of Life Questionnaire (WAA‐QOL) (Biondo 2010; Dolte 2000). Standardised and uniform scales should be developed and used for physicians' assessments, and these should reliably reflect proportion of participants with investigator‐rated clinically significant hair regrowth and mean change in total hair count from baseline to the end of the study. Studies should address the sustainability of hair regrowth after discontinuation of treatment. An important patient‐reported outcome should be the impact of the hair regrowth reflected by the time spent by women with FPHL on hair styling, including the use of wigs. |
| Time stamp (T) | Date of literature search or recommendation | 7 July 2015 |
| Study type | What is the most appropriate study design to address the proposed question? |
|
Abbreviations: FPHL: female pattern hair loss.
Feedback
Studies that examined finasteride, 22 July 2016
Summary
A comment was received from Erik von Elm, Co‐director Cochrane Switzerland, that two additional studies that examined finasteride, Keene 2011 and Mazzarella 1997, were not mentioned in the results section (Description of studies: Characteristics of the interventions). The authors agree that this was an omission but stress that the data was nevertheless reported on in the review.
Reply
Both studies should have been listed under Characteristics of the interventions, this is an omission. Thank you for picking this up. They are added now.
However, the study of Mazzarella did not have separate data for women and 61.5% drop‐out in placebo group and is therefore listed in Table 8 (table with no usable or irretrievable data). We e‐mailed principal investigators several times but received no answer. Under effects of interventions first paragraph it is reported that 15 studies did not provide usable data, as well as being listed in Notes section of COI table. So it is clear why we did not report on data of Mazzarella.
Keene is reported in comparison 17 so data are included.
In conclusion, we did not fail to report data on studies where we should have reported data, but it was incorrect not to mention those two studies under characteristics of interventions.
Contributors
Our Co‐ordinating Editor Hywel Williams, our Feedback Editor Urbà González and the lead author Esther van Zuuren.
What's new
| Date | Event | Description |
|---|---|---|
| 10 October 2016 | Feedback has been incorporated | In response to feedback, minor correction in results section; please refer to Feedback section for details. |
History
Protocol first published: Issue 1, 2009 Review first published: Issue 5, 2012
| Date | Event | Description |
|---|---|---|
| 24 May 2016 | New search has been performed | We updated the review; we have added 25 randomised controlled trials |
| 24 May 2016 | New citation required but conclusions have not changed | There has been no significant alteration to the conclusions of the original review |
Acknowledgements
The review authors thank the former authors of this Cochrane review for their contribution to an earlier version: Carlos Cesar Cusmanich, Virginia Fernandes Moça Trevisani, C Whitney Hannon, Hermenio C Lima, Regis Bruni Andriolo, Ben Carter, and Rod Sinclair. We are indebted to the following colleagues for their help with translating studies: Ching‐Chi Chi, Department of Dermatology & Centre for EBM, Chang Gung Memorial Hospital, Puzih, Taiwan and Yu‐Shiun Tsai, Chang Gung Memorial Hospital, Puzih, Taiwan (Chinese into English); Dr Hyemin Pomerantz, Center for Dermatoepidemiology, Department of Veterans Affairs Medical Center, Providence, Rhode Island; Department of Dermatology, Brown University, Providence, Rhode Island for translating three articles from Korean into English. ZF thanks Armin van Buuren for providing the creative atmosphere that was essential to the writing up phase of this review.
The Cochrane Skin Group editorial base wishes to thank Sue Jessop, who was the Dermatology Editor for this review; Thomas Chu who was the Statistical Editor; Ching‐Chi Chi, who was Methods Editor; the clinical referees, Abby Macbeth and Hélio Miot; and the consumer referee, Denise Hayes.
Appendices
Appendix 1. CENTRAL (the Cochrane Library) search strategy
#1 (androgenic alopecia) or (androgenetic alopecia) or (female pattern hair loss) or (female baldness) #2 MeSH descriptor Alopecia explode all trees #3 (androgen*) #4 (#2 AND #3) #5 (#1 OR #4) #6 SR‐SKIN #7 (#5 AND NOT #6)
Appendix 2. MEDLINE (Ovid) search strategy
This strategy also used for AMED and PsycINFO
1. randomized controlled trial.pt. 2. controlled clinical trial.pt. 3. randomized.ab. 4. placebo.ab. 5. clinical trials as topic.sh. 6. randomly.ab. 7. trial.ti. 8. 1 or 2 or 3 or 4 or 5 or 6 or 7 9. (animals not (human and animals)).sh. 10. 8 not 9 11. androgenic alopecia.mp. 12. androgenetic alopecia.mp. 13. (female pattern hair loss or female baldness).mp. 14. exp Alopecia/ 15. androgen$.mp. or exp Androgens/ 16. 14 and 15 17. 11 or 12 or 13 or 16 18. 10 and 17
Appendix 3. EMBASE (Ovid) search strategy
1. random$.mp. 2. factorial$.mp. 3. (crossover$ or cross‐over$).mp. 4. placebo$.mp. or PLACEBO/ 5. (doubl$ adj blind$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name] 6. (singl$ adj blind$).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name] 7. (assign$ or allocat$).mp. 8. volunteer$.mp. or VOLUNTEER/ 9. Crossover Procedure/ 10. Double Blind Procedure/ 11. Randomized Controlled Trial/ 12. Single Blind Procedure/ 13. 1 or 2 or 3 or 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 14. androgenic alopecia.mp. 15. androgenetic alopecia.mp. 16. (female adj pattern adj hair adj loss).mp. [mp=title, abstract, subject headings, heading word, drug trade name, original title, device manufacturer, drug manufacturer name] 17. female baldness.mp. 18. alopecia.mp. or exp Alopecia/ 19. androgens.mp. or exp Androgen/ 20. 18 and 19 21. 16 or 17 or 20 or 15 or 14 22. 21 and 13
Appendix 4. PubMed search strategy
("androgenic alopecia" OR "androgenetic alopecia" OR "alopecia androgenetica" OR ((hair loss OR baldness OR alopecia) AND (androgen OR androgens)) OR "female pattern hair loss" OR "female baldness" OR ("female pattern" AND hairloss)) AND ("Randomized Controlled Trial"[Publication Type] OR "Randomized Controlled Trials as Topic"[Mesh] OR "Controlled Clinical Trial"[Publication Type] OR "Controlled Clinical Trials as Topic"[Mesh] OR randomized OR random* OR "Random Allocation"[mesh] OR placebo OR placebo* OR "Clinical Trials as Topic"[Mesh] OR RCT OR randomly OR factorial OR factorial* OR crossover OR crossover* OR cross‐over OR cross‐over* OR "double blind" OR "double blinded" OR "Double‐Blind Method"[mesh] OR "Single‐Blind Method"[mesh] OR "single blind" OR "single blinded" OR assign* OR allocat* OR volunteer OR volunteer* OR "Clinical Trial"[Publication Type] OR trial OR trials) NOT (animals NOT (human AND animals))
Appendix 5. Web of Science search strategy
TS=((androgenic alopecia OR androgenetic alopecia OR alopecia androgenetica OR ((hair loss OR baldness OR alopecia) AND androgen*) OR female pattern hair loss OR female baldness OR female pattern hairloss) AND (Random* OR Controlled OR Trial* OR placebo* OR RCT OR factorial* OR crossover* OR "cross‐over*" OR "double blind*" OR "Single Blind*" OR assign* OR allocat*))
Appendix 6. AMED (Ovid) search strategy
1. alopecia/ 2. exp Androgens/ 3. alopecia.mp. 4. androgen$.mp. 5. 1 or 3 6. 2 or 4 7. 5 and 6 8. (female and pattern and hair and loss).mp. 9. (female and baldness).mp. 10. (female and pattern and alopecia).mp. 11. or/7‐10 12. randomized controlled trial$/ 13. random allocation/ 14. double blind method/ 15. single blind method.mp. 16. exp Clinical trials/ 17. (clin$ adj25 trial$).mp. 18. ((singl$ or doubl$ or trebl$ or tripl$) adj25 (blind$ or mask$ or dummy)).mp. 19. (placebo$ or random$).mp. 20. research design/ or clinical trials/ or comparative study/ or double blind method/ or random allocation/ 21. prospective studies.mp. 22. cross over studies.mp. 23. Follow up studies/ 24. control$.mp. 25. (multicent$ or multi‐cent$).mp. 26. ((stud or design$) adj25 (factorial or prospective or intervention or crossover or cross‐over or quasi‐experiment$)).mp. 27. 12 or 13 or 14 or 15 or 16 or 17 or 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 28. 11 and 27
Appendix 7. LILACS search strategy
((hair loss OR baldness or alopecia) AND (androgen$)) OR (female and pattern and hair and loss) OR (female and baldness) or (female and pattern and alopecia) These terms were combined with the Controlled clinical trials topic‐specific query filter.
Data and analyses
Comparison 1. Minoxidil versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Proportion of participants with self‐rated at least moderate hair regrowth | 6 | 1148 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.93 [1.51, 2.47] |
| 2 Proportion of participants with adverse events | 6 | 1301 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.34 [0.98, 1.83] |
| 2.1 Topical minoxidil solution (1%) versus placebo | 1 | 280 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.12 [0.61, 2.06] |
| 2.2 Topical minoxidil solution (2%) versus placebo | 4 | 727 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.24 [0.82, 1.87] |
| 2.3 Topical minoxidil solution (5%) versus placebo | 2 | 294 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.05 [0.96, 4.37] |
| 3 Proportion of participants with investigator‐rated at least moderate hair regrowth | 7 | 1181 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.35 [1.68, 3.28] |
| 4 Mean increase in total hair count from baseline | 8 | 1242 | Mean Difference (IV, Fixed, 95% CI) | 13.18 [10.92, 15.44] |
| 4.1 Topical (1% to 2%) minoxidil versus placebo | 8 | 1116 | Mean Difference (IV, Fixed, 95% CI) | 12.97 [10.58, 15.35] |
| 4.2 Topical (5%) minoxidil versus placebo | 1 | 126 | Mean Difference (IV, Fixed, 95% CI) | 15.1 [7.96, 22.24] |
| 5 Mean increase in total hair count from baseline (sensitivity analysis) | 7 | 1234 | Mean Difference (IV, Fixed, 95% CI) | 12.96 [10.69, 15.24] |
| 5.1 Topical (1% to 2%) minoxidil versus placebo | 7 | 1108 | Mean Difference (IV, Fixed, 95% CI) | 12.72 [10.33, 15.12] |
| 5.2 Topical (5%) minoxidil versus placebo | 1 | 126 | Mean Difference (IV, Fixed, 95% CI) | 15.1 [7.96, 22.24] |
Comparison 2. Minoxidil 2% versus minoxidil 5%.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number of adverse events | 4 | 1006 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.91, 1.15] |
| 2 Number of adverse events (sensitivity analysis) | 3 | 699 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.10 [0.99, 1.23] |
| 3 Mean increase in total hair count from baseline | 3 | 631 | Mean Difference (IV, Fixed, 95% CI) | ‐2.12 [‐5.47, 1.23] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Blume‐Peytavi 2007.
| Methods | This was a randomised, parallel group comparative phase IV study for the first 6 months, then those in the comparator group were crossed over to minoxidil for months 7 to 12 Setting Multicentre (4), Germany Date of study Unspecified (12‐month duration) |
|
| Participants | 103 women Mean age = 50.7 years in group I, 45.6 years in group II Inclusion criteria
Exclusion criteria Nothing was reported Randomised 103 participants were randomised (minoxidil 2% group = 52, alfatradiol 0.025% = 51) Withdrawals/losses to follow‐up
Non‐compliant participants were excluded from the analysis Baseline data There was a minimal data set, and baseline data for early withdrawals were unreported |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (4): at baseline 3‐, 6‐, 12‐month recall of the central parietal region of the scalp, defined and marked with a semipermanent tattoo. The area was shaved and assessed by TrichoScan® (Hoffmann 2002), epiluminescence microscopy and digital image analysis. Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared. Although they did not confirm what, if any, support was provided, the intervention under investigation was Regaine ® Frauen (Pfizer Consumer Healthcare). | |
| Declaration of interest | The Principal Investigator, Ulrike Blume‐Peytavi, was an advisor for Pfizer and Galderma R&D. Three investigators; Christian Kunte, Natalie Garcia Bartels, and Rolf Hoffmann were advisors for Pfizer. | |
| Notes | This is a comparative study of minoxidil versus alfatradiol alone for 6 months, with a cross‐over to minoxidil alone for 6 months. No wash‐out period was reported. We only included the first 6 months. It was unclear whether "tolerability of treatment" referred to the satisfaction of the participant, physician, or both. It was rated on a scale of 1 (excellent) to 5 (unsatisfactory). E‐mail contact with the Principal Investigator (PI) suggests that this outcome refers to adverse/side‐effects, rather than satisfaction. We requested that the investigators provide individual patient data, but none were unavailable. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 392): "were randomized online into two treatment groups." Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | The method used to generate the sequence would appear to indicate that intervention allocations could not have been foreseen in advance of, or during, enrolment. Comment: this was probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote (page 392): "open randomized study" Comment: the outcome is likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote (page 392): "open randomized study" Comment: the outcome measurement is likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote (page 392): "Included in the statistical analysis were all patients who appeared at least at visit 1 (baseline) and visit 3 (6 months)" There was a significant amount of incomplete and missing outcome data; it was unclear if these were withdrawals or losses to follow‐up.
Comment: the analysis did not account for the large number of postrandomisation losses of participants, nor the potential carry‐over and period effects due to the cross‐over design in 1 treatment arm. The high attrition rate and the per‐protocol analysis of these data may raise concerns about the reliability of the data as reported. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Blume‐Peytavi 2011a.
| Methods | This was a randomised, investigator‐blind, active‐controlled trial Setting Departments of Dermatology and Allergy, Clinical Research Center for Hair and Skin Science, Charité‐Universitätsmedizin Berlin, Germany Date of study June 2008 to January 2009 (24‐week duration) |
|
| Participants | 114 women Mean age (range) = 49.9 years (23 to 75 years) Inclusion criteria
Exclusion criteria
Randomised 113 participants were randomised (minoxidil 5% group = 56, minoxidil 2% group = 57) Withdrawals/losses to follow‐up There were 13/113 (11.5%) withdrawals/losses to follow‐up: 6/56 (10.7%) in the 5% minoxidil group, and 7/57 (12.3%) in the 2% minoxidil group
Baseline data The mean Savin hair density score was 4.13 in the minoxidil 5% group, and 3.84 in the minoxidil group 2%. There was a higher proportion of participants with more extensive hair thinning (Savin scores of D5 or D6) in the minoxidil 5% group (N = 19) compared to the minoxidil 2% group (N = 9). |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, week 12 and 24 Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review. |
|
| Funding source | Quote (page 1126): "Supported by a medical grant application, Johnson & Johnson Consumer Co Inc." | |
| Declaration of interest | Quote (page 1126): "Dr Blume‐Peytavi is a consultant for Johnson & Johnson Consumer Co Inc. Dr Garcia Bartels was a consultant for Pfizer GmbH Germany until 2008." | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 1127): "24‐week, randomized, investigator initiated and ‐blinded, 2‐arm comparative study" "Participants were randomized (1:1) to treatment with either half a capful of 5% MTF applied once daily or 1 mL of 2% MTS applied twice daily." After e‐mail communication with investigators: the allocation was performed using block randomisation (27 blocks, sequences 4 and 6). Comment: we judged this as adequate. |
| Allocation concealment (selection bias) | Low risk | The method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment, was not reported. Comment: There was insufficient information to permit a clear judgement. After e‐mail communication with investigators: in this study, the allocation concealment was "performed using sequentially numbered, sealed, opaque envelopes, and kept by the project manager of the CRC." Comment: we judged this as adequate. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 1128): "investigator‐blinded." "To ensure investigator blinding, participants were instructed to speak in the presence of an investigator only about 'the product' and not to use the terms 'foam' or 'solution' or to mention how many times per day they used the study product. In addition, each participant was instructed to wash their hair before each study visit to avoid providing the study investigators with any indication as to which product they were using." Comment: the blinding of investigators appeared to have been adequate, but the impact of lack of blinding of participants was unclear. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 1127): "investigator‐blinded." Both investigator and participants were the outcomes assessors. Quote (page 1128): "To ensure investigator blinding, participants were instructed to speak in the presence of an investigator only about 'the product' and not to use the terms 'foam' or 'solution' or to mention how many times per day they used the study product. In addition, each participant was instructed to wash their hair before each study visit to avoid providing the study investigators with any indication as to which product they were using." Comment: reasonable attempts were made to blind outcomes assessors (personnel), but it was not possible to blind participants. It's unclear to what extent the lack of blinding had any impact on the participant‐assessed outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The reasons and number of dropouts/withdrawals (13/113 = 11%) from each group were reported and balanced across both active intervention groups. The data analysis was per‐protocol. Comment: although there was per‐protocol analysis, the low percentage of dropouts posed a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was available on clinicaltrials.gov (NCT00958750 and EUCTR2008‐001770‐33‐DE and MINALO3005). The prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Unclear risk | There was baseline imbalance, with a higher proportion of participants with more extensive hair thinning in the minoxidil (5%) group. We cannot exclude a potential risk of bias. |
Bureau 2003.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Multicentre (4), France Date of study Unspecified (6‐month duration) |
|
| Participants | 93 men/women Mean age = 38.6 ± 8.14 years (standard deviation (SD)) in group I, 40.6 ± 9.32 years (SD) in group II Inclusion criteria
Exclusion criteria Nothing was reported Randomised 93 participants were randomised, data on 69 participants (group I = 31 men/9 women, group II = 21 men/8 women) Withdrawals/losses to follow‐up There were 24/93 (26%) withdrawals/losses to follow‐up.
The losses in each group and number of men/women were unreported Baseline data Average hair density: group I = 152 h/cm², group II = 165 h/cm² |
|
| Interventions |
Intervention
(E2F7 essential oil solution contains:Pimenta racemosa, Rosmarinus officinalis, Myrtus communis, Salvia officinalis, Cedrus atlantica, Salvia sclarea, Laurus nobilis, Thymus satureioides, Pogostemon patchouli, Cananga odorata) Comparator
|
|
| Outcomes | Assessments were monthly (6) Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | Data were not stratified for gender. We sent e‐mails to the PI, but unfortunately they were unable to help us. See Table 8 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 221): "were placed into either group A (treatment, n=40) or group B (control placebo, n=29) according to a statistical randomization plan". Comment: the study authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment, was not reported. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 221): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit clear judgement of risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There were 24/93 (26%) withdrawals/losses to follow‐up, with reasons reported. The data analysis was per‐protocol. Comment: the large number of dropouts (26%), incomplete outcome data, and inappropriate analysis were potential sources of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Carmina 2003.
| Methods | This was a randomised "unmasked" study with 2 active control groups and an observation/no treatment group Setting Endocrinology outpatient practice in Italy Date of study Unspecified (12 month‐duration) |
|
| Participants | 48 hyperandrogenic women with alopecia Mean age = 25 ± 2 years Inclusion criteria
Exclusion criteria The trial did not report any exclusion criteria Randomised 36 participants were randomised (group I = 12, group II = 12, group III = 12) (Untreated controls (12), these were enrolled, not randomised, but refused treatment and served as an observation group) Withdrawals/losses to follow‐up None were reported Baseline data Ludwig scale (mean)
|
|
| Interventions |
Intervention
Comparator 1
Comparator 2
Comparator 3
The duration of treatment for groups I, II, and III was 1 year |
|
| Outcomes | There was inadequate and unclear information on the frequency and timing of the following assessments Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 92): "Thirty‐six women were randomized to one of three treatments, each composed of 12 subjects." Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote (page 91): "unmasked trial of three treatments." Comment: the outcome was likely to be influenced by the lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote (page 91): "unmasked trial of three treatments." Quote (page 92): "All assessments were carried out by one of the authors." Comment: the outcome measurement was likely to have been influenced by the lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No dropouts, withdrawals, or missing outcome data were reported. The time points of outcome assessments were unclear, and only end of study data were reported. Comment: There was insufficient information to permit a clear judgement of the risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was not available, but the prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
DeVillez 1994.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Multicentre (11), USA, but no further details reported Date of study Unspecified (32‐week duration) |
|
| Participants | 308 women Mean age (SD) = 33.6 years (6.67) in the minoxidil group, 34.4 years (6.32) in the placebo group Inclusion criteria
Exclusion criteria
Randomised 308 participants were randomised (minoxidil group = 157, placebo group = 151) Withdrawals/losses to follow‐up There were 52/308 (17%) withdrawals/losses to follow‐up: 27/157 (16.6%) in the minoxidil group, and 25/151 (14.8%) in the placebo group
Baseline data Duration of hair loss (SD): minoxidil group = 9.5 years (6.67), placebo group = 9.0 years (6.68) Age at onset (SD) : minoxidil group = 24.1 years (7.26), placebo group = 25.4 years (7.14) Degree of thinning, Ludwig scale (% of participants by grade and group)
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (9): at baseline and every 4 weeks Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | The report was unclear about the extent and level of any funding or support, but as three of the investigators were employed by the manufacturer, a level of support and possibly funding is most probable. | |
| Declaration of interest | Three of the four investigators were from the Dermatology Division of Upjohn Laboratories, the manufacturer of the intervention under investigation | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 304): "randomized to receive either". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 304): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 304): "Both the investigator and the patient assessed visible new hair growth." Comment: there was uncertainty with effective blinding of outcomes assessors (participants/healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 52/308 participants "discontinued"; most were voluntary withdrawals and were balanced across both groups. The data analysis was per‐protocol. Comment: the number of dropouts (17%) and incomplete outcome data, combined with per‐protocol analysis were potential sources of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Draelos 2005.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting A "research center" in the USA Date of study Unreported (6‐month duration) |
|
| Participants | 60 women Inclusion criteria
Exclusion criteria Nothing was reported Randomised 60 participants were randomised (active intervention group = 40, placebo group = 20) Withdrawals/losses to follow‐up There were 8/40 (20%) withdrawals/losses to follow‐up in the active intervention group, and 8/20 (40%) in the placebo group.
Baseline data The duration and extent of thinning was unreported |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (4): at baseline, 2, 4, and 6 months Outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 261): "This research was supported in part by NIH grantR43CA90085 and a grant from Niadyne Inc." | |
| Declaration of interest | Quote (page 261): "MKJ and ELJ are principals in Niadyne Inc., whose sponsored research is managed in accordance with the University of Arizona conflict‐of‐interest policies. Dr Draelos owns no stock in Niadyne and has no other financial interest in the corporation." | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 259): "Subjects were assigned randomly to the placebo (20, vehicle only) or active groups." Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 259): "Dispensed products were packaged in identical containers." Comment: the report provided sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes were participant‐ and investigator‐assessed. Blinding of participants and key study personnel was ensured, and it was unlikely that the blinding could have been broken. Comment: we judged this as at a low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There were 8/40 (20%) withdrawals/losses to follow‐up in the active intervention group, and 8/20 (40%) in the placebo group. Incomplete outcome data were not adequately addressed; timing of, and reasons for, withdrawal were unreported; and there were substantial differences in attritional losses between the 2 groups. Comment: we judged this as at a high risk of bias. |
| Selective reporting (reporting bias) | Low risk | Although only minimal data were reported, the outcomes listed in the 'Methods' section were comparable to the reported results. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Fischer 2004.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Friedrich‐Schiller‐University, Jena, Germany Date of study Unspecified (6‐month duration) |
|
| Participants | 40 women (28 with diffuse alopecia, 12 with androgenetic alopecia) Age = 20 to 70 years Inclusion criteria
Exclusion criteria
Randomised 40 participants were randomised (melatonin group = 20, placebo group = 20) Withdrawals/losses to follow‐up None were reported Baseline data % anagen hairs, trichogram‐assessed
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, 3, and 6 months Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 344): "This study was performed with kind support of ASAT Applied Science and Technology, Zug, Switzerland. Special thanks are given to Dr D.Menne, BiomedicalSoftware, Tübingen, Germany, for the statistical analysis." | |
| Declaration of interest | None declared | |
| Notes | There was separate analysis for AGA and diffuse alopecia | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (pg 342): "double‐blind randomized". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. After e‐mail communication with investigators: "Randomisation was performed in two groups (placebo/verum) of 20 cards by drawing the cards and allocating them to the numeric numbers 1 to 40." Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment.
Comment: there was insufficient information to permit a clear judgement. After e‐mail communication with investigators: "The bottles in which the test solutions were filled up were numbered and randomly allocated by the producer/sponsor to verum and placebo." "The patients received the test numbers in order of their recruitment." Comment: as a form of central randomisation, this was probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. After e‐mail communication with investigators: "The bottles in which the test solutions were filled up were numbered and randomly allocated by the producer/sponsor to verum and placebo. The study was double‐blind, so there was no code except the emergency code to identify the numbers with their respective ingredients." Comment: it appears that reasonable attempts were made to blind participants and personnel from knowledge of which intervention a participant received. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | There was insufficient information in the report to permit a clear judgement. After e‐mail contact with the investigators (see above), we judged the blinding to be adequate. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No dropouts, withdrawals, or missing outcome data were reported. Comment: there was insufficient information to permit a clear judgement of the risk of bias. |
| Selective reporting (reporting bias) | Low risk | Although only minimal data were reported, the outcomes listed in the 'Methods' section were comparable to the reported results. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Gassmueller 2008.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial (please see the Notes section) Setting 2 centres in Germany Date of study Unspecified (16‐week duration) |
|
| Participants | 70 women Age = 49 to 72 years Inclusion criteria
Exclusion criteria
Randomised 70 participants were randomised (topical fulvestrant group = 34, vehicle only group = 36) Withdrawals/losses to follow‐up 3/70 (4%) withdrawals/losses to follow‐up. It was unclear from which of the groups the losses were: 2 for personal reasons (on days 15 & 17), and 1 for protocol deviation (use of prohibited concomitant medication). Baseline data Mean hair density as hairs per cm² (range)
Mean cumulative hair thickness in mm per cm² (range)
Mean hair growth rate in mm per day (range)
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (5): at baseline, day 29, 57, 85, and 113 Outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 115): "This study (including editorial assistance provided by Dr Keri Wellington of Mudskipper Bioscience) was supported financially by AstraZeneca." | |
| Declaration of interest | None declared. One investigator (A. Webster) was associated with AstraZeneca | |
| Notes | Of two phase II randomised controlled trials (RCTs) of men and women with AGA, we only considered the trial that included women | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 110): "randomized (via a randomization list generated by AstraZeneca)." Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | The method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment, was not reported. After e‐mail contact with the investigators: "Randomization was performed centrally by the sponsor of the study in a balanced manner." "The random assignment for each subject was kept in a sealed envelope at the site which was only to be opened in case of an emergency." Comment: although the sponsor generated the sequence, this was a form of central randomisation. This was probably done; therefore, we judged this as at a low risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 110): "The female study was double blind." The report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. After e‐mail contact with the investigators: "The study medication was labelled with the respective subject (randomisation) number by the sponsor, before delivery to the test sites." Comment: we judged this as at a low risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote (page 111): "TrichoScan analysis...images analysed at the end of the study by an independent observer who was blinded to the treatment received and who was also unaware of the time point in the study for each image". Comment: the measures used to blind the outcome assessor from knowledge of which intervention a participant received was adequately reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 3/70 participants dropped out (unclear which group); the reasons for withdrawal were reported. There was an intention‐to‐treat (ITT) analysis. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Gehring 2000.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Dermatology Clinic in Karlsruhe, Germany Date of study Unspecified (6‐month duration ‐ late autumn until summer) |
|
| Participants | 41 women Mean age (range) = 38.1 years (19 to 57) in the active treatment group, 39.2 years (23 to 54) in the placebo group Inclusion criteria
Exclusion criteria
Randomised 41 participants were randomised (active treatment group = 21, placebo group = 20) Withdrawals/losses to follow‐up There was 1 in the placebo group; the reason was unreported Baseline data Anagen hairs (%)
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, 3, and 6 months Outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared. Quote (page 420): "Medication provided by Company Roche Nicholas, Germany" | |
| Declaration of interest | None declared | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 419): "randomized, double‐blind". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 419): "double‐blind". The report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 419): "double‐blind". Comment: uncertainty with the effectiveness of blinding of outcomes assessors (healthcare providers) during the study. Insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was 1 dropout in the placebo group; the reason was unreported. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Georgala 2004.
| Methods | This was a randomised, placebo‐controlled trial Setting Hospital in Athens, Greece Date of study 1998 to 2000 (the duration in group I was 12 weeks, in group II & III it was 24 weeks) |
|
| Participants | 75 women Age = 48 to 71 years Inclusion criteria
Exclusion criteria
Randomised 75 participants were randomised into 3 treatment groups (group I = 25, group II = 25, group III = 25) Withdrawals/losses to follow‐up There were 13/75 (17%) withdrawals/losses to follow‐up: 3 in group I, 5 in group II, and 5 in group III.
Baseline data The duration and extent of thinning was unreported. Anagen/telogen ratio at baseline.
|
|
| Interventions |
Intervention
Comparator 1
Comparator 2
|
|
| Outcomes | Assessments (3): at baseline, 3, and 6 months Outcomes of the trial (as reported) Trichograms were taken at baseline and the completion of the study.
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | This report was a Letter to "Dermatology". Data reporting were inadequate | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 178): "Patients were randomised into three treatment groups." Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of the risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement of the risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit a clear judgement of risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | The ratio of dropouts/withdrawals was as follows: 13/75 (17%). The reasons and number from each group were reported and balanced across active intervention groups only. The data analysis was per‐protocol. Comment: although the numbers of dropouts were balanced between the groups, the percentage of dropouts and subsequent per‐protocol analysis poses an unclear risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Guerrero 2009.
| Methods | This was a randomised, active‐controlled trial Setting 3 centres in Chile Date of study November 2007 to April 2008 (12‐week duration) |
|
| Participants | 40 men and women (22 men, 18 women) Mean age = 43.7 years (range 20 to 69) Inclusion criteria
Exclusion criteria
Randomised 40 participants were randomised (minoxidil group = 15, estradiol group 18, unclear = 7) Withdrawals/losses to follow‐up 7/40 (18%) withdrawals/losses to follow‐up: unclear how many from each group
Baseline data Most men had Hamilton II/III and most women Ludwig II |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (4): at baseline, days 30, 60, and 90 Outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | The sample comprised of participants of both genders, and the sequence was generated according to simple randomisation (specific method unreported, but without stratification). The results did not consider gender as a factor or covariate, and the data reported and subsequent analysis is not gender‐specific. We e‐mailed the PI, but received no response. See Table 8 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 22): "un estudio randomizado" and "en forma aleatoria mediante randomización simple en dos grupos" (randomised through simple randomisation). Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment, was not reported. Comment: there was insufficient information to permit a clear judgement of the risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 22): "triple ciego" (triple‐blind). Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 22): "triple ciego" (triple‐blind). Comment: uncertainty with the effectiveness of blinding of outcomes assessors (participants, healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 7/40 (18%) withdrawals/losses to follow‐up: unclear how many from each group. The data analysis was per‐protocol. Comment: although the numbers of dropouts appear balanced between the groups, the percentage of dropouts and subsequent per‐protocol analysis poses an unclear risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the trial appears to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Hong 2007.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Dermatology Department at Chung‐Ang University Medical Center, Seoul, Korea Date of study Unspecified (16‐week duration) |
|
| Participants | 95 men and women (74 men, 21 women) Mean (SD) age = 38.8 (8.9) years in active treatment group, 41.7 (8.9) years in placebo group Inclusion criteria
Exclusion criteria
Randomised 95 participants were randomised (active treatment group = 33, placebo group 40, unclear = 22) Withdrawals/losses to follow‐up 22/95 (23.2%) withdrawals/losses to follow‐up: unclear how many from each group Baseline data Hair shed per (mean): active treatment group 250, placebo group 300 |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, week 8, and week 16 Outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | The trial did not stratify data by gender. See Table 8 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 1233): "randomized". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. After e‐mail communication: "We used computer software program that generates the random sequence" Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | The trial did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of the risk of bias. After e‐mail communication: "We used sequentially numbered, opaque, sealed envelopes". |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 1233): "double‐blind". The report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 1233): "double‐blind". Comment: uncertainty with the effectiveness of blinding of outcomes assessors (healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 22/95 (23.2%) withdrawals/losses to follow‐up: unclear how many from each group. Data analysis was per‐protocol. Comment: we judged this as at a high risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the trial appear to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Jacobs 1993.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Multicentre (10), Europe (France/Belgium/UK/Holland/Switzerland) Date of study Unspecified (32‐week duration) |
|
| Participants | 346 women Mean age (SD) = 33.1 years (6.93) in the minoxidil group, 34.2 years (6.35) in the placebo group Inclusion criteria
Exclusion criteria
Randomised 346 participants were randomised (minoxidil group = 176, placebo group = 170) Withdrawals/losses to follow‐up There were 52/346 (15%) withdrawals/losses to follow‐up: 21/176 (11.9%) in the minoxidil group, and 31/170 (18.2%) in the placebo group Baseline data Duration hair loss (SD): minoxidil group = 8.0 years (6.31), placebo group = 8.6 years (5.91) Age at onset of hair loss (SD): minoxidil group = 25.1 years (7.19), placebo group = 25.6 years (6.99) Degree of thinning Ludwig scale (% of participants by grade and group)
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (9): at baseline and every 4 weeks Primary outcomes (as reported)
Secondary outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared, but the three trial authors are employed by Upjohn Laboratories, the manufacturer of minoxidil. | |
| Declaration of interest | The three PIs were from the Dermatology Division of Upjohn Laboratories, Kalamazoo | |
| Notes | Although adverse events are not mentioned as an outcome by the investigators, 2 adverse events were reported regarding withdrawals in the minoxidil group | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 759): "randomized to receive either". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment, was not reported. Comment: there was insufficient information to permit a clear judgement of the risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 758): "double‐blind trial." Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 758): "double‐blind trial." Comment: there was uncertainty with the effectiveness of blinding of outcomes assessors (participants, healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was a 15% rate of withdrawals and losses to follow‐up: 21/176 withdrew in the minoxidil group, and 31/170 in the placebo group. The trial reported the reasons for withdrawal, and the numbers were reasonably balanced across the groups. The data analysis was per‐protocol. Comment: although the numbers of dropouts appear balanced between the groups, the percentage of dropouts and subsequent per‐protocol analysis poses an unclear risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Jimenez 2014a.
| Methods | This was a randomised, double‐blind, sham‐controlled trial Setting Multicentre (5), Europe and USA Date of study The recruitment period was from 29 October 2008 to 2 March 2009 (26‐week duration) |
|
| Participants | 78 women Mean age (SD) = 49.3 years (9.1) in the laser comb group, 49.8 years (7.3) in the sham group Inclusion criteria
Exclusion criteria
Randomised 78 participants were randomised (laser comb group = 53, sham group = 25) Withdrawals/losses to follow‐up There were 15/78 (19.2%) withdrawals/losses to follow‐up: 11/53 (21%) in the laser comb group, and 4/25 (16%) in the placebo group Baseline data
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, week 16, and week 26 Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 126): "Lexington International, LLC partially funded the study, and provided the treatment and sham devices and equipment (including the digital imaging system)". | |
| Declaration of interest | Quote (page 126): "M.R. Hamblin has received honorarium/consulting fees, and L.A. Schachner has received fees for participation from Lexington International, LLC. M. Hordinsky has received, on behalf of the Department of Dermatology at the University of Minnesota, a grant to conduct part of this study. J.J. Jimenez, T.C. Wikramanayake, W.F. Bergfeld, and J.G.Hickman have no conflicts of interest that are directly relevant to this study. M.R. Hamblin was supported by a NIH grant R01AI050875". | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 118): "Randomization was generated by EugeneR. Heyman (http://www.erhstats.com) using the SASPROC RAND method...randomization was 2:1 with a block size of 3." Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | Quote (page 118); "The lasercomb and sham devices, along with instructions, were provided to the site investigator in sealed, sequentially numbered opaque packets in a blinded manner, and were dispensed sequentially. Both the site investigators and the subjects remain blinded to the type of device they dispensed/received throughout the trial." Comment: the report provides sufficient detail and reassurance that participants and investigators enrolling participants could not foresee the upcoming assignment. This was probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 118): "An independent evaluator not connected to the clinical trials analyzed the uploaded images and performed computer‐assisted hair counts…The evaluator…… was blinded to which trial arm the subject belonged, as well as which images were from baseline and which were from follow‐up." After e‐mail contact: "the study site evaluators never observed or knew which device the subject was using at any evaluation point, only knew by a randomization code numbers which device each subject was using. The sealed bag containing the device the subject were to use, was only opened at the subject’s home and was never brought to the study center during the course of the study. Since the subject used the device assigned and had no comparator, they did not have any way of knowing if the device was active or not, since both the active and sham device emitted light." Comment: the report provided sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes were investigator and participant assessed. Blinding of participants and key study personnel was ensured, and it is unlikely that the blinding could have been broken. Comment: we judged this as at a low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 15/78 (19.2%) unspecified withdrawals/losses to follow‐up: 11/53 in the laser comb group (21%), and 4/25 in the placebo group (16%). The data analysis was per‐protocol. Comment: we judged this as at high risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was available on clinicaltrials.gov (NCT00981461). The prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Jimenez 2014b.
| Methods | This was a randomised, double‐blind, sham‐controlled trial Setting Multicentre (5), Europe and USA Date of study The recruitment period was from 1 February to 28 September 2010 (26‐week duration) |
|
| Participants | 63 women Mean age (SD) = 48.7 years (10.2) in the laser comb group, 49.1 years (8.3) in the sham group Inclusion criteria
Exclusion criteria
Randomised 63 participants were randomised (laser comb group = 42, sham group = 21) Withdrawals/losses to follow‐up There were 6/63 (9.5%) withdrawals/losses to follow‐up: 3/42 (7.1%) in the laser comb group, and 3/21 (14.3%) in the placebo group (14.3%) Baseline data
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, week 16, and week 26 Primary outcomes (as reported)
Secondary outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 126): "Lexington International, LLC partially funded the study, and provided the treatment and sham devices and equipment (including the digital imaging system)". | |
| Declaration of interest | Quote (page 126): "M.R. Hamblin has received honorarium/consulting fees, and L.A. Schachner has received fees for participation from Lexington International, LLC. M. Hordinsky has received, on behalf of the Department of Dermatology at the University of Minnesota, a grant to conduct part of this study. J.J. Jimenez, T.C. Wikramanayake, W.F. Bergfeld, and J.G.Hickman have no conflicts of interest that are directly relevant to this study. M.R. Hamblin was supported by a NIH grant R01AI050875". | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 118): "Randomization was generated by Eugene R. Heyman (http://www.erhstats.com) using the SASPROC RAND method...randomization was 2:1 with a block size of 3." Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | Quote (page 118): "The lasercomb and sham devices, along with instructions, were provided to the site investigator in sealed, sequentially numbered opaque packets in a blinded manner, and were dispensed sequentially. Both the site investigators and the subjects remain blinded to the type of device they dispensed/received throughout the trial." Comment: the report provides sufficient detail and reassurance that participants and investigators enrolling participants could not foresee the upcoming assignment. This was probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 118): "An independent evaluator not connected to the clinical trials analyzed the uploaded images and performed computer‐assisted hair counts…The evaluator…… was blinded to which trial arm the subject belonged, as well as which images were from baseline and which were from follow‐up." After e‐mail contact: "the study site evaluators never observed or knew which device the subject was using at any evaluation point, only knew by a randomization code numbers which device each subject was using. The sealed bag containing the device the subject were to use, was only opened at the subject’s home and was never brought to the study center during the course of the study. Since the subject used the device assigned and had no comparator, they did not have any way of knowing if the device was active or not, since both the active and sham device emitted light." Comment: the report provided sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes were investigator and participant assessed. Blinding of participants and key study personnel was ensured, and it is unlikely that the blinding could have been broken. Comment: we judged this as at a low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were 6/63 (9.5%) unspecified and unbalanced withdrawals/losses to follow‐up: 3/42 in the laser comb group (7.1%), and 3/21 in the placebo group (14.3%). The data analysis was per‐protocol. Comment: we judged this as at unclear risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was available on clinicaltrials.gov (NCT01016964). The trial appears to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Keene 2011.
| Methods | This was a randomised, blinded, sham‐controlled trial Setting Unspecified USA Date of study December 2008 to December 2009 (6‐month duration) |
|
| Participants | 13 women Mean age (SD) = not reported Inclusion criteria
Exclusion criteria
Randomised 13 participants were randomised (finasteride group = 8, placebo group = 5) Withdrawals/losses to follow‐up None were reported Baseline data None reported |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (7): at baseline and every month Outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 268): "study was funded by DermaGenoma, Inc" | |
| Declaration of interest | Quote (page 268): "Both authors are employed by DermaGenoma, Inc" | |
| Notes | Study participants used buccal swabs to obtain DNA for identification of their AR gene polymorphism. This involved evaluation of the number of CAG nucleotide repeats in the first exon of the AR gene in each X chromosome. Subsequently, each participant who was not homozygous in the AR‐CAG allele underwent determination of percentage of X inactivation for each allele based on the method of quantitative polymerase chain reaction (PCR) product (inactive allele) following digestion of unmethylated (activated) DNA | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 297): "Patients who met the inclusion criteria were randomized to either placebo or 1‐mg finasteride". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. After e‐mail communication: "For each subject, the site supervisor picked from a bowl a random letter A or B". Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | The method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment, was not reported.
Comment: there was insufficient information to permit a clear judgement. After e‐mail communication: "The study was conducted for a sponsor. The sponsor recruited an independent site monitor (Registered Nurse) who had the sole access to the information during the course of the study". Comment: this is a form of central allocation, which was probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 297): "6‐month blinded". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. After e‐mail communication: "Tablets were compounded at a pharmacy to look exactly the same". |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote (page 297): "6‐month blinded". Comment: there was uncertainty with effective blinding of outcomes assessors (participants/healthcare providers) during the study. There was insufficient information to permit a clear judgement. After e‐mail communication: "Tablets were compounded at a pharmacy to look exactly the same". Comment: it was unlikely that the blinding was broken. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts were reported and data are available for all individual participants. Comment: we judged this as at low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was available on clinicaltrials.gov (NCT01052870). The trial appeared to report the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Kim 2009.
| Methods | This was a randomised, placebo‐controlled trial Setting Department of Dermatology, College of Medicine, Korea University, Seoul, Korea Date of study Unspecified (24‐week duration) |
|
| Participants | 40 men and women (28 men, 12 women) Mean age = 43.1 years Inclusion criteria
Exclusion criteria
Randomised 40 men and women were randomised (ginseng group = 20, placebo group = 20) Withdrawals/losses to follow‐up 8/40 (20%) withdrawals/losses to follow‐up, 3/20 (15%) in ginseng group and 5/20 (25%) in placebo group, reasons unreported Baseline data of the females 7 had Ludwig Grade I female pattern alopecia, and 5 had Grade II. None had Grade III |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, 12 weeks, and 24 weeks Outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | The Korean Ginseng Corporation funded the study and provided the medications used in the intervention group | |
| Declaration of interest | None declared | |
| Notes | The trial did not stratify data by gender. We sent e‐mails were sent to the PI, but received no reply. See Table 8 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 223): "randomly". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of the risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 223): "double‐blind trial." Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 758): "double‐blind trial." Comment: there was uncertainty regarding the effectiveness of blinding of outcomes assessors (participants, healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 8/40 (20%) withdrawals/losses to follow‐up, 3/20 (15%) in ginseng group and 5/20 (25%) in placebo group (unbalanced), but the reasons were unreported. Comment: we judged this as at a high risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Kim 2013.
| Methods | This was a randomised, double‐blind, sham device‐controlled trial Setting Multicentre (2), Korea Date of study Unspecified (24‐week duration) |
|
| Participants | 40 men and women (26 men, 14 women) Mean age (SD) = 43.9 years (12.2) in the low level light therapy group, 44.5 years (11.4) in the sham device group Inclusion criteria
Exclusion criteria
Randomised 40 participants were randomised (low level light therapy group = 20, sham group = 20) Withdrawals/losses to follow‐up There were 11/40 (27.5%) withdrawals/losses to follow‐up: 5/20 (25%) in the low‐level light therapy group, and 6/20 (30%) in the sham group
Baseline data Duration hair loss (SD): low level light therapy group = 114.3 months (86.2), sham group = 100.55 months (84.8)
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, week 12, and week 24 Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 1177): "This study was supported by Won Technology, Daejeon, Republic of Korea." | |
| Declaration of interest | Quote (page 1177): "The authors have indicated no significant interest with commercial supporters" | |
| Notes | The trial did not stratify data by gender. We sent e‐mails to the PI, but received no reply. See Table 8 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 1177): "randomized" and "We randomly assigned". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. After e‐mail communication: "coin throw method for the allocation" Comment: this was probably done. |
| Allocation concealment (selection bias) | Unclear risk | The trial did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 1177): "double‐blind" and "The sham device was identical in appearance and its regulator operated, although it emitted no light", and (page 1179) " they were totally blinded". Comment: we judged this as at an unclear risk of bias in view of the fact that the helmet did not emit light. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 1177): "double‐blind", (page 1179 " the investigators in charge of efficacy assessment were totally blinded". Comment: both investigators and participants were outcome assessors. We judged this as at an unclear risk of bias as the helmet did not emit light and might have influenced the outcome assessment by the participants. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There were 11/40 (27.5%) withdrawals/losses to follow‐up: 5/20 in the low‐level light therapy group (25%), and 6/20 in the sham group (30%). The data analysis was per‐protocol. Comment: the large number of dropouts (26%), incomplete outcome data, and inappropriate analysis were potential sources of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the trial appeared to report the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Lanzafame 2014.
| Methods | This was a randomised, double‐blind, sham‐controlled trial Setting Multicentre (2), USA Date of study Unspecified (16‐week duration) |
|
| Participants | 47 women Mean age (SD) = 46.3 years (9.2) in the visible red light laser and LED sources group, 51.0 years (7.05) in the sham device group Inclusion criteria
Exclusion criteria
Randomised 47 participants were randomised (unclear how many to each arm) Withdrawals/losses to follow‐up There were 5/47 (0.6%%) withdrawals/losses to follow‐up: prior to treatment, reasons unknown Baseline data Duration hair loss (SD): low level light therapy group = 114.3 months (86.2), sham group = 100.55 months (84.8)
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (2): at baseline and week 16 Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 601): "This study was funded by Apira Science, Inc" | |
| Declaration of interest | RP Chiacchierini, E Kazmirek and JA Sklar have no disclosures. RR Blanche received consulting fees, had study‐related travel expenses paid and has ownership interest in Apira Science. RJ Lanzafame received consulting fees, fees for manuscript preparation, and has ownership interest in Apira Science. | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 602): "Subjects were randomly assigned". Comment:the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. After e‐mail communication: "Central allocation, blocks of six." Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | Quote (page 602): "A serial number was assigned to each helmet, which was recorded in a device log that contained the reference code for placebo and actual test unit. This log was not revealed to any investigator, subject, office staff, hair counter or sponsor employee." Comment: the method used to generate the sequence would appear to indicate that intervention allocations could not have been foreseen in advance of, or during, enrolment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 602): "The sham group received a unit that was identical in appearance and function to the laser group devices, with the exception that the light sources were incandescent wheat lights that were painted red to mimic the appearance and configuration of the functioning device". Comment: the report provided sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote (page 601): "These baseline images were coded and then forwarded to the photographic consultant. The photographic consultant verified that the images were of acceptable quality and processed the images for transmission to the investigator responsible for conducting the hair counts. The transmitted images were masked using a black mask to produce a 1.9 cm diameter circle centered on the tattoo, which provided a consistent 2.85 cm2 area for hair counts. Neither the photographic consultant nor the investigator performing the hair counts was aware of the identity of the subject or the subjects’ study group assignment". Comment: the measures used to blind the outcome assessor from knowledge of which intervention a participant received were adequately reported. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 5/47 (10.6%) withdrawals/losses to follow‐up, but it was unclear from which group. The data analysis was per‐protocol. Comment: although unclear from which group the low number of drop‐outs occurred, we judged it as at low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was available (NCT01437163), and the prespecified outcomes and those mentioned in the methods section appeared to have been reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Le Floc'h 2015.
| Methods | This was a RCT Setting One centre in Italy Date of study January to September 2012 (6‐month duration) |
|
| Participants | 120 women Mean age (SD) = 48.6 years (11.2) in supplement group, 46.0 years (14.9) in the control group Inclusion criteria
Exclusion criteria
Randomised 120 participants were randomised (nutritional supplement = 80, control group = 40) Withdrawals/losses to follow‐up There were 2/120 (1.6%) withdrawals/losses to follow‐up: one in each group for personal reasons Baseline data
|
|
| Interventions |
Intervention
Comparator
Each participant was provided with a neutral shampoo to be used every other day |
|
| Outcomes | Assessments (2): at baseline and month 6 Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None reported, but the first author is employed by Innéov (L'Oréal) the manufacturer of the supplement | |
| Declaration of interest | None reported, but the first author is employed by Innéov (Loreal) the manufacturer of the supplement | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 77): "were randomly assigned". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 77): "expert‐blinded". Comment: the report did not provide sufficient detail about the measures used to blind study personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Both investigators and participants were the outcomes assessors. Participants did not receive a substitute for the nutritional supplement, which is likely to influence their outcome assessment. Comment: we judged this as at high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 2/120 (1.6%%) withdrawals/losses to follow‐up: one in each group for personal reasons. The data analysis was per‐protocol. Comment: we judged this as at low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was available (NCT01437163), and the trial appeared to report the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Lucky 2004.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Multicentre (9), USA Date of study May 1992 to 1993 (48‐week duration) |
|
| Participants | 381 women Inclusion criteria
Exclusion criteria
Randomised 381 participants were randomised (minoxidil 5% group = 153, minoxidil 2% = 154, placebo group = 74) Withdrawals/losses to follow‐up There were 121/381 (32%) withdrawals/losses to follow‐up: 52/153 (33.9%) in the minoxidil 5% group, 46/154 (29.8%) in the minoxidil 2% group, and 23/74 (31.1%) in the placebo group
Baseline data Degree of thinning Ludwig scale (% of participants by grade and group)
Hair density Savin female density scale (% of participants by score and group)
|
|
| Interventions |
Intervention
Comparator
Placebo
|
|
| Outcomes | Assessments (2): at baseline and week 48 Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 541): "Supported by Pfizer Inc (formerly Pharmacia Corporation, formerly The Upjohn Company)." | |
| Declaration of interest | Quote (page 541): "Disclosure: All authors were the clinical investigators involved in the conduct of the trial." | |
| Notes | The Savin female density scale appears to be validated. It was developed by Dr Trancik of Upjohn Laboratories. There were concomitant prohibited medications in 13/153 in the 5% topical minoxidil group; 5/154 in the 2% topical minoxidil group; and 3/74 participants in the placebo group. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 542): "Randomization occurred in a 2:2:1 design...according to a predetermined, computerized randomization plan." Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | Quote (page 542): "Each trial site was provided with a unique list of randomization code numbers". Comment: the report provides sufficient detail and reassurance that participants and investigators enrolling participants could not foresee the upcoming assignment. This was probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 542): "double blind". "The investigational medications were provided to each trial site in identically appearing, prepackaged, and pre‐labelled bottles" Comment: the report provided sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes were participant‐ and investigator‐assessed. Blinding of participants and key study personnel was ensured, and it was unlikely that the blinding could have been broken. Comment: we judged this as at a low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Although a flow chart tracked participants through the study, losses to follow‐up/withdrawals were substantial (> 30%) in all treatment groups. Quote (page 544): "261 patients were included in the efficacy evaluable population." The data analysis was per‐protocol (261/381). Comment: we judged this as at a high risk of bias. |
| Selective reporting (reporting bias) | Low risk | Although the study protocol was unavailable, the outcomes listed in the 'Methods' section were comparable to the reported results. Comment: we judged this as at a low risk of bias. |
| Other bias | High risk | There was an influence of co‐interventions as effect modifiers. Quote (page 545): "21 patients used protocol‐prohibited concomitant medications (systemic corticosteroids)." Comment: potential effects of co‐interventions represented a possible risk of bias. |
Mazzarella 1997.
| Methods | This was a randomised, single‐blind, placebo‐controlled study Setting Department of Dermatology, University of Bari, Bari, Italy Date of study Unspecified (16‐month duration) |
|
| Participants | 52 men and women (28 male and 24 women) Mean age (range) = 28 years (18 to 38) Inclusion criteria
Exclusion criteria
Randomised 52 participants were randomised (finasteride group = 26, vehicle group = 26) Withdrawals/losses to follow‐up There were 16/52 (30.7%) withdrawals/losses to follow‐up: 0/26 (0%) in the finasteride group, 16/26 (61.5%) in the vehicle group (61.5%), reasons and gender not reported Baseline data Hamilton stage II: 13 male; stage III 15 male Ludwig stage I: 11 female; stage II 13 female |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (5): at baseline, week 4, 8, 12, and 16 Outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | There were no separate data for women. There were 16 dropouts (61.5%) in the placebo group only. We e‐mailed the PI, but received no response. See Table 8 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 189): "were randomly allocated". Comment: the trial did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: There was insufficient information to permit a clear judgement of the risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 189): "single‐blind trial" Comment: the investigators were not blinded. The report did not provide sufficient detail about the measures used to blind study participants from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote (page 189): "single‐blind trial". Both investigators and participants were outcome assessors and the investigators were not blinded. Comment: the outcome measurement by the investigators is likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There were 16/52 (30.7%) withdrawals/losses to follow‐up: 0/26 in the finasteride group (0%), 16/26 in the vehicle group (61.5%), reasons were not reported. There were unbalanced withdrawals. The data analysis was per‐protocol. Comment: we judged this as at a high risk of bias |
| Selective reporting (reporting bias) | Low risk | Although the study protocol was unavailable, the outcomes listed in the 'Methods' section were comparable to the reported results. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Minozzi 1997.
| Methods | This was a randomised, active‐controlled trial Setting Center for Climacteric and Menopause of the Institute of Obstetrics and Gynecology, Policlinico Umberto l, Rome, Italy Date of study Unspecified (12‐month duration) |
|
| Participants | 63 women Age = 52 to 63 years Inclusion criteria
Exclusion criteria
Randomised 63 participants were randomised (group I = 21, group II = 21, group III = 21) Withdrawals/losses to follow‐up None were reported Baseline data Minimal data, blood tests: routine blood tests, serum follicle‐stimulating hormone (FSH), LH, estradiol, testosterone, free testosterone, dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulphate (DHEAS), delta‐4‐androstenedione, dihydrotestosterone, SHBG. Hormonal status consistent with menopause |
|
| Interventions |
Intervention
Comparator 1
Comparator 2
|
|
| Outcomes | Assessments (2): at baseline and month 12 Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | Diagnosis of FPHL was not clearly defined/stated We sent several e‐mails to the PI but received no response. None of our outcomes were assessed. See Table 8 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 341): "The patients have been randomized in three equal groups to which a different treatment had been administered." Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment, was not reported. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study was open. The nature of the treatment interventions precludes any possibility of blinding of participants and personnel. Comment: the outcome is likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study was open. The nature of the treatment interventions precludes any possibility of blinding of participants and personnel. Comment: the outcome measurement is likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No dropouts were reported. It was unclear if data analysis was per‐protocol or intention‐to‐treat. Comment: there was insufficient information to permit a clear judgement of risk of bias. |
| Selective reporting (reporting bias) | Unclear risk | The investigators did not report all of their prespecified outcomes of the hormonal screening (only SHBG), but it was uncertain to what extent the lack of data for other than SHBG had any impact on their reported results. Therefore, we judged this domain as at an unclear risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Morganti 1998.
| Methods | This was a randomised, double‐blind, placebo‐controlled study Setting Department of Cosmetic Dermatology, Accademia di Storia dell'Arte Sanitaria, Rome, Italy Date of study Unspecified (50‐week duration) |
|
| Participants | 60 men and women (at least 24 men and 24 women; for 12, the gender was not reported). Age = 21 to 38 years Inclusion criteria
Exclusion criteria
Randomised 60 participants were randomised (12 to each group) Withdrawals/losses to follow‐up None were reported Baseline data Nothing reported |
|
| Interventions |
Intervention
Comparator 1
Comparator 2
Comparator 3
Comparator 4
The lotion was applied twice a day, and the pills were administered 4 times per day |
|
| Outcomes | Assessments (6): at baseline and every 10 weeks Outcomes of the trial (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | There were no separate data for women. Dropouts were not reported. We were unable to contact the investigators. See Table 8 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 59): "assigned in a randomized double‐blind manner". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 59): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 59): "double‐blind". Comment: there was uncertainty with effective blinding of outcomes assessors (healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No dropouts, withdrawals, or missing outcome data were reported. The reporting was overall incomplete and inconsistent. Comment: there was insufficient information to permit a clear judgement of the risk of bias. |
| Selective reporting (reporting bias) | Low risk | Although the study protocol was unavailable, the outcomes listed in the 'Methods' section were comparable to the reported results. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
NCT01145625.
| Methods | This is a randomised, single‐blind, active‐controlled trial Setting Multicentre (UK, USA, and Canada) Date of study June 2010 to February 2012 (52‐week duration) |
|
| Participants | 322 women Inclusion criteria
Exclusion criteria
Randomised 322 participants were randomised (minoxidil 2% group = 161, minoxidil 5% group = 161) Withdrawals/losses to follow‐up There were 55/322 (17.1%) withdrawals/losses to follow‐up: 24/161 (14.9%) minoxidil 2% group, 31/161 (19.3%) in the minoxidil 5% group
Baseline data Target area hair count hairs/cm² (SD): minoxidil 2% group 167.3 (55.0), minoxidil 5% group 169.7 (58.6) |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (4): at baseline, week 12, 24, and 52. Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Sponsor: Johnson & Johnson Healthcare Products Division of McNEIL‐PPC, Inc | |
| Declaration of interest | This was unclear from clinicaltrials.gov, but some investigators appear to be employees of Johnson & Johnson Consumer Companies Inc | |
| Notes | We accessed the website on 17 July 2015 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (clinicaltrials.gov): "randomized". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (clinicaltrials.gov): "single‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (clinicaltrials.gov): "single‐blind". Comment: there was uncertainty with effective blinding of outcomes assessors (healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were 55/322 (17.1%) withdrawals/losses to follow‐up: 24/161 minoxidil 2% group (14.9%), 31/161 in the minoxidil 5% group (19.3%). Data analysis was intention‐to‐treat. Comment: we judged this as at an unclear risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was available (NCT01145625), and the trial reported all outcomes listed. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
NCT01189279.
| Methods | This is a randomised active‐controlled trial Setting Unspecified, Tempe, Arizona, USA Date of study October 2010 to January 2011 (17‐day duration) |
|
| Participants | 42 men and women (21 men/21 women) Age = 18 to 64 years Inclusion criteria
Exclusion criteria
Randomised 42 participants were randomised (bimatoprost formulation A group = 14, bimatoprost formulation B group = 14, bimatoprost formulation C group = 14) Withdrawals/losses to follow‐up There were 2/42 (4.8%) withdrawals/losses to follow‐up: 1/14 (7.1%) in the bimatoprost formulation A group, 1/14 (7.1%) in the bimatoprost formulation C group Baseline data Nothing reported |
|
| Interventions |
Intervention
Comparator
Comparator 2
|
|
| Outcomes | Assessments (2): at day 1 and 17 Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Sponsor is Allergan | |
| Declaration of interest | No information, probably Allergan | |
| Notes | None of our outcomes were addressed, and there were no separate data for men and women. See Table 8. We e‐mailed Allergan several times, without any response | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (clinicaltrials.gov): "randomized". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment, was not reported. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (clinicaltrials.gov): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (clinicaltrials.gov): "double‐blind". Comment: there was uncertainty with effective blinding of outcomes assessors (healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 2/42 (4.8%) withdrawals/losses to follow‐up: 1/14 in the bimatoprost formulation A group (7.1%), 1/14 in the bimatoprost formulation C group (7.1%), Data analysis was per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was available (NCT01189279), and the outcomes listed were all reported. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
NCT01226459.
| Methods | This is a randomised double‐blind placebo‐controlled trial Setting Multicentre (17) in USA, UK, France and Germany Date of study September 2010 to August 2011 (24‐week duration) |
|
| Participants | 404 women Inclusion criteria
Exclusion criteria
Randomised 404 participants were randomised (minoxidil 5% foam group = 203, vehicle foam group = 201) Withdrawals/losses to follow‐up There were 53/404 (13.1%) withdrawals/losses to follow‐up: 32/203 in the minoxidil 5% foam group (15.8%), 21/201 in the vehicle foam group (10.4%)
Baseline data Target area hair count hairs/cm² (SD): minoxidil 5% foam group 158.6 (61.6), vehicle foam group 152.7 (59.7) |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, week 12, and week 24 Primary outcomes (as reported)
Secondary outcomes (as reported)
|
|
| Funding source | Johnson & Johnson Healthcare Products Division of McNEIL‐PPC, Inc | |
| Declaration of interest | This was unclear from clinicaltrials.gov, but collaborators are Johnson & Johnson Consumer and Personal Products Worldwide | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (clinicaltrials.gov): "randomly assigned in a 1:1 ratio to use either 5% MTF OD or foam vehicle OD". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (clinicaltrials.gov): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (clinicaltrials.gov): "double‐blind". Comment: there was uncertainty with effective blinding of outcomes assessors (healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were 53/404 (13.1%) withdrawals/losses to follow‐up: 32/203 in the minoxidil 5% foam group (15.5%), 21/201 in the vehicle foam group (10.4%). Data analysis was per‐protocol. Comment: we judged this as at an unclear risk of bias |
| Selective reporting (reporting bias) | Low risk | The study protocol was available (NCT01226459 as well as MINALO3005, EudraCT 2010‐019881‐96), and the trial reported all outcomes listed. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
NCT01325350.
| Methods | This was a randomised, double‐blind, placebo and active‐controlled study Setting Multicentre (2) USA and Germany Date of study June 2011 to July 2012 (6‐month duration) |
|
| Participants | 306 women Age = 18 to 59 years Inclusion criteria
Exclusion criteria
Randomised 306 participants were randomised (bimatoprost A = 61, bimatoprost B = 61, bimatoprost C = 61, bimatoprost vehicle = 61, minoxidil 2% = 62) Withdrawals/losses to follow‐up There were 49/306 (16.0%) withdrawals/losses to follow‐up: 6/61 (9.8%) bimatoprost A group, 5/61 (8.2%) bimatoprost B group, 17/61 (27.9%) bimatoprost C group, 9/61 (14.8%) bimatoprost vehicle group, 12/62 (19.4%) minoxidil 2% group.
Baseline data Target area hair count hairs/cm² (SD): bimatoprost 153.1 (54.78), bimatoprost B 161.1 (63.85), bimatoprost C 145.2 (63.42), bimatoprost vehicle 163.0 (57.28), minoxidil 2% 156.3 (55.46) |
|
| Interventions |
Intervention
Comparator
Comparator 2
Comparator 3
Comparator 4
|
|
| Outcomes | Assessments (2): at baseline and month 6 Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Sponsor is Allergan | |
| Declaration of interest | No information, probably Allergan | |
| Notes | We e‐mailed Allergan several times, without any response | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (clinicaltrials.gov): "randomized". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (clinicaltrials.gov): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (clinicaltrials.gov): "double‐blind". Comment: there was uncertainty with effective blinding of outcomes assessors (healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were 49/306 (16.0%) withdrawals/losses to follow‐up: 6/61 (9.8%) bimatoprost A group, 5/61 (8.2%) bimatoprost B group, 17/61 (27.9%) bimatoprost C group, 9/61 (14.8%) bimatoprost vehicle group, 12/62 (19.4%) minoxidil 2% group. Reasons were reported, and there was an unbalanced number of drop‐outs. However, the higher percentage of drop‐outs in bimatoprost C group was unrelated to adverse events or lack of effect. Data analysis was per‐protocol. Comment: we judged this as at unclear risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was available (NCT01325350 as well as EudraCT 2011‐000380‐27), and trial reported all outcomes listed. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
NCT01655108.
| Methods | This is a double‐blind randomised active‐controlled trial Setting Brasilia University Hospital, Brasilia, Brazil Date of study March 2012 to October 2013 (10‐week duration) |
|
| Participants | 54 women Inclusion criteria
Exclusion criteria
Randomised 54 participants were randomised (minoxidil group = 27, placebo group = 27) Withdrawals/losses to follow‐up There were 4/54 (7.4%) withdrawals/losses to follow‐up: 3/27 (11.1%) minoxidil group, 1/27 (3.7%) placebo group
Baseline data Not clearly reported |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, week 10, and week 18 Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Unclear | |
| Declaration of interest | Unclear | |
| Notes | We last accessed the website on 29 July 2015, and the study was completed. We received data from the PI | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (clinicaltrials.gov): "randomized". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. After e‐mail contact: "We made the allocation using the random function of Microsoft Excel, version 6.1.7601 (Microsoft Corporation, Santa Rosa, U.S.A)...The clinical record of the patient was developed / created on a computer, where it was connected to the randomization Excel spreadsheet described above so that upon being selected for the study (control group?) , the patient would be randomized by the system itself, therefore eliminating the possibility of interference from the investigators / researchers". Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. After e‐mail contact: central allocation, which was probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (clinicaltrials.gov): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. After e‐mail contact: "The medications utilized in the study (minoxidil 0,5%) and placebo (0,9% saline) were produced by a compounding pharmacy (Health Tech Farmácia de Manipulação LTDA ‐ Rusa Teresina 208/210, Vila Bertioga, São Paulo ‐ SP) in identical bottles. The bottles were labelled with number only: 1 (0,9% Saline) and 2 (Minoxidil 0,5%) so that identification of the original medication was not possible". Comment: we judged this as at low risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote (clinicaltrials.gov): "double‐blind". Comment: there was uncertainty with effective blinding of outcomes assessors (healthcare providers) during the study. There was insufficient information to permit a clear judgement. After e‐mail contact (see above): the trial authors adequately reported the measures used to blind the outcome assessor from knowledge of which intervention a participant received. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 4/54 (7.4%) withdrawals/losses to follow‐up: 3/27 (11.1%) minoxidil group, 1/27 (3.7%) placebo group). Data analysis was per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was available (NCT01655108), and the trial reported all outcomes listed. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
NCT01900041.
| Methods | This is a open‐label randomised active‐controlled trial Setting Multicentre, Moscow Scientific Clinical Center of Dermatology and Cosmetology, Moscow, Russian Federation Date of study April 2012 to July 2013 (26‐week duration) |
|
| Participants | 74 women Age = 18 to 45 years Inclusion criteria
Exclusion criteria
Randomised 74 participants were randomised (37 in each treatment arm) Withdrawals/losses to follow‐up There were 13/74 (17.6%) withdrawals/losses to follow‐up: 7/37 (18.9%) Pantovigar + minoxidil group, 6/37 (16.2%) minoxidil group)
Baseline data Nothing reported |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (4): at baseline, 4, 6, and 9 months Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Merz Pharmaceuticals GmbH, Frankfurt, Germany | |
| Declaration of interest | Unclear, not full publication | |
| Notes | Website accessed 3 August 2015, and last updated December 2013. Pantovigar consists of medicinal yeast 100 mg, thiamine mononitrate 60 mg, calcium pantothenate 60 mg, cystine 20 mg, para‐aminobenzoic acid 20 mg, and keratin 20 mg | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (clinicaltrials.gov): "randomized". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. After e‐mail communication: "The patients were randomized by the block method. The randomization sequence was formed using computer." Comment: this was probably done. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote (clinicaltrials.gov): "open label". Comment: the outcome is likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote (clinicaltrials.gov): "open label". Comment: the outcome measurement is likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There were 13/74 (17.6%) withdrawals/losses to follow‐up: 7/37 (18.9%) Pantovigar + minoxidil group, 6/37 (16.2%) minoxidil group. Data analysis was per‐protocol. Comment: we judged this as at an unclear risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was available (NCT01900041), and the trial reported all outcomes listed. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
NCT01967277.
| Methods | This is a double‐blind randomised active‐controlled trial Setting Two centres USA Date of study March 2014 to August 2014 (16‐week duration) |
|
| Participants | 44 women Mean age = 49 years Inclusion criteria
Exclusion criteria
Randomised 44 participants were randomised (Handi‐Dome Laser One group = 22, Incandescent Red Light Source group = 22) Withdrawals/losses to follow‐up There were 4/44 (9%) withdrawals/losses to follow‐up: 3/22 (13.6%) Handi‐Dome Laser One group, 1/22 (4.5%) Incandescent Red Light Source group)
Baseline data Hair count on 25 mm area (SD): Handi‐Dome Laser One group 189.3 (85.8), Incandescent Red Light Source group 216.9 (109.1) |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (2): at baseline and week 16 Primary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Study is sponsored by Capillus LLC | |
| Declaration of interest | Unclear | |
| Notes | We last accessed the website on 4 August 2015, which was last updated on 5 May 2015. The study is completed | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (clinicaltrials.gov): "randomized". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. After e‐mail communication: we did not receive any additional information to enable a clear judgement. |
| Allocation concealment (selection bias) | Low risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. After e‐mail communication: "These devices were then logged in, tested for function and segregated by actual and sham models. A serial number was written on each helmet top with a permanent marker. This procedure was performed by a technical assistant who had no contact with any other member of the clinical trial, subject or investigator and staff. The helmets were then packed in boxes of twelve (12) and delivered to each site, as required by appointment". Comment: this was probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (clinicaltrials.gov): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. After e‐mail communication: "All test devices, per the IRB approved protocol were unmarked, unadorned with labels (unless required by law), paints or signage. The devices were differentiated from internationally available similar devices by assigning the fictitious name of Handi‐Dome 650...The helmets were then packed in boxes of twelve (12) and delivered to each site, as required by appointment". Comment: it appears that the trial made reasonable attempts to blind participants and personnel from knowledge of which intervention a participant received. We judged this as at a low risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote (clinicaltrials.gov): "double‐blind". Comment: there was uncertainty with effective blinding of outcomes assessors (healthcare providers) during the study. There was insufficient information to permit a clear judgement. After e‐mail communication: see above. We judged this as at a low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 4/44 (9%) withdrawals/losses to follow‐up: 3/22 (13.6%) Handi‐Dome Laser One group, 1/27 (4.5%) Incandescent Red Light Source group). The trial reported the reasons, and performed data analysis per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was available (NCT01967277), and the trial reported all outcomes listed. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Olsen 1991.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Duke University Medical Center, Durham, USA Date of study Not reported (32‐week duration) |
|
| Participants | 30 women Mean age (range) = 36.0 years (19 to 45) in the minoxidil group, 38.9 years (33 to 43) in the placebo group Inclusion criteria
Exclusion criteria
Randomised 30 participants were randomised (15 to each of 2 groups) Withdrawals/losses to follow‐up There were 2/30 (6.7%) (1/group) withdrawals/losses to follow‐up. The time and reasons were unreported Baseline data Duration of hair thinning in mean (SD) years
Degree of thinning Ludwig scale (participants by grade and group)
Number of non‐vellus hairs in the target area, mean (SD)
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (9): at baseline, 4, 8, 12, 16, 20, 24, 28, and 32 weeks
Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 248): "This work was supported in part by a grant from the Upjohn Company" | |
| Declaration of interest | None declared | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 243): "were randomly assigned to apply". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of the risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 244): "Both subjects and investigators remained blinded during the entire study." Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote (page 245): "One technician at Duke University Medical Center blinded as to treatment counted the nonvellus target areas hairs on each set of before and after photographs." Comment: this was probably done. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was a balanced and low number (1 in each group) of losses to follow‐up. The data analysis was per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the trial appears to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Oura 2008.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Department of Dermatology, University of Tokushima, Japan Date of study Unreported (12‐month duration) |
|
| Participants | 30 women Mean age (range) = 38.9 years (22 to 53) Inclusion criteria
Exclusion criteria Nothing was reported Randomised 30 women participants were randomised into 2 equal groups Withdrawals/losses to follow‐up There were 3/30 (10%) withdrawals/losses to follow‐up: adenosine group 2/15, placebo group 1/15: 1 in each group before intervention, and voluntary withdrawal in the adenosine group (1) Baseline data Participants had a clinical diagnosis of FPHL that was rated > 1.5 (6‐point scale 1 = no hair loss, to 6 = detectable hair loss) (Tajima 2007) |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, 6, and 12 months Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 767): "Shiseido Research Centre were cooperative investigators." | |
| Declaration of interest | None declared | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 764): "Randomization was carried out to divide the volunteers into two groups". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 764): "a double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement of risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit a clear judgement of risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was a low number of withdrawals (3/30, 10%): 1 in each group before using the test lotion. There was 1 voluntary withdrawal from the adenosine group at month 12. The data analysis was per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the trial appears to have reported all prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Pazoki‐Toroudi 2012.
| Methods | This was a randomised placebo‐controlled trial Setting Iran University of Medical Sciences, Tehran, Iran Date of study Unspecified (24‐week duration) |
|
| Participants | 124 men and women (62 men, 62 women) Mean age (SD) = 33.63 (7.32) minoxidil 5% group, 32.81 (8.24) in minoxidil high extra combination (MHEC) 37.17 (6.40) years in placebo group Inclusion criteria
Exclusion criteria
Randomised 124 participants were randomised (minoxidil group = 49, MHEC group = 57, placebo group = 18) There were 8/124 (6.5%) withdrawals/losses to follow‐up: 6/49 in minoxidil group and 2/57 in MHEC group, all due to adverse events Baseline data Hamilton scale II: minoxidil group 2, MHEC group 1, placebo group 0 Hamilton scale IIa: minoxidil group 3, MHEC group 6, placebo group 1 Hamilton scale III: minoxidil group 5, MHEC group 4, placebo group 2 Hamilton scale IIIa: minoxidil group 3, MHEC group 4, placebo group 1 Hamilton scale III vertex: minoxidil group 6, MHEC group 5, placebo group 3 Hamilton scale IV: minoxidil group 3, MHEC group 3, placebo group 1 Hamilton scale IVa: minoxidil group 2, MHEC group 1, placebo group 1 Ludwig scale I: minoxidil group 4, MHEC group 2, placebo group 2 Ludwig scale II: minoxidil group 13, MHEC group 28, placebo group 7 Ludwig scale III: minoxidil group 2, MHEC group 1, placebo group 0 |
|
| Interventions |
Intervention
Comparator
Comparator 2
|
|
| Outcomes | Assessments (5): at baseline, week 6, 12, 18, and 24 Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared but one PI is employed by Nano Vichar Pharmaceutical Co.Ltd | |
| Declaration of interest | None declared | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 277): "were randomly allocated". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 277): "blinded investigators (specialists) made observations; therefore neither investigators nor patients knew about the group of study". The report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement of risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Investigators and participants were both outcome assessors. MHEC treatment was applied once a day versus minoxidil and placebo twice a day. Comment: there was uncertainty with effective blinding of outcomes assessors (healthcare providers) during the study. There was insufficient information to permit a clear judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 8/124 (6.5%) withdrawals/losses to follow‐up: 6/49 in minoxidil group and 2/57 in MHEC group, all due to adverse events. Data analysis was per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the trial appears to have reported all prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Policarpi 1993.
| Methods | This was a randomised controlled (sham treatment) trial Setting Departments of Dermatology, University of Florence, Italy Date of study Unreported (36‐week duration) |
|
| Participants | 30 (24 male, 6 female) Mean age (range) = 29.1 years (17 to 58) Inclusion criteria
Exclusion criteria
Randomised Participants were randomised into 2 groups (active intervention group = 20, sham intervention group = 10) Withdrawals/losses to follow‐up There were 6/30 (20%) withdrawals/losses to follow‐up: 5/20 (25%) in the active intervention group, and 1/10 (10%) in the sham intervention group
Baseline data Degree of thinning Ludwig scale
|
|
| Interventions |
Intervention
Comparator
Very limited information was reported |
|
| Outcomes | Assessments (3): at baseline, 8, and 16 weeks Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | Individual patient data were available | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 228): "in modo random in 2 gruppi." Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. Comment: there was insufficient information to permit judgement of whether there was low or high risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit a clear judgement of the risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no missing outcome data for women. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the trial appears to have reported all prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Price 1990.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Departments of Dematology of Kaiser Permanente Medical Center and University, San Francisco, and Trichos Research, Richmond, USA Date of study Unreported (40‐week duration) |
|
| Participants | 9 women Age = 22 to 41 years Inclusion criteria
Exclusion criteria
Randomised 9 participants were randomised (minoxidil group = 5, placebo group = 4) Withdrawals/losses to follow‐up There was 1 withdrawal in the minoxidil group due to hyperprolactinaemia Baseline data Degree of thinning Ludwig scale (participants by grade, intervention group)
|
|
| Interventions |
Intervention
Comparator
The study duration was 40 weeks, and treatment was started after the 2nd visit at 4 weeks from baseline |
|
| Outcomes | Assessments (6): at baseline and at 8‐week intervals Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 683): "The Upjohn Company provided support and encouragement of this research." | |
| Declaration of interest | None declared | |
| Notes | Individual patient data were reported, but there were small sample sizes | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 684): "The subjects were given test solutions in a random, double‐blind manner." Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 683): "double‐blind protocol". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement of risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit a clear judgement of the risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was a small number of withdrawals: 1/9 in the minoxidil group (hyperprolactinaemia). Individual patient data were reported. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The protocol for the study was unavailable, but the trial appears to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Price 2000.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting 8 investigational sites in the USA Date of study Unreported (12‐month duration) |
|
| Participants | 137 women Mean age (range) = 53 years (41 to 60) Inclusion criteria
Exclusion criteria Nothing was reported Randomised 137 participants were randomised (finasteride group = 67, placebo group = 70) Withdrawals/losses to follow‐up There were 12/137 (8.8%) withdrawals/losses to follow‐up: 5/67 (7.5%) in the finasteride group, and 7/70 (10%) in the placebo group
Baseline data Mean baseline hair count measured in a 1 cm² circular area at the anterior/mid area of the scalp ± SD
Savin score (number [%] of women)
Ludwig scale (number (%) of women)
Concomitant hormone replacement therapy (number (%) of women)
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (6): at baseline, 1, 3, 6, 9, and 12 months Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | Quote (page 768): "supported by Merck Research Laboratories." | |
| Declaration of interest | None declared but almost half of the investigators indicated an affiliation with Merck Research Laboratories | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 769): "randomized to receive either". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 769): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement of risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 770): "At the end of the study, an expert panel of 3 dermatologists (E. Olsen, R. Savin, and D. Whiting), blinded as to treatment, independently evaluated hair growth or loss by comparing baseline photographs." Comment: participants and the 3 dermatologists (investigators) were assessors for several outcomes, and, although stated to be "blinded", the measures used were not reported. There was insufficient information to permit clear judgement of bias across all outcomes. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 12/137 (8.8%) dropouts; and the reasons were reported. Intention‐to‐treat analysis (ITT) analysis was done. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was unavailable, but the study appeared to have reported the prespecified outcomes and those mentioned in the methods section. Comment: We judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Rietschel 1987.
| Methods | This was a randomised, double‐blind, controlled study Setting 2 centres in the USA Date of study Unspecified (2‐year duration) |
|
| Participants | 149 with men and women (142 men and 7 women) Age (range) = 34.1 years (18 to 49) Inclusion criteria
Exclusion criteria
Randomised 149 participants (minoxidil 2% = 48, minoxidil 3% = 51, placebo to 3% minoxidil = 50) Withdrawals/losses to follow‐up There were 47/149 (31.9%) withdrawals/losses to follow‐up: 14/48 (29.2%) minoxidil 2% group, 18/51 (35.3%) minoxidil 3%, 15/50 (30%) placebo to 3% minoxidil, reasons unreported Baseline data Duration of baldness ranged from 1 to 32 years, averaging 10.2 years. The average diameter of the vertex bald spot at its widest measurement was 10.7 cm (range = 3.81 to 24.13). All participants had a receding hairline and bitemporal recession |
|
| Interventions |
Intervention
Comparator
Comparator 2
At the end of 4 months, the placebo group switched to a 3% minoxidil solution for the duration of the study. At 12 months, the 2% minoxidil group also switched to a 3% solution. Thus, all participants that continued past 12 months were using the 3% solution. Participants applied 1 mL of solution to the balding area of the scalp in the morning and in the evening. |
|
| Outcomes | Assessments: at baseline, week 2, and then at monthly intervals in the first year and after a year at 3 monthly intervals Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | There were no separate data for the 7 women. We were unable to contact the trial investigators. See Table 8 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 678): "Subjects were randomly assigned to use one of three topical solutions" Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 678): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement of risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit a clear judgement of the risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There were 47/149 (31.9%) withdrawals/losses to follow‐up: 14/48 (29.2%) minoxidil 2% group, 18/51 (35.3%) minoxidil 3%, 15/50 (30%) placebo to 3% minoxidil, reasons unreported. Comment: we judged this as at a high risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was unavailable, but the trial appeared to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Sheng 2014.
| Methods | This was a randomised, double‐blind, active‐controlled study Setting Department of Dermatology, Huashan Hospital, Fudan University, Shanghai, China Date of study Unspecified (6‐month duration) |
|
| Participants | 264 women Mean age (SD) = 32.16 (7.07) years in 2% minoxidil group and 33.08 (2.68) in 5% minoxidil group Inclusion criteria
Exclusion criteria
Randomised 264 participants (minoxidil 2% = 132, minoxidil 5% = 132) Withdrawals/losses to follow‐up There were 13/264 (4.9%) withdrawals/losses to follow‐up: 1/132 (<1%) minoxidil 2% group, 11/132 (8.3%), minoxidil 5% group Baseline data Ludwig grade II: minoxidil 2% group (103), minoxidil 5% group (85) Ludwig grade III: minoxidil 2% group (28), minoxidil 5% group (36) |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, month 3, and month 6 Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (translated): "Using randomisation, 264 patients with FPHL were allocated to 2% and 5% minoxidil solution." Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. After e‐mail communication: "In the study design, randomization (completely randomized design) was done with SPSS. Since FPHL patients were not enrolled simultaneously, they were sorted by the enrolment time (1st, 2nd, 3rd, …, 264th). Then, each subject was allocated according to the random assignment table generated by SPSS." Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. After e‐mail communication: "Random assignment table from SPSS was generate and kept by a separate colleague, and therefore, other participants and investigators enrolling patients could not foresee the assignment." Comment: this was probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 583): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement of risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit a clear judgement of the risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 13/264 (4.9%) withdrawals/losses to follow‐up: 1/132 (< 1%) minoxidil 2% group, 11/132 (8.3%), minoxidil 5% group. Data analysis was per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was unavailable, but the trial appeared to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Shin 2007.
| Methods | This was a randomised, double‐blind,placebo‐controlled study Setting Department of Dermatology, Seoul National University College of Medicine, Seoul, Korea Date of study Unspecified (18‐week duration) |
|
| Participants | 36 women Mean age (SD) = 34.1 (4.8) years in the AP‐FHG0604T group 32.2 (7.3) years in placebo group Inclusion criteria
Exclusion criteria
Randomised 36 participants (AP‐FHG0604T group = 17, placebo group = 16, 3 unclear) Withdrawals/losses to follow‐up There were 3/36 (8.3%) withdrawals/losses to follow‐up: lost to follow‐up, did not want to shave, unclear from which group Baseline data Total hair count (n/cm2(SD)): 100.2 (5.4) in AP‐FHG0604T group, 100.8 (4.1) |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): baseline, week 9, and week 18 Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 119): "randomized." Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. After e‐mail communication: "We used a block randomization method. Random numbers were generated by SAS program" Comment: this was probably done. |
| Allocation concealment (selection bias) | Low risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. After e‐mail communication: "The allocation sequence was kept in an envelop secretly by a third staff (a pharmacist who was not involved in the intervention and analysis). This concealment was maintained until the completion of the last follow up and evaluation of all results." Comment: this was probably done. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 119): "double‐blind". Comment: the report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement of risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit a clear judgement of the risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 3/36 (8.3%) withdrawals/losses to follow‐up: lost to follow‐up, did not want to shave, unclear from which group. Data analysis per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was unavailable, but the trial appears to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Thom 2001.
| Methods | This was a randomised, placebo‐controlled, double‐blind study Setting Norway Date of study Unspecified (6‐month duration) |
|
| Participants | 60 (55 men and 5 women) Mean age (SD) = 37.8 (3.9) years in Hairgain® group and 38.6 (3.4) years in placebo group Inclusion criteria
Exclusion criteria
Randomised 60 participants (30 in both groups) Withdrawals/losses to follow‐up There were no withdrawals/losses to follow‐up Baseline data AGA (56), alopecia totalis (4) Duration of hair loss: 18.6 months in the Hairgain® group, 20.4 in the placebo group |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (4): baseline, month 2, 4, and 6 Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared, but PI is employed by PAREXEL Medstat AS, Lillestrøm, Norway | |
| Notes | The 6‐month blinded phase was followed by an open phase on active treatment. There were no separate data for women. We e‐mailed the PI but received no response. See Table 8 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 2 to 3): "randomized" "simple block‐randomization procedure (blocks of six)" Comment: this was probably done. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 3): "Hairgain® and placebo capsules had the same appearance and were packed in
similar plastic bottles in order to keep the study blind. The report provided sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators and participants were outcome assessors. Blinding of participants and key study personnel was ensured, and it is unlikely that the blinding could have been broken Comment: we judged this as at a low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no losses to follow‐up. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was unavailable, but the trial appears to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Thom 2006.
| Methods | This was a randomised, placebo‐controlled, double‐blind study Setting Norway Date of study Unspecified (6‐month duration) |
|
| Participants | 60 (51 men and 4 women, 5 gender unclear) Mean age (SD) = 40.4 (5.4) years in Hairgain® group and 41.7 (6.0) years in placebo group Inclusion criteria
Exclusion criteria
Randomised 60 participants (30 in both groups) Withdrawals/losses to follow‐up There were 5/60 (8.3%) withdrawals/losses to follow‐up: 3/30 (10%) in Nourkrin® group, 2/30 (6.7%) placebo group for not attending follow‐up visits not related to side effects Baseline data Duration of hair loss in months (SD): 20.2 (6.7) in Nourkrin® group and 18.7 (5.6) in placebo group |
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (4): at baseline, month 2, 4, and 6 Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | The 6‐month blinded phase was followed by an open phase on active treatment. There were no separate data for women. We e‐mailed the PI but received no response. See Table 8 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 515): "The subjects were first randomized to receive". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 515): "Nourkrin® and placebo capsules had the same appearance and were packed in similar plastic bottles". Comment: the report provided sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. We judged this as at a low risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Investigators and participants were outcome assessors. Blinding of participants and key study personnel was ensured, and it is unlikely that the blinding could have been broken Comment: we judged this as at a low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were 5/60 (8.3%) withdrawals/losses to follow‐up: 3/30 (10%) in Nourkrin® group, 2/30 (6.7%) placebo group. Data analysis was per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was unavailable, but the trial appears to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Tsuboi 2007.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Multicentre, Japan Date of study January 2001 to January 2002 (24‐week duration) |
|
| Participants | 280 women Mean age (SD) = 56.3 years (10.4) in the minoxidil group, 57.2 years (9.7) in the placebo group Inclusion criteria
Exclusion criteria
Randomised 280 participants were randomised (minoxidil group = 140, placebo group = 140). Withdrawals/losses to follow‐up There were 25/280 (8.9%) withdrawals/losses to follow‐up: 11/140 in the minoxidil group (7.8%), and 14/140 in the placebo group (10%).
3 participants in the minoxidil group and 4 in the placebo group had concomitant or suspected thyroid disease and were considered ineligible for efficacy analyses. Baseline data History of hair loss (years)
Degree of thinning Ludwig scale (% of participants by grade and group)
Non‐vellus hair count (mean ± SD)
Vellus hair count (mean ± SD)
Total hair count (mean ± SD)
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessment was every 4 weeks. Outcomes (as reported)
¹Denotes outcomes prespecified for this review. |
|
| Funding source | Quote (page 43): "The authors received financial support from: Taisho Pharmaceutical Co.,ltd." | |
| Declaration of interest | Quote (page 43): "Conflict of interest: None" | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 38): "were randomly allocated to either of two groups (n = 140 in each group)." "The person responsible for study drug allocation assigned patients to either the TMS or PBO group at a ratio of 1:1, and disclosed the allocation codes to no one until the end of the trial." After e‐mail communication with investigators: in order to obtain the random allocation sequence for making intervention assignments, a computerised random‐number generator was used. Blocked randomisation was used for the generation of the allocation sequence. Comment: we judged this as adequate. |
| Allocation concealment (selection bias) | Low risk | Quote (page 38): "The person responsible for study drug allocation assigned patients to either the TMS or PBO group at a ratio of 1:1, and disclosed the allocation codes to no one until the end of the trial." After e‐mail communication with investigators: a third party, who was independent of the investigator and the sponsor, assigned drugs to either the TMS or PBO group at a ratio of 1:1. The study drug was indistinguishable in appearance and had indistinguishable packaging, and a third party identified indistinguishably in appearance at the time of drug allocation and the end of the trial. A third party disclosed the allocation tables to no one until the end of data lock for analysis, and identified that it had been unopened. Therefore, the allocation sequence was kept blinded to participants, investigators, and sponsor staff who were involved in the treatment or clinical evaluation, until the end of data lock. Comment: this was probably done; we judged this as at a low risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote (page 37): "double‐blind". Quote (page 38): "The active drug and placebo were indistinguishable in appearance and had indistinguishable packaging." Comment: the report provided sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. We judged this as a low risk of bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcomes were participant‐ and investigator‐assessed. Blinding of participants and key study personnel was ensured, and it was unlikely that the blinding could have been broken. Comment: we judged this as at a low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | The incomplete outcome data was as follows:17/140 (12.1%) in the minoxidil group, and 18/140 (12.9%) in the placebo group. Reasons were stated and equally balanced. The data analysis was per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was unavailable, but the trial appears to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Ukşal 1999.
| Methods | This was a randomised, active‐controlled trial Setting Departments of Dermatology and Endocrinology, Kayseri, Turkey Date of study Unspecified (3‐month duration) |
|
| Participants | Number unclear Age = unclear Inclusion criteria
Exclusion criteria. Nothing was reported Randomised It was unclear how many participants were randomised Withdrawals/losses to follow‐up This was unclear Baseline data This was unclear |
|
| Interventions |
Intervention
Comparator
Comparator 2
|
|
| Outcomes | The trial authors did not state these | |
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | The poster abstract had minimal reporting of trial details. We sent 3 e‐mails to the PI, but received no response. See Table 8 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page S238): "Patients were randomly divided into three groups." Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of the risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit a clear judgement of the risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | There was insufficient information to permit a clear judgement of the risk of bias. |
| Selective reporting (reporting bias) | Unclear risk | There was insufficient information to permit a clear judgement of the risk of bias. |
| Other bias | Unclear risk | There was insufficient information to permit a clear judgement of the risk of bias. |
Vexiau 2002.
| Methods | This was a randomised, active‐controlled trial Setting Endocrinology Department, Hôpital Saint‐Louis, Paris, France Date of study July 1993 to November 1995 (6‐month duration) |
|
| Participants | 66 women Mean age = 26.4 years (range 18 to 34) (25.7 years in the cyproterone acetate group (CPA), 27.1 years in the minoxidil group) Inclusion criteria
Exclusion criteria
Randomised 66 participants were randomised (minoxidil group = 33, CPA group = 33) Withdrawals/losses to follow‐up There were 14/66 (21.2%) withdrawals/losses to follow‐up: 6/33 (18%) in the minoxidil group, and 8/33 (24.2%) in the CPA group
Baseline data Degree of thinning Ludwig scale (participants by grade and group)
Mean duration of alopecia
Presence of acne, hirsutism, or both
Menstrual cycle irregularities
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (3): at baseline, 6 months, and 12 months Primary outcomes (as reported)
Secondary outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared, after e‐mail contact. "The study was sponsored by Schering Laboratories." | |
| Declaration of interest | None declared | |
| Notes | The trial authors did not provide baseline data for all randomised participants. women with hyperandrogenic profile included. The trial authors provided intergroup data as well as intragroup (women with versus women without hyperandrogenism), although they did not specify this in the methods section. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote (page 993): "Patients were randomly assigned to one of two groups with stratification every six patients." Comment: this appeared to be block (6) randomisation. This was probably done. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement of risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | The study was open. The nature of the treatment interventions precludes any possibility of blinding of participants and personnel. Comment: the outcome or outcome measurement is likely to be influenced by lack of blinding. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Quote (page 993): "Upon completion of the study, all phototrichograms were read in a blind manner by two independent dermatologists. Conflicting results between the two primary dermatologists were agreed with a third dermatologist." However, participant assessments are likely to be influenced by lack of blinding. Comment: we judged this as at an unclear risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote (page 993): "We had intended to analyse only the results of patients who fully completed the study; however, in cases in which the final measurements at M12 were not carried out, the last documented measurement after day zero (M6) was taken as the final measurement." Quote (page 993): "We were able to analyse the results obtained from the last measurement in 58 of the 66 patients (30 in the CPA group and 28 in the minoxidil group), who were evaluated at least once in addition to d0. A total of 12 patients left the study after the beginning of the treatment, 7 in the CPA group and 5 in the minoxidil group." Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was unavailable, but the trial authors appear to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Whiting 1992.
| Methods | This was a randomised, double‐blind, placebo‐controlled trial Setting Departments of Dermatology and Pediatrics, University of Texas, Dallas, Texas; and the Baylor Hair Research and Treatment Center, Baylor University Medical Center, Dallas, Texas, USA Date of study Unspecified (32‐week duration) |
|
| Participants | 33 women Mean age (range) = 34 years (20 to 44) Inclusion criteria
Exclusion criteria
Randomised 33 participants were randomised (minoxidil group = 17, placebo group = 16) Withdrawals/losses to follow‐up Because of other health problems, relocation, or noncompliance with follow‐up 2 withdrew/were lost to follow‐up in the minoxidil group and 3 in the placebo group Baseline data Mean duration of hair loss
Degree of thinning Ludwig scale (participants by grade and group)
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessment was every 4 weeks. Outcomes (as reported)
¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | Adverse events were not an outcome, but the trial reported that local side‐effects were not severe, and no participants stopped using the medication because of irritation (page 803) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 801): "randomized". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote (page 801): "double‐blind". The report did not provide sufficient detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit clear a judgement of the risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There was a low number (5/33) of dropouts: 2 in the minoxidil group and 3 in the placebo group. Reasons were reported and balanced across groups. The data analysis was per‐protocol. Comment: we judged this as at a low risk of bias. |
| Selective reporting (reporting bias) | Unclear risk | The study protocol was unavailable. Quote (page 802): "The investigator and patient subjectively evaluated visible hair regrowth." Comment: No data were reported for these participant‐ and investigator‐subjective assessments of hair, only that these "correlated poorly with the actual hair counts in the test area." The primary outcomes for this review were under‐reported, so judged this as at unclear risk of bias. |
| Other bias | Unclear risk | The potential impact of the wide range in duration (6 months to 25 years) of hair loss at baseline was unclear. Comment: there was insufficient information to permit a clear judgement of the risk of bias. |
Whiting 1999.
| Methods | This was a randomised, placebo‐controlled trial (phase II study) Setting Multicentre, USA Date of study Unspecified (12‐month duration) |
|
| Participants | 137 women Age = 41 to 60 years Inclusion criteria
Exclusion criteria
Randomised 137 participants were randomised. Data including information on allocation was only available for 94 (finasteride group = 44, placebo group = 50) participants who underwent biopsy at baseline and 12 months Withdrawals/losses to follow‐up 43/137 (31.3%) participants were not analysed, and the reasons for why there was no biopsy (baseline, 12 months) were unreported Baseline data Total terminal anagen hairs (SD)
Total terminal telogen hairs (SD)
Total terminal hairs (SD)
Total vellus or miniaturised hairs (SD)
Total terminal and vellus hairs (SD)
Ratio (± SE) anagen/telogen
Ratio (± SE) terminal/vellus
|
|
| Interventions |
Intervention
Comparator
|
|
| Outcomes | Assessments (2): at baseline and 12 months Outcomes (as reported) 1. Scalp biopsy: all terminal hair bulbs; terminal anagen, catagen, and telogen hairs; vellus hairs and vellus‐like hairs (miniaturised); stelae (streamers); and follicular units counted.¹ ¹Denotes outcomes prespecified for this review |
|
| Funding source | None declared | |
| Declaration of interest | None declared | |
| Notes | — | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote (page 282): "137 patients randomized". Comment: the trial authors did not report the method used to generate the allocation sequence in sufficient detail to allow a clear assessment of whether it would produce comparable groups. |
| Allocation concealment (selection bias) | Unclear risk | The trial authors did not report the method used to conceal the allocation sequence, that is to determine whether intervention allocations could have been foreseen in advance of, or during, enrolment. Comment: there was insufficient information to permit a clear judgement. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The report did not provide any detail about the measures used to blind study participants and personnel from knowledge of which intervention a participant received, to permit a clear judgement. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote (page 282): "Horizontal sections of reticular and papillary dermis were read by one observer blinded to patient, treatment and time." Comment: this was probably done. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The dropout rate was > 30%; the reasons were not stated. The data analysis was per‐protocol. Comment: the large number of dropouts (> 30%), incomplete outcome data, and inappropriate analysis were potential sources of bias. |
| Selective reporting (reporting bias) | Low risk | The study protocol was unavailable, but the study appears to have reported the prespecified outcomes and those mentioned in the methods section. Comment: we judged this as at a low risk of bias. |
| Other bias | Low risk | Comment: the study appeared to be free of other forms of bias. |
Abbreviations: AGA: androgenetic alopecia; CPA: cyproterone acetate; RCT: randomised controlled trial; FPHL: female pattern hair loss.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ahn 2006 | This was a non‐randomised controlled trial (RCT). |
| Bazzano 1986 | This was a controlled clinical trial (CCT). |
| Bezzola 2009 | This was a quasi‐randomised CCT study (3 groups, each with 15 men and 15 women). In van Zuuren 2012, the former version of the review, we excluded the study because there were no separate data for men and women, but should excluded it for being a CCT. |
| Califano 1991 | This was published in Italian; the language abstract was in English, and the study only included male participants. |
| Caserini 2013 | The study only included male participants. |
| DDI 2008 | This was a non‐RCT. All women received the same treatment. |
| Dhurat 2013 | The study only included male participants. |
| Enshaieh 2005 | Mona Nasser translated this study from Farsi into English (see Acknowledgements). The full study is available in Farsi: Journal of Arak University of Medical Sciences 2003; 6(23)):1‐6.IRANMEDEX http://www.iranmedex.com/English/ (accessed 29 March 2011). The study only included male participants. |
| Farella 1991 | The Italian Cochrane Centre translated and assessed this study, but it was a CCT, so we excluded it. |
| Fisher 2012 | The study describes five studies, but none is a RCT. |
| Golpour 2013 | ZF translated this study, which only included male participants. |
| Greenberg 1996 | This study only included male participants. |
| Gómez Grau 2015 | This was a non‐RCT. |
| Inui 2007 | This study only included male participants. |
| Kohler 2007 | This was a non‐RCT (retrospective study). |
| Lee 2013a | This was a non‐RCT (case‐series). |
| Lee 2015 | This was a non‐RCT. |
| Li 1996 | This was a quasi‐randomised (CCT) study, which assigned participants into two treatment groups by odd‐even visit number. |
| Moftah 2013 | This was a CCT (quasi‐randomised). |
| Navadeh 2002 | This was a CCT (quasi‐randomised). |
| Orfanos 1980 | This study included both male and female participants (9), but there was no separate analysis. The study is more than 35 years old, so it was unlikely that we would receive individual patient data. |
| Panahi 2015 | This study only included male participants. |
| Peereboom‐Wynia 1989 | This was a non‐RCT. |
| Piérard 1996 | This was a non‐RCT. |
| Piérard‐Franchimont 1998 | This study only included male participants. |
| Prager 2002 | This study only included male participants. |
| Rinaldi 2006 | Allocation was by alternation on arrival. There was an inadequate method of sequence generation, which allows for knowledge of intervention assignment among those recruiting participants to the study. It was quasi‐randomised. |
| Roberts 1987 | Although this was a RCT, the study randomised 60 participants, but only included 1 woman (with male pattern baldness). |
| Satino 2003 | This was a CCT. |
| Sinclair 2002 | Allocation was according to month of birth, so the study was a CCT (quasi‐randomised). The study used an inadequate method of sequence generation. |
| Sinclair 2005 | This study used the same data set as Sinclair 2002. It was a non‐RCT. |
| Sisto 2013 | The study only included male participants. |
| Slaught 2013 | This was a non‐RCT (case‐series). Retrospective cohort, chart review, study. |
| Takeda 2014 | The study only included male participants. |
| Yang 2002 | The study only included male participants. |
Abbreviations: RCT: randomised controlled trial; CCT: controlled clinical trial (quasi‐randomised).
Characteristics of studies awaiting assessment [ordered by study ID]
NCT01292746.
| Methods | This is a RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Notes | We accessed the website on 24 July 2015. Last updated April 2013. This study has been terminated. (Recruitment and participant study compliance was difficult) |
NCT01451021.
| Methods | This is a investigator‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Notes | We accessed the website on 4 August 2015, which was last updated on 28 February 2012. The study has been completed |
NCT01451047.
| Methods | This is a investigator‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Notes | We accessed the website on 4 August 2015, which was last updated on 28 February 2012. The study has been completed |
NCT01451073.
| Methods | This is a investigator‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Notes | We accessed the website on 4 August 2015, which was last updated on 28 February 2012. The study has been completed |
NCT01451099.
| Methods | This is a investigator‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Notes | We accessed the website on 4 August 2015, which was last updated on 28 February 2012. The study has been completed |
NCT01451112.
| Methods | This is a investigator‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Notes | We accessed the website on 4 August 2015, which was last updated in July 2013. The study has been completed |
NCT01451125.
| Methods | This is a investigator‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Notes | We accessed the website on 4 August 2015, which was last updated in July 2013. This study has been completed |
NCT01451138.
| Methods | This is a investigator‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Notes | We accessed the website on 4 August 2015, which was last updated in July 2013. This study has been completed |
NCT01451151.
| Methods | This is a investigator‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Notes | We accessed the website on 4 August 2015, which was last updated in July 2013. This study has been completed |
NCT01451177.
| Methods | This is a investigator‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Notes | We accessed the website on 4 August 2015, which was last updated in July 2013. This study has been completed |
NCT01451190.
| Methods | This is a investigator‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Notes | We accessed the website on 4 August 2015, which was last updated on July 2013. This study has been completed |
Characteristics of ongoing studies [ordered by study ID]
ACTRN12607000027415.
| Trial name or title | Double blind placebo controlled trial into the treatment of female pattern hair loss with spironolactone and minoxidil |
| Methods | This is a randomised controlled trial (RCT) |
| Participants |
Inclusion criteria
|
| Interventions |
Both groups are treated for 12 months. |
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 1 February 2007 |
| Contact information | Dr. A Yazdabadi (yazdaa27@gmail.com) Department of Dermatology St Vincent's Hospital Fitzroy Australia Telephone: (03) 9288 2211, yazdaa27@gmail.com |
| Notes | At 24 July 2015, the trial was "not yet recruiting". Contact: Rod.SINCLAIR@svhm.org.au |
EUCTR2013‐002740‐85‐ES.
| Trial name or title | A Pilot Randomized, Double‐blind, Placebo‐controlled Clinical Trial to Obtain Preliminary Data on Efficacy and Safety in the Application of PRGF‐Endoret by Mesotherapy, in the Treatment of Male and Female Androgenetic Alopecia of Over 6 Months Duration |
| Methods | This is a double‐blind randomised placebo‐controlled trial |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | 11 February 2014 |
| Contact information | virginia.cuadrado@bti‐implant.es |
| Notes | It looks exactly like NCT01885676 and has the same sponsor, but has different contact people |
NCT00175617.
| Trial name or title | Efficacy of Therapy With the Anti‐androgen Spironolactone Compared to Topical Minoxidil in Female Pattern Hair Loss |
| Methods | This is a open‐label RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | September 2005 |
| Contact information | Andreas Finner UBC Division of Dermatology Hair Research and Treatment Centre Vancouver British Columbia Canada V6G 1Y6 Telephone: 604 875 4747, e‐mail andreas.finner@vch.ca and Jerry Shapiro jerry.shapiro@vch.ca |
| Notes | Recruitment status unknown because information has not been verified recently (website accessed 17 July 2015) |
NCT00197379.
| Trial name or title | The Study for New Effect of Roxithromycin on Androgenetic Alopecia |
| Methods | This is a cross‐over, open‐label RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | May 2005 |
| Contact information | Department of Dermatology Hamamatsu University School of Medicine Hamamatsu Japan 431‐3192 E‐mail: itoutai@hama‐med.ac.jp |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently; it was last updated in July 2010 (website accessed 17 July 2015). See Table 6 |
NCT00418249.
| Trial name or title | Topical AS101 for Treatment of FAGA (Female Androgenetic Alopecia) in Menopause Women |
| Methods | This is a randomised placebo‐controlled trial |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | January 2007 |
| Contact information | Raziel Lurie, MD, rlurie@bezeqint.net or Danny Ben Amitai danb@clalit.org.il |
| Notes | The recruitment status of this study is unknown because the information has not been verified recently (website accessed 17 July 2015). We received no response from the contact person |
NCT01286649.
| Trial name or title | Safety and Efficacy Study of Human Autologous Hair Follicle Cells to Treat Androgenetic Alopecia |
| Methods | This is a double‐blind randomised placebo‐controlled trial |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | December 2010 |
| Contact information | RepliCel Life Sciences, Inc. Principal investigator Nino Lortkipanidze, Scientific Research Institute for Skin and Venereal Diseases, Tbilisi Georgia |
| Notes | This trial is expected to be completed October 2016. We accessed the website on 29 July 2015. No e‐mail address |
NCT01662089.
| Trial name or title | The Efficacy in Treatment of Female Pattern Hair Loss Using 5% Minoxidil Solution Combined With Zinc Supplement |
| Methods | This is a double‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | January 2012 |
| Contact information | Rattapon Thuangtong, MD, rattapongthuangtong@yahoo.com Siriraj Hospital Bangkok Thailand |
| Notes | We accessed the website on 4 August 2015, which was last updated on 7 August 2012 |
NCT01686295.
| Trial name or title | 24 Week Clinical Trial to Evaluate Safety and Effectiveness of a Hair Growth System to Treat Male and Female Baldness |
| Methods | This is a double‐blind RCT |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | July 2012 |
| Contact information | C Andresen, candresen@tklresearch.com J Dosik Jdosik@tklresearch.com |
| Notes | We accessed the website on 4 August 2015, which was last updated on 12 September 2012 |
NCT01885676.
| Trial name or title | Plasma Rich in Growth Factors (PRGF‐Endoret) in the Treatment of Androgenetic Alopecia |
| Methods | This is a double‐blind randomised placebo‐controlled trial |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | Not stated |
| Contact information | Eduardo Anitua, MD, DDS, PhD, eduardoanitua@eduardoanitua.com |
| Notes | We accessed the website on 3 August 2015, which was last updated on 24 December 2013. This is same as EUCTR2013‐002740‐85‐ES above. Both are known under 'BTI‐01D‐EC/12/ALO', but with another contact name virginia.cuadrado@bti‐implant.es |
NCT02074943.
| Trial name or title | Efficacy and Safety of Platelet Rich Plasma in Androgenetic Alopecia |
| Methods | This is a double‐blind randomised placebo‐controlled trial |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | April 2014. Currently recruiting |
| Contact information | Jerry Shapiro, MD Jerry.Shapiro@vch.ca |
| Notes | We accessed the website on 29 July 2015, which was last updated on 10 December 2014 |
NCT02483195.
| Trial name or title | The Use of 5mg Finasteride Versus 200mg Spironolactone and Topical 5% Minoxidil in Treating Postmenopausal Female Androgenetic Alopecia |
| Methods | This is a double‐blind randomised active‐controlled trial |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
|
| Starting date | Not yet recruiting |
| Contact information | Andrea L Taylor, MD andrea.taylor@medicine.ufl.edu and Mark Correa mcorrea@ufl.edu |
| Notes | We accessed the website on 3 August 2015, which was last updated on 24 June 2015 |
NCT02486848.
| Trial name or title | Minoxidil Dose Response Study in Females Identified Through IVD Testing as Non‐Responders to 5% Topical Minoxidil |
| Methods | This is a double‐blind randomised active‐controlled trial |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
|
| Outcomes |
Primary outcomes
Secondary outcomes
|
| Starting date | June 2015. Not yet recruiting |
| Contact information | Alessandra Palmieri, proricerca@hotmail.it |
| Notes | We accessed the website on 3 August 2015, which was last updated on 29 June 2015 |
Abbreviations: AGA: androgenetic alopecia; CPA: cyproterone acetate; CNS: central nervous system; FPHL: female pattern hair loss; RCT: randomized controlled trial
Differences between protocol and review
After consultation and with the direct agreement of the Cochrane Skin Group editorial base, we made substantial changes to the published protocol during the preparation of the 2012 published version of this review (van Zuuren 2012). This was partly due to almost the whole review author team on the protocol being replaced for the review, and due to changes in advice from Cochrane including the introduction of Cochrane’s Methodological Expections of Cochrane Intervention Reviews (MECIR) standards. Changes to the 2012 review which remain the same for this review update include rewriting the Background and Methods sections, clarification of the types of participants, and inclusion of a broader spread of interventions to be considered in this review. In this review update we revised search strategies in line with current practices.
Subgroup analysis and investigation of heterogeneity: the only subgroup we investigated in this update was dose, which was not planned in the protocol or review.
Incomplete outcome data (attrition bias): regarding attrition bias, we are now consistent in all our reviews that we consider the following: up to 10% attrition (low risk); 10% to 20% (unclear risk); and greater than 20% (high risk).
Contributions of authors
JS and the Cochrane Skin Group performed the literature searches. EvZ and ZF identified the relevant titles and abstracts from searches. EvZ obtain the full‐text articles of trials. EvZ and ZF selected trials for inclusion. EvZ translated two Italian studies, one Spanish study, and one German study. ZF translated one Farsi study. EvZ and ZF extracted data from trials, entered data into RevMan (Review Manager (RevMan) 2014), performed data analyses, interpreted the data, drafted the final review, and updated the review.
Disclaimer
This project was supported by the National Institute for Health Research (NIHR), via Cochrane Infrastructure funding to the Cochrane Skin Group. The views and opinions expressed therein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, the NIHR, the NHS, or the Department of Health.
Sources of support
Internal sources
No sources of support supplied
External sources
-
The National Institute for Health Research (NIHR), UK.
The NIHR, UK, is the largest single funder of the Cochrane Skin Group.
Declarations of interest
Evz has no know conflicts of interest. ZF has no known conflict of interest, JS has no known conflict of interest. There are no financial conflicts of interest; the review authors declare that they do not have any associations with any parties who may have vested interests in the results of this Cochrane review.
Edited (no change to conclusions), comment added to review
References
References to studies included in this review
Blume‐Peytavi 2007 {published data only}
- Blume‐Peytavi U, Kunte C, Krisp A, Garcia Bartels N, Ellwanger U, Hoffmann R. Comparison of the efficacy and safety of topical minoxidil and topical alfatradiol in the treatment of androgenetic alopecia in women [Vergleich der Wirksamkeit und Sicherheit von Minoxidil und Alfatradiol bei der topischen Behandlung der androgenetischen Alopezie der Frau]. Journal der Deutschen Dermatologischen Gesellschaft 2007;5(5):391‐5. [PUBMED: 17451383] [DOI] [PubMed] [Google Scholar]
Blume‐Peytavi 2011a {published data only}
- Blume‐Peytavi U, Hillmann K, Dietz E, Canfield D, Garcia Bartels N. A randomized, single‐blind trial of 5% minoxidil foam once daily versus 2% minoxidil solution twice daily in the treatment of androgenetic alopecia in women. Journal of the American Academy of Dermatology 2011;65(6):1126‐34.e.2. [PUBMED: 21700360] [DOI] [PubMed] [Google Scholar]
Bureau 2003 {published data only}
- Bureau JP, Ginouves P, Guilbaud J, Roux ME. Essential oils and low‐intensity electromagnetic pulses in the treatment of androgen‐dependent alopecia. Advances in Therapy 2003;20(4):220‐9. [PUBMED: 14669818] [DOI] [PubMed] [Google Scholar]
Carmina 2003 {published data only}
- Carmina E, Lobo RA. Treatment of hyperandrogenic alopecia in women. Fertility and Sterility 2003;79(1):91‐5. [PUBMED: 12524069] [DOI] [PubMed] [Google Scholar]
DeVillez 1994 {published data only}
- DeVillez RL, Jacobs JP, Szpunar CA, Warner ML. Androgenetic alopecia in the female. Treatment with 2% topical minoxidil solution. Archives of Dermatology 1994;130(3):303‐7. [PUBMED: 8129407] [PubMed] [Google Scholar]
Draelos 2005 {published data only}
- Draelos ZD, Jacobson EL, Kim H, Kim M, Jacobson MK. A pilot study evaluating the efficacy of topically applied niacin derivatives for treatment of female pattern alopecia. Journal of Cosmetic Dermatology 2005;4(4):258‐61. [PUBMED: 17168873] [DOI] [PubMed] [Google Scholar]
Fischer 2004 {published data only}
- Fischer TW, Burmeister G, Schmidt HW, Elsner P. Melatonin increases anagen hair rate in women with androgenetic alopecia or diffuse alopecia: results of a pilot randomized controlled trial. British Journal of Dermatology 2004;150(2):341‐5. [PUBMED: 14996107] [DOI] [PubMed] [Google Scholar]
Gassmueller 2008 {published data only}
- Gassmueller J, Hoffmann R, Webster A. Topical fulvestrant solution has no effect on male and postmenopausal female androgenetic alopecia: results from two randomized, proof‐of‐concept studies. British Journal of Dermatology 2008;158(1):109‐15. [PUBMED: 17986309] [DOI] [PubMed] [Google Scholar]
Gehring 2000 {published data only}
- Gehring W, Gloor M. Use of the phototrichogram to assess the stimulation of hair growth ‐ An in vitro study of women with androgenetic alopecia [Das Phototrichogramm als Verfahren zur Beurteilung haarwachstumsfördernder Präparate am beispiel einer Kombination von Hirsefruchtextrakt, L‐Cystin und Calcium panthonthenat‐Ergebnisse einer in vivo Untersuchung bei Frauen mit androgenetischem Haarausfall]. H+G Zeitschrift fur Hautkrankheiten 2000;75(7‐8):419‐23. [EMBASE: 2000344799] [Google Scholar]
Georgala 2004 {published data only}
- Georgala S, Katoulis AC, Georgala C, Moussatou V, Bozi E, Stavrianeas NG. Topical estrogen therapy for androgenetic alopecia in menopausal females. Dermatology 2004;208(2):178‐9. [PUBMED: 15057016] [DOI] [PubMed] [Google Scholar]
Guerrero 2009 {published data only}
- Guerrero AR, Medina KR, Kahn MC, Guerrero MJ. Usefulness and safety of 0.025 percent 17‐alpha‐estradiol versus 2 percent minoxidil in the treatment of androgenetic alopecia [Utilidad y seguridad del 17‐alfa‐estradiol 0,025 por ciento versus minoxidil 2 por ciento en el tratamiento de la alopecia androgenetica]. Revista Chilena de Dermatologia 2009;25(1):21‐5. [Google Scholar]
Hong 2007 {published data only}
- Hong CK, Han TY, Kang HC, Ahn SW, Ahn JY, Seo SJ. The efficacy and safety of a combination cytopurine/pentadecanoic glyceride topical solution on androgenetic alopecia: A randomized double‐blind placebo‐controlled clinical trial. Korean Journal of Dermatology 2007;45(12):1233‐9. [Google Scholar]
Jacobs 1993 {published data only}
- Jacobs JP, Szpunar CA, Warner ML. Use of topical minoxidil therapy for androgenetic alopecia in women. International Journal of Dermatology 1993;32(10):758‐62. [PUBMED: 8225725] [DOI] [PubMed] [Google Scholar]
Jimenez 2014a {published data only}
- Jimenez J, Wikramanayake TC, Bergfeld W, Hordinsky M, Hickman J, Hamblin M, et al. Efficacy and safety of a low‐level laser device in the treatment of male and female pattern hair loss. Journal of Investigative Dermatology 2014;134(Suppl 1):S100. [EMBASE: 71440974] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez JJ, Wikramanayake TC, Bergfeld W, Hordinsky M, Hickman JG, Hamblin MR, et al. Efficacy and safety of a low‐level laser device in the treatment of male and female pattern hair loss: a multicenter, randomized, sham device‐controlled, double‐blind study. American Journal of Clinical Dermatology 2014;15(2):115‐27. [PUBMED: 24474647] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jimenez 2014b {published data only}
- Jimenez J, Wikramanayake TC, Bergfeld W, Hordinsky M, Hickman J, Hamblin M, et al. Efficacy and safety of a low‐level laser device in the treatment of male and female pattern hair loss. Journal of Investigative Dermatology 2014;134(Suppl 1):S100. [EMBASE: 71440974] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez JJ, Wikramanayake TC, Bergfeld W, Hordinsky M, Hickman JG, Hamblin MR, et al. Efficacy and safety of a low‐level laser device in the treatment of male and female pattern hair loss: a multicenter, randomized, sham device‐controlled, double‐blind study. American Journal of Clinical Dermatology 2014;15(2):115‐27. [PUBMED: 24474647] [DOI] [PMC free article] [PubMed] [Google Scholar]
Keene 2011 {published data only}
- Goren AO, Tam PY, Frie D, Keene SA. Epigenetic markers predict treatment response to finasteride in female androgenetic alopecia: Poster session. Experimental Dermatology 2010;19(6):584. [EMBASE: 70193861] [Google Scholar]
- Keene S, Goren A. Therapeutic hotline. Genetic variations in the androgen receptor gene and finasteride response in women with androgenetic alopecia mediated by epigenetics. Dermatologic Therapy 2011;24(2):296‐300. [PUBMED: 21410621] [DOI] [PubMed] [Google Scholar]
Kim 2009 {published data only}
- Kim JH, Y SM, Choi JE, Son SW. Study of the efficacy of Korean red ginseng in the treatment of androgenic alopecia. Journal of Ginseng Research 2009;33(3):223‐8. [Google Scholar]
Kim 2013 {published data only}
- Kim H, Choi JW, Kim JY, Shin JW, Lee SJ, Huh CH. Low‐level light therapy for androgenetic alopecia: a 24‐week, randomized, double‐blind, sham device‐controlled multicenter trial. Dermatologic Surgery 2013;39(8):1177‐83. [PUBMED: 23551662] [DOI] [PubMed] [Google Scholar]
Lanzafame 2014 {published data only}
- Lanzafame RJ, Blanche RR, Chiacchierini RP, Kazmirek ER, Sklar JA. The growth of human scalp hair in females using visible red light laser and LED sources. Lasers in Surgery and Medicine 2014;46(8):601‐7. [PUBMED: 25124964] [DOI] [PMC free article] [PubMed] [Google Scholar]
Le Floc'h 2015 {published data only}
- Floc'h C, Cheniti A, Connétable S, Piccardi N, Vincenzi C, Tosti A. Effect of a nutritional supplement on hair loss in women. Journal of Cosmetic Dermatology 2015;14(1):76‐82. [PUBMED: 25573272] [DOI] [PubMed] [Google Scholar]
Lucky 2004 {published data only}
- Lucky AW, Piacquadio DJ, Ditre CM, Dunlap F, Kantor I, Pandya AG, et al. A randomized, placebo‐controlled trial of 5% and 2% topical minoxidil solutions in the treatment of female pattern hair loss. Journal of the American Academy of Dermatology 2004;50(4):541‐53. [PUBMED: 15034503] [DOI] [PubMed] [Google Scholar]
Mazzarella 1997 {published data only}
- Mazzarella F, Loconsole F, Cammisa A, Mastrolonardo M, Vena GA. Topical finasteride in the treatment of androgenic alopecia. Preliminary evaluations after a 16‐month therapy course. Journal of Dermatological Treatment 1997;8(3):189‐92. [EMBASE: 1997295968] [Google Scholar]
Minozzi 1997 {published data only}
- Minozzi M, Unfer V, Constabile L. Androgenetic alopecia in postmenopausal women [Alopecia androgenetica in post‐menopausa]. Giornale Italiano di Ostetricia e Ginecologia 1997;19(6):341‐4. [EMBASE: 1997225861] [Google Scholar]
Morganti 1998 {published data only}
- Morganti P, Fabrizi G, James B, Bruno C. Effect of gelatin‐cystine and serenoa repens extract on free radicals level and hair growth. Journal of Applied Cosmetology 1998;16(3):57‐64. [EMBASE: 1998406625] [Google Scholar]
NCT01145625 {unpublished data only}
- Doshi U, Israel M, Kendall C, Quiza C, Sacavage S, Tyndall D, et al. Effectiveness of once daily 5% minoxidil topical foam in the treatment of women with thinning hair due to female pattern hair loss (FPHL). Journal of the American Academy of Dermatology 2015;72(5 Suppl 1):AB112. [EMBASE: 71895204] [Google Scholar]
- NCT01145625. A Phase 3 multi‐center parallel design clinical trial to compare the efficacy and safety of 5% Minoxidil Foam vs. 2% Minoxidil Solution in females for the treatment of female pattern hair loss ‐ androgenetic alopecia (MINALO3004, NCT01145625). clinicaltrials.gov/ct2/show/NCT01145625 (accessed 14 January 2016).
NCT01189279 {unpublished data only}
- NCT01189279. Safety and pharmacokinetics study of new formulation of Bimatoprost in patients with alopecia. clinicaltrials.gov/ct2/show/NCT01189279 (accessed 14 January 2016).
NCT01226459 {unpublished data only}
- NCT01226459. A Phase 3 Multi‐Center Parallel Design Clinical Trial to Compare the Efficacy and Safety of 5% Minoxidil Foam vs. Vehicle in Females for the Treatment of Female Pattern Hair Loss (Androgenetic Alopecia). clinicaltrials.gov/ct2/show/NCT01226459 (accessed 14 January 2016).
NCT01325350 {unpublished data only}
- NCT01325350. Safety and Efficacy Study of Bimatoprost in the Treatment of Women With Female Pattern Hair Loss. clinicaltrials.gov/ct2/show/NCT01325350 (accessed 14 January 2016).
NCT01655108 {unpublished data only}
- NCT01655108. Efficacy and Safety of Mesotherapy With Minoxidil 0.5%/2ml for Androgenetic Alopecia in Female Patients. clinicaltrials.gov/ct2/show/NCT01655108 (accessed 14 January 2016).
NCT01900041 {published and unpublished data}
- NCT01900041. A Study to Evaluate the Superiority, Efficacy and Tolerability of Combination Pantovigar With 2% Minoxidil vs 2% Minoxidil in Women With Female Pattern Hair Loss. clinicaltrials.gov/ct2/show/NCT01900041 (accessed 14 January 2016).
NCT01967277 {unpublished data only}
- NCT01967277. A Novel Approach to Treating Androgenetic Alopecia in Females With Low Level Laser Therapy. clinicaltrials.gov/ct2/show/NCT01967277 (accessed 14 January 2016).
Olsen 1991 {published data only}
- Olsen EA. Topical minoxidil in the treatment of androgenetic alopecia in women. Cutis 1991;48(3):243‐8. [PUBMED: 1935254] [PubMed] [Google Scholar]
Oura 2008 {published data only}
- Oura H, Iino M, Ideta R, Nakazawa Y, Kishimoto J, Arase S. Efficacy of adenosine on female pattern hair loss and male androgenetic alopecia. Journal of Investigative Dermatology 2008;128(Suppl 1):S151. [Google Scholar]
- Oura H, Iino M, Nakazawa Y, Tajima M, Ideta R, Nakaya Y, et al. Adenosine increases anagen hair growth and thick hairs in Japanese women with female pattern hair loss: a pilot, double‐blind, randomized, placebo‐controlled trial. Journal of Dermatology 2008;35(12):763‐7. [PUBMED: 19239555] [DOI] [PubMed] [Google Scholar]
Pazoki‐Toroudi 2012 {published data only}
- Pazoki‐Toroudi H, Babakoohi S, Nilforoushzadeh MA, Nassiri‐Kashani M, Shizarpour M, Ajami M, et al. Therapeutic effects of minoxidil high extra combination therapy in patients with androgenetic alopecia. Skinmed 2012;10(5):276‐82. [PUBMED: 23163069] [PubMed] [Google Scholar]
Policarpi 1993 {published data only}
- Policarpi F, Giorgini S, Farella V, Melli MC, Campolmi P, Panconesi E. Effects of application of a pulsed electrostatic field in male and female androgenic alopecia [Efficacia dell'applicazione di un campo elettrostatico pulsato nell'alopecia androgenetica maschile e femminile]. Annali Italiani di Dermatologia Clinica e Sperimentale 1993;47(3):227‐32. [EMBASE: 1993274287] [Google Scholar]
Price 1990 {published data only}
- Price VH, Menefee E. Quantitative estimation of hair growth. I. androgenetic alopecia in women: effect of minoxidil. Journal of Investigative Dermatology 1990;95(6):683‐7. [PUBMED: 2250110] [DOI] [PubMed] [Google Scholar]
Price 2000 {published data only}
- Price VH, Roberts JL, Hordinsky M, Olsen EA, Savin R, Bergfeld W, et al. Lack of efficacy of finasteride in postmenopausal women with androgenetic alopecia. Journal of the American Academy of Dermatology 2000;43(5 Pt 1):768‐76. [PUBMED: 11050579] [DOI] [PubMed] [Google Scholar]
Rietschel 1987 {published data only}
- Rietschel ML, Duncan SH. Safety and efficacy of topical minoxidil in the management of androgenetic alopecia. Journal of the American Academy of Dermatology 1987;16(3 Pt 2):677‐85. [EMBASE: 1987099274] [DOI] [PubMed] [Google Scholar]
Sheng 2014 {published data only}
- Sheng Y‐Y, Rui W‐L, Xu F, Qi S‐S, Hu R‐M, Miao Y, et al. A randomized controlled clinical study on 2% and 5% topical minoxidil solution in the treatment of female pattern hair loss [Chinese]. Journal of Clinical Dermatology 2014;43(10):583‐5. [EMBASE: 2015699887] [Google Scholar]
Shin 2007 {published data only}
- Shin HS, Lee SH, Kim DH, An JS, Kwon OS, Eun HC. The efficacy and safety of AP‐FHG0604T on female pattern hair loss: A randomized double‐blind placebo‐controlled clinical trial. Korean Journal of Dermatology 2007;45(2):119‐26. [Google Scholar]
Thom 2001 {published data only}
- Thom E. Efficacy and tolerability of Hairgain® in individuals with hair loss: a placebo‐controlled, double‐blind study. Journal of International Medical Research 2001;29(1):2‐6. [EMBASE: 2001114261] [DOI] [PubMed] [Google Scholar]
Thom 2006 {published data only}
- Thom E. Nourkrin: objective and subjective effects and tolerability in persons with hair loss. Journal of International Medical Research 2006;34(5):514‐9. [EMBASE: 2006563255] [DOI] [PubMed] [Google Scholar]
Tsuboi 2007 {published data only}
- Tsuboi R, Tanaka T, Nishikawa T, Ueki R, Yamada H, Katsuoka K, et al. A randomized, placebo‐controlled trial of 1% topical minoxidil solution in the treatment of androgenetic alopecia in Japanese women. European Journal of Dermatology 2007;17(1):37‐44. [PUBMED: 17324826] [DOI] [PubMed] [Google Scholar]
Ukşal 1999 {published data only}
- Ukşal Ü, Bayram F, Utaş S, Çolak R, Borlu M. Comparision of low dose finasteride, flutamide and spirolactone in female androgenetic alopecia. Journal of the European Academy of Dermatology & Venereology 1999;12(Suppl 2):S238. [Google Scholar]
Vexiau 2002 {published data only}
- Vexiau P, Chaspoux C, Boudou P, Fiet J, Jouanique C, Hardy N, et al. Effects of minoxidil 2% vs. cyproterone acetate treatment on female androgenetic alopecia: a controlled, 12‐month randomized trial. British Journal of Dermatology 2002;146(6):992‐9. [PUBMED: 12072067] [DOI] [PubMed] [Google Scholar]
Whiting 1992 {published data only}
- Whiting DA, Jacobson C. Treatment of female androgenetic alopecia with minoxidil 2%. International Journal of Dermatology 1992;31(11):800‐4. [PUBMED: 1428436] [DOI] [PubMed] [Google Scholar]
Whiting 1999 {published data only}
- Whiting DA, Waldstreicher J, Sanchez M, Kaufman KD. Measuring reversal of hair miniaturization in androgenetic alopecia by follicular counts in horizontal sections of serial scalp biopsies: Results of finasteride 1 mg treatment of men and postmenopausal women. Journal of Investigative Dermatology. Symposium Proceedings 1999;4(3):282‐4. [PUBMED: 10674382] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Ahn 2006 {published data only}
- Ahn JY, Cho SH, Kim MN, Ro BI. Finasteride treatment of female patterned hair loss in postmenopausal women. Korean Journal of Dermatology 2006;44(9):1094‐7. [EMBASE: 2006621285] [Google Scholar]
Bazzano 1986 {published data only}
- Bazzano GS, Terezakis N, Galen W. Topical tretinoin for hair growth promotion. Journal of the American Academy of Dermatology 1986;15(4 Pt 2):880‐3, 890‐3. [EMBASE: 1987002649] [DOI] [PubMed] [Google Scholar]
Bezzola 2009 {published data only}
- Bezzola P, Sorbellini E. The role of microinflammation and apoptosis in androgenic alopecia: New therapeutical strategies [Il ruolo della microinfiammazione e dell' apoptosi nella alopecia androgenetica: Nouve strategie terapeutiche]. Journal of Plastic Dermatology 2009;5(1):47‐58. [EMBASE: 2009226819] [Google Scholar]
Califano 1991 {published data only}
- Califano A, Virgili A, Caputo R, Cavalieri R, Celleno L, Finzi A, et al. 2% Minoxidil topical solution in the treatment of alopecia androgenetica. A controlled clinical study [Minoxidil topico al 2% nel trattamento dell' alopecia androgenetica. Studio clinico controllato]. Giornale Italiano di Dermatologia e Venereologia 1991;126(3):263‐9. [EMBASE: 1991186738] [Google Scholar]
Caserini 2013 {published data only}
- Caserini M, Mailland F, Radicioni M, Annoni O, Palmieri R. Single and repeated dose of finasteride topical solution in subjects with androgenetic alopecia: A pharmacokinetic and pharmacodynamic study. Journal of the American Academy of Dermatology 2013;68(4 Suppl 1):AB108. [EMBASE: 70997515] [Google Scholar]
- Caserini M, Palmieri R, Radicioni M, Terragni E. Pharmacodynamic of P‐3074 (finasteride 0.25% topical solution) in subjects with androgenetic alopecia. Journal of Investigative Dermatology 2013;133(5):1397. [EMBASE: 71059515] [Google Scholar]
- Caserini M, Radicioni M, Annoni O, Palmieri R. Pharmacokinetics and pharmacodynamics of finasteride topical solution after single and repeated dose in subjects with androgenetic alopecia. Journal of Investigative Dermatology 2012;132(Suppl 1):S110. [EMBASE: 71015362] [Google Scholar]
DDI 2008 {published data only}
- Donne Dermatologhe Italia. Clinical investigation of the effects of topical lotion and shampoo containing synthetic thymus peptides on androgenetic alopecia and chronic telogen effluvium in women [Studio prospettico a sei mesi sugli effetti di un prodotto topico in lozione e shampoo a base di timo‐peptididi sintesi a basso peso molecolare nel telogen effluvium cronico e nell’AGA iniziale della donna]. Journal of Plastic Dermatology 2008;4(2):199‐205. [EMBASE: 2008520520] [Google Scholar]
Dhurat 2013 {published data only}
- Dhurat R, Sukesh M, Avhad G, Dandale A, Pal A, Pund P. A randomized evaluator blinded study of effect of microneedling in androgenetic alopecia: a pilot study. International Journal of Trichology 2013;5(1):6‐11. [PUBMED: 23960389] [DOI] [PMC free article] [PubMed] [Google Scholar]
Enshaieh 2005 {published data only}
- Enshaieh S, Siadat AH, Shariat S, Iraji F, Nilforoushzadeh MA, Asilian A. Comparison between an herbal drug and 2% minoxidil solution in the treatment of the androgenetic alopecia [Abstract P03.30]. The 14th Congress of the European Academy of Dermatology and Venereology; 2005 Oct 12‐15; London. Journal of the European Academy of Dermatology & Venereology 2005;19(Suppl 2):74‐5. [Google Scholar]
Farella 1991 {published data only}
- Farella V, Melli MC, Giorgini S. Mucopolysaccharides and depolymerized nucleic acids in the treatment of androgenic alopecia [Mucopolisaccaridi e acidi nucleici depolimerizzati nella alopecia androgenetica, studio clinico in doppio cieco]. Dermatologia Oggi 1991;6(3):39‐46. [EMBASE: 1992071835] [Google Scholar]
Fisher 2012 {published data only}
- Fischer TW, Trueb RM, Hänggi G, Innocenti M, Elsner P. Topical melatonin for treatment of androgenetic alopecia [Melatonin in der topischen Behandlung der androgenetischen Alopezie]. Aktuelle Dermatologie 2011;37(11):410‐8. [EMBASE: 2011615861] [Google Scholar]
- Fischer TW, Trueb RM, Hänggi G, Innocenti M, Elsner P. Topical melatonin for treatment of androgenetic alopecia. International Journal of Trichology 2012;4(4):236‐45. [PUBMED: 23766606] [DOI] [PMC free article] [PubMed] [Google Scholar]
Golpour 2013 {published data only}
- Golpour M, Rabbani H, Farzin D, Azizi F. Comparing the effectiveness of local solution of minoxidil and caffeine 2.5% with local solution of minoxidil 2.5% in treatment of androgenetic alopecia. Journal of Mazandaran University of Medical Sciences 2013;23(106):29‐36. [EMBASE: 2014593395] [Google Scholar]
Gómez Grau 2015 {published data only}
- Gómez Grau E, Lladós Sevilla M, Mira J, Vivancos F. Effectiveness of a dietary supplement with serenoa serrulata and tocotrienol‐tocopherol against female androgenetic alopecia and telogen effluvium. Report of a pilot study [Eficacia de un complemento alimenticio con serenoa serrulata y tocotrienol‐tocoferol frente a la alopecia androgenética y el efluvio telógeno femeninos. A propósito de un estudio piloto]. Revista Argentina de Dermatología 2015;96(1):43‐55. [EMBASE: 2015058643] [Google Scholar]
Greenberg 1996 {published data only}
- Greenberg JH, Katz M, Eliaz I. Treatment of androgenetic alopecia with HairPrime ‐ a 7.5% herbal preparation. Townsend Letter 1996;160:68‐72. [Google Scholar]
Inui 2007 {published data only}
- Inui S, Itami S. Reversal of androgenetic alopecia by topical ketoconzole: Relevance of anti‐androgenic activity. Journal of Dermatological Science 2007;45(1):66‐8. [EMBASE: 2006629196] [DOI] [PubMed] [Google Scholar]
Kohler 2007 {published data only}
- Kohler C, Tschumi K, Bodmer C, Schneiter M, Birkhaeuser M. Effect of finasteride 5 mg (Proscar) on acne and alopecia in female patients with normal serum levels of free testosterone. Gynecological Endocrinology 2007;23(3):142‐5. [EMBASE: 2007177333] [DOI] [PubMed] [Google Scholar]
Lee 2013a {published data only}
- Lee YB, Eun YS, Lee JH, Cheon MS, Park YG, Cho BK, et al. Effects of topical application of growth factors followed by microneedle therapy in women with female pattern hair loss: A pilot study. Journal of Dermatology 2013;40(1):81‐3. [PUBMED: 23039201] [DOI] [PubMed] [Google Scholar]
Lee 2015 {published data only}
- Lee S‐H, Zheng Z, Kang J‐S, Kim D‐Y, Oh SH, Cho SB. Therapeutic efficacy of autologous platelet‐rich plasma and polydeoxyribonucleotide on female pattern hair loss. Wound Repair & Regeneration 2015;23(1):30‐6. [PUBMED: 25524027] [DOI] [PubMed] [Google Scholar]
Li 1996 {published data only}
- Li MC, Shu W, Wang HY, Wang XS. Randomized double‐blind placebo‐controlled evaluation of topical minoxidil in the treatment of female androgonetic alopecia. Chinese Journal of Dermatology 1996;29:223‐5. [Google Scholar]
Moftah 2013 {published data only}
- Moftah N, Moftah N, Abd‐Elaziz G, Ahmed N, Hamed Y, Ghannam B, et al. Mesotherapy using dutasteride‐containing preparation in treatment of female pattern hair loss: Photographic, morphometric and ultrustructural evaluation. Journal of the European Academy of Dermatology and Venereology 2013;27(6):686‐93. [PUBMED: 22486925] [DOI] [PubMed] [Google Scholar]
Navadeh 2002 {published data only}
- Navadeh A. Treatment of female androgenetic alopecia with finasteride. NederlandsTijdschrift voor Dermatologie en Venereologie 2002;12:119‐21. [Google Scholar]
Orfanos 1980 {published data only}
- Orfanos CE, Vogels L. Local therapy of androgenetic alopecia with 17 alpha‐estradiol. A controlled, randomized double‐blind study [Lokaltherapie der Alopecia androgenetica mit 17 alpha‐Ostradiol. Eine kontrollierte, randomisierte Doppelblindstudie]. Dermatologica 1980;161(2):124‐32. [EMBASE: 1980152416] [PubMed] [Google Scholar]
Panahi 2015 {published data only}
- Panahi Y, Taghizadeh M, Marzony ET, Sahebkar A. Rosemary oil vs minoxidil 2% for the treatment of androgenetic alopecia: a randomized comparative trial. Skinmed 2015;13(1):15‐21. [PUBMED: 25842469] [PubMed] [Google Scholar]
Peereboom‐Wynia 1989 {published data only}
- Peereboom‐Wynia JDR, Willigen AH, Joost T, Stolz E. The effect of cyproterone acetate on hair roots and hair shaft diameter in androgenetic alopecia in females. Acta Dermato‐Venereologica 1989;69(5):395‐8. [EMBASE: 1989226146] [PubMed] [Google Scholar]
Piérard 1996 {published data only}
- Piérard GE, Piérard‐Franchimont C, Nikkels‐Tassoudji N, Nikkels AF, Saint Léger D. Improvement in the inflammatory aspect of androgenetic alopecia. A pilot study with an antimicrobial lotion. Journal of Dermatological Treatment 1996;7(3):153‐7. [EMBASE: 1996320919] [Google Scholar]
Piérard‐Franchimont 1998 {published data only}
- Piérard‐Franchimont C, Doncker P, Cauwenbergh G, Piérard GE. Ketoconazole shampoo: effect of long‐term use in androgenic alopecia. Dermatology 1998;196(4):474‐7. [EMBASE: 1998207910] [DOI] [PubMed] [Google Scholar]
Prager 2002 {published data only}
- Prager N, Bickett K, French N, Marcovici G. A randomized, double‐blind, placebo‐controlled trial to determine the effectiveness of botanically derived inhibitors of 5‐alpha‐reductase in the treatment of androgenetic alopecia. Journal of Alternative and Complementary Medicine 2002;8(2):143‐52. [EMBASE: 2002152771] [DOI] [PubMed] [Google Scholar]
Rinaldi 2006 {published data only}
- Rinaldi F. Evaluation of the efficacy of an extract of Cichorium intybus against hair loss [Avaliação da eficácia de um extrato de Cichorium intybus contra a queda dos cabelos]. Revista Brasileira De Medicina 2006;63(6):271‐3. [Google Scholar]
Roberts 1987 {published data only}
- Roberts JL. Androgenetic alopecia: treatment results with topical minoxidil. Journal of the American Academy of Dermatology 1987;16(3 Pt 2):705‐10. [EMBASE: 1987099278] [DOI] [PubMed] [Google Scholar]
Satino 2003 {published data only}
- Satino JL, Markou M. Hair regrowth and increased hair tensile strength using the HairMax LaserComb for Low‐Level Laser Therapy. International Journal of Cosmetic Surgery and Aesthetic Dermatology 2003;5(2):113‐7. [DOI: 10.1089/153082003769591209; EMBASE: 2003431467] [DOI] [Google Scholar]
Sinclair 2002 {published data only}
- Sinclair R. Treatment of androgenetic alopecia in women with oral antiandrogen therapy. Annales de Dermatologie et de Venereologie 2002;129(Suppl 1 Pt 1):1S324. [Google Scholar]
Sinclair 2005 {published data only}
- Sinclair R, Wewerinke M, Jolley D. Treatment of female pattern hair loss with oral antiandrogens. British Journal of Dermatology 2005;152(3):466‐73. [EMBASE: 2005182968] [DOI] [PubMed] [Google Scholar]
Sisto 2013 {published data only}
- Sisto T, Bussoletti C, Celleno L. Efficacy of a cosmetic caffeine shampoo in androgenetic alopecia management. II Note. Journal of Applied Cosmetology 2013;31(1):57‐66. [EMBASE: 2013520562] [Google Scholar]
Slaught 2013 {published data only}
- Slaught C, Goh C. Effectiveness of spironolactone in reducing hair loss in women with androgenetic alopecia. Journal of Investigative Medicine 2013;61(1):201. [EMBASE: 70993769] [Google Scholar]
Takeda 2014 {published data only}
- Takeda A, Sato A, Cauwenbergh G, Liu JW, Zhang L, Harti S. Enabling finasteride 1 mg users to further improve hair pattern: A randomized, double‐blind, placebo‐controlled trial of a novel product. Journal of the American Academy of Dermatology 2014;70(5 Suppl 1):AB90. [EMBASE: 71390449] [Google Scholar]
Yang 2002 {published data only}
- Yang SX, Ji SZ, Chen W, Wang D, Wang K, Wang H, et al. A double blind randomized placebo controlled clinical trial of finasteride in the treatment of androgenetic alopecia. Chinese Journal of Clinical Pharmacology 2002;18(1):3‐6. [Google Scholar]
References to studies awaiting assessment
NCT01292746 {unpublished data only}
- NCT01292746. Study of the Effect of Low Level Laser Light on Hair Growth on the Female Human Scalp. clinicaltrials.gov/ct2/show/NCT01292746 (accessed 14 January 2016).
NCT01451021 {unpublished data only}
- NCT01451021. A Study to Evaluate and Compare Injections of Autologous Dermal and Epidermal Cells Into the Balding Scalp of Subjects With Hair Loss. clinicaltrials.gov/ct2/show/NCT01451021 (accessed 14 January 2016).
NCT01451047 {unpublished data only}
- NCT01451047. A Study to Evaluate and Compare Injections of Autologous Dermal and Epidermal Cells Into the Balding Scalp of Subjects With Hair Loss (CA‐0002012). clinicaltrials.gov/ct2/show/NCT01451047 (accessed 14 January 2016).
NCT01451073 {unpublished data only}
- NCT01451073. A Study to Evaluate and Compare Injections of Autologous Dermal and Epidermal Cells Into the Balding Scalp of Subjects With Hair Loss (CA‐0002013). clinicaltrials.gov/ct2/show/NCT01451073 (accessed 14 January 2016).
NCT01451099 {unpublished data only}
- NCT01451099. A Study to Evaluate and Compare Injections of Autologous Dermal Cells Into the Balding Scalp of Subjects With Hair Loss. clinicaltrials.gov/ct2/show/NCT01451099 (accessed 14 January 2016).
NCT01451112 {unpublished data only}
- NCT01451112. A Study to Evaluate and Compare Injections of Autologous Dermal and Epidermal Cells Into the Balding Scalp of Subjects With Hair Loss (CA‐0002899). clinicaltrials.gov/ct2/show/NCT01451112 (accessed 14 January 2016).
NCT01451125 {unpublished data only}
- NCT01451125. A Study to Evaluate and Compare Injections of Autologous Dermal and Epidermal Cells Into the Balding Scalp of Subjects With Hair Loss (CA‐0004512). clinicaltrials.gov/ct2/show/NCT01451125 (accessed 14 January 2016).
NCT01451138 {unpublished data only}
- NCT01451138. A Study to Evaluate and Compare Injections of Autologous Dermal and Epidermal Cells Into the Balding Scalp of Subjects With Hair Loss (CA‐0004541). clinicaltrials.gov/ct2/show/NCT01451138 (accessed 14 January 2016).
NCT01451151 {unpublished data only}
- NCT01451151. A Study to Evaluate and Compare Injections of Autologous Mixed Population of Dermal Cells Into the Balding Scalp of Subjects With Hair Loss (CA‐0004542). clinicaltrials.gov/ct2/show/NCT01451151 (accessed 14 January 2016).
NCT01451177 {unpublished data only}
- NCT01451177. A Study to Evaluate and Compare Injections of Autologous Dermal and Epidermal Cells Into the Balding Scalp of Subjects With Hair Loss (CA‐0004669). clinicaltrials.gov/ct2/show/NCT01451177 (accessed 14 January 2016).
NCT01451190 {unpublished data only}
- NCT01451190. A Study to Evaluate and Compare Injections of Autologous Mixed Population of Dermal Cells Into the Balding Scalp of Subjects With Hair Loss (CA‐0005995). clinicaltrials.gov/ct2/show/NCT01451190 (accessed 14 January 2016).
References to ongoing studies
ACTRN12607000027415 {unpublished data only}
- ACTRN12607000027415. Double blind placebo controlled trial into the treatment of female pattern hair loss with spironolactone and minoxidil. www.anzctr.org.au/Trial/Registration/TrialReview.aspx?id=81787 (accessed 14 January 2016).
EUCTR2013‐002740‐85‐ES {unpublished data only}
- EUCTR2013‐002740‐85‐ES. A Pilot Randomized, Double‐blind, Placebo‐controlled Clinical Trial to Obtain Preliminary Data on Efficacy and Safety in the Application of PRGF‐Endoret by Mesotherapy, in the Treatment of Male and Female Androgenetic Alopecia of Over 6 Months Duration. www.clinicaltrialsregister.eu/ctr‐search/search?query=EUCTR2013‐002740‐85‐ES (accessed 16 January 2016).
NCT00175617 {unpublished data only}
- NCT00175617. Efficacy of Therapy With the Spironolactone Pills Compared to Minoxidil Lotion in Female Pattern Hair Loss. clinicaltrials.gov/ct2/show/NCT00175617 (accessed 14 January 2016).
NCT00197379 {unpublished data only}
- NCT00197379. The Effect of 0.5% Roxithromycin Lotion for Androgenetic Alopecia. clinicaltrials.gov/ct2/show/NCT00197379 (accessed 14 January 2016).
NCT00418249 {unpublished data only}
- NCT00418249. Topical AS101 for Treatment of FAGA (Female Androgenetic Alopecia) in Menopause Women. clinicaltrials.gov/ct2/show/NCT00418249 (accessed 14 January 2016).
NCT01286649 {unpublished data only}
- NCT01286649. Safety and Efficacy Study of Human Autologous Hair Follicle Cells to Treat Androgenetic Alopecia. clinicaltrials.gov/ct2/show/NCT01286649 (accessed 14 January 2016).
NCT01662089 {unpublished data only}
- NCT01662089. The Efficacy in Treatment of Female Pattern Hair Loss Using 5% Minoxidil Solution Combinded With Zinc Supplement. clinicaltrials.gov/ct2/show/NCT01662089 (accessed 14 January 2016).
NCT01686295 {unpublished data only}
- NCT01686295. 24 Week Clinical Trial to Evaluate Safety and Effectiveness of a Hair Growth System to Treat Male and Female Baldness. clinicaltrials.gov/ct2/show/NCT01686295 (accessed 14 January 2016).
NCT01885676 {unpublished data only}
- NCT01885676. Plasma Rich in Growth Factors (PRGF‐Endoret) in the Treatment of Androgenetic Alopecia. clinicaltrials.gov/ct2/show/NCT01885676 (accessed 14 January 2016).
NCT02074943 {unpublished data only}
- NCT02074943. Efficacy and Safety of Platelet Rich Plasma in Androgenetic Alopecia. clinicaltrials.gov/ct2/show/NCT02074943 (accessed 14 January 2016).
NCT02483195 {unpublished data only}
- NCT02483195. The Use of 5mg Finasteride Versus 200mg Spironolactone and Topical 5% Minoxidil in Treating Postmenopausal FemaleAndrogenetic Alopecia. clinicaltrials.gov/ct2/show/NCT02483195 (accessed 14 January 2016).
NCT02486848 {unpublished data only}
- NCT02486848. Minoxidil Dose Response Study in Females Identified Through IVD Testing as Non‐Responders to 5% Topical Minoxidil. clinicaltrials.gov/ct2/show/NCT02486848 (accessed 14 January 2016).
Additional references
Ali 2008
- Ali I, Dawber RPR, Wojnarowska FT. The role of the androgen receptor gene CAG repeat polymorphism and X‐chromosome inactivation pattern in postmenopausal female pattern hair loss. British Journal of Dermatology 2008;159(Suppl 1):8‐9. [Google Scholar]
Atanaskova Mesinkovska 2013
- Atanaskova Mesinkovska N, Bergfeld WF. Hair: What is new in diagnosis and management? Female pattern hair loss update: Diagnosis and treatment. Dermatologic Clinics 2013;31(1):119‐27. [PUBMED: 23159181] [DOI] [PubMed] [Google Scholar]
Avci 2013
- Avci P, Gupta GK, Clark J, Wikonkal N, Hamblin MR. Low‐level laser (light) therapy (LLLT) for treatment of hair loss. Lasers in Surgery and Medicine 2014;46(2):144‐51. [PUBMED: 23970445] [DOI] [PMC free article] [PubMed] [Google Scholar]
Barber 1998
- Barber BL, Kaufman KD, Kozloff RC, Girman CJ, Guess HA. A hair growth questionnaire for use in the evaluation of therapeutic effects in men. Journal of Dermatological Treatment 1998;9(3):181‐6. [EMBASE: 1998346166] [Google Scholar]
Bienová 2005
- Bienová M, Kucerová M, Fiurasková M, Hajdúch M, Kolár Z. Androgenetic alopecia and current methods of treatment. Acta Dermatovenerologica Alpina, Pannonica, et Adriatica 2005;14(1):5‐8. [PUBMED: 15818439] [PubMed] [Google Scholar]
Biondo 2004
- Biondo S, Goble D, Sinclair R. Women who present with female pattern hair loss tend to underestimate the severity of their hair loss. British Journal of Dermatology 2004;150(4):750‐2. [PUBMED: 15099373] [DOI] [PubMed] [Google Scholar]
Biondo 2010
- Biondo S, Sinclair R. Quality of life in Australian women with female pattern hair loss. The Open Dermatology Journal 2010;4:90‐4. [Google Scholar]
Birch 2002
- Birch MP, Lalla SC, Messenger AG. Female pattern hair loss. Clinical and Experimental Dermatology 2002;27(5):383‐8. [PUBMED: 12190638] [DOI] [PubMed] [Google Scholar]
Blume‐Peytavi 2011b
- Blume‐Peytavi U, Blumeyer A, Tosti A, Finner A, Marmol V, Trakatelli M, et al. S1 guideline for diagnostic evaluation in androgenetic alopecia in men, women and adolescents. British Journal of Dermatology 2011;164(1):5‐15. [PUBMED: 20795997] [DOI] [PubMed] [Google Scholar]
Blumeyer 2011
- Blumeyer A, Tosti A, Messenger A, Reygagne P, Marmol V, Spuls PI, et al. Evidence‐based (S3) guideline for the treatment of androgenetic alopecia in women and in men. Journal of the German Society of Dermatology 2011;9(Suppl 6):S1‐57. [PUBMED: 21980982] [DOI] [PubMed] [Google Scholar]
Brown 2006
- Brown P, Brunnhuber K, Chalkidou K, Chalmers I, Clarke M, Fenton M, et al. How to formulate research questions. BMJ 2006;333(7572):804‐6. [PUBMED: 17038740 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Camacho‐Martínez 2009
- Camacho‐Martínez FM. Hair loss in women. Seminars in Cutaneous Medicine and Surgery 2009;28(1):19‐32. [PUBMED: 19341939] [DOI] [PubMed] [Google Scholar]
Cash 2001
- Cash TF. The psychology of hair loss and its implications for patient care. Clinics in Dermatology 2001;19(2):161‐6. [PUBMED: 11397595 ] [DOI] [PubMed] [Google Scholar]
Dinh 2007
- Dinh QQ, Sinclair R. Female pattern hair loss: current treatment concepts. Clinical Interventions in Aging 2007;2(2):189‐99. [PUBMED: 18044135] [PMC free article] [PubMed] [Google Scholar]
Dolte 2000
- Dolte KS, Girman CJ, Hartmaier S, Roberts J, Bergfeld W, Waldstreicher J. Development of a health‐related quality of life questionnaire for women with androgenetic alopecia. Clinical and Experimental Dermatology 2000;25(8):637‐42. [PUBMED: 11167980] [DOI] [PubMed] [Google Scholar]
Drake 1996
- Drake LA, Dinehart SM, Farmer ER, Goltz RW, Graham GF, Hordinsky MK, et al. Guidelines of care for androgenetic alopecia. American Academy of Dermatology. Journal of the American Academy of Dermatology 1996;35(3 Pt 1):465‐9. [PUBMED: 8784287] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [PUBMED: 9310563] [DOI] [PMC free article] [PubMed] [Google Scholar]
El‐Samahy 2009
- El‐Samahy MH, Shaheen MA, Saddik DE, Abdel Fattah NS, El‐Sawi MA, Mahran MZ, et al. Evaluation of androgen receptor gene as a candidate gene in female androgenetic alopecia. International Journal of Dermatology 2009;48(6):584‐7. [PUBMED: 19538365] [DOI] [PubMed] [Google Scholar]
Ellis 2002
- Ellis JA, Sinclair R, Harrap SB. Androgenic alopecia: pathogenesis and potential for therapy. Expert Reviews in Molecular Medicine 2002;4(22):1‐11. [PUBMED: 14585162] [DOI] [PubMed] [Google Scholar]
Gan 2005
- Gan DC, Sinclair RD. Prevalence of male and female pattern hair loss in Maryborough. Journal of Investigative Dermatology. Symposium Proceedings 2005;10(3):184‐9. [PUBMED: 16382660] [DOI] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck‐Ytter Y, Alonso‐Coello P, et al. GRADE; an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924‐6. [PUBMED: 18436948] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hadshiew 2004
- Hadshiew IM, Foitzik K, Arck PC, Paus R. Burden of hair loss: stress and the underestimated psychosocial impact of telogen effluvium and androgenetic alopecia. Journal of Investigative Dermatology 2004;123(3):455‐7. [PUBMED: 15304082] [DOI] [PubMed] [Google Scholar]
Hassani 2012
- Hassani M, Gorouhi F, Babakoohi S, Moghadam‐Kia S, Firooz A. Treatment of female pattern hair loss. Skinmed 2012;10(4):218‐27. [PUBMED: 23008939] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org. Chichester, UK: John Wiley & Sons, Ltd.
Hoffmann 2000
- Hoffmann R, Happle R. Current understanding of androgenetic alopecia. Part I: etiopathogenesis. European Journal of Dermatology 2000;10(4):319‐27. [PUBMED: 10846263] [PubMed] [Google Scholar]
Hoffmann 2002
- Hoffmann R. TrichoScan. A new instrument for digital hair analysis [Ein neues Werkzeug für die digitale Haarzählung]. Hautarzt 2002;53(12):798‐804. [DOI: 10.1007/s00105-002-0421-1; EMBASE: 2003025571] [DOI] [PubMed] [Google Scholar]
Inui 2013
- Inui S, Inoue T, Itami S. Effect of hairpieces on perceived quality of life in female pattern hair loss patients: questionnaire based study. Journal of Dermatology 2013;40(8):671. [PUBMED: 23734854 ] [DOI] [PubMed] [Google Scholar]
Kantor 2003
- Kantor J, Kessler LJ, Brooks DG, Cotsarelis G. Decreased serum ferritin is associated with alopecia in women. Journal of Investigative Dermatology 2003;121(5):985‐8. [PUBMED: 14708596] [DOI] [PubMed] [Google Scholar]
Leavitt 2008
- Leavitt M. Understanding and management of female pattern alopecia. Facial Plastic Surgery 2008;24(4):414‐27. [PUBMED: 19034818] [DOI] [PubMed] [Google Scholar]
Lee 2013b
- Lee WS, Lee HJ, Choi GS, Cheong WK, Chow SK, Gabriel MT, et al. Guidelines for management of androgenetic alopecia based on BASP classification‐‐the Asian Consensus Committee guideline. Journal of the European Academy of Dermatology & Venereology : JEADV 2013;27(8):1026‐34. [PUBMED: 23176122] [DOI] [PubMed] [Google Scholar]
Levy 2013
- Levy LL, Emer JJ. Female pattern alopecia: current perspectives. International Journal of Women's Health 2013;5:541‐56. [PUBMED: 24039457] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ludwig 1977
- Ludwig E. Classification of the types of androgenetic alopecia (common baldness) occurring in the female sex. British Journal of Dermatology 1977;97(3):247‐54. [PUBMED: 921894] [DOI] [PubMed] [Google Scholar]
Martin 1998
- Martin A (editor). Concise Colour Medical Dictionary. 2nd edition. Oxford University Press, 1998. [Google Scholar]
Messenger 2000
- Messenger AG. Thyroid hormone and hair growth. British Journal of Dermatology 2000;142(4):633‐4. [PUBMED: 10792210] [DOI] [PubMed] [Google Scholar]
Messenger 2006
- Messenger AG, Sinclair R. Follicular miniaturization in female pattern hair loss: clinicopathological correlations. British Journal of Dermatology 2006;155(5):926‐30. [PUBMED: 17034520] [DOI] [PubMed] [Google Scholar]
Nasser 2011
- Nasser M, Fedorowicz Z. Grading the quality of evidence and strength of recommendations: the GRADE approach to improving dental clinical guidelines. Journal of Applied Oral Science 2011;19(1):pii: S1678‐77572011000100001. [PUBMED: 21437460] [DOI] [PMC free article] [PubMed] [Google Scholar]
Olsen 2001
- Olsen EA. Female pattern hair loss. Journal of the American Academy of Dermatology 2001;45(3 Suppl):S70‐80. [PUBMED: 11511856] [DOI] [PubMed] [Google Scholar]
Olsen 2005
- Olsen EA, Messenger AG, Shapiro J, Bergfeld WF, Hordinsky MK, Roberts JL, et al. Evaluation and treatment of male and female pattern hair loss. Journal of the American Academy of Dermatology 2005;52(2):301‐11. [PUBMED: 15692478] [DOI] [PubMed] [Google Scholar]
Olsen 2008
- Olsen EA. Female pattern hair loss. In: Blume‐Peytavi U, Tosti A, Whiting DA, Trüeb RM editor(s). Hair Growth and Disorders. Berlin, Heidelberg: Springer Verlag, 2008:171‐86. [Google Scholar]
Olsen 2010
- Olsen AE, Reed KB, Cacchio PB, Caudill L. Iron deficiency in female pattern hair loss, chronic telogen effluvium, and control groups. Journal of the American Academy of Dermatology 2010;63(6):991‐9. [PUBMED: 20947203] [DOI] [PubMed] [Google Scholar]
Price 2003
- Price VH. Androgenetic alopecia in women. Journal of Investigative Dermatology. Symposium Proceedings 2003;8(1):24‐7. [PUBMED: 12894991 ] [DOI] [PubMed] [Google Scholar]
Reid 2011
- Reid EE, Haley AC, Borovicka JH, Rademaker A, West DP, Colavincenzo M, et al. Clinical severity does not reliably predict quality of life in women with alopecia areata, telogen effluvium, or androgenic alopecia. Journal of the American Academy of Dermatology 2011;66(3):e97‐102. [PUBMED: 21601948] [DOI] [PubMed] [Google Scholar]
Review Manager (RevMan) 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Richeti 2013
- Richeti F, Kochi C, Rocha MN, Sant'Anna Corrêa C, Lazzarini R, Guazzelli RM, et al. Increased androgen receptor messenger RNA in frontal‐parietal hair follicles of women with androgenetic alopecia. Genetics and Molecular Research 2013;12(2):1834‐40. [PUBMED: 23479164 ] [DOI] [PubMed] [Google Scholar]
Rogers 2008
- Rogers NE, Avram MR. Medical treatments for male and female pattern hair loss. Journal of the American Academy of Dermatology 2008;59(4):547‐66. [PUBMED: 18793935] [DOI] [PubMed] [Google Scholar]
Schulz 1995
- Schulz KF, Chalmers I, Hayes RJ, Altman DG. Empirical evidence of bias. Dimensions of methodological quality associated with estimates of treatment effects in controlled trials. JAMA 1995;273(5):408‐12. [PUBMED: 7823387] [DOI] [PubMed] [Google Scholar]
Shorter 2008
- Shorter K, Farjo NP, Picksley SM, Randall VA. Human hair follicles contain two forms of ATP‐sensitive potassium channels, only one of which is sensitive to minoxidil. FASEB Journal 2008;22(6):1725‐36. [PUBMED: 18258787] [DOI] [PubMed] [Google Scholar]
Sinclair 2004
- Sinclair R, Jolley D, Mallari R, Magee J. The reliability of horizontally sectioned scalp biopsies in the diagnosis of chronic diffuse telogen hair loss in women. Journal of the American Academy of Dermatology 2004;51(2):189‐99. [PUBMED: 15280836] [DOI] [PubMed] [Google Scholar]
Sinclair 2011
- Sinclair R, Patel M, Dawson TL Jr, Yazdabadi A, Yip L, Perez A, et al. Hair loss in women: medical and cosmetic approaches to increase scalp hair fullness. British Journal of Dermatology 2011;165(Suppl 3):12‐8. [PUBMED: 22171680] [DOI] [PubMed] [Google Scholar]
Tajima 2007
- Tajima M, Hamada C, Arai T, Miyazawa M, Shibata R, Ishino A. Characteristic features of Japanese women’s hair with aging and with progressing hair loss. Journal of Dermatological Science 2007;45(2):93‐103. [PUBMED: 17161939] [DOI] [PubMed] [Google Scholar]
Torres 2015
- Torres F, Tosti A. Female pattern alopecia and telogen effluvium: figuring out diffuse alopecia. Seminars in Cutaneous Medicine and Surgery 2015;34(2):67‐71. [PUBMED: 26176282] [DOI] [PubMed] [Google Scholar]
Trancik 1996
- Trancik RJ, Savin RC. Classification scale for male and female androgenetic alopecia. Clinical Pharmacology & Therapeutics 1996;59(2):166. [DOI: 10.1038/sj.clpt.1996.164] [DOI] [Google Scholar]
Treadwell 2006
- Treadwell JR, Tregear SJ, Reston JT, Turkelson CM. A system for rating the stability and strength of medical evidence. BMC Medical Research Methodology 2006;6:52. [PUBMED: 17052350] [DOI] [PMC free article] [PubMed] [Google Scholar]
Trüeb 2002
- Trüeb RM. Molecular mechanisms of androgenetic alopecia. Experimental Gerontology 2002;37(8‐9):981‐90. [PUBMED: 12213548] [DOI] [PubMed] [Google Scholar]
Trüeb 2010
- Trüeb RM. Systematic approach to hair loss in women [Systematisches Vorgehen bei Frauen mit Haarausfall]. Journal der Deutschen Dermatologischen Gesellschaft 2010;8(4):284‐97. [PUBMED: 20105246] [DOI] [PubMed] [Google Scholar]
van der Donk 1991
- Donk J, Passchier J, Knegt‐Junk C, Wegen‐Keijser MH, Nieboer C, Stolz E, et al. Psychological characteristics of women with androgenetic alopecia: a controlled study. British Journal of Dermatology 1991;125(3):248‐52. [PUBMED: 1911317] [DOI] [PubMed] [Google Scholar]
van Zuuren 2015
- Zuuren EJ, Fedorowicz Z, Carter B, Pandis N. Interventions for hirsutism (excluding laser and photoepilation therapy alone). Cochrane Database of Systematic Reviews 2015, Issue 4. [DOI: 10.1002/14651858.CD010334.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Varothai 2014
- Varothai S, Bergfeld WF. Androgenetic alopecia: an evidence‐based treatment update. American Journal of Clinical Dermatology 2014;15(3):217‐30. [PUBMED: 24848508] [DOI] [PubMed] [Google Scholar]
Westberg 2001
- Westberg L, Baghaei F, Rosmond R, Hellstrand M, Landén M, Jansson M, et al. Polymorphisms of the androgen receptor gene and the estrogen receptor beta gene are associated with androgen levels in women. Journal of Clinical Endocrinology & Metabolism 2001;86(6):2562‐8. [PUBMED: 11397855] [DOI] [PubMed] [Google Scholar]
Yazdabadi 2011
- Yazdabadi A, Sinclair R. Treatment of female pattern hair loss with the androgen receptor antagonist flutamide. Australasian Journal of Dermatology 2011;52(2):132‐4. [PUBMED: 21605098] [DOI] [PubMed] [Google Scholar]
Yip 2011
- Yip L, Rufaut N, Sinclair R. Role of genetics and sex steroid hormones in male androgenetic alopecia and female pattern hair loss: An update of what we now know. Australasian Journal of Dermatology 2011;52(2):81‐8. [PUBMED: 21605090] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Cusmanich 2009
- Cusmanich CC, Hannon CW, Andriolo RB, Lima HC. Interventions for androgenic alopecia in women. Cochrane Database of Systematic Reviews 2009, Issue 2. [DOI: 10.1002/14651858.CD007628.pub2] [DOI] [Google Scholar]
van Zuuren 2012
- Zuuren EJ, Fedorowicz Z, Carter B, Andriolo RB, Schoones J. Interventions for female pattern hair loss. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD007628.pub3] [DOI] [PubMed] [Google Scholar]