

Abstract
A general method for a new, hindered lithium diadamantylamide (LDAM) base-promoted insertion of arynes into Si–P, Si–S, Si–N, and C–C bonds is described. Arynes are generated from easily available aryl triflates and halides. Subsequent reaction of the aryne with silylated phosphines, sulfides, or amines affords the insertion products. Furthermore, a one-step synthesis of anthracenes from aryl halides and aryl ketones is also demonstrated. Cyano, aryl, alkyl, trifluoromethyl, vinyl, methoxy, chloro, fluoro, and even formyl moieties are compatible with the reaction conditions. The new lithium amide affords higher yields compared with lithium tetramethylpiperidide (LiTMP)-promoted reactions. Furthermore, the bulkiness of LDAM base essentially suppresses aryne reaction with base, allowing use of aryl halides and triflates as the limiting reagents.
Graphical Abstract
1. INTRODUCTION
Aryne intermediates are important in synthesis of bioactive molecules, natural products, catalysts, biaryls, agricultural chemicals, and organic materials.1 Several methods have been used for aryne generation (Scheme 1).2 Pathway A involves lithium-halogen exchange followed by LiX elimina tion. This methodology is limited by alkyllithium reactivity that precludes use of many common functionalities. Furthermore, the availability of 1,2-dihaloarenes is limited. Pathway B involves deprotonation of aryl halides or triflates by a strong base, with rapid (or concerted) loss of a labile group, resulting in aryne formation. The starting aryl halides are among the most available aryne precursors. Early investigations involved use of hydroxide, alkoxide, or amide (NaNH2) bases which trapped the formed arynes.2c,d Deprotonation of aryl fluorides with a BuLi/NaOtBu mixture or aryl chlorides with BuLi allowed reactions to proceed at low temperatures; however, this method for aryne generation is incompatible with many functional groups due to high reactivity of alkyllithium reagents.2f,g Lithium amide bases such as LDA or lithium tetramethylpiperidide could be used at low temperatures as well.2a,b,h,i However, the amide base reaction with generated aryne decreases product yields and necessitates use of excess aryne source, which may be problematic for complex molecule synthesis. Pathway C, reaction of silyl aryl triflates or halides, is the mildest method for aryne generation.2p,q The main disadvantage of this method is requirement for the synthesis of aryne sources. Pathways D and E use precursors which are either unstable or are not readily available. Hexadehydro Diels–Alder reactions were recently used to generate arynes as well.2n,o
Scheme 1.

Methods for Aryne Generation
The analysis of the above methods for aryne generation shows that Pathway B holds the most promise due to wide commercial availability of aryl halides. However, the side reaction of the formed benzyne with base has to be suppressed. Furthermore, the base should be unreactive with reagent used for aryne functionalization.
Base reaction with arynes can be suppressed by increasing its steric hindrance. The ideal base candidate has to be (1) bulkier than lithium tetramethylpiperidide which reacts with arynes2b and (2) the parent amine should be prepared in one step on a large scale from commercially available starting materials. While di-t-butylamine, t-butyl-t-octylamine, and dispiro[cyclo-hexane-2,2′-piperidine-6′,2″-cyclohexane] (CPC-H) have been reported previously, their syntheses are rather lengthy and difficult to accomplish on a large scale.3 Amine CPC-H has been prepared on a 45 g scale in two steps (16% and 75%).3c We report here the synthesis of lithium di-1-adamantylamide, arynes for reactions with silicon–phosphorus, sulfur, nitrogen, as well as carbon–carbon bonds. We believe that lithium di-1-adamantylamide will be useful as a more hindered substitute for lithium tetramethylpiperidide and lithium diisopropylamide in many other types of reactions as well.4
RESULTS AND DISCUSSION
2.1. Synthesis and Characterization of the New Base.
Following a modified reported procedure, reaction of 1-adamantylamine and 1-bromoadamantane afforded di-1-adamantylamine in 75% yield (Scheme 2).5a The amine synthesis was performed on several hundred gram scale and has been repeated many times. Both starting materials are commercially available on a kilogram scale. Deprotonation of amine with n-BuLi gave the lithium di-1-adamantylamide 1(LDAM) in 86% isolated yield. Amide 1 was fully characterized by 1H and 13C NMR as well as X-ray diffraction analysis. It exists as a highly crystalline colorless solid, sparingly soluble in pentane and toluene and slowly decomposing THF at room temperature.
Scheme 2.

Base Synthesis
Crystals of 1 suitable for X-ray analysis were grown by dropwise addition of n-butyl lithium in hexane to a suspension of amine in pentane at room temperature. Amide 1 crystallizes as colorless plates in triclinic space group P-1. The ORTEP diagram of lithium di-1-adamantylamide 1 is shown in Figure 1. A space-filling diagram of 1 can be found in Supporting Information. The solid-state structure of the base shows a tetramer adopting a puckered eight-membered (Li–N)4 ring, with N1 and N3 positioned below and N2 and N4 above the plane of lithium atoms. In contrast, LiTMP tetramer adopts a planar conformation, while unsolvated LDA crystallizes in an infinite helical arrangement.5b,c Lithiated dispiro[cyclohexane-2,2′-piperidine-6′,2″-cyclohexane] crystallizes as a cyclic trimer.3c Average Li–N bond length in 1 is 2.043(3) Å, while the corresponding distance in LiTMP tetramer is 2.00(2) Å. Longer Li–N bonds and eight-membered ring distortion can be explained by higher steric congestion in 1. Furthermore, average Li–N–Li angle in LDAM (98.35(13)°) is smaller than that in LiTMP (101.5(3)°). The average N–Li–N angle in LDAM is 169.52(18)°.
Figure 1.

ORTEP view of the molecular structure of 1. Thermal ellipsoids are drawn to encompass 50% probability. Hydrogens are omitted for clarity. Selected bond distances (Å) and angles (deg): N(1)–Li(1) 2.054(3); N(2)–Li(2) 2.030(3); N(3)–Li(3) 2.041(3); N4–Li(4) 2.036(3); Li(4)–N(1)–Li(1) 98.14(13); Li(2)–N(2)–Li(1) 96.28(14); Li(3)–N(3)–Li(2) 99.97(13).
2.2. Insertion Reaction Optimization.
Several aryne insertions in element–element σ-bonds have been reported.6 However, these reactions require silyl aryl triflate aryne precursors. Our goal was to (1) generate arynes from commercially available aryl halides or triflates at synthetically convenient (0 °C or RT) temperatures by using the new LDAM base and (2) develop new reactions of the generated arynes with element–element σ-bonds.
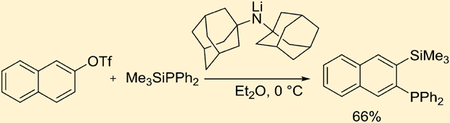
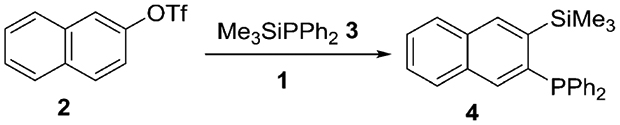
We have previously reported that LiTMP-promoted aryne generation from aryl triflates can be performed at −78 °C.2b One of the reasons for low-temperature requirement is to minimize the nucleophilic attack of LiTMP on the aryne. A bulkier base used in this study should retard the nucleophilic attack of the base on the aryne and the reaction could potentially be performed at more convenient 0 °C or even under ambient conditions. The optimization reactions for aryne insertion into phosphorus–silicon bonds are summarized in Table 1. The detailed optimization of the other reactions can be found in the Supporting Information. Pure ether is the best solvent (entry 3), while reactions in THF (entry 1) or solvent mixtures (entry 2) afford lower yields. Use of 3 equiv of base at 0 °C gave the highest yield of the product (entry 6), while reactions at 25 or −78 °C were less efficient. Addition of DMPU resulted in substantially lower conversion and multiple side products were observed in crude reaction mixture. Regioisomers of 4 were not observed in crude reaction mixtures showing selective formation of one aryne isomer, presumably due to steric factors in deprotonation of 2 by hindered amide 1.
Table 1.
Reaction Condition Optimizationa
| entry | (2/3/1) | solvent | T (°C), t (h) | yield (%)b |
|---|---|---|---|---|
| 1 | 1/2/2 | THF | 0, 30 | 44 |
| 2 | 1/2/2 | Et2O/THF (20/1) | 0, 30 | 29 |
| 3 | 1/2/2 | Et2O | 0, 30 | 48 |
| 4 | 1/2.5/2 | Et2O | 0, 30 | 40 |
| 5 | 1/1.5/2 | Et2O | 0, 30 | 41 |
| 6 | 1/2/3 | Et2O | 0, 30 | 67 |
| 7 | 1/2/3 | Et2O | 25, 30 | 61 |
| 8 | 1/2/3 | Et2O | −78, 30 | trace |
Aryne precursor (0.25 mmol), solvent (1.5 mL).
Yields determined by GC analysis with n-decane as an internal standard.
2.3. Insertion of Aryne into the Silicon–Phosphorus Bonds.
Triarylphosphines have been employed as ligands in many transition-metal-catalyzed reactions.7 Specifically, Peng and co-workers showed that diphenyl(2-(trimethylsilyl)-phenyl)phosphine ligand gives high conversion and selectivity in hydrosilylation of alkenes.8 2-Silyl substituted phosphines are usually prepared by lithium-halogen exchange in (2-bromophenyl)diphenylphosphine followed by reaction with trimethylsilyl chloride.9
Our method employs commercially available aryl halides and triflate starting materials and thus greater diversity of products can be easily obtained. Examples of aryne insertion into Me3SiPPh2 silicon–phosphorus bond are presented in Table 2. All reactions were carried out at 0 °C in either diethyl ether (for ArOTf) or pentane/tetrahydrofuran (for aryl halides) solvent. Substrates possessing both electron-donating and electron-withdrawing functionalities are reactive. Aryl halides carrying trifluoromethyl functionality gave the product in 91% and 64% isolated yields (entries 1 and 2). (Trimethylsilylphenyl)diphenylphosphine can be synthesized from both chlorobenzene and phenyl triflate in 84% and 82% isolated yields, respectively (entries 3 and 7). 2-Biphenyl triflate and 2-naphthyl triflate reacted efficiently to give the product in 62% and 66% isolated yields (entries 4 and 5). 3,4,5-Trimethoxyphenyl triflate afforded the product in 47% isolated yield (entry 6). In all cases except that of entry 2, products were isolated as single isomers.
Table 2.
Reaction Scope with Respect to Aryne Precursorsa
 |
ArX (1 equiv), Me3SiPPh2 (2 or 2.5 equiv), base (1.5 or 3 equiv), diethyl ether for ArOTf and pentane/THF for ArCl, 0.5 mmol scale, yields are isolated yields. See the Supporting Information for details.
Product contains 7% of isomeric impurity.
Reaction scope with respect to silyl phosphines is shown in Table 3. Trifluoromethyl substituent on aryl bromide and triflate is tolerated and reactions gave the products in 64% and 48% isolated yields, respectively (entry 1). If a bulkier silyl substituent was used, the product was isolated in a somewhat reduced 63% yield (entry 2). Tolyl substituents on phosphorus are tolerated and reaction with arynes generated from chlorobenzene and phenyl triflate gave products in 89% and 86% isolated yields (entry 3). Slightly lower yields were obtained when employing p-methoxyphenylphosphine derivatives. The product was formed in 41% and 44% yields from aryl chloride and triflate aryne precursors (entry 4). Mechanistically, the reaction is likely triggered by a nucleophilic attack of a phosphorus atom of silyl phosphine on aryne triple bond followed by an intramolecular nucleophilic substitution as suggested in similar reactions.6e
Table 3.
Reaction Scope with Respect to Silylated Phosphinesa
 |
ArX (1 equiv), R3SiPAr2 (2 or 2.5 equiv), base (1.5 or 3 equiv), diethyl ether for ArOTf and pentane/THF for ArHal, 0.5 mmol scale, yields are isolated yields. See the Supporting Information for more details. R = H for entries 2–4, R = 3,5-(CF3)2 for entry 1.
2.4. Insertion of Arynes into the Silicon–Sulfur Bonds.
Insertion of arynes into silicon–sulfur bonds is summarized in Table 4. Both aryl triflates and halides can be used for aryne generation. 2-Naphthyl triflate gives single regioisomer of the product in 81% yield (entry 1). Phenyl functionality on aryne is compatible with the reaction conditions and the product was isolated in 71% yield (entry 2). 1,4-Dichlorophenyl triflate afforded the product in 52% isolated yield showing that some halide substituents are compatible with reaction conditions (entry 3). Alkoxy functionalities on phenyl and naphthyl rings are also tolerated and the products were isolated in 61% and 66% yields, respectively (entries 4 and 6). In entry 5, 1,5-bis-(trifuoromethyl)-3-(ethylthio)-2-(trimethylsilyl)benzene was synthesized in 67% isolated yield. 1,2,3,4-Tetrahydro-6-naphthyl triflate exclusively gave one isomer of the product, which is consistent with the result observed for 2-naphthyl triflate (entry 7). 1-Naphthyl triflate gave a 1.6:1 mixture of 2-(ethylthio)-1-(trimethylsilyl)naphthalene and 1-(ethylthio)-2-(trimethylsilyl)-naphthalene in 69% overall yield (entry 8). Entries 9–16 in Table 4 show the use of aryl halides as aryne precursors. The reaction conditions are similar to the ones used for aryl triflates. Aryl halides equipped with methoxy functionality are compatible with the reaction conditions, and the products were obtained in 63%, 74%, and 68% isolated yields, respectively (entries 9, 11, and 15). 9-Bromophenan threne reacted to give the product in 78% isolated yield (entry 10). Aryne precursors possessing electron-withdrawing tri-fluoromethyl substituents gave 74% and 75% isolated yields (entries 12 and 13). Entry 14 shows the formation of two isomeric products from 1-naphthyl chloride. They were isolated in overall yield of 70%, and 1.9:1 crude isomer ratio was observed. 2-Chlorostyrene is reactive, and the product was isolated in 51% yield (entry 16).
Table 4.
Insertion of Arynes into Si–S Bondsa
 |
ArX (1 equiv), Me3SiSEt (3 equiv), base (2 equiv), diethyl ether/C6H12 (1:1, 3 mL) for ArOTf, diethyl ether (3 mL) for ArHal, 0.5 mmol scale, yields are isolated yields. See the Supporting Information for details.
Product contains 4% of an isomeric impurity.
Crude mixture contains another isomer (4.2:1), which was separated during chromatography.
Yields are of pure isomers, separated by column chromatography.
Reaction mixture contained another isomer (16:1), which was separated by column chromatography.
Yields are of pure isomers, separated by column chromatography. Crude isomer ratio 1.9:1.
The scope of reactions with respect to the thio substituents and heterocycles is shown in Scheme 3. If isopropyl and tert-butyl trimethylsilyl sulfides were employed, 2-(isopropylthio)-3-(trimethylsilyl)-naphthalene, 5, and 9-(tert-butylthio)-10-(trimethylsilyl)phenanthrene, 6, were isolated in 47% and 61% yields, respectively. Aromatic sulfides such as phenyl trimethylsilyl sulfide react as well and product 7 was isolated in 46% yield. Interestingly, 2-pyridyl triflate and 2-bromopyr idine gave 2-(ethylthio)-3-(trimethylsilyl)pyridine 8 in 32% and 31% isolated yields, respectively. 7-(Ethylthio)-8-(trimethylsilyl)quinoline 9 was isolated in 26% yield.
Scheme 3.

Reaction Scope with Respect to Silyl Sulfides
2.5. Insertion of Arynes into the Silicon–Nitrogen Bonds.
Aryne insertion into the silicon–nitrogen bonds was also successful. This methodology provides an efficient synthesis of aniline derivatives. Silyl aryl triflate reaction with aminosilanes has been described.10 However, as discussed previously, most of silyl aryl triflates are not commercially available and no reactions with aromatic silylamines were reported. Few functional groups on aryne reaction component were studied. We show here that commercially available aryl halides and triflates can be employed as aryne precursors.
Table 5 illustrates the scope of aryne insertion into the silicon–nitrogen bonds. Formyl, cyano, chloro, trifluoromethyl, methoxy, trifluoromethoxy, and 1,3-dioxole functionalities are compatible with the reaction conditions. Methoxy substituents on aryl chloride are tolerated affording nearly quantitative yields of the product. Reaction can be scaled up to 6.0 mmol with little loss of yield (entry 1). Penta- and tetrachlorinated substrates give 3,4,5,6-tetrachloro and 3,4,5-trichloro-2-(trimethylsilyl)-N,N-dimethylanilines in 76% and 57% isolated yields (entries 2 and 3). Aryl halide possessing a trifluoromethoxy functionality reacted to afford the product in 46% isolated yield (entry 4). Aryl triflates such as 3,5-bis(trifluoromethyl)phenyl triflate and 4-formylphenyl triflate provided the product in 70% and 87% isolated yields, respectively (entries 5 and 6). The last example shows that, quite unexpectedly, formyl group is compatible with reaction conditions employing strong base, presumably due to extreme bulkiness of LDAM. Cyano-substituted phenyl triflate is reactive as well, giving 3-dimethylamino-2-(trimethylsilyl)-benzonitrile in 61% isolated yield (entry 7). Entries 8 and 9 show that trifluoromethoxy and 1,4-dichloro substituents are tolerated. Isolated yields of 58% and 55% were obtained, respectively. 6-(N,N-Dimethylamino)-5-(trimethylsilyl)-1,3-benzodioxole was formed by reacting triflated 5-hydroxy-1,3-benzodioxole with (trimethylsilyl)dimethylamine. A 64% isolated yield was obtained (entry 10).
Table 5.
Insertion of Arynes into Si–N Bondsa
 |
ArX (1 equiv), Me3SiNMe2 (4 equiv), base (1.5 or 2 equiv), diethyl ether/C6H12 (1:1), 0.3 or 0.5 mmol scale, yields are isolated yields.
Scale: 6 mmol. See the Supporting Information for details.
Isomers separated by column chromatography. Crude ratio 2:1.
The reaction scope with respect to silylamines is shown in Scheme 4. Ethyl substituents on nitrogen are tolerated, and product 10 is formed in an acceptable 47% yield. A silylated morpholine gave 11 in 81% isolated yield. Aromatic silyl amines afforded 12 and 13 in 44% and 46% isolated yields, respectively.
Scheme 4.

Reactions with Other Aminosilanes
Heterocyclic and phosphorus-containing aryne precursors can be used as well. An efficient synthesis of a pincer type ligand motif 14 in a one-step reaction from simple and readily accessible starting materials is possible (Scheme 5). The product partially decomposes by hydrodesilylation during column chromatography on silica gel, affording 58% yield of product. The NMR yield was 79%. 2-Pyridyl triflate and 2-bromopyridine gave 15 in 31% and 46% yields, respectively.
Scheme 5.

Multidentate Ligand Synthesis
2.6. Insertion of Arynes into C–C Bonds.
Two reactions involving aryne reactions with ketone enolates were performed as well. An efficient one-step synthesis of substituted anthracenes was achieved by treatment of chlorobenzene and aryl alkyl ketones with 1 at room temperature (Scheme 6). Fleming and Mah have shown that arynes react with acetaldehyde enolate affording anthracenes.11a A related method using LiTMP base has been described by employing benzocyclobutenoxide starting materials.11b,c
Scheme 6.

Anthracene Synthesis
2.7. Comparison of LiTMP and LDAM Bases.
The performance of two bases in aryne reaction with silicon–nitrogen and silicon–sulfur bonds was studied (Scheme 7). Identical conditions were used for comparing LiTMP and lithium di-1-adamantylamide (LDAM, 1) bases. In the first reaction, triflate 18 was reacted with Me3SiNMe2 in the presence of LiTMP or LDAM. The crude NMR yields were measured by using benzotrifluoride internal standard. Reaction promoted by LiTMP afforded 51% NMR yield of 19. Additionally, 8% of 20 was observed. Compound 20 is produced by the side-reaction of base with aryne. Reaction promoted by LDAM gave 66% NMR and 59% isolated yield of 19. Byproduct 21, which would result from reaction of LDAM with aryne, was not observed in the reaction mixture. In the second reaction, bromide 22 was reacted with Me3SiSEt in the presence of either LiTMP or LDAM bases. The results are similar to ones obtained with 18. A lower yield of product 23 was obtained if LiTMP base was employed. Additionally, 13% of 20 was observed in a crude reaction mixture. About 3% of 21, which is produced in reaction of LDAM with aryne, was observed in crude reaction mixture. As expected, increasing steric bulk of the base nearly eliminates byproduct formation. Consequently, a lower excess of aryl halide can be used for aryne generation which may be important if complex or expensive substrates are used. Overall, bulky base affords the desired products in higher yields.
Scheme 7.

Comparison of LiTMP and LDAM Bases
3. SUMMARY
This paper reports the synthesis and use of a new bulky lithium di-1-adamantylamide base. Parent amine was synthesized in one-step on the large scale from cheap and commercially available 1-bromoadamantane and 1-adamantanamine. After lithiation, lithium di-1-adamantylamide was obtained in high yield, characterized by X-ray crystallography, and used in aryne generation from aryl halides and triflates. Several new aryne reactions with silicon–phosphorus, sulfur, and nitrogen bonds were developed. Furthermore, a one-step synthesis of anthracenes from aryl halides and aryl ketones was also demonstrated. The reactions are remarkably functional group tolerant. Formyl, cyano, chloro, tertiary phosphine, trifluor-omethyl, methoxy, alkyl, aryl, vinyl, and 1,3-dioxole function-alities are compatible with reaction conditions. Furthermore, we have shown that new base is superior to LiTMP in aryne generation, affording higher yields of products. Nucleophilic attack of the base on aryne intermediates is essentially suppressed.
4. EXPERIMENTAL SECTION
General Procedure for Reactions.
Outside the glovebox a 2- dram vial was equipped with two magnetic stirring bars (size, 5 mm × 1 mm × 1 mm). The vial was placed inside the glovebox. To the vial was added solid LDAM (0.29 g, 1.0 mmol). The sealed vial was then taken out of the glovebox and placed into oil bath/cooling bath at the reaction temperature. Two thirds of solvent or solvent mixture (2.0 mL) was added via a syringe to the reaction vial. The vial was stirred for 5–10 min at reaction temperature. In another vial, haloarene or aryl triflate (0.5 mmol) was mixed with silyl compound (2–4 equiv). Subsequently, one-third of reaction solvent was added to this vial. The vial with reactants was kept at the reaction temperature for 5–10 min. Subsequently, solution of reactants was added to the reaction vial containing base in 1 min by syringe. After stirring at indicated temperature for indicated time, reactions were quenched by adding 2-methyl-2-butanol and then methanol (0.5 mL, unless otherwise stated), followed by dilution with dichloromethane (0.5 mL). To the diluted reaction mixture was added silica gel, mixture was dried on rotary evaporator, and subjected to flash chromatography in hexanes followed by appropriate solvent to elute the products. After concentrating the fractions containing the product, the residue was dried under reduced pressure to yield pure product.
Supplementary Material
ACKNOWLEDGMENTS
We thank the Welch Foundation (Chair No. E-0044) and NIGMS (Grant No. R01GM077635) for supporting this research. We are grateful to Dr. Xiqu Wang for collecting and solving of the X-ray structure of 1 and to Dr. Scott Smith for help with low-temperature NMR experiments.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b07064.
Crystallographic data for LDAM 1 CIF)
Detailed experimental procedures and characterization data for new compounds (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).(a) Tadross PM; Stoltz BM A Comprehensive History of Arynes in Natural Product Total Synthesis. Chem. Rev 2012, 112, 3550–3577. [DOI] [PubMed] [Google Scholar]; (b) Bronner SM; Goetz AE; Garg NK Overturning Indolyne Regioselectivities and Synthesis of Indolactam V. J. Am. Chem. Soc 2011, 133, 3832–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huters AD; Quasdorf KW; Styduhar ED; Garg NK Total Synthesis of (−)-N-Methylwelwitindolinone C Isothiocyanate. J. Am. Chem. Soc 2011, 133, 15797–15799. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Day JJ; McFadden RM; Virgil SC; Kolding H; Alleva JL; Stoltz BM The Catalytic Enantioselective Total Synthesis of (+)-Liphagal. Angew. Chem., Int. Ed 2011, 50, 6814–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Larrosa I; Da Silva MI; Gómez PM; Hannen P; Ko E; Lenger SR; Linke SR; White AJP; Wilton D; Barrett AGM Highly Convergent Three Component Benzyne Coupling: The Total Synthesis of ent-Clavilactone B. J. Am. Chem. Soc 2006, 128, 14042–14043. [DOI] [PubMed] [Google Scholar]; (f) Okano K; Fujiwara H; Noji T; Fukuyama T; Tokuyama H Total Synthesis of Dictyodendrin A and B. Angew. Chem., Int. Ed 2010, 49, 5925–5929. [DOI] [PubMed] [Google Scholar]; (g) Wu D; Ge H; Liu SH; Yin J Arynes in the synthesis of polycyclic aromatic hydrocarbons. RSC Adv. 2013, 3, 22727–22738. [Google Scholar]; (h) Pérez D; Peña D; Guitián E Aryne Cycloaddition Reactions in the Synthesis of Large Polycyclic Aromatic Compounds. Eur. J. Org. Chem 2013, 2013, 5981–6013. [Google Scholar]
- (2).(a) Truong T; Daugulis O Divergent Reaction Pathways for Phenol Arylation by Arynes: Synthesis of Helicenes and 2-Arylphenols. Chem. Sci 2013, 4, 531–535. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Truong T; Mesgar M; Le KKA; Daugulis O A General Method for Functionalized Polyaryl Synthesis via Aryne Intermediates. J. Am. Chem. Soc 2014, 136, 8568–8576. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stoermer R; Kahlert B Ueber das 1- und 2-Brom-cumaron. Ber. Dtsch. Chem. Ges 1902, 35, 1633–1640. [Google Scholar]; (d) Gilman H; Avakian S Dibenzofuran. XXIII. Rearrangement of halogen compounds in amination by sodamide. J. Am. Chem. Soc 1945, 67, 349–351. [Google Scholar]; (e) Huisgen R; Sauer J; Hauser A Nucleophile aromatische Substitutionen, VI. Katalytische Arylierung der Chlorar omaten. Chem. Ber 1958, 91, 2366–2374. [Google Scholar]; (f) Meyers AI; Pansegrau PD Tandem additions of cuprates to benzynes. A regioselective synthesis of 3-alkyl or aryl-2-substituted benzoic acids. J. Chem. Soc., Chem. Commun 1985, 690–691. [Google Scholar]; (g) Fossatelli M; Brandsma L An Efficient Synthesis of Triphenylene. Synthesis 1992, 1992, 756. [Google Scholar]; (h) Wickham PP; Hazen KH; Guo H; Jones G; Reuter KH; Scott WJ Benzyne generation from aryl triflates. J. Org. Chem 1991, 56, 2045–2050. [Google Scholar]; (i) Tripathy S; LeBlanc R; Durst T Formation of 2-Substituted Iodobenzenes from Iodobenzene via Benzyne and Ate Complex Intermediates. Org. Lett 1999, 1, 1973–1975. [Google Scholar]; (j) Hart H; Harada K; Du C-JF Synthetically useful aryl aryl bond formation via Grignard generation and trapping of arynes. A one-step synthesis of p-terphenyl and unsymmetrical biaryls. J. Org. Chem 1985, 50, 3104–3110. [Google Scholar]; (k) Campbell CD; Rees CW Reactive intermediates. I. Synthesis and oxidation of 1- and 2- aminobenzotriazole. J. Chem. Soc. C 1969, 742–747. [Google Scholar]; (l) Wittig G; Hoffmann RW Dehydrobenzol aus 1.2.3-Benzothiadiazol-1.1-dioxyd. Chem. Ber 1962, 95, 2718–2728. [Google Scholar]; (m) Stiles M; Miller RG; Burckhardt U Reactions of benzyne intermediates in nonbasic media. J. Am. Chem. Soc 1963, 85, 1792–1797. [Google Scholar]; (n) Niu D; Willoughby PH; Woods BP; Baire B; Hoye TR Alkane desaturation by concerted double hydrogen atom transfer to benzyne. Nature 2013, 501, 531–534. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Yun SY; Wang K-P; Lee N-K; Mamidipalli P; Lee D Alkane C-H Insertion by Aryne Intermediates with a Silver Catalyst. J. Am. Chem. Soc 2013, 135, 4668–4671. [DOI] [PubMed] [Google Scholar]; (p) Himeshima Y; Sonoda T; Kobayashi H Fluoride-induced 1,2-elimination of o-(trimethylsilyl)phenyl triflate to benzyne under mild conditions. Chem. Lett 1983, 12, 1211–1214. [Google Scholar]; (q) Mesgar M; Daugulis O Silylaryl Halides Can Replace Triflates as Aryne Precursors. Org. Lett 2016, 18, 3910–3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Corey EJ; Gross AW Synthesis of di-tert-alkylamines. Tetrahedron Lett. 1984, 25, 491–494. [Google Scholar]; (b) Gajda T; Koziara A; Zawadzki S; Zwierzak A Phase-transfer-catalyzed N-alkylation of N-substituted formamides; an alternative procedure for the Ritter reaction. Synthesis 1979, 1979, 549–552. [Google Scholar]; (c) Morisako S; Shang R; Yamamoto Y Synthesis of a Sterically Demanding Dispiropiperidine and Its Application in Monoamidodialkyl Zincate Complexes. Inorg. Chem 2016, 55, 10767–10773. [DOI] [PubMed] [Google Scholar]
- (4).(a) Olofson RA; Dougherty CM Lithium 2,2,6,6-tetramethylpiperidide and related, strong, proton-specific bases. Evaluation in synthesis. J. Am. Chem. Soc 1973, 95, 582–584. [Google Scholar]; (b) Khanapure SP; Reddy RT; Biehl ER The preparation of anthraquinones and anthracyclinones via the reaction of haloarenes and cyanophthalides under aryne-forming conditions. J. Org. Chem 1987, 52, 5685–5690. [Google Scholar]; (c) Abboud M; Mamane V; Aubert E; Lecomte C; Fort Y Synthesis of Polyhalogenated 4,4’-Bipyridines via a Simple Dimerization Procedure. J. Org. Chem 2010, 75, 3224–3231. [DOI] [PubMed] [Google Scholar]; (d) Haag B; Mosrin M; Ila H; Malakhov V; Knochel P Regio- and Chemoselective Metalation of Arenes and Heteroarenes Using Hindered Metal Amide Bases. Angew. Chem., Int. Ed 2011, 50, 9794–9824. [DOI] [PubMed] [Google Scholar]; (e) d’Angelo J Ketone enolates: regiospecific prepara tion and synthetic uses. Tetrahedron 1976, 32, 2979–2990. [Google Scholar]
- (5).(a) Krumkalns EV; Pfeifer W Adamantylamines by direct amination of 1-bromoadamantane. J. Med. Chem 1968, 11, 1103. [DOI] [PubMed] [Google Scholar]; (b) Lappert MF; Slade MJ; Singh A; Atwood JL; Rogers RD; Shakir R Structure and reactivity of sterically hindered lithium amides and their diethyl etherates: crystal and molecular structures of [Li{N(SiMe3)2}(OEt2)]2 and tetrakis(2,2,6,6-tetramethylpiperidina tolithium). J. Am. Chem. Soc 1983, 105, 302–304. [Google Scholar]; (c) Barnett NDR; Mulvey RE; Clegg W; O’Neil PA Crystal structure of lithium diisopropylamide (LDA): an infinite helical arrangement composed of near-linear nitrogen-lithium-nitrogen units with four units per turn of helix. J. Am. Chem. Soc 1991, 113, 8187–8188. [Google Scholar]
- (6).(a) Tambar UK; Stoltz BM The Direct Acyl-Alkylation of Arynes. J. Am. Chem. Soc 2005, 127, 5340–5341. [DOI] [PubMed] [Google Scholar]; (b) Yoshida H; Watanabe M; Ohshita J; Kunai A Carbophosphinylation of arynes with cyanomethyldiphenylphosphine oxide. Chem. Lett 2005, 34, 1538–1539. [Google Scholar]; (c) Yoshida H; Shirakawa E; Honda Y; Hiyama T Addition of ureas to arynes: straightforward synthesis of benzodia zepine and benzodiazocine derivatives. Angew. Chem., Int. Ed 2002, 41, 3247–3249. [DOI] [PubMed] [Google Scholar]; (d) Rodríguez-Lojo D; Cobas A; Peña D; Perez D; Guitián E Aryne Insertion into I-I σ-Bonds. Org. Lett 2012, 14, 1363–1365. [DOI] [PubMed] [Google Scholar]; (e) Yoshida H; Terayama T; Ohshita J; Kunai A Thiostannylation of arynes with stannyl sulfides: synthesis and reaction of 2-(arylthio)arylstannanes. Chem. Commun 2004, 1980–1981. [DOI] [PubMed] [Google Scholar]; (f) Liu Z; Larock RC Intermolecular C-N Addition of Amides and S-N Addition of Sulfinamides to Arynes. J. Am. Chem. Soc 2005, 127, 13112–13113. [DOI] [PubMed] [Google Scholar]
- (7).(a) Baba S; Negishi E A novel stereospecific alkenyl-alkenyl cross-coupling by a palladium- or nickel-catalyzed reaction of alkenylalanes with alkenyl halides. J. Am. Chem. Soc 1976, 98, 6729–6731. [Google Scholar]; (b) Miyaura N; Yamada K; Suzuki A A new stereospecific cross-coupling by the palladium-catalyzed reaction of 1-alkenylboranes with 1-alkenyl or 1-alkynyl halides. Tetrahedron Lett. 1979, 20, 3437–3440. [Google Scholar]; (c) Stille JK Palladium-catalyzed coupling reactions of organic electrophiles with organic tin compounds. Angew. Chem., Int. Ed. Engl 1986, 25, 508–524. [Google Scholar]; (d) Heck RF; Nolley JP Palladium-catalyzed vinylic hydrogen substitution reactions with aryl, benzyl, and styryl halides. J. Org. Chem 1972, 37, 2320–2322. [Google Scholar]; (e) Surry DS; Buchwald SL Dialkylbiaryl phosphines in Pd catalyzed amination: a user’s guide. Chem. Sci 2011, 2, 27–50. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Breit B; Winde R; Mackewitz T; Paciello R; Harms K Phosphabenzenes as monodentate π-acceptor ligands for rhodium catalyzed hydroformylation. Chem. - Eur. J 2001, 7, 3106–3121. [DOI] [PubMed] [Google Scholar]
- (8).Xue M; Li J; Peng J; Bai Y; Zhang G; Xiao W; Lai G Effect of triarylphosphane ligands on the rhodium-catalyzed hydro-silylation of alkene. Appl. Organomet. Chem 2014, 28, 120–126. [Google Scholar]
- (9).Kawachi A; Yoshioka T; Yamamoto Y Anionic 1,4-Silyl Migration in (2-(Trimethylsilyl)phenyl)phosphonium Methylides. Organometallics 2006, 25, 2390–2393. [Google Scholar]
- (10).Yoshida H; Minabe T; Ohshita J; Kunai A Aminosilylation of arynes with aminosilanes: synthesis of 2-silylaniline derivatives. Chem. Commun 2005, 3454–3456. [DOI] [PubMed] [Google Scholar]
- (11).(a) Fleming I; Mah T A Simple Synthesis of Anthracenes. J. Chem. Soc., Perkin Trans. 1 1975, 964–965. [Google Scholar]; (b) Fitzgerald JJ; Drysdale NE; Olofson RA A Rapid, Convergent, and Regioselective Synthesis of Anthracenes. J. Org. Chem 1992, 57, 7122–7126. [Google Scholar]; (c) Tripathy S; Reddy R; Durst T Preparation of benzocyclobutenols by low temperature reaction of ketone enolates with benzynes. Can. J. Chem 2003, 81, 997–1002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.