

Abstract
Precise genetic and epigenetic spatiotemporal regulation of gene expression is critical for proper brain development, function and circuitry formation in the mammalian central nervous system. Neuronal differentiation processes are tightly regulated by epigenetic mechanisms including DNA methylation, histone modifications, chromatin remodelers and non-coding RNAs. Dysregulation of any of these pathways is detrimental to normal neuronal development and functions, which can result in devastating neuropsychiatric disorders, such as depression, schizophrenia and autism spectrum disorders. In this review, we focus on the current understanding of epigenetic regulations in brain development and functions, as well as their implications in neuropsychiatric disorders.
Keywords: epigenetics, neuropsychiatric disorders, DNA methylation, Rett syndrome, Fragile X syndrome, autism spectrum disorders, schizophrenia, major depressive disorders
Introduction
The concept of epigenetics was first proposed in 1939 by Conrad Waddington to describe early embryonic development (Waddington, 1939). He proposed that development originates from the interactions of the starting material in the fertilized egg, and that the interactions give rise to something new. He further postulated that this process cycles, leading to the formation of a whole organism. Today, the accepted definition of epigenetics is the study of modifications that directly affect the expression of a gene, but do not change the underlying DNA sequence (Goldberg et al., 2007; Allis and Jenuwein, 2016; Gayon, 2016). There are several major epigenetic mechanisms that are extensively studied including DNA modifications, histone modifications, chromosome remodeling and RNA regulation via non-coding RNAs such as microRNA (miRNA) and long non-coding RNA (lncRNA) (Figure 1). Modifications can be added, removed and interpreted by various classes of proteins collectively known as ‘writers,’ ‘erasers’ and ‘readers,’ respectively. Disruption of these epigenetic mechanisms and their molecular machinery can have catastrophic consequences in the mammalian central nervous system (CNS).
FIGURE 1.
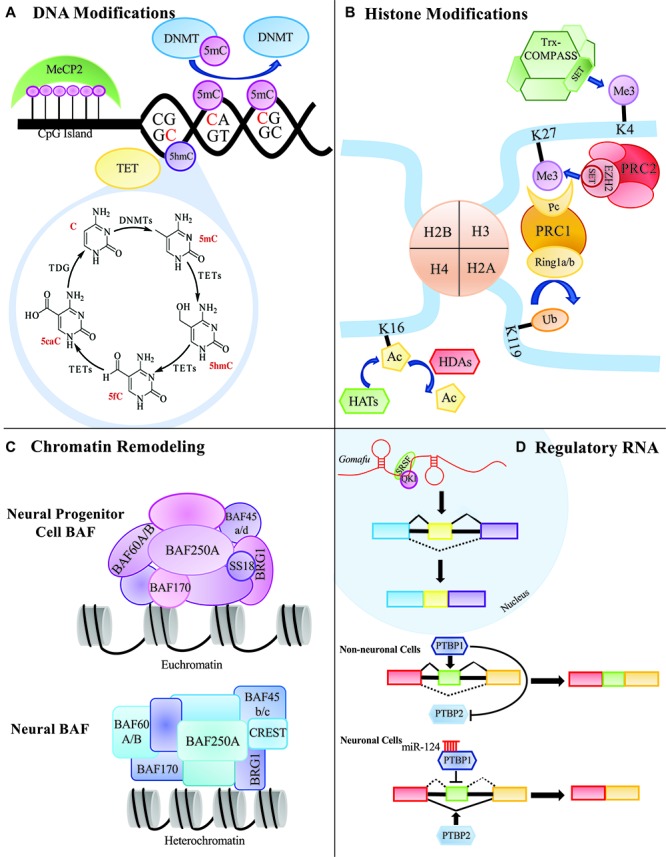
Summary of epigenetic processes that can occur in the mammalian central nervous system. (A) DNA modifying proteins can methylate CG of CH dinucleotides. Methylated cytosines can be further modified to 5hmC, 5fC, or 5caC to be replaced with an unmodified cytosine through thymine DNA glycosylase, TDG. (B) Histone modifiers can add various groups to the tails of the histone proteins that can affect the expressivity of a gene’s transcript. (C) Chromatin remodeling proteins can remodel the chromatin environment by affecting how tightly and loosely packed histones are and ultimately contribute to the gene’s expression. (D) Regulatory RNAs, such as long non-coding RNAs and microRNAs, can affect alternative splicing and protein expression.
Both nervous system development and function can be affected by epigenetic spatiotemporal regulation of gene expression. In the mammalian CNS, epigenetic dysregulation is associated with neuropsychiatric diseases such as major depressive disorder (MDD), autism spectrum disorders (ASDs), Fragile X, Rett syndrome and schizophrenia. Epigenetic studies are actively trying to identify biomarkers that could be associated with diseases to aid in our development of novel therapeutics. This information is critical, as the prevalence of neuropsychiatric diseases is on the rise (Atladottir et al., 2015). Here, we review the current understanding of epigenetic regulation in brain development and functions, with a focus on DNA methylation, as well as their implications in psychiatric diseases.
DNA Methylation
Functional Roles of DNA Methylation
DNA methylation is one of the best characterized epigenetic marks studied and has been regarded as a highly stable mark found in differentiated cells (Reik, 2007; Suzuki and Bird, 2008). It involves the covalent methylation of the fifth position in the cytosine ring, generating 5-methylcytosine (5mC) (Figure 1A). DNA methylation largely occurs at CpG dinucleotides (Bird, 1986). Accumulation of short, unmethylated CpG-rich clusters known as CpG islands occurs in the promoter regions of most genes (Jones, 2012). Genome-wide studies have implicated that the distribution of 5mC in transcripts could have differential roles in gene expression. For example, methylation status of the CpG islands helps to determine whether the corresponding gene will be expressed, whereas gene body methylation has been proposed to promote transcriptional elongation (Neri et al., 2017) and affect splicing (Maunakea et al., 2013). In addition, the methylation status of CpG islands can be influenced spatially based on tissue and cell type (Illingworth and Bird, 2009). For instance, the gene HTR2A, which has been implicated in many neuropsychiatric disorders (Norton and Owen, 2005), shows differential expression in the cerebellum and the cortex and is regulated by DNA methylation (Ladd-Acosta et al., 2007). Strikingly, the methylated CpG loci regulating HTR2A expression is over 1 Kb upstream of the promoter rather than being in the promoter region, illustrating that methylation can regulate genes across long distances. Thus, DNA methylation has important roles for brain region-specific transcriptome profiles.
Not only can DNA methylation regulate protein coding genes, it can also regulate non-protein coding RNA like lncRNAs. Random X-inactivation, an essential embryonic event, is triggered by the production of Xist, a lncRNA that coats the X chromosome destined to be inactivated (Borsani et al., 1991; Brown et al., 1992). The promoter of the Xist gene contains a CpG island whose methylation status ultimately dictates whether the X chromosome is active (Beard et al., 1995). How DNA methylation regulates lncRNA in the brain is still unclear. One study compared the DNA methylation patterns around the transcription start sites (TSSs) of protein coding genes and lncRNA loci (Sati et al., 2012). Surprisingly, a sharp increase in DNA methylation immediately downstream of the TSS was associated with lncRNA loci, but did not correlate with expression of the lncRNA. While this finding suggests that DNA methylation may not play an essential role in lncRNA expression, it would be interesting to investigate if blocking methylation at these sites influenced lncRNA expression.
In addition to its roles in gene regulation, DNA methylation also maintains genomic stability by controlling the expression of highly repetitive regions in the genome such as retrotransposons and satellite DNA (Liu et al., 1994; Woodcock et al., 1997; Walsh et al., 1998). In general, long interspersed nuclear element-1 (LINE 1) is only active in the germline and during early development (Ma et al., 2010). During somatic cell differentiation, DNA methylation silences LINE 1. Interestingly, studies have suggested that LINE 1 may be active during human and rodent neuronal differentiation and influence neuronal gene expression to create cell heterogeneity in the adult brain (Muotri et al., 2005; Muotri and Gage, 2006; Coufal et al., 2009). Indeed, LINE 1 has been shown to be more active in the brain compared to other tissues (Coufal et al., 2009). Increases in LINE 1 and other repetitive elements have been associated with the neuropsychiatric disorder Rett syndrome (Muotri et al., 2010). Suppression of LINE 1 requires methylation of its promoter and binding of the methyl-binding protein MeCP2, which plays a causal role in Rett syndrome.
Suppressing the expression of repetitive elements is one way by which DNA methylation maintains genomic stability and integrity. Genome instability has been shown to be highly correlated with many neuropsychiatric diseases such as schizophrenia, autism, Rett syndrome and several others (Smith et al., 2010). Numerous genes associated with these disorders, particularly schizophrenia and autisms, co-localize with regions of the genome that are more susceptible to mutations, or epigenetic alterations known as fragile sites. The most studied fragile site is associated with Fragile X syndrome and will be discussed later in this review.
Finally, DNA methylation has important roles in early developmental processes such as gene imprinting. Often, the “imprint” is methylation of a long-range control element called an imprint control element (ICE) (also referred to as imprint control region, ICR, or imprint center, IC) (Li et al., 1993; Barlow, 2011). Parental specific methylation of the ICE is established by the DNA methyltransferase (DNMT) complex DNMT3A/3L during gamete development (Bourc’his et al., 2001; Kaneda et al., 2004). Of the approximately 100 imprinted genes currently known, the majority of them are expressed in brain tissues, though not always exclusively, and have been reviewed previously (Wilkinson et al., 2007). One of the more extensively studied imprinted genes, specifically in the CNS of mammals, is the paternally expressed gene Necdin (Ndn) (Aizawa et al., 1992). Ndn regulates neuronal differentiation and axonal outgrowth. Also, Ndn is most highly expressed during mouse neuronal generation and between postnatal days 1–4.
DNA Methylation in the Brain
DNA methylation in the brain is required for brain development and function throughout all stages in life. Dynamic regulation of DNA methylation is critical for cellular differentiation. One study compared the changes in DNA methylation patterns between two differentiation phases: the transition of embryonic stem cells (ESCs) to neuronal progenitor cells (NPCs), and the transition of NPCs to differentiated neurons (Mohn et al., 2008). The most dynamic changes in DNA methylation patterns were found when ESCs lost their pluripotency and became NPCs. In fact, ESCs were nearly devoid of DNA methylation marks except at the promoters of genes that were germline specific. In contrast, during the differentiation of NPCs to mature neurons, only 2.3% of the analyzed promoters gained de novo methylation and only 0.1% of promoters were demethylated, suggesting that the majority of DNA methylation dynamics do not occur in this phase. Similar to neurogenesis, astrocytogenesis is tightly controlled by DNA methylation. In mouse, astrocyte differentiation from neuroepithelial cells requires that the promoter of the GFAP gene be demethylated on embryonic day 14.5, allowing for the transcription factor STAT3 to bind and activate GFAP expression (Teter et al., 1994; Takizawa et al., 2001).
Very few studies have focused on how DNA methylation regulates other brain developmental features, such as neural migration and axonal/dendritic outgrowth. Two recent studies have demonstrated that the DNA methyltransferase, DNMT1, as having putative regulatory roles in immature GABAergic interneuron migration (Pensold et al., 2017; Symmank et al., 2018). They found that Dnmt1 promotes the migration and survival of immature migratory GABAergic interneurons that derive from the embryonic preoptic area (POA) by repressing Pak6 expression (Pensold et al., 2017). p21-active kinases (PAKs) are known for their roles in cytoskeletal organization (Kumar et al., 2017), and Pak6 has previously been shown to stimulate neurite outgrowth in post-migratory neurons derived from POA (Civiero et al., 2015; Pensold et al., 2017). De novo methylation by Dnmt3b in early embryonic neurodevelopmental processes has been shown to be critical in regulating the clustered protocadherins (Pcdhs) genes (Toyoda et al., 2014). Protocadherins are cell-surface adhesion proteins that are predominantly expressed in the nervous system (Sano et al., 1993), and have critical functions in neurite self-avoidance (Lefebvre et al., 2012), neuronal survival (Wang et al., 2002b), and dendritic patterning (Garrett et al., 2012). In mammals, they are found in three closely linked gene clusters call α (Pcdha), β (Pcdhb), and γ (Pcdhg) (Kohmura et al., 1998; Wu and Maniatis, 1999). Interestingly, the Pcdhs are stochastically expressed by alternative promoters in individual neurons generating single cell diversity of isoforms in the brain (Wang et al., 2002a). This stochastic expression is regulated by methylation of variable exons and this has been thoroughly reviewed elsewhere (Hirayama and Yagi, 2017). Protocadherins have critical roles in neural development and are starting to be implicated in neuropsychiatric disorders such as ASDs, depression and schizophrenia (Redies et al., 2012; El Hajj et al., 2017).
DNA methylation also has roles in brain function such as memory processing. In the mammalian brain, the hippocampus and the cortex are largely responsible for memory formation and storage (Morris et al., 1982; Squire, 1986; Miller et al., 2010). In the hippocampus, contextual fear conditioning induced changes in DNA methylation during memory formation in rats. When DNMTs were inhibited by either zebularine or 5-aza-2′-deoxycytidine, neuronal plasticity-promoting genes Bdnf and Reelin demonstrated altered methylation patterns (Levenson et al., 2006). After contextual fear conditioning, Dnmt3a and Dnmt3b mRNA were highly upregulated in the brain; however, when DNMT inhibitors, zebularine or 5-aza-2′-deoxycytidine, were injected into the hippocampus immediately after contextual fear conditioning, the fear response was eliminated, suggesting that DNA methylation is required for memory formation (Miller and Sweatt, 2007). Importantly, when the memory suppressor gene Pp1 was examined after fear conditioning, there was an increase in methylation at the CpG island upstream of the Pp1 transcriptional start site. It was postulated that the increase in de novo Dnmts may be necessary to transcriptionally silence memory suppressor genes after fear conditioning training to allow for memory formation and consolidation. In addition to the formation of memories, DNA methylation also has putative roles in long-term memory storage. Contextual fear conditioning was found to disrupt DNA methylation at three genes associated with memory, Egr1, reelin, and calcineurin, which also happen to have large promoter CpG islands (Miller et al., 2010). Both reelin and calcineurin were hypermethylated; however, only calcineurin maintained this hypermethylated state for 30 days, suggesting that DNA methylation might be required for long term memory storage.
Worth noting is that DNA methylation patterns in the brain can be affected by external stimuli in one’s environment. Interestingly, a study found that in mature neuronal cells, CpGs in low density regions compared to CpG islands undergo dynamic DNA methylation changes in response to electroconvulsive stimulation (Guo et al., 2011a). Numerous studies have shown that maternal care during childhood (Weaver et al., 2004), early life stressors including abuse (McGowan et al., 2009), parental separation and social defeat stressors can alter DNA methylation patterns in the brain and have been reviewed elsewhere (Yu et al., 2011).
DNA Methyltransferases
DNA methylation is generated by a group of DNMTs, also regarded as 5mC enzymatic “writers” (Figure 1A). Each Dnmt (Dnmt1, 3a, 3b, 2, and 3L) has evolved to have its own specialized regulatory functions. These specialized functions could be attributed to the lack of sequence homology seen in the N-terminal regulatory domains of the Dnmts (Bestor and Verdine, 1994). All of the Dnmts contain some version of a cysteine rich domain that further define their functions. The most conserved region between the DNMTs is the C-terminal catalytic domain, which is characteristic of all enzymes that modify pyrimidines at the fifth position (Figure 2A).
FIGURE 2.
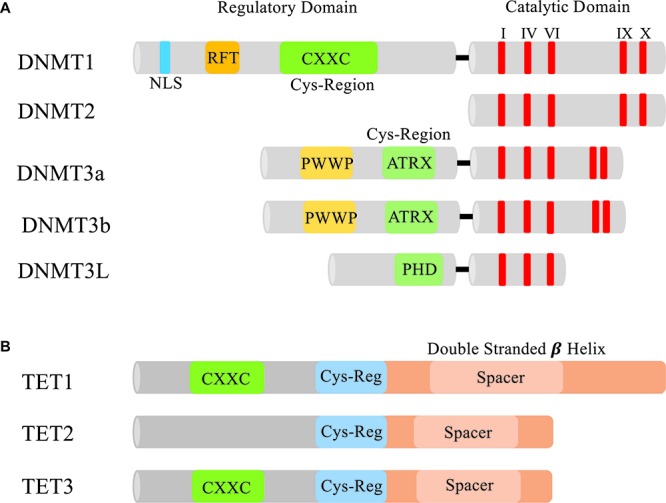
Domains of DNMTs and TETs. (A) The N-terminal and C-terminal domains of DNMTs. In the N-terminus of each Dnmt is a cysteine rich region. In Dnmt1 this region contains a CXXC zinc finger which is thought to aid in DNA binding (Frauer et al., 2011). Dnmt3a and 3b both contain a PWWP domain that specifically recognizes the repressive histone 3 lysine 36 trimethylation mark (H3K36me3) found in heterochromatin (Dhayalan et al., 2010). Dnmt3L contains a PHD-like cysteine rich domain that closely resembles the PHD domain encoded in Dnmt3a and 3b’s ATRX domain (Hata et al., 2002). All the DNMTs have a conserved C-terminal catalytic domain (I, IV, VI, IX, and X are the most conserved motifs in cytosine methyltransferases) responsible for modifying pyrimidines. NLS, nuclear localization signal; RFT, replication foci-targeting domain. (B) Domains of TET1, TET2, and TET3. Each TET protein has a core catalytic domain structured as a double stranded beta helix and a cys-regulatory region. Only TET1 and TET3 contain a CXXC domain to facilitate chromatin binding.
The first Dnmt purified was Dnmt1 back in 1983, and was found to be responsible for maintaining methylated CpG sites during DNA replication (Bestor and Ingram, 1983). Dnmt1 interacts with replication machinery, such as proliferating cell nuclear antigen (PCNA). Maintenance of the genomic methylation pattern requires that unmethylated regions also be maintained during replication. During the S phase, the transcription factor p21 blocks Dnmt1 from interacting with PCNA, which ensures that unmethylated regions maintain their original state (Chuang et al., 1997). This regulation of Dnmt1 plays an important role in asynchronous replication, specifically at replication origins that include CpG islands (Delgado et al., 1998). Mutation and loss-of-function studies have demonstrated the necessity of Dnmt1 during embryonic development. By gestational day 9.5, Dnmt1-null mouse embryos failed to develop and died by gestational day 11 (Li et al., 1992). In addition, overall global methylation levels decreased by threefold in the Dnmt1-null embryos.
Nearly 15 years later, two additional Dnmts were discovered, Dnmt3a and Dnmt3b. Both Dnmt3a and Dnmt3b are responsible for de novo methylation, which is also critical during early embryogenesis (Okano et al., 1998a). When either Dnmt3a or Dnmt3b are deleted during embryogenesis, severe developmental defects or embryonic lethality are observed, respectively (Okano et al., 1999). Mouse embryos with Dnmt3a depletion appear normal at birth, but die around 4 weeks of age. In contrast, embryos null for Dnmt3b were not viable and had growth retardation and neural tube defects. In addition to embryonic development, the de novo methyltransferases work in conjunction with Dnmt1 to regulate genome stability and imprinted genes. At a global level, deletion of Dnmt3a and/or Dnmt3b results in slight demethylation at repetitive sequences, but not to the same extent observed in Dnmt1 gene deletion. This indicates that Dnmt1 is more important for the maintenance of methylation at repetitive sequences. At a loci-specific level, deletion of Dnmt3a and/or Dnmt3b has varied effects. For example, at several imprinted gene loci, Igf2r and H19, neither single nor dual gene disruption of Dnmt3a or Dnmt3b resulted in the demethylation pattern observed in Dnmt1 gene disruption. However, at another imprinted loci, Igf2, dual deletion of Dnmt3a/Dnmt3b showed demethylation levels comparable to Dnmt1 loss, whereas single gene disruption had no effect on demethylation. This indicates that there is some overlap in the roles of the Dnmts at certain gene sites.
Lesser known methyltransferases include Dnmt2 and Dnmt3L that were identified by sequence homology studies. Dnmt2 contains all of the C-terminal catalytic domains necessary to act as a methyltransferase; however, it was found to be non-essential for maintenance or de novo methylation (Okano et al., 1998b), but rather responsible for tRNA methylation (Goll et al., 2006; Schaefer et al., 2010). Dnmt3L demonstrates homology with Dnmt3a and Dnmt3b, but lacks the enzymatic activity required to generate de novo methylation (Bourc’his et al., 2001; Hata et al., 2002). Instead, Dnmt3L is essential in the establishment of maternal imprints and co-localizes with Dnmt3a/3b to regulate imprinting. Furthermore, in the male germ line, loss of Dnmt3L resulted in the reactivation of retrotransposons and meiotic failure in spermatocytes (Bourc’his and Bestor, 2004), suggesting a role in genomic stability.
DNA Methyltransferases in the CNS
As writers of DNA methylation, Dnmts play critical roles in the mammalian CNS. Studies conducted on embryonic and adult mice revealed that Dnmts are highly expressed in neural progenitor cells, but are maintained at substantially lower levels in most differentiated neurons (Goto et al., 1994). Furthermore, mouse studies revealed that in the CNS, Dnmt3a is detected as early as embryonic day (E) E10.5 in the ventricular and subventricular zones, but its expression is predominantly in adult post-mitotic neurons (Feng et al., 2005). In contrast, Dnmt3b could only be detected during early neurogenesis. These specific time points of expression suggest that Dnmt3b may be important during the early stages of brain development, whereas Dnmt3a is more crucial to mature neurons. Further supporting different spatiotemporal roles for the de novo methyltransferases, it was shown that Dnmt3b is required for methylation at centromeric minor satellite repeats during embryonic brain development, whereas Dnmt3a is not (Okano et al., 1999).
Targeted mutagenesis studies revealed how critical the Dnmts are in the CNS. Conditional deletion of Dnmt1 in CNS precursor cells, but not post-mitotic neurons, caused daughter cells to be severely hypomethylated (Fan et al., 2001). Interestingly, mice that had 30% of their CNS cells mutated showed selective pressure against the Dnmt-knockout cells in their brain. Three weeks after birth, all Dnmt-knockout cells were abolished. In adult forebrain neurons, double knockout of both Dnmt1 and Dnmt3a (but neither gene by itself) resulted in significantly smaller hippocampi and dentate gyrus brain regions, due to smaller neurons (Feng et al., 2010). These mice also showed impairments in learning and memory as well as inappropriate upregulation of immune genes associated with demethylation. These results suggest that Dnmt1 and Dnmt3a may have redundant roles in post-mitotic neurons.
To further enhance the elaborate network of DNA methylation in the mammalian CNS, non-CpG dinucleotide methylation (CpH) has surfaced and shown to be highly enriched and have critical roles in the brain (Figure 1A). CpG dinucleotides make up around 75% of total cytosine methylation, whereas CpH dinucleotides (‘H’ could be adenosine, thymine or cytosine) make up the remaining 25% (Guo et al., 2014). Interestingly, CpH methylation is enriched in low CpG dense regions, is associated with repressed gene expression, but is unassociated with protein–DNA interaction sites. As previously mentioned, Dnmt1 preferentially associates with CpG dinucleotides, and maintains symmetric CpG methylation on both strands of DNA during replication. This symmetric balance is further facilitated by the complimentary base pairing (GpC). CpH methylation does not maintain the sequence symmetry and consequently during replication, CpH methylation is not conserved. This requires the re-establishment of CpH methylations after each cell division (Shirane et al., 2013). Re-establishment of CpH methylation has been linked to Dnmt3a gene expression (Xie et al., 2012; Shirane et al., 2013; Varley et al., 2013). In knockdown experiments, loss of Dnmt3a, but not Dnmt1 or Dnmt3b, resulted in reduced CpH methylation with no effect on CpG methylation (Guo et al., 2014).
Like CpG methylation dynamics in early development, CpH methylation levels change during development. CpH methylation has been shown in relatively high abundance in stem cells (Lister et al., 2009; Laurent et al., 2010) and found to be enriched in both adult mouse and human brain tissues (Xie et al., 2012; Lister et al., 2013; Varley et al., 2013). A recent study showed that CpH methylation accumulates in the frontal cortex of the brain early after birth through adolescence and then slightly diminishes during aging (Lister et al., 2013). Different subclasses of neurons have unique CpH and CpG methylomes and CpH methylation may correlate more robustly with gene expression as compared to CpG methylation (Mo et al., 2015).
Methyl-Binding Proteins
After the establishment of DNA methylation marks by “writers,’ a subset of proteins with methyl binding abilities known as “readers” can bind, protect and interpret these marks and facilitate function (Figure 1A). There are two main classes of methyl-CpG-binding proteins that have been thoroughly reviewed elsewhere (Ballestar and Wolffe, 2001), so this review will briefly discuss methyl-CpG-binding domain (MBD) proteins and MeCP2. Both protein families, for the most part, selectively bind to methylated DNA and aid in transcriptional repression (Hendrich and Bird, 1998). MeCP2 can facilitate gene repression by recruiting histone deacetylase (HDAC) machinery that further remodel the chromatin environment, facilitating a repressed state (Jones et al., 1998; Nan et al., 1998; Fuks et al., 2003). Later it was found that MeCP2 could also bind to non-CpG methylation modifications (Mellen et al., 2012; Guo et al., 2014; Gabel et al., 2015).
Methyl-binding proteins are ubiquitously expressed in somatic cells, but are particularly enriched in the mammalian CNS (Hendrich and Bird, 1998; Nan et al., 1998; Shahbazian et al., 2002; Cassel et al., 2004; Mullaney et al., 2004). Several studies have found that MeCP2 is involved in the regulation of brain-derived neurotrophic factor (BDNF), which promotes neuronal maturation (Chen et al., 2003; Martinowich et al., 2003). Additionally, MeCP2 was found to regulate a maternally imprinted gene called Dlx5 that is part of the gamma-aminobutyric acid (GABA) pathway for inhibitory GABAergic neurons (Horike et al., 2005). Importantly, mutations in the MBD of MeCP2 have been implicated in the X-linked, neurodevelopment disorder known as Rett syndrome (Amir et al., 1999).
In addition to MeCP2, there are four other mammalian MBD proteins. MBD1-3 are known for their roles in transcriptional repression, whereas MBD4 functions as a thymine glycosylase in the mismatch repair pathway (Fujita et al., 2003). The MBD proteins can repress gene expression in several ways. One is through the recruitment of the H3K9 methyltransferase Suv39h1 and heterochromatin protein 1 (HP1). Both Suv39h1 and HP1 interact with MBD1 and aid in the establishment and maintenance of a repressive chromatin state which is further facilitated by the recruitment of both HDAC1 and HDAC2 (Fujita et al., 2003). During the S phase of DNA replication, regions of the chromosome that are repressed by DNA methylation, or histone modifications must be maintained. MBD1 forms an S phase specific complex with the H3K9 methyltransferase SETDB1, and then associates with chromatin assembly factor (CAF-1) to help maintain a repressed chromatin state (Sarraf and Stancheva, 2004). MBD2 and 3 were found to be in the nucleosome remodeling and histone deacetylation (NURD) complex, further associating the cross-talk of DNA methylation with histone modifications and chromatin remodeling enzymes (Zhang et al., 1999). Although MBD3 cannot bind methylated DNA, it was found to mediate the association between metastasis-associated protein 2 (MTA2), a MBD-containing protein, and the HDAC core of the NuRD complex. MBD2 is thought to direct the NuRD complex to methylated DNA and aid in the maintenance of a repressed environment.
Very little work has been done to identify functions of MBD1-3 in the CNS. Mice with a loss-of-function MBD1 gene showed normal development, but as adults exhibited deficits in neurogenesis, impaired spatial learning and reduced long-term potentiation in the dentate gyrus (Zhao et al., 2003). Additionally, MBD1 was most enriched in hippocampus. During early embryogenesis, MBD3 was found to be highly expressed in the developing brain compared to MBD2 expression (Jung et al., 2003). In addition, in the adult brain, MBD3 is highly expressed in hippocampal and cortex neurons, but has very little expression in the outer cortical layer. Based on overall brain region enrichment patterning, it appears that the MBD proteins have some role in adult neurogenesis, but to what extent is unknown.
DNA Demethylation
Mechanism of DNA Demethylation
The mammalian genome undergoes genome-wide passive and active DNA demethylation processes during early embryogenesis and in the germline (Monk et al., 1987; Kafri et al., 1992; Tada et al., 1998). During passive demethylation, there is either a lack of, or inhibition of Dnmt1 preventing the replacement of methyl marks (Howlett and Reik, 1991; Mertineit et al., 1998; Rougier et al., 1998; Howell et al., 2001). Furthermore, Dnmt1 is unable to recognize and bind to unmethylated DNA (Valinluck and Sowers, 2007), rather it prefers to bind to hemi-methylated DNA. The precise molecular events of active DNA demethylation were not elucidated until 2009 when two seminal studies identified the presence of 5-hydroxymethylcytosine (5hmC) in the mammalian genome (Kriaucionis and Heintz, 2009; Tahiliani et al., 2009). Tahiliani et al. (2009) discovered that Ten-Eleven Translocation 1 (TET1) could oxidize the methyl group on 5mC to generate 5hmC (Figure 1A). Subsequent studies further identified TET2 and TET3 proteins as additional “erasers” of 5mC (Ito et al., 2010). 5hmC can be furthered catalyzed by all TETs to form 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (He et al., 2011; Ito et al., 2011). In addition, 5hmC can be converted to 5-hydroxymethyluracil (5hmU) via the activation-induced cytidine deaminase (AID) and apolipoprotein B mRNA-editing catalytic polypeptides (APOBEC) enzymes (Bhutani et al., 2011). All three of these derivatives (5fC, 5caC, and 5hmU) can be cleaved by thymine-DNA glycosylase (TDG), which excises the modified cytosine base allowing for the base excision repair (BER) pathway to return it to an unmodified cytosine base (Bhutani et al., 2011; He et al., 2011). Contrary to previous belief that the accumulation of 5hmC was solely dependent on TET activity on 5mC, recent work has suggested that Dnmt1 and Dnmt3a, drive the initial accumulation of 5hmC in the early mouse zygote stage (Amouroux et al., 2016). Knockout models and small molecule inhibitor studies were able to uncouple the formation of 5hmC from 5mC in the paternal pronucleus. This suggests that 5hmC could itself be an independent epigenetic modification.
TET Enzymes
TET enzymes catalytically oxidize the methyl group on 5mC to form 5hmC. The TET protein family is made up of three members: TET1, 2 and 3 (Figure 2B). Each contains a core catalytic domain structured as a double-stranded β-helix (DSBH) fold (Iyer et al., 2009; Tahiliani et al., 2009). Distinguishing the TET proteins from other related TET J-binding proteins (TET-JBP) families is the presence of a Cys domain located in the N-terminus of the DSBH domain that is thought to be essential for the catalytic activity. Also contained in TET1 and TET3 is a CXXC domain allowing the TET proteins to associate with chromatin through its binding to methylated cytosines. During development, the TET proteins can elect both an activating and repressive response from the genes they control based on what cofactors associate with them. In ES cells, TET1 has a repressive role when bound to the promoter region because it recruits MBD3-NURD (Yildirim et al., 2011) and SIN3A (Deplus et al., 2013). On the other hand, TET2 is not able to recruit either repressive component and has been associated with active cofactors such as Nanog and OGT (O-GlcNAc transferase) (Costa et al., 2013; Vella et al., 2013). In the male pronucleus, TET3 is responsible for the complete loss of 5mC and the accumulation of 5hmC, as shown by antibody staining and TET3 knockdown studies (Gu et al., 2011; Iqbal et al., 2011; Wossidlo et al., 2011).
TET Enzymes in the CNS
Once it was discovered that TET enzymes were the long sought-after DNA demethylases, (Iyer et al., 2009; Tahiliani et al., 2009) extensive efforts were made to understand the dynamics of the global demethylation events observed in early embryogenesis. The catalytic function of the TET enzyme family and their putative novel roles were yet to be discovered. Even after all the advancements made in the past 9 years, very little is known about the function of TET enzymes in the mammalian CNS. Although all three TET proteins are expressed in the brain, Tet2 and Tet3 have higher expression compared to Tet1 (Kriaucionis and Heintz, 2009; Szulwach et al., 2011; Hahn et al., 2013). When Tet2 and Tet3 are overexpressed, premature neuronal differentiation was observed, whereas knockdown caused defects in differentiation progression (Hahn et al., 2013). Tet1 knockout studies have identified several neural activity-regulated genes that are downregulated. Animals with this knockout display abnormal hippocampal synaptic plasticity and impaired memory extinction (Rudenko et al., 2013). Intriguingly, Tet1 deletion did not appear to affect anxiety or depression related behaviors. Due to the embryonic lethality of Tet3 deletion in mice, determining its function in the adult brain has been challenging. Instead of knockout studies, several groups have utilized small hairpin RNAs (shRNAs) to conditionally inhibit Tet3 expression. A recent study demonstrated that deletion of Tet3, and not Tet1, in mouse infralimbic prefrontal cortex (ILPFC), a region of the brain associated with fear extinction learning, impaired their ability to reverse a previously learned fear response (Li et al., 2014). Importantly, it was found that Tet3 mediates the drastic genome-wide redistribution of 5hmC in the ILPFC in response to extinction learning. Furthermore, posttraumatic stress disorders and phobias have been associated with impairments in fear extinction learning (Orsini and Maren, 2012).
Roles of 5hmC, 5fC, and 5caC in the CNS
As previously discussed, 5hmC is the immediate product of TET enzymes’ in the demethylation of 5mC. Relative to other tissue types, 5hmC is found to be approximately 10 times higher in the brain compared to ESCs (Tahiliani et al., 2009; Globisch et al., 2010; Song et al., 2011). Genome-wide analysis studies have demonstrated that 5hmC is dynamically regulated in human (Wang et al., 2012) and mouse brains during neurodevelopment and aging (Szulwach et al., 2011). Dot blot analysis on cerebellum DNA showed 5hmC increased roughly 42% from fetal to adult brains. Furthermore, human 5hmC modifications were enriched at CpG islands and shores, exons and untranslated regions, consistent with 5hmC being associated with active genes. Notably, 5hmC has been found to be enriched at genes that are associated with ASDs. Differential hydroxymethylated regions found in human fetal and adult cerebellum were more likely to localize on Fragile X mental retardation protein (FMRP) target genes (Wang et al., 2012). These pieces of evidence clearly indicate the key roles of 5hmC in mammalian CNS. In addition to brain regions, some neurons have been found to contain high levels of 5hmC. For example, Purkinje neurons in the cerebellum were found to have roughly 40% more 5hmC relative to 5mC (Kriaucionis and Heintz, 2009). The enrichment of 5hmC in Purkinje neurons could account for its active biological functions as motor neurons that require an active transcriptome. Locus specific demethylation has been observed at the Bdnf loci. Bdnf is involved in adult neural plasticity and learning and memory (West et al., 2001). When cortical and hippocampal neurons experience a depolarization event, the Bdnf promoter is activated, enhancing its transcription (Shieh et al., 1998; Tao et al., 1998). The depolarization was also found to correlate with a decrease in CpG methylation in the Bdnf regulatory region (Martinowich et al., 2003; Guo et al., 2011b).
Very little is known about the functional roles of 5fC and 5caC other than their roles in active demethylation and conversion back to an unmodified cytosine. Genome-wide profiling studies found an enrichment of 5fC at poised and active enhancers, but with a clear preference for poised enhancers (Song et al., 2013). A recent study examined the dynamics of 5fC and 5caC in embryonic day 11.5 mice through 15-week-old adult mice (Bachman et al., 2015). They found that 5fC could be detected throughout all of the developmental time points, while 5caC could not be detected. Interestingly, both 5fC and 5caC were found to induce pausing of RNA Pol II during elongation, where this effect was not observed at C, 5mC nor 5hmC bases (Kellinger et al., 2012). It is possible that TDG could be recruited to sites of paused RNA Pol II to initiate the BER mechanism. Interestingly, TDG is the only glycosylase that is required for embryonic development (Cortazar et al., 2011; Cortellino et al., 2011). Even more intriguing is that in ESCs, both 5fC and 5caC recruit more proteins than either 5mC or 5hmC (Spruijt et al., 2013). The recruited proteins mostly had functional roles in DNA damage response (such as Tdg and p53), and proteins involved in chromatin remodeling (such as BAF170) were also found to interact with them.
Histone Modifications
DNA is wrapped around a core histone octamer containing two copies each of the histone variants H2A, H2B, H3 and H4 forming a chromatin structure (Kornberg, 1974). The amino acids that make up the amino-terminal ‘histone tails,’ specifically lysines and arginines, are subject to modifications, such as methylation and acetylation, that can affect transcription (Figure 1B). Unlike DNA methylation which only has three major methyltransferases, there have been numerous histone methyltransferases and demethylases identified for histones (Hyun et al., 2017). The potential crosstalk between histone methylations and DNA modifications and chromatin remodelers and regulatory RNAs add another layer of complexity. These crosstalk events are thought to establish and maintain the local chromatin environment as well as help cells “remember” their differentiated state (Cedar and Bergman, 2009; Jobe et al., 2012). Several mechanisms facilitate this cross-talk such as DNMT3L and methyl-binding proteins like MeCP2 and MBD2, but we will focus in detail on the Polycomb (PcG) repressive proteins and the Trithorax (TrxG) activating proteins (Figure 1B). These two groups of proteins antagonistically regulate genes that are critical for development and cell differentiation pathways (Schwartz and Pirrotta, 2008). The proteins encoded by PcG and TrxG form large complexes to maintain the local chromatin environment in either a repressed or active state, respectively (Locke et al., 1988; Franke et al., 1992).
Polycomb Group Proteins
The PcG proteins are divided into two major multiprotein complexes: polycomb repressive complexes 1 and 2 (PRC1 and PRC2) (Shao et al., 1999). Both complexes contain a core set of proteins critical for their basic function and can incorporate accessory proteins, permitting the complex to act in a spatiotemporal manner. There are four core proteins that are present in all PRC2 complexes: the SET domain contained in the enhancer of zeste [E(z), EZH1, and EZH2] protein, extra sex combs (Esc, EED) proteins, suppressor of zeste 12 [Su(z)12, SU(Z)12] and the histone binding protein p55 (RBAP48 and RBAP46) (Ng et al., 2000; Tie et al., 2001; Kuzmichev et al., 2002). The SET domain within E(Z) is responsible for the lysine methyltransferase activity specifically occurring on histone 3 at lysine 27 (H3K27) (Cao et al., 2002). PRC1 is also composed of a set of four major core proteins including polycomb (Pc), polyhomeotic (Ph), posterior sex combs (Psc) and Sex combs extra (Sce/dRing 1) (Shao et al., 1999). The chromodomain in Pc is responsible for recognizing and binding trimethylated H3K27 (H3K27me3) and upon binding will induce structural changes in the chromatin (Fischle et al., 2003; Min et al., 2003). In addition, PRC1 is also responsible for the monoubiquitination of lysines on histone H2A via the proteins Ring1A/B (de Napoles et al., 2004).
Trithorax Group Proteins
Antagonistic to the PcG proteins, the TrxG proteins are recognized for their activating mechanisms and addition of histone 3 lysine 4 trimethylation (H3K4me3). TrxG proteins are also evolutionarily conserved and are categorized into three groups based on their function. Group one is composed of the SET-domain-containing proteins that methylate histone tails, group two contains ATP-dependent chromatin remodeling proteins and finally group three contains the TrxG proteins that can bind DNA in a sequence specific manner. Each of these groups are thoroughly reviewed elsewhere (Schuettengruber et al., 2011). One of the first SET-domain-containing histone modifying complexes identified that could catalyze mono-, di-, and trimethylation on H3K4 was a complex called COMPASS in yeast (Miller et al., 2001; Roguev et al., 2001). Mammals have six COMPASS-like complexes that have been shown to facilitate most H3K4me3 present, indicating that they are likely involved in global gene activation (Wu et al., 2008).
PcG and TrxG Proteins in the CNS
In the mammalian CNS, both PcG and TrxG proteins help to regulate the differentiation process of neuronal cells. In ESCs, polycomb proteins prevent neuronal differentiation by adding H3K27me3 repressive marks at neuronal specific genes such as Ngns, Pax6, Sox1 (Bernstein et al., 2006; Mikkelsen et al., 2007). However, these genes simultaneously contain the active trithorax H3K4me3 mark, making these promoters bivalent. As ESCs differentiate into NPCs, the H3K27me3 polycomb mark is removed specifically by the histone demethylase Jmjd3 to further commit them to a neural lineage (Burgold et al., 2008). In addition to histone demethylation, activation of the TrxG COMPASS-like complex proteins RBBP5 and DBY30 are essential for the differentiation of ESCs into NPCs (Jiang et al., 2011). In NPCs, the PRC2 subunit Ezh2 is initially highly expressed, but declines during cortical neuron differentiation (Pereira et al., 2010). The loss of Ezh2 was shown to augment neurogenesis and neuronal differentiation. PcG complexes have also been associated with differentiation of NPCs to astrocytes (Hirabayashi et al., 2009) and oligodendrocytes (Sher et al., 2008). As the brain develops, NPCs can travel up and outward to form the outer layers of the brain. A study demonstrated that Ezh2 silences genes associated with neuron migration, such as Netrin1, to maintain correct migration patterns throughout the brain (Di Meglio et al., 2013).
Furthermore, several studies have demonstrated the importance of cross-talk between DNA methylation and histone modifications during mammalian brain development (Wu et al., 2010a; Hahn et al., 2013). As previously described, during neurogenesis as NPCs begin to differentiate, there is an increase in 5hmC specifically in gene bodies of developmentally active genes with little change in 5mC. Accompanying this increase, there is also a decrease in Polycomb-mediated repression and H3K27me3 formation (Hahn et al., 2013). Overexpression of Tet2 and Tet3, both of which are highly expressed in the embryonic cortex, prompted early differentiation of NPCs. An analogous and more obvious transition was seen when Ezh2 was also depleted. Moreover, when Tet proteins were inhibited and Ezh2 overexpressed, NPCs failed to differentiate. This suggests that Polycomb may regulate the transition of NPCs differentiation, and Tet proteins putatively maintain the differentiated state. Additionally, it has been demonstrated that there is an inverse association of Dnmt3a de novo methylation on non-promoter CpGs and H3K27me3 formation in the mouse brain (Wu et al., 2010a). Mice deficient for Dnmt3a had an increase of H3K27me3 as well as increases of PRC2 components Suz12 and Ezh2 at Dnmt3a targets. As previously discussed, Dnmt3a has more of a role in DNA methylation maintenance in postnatal development. The proposed cross-talk suggests that in addition to methylating promoters of self-renewal genes in NPCs, Dnmt3a also has an activating function by inducing transcription of mature neural genes by down regulating H3K27me3 and antagonizing PRC2 binding.
Histone Acetylation
Methylation is just one type of modification that can be present on histone tails; acetylation is a second type of modification that also regulates chromatin dynamics. Histone acetyltransferases (HATs) and HDACs are enzymatic proteins that either add or remove acetylation residues on lysines, respectively (Inoue and Fujimoto, 1970; Racey and Byvoet, 1971) (Figure 1B). Core histones are acetylated by transcriptional coactivators like CBP/p300 that are ubiquitously expressed and involved in cell cycle control, differentiation and apoptosis (Yang et al., 1996). HATs can be divided into three families based on the structure of their catalytic domains: GNAT, MYST and CBP/p300 which are reviewed elsewhere (Sterner and Berger, 2000; Kouzarides, 2007). Supportive of their activating role, HATs will interact with various transcription factors to promote many signaling cascades (Saha and Pahan, 2006). Similar to methylation, acetylation is reversible and removed by HDACs that silence gene expression. HDACs can also be categorized into four distinct classes where class 1 and class 2 HDACs seem to have important roles in the nervous system (Gray and Ekstrom, 2001; Abel and Zukin, 2008). Inhibitors of HDACs have shown promising effects in treating both neurodegenerative and neuropsychiatric diseases. It has been demonstrated that HDAC inhibitors could re-establish histone acetylation that is potentially lost due to dysregulation of the HAT, Tip60 (Cao and Sudhof, 2001). Furthermore, inhibition of HDACs restored learning and memory in a mouse model of neurodegeneration (Fischer et al., 2007). In Fragile X studies, combined administration of 5-azadeoxycytidine and various HDAC inhibitors cause reactivation of FMR1 gene expression (Chiurazzi et al., 1998). In the mouse brain, Hdac3 deletion provoked abnormal locomotor coordination, sociability and cognition (Nott et al., 2016). Interestingly, a cross-talk between HDAC3 and MeCP2 was shown to positively regulate neuronal genes by deacetylating FOXO, a transcription factor that is highly expressed in the hippocampus. A putative link for this cross-talk in relation to Rett syndrome is discussed below.
Chromatin Remodeling
The total length of DNA in one mammalian cell is on average 2 meters, yet the size of the nucleus is only 6 μm. In order to fit the entire genome into such a limited space, DNA molecules have to undergo extraordinary consolidation by a process termed chromatin remodeling. In addition to histones, a major contributor to chromatin compaction is a family of ATP-dependent remodeling proteins. The BAF (mammalian SWI/SNF) complex is a chromatin remodeling multiplex that uses ATP-dependent energy to modify the chromatin landscape to promote cell differentiation (Son and Crabtree, 2014) (Figure 1C). BAF complexes exist in a very spatiotemporal specific fashion. For example, in the mammalian CNS, there are developmental stage-specific BAF complexes in ESCs (Kaeser et al., 2008), NPCs and in post-mitotic neurons (Lessard et al., 2007). A unique feature to BAF complexes is that the alternative subunits that make up the various stage-specific complexes are not interchangeable, indicating their functions are non-overlapping (Wang et al., 1996a,b). Interestingly, BAF complexes are being increasingly associated with neuropsychiatric diseases such as ASD (Neale et al., 2012; O’Roak et al., 2012) and schizophrenia (Koga et al., 2009).
BAF Chromatin Remodelers
The ESC specific BAF (esBAF) contains the ATPase BRG1, BAF250a, BAF60a/b and BAF155 (Kaeser et al., 2008). Deletion of any of the core subunits results in a lethal phenotype (Bultman et al., 2000). For example, shRNA depletion of Brg1 impairs self-renewal properties of ESCs and results in loss of key ESC markers such as Oct4, Sox2 and Nanog (Ho et al., 2009). In addition, deletion of Brg1 also resulted in an increase of the PRC2 recruitment and subsequently, H3K27me3 repression at active ESC genes (Ho et al., 2011). All this evidence suggests that esBAF maintains a euchromatic environment that is required to maintain the pluripotency of ESCs.
The transition from esBAF to neural progenitor BAF (npBAF) is associated with the replacement of esBAF155 with npBAF170 (Ho et al., 2009; Tuoc et al., 2013). npBAF is composed of a combination of either ATPase BRG1 or BRM along with several other BAF subunits. Similar to esBAF, npBAF are critical for the self-renewal properties of NPCs and loss of Brg1 shows similar phenotypes as those seen in esBAF. Interestingly, BAF170 was shown to interact with the transcription factor Pax6 whose primary function is to regulate neural progenitor division during early cortical development (Gotz et al., 1998). Upon BAF170 binding to Pax6, the transcriptional repressor REST (RE1-silencing transcription factor, also known as NRSF) is recruited, and represses Pax6 in non-neuronal radial glia cells (Tuoc et al., 2013). A conserved, 23 base pair sequence known as RE1 (repressor element 1, also known as NRSE) acts as the binding site for REST (Chong et al., 1995; Schoenherr and Anderson, 1995; Chen et al., 1998). Two corepressors are required for REST mediated silencing, Sin3-HDAC and the CoREST protein complex that contains HDACs (Andres et al., 1999; Grimes et al., 2000). Additionally, it was shown that CoREST interacts with BAF57, a subunit present in all stage-specific complexes, to induce long term silencing (Battaglioli et al., 2002). BAF170 is present in the subset of radial glia cells that are destined to be non-neuronal, and absent in radial glia cells destined to become intermediate progenitors that migrate outward to form the outer cortex layer (Andres et al., 1999; Grimes et al., 2000; Tuoc et al., 2013).
The substitutions of BAF53a for BAF53b, SS18 for CREST and BAF45a/d for BAF45b/c marks the transition from npBAF to the mature neuron (nBAF) complex (Olave et al., 2002). Importantly, the nBAF subunits are exclusive to neuronal cells and maintain the chromatin environment of post-mitotic neurons (Olave et al., 2002; Naik et al., 2007). nBAF, in complex with CREST, is essential in regulating dendritic outgrowth (Wu et al., 2007). Normal brain function depends on the correct wiring and synaptic function controlled by adequate dendritic outgrowth. Calcium regulation in the CNS can activate calcium mediated transcription factors, such as CREST, to promote the activation of genes required for dendrite growth (Aizawa et al., 2004).
Regulatory RNA
An emerging field in epigenetics is focusing on debunking the large amount of non-protein coding DNA contained in the mammalian genome. Over the past 20 years, scientists have begun to discover that non-coding is not equivalent to non-functional. When transcribed, these regions generate non-coding RNA (ncRNA) that can range in size from just ∼21 nucleotides to 100,000 nucleotides and can post-transcriptionally regulate mRNA. Many flavors of ncRNAs have been identified (Cech and Steitz, 2014); however, this review will briefly cover miRNA and lncRNA and the putative functions they may serve in the mammalian CNS.
MicroRNAs
MicroRNAs are roughly 22 nucleotides in length and have major roles in post-transcriptionally regulating gene expression by destabilizing their target mRNA (Bartel, 2004). Partial sequence complementarity to the 3′ untranslated region (3′UTR) of the target is adequate for gene downregulation (Lewis et al., 2005). Perfect complementarity is required at what is called the “seed sequence” in the 5′UTR of the miRNA. Interestingly, a single miRNA can target hundreds of different mRNA and that a single mRNA can be targeted by more than one miRNA (Lim et al., 2005). Determining functional roles for the hundreds of miRNAs discovered has eluded scientists for years. Early studies proposed that miRNA had extensive roles during mammalian brain development and several of these studies identified neural-specific miRNA (Krichevsky et al., 2003; Kim et al., 2004; Miska et al., 2004; Sempere et al., 2004). Of the neural-specific miRNA identified, one in particular stands out, miR-124. miR-124 is the most abundant and highly conserved miRNA found in the mammalian brain (Lagos-Quintana et al., 2002). Accounting for nearly 25–48% of all the miRNA in the brain, miR-124 has been implicated as a major contributor in neuronal differentiation and maturation (Krichevsky et al., 2006; Makeyev et al., 2007). For example, the direct targeting and repression of the RNA binding protein, PTBP1 by miR-124 has critical roles in non-neuronal cell development (Makeyev et al., 2007) (Figure 1D). PTBP1 is highly expressed in non-neuronal cells and inhibits alternative splicing of neuron-specific genes (Wagner and Garcia-Blanco, 2001; Sharma et al., 2005). In cells destined to become neurons, miR-124 binds and represses PTBP1, resulting in an increase of PTBP1’s neuronal homolog, PTBP2 protein expression, inducing neuron-specific alternative splicing.
Another brain enriched miRNA, miR-137, is thought to have roles in both adult neurogenesis and neuronal maturation. During adult neurogenesis, miR-137 regulation of proliferation versus differentiation is coupled with its ability to cross-talk with MeCP2 and Ezh2 (Szulwach et al., 2010). Roughly 2–4 Kb upstream of miR-137, methylated CpGs were found as well as a threefold enrichment of MeCP2 binding. Subsequently, it was found that Sox2 also binds upstream of miR-137, and concurrent binding of Sox2 with MeCP2 inhibited miR-137. When miR-137 expression is reduced, there is an increase in neuronal differentiation and a decrease in adult neural stem cell proliferation. This is concurrent with a previous observation that miR-137 expression increases during neuronal differentiation (Silber et al., 2008). The polycomb protein Ezh2, was found to be a direct target of miR-137 in vitro (Szulwach et al., 2010). MiR-137 reduces the expression of Ezh2 and consequently there is also a decrease in H3K27me3. Loss of H3K27me3 encourages adult stem cells to begin to differentiate rather than proliferate. With regards to neuropsychiatric disorders, Genome wide association studies (GWAS) identified miR-137 as one of the strongest associated factors with schizophrenia (Schizophrenia Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium, 2011; Kwon et al., 2013; Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014). Intriguingly, four targets of miR-137 were also found to be highly associated with schizophrenia (Kwon et al., 2013); however, the biological impact of miR-137 in schizophrenia still remains to be explored.
Long Non-coding RNAs
Long non-coding RNAs are classified as having at least 200 nucleotides and non-protein coding abilities (Kapranov et al., 2007). They are also one of the least well understood class of ncNRAs because of the difficulty in distinguishing them from transcription by-products. Compositionally, lncRNA do not appear to be very well conserved between mouse and human (Pang et al., 2006). In the mouse genome, the vast majority of lncRNAs do not contain an open reading frame (Ravasi et al., 2006). In addition, compared to protein coding transcripts, lncRNA tend to be shorter and contain fewer introns. Unusually, some lncRNA such as the paternally imprinted lncRNA H19, are polyadenylated, spliced and exported to the cytoplasm just like protein coding transcripts (Brannan et al., 1990). Functional roles of lncRNA may depend on where in the genome they are located. Those that are transcribed near expressed genes have the potential to regulate the expression of that gene in cis. One of the most well studied lncRNAs is Xist, which functions in cis and is critical for inactivating one of the X chromosomes in mammalian females (Brockdorff et al., 1991). As Xist coats the X chromosome, other repressive factors are recruited, such as Polycomb repressive complexes PRC1 and PRC2 and other histone modifying enzymes (Plath et al., 2003; Silva et al., 2003; de Napoles et al., 2004). lncRNAs have also been demonstrated to regulate transcriptional repressors and activators from a distance (in trans). The HOTAIR lncRNA is 2.2 Kb in length, and was shown to repress the transcription of 40 Kb of the HOXD locus (Rinn et al., 2007). It is proposed that HOTAIR interacts with PRC2 to facilitate H3K27me3 of the HOXD locus because siRNA mediated knockdown of HOTAIR resulted in the loss of H3K27me3 marks specifically at HOXD. Beyond chromatin remodeling, other putative functions for lncRNAs have been suggested, such as transcriptional control and post-transcriptional processing, which are reviewed in detail elsewhere (Mercer et al., 2009; Ponting et al., 2009).
The role of lncRNAs in chromatin remodeling has been extensively studied and the scientific community is just starting to make strides in investigating their roles in the brain (reviewed by Ng et al., 2013). lncRNAs have been found in many tissues (Iyer et al., 2015), but are strikingly enriched in the mammalian brain. One study identified over 800 lncRNAs in the mouse brain, and found that most were associated with specific brain regions, cell types or subcellular compartments, suggesting some putative function (Mercer et al., 2008). One of the better studied lncRNAs in the brain is Malat1 (also known as NEAT2), which is particularly enriched in neurons (Bernard et al., 2010; Lipovich et al., 2012). Malat1 localizes to nuclear speckles which are storage/assembly sites for processing factors involved in pre-mRNA splicing (Lamond and Spector, 2003; Hutchinson et al., 2007; Clemson et al., 2009). Studies demonstrated that Malat1 recruits SR splicing factors in the nuclear spectacle and can regulate genes involved in neural processes and synaptic function (Bernard et al., 2010). Importantly, Malat1 was shown to have 90% conservation between human and mouse, suggesting maintenance of a critical function. A recent computational study utilized RNA-seq data from mouse embryonic brains to identify temporally regulated lncRNAs in brain development. Interestingly, lncRNAs specifically expressed in embryonic brains were no longer expressed in adult brains (Lv et al., 2013). Another study employed RNA-seq on human iPSCs to investigate the expression of lncRNAs during their differentiation into mature neurons (Lin et al., 2011). Several of the lncRNAs that were aberrantly regulated during differentiation were associated with candidate genes of neuropsychiatric disorders, such as ASDs, bipolar disorder and schizophrenia. Much research is being conducted on identifying and determining functional roles of the ever-growing list of lncRNAs; however, more work remains to be done.
To add another layer of complexity, different groups of non-coding RNAs have been found to cross-talk with each other and form regulatory networks in the brain (Kleaveland et al., 2018). A recent study found that in mouse brain, the lncRNA Cyrano destabilizes miR-7 through its highly complementary site for miR-7. Degradation of miR-7 promoted the accumulation of a circular RNA Cdr1as, which is known to dampen neuronal activity (Memczak et al., 2013; Piwecka et al., 2017). Interestingly, Cdr1as contains an inherent destruction mechanism where binding of miR-671 induces its slicing (Kleaveland et al., 2018). It has been proposed that because the binding sites for miR-7 and miR-671 are so close on Cdr1as, cooperative binding could recruit a silencing complex and control the accumulation of Cdr1as in the brain (Grimson et al., 2007; Saetrom et al., 2007).
Proper epigenetic regulations are critical for normal brain development and functions. Numerous evidences suggest that their dysregulation could serve as causal roles in the onset of neurological, neurodegenerative and neuropsychiatric disorders. In the following sections, we will focus on several neuropsychiatric disorders with known roles of epigenetic regulation in their etiology and progression (Table 1).
Table 1.
Summary table of epigenetic processes that can occur in various neuropsychiatric diseases.
| Disease | Epigenetic modifications | Description | Reference |
|---|---|---|---|
| Major depressive disorder | DNA modifications | • Hypermethylation at BDNF locus • CpG methylation in exon 1 biomarker for depressed patients • Patients may not respond to antidepressants • Antidepressants phosphorylate MeCP2 causing it to disassociate from methylated DNA •SLC6A4 methylation may be related to depression heritability • Environmental stressors disrupt methylation • Child-hood stressors associated with increased methylation at exon 1F of NR3C1 • Early life adversity changes methylation of CpG sites in FKBP5 • Pcdh genes show reduced promoter methylation in response to good maternal care |
Angelucci et al., 2005; Fuchikami et al., 2011 Tadic et al., 2014 Hutchinson et al., 2012 Mendonca et al., 2019 Farrell et al., 2018; Efstathopoulos et al., 2018 Farrell et al., 2018 McGowan et al., 2011 |
| Histone modifications | • HDAC2 reduced in nucleus accumbens and postmortem brains •Hdac5 knockout (mouse) • Sensitive to depressive like behaviors • Hdac5 increased in hippocampus of chronically stressed mice • Hdac5 levels reversed with antidepressant treatment • HDAC inhibitors used as antidepressants • MS-275 reverses depressive behaviors |
Covington et al., 2009 Renthal et al., 2007 Tsankova et al., 2006 Eckschlager et al., 2017;Covington et al., 2011 |
|
| Regulatory RNA | • miR-132 and miR-124 regulate BDNF • miR-132 potential biomarker for MDD • miR-124 not a reliable biomarker • miR-132 consistently identified in MDD studies • SSRI treatment increases miR-16 expression and regulates SERT uptake of serotonin • Patients with low miR-1202 levels predicted to respond better to serotonin-based drugs |
Fang et al., 2018 Bocchio-Chiavetto et al., 2013; He et al., 2016 Baudry et al., 2010; Yuan et al., 2018,Lopez et al., 2014;Fiori et al., 2017 |
|
| Autism spectrum disorders | DNA modifications | • Hypermethylation of CpG islands at SHANK3 | Zhu et al., 2014 |
| Histone modifications | • AUTS2 protein functioning in complex with PRC1 to promote gene activation • AUTS2 knockout (mouse) • Impaired developmental phenotypes seen in humans |
Gao et al., 2012, 2014 Gao et al., 2014 |
|
| Regulatory RNA | • Found 28 differentially expressed miRNAs in cortex of ASD patients • Over 200 differentially expressed lncRNA in ASD brains |
Abu-Elneel et al., 2008 Ziats and Rennert, 2013 |
|
| Fragile X | DNA modifications | • Loss of FMR1 results in hypermethylation of CGG repeat | Bell et al., 1991; Pieretti et al., 1991; Sutcliffe et al., 1992; Orsini and Maren, 2012 |
| Regulatory RNA | • Mutated FMR1 associates with the RNA interference pathway | Jin et al., 2004a | |
| Rett syndrome | DNA modifications | • Missense mutation in MeCP2 •Mecp2 knockout (mouse) • Impaired motor coordination; increase in anxiety; abnormal social behavior • Genes that acquire mCH more likely to be dysregulated in RTT mouse model •MeCP2 duplication syndrome • Hypersynchrony in hippocampal neurons |
Mellen et al., 2012; Lyst et al., 2013 Gemelli et al., 2006 Chen et al., 2015 Van Esch et al., 2005 Lu et al., 2016 |
| Histone modifications | • Lose MeCP2 interaction with NCoR/HDAC3 • Conditional knockout of Hdac3 (mouse) • Lose Hdac3 and FOXO at promoters |
Nan et al., 1998; Ebert et al., 2013; Lyst et al., 2013 Nott et al., 2016 |
|
| Regulatory RNA | • Downregulation of miR-146a and miR-146b in Mecp2-null mouse putatively upregulated Irak1 in RTT brain • Bdnf gene contains multiple miRNA binding sites |
Taganov et al., 2006; Urdinguio et al., 2008; Nahid et al., 2009 Wu et al., 2010b |
|
| Schizophrenia | DNA modifications | • 50% increase in DNA methylation at RELN promoter • Transcription factor Sp1 signals for demethylation and prevents de novo methylation, possibly regulates reelin • Olanzapine treatment alters methylation at Pcdha11, Pcdha9, and Pcdhga5 |
Impagnatiello et al., 1998 Han et al., 2001; Chen et al., 2002 Melka et al., 2014 |
| Regulatory RNA | • miR-132 is downregulated and is associated with cognitive and behavioral impairments • miR-132 targets Dnmt3a • miR-195 upregulated and targets BDNF, RELN and DRD1 • High risk SNP in miR-137 is most common among SZ patients • Earlier age of onset, abnormal neurodevelopment • lncRNA GOMAFU is reduced in SZ patients • QKI and SRSF interact with GOMAFU to regulate alternative splicing • Disruption of QKI could account for decreased myelin-related genes expression |
Moreau et al., 2011; Miller et al., 2012 Cannon, 1996 Beveridge et al., 2010 Hamshere et al., 2013; Guan et al., 2014 Lett et al., 2013; van Erp et al., 2014 Barry et al., 2014 Aberg et al., 2006a |
|
Major Depressive Disorder
Individuals with MDD present clinically with not only a depressed mood, but can also suffer from anhedonia, dysregulated appetite and sleep, fatigue, poor concentration and suicidal ideations or acts (Belmaker and Agam, 2008). In the United States, the incidence of depression in women is greater than 20%, nearly twice that of men (Kessler et al., 2003). Twin studies have suggested that MDD has a high heritability rate of about 37% (Sullivan et al., 2000). However, MDD is not monogenic, but rather caused by many genes each contributing only a small proportion. Environment, such as early life stress or trauma, is a major risk factor. Many studies have tried to identify biomarkers to assess a patient’s predisposition for MDD; however, no useful biomarkers have yet been identified. Furthermore, many individuals with MDD are resistant to treatments, and so developing a greater understanding of the neurological facets of MDD has become paramount to the creation of efficacious therapies.
Differential DNA Methylation
A major candidate gene for MDD is BDNF. Individuals with MDD show reduced BDNF protein, and multiple studies have associated this reduction with increased methylation of the BDNF promoter in peripheral blood cells (Angelucci et al., 2005). BDNF has two small CpG islands upstream of exons 1 and 4. One study found that the methylation status of exon 1 in BDNF could be used to accurately distinguish between MDD patients and healthy controls. Remarkably, the depressed patients consistently showed a complete absence of methylation at certain CpG sites in exon 1 (Fuchikami et al., 2011). Although this study was only based on a small number of participants, it would be worth investigating whether these findings could be replicated in larger populations. Absence of methylation at one particular CpG site in exon 4 of BDNF has been associated with reduced response to antidepressant drugs (Tadic et al., 2014). While antidepressants showed no effect on exon 4 methylation, in vitro experiments established that antidepressants could regulate the promoter activity of BDNF. Furthermore, antidepressants have been shown to increase BDNF expression in mice by phosphorylation of MeCP2, which causes the removal of MeCP2 from the DNA (Hutchinson et al., 2012). BDNF exon 4 methylation levels and circulating BDNF protein together may predict a patient’s treatment response (Lieb et al., 2018). These findings collectively suggest that BDNF methylation levels may be a useful biomarker and tool to make more informed choices about individual therapies. Another well-studied biological factor in MDD is the serotonin transporter gene SLC6A4. SLC6A4 methylation correlates with depression in a variety of ways. For example, in an analysis of individuals with MDD, those who had a family member with depression showed a higher percentage of SLC6A4 methylation, indicating that epigenetic regulation of this loci may be related to depression heritability. In mother–child pairs that were concordant for depression, increased methylation of the SLC6A4 promoter was seen in both mother and child (Mendonca et al., 2019).
Disruption of DNA Methylation From Environmental Stressors
Stressful, traumatic events in early life are a major environmental risk factor for MDD, and changes in stress-related genes may be part of the mechanism of depression for some individuals. The glucocorticoid receptor gene, NR3C1, plays an important role in the hypothalamic–pituitary–adrenal (HPA) axis, a stress response system that becomes dysregulated in MDD. Exon 1F of NR3C1 has been extensively studied with regards to its role in early life adversity (Daskalakis and Yehuda, 2014), and has been the target of focus for many depression studies as well. Individuals with MDD show hypermethylation of NR3C1 exon 1F, which correlated with morning cortisol levels (Farrell et al., 2018). In adolescent males, increased NR3C1 exon 1F methylation was associated with stressful experiences such as being bullied, lacking friends and internalizing symptoms, as assessed by a depression scale (Efstathopoulos et al., 2018). Polymorphisms of the glucocorticoid receptor co-chaperone protein, FK506 binding protein 5 (FKBP5), have also been associated with MDD. Interestingly, methylation of certain CpG sites of FKBP5 intron 7 significantly correlated with early life adversity in MDD patients (Farrell et al., 2018). Thus, the connection between many MDD cases and early life trauma involves disruption to the stress response system at an epigenetic level. It is plausible to imagine potential pharmacotherapies that could target methylation of key genes in this system to help restore balance in the HPA axis, and thus attenuate MDD symptoms. Whether targeting HPA axis genes alone would be enough to improve MDD, remains to be understood.
Several studies have linked clustered Pcdhs to depression-like behaviors. A rat model of depression revealed that Pcdhga11 expression levels were increased in the hippocampus (Garafola and Henn, 2014), suggesting Pcdhga11 could be used as a putative biomarker. In contrast to early life stressors, which epigenetically alter the HPA axis, positive early-life parental interactions can epigenetically alter genes that promote neuronal function. For example, adult mice that receive good maternal care (high licking), showed increased histone acetylation and DNA methylation in exons of Pcdh genes. Also, there was reduced methylation at their promoter, increasing over all expression of Pcdh genes (McGowan et al., 2011).
HDAC Inhibitors as a Putative Antidepressant
Histone deacetylases are a promising target for MDD therapies. Mouse behavioral paradigms, such as chronic social defeat stress, have been relied upon as a way to measure antidepressant efficacy (Yin et al., 2016). In mice that have experienced chronic social defeat stress and in postmortem brains from humans with clinical depression, HDAC2 protein is reduced in the nucleus accumbens (NAc; a brain region associated with reward) (Covington et al., 2009). Hdac5 expression is also reduced in the NAc of chronically stressed mice, and this expression is restored and further increased with antidepressant treatment. Consistent with this, mice lacking Hdac5 exhibit enhanced depressive-like behaviors in response to chronic stress (Renthal et al., 2007). In the hippocampus, however, chronically stressed mice have increased Hdac5, and this can be reversed by antidepressant administration (Tsankova et al., 2006). It is no surprise then that HDAC inhibitors, which have been commonly used as anti-cancer agents, are now also being studied for their antidepressant actions (Eckschlager et al., 2017). For example, MS-275 delivery to the hippocampus reverses anhedonia and reduces social avoidance in mice that experienced continuous social defeat stress (Covington et al., 2011). While HDAC inhibitors remain strong candidates for potential therapeutics in humans, translatability from mouse studies is currently lacking. A gap in this research includes determining whether HDAC expression in one particular brain region may drive MDD; and if so, whether there are therapeutics that may regulate this.
MicroRNAs in MDD
Several studies have begun to look at miRNAs as a putative peripheral biomarker for MDD. Remarkably, evidence supports that under certain conditions, miRNAs expressed in the brain can cross through the blood–brain barrier and circulate in the plasma (Sheinerman and Umansky, 2013). In patients with MDD, BDNF levels were found to be decreased in plasma (Molendijk et al., 2014). More importantly, two miRNAs known to interact with BDNF have also been found in plasma of MDD individuals (Fang et al., 2018). This study compared the levels of BDNF, miR-132 and miR-124 in MDD patients that were either treated or not treated with citalopram to healthy control patients. It was found that miR-132 was highest in non-treated MDD patients relative to treated patients and controls, suggesting that miR-132 could be used as a potential biomarker for MDD individuals. Notably, miR-132 is the only miRNA that has been consistently identified in several MDD studies (Yuan et al., 2018). Additionally, MDD patients had higher levels of miR-124, with citalopram treated patients having the largest increase (Fang et al., 2018). Conflicting evidence has been reported with regard to how reliable miR-124 plasma expression levels are for being used as an MDD biomarker (Bocchio-Chiavetto et al., 2013; He et al., 2016). Many other prospective miRNA biomarkers have been proposed (Lopez et al., 2018; Yuan et al., 2018), however; much work remains in validating if any of these biomarkers can be used reliably.
Antidepressant drugs are the most common treatment for individuals with MDD, however; many patients do not respond to them. An interesting area of research is focusing on how miRNAs can help predict patient response to antidepressants. Selective serotonin reuptake inhibitors (SSRIs) are a commonly prescribed class of antidepressants that target the serotonin transporter (SERT). Interestingly, it was found that long term treatment of MDD with SSRIs increases the expression of miR-16, which serendipitously also directly targets SERT (Baudry et al., 2010). Subsequently, SSRI promotes the conversion of precursor miR-16 into its mature form to regulate SERT uptake of serotonin. Another study examined the expression of three miRNAs: miR-1202, miR-135a and miR-16, of MDD patients and controls from two independent cohorts and compared miRNA expression between antidepressant responders and non-responders (Fiori et al., 2017). In both cohorts, decreased levels of miR-1202 correlated with patients responding to either an SSRI or a serotonin-norepinephrine reuptake inhibitor (SNRI). After 8 weeks of antidepressant treatment, the responders’ miR-1202 expression levels increased and were indistinguishable from non-responders and the healthy controls. Importantly, in vitro studies demonstrated a similar result, where NPCs treated with SSRI drugs had an increase in miR-1202; however, miR-1202 expression did not increase when NPCs were treated with non-serotonergic drugs (Lopez et al., 2014). This suggests that MDD patients with low miR-1202 may be more likely to respond to serotonin-based antidepressants. With continued research, miRNAs may become valuable tools for developing a personalized treatment plan, increasing the chances of patients receiving the most appropriate antidepressant the first time.
In summary, epigenetic studies will be highly beneficial in the development of individualized MDD therapeutics, categorization of MDD subtypes and for enhancing efficacy of currently existing treatments.
Autism Spectrum Disorders
Autism spectrum disorders are characterized as heritable neurodevelopmental disorders in which affected individuals have deficits in social interactions, communication and behaviors (American Psychiatric Association, 2013). Over the years, genomic studies have identified genes that seem to contribute to the condition (Abrahams and Geschwind, 2008); however, none significantly stand out as a major contributor to ASD. Rather, it appears that much of the heritability is polygenic with each gene only contributing a very small portion. Recent studies are beginning to suggest that in addition to genetics, ASD may also have an epigenetic component.
Putative Role for Polycomb Repressive Complex 1 in ASD
Several putative genes have been proposed for contributing to ASD, one of which is autism susceptibility candidate 2 (AUTS2) (Sultana et al., 2002; Oksenberg and Ahituv, 2013). Surprisingly, recent studies have demonstrated that AUTS2 can be in complex with PRC1 and function in gene promotion contrary to PRC1’s traditional repressive role (Gao et al., 2012; Gao et al., 2014). It is proposed that the PRC1-AUTS2 complex can promote gene expression through the recruitment of CK2 and the co-activator P300 protein. CK2 inhibits monoubiquitination of lysine 119 on histone H2A by phosphorylating RING1B. Further supporting the role of AUTS2 in gene activation, ChIP-seq analysis has localized AUTS2 predominantly near TSSs in the mouse brain. These binding sites also possess active histone marks such as histone 3 lysine 27 acetylation (H3K27ac) and H3K4me3 and were reduced for repressive histone mark H3K27me3. Furthermore, gene ontology analysis of PRC1-AUTS2 targets identified functional terms that were associated with CNS transcriptional programming. All of this evidence supports the PRC1-AUTS2 complex as being involved in promoting gene expression. Behavioral and developmental analysis of AUTS2 knockout mice also showed similar impaired developmental phenotypes as observed in humans with a disruption in AUTS2 (Gao et al., 2014). The interaction between AUTS2 and epigenetic machinery could be a rich area to investigate to uncover potential therapeutic targets for individuals with AUTS2 polymorphisms.
Differential DNA Methylation
The SHANK3 gene has been identified as a strong contributing factor to ASDs (Durand et al., 2007; Moessner et al., 2007; Gauthier et al., 2009). In neuronal synapses, SHANK3 acts as a scaffolding protein with critical roles in the formation, maturation and maintenance of synapses (Du et al., 1998; Boeckers et al., 1999). The SHANK3 gene contains 5 CpG islands at putative intragenic promoters whose methylation status has been associated with alternative splice variants (Zhu et al., 2014). In postmortem ASD brains, there was a significant increase in DNA methylation at the CpG islands 2, 3, and 4 of SHANK3. In addition, the methylation at these islands was associated with decreased expression and decreased alternative splicing of SHANK3, suggesting DNA methylation regulates the expression of the splice variants. This evidence introduces the possibility that the methylation status of SHANK3 could serve as a putative predictor for ASD.
Dysregulation of Non-coding RNAs
Although very little is known about the contributions miRNA and lncRNA make in ASD, several studies have investigated non-coding RNAs in this disorder. One study identified 28 differentially expressed miRNAs in ASD cerebellar cortex tissue using qPCR (Abu-Elneel et al., 2008). Interestingly, 7 of the identified miRNAs were predicted to target autism-associated genes NEUREXIN and SHANK3. Another study that looked at lncRNA detected over 200 differentially expressed lncRNAs in ASD (Ziats and Rennert, 2013). Of those identified, more than 90% mapped within 500 Kb of a known gene, many of which were genes with functional roles in neurodevelopment and psychiatric diseases. These findings imply that lncRNAs could be part of the mechanism that regulates genes contributing to ASD. This study was also able to compare the expression of lncRNAs in in the cerebellum and cortex from the same patient of healthy and ASD diseased brains. Between brain regions, the ASD brains had significantly less differentially expressed genes and lncRNAs compared to the control brains. This finding is consistent with imaging studies that show autistic brains have less specialized, less distinct regions as compared to healthy brains (Minshew and Keller, 2010). In summary, because ASD lacks a strong heritability factor, epigenetic studies will likely fill in many gaps of mechanisms and risk factors contributing to ASD.
Hundreds of genes have been found to be associated with ASD including Pcdh genes. A GWAS study identified 5 SNPs in the PCDHA gene that were significantly associated with ASD (Anitha et al., 2013). Interestingly, deletions near PCDH10 have consistently been found in families with autism (Morrow et al., 2008; Bucan et al., 2009). Further supporting roles for protocadherins in ASD was the finding that ASD brains have increased dendritic spine densities compared to controls (Hutsler and Zhang, 2010). Pcdh genes are renowned for their roles in dendritogenesis, dendrite arborization and dendritic spine regulation (Keeler et al., 2015) making them perfect candidate genes for autism studies. Studying Pcdh genes in neuropsychiatric diseases has become a hot topic for the field, and it would be interesting to see how epigenetic regulation of them also contributes to disease pathology.
Fragile X Syndrome
Fragile X syndrome is the most commonly inherited form of mental retardation, and is caused by a trinucleotide repeat in the 5′UTR of the FMR1 gene, which encodes the RNA binding protein FMRP (Webb et al., 1986; Verkerk et al., 1991; Ashley et al., 1993). FMRP is widely expressed in fetal and adult tissues with the highest enrichment in the brain and testes (Devys et al., 1993). It predominantly localizes in the cytoplasm; however, it can be transported to the nucleus via its nuclear localization signal (Devys et al., 1993; Eberhart et al., 1996). As an RNA binding protein, FMRP appears to have several functions ranging from translation regulation, miRNA-mediated translation suppression and neuronal synaptic plasticity (Jin et al., 2004a). Currently, the precise mechanisms by which FMRP regulates transcription/translation as well as its target RNAs are still under rigorous investigation. Recent work has started to unveil putative functional roles of FMRP as well as potential regulatory targets of FMRP in FXS and other intellectual disabilities (Nelson et al., 2013).
Hypermethylation of FMR1 Putatively Mediated by RNAi
Fragile X has been shown to be caused by the loss of FMR1 gene expression in conjunction with the hypermethylation of the cytosines in the CGG trinucleotide repeat (Bell et al., 1991; Pieretti et al., 1991; Sutcliffe et al., 1992; Orsini and Maren, 2012). Methylation of the CGG repeats was identified in human fetal tissue, suggesting that the methylation is acquired after fertilization, or is already present in the carrier female’s oocytes (Sutcliffe et al., 1992). Remarkably, FMR1 gene expression could be rescued in vitro by utilizing DNMT inhibitors and CRISPR/Cas9 to remove the DNA methylation (Bar-Nur et al., 2012; Park et al., 2015; Liu et al., 2018). The question that remains at large is what initiates or causes the hypermethylation of the expanded repeats seen at the CpG island? One model proposes that the RNA interference (RNAi) pathway may be involved (Jin et al., 2004a). This model suggests that the mRNA produced from the expanded FMR1 gene can fold back on itself, generating a hairpin-like structure and be processed by the RNAi machinery. Ultimately, targeting of the RNAi complex is thought to recruit de novo DNMTs and histone methyltransferases (HMTs) to the expanded FMR1 sequence. This model is supported by the initial finding that the mutant FMR1 RNA sequence forms different hairpin structures with the prominent structure forming in the 3′UTR of the transcript (Handa et al., 2003).
MicroRNA Pathway in Fragile X
FMRP has been shown to function as a translational repressor through its RNA binding properties (Laggerbauer et al., 2001; Li et al., 2001). In the brain, FMRP bound to mRNA has been found at dendritic spines associated with polyribosomes, suggesting some involvement in protein synthesis at synapses (Feng et al., 1997). Also, in human brains of fragile X patients there is abnormal dendritic spine growth (Hinton et al., 1991). Recent work has prompted a model where FMRP regulates its mRNA expression through the miRNA pathway. Immunoprecipitation studies demonstrated that mammalian wildtype FMRP, but not mutant FMRP, could associate with miRNA and miRNA pathway proteins Dicer, eIF2C2 and the mammalian Argonaute (AGO) protein (Jin et al., 2004b). This study also determined that in fly, AGO1 is required for dFmr1, the fly ortholog of FMR1, regulation of synaptic plasticity. These observations are supported from previous studies in Drosophila that showed dFmr1 associated with AGO2 and the RNA inducing silencing complex (RISC) (Caudy et al., 2002; Ishizuka et al., 2002). In vitro rescue studies of FMR1 have demonstrated that current technologies, such as DNMT inhibitors and CRISPR/Cas9, can be applied as putative therapeutics. The next step is to conduct translational studies to test whether FMR1 expression can be rescued in mammals. A possible place to begin would be in vitro fertilization experiments. Hypermethylation of FMR1 is observed either after fertilization or is already in the oocyte. It would be interesting to explore the effects of CRISPR technology on FMR1 expression at these early stages in development.
Rett Syndrome
MeCP2 Dysregulation
Rett syndrome (RTT) is a rare disease that was first described in 1966 although the criteria for diagnosing patients did not become available until the 1980s (Rett, 1966; Hagberg et al., 1985). Described as a progressive neurodevelopment disorder, Rett syndrome is most common in females and symptoms, such as autistic behavior, stereotypic hand wringing and loss of facial expression, begin to appear around 18 months of age (Hagberg et al., 1983). Later clinical presentations can include difficulty with motor control, breathing, communication, small head size, muscle wasting and seizures (Gold et al., 2018). The Rett loci was mapped to a region on the X chromosome (Xq28) (Sirianni et al., 1998). Further mapping studies found that the MeCP2 gene also mapped to this region, and that mutations in the methyl binding domain and transcription of repression domain (TRD) of MeCP2 caused RTT (Amir et al., 1999). The MeCP2 missense mutation R133C results in the abolishment of any methyl binding ability of the MBD (Mellen et al., 2012). Many other RTT-associated missense mutations in the MBD and TRD also have been shown to prevent MeCP2’s ability to interact with complexes and methylated DNA (Lyst et al., 2013). MeCP2 is essential to normal brain morphology and consequently, individuals with RTT have more densely packed, shorter neurons with dendrites that are less dense and less complex (Armstrong et al., 1995). Conditional deletion of Mecp2 in postnatal mice produced similar phenotypes as those observed in RTT patients (Gemelli et al., 2006). These mice had impaired motor coordination, increased anxiety and abnormal social behavior.
The mechanism by which MeCP2 mutation (or loss of function) causes Rett is not known; although several studies have tried to identify dysregulated genes in RTT that are direct targets of MeCP2 (Colantuoni et al., 2001; Peddada et al., 2006; Jordan et al., 2007). One study determined that MeCP2 deficient mice and RTT human brains showed significant upregulation of inhibitors of differentiation genes (ID1-4), which are targets of MeCP2 (Peddada et al., 2006). In vitro studies demonstrated that MeCP2 normally downregulates the protocadherin genes PCDHB1 and PCDH7 (Miyake et al., 2011). Because protocadherins are critical for proper brain development, aberrant expression of these genes could contribute to the pathogenesis of RTT. Furthermore, a study involving four independent cDNA microarrays demonstrated that the majority of differentially expressed genes were downregulated in human RTT postmortem brains, but they failed to investigate whether any of these genes were associated with MeCP2 (Colantuoni et al., 2001). Another study that used microarrays to identify differentially expressed genes in a MeCP2-null mouse model looked at several brain regions (cortex, midbrain and cerebellum) to determine if certain regions were more sensitive to loss of MeCP2 (Urdinguio et al., 2008). Although no significant differences were found between brain regions, the study did identify several genes that are direct binding targets of MeCP2. Importantly, these genes (Fkbp5, Mobp, Plagl1, Ddc, Mllt2h, Eya2, and S100a9) were found to be upregulated in the RTT mouse model, and their functions are associated with neural function. Identifying candidate genes in RTT is important for developing a greater understanding of the underlying mechanism.
While Rett syndrome is the result of MeCP2 loss of function, MeCP2 duplication syndrome is the result of MeCP2 overexpression, and mimics some of the symptoms of Rett (Van Esch et al., 2005). Rodent models have clearly demonstrated that having a balance of MeCP2 expression and function is absolutely essential to normal brain activity. Mice that have either overexpression or deletion of MeCP2 show disrupted neuronal activity in the hippocampus (Lu et al., 2016). Both mouse models exhibit neuronal hypersynchrony, which is an aberration from the normal asynchrony typically present at baseline. Importantly, this phenotype could be observed several months before the animals started to have seizures. Deep brain stimulation therapy rescued the abnormal synchrony in both mouse models. Thus, proper MeCP2 expression levels are required for stable neuronal activity.
DNA Methylation Affects MeCP2 Binding
Several studies have looked at how dynamic changes in DNA methylation, of both CpG and CpH, could correlate to Rett syndrome pathology, and have even speculated as to how they could contribute to the delayed onset of RTT symptoms. In addition to mCG, MeCP2 also binds to mCH, preferentially to mCA (Guo et al., 2014; Gabel et al., 2015). Accumulation of MeCP2 in mammalian neurons occurs early after birth when mCH starts to accumulate (Shahbazian et al., 2002; Ballas et al., 2009; Lister et al., 2013). Interestingly, in maturing neurons, those genes that acquired mCH marks were more likely to be dysregulated in the RTT mouse model (Chen et al., 2015). This evidence advocates that early in brain development, MeCP2 initially binds to mCG and then to mCH as it accumulates around a subgroup of neuronal genes (such as Bdnf) to influence gene transcription. This epigenetic mechanism could contribute to why a genetic disease like Rett syndrome could have a delayed onset.
Important for brain development is the proper regulation of LINE 1 retrotransposon. MeCP2 directly targets the 5′UTR of LINE 1 in the brain to regulate LINE 1 mobility (Muotri et al., 2010). Mutations in MeCP2, as seen in RTT, prevent its binding to LINE 1 resulting in increased expression of LINE 1 in both in vitro and in vivo models of RTT. Whether or not the increase in neuronal retrotransposition contributes to the cause of RTT, or is simply an effect is not clear. These findings warrant further investigation of LINE 1’s contribution to RTT. It could also be worth investigating global methylation profiles of developing mouse embryos to determine if DNA methylation patterns are also disrupted, contributing to aberrant LINE 1 expression.
MeCP2 Interacts With HDACs
A possible epigenetic mechanism to investigate for RTT is the interaction of MeCP2 with HDAC3. MeCP2 associates with HDAC3 as part of the NCoR/SMRT co-suppressor complex (Lyst et al., 2013), and MeCP2 missense mutations that occur in Rett Syndrome prevent this interaction (Nan et al., 1998; Ebert et al., 2013; Lyst et al., 2013). Furthermore, in mice, HDAC3 binds near transcriptional start sites of active gene promoters, including the Bdnf gene promoter in the brain (Nott et al., 2016). In Rett syndromic mice, MeCP2 mutations prevent the recruitment of HDAC3 and FOXO to gene promoters. FOXO is a transcription factor that when acetylated has reduced binding affinity to DNA (Daitoku et al., 2004; Matsuzaki et al., 2005; Hatta et al., 2009). Recruitment of HDAC3 to active gene promoters through MeCP2 regulates the deacetylation of FOXO, and promotes gene expression of neuronal genes (Nott et al., 2016). The gene targets of this complex might yield insightful avenues for developing site-directed therapeutics for Rett patients.
Dysregulation of miRNAs
Recently, miRNAs have been suggested to interact with MeCP2 and potentially contribute to RTT. Using a mouse RTT model, one study found that just over one-fourth of the miRNAs analyzed showed different expression patterns in Mecp2-null brains compared to wildtype, most of which were downregulated (Urdinguio et al., 2010). Additionally, they found that MeCP2 associated with the miRNAs that had 5′UTR hypermethylation. Interestingly, two of the downregulated miRNA, miR-146a and miR-146b, base pair to the 3′UTR of IL-1 receptor-associated kinase 1 (Irak1), which is upregulated in RTT mouse brains (Taganov et al., 2006; Urdinguio et al., 2008). It was then shown that both miR-146a and miR-146b could downregulate IRAK1 expression in vitro (Nahid et al., 2009) and it was proposed that in Rett syndrome the downregulation of miR-146a/b contributes to the overexpression of IRAK1 (Urdinguio et al., 2010). Another study identified altered expression of miRNA in the cerebellum of Mecp2-null mice (Wu et al., 2010b). They showed that the promoters of the dysregulated miRNAs were methylated and bound by MeCP2, downregulating their expression. Furthermore, the 3′UTR of the Bdnf transcript contained multiple miRNA binding sites for miRNA that were upregulated, providing mechanistic evidence to explain reduced Bdnf expression in RTT. MeCP2 and its interactions with epigenetic factors play major roles in Rett syndrome, yet why there is a delay in disease onset is not fully elucidated. It may be worthwhile to investigate a spectrum of early developmental stages to determine what epigenetic changes are occurring before the onset of disease, and how these changes could contribute to the delayed onset of RTT.
Schizophrenia
Schizophrenia (SZ) is a mental illness with clinical phenotypes such as dissociation of thought, ideas, identity and emotion (Moskowitz and Heim, 2011). SZ has a wide range of first episodic onset, with early onset occurring in adolescence and late onset being in the mid-50s (Jablensky et al., 1992). Interestingly, males appear to experience their first episodic event 4–5 years earlier than females, on average (Hafner et al., 1998). Like ASD, SZ lacks a single causal gene (International Schizophrenia et al., 2009); however, epigenetic factors are a promising area of research.
Aberrant DNA Methylation
In all postmortem brains of schizophrenic individuals, studies have found ∼50% increase in DNA methylation at the Reelin gene (RELN) promoter (Impagnatiello et al., 1998). Reelin is an extracellular matrix protein highly expressed in GABAergic neurons (Pesold et al., 1999). Functionally, RELN has been shown to be essential in brain development contributing to neuronal migration, axonal branching and synaptogenesis. Upstream of the promoter region is a CpG island, suggesting that inappropriate methylation could regulate RELN (Royaux et al., 1997). One study demonstrated that hypermethylation of the RELN promoter was associated with a decrease of RELN expression found in the brains of schizophrenic patients (Abdolmaleky et al., 2005). It was also established that the transcription factors Sp1 and Tbr1 have binding sites upstream of the RELN promoter and induce gene expression (Chen et al., 2002). Interestingly, Sp1 regulation of the adenine ribosyltransferase gene triggers demethylation and prevents de novo methylation, and it is proposed that Sp1 could similarly regulate RELN (Han et al., 2001; Chen et al., 2002). Additionally, prevention of DNA methylation with 5-aza-2′-deoxycytidine (5-azadC) at the CpG island increased gene expression of RELN more than 50-fold (Chen et al., 2002). This evidence indicates that methylation at the RELN gene likely plays a major role in SZ.
Very little research has been done to link how aberrant epigenetic modifications can affect the expression of protocadherins in SZ. Interestingly, olanzapine, a common antipsychotic drug often prescribed to SZ patients, is proposed to induce its effect by causing DNA methylation changes throughout the brain (Melka et al., 2014). Importantly, several protocadherin genes (Pcdha11, Pcdha9, and Pcdhga5) had altered promoter methylation in the cerebellum, whereas hypomethylation of the Pcdhga8 promoter was observed in the hippocampus. Regions of the genome that are thought to contribute to SZ susceptibility appear to overlap with cadherin superfamily genes (Pedrosa et al., 2008). Polymorphisms in PCDH12 and PCDH15 were found to be in association (Gregorio et al., 2009; Narayanan et al., 2015), and linkage studies found the CTNNA2 gene in sibling pairs with SZ (DeLisi et al., 2002; Chu and Liu, 2010). Protocadherin gene expression clearly has important roles in SZ, but to what extent they are affected by epigenetic changes is unclear. It would be interesting to test whether manipulation of methylation at various protocadherin genes could significantly impact brain development and function in neuropsychiatric diseases.
Non-coding RNAs
The contribution of miRNAs to cognitive disorders has been best characterized in SZ. In postmortem SZ brains, miR-132 was found to be dysregulated, and has been associated with cognitive and behavioral impairments (Moreau et al., 2011; Miller et al., 2012). In the prefrontal cortex of SZ brains, miR-132 was significantly downregulated while its target mRNAs were all upregulated (Miller et al., 2012). Some of the identified targets were associated with synaptic long-term potentiation and depression, neuronal CREB signaling and DNA methylation. Interestingly, Dnmt3a was found to be a putative target of miR-132; however, the expression patterns of Dnmt3a and miR-132 at early developmental stages are opposite. It is not until later in development when miR-132 expression drastically increases that it would have the potential to target Dnmt3a. One could speculate that this temporal expression of miR-132 and Dnmt3a prevents their dysregulation during early development, consistent with SZ requiring an ongoing and prolonged accumulation of dysregulated events that must reach a threshold for symptoms to develop (Cannon, 1996). Another miRNA found in SZ brains was miR-195, which targets several genes (BDNF, RELN, DRD1) implicated in SZ (Beveridge et al., 2010). GWAS for SZ have identified a locus on chromosome 1p21.3 that is highly associated with miR-137 (Schizophrenia Working Group of the Psychiatric Genomics Consortium, 2014; Gianfrancesco et al., 2017). Several independent GWAS studies have identified a single nucleotide polymorphism (SNP) within the miR-137 gene that is common amongst schizophrenic patients (Hamshere et al., 2013; Guan et al., 2014). Patients with this high-risk SNP had earlier age of onset (Lett et al., 2013), abnormal development of brain structure and lower prefrontal cortex activity during working memory (van Erp et al., 2014). In vitro analysis identified a novel lncRNA whose expression pattern is very comparable to miR-137 (Gianfrancesco et al., 2017). The lncRNA was found to be highly expressed, specifically in the prefrontal cortex, and transcriptionally induced by psychoactive drugs, suggesting that there might be a potential connection with the hallucinations that many SZ patients experience. Although further work still needs to be done to better understand the functional role of this lncRNA, it could be postulated that miR-137 and the lncRNA could regulate each other.
Over 200 lncRNAs have been found in the brains of individuals with psychiatric disorders such as SZ (Ziats and Rennert, 2013). The lncRNA GOMAFU in humans is involved in brain development (Mercer et al., 2010) and post-mitotic neuronal function (Sone et al., 2007). In addition, it has been found that SZ patients have reduced GOMAFU expression, which was found to be important for cognitive function (Barry et al., 2014). Interestingly, GOMAFU can directly interact with the splicing factors quaking (QKI) and SRSF1 (serine/arginine-rich splicing factor 1), and when GOMAFU is dysregulated, the alternative splicing resembles that seen in schizophrenia-associated genes DISC1 and ERBB4 (Figure 1D). QKI was identified as a potential SZ gene because it is the only gene located in the chromosome susceptibility locus, 6q25-6q27, in a schizophrenia pedigree (Aberg et al., 2006b). mRNA expression analysis revealed that two QKI splice variants were significantly down regulated in SZ patient brains, suggesting that the splice variants could increase the susceptibility of SZ. Moreover, disrupted QKI splicing could account for the decreased expression of myelin-related genes associated with SZ (Aberg et al., 2006a). Interestingly, most of the myelin-related gene repression was explained by the splice variant QKI-7kb, and putative QKI-binding sites were identified in five myelin gene’s mRNA.
In summary, non-coding RNAs as well as methylation of the RELN gene have been implicated as epigenetic research areas that may hold potential therapeutic targets for SZ. Future studies should aim to further elucidate the role of miRNAs in the SZ brain, in order to pinpoint certain miRNAs that may be pivotal in SZ symptoms. Because SZ has so many genetic variants that only contribute a small portion to the overall increased risk, identifying global epigenetic dysregulation patterns may be more promising. Additionally, there is a lack of studies looking at how epigenetic patterning in the brain changes due to environmental risk factors such as drug use, birth complications and childhood adversity (Neilson et al., 2017). All of these environmental risk factors have been highly correlated with SZ and shown to impact brain development.
Conclusion and Outlook
Genetic and epigenetic regulations are critical for brain development, function and prevention of neurological diseases. Currently, the field lacks clear molecular mechanisms underlying neuropsychiatric diseases and effective treatment options. Epigenetics provides a whole new dimension for therapeutic treatments because so many of these diseases are not monogenic and likely have a significant environmental contribution. The epigenome is greatly influenced by environmental factors such as nutrition, chemical pollutants, traumatic early life experiences, temperature changes and exercise (Roth and Sweatt, 2011; Feil and Fraga, 2012), but how they affect brain development is poorly understood. Importantly, the effect of the environment on epigenetics is not limited to development after birth, but can also affect development in utero. Recent work hypothesized that early life stressors that cause long-lasting epigenetic changes may be due to cellular epigenetic “priming.” Similar to the immune system, once a particular environmental exposure is experienced and alters the epigenetic state of a gene, that gene now remains in a state of “primed responsiveness,” and will have a quicker response if that same environmental exposure is experienced again (Vineis et al., 2017). This concept of epigenetic memory in response to environmental stimuli could serve as a way to identify individuals predisposed to developing neuropsychiatric diseases.
For several of the monogenic neuropsychiatric diseases, such as Rett syndrome and Fragile X, exploring epigenetic mechanisms may lead to understanding whether or not there could be early intervention treatment that could attenuate the disease prior to its onset. Prenatal genome sequencing could be implemented to look for mutations in specific genes as the cost of sequencing continuously decreases. If it is known ahead of time that a child is predisposed, early intervention treatments could be started to slow or prevent disease progression. Possible directions for treatment development could include the use of CRISPR editing to fix missenses mutations in MeCP2 of Rett patients, or developing DNMT based drugs to remove the methylation on the CGG expanded repeat in Fragile X. Additionally, it could be useful to look at developmental time points to identify what epigenetic changes are occurring just before the onset of disease. This could shed light on when key epigenetic remodeling events take place and when potential interventions could be tested.
Treatments for polygenic neuropsychiatric diseases, such as MDD, could benefit the most from epigenetic treatments because there is no clear-cut mechanism to explain disease development. The field is currently focusing on exploring two approaches for developing HDAC inhibitor treatments. The first approach combines HDAC inhibitors with antidepressant drugs (Fuchikami et al., 2016). In this method, HDACs are thought to promote the condensation of the chromatin and prevent transcription factors from binding, regardless of whether the antidepressant is able to increase the levels of the transcription factor. Administration of both HDAC inhibitors and antidepressant could make both drugs work better. The second approach addresses the problem of low specificity of current HDAC inhibitors. The goal of this approach is to synthesize new, highly selective compounds/analogs that can cross the blood brain barrier and be administered acutely instead of chronically (Misztak et al., 2018). Additionally, identifying more reliable biomarkers, such as miR-1202, that can help predict a patient’s likelihood to respond to antidepressants could eliminate much of the guess work in finding a drug that will best treat a patient.
In summary, this review has discussed several epigenetic processes and how dysregulation of any of them can affect brain development, function and disease. An important topic not covered in this review is that dysregulation of DNA methylation, histone modifications, chromatin remodeling, and regulatory RNA also contribute to neurodegenerative diseases such as Huntington’s, Parkinson’s and Alzheimer’s. Several model systems, such as mice and postmortem human brains, have been used to generate the current knowledge bank available. A promising new model system, the organoid, can help evolve our understanding of genetics and epigenetics in neuropsychiatric disorders.
Currently, a major challenge in studying neuropsychiatric diseases is the limitations of the model systems available. Mouse models and human postmortem brains have been heavily relied upon to provide insight into neuropsychiatric disease pathology and etiology. However, both options have their limitations. Although mouse and human brains are highly similar at genetic, structural and general circuitry levels, key differences limit them as models of human diseases that are characterized by complex dysfunction of behavior and thought. For example, human brains have evolved to contain the granular prefrontal cortex, which is absent in mouse brains (Passingham and Wise, 2012). This portion of the cortex is thought to have emerged in relation to increasing brain size, and have roles in comprehension, planning and perception (Goldman-Rakic, 1996; Barbas, 2000; Rolls, 2000; Miller and Cohen, 2001). Human brain samples are obtained postmortem, and thus can never fully recapitulate the epigenetic landscape of a living brain. Postmortem brains only provide a snap shot in the timeline of the disease, and this snapshot is usually biased toward the state of death. Thus, postmortem human brains fail to provide data regarding disease initiation and progression over time.
A new and promising model system that can compensate for animal models and postmortem brains are organoids. Organoids are 3-dimensional cultures that model whole developing organs (Itskovitz-Eldor et al., 2000). This system evolved from embryoid cultures, which are 3D aggregates of stem cells that are grown in a suspension that will induce their differentiation. When organoids are used to generate neuronal lineages, they can recapitulate human brain development in vitro. Morphological studies have further confirmed that forebrain organoids have similar developmental patterns as the developing human cortex (Qian et al., 2016; Zhang et al., 2016). For example, developing organoids can undergo neural differentiation, form multi-layer progenitor zones, form discrete brain regions and portray typical neuron morphologies such as spine-like structures (Lancaster et al., 2013; Qian et al., 2016). Epigenomic studies have also confirmed that brain organoids recapitulate the fetal brain epigenome (Luo et al., 2016). Whole-genome methylome profiling revealed that mCH accumulation in both fetal brain and cerebral organoid occurred at super-enhancers that are specifically active during fetal development, and later became repressed. Additionally, organoid mCG signatures at DNA methylated valleys, large domains depleted of mCG, were comparable to fetal cortex and localized to genes with roles in brain development. Organoids are cultured from mature epithelial cells that are reverted back to induced pluripotent stem cells. The mature epithelial cells can be obtained non-invasively from an individual affected by a neurological disease, allowing researchers to use a model that is genetically identical to the patient. This provides the field with the ability to develop unique therapeutic options specific to each patient.
In conclusion, the study of epigenetics, along with the exploitation of organoid models, can accelerate our understanding of neuropsychiatric diseases to better develop enhanced treatments.
Author Contributions
JK and BY wrote the review together. EB and ZW helped with revisions. EB contributed to the MDD section.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Abdolmaleky H. M., Cheng K. H., Russo A., Smith C. L., Faraone S. V., Wilcox M., et al. (2005). Hypermethylation of the reelin (RELN) promoter in the brain of schizophrenic patients: a preliminary report. Am. J. Med. Genet. B Neuropsychiatr. Genet. 134B 60–66. 10.1002/ajmg.b.30140 [DOI] [PubMed] [Google Scholar]
- Abel T., Zukin R. S. (2008). Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr. Opin. Pharmacol. 8 57–64. 10.1016/j.coph.2007.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberg K., Saetre P., Jareborg N., Jazin E. (2006a). Human QKI, a potential regulator of mRNA expression of human oligodendrocyte-related genes involved in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 103 7482–7487. 10.1073/pnas.0601213103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aberg K., Saetre P., Lindholm E., Ekholm B., Pettersson U., Adolfsson R., et al. (2006b). Human QKI, a new candidate gene for schizophrenia involved in myelination. Am. J. Med. Genet. B Neuropsychiatr. Genet. 141B 84–90. 10.1002/ajmg.b.30243 [DOI] [PubMed] [Google Scholar]
- Abrahams B. S., Geschwind D. H. (2008). Advances in autism genetics: on the threshold of a new neurobiology. Nat. Rev. Genet. 9 341–355. 10.1038/nrg2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abu-Elneel K., Liu T., Gazzaniga F. S., Nishimura Y., Wall D. P., Geschwind D. H., et al. (2008). Heterogeneous dysregulation of microRNAs across the autism spectrum. Neurogenetics 9 153–161. 10.1007/s10048-008-0133-5 [DOI] [PubMed] [Google Scholar]
- Aizawa H., Hu S. C., Bobb K., Balakrishnan K., Ince G., Gurevich I., et al. (2004). Dendrite development regulated by CREST, a calcium-regulated transcriptional activator. Science 303 197–202. 10.1126/science.1089845 [DOI] [PubMed] [Google Scholar]
- Aizawa T., Maruyama K., Kondo H., Yoshikawa K. (1992). Expression of necdin, an embryonal carcinoma-derived nuclear protein, in developing mouse brain. Brain Res. Dev. Brain Res. 68 265–274. 10.1016/0165-3806(92)90069-9 [DOI] [PubMed] [Google Scholar]
- Allis C. D., Jenuwein T. (2016). The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 17 487–500. 10.1038/nrg.2016.59 [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders. Washington, DC: APA; 10.1176/appi.books.9780890425596 [DOI] [Google Scholar]
- Amir R. E., Van den Veyver I. B., Wan M., Tran C. Q., Francke U., Zoghbi H. Y. (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 23 185–188. 10.1038/13810 [DOI] [PubMed] [Google Scholar]
- Amouroux R., Nashun B., Shirane K., Nakagawa S., Hill P. W., D’Souza Z., et al. (2016). De novo DNA methylation drives 5hmC accumulation in mouse zygotes. Nat. Cell Biol. 18 225–233. 10.1038/ncb3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres M. E., Burger C., Peral-Rubio M. J., Battaglioli E., Anderson M. E., Grimes J., et al. (1999). CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. U.S.A. 96 9873–9878. 10.1073/pnas.96.17.9873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelucci F., Brene S., Mathe A. A. (2005). BDNF in schizophrenia, depression and corresponding animal models. Mol. Psychiatry 10 345–352. 10.1038/sj.mp.4001637 [DOI] [PubMed] [Google Scholar]
- Anitha A., Thanseem I., Nakamura K., Yamada K., Iwayama Y., Toyota T., et al. (2013). Protocadherin alpha (PCDHA) as a novel susceptibility gene for autism. J. Psychiatry Neurosci. 38 192–198. 10.1503/jpn.120058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong D., Dunn J. K., Antalffy B., Trivedi R. (1995). Selective dendritic alterations in the cortex of Rett syndrome. J. Neuropathol. Exp. Neurol. 54 195–201. 10.1097/00005072-199503000-00006 [DOI] [PubMed] [Google Scholar]
- Ashley C. T., Jr., Wilkinson K. D., Reines D., Warren S. T. (1993). FMR1 protein: conserved RNP family domains and selective RNA binding. Science 262 563–566. 10.1126/science.7692601 [DOI] [PubMed] [Google Scholar]
- Atladottir H. O., Gyllenberg D., Langridge A., Sandin S., Hansen S. N., Leonard H., et al. (2015). The increasing prevalence of reported diagnoses of childhood psychiatric disorders: a descriptive multinational comparison. Eur. Child Adolesc. Psychiatry 24 173–183. 10.1007/s00787-014-0553-8 [DOI] [PubMed] [Google Scholar]
- Bachman M., Uribe-Lewis S., Yang X., Burgess H. E., Iurlaro M., Reik W., et al. (2015). 5-Formylcytosine can be a stable DNA modification in mammals. Nat. Chem. Biol. 11 555–557. 10.1038/nchembio.1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N., Lioy D. T., Grunseich C., Mandel G. (2009). Non-cell autonomous influence of MeCP2-deficient glia on neuronal dendritic morphology. Nat. Neurosci. 12 311–317. 10.1038/nn.2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballestar E., Wolffe A. P. (2001). Methyl-CpG-binding proteins. Targeting specific gene repression. Eur. J. Biochem. 268 1–6. 10.1046/j.1432-1327.2001.01869.x [DOI] [PubMed] [Google Scholar]
- Barbas H. (2000). Connections underlying the synthesis of cognition, memory, and emotion in primate prefrontal cortices. Brain Res. Bull. 52 319–330. 10.1016/S0361-9230(99)00245-2 [DOI] [PubMed] [Google Scholar]
- Barlow D. P. (2011). Genomic imprinting: a mammalian epigenetic discovery model. Annu. Rev. Genet. 45 379–403. 10.1146/annurev-genet-110410-132459 [DOI] [PubMed] [Google Scholar]
- Bar-Nur O., Caspi I., Benvenisty N. (2012). Molecular analysis of FMR1 reactivation in fragile-X induced pluripotent stem cells and their neuronal derivatives. J. Mol. Cell Biol. 4 180–183. 10.1093/jmcb/mjs007 [DOI] [PubMed] [Google Scholar]
- Barry G., Briggs J. A., Vanichkina D. P., Poth E. M., Beveridge N. J., Ratnu V. S., et al. (2014). The long non-coding RNA Gomafu is acutely regulated in response to neuronal activation and involved in schizophrenia-associated alternative splicing. Mol. Psychiatry 19 486–494. 10.1038/mp.2013.45 [DOI] [PubMed] [Google Scholar]
- Bartel D. P. (2004). MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116 281–297. 10.1016/S0092-8674(04)00045-5 [DOI] [PubMed] [Google Scholar]
- Battaglioli E., Andres M. E., Rose D. W., Chenoweth J. G., Rosenfeld M. G., Anderson M. E., et al. (2002). REST repression of neuronal genes requires components of the hSWI.SNF complex. J. Biol. Chem. 277 41038–41045. 10.1074/jbc.M205691200 [DOI] [PubMed] [Google Scholar]
- Baudry A., Mouillet-Richard S., Schneider B., Launay J. M., Kellermann O. (2010). miR-16 targets the serotonin transporter: a new facet for adaptive responses to antidepressants. Science 329 1537–1541. 10.1126/science.1193692 [DOI] [PubMed] [Google Scholar]
- Beard C., Li E., Jaenisch R. (1995). Loss of methylation activates Xist in somatic but not in embryonic cells. Genes Dev. 9 2325–2334. 10.1101/gad.9.19.2325 [DOI] [PubMed] [Google Scholar]
- Bell M. V., Hirst M. C., Nakahori Y., MacKinnon R. N., Roche A., Flint T. J., et al. (1991). Physical mapping across the fragile X: hypermethylation and clinical expression of the fragile X syndrome. Cell 64 861–866. 10.1016/0092-8674(91)90514-Y [DOI] [PubMed] [Google Scholar]
- Belmaker R. H., Agam G. (2008). Major depressive disorder. N. Engl. J. Med. 358 55–68. 10.1056/NEJMra073096 [DOI] [PubMed] [Google Scholar]
- Bernard D., Prasanth K. V., Tripathi V., Colasse S., Nakamura T., Xuan Z., et al. (2010). A long nuclear-retained non-coding RNA regulates synaptogenesis by modulating gene expression. EMBO J. 29 3082–3093. 10.1038/emboj.2010.199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein B. E., Mikkelsen T. S., Xie X., Kamal M., Huebert D. J., Cuff J., et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125 315–326. 10.1016/j.cell.2006.02.041 [DOI] [PubMed] [Google Scholar]
- Bestor T. H., Ingram V. M. (1983). Two DNA methyltransferases from murine erythroleukemia cells: purification, sequence specificity, and mode of interaction with DNA. Proc. Natl. Acad. Sci. U.S.A. 80 5559–5563. 10.1073/pnas.80.18.5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bestor T. H., Verdine G. L. (1994). DNA methyltransferases. Curr. Opin. Cell Biol. 6 380–389. 10.1016/0955-0674(94)90030-2 [DOI] [PubMed] [Google Scholar]
- Beveridge N. J., Gardiner E., Carroll A. P., Tooney P. A., Cairns M. J. (2010). Schizophrenia is associated with an increase in cortical microRNA biogenesis. Mol. Psychiatry 15 1176–1189. 10.1038/mp.2009.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhutani N., Burns D. M., Blau H. M. (2011). DNA demethylation dynamics. Cell 146 866–872. 10.1016/j.cell.2011.08.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. P. (1986). CpG-rich islands and the function of DNA methylation. Nature 321 209–213. 10.1038/321209a0 [DOI] [PubMed] [Google Scholar]
- Bocchio-Chiavetto L., Maffioletti E., Bettinsoli P., Giovannini C., Bignotti S., Tardito D., et al. (2013). Blood microRNA changes in depressed patients during antidepressant treatment. Eur. Neuropsychopharmacol. 23 602–611. 10.1016/j.euroneuro.2012.06.013 [DOI] [PubMed] [Google Scholar]
- Boeckers T. M., Kreutz M. R., Winter C., Zuschratter W., Smalla K. H., Sanmarti-Vila L., et al. (1999). Proline-rich synapse-associated protein-1/cortactin binding protein 1 (ProSAP1/CortBP1) is a PDZ-domain protein highly enriched in the postsynaptic density. J. Neurosci. 19 6506–6518. 10.1523/JNEUROSCI.19-15-06506.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsani G., Tonlorenzi R., Simmler M. C., Dandolo L., Arnaud D., Capra V., et al. (1991). Characterization of a murine gene expressed from the inactive X chromosome. Nature 351 325–329. 10.1038/351325a0 [DOI] [PubMed] [Google Scholar]
- Bourc’his D., Bestor T. H. (2004). Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature 431 96–99. 10.1038/nature02886 [DOI] [PubMed] [Google Scholar]
- Bourc’his D., Xu G. L., Lin C. S., Bollman B., Bestor T. H. (2001). Dnmt3L and the establishment of maternal genomic imprints. Science 294 2536–2539. 10.1126/science.1065848 [DOI] [PubMed] [Google Scholar]
- Brannan C. I., Dees E. C., Ingram R. S., Tilghman S. M. (1990). The product of the H19 gene may function as an RNA. Mol. Cell. Biol. 10 28–36. 10.1128/MCB.10.1.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockdorff N., Ashworth A., Kay G. F., Cooper P., Smith S., McCabe V. M., et al. (1991). Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature 351 329–331. 10.1038/351329a0 [DOI] [PubMed] [Google Scholar]
- Brown C. J., Hendrich B. D., Rupert J. L., Lafreniere R. G., Xing Y., Lawrence J., et al. (1992). The human XIST gene: analysis of a 17 kb inactive X-specific RNA that contains conserved repeats and is highly localized within the nucleus. Cell 71 527–542. 10.1016/0092-8674(92)90520-M [DOI] [PubMed] [Google Scholar]
- Bucan M., Abrahams B. S., Wang K., Glessner J. T., Herman E. I., Sonnenblick L. I., et al. (2009). Genome-wide analyses of exonic copy number variants in a family-based study point to novel autism susceptibility genes. PLoS Genet. 5:e1000536. 10.1371/journal.pgen.1000536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultman S., Gebuhr T., Yee D., La Mantia C., Nicholson J., Gilliam A., et al. (2000). A Brg1 null mutation in the mouse reveals functional differences among mammalian SWI/SNF complexes. Mol. Cell 6 1287–1295. 10.1016/S1097-2765(00)00127-1 [DOI] [PubMed] [Google Scholar]
- Burgold T., Spreafico F., De Santa F., Totaro M. G., Prosperini E., Natoli G., et al. (2008). The histone H3 lysine 27-specific demethylase Jmjd3 is required for neural commitment. PLoS One 3:e3034. 10.1371/journal.pone.0003034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon T. D. (1996). Abnormalities of brain structure and function in schizophrenia: implications for aetiology and pathophysiology. Ann. Med. 28 533–539. 10.3109/07853899608999117 [DOI] [PubMed] [Google Scholar]
- Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., et al. (2002). Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 298 1039–1043. 10.1126/science.1076997 [DOI] [PubMed] [Google Scholar]
- Cao X., Sudhof T. C. (2001). A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293 115–120. 10.1126/science.1058783 [DOI] [PubMed] [Google Scholar]
- Cassel S., Revel M. O., Kelche C., Zwiller J. (2004). Expression of the methyl-CpG-binding protein MeCP2 in rat brain. An ontogenetic study. Neurobiol. Dis. 15 206–211. 10.1016/j.nbd.2003.10.011 [DOI] [PubMed] [Google Scholar]
- Caudy A. A., Myers M., Hannon G. J., Hammond S. M. (2002). Fragile X-related protein and VIG associate with the RNA interference machinery. Genes Dev. 16 2491–2496. 10.1101/gad.1025202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cech T. R., Steitz J. A. (2014). The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157 77–94. 10.1016/j.cell.2014.03.008 [DOI] [PubMed] [Google Scholar]
- Cedar H., Bergman Y. (2009). Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 10 295–304. 10.1038/nrg2540 [DOI] [PubMed] [Google Scholar]
- Chen L., Chen K., Lavery L. A., Baker S. A., Shaw C. A., Li W., et al. (2015). MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 112 5509–5514. 10.1073/pnas.1505909112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W. G., Chang Q., Lin Y., Meissner A., West A. E., Griffith E. C., et al. (2003). Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science 302 885–889. 10.1126/science.1086446 [DOI] [PubMed] [Google Scholar]
- Chen Y., Sharma R. P., Costa R. H., Costa E., Grayson D. R. (2002). On the epigenetic regulation of the human reelin promoter. Nucleic Acids Res. 30 2930–2939. 10.1093/nar/gkf401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. F., Paquette A. J., Anderson D. J. (1998). NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 20 136–142. 10.1038/2431 [DOI] [PubMed] [Google Scholar]
- Chiurazzi P., Pomponi M. G., Willemsen R., Oostra B. A., Neri G. (1998). In vitro reactivation of the FMR1 gene involved in fragile X syndrome. Hum. Mol. Genet. 7 109–113. 10.1093/hmg/7.1.109 [DOI] [PubMed] [Google Scholar]
- Chong J. A., Tapia-Ramirez J., Kim S., Toledo-Aral J. J., Zheng Y., Boutros M. C., et al. (1995). REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 80 949–957. 10.1016/0092-8674(95)90298-8 [DOI] [PubMed] [Google Scholar]
- Chu T. T., Liu Y. (2010). An integrated genomic analysis of gene-function correlation on schizophrenia susceptibility genes. J. Hum. Genet. 55 285–292. 10.1038/jhg.2010.24 [DOI] [PubMed] [Google Scholar]
- Chuang L. S., Ian H. I., Koh T. W., Ng H. H., Xu G., Li B. F. (1997). Human DNA-(cytosine-5) methyltransferase-PCNA complex as a target for p21WAF1. Science 277 1996–2000. 10.1126/science.277.5334.1996 [DOI] [PubMed] [Google Scholar]
- Civiero L., Cirnaru M. D., Beilina A., Rodella U., Russo I., Belluzzi E., et al. (2015). Leucine-rich repeat kinase 2 interacts with p21-activated kinase 6 to control neurite complexity in mammalian brain. J. Neurochem. 135 1242–1256. 10.1111/jnc.13369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemson C. M., Hutchinson J. N., Sara S. A., Ensminger A. W., Fox A. H., Chess A., et al. (2009). An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell 33 717–726. 10.1016/j.molcel.2009.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C., Jeon O. H., Hyder K., Chenchik A., Khimani A. H., Narayanan V., et al. (2001). Gene expression profiling in postmortem Rett Syndrome brain: differential gene expression and patient classification. Neurobiol. Dis. 8 847–865. 10.1006/nbdi.2001.0428 [DOI] [PubMed] [Google Scholar]
- Cortazar D., Kunz C., Selfridge J., Lettieri T., Saito Y., MacDougall E., et al. (2011). Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 470 419–423. 10.1038/nature09672 [DOI] [PubMed] [Google Scholar]
- Cortellino S., Xu J., Sannai M., Moore R., Caretti E., Cigliano A., et al. (2011). Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146 67–79. 10.1016/j.cell.2011.06.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa Y., Ding J., Theunissen T. W., Faiola F., Hore T. A., Shliaha P. V., et al. (2013). NANOG-dependent function of TET1 and TET2 in establishment of pluripotency. Nature 495 370–374. 10.1038/nature11925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coufal N. G., Garcia-Perez J. L., Peng G. E., Yeo G. W., Mu Y., Lovci M. T., et al. (2009). L1 retrotransposition in human neural progenitor cells. Nature 460 1127–1131. 10.1038/nature08248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington H. E., III, Maze I., LaPlant Q. C., Vialou V. F., Ohnishi Y. N., Berton O., et al. (2009). Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 29 11451–11460. 10.1523/JNEUROSCI.1758-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington H. E., III, Vialou V. F., LaPlant Q., Ohnishi Y. N., Nestler E. J. (2011). Hippocampal-dependent antidepressant-like activity of histone deacetylase inhibition. Neurosci. Lett. 493 122–126. 10.1016/j.neulet.2011.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daitoku H., Hatta M., Matsuzaki H., Aratani S., Ohshima T., Miyagishi M., et al. (2004). Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc. Natl. Acad. Sci. U.S.A. 101 10042–10047. 10.1073/pnas.0400593101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daskalakis N. P., Yehuda R. (2014). Site-specific methylation changes in the glucocorticoid receptor exon 1F promoter in relation to life adversity: systematic review of contributing factors. Front. Neurosci. 8:369. 10.3389/fnins.2014.00369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Napoles M., Mermoud J. E., Wakao R., Tang Y. A., Endoh M., Appanah R., et al. (2004). Polycomb group proteins Ring1A/B link ubiquitylation of histone H2A to heritable gene silencing and X inactivation. Dev. Cell 7 663–676. 10.1016/j.devcel.2004.10.005 [DOI] [PubMed] [Google Scholar]
- Delgado S., Gomez M., Bird A., Antequera F. (1998). Initiation of DNA replication at CpG islands in mammalian chromosomes. EMBO J. 17 2426–2435. 10.1093/emboj/17.8.2426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLisi L. E., Shaw S. H., Crow T. J., Shields G., Smith A. B., Larach V. W., et al. (2002). A genome-wide scan for linkage to chromosomal regions in 382 sibling pairs with schizophrenia or schizoaffective disorder. Am. J. Psychiatry 159 803–812. 10.1176/appi.ajp.159.5.803 [DOI] [PubMed] [Google Scholar]
- Deplus R., Delatte B., Schwinn M. K., Defrance M., Mendez J., Murphy N., et al. (2013). TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 32 645–655. 10.1038/emboj.2012.357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devys D., Lutz Y., Rouyer N., Bellocq J. P., Mandel J. L. (1993). The FMR-1 protein is cytoplasmic, most abundant in neurons and appears normal in carriers of a fragile X premutation. Nat. Genet. 4 335–340. 10.1038/ng0893-335 [DOI] [PubMed] [Google Scholar]
- Dhayalan A., Rajavelu A., Rathert P., Tamas R., Jurkowska R. Z., Ragozin S., et al. (2010). The Dnmt3a PWWP domain reads histone 3 lysine 36 trimethylation and guides DNA methylation. J. Biol. Chem. 285 26114–26120. 10.1074/jbc.M109.089433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meglio T., Kratochwil C. F., Vilain N., Loche A., Vitobello A., Yonehara K., et al. (2013). Ezh2 orchestrates topographic migration and connectivity of mouse precerebellar neurons. Science 339 204–207. 10.1126/science.1229326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y., Weed S. A., Xiong W. C., Marshall T. D., Parsons J. T. (1998). Identification of a novel cortactin SH3 domain-binding protein and its localization to growth cones of cultured neurons. Mol. Cell. Biol. 18 5838–5851. 10.1128/MCB.18.10.5838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand C. M., Betancur C., Boeckers T. M., Bockmann J., Chaste P., Fauchereau F., et al. (2007). Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 39 25–27. 10.1038/ng1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhart D. E., Malter H. E., Feng Y., Warren S. T. (1996). The fragile X mental retardation protein is a ribonucleoprotein containing both nuclear localization and nuclear export signals. Hum. Mol. Genet. 5 1083–1091. 10.1093/hmg/5.8.1083 [DOI] [PubMed] [Google Scholar]
- Ebert D. H., Gabel H. W., Robinson N. D., Kastan N. R., Hu L. S., Cohen S., et al. (2013). Activity-dependent phosphorylation of MeCP2 threonine 308 regulates interaction with NCoR. Nature 499 341–345. 10.1038/nature12348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckschlager T., Plch J., Stiborova M., Hrabeta J. (2017). Histone deacetylase inhibitors as anticancer drugs. Int. J. Mol. Sci. 18:E1414. 10.3390/ijms18071414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efstathopoulos P., Andersson F., Melas P. A., Yang L. L., Villaescusa J. C., Ruegg J., et al. (2018). NR3C1 hypermethylation in depressed and bullied adolescents. Transl. Psychiatry 8:121. 10.1038/s41398-018-0169-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hajj N., Dittrich M., Haaf T. (2017). Epigenetic dysregulation of protocadherins in human disease. Semin. Cell Dev. Biol. 69 172–182. 10.1016/j.semcdb.2017.07.007 [DOI] [PubMed] [Google Scholar]
- Fan G., Beard C., Chen R. Z., Csankovszki G., Sun Y., Siniaia M., et al. (2001). DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J. Neurosci. 21 788–797. 10.1523/JNEUROSCI.21-03-00788.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y., Qiu Q., Zhang S., Sun L., Li G., Xiao S., et al. (2018). Changes in miRNA-132 and miR-124 levels in non-treated and citalopram-treated patients with depression. J. Affect. Disord. 227 745–751. 10.1016/j.jad.2017.11.090 [DOI] [PubMed] [Google Scholar]
- Farrell C., Doolin K., O’ Leary N., Jairaj C., Roddy D., Tozzi L., et al. (2018). DNA methylation differences at the glucocorticoid receptor gene in depression are related to functional alterations in hypothalamic-pituitary-adrenal axis activity and to early life emotional abuse. Psychiatry Res. 265 341–348. 10.1016/j.psychres.2018.04.064 [DOI] [PubMed] [Google Scholar]
- Feil R., Fraga M. F. (2012). Epigenetics and the environment: emerging patterns and implications. Nat. Rev. Genet. 13 97–109. 10.1038/nrg3142 [DOI] [PubMed] [Google Scholar]
- Feng J., Chang H., Li E., Fan G. (2005). Dynamic expression of de novo DNA methyltransferases Dnmt3a and Dnmt3b in the central nervous system. J. Neurosci. Res. 79 734–746. 10.1002/jnr.20404 [DOI] [PubMed] [Google Scholar]
- Feng J., Zhou Y., Campbell S. L., Le T., Li E., Sweatt J. D., et al. (2010). Dnmt1 and Dnmt3a maintain DNA methylation and regulate synaptic function in adult forebrain neurons. Nat. Neurosci. 13 423–430. 10.1038/nn.2514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Gutekunst C. A., Eberhart D. E., Yi H., Warren S. T., Hersch S. M. (1997). Fragile X mental retardation protein: nucleocytoplasmic shuttling and association with somatodendritic ribosomes. J. Neurosci. 17 1539–1547. 10.1523/JNEUROSCI.17-05-01539.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiori L. M., Lopez J. P., Richard-Devantoy S., Berlim M., Chachamovich E., Jollant F., et al. (2017). Investigation of miR-1202, miR-135a, and miR-16 in Major depressive disorder and antidepressant response. Int. J. Neuropsychopharmacol. 20 619–623. 10.1093/ijnp/pyx034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A., Sananbenesi F., Wang X., Dobbin M., Tsai L. H. (2007). Recovery of learning and memory is associated with chromatin remodelling. Nature 447 178–182. 10.1038/nature05772 [DOI] [PubMed] [Google Scholar]
- Fischle W., Wang Y., Jacobs S. A., Kim Y., Allis C. D., Khorasanizadeh S. (2003). Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev. 17 1870–1881. 10.1101/gad.1110503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A., DeCamillis M., Zink D., Cheng N., Brock H. W., Paro R. (1992). Polycomb and polyhomeotic are constituents of a multimeric protein complex in chromatin of Drosophila melanogaster. EMBO J. 11 2941–2950. 10.1002/j.1460-2075.1992.tb05364.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauer C., Rottach A., Meilinger D., Bultmann S., Fellinger K., Hasenoder S., et al. (2011). Different binding properties and function of CXXC zinc finger domains in Dnmt1 and Tet1. PLoS One 6:e16627. 10.1371/journal.pone.0016627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchikami M., Morinobu S., Segawa M., Okamoto Y., Yamawaki S., Ozaki N., et al. (2011). DNA methylation profiles of the brain-derived neurotrophic factor (BDNF) gene as a potent diagnostic biomarker in major depression. PLoS One 6:e23881. 10.1371/journal.pone.0023881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchikami M., Yamamoto S., Morinobu S., Okada S., Yamawaki Y., Yamawaki S. (2016). The potential use of histone deacetylase inhibitors in the treatment of depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 64 320–324. 10.1016/j.pnpbp.2015.03.010 [DOI] [PubMed] [Google Scholar]
- Fujita N., Watanabe S., Ichimura T., Tsuruzoe S., Shinkai Y., Tachibana M., et al. (2003). Methyl-CpG binding domain 1 (MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for DNA methylation-based transcriptional repression. J. Biol. Chem. 278 24132–24138. 10.1074/jbc.M302283200 [DOI] [PubMed] [Google Scholar]
- Fuks F., Hurd P. J., Wolf D., Nan X., Bird A. P., Kouzarides T. (2003). The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J. Biol. Chem. 278 4035–4040. 10.1074/jbc.M210256200 [DOI] [PubMed] [Google Scholar]
- Gabel H. W., Kinde B., Stroud H., Gilbert C. S., Harmin D. A., Kastan N. R., et al. (2015). Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature 522 89–93. 10.1038/nature14319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z., Lee P., Stafford J. M., von Schimmelmann M., Schaefer A., Reinberg D. (2014). An AUTS2-Polycomb complex activates gene expression in the CNS. Nature 516 349–354. 10.1038/nature13921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z., Zhang J., Bonasio R., Strino F., Sawai A., Parisi F., et al. (2012). PCGF homologs, CBX proteins, and RYBP define functionally distinct PRC1 family complexes. Mol. Cell 45 344–356. 10.1016/j.molcel.2012.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garafola C. S., Henn F. A. (2014). A change in hippocampal protocadherin gamma expression in a learned helpless rat. Brain Res. 1593 55–64. 10.1016/j.brainres.2014.08.071 [DOI] [PubMed] [Google Scholar]
- Garrett A. M., Schreiner D., Lobas M. A., Weiner J. A. (2012). gamma-protocadherins control cortical dendrite arborization by regulating the activity of a FAK/PKC/MARCKS signaling pathway. Neuron 74 269–276. 10.1016/j.neuron.2012.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J., Spiegelman D., Piton A., Lafreniere R. G., Laurent S., St-Onge J., et al. (2009). Novel de novo SHANK3 mutation in autistic patients. Am. J. Med. Genet. B Neuropsychiatr. Genet. 150B 421–424. 10.1002/ajmg.b.30822 [DOI] [PubMed] [Google Scholar]
- Gayon J. (2016). From Mendel to epigenetics: history of genetics. C. R. Biol. 339 225–230. 10.1016/j.crvi.2016.05.009 [DOI] [PubMed] [Google Scholar]
- Gemelli T., Berton O., Nelson E. D., Perrotti L. I., Jaenisch R., Monteggia L. M. (2006). Postnatal loss of methyl-CpG binding protein 2 in the forebrain is sufficient to mediate behavioral aspects of Rett syndrome in mice. Biol. Psychiatry 59 468–476. 10.1016/j.biopsych.2005.07.025 [DOI] [PubMed] [Google Scholar]
- Gianfrancesco O., Warburton A., Collier D. A., Bubb V. J., Quinn J. P. (2017). Novel brain expressed RNA identified at the MIR137 schizophrenia-associated locus. Schizophr. Res. 184 109–115. 10.1016/j.schres.2016.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globisch D., Munzel M., Muller M., Michalakis S., Wagner M., Koch S., et al. (2010). Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 5:e15367. 10.1371/journal.pone.0015367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gold W. A., Krishnarajy R., Ellaway C., Christodoulou J. (2018). Rett syndrome: a genetic update and clinical review focusing on comorbidities. ACS Chem. Neurosci. 9 167–176. 10.1021/acschemneuro.7b00346 [DOI] [PubMed] [Google Scholar]
- Goldberg A. D., Allis C. D., Bernstein E. (2007). Epigenetics: a landscape takes shape. Cell 128 635–638. 10.1016/j.cell.2007.02.006 [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic P. S. (1996). The prefrontal landscape: implications of functional architecture for understanding human mentation and the central executive. Philos. Trans. R. Soc. Lond. B Biol. Sci. 351 1445–1453. 10.1098/rstb.1996.0129 [DOI] [PubMed] [Google Scholar]
- Goll M. G., Kirpekar F., Maggert K. A., Yoder J. A., Hsieh C. L., Zhang X., et al. (2006). Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science 311 395–398. 10.1126/science.1120976 [DOI] [PubMed] [Google Scholar]
- Goto K., Numata M., Komura J. I., Ono T., Bestor T. H., Kondo H. (1994). Expression of DNA methyltransferase gene in mature and immature neurons as well as proliferating cells in mice. Differentiation 56 39–44. 10.1046/j.1432-0436.1994.56120039.x [DOI] [PubMed] [Google Scholar]
- Gotz M., Stoykova A., Gruss P. (1998). Pax6 controls radial glia differentiation in the cerebral cortex. Neuron 21 1031–1044. 10.1016/S0896-6273(00)80621-2 [DOI] [PubMed] [Google Scholar]
- Gray S. G., Ekstrom T. J. (2001). The human histone deacetylase family. Exp. Cell Res. 262 75–83. 10.1006/excr.2000.5080 [DOI] [PubMed] [Google Scholar]
- Gregorio S. P., Sallet P. C., Do K. A., Lin E., Gattaz W. F., Dias-Neto E. (2009). Polymorphisms in genes involved in neurodevelopment may be associated with altered brain morphology in schizophrenia: preliminary evidence. Psychiatry Res. 165 1–9. 10.1016/j.psychres.2007.08.011 [DOI] [PubMed] [Google Scholar]
- Grimes J. A., Nielsen S. J., Battaglioli E., Miska E. A., Speh J. C., Berry D. L., et al. (2000). The co-repressor mSin3A is a functional component of the REST-CoREST repressor complex. J. Biol. Chem. 275 9461–9467. 10.1074/jbc.275.13.9461 [DOI] [PubMed] [Google Scholar]
- Grimson A., Farh K. K., Johnston W. K., Garrett-Engele P., Lim L. P., Bartel D. P. (2007). MicroRNA targeting specificity in mammals: determinants beyond seed pairing. Mol. Cell 27 91–105. 10.1016/j.molcel.2007.06.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu T. P., Guo F., Yang H., Wu H. P., Xu G. F., Liu W., et al. (2011). The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477 606–610. 10.1038/nature10443 [DOI] [PubMed] [Google Scholar]
- Guan F., Zhang B., Yan T., Li L., Liu F., Li T., et al. (2014). MIR137 gene and target gene CACNA1C of miR-137 contribute to schizophrenia susceptibility in Han Chinese. Schizophr. Res. 152 97–104. 10.1016/j.schres.2013.11.004 [DOI] [PubMed] [Google Scholar]
- Guo J. U., Ma D. K., Mo H., Ball M. P., Jang M. H., Bonaguidi M. A., et al. (2011a). Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 14 1345–1351. 10.1038/nn.2900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. U., Su Y., Shin J. H., Shin J., Li H., Xie B., et al. (2014). Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 17 215–222. 10.1038/nn.3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J. U., Su Y., Zhong C., Ming G. L., Song H. (2011b). Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145 423–434. 10.1016/j.cell.2011.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner H., an der Heiden W., Behrens S., Gattaz W. F., Hambrecht M., Loffler W., et al. (1998). Causes and consequences of the gender difference in age at onset of schizophrenia. Schizophr. Bull. 24 99–113. 10.1093/oxfordjournals.schbul.a033317 [DOI] [PubMed] [Google Scholar]
- Hagberg B., Aicardi J., Dias K., Ramos O. (1983). A progressive syndrome of autism, dementia, ataxia, and loss of purposeful hand use in girls: Rett’s syndrome: report of 35 cases. Ann. Neurol. 14 471–479. 10.1002/ana.410140412 [DOI] [PubMed] [Google Scholar]
- Hagberg B., Goutières F., Hanefeld F., Rett A., Wilson J. (1985). Rett syndrome: criteria for inclusion and exclusion. Brain. Dev. 7 372–373. [DOI] [PubMed] [Google Scholar]
- Hahn M. A., Qiu R., Wu X., Li A. X., Zhang H., Wang J., et al. (2013). Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep. 3 291–300. 10.1016/j.celrep.2013.01.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamshere M. L., Walters J. T., Smith R., Richards A. L., Green E., Grozeva D., et al. (2013). Genome-wide significant associations in schizophrenia to ITIH3/4, CACNA1C and SDCCAG8, and extensive replication of associations reported by the Schizophrenia PGC. Mol. Psychiatry 18 708–712. 10.1038/mp.2012.67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L., Lin I. G., Hsieh C. L. (2001). Protein binding protects sites on stable episomes and in the chromosome from de novo methylation. Mol. Cell. Biol. 21 3416–3424. 10.1128/MCB.21.10.3416-3424.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa V., Saha T., Usdin K. (2003). The fragile X syndrome repeats form RNA hairpins that do not activate the interferon-inducible protein kinase, PKR, but are cut by Dicer. Nucleic Acids Res. 31 6243–6248. 10.1093/nar/gkg818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata K., Okano M., Lei H., Li E. (2002). Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 129 1983–1993. [DOI] [PubMed] [Google Scholar]
- Hatta M., Liu F., Cirillo L. A. (2009). Acetylation curtails nucleosome binding, not stable nucleosome remodeling, by FoxO1. Biochem. Biophys. Res. Commun. 379 1005–1008. 10.1016/j.bbrc.2009.01.014 [DOI] [PubMed] [Google Scholar]
- He S., Liu X., Jiang K., Peng D., Hong W., Fang Y., et al. (2016). Alterations of microRNA-124 expression in peripheral blood mononuclear cells in pre- and post-treatment patients with major depressive disorder. J. Psychiatr. Res. 78 65–71. 10.1016/j.jpsychires.2016.03.015 [DOI] [PubMed] [Google Scholar]
- He Y. F., Li B. Z., Li Z., Liu P., Wang Y., Tang Q., et al. (2011). Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science 333 1303–1307. 10.1126/science.1210944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich B., Bird A. (1998). Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol. Cell. Biol. 18 6538–6547. 10.1128/MCB.18.11.6538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinton V. J., Brown W. T., Wisniewski K., Rudelli R. D. (1991). Analysis of neocortex in three males with the fragile X syndrome. Am. J. Med. Genet. 41 289–294. 10.1002/ajmg.1320410306 [DOI] [PubMed] [Google Scholar]
- Hirabayashi Y., Suzki N., Tsuboi M., Endo T. A., Toyoda T., Shinga J., et al. (2009). Polycomb limits the neurogenic competence of neural precursor cells to promote astrogenic fate transition. Neuron 63 600–613. 10.1016/j.neuron.2009.08.021 [DOI] [PubMed] [Google Scholar]
- Hirayama T., Yagi T. (2017). Regulation of clustered protocadherin genes in individual neurons. Semin. Cell Dev. Biol. 69 122–130. 10.1016/j.semcdb.2017.05.026 [DOI] [PubMed] [Google Scholar]
- Ho L., Miller E. L., Ronan J. L., Ho W. Q., Jothi R., Crabtree G. R. (2011). esBAF facilitates pluripotency by conditioning the genome for LIF/STAT3 signalling and by regulating polycomb function. Nat. Cell Biol. 13 903–913. 10.1038/ncb2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho L., Ronan J. L., Wu J., Staahl B. T., Chen L., Kuo A., et al. (2009). An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. U.S.A. 106 5181–5186. 10.1073/pnas.0812889106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horike S., Cai S., Miyano M., Cheng J. F., Kohwi-Shigematsu T. (2005). Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat. Genet. 37 31–40. 10.1038/ng1491 [DOI] [PubMed] [Google Scholar]
- Howell C. Y., Bestor T. H., Ding F., Latham K. E., Mertineit C., Trasler J. M., et al. (2001). Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 104 829–838. 10.1016/S0092-8674(01)00280-X [DOI] [PubMed] [Google Scholar]
- Howlett S. K., Reik W. (1991). Methylation levels of maternal and paternal genomes during preimplantation development. Development 113 119–127. [DOI] [PubMed] [Google Scholar]
- Hutchinson A. N., Deng J. V., Cohen S., West A. E. (2012). Phosphorylation of MeCP2 at Ser421 contributes to chronic antidepressant action. J. Neurosci. 32 14355–14363. 10.1523/JNEUROSCI.2156-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson J. N., Ensminger A. W., Clemson C. M., Lynch C. R., Lawrence J. B., Chess A. (2007). A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics 8:39. 10.1186/1471-2164-8-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutsler J. J., Zhang H. (2010). Increased dendritic spine densities on cortical projection neurons in autism spectrum disorders. Brain Res. 1309 83–94. 10.1016/j.brainres.2009.09.120 [DOI] [PubMed] [Google Scholar]
- Hyun K., Jeon J., Park K., Kim J. (2017). Writing, erasing and reading histone lysine methylations. Exp. Mol. Med. 49 e324. 10.1038/emm.2017.11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth R. S., Bird A. P. (2009). CpG islands–’a rough guide’. FEBS Lett. 583 1713–1720. 10.1016/j.febslet.2009.04.012 [DOI] [PubMed] [Google Scholar]
- Impagnatiello F., Guidotti A. R., Pesold C., Dwivedi Y., Caruncho H., Pisu M. G., et al. (1998). A decrease of reelin expression as a putative vulnerability factor in schizophrenia. Proc. Natl. Acad. Sci. U.S.A. 95 15718–15723. 10.1073/pnas.95.26.15718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A., Fujimoto D. (1970). Histone deacetylase from calf thymus. Biochim. Biophys. Acta 220 307–316. 10.1016/0005-2744(70)90015-X [DOI] [PubMed] [Google Scholar]
- International Schizophrenia C., Purcell S. M., Wray N. R., Stone J. L., Visscher P. M., O’Donovan M. C., et al. (2009). Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 460 748–752. 10.1038/nature08185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K., Jin S. G., Pfeifer G. P., Szabo P. E. (2011). Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci. U.S.A. 108 3642–3647. 10.1073/pnas.1014033108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka A., Siomi M. C., Siomi H. (2002). A Drosophila fragile X protein interacts with components of RNAi and ribosomal proteins. Genes Dev. 16 2497–2508. 10.1101/gad.1022002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S., D’Alessio A. C., Taranova O. V., Hong K., Sowers L. C., Zhang Y. (2010). Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466 1129–1133. 10.1038/nature09303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S., Shen L., Dai Q., Wu S. C., Collins L. B., Swenberg J. A., et al. (2011). Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333 1300–1303. 10.1126/science.1210597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itskovitz-Eldor J., Schuldiner M., Karsenti D., Eden A., Yanuka O., Amit M., et al. (2000). Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 6 88–95. 10.1007/BF03401776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer L. M., Tahiliani M., Rao A., Aravind L. (2009). Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 8 1698–1710. 10.4161/cc.8.11.8580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer M. K., Niknafs Y. S., Malik R., Singhal U., Sahu A., Hosono Y., et al. (2015). The landscape of long noncoding RNAs in the human transcriptome. Nat. Genet. 47 199–208. 10.1038/ng.3192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablensky A., Sartorius N., Ernberg G., Anker M., Korten A., Cooper J. E., et al. (1992). Schizophrenia: manifestations, incidence and course in different cultures. A World Health Organization ten-country study. Psychol. Med. Monogr. Suppl. 20 1–97. 10.1017/S0264180100000904 [DOI] [PubMed] [Google Scholar]
- Jiang H., Shukla A., Wang X., Chen W. Y., Bernstein B. E., Roeder R. G. (2011). Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell 144 513–525. 10.1016/j.cell.2011.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P., Alisch R. S., Warren S. T. (2004a). RNA and microRNAs in fragile X mental retardation. Nat. Cell Biol. 6 1048–1053. 10.1038/ncb1104-1048 [DOI] [PubMed] [Google Scholar]
- Jin P., Zarnescu D. C., Ceman S., Nakamoto M., Mowrey J., Jongens T. A., et al. (2004b). Biochemical and genetic interaction between the fragile X mental retardation protein and the microRNA pathway. Nat. Neurosci. 7 113–117. 10.1038/nn1174 [DOI] [PubMed] [Google Scholar]
- Jobe E. M., McQuate A. L., Zhao X. (2012). Crosstalk among epigenetic pathways regulates neurogenesis. Front. Neurosci. 6:59. 10.3389/fnins.2012.00059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P. A. (2012). Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 13 484–492. 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- Jones P. L., Veenstra G. J., Wade P. A., Vermaak D., Kass S. U., Landsberger N., et al. (1998). Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat. Genet. 19 187–191. 10.1038/561 [DOI] [PubMed] [Google Scholar]
- Jordan C., Li H. H., Kwan H. C., Francke U. (2007). Cerebellar gene expression profiles of mouse models for Rett syndrome reveal novel MeCP2 targets. BMC Med. Genet. 8:36. 10.1186/1471-2350-8-36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung B. P., Zhang G., Nitsch R., Trogadis J., Nag S., Eubanks J. H. (2003). Differential expression of methyl CpG-binding domain containing factor MBD3 in the developing and adult rat brain. J. Neurobiol. 55 220–232. 10.1002/neu.10199 [DOI] [PubMed] [Google Scholar]
- Kaeser M. D., Aslanian A., Dong M. Q., Yates J. R., III, Emerson B. M. (2008). BRD7, a novel PBAF-specific SWI/SNF subunit, is required for target gene activation and repression in embryonic stem cells. J. Biol. Chem. 283 32254–32263. 10.1074/jbc.M806061200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kafri T., Ariel M., Brandeis M., Shemer R., Urven L., McCarrey J., et al. (1992). Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev. 6 705–714. 10.1101/gad.6.5.705 [DOI] [PubMed] [Google Scholar]
- Kaneda M., Okano M., Hata K., Sado T., Tsujimoto N., Li E., et al. (2004). Essential role for de novo DNA methyltransferase Dnmt3a in paternal and maternal imprinting. Nature 429 900–903. 10.1038/nature02633 [DOI] [PubMed] [Google Scholar]
- Kapranov P., Cheng J., Dike S., Nix D. A., Duttagupta R., Willingham A. T., et al. (2007). RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 316 1484–1488. 10.1126/science.1138341 [DOI] [PubMed] [Google Scholar]
- Keeler A. B., Molumby M. J., Weiner J. A. (2015). Protocadherins branch out: Multiple roles in dendrite development. Cell Adh. Migr. 9 214–226. 10.1080/19336918.2014.1000069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellinger M. W., Song C. X., Chong J., Lu X. Y., He C., Wang D. (2012). 5-formylcytosine and 5-carboxylcytosine reduce the rate and substrate specificity of RNA polymerase II transcription. Nat. Struct. Mol. Biol. 19 831–833. 10.1038/nsmb.2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler R. C., Berglund P., Demler O., Jin R., Koretz D., Merikangas K. R., et al. (2003). The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R). JAMA 289 3095–3105. 10.1001/jama.289.23.3095 [DOI] [PubMed] [Google Scholar]
- Kim J., Krichevsky A., Grad Y., Hayes G. D., Kosik K. S., Church G. M., et al. (2004). Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc. Natl. Acad. Sci. U.S.A. 101 360–365. 10.1073/pnas.2333854100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleaveland B., Shi C. Y., Stefano J., Bartel D. P. (2018). A network of noncoding regulatory RNAs acts in the mammalian brain. Cell 174 350.e–362.e. 10.1016/j.cell.2018.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga M., Ishiguro H., Yazaki S., Horiuchi Y., Arai M., Niizato K., et al. (2009). Involvement of SMARCA2/BRM in the SWI/SNF chromatin-remodeling complex in schizophrenia. Hum. Mol. Genet. 18 2483–2494. 10.1093/hmg/ddp166 [DOI] [PubMed] [Google Scholar]
- Kohmura N., Senzaki K., Hamada S., Kai N., Yasuda R., Watanabe M., et al. (1998). Diversity revealed by a novel family of cadherins expressed in neurons at a synaptic complex. Neuron 20 1137–1151. 10.1016/S0896-6273(00)80495-X [DOI] [PubMed] [Google Scholar]
- Kornberg R. D. (1974). Chromatin structure: a repeating unit of histones and DNA. Science 184 868–871. 10.1126/science.184.4139.868 [DOI] [PubMed] [Google Scholar]
- Kouzarides T. (2007). Chromatin modifications and their function. Cell 128 693–705. 10.1016/j.cell.2007.02.005 [DOI] [PubMed] [Google Scholar]
- Kriaucionis S., Heintz N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324 929–930. 10.1126/science.1169786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krichevsky A. M., King K. S., Donahue C. P., Khrapko K., Kosik K. S. (2003). A microRNA array reveals extensive regulation of microRNAs during brain development. RNA 9 1274–1281. 10.1261/rna.5980303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krichevsky A. M., Sonntag K. C., Isacson O., Kosik K. S. (2006). Specific microRNAs modulate embryonic stem cell-derived neurogenesis. Stem Cells 24 857–864. 10.1634/stemcells.2005-0441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R., Sanawar R., Li X., Li F. (2017). Structure, biochemistry, and biology of PAK kinases. Gene 605 20–31. 10.1016/j.gene.2016.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmichev A., Nishioka K., Erdjument-Bromage H., Tempst P., Reinberg D. (2002). Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 16 2893–2905. 10.1101/gad.1035902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon E., Wang W., Tsai L. H. (2013). Validation of schizophrenia-associated genes CSMD1, C10orf26, CACNA1C and TCF4 as miR-137 targets. Mol. Psychiatry 18 11–12. 10.1038/mp.2011.170 [DOI] [PubMed] [Google Scholar]
- Ladd-Acosta C., Pevsner J., Sabunciyan S., Yolken R. H., Webster M. J., Dinkins T., et al. (2007). DNA methylation signatures within the human brain. Am. J. Hum. Genet. 81 1304–1315. 10.1086/524110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggerbauer B., Ostareck D., Keidel E. M., Ostareck-Lederer A., Fischer U. (2001). Evidence that fragile X mental retardation protein is a negative regulator of translation. Hum. Mol. Genet. 10 329–338. 10.1093/hmg/10.4.329 [DOI] [PubMed] [Google Scholar]
- Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., Tuschl T. (2002). Identification of tissue-specific microRNAs from mouse. Curr. Biol. 12 735–739. 10.1016/S0960-9822(02)00809-6 [DOI] [PubMed] [Google Scholar]
- Lamond A. I., Spector D. L. (2003). Nuclear speckles: a model for nuclear organelles. Nat. Rev. Mol. Cell Biol. 4 605–612. 10.1038/nrm1172 [DOI] [PubMed] [Google Scholar]
- Lancaster M. A., Renner M., Martin C. A., Wenzel D., Bicknell L. S., Hurles M. E., et al. (2013). Cerebral organoids model human brain development and microcephaly. Nature 501 373–379. 10.1038/nature12517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent L., Wong E., Li G., Huynh T., Tsirigos A., Ong C. T., et al. (2010). Dynamic changes in the human methylome during differentiation. Genome Res. 20 320–331. 10.1101/gr.101907.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre J. L., Kostadinov D., Chen W. V., Maniatis T., Sanes J. R. (2012). Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature 488 517–521. 10.1038/nature11305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J., Wu J. I., Ranish J. A., Wan M., Winslow M. M., Staahl B. T., et al. (2007). An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron 55 201–215. 10.1016/j.neuron.2007.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lett T. A., Chakravarty M. M., Felsky D., Brandl E. J., Tiwari A. K., Goncalves V. F., et al. (2013). The genome-wide supported microRNA-137 variant predicts phenotypic heterogeneity within schizophrenia. Mol. Psychiatry 18 443–450. 10.1038/mp.2013.17 [DOI] [PubMed] [Google Scholar]
- Levenson J. M., Roth T. L., Lubin F. D., Miller C. A., Huang I. C., Desai P., et al. (2006). Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J. Biol. Chem. 281 15763–15773. 10.1074/jbc.M511767200 [DOI] [PubMed] [Google Scholar]
- Lewis B. P., Burge C. B., Bartel D. P. (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120 15–20. 10.1016/j.cell.2004.12.035 [DOI] [PubMed] [Google Scholar]
- Li E., Beard C., Jaenisch R. (1993). Role for DNA methylation in genomic imprinting. Nature 366 362–365. 10.1038/366362a0 [DOI] [PubMed] [Google Scholar]
- Li E., Bestor T. H., Jaenisch R. (1992). Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 69 915–926. 10.1016/0092-8674(92)90611-F [DOI] [PubMed] [Google Scholar]
- Li X., Wei W., Zhao Q. Y., Widagdo J., Baker-Andresen D., Flavell C. R., et al. (2014). Neocortical Tet3-mediated accumulation of 5-hydroxymethylcytosine promotes rapid behavioral adaptation. Proc. Natl. Acad. Sci. U.S.A. 111 7120–7125. 10.1073/pnas.1318906111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z., Zhang Y., Ku L., Wilkinson K. D., Warren S. T., Feng Y. (2001). The fragile X mental retardation protein inhibits translation via interacting with mRNA. Nucleic Acids Res. 29 2276–2283. 10.1093/nar/29.11.2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieb K., Dreimuller N., Wagner S., Schlicht K., Falter T., Neyazi A., et al. (2018). BDNF plasma levels and BDNF exon IV promoter methylation as predictors for antidepressant treatment response. Front. Psychiatry 9:511. 10.3389/fpsyt.2018.00511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L. P., Lau N. C., Garrett-Engele P., Grimson A., Schelter J. M., Castle J., et al. (2005). Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433 769–773. 10.1038/nature03315 [DOI] [PubMed] [Google Scholar]
- Lin M., Pedrosa E., Shah A., Hrabovsky A., Maqbool S., Zheng D., et al. (2011). RNA-Seq of human neurons derived from iPS cells reveals candidate long non-coding RNAs involved in neurogenesis and neuropsychiatric disorders. PLoS One 6:e23356. 10.1371/journal.pone.0023356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipovich L., Dachet F., Cai J., Bagla S., Balan K., Jia H., et al. (2012). Activity-dependent human brain coding/noncoding gene regulatory networks. Genetics 192 1133–1148. 10.1534/genetics.112.145128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R., Mukamel E. A., Nery J. R., Urich M., Puddifoot C. A., Johnson N. D., et al. (2013). Global epigenomic reconfiguration during mammalian brain development. Science 341:1237905. 10.1126/science.1237905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R., Pelizzola M., Dowen R. H., Hawkins R. D., Hon G., Tonti-Filippini J., et al. (2009). Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462 315–322. 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W. M., Maraia R. J., Rubin C. M., Schmid C. W. (1994). Alu transcripts: cytoplasmic localisation and regulation by DNA methylation. Nucleic Acids Res. 22 1087–1095. 10.1093/nar/22.6.1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X. S., Wu H., Krzisch M., Wu X., Graef J., Muffat J., et al. (2018). Rescue of Fragile X Syndrome Neurons by DNA Methylation Editing of the FMR1 Gene. Cell 172 979.e–992.e. 10.1016/j.cell.2018.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locke J., Kotarski M. A., Tartof K. D. (1988). Dosage-dependent modifiers of position effect variegation in Drosophila and a mass action model that explains their effect. Genetics 120 181–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez J. P., Kos A., Turecki G. (2018). Major depression and its treatment: microRNAs as peripheral biomarkers of diagnosis and treatment response. Curr. Opin. Psychiatry 31 7–16. 10.1097/YCO.0000000000000379 [DOI] [PubMed] [Google Scholar]
- Lopez J. P., Lim R., Cruceanu C., Crapper L., Fasano C., Labonte B., et al. (2014). miR-1202 is a primate-specific and brain-enriched microRNA involved in major depression and antidepressant treatment. Nat. Med. 20 764–768. 10.1038/nm.3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H., Ash R. T., He L., Kee S. E., Wang W., Yu D., et al. (2016). Loss and gain of MeCP2 cause similar hippocampal circuit dysfunction that is rescued by deep brain stimulation in a rett syndrome mouse model. Neuron 91 739–747. 10.1016/j.neuron.2016.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C., Lancaster M. A., Castanon R., Nery J. R., Knoblich J. A., Ecker J. R. (2016). cerebral organoids recapitulate epigenomic signatures of the human fetal brain. Cell Rep. 17 3369–3384. 10.1016/j.celrep.2016.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv J., Cui W., Liu H., He H., Xiu Y., Guo J., et al. (2013). Identification and characterization of long non-coding RNAs related to mouse embryonic brain development from available transcriptomic data. PLoS One 8:e71152. 10.1371/journal.pone.0071152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyst M. J., Ekiert R., Ebert D. H., Merusi C., Nowak J., Selfridge J., et al. (2013). Rett syndrome mutations abolish the interaction of MeCP2 with the NCoR/SMRT co-repressor. Nat. Neurosci. 16 898–902. 10.1038/nn.3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma D. K., Marchetto M. C., Guo J. U., Ming G. L., Gage F. H., Song H. (2010). Epigenetic choreographers of neurogenesis in the adult mammalian brain. Nat. Neurosci. 13 1338–1344. 10.1038/nn.2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makeyev E. V., Zhang J., Carrasco M. A., Maniatis T. (2007). The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 27 435–448. 10.1016/j.molcel.2007.07.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K., Hattori D., Wu H., Fouse S., He F., Hu Y., et al. (2003). DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 302 890–893. 10.1126/science.1090842 [DOI] [PubMed] [Google Scholar]
- Matsuzaki H., Daitoku H., Hatta M., Aoyama H., Yoshimochi K., Fukamizu A. (2005). Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 102 11278–11283. 10.1073/pnas.0502738102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea A. K., Chepelev I., Cui K., Zhao K. (2013). Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 23 1256–1269. 10.1038/cr.2013.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan P. O., Sasaki A., D’Alessio A. C., Dymov S., Labonte B., Szyf M., et al. (2009). Epigenetic regulation of the glucocorticoid receptor in human brain associates with childhood abuse. Nat. Neurosci. 12 342–348. 10.1038/nn.2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGowan P. O., Suderman M., Sasaki A., Huang T. C., Hallett M., Meaney M. J., et al. (2011). Broad epigenetic signature of maternal care in the brain of adult rats. PLoS One 6:e14739. 10.1371/journal.pone.0014739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melka M. G., Castellani C. A., Rajakumar N., O’Reilly R., Singh S. M. (2014). Olanzapine-induced methylation alters cadherin gene families and associated pathways implicated in psychosis. BMC Neurosci. 15:112. 10.1186/1471-2202-15-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellen M., Ayata P., Dewell S., Kriaucionis S., Heintz N. (2012). MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 151 1417–1430. 10.1016/j.cell.2012.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memczak S., Jens M., Elefsinioti A., Torti F., Krueger J., Rybak A., et al. (2013). Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495 333–338. 10.1038/nature11928 [DOI] [PubMed] [Google Scholar]
- Mendonca M. S., Mangiavacchi P. M., De Sousa P. F., Crippa J. A. S., Mendes A. V., Loureiro S. R., et al. (2019). Epigenetic variation at the SLC6A4 gene promoter in mother-child pairs with major depressive disorder. J. Affect. Disord. 245 716–723. 10.1016/j.jad.2018.10.369 [DOI] [PubMed] [Google Scholar]
- Mercer T. R., Dinger M. E., Mattick J. S. (2009). Long non-coding RNAs: insights into functions. Nat. Rev. Genet. 10 155–159. 10.1038/nrg2521 [DOI] [PubMed] [Google Scholar]
- Mercer T. R., Dinger M. E., Sunkin S. M., Mehler M. F., Mattick J. S. (2008). Specific expression of long noncoding RNAs in the mouse brain. Proc. Natl. Acad. Sci. U.S.A. 105 716–721. 10.1073/pnas.0706729105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer T. R., Qureshi I. A., Gokhan S., Dinger M. E., Li G., Mattick J. S., et al. (2010). Long noncoding RNAs in neuronal-glial fate specification and oligodendrocyte lineage maturation. BMC Neurosci. 11:14. 10.1186/1471-2202-11-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertineit C., Yoder J. A., Taketo T., Laird D. W., Trasler J. M., Bestor T. H. (1998). Sex-specific exons control DNA methyltransferase in mammalian germ cells. Development 125 889–897. [DOI] [PubMed] [Google Scholar]
- Mikkelsen T. S., Ku M., Jaffe D. B., Issac B., Lieberman E., Giannoukos G., et al. (2007). Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448 553–560. 10.1038/nature06008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller B. H., Zeier Z., Xi L., Lanz T. A., Deng S., Strathmann J., et al. (2012). MicroRNA-132 dysregulation in schizophrenia has implications for both neurodevelopment and adult brain function. Proc. Natl. Acad. Sci. U.S.A. 109 3125–3130. 10.1073/pnas.1113793109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. A., Gavin C. F., White J. A., Parrish R. R., Honasoge A., Yancey C. R., et al. (2010). Cortical DNA methylation maintains remote memory. Nat. Neurosci. 13 664–666. 10.1038/nn.2560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. A., Sweatt J. D. (2007). Covalent modification of DNA regulates memory formation. Neuron 53 857–869. 10.1016/j.neuron.2007.02.022 [DOI] [PubMed] [Google Scholar]
- Miller E. K., Cohen J. D. (2001). An integrative theory of prefrontal cortex function. Annu. Rev. Neurosci. 24 167–202. 10.1146/annurev.neuro.24.1.167 [DOI] [PubMed] [Google Scholar]
- Miller T., Krogan N. J., Dover J., Erdjument-Bromage H., Tempst P., Johnston M., et al. (2001). COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc. Natl. Acad. Sci. U.S.A. 98 12902–12907. 10.1073/pnas.231473398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J., Zhang Y., Xu R. M. (2003). Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev. 17 1823–1828. 10.1101/gad.269603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minshew N. J., Keller T. A. (2010). The nature of brain dysfunction in autism: functional brain imaging studies. Curr. Opin. Neurol. 23 124–130. 10.1097/WCO.0b013e32833782d4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miska E. A., Alvarez-Saavedra E., Townsend M., Yoshii A., Sestan N., Rakic P., et al. (2004). Microarray analysis of microRNA expression in the developing mammalian brain. Genome Biol. 5:R68. 10.1186/gb-2004-5-9-r68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misztak P., Panczyszyn-Trzewik P., Sowa-Kucma M. (2018). Histone deacetylases (HDACs) as therapeutic target for depressive disorders. Pharmacol. Rep. 70 398–408. 10.1016/j.pharep.2017.08.001 [DOI] [PubMed] [Google Scholar]
- Miyake K., Hirasawa T., Soutome M., Itoh M., Goto Y., Endoh K., et al. (2011). The protocadherins, PCDHB1 and PCDH7, are regulated by MeCP2 in neuronal cells and brain tissues: implication for pathogenesis of Rett syndrome. BMC Neurosci. 12:81. 10.1186/1471-2202-12-81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A., Mukamel E. A., Davis F. P., Luo C., Henry G. L., Picard S., et al. (2015). Epigenomic signatures of neuronal diversity in the mammalian brain. Neuron 86 1369–1384. 10.1016/j.neuron.2015.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moessner R., Marshall C. R., Sutcliffe J. S., Skaug J., Pinto D., Vincent J., et al. (2007). Contribution of SHANK3 mutations to autism spectrum disorder. Am. J. Hum. Genet. 81 1289–1297. 10.1086/522590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohn F., Weber M., Rebhan M., Roloff T. C., Richter J., Stadler M. B., et al. (2008). Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 30 755–766. 10.1016/j.molcel.2008.05.007 [DOI] [PubMed] [Google Scholar]
- Molendijk M. L., Spinhoven P., Polak M., Bus B. A., Penninx B. W., Elzinga B. M. (2014). Serum BDNF concentrations as peripheral manifestations of depression: evidence from a systematic review and meta-analyses on 179 associations (N = 9484). Mol. Psychiatry 19 791–800. 10.1038/mp.2013.105 [DOI] [PubMed] [Google Scholar]
- Monk M., Boubelik M., Lehnert S. (1987). Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development 99 371–382. [DOI] [PubMed] [Google Scholar]
- Moreau M. P., Bruse S. E., David-Rus R., Buyske S., Brzustowicz L. M. (2011). Altered microRNA expression profiles in postmortem brain samples from individuals with schizophrenia and bipolar disorder. Biol. Psychiatry 69 188–193. 10.1016/j.biopsych.2010.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris R. G., Garrud P., Rawlins J. N., O’Keefe J. (1982). Place navigation impaired in rats with hippocampal lesions. Nature 297 681–683. 10.1038/297681a0 [DOI] [PubMed] [Google Scholar]
- Morrow E. M., Yoo S. Y., Flavell S. W., Kim T. K., Lin Y., Hill R. S., et al. (2008). Identifying autism loci and genes by tracing recent shared ancestry. Science 321 218–223. 10.1126/science.1157657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskowitz A., Heim G. (2011). Eugen Bleuler’s Dementia praecox or the group of schizophrenias 1911): a centenary appreciation and reconsideration. Schizophr. Bull. 37 471–479. 10.1093/schbul/sbr016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullaney B. C., Johnston M. V., Blue M. E. (2004). Developmental expression of methyl-CpG binding protein 2 is dynamically regulated in the rodent brain. Neuroscience 123 939–949. 10.1016/j.neuroscience.2003.11.025 [DOI] [PubMed] [Google Scholar]
- Muotri A. R., Chu V. T., Marchetto M. C., Deng W., Moran J. V., Gage F. H. (2005). Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature 435 903–910. 10.1038/nature03663 [DOI] [PubMed] [Google Scholar]
- Muotri A. R., Gage F. H. (2006). Generation of neuronal variability and complexity. Nature 441 1087–1093. 10.1038/nature04959 [DOI] [PubMed] [Google Scholar]
- Muotri A. R., Marchetto M. C., Coufal N. G., Oefner R., Yeo G., Nakashima K., et al. (2010). L1 retrotransposition in neurons is modulated by MeCP2. Nature 468 443–446. 10.1038/nature09544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahid M. A., Pauley K. M., Satoh M., Chan E. K. (2009). miR-146a is critical for endotoxin-induced tolerance: IMPLICATION IN INNATE IMMUNITY. J. Biol. Chem. 284 34590–34599. 10.1074/jbc.M109.056317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik S. H., Sathe P., Park H. Y., Metcalf D., Proietto A. I., Dakic A., et al. (2007). Development of plasmacytoid and conventional dendritic cell subtypes from single precursor cells derived in vitro and in vivo. Nat. Immunol. 8 1217–1226. 10.1038/ni1522 [DOI] [PubMed] [Google Scholar]
- Nan X., Ng H. H., Johnson C. A., Laherty C. D., Turner B. M., Eisenman R. N., et al. (1998). Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature 393 386–389. 10.1038/30764 [DOI] [PubMed] [Google Scholar]
- Narayanan B., Soh P., Calhoun V. D., Ruano G., Kocherla M., Windemuth A., et al. (2015). Multivariate genetic determinants of EEG oscillations in schizophrenia and psychotic bipolar disorder from the BSNIP study. Transl. Psychiatry 5:e588. 10.1038/tp.2015.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale B. M., Kou Y., Liu L., Ma’ayan A., Samocha K. E., Sabo A., et al. (2012). Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485 242–245. 10.1038/nature11011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilson E., Bois C., Gibson J., Duff B., Watson A., Roberts N., et al. (2017). Effects of environmental risks and polygenic loading for schizophrenia on cortical thickness. Schizophr. Res. 184 128–136. 10.1016/j.schres.2016.12.011 [DOI] [PubMed] [Google Scholar]
- Nelson D. L., Orr H. T., Warren S. T. (2013). The unstable repeats–three evolving faces of neurological disease. Neuron 77 825–843. 10.1016/j.neuron.2013.02.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neri F., Rapelli S., Krepelova A., Incarnato D., Parlato C., Basile G., et al. (2017). Intragenic DNA methylation prevents spurious transcription initiation. Nature 543 72–77. 10.1038/nature21373 [DOI] [PubMed] [Google Scholar]
- Ng J., Hart C. M., Morgan K., Simon J. A. (2000). A Drosophila ESC-E(Z) protein complex is distinct from other polycomb group complexes and contains covalently modified ESC. Mol. Cell. Biol. 20 3069–3078. 10.1128/MCB.20.9.3069-3078.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S. Y., Lin L., Soh B. S., Stanton L. W. (2013). Long noncoding RNAs in development and disease of the central nervous system. Trends Genet. 29 461–468. 10.1016/j.tig.2013.03.002 [DOI] [PubMed] [Google Scholar]
- Norton N., Owen M. J. (2005). HTR2A: association and expression studies in neuropsychiatric genetics. Ann. Med. 37 121–129. 10.1080/07853890510037347 [DOI] [PubMed] [Google Scholar]
- Nott A., Cheng J., Gao F., Lin Y. T., Gjoneska E., Ko T., et al. (2016). Histone deacetylase 3 associates with MeCP2 to regulate FOXO and social behavior. Nat. Neurosci. 19 1497–1505. 10.1038/nn.4347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M., Bell D. W., Haber D. A., Li E. (1999). DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 99 247–257. 10.1016/S0092-8674(00)81656-6 [DOI] [PubMed] [Google Scholar]
- Okano M., Xie S., Li E. (1998a). Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat. Genet. 19 219–220. 10.1038/890 [DOI] [PubMed] [Google Scholar]
- Okano M., Xie S., Li E. (1998b). Dnmt2 is not required for de novo and maintenance methylation of viral DNA in embryonic stem cells. Nucleic Acids Res. 26 2536–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oksenberg N., Ahituv N. (2013). The role of AUTS2 in neurodevelopment and human evolution. Trends Genet. 29 600–608. 10.1016/j.tig.2013.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olave I., Wang W., Xue Y., Kuo A., Crabtree G. R. (2002). Identification of a polymorphic, neuron-specific chromatin remodeling complex. Genes Dev. 16 2509–2517. 10.1101/gad.992102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Roak B. J., Vives L., Girirajan S., Karakoc E., Krumm N., Coe B. P., et al. (2012). Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature 485 246–250. 10.1038/nature10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsini C. A., Maren S. (2012). Neural and cellular mechanisms of fear and extinction memory formation. Neurosci. Biobehav. Rev. 36 1773–1802. 10.1016/j.neubiorev.2011.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang K. C., Frith M. C., Mattick J. S. (2006). Rapid evolution of noncoding RNAs: lack of conservation does not mean lack of function. Trends Genet. 22 1–5. 10.1016/j.tig.2005.10.003 [DOI] [PubMed] [Google Scholar]
- Park C. Y., Halevy T., Lee D. R., Sung J. J., Lee J. S., Yanuka O., et al. (2015). Reversion of FMR1 methylation and silencing by editing the triplet repeats in fragile X iPSC-derived neurons. Cell Rep. 13 234–241. 10.1016/j.celrep.2015.08.084 [DOI] [PubMed] [Google Scholar]
- Passingham R. E., Wise S. P. (2012). The Neurobiology of the Prefrontal Cortex : Anatomy, Evolution, and the Origin of Insight. Oxford: Oxford University Press; 10.1093/acprof:osobl/9780199552917.001.0001 [DOI] [Google Scholar]
- Peddada S., Yasui D. H., LaSalle J. M. (2006). Inhibitors of differentiation (ID1, ID2, ID3 and ID4) genes are neuronal targets of MeCP2 that are elevated in Rett syndrome. Hum. Mol. Genet. 15 2003–2014. 10.1093/hmg/ddl124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrosa E., Stefanescu R., Margolis B., Petruolo O., Lo Y., Nolan K., et al. (2008). Analysis of protocadherin alpha gene enhancer polymorphism in bipolar disorder and schizophrenia. Schizophr. Res. 102 210–219. 10.1016/j.schres.2008.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pensold D., Symmank J., Hahn A., Lingner T., Salinas-Riester G., Downie B. R., et al. (2017). The DNA methyltransferase 1 (DNMT1) controls the shape and dynamics of migrating POA-derived interneurons fated for the murine cerebral cortex. Cereb. Cortex 27 5696–5714. 10.1093/cercor/bhw341 [DOI] [PubMed] [Google Scholar]
- Pereira J. D., Sansom S. N., Smith J., Dobenecker M. W., Tarakhovsky A., Livesey F. J. (2010). Ezh2, the histone methyltransferase of PRC2, regulates the balance between self-renewal and differentiation in the cerebral cortex. Proc. Natl. Acad. Sci. U.S.A. 107 15957–15962. 10.1073/pnas.1002530107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesold C., Liu W. S., Guidotti A., Costa E., Caruncho H. J. (1999). Cortical bitufted, horizontal, and Martinotti cells preferentially express and secrete reelin into perineuronal nets, nonsynaptically modulating gene expression. Proc. Natl. Acad. Sci. U.S.A. 96 3217–3222. 10.1073/pnas.96.6.3217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pieretti M., Zhang F. P., Fu Y. H., Warren S. T., Oostra B. A., Caskey C. T., et al. (1991). Absence of expression of the FMR-1 gene in fragile X syndrome. Cell 66 817–822. 10.1016/0092-8674(91)90125-I [DOI] [PubMed] [Google Scholar]
- Piwecka M., Glazar P., Hernandez-Miranda L. R., Memczak S., Wolf S. A., Rybak-Wolf A., et al. (2017). Loss of a mammalian circular RNA locus causes miRNA deregulation and affects brain function. Science 357:eaam8526. 10.1126/science.aam8526 [DOI] [PubMed] [Google Scholar]
- Plath K., Fang J., Mlynarczyk-Evans S. K., Cao R., Worringer K. A., Wang H., et al. (2003). Role of histone H3 lysine 27 methylation in X inactivation. Science 300 131–135. 10.1126/science.1084274 [DOI] [PubMed] [Google Scholar]
- Ponting C. P., Oliver P. L., Reik W. (2009). Evolution and functions of long noncoding RNAs. Cell 136 629–641. 10.1016/j.cell.2009.02.006 [DOI] [PubMed] [Google Scholar]
- Qian X., Nguyen H. N., Song M. M., Hadiono C., Ogden S. C., Hammack C., et al. (2016). Brain-region-specific organoids using mini-bioreactors for modeling ZIKV exposure. Cell 165 1238–1254. 10.1016/j.cell.2016.04.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racey L. A., Byvoet P. (1971). Histone acetyltransferase in chromatin. Evidence for in vitro enzymatic transfer of acetate from acetyl-coenzyme A to histones. Exp. Cell Res. 64 366–370. 10.1016/0014-4827(71)90089-9 [DOI] [PubMed] [Google Scholar]
- Ravasi T., Suzuki H., Pang K. C., Katayama S., Furuno M., Okunishi R., et al. (2006). Experimental validation of the regulated expression of large numbers of non-coding RNAs from the mouse genome. Genome Res. 16 11–19. 10.1101/gr.4200206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redies C., Hertel N., Hubner C. A. (2012). Cadherins and neuropsychiatric disorders. Brain Res. 1470 130–144. 10.1016/j.brainres.2012.06.020 [DOI] [PubMed] [Google Scholar]
- Reik W. (2007). Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 447 425–432. 10.1038/nature05918 [DOI] [PubMed] [Google Scholar]
- Renthal W., Maze I., Krishnan V., Covington H. E., III, Xiao G., Kumar A., et al. (2007). Histone deacetylase 5 epigenetically controls behavioral adaptations to chronic emotional stimuli. Neuron 56 517–529. 10.1016/j.neuron.2007.09.032 [DOI] [PubMed] [Google Scholar]
- Rett A. (1966). On a unusual brain atrophy syndrome in hyperammonemia in childhood. Wien. Med. Wochenschr. 116 723–726. [PubMed] [Google Scholar]
- Rinn J. L., Kertesz M., Wang J. K., Squazzo S. L., Xu X., Brugmann S. A., et al. (2007). Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 129 1311–1323. 10.1016/j.cell.2007.05.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roguev A., Schaft D., Shevchenko A., Pijnappel W. W., Wilm M., Aasland R., et al. (2001). The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J. 20 7137–7148. 10.1093/emboj/20.24.7137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolls E. T. (2000). The orbitofrontal cortex and reward. Cereb. Cortex 10 284–294. 10.1093/cercor/10.3.284 [DOI] [PubMed] [Google Scholar]
- Roth T. L., Sweatt J. D. (2011). Annual research review: epigenetic mechanisms and environmental shaping of the brain during sensitive periods of development. J. Child Psychol. Psychiatry 52 398–408. 10.1111/j.1469-7610.2010.02282.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rougier N., Bourc’his D., Gomes D. M., Niveleau A., Plachot M., Paldi A., et al. (1998). Chromosome methylation patterns during mammalian preimplantation development. Genes Dev. 12 2108–2113. 10.1101/gad.12.14.2108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royaux I., Lambert de Rouvroit C., D’Arcangelo G., Demirov D., Goffinet A. M. (1997). Genomic organization of the mouse reelin gene. Genomics 46 240–250. 10.1006/geno.1997.4983 [DOI] [PubMed] [Google Scholar]
- Rudenko A., Dawlaty M. M., Seo J., Cheng A. W., Meng J., Le T., et al. (2013). Tet1 is critical for neuronal activity-regulated gene expression and memory extinction. Neuron 79 1109–1122. 10.1016/j.neuron.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saetrom P., Heale B. S., Snove O., Jr., Aagaard L., Alluin J., Rossi J. J. (2007). Distance constraints between microRNA target sites dictate efficacy and cooperativity. Nucleic Acids Res. 35 2333–2342. 10.1093/nar/gkm133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha R. N., Pahan K. (2006). HATs and HDACs in neurodegeneration: a tale of disconcerted acetylation homeostasis. Cell Death Differ. 13 539–550. 10.1038/sj.cdd.4401769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano K., Tanihara H., Heimark R. L., Obata S., Davidson M., St John T., et al. (1993). Protocadherins: a large family of cadherin-related molecules in central nervous system. EMBO J. 12 2249–2256. 10.1002/j.1460-2075.1993.tb05878.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarraf S. A., Stancheva I. (2004). Methyl-CpG binding protein MBD1 couples histone H3 methylation at lysine 9 by SETDB1 to DNA replication and chromatin assembly. Mol. Cell 15 595–605. 10.1016/j.molcel.2004.06.043 [DOI] [PubMed] [Google Scholar]
- Sati S., Ghosh S., Jain V., Scaria V., Sengupta S. (2012). Genome-wide analysis reveals distinct patterns of epigenetic features in long non-coding RNA loci. Nucleic Acids Res. 40 10018–10031. 10.1093/nar/gks776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer M., Pollex T., Hanna K., Tuorto F., Meusburger M., Helm M., et al. (2010). RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 24 1590–1595. 10.1101/gad.586710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Psychiatric Genome-Wide Association Study (GWAS) Consortium (2011). Genome-wide association study identifies five new schizophrenia loci. Nat. Genet. 43 969–976. 10.1038/ng.940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium (2014). Biological insights from 108 schizophrenia-associated genetic loci. Nature 511 421–427. 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenherr C. J., Anderson D. J. (1995). The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science 267 1360–1363. 10.1126/science.7871435 [DOI] [PubMed] [Google Scholar]
- Schuettengruber B., Martinez A. M., Iovino N., Cavalli G. (2011). Trithorax group proteins: switching genes on and keeping them active. Nat. Rev. Mol. Cell Biol. 12 799–814. 10.1038/nrm3230 [DOI] [PubMed] [Google Scholar]
- Schwartz Y. B., Pirrotta V. (2008). Polycomb complexes and epigenetic states. Curr. Opin. Cell Biol. 20 266–273. 10.1016/j.ceb.2008.03.002 [DOI] [PubMed] [Google Scholar]
- Sempere L. F., Freemantle S., Pitha-Rowe I., Moss E., Dmitrovsky E., Ambros V. (2004). Expression profiling of mammalian microRNAs uncovers a subset of brain-expressed microRNAs with possible roles in murine and human neuronal differentiation. Genome Biol. 5:R13. 10.1186/gb-2004-5-3-r13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian M. D., Antalffy B., Armstrong D. L., Zoghbi H. Y. (2002). Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum. Mol. Genet. 11 115–124. 10.1093/hmg/11.2.115 [DOI] [PubMed] [Google Scholar]
- Shao Z., Raible F., Mollaaghababa R., Guyon J. R., Wu C. T., Bender W., et al. (1999). Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell 98 37–46. 10.1016/S0092-8674(00)80604-2 [DOI] [PubMed] [Google Scholar]
- Sharma S., Falick A. M., Black D. L. (2005). Polypyrimidine tract binding protein blocks the 5’ splice site-dependent assembly of U2AF and the prespliceosomal E complex. Mol. Cell 19 485–496. 10.1016/j.molcel.2005.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheinerman K. S., Umansky S. R. (2013). Circulating cell-free microRNA as biomarkers for screening, diagnosis and monitoring of neurodegenerative diseases and other neurologic pathologies. Front. Cell. Neurosci. 7:150. 10.3389/fncel.2013.00150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sher F., Rossler R., Brouwer N., Balasubramaniyan V., Boddeke E., Copray S. (2008). Differentiation of neural stem cells into oligodendrocytes: involvement of the polycomb group protein Ezh2. Stem Cells 26 2875–2883. 10.1634/stemcells.2008-0121 [DOI] [PubMed] [Google Scholar]
- Shieh P. B., Hu S. C., Bobb K., Timmusk T., Ghosh A. (1998). Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 20 727–740. 10.1016/S0896-6273(00)81011-9 [DOI] [PubMed] [Google Scholar]
- Shirane K., Toh H., Kobayashi H., Miura F., Chiba H., Ito T., et al. (2013). Mouse oocyte methylomes at base resolution reveal genome-wide accumulation of non-CpG methylation and role of DNA methyltransferases. PLoS Genet. 9:e1003439. 10.1371/journal.pgen.1003439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber J., Lim D. A., Petritsch C., Persson A. I., Maunakea A. K., Yu M., et al. (2008). miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 6:14. 10.1186/1741-7015-6-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva J., Mak W., Zvetkova I., Appanah R., Nesterova T. B., Webster Z., et al. (2003). Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev. Cell 4 481–495. 10.1016/S1534-5807(03)00068-6 [DOI] [PubMed] [Google Scholar]
- Sirianni N., Naidu S., Pereira J., Pillotto R. F., Hoffman E. P. (1998). Rett syndrome: confirmation of X-linked dominant inheritance, and localization of the gene to Xq28. Am. J. Hum. Genet. 63 1552–1558. 10.1086/302105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith C. L., Bolton A., Nguyen G. (2010). Genomic and epigenomic instability, fragile sites, schizophrenia and autism. Curr. Genomics 11 447–469. 10.2174/138920210793176001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son E. Y., Crabtree G. R. (2014). The role of BAF (mSWI/SNF) complexes in mammalian neural development. Am. J. Med. Genet. C Semin. Med. Genet. 166C 333–349. 10.1002/ajmg.c.31416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone M., Hayashi T., Tarui H., Agata K., Takeichi M., Nakagawa S. (2007). The mRNA-like noncoding RNA Gomafu constitutes a novel nuclear domain in a subset of neurons. J. Cell Sci. 120(Pt 15) 2498–2506. 10.1242/jcs.009357 [DOI] [PubMed] [Google Scholar]
- Song C. X., Szulwach K. E., Dai Q., Fu Y., Mao S. Q., Lin L., et al. (2013). Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell 153 678–691. 10.1016/j.cell.2013.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C. X., Szulwach K. E., Fu Y., Dai Q., Yi C., Li X., et al. (2011). Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol. 29 68–72. 10.1038/nbt.1732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruijt C. G., Gnerlich F., Smits A. H., Pfaffeneder T., Jansen P. W., Bauer C., et al. (2013). Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell 152 1146–1159. 10.1016/j.cell.2013.02.004 [DOI] [PubMed] [Google Scholar]
- Squire L. R. (1986). Mechanisms of memory. Science 232 1612–1619. 10.1126/science.3086978 [DOI] [PubMed] [Google Scholar]
- Sterner D. E., Berger S. L. (2000). Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64 435–459. 10.1128/MMBR.64.2.435-459.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan P. F., Neale M. C., Kendler K. S. (2000). Genetic epidemiology of major depression: review and meta-analysis. Am. J. Psychiatry 157 1552–1562. 10.1176/appi.ajp.157.10.1552 [DOI] [PubMed] [Google Scholar]
- Sultana R., Yu C. E., Yu J., Munson J., Chen D., Hua W., et al. (2002). Identification of a novel gene on chromosome 7q11.2 interrupted by a translocation breakpoint in a pair of autistic twins. Genomics 80 129–134. 10.1006/geno.2002.6810 [DOI] [PubMed] [Google Scholar]
- Sutcliffe J. S., Nelson D. L., Zhang F., Pieretti M., Caskey C. T., Saxe D., et al. (1992). DNA methylation represses FMR-1 transcription in fragile X syndrome. Hum. Mol. Genet. 1 397–400. 10.1093/hmg/1.6.397 [DOI] [PubMed] [Google Scholar]
- Suzuki M. M., Bird A. (2008). DNA methylation landscapes: provocative insights from epigenomics. Nat. Rev. Genet. 9 465–476. 10.1038/nrg2341 [DOI] [PubMed] [Google Scholar]
- Symmank J., Bayer C., Schmidt C., Hahn A., Pensold D., Zimmer-Bensch G. (2018). DNMT1 modulates interneuron morphology by regulating Pak6 expression through crosstalk with histone modifications. Epigenetics 13 536–556. 10.1080/15592294.2018.1475980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach K. E., Li X., Li Y., Song C. X., Wu H., Dai Q., et al. (2011). 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 14 1607–1616. 10.1038/nn.2959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach K. E., Li X., Smrt R. D., Li Y., Luo Y., Lin L., et al. (2010). Cross talk between microRNA and epigenetic regulation in adult neurogenesis. J. Cell Biol. 189 127–141. 10.1083/jcb.200908151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tada T., Tada M., Hilton K., Barton S. C., Sado T., Takagi N., et al. (1998). Epigenotype switching of imprintable loci in embryonic germ cells. Dev. Genes Evol. 207 551–561. 10.1007/s004270050146 [DOI] [PubMed] [Google Scholar]
- Tadic A., Muller-Engling L., Schlicht K. F., Kotsiari A., Dreimuller N., Kleimann A., et al. (2014). Methylation of the promoter of brain-derived neurotrophic factor exon IV and antidepressant response in major depression. Mol. Psychiatry 19 281–283. 10.1038/mp.2013.58 [DOI] [PubMed] [Google Scholar]
- Taganov K. D., Boldin M. P., Chang K. J., Baltimore D. (2006). NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U.S.A. 103 12481–12486. 10.1073/pnas.0605298103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M., Koh K. P., Shen Y., Pastor W. A., Bandukwala H., Brudno Y., et al. (2009). Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 324 930–935. 10.1126/science.1170116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takizawa T., Nakashima K., Namihira M., Ochiai W., Uemura A., Yanagisawa M., et al. (2001). DNA methylation is a critical cell-intrinsic determinant of astrocyte differentiation in the fetal brain. Dev. Cell 1 749–758. 10.1016/S1534-5807(01)00101-0 [DOI] [PubMed] [Google Scholar]
- Tao X., Finkbeiner S., Arnold D. B., Shaywitz A. J., Greenberg M. E. (1998). Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 20 709–726. 10.1016/S0896-6273(00)81010-7 [DOI] [PubMed] [Google Scholar]
- Teter B., Osterburg H. H., Anderson C. P., Finch C. E. (1994). Methylation of the rat glial fibrillary acidic protein gene shows tissue-specific domains. J. Neurosci. Res. 39 680–693. 10.1002/jnr.490390609 [DOI] [PubMed] [Google Scholar]
- Tie F., Furuyama T., Prasad-Sinha J., Jane E., Harte P. J. (2001). The Drosophila Polycomb Group proteins ESC and E(Z) are present in a complex containing the histone-binding protein p55 and the histone deacetylase RPD3. Development 128 275–286. [DOI] [PubMed] [Google Scholar]
- Toyoda S., Kawaguchi M., Kobayashi T., Tarusawa E., Toyama T., Okano M., et al. (2014). Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron 82 94–108. 10.1016/j.neuron.2014.02.005 [DOI] [PubMed] [Google Scholar]
- Tsankova N. M., Berton O., Renthal W., Kumar A., Neve R. L., Nestler E. J. (2006). Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 9 519–525. 10.1038/nn1659 [DOI] [PubMed] [Google Scholar]
- Tuoc T. C., Boretius S., Sansom S. N., Pitulescu M. E., Frahm J., Livesey F. J., et al. (2013). Chromatin regulation by BAF170 controls cerebral cortical size and thickness. Dev. Cell 25 256–269. 10.1016/j.devcel.2013.04.005 [DOI] [PubMed] [Google Scholar]
- Urdinguio R. G., Fernandez A. F., Lopez-Nieva P., Rossi S., Huertas D., Kulis M., et al. (2010). Disrupted microRNA expression caused by Mecp2 loss in a mouse model of Rett syndrome. Epigenetics 5 656–663. 10.4161/epi.5.7.13055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urdinguio R. G., Lopez-Serra L., Lopez-Nieva P., Alaminos M., Diaz-Uriarte R., Fernandez A. F., et al. (2008). Mecp2-null mice provide new neuronal targets for Rett syndrome. PLoS One 3:e3669. 10.1371/journal.pone.0003669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valinluck V., Sowers L. C. (2007). Endogenous cytosine damage products alter the site selectivity of human DNA maintenance methyltransferase DNMT1. Cancer Res. 67 946–950. 10.1158/0008-5472.CAN-06-3123 [DOI] [PubMed] [Google Scholar]
- van Erp T. G., Guella I., Vawter M. P., Turner J., Brown G. G., McCarthy G., et al. (2014). Schizophrenia miR-137 locus risk genotype is associated with dorsolateral prefrontal cortex hyperactivation. Biol. Psychiatry 75 398–405. 10.1016/j.biopsych.2013.06.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Esch H., Bauters M., Ignatius J., Jansen M., Raynaud M., Hollanders K., et al. (2005). Duplication of the MECP2 region is a frequent cause of severe mental retardation and progressive neurological symptoms in males. Am. J. Hum. Genet. 77 442–453. 10.1086/444549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varley K. E., Gertz J., Bowling K. M., Parker S. L., Reddy T. E., Pauli-Behn F., et al. (2013). Dynamic DNA methylation across diverse human cell lines and tissues. Genome Re. 23 555–567. 10.1101/gr.147942.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vella P., Scelfo A., Jammula S., Chiacchiera F., Williams K., Cuomo A., et al. (2013). Tet proteins connect the O-linked N-acetylglucosamine transferase Ogt to chromatin in embryonic stem cells. Mol. Cell 49 645–656. 10.1016/j.molcel.2012.12.019 [DOI] [PubMed] [Google Scholar]
- Verkerk A. J., Pieretti M., Sutcliffe J. S., Fu Y. H., Kuhl D. P., Pizzuti A., et al. (1991). Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65 905–914. 10.1016/0092-8674(91)90397-H [DOI] [PubMed] [Google Scholar]
- Vineis P., Chatziioannou A., Cunliffe V. T., Flanagan J. M., Hanson M., Kirsch-Volders M., et al. (2017). Epigenetic memory in response to environmental stressors. FASEB J. 31 2241–2251. 10.1096/fj.201601059RR [DOI] [PubMed] [Google Scholar]
- Waddington C. H. (1939). An Introduction to Modern Genetics. London: G. Allen & Unwin ltd. [Google Scholar]
- Wagner E. J., Garcia-Blanco M. A. (2001). Polypyrimidine tract binding protein antagonizes exon definition. Mol. Cell. Biol. 21 3281–3288. 10.1128/MCB.21.10.3281-3288.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh C. P., Chaillet J. R., Bestor T. H. (1998). Transcription of IAP endogenous retroviruses is constrained by cytosine methylation. Nat. Genet. 20 116–117. 10.1038/2413 [DOI] [PubMed] [Google Scholar]
- Wang T., Pan Q., Lin L., Szulwach K. E., Song C. X., He C., et al. (2012). Genome-wide DNA hydroxymethylation changes are associated with neurodevelopmental genes in the developing human cerebellum. Hum. Mol. Genet. 21 5500–5510. 10.1093/hmg/dds394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Cote J., Xue Y., Zhou S., Khavari P. A., Biggar S. R., et al. (1996a). Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 15 5370–5382. [PMC free article] [PubMed] [Google Scholar]
- Wang W., Xue Y., Zhou S., Kuo A., Cairns B. R., Crabtree G. R. (1996b). Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 10 2117–2130. [DOI] [PubMed] [Google Scholar]
- Wang X., Su H., Bradley A. (2002a). Molecular mechanisms governing Pcdh-gamma gene expression: evidence for a multiple promoter and cis-alternative splicing model. Genes Dev. 16 1890–1905. 10.1101/gad.1004802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Weiner J. A., Levi S., Craig A. M., Bradley A., Sanes J. R. (2002b). Gamma protocadherins are required for survival of spinal interneurons. Neuron 36 843–854. 10.1016/S0896-6273(02)01090-5 [DOI] [PubMed] [Google Scholar]
- Weaver I. C., Cervoni N., Champagne F. A., D’Alessio A. C., Sharma S., Seckl J. R., et al. (2004). Epigenetic programming by maternal behavior. Nat. Neurosci. 7 847–854. 10.1038/nn1276 [DOI] [PubMed] [Google Scholar]
- Webb T. P., Bundey S., Thake A., Todd J. (1986). The frequency of the fragile X chromosome among schoolchildren in Coventry. J .Med. Genet. 23 396–399. 10.1136/jmg.23.5.396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A. E., Chen W. G., Dalva M. B., Dolmetsch R. E., Kornhauser J. M., Shaywitz A. J., et al. (2001). Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. U.S.A. 98 11024–11031. 10.1073/pnas.191352298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson L. S., Davies W., Isles A. R. (2007). Genomic imprinting effects on brain development and function. Nat. Rev. Neurosci. 8 832–843. 10.1038/nrn2235 [DOI] [PubMed] [Google Scholar]
- Woodcock D. M., Lawler C. B., Linsenmeyer M. E., Doherty J. P., Warren W. D. (1997). Asymmetric methylation in the hypermethylated CpG promoter region of the human L1 retrotransposon. J. Biol. Chem. 272 7810–7816. 10.1074/jbc.272.12.7810 [DOI] [PubMed] [Google Scholar]
- Wossidlo M., Nakamura T., Lepikhov K., Marques C. J., Zakhartchenko V., Boiani M., et al. (2011). 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun. 2:241. 10.1038/ncomms1240 [DOI] [PubMed] [Google Scholar]
- Wu H., Coskun V., Tao J., Xie W., Ge W., Yoshikawa K., et al. (2010a). Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science 329 444–448. 10.1126/science.1190485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Tao J., Chen P. J., Shahab A., Ge W., Hart R. P., et al. (2010b). Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc. Natl. Acad. Sci. U.S.A. 107 18161–18166. 10.1073/pnas.1005595107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J. I., Lessard J., Olave I. A., Qiu Z., Ghosh A., Graef I. A., et al. (2007). Regulation of dendritic development by neuron-specific chromatin remodeling complexes. Neuron 56 94–108. 10.1016/j.neuron.2007.08.021 [DOI] [PubMed] [Google Scholar]
- Wu M., Wang P. F., Lee J. S., Martin-Brown S., Florens L., Washburn M., et al. (2008). Molecular regulation of H3K4 trimethylation by Wdr82, a component of human Set1/COMPASS. Mol. Cell. Biol. 28 7337–7344. 10.1128/MCB.00976-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q., Maniatis T. (1999). A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell 97 779–790. 10.1016/S0092-8674(00)80789-8 [DOI] [PubMed] [Google Scholar]
- Xie W., Barr C. L., Kim A., Yue F., Lee A. Y., Eubanks J., et al. (2012). Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148 816–831. 10.1016/j.cell.2011.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. J., Ogryzko V. V., Nishikawa J., Howard B. H., Nakatani Y. (1996). A p300/CBP-associated factor that competes with the adenoviral oncoprotein E1A. Nature 382 319–324. 10.1038/382319a0 [DOI] [PubMed] [Google Scholar]
- Yildirim O., Li R., Hung J. H., Chen P. B., Dong X., Ee L. S., et al. (2011). Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell 147 1498–1510. 10.1016/j.cell.2011.11.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X., Guven N., Dietis N. (2016). Stress-based animal models of depression: Do we actually know what we are doing? Brain Res. 1652 30–42. 10.1016/j.brainres.2016.09.027 [DOI] [PubMed] [Google Scholar]
- Yu N. K., Baek S. H., Kaang B. K. (2011). DNA methylation-mediated control of learning and memory. Mol. Brain 4:5. 10.1186/1756-6606-4-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H., Mischoulon D., Fava M., Otto M. W. (2018). Circulating microRNAs as biomarkers for depression: many candidates, few finalists. J. Affect. Disord. 233 68–78. 10.1016/j.jad.2017.06.058 [DOI] [PubMed] [Google Scholar]
- Zhang Y., Ng H. H., Erdjument-Bromage H., Tempst P., Bird A., Reinberg D. (1999). Analysis of the NuRD subunits reveals a histone deacetylase core complex and a connection with DNA methylation. Genes Dev. 13 1924–1935. 10.1101/gad.13.15.1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Sloan S. A., Clarke L. E., Caneda C., Plaza C. A., Blumenthal P. D., et al. (2016). Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89 37–53. 10.1016/j.neuron.2015.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X., Ueba T., Christie B. R., Barkho B., McConnell M. J., Nakashima K., et al. (2003). Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc. Natl. Acad. Sci. U.S.A. 100 6777–6782. 10.1073/pnas.1131928100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L., Wang X., Li X. L., Towers A., Cao X., Wang P., et al. (2014). Epigenetic dysregulation of SHANK3 in brain tissues from individuals with autism spectrum disorders. Hum. Mol. Genet. 23 1563–1578. 10.1093/hmg/ddt547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziats M. N., Rennert O. M. (2013). Aberrant expression of long noncoding RNAs in autistic brain. J. Mol. Neurosci. 49 589–593. 10.1007/s12031-012-9880-8 [DOI] [PMC free article] [PubMed] [Google Scholar]